Lisosomas
Anuncio

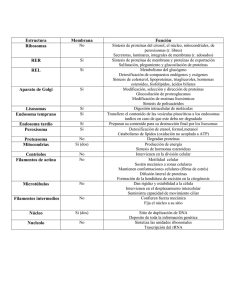
Introducción Los lisosomas fueron descubiertos por casualidad en los años ‘50 por Christian De Duve y colaboradores, quienes aplicando técnicas de fraccionamiento celular para ubicar la distribución de diversas enzimas, encontraron que la fosfatasa ácida se encontraba en un grupo especial de partículas membranosas de tamaño intermedio entre las mitocondrias y los microsomas. Desde entonces hasta ahora se han identificado dentro de los lisosomas unas 50 enzimas diferentes que degradan una amplia gama de compuestos biológicos en un ambiente ácido (pH 4.6). Todas las células eucarióticas poseen lisosomas, tanto en animales como en vegetales. Las bacterias (procarionte) carecen de lisosomas, pero el llamado espacio periplasmático (entre la membrana plasmática y la pared celular) actúa como un espacio lisosómico. Las funciones de estos organelos repletos de enzimas y limitados por membrana, pueden resumirse en: (a) digestión de materiales incorporados por endocitosis (fagocitosis, pinocitosis y endocitosis mediada por receptor); (b) digestión de compuestos de la propia célula (autofagia); (c) digestión de materiales extracelulares mediante liberación de enzimas al medio circundante (caso del osteoclasto, en el tejido óseo). Micrografía electrónica que muestra un grupo de lisosomas electrondensos cerca del Golgi y rodeados por mitocondrias, en una célula de corteza adrenal. Lisosomas Clasificación Morfológicamente, los lisosomas presentan un amplio polimorfismo, sin embargo, pueden reconocerse dos tipos fundamentales de lisosomas: (b-3) autolisosomas, se llaman también vacuolas autofágicas o citolisosomas, pues son vacuolas que contienen restos de organelos celulares (RE, mitocondrias, etc.) (a) lisosomas primarios, son las vesículas de almacenamiento de enzimas recién sintetizadas, liberadas desde el retículo trans del Golgi, de unos 0.4 µm de diámetro limitados por una membrana; (b) lisosomas secundarios, son las vesículas resultantes de la fusión de los lisosomas primarios con vacuolas que contienen material fagocitado. Estos lisosomas secundarios pueden a su vez clasificarse en: (b-1) heterofagosomas o vacuolas digestivas que se producen por fusión de lisosomas primarios con vacuolas de fagocitosis o pinocitosis, las cuales contienen material extracelular incorporado por endocitosis inespecífica; (b-2) cuerpos residuales, vacuolas conteniendo material no digerido, el cual puede ser eliminado fuera de la célula o permanecer en el interior como inclusión (caso de lipofucsina en las neuronas); y Micrografía electrónica que muestra un lisosoma que contiene una mitocondria y un peroxisoma. Biogénesis de las enzimas lisosomales. Se considera que los lisosomas primarios son productos de secreción igual que otras secreciones, con la diferencia que no abandonan la célula. Entonces, las proteínas integrantes de ambos sistemas que fueron sintetizadas en el RER y pasaron al Golgi para su segregación final, deben tener en algún punto un tratamiento especial que les permitan seguir una vía en particular. Lisosomas Una proteína destinada a los lisosomas, sintetizada en los ribosomas es secretada a la luz del RER donde es glucosilada. Pasa al aparato de Golgi, en cuya cisterna cis existe una enzima llamada NAcGlc-fosfotransferasa que tiene doble función: Al llegar al lisosoma, se encuentran en un lumen con pH ácido (cerca de 5.0), ambiente en el cual la enzima se disocia de su receptor, quedando así lista para actuar y el receptor retorna mediante vesículas hacia las cisternas retículo trans del Golgi para ser utilizados de nuevo. (a) reconoce la secuencia proteica de una enzima lisosomal (parche señal), y La existencia de algunos receptores de M–6–P en la membrana plasmática, permiten recuperar, mediante endocitosis, cualquier enzima lisosómica que haya equivocado su destino normal. (b) añade un residuo de manosa-6fosfato (Man-6-P). Estas proteínas lisosomales marcadas con PO4 en sus manosas son movilizadas a través de las cisternas cis, medial y trans del Golgi sin sufrir modificaciones en su estructura. Al llegar al retículo trans se encuentran con receptores de membrana que reconocen las M–6– P, son los receptores M–6–P. Las enzimas lisosomales unidas a su receptor son movilizadas hacia los lisosomas a través de vesículas cubiertas con clatrina. Bomba protónica Experimentos fisiológicos sugieren que un sistema de transporte de protones dependiente de ATP esté presente en la membrana de los lisosomas para mantener ácido el interior de este organelo (pH ≈ 4.5). Esta ATPasa de H+ está integrada por varias cadenas polipeptídicas. Unión parche señal al sitio renocimiento Enzima lisosomal Unión NAcGlc-UDP al sitio catalítico NAcGlc-fosfotransferasa Sitio catalítico Sitio reconocimiento Lisosomas La llamada crinofagia es una variante de la autofagia y se aplica a la remoción del exceso de gránulos de secreción. Caso de las células mamotropas de la adenohipófisis de una mujer después del destete. En la tiroides, la hormona se acumula en el lumen folicular, de allí ingresa a las células foliculares mediante fagocitosis. La tiroglobulina contenida en los endosomas debe degradarse para liberar la hormona tiroidea. Dicha liberación ocurre cuando los endosomas se unen a los lisosomas que contienen las enzimas específicas para tal función. Algunas funciones específicas de los lisosomas. En condiciones normales, las enzimas lisosomales intervienen en una amplia variedad de fenómenos celulares, todos relacionados con la digestión de materiales. La autofagia es importante en el desarrollo prenatal y postnatal. Durante el desarrollo embrionario y fetal existen muchas estructuras que debe desaparecer para dejar paso a otras que serán definitivas. Por ejemplo, en un embrión de sexo masculino los conductos paramesonéfricos propios del sexo femenino deben desaparecer. La remodelación del tejido óseo se logra por acción de los osteoclastos, cuyas enzimas lisosomales son liberadas al exterior degradando la matriz ósea. Esta acción es estimulada por la parathormona, una hormona producida por las glándulas paratiroides. En la cabeza del espermatozoide se encuentra el acrosoma, una modificación del Golgi, el cual puede considerarse funcionalmente como un lisosoma, debido al contenido de importantes enzimas liberadas durante la fecundación. El espermatozoide debe atravesar dos barreras: una está conformada por varias capas de células llamada corona radiada y para atravesarla libera hialuronidasa; y la otra lo constituye la zona pelúcida, para lo cual libera acrosina y neuraminidasa Lisosomas Enfermedades relacionadas con los lisosomas La importancia de los lisosomas en la degradación de constituyentes de la membrana celular se demuestra por la existencia de mutaciones en ciertas hidrolasas lisosomales. Se ha caracterizados unas 30 diferentes enfermedades genéticas humanas, denominadas en conjunto enfermedades de almacenamiento El nombre se debe a que un cierto material, que debía ser degradado, se acumula dentro de los lisosomas por carencia de la enzima encargada de su degradación. Una de estas enfermedades es la llamada enfermedad de Tay-Sachs que se hereda como un gen recesivo, caracterizándose por un retardo mental, deterioro del sistema nervioso central y muerte.En personas normales, un constituyente de la membrana plasmática de las células nerviosas es el gangliósido GM2, el cual está continuamente sintetizándose y degradándose por acción de una enzima lisosomal: la hidrolasa β-N-hexosaminidasa A. Los pacientes con esta enfermedad acumulan grandes cantidades del gangliósido GM2 debido a la ausencia hereditaria de la enzima y es la acumulación de este gangliósido el responsable de toda la sintomatología de la enfermedad. Otra transtorno lisosomal es la enfermedad de Gaucher. Esta enfermedad es frecuente en la población judía (1:2500 individuos). Hay 3 tipos de severidad: el tipo I es el hígado y el bazo los órganos afectados, además de transtornos óseos; el tipo II manifiesta severas alteraciones nerviosas y los pacientes mueren a temprana edad; y el tipo III posee una severidad intermedia entre los dos tipos anteriores. La enfermedad es causada por la deficiencia de la glucocerebrosidasa enzima que transforma los glucocerebrósidos en glucosa y ceramida. La deficiencia de esta enzima es evidente en los macrófagos hepáticos y esplénicos, lo que explica las manifestaciones del tipo I. En el tipo I se ha determinado en la enzima una sustitución de la serina por la asparagina; mientras que en los tipos II y III es la prolina que fue sustituída por la leucina. Desde hace años se ha intentado un tratamiento para esta enfermedad mediante el uso de glucocerebrosidasa exógena obtenida de placenta humana. Pero esto es muy caro. Otra alternativa actual es el uso del método del ADN recombinante el cual puede introducirse en la médula ósea.