lecciones y conferencias
Anuncio

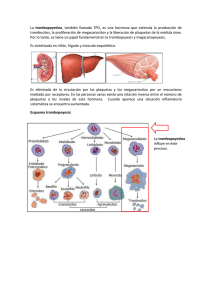
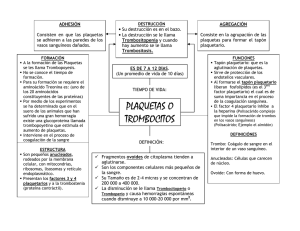
Rev. Med. Univ. Navarra X; 225, 1966 LECCIONES Y CONFERENCIAS Mecanismo de la detención espontánea de la hemorragia P. A. Owrm 'k El que los clínicos se enfrenten a menudo con problemas relacionados con la coagulación sanguínea, las plaquetas y la hemostasia, nos ha animado a estudiar brevemente este campo de la hematología, a fin de sintetizarlo y ordenarlo lo más posible, de forma tal que su conocimiento pueda ser útil al que debe desempeñar su función al servicio del enfermo en la Clínica Médica. Es sabido que en los hemofílicos se dan con frecuencia graves hemorragias, que inclu_so pueden terminar con la vida del paciente. Esto aboga en favor de que una anomalía en algunos de los tres campos anteriormente señalados, plaquetas, pared vascular o coagulación sanguínea, sea suficiente para desequilibrar el sistema hemostático. Por otra parte, junto a hemofílicos con un tiempo de coagulación normal y que no tienen hemorragias, existen pacientes con el defecto Hageman que, teniendo un tiempo de coagulación más alargado, difícilmente presentan hemorragias espontáneas. Asimismo, con la introducción de la te- * Instituto para la Investigación de la Trombosis. Universidad de Oslo. rapéutica anticoagulante, se han presentado nuevos problemas, que al resolverse, han contribuido al mejor conocimiento del sistema hemostático. En efecto, cuando se administran anticoagulantes orales, se pueden producir severas hemorragias en pacientes con un tiempo de coagulación casi normal, mientras que otros que han sido heparinizados, y que tienen un tiempo de coagulación de hasta 6 horas y más, cursan con un tiempo de hemorragia normal. También se ha hecho hincapié en que la trombosis y la hemostasia son fundamentalmente procesos idénticos, y que la trombosis arterial no es sino una reacción hemostática ante una lesión arteriosclérotica. A fin de esclarecer estas observaciones, daremo? algunas ideas sobre el mecanismo básico de la hemostasia y de la trombosis, y del exacto papel que el proceso hemocoagulativo desempeña en la misma, ya que nos parece que ello puede ser de interés, no sólo para el fisiólogo, sino también para el médico general, bien sea en el campo del tratamiento de la hemorragia o en el de la 226 P. A. OWREN instauración de una terapéutica anticoagulante eficiente. En los capilares, las hemorragias se pueden detener por la adhesión de sus pa redes o por el colapso de las mismas. Cuando la hemorragia se produce en un vaso de mayor calibre es imprescindible que se forme el tapón plaquetar para que la hemotasia se consolide. La formación de dicho tapón ha podido ser estudiada por micrnscopía in vivo utilizando las adecuadas técnicas, habiéndose podido separar en el proceso, las siguientes fases : l. Dentro de los tres primeros segundos, después de que un pequeño vaso ha sido traumatizado. las plaquetas comienzan a adherirse a las células endoteliales del borde de la lesión vascular, principalmente a las fibras colágenas. 2. Sobre las primeras plaquetas adheridas se suman otras nuevas, formándose así agregados plaquetares independientes, los cuales al ponerse en contacto cubren la lesión vascular. En un principio, estos agregados son permeables a los elementos formes sanguíneos, merced a una serie de pequeños canales que los atraviesan. 3. Transcurridos algunos minutos, estos canales se taponan y, en consecuencia, la hemorragia cesa. En este momento se puede comprobar por microscopía que las plaquetas que forman un agregado se han fusionado y que sus relieves han desaparecido, habiéndose perdido por tanto la individualidad de los trombocitos. Este proceso es conocido como metamorfosis viscosa de las plaquetas. Por microscopía electrónica se ha podido comprobar cómo, paralelamente a la formación del conglomerado plaquetar, algunos de sus elementos subcelulares sufren modificaciones, desapareciendo sus gránulos y mitocundrias, pero conser- Vol. X vando la membrana en muchas de las plaquetas. 4. El último paso en la formación del trombo es la constitución de la red de fibrina, como consecuencia de la coagulación plasmática. La fibrina refuerza y consolida el tapón hemostático, pero no parece que sea primordial para que la hemo;>tasia primaria se lleve a efecto. De estos cuatro apartados anteriores, vamos a estudiar detenidamente los tres primeros, para, de esta forma, comprender mejor el papel que la plaqueta juega en el conjunto hemostático. l. Adhesión de las plaquetas a la superficie de la herida. El mecanismo de adhesión de las plaquetas a la$ fibras colágenas parece obedecer más a fuerzas físicas que químicas, aunque su verdadero fundamento es por el momento desconocido. Dicho proceso parece independiente del ión calcio, ya que no es inhibido por soluciones concentradas de citrato sódico o EDTA. Las propiedades peculiares que debe tener una superficie para atraer a las plaquetas s:::m por el momento desconocidas, así como tamuoco se han identificado los grupos activos en la superficie de éstas, conociéndose únicamente que estos grupos son deficientes en las trom boa;:;tenias. 2. Agregación irreversible. En nuestro laboratorio, fue descubierto por Hellem, en los hematíes, un factor dializable y estable al calor, que cuando se añade en pequeñas cantidades a un concentrado de plaquetas desencadena la agregación de las mismas. Este factor fue más tarde identificado por Gaarder y col., como el adenosin-difosfato. Para que la agregación por ADP se produzca, es necesaria la presencia de iones calcio, aunque sea en pequeña cantidad. El mecanismo de actuación del ADP, hasta el momento, es desconocido. Una posible vía de acción es la sugerida por Laaland, en la que el ADP se une a la Septfambre 1966 MECANISMO DE LA DETENCION ESPONTANEA DE LA HEMORRAGIA superficie de la plaqueta, sirviendo el ion calcio como puente de unión entre dos ADP y, por tanto, entre dos plaquetas. Para desarrollar esta acción puente entre plaqueta y el calcio, únicamente pueden ser utilizados aquellos nucleótidos que tengan número impar de valencias negativas libres. De esta forma, el ADP, que tiene tres, se uniría con dos de ellas a un ión calcio y le quedaría otra valencia libre para unirse a otro ión caldo, que sirve de puente. Este mecanismo explica la falta de capacidad agregante del AMP y ATP, ya que el primero únicamente tiene dos valencias negativas libres, y el ATP, cuatro, neutralizándo¡>e, por tanto, cada uno de ellos con uno o dos iones calcio, respectivamente, y no restándoles ninguna valencia libre para la formación del puente cálcico. Si esta teoría es correcta, el adenosin - tetrafosfato, que tiene cinco valencias libres, actuará de forma similar al ADP. Nosotros hemos comprobado recientemente que esta sustancia agrega las plaqueta¡>, de la misma manera que el ADP, lo cual apoya fuertemente la teoría del puente cálcico sugerida por Laaland. Para que el proceso de agregación se lleve a cabo, es asimismo condición indispensable que las plaquetas sean viables y que tengan un sistema glicolítico normal. Este aserto se basa en las siguientes observaciones experimentales: Por repetidos lavados, o prolongado almacenamiento, las plaquetas no reaccionan tras la adición del ADP. La agregación es asimismo impedida por el iodoacetato, el cual bloquea la glicolisis. En cambio, la adición de fluoruro, aumenta la reactividad al ADP. Como quiera que el oidoaoetato y el fluoruro actuan en fases diferentes de la glicolisis, en aldolasa y enolasa, respectivamente, es razonable creer que, o bien los ácidos fosfoglicéricos u otras reacciones entre estas dos fases, son necesarios para mantener la superficie de las plaquetas en 227 condiciones adecuadas para que se les pueda unir el ADP. El que el ADP esté unido a la superficie de las plaquetas parece probable, ya que la reacción se inhibe por la adición de AMP y ATP. Nosotros hemos hallado que la reacción es bloqueada también por adenina y adenosina. La inhibición por estas sustancias parece competitiva, ya que una nueva adición de ADP en mayores cantidades invierte la reacción. La reacción ADP-plaquetas es también inhibida por la cisteína, ethyl-mercaptano, cisteamina y otras sustancias que contienen grupos SH libres. Todas estas sustancias prolongan asimismo el tiempo de hemorragia, si la herida se lava con soluciones de alguna de ellas. La tromboastenia es el único síndrome clínico en el cual la adición de ADP al plasma citratado rico en plaquetas no produce la agregación de las mismas, lo cual puede ser utilizado como un simple test diagnóstico, ya que en el plasma normal la agregación es muy marcada 30 segundos después de la adición del nucleótido, mientras que en el plasma tromboasténico no se produce ningún tipo de agregación. La fuente de ADP necesaria para la puesta en marcha del proceso de agregación plaquetar parece que es la misma plaqueta, ya que, rpor una parte, la plaqueta es muy rica en ADP y, por otra, poniendo en contacto plaquetas con fibras colágenas, se libera a partir de éstas, gran cantidad de ADP, suficiente para que su agregación se lleve a cabo. Otra enfermedad en la que la agregación plaquetar parece estar afectada es la enfermedad de Willebrand. En la misma se encuentran generalmente tiempos de hemorragia alargados y la adhesión de las plaquetas a la superficie de la herida es defectuosa. In vitro, la adición de grandes cantidades de ADP a plasma de estos pacientes rico en plaquetas produce la agregación normal de las mismas; pero cuando se 228 P. A. OWREN añaden pequeñas cantidades la agregación obtenida es deficiente. Nosotros hemos confirmado estos hallazgos. En efecto, añadiendo ADP a una concentración final de 0,05 mg por ml de plasma, no se produce agregación plaquetar en el plasma de estos pacientes, en oposición a la agregación conseguida en plasmas normales y en plasmas de pacientes con diferentes coagulopatías. La respuesta anormal de las plaquetas de estos pacientes al ADP se corrige in vitro, tras la adición de plasma de individuos normales pobre en plaquetas; especialmente si el donante, previamente, había realizado algún ejercicio muscular. Estos hallazgos sustentan fuertemente la teoría de Nilson y col., según la cual, un factor plasmático es asimismo nec·esario para una hemostasia normal. Egebert, en nuestro laboratorio, demostró que el ejercicio muscular en los enfermos de V. Willebrand, no solamente ocasiona un incremento de la actividad del factor VIII, sino también una disminución del tiempo de hemorragia. Asimismo, se comprobó que la transfusión de sangre normal a tales pacientes, sólo tiene un efecto moderado sobre el tiempo de sangría, mientras que la transfusión de sangre extraída después que el donante ha realizado un ejercicio fuerte durante algunos minutos puede restaurar completamente el tiempo de sangría a sus límites normales. Esta observación es de valor para el tratamiento de episodios hemorrágicos en tales pacientes, pudiendo éstos ser incluso operados sin hemorragias anormales, después de administrarles una transfusión con las características citadas. 3. La tercera fase en la reacción hemostática es la agregación irreversible de las plaquetas y su posterior metamorfosis viscosa. Como consecuencia de ello se produce la impermeabilidad del tapón plaquetar y la detención de la hemorragia. Esta reacción requiere trombina, la cual, a nuestro juicio, es proporcionada por un proceso de micro- Vol. X coagulación que se lleva a cabo en la atmósfera y no por la coagulación del plasma. El concepto de que la capa de plasma que rodea a las plaquetas, atmósfera plasmática de las plaquetas, puede jugar un papel en la hemostasia, fue primeramente advertido por Roskam en 1922. La naturaleza de la atmósfera plaquetar no está totalmente esclarecida. En 1955, Hjort, Rapaport y Owren, demostraron que el factor V del plasma está fuertemente adsorbido a las plaquetas y no puede ser separado de las mismas por sucesivos lavados. En efecto, el llamado factor I plaquetar, es el factor V mático adsorbido sobre la plaqueta. Bounameaux demostró que el factor VIII también se encuentra en la atmósfera periplaquetar, mientras que los factores VII y IX, se adhieren con menor fuerza y pueden ser eludidos con facilidad. A nosotros nos parece que la protrombina y el factor X se conducen de manera análoga a los factores VII IX. Así, pues, tanto el sistema extrínseco como el intrínseco están representados sobre la superficie de la plaqueta y, como consecuencia, sobre la misma se puede llevar a cabo un proceso de coagulación normal. El problema es saber si estos sistemas plaquetares tienen algún papel en la hemostasia. Yo he tratado de contestar esta pregunta analizando el efecto hemostásico de la trasfusión de plaquetas en pacientes con deficiencia de los factores de la coagulación. Para estudiar la tendencia que tienen los hemofílicos a las hemorragias tardías se introdujo en nuestro laboratorio el tiempo de hemorragia secundario, pudiéndose demostrar que los hemofílicos tienen un tiempo de hemorragia alargado. Pues bien, cuando a un hemofílico se le administra un concentrado de plaquetas normales, se normaliza su tiempo de hemorragia secundario, volviendo a su nivel pretrasfusional en seis días aproximadamente. Los niveles plasmáticos del factor VIII Septiembre 1966 MECANISMO DE LA DETENCION ESPONTANEA DE LA HEMORRAGIA permanecieron inferiores al l por 100 durante toda la experiencia, lo cual era lógico, sabiendo que la suspensión de plaquetas utilizadas contenía muy pequeña cantidad de plasma. Por otro lado, la trasfusión de una cantidad idéntica del mismo pla$ma libre de plaquetas no tuvo efectos sobre el tiempo de hemorragia secundario. En consecuencia, el efecto hemostático debe achacarse al factor adsorbido sobre las plaquetas trasfundidas. El que el efecto sea tan prolongado indica además que el factor VIII, que está adsorbido $Obre las plaquetas, tiene un tiempo de supervivencia más largo que el factor VIII del plasma, lo cual sugiere que una elución mm1ma o nula del factor VIII de las plaquetas tiene lugar en la circulación. Esta conclusión es consistenk con el hallazgo experimental de que las plaquetas de los hemofílico$, incubadas con plasma normal libre de plaquetas, adsorben factor VIII tan fuertemente que no es eluído por lavado. En las deficiencias del factor V, parahemofilia, los resultados obtenidos en plaquetas normales son similares. El paciente con deficiencia de factor V, que nosotros diagnosticamos por primera vez en 1947, fue trasfundido coa un concentrado de plaquetas normales, y el tiempo de hemorragia secundario volvió inmediatament,e a la normalidad. Como ocurría en la hemofilia A, retornó lentamente al nivel pretrasfusional en 5 ó 6 días, y, asimismo, la concentración de factor V en el plasma permaneció alrededor del 1 por 100. Igualmente pudimos demostrar que las plaquetas del paciente eran deficientes en factor V. Todo ello aboga en favor de que el factor V se adsorbe sobre las plaquetas y de que éstas sirven de vehículo transportador desde el donante al paciente. En contraste con los factores V y VIII, el IX y X, son adsorbidos con menor intensidad sobre las plaquetas y diluídos más fácilmente, lo cual puede ser demostrado porque en transfusiones de 229 plaquetas normales a pacientes con deficiencia de factor IX se acorta igualmente su tiempo de hemorragia secundario, pero que esta acción terapéutica desaparece mucho antes que en la hemofilia A o en la parahemofilia. Esto pueser debido a que el factor IX es rápidamente eluido en el plasma y que, en consecuencia, el sistema intrínseco de coagulación se vuelve de nuevo deficiente. En la carencia grave del factor X, no sólo el tiempo de hemorragia secundario, sino también el primario, están prolongados. La transfusión de plaquetas a estos enfermos tiene un efecto muy marcado, como era de esperar, pero únicamente visible sobre el tiempo de hemorragia primario. Todos estos hallazgos apoyan fuertemente la hipótesis de que las reacciones de coagulación, que son responsables del efecto hemostático, cuando se miden por el test del tiempo de hemorragia primario y secundario, se realizan en la superficie de las plaquetas trasfundidas. Nosotros por consiguiente creemos que los sistemas de coagulación de la superficie plaquetar tienen un papel clave en la hemostasia y que desde este punto de vista deben ser considerados como sistemas de coagulación independientes de los sistemas plasmáticos. Esto también explica que la hemostasia provisional plaquear puede ser normal a pesar de que el plasma del paciente sea incoagulable, como sucede en la deficiencia congénita del fibrinógeno y después de la administración de grandes dosis de heparina. Como conclusión de todo lo anterior, nuestro actual concepto del mecanismo hemostático puede ser resumido como sigue: l. Inmediata adhesión de las plaquetas a las células endotelíales dañadas. Esto puede estar condicíon2.do, posiblemente, a la liberación de ADP por estas células. 230 P. A. OWHEN Esta reacción no se produce en la tromboastenia. 2. Posterior adhesión de las plaquetas a las fibras colágenas que han quedado libres en la superficie de la herida. La reacción es independiente del calcio y del adenosindifosfato. Los grupos activos en la superficie dél fejido colágeno que sirven para atraer las plaquetas, no han sido todavía idefífificados, ni tampoco los grupos activos de la superficie plaquetar. Para esta reacción no es necesario ningún componente plasmático. La reacción puede ser bloqueada por varias su,stancias, tales como la cocaína y los antihistamínicos. Clínicamente esta reacción es únicamente defectuosa en la tromboastenia. 3. El contacto de las plaquetas con las fibras colágenas es seguido de la liberación de ADP de las propias plaquetas, ya que ellas mismas son portadoras del ADP necesario para su propia reacción de agregación. Estudios preliminares parecen indicar que la liberación de ADP a partir de las plaquetas es deficiente en ciertos estados patológicos, en los que las plaquetas parece se recubren de proteínas anormal·es (isoanticuerpos y paraproteínas), siendo ésta la causa por la que el tiempo de hemorragia sea prolongado en dichas afecciones. 4. El ADP que es liberado de las plaquetas las convierte en plaquetas adhesivas, las cuales se adhieren a diversos tipos de superficies y entre sí, produciendo de esta forma la agregación reversible, con formación de tapones plaquetares que todavía son permeables a la corriente sanguínea. Dicha reacción de agregación depende de que las plaquetas Vol. X sean viables y de que tengan un sistema glicolítico normal, el cual es deficiente en la tromboastenia; de la presencia de un factor plasmático que no ha sido todavía identificado, y que está ausente en el plasma de V. Willebrand; y de que el ión calcio circulante se halle en la proporción adecuada. La agregación irreversible depende fundamentalmente de la formación de trombina en la atmósfera periplaquetar por factores de la coagulación adsorbidos sobre la superficie de la plaqueta. Esta fase puede 5er deficiente en afecciones graves de la coagulación, tales como la sobredosis de anticoagulantes o en la hemofilia. El mecanismo bioquímico, por el cual la trombina produce la llamada metamorfosis viscosa y la agregación irreversible, ha sido estudiado por muchos investigadores. Recientes trabajos parecen indicar que la trombina actúa sobre una proteína plaquetar similar al fibrinógeno situada sobre la superficie de la plaqueta o bien en la propia membrana, ya que alterando la antitrombina y el fibrinógeno absorbidos sobre la superficie plaquetar se daña seriamente la membrana de la célula y, como consecuencia de ello, se producen cambios metabólicos intracelulares, como son la estimulación de la actividad glicolítica y el aumento de la síntesis de ATP, seguido de su transformación en ADP y fosfato inorgánico. 6. Por último, se lleva a cabo la coagulación plasmática, con la formación de fibrina a partir del fibrinógeno, reacción que sirve para consolidar el tapón hemostático, pero que no es necesaria para la hemostasia provisional. ASMO - HUBBER COMPRIMIDOS MODERNA TERAPEUTICA DEL ASMA BRONQUIAL Paro obtener un efecto broncodilatador persistente, de aumento de las excursiones respiratorias y de la capacidad vital, se asocian en esta especialidad las acc iones farmacológicas de la prednisolona, teofilinato de colina y efedrina con el meprobamato, de acción tranquilizante y sedante, dada la extraordinaria importancia que en los pacientes afectos de asma bronquial tiene el lograr una sedación de su psiquismo. INDICACIONES Las indicaciones de ASMO-HUBBER son fundamentalmente la terapéutica del asl'na bronquial, tanto de etiología alérgica como bacteriana . Estó indicado también en el enfisema obstructivo, bronquitis asmática, etc. DOSIFICACION Se recomienda una dosis de inicio de 4-6 comprimidos diarios, que se irán disminuyendo a juicio del facultativo, en relación al regreso de la sintomotología. Como dosis de sostén , son suficientes generalmente de 1 a 2 comprimidos diarios, distribuidos en las principales comidas. PRESENT ACION Y FORMULA Frascos de 12 y 50 comprimidos, conteniendo cada uno: Clorhidrato de Efedrina Teofilinato de Colina Meprobamato Prednisolona Excipiente LABORATORIOS 0,020 grs . 0,200 • 0,200 0,0025 • c. s. HUBBER, S. A. Fábrica y Laboratorios: Berlin, 38, 40 y 42 - BARCELONA (15) - Teléfonos 230 72 00 - 230 72 08 - 230 72 09