PROTOCOLO DE DIAGNOSTICO Y TRATAMIENTO DE EPILEPSIA
Anuncio

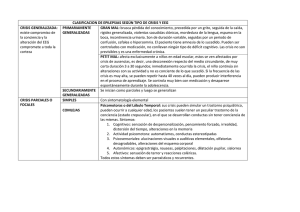
PROTOCOLO DE DIAGNOSTICO Y TRATAMIENTO DE EPILEPSIA SOCIEDAD BOLIVIANA DE NEUROLOGIA Presidente Dr. Mario Camargo Vicepresidente Dr. Alejandro Peralta Tesorero Dr. Walter Arias Secretario Dr. Carlos Mercado Vocales Dra. Yanet Laguna Dr. Jose Cuellar Comisión de Redacción Dr. Mario Camargo Dr. Alejandro Peralta Dr. Walter Arias Dr. Carlos Mercado Dra. Yanet Laguna Dr. Jose Cuellar Dr. Carlos Laforcada Dr. Gunter Paz Dr. Juan Carlos Duran Dr. Marcelo Aramayo Dr. Federico Fortún Dr. Henry Nuñez INDICE • • • • • • Protocolo de Actuación ante una primera crisis Epiléptica. Protocolo de Seguimiento del Paciente Epiléptico. Pacientes Refractarios y no Refractarios. Protocolo de Diagnostico y Actuación ante un estado de mal Epiléptico. Situaciones Especiales o Crisis Epilépticas Post o Trauma Encéfalo Craneano o Epilepsia Durante Embarazo Crisis Epilépticas post Cirugía de Cráneo Tratamiento Quirúrgico de la Epilepsia 1 PROTOCOLO DE ACTUACIÓN ANTE UNA PRIMERA CRISIS EPILÉPTICA Una crisis epiléptica es la manifestación clínica resultante de una descarga neuronal anómala y excesiva. La epilepsia es una enfermedad caracterizada por crisis epilépticas recurrentes (dos o más) no provocadas por una causa inmediata identificable. Por tanto, la ocurrencia de una única crisis no permite el diagnóstico de epilepsia. Múltiples crisis epilépticas en un período único se consideran como un evento único. • Actitud diagnóstica ante una primera crisis epiléptica El diagnóstico de epilepsia es fundamentalmente clínico. La descripción clínica del episodio paroxístico por parte del paciente y en la mayoría de los casos por un testigo es lo que va a permitir la caracterización del episodio como epiléptico. La epilepsia es un diagnóstico positivo, no de exclusión, ya que las pruebas complementarias pueden resultar normales. • Clasificación de las crisis epilépticas Clasificación internacional de crisis epilépticas (1981) I A. 1. a) b) c) d) e) 2. a) b) c) d) e) 3. 4. a) b) c) d) e) f) B. CRISIS PARCIALES Crisis Parciales simples (sin alteración de conciencia) Con signos motores: sin marcha con marcha (jacksoniana) versiva postural fonatoria (vocalización o detención del lenguaje) Con síntomas somatosensitivos o sensoriales especiales (alucinaciones simples): visual auditiva olfatoria gestatoria vertiginosa Con síntomas autonómicos (incluyendo sensación epigástrica, palidez, sudoración, rubefacción, piloerección y dilatación pupilar). Con síntomas psíquicos (trastornos en las funciones cerebrales superiores). Estos síntomas raramente ocurren sin trastorno de conciencia, y son vivenciados mucho más frecuentemente como crisis parciales complejas. disfásica dismnésica (por ej. déjá vu) cognitiva (por ej. estados de ensoñación, distorsiones del sentido del tiempo) afectiva (miedo, enojo, etc.) ilusiones (por ej. macropsia) alucinaciones estructuradas (por ej. música, escenas) Crisis Parciales complejas (con trastorno de conciencia, a veces pueden iniciar con sintomatología simple) 2 1. Inicio parcial simple seguido por trastorno de conciencia a) con manifestaciones parciales simples (A1-A4), seguidos de trastorno de conciencia b) con automatismos 2. Con trastorno inicial de conciencia a) con trastorno de conciencia únicamente b) con automatismos C. Crisis parciales que evolucionan a crisis secundariamente generalizadas (estas pueden ser generalizadas tónico-clónicas, tónicas o clónicas) Crisis parciales simples (A) que evolucionan a crisis generalizadas Crisis parciales complejas (B) que evolucionan a crisis generalizadas Crisis parciales simples que evolucionan a crisis parciales complejas, que evolucionan a crisis generalizadas. 1. 2. 3. II 1. A. a) b) c) d) e) f) g) B. a) b) 2. 3. 4. 5. 6. III CRISIS GENERALIZADAS Ausencias Ausencias con trastornos de conciencia únicamente con componentes clónicos leves con componente atómicos con componentes tónicos con automatismos con componentes autonómicos a (f) pueden ser usados solos o en combinación Ausencias típicas: con cambios en el tono más pronunciados que en A1 inicio y/o cese que no es brusco Crisis mioclónicas, sacudidas mioclónicas (única o múltiple) Crisis clónicas Crisis tónicas Crisis tónico-clónicas Crisis atómicas CRISIS EPILEPTICAS NO CLASIFICABLES Incluye aquellas crisis que no pueden ser clasificadas debido a: (a) datos inadecuados o incompletos (b) cuya semiología no está contemplada en las categorías hasta ahora descritas. 3 Clasificación internacional de epilepsias y síndromes epilépticos (1989) 1. 1.1 1.2 - Epilepsia y síndromes epilépticos parciales Idiopáticos (con inicio vinculado a la edad) Epilepsia benigna de la infancia con puntas centro-temporales Epilepsia de la infancia con paroxismos occipitales Epilepsia primaria de la lectura Sintomáticos Epilepsia parcial continua progresiva crónica de la infancia (síndrome de Kojewnikow) - Síndromes caracterizados por crisis con modos específicos de precipitación - Epilepsias del lóbulo temporal - Epilepsias del lóbulo frontal - Epilepsias del lóbulo parietal - Epilepsias del lóbulo occipital 1.3 Criptogenéticos (presumiblemente sintomáticos, sin evidencia etiológica) 2. Epilepsias y síndromes epilépticos generalizados 2.1 Idiopáticos (con inicio vinculado a la edad-enumerados en orden de edad) - Convulsiones neonatales benignas familiares - Convulsiones neonatales benignas - Epilepsia mioclónica benigna de la infancia - Epilepsia con ausencias de la infancia (picnolesia) - Epilepsia con ausencias de la juventud - Epilepsia mioclónica juvenil - Epilepsia con crisis de gran mal (CTCG) del despertar - Otras epilepsias generalizadas idiomáticas no definidas anteriormente - Epilepsias con crisis precipitadas por modos específicos de activación 2.2 Criptogenéticos o sintomáticos (en orden de edad) - Espasmos infantiles (síndrome de West) - Síndrome de Lenonox-Gastaut - Epilepsia con crisis mioclónico-astáticas - Epilepsia con ausencias mioclónicas 2.3 Sintomáticos 2.3.1. Etiología inespecífica - Encefalopatía mioclónica temprana - Encefalopatía epiléptica infantil temprana con salvas-supresión - Otras epilepsias generalizadas sintomáticas no definidas anteriormente 2.3.2. Síndromes específicos - Las crisis epilépticas pueden complicar varios estados patológicos. Bajo este encabezamiento se incluyen las patologías en que las crisis constituyen una forma de inicio o un hecho predominante. 3. Epilepsias y síndromes indeterminados (si son focales o generalizados) 3.1 Con crisis tanto parciales como generalizadas - Crisis neonatales - Epilepsia mioclónica severa de la infancia - Epilepsia con punta-onda lenta continua durante el sueño lento - Afasia epiléptica adquirida (síndrome de Landau-Kleffner) - Otras epilepsias indeterminadas no definidas anteriormente 4 3.2 4. 4.1 - Sin características inequívocamente generalizadas o focales. Todos los casos con crisis tónico-clónicas en que los hallazgos clínicos y EEG no permiten la clasificación como claramente generalizadas o parciales, como muchos casos de crisis de gran mal hípnicas, son considerados en este grupo. Síndromes especiales Crisis relacionadas a situaciones especiales Convulsiones febriles Crisis aisladas o estados epilépticos aislados Crisis que ocurren solamente cuando hay un evento tóxico o metabólico agudo, debido a factores como alcohol, drogas, eclampsia e hiperglicemia no cetósica. • Utilidad del electroencefalograma Las alteraciones del EEG por sí solas de forma aislada no permiten el diagnóstico de epilepsia. La detección de anomalía en el EEG (ondas lentas, ondas agudas o puntas solas o combinadas) durante la crisis permite, en general, el diagnóstico de certeza. El EEG convencional (20 minutos, sistema internacional 10-20) detecta únicamente anomalías en el 50% de los pacientes. La repetición de los registros de rutina y el de aumentar la sensibilidad hasta el 85% - 90%. La estimulación luminosa intermitente es especialmente útil para provocación de mioclonías, y la hiperventilación para provocar ausencias típicas en los niños. En los casos en los que las crisis tengan un modo específico de provocación (comida, lectura, etc.), éste ha de ser utilizado durante el registro. La privación del sueño es útil para poner de manifiesto la actividad epiléptica en el caso de las crisis generalizadas, y el sueño fisiológico para las crisis parciales complejas. • Utilidad de la neuroimagen Es deseable que a todos los pacientes con una crisis se les realice una tomografía axial computarizada (TAC) o una resonancia magnética (RM). La utilidad de la TAC se encuentra relegada a las situaciones de urgencia. La RM es superior a la TAC para detectar pequeños tumores, malformaciones corticales, malformaciones vasculares, trastornos de la migración e hipotrofias o esclerosis del hipocampo. Se encuentra indicada cuando las crisis comienzan en la edad adulta y si existen signos focales en la exploración neurológica o en el EEG. • Actitud terapéutica ante una primera crisis epiléptica Habitualmente no se administra tratamiento con fármacos antiepilépticos (FAE) tras una primera crisis. Si se produce una segunda, el riesgo de recidiva es superior al 65%, por lo que en general el tratamiento esta indicado. Crisis única Norma General No iniciar medicación Crisis repetidas Iniciar medicación Excepciones Lesión orgánica cerebral Síndrome epiléptico definido Criterio en contra del paciente Factores desencadenantes Identificados y evitables Intervalo ínter crisis largo (más de 4-5 años) Epilepsia benigna de la infancia 5 • Modo de inicio, ascenso y dosis de mantenimiento Una vez decidido el inicio del tratamiento farmacológico, debe indicarse solo una de las drogas antiepilépticas de primera línea y pretendidamente eficaz para la forma clínica .Comenzando con dosis bajas (a poder ser tomas nocturnas) y aumentándolas progresivamente hasta alcanzar la mínima dosis eficaz, es decir aquella en la que el paciente permanezca libre de crisis. Así, la carbamacepina conviene aumentarla en niños en el curso de dos a tres semanas y en adultos a razón de 200 mg. cada tres días. Tanto el fenobarbital como la primidona se sugiere incrementarlos con intervalos de 7-10 días, debido principalmente a sus efectos sedativos y a su larga vida media. El valproato se recomienda ascenderlo en niños durante al menos dos semanas. En adultos, si bien este fármaco así como la fenitoina pueden administrarse a una dosis plena desde el primer día, también se reducirán al máximo los posibles efectos colaterales con la introducción gradual. Entre las drogas nuevas la precaución de incremento paulatino debe tenerse sobre todo con oxcarbacepina y con lamotrigina, en el último caso más aún si se combina con ácido valproico. La dosis correcta teóricamente es la menor posible capaz de controlar las crisis sin ocasionar efectos secundarios. Es entonces que se recurre a promedios o rangos de dosis sugeridas, válidas para la mayoría de los pacientes, modificables según la respuesta individual. En aquellos pacientes en los que la primera droga indicada no resultó efectiva, habiéndola utilizado a dosis adecuadas, antes de pasar a una biterapia, debe recurrirse a otra droga de primera línea, también en monoterapia. La indicación de politerapia encuentra recién su lugar en situaciones especiales, tales como casos refractarios al tratamiento con varias drogas de primera línea utilizadas como medicación única, lo que suele ocurrir más frecuentemente en epilepsias parciales sintomáticas o criptogenéticas y en aquellas generalizadas sintomáticas Antiepiléptico Carbamacepina Fenitoina Valproato Fenobarbital Primidona Clonazepam Etosuximida Oxcarbacepina Vigabatrina Gabapentina Lamotrigina Topiramato Adultos (mg/día). 600 a 1800 200 a 400 600 a 2000 SO a 250 500 a 1000 2a6 1000 a 1500 900 a 2400 1500 a 3000 900 a 3600 200 a 400 200 a 400 Niños (mg/kg/día) 10 a 30 S a 10 30 a 200 2a8 10 a15 0.05 a 0.1 15 a 30 20 a 40 SO a 1 SO Est. no completados Sa1S Est. no completados Número de dosis diarias 3a2 1a2 3 1a2 3 2a3 1a2 2 2 3 2 2 6 • Elección de la droga apropiada Forma clínica E.G.I. con ausencias E.G.I. con mioclonias Antiepiléptico (primera línea) Valoroato / Etosuximida Valproato Antiepiléptico (opción) Clonazepam / Lamotrigina Lamotrigina / Primidona Lamotrigina/Carbamacepina/ E.G.I. con crisis tón.-clón. Valproato Fenitoina E.P. sintomática o Carbamacepina / Fenitoina Vigabatrina / Lamotrigina / Criptogentica Oxcarbacepina / Gabapentina T opiramato / Fenobarbital Síndr. de Lennox-Gastaut VaIproato/Clobazam/Lamotr./Felba Clonazepam Espasmos infantiles ACTH / Prednisona / Valproato Clobazam (Síndr. de West) Vigabatrina 7 PROTOCOLO DE SEGUIMIENTO DEL PACIENTE EPILÉPTICO. PACIENTES REFRACTARIOS Y NO REFRACTARIOS • Objetivos del seguimiento del paciente epiléptico Los objetivos que debemos plantearnos en el seguimiento de los pacientes epilépticos son los que se exponen a continuación: • Controles analíticos. Niveles farmacológicos Previamente a la instauración del tratamiento deberíamos disponer en todos los pacientes de una analítica básica de control (Hemograma y pruebas de función hepática, renal y pancreática) que nos permita no sólo ajustar las dosis de medicación en función de si existen o no alteraciones previas, sino también por la posibilidad de que sean los propios fármacos los que induzcan estas alteraciones. El primer control analítico con niveles hemáticos se debería llevar a cabo en todos los pacientes al mes de recibir la dosis total del fármaco, tiempo en que se considera que todos los FAE han alcanzado ya niveles plasmáticos estables. La medición de los niveles plasmáticos de los fármacos sólo se ha demostrado eficaz con algunos de los FAE clásicos, y sigue siendo motivo de controversia. A pesar de que lo ideal es que los niveles estén dentro del rango terapéutico, si la tolerancia y la eficacia son buenas se aceptan niveles discretamente altos o bajos de medicación. No es necesaria la determinación de niveles plasmáticos a largo plazo en pacientes con buen control de las crisis y con buena tolerancia de la medicación, ni en pacientes que se encuentren en monoterapia con nos nuevos FAE dado que no existe una relación adecuada entre los niveles plasmáticos y los efectos clínicos favorables o adversos. • Periodicidad de las visitas y electroencefalograma Las visitas de seguimiento posteriores dependerán del grado de control que se haya conseguido con la medicación, intentando siempre la supresión total de las crisis sin ocasionar efectos secundarios, e insistiendo repetidamente al paciente en la necesidad de tomar correctamente la medicación. Si las crisis se controlan totalmente, como suele ocurrir en muchos síndromes epilépticos idiomáticos y con crisis generalizadas, y el paciente tolera perfectamente la medicación, son suficientes controles clínicos y analíticos con una periodicidad anual o bianual por parte del neurólogo. El principal problema lo plantean los pacientes refractarios desde el inicio, lo cual es bastante frecuente en determinados síndromes epilépticos generalmente de origen sintomático o criptogénico. En estos casos ha de valorarse periódicamente tanto la eficacia del tratamiento mediante el registro del número y características de las crisis, como pos posibles efectos secundarios de la medicación mediante la anamnesis, la exploración física y analíticas de control con determinación de niveles plasmáticos en los supuestos descritos anteriormente. En general, los controles hematológicos y bioquímicos de rutina se realizarán con una periodicidad semestral o anual, o en cualquier momento si aparecen efectos. Esta vigilancia analítica periódica tiene un efecto psicológico beneficioso en el paciente, le tranquiliza sobre el riesgo de toxicidad y aumenta el cumplimiento terapéutico. Como parte del control clínico, interesa además pesquisar posibles efectos colaterales, tanto en el interrogatorio como en el examen físico. Tomadas en conjunto, las reacciones adversas pueden clasificarse en idiosincráticas y dependientes de la dosis. Las primeras son menos frecuentes, potencialmente provocables por cualquier antiepiléptico, pueden ser muy severas, e incluyen agranulocitosis, anemia aplásica, 8 insuficiencia hepática, dermatitis alérgica y pancreatitis. Los efectos secundarios relacionados con la dosis son más frecuentes, en general menos severos y en muchos casos comunes a varios fármacos. El electroencefalograma (EEG) de vigilia puede ser normal al inicio de la enfermedad en casi la mitad de los pacientes, dependiendo del tipo de crisis o de síndrome antiepiléptico, pero si se realizan estudios más prolongados (EEG de sueño o de monitorización continua) o con otras activaciones (EEG de privación de sueño) el porcentaje de normalidad no alcanza el 20%. No se deben realizar ajustes o modificaciones de la medicación en función únicamente de criterios electroencefalográficos. Se han de considerar en algunos síndromes epilépticos definidos, como por ejemplo el síndrome de West, en el que la evolución clínica es paralela a los hallazgos el EEG. En el caso de las epilepsias refractarias el objetivo debe ser el registro de las crisis, con técnicas más sofisticadas de monitorización continua con video – EEG, ya que se ha observado que hasta un 20% de los pacientes considerados refractarios al tratamiento tienen en realidad pseudocricis epilépticas, y no una verdadera epilepsia. • Suspensión programada de la medicación En los pacientes que han evolucionado favorablemente y han estado libres de crisis durante un período que oscila según la mayoría de los autores entre 2 y 3 años, se puede plantear discontinuar gradualmente la medicación. Dicha actitud se basa en el hecho de que se evitarían así los posibles efectos colaterales vinculados con la medicación antiepiléptica a largo plazo. 9 PROTOCOLO DE DIAGNOSTICO Y ACTUACIÓN ANTE UN ESTADO DE MAL EPILEPTICO. • Concepto de estatus epiléptico El estatus epiléptico (EE) constituye una emergencia médica y tiene una morbilidad y mortalidad elevada si no se trata de forma rápida y eficaz. La comisión de clasificación internacional de las crisis epilépticas lo definió como “una crisis que persiste durante un tiempo suficientemente largo o se repite con la suficiente frecuencia como para crear una condición epiléptica fija y duradera”. La fundación de epilepsia del America’s Working Group definió el EE como un episodio de más de 30 minutos de duración de actividad epiléptica continua, o dos o más crisis sucesivas sin una recuperación total de la conciencia; esta definición es la utilizada de forma mayoritaria, aunque recientemente otros trabajos han propuesto la reducción de la duración a 10 minutos. El Estatus epiléptico convulsivo generalizado: es el más común y grave de todos los tipos de estatus. Puede ocurrir como inicio de una epilepsia o en un paciente epiléptico ya conocido. Se caracteriza por movimientos tónicos o clónicos continuos asociados a afectación importante del nivel de consciencia o coma y descargas bilaterales paroxisticas en el electroencefalograma (EEG). Puede tener repercusiones generales graves como hipertermia, edema pulmonar, rabdomiolisis y otras Estatus epiléptico focal o parcial simple: Se caracteriza por crisis focales repetidas durante al menos 30 minutos sin deterioro del nivel de consciencia. El patrón EEG puede mostrar descargas irritativas focales unilaterales o ser normal si el área crítica cortical es muy pequeña. La forma más grave de este tipo de estatus es la epilepsia parcial continua. El Estatus epiléptico parcial complejo se caracteriza por la aparición de crisis parciales complejas repetidas, y se manifiesta por disminución del nivel de conciencia, confusión, mental, amnesia y alteraciones en la conducta con o sin automatismos que dan lugar a errores de diagnóstico. Su origen más frecuente es la región mesial temporal, aunque también puede originarse en la región frontal. En el Estatus epiléptico de ausencias, hay que diferenciar varios subtipos: a. El que se presenta en niños con crisis de ausencias típicas (muy raramente en un adulto). b. El que se presenta en niños con encefalopatías epilépticas y crisis de ausencia atípicas epilépticas y crisis de ausencia atípicas. c. El que aparece en un adulto no epiléptico conocido y que denominamos estado de mal no convulsivo con punta-onda del adulto (cuyo diagnóstico diferencial con los estatus fiscales frontales resulta en ocasiones difícil). El Estatus epilépticos mioclónicos generalizados, ocurren sobre todo en niños con encefalopatías epilépticas o en casos de encefalopatías postanóxicas. El patrón EEG es bilateral y simétrico con descargas de polipunta que coinciden con las contracciones mioclónicas. 10 • Medidas terapéuticas en estatus epiléptico A. MEDIDAS INCIALES 1. Mantener vía aérea permeable colocando al paciente en decúbito prono con la cabeza más baja que el cuerpo para prevenir aspiración. Si es necesario, colocar tubo oral o intubación endotraqueal. 2. Administrar O2 al 100%. 3. Determinación de glucemia capilar inmediata. 4. Iniciar tratamiento farmacológico. 5. Obtener muestra de sangre para analítica. a) Hemograma, Na, K, Mg, Ca, glucosa, creatinina, GOT, GPT, estudio de coagulación. b) Determinación de tóxicos en sangre y orina. 6. Medir temperatura, oximetría continua, tensión arterial (TA) continua y ECG. 7. Colocar vía IV con: a) Dextrosa 40% 250 cm3 en adultos; 25% 2 a 4 mL/kg en niños. b) Tiamina 100 mg. c) Continuar con suero glucosado y salino 50 a 100 cc/hora. 8. Ingreso en UTI. 9. Monitorización continua EEG o EEG seriados. 10. Estudio de imagen cerebral (TAC o RNM) cuando el paciente está estabilizado. B. TRATAMIENTO FARMACOLOGICO 1 En todos los pacientes: a. Diacepam (ampollas de 10 mg) 0,2 a 0,5 mg/kg, 1 a 4 mg/min IV o doble dosis por tubo endotraqueal si no se consigue una vía IV. Controlar función respiratoria y estar preparado para intubar.. b. Fenitoína (ampollas de 250 mg) 15 a 20 mg/kg, 20 a 50 mg/min IV. Mantenimiento 5 a 10 mg/kg cada 24 h dividido en dos dosis. Se puede administrar de forma directa o disuelta en 250 cm3 de suero fisiológico, por vía IV periférica. Controlar ECG y TA: riesgo de hipotensión (hasta 50%) y bradiarritmias (hasta 2%) sobre todo en >50 años y cardiópatas. Si ocurre, ir más despacio en la infusión o suspenderla. Atención a la vía IV, debe ser vigilada permanentemente debido al riesgo de necrosis tisular. En pacientes que no están tomando fenitoína oral crónica o que la están tomando y el nivel plasmático se encuentra por debajo de 10 mg/l, si persisten las crisis se puede añadir fenitoína 10 mg/kg, 20 a 50 mg/min IV, hasta completar 30 mg/kg. 2. Si persiste el status: El paciente debe estar intubado, con oximetría y control de EEG. Las dosis se pueden titular hasta que desaparecen las puntas en el registro electroencefalográfico o se alcance un patrón de brote-supresión, con períodos de brote <1 s o aproximadamente 80% de supresión y 20% de brote. Cada 12 h disminuir la dosis y examinar el EEG, reiniciar tratamiento si persiste la actividad epileptiforme. Continuar los estudios para identificar la causa del status y corregirla. Corregir trastornos metabólicos e hidroelectrolíticos. a) Fenobarbital (ampollas de 200 mg) 20 mg/kg IV, 50 a 75 mg/min. Acción en 15 a 60 min, duración 24 a 96 h. Intubar al paciente siempre y tratar la hipotensión con fármacos vasoactivos (dopamina y dobutamina). b) Midazolam (ampollas de 5 y 15 mg) 0,05 a 2 mg/kg en bolo IV lento, 11 mantenimiento de 0,75 a 10 mg/kg/min. Efecto dura 1 a 5 h, causa menos hipotensión que los barbitúricos, no se distribuye en tejido graso. c) Propofol (viales de 10 mg/mL de 20, 50 y 100 mU 1 a 2 mg/kg IV, mantenimiento de 2 a 10 mg/kg/h. Riesgo de pancreatitis química en tratamientos prolongados. d) Pentobarbital 10 a 15 mg/kg IV en 60 min, mantenimiento 0,5 a 1 mg/kg/h. Produce hipotensión grave (tratar con dopamina y dobutamina) y edema (principalmente edema de glotis). e) Lidocaína: su utilidad es limitada porque puede causar crisis en dosis altas y la distribución cerebral es variable. 12 SITUACIONES ESPECIALES • CRISIS EPILÉPTICAS POST-TRAUMA ENCEFALO CRANEANO 1. Se reserva el uso profiláctico de drogas anti-epilépticas para prevenir crisis postTEC precoces, únicamente en pacientes con alto riesgo: Cuando mas severo el TEC, más probable que ocurran crisis epilépticas Perdida de conocimiento prolongada o amnesia postraumática mayor de 24 horas Puntaje e la escala de coma de G1asgow < 10. Déficit focal neuro1ógico, hematoma cerebral y/o contusión cortical Hematoma subdural o extradura1 de entidad. Traumatismo abierto con herida penetrante o fractura de cráneo con hundimiento. Crisis inmediatas (en las primeras 24 horas del TEC). 2. La droga de elección es la fenitoina. Se administra una dosis carga de 15 mg/kg iv., a pasar en 20 minutos (diluyendo 1 gramo de PHT en 200 cc de suero fisiológico). Se continúa con una dosis de mantenimiento de 125 mg i.v. cada 8 horas. Se debe pasar lo antes posible a la vía oral: 300 mg/día (o administrarla por sonda naso gástrica), superponiendo a la administración iv. durante 24 horas. 3. Debe utilizarse la droga anti-epiléptica a dosis suficientes para mantener rangos terapéuticos y únicamente durante 2 semanas luego del TEC en pacientes no operados. Suspensión rápida de la droga. 4. No se justifica la profilaxis antiepiléptica mas allá de este periodo en pacientes que no tuvieron crisis, dado que aun no es posible disminuir el riesgo de CPT tardías. 5. Los pacientes operados o aquellos que presentaron una o mas crisis precoces, deben recibir drogas anti-epilépticas durante 3 meses. Suspensión gradual en 2 meses. • CRISIS EPILÉPTICAS POST CIRUGIA DE CRANEO 1. La profilaxis con DAE en cirugía de cráneo esta indicada ante todo procedimiento supratentorial. 2. La droga de elección es la fenitoina. 3. Debe utilizarse la DAE a dosis suficientes para mantener rangos terapéuticos. 4. La duración del tratamiento dependerá del riesgo de CPO: - Con bajo riesgo: 2 semanas, suspensión en 3 días. - Con alto riesgo: 3 meses, suspensión en 2 meses. 5. La aparición de crisis tardías determina que el paciente sea tratado como todo paciente con epilepsia sintomática. 13 • EPILEPSIA DURANTE EMBARAZO 1. La gestación en pacientes con epilepsia se considera de alto riesgo, dada la mayor incidencia de complicaciones obstétricas, como prematuridad y muerte neonatal. 2. Durante el embarazo, la frecuencia de las crisis puede permanecer inalterada (61 %), disminuir (24%) o incrementarse (15%). 3. Las crisis durante el embarazo no se han relacionado con una mayor incidencia de malformaciones, pero las crisis generalizadas tónico clónicas pueden producir hipoxia fetal y el traumatismo abdominal de la madre puede afectar al feto. 4. Para reducir en lo posible los riesgos se recomienda: a) Transmitir a la paciente la conveniencia de una maternidad planificada. b) Considerar la retirada progresiva de la medicación en pacientes libres de crisis por un período superior a dos años. c) Si el tratamiento es aconsejable, se tratará con el fármaco más eficaz para el paciente, en lo posible evitando la poli terapia. Puede ser útil incrementar el número de dosis diarias para reducir la presencia de niveles pico demasiado elevados. d) El ácido fólico disminuye la incidencia de defectos del tubo neural en la población general. Para que sea eficaz debe iniciarse antes de la concepción, por lo que se recomienda administrar 0,4 mgr a todas las mujeres epilépticas en edad fértil, en mujeres sexualmente activas que pueden quedar embarazadas y así como en aquellas con déficit de folato o con antecedentes de defectos del tubo neural. e) Realizar una determinación de alfa-feto proteína en sangre y una ecografía de alta resolución en la semana 18 de gestación. Estas pruebas permiten detectar la mayoría de las malformaciones del tubo neural. I. TRATAMIENTO QUIRURGICO DE LA EPILEPSIA Es un hecho conocido que aproximadamente un 20 % de los pacientes epilépticos no se controlan adecuadamente con los fármacos antiepilépticos disponibles en la actualidad. Un porcentaje de éstos (alrededor del 2 % - 3 %) serán candidatos a tratamiento quirúrgico de la epilepsia, en cuyo campo existen diferentes técnicas. El objetivo de la cirugía de la epilepsia es resecar tanto tejido epileptogénico como sea posible, respetando el tejido normal y evitando la resección del tejido funcionalmente importante. La cirugía de la epilepsia más extendida es la de la epilepsia del lóbulo temporal. Su causa más frecuente es la esclerosis mesial del hipocampo que se objetiva por técnicas de neuroimagen en el 60 % de los pacientes. La resección puede limitarse al hipocampo y amígdala (amigdalo-hipocampectromía selectiva) o extenderse a la neocorteza e incluir la parte anterior del lóbulo temporal84. La amígdalo hipocampectomía presenta menores riesgos de defectos neuropsicológicos. Las complicaciones graves de esta cirugía son poco frecuentes y los resultados buenos con ausencia de crisis total en el 70 % de los pacientes intervenidos. Otro 30 % de los pacientes presentan un número de crisis menor que antes de la intervención y se mantienen con bajas dosis de medicación. Indicaciones de neurocirugía en epilepsia • Falta de respuesta al tratamiento médico durante 2 anos, que incluya carbamacepina y fenitoina • Crisis incapacitantes • Repercusión socio-laboral importante 14 • • • EEG patológico RM y SPECT con patología focal congruentes con la clínica Epilepsia sintomática II. DIETA CETOGENA El uso clínico de la dieta cetogena se remonta a los años veinte. En los últimos anos ha habido un resurgimiento de este tipo de tratamiento88 y se han publicado ensayos clínicos demostrando que tienen un efecto beneficioso en niños con epilepsias de difícil control. 15