1- Endocrinología
Anuncio

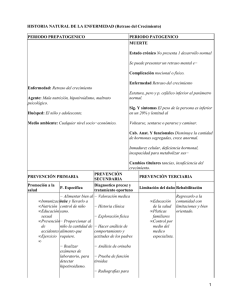
1- Endocrinología Retraso o ausencia de pubertad en la niña: ¿qué actitud adoptar? ¿Cuándo hay que inquietarse ante un retraso puberal en la niña? ¿Cuáles son los signos sugestivos de un retraso de origen orgánico? ¿Cuáles son los exámenes complementarios que permiten determinar su etiología? Estas son las preguntas a las que este artículo quiere responder. D. SAMARA-BOUSTANI C. DUFLOS-COHADE E. THIBAUD Unidad de endocrinología y ginecología pediátricas, hospital Necket de Niños Enfermos, París El retraso puberal en la niña se define por la ausencia de desarrollo de las mamas después de los 13 años o la ausencia de aparición de las menstruaciones después de los 16 años o 4 años después del inicio del desarrollo mamario. Existen pues dos situaciones: el impuberismo (ausencia de desarrollo de las mamas) y la amenorrea primaria (desarrollo de las mamas pero ausencia de reglas). Impuberismo Ante todo retraso puberal en la niña, hay que buscar signos a favor de una etiología orgánica «general» o endocrina. El retraso puberal simple es un diagnóstico de eliminación. Es raro en la niña. Insuficiencia gonádica (o hipogonadismo hipergonadotropo) La insuficiencia gonádica es responsable de alrededor de un 40% de los retrasos de la pubertad en la niña. Puede ser congénita o adquirida. Insuficiencia gonádica congénita - Síndrome de Turner: es la etiología más frecuente. Hay que pensar en ella ante un retraso de crecimiento intrauterino, una estatura baja (estatura inferior a -2 DE y/o una ralentización de la velocidad de crecimiento) y signos dismórficos, que pueden ser discretos. El cariotipo es 45 XO o en mosaico, o comporta una anomalía de estructura de un cromosoma X. - Insuficiencia ovárica con cariotipo normal 46 XX. La estatura es normal y los ovarios son de pequeño tamaño. Se han podido identificar algunas anomalías genéticas: microdelección del cromosoma X, mutación del gen del receptor de la FSH, mutación del gen Foxl2 (síndrome ptosis-blefarofimosis) y premutación del gen X Fra. Pero en la mayoría de los casos, la causa no se encuentra. - Disgenesia gonádica pura 46 XY (fenotipo femenino, órganos genitales internos femeninos, gónadas de pequeño tamaño). La insuficiencia gonádica es responsable de alrededor del 40% de los retrasos de la pubertad en la niña. Insuficiencia gonádica adquirida - Secundaria a una radioterapia: la insuficiencia ovárica es casi constante si la dosis de irradiación ovárica es de más de 20 grays por irradiación pelviabdominal y en caso de irradiación corporal total (preparación para un injerto de médula ósea). - Secundaria a una quimioterapia con agentes alquilantes y especialmente después de intensificación con busulfán o dosis elevadas de ciclofosfamida. - Autoinmune: generalmente asociada a otras afecciones autoinmunes, sobre todo en el marco de las poliendocrinopatias autoinmunes múltiples (candidosis, hipotiroidismo, hipoparatiroidismo, insuficiencia suprarrenal, diabetes tipo 1). - Galactosemia: acumulación intracelular de galactosa y sus metabolitos. Insuficiencia gonadotropa (hipogonadismo hipogonadotropo) Puede ser permanente o transitoria Hipogonadismo hipogonadotropo permanente - Deficiencia gonadotropa aislada o asociada a trastornos de la olfacción (anosmia o hiposmia) en el marco de un síndrome de kallmann (anomalías de migración de las células LRRH y de las células olfativas). - Síndromes malformativos: síndrome de Prader-Willi (obesidad, retraso mental, retraso estatural, dismorfia), síndrome de Lawrence-Moon-Bardet-Biedl (obesidad, retraso mental, polidactilia, retinitis pigmentaria). - Insuficiencia hipofisaria global con deficiencia somatotropa (GH) y a veces tireotropa (TSH) o corticotropa (ACTH); puede ser: - tumoral: craneofaringioma (tumor más frecuente de la región hipotalamohipofisaria responsable de una hipertensión endocraneal y de un retraso estatural), germinona, astrocitoma y más raramente adenoma productor de prolactina; - congénita: síndrome de interrupción del tallo pituitario, generalmente con una deficiencia de GH conocida y tratada desde la infancia; - secuela de cirugía o radioterapia (irradiación hipofisaria > 30 Gy) por tumor de la región hipotalamohipofisaria. Hipogonadismo hipogonadotropo funcional transitorio Todas las enfermedades crónicas con repercusión nutricional o síndrome inflamatorio que evolucionan desde la infancia pueden ser la causa de un retraso puberal. Pero este retraso a veces puede poner de manifiesto la enfermedad crónica; ocurre en especial en la enfermedad de Crohn o la enfermedad celiaca. - La anorexia mental Retraso puberal simple El retraso puberal simple es un diagnóstico de eliminación. A menudo, existe una ralentización estatural asociada, un retraso de la edad ósea y antecedentes de pubertad tardía en la familia. El retraso puberal simple es un diagnóstico de eliminación Conducta a seguir en la práctica El retraso puberal es esperado Existe una de las enfermedades o uno de los tratamientos citados anteriormente que pueden ser responsables de un hipogonadismo. La determinación de gonadotropinas FSH y LH basales o la prueba de la LHRH permitirán confirmar su origen central o periférico. El retraso puberal es el motivo de consulta (figura 1) Hay que buscar: - en el interrogatorio: antecedentes familiares de retraso puberal, un trastorno de la olfacción, signos digestivos, trastornos del comportamiento alimentario; - en el examen clínico: un retraso pondoestatural, signos generales (palidez, adenopatías, etc.), signos dismórficos a favor de un síndrome de Turner, cefalea, trastornos visuales sugestivos de una hipertensión endocraneal. Los primeros exámenes que deben realizarse son: - una edad ósea, - una determinación de FSH y de LH, que permitirá saber si el hipogonadismo es de origen periférico o central. La concentración de FSH y LH está elevada: se trata de una insuficiencia ovárica. El cariotipo se impone, porque la primera causa que hay que buscar es el síndrome de Turner (sobre todo si se asocia un retraso de crecimiento). Si el cariotipo es normal, hay que buscar una autoinmunidad a través de la búsqueda de una afección autoinmune asociada; la determinación de anticuerpos antiováricos es inespecífica. También hay que realizar una ecografía pélvica para evaluar el tamaño de los ovarios. Si el estudio de autoinmunidad es negativo, se propone la búsqueda de una causa genética más rara. La concentración de LH y FSH es normal; entonces hay que evaluar la edad ósea. - Si la edad ósea es ≥ 12 años, hay que buscar una deficiencia gonadotropa mediante una prueba de la LHRH y eventualmente otras deficiencias hipofisarias asociadas (GH, TSH y ACTH). Si la deficiencia gonadotropa se confirma, debe realizarse una RM cerebral para buscar un tumor o una anomalía de los bulbos olfatorios (a favor de un síndrome de Kallmann). - Si la edad ósea es < 12 años, la deficiencia gonadotropa es difícil de diagnosticar, porque la prueba de la LHRH es difícilmente interpretable y, por lo tanto, no debe realizarse. A menudo, este cuadro se asocia a un retraso estatural que requiere el estudio de una deficiencia de hormona de crecimiento (GH) o de un hipotiroidismo. Si este estudio es normal, el diagnóstico más probable es un retraso puberal simple, que confirmará la evolución clínica. Amenorrea primaria (Desarrollo normal de los caracteres sexuales secundarios) Ante toda amenorrea primaria, en primer lugar hay que buscar una malformación de los órganos genitales mediante un examen de la vulva y signos de hiperandrogenia. La ecografía pelviana siempre es necesaria. El estudio hormonal se orienta por la clínica (figura 2). Ante toda amenorrea primaria, la ecografía pelviana es siempre necesaria. Si no hay signos de hiperandrogenia y el desarrollo de los caracteres sexuales secundarios es normal y completo (estadio Tanner S5 P5), hay que buscar una anomalía de los órganos genitales: - La imperforación del himen se manifiesta generalmente por un dolor abdominal cíclico y la constitución de un hematocolpos que provoca un abombamiento azulado del himen y una masa pelviabdominal, a veces palpable por vía abdominal y siempre por tacto rectal. - El síndrome de Rokitanski es una aplasia uterovaginal. La vulva es normal, pero la vagina es corta. La aplasia uterina es completa, pero puede ser parcial. El diagnóstico es clínico y ecográfico. No hay signos de hiperandrogenia y el desarrollo de los caracteres sexuales secundarios es incompleto con una pilosidad pubiana ausente o escasa que contrasta con un desarrollo normal de las mamas (estadio Tanner S5 P1-2). Se sospecha entonces una resistencia completa a los andrógenos y hay que buscar unas gónadas en posición inguinal y un antecedente de hernia inguinal en periodo neonatal. La ecografía no muestra útero y se observan unas gónadas (testículos) en posición inguinal. El cariotipo es 46 XY. El estudio muestra una concentración de testosterona muy elevada y una concentración de LH elevada. Existe una mutación del gen del receptor de los andrógenos, localizado en el cromosoma X. La presencia de una hiperandrogenia clínica (hirsutismo, acné) debe inducir a buscar: - Un trastorno de la hormonosíntesis suprarrenal de presentación tardía por: - un bloqueo en 21-hidroxilasa (hirsutismo en el 85% de los casos, amenorrea en el 15% de los casos). Biológicamente, la concentración de 17-OHP está elevada, sobre todo después de la estimulación mediante el sinacteno (ACTH); - bloqueo en 11-β-hidroxilasa, más raro y muy a menudo asociado a una hipertensión arterial. El diagnóstico se hace por la elevación del compuesto S de base y después de la estimulación mediante el sinacteno. - Un síndrome del ovario poliquístico, que asocia hirsutismo, a menudo obesidad y a veces acantosis nigricans. Los andrógenos (testosterona, delta-4-androstendiona) están elevados. La SHBG (sex hormone binding globulin) a menudo está disminuida. En un gran número de casos, existe una resistencia a la insulina, con hiperinsulinemia y a veces intolerancia a la glucosa. En la ecografía pelviana, los ovarios están aumentados de tamaño, son globulosos y con un estroma hipertrofiado y una ecoestructura multifolicular, pero estos diferentes criterios no siempre se reúnen. Todas las causa de hipogonadismo responsables de un retraso puberal pueden producir una amenorrea primaria cuando el hipogonadismo es incompleto o aparece en el curso de la pubertad. En la práctica, recordar • Excepto en las situaciones en que el retraso puberal es esperado, el retraso puberal en la niña debe considerarse como una enfermedad mientras las principales etiologías orgánicas no se hayan descartado. • Conviene orientar el diagnóstico por el interrogatorio, la clínica y la concentración de gonadotropinas. • Si las gonadotropinas están elevadas, existe una insuficiencia ovárica, cuya primera causa es el síndrome de Turner. • Si las gonadotropinas son normales, existe un hipogonadismo hipogonadotropo transitorio o definitivo que debe confirmarse mediante una prueba de la LHRH según la edad ósea. Las principales etiología que hay que buscar son el tumor hipotalamohipofisario y el síndrome de Kallmann (de Morsier) mediante la realización de una RM cerebral. • Si el estudio es normal, el diagnóstico más probable es un retraso puberal simple o agravado por trastornos del comportamiento alimentario. • La conducta a seguir ante una amenorrea primaria es esencialmente clínico y ecográfico, mediante la búsqueda, en primer lugar, de una anomalía de los órganos genitales externos. Pies de foto: Preguntas ¿Ausencia de pubertad o ¿Retraso puberal ¿Hay que considerar un inicio de pubertad? patológico o simple? tratamiento? Interrogatorio Estaturas - Antecedentes Aporte Trastornos Contexto edades personales alimentario funcionales: psicológico puberales en la cefalea, familia digestivos, olfato Examen clínico Curva de crecimiento Estadio puberal Búsqueda elementos pondoestatural dismórficos Exámenes complementarios Edad ósea Concentraciones Según clínica: T4-TSH, plasmáticas estradiol, LH. recuento y fórmula, FSH creatinina, VSG, ac. transglutaminasa Concentración LH, FSH elevada Concentración LH, FSH normal → cariotipo, eco pelviana → edad ósea Edad ósea < 12 años Edad ósea ≥ 12 años T4-TSH, prueba GH, prolactina por prueba prueba LHRH, prolactina, RM hipofisaria, LHRH olfatometría ± T4, TSH, IGF1 Figura 1. Conducta a seguir ante un retraso puberal en la niña. S5P5 Estadio puberal Hiperandrogenia Resistencia a los andrógenos no sí Imperforación HCS SOPQ himeneal Testosterona Cariotipo Síndrome de Rokitanski Clínica eco S5P1-2 17-OHP compuesto Obesidad S Andrógenos SHBG Insulina HCS: hiperplasia congénita de las suprarrenales SOPQ: síndrome del ovario poliquístico Figura 2. Conducta a seguir ante una amenorrea primaria.