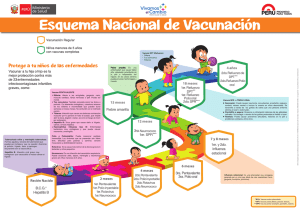
Vademécum Vacunas
Anuncio

Vademécum Vacunas Sanofi Pasteur Un mundo en que nadie sufra ni muera a causa de una enfermedad que puede ser evitada mediante la vacunación. 1 2 Indice Vademécum Sanofi Pasteur Pág.: Nuestras Raices 5 La Vacunación Salva Vidas 9 ¿Qué es la vacunación? 10 Las Enfermedades Infecciosas 11 Acerca de la Inmunidad 11 Las Vacunas estimulan el sistema inmune 13 Principales tipos de vacunas 16 Ciclo de desarrollo de una vacuna 17 Va c u n a s ACTACEL21 ACT-HIB27 ADACEL31 AVAXIM 80U 37 AVAXIM 160U 39 HEXAXIM43 IPV51 MENACTRA55 PNEUMO2362 RECOMVAX-B65 STAMARIL68 TETAVAX71 TYPHIM VI 75 VAXIGRIP78 VERORAB82 3 4 NUESTRAS RAICES...... Sanofi Pasteur no existiría, si en los años treinta el Dr. Charles Mérieux no hubiera recuperado el laboratorio artesanal de su padre, Marcel Mérieux, en Lyon para hacer de él, bajo el nombre de Institut Mérieux el líder de la industria de la vacuna humana y animal. Dr. Charles Mérieux tenía una visión unificadora sin fronteras, ya que integraba a los países más pobres en todas sus acciones, la que le permitió federar la industria francesa de la vacuna con la fusión del Institut Mérieux y de Pasteur Vaccins bajo el nombre de Pasteur Mérieux Sérums et Vaccins, y formar luego una sociedad verdaderamente mundial con la compra de los Laboratorios Connaught. La nueva sociedad tomó entonces el nombre de Pasteur Mérieux Connaught, antes de volverse Aventis Pasteur, luego la creación del Grupo Aventis nació de la fusión de Rhône-Poulenc y de Hoechst en 1999. Verdadero inventor de la virología industrial, el Institut Mérieux hoy Sanofi Pasteur, desarrolló la gama de vacunas más amplia. Es uno de los principales proveedores de vacunas de la Unicef, en particular para el programa de la erradicación de la poliomielitis, erradicación que es hoy un objetivo realizable. El crecimiento de la sociedad se debió a la visión de proteger al mundo de serias enfermedades y a la pasión del Doctor Charles Mérieux por la Salud Pública, mantenidas por su dinamismo, su apertura hacia todos y su convicción de que todo era posible. 5 Nuestras vacunas protegen 6 la vida Nuestro compromiso: Contribuir a salvar millones de vidas 7 Vacunas para proteger vidas La Organización Mundial de la Salud estima que la vacunación salva, por año, 3 millones de vidas en todo el mundo... Sin embargo, al mismo tiempo, más de 3 millones de personas mueren por no poder acceder a las vacunas existentes. 8 Exceptuando la generalización del agua potable y la mejora de condiciones de saneamiento y de higiene, ninguna acción humana tuvo un impacto comparable al de la vacunación en la lucha contra enfermedades infecciosas y en la reducción de la tasa de mortalidad. La vacunación salva vidas Hace cien años, las enfermedades infecciosas eran la principal causa de muerte en todo el mundo, incluso en los países más desarrollados. Hoy en día, la vacunación permite prevenir numeroras enfermedades infecciosas y muy pronto, nuevas vacunas ampliarán el espectro de la prevención. Los programas de vacunación en masa demostraron su eficacia en la lucha contra las enfermedades e incluso, en el logro de su erradicación. Antes de que una campaña de vacunación permitiera erradicar la viruela, en 1980, la enfermedad amenazaba el 60% de la población mundial y producía la muerte de una de cada cuatro personas. Entre 2000 y 2008, la cantidad de muertes causadas por sarampión disminuyó un 78% en el mundo; la lucha por erradicar la enfermedad continúa. La erradicación de la poliomielitis está al alcance de la mano. Desde el lanzamiento por la OMS y de sus colaboradores de la iniciativa mundial para erradicación de la poliomielitis, en 1988, la infecciones se redujeron un 99%, y se estima que se evitó la parálisis en 5 millones de personas. 9 ¿Qué es la vacunación? La vacunación consiste en introducir en el organismo un agente (virus, bacteria, molécula, etc.) desprovisto de propiedades patógenas, pero capaz de producir una respuesta inmune. Cuando el sistema inmune vuelva a tener un contacto con el agente patógeno, será capaz de defenderse y de proteger, así, a la persona vacunada. 10 Las enfermedades infecciosas Las enfermedades infecciosas representan una causa importante de mortalidad, en particular de niños y jóvenes. Las enfermedades infecciosas son provocadas por microorganismos patógenos, como los virus, las bacterias, los parásitos o los hongos. Omnipresentes, los microorganismos pueden sobrevivir en numerosos lugares (tierra, agua, alimentos) o seres vivos (humanos, animales). Acerca de la inmunidad El hombre está protegido de los microorganismos que lo rodean por un sistema de defensa extremadamente eficaz; el sistema inmune, un verdadero ejército interno, compuesto por miles de millones de células y moléculas. Una de las propiedades más notables del sistema inmune es su capacidad de distinguir los elementos del cuerpo que defiende (células, moléculas) de cualquier elemento “extraño”. Cuando un organismo se ve atacado por un agente infeccioso, su sistema inmune aprende a reconocerlo y a defenderse produciendo células y moléculas, a menudo muy longevas, dirigidas, específicamente, contra el agente extraño. 11 La primera acción defensiva de nuestro organismo consiste en una respuesta inmune general. La segunda acción defensiva es más específica: se basa en el reconocimiento del agente a atacar y en su eliminación. Nuestro sistema inmune memoriza las bacterias y virus particulares con los que se va encontrando y prepara, así, una respuesta mucho más eficaz y rápida para el segundo encuentro. Este fenómeno explica, por ejemplo, que no se pueda padecer el sarampión dos veces. El efecto de la vacunación es inducir a la memoria genética, a partir de ese primer encuentro con el agente patógeno. 12 Un antígeno es una sustancia externa para el organismo, capaz de provocar una reacción determinada en el sistema inmune que lo eliminará. Por lo general, se trata de proteínas, polisacáridos o sus derivados lipídicos, correspondientes, por ejemplo, a la capa externa o pared de un virus o de una bacteria. La respuesta inmune está a cargo, en particular, de los glóbulos blancos de la sangre, de los linfocitos T y de los anticuerpos, que son moléculas producidas por otros glóbulos blancos, los linfocitos B. Los anticuerpos circulan en el organismo y son capaces de neutralizar sustancias biológicamente activas, como por ejemplo toxinas, adherirse a microorganismos y permitir su eliminación por medio de células del sistema inmune. Las vacunas estimulan el sistema inmune Por medio de las vacunas, se busca la prevención y la disposición más rápida de medios de defensa específicos (anticuerpos específicos, reacciones celulares adaptadas, etc.) para evitar el desarrollo de la infección y proteger, así, al individuo. Una vacuna es un producto biológico que contiene uno o varios antígenos y está fabricada a partir de bacterias o virus completos, de sus componentes (polisacáridos, proteínas, etc.) o del producto secretado (toxinas); mediante diversos procedimientos, se toma de dichos elementos la capacidad de producir una enfermedad, conservando la de inducir respuesta inmune. 13 14 Principales tipos de vacunas Vacunas vivas atenuadas Se atenúa el poder patógeno mediante sucesivos pasajes del microorganismo por cultivos celulares. Ejemplos: parotiditis, sarampión, rubéola, polio (oral), fiebre amarilla, BCG. Vacunas inactivadas (o inertes) Se obtienen exponiendo al agente patógeno a un agente físico o químico que produce pérdida de la virulencia sin desnaturalizar el poder inmunogénico. Las vacunas inactivadas pueden producirse a partir de un microoganismo completo o de una parte de él: - vacunas a germen entero, constituidas por la totalidad del microorganismo. - vacunas fabricadas de proteínas (vacuna acelular/subunitaria), toxinas o polisacáridos (conjugados o no) extraídos del microorganismo. Ejemplos: difteria, tétanos, tos ferina, Hib, meningococo, fiebre tifoídea, neumococo, influenza, rabia, polio (inyectable), hepatitis A. Vacunas recombinadas Se producen por ingeniería genética. Utilizan una célula animal o una levadura para la síntesis del antígeno de vacuna. Ejemplos: hepatitis B. 15 “La tareas son innumerables, pero todas convergen en la realización del mismo ideal: liberar a la humanidad de esas epidemias que la diezman.” Charles Mérieux, Médico francés (1907 - 2001) 16 El ciclo de desarrollo de una vacuna Duración media de desarrollo de una vacuna: Inversión media global para el desarrollo de una vacuna: 12 años más de 500 millones de euros 70% del tiempo de producción de una vacuna está consagrado al control de calidad INVESTIGACIÓN Y DESARROLLO Fases de desarrollo clínico 3 Fase de exploración 1 Fase preclínica 2 Fase I Fase II Fase III - Apoyo continuo a procedimiento industrial - Gestión de etapas de los expedientes regulatorios Entre 9 y 14 años OPERACIONES INDUSTRIALES Producción a granel Estudio de posibilidades de realización Puesta a punto de procedimiento de fabricación Desarrolo de procedimiento de fabricación industrial Producción de primeros lotes 4 Registro 5 Lanzamiento del producto Cultivos de gérmenes Cosecha Purificación Inactivación Unión de valencias Formulación Distribución en la línea Liofilización Acondicionamiento Liberación de los lotes Distribución de 6 a 22 meses 6 OPERACIONES COMERCIALES Evaluación de necesidades y estudios de mercado Perfil elegido del producto: - población escogida - características de la vacuna candidata Comercialización de lotes: - Mercado privado - Mercado Internacional - Socios en el ámbito de la salud (Unicef. OMS, etc.) Calidad: Control de calidad, garantía de calidad Farmacovigilancia Comentarios 1 Fase de exploración: 2 a 4 años Identificación de antígenos para la selección de vacunas candidatas que seguirán el proceso 2 Fase preclínica: entre 1 y 2 años Evaluación de antígenos en animales y selección de la mejor vacuna candidata. 3 Fase de desarrollo clínico: de 6 a 8 años. Test de la vacuna candidata en el ser humano. Fase I: test de inocuidad en 10 a 100 individuos. Fase II: respuesta inmune en 100 a 3.000 individuos. Fase III: estudio de eficacia, de tolerancia a gran escala y de consistencia en 3.000 a 40.000 individuos. 4 Los primeros lotes son lotes clínicos y lotes industriales de conformidad. 5 Registro: fase de síntesis de 12 a 18 meses. Se recopila todos los datos de las etapas anteriores y se agrupan en un sólo documento, que se presenta a las autoridades de salud para obtener una Autorización para la comercialización 6 Se cultivan, cosechan y purifican los gérmenes infecciosos. Luego de la formulación de la liofilización (que estabiliza las vacunas más frágiles) y de la distribución, desde el despacho principalmente, en hasta la administración frascos y jeringas, se de las vacunas a los procede al envasado pacientes. de las vacunas. Al final del proceso de fabricación, debe mantenerse la cadena de frío en todas las etapas, La puesta a punto de una vacuna es un proceso largo, complejo y El ciclo de desarrollo de una vacuna costoso que debe superar diversas etapas desde la comprensión de la enfermedad y de los datos epidemiológicos hasta la comercialización de una nueva vacuna El ciclo de desarrollo de una vacuna La elaboración de las vacunas es muy diferente a la de los medicamentos. Se trata de una producción biológica a partir de microorganismos vivos. Según la vacuna, son necesarios de 6 a 22 meses para lograr un lote de vacunas. Este ciclo de producción es mucho más prolongado que el de un medicamento; este hecho se debe, principalmente, a la necesidad de controles extensos y exigentes que garanticen la calidad y la eficacia del producto final. 19 Bacteria Infecciones por Haemophilus Influenzae tipo b “Crear vacunas, es proteger la vida” 20 ACTACEL Vacuna Conjugada de Haemophilus tipo b (conjugada a la proteína tetánica) reconstituida con toxoides diftérico y tetánico y Vacuna antipertussis acelular adsorbidos. FORMA FARMACÉUTICA: Producto reconstituido inyectable ACTacel (Act-HIB reconstituido con TRIACEL) se presenta en un vial monodosis como un polvo liofilizado estéril de color blanco que debe reconstituirse con una suspensión estéril, uniforme y turbia de color blanco a blancuzco que se presenta en otro vial monodosis. Tras la reconstitución, ACTacel es una suspensión estéril, uniforme y turbia de color blanco a blancuzco, para ser administrada por vía intramuscular. COMPOSICION: 1 dosis - aproximadamente 0,5 ml de suspensión reconstituida. La composición de una dosis inmunizante de Vacuna de componentes antipertúsicos, toxoides diftérico y tetánico adsorbidos (suspensión inyectable) es: Toxoide pertúsico (TP) 10 μg Hemaglutinina filamentosa (HAF) 5 μg 5 μg Fimbrias (AGG 2+3) Pertactina (proteína 69kDa) 3 μg Toxoide diftérico ≥30 UI (2.0 unidades / mL) Toxoide tetánico ≥40 UI (2.0 unidades / mL) Excipientes: según fórmula aprobada en el registro sanitario. La composición de una dosis inmunizante de Vacuna Conjugada de Haemophilus tipo b (conjugada a la proteína tetánica) (liofilizado) es: Polisacárido de Haemophilus influenzae tipo b conjugado a 20 μg de proteína tetánica 10 μg. Excipientes: según fórmula aprobada en el registro sanitario. DESCRIPCION: ACTacel [Vacuna conjugada anti-Haemophilus tipo b (conjugada con proteína tetánica) reconstituida con toxoides diftérico y tetánico y vacuna antipertussis acelular adsorbidos] se presenta en dos viales: uno que contiene la vacuna conjugada anti-Haemophilus tipo b liofilizada, formada por el polisacárido capsular PRP de Haemophilus influenzae tipo b unido por enlace covalente a la proteína tetánica (Act-HIB), y otro que contiene una suspensión de los toxoides diftérico y tetánico y la vacuna antipertussis acelular adsorbidos sobre fosfato de aluminio y suspendidos en agua para inyectables (TRIACEL). La vacuna antipertussis acelular está compuesta por 5 antígenos de pertussis purificados (Tp, HAF, PRN y FIM). INDICACIONES Y USO CLINICO: ACTacel está indicado para la vacunación primaria contra la infección invasora por Haemophilus influenzae tipo b y la difteria, el tétanos y la coqueluche o tos ferina (pertussis) (DTP), en lactantes a partir de los 2 meses de edad y en niños de hasta 6 años (antes del séptimo cumpleaños). (Véase POSOLOGÍA Y ADMINISTRACIÓN). En la actualidad no se recomienda la administración de vacunas conjugadas anti-Haemophilus tipo b en lactantes de menos de 2 meses. Los niños que hayan padecido una infección invasora por H. influenzae tipo b (Hib), difteria, tétanos o pertussis deberán ser vacunados también, ya que estas infecciones clínicas no siempre confieren inmunidad. Las personas infectadas por el virus de la inmunodeficiencia humana (VIH), tanto asintomáticas como sintomáticas, deberán ser vacunadas contra H. influenzae tipo b, la difteria, el tétanos y la coqueluche conforme a los calendarios de vacunación estándar. ACTacel no debe utilizarse para el tratamiento de infecciones causadas por H. influenzae tipo b, Corynebacterium diphtheriae, Clostridium tetani o Bordetella pertussis. Uso en pediatría: ACTacel no está indicado en personas de menos de 2 meses de edad ni de 7 años en adelante. Uso en geriatría: ACTacel no está indicado en adultos ni ancianos. 21 CONTRAINDICACIONES: Hipersensibilidad. La vacunación está contraindicada en personas con antecedentes de reacción sistémica de hipersensibilidad a cualquiera de los componentes de ACTacel o de reacción potencialmente mortal tras una administración previa de la vacuna o de una vacuna que contuviera uno o varios de los componentes de ACTacel. Dada la incertidumbre sobre qué componente de la vacuna puede ser el causante, no debe administrarse ninguno de los componentes. Si se está considerando la posibilidad de administrar más vacunas, se puede remitir a dichas personas a un alergólogo para que realice una evaluación. Trastornos neurológicos. La administración de cualquier vacuna antipertussis, incluida ACTacel, está contraindicada en los siguientes casos: Encefalopatía (por ej., coma, disminución del grado de conciencia, convulsiones prolongadas) de etiología desconocida que haya aparecido en los 7 días posteriores a la administración de una vacuna antipertussis. Trastorno neurológico progresivo, como los espasmos infantiles, la epilepsia no controlada y la encefalopatía progresiva. La vacuna antipertussis no debe administrarse a personas que padezcan dichos trastornos hasta que se haya instaurado un régimen terapéutico y el proceso se haya estabilizado. ADVERTENCIAS Y PRECAUCIONES ESPECIALES: Generalidades. Antes de administrar ACTacel, los profesionales de la salud deberán informar al padre, la madre o el tutor de la persona que va a ser vacunada sobre los beneficios y los riesgos de la vacunación, preguntar por el estado de salud reciente del receptor, repasar sus antecedentes en busca de posibles casos de hipersensibilidad a la vacuna o a vacunas similares, revisar los antecedentes de vacunaciones, descartar la presencia de cualquier contraindicación a la vacunación, y cumplir con todos los requisitos locales relativos a la información que se haya de dar al padre, la madre o el tutor antes de la vacunación y sobre la importancia de completar la serie de vacunaciones en su totalidad. Es sumamente importante preguntar al padre, la madre o el tutor por cualquier síntoma y/o signo de reacción adversa que pueda haberse presentado después de una dosis previa de la vacuna. (Véase CONTRAINDICACIONES y REACCIONES ADVERSAS). En las tasas y la intensidad de los episodios adversos que aparecen en los receptores del toxoide tetánico influyen la cantidad de dosis previas y el nivel de antitoxinas preexistentes. Como ocurre con cualquier vacuna, ACTacel puede no proteger al 100% de las personas vacunadas. Durante los brotes de poliomielitis la inmunización electiva de los niños mayores de 6 meses de edad debe postergarse. Las vacunas que contienen el antígeno del Hib no protegen contra las infecciones causadas por otros tipos de H. influenzae ni contra las meningitis de otro origen. La proteína tetánica presente en las vacunas conjugadas que contienen toxoide tetánico como portador de proteínas no puede usarse en ningún caso como sustituta de la vacunación antitetánica habitual. Tras la administración de vacunas con Haemophilus influenzae tipo b se han producido casos de reacciones edematosas en una de las extremidades inferiores o en ambas. Esta reacción se produce principalmente después de la inyección inicial y se observa en las primeras horas siguientes a la vacunación. Puede ir acompañada de síntomas como cianosis, enrojecimiento, púrpura transitoria y llanto intenso. Todos los casos notificados se resolvieron por sí solos y sin secuelas en un plazo de 24 horas. Se han observado casos de celulitis inflamatoria sin infección bacteriana, somnolencia y fiebre alta (>40,5° C/105° F) asociados con vacunas que contienen un antígeno similar. Precauciones relacionadas con la vía de administración: No administre ACTacel por inyección intravascular; asegúrese de que la aguja no penetre en un vaso sanguíneo. No debe administrarse por vía intradérmica ni subcutánea. ACTacel no debe administrarse en las nalgas. Enfermedades febriles o agudas: En caso de enfermedad aguda o febril deberá posponerse la vacunación, pero, en general, la presencia de enfermedades con febrícula no debe ser motivo para posponer la vacunación. Si después de administrar una vacuna antipertussis celular o una vacuna que contenga un componente de pertussis acelular aparece, dentro del periodo especificado, uno de los episodios que se indican a continuación, la decisión de administrar ACTacel deberá basarse en un análisis minucioso de los posibles beneficios y riesgos. • Temperatura ≥40,5 °C (105° F) de etiología desconocida en un periodo de 48 horas; • Colapso o estado de choque (episodio hipotónico-hiporreactivo) en un periodo de 48 horas; • Llanto persistente de ≥3 horas de duración en un periodo de 48 horas; • Convulsiones febriles o afebriles en un periodo de 3 días. Alteraciones hematológicas. Debido a que toda inyección intramuscular puede causar un hematoma en el lugar de la aplicación en personas con trastornos hemorrágicos (como la hemofilia o la trombocitopenia) o en personas sometidas a tratamiento anticoagulante, no deben administrarse inyecciones intramusculares de ACTacel a dichas personas a menos que los posibles beneficios sean mayores que el riesgo de la administración. Si se decide administrar a dichas personas cualquier producto por vía intramuscular, deberá hacerse con precaución y tomando las medidas necesarias para evitar el riesgo de que se formen hematomas después de la inyección. Alteraciones inmunológicas. Se deberá evaluar la posibilidad de que aparezcan reacciones alérgicas en personas sensibles a los componentes de la vacuna. Tras la administración de ACTacel® pueden presentarse reacciones de hipersensibilidad incluso en personas sin antecedentes de hipersensibilidad a los componentes del producto. Se han notificado casos de reacciones alérgicas o anafilácticas tras la administración de algunos preparados que contenían 22 toxoides diftérico y tetánico y/o antígenos de pertussis. Se deberán tener a mano una solución de clorhidrato de epinefrina (1:1.000) y otros fármacos apropiados para su uso inmediato si se produjeran reacciones anafilácticas o de hipersensibilidad aguda. Los profesionales de la salud deben estar familiarizados con las recomendaciones vigentes sobre el tratamiento inicial de la anafilaxia en entornos no hospitalarios, incluido el manejo adecuado de las vías respiratorias. En las personas inmunocomprometidas (ya sea por enfermedad o por tratamiento) puede no obtenerse la respuesta inmune esperada. Si es factible, deberá considerarse la posibilidad de posponer la vacunación hasta que hayan concluido los tratamientos inmunosupresores. No obstante, en personas con inmunodeficiencia crónica (p. ej., en casos de infección por VIH) se recomienda la vacunación, aunque la formación de anticuerpos podría ser limitada. Se han observado casos de reacción anafiláctica, ronchas y urticaria asociados con vacunas que contienen un antígeno similar. Alteraciones neurológicas. Un estudio realizado por el Instituto de Medicina (IOM) de los Estados Unidos descubrió pruebas de una relación causal entre el toxoide tetánico y la neuritis braquial y el síndrome de Guillain- Barré (SGB). Cuando se haya presentado un SGB dentro de las 6 semanas posteriores a la administración previa de una vacuna que contuviera toxoide tetánico, la decisión de administrar ACTacel o cualquier vacuna que contenga toxoide tetánico deberá basarse en un análisis minucioso de los posibles beneficios y riesgos. En lactantes o niños cuyo riesgo de sufrir convulsiones sea mayor que el de la población general, en el momento de la administración de vacunas que contengan un componente de pertussis acelular (incluida ACTacel) y durante las 24 horas siguientes se puede administrar un antipirético apropiado (con la posología recomendada en su ficha técnica) para reducir la probabilidad de que aparezca fiebre. Los episodios hipotónicos-hiporreactivos son raros tras la administración de vacunas DTP con elementos de pertussis celulares, y son aun más infrecuentes tras la administración de vacunas DTP y DT con elementos de pertussis acelulares. En personas con antecedentes de episodios hipotónicos-hiporreactivos no está contraindicado el uso de vacunas antipertussis acelulares, pero se recomienda precaución. Se han observado casos de convulsiones febriles, convulsiones (convulsiones parciales, convulsiones tonicoclónicas), polirradiculopatías y enfermedades desmielinizantes (como el síndrome de Guillain-Barré) asociados con vacunas que contienen un antígeno similar. Mujeres embarazadas. Esta vacuna no está indicada en personas de 7 años de edad en adelante. Madres lactantes. Esta vacuna no está indicada en personas de 7 años de edad en adelante. REACCIONES ADVERSAS Reacciones adversas en los ensayos clínicos. Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de una vacuna no pueden compararse directamente con las obtenidas en los ensayos clínicos de otra vacuna y puede que no reflejen las tasas observadas en la práctica. Sin embargo, la información sobre reacciones adversas procedente de los ensayos clínicos sirve de base para identificar los episodios adversos que parecen estar relacionados con el uso de la vacuna y para estimar las tasas de dichos episodios. A continuación se indica la frecuencia de reacciones adversas sistémicas y en el punto de inyección (respuestas obtenidas mediante interrogatorio) que se observaron en ensayos clínicos realizados en Canadá y Taiwán (n = 241) entre las 48 y las 72 horas posteriores a la administración de cualquier dosis de ACTacel® a los 2, 4, 6 y 17 a 21 meses de edad. Muy frecuentes: ≥10% Frecuentes: ≥1% y <10% Alteraciones en el lugar de administración Muy frecuentes: Sensibilidad al tacto en el lugar de la inyección, inflamación, enrojecimiento. Alteraciones sistémicas Muy frecuentes: irritabilidad, inapetencia, llanto Frecuentes: decaimiento, vómitos, diarrea, fiebre. Datos de la experiencia posterior a la comercialización Durante el uso internacional posterior a la comercialización de ACTacel se han notificado espontáneamente otros episodios adversos, que se indican a continuación. Debido a que estos episodios se han notificado de forma voluntaria y proceden de una población de tamaño indeterminado, no es posible calcular con fiabilidad su frecuencia o establecer una relación causal con la exposición a la vacuna. Trastornos psiquiátricos: Irritabilidad, gritos, llanto prolongado o de tono inusualmente agudo, inquietud. Trastornos del sistema nervioso: Episodios hipotónicos-hiporreactivos, hipotonía, encefalopatía. Trastornos vasculares: Palidez. Trastornos gastrointestinales: Náuseas. Trastornos de la piel y del tejido subcutáneo: Eritema, exantema, prurito Trastornos generales y alteraciones en el lugar de administración: Reacciones en el lugar de la inyección, incluidos dolor e inflamación; reacciones extensas en el lugar de la inyección, incluida la inflamación de la extremidad con posible extensión desde el lugar de la inyección hasta más allá de una o ambas articulaciones. Somnolencia, adormecimiento. 23 INTERACCIONES MEDICAMENTOSAS: Interacciones de la vacuna con otros medicamentos Los tratamientos inmunosupresores pueden interferir en el desarrollo de la respuesta inmune esperada. (Véase ADVERTENCIAS Y PRECAUCIONES). Administración simultánea de otras vacunas: La administración al paciente, durante una misma visita, de las vacunas de organismos vivos e inactivados más utilizadas ha producido tasas de seroconversión y de reacciones adversas parecidas a las observadas cuando las vacunas se administran por separado. Las vacunas administradas simultáneamente deben inyectarse con jeringas distintas y en lugares diferentes. Se recomienda la administración simultánea, en particular cuando se tema que la persona no vaya a regresar para una vacunación posterior. Se sugiere la administración simultánea de vacunas infantiles (como ACTacel, la triple vírica, la vacuna contra la varicela, la antineumocócica conjugada y la vacuna contra la hepatitis B) en niños que tengan la edad recomendada para recibir dichas vacunas y no presenten contraindicaciones. La vacuna puede ser administrada concomitantemente con la vacuna antipolio inactivada (IPV) y la vacuna antipolio oral. Si la vacuna se administra junto con IPV, se deben usar jeringas separadas y se deben aplicar en sitios diferentes. ACTacel no debe mezclarse en una misma jeringa con otros productos de administración parenteral. Interacciones entre la vacuna y los análisis clínicos: Se han detectado algunos casos de antigenuria tras la administración de vacunas que contenían el antígeno del Hib, por lo que la detección urinaria del antígeno puede carecer de valor diagnóstico firme cuando se sospeche una infección por Haemophilus influenzae tipo b en las dos semanas posteriores a la vacunación. POSOLOGÍA Y ADMINISTRACIÓN Dosis recomendada: 1 dosis = 0,5 ml El calendario de vacunación estándar con ACTacel deberá seguir las recomendaciones locales. Como guía, ACTacel se puede administrar en una serie de tres dosis a intervalos de 2 meses entre sí, seguidas de una cuarta dosis administrada aproximadamente de 6 a 12 meses después de la tercera dosis. Siempre que sea factible, se deberá usar ACTacel para las cuatro dosis de la serie de vacunación, ya que no hay datos clínicos que apoyen el uso de ACTacel en una secuencia mixta con otras vacunas antipertussis acelulares de combinación autorizadas. Con independencia de su peso al nacer, los niños prematuros cuyo estado clínico sea satisfactorio deberán ser vacunados con dosis completas de la vacuna a la misma edad cronológica y conforme al mismo calendario que los niños nacidos a término. No deben administrarse dosis fraccionarias (<0,5 ml). No se ha determinado el efecto de las dosis fraccionarias en la seguridad y la eficacia de ACTacel. ACTacel no debe administrarse a personas de menos de 2 meses de edad ni de 7 años en adelante. (Véase INDICACIONES Y USO CLÍNICO). Administración: Inspeccione el producto antes de su utilización para descartar la presencia de partículas extrañas o de decoloración. El producto no debe administrarse si presenta tales condiciones. Reconstitución del producto liofilizado y extracción del vial taponado: Act-HIB debe reconstituirse con la vacuna TRIACEL. Antes de la reconstitución, limpie el tapón de los viales de TRIACEL y Act-HIB con un germicida adecuado. No retire el tapón de ninguno de los viales ni el sello metálico que lo fija. Agite bien pero con suavidad el vial de TRIACEL, extraiga todo el contenido de la vacuna líquida e inyéctelo lentamente en el vial de Act-HIB liofilizado. Agite bien el vial que ahora contiene ACTacel hasta obtener una suspensión uniforme y turbia de color blanco a blancuzco. Extraiga el volumen íntegro de la vacuna combinada reconstituida. ACTacel debe usarse inmediatamente después de su reconstitución. Es indispensable utilizar una técnica aséptica. Con el fin de evitar la transmisión de enfermedades, use para cada paciente jeringas y agujas estériles distintas o una unidad desechable estéril. No se debe colocar de nuevo el capuchón a las agujas, que deberán desecharse según las directrices sobre residuos biopeligrosos. Antes de la inyección, la piel donde vaya a aplicarse deberá limpiarse con un germicida adecuado. Administre por vía intramuscular (IM) el volumen total de la vacuna reconstituida. En niños menores de un año, la cara anterolateral del muslo presenta el músculo más grande y es el lugar de inyección preferible. En niños de más edad, el deltoides suele ser suficientemente grande para la inyección. CONSERVACIÓN Y ESTABILIDAD Consérvese a una temperatura de entre 2 y 8 °C (35 a 46° F). No congelar. Desechar el producto si ha estado expuesto a la congelación. La vacuna debe usarse inmediatamente después de su reconstitución. No usar después de la fecha de caducidad. Productos residuales del proceso de fabricación: Contiene cantidades residuales de formaldehído y glutaraldehído, según las indicaciones del doctor. Como referencia: la inmunización primaria se empieza a los 2 meses como mínimo y consiste en 3 dosis administradas con un intervalo de uno o dos meses seguido por una dosis de refuerzo administrada un año después de la tercera dosis. Administre la vacuna por vía intramuscular. El área preferida para la inyección es el músculo deltoides o la parte anterolateral del 24 muslo (músculo vastus lateralis), En los niños mayores de 1 año de edad se prefiere inyectar la vacuna en el músculo deltoides ya que la inyección en el muslo ha resultado en reportes frecuentes de cojeras debido al dolor muscular. No inyectar por vía intravascular. Envasado. ACTacel (Act-HIB reconstituido con TRIACEL) se presenta en un vial monodosis de 3 ml y un vial monodosis de 2 ml (diluyente). Los viales son de vidrio tipo 1. Los tapones de los viales no contienen látex (goma natural). ACTacel se presenta en envases de: 1 vial monodosis de Act-HIB y 1 vial monodosis de TRIACEL (diluyente) 5 viales monodosis de Act-HIB y 5 viales monodosis de TRIACEL (diluyente) Documento revisado y basado en aprobación por ISP 2012 25 Bacteria Infecciones por Haemophilus Influenzae tipo b “Crear vacunas, es proteger la vida” 26 ACT-HIB Vacuna Anti-Haemophilus Tipo b, Conjugado 1. DENOMINACIÓN DEL MEDICAMENTO ACT-HIB 10 microgramos/0,5 mL, polvo y disolvente para solución inyectable en jeringa prellenada. Vacuna Haemophilus influenzae tipo b (conjugada). 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Poliósido de Haemophilus influenzae tipo b 10 microgramos 18-30 microgramos Conjugado con la proteína tetánica Para una dosis de 0,5 mL de solución reconstituida. 3. Forma farmacéutica Polvo y disolvente para solución inyectable en jeringa prellenada. 4. Datos clínicos 4.1. Indicaciones terapéuticas Esta vacuna está indicada para la prevención de las infecciones invasivas provocadas por Haemophilus influenzae tipo b (meningitis, septicemias, celulitis, artritis, epiglotitis, etc.) en el niño a partir de 2 meses de edad. Esta vacuna no protege contra las infecciones provocadas por otros tipos de Haemophilus influenzae ni contra las meningitis de otros orígenes. La proteína tetánica contenida en esta vacuna no reemplaza la vacunación antitetánica habitual. 4.2. Posología y forma de administración Posología • Antes de los 6 meses de edad, 3 dosis sucesivas de 0,5 mL con uno o dos meses de intervalo, seguidas de una inyección de refuerzo (4ª dosis) un año después de la 3ª inyección. • Entre 6 y 12 meses de edad, 2 dosis de 0,5 mL con un mes de intervalo, seguidas de una inyección de refuerzo (0,5 mL) a los 18 meses de edad. • Entre 1 y 5 años, 1 única dosis de 0,5 mL En caso de contacto: cuando ha habido contacto con un caso de infección invasiva por Haemophilus influenzae (en la familia o la guardería), la vacunación debe realizarse siguiendo el esquema adecuado a la edad. También se debe vacunar al caso índice Forma de administración Vía intramuscular o subcutánea profunda Las zonas recomendadas para la inyección son la cara anterolateral del muslo (tercio medio) en el lactante y la región del deltoides en el niño. No inyectar por vía intravascular. Para las instrucciones de reconstitución, ver sección 6.6. 4.3. Contraindicaciones Hipersensibilidad conocida a uno de los componentes de la vacuna, en particular a la proteína tetánica o bien aparecida después de una inyección anterior de una vacuna Haemophilus influenzae tipo b conjugada. 4.4. Advertencias y precauciones especiales de empleo Advertencias. No inyectar por vía intravascular, asegurándose de que la aguja no penetre en un vaso sanguíneo. En caso de fiebre o enfermedad aguda, es preferible diferir la vacunación. Al igual que con todas las vacunas inyectables susceptibles de inducir una reacción anafiláctica inmediata, se recomienda tener previsto el tratamiento médico oportuno. La vacunación puede realizarse en niños que presentan un estado de inmunodepresión congénito o adquirido, teniendo 27 en cuenta que, en función del estado del sistema inmunitario, la respuesta a la vacuna será menor o mayor. En el caso de niños bajo tratamiento con inmunodepresores (corticoterapia, quimioterapia antimitótica, etc.), se recomienda vacunar una vez finalizado el tratamiento. Debe tenerse muy en cuenta el riesgo potencial de apnea y la necesidad de vigilancia respiratoria durante 48-72 horas cuando se administren las dosis de primovacunación en los muy prematuros (nacidos < 28 semanas de embarazo) y particularmente aquellos que tienen antecedentes de inmadurez respiratoria. Debido al beneficio considerable de la vacunación en estos lactantes, la administración no debe suspenderse ni aplazarse. 4.5. Interacción con otros medicamentos y otras formas de interacción Esta vacuna puede administrarse simultáneamente, practicando las inyecciones en 2 lugares diferentes, en particular con las otras vacunaciones recomendadas: difteria, tétanos, tos ferina, poliomielitis, sarampión, paperas, rubéola. 4.6. Embarazo y lactancia. No aplicable. 4.7. Efectos sobre la capacidad para conducir y utilizar máquinas. No aplicable. 4.8. Reacciones adversas. De acuerdo con los calendarios vacunales pediátricos, las recomendaciones de la OMS (Organización Mundial de la Salud) y del Comité Asesor de Prácticas de Inmunización (ACIP), ACT-HIB raramente se administra sola, sino que a menudo se administra en asociación o en combinación con otras vacunas concomitantes, como las vacunas que contienen las valencias diftérica, tetánica y pertúsica (de células enteras o acelular). El perfil de tolerancia de ACT-HIB reflejará pues, esta utilización conjunta. A continuación se presenta la lista de las reacciones adversas informadas durante los ensayos clínicos o desde la comercialización, según la terminología MedDRA (por sistemas de órganos y por frecuencia), y para todos los grupos de edad. La frecuencia se define como: muy frecuentes (>10%), frecuentes (>1% y <10%), poco frecuentes (> 0,1% y <1%), raras (>0,01% y <0,1%), muy raras (<0,01%) incluyendo notificaciones aisladas. Reacciones adversas observadas durante los ensayos clínicos Se ha evaluado la tolerabilidad de la vacuna durante los diferentes estudios clínicos controlados mediante una vigilancia activa de las reacciones adversas, y durante los cuales, más de 7.000 niños de menos de 2 años con buena salud han recibido una inyección de ACT-HIB, inyección casi siempre combinada con una vacuna de células enteras o acelular diftérica-tetánica-pertúsica (DTP). En los estudios controlados, cuando ACT-HIB ha sido administrada en combinación con vacunas DTP, la frecuencia y el tipo de reacciones sistémicas postvacunales observadas no han diferido de lo observado con una vacuna DTP administrada sola. Las reacciones adversas que pueden estar relacionadas con la vacuna, observadas con una frecuencia >1% han aparecido, en general, entre 6 y 24 horas siguientes a la vacunación y han sido, en su mayor parte, transitorias y de una intensidad entre leve y moderada. No se ha observado ningún aumento de la incidencia o de la gravedad de las reacciones locales o sistémicas tras la administración de las dosis sucesivas del esquema de primovacunación. Trastornos generales y alteraciones en el lugar de administración De muy frecuentes a frecuentes: reacciones en el lugar de la inyección como dolor, eritema, hinchazón y/o inflamación, induración. Poco frecuentes: fiebre (> de 39°C). Trastornos psiquiátricos. Muy frecuentes: irritabilidad. De frecuentes a poco frecuentes: llantos (incontrolables o anormales). Reacciones adversas observadas después de la comercialización Durante la vigilancia después de la comercialización basada en una amplia experiencia (varios millones de dosis administradas en el mundo), se han informado otras reacciones en asociación temporal con esta vacuna. Ninguna de las reacciones adversas a continuación ha sido informada con una frecuencia superior a 0,01% (muy raras). Las frecuencias están basadas en los índices de notificaciones espontáneas y calculadas a partir del número de notificaciones y del número de dosis distribuidas en un mismo periodo. Trastornos generales y alteraciones en el lugar de administración. Muy raras: edema de los miembros inferiores con cianosis o púrpura transitorias que se producen en las primeras horas siguientes a la vacunación y que desaparecen rápidamente y de manera espontánea sin secuelas. Estas reacciones no van acompañadas de signos cardiorrespiratorios. Éstas han sido informadas principalmente cuando la vacuna fue administrada simultáneamente o en combinación con otras vacunas (como las vacunas que contienen las valencias diftérica, tetánica y pertúsica). Trastornos del sistema inmunológico. Muy raras: reacciones de hipersensibilidad. Trastornos del sistema nervioso. Muy raras: convulsiones asociadas o no a fiebre. Trastornos de la piel y del tejido subcutáneo. Muy raras: urticaria, erupción, prurito. Apnea en los muy prematuros (nacidos < 28 semanas de embarazo) (ver sección 4.4). 4.9 Sobredosis. No aplicable. 5. Propiedades farmacológicas 5.1. Propiedades farmacodinámicas Vacuna contra las infecciones por Haemophilus influenzae tipo b. La vacuna Haemophilus influenzae tipo b aporta inmunidad contra las infecciones invasivas por Haemophilus influenzae tipo b. El poliósido capsular (polirribosil ribitol fosfato: PRP) induce en el hombre una respuesta serológica anti PRP. Sin embargo, 28 al igual que con todos los antígenos de polisacárido, la naturaleza de la respuesta inmunitaria es timo independiente, caracterizada por la ausencia de un efecto de refuerzo durante repetidas inyecciones y por una baja inmunogenicidad en los lactantes. La unión covalente del poliósido capsular de Haemophilus influenzae tipo b de una proteína tetánica, le permite a la vacuna conjugada comportarse como un antígeno timo dependiente provocando una respuesta serológica anti PRP específica en los lactantes con inducción de IgG específicas y creación de una memoria inmunitaria. El estudio de la actividad funcional de los anticuerpos específicos anti PRP inducidos por la vacuna Haemophilus influenzae tipo b conjugada en los lactantes y los niños, ha demostrado tanto su actividad bactericida como su actividad opsonizante. Los estudios de inmunogenicidad en los lactantes vacunados desde los 2 meses de edad han demostrado que prácticamente todos los niños tenían un título de anticuerpos anti PRP >0,15 mg/mL después de la 3ª dosis, y >1 mg/mL aproximadamente el 90 % de ellos. En los lactantes menores de 6 meses que hayan recibido tres dosis de vacuna Haemophilus influenzae tipo b conjugada, la administración de una inyección de refuerzo entre 8 y 12 meses más tarde dio lugar a un aumento muy significativo del título medio de los anticuerpos anti PRP. 5.2. Propiedades farmacocinéticas: No aplicable. 5.3. Datos preclínicos sobre seguridad: No aplicable. 6. Datos farmacéuticos. Lista de excipientes. Polvo: trometamol, sacarosa. Disolvente: cloruro de sodio, agua para preparaciones inyectables. 6.1. Incompatibilidades. Este medicamento no debe mezclarse con otros. 6.2. Periodo de validez. 3 años. 6.3. Precauciones especiales de conservación. Conservar en nevera (entre 2°C y 8°C). No congelar. 6.4. Instrucciones de uso, manipulación y eliminación. Reconstituir la solución, inyectando el contenido de la jeringa de disolvente en el frasco de polvo. Agitar hasta la disolución completa del polvo. Para las jeringas sin aguja acoplada, la aguja separada debe montarse firmemente sobre la jeringa efectuando un movimiento rotatorio de un cuarto de vuelta. 7. Titular de la autorización de comercialización SANOFI PASTEUR S.A. 2, avenue Pont Pasteur 69007 LYON, FRANCIA 29 Bacteria Bordetella Pertussis “Crear vacunas, es proteger la vida” 30 ADACEL Toxoides tetánico y diftérico reducido y vacuna antipertussis acelular adsorbidos. Inyección intramuscular DESCRIPCIÓN ADACEL® [Toxoides tetánico y diftérico reducido y vacuna antipertussis acelular adsorbidos], es una suspensión estéril, uniforme, turbia, de color blanco, que contiene los toxoides tetánico y diftérico adsorbidos por separado sobre fosfato de aluminio, combinados con la vacuna antipertussis acelular y suspendidos en agua para inyectables. La vacuna antipertussis acelular está compuesta por 5 antígenos de pertussis purificados (TP, HAF, PRN y FIM). FORMA FARMACÉUTICA: ADACEL® se presenta en un vial en forma de suspensión estéril, uniforme, turbia y de color blanco. COMPOSICIÓN Cada dosis (0,5 ml) contiene: Ingredientes activos 5 Lf Toxoide tetánico Toxoide diftérico 2 Lf Elemento de pertussis acelular Toxoide de pertussis (TP) 2,5 μ5 g Hemaglutinina filamentosa (HAF) 5 μ5 g 3 μ5g Pertactina (PRN) Fimbrias de tipo 2 y 3 (FIM) 5 μ5g Otros ingredientes Excipientes 1,5 mg Fosfato de aluminio (adyuvante) 0,6% v/v 2-fenoxietanol Productos residuales del proceso de fabricación Puede contener trazas de formaldehído y glutaraldehído. INDICACIONES: ADACEL® está indicada en la inmunización activa de refuerzo para prevenir el tétanos, la difteria y la pertussis (tos ferina) en personas de 4 años o más de edad. Conforme a las recomendaciones locales, ADACEL® puede considerarse una alternativa de la quinta dosis de la vacuna antitetánica, antidiftérica y antipertussis acelular (DTaP) en niños de edades comprendidas entre los 4 y los 6 años, administrada simultáneamente con la vacuna antipoliomielítica inactivada (IPV) en lugares distintos para completar la serie de vacunación para esta edad, cuando se indique. Las personas que hayan padecido tétanos, difteria o tos ferina deben ser vacunadas también, ya que estas infecciones clínicas no siempre confieren inmunidad. Las personas infectadas por el virus de la inmunodeficiencia humana (VIH), tanto asintomáticas como sintomáticas, deben ser vacunadas contra el tétanos, la difteria y la tos ferina conforme a los calendarios de vacunación estándar. ADACEL® no debe utilizarse para el tratamiento de infecciones causadas por Bordetella pertussis, Corynebacterium diphtheriae o Clostridium tetani. Uso en pediatría ADACEL® no está indicado para la vacunación de niños menores de 4 años. POSOLOGÍA Y ADMINISTRACIÓN: Dosis recomendada. El calendario de vacunación con ADACEL® deberá seguir las recomendaciones locales. ADACEL® (0,5 ml) deberá administrarse como una dosis de refuerzo por vía intramuscular. Puede administrarse otra dosis de ADACEL® para reforzar la inmunidad contra la difteria, el tétanos y la tos ferina en intervalos de 5 a 10 años. El lugar preferible es el músculo deltoides. No deben administrarse dosis fraccionarias (dosis <0,5 ml). No se ha determinado el efecto de las dosis fraccionarias 31 en cuanto a la seguridad y la eficacia. El uso de ADACEL® en el tratamiento de heridas propensas al tétanos debe seguir las recomendaciones locales. Como se muestra en la Tabla 3, el Comité Asesor Nacional de Inmunización (NACI) de Canadá y el Comité Asesor de Prácticas de Inmunización (ACIP) de Estados Unidos han emitido directrices para la profilaxis del tétanos en el tratamiento rutinario de heridas. Tabla 3: Uso recomendado por el NACI de agentes inmunizantes en el tratamiento de heridas. * Toxoides tetánico y diftérico tipo adulto. † Inmunoglobulina tetánica, administrada en lugar distinto del Td. ‡ La vacunación primaria es como mínimo 3 dosis a intervalos de edad apropiados. § Sí, si han pasado >10 años desde la última dosis de refuerzo. ** Sí, si han pasado >5 años desde la última dosis de refuerzo. †† Sí, si se sabe que las personas tienen un estado de inmunodeficiencia humoral considerable (p.ej., VIH, agammaglobulinemia) ya que la respuesta inmunitaria al toxoide tetánico puede ser subóptima. Debe realizarse un intento riguroso para determinar si un paciente ha concluido la vacunación primaria. Las personas que han concluido la vacunación primaria contra el tétanos y que tienen heridas leves y no contaminadas, deben recibir una dosis de refuerzo de una preparación con toxoide tetánico si no han recibido toxoide tetánico en los 10 años anteriores. En el caso de heridas propensas al tétanos (p.ej., heridas contaminadas con suciedad, heces, tierra y saliva, heridas punzantes, avulsiones y heridas ocasionadas por misiles, aplastamiento, quemaduras o congelación), es apropiada una dosis de refuerzo si el paciente no ha recibido una preparación con toxoide tetánico en los 5 años anteriores. Administración. Inspeccione el producto antes de su utilización para descartar la presencia de partículas extrañas y/o decoloraciones. (Véase DESCRIPCIÓN.) El producto no debe administrarse si presenta tales condiciones. Agite bien el vial hasta obtener una suspensión uniforme y turbia. Antes de extraer la dosis, limpie el tapón del vial con un germicida adecuado. No retire el tapón ni quite el sello metálico que lo fija. Es indispensable utilizar una técnica aséptica. Con el fin de evitar la transmisión de enfermedades, use para cada paciente jeringas y agujas estériles distintas o una unidad desechable estéril. No se debe colocar de nuevo el capuchón a las agujas, que deberán desecharse según las directrices sobre residuos biopeligrosos. (Véase ADVERTENCIAS Y PRECAUCIONES.) Antes de la inyección, la piel donde vaya a aplicarse deberá limpiarse con un germicida adecuado. Administre el volumen total de 0,5 ml por vía intramuscular (IM). El lugar de inyección preferible es el músculo deltoides. CONTRAINDICACIONES: Hipersensibilidad. La vacunación está contraindicada en personas con antecedentes de reacción sistémica de hipersensibilidad a cualquiera de los componentes de ADACEL® o de reacción potencialmente mortal tras una administración previa de la vacuna o de una vacuna que contenga uno o varios de los componentes de ADACEL®. (Véase FORMAS FARMACÉUTICAS, COMPOSICIÓN Y ENVASADO.) Dada la incertidumbre sobre qué componente de la vacuna puede ser el causante, no debe administrarse ninguno de los componentes. Si se está considerando la posibilidad de administrar más vacunas, se puede remitir a dichas personas a un alergólogo para que realice una evaluación. Trastornos neurológicos agudos. La administración de cualquier vacuna antipertussis, incluida ADACEL®, está contraindicada en casos de encefalopatía (p.ej., coma, disminución del grado de consciencia, convulsiones prolongadas) de etiología desconocida que haya aparecido en los 7 días posteriores a la administración de una vacuna antipertussis. ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO Generalidades. Antes de administrar ADACEL®, los profesionales de la salud deberán informar al receptor o al padre, madre o tutor de éste sobre los beneficios y los riesgos de la vacunación, preguntar sobre el estado de salud reciente del receptor, repasar sus antecedentes en busca de posibles casos de hipersensibilidad a la vacuna o a vacunas similares, revisar los antecedentes de vacunaciones, descartar la presencia de cualquier contraindicación a la vacunación y cumplir con todos los requisitos locales relativos a la información que se haya de dar al receptor o al tutor antes de la vacunación. Es sumamente importante preguntar al receptor, al padre, la madre o el tutor por cualquier síntoma o signo de reacción adversa que pueda haberse presentado después de una dosis previa de la vacuna. (Véanse CONTRAINDICACIONES y REACCIONES ADVERSAS.) En las tasas y la intensidad de los episodios adversos que aparecen en los receptores del toxoide tetánico influyen la cantidad de dosis previas y el nivel de antitoxinas preexistentes. Como ocurre con cualquier vacuna, ADACEL® puede no proteger al 100% de las personas vacunadas. Precauciones relacionadas con la vía de administración: No administre ADACEL® por inyección intravascular: asegúrese de que la aguja no penetre en un vaso sanguíneo. La vacuna no debe administrarse por vía intradérmica ni subcutánea. ADACEL® no debe administrarse en las nalgas. Enfermedades febriles o agudas: En casos de enfermedad aguda o febril, deberá posponerse la vacunación, pero, en general, la presencia de enfermedades con febrícula no debe ser motivo para posponer la vacunación. Alteraciones hematológicas. Debido a que toda inyección intramuscular puede causar un hematoma en el lugar de la aplicación en personas con trastornos hemorrágicos (como la hemofilia o la trombocitopenia) o en personas sometidas a tratamiento anticoagulante, no deben administrarse inyecciones intramusculares de ADACEL® a dichas personas a menos que los posibles beneficios sean mayores que el riesgo de la administración. Si se decide administrar a dichas personas cualquier producto por vía intramuscular, deberá hacerse con precaución y tomando las medidas necesarias para evitar el riesgo de que se formen hematomas después de la inyección. 32 Alteraciones inmunitarias. Se deberá evaluar la posibilidad de que aparezcan reacciones alérgicas en personas sensibles a los componentes de la vacuna. Tras la administración de ADACEL® pueden presentarse reacciones de hipersensibilidad, incluso en personas sin antecedentes de hipersensibilidad a los componentes del producto. Como sucede con otros productos, se deberán tener a mano una solución de clorhidrato de epinefrina (1:1.000) y otros fármacos apropiados para su uso inmediato si se produjeran reacciones anafilácticas o de hipersensibilidad aguda. Los profesionales de la salud deben estar familiarizados con las recomendaciones vigentes sobre el tratamiento inicial de la anafilaxis en entornos no hospitalarios, incluido el manejo adecuado de las vías respiratorias. En las personas inmunocomprometidas (ya sea por enfermedad o por tratamiento) puede no obtenerse la respuesta inmunitaria esperada. Si es factible, deberá considerarse la posibilidad de demorar la vacunación hasta que hayan concluido los tratamientos inmunosupresores. No obstante, en las personas con inmunodeficiencia crónica (p.ej., en casos de infección por VIH) se recomienda la vacunación, aunque la formación de anticuerpos podría ser limitada. Alteraciones neurológicas. No debe administrarse ADACEL® a personas con trastornos neurológicos progresivos o inestables, epilepsia incontrolada o encefalopatía progresiva, hasta que se haya establecido un régimen de tratamiento, se haya estabilizado la afección y los beneficios sean claramente mayores que los riesgos. Un estudio realizado por el Instituto de Medicina (IOM) de los Estados Unidos descubrió pruebas de una relación causal entre el toxoide tetánico y la neuritis braquial y el síndrome de Guillain-Barré (SGB). Cuando se haya presentado un SGB dentro de las 6 semanas posteriores a la administración previa de una vacuna que contenga toxoide tetánico, la decisión de administrar ADACEL® o cualquier vacuna que contenga toxoide tetánico deberá basarse en un análisis minucioso de los posibles beneficios y riesgos. Se han notificado algunos casos de enfermedades desmielinizantes del sistema nervioso central,mononeuropatías periféricas y mononeuropatías de pares craneales tras la administración de vacunas que contenían toxoide tetánico y/o diftérico, aunque el IOM llegó a la conclusión de que los datos no eran adecuados para confirmar o descartar la existencia de una relación causal entre esos trastornos y la vacunación. Mujeres embarazadas. No se ha evaluado el efecto de ADACEL® en el desarrollo del embrión y del feto. No se recomienda la vacunación durante el embarazo a menos que exista un riesgo seguro de contraer tos ferina. Dado que la vacuna está inactivada, el riesgo para el embrión o el feto es improbable. Deberán evaluarse minuciosamente los beneficios frente a los riesgos de administrar ADACEL® durante el embarazo cuando haya un probable riesgo de exposición a un contacto familiar o durante un brote en la comunidad. Madres lactantes. No se ha evaluado el efecto de la administración de ADACEL® durante la lactancia. Ya que ADACEL® está inactivada, es improbable cualquier riesgo para la madre o el lactante. Sin embargo, no se ha estudiado el efecto sobre los lactantes de la administración de ADACEL® a sus madres. Deben evaluarse los riesgos y los beneficios de la vacunación antes de tomar una decisión con respecto a la vacunación de una madre lactante. INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN Interacciones de la vacuna con otros medicamentos. Los tratamientos inmunosupresores pueden interferir en el desarrollo de la respuesta inmunitaria esperada. (Véase ADVERTENCIAS Y PRECAUCIONES.) Administración simultánea de otras vacunas ADACEL® puede administrarse en adolescentes de 11 a 12 años simultáneamente con una dosis de vacuna antigripal inactivada trivalente y con una dosis de vacuna contra la hepatitis B. El uso simultáneo de ADACEL® y de la vacuna antigripal inactivada trivalente fue evaluado en un ensayo clínico en el que participaron 696 adultos de edades comprendidas entre los 19 y los 64 años. Los perfiles de inocuidad e inmunogenicidad en los adultos que recibieron las vacunas simultáneamente fueron similares a los observados cuando las vacunas se administraron en distintas ocasiones con un mes de diferencia. El uso simultáneo de ADACEL® y de la vacuna contra la hepatitis B se evaluó en un ensayo clínico en el que participaron 269 adolescentes de 11 a 12 años. Los perfiles de inocuidad e inmunogenicidad en los adolescentes que recibieron las vacunas simultáneamente fueron similares a los observados cuando las vacunas se administraron en distintas ocasiones con un mes de diferencia. No se observó interferencia alguna en las respuestas inmunitarias de cualquiera de los antígenos de la vacuna cuando se administraron ADACEL® y la vacuna contra la hepatitis B simultáneamente o por separado. Las vacunas administradas simultáneamente deben inyectarse con jeringas distintas y en lugares diferentes, preferiblemente en distintas extremidades. ADACEL® no debe mezclarse en una misma jeringa con otros productos de administración parenteral. REACCIONES ADVERSAS: Reacciones adversas en los ensayos clínicos. Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de una vacuna no pueden compararse directamente con las obtenidas en los ensayos de otra vacuna y puede que no reflejen las tasas observadas en la práctica. Sin embargo, la información sobre reacciones adversas procedente de los ensayos clínicos sirve de base para identificar los episodios adversos que parecen estar relacionados con el uso de la vacuna y para estimar las tasas de dichos episodios. La inocuidad de ADACEL® se evaluó en un total de 5.818 sujetos que recibieron una sola dosis de ADACEL® en 6 ensayos clínicos (298 niños de 4 o más años de edad, 1.508 adolescentes, 2.842 adultos de menos de 65 años de edad y 1.170 adultos de 65 o más años de edad). La reacción más común en el lugar de la inyección (respuesta obtenida mediante interrogatorio) fue el dolor. La mayoría de las reacciones en el lugar de la inyección se produjeron en los 3 días siguientes a la vacunación y su duración media fue inferior a 3 días. La reacción sistémica más frecuente fue el cansancio en los niños y el dolor de cabeza en los adolescentes y adultos (18 a 64 años). La reacción sistémica informada con más frecuencia fue mialgia en adultos de 65 o más años de edad. Se indicó la presencia de fiebre en menos del 10% de los vacunados. Estas reacciones fueron por lo general transitorias y de intensidad leve a moderada. Además, entre los adolescentes y adultos, la incidencia de reacciones sistémicas y en el lugar de la inyección tras la administración de ADACEL® fue similar a la observada tras administrar una dosis de refuerzo de la vacuna Td. En los niños, la frecuencia de reacciones observadas en el lugar de la 33 inyección y de la presencia de fiebre tras la administración de ADACEL® fue considerablemente inferior a la observada tras administrar QUADRACEL® (DTaP-IPV) como dosis de refuerzo a niños de edades comprendidas entre los 4 y los 6 años. Excepto en el caso de la fiebre, las tasas de reacciones sistémicas observadas fueron similares en ambas vacunas. En la Tabla 1 se muestra la frecuencia de las reacciones sistémicas y en el lugar de la inyección (respuestas obtenidas mediante interrogatorio) en dos ensayos clínicos. Se informaron dos episodios adversos graves durante el estudio Td506 que se consideraron relacionados con la vacunación: un caso de migraña intensa con parálisis facial unilateral y un diagnóstico de compresión nerviosa en el cuello y el brazo izquierdo. Ambas afecciones se resolvieron de forma espontánea o con tratamiento. Tabla 1: Frecuencia (%) de reacciones (respuestas obtenidas mediante interrogatorio) observadas en ensayos clínicos con niños, adolescentes y adultos entre 0 y 14 días después de recibir una única dosis de ADACEL® * No se solicitó mediante interrogatorio. † Dolor corporal o debilidad muscular fue el término usado en el interrogatorio en los ensayos realizados en niños, adolescentes y adultos de 18 a 64 años de edad. ‡ Mialgia fue el término usado en el interrogatorio en el ensayo realizado en adultos de 65 o más años de edad. § Cansancio fue el término usado en el interrogatorio en los ensayos realizados en niños, adolescentes y adultos de 18 a 64 años de edad. ** Malestar fue el término usado en el interrogatorio en el ensayo realizado en adultos de 65 o más años de edad. Tabla 2: Frecuencia (%) de reacciones (respuestas obtenidas mediante interrogatorio) observadas en adolescentes y adultos tras una nueva administración de ADACEL® a los 5 y 10 años, respectivamente. * Reacciones adversas observadas entre 0 y 14 días después de la vacunación. † Reacciones adversas observadas entre 0 y 7 días después de la vacunación. Datos de la experiencia posterior a la comercialización: Durante el uso posterior a la comercialización de ADACEL® se han notificado espontáneamente otros episodios adversos, que se indican a continuación. Debido a que estos episodios se han informado de forma voluntaria y proceden de una población de tamaño indeterminado, no siempre es posible calcular con fiabilidad su frecuencia o establecer una relación causal con la exposición a la vacuna. Se decidió incluir estos episodios en el prospecto teniendo en cuenta uno o más de los factores siguientes: 1) gravedad del episodio, 2) frecuencia con que se comunicó, o 3) grado de relación causal con ADACEL®. Trastornos del sistema inmunitario: Reacción de hipersensibilidad (anafiláctica) (angioedema, edema, erupción cutánea, hipotensión) Trastornos del sistema nervioso: Parestesia, hipoestesia, síndrome de Guillain-Barré, neuritis braquial, parálisis facial, convulsiones, síncope, mielitis Trastornos cardíacos: Miocarditis Trastornos de la piel y el tejido subcutáneo: Prurito, urticaria Trastornos musculoesqueléticos y del tejido conjuntivo: Miositis, espasmo muscular 34 Trastornos generales y alteraciones en el lugar de administración Tras la administración de ADACEL® a adolescentes y adultos se han notificado reacciones extensas (>50 mm) en el lugar de la inyección, y la inflamación de la extremidad con extensión desde el lugar de la inyección hasta más allá de una o ambas articulaciones. Estas reacciones suelen comenzar dentro de las 24 a 72 horas posteriores a la vacunación, y pueden cursar con eritema, calor, sensibilidad al tacto o dolor en el lugar de la inyección, y se resuelven por sí solas en un plazo de 3 a 5 días. El riesgo parece depender de la cantidad de dosis previas de vacuna antipertussis acelular recibidas. Moretones y abscesos estériles en el lugar de la inyección. ACCIÓN Y FARMACOLOGÍA CLÍNICA Tétanos y difteria: el tétanos es una enfermedad aguda y con frecuencia mortal provocada por una neurotoxina extremadamente potente producida por el C. tetani. La toxina causa disfunción neuromuscular, y rigidez y espasmos de los músculos esqueléticos. La protección contra la enfermedad provocada por el C. tetani se debe al desarrollo de anticuerpos neutralizantes de la toxina tetánica. Se considera que un nivel sérico de antitoxina tetánica de al menos 0,01 UI/ml, según la medición del ensayo de neutralización, es el nivel de protección mínima. Asimismo, se considera que el nivel de antitoxina tetánica de al menos 0,1 UI/ml, según la medición del ensayo ELISA usado en estudios clínicos con ADACEL®, brinda protección contra el tétanos. Los niveles de 1,0 UI/ml se relacionaron con la protección prolongada. Las cepas de C. diphtheriae que producen la toxina diftérica pueden provocar enfermedades graves o mortales caracterizadas por la inflamación de la membrana de las vías respiratorias superiores y daño inducido por la toxina en el miocardio y el sistema nervioso. La protección contra la enfermedad provocada por la C. diphtheriae se debe al desarrollo de anticuerpos neutralizantes de la toxina diftérica. El nivel sérico de antitoxina diftérica de 0,01 UI/ml es el nivel más bajo que brinda algún grado de protección. En general, se considera que los niveles de antitoxina de al menos 0,1 UI/ml brindan protección. Los niveles de 1,0 UI/ml se relacionaron con la protección prolongada. Tos Ferina o Coqueluche: la Tos ferina es una enfermedad respiratoria provocada por la B. pertussis. Este cocobacilo Gram negativo produce diversos componentes biológicamente activos, aunque su función en la patogénesis de la pertussis o en su inmunidad no se ha definido con claridad. No se comprende muy bien el mecanismo de protección contra la infección por B pertussis. Sin embargo, en un ensayo clínico realizado en Suecia (Ensayo de eficacia Sweden I), se ha demostrado que los mismos componentes de la pertussis presentes en ADACEL® (es decir, TP, HAF, PRN y FIM) previenen la tos ferina o coqueluche en lactantes, y su eficacia de protección es del 85,2%, según la definición de caso de la Organización Mundial de la Salud (OMS) (≥21 días consecutivos de accesos de tos paroxística con confirmación por serología o cultivo, o relación epidemiológica con un caso confirmado). En el mismo estudio, la eficacia de la protección contra la enfermedad leve fue del 77,9%. Un estudio de contacto con la enfermedad en el hogar incluido en este ensayo de eficacia mostró que hubo correlaciones estadísticamente significativas entra la protección clínica y la presencia de anticuerpos contra la TP, PRN y FIM en los sueros previos a la exposición. No se han identificado los niveles séricos mínimos de anticuerpos contra componentes específicos de la vacuna contra B. pertussis que brinden protección contra el desarrollo de la tos ferina clínica. Sin embargo, diversos estudios han demostrado que existe una correlación entre la presencia de respuestas de anticuerpos séricos contra los componentes de la vacuna contra la pertussis y la protección contra la enfermedad clínica. En los ensayos clínicos con ADACEL® realizados en niños, adolescentes y adultos menores de 65 años de edad, las Concentraciones Medias Geométricas (CMG), posteriores a la vacunación, de todos los anticuerpos contra la pertussis estuvieron sistemáticamente por encima de las observadas con TRIPACEL® en el Ensayo de eficacia Sweden I. Los adultos mayores (de 65 o más años de edad) que fueron vacunados con una dosis única de ADACEL® tuvieron CMG más bajas respecto de algunos de los anticuerpos contra la pertussis que las observadas en los lactantes que habían recibido 3 o 4 dosis de TRIPACEL®. Sin embargo, sus niveles de anticuerpos contra la pertussis posteriores a la inmunización fueron 4,4 a 15,1 veces superiores a los niveles observados antes de la inmunización, lo cual sugirió un mejor grado de protección contra la pertussis. Duración del efecto. El seguimiento prolongado de los niveles de anticuerpos séricos de los adolescentes y adultos que recibieron una dosis única de ADACEL® muestra que los niveles de protección de la antitoxina tetánica (≥0,01 UE/ml) y la antitoxina diftérica (≥0,01 UI/ml) persisten en el 99,2% y el 92,6% de los sujetos, respectivamente, después de 10 años de haber recibido la vacuna. Si bien los niveles de protección contra la pertussis todavía no se han definido con claridad, los niveles de anticuerpos contra la B. pertussis siguen siendo 2 a 9 veces superiores a los niveles observados antes de la inmunización al cabo de 5 años. Sin embargo, 10 años después de la vacunación, se observó que los niveles de anticuerpos contra la pertussis disminuyeron a los niveles registrados antes de la vacunación. Se recomienda administrar refuerzos de toxoides tetánico y diftérico cada 10 años. Los datos obtenidos a partir del seguimiento de serología y la readministración de ADACEL® sugieren que esta vacuna puede utilizarse en lugar de la vacuna de toxoides tetánico y diftérico para brindar refuerzo con intervalos de 10 años en adultos. CONSERVACIÓN: Consérvese a una temperatura de entre 2° y 8°C. No congelar. Desechar el producto si ha estado expuesto a la congelación. No usar después de la fecha de caducidad. ENVASADO: ADACEL® se suministra en viales monodosis de vidrio de 0,5 ml. Los viales son de vidrio de tipo 1. El sistema de cierre del envase de ADACEL® no contiene látex (goma natural). ADACEL® se presenta en envases de: 1 vial monodosis, 5 viales monodosis Fabricado por: Sanofi Pasteur Limited, Toronto, Ontario, Canadá Documento revisado y aprobado por ISP el 27 de Febrero de 2013 35 Virus Hepatitis A “Crear vacunas, es proteger la vida” 36 AVAXIM 80 Vacuna AVAXIM Pediátrico Inactivada antihepatitis A DENOMINACIÓN DEL MEDICAMENTO: AVAXIM pediátrico vacuna inactivada antihepatitis A, suspensión inyectable 80 U. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Virus de la hepatitis A (cepa GBM)* inactivado** 80 U***.Para una dosis de 0,5 mL. * cultivada sobre células diploides humanas MRC-5. ** adsorbido en hidróxido de aluminio (cantidad que corresponde a 0,15 mg de aluminio). *** unidades antigénicas medidas según el método interno del fabricante. FORMA FARMACEUTICA: Suspensión inyectable en jeringa precargada. Suspensión inyectable en frasco multidosis. DATOS CLINICOS: Indicaciones terapéuticas: AVAXIM 80 U pediátrico está indicado para la inmunización activa contra la infección causada por el virus de la hepatitis A en el niño de 12 meses de edad hasta los 15 años, inclusive. La transmisión del virus de la hepatitis A se realiza en general por ingestión de agua o alimentos contaminados. Las personas en contacto con individuos contaminados se infectan en general por vía oro – fecal. También ha sido demostrada la posibilidad de transmisión por vía sanguínea o sexual (relaciones oro – anales). Posología y forma de administración. Posología: La primovacunación queda garantizada por una sola dosis de vacuna. La dosis recomendada es de 0,5 mL por cada inyección. Para asegurar una protección a largo plazo, está recomendado administrar una dosis de refuerzo 6 meses después de la primera inyección. La persistencia de los anticuerpos anti – VHA después de la vacunación no se conoce. Los datos disponibles sugieren la persistencia de anticuerpos anti-VHA a un nivel protector hasta los 10 años después de la primo – inmunización. Método de administración: Se tiene que inyectar la vacuna por vía intramuscular en el músculo deltoides. Excepcionalmente, la vacuna puede ser administrada por vía subcutánea a pacientes con trombocitopenia o con riesgo de hemorragia. Contraindicaciones: Contraindicaciones habituales de toda vacunación: en caso de fiebre, enfermedad aguda o enfermedad crónica evolutiva, es preferible aplazar la vacunación. Hipersensibilidad a la sustancia activa, a uno de los excipientes, a la neomicina, al polisorbato o como consecuencia de una inyección anterior de esta vacuna. Advertencias y precauciones especiales de empleo: No inyectar por vía intravascular: asegurarse de que la aguja no penetre en un vaso sanguíneo. La vacuna no debe inyectarse en el músculo glúteo debido a la variabilidad de este sitio anatómico (presencia más o menos importante de tejido adiposo) ni por vía intradérmica, ya que estos modos de administración pueden inducir una menor respuesta inmunitaria. Como en el caso de cualquier vacuna inyectable, se recomienda disponer un tratamiento médico apropiado después de la administración de la vacuna en caso de una eventual reacción anafiláctica. Un tratamiento inmunosupresor o un estado de inmunodeficiencia pueden inducir una disminución de la respuesta inmunitaria a la vacuna. Se recomienda por lo tanto esperar el fin de tratamiento para vacunar o verificar la adecuada protección de la persona. Sin embargo, se recomienda la vacunación de personas que presentan inmunodepresión crónica, tal como una infección con VIH, si la enfermedad subyacente permite una respuesta en anticuerpos, incluso limitada. Dado el tiempo de incubación de la enfermedad, es posible que la infección no reconocida se encuentre ya presente en el momento de la vacunación. En este caso, puede ocurrir que la vacunación no tenga efecto sobre el desarrollo de la hepatitis A. La utilización de esta vacuna en personas que tienen una afección hepática deberá ser considerada con atención, dado que no se ha efectuado hasta ahora ningún estudio en estos individuos. No existe ningún estudio concerniente a la administración de esta vacuna en pacientes que 37 padecen inmunodepresión. Esta vacuna no protege contra la infección causada por los virus de la hepatitis B, hepatitis C, hepatitis E o por otros agentes patógenos conocidos del hígado. Interacción con otros medicamentos y otras formas de interacción: Se puede practicar la administración simultánea de inmunoglobulinas con esta vacuna en dos sitios distintos. No se modifican las tasas de seroprotección, pero los títulos de anticuerpos pueden ser inferiores a los obtenidos cuando se administra esta vacuna sola. Esta vacuna se puede administrar simultáneamente, pero no debe ser mezclada con otras vacunas en la misma jeringa, y las otras vacunas tienen que ser administradas en sitios distintos con jeringas y agujas diferentes. Esta vacuna puede ser utilizada como refuerzo en personas que hayan sido primovacunadas con otra vacuna inactivada contra la hepatitis A. Embarazo y lactancia: No hay datos fiables de teratogénesis en animales. En clínica, no existen actualmente datos que permitan descartar un eventual efecto mal formativo o fototóxico de la vacuna contra la hepatitis A cuando se administra durante el embarazo. No utilizar esta vacuna durante el embarazo, excepto en caso de situación de riesgo inminente evaluada por el médico. La utilización de esta vacuna no se recomienda durante la lactancia. Efectos sobre la capacidad para conducir y utilizar máquinas: Es improbable que la vacunación produzca un efecto sobre la capacidad a conducir vehículos o utilizar máquinas. Reacciones adversas: Durante el desarrollo clínico, más de 3.500 niños de 12 meses a 15 años de edad fueron vacunados con esta vacuna (unas 7.000 dosis fueron administradas). Todas las reacciones adversas se revelan moderadas y limitadas a los primeros días después de la vacunación con regresión espontánea. Las reacciones han sido más raramente reportadas después de la administración de la dosis de refuerzo que después de la primera dosis. Sin embargo, como para toda especialidad farmacéutica, puede que las reacciones adversas más raras sean comunicadas durante una utilización más amplia de la vacuna. Las reacciones más frecuentes con una incidencia de 1 a 10% son reacciones locales al punto de inyección, tal como dolor, enrojecimiento, edema o induración y reacciones generales como cefalea, trastornos gastrointestinales (dolor abdominal, diarrea, náusea, vómitos), mialgias o artralgias, trastornos transitorios del comportamiento (disminución del apetito, insomnio, irritabilidad), fiebre, astenia. Las reacciones menos frecuentes con una incidencia inferior a 1% son manifestaciones cutáneas (rash, urticaria). En raras ocasiones se ha observado elevaciones leves de las transaminasas. Sobredosis: Parece improbable que una sobredosis provoque un efecto nocivo. PROPIEDADES FARMACOLOGICAS: Propiedades farmacodinámicas: VACUNA CONTRA LA HEPATITIS A (J: Antiinfeccioso). Esta vacuna se prepara a partir del virus de la hepatitis A cultivado, purificado e inactivado con formaldehído. Confiere inmunidad frente al virus de la hepatitis A, induciendo títulos de anticuerpos más durables y más elevados que los obtenidos después de una inmunización pasiva con inmunoglobulinas. Esta vacuna confiere títulos de anticuerpos protectores frente al virus de la hepatitis A (≥20 MUI/mL) en las dos semanas siguientes a la inyección en más de 95% de los sujetos y en 100% de los sujetos antes de la administración de la dosis de refuerzo. La inmunidad persiste 6 meses y está reforzada por una dosis de refuerzo. Los datos de persistencia a largo plazo de los anticuerpos del virus de la hepatitis A después de una dosis de refuerzo, no están aún disponibles. Sin embargo, los títulos de anticuerpos obtenidos después del primer refuerzo son compatibles con una protección a largo plazo (al menos 10 años). Propiedades farmacocinéticas: Sin objeto. Datos preclínicos sobre seguridad: Sin objeto. DATOS FARMACÉUTICOS. Lista de excipientes: 2-Fenoxietanol, formaldehído, hidróxido de aluminio, medio 199 Hanks que contiene principalmente una combinación compleja de aminoácidos, polisorbato 80, de sales minerales y de vitaminas, ácido clorhídrico o hidróxido de sodio para el ajuste del pH entre 6,8 y 7,8 y agua para preparados inyectables. Incompatibilidades. Por falta de estudio de compatibilidad, no debe mezclar esta vacuna con otros medicamentos. Período de validez: 3 años. Precauciones especiales de conservación. Presentación monodosis. Presentación multidosis. Conservar entre +2 ºC y +8 ºC (en el refrigerador) y al abrigo de la luz. No congelar. Una vez abierto, se recomienda utilizar la vacuna inmediatamente. Naturaleza y contenido del recipiente: Presentación monodosis. 0,5 mL de suspensión en jeringa precargada (vidrio tipo 1) con o sin aguja – caja de 1, de 10, ó de 20. Presentación multidosis. 5 mL de suspensión en frasco (vidrio tipo 1) provista de un tapón (clorobutilo) – caja de 1, 10 ó 20 frascos de 10 dosis. 10 mL de suspensión en frasco (vidrio tipo 1) provista de un tapón (clorobulilo) – caja de 1, 10 ó 20 frascos de 20 dosis. Instrucciones de uso, manipulación y eliminación: Agitar antes de inyectar para obtener una suspensión homogénea. Documento revisado y actualizado febrero de 2013 38 AVAXIM 160 U Vacuna inactivada contra la hepatitis A, adsorbida Suspensión inyectable (IM) en jeringa precargada 1.NOMBRE DEL MEDICAMENTO AVAXIM 160U, suspensión inyectable en jeringa precargada Vacuna contra la hepatitis A (inactivada, adsorbida) 2.COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Una dosis (0,5 mL) contiene: Virus de la hepatitis A, cepa GBM* (inactivado**) 160 unidades*** *cultivado sobre células diploides humanas MRC-5. **adsorbido en hidróxido de aluminio (cantidad equivalente a 0,3 mg de aluminio). *** unidades antigénicas expresadas mediante una referencia interna del fabricante. Para la lista de excipientes, ver sección 6.1. 3.FORMA FARMACÉUTICA Suspensión inyectable en jeringa precargada. 4.DATOS CLÍNICOS 4.1Indicaciones terapéuticas - Esta vacuna está indicada para la inmunización activa contra la infección causada por el virus de la hepatitis A en adolescentes a partir de los 16 años y adultos. - La vacuna no protege contra la infección provocada por el virus de la hepatitis B, de la hepatitis C, de la hepatitis E o por otros agentes patógenos conocidos del hígado. - La transmisión del virus de la hepatitis A se realiza en general por ingestión de agua o alimentos contaminados. Las personas en contacto con individuos contaminados se infectan en general por vía oro-fecal. - También ha sido demostrada la posibilidad de transmisión por vía sanguínea o sexual (relaciones oro-anales). - Esta vacuna debe ser administrada en conformidad con las recomendaciones oficiales. 4.2 Posología y modo de administración. Posología - La dosis recomendada por cada inyección es de 0,5 ml. - La protección inicial se obtiene tras una sola inyección. Para obtener una protección a largo plazo contra las infecciones causadas por el virus de la hepatitis A en adolescentes a partir de los 16 años y adultos, debe administrarse una dosis de refuerzo, preferentemente de 6 a 12 meses después de la primera vacunación, y puede hacerse hasta 36 meses después de la primera vacunación (ver rúbrica 5.1). Se calcula que los anticuerpos anti-VHA tiene una persistencia de varios años (al menos 10 años) tras la segunda dosis (refuerzo). Esta vacuna puede administrarse igualmente en dosis de refuerzo de la vacunación contra la hepatitis A en personas a partir de los 16 años de edad que hayan recibido una primera inyección con la vacuna combinada tifoidea (poliosídica Vi purificada) y hepatitis A (inactivada) de 6 a 36 meses antes. Forma de administración. Dado el carácter adsorbido de la vacuna, se recomienda administrarla por vía intramuscular (I.M.), con el fin de minimizar las reacciones locales. El sitio de inyección recomendado es la región deltoidea. 39 Excepcionalmente, se puede administrar la vacuna por vía subcutánea en pacientes con trombocitopenia o con riesgo de hemorragia. La vacuna no será administrada en el glúteo debido a la variabilidad de este sitio anatómico (presencia de más o menos importante de tejido adiposo), ni por vía intradérmica ya que estos modos de administración pueden dar lugar a una disminución de la respuesta inmunitaria. No inyectar por vía intravascular: asegurarse de que la vacuna no penetre en un vaso sanguíneo. Esta vacuna no debe ser mezclada con otras vacunas en la misma jeringa. 4.Contraindicaciones. Las contraindicaciones habituales de toda vacunación: en caso de fiebre, enfermedad aguda o enfermedad crónica evolutiva, es preferible deferir la vacunación. En caso de hipersensibilidad a cualquiera de los componentes de la vacuna o después de una inyección previa de la misma. 4.4 Advertencias y precauciones especiales de empleo - No inyectar por vía intravascular: asegurarse de que la aguja no penetre en un vaso sanguíneo. - Como en el caso de cualquier vacunación, se recomienda disponer de una solución de adrenalina inyectable en caso de una eventual reacción anafiláctica. - La vacuna no debe inyectarse en el músculo glúteo debido a la variabilidad de este sitio anatómico (presencia de más o menos importante de tejido adiposo), ni por vía intradérmica ya que estos modos de administración pueden dar lugar a una disminución de la respuesta inmunitaria. - Un tratamiento inmunosupresor o un estado de inmunodeficiencia pueden inducir una disminución de la respuesta inmunitaria a la vacuna. Se recomienda por lo tanto, esperar la finalización del tratamiento para vacunar o bien, asegurarse de la buena protección del sujeto. Sin embargo se recomienda la vacunación de sujetos que presentan inmunodepresión crónica, tales como los infectados por el VIH, cuando la enfermedad subyacente permita una respuesta de anticuerpos, así sea limitada. - Dado el tiempo de incubación de la enfermedad, es posible que la infección no reconocida se encuentre ya presente en el momento de la vacunación. En este caso, puede ocurrir que la vacunación no tenga efecto sobre el desarrollo de la hepatitis A. - La utilización de esta vacuna en sujetos que tienen afección hepática deberá ser considerada con atención, dado que no se ha efectuado hasta ahora ningún estudio en los sujetos. - Excepcionalmente, la vacuna puede administrarse por vía subcutánea en pacientes con trombocitopenia o con riesgo de hemorragia. - Dado que cada dosis contiene trazas de neomicina, es conveniente utilizar esta vacuna con precaución en las personas que presentan hipersensibilidad a este antibiótico. 4.5 Interacción con otros medicamentos y otras formas de interacción. Esta vacuna se puede administrar simultáneamente con inmunoglobulinas cuando se usen dos sitios diferentes para la inyección. Las tasas de seroprotección no se modifican, pero los títulos de anticuerpos pueden ser inferiores a los obtenidos cuando esta vacuna es administrada sola. Dado que esta vacuna está inactivada, la asociación con otra(s) vacuna(s) inactivada(s) utilizando un sitio de inyección diferente no provoca en general interferencia. Esta vacuna puede administrarse simultáneamente, pero en dos sitios separados, con una vacuna tifoidea polisacárida (Typhim Vi) o con una vacuna recombinante contra la hepatitis B obtenida por clonación y expresión del gen viral en las levaduras Saccharomyces cerevisiae, sin que la respuesta inmunitaria de los antígenos de una u otra sea modificada. Esta vacuna puede ser administrada simultáneamente, pero en dos sitios separados, con una vacuna viva estabilizada contra la fiebre amarilla. Esta vacuna puede ser utilizada como refuerzo en personas que hayan sido primovacunadas con otra vacuna inactivada contra la hepatitis A. 4.6 Embarazo y lactancia. Embarazo No hay datos fiables de teratogénesis en animales. Tampoco hay disponible información clínica suficientemente relevante que permita la evaluación de posibles malformaciones fetales o fetotoxicidad de la vacuna contra la hepatitis A cuando se administra durante el embarazo. Como medida de precaución, no se recomienda el uso de esta vacuna durante el embarazo, excepto cuando existe un riesgo elevado de contaminación. Lactancia. No existen datos sobre el efecto de la administración de AVAXIM 160U durante la lactancia, por lo que no se recomienda su uso en este periodo. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas. No procede. 4.8 Reacciones adversas. Las reacciones adversas informadas durante los estudios clínicos fueron generalmente leves, de corta duración y evolucionaron favorablemente sin tratamiento. - Reacciones locales en el sitio de inyección La reacción local más frecuente fue el dolor local asociado a veces con un eritema. La aparición de un nódulo en el sitio de la inyección ha sido observada en muy raras ocasiones. 40 - Reacciones generales Fiebre moderada, astenia, cefalea, mialgias o artralgias y trastornos gastrointestinales han sido informados con más frecuencia. En raras ocasiones ha sido observada una elevación leve y reversible de las transaminasas séricas. Excepcionalmente se han observado reacciones locales tales como prurito, rash o urticaria. Las reacciones fueron menos frecuentes después de la dosis de refuerzo que después de la primera dosis. Esta vacuna es igualmente bien tolerada tanto por individuos seropositivos contra el VHA, como por individuos seronegativos. 4.9 Sobredosis . No procede. 5.PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Vacuna contra la hepatitis A Esta vacuna se prepara a partir del virus de la hepatitis A cultivado, purificado y luego inactivado con formaldehído. Confiere inmunidad frente al virus de la hepatitis A, induciendo una respuesta de anticuerpos superiores a la obtenida después de la inmunización pasiva con inmunoglobulinas. La inmunidad aparece poco después de la primera inyección, y 14 días después de la vacunación más del 90% de los sujetos inmunocompetentes presentan una seroconversión con títulos protectores (título superior a 20 mUI/ml). Un mes después de la primera inyección, casi 100% de los sujetos se encuentran protegidos. La inmunidad puede persistir hasta el mes 36 y se incrementa después de una inyección de refuerzo. 5.2 Propiedades farmacocinéticas. No procede. 5.3 Datos de seguridad preclínicas. No procede. 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Fenoxietanol, formaldehído, medio 199 Hanks (contiene principalmente una mezcla compleja de aminoácidos, sales minerales, vitaminas, ácido clorhídrico o hidróxido de sodio para ajuste del pH y agua para preparaciones inyectables). 6.2 Incompatibilidades. En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos. 6.3 Periodo de validez. 3 años. 6.4 Precauciones especiales de conservación Conservar en refrigerador (entre 2°C y 8°C). No congelar. 6.5 Naturaleza y contenido del envase externo 0,5 ml de suspensión en jeringa precargada (vidrio tipo I), provista de un tapón émbolo (clorobromobutilo). 6.6 Instrucciones de uso y de manipulación Agitar antes de la inyección, hasta tener una suspensión homogénea. En las jeringas sin aguja unida, la aguja separada se debe montar firmemente en la jeringa efectuando una rotación de un cuarto de vuelta. La eliminación de los productos no utilizados o de los desechos se establecerá de acuerdo con las regulaciones vigentes. 7.TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN Sanofi Pasteur S.A. 2, avenue Pont Pasteur 69007 Lyon – Francia 41 Difteria Bacteria Tétanos Bacteria Pertusiss Bacteria Haemophilus Influenzae tipo b Bacteria Poliomielitis Virus Hepatitis A Virus “Crear vacunas, es proteger la vida” 42 HEXAXIM Vacuna Hexavalente Suspensión inyectable adsorbida Vacuna contra la difteria, el tétanos, la tos ferina (acelular, compuesta), la hepatitis B (ADN recombinante), la poliomielitis (inactivada), y Haemophilus influenzae de tipo b (conjugada), adsorbida. 1. NOMBRE DEL MEDICAMENTO Hexaxim, suspensión inyectable en jeringa prellenada o en vial. Vacuna contra la difteria, el tétanos, la tos ferina (acelular, compuesta), la hepatitis B (ADN recombinante), la poliomielitis (inactivada), y Haemophilus influenzae de tipo b (conjugada), adsorbida. 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Una dosis1 (0,5 ml) contiene: Polisacárido de Haemophilus influenzae tipo b 12 microgramos conjugado con la proteína tetánica *Antígeno de superficie recombinante del virus de la hepatitis B, DNAr 10 microgramos 25 microgramos Hemaglutinina filamentosa Toxoide pertúsico 25 microgramos no menos de 20 UI (30Lf) Toxoide diftérico Toxoide tetánico no menos de 40 UI (10Lf) **Poliovirus Tipo 1 (Mahoney), inactivado 40 UD*** 8 UD*** ** Poliovirus Tipo 2 (MEF-1), inactivado ** Poliovirus Tipo 3 (Saukett), inactivado 32 UD*** 1 Adsorbido en hidróxido de aluminio, dihidratado (0,6 mg Al 3+) * Obtenido por técnica de ADN recombinante en células de levadura Hansenula polymorpha ** Obtenido en células Vero *** Unidad antigénica D La vacuna puede contener restos de glutaraldehído, formaldehído, neomicina, estreptomicina y polimixina B que se utilizan durante el proceso de fabricación (ver sección 4.3). Para consultar la lista completa de excipientes, ver sección 6.1. 3. Forma farmacéutica Suspensión inyectable en jeringa prellenada o en vial. Hexaxim es una suspensión turbia blancuzca. 4. DATOS clínicos 4.1 Indicaciones terapéuticas Hexaxim está indicada para la primovacunación y la vacunación de refuerzo en lactantes y niños pequeños desde las 43 6 semanas hasta 24 meses de edad contra la difteria, tétanos, tos ferina, hepatitis B, poliomielitis y las enfermedades invasivas causadas por Haemophilus influenzae tipo b. El uso de esta vacuna debe ser según las recomendaciones oficiales. 4.2 Posología y forma de administración Posología Vacunación primaria: La vacunación primaria consiste en tres dosis de 0,5 ml para administrarse en intervalos de al menos cuatro semanas según el esquema 6, 10, 14 semanas; 2, 3, 4 meses; 3, 4, 5 meses; 2, 4, 6 meses. Todos los esquemas de vacunación, incluyendo el del Programa Ampliado de Inmunización (PAI) de la OMS a las 6, 10, 14 semanas de edad pueden aplicarse independientemente de si se ha administrado o no una dosis de vacuna contra la hepatitis B al nacimiento. Cuando se ha administrado una dosis de vacuna contra la hepatitis B al nacimiento, Hexaxim se puede utilizar como dosis adicional de vacuna contra la hepatitis B a partir de las seis semanas de edad. Si se requiere una segunda dosis de vacuna contra la hepatitis B antes de esta edad, se debe utilizar una vacuna monovalente contra la hepatitis B. El uso de esta vacuna debe ser según las recomendaciones oficiales. Vacunación de refuerzo: Después de las tres dosis de vacunación primaria con Hexaxim, debe administrarse una dosis de refuerzo preferentemente durante el segundo año de vida, al menos 6 meses después de la última dosis de primovacunación. Las dosis de refuerzo se deben administrar según las recomendaciones oficiales. Debe administrarse al menos, una dosis contra Hib. Después de las tres dosis de vacunación primaria con Hexaxim (2, 3, 4 meses; 3, 4, 5 meses; 2, 4, 6 meses) y en ausencia de la vacuna contra la hepatitis B al nacimiento, es necesario administrar una dosis de refuerzo de una vacuna contra la hepatitis B. Hexaxim puede utilizarse como dosis de refuerzo. Después de las tres dosis del esquema de vacunación del PAI de la OMS con Hexaxim (6, 10, 14 semanas) y en ausencia de la vacuna contra la hepatitis B al nacimiento, debe administrarse una dosis de refuerzo de la vacuna contra la hepatitis B. Debería administrarse al menos, una dosis de refuerzo de la vacuna contra la poliomielitis. Hexaxim puede utilizarse como dosis de refuerzo. Cuando se administra una vacuna contra la hepatitis B en el nacimiento, Hexaxim o una vacuna pentavalente DTaP-IPV/ Hib se puede administrar como dosis de refuerzo después de tres dosis de vacunación primaria. Hexaxim puede utilizarse como dosis de refuerzo en individuos que hayan sido vacunados previamente con otra vacuna hexavalente o una vacuna pentavalente DTaP-IPV/Hib asociada con una vacuna monovalente contra la hepatitis B. Otra población pediátrica No se ha establecido la seguridad y eficacia de Hexaxim en niños mayores de 24 meses de edad. Forma de administración La vacunación debe realizarse por vía intramuscular (IM). Los lugares de inyección recomendados son preferentemente el área anterolateral de la parte superior del muslo y músculo deltoides en niños mayores (posiblemente a partir de los 15 meses de edad). Para consultar las instrucciones de manipulación del medicamento antes de la administración, ver sección 6.6. 4.3 Contraindicaciones Antecedente de reacción anafiláctica después de la administración previa de Hexaxim. Hipersensibilidad a los principios activos, a alguno de los excipientes incluidos en la sección 6.1, a residuos en cantidades de trazas (glutaraldehído, formaldehído, neomicina, estreptomicina y polimixina B) o a la vacuna contra la tos ferina o tras una administración previa de Hexaxim o una vacuna que contenga los mismos componentes. La vacunación con Hexaxim está contraindicada si el individuo ha experimentado una encefalopatía de etiología desconocida, en los 7 días posteriores a una vacunación previa con una vacuna que contenga el componente pertúsico (vacuna contra la tos ferina de células completas o acelular). En estas circunstancias la vacunación contra la tos ferina debe suspenderse y la serie de vacunación debe continuarse con vacunas contra difteria, tétanos, hepatitis B, poliomielitis y Hib. No deberían administrarse vacunas contra la tos ferina a individuos con trastorno neurológico no controlado o epilepsia no controlada hasta que se haya establecido el tratamiento contra la afección, y el beneficio sea evidentemente mayor que el riesgo. 4.4 Advertencias y precauciones especiales de empleo Hexaxim no prevendrá la enfermedad causada por patógenos distintos a Corynebacterium diphtheriae, Clostridium tetani, Bordetella pertussis, virus de hepatitis B, poliovirus o Haemophilus influenzae tipo b. Sin embargo, se puede esperar que prevenga la hepatitis D (causada por el agente delta) no ocurre si no hay infección por hepatitis B. Hexaxim no protegerá contra una infección de hepatitis provocada por otros agentes como la hepatitis A, hepatitis C y hepatitis E o por otros patógenos del hígado. Debido al largo periodo de incubación de la hepatitis B, es posible que una infección no identificada de hepatitis B esté presente en el momento de la vacunación. En dichos casos, es posible que la vacuna no evite la infección por hepatitis B. Hexaxim no protege contra enfermedades infecciosas causadas por otros tipos de Haemophilus influenzae ni contra las meningitis de otro origen. Antes de la vacunación. La inmunización debe posponerse en individuos que padezcan enfermedad o infección febril aguda moderada a severa. La presencia de una infección leve y/o fiebre de bajo grado no debe dar lugar al aplazamiento 44 de la vacunación. La vacunación debe ir precedida por una revisión de la historia clínica del individuo (en particular las vacunaciones previas y posibles reacciones adversas). Se debe evaluar cuidadosamente la administración de la vacuna Hexaxim en individuos que tengan antecedentes de reacciones serias o graves en las 48 horas posteriores a la administración de una vacuna que contenga componentes similares. Antes de la inyección de cualquier producto biológico, la persona responsable de la administración debe tomar todas las precauciones conocidas para prevenir reacciones alérgicas o de cualquier otro tipo. Como todas las vacunas inyectables, deberá estar fácilmente disponible el tratamiento y la supervisión médica adecuados para su uso inmediato en el caso de que se produzca una reacción anafiláctica después de la administración de la vacuna. Si se conoce que alguno de los siguientes eventos adversos se ha producido tras la administración de una vacuna que contenga un componente pertusico, deberá evaluarse cuidadosamente la decisión de administrar dosis adicionales de vacunas que contengan dicho componente: • Fiebre ≥ 40º C en las siguientes 48 horas, no atribuible a otra causa identificable. • Síncope o estado que recuerde una situación de “shock” (episodio de hipotonía – hiporreactividad) en las 48 horas siguientes a la vacunación. • Llanto inconsolable, persistente ≥ 3 horas de duración, producido en las 48 horas siguientes a la vacunación. • Convulsiones con o sin fiebre, producidas en los 3 días siguientes a la vacunación. Puede haber algunas circunstancias, tales como una alta incidencia de la tos ferina, en las que los beneficios potenciales superen los posibles riesgos. Un antecedente de convulsiones febriles, antecedentes familiares de convulsiones o síndrome de muerte súbita del lactante (SMSL) no constituye una contraindicación para el uso de Hexaxim. Se debe realizar un seguimiento estrecho de los individuos que presenten un antecedente de convulsiones febriles ya que estos eventos adversos pueden ocurrir en los 2-3 días posteriores a la vacunación. Si se ha producido síndrome de Guillain Barré o neuritis braquial después de recibir una vacuna previa que contenga toxoide tetánico, la decisión de administrar cualquier vacuna que contenga toxoide tetánico deberá basarse en una cuidadosa evaluación de los beneficios potenciales y los posibles riesgos, como por ejemplo, si se ha completado el calendario primario de inmunización. Normalmente, la vacunación está justificada en individuos que no hayan completado la serie primaria (es decir, que han recibido menos de tres dosis). La inmunogenicidad de la vacuna puede verse reducida por un tratamiento inmunosupresor o una inmunodeficiencia. Se recomienda posponer la vacunación hasta el final de dicho tratamiento o enfermedad. No obstante, se recomienda la vacunación de individuos con una inmunodeficiencia crónica tal como una infección por el VIH, incluso la respuesta de anticuerpos puede ser limitada. Poblaciones especiales: No hay datos disponibles para lactantes prematuros. Sin embargo, podría observarse una respuesta inmunitaria menor y se desconoce el nivel de protección clínica. Las respuestas inmunitarias a la vacuna no se han estudiado en el contexto del polimorfismo genético. En los individuos con insuficiencia renal crónica, se observa una respuesta afectada contra la hepatitis B y se debe considerar la administración de dosis adicionales de la vacuna contra la hepatitis B según el nivel de anticuerpos contra el antígeno de superficie del virus de la hepatitis B (anti-HBsAg). Precauciones de empleo No administrar por vía intravascular, intradérmica o subcutánea. Como todas las vacunas inyectables, la vacuna debe administrarse con precaución en individuos con trombocitopenia o con trastornos hemorrágicos ya que puede producirse una hemorragia tras la administración intramuscular. Cuando se administre la serie de inmunización primaria en lactantes muy prematuros (nacidos con ≤ 28 semanas de gestación) y especialmente en aquellos con un antecedente de inmadurez respiratoria, debe evaluarse tanto el riesgo potencial de apnea como la necesidad de monitorización respiratoria durante 48 a 72 horas. Como el beneficio de la vacunación es alto en este grupo de lactantes, la vacunación no se debe impedir ni retrasar. Interferencia con pruebas de laboratorio Puesto que el antígeno polisacárido capsular de Hib se excreta en la orina, se puede observar una prueba en orina positiva entre 1 y 2 semanas después de la vacunación. Se deben realizar otras pruebas para confirmar la infección por Hib durante este período. 4.5 Interacción con otros medicamentos y otras formas de interacción Los datos relativos a la administración concomitante de Hexaxim con la vacuna conjugada de polisacáridos neumocócicos no han demostrado ninguna interferencia clínicamente relevante en la respuesta de anticuerpos a cada uno de los antígenos. Los datos relativos a la administración concomitante de una dosis de refuerzo de Hexaxim con la vacuna combinada contra sarampión-parotiditis y rubéola no han demostrado ninguna interferencia clínicamente relevante en la respuesta de anticuerpos a cada uno de los antígenos. Es posible que exista una interferencia clínicamente relevante en la respuesta de anticuerpos de Hexaxim y una vacuna contra la varicela, por lo tanto, estas vacunas no se deben administrar al mismo tiempo. Los datos relativos a la administración concomitante de vacunas contra el rotavirus no han demostrado ninguna interfe- 45 rencia clínicamente relevante en la respuesta de anticuerpos a cada uno de los antígenos. No hay datos disponibles relativos a la administración concomitante de Hexaxim con vacunas antimeningocócicas. En caso de administración concomitante con otras vacunas, se deben administrar en lugares de inyección separados. Hexaxim no debe mezclarse con otras vacunas u otros medicamentos administrados por vía parenteral. Excepto en el caso de terapia inmunosupresiva (ver sección 4.4), no se ha notificado interacciones clínicamente significativas con otros tratamientos o productos biológicos. nterferencia con las pruebas de laboratorio: ver sección 4.4. 4.6 Fertilidad, embarazo y lactancia No procede. Esta vacuna no está destinada para administración a mujeres en edad fértil. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas No procede. 4.8 Reacciones adversas a- Resumen del perfil de seguridad En estudios clínicos en individuos que recibieron Hexaxim, las reacciones más frecuentemente notificadas incluyen dolor en el lugar de la inyección, irritabilidad, llanto y eritema en el lugar de la inyección. Se ha observado una reactogenicidad ligeramente más alta después de la primera dosis en comparación con las subsiguientes dosis. b- Lista tabulada de reacciones adversas Se ha utilizado la siguiente convención para clasificar las reacciones adversas: (≥1/10) Muy frecuentes (≥1/100 a <1/10) Frecuentes Poco frecuentes (≥1/1.000 a <1/100) Raras (≥1/10.000 a <1/1000) Muy raras (<1/10.000) Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) Tabla 1: Eventos adversos en los estudios clínicos. c- Descripción de las reacciones adversas seleccionadas Hinchazón extensa de un miembro: Se han informado en niños reacciones extensas en el lugar de inyección (>50 mm), incluyendo hinchazón extensa de la extremidad que se extiende desde el lugar de la inyección hasta más allá de una o ambas articulaciones. Estas reacciones comienzan en las 24-72 horas posteriores a la vacunación y pueden estar asociadas con eritema, calor, sensibilidad y dolor en el lugar de inyección y remiten espontáneamente en el plazo de 3-5 días. El riesgo parece depender del número de dosis anteriores de vacunas que contengan tos ferina acelular, con un mayor riesgo tras las dosis 4ª y 5ª. d- Reacciones adversas potenciales (es decir reacciones adversas que han sido informadas con otras vacunas que contenían uno o varios de los componentes o constituyentes de Hexaxim y no directamente con Hexaxim). Trastornos del sistema inmunológico - Reacción anafiláctica 46 Trastornos del sistema nervioso - Convulsiones con o sin fiebre - Se ha informado neuritis braquial y Síndrome de Guillain-Barré después de la administración de una vacuna que contenga toxoide tetánico - Se ha notificado después de la vacunación con vacunas que contengan el antígeno de la hepatitis B neuropatía periférica (polirradiculoneuritis, parálisis facial), neuritis óptica, desmielinización del sistema nervioso central (esclerosis múltiple) - Encefalopatía/encefalitis Trastornos respiratorios, torácicos y mediastínicos Apnea en niños muy prematuros (≤28 semanas de gestación) (ver sección 4.4) Trastornos generales y alteraciones en el lugar de administración Una reacción edematosa simple o bilateral de los miembros inferiores puede ocurrir tras la inmunización con vacunas que contengan Haemophilus influenzae tipo b. Si esta reacción se produce, sucede principalmente después de las inyecciones primarias y se observa en las primeras horas siguientes a la vacunación. Los síntomas asociados pueden incluir cianosis, enrojecimiento, púrpura transitoria y llanto severo. Todos estos eventos desaparecen espontáneamente sin dejar secuelas en las 24 horas siguientes. 4.9 Sobredosis No se han informado casos de sobredosis. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Grupo farmacoterapéutico: Vacunas combinadas bacterianas y virales, código ATC: J07CA09. Los esquemas de vacunación primaria que han sido utilizados son: 6, 10, 14 semanas con y sin vacunación contra la hepatitis B al nacimiento; 2, 3, 4 meses sin vacunación contra la hepatitis B al nacimiento; 2, 4, 6 meses con y sin vacunación contra la hepatitis B al nacer. Los resultados obtenidos para cada uno de los componentes se resumen en las siguientes tablas: Tabla 2: Porcentaje de individuos con títulos de anticuerpos ≥ tasas* de seroprotección/seroconversión un mes después de la vacunación primaria con 3 dosis de Hexaxim * aceptable como correlaciones o sustitutos de protección † 6, 10, 14 semanas con y sin vacunación contra hepatitis B al nacimiento (República de Sudáfrica); 2, 3, 4 meses sin vacunación contra hepatitis B al nacimiento (Turquía); 2, 4, 6 meses sin vacunación contra hepatitis B al nacimiento (Argentina, México, Perú); 2, 4, 6 meses con vacunación contra hepatitis B al nacimiento (Costa Rica y Colombia) †† Número de individuos analizados (según el conjunto de análisis por protocolo) 47 Tabla 3: Porcentaje de individuos con títulos de anticuerpos ≥ tasas* de seroprotección/seroconversión un mes después del refuerzo con Hexaxim * Aceptable como correlaciones o sustitutos de protección † 6, 10, 14 semanas con y sin vacunación contra hepatitis B al nacimiento (República de Sudáfrica); 2, 3, 4 meses sin vacunación contra hepatitis B al nacimiento (Turquía); 2, 4, 6 meses sin vacunación contra hepatitis B al nacimiento (México) †† Número de individuos analizados (según el conjunto de análisis por protocolo) La eficacia de los antígenos pertúsicos acelulares (aP) contenidos en la vacuna Hexaxim contra la tos ferina típica definida por la OMS como más grave ( 21 días de tos paroxística) se ha documentado en un estudio aleatorizado, doble ciego en niños con una serie primaria de tres dosis utilizando una vacuna DTaP en un país altamente endémico (Senegal). En este estudio se observó la necesidad de administrar una dosis de refuerzo en niños pequeños. La capacidad a largo plazo de los antígenos pertúsicos acelulares (aP) que contiene Hexaxim ha demostrado que se reduce la incidencia de la tos ferina y se controla la enfermedad en un estudio nacional de vigilancia de la tos ferina en Suecia de 10 años de duración con la vacuna pentavalente DTaP-IPV/Hib utilizando un esquema de vacunación de 3, 5, 12 meses. Los resultados del seguimiento a largo plazo demostraron una reducción drástica de la incidencia de la tos ferina después de la segunda dosis, independientemente de la vacuna utilizada. Se ha demostrado la efectividad contra la enfermedad invasiva por Hib de las vacunas combinadas DTaP y Hib (pentavalentes y hexavalentes incluidas las vacunas que contienen antígenos contra Hib provenientes de Hexaxim) mediante un extenso estudio de vigilancia de la enfermedad posterior a la comercialización (periodo de seguimiento de más de 5 años) en Alemania. La efectividad de la vacuna fue de 96,7 % para las primovacunaciones completas y de 98,5% para las dosis de refuerzo (independientemente de la primovacunación). 5.2 Propiedades farmacocinéticas. No se han realizado estudios farmacocinéticos. 5.3 Datos preclínicos sobre seguridad. Los datos no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad a dosis repetidas y los estudios de tolerancia local. 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Fosfato disódico de hidrógeno dodecahidratado Dihidrogenofosfato de potasio Trometamol Sacarosa Aminoácidos esenciales Hidróxido de sodio y/o ácido acético glacial y/o ácido clorhídrico concentrado (para ajustar el pH) Agua para inyectables. Para el adsorbente: ver sección 2. 6.2 Incompatibilidades. En ausencia de estudios de compatibilidad, esta vacuna no debe mezclarse con otras vacunas o medicamentos. 6.3 Periodo de validez. 3 años. 6.4 Precauciones especiales de conservación. Conservar en refrigerador (entre 2°C y 8 C). No congelar. 48 Conservar el envase en el embalaje exterior para protegerlo de la luz. 6.5 Naturaleza y contenido del envase 0,5 ml de suspensión en jeringa prellenada (vidrio de tipo I) con tapón émbolo (halobutilo) y capuchón (halobutilo), sin aguja. 0,5 ml de suspensión en jeringa prellenada (vidrio de tipo I) con tapón émbolo (halobutilo) y capuchón (halobutilo), con 1 aguja separada. 0,5 ml de suspensión en jeringa prellenada (vidrio de tipo I) con tapón émbolo (halobutilo) y capuchón (halobutilo), con 2 agujas separadas. 0,5 ml de suspensión en vial (vidrio de tipo I) con un tapón (halobutilo) Tamaño de envase de 1 ó 10 ó 20 ó 50. Puede que solamente estén comercializados algunos tamaños de envases. 6.6 Precauciones especiales de eliminación y otras manipulaciones Antes de la administración, la jeringa prellenada o vial deberá agitarse para obtener una suspensión turbia blanquecina homogénea. Debe inspeccionarse visualmente la suspensión antes de su administración. En caso de detectar cualquier partícula extraña y/o variación del aspecto físico, desechar la jeringa prellenada o el vial. Para los viales se extrae una dosis de 0,5 ml utilizando una jeringa. Para las jeringas sin aguja acoplada, la aguja debe ajustarse firmemente sobre la jeringa, girándola un cuarto de vuelta. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. 7. TITULAR DE LA OPINIÓN CIENTÍFICA Sanofi Pasteur SA 2, avenue Pont Pasteur F-69007 Lyon Francia 49 Virus Poliomielitis “Crear vacunas, es proteger la vida” 50 IPV Vacuna Antipoliomielitica Inactivada FORMA FARMACÉUTICA Y FORMULACIÓN. Solución inyectable. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA. Una dosis (0,5 ml contiene): Virus poliomielítico tipo 1 (inactivado) 40 UD*† Virus poliomielítico tipo 2 (inactivado) 8 UD*† Virus poliomielítico tipo 3 (inactivado) 32 UD*† Esta vacuna está en conformidad con las especificaciones de la Farmacopea Europea y con las recomendaciones de la OMS. * UD: unidad antígeno D † O cantidad equivalente de antígenos, determinada según un método inmunoquímico apropiado. INDICACIONES TERAPÉUTICAS. Esta vacuna está indicada para la prevención de la poliomielitis en lactantes, el niño y el adulto, tanto en primovacunación como en refuerzo. POSOLOGIA Y FORMA DE ADMINISTRACION Posología Primovacunación: A partir de los 2 meses de edad, es conveniente administrar 3 dosis sucesivas de 0,5 ml con uno o dos meses de intervalo. En el adulto no vacunado, es conveniente administrar 2 dosis sucesivas de 0,5 ml con uno o, preferentemente, dos meses de intervalo. Refuerzo: En los niños en el transcurso del segundo año, se administra una 4ª dosis (1er refuerzo) un año después de la 3ª inyección. En el adulto, se administra una 3ª dosis (1er refuerzo) entre 8 y 12 meses después de la 2ª inyección. Para los refuerzos posteriores, se le aplica una inyección de refuerzo cada 5 años al niño y al adolescente, y cada 10 años al adulto. Forma de administración. La administración se realiza por vía intramuscular (vía recomendada), o por vía subcutánea. La inyección intramuscular se realizará preferentemente en la cara anterolateral del muslo en el niño pequeño y en el deltoides en el niño mayor, el adolescente y el adulto. CONTRAINDICACIONES La hipersensibilidad grave conocida a uno de los componentes de la vacuna, a una vacuna que contenga las mismas sustancias, a uno de los excipientes, a la neomicina, a la estreptomicina o a la polimixina B. Contraindicaciones transitorias típicas de cualquier vacunación: en caso de fiebre o de enfermedad aguda, es preferible aplazar la vacunación. ADVERTENCIAS Y PRECAUCIONES DE EMPLEO No inyectar por vía intravascular: asegurarse de que la aguja no penetre en un vaso sanguíneo. Al igual que con todas las vacunas inyectables, VACUNA ANTIPOLIOMIELÍTICA INACTIVADA debe administrarse con precaución en caso de trombocitopenia o de trastornos de coagulación debido a que la inyección intramuscular puede provocar una hemorragia en estos sujetos. Al igual que con todas las vacunas inyectables, se debe tener previsto inmediatamente un tratamiento médico oportuno 51 y debe realizarse una vigilancia en caso de aparición de una reacción anafiláctica (aunque esto ocurre raramente) que acontezca después de la administración de la vacuna. Un tratamiento inmunosupresor o un estado de inmunodeficiencia puede inducir una disminución de la respuesta inmunitaria a la vacuna. En este caso, se recomienda esperar al final del tratamiento para vacunar o asegurarse de la buena protección del sujeto. Sin embargo, se recomienda la vacunación de sujetos que presentan una inmunodepresión crónica, como una infección por VIH, si la enfermedad subyacente permite una respuesta de anticuerpos aunque ésta sea limitada. VACUNA ANTIPOLIOMIELÍTICA INACTIVADA puede recomendarse igualmente en sujetos para los cuales la vacuna oral está contraindicada, al igual que en refuerzo para los sujetos previamente con la vacuna oral. Debe tenerse muy en cuenta el riesgo potencial de apnea y la necesidad de vigilancia respiratoria durante 48-72 horas cuando se administren las dosis de primovacunación en los muy prematuros (nacidos < 28 semanas de embarazo) y particularmente aquellos que tienen antecedentes de inmadurez respiratoria. Debido al beneficio considerable de la vacunación en estos lactantes, la administración no debe suspenderse ni aplazarse. Interacciones con otros medicamentos y otras formas de interacción No hay inconveniente conocido en administrar VACUNA ANTIPOLIOMIELÍTICA INACTIVADA en el transcurso del mismo acto de vacunación, con otras vacunas habituales. En caso de administración concomitante, deben utilizarse jeringas diferentes y lugares de inyección separados. FARMACOCINÉTICA Y FARMACODINAMIA. VACUNA ANTIPOLIOMIELITICA INACTIVADA es una vacuna indicada para la prevención de la poliomielitis. Al compilar varios estudios clínicos llevados a cabo en lactantes, con un esquema de dos dosis, los porcentajes de seropositividad observados fueron de un rango entre 97.8% a 100% para el poliovirus tipo 1, de 98.4% a 100% para el poliovirus tipo 2 y de 93.5% a 100% para el poliovirus tipo 3. Estos resultados indican que se obtiene una respuesta de anticuerpos elevada con dos inyecciones del esquema primario y que la inmunogenicidad se refuerza después de la tercera inyección. Un estudio que midió los anticuerpos antipoliomielitis en niños previamente vacunados con VACUNA ANTIPOLIOMIELITICA INACTIVADA, con una media de edad de 11 años, y que habían sido vacunados hace algunos años (media de 5.4 años), mostró que 95.6% todavía presentaban anticuerpos protectores para el poliovirus tipo 1, 100% para el tipo 2 y 95.6% para el tipo 3. Después de una dosis de refuerzo, el 100% presentó niveles protectores contra los 3 tipos de poliovirus, con un incremento elevado en los títulos de anticuerpos (GMT). Varios ensayos clínicos en adultos han demostrado que la revacunación con VACUNA ANTIPOLIOMIELITICA INACTIVADA produce un nivel de anticuerpos protector en el 100% de los vacunados, contra los 3 tipos de poliovirus. Embarazo y lactancia Teniendo en cuenta los datos clínicos, esta vacuna puede prescribirse durante el embarazo, sólo en caso de necesidad. Esta vacuna puede ser utilizada durante la lactancia. Reacciones adversas Experiencia adquirida en el transcurso de los ensayos clínicos La reactogenicidad local fue evaluada en el transcurso de dos ensayos clínicos multicéntricos randomizados que incluyeron 395 pacientes en total. En el lugar de la inyección, las reacciones locales fueron informadas de manera frecuente a muy frecuente: enrojecimiento (entre 0,7 y 2,4% de los sujetos en cada ensayo), dolor (entre 0,7 y 34%), induración (0,4%). La incidencia y la gravedad de las reacciones locales pueden estar influenciadas por el lugar, la vía y la forma de inyección, así como por el número de inyecciones previamente recibidas. Durante un estudio multicéntrico randomizado de fase III que incluía 205 niños, se informaron casos de fiebre superior a 38,1ºC (en el 10% de los niños después de la primera dosis, en el 18% después de la segunda dosis y en el 7% después de la tercera dosis). Otras reacciones adversas advertidas después de la comercialización En base a los datos de declaraciones espontáneas, los siguientes acontecimientos se informaron muy raramente (< 0,01%), después de la comercialización. Sin embargo la frecuencia exacta no puede calcularse de una manera precisa. Teniendo en cuenta el calendario de vacunación para el niño, VACUNA ANTIPOLIOMIELÍTICA INACTIVADA raramente se inyecta sola. Trastornos generales y reacciones locales Pueden ocurrir reacciones locales en el lugar de la inyección, tales como edema, dentro de las 48 horas siguientes a la vacunación y persistir uno o dos días. Linfadenopatías. Trastornos del sistema inmunitario Reacción de hipersensibilidad tipo I a uno de los componentes de la vacuna, como urticarias, angioedemas, reacciones anafilácticas o choques anafilácticos. Músculo esquelético y tejido conjuntivo Se han informado artralgias moderadas y transitorias y mialgias en los días siguientes a la vacunación. Trastorno del sistema nervioso Convulsiones (asociadas o no a fiebre) en los días siguientes a la vacunación, cefaleas, parestesias moderadas y transitorias (principalmente de los miembros inferiores) que acontecen dentro de las dos semanas siguientes a la 52 vacunación. Trastornos psiquiátricos Agitación, somnolencia e irritabilidad en las primeras horas o en los días siguientes a la vacunación y que desaparecen rápidamente. Trastornos de la piel y del tejido subcutáneo Rash Apnea en los muy prematuros (nacidos < 28 semanas de embarazo) PROPIEDADES FARMACOLÓGICAS Propiedades farmacodinámicas La vacuna está preparada a partir de los virus poliomielíticos tipos 1, 2 y 3 cultivados en células Vero, purificados e inactivados con formaldehído. Un mes después de la primovacunación (3 dosis), los índices de seroprotección eran de 100% para las vacunas poliomielíticas de tipos 1 y 3 y del 99% al 100% para el tipo 2. En el lactante, la dosis de refuerzo (4ª dosis) provocó un aumento importante en los títulos con índices de seroprotección del 97.5% al 100% para los 3 tipos de virus poliomielíticos. Entre cuatro y cinco años después de la dosis de refuerzo, entre un 94% y un 99% de los sujetos tenían títulos protectores. En el adulto primovacunado, una inyección de refuerzo estuvo seguida de una respuesta anamnésica. Estos datos, proceden en su mayoría, de estudios realizados con vacunas combinadas que contienen las vacunas poliomielíticas. La inmunidad permanece durante al menos 5 años después de la 4ª inyección. DATOS FARMACÉUTICOS Lista de excipientes 2-fenoxietanol, formaldehído, medio 199 de Hanks, ácido clorhídrico o hidróxido de sodio para ajuste de pH. El 2-fenoxietanol se presenta en forma de una solución de 2-fenoxietanol al 50% en el etanol. El medio 199 de Hanks (sin rojo de fenol) es una mezcla compleja de aminoácidos (entre ellos la fenilalanina), de sales minerales, de vitaminas y de otros componentes (como la glucosa), suplementado con polisorbato 80, diluido en agua para preparaciones inyectables. Incompatibilidades En ausencia de estudios de compatibilidad esta vacuna no debe mezclarse con otras. Período de validez 30 meses Precauciones especiales de conservación Consérvese en el refrigerador (entre +2ºC y +8ºC) y al abrigo de la luz. No congelar. Instrucciones de uso y manipulación Comprobar el aspecto límpido e incoloro de la vacuna. No utilizar si el producto presenta un aspecto turbio. Para las jeringas sin aguja acoplada, la aguja debe montarse firmemente sobre la jeringa efectuando un movimiento rotatorio de un cuarto de vuelta. Documento revisado y basado en aprobación por ISP 53 Bacteria Infecciones por meningococo “Crear vacunas, es proteger la vida” 54 MENACTRA Vacuna Antimeningocóccica Polisacárida para serogrupos A, C, Y y W Conjugada con Toxoide Diftérico PARA INYECCIÓN INTRAMUSCULAR INDICACIONES Y USO Menactra®, la vacuna antimeningocóccica polisacárida para serogrupos A, C, Y y W-135 conjugada con toxoide diftérico está indicada para la inmunización activa para la prevención de la enfermedad meningocóccica invasiva causada por los serogrupos A, C, Y y W-135 de Neisseria meningitidis. Menactra está indicada para ser usada en personas de 9 meses a 55 años de edad. DOSIFICACIÓN Y ADMINISTRACIÓN La vacuna Menactra debe administrarse como una inyección única de 0,5 ml por vía intramuscular, preferentemente en la región del deltoides o del muslo anterolateral, según la edad y la masa muscular del individuo. En niños de 9 a 23 meses de edad, Menactra se administra en una serie de 2 dosis con una separación de al menos tres meses. Las personas de 2 a 55 años de edad reciben una dosis única. No administre este producto por vía intravenosa, subcutánea ni intradérmica. Aún no se ha determinado la necesidad, o el momento adecuado, de una dosis de refuerzo de la vacuna Menactra. Los medicamentos parenterales deben ser revisados de forma visual para verificar la integridad del recipiente, la presencia de partículas y la decoloración antes de la administración, cuando la solución o el recipiente lo permitan. CONTRAINDICACIONES Hipersensibilidad Reacción alérgica grave (p. ej., anafilaxia) luego de una dosis previa de una vacuna antimeningocóccica que contiene la proteína CRM197, toxoide diftérico o polisacáridos capsulares, o a cualquier componente de la vacuna Menactra (consulte DESCRIPCIÓN). Síndrome de Guillain-Barré. Tener antecedentes conocidos del síndrome de Guillain-Barré (SGB) es una contraindicación para la administración de la vacuna (consulte ADVERTENCIAS Y PRECAUCIONES). Enfermedad aguda o febril. Debe postergarse la vacunación en caso de padecer una enfermedad aguda o febril. No obstante, una enfermedad leve con o sin fiebre, como una infección leve de las vías respiratorias superiores, generalmente no es motivo para postergar la vacunación. Embarazo. Consulte la sección Embarazo. ADVERTENCIAS Y PRECAUCIONES Síndrome de Guillain-Barré Se informó la presencia del SGB en una relación temporal luego de la administración de la vacuna Menactra (consulte Reportes Post- Comercialización). Una evaluación temprana de los eventos adversos posteriores a la comercialización sugirió una posibilidad de que se produzca un mayor riesgo de padecer el SGB luego de la vacunación con Menactra. Sin embargo, en un estudio reciente de control de casos localizados y de cohorte, retrospectivo y multicéntrico, en el que participaban más de 12 millones de adolescentes, de los cuales 1,4 millones recibieron la vacuna Menactra, no se descubrió ninguna evidencia de un mayor riesgo de padecer el SGB en relación con el uso de la vacuna Menactra. No obstante, las personas con un diagnóstico previo de SGB no deben recibir la vacuna Menactra (consulte CONTRAINDICACIONES). Prevención y control de las reacciones alérgicas provocadas por la vacuna. Antes de la administración, el proveedor de atención médica debe revisar el historial de vacunación para detectar una 55 posible sensibilidad a la vacuna y reacciones adversas relacionadas con la vacunación anterior para poder evaluar los riesgos y beneficios. La epinefrina y otros agentes adecuados que se utilizan para controlar las reacciones alérgicas urgentes deben estar disponibles de forma inmediata en caso de que ocurra una reacción anafiláctica aguda. Trombocitopenia o trastornos hemorrágicos. No se ha evaluado la vacuna Menactra en personas con trombocitopenia o trastornos hemorrágicos. Al igual que con cualquier otra vacuna que se administra por vía intramuscular, debe evaluarse la relación entre los riesgos y beneficios de la vacuna para personas con riesgo de padecer una hemorragia luego de la inyección intramuscular. Alteración de la inmunocompetencia. Las personas inmunodeprimidas, incluidas las personas que reciben tratamiento inmunodepresor, pueden tener una menor respuesta inmunológica a la vacuna Menactra. Limitaciones de la eficacia de la vacuna. Es posible que la vacuna Menactra no proteja a todos los receptores contra los serogrupos de la vacuna. La vacuna Menactra no está indicada para la prevención de la meningitis causada por otros microorganismos o para la prevención de la enfermedad meningocóccica invasiva causada por el serogrupo B de N. meningitidis. REACCIONES ADVERSAS Reacciones adversas del estudio clínico Debido a que los estudios clínicos se realizan en condiciones que varían en gran medida, los índices de reacciones adversas observados en los estudios clínicos de una vacuna no pueden compararse directamente con los índices en los estudios clínicos de otra vacuna, y posiblemente no reflejen los índices observados en la práctica. Niños de 9 a 23 meses de edad. Se evaluó la seguridad de la vacuna Menactra en cuatro estudios clínicos en los que se incluyeron 3.721 participantes que recibieron la vacuna Menactra a los 9 y 12 meses de edad. A los 12 meses de edad, estos niños también recibieron una o más vacunas de otro tipo [Measles, Mumps, Rubella and Varicella Virus Vaccine Live (vacuna cuádruple vírica sarampión-rubéola-parotiditis-varicela) MMRV o Measles, Mumps, and Rubella (vacuna triple vírica sarampión-paroditis-rubéola) MMR y vacuna atenuada contra el virus de la varicela (V); vacuna conjugada antineumocócica heptavalente (proteína CRM197 diftérica) (PCV7); vacuna contra la hepatitis A (HepA)]. Se incluyó un grupo de control de 997 niños a los 12 meses de edad que recibió dos o más vacunas de la infancia [MMRV (o MMR + V), PCV7, HepA] a los 12 meses de edad (consulte ESTUDIOS CLÍNICOS: Administración de vacunas concomitantes). El tres por ciento de las personas recibió las vacunas MMR y V, en lugar de la vacuna MMRV, a los 12 meses de edad. El estudio de seguridad primario fue un estudio controlado en el que se incluyeron 1.256 niños que recibieron la vacuna Menactra a los 9 y 12 meses de edad. A los 12 meses de edad, estos niños recibieron las vacunas MMRV (o MMR + V), PCV7 y HepA. Un grupo de control de 522 niños recibió las vacunas MMRV, PCV7 y HepA. De los 1.778 niños, el 78% de los participantes (vacuna Menactra, N=1.056; grupo de control, N=322) se incluyeron en centros de los Estados Unidos (EE. UU.) y el 22% en un centro chileno (vacuna Menactra, N=200; grupo de control, N=200). Personas de 2 a 55 años de edad. Se evaluó la seguridad de la vacuna Menactra en ocho estudios clínicos en los que se incluyeron 10.057 participantes de 2 a 55 años de edad que recibieron la vacuna Menactra y 5.266 participantes que recibieron la vacuna Menomune – A/C/Y/W-135. Los tres estudios de seguridad primarios fueron estudios, aleatorizados, con un grupo control activo, en los que se incluyeron participantes de 2 a 10 años de edad (vacuna Menactra, N=1.713; vacuna Menomune – A/C/Y/W-135, N=1.519), de 11 a 18 años de edad (vacuna Menactra, N=2.270; vacuna Menomune – A/C/Y/W-135, N=972), y de 18 a 55 años de edad (vacuna Menactra, N=1.384; vacuna Menomune – A/C/Y/W-135, N=1.170), respectivamente. Eventos adversos serios en todos los estudios de seguridad. Se informaron eventos adversos serios (EAS) durante un período de seis meses posterior a las vacunaciones en personas de 9 meses a 55 años de edad. En niños que recibieron la vacuna Menactra a los 9 meses y a los 12 meses de edad, los EAS ocurrieron con un índice del 2,0% al 2,5%. En participantes que recibieron una o más vacunas de la infancia (sin la administración conjunta de la vacuna Menactra) a los 12 meses de edad, los EAS ocurrieron con un índice del 1,6% al 3,6%, según la cantidad y el tipo de vacunas recibidas. En niños de 2 a 10 años de edad, los EAS ocurrieron con un índice del 0,6% luego de la vacuna Menactra y con un índice del 0,7% luego de la vacuna Menomune – A/C/Y/W-135. En adolescentes de 11 a 18 años de edad y adultos de 18 a 55 años de edad, los EAS ocurrieron con un índice del 1,0% luego de la vacuna Menactra y con un índice del 1,3% luego de la vacuna Menomune – A/C/Y/W-135. Eventos adversos solicitados en los estudios de seguridad primarios. Las reacciones adversas solicitadas generales y en el lugar de la inyección que se reportaron con mayor frecuencia dentro de los siete días posteriores a la vacunación en niños de 9 meses a 12 meses de edad fueron irritabilidad y dolor con la palpación en el lugar de la inyección. Las reacciones adversas solicitadas generales y locales que se reportaron con mayor frecuencia en niños de 2 a 10 años de edad fueron dolor en el lugar de la inyección, irritabilidad, diarrea, somnolencia y anorexia. En adolescentes de 11 a 18 años y adultos de 18 a 55 años, las reacciones que se reportaron con mayor frecuencia fueron dolor en el lugar de la inyección, cefalea y fatiga. A excepción del enrojecimiento en adultos, las reacciones en el lugar de la inyección se informaron con mayor frecuencia luego de la administración de la vacuna Menactra que de la vacuna 56 Menomune – A/C/Y/W-135. Eventos adversos en estudios de vacunas concomitantes. Reacciones solicitadas generales y en el lugar de la inyección cuando se administra la vacuna Menactra con otras vacunas pediátricas. En el estudio de seguridad primario, se inscribieron 1.378 niños de EE.UU. para recibir la vacuna Menactra sola a los 9 meses de edad y la vacuna Menactra más una o más vacunas de otro tipo que se administran habitualmente (MMRV, PCV7 y HepA) a los 12 meses de edad (N=961). Otro grupo de niños recibió dos o más vacunas administradas (vacunas MMRV, PCV7 y HepA) (grupo de control, N=321) a los 12 meses de edad. Los participantes que recibieron la vacuna Menactra y las vacunas concomitantes a los 12 meses de edad antes mencionadas informaron dolor con la palpación, enrojecimiento e hinchazón en el lugar de la inyección de la vacuna Menactra y en los lugares de las inyecciones de vacunas concomitantes con frecuencias similares. El dolor con la palpación fue la reacción más frecuente en el lugar de la inyección (48%, 39%, 46% y 43% en los lugares de las vacunas Menactra, MMRV, PCV7 y HepA, respectivamente). La irritabilidad fue la reacción general más frecuente, reportada en el 62% de aquellos que recibieron la vacuna Menactra más las vacunas concomitantes, y el 65% del grupo de control. (Consulte ESTUDIOS CLÍNICOS: Administración de vacunas concomitantes). Reacciones solicitadas generales y en el lugar de la inyección cuando se administra la vacuna Menactra con la vacuna de toxoides tetánico y diftérico absorbidos (Td) El dolor en el lugar de la inyección se informó con más frecuencia luego de una vacuna Td que de una vacuna Menactra (71% frente a 53%). El índice global de eventos adversos generales fue superior cuando se administraron las vacunas Menactra y Td de manera concomitante que cuando se administró la vacuna Menactra 28 días después de la vacuna Td (59% frente a 36%). En ambos grupos, las reacciones más frecuentes fueron cefalea (vacuna Menactra + Td, 36%; Td + placebo, 34%; vacuna Menactra sola, 22%) y fatiga (vacuna Menactra + Td, 32%; Td + placebo, 29%; vacuna Menactra sola, 17%). Se produjo fiebre ≥40,0ºC en ­0,5% de todos los grupos. Reacciones solicitadas generales y en el lugar de la inyección cuando se administra la vacuna Menactra con la vacuna antitifoidea de polisacárido Vi. Más participantes experimentaron dolor luego de la vacuna antitifoidea que de la vacuna Menactra (antitifoidea + placebo, 76% frente a la vacuna Menactra + antitifoidea, 47%). La mayoría (del 70% al 77%) de las reacciones solicitadas en el lugar de la inyección para ambos grupos en cualquier lugar de la inyección se informó como reacciones de Grado 1 y se solucionó dentro de los tres días posteriores a la vacunación. En ambos grupos, la reacción general más frecuente fue cefalea (vacuna Menactra + antitifoidea, 41%; antitifoidea + placebo, 42%; vacuna Menactra sola, 33%) y fatiga (vacuna Menactra + antitifoidea, 38%; antitifoidea + placebo, 35%; vacuna Menactra sola, 27%). No se informó la presencia de fiebre ≥40,0ºC ni convulsiones en ningún grupo. Reportes Post-Comercialización. Además de los reportes en los estudios clínicos, a continuación se mencionan los reportes voluntarios de eventos adversos a nivel mundial que se recibieron desde la introducción de la vacuna Menactra en el mercado. Debido a que estos eventos fueron informados de manera voluntaria por parte de una población de un tamaño indeterminado, no siempre es posible calcular su frecuencia de un modo fiable o establecer una relación causal con la exposición a la vacuna Menactra. Trastornos en el sistema inmunitario: reacciones de hipersensibilidad como anafilaxia/reacción anafiláctica, sibilancias, dificultad para respirar, hinchazón de las vías respiratorias superiores, urticaria, eritema, prurito, hipotensión. Trastornos en el sistema nervioso: síndrome de Guillain-Barré, parestesia, síncope vasovagal, mareos, convulsiones, parálisis facial, encefalomielitis diseminada aguda, mielitis transversa. Trastornos en el sistema músculo-esquelético y tejido conjuntivo: mialgia INTERACCIONES MEDICAMENTOSAS Administración concomitante con otras vacunas La vacuna Menactra se administró de manera concomitante con la vacuna Typhim Vi® [vacuna antitifoidea de polisacárido vi] (antitifoidea) y la vacuna de toxoides tetánico y diftérico absorbidos, para uso en adultos (Td), en personas de 18 a 55 y de 11 a 17 años de edad, respectivamente. En niños menores de 2 años de edad, la vacuna Menactra se administró de manera conjunta con una o más de las siguientes vacunas: vacuna PCV7, MMR, V, MMRV, HepA o Hib (consulte ESTUDIOS CLÍNICOS y REACCIONES ADVERSAS). Luego de la administración conjunta de la vacuna Menactra y la PCV7, disminuyeron las respuestas de los anticuerpos neumocócicos a algunos serotipos en la PCV7. Dado los altos índices de respuesta de los anticuerpos mediante una evaluación con el método ELISA u OPA, es poco probable que se produzca algún impacto en la eficacia clínica de cualquiera de estas vacunas cuando se administran de manera concomitante (consulte ESTUDIOS CLÍNICOS: Administración de vacunas concomitantes). No mezcle la vacuna Menactra con otras vacunas en la misma jeringa. Cuando se administra la vacuna Menactra de manera concomitante con otras vacunas inyectables, las vacunas deben administrarse con jeringas diferentes y deben aplicarse en lugares separados. Los tratamientos i nmunodepresores, incluida la irradiación, los antimetabolitos, los agentes alquilantes, los medicamentos citotóxicos y los corticoesteroides (utilizados en dosis mayores a las fisiológicas) pueden reducir la respuesta inmunológica a las vacunas. 57 USO EN POBLACIONES ESPECÍFICAS Embarazo Los estudios sobre la reproducción animal no han demostrado un riesgo con respecto a los efectos en el embarazo y el desarrollo del embrión o el feto, en el parto y en el desarrollo posnatal. Sin embargo, debido a que no hay ningún dato sobre el uso de esta vacuna en mujeres embarazadas, la vacuna Menactra debe administrarse a una mujer embarazada únicamente si es claramente necesario, como por ejemplo, durante un brote o antes de un viaje necesario a un área endémica, y solamente luego de una evaluación de los riesgos y beneficios, en la que participen el profesional de atención médica y la paciente. Teniendo en cuenta la gravedad de la enfermedad meningocóccica, el embarazo no debe impedir la vacunación cuando el riesgo esté claramente identificado. Madres lactantes Se desconoce si la vacuna Menactra se excreta por la leche materna. Debido a que muchos medicamentos se excretan por la leche materna, debe tenerse cuidado cuando se administre la vacuna Menactra a una mujer lactando. Uso pediátrico No se ha establecido la seguridad y eficacia de la vacuna Menactra en lactantes menores de 9 meses de edad. Uso geriátrico No se ha establecido la seguridad y eficacia de la vacuna Menactra en adultos mayores de 55 años de edad. DESCRIPCIÓN Menactra es una vacuna estéril, para la administración por vía intramuscular, que contiene antígenos de polisacáridos capsulares del serogrupo A, C, Y y W-135 de N. meningitidis conjugados de manera individual con la proteína del toxoide diftérico. Las cepas A, C, Y y W- 135 de N. meningitidis se cultivan en agar Mueller Hinton y en medios Watson Scherp. Los polisacáridos se extraen de las células de N. meningitidis y se purifican mediante centrifugación, precipitación con detergente, precipitación con alcohol, extracción con disolventes y diafiltración. A fin de preparar los polisacáridos para su conjugación, se los despolimeriza, derivatiza y purifica mediante diafiltración. Los cultivos de Corynebacterium diphtheriae crecen en un medio Mueller y Miller modificado y se desintoxican con formaldehído. La proteína del toxoide diftérico se purifica mediante diafiltración y fraccionamiento con sulfato de amonio. Los polisacáridos derivatizados se unen de forma covalente con el toxoide diftérico y se purifican mediante diafiltración en serie. Los cuatro componentes meningocócicos, presentes como glucoconjugados individuales específicos del serogrupo, componen la vacuna formulada final. Durante la elaboración, no se agrega ningún conservante o adjuvante. Cada dosis de 0,5 ml puede contener cantidades residuales de formaldehído de menos de 2,66 mcg (0,000532%), por cálculo. La potencia de la vacuna Menactra se determina al cuantificar la cantidad de cada antígeno polisacárido que se conjuga con la proteína del toxoide diftérico y la cantidad de polisacárido no conjugado presente. La vacuna Menactra se elabora en forma de líquido estéril, de aspecto transparente a levemente turbio. Cada dosis de 0,5 ml de la vacuna se formula en solución isotónica de cloruro de sodio amortiguada con fosfato de sodio para que contenga 4 mcg de polisacáridos A, C, Y y W-135 meningocóccicos conjugados con aproximadamente 48 mcg de proteína portadora de toxoide diftérico. Ningún componente del frasco ampolla contiene látex. FARMACOLOGÍA CLÍNICA Mecanismo de acción La presencia de anticuerpos meningocóccicos anticapsulares bactericidas se ha asociado con la protección frente a la enfermedad meningocóccica invasiva. La vacuna Menactra induce la producción de anticuerpos bactericidas específicos de los polisacáridos capsulares de los serogrupos A, C, Y y W-135. TOXICOLOGÍA NO CLÍNICA Carcinogenia, mutagenia, deterioro de la fertilidad La vacuna Menactra no ha sido evaluada para determinar su potencial cancerígeno o mutágeno, o para determinar el deterioro de la fertilidad. ESTUDIOS CLÍNICOS. Eficacia El ensayo bactericida del suero (Serum Bactericidal Assay, SBA) utilizado para evaluar el suero contenía una fuente exógena de complemento que era humano (SBA-H) o de cría de conejo (SBA-BR). La respuesta a la vacuna Menactra administrada a niños de 9 meses a 10 años de edad se evaluó según la proporción de sujetos con un valor de anticuerpo SBA-H de 1:8 o mayor, para cada serogrupo. En personas de 11 a 55 años de edad, la respuesta a la vacuna Menactra se evaluó según la proporción de sujetos con un aumento de cuatro veces o mayor de anticuerpo bactericida de referencia para cada serogrupo, según lo midió el SBA-BR. Para personas de 2 a 55 años de edad, la eficacia de la vacuna se dedujo a partir de la demostración de la equivalencia inmunológica con una vacuna polisacárida meningocóccica aprobada por los EE. UU., la vacuna Menomune – A/C/Y/W-135, según se evaluó con el ensayo bactericida del suero (SBA). Inmunogenicidad Niños de 9 a 23 meses de edad En un estudio aleatorizado, multicéntrico, en los EE.UU., los niños recibieron la vacuna Menactra a los 9 meses y 12 meses de edad. La primera dosis de la vacuna Menactra se administró sola, seguida de una segunda dosis de la vacuna Menactra administrada sola (N=404), o con la vacuna MMRV (N=302), o con la PCV7 (N=422). Para todos los 58 participantes, se obtuvo suero aproximadamente 30 días después de la última vacunación. No hubo ninguna diferencia importante en las características demográficas entre los grupos de vacuna. El rango de mediana edad para la administración de la primera dosis de la vacuna Menactra fue aproximadamente a los 9 meses de edad. En estudios estadounidenses, se evaluó la administración de dos dosis de la vacuna Menactra para niños de 9 meses a 15 meses de edad. En el estudio de inmunogenicidad primaria, los niños recibieron la vacuna Menactra a los 9 y 12 meses de edad, la mayoría de los participantes en grupos que recibieron la segunda dosis de la vacuna Menactra sola o con vacunas pediátricas concomitantes alcanzaron valores de SBA-HC ≥ 1:8 para todos los serogrupos. Los grupos que recibieron la segunda dosis de la vacuna Menactra sola, tenían ≥ 91% de sujetos que alcanzaron un valor de SBA-HC ≥ 1:8 para los serogrupos A, C e Y y ≥ 86% para el serogrupo W-135. Cuando se administró la segunda dosis de la vacuna. Menactra de forma concomitante con MMRV (o MMRV + Hib) o con la PCV, los porcentajes de sujetos con valores de SBA-HC ≥ 1:8 fueron altos (>90% para los serogrupos A, C e Y y >81% para el serogrupo W-135). Los GMT (valores de la media geométrica de los títulos de anticuerpos) del SBA-HC fueron altos para todos los serogrupos. Luego de la segunda dosis de la vacuna Menactra en niños que recibieron la vacuna Menactra a los 9 y 15 meses de edad, el porcentaje de participantes con un valor de hSBA >1:8 fue alto para todos los serogrupos (>96% para C, Y y W-135 y >85,2% para el serogrupo A). Personas de 2 a 55 años de edad Se evaluó la inmunogenicidad en tres estudios clínicos comparativos, aleatorizados, multicéntricos, en los EE.UU., con un grupo control activo, en los que se incluyeron niños (de 2 a 10 años de edad), adolescentes (de 11 a 18 años de edad) y adultos (de 18 a 55 años de edad). Los participantes recibieron una dosis única de la vacuna Menactra (N=2.526) o la vacuna Menomune – A/C/Y/W-135 (N=2.317). Para los grupos de todas las edades estudiados, se obtuvo suero antes y aproximadamente 28 días después de la vacunación. (Los procedimientos de enmascaramiento para las evaluaciones de seguridad se describen en la sección REACCIONES ADVERSAS.) En cada uno de los estudios, no hubo ninguna diferencia importante en las características demográficas entre los grupos de vacuna, entre los subconjuntos de inmunogenicidad o la población global del estudio. Inmunogenicidad en niños de 2 a 10 años de edad De los 1.408 niños inscritos de 2 a 10 años de edad, las respuestas inmunológicass evaluadas por el hSBA en un subconjunto de participantes que recibieron la vacuna Menactra (de 2 a 3 años de edad, N=52; de 4 a 10 años de edad, N=84) y participantes que recibieron la vacuna Menomune – A/C/Y/W-135 (de 2 a 3 años de edad, N=53; de 4 a 10 años de edad, N=84), los porcentajes de sujetos con un valor ≥ 1:8 fueron constantemente más altos en el grupo de Menactra para los cuatro serogrupos. En el subconjunto evaluado de participantes de 2 a 3 años de edad, el porcentaje de participantes con un valor del hSBA ³1:8 en el día 28 fue del 73%, serogrupo A; 63%, serogrupo C; 88%, serogrupo Y; 63%, serogrupo W-135 en el grupo de Menactra y 64%, serogrupo A; 38%, serogrupo C; 73%, serogrupo Y; y 33%, serogrupo W-135 en el grupo de Menomune. En el subconjunto evaluado de participantes de 4 a 10 años de edad, el porcentaje de participantes con un valor del hSBA ≥ 1:8 en el día 28 fue del 81%, serogrupo A; 79%, serogrupo C; 99%, serogrupo Y; 85%, serogrupo W-135 en el grupo de Menactra y 55%, serogrupo A; 48%, serogrupo C; 92%, serogrupo Y; y 79%, serogrupo W-135 en el grupo de Menomune. Inmunogenicidad en adolescentes de 11 a 18 años de edad Los resultados del estudio clínico comparativo realizado en 881 adolescentes (de 11 a 18 años de edad) demostraron que las respuestas inmunológicas que midió el SBA-BR con respecto a la vacuna Menactra y la vacuna Menomune – A/C/Y/W-135 fueron similares para los cuatro serogrupos. El porcentaje de participantes con un valor del SBA-BR con un aumento cuatro veces o mayor respecto del valor de referencia fue del 93%, serogrupo A; 92%, serogrupo C; 82%, serogrupo Y; 97%, serogrupo W-135 en el grupo de Menactra y 92%, serogrupo A; 89%, serogrupo C; 80%, serogrupo Y; y 95%, serogrupo W-135 en el grupo de Menomune. En participantes con valores anteriores a la vacunación imperceptibles (es decir, menos de 1:8 en el día 0), los índices de seroconversión (definidos como un aumento cuatro veces o mayor en el día 28 en los valores del SBA-BR) fueron similares entre los que recibieron la vacuna Menactra y la vacuna Menomune – A/C/Y/W-135. Los participantes que recibieron la vacuna Menactra alcanzaron índices de seroconversión de: 100%, serogrupo A; 99%, serogrupo C; 98%, serogrupo Y; 99%, serogrupo W-135. Los índices de seroconversión para los receptores de la vacuna Menomune – A/C/Y/W-135 fueron: 100%, serogrupo A; 99%, serogrupo C; 100%, serogrupo Y; 99%, serogrupo W-135. Inmunogenicidad en adultos de 18 a 55 años de edad Los resultados del estudio clínico comparativo realizado en 2.554 adultos de 18 a 55 años de edad demostraron que las respuestas inmunológicas que midió el SBA-BR con respecto a la vacuna Menactra y la vacuna Menomune – A/ C/Y/W-135 fueron similares para los cuatro serogrupos. El porcentaje de participantes con un valor del SBA-BR con un aumento cuatro veces o mayor respecto del valor de referencia fue del 81%, serogrupo A; 89%, serogrupo C; 74%, serogrupo Y; y 89%, serogrupo W-135 en el grupo de Menactra y 85%, serogrupo A; 90%, serogrupo C; 79%, serogrupo Y; y 94%, serogrupo W-135 en el grupo de Menomune. 59 En participantes con valores anteriores a la vacunación imperceptibles (es decir, menos de1:8 en el día 0), los índices de seroconversión (definidos como un aumento cuatro veces o mayor en el día 28 en los valores del SBA-BR) fueron similares entre los que recibieron la vacuna Menactra y la vacuna Menomune – A/C/Y/W-135. Los participantes que recibieron la vacuna Menactra alcanzaron índices de seroconversión de: 100%, serogrupo A; 99%, serogrupo C; 91%, serogrupo Y; y 97%, serogrupo W-135. Los índices de seroconversión para los receptores de la vacuna Menomune – A/C/Y/W-135 fueron: 99%, serogrupo A; 98%, serogrupo C; 97%, serogrupo Y; y 99%, serogrupo W-135. Administración de vacunas concomitantes MMRV (o MMR + V) o PCV7 En un ensayo en los EE.UU., con un grupo control activo, 1.179 niños recibieron la vacuna Menactra a los 9 meses y 12 meses de edad. A los 12 meses de edad, estos niños recibieron la vacuna Menactra de forma concomitante con MMRV (N=616), o MMR + V (N=48), o PCV7 (N=250). Otro grupo de niños de 12 meses de edad recibió MMRV + PCV7 (N=485). Se obtuvo suero aproximadamente 30 días después de las últimas vacunaciones. Las respuestas de anticuerpos frente al sarampión, la parotiditis, la rubéola y la varicela en niños que recibieron la vacuna Menactra y MMRV (o MMR y V) fueron equiparables a las respuestas de anticuerpos correspondientes en niños que recibieron MMRV y PCV7. Cuando se administró la vacuna Menactra de forma concomitante con la PCV7, no se alcanzaron los criterios de no inferioridad para comparaciones de concentraciones medias geométricas (GMC) de inmunoglobulina G (IgG) contra el neumococo (límite superior del intervalo de confianza [confidence interval, CI] bilateral del 95% de la proporción de GMC­2) para 3 de 7 serotipos (4, 6B, 18C). En un subconjunto de 196 sujetos (todos con suero disponible) que recibieron la vacuna Menactra y PCV7 de forma concomitante, se evaluó el suero mediante el ensayo de la actividad opsonofagocítica (opsonophagocytic assay, OPA) contra el neumococo y >99% de los sujetos tenía un valor muy por encima del nivel protector de ≥1:8. Td En un ensayo doble ciego, aleatorizado, controlado, 1.021 participantes de 11 a 17 años recibieron las vacunas Td y Menactra de forma concomitante (N=509), o Td seguida un mes después por una vacuna Menactra (N=512). Se obtuvo suero aproximadamente 28 días después de cada respectiva vacunación. La proporción de participantes con un aumento de cuatro veces o mayor en el valor del SBA-BR para los serogrupos meningocócicos C, Y y W-135 fue superior cuando se administró la vacuna Menactra de forma concomitante con Td (86 a 96%) que cuando se administró la vacuna Menactra un mes después de la Td (65 a 91%). Las respuestas de anticuerpos frente al tétano y la difteria fueron similares en ambos grupos del estudio. Typhim Vi (vacuna antitifoidea de polisacárido vi) En un ensayo doble ciego, aleatorizado, controlado, 945 participantes de 18 a 55 años recibieron las vacunas Typhim Vi y Menactra de forma concomitante (N=469), o la vacuna Typhim Vi seguida un mes después por la vacuna Menactra (N=476). Se obtuvo suero aproximadamente 28 días después de cada respectiva vacunación. Las respuestas de anticuerpos frente a los componentes de la vacuna Menactra y de la vacuna Typhim Vi fueron similares en ambos grupos del estudio. PRESENTACIÓN Frasco ampolla monodosis (estuche con cinco frascos ampolla) Frasco ampolla monodosis (estuche con un frasco ampolla) CONSERVACIÓN Conservar el medicamento a una temperatura entre 35 y 46°F (entre 2 y 8°C). NO CONGELAR. No debe utilizarse el producto si se expuso a congelación. No utilizar el medicamento luego de la fecha de vencimiento. INFORMACIÓN PARA PACIENTES Antes de la administración de la vacuna Menactra, el profesional de atención médica debe informar al paciente, padre, tutor u otro adulto responsable acerca de los riesgos y beneficios potenciales para el paciente (consulte REACCIONES ADVERSAS y ADVERTENCIAS Y PRECAUCIONES). Se debe indicar a los pacientes, padres o tutores que deben informar cualquier reacción adversa sospechada al profesional de atención médica, quien debe informar estos eventos a Sanofi Pasteur Inc. MENACTRA es una marca comercial registrada de sanofi pasteur y sus subsidiarias. Fabricado por: Sanofi Pasteur Inc. Swiftwater PA 18370 EE.UU. 60 Bacteria Infecciones por neumococo “Crear vacunas, es proteger la vida” 61 PNEUMO 23 Vacuna Antineumocócica polisacárida 23-valente (contra Streptococcus pneumoniae serotipos 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19f, 20, 22F, 23F, 33F) DENOMINACIÓN. PNEUMO 23, solución inyectable en jeringa precargada. Vacuna neumocócica polisacarídica. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA. Polisacáridos de Streptococcus pneumoniae serotipos 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, 33F 25 mg de cada uno de los 23 serotipos para 0,5 ml. FORMA FARMACÉUTICA. Solución inyectable en jeringa precargada. DATOS CLÍNICOS. Indicaciones terapéuticas. Vacuna indicada para la prevención de las infecciones por neumococo, en particular de las neumonías debidas a los serotipos contenidos en la vacuna, en los sujetos de alto riesgo a partir de la edad de 2 años. Grupos identificados en alto riesgo: • Personas mayores de 65 años, especialmente las que viven en instituciones. • Pacientes inmunocompetentes debilitados o susceptibles de hospitalización frecuente (diabetes, bronquitis, crónica, insuficiencia respiratoria, insuficiencia cardíaca, antecedentes de alcoholismo – tabaquismo); • Pacientes inmunodeprimidos: esplenectomizados, drepanocitarios, con síndrome nefrótico, linfoma, mieloma múltiple; • Pacientes con herida ósteo – meníngea. Conviene añadir que las infecciones reincidentes de las vías aéreas superiores, especialmente otitis media y sinusitis, no constituyen una indicación de la vacunación. Posología y método de administración. Posología: • Primovacunación: una inyección 0,5 ml. • Revacunación: una inyección de 0,5 ml.,se recomienda cada 3 a 5 años en los individuos con riesgo alto de infección por neumococo. Método de Administración: Administrar con preferencia por vía intramuscular (IM) o subcutánea (SC). Contraindicaciones. - Hipersensibilidad comprobada a uno de los componentes de la vacuna. - Contraindicaciones normales de cualquier otra vacunación: en caso de fiebre, enfermedad aguda o recidiva de enfermedad crónica, conviene aplazar la vacunación. - Se desaconseja la utilización de este medicamento durante el primer trimestre de embarazo. - No se recomienda la vacunación para los sujetos vacunados durante los 3 últimos años, salvo indicaciones particulares. La vacunación de los sujetos que han presentado una infección por neumococo, real o no, no constituye una contraindicación y se debe considerar según los antecedentes (véase Reacciones adversas). Advertencias y precauciones especiales de uso. Se debe efectuar la revacunación estrictamente de acuerdo con el folleto. Posología y Método de Administración. Se aconseja vacunar al menos 2 semanas antes de intervención quirúrgica para esplenectomia. Interacciones con otros medicamentos y otras formas de interacciones. Se puede administrar la vacuna al mismo tiempo que una vacuna antigripal practicando las inyecciones en dos sitios diferentes. Embarazo y lactancia. Embarazo: No hay datos fiables de teratogénesis en animales. Actualmente no existen datos clínicos concluyentes para calibrar el eventual efecto malformativo o fetotóxico de la vacuna cuando se administra durante el primer trimestre de embarazo. Un estudio realizado sobre una muestra muy limitada ha mostrado una protección ligeramente aumentada durante los 6 primeros meses de vida en lactantes cuya madre había sido vacunada. En consecuencia, se desaconseja la utilización de esta vacuna 62 durante el primer trimestre de embarazo. Durante el tercer trimestre sólo se planteará su uso si es necesario. Lactancia: La administración de esta vacuna es posible durante la lactancia. Reacciones adversas. Reacciones locales en el sitio de inyección: dolor, eritema, induración, edema pueden sobrevenir; estas reacciones son benignas y transitorias. Raras reacciones de tipo fenómeno de Arthus, reversibles sin secuelas, han sido notificadas. Sobrevienen principalmente en los sujetos cuyo título de anticuerpos antineumocócicos es inicialmente alto. Reacciones generales: hipertermia moderada y transitoria en aproximadamente el 2% de los casos. Pocos casos de hipertermia superior a 39º C pueden observarse. Sobrevienen la mayoría de las veces muy rápidamente después de la vacunación y desaparecen en 24 horas. Excepcionalmente otras reacciones generales han sido relatadas: adenopatías, erupciones, artralgias y reacciones alérgicas (urticaria, edema de Quincke, reacciones anafilactoides). PROPIEDADES FARMACOLOGICAS VACUNA ANTI – NEUMOCÓCICA Propiedades farmacodinámicas. Esta es una vacuna preparada a partir de antígenos capsulares polisacarídicos purificados de Streptococcus pneumoniae y contiene 23 serotipos diferentes (responsable del 90% de las infecciones invasivas neumocócicas). La inmunidad aparece 2 ó 3 semanas después de la inyección. DATOS FARMACÉUTICOS. Excipientes. Solución tamponada con fenol (csp pH 6.9) que contiene fenol, cloruro de sodio, fosfato disódico dihidratado, fosfato monosódico dihidratado, agua para preparaciones inyectables. Período de validez. 2 años. Precauciones especiales de conservación. Conservar entre + 2° C y + 8° C. No congelar. Naturaleza y contenido del recipiente: jeringa precargada (vidrio) de 0,5 ml con tapa-pistón de elastómero (clorobromobutil). 63 Virus Heptatitis B “Crear vacunas, es proteger la vida” 64 RECOMVAX-B Vacuna contra la Hepatitis B, Recombinante. Vacuna contra la Hepatitis B, Recombinante. Recomvax B está formada por partículas altamente purificadas no infecciosas de antígeno de superficie de la hepatitis B (HBsAg) adsorbidas en sales de aluminio como adyuvante. Es una vacuna de ADN recombinante contra hepatitis B derivada del HBsAg, producida por una tecnología de ADN recombinante aplicada sobre células de levadura (Saccharomyces cerevisiae). La vacuna cumple con las exigencias de la O.M.S. para las vacunas recombinantes contra hepatitis B. En su elaboración no se utilizan sustancias de origen humano. COMPOSICIÓN: Cada dosis de 1 mL de suspensión inyectable contienen: Antígeno de superficie purificado del virus hepatitis B recombinante (HBsAg) 20,00 mcg. Hidróxido de aluminio gel 0,50 mg. Cloruro de sodio 8,50 mg. Fosfato de potasio monobásico 1,20 mg. Fosfato de sodio dibásico 0,45 mg. Agua para inyectable c.s.p. 1 mL. DESCRIPCIÓN: Recomvax B es una suspensión blanca levemente opalescente. INDICACIONES Y USO: Inmunización contra la infección causada por todos los subtipos conocidos del virus de la Hepatitis B. POSOLOGÍA Y ADMINISTRACIÓN: Recomvax B está destinada exclusivamente al uso intramuscular. - Una dosis pediátrica (recién nacidos, lactantes y niños de hasta 15 años de edad) es 0,5 ml, que contiene 10 ug de HbsAg. Una dosis adulta (a partir de 16 años) es 1,0 ml, que contiene 20 ug de HbsAg. El régimen de inmunización consiste en 3 dosis de vacuna administradas en el siguiente calendario: 1ª dosis: en la fecha elegida. 2ª dosis: 1 mes después de la primera dosis. 3ª dosis: 6 meses después de la primera dosis. Revacunación: la OMS no recomienda una vacunación de refuerzo, puesto que ha sido demostrado que una serie primaria de 3 dosis de la vacuna de hepatitis B, protege por lo menos durante 15 años y que además, existe una respuesta anamnésica, luego de una exposición al VHB, aunque los anticuerpos protectores se hayan perdido durante ese lapso de tiempo. Se debe tener en cuenta que algunos programas locales de vacunación, incluyen la recomendación de una dosis de refuerzo y esto debe ser respetado. Un calendario alternativo a los 0, 1 y 2 meses con revacunación a los 12 meses puede ser utilizada en determinadas poblaciones (p. ej., recién nacidos de madres contagiadas con hepatitis B, personas que hayan estado o puedan haber estado recientemente expuestas al virus, viajeros a zonas de alto riesgo). Una dosis adicional de vacuna puede ser necesaria en pacientes sometidos a hemodiálisis o inmunodeficientes cuando no sea posible alcanzar títulos de anticuerpos protectores (>10 UI/l) después de un ciclo de inmunización primaria. CONTRAINDICACIONES: La vacuna contra la hepatitis B está contraindicada para personas con hipersensibilidad a la levadura o a cualquier componente de Recomvax B. ADVERTENCIAS Y PRECAUCIONES: Precauciones generales: - La administración de Recomvax B debe ser postergada en pacientes que sufran de una enfermedad febril severa aguda. - En pacientes que sufren de esclerosis múltiple cualquier estímulo del sistema inmunológico puede inducir la exacerbación de su sintomatología. Por lo tanto, en estos pacientes los beneficios de la vacunación contra la Hepatitis B deben ser contrastados con los riesgos de exacerbación de la esclerosis múltiple (ver Reacciones adversas). Se considera que la protección no puede alcanzarse con la vacunación de pacientes con un estado latente o progresivo de la hepatitis B. - Como siempre en el caso de vacunas inyectables, debe tenerse a mano un tratamiento médico apropiado en caso de producirse una de las raras reacciones anafilácticas 65 que pueden seguir a la administración de la vacuna. Precauciones de uso: - Agitar antes de la administración por cuanto puede formarse durante el almacenamiento un fino depósito blanco con un sobrenadante transparente incoloro. - Recomvax B no debe ser administrado en la región glútea ni debe ser administrado por vía intravenosa. Embarazo y lactancia: - El efecto de HBsAg sobre el desarrollo fetal no ha sido evaluado. Sin embargo, como en todas las vacunas antivirales inactivadas, los riesgos para el feto deben ser considerados insignificantes. Recomvax B debe ser utilizada durante el embarazo sólo en caso de ser claramente necesario, no recomendándose la vacunación de estas mujeres. - El efecto de administrar Recomvax B a las madres sobre sus hijos lactantes no ha sido aún evaluado en estudios clínicos. REACCIONES ADVERSAS: Comunes: - Reacciones locales tales como eritema, dolor, hinchazón o fiebre menor pueden ocurrir en raras ocasiones; estos síntomas desaparecen en 2 días. Raras: - Hipertermia (por sobre 38ºC) - Reacciones sistémicas tales como desfallecimiento, astenia, náuseas, vómitos, mareo, mialgia, artritis. - Rash cutáneo y aumento transitorio de las transaminasas. Muy rara vez: - No puede establecerse una secuencia de causa y efecto para neuritis múltiple, neuritis óptica, parálisis facial, exacerbación de la esclerosis múltiple y síndrome de Guillain-Barré. CONDICIONES DE ALMACENAMIENTO: No sobrepasar la fecha límite de utilización que figura en el envase exterior. Almacenar entre +2ºC y 8°C (en refrigerador). No congelar. PRESENTACIONES: Frasco 0,5 ml x 1 frasco. Frasco 1,0 ml x 1 frasco 66 Virus Fiebre Amarilla “Crear vacunas, es proteger la vida” 67 STAMARIL Vacuna contra la Fiebre Amarilla COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Después de la reconstitución, 1 dosis (0,5 mL) contiene: Virus de fiebre amarilla1 cepa 17D-204 (vivos, atenuados)……no menos de 1.000 unidades DL502 1 propagados en embriones de pollo libres de patógenos 2 dosis letal determinada estadísticamente en el 50% de los animales analizados PREPARACIÓN: La vacuna STAMARIL es una vacuna viva atenuada contra la fiebre amarilla, termoestable y liofilizada. De acuerdo con las normas de la O.M.S, se obtiene por multiplicación del virus de la fiebre amarilla, cepa 17 D (Fundación Rockefeller), sobre embriones de pollo, exentos de leucosis aviar. FORMA FARMACÉUTICA: Polvo y disolvente para suspensión inyectable. Antes de la reconstitución, el polvo es de color beige a beige-anaranjado; el disolvente es transparente e incoloro. INDICACIONES: • Prevención de la fiebre amarilla. • Se recomienda vacunar a: • Personas que viajen, aunque sea por poco tiempo, a zonas endémicas o que vivan en ellas. • Personas no vacunadas que se desplacen de una zona endémica a una zona no endémica • Personas expuestas profesionalmente • Personas infectadas por VIH asintomáticas y que presenten riesgo. • Para que la vacunación contra la fiebre amarilla sea oficialmente reconocida, deberá estar registrada en un certificado internacional, que será firmado y aprobado por un centro de vacunación autorizado. Dicho certificado tendrá una validez de diez años a partir del décimo día siguiente a la fecha de vacunación. CONTRAINDICACIONES: Las contraindicaciones de la vacuna contra la fiebre amarilla son las de cualquier vacuna: - enfermedades infecciosas en fase evolutiva. - afecciones evolutivas malignas. - terapias inmunodepresoras en curso. Las contraindicaciones específicas son: - alergia verdadera a las proteínas del huevo. - déficit inmunitario congénito o adquirido. Las mujeres embarazadas y los niños menores de 6 meses no deben ser vacunados. No obstante, en caso de epidemia, podrán vacunarse las mujeres embarazadas y los niños a partir de 4 meses. En caso de duda, se consultará obligatoriamente con el médico. PRECAUCIONES DE EMPLEO: En el caso de personas alérgicas , puede realizarse una prueba de tolerancia que consiste en una inyección intradérmica de 0,1 ml de vacuna. Si no se produce ninguna reacción en un plazo de 10 a 15 minutos, se les administrará por vía subcutánea el resto de la dosis, es decir, 0,4 ml. Si se prescribe la vacunación a enfermos que han estado sometidos a un tratamiento inmunosupresor, conviene dejar transcurrir un mes entre el final del tratamiento y la administración de la vacuna, verificando en todos los casos que los parámetros biológicos son normales. INTERACCIONES CON OTROS MEDICAMENTOS: Se puede asociar la vacuna STAMARIL , con la vacuna poliomielítica oral o inyectable, con la vacuna antisarampión, con la 68 vacuna Typhim Vi y con la vacuna antidiftérica, antitetánica y anti-tos ferina sin que resulte afectada la inmunogenicidad de dichas vacunas.Sin embargo, la vacunación simultánea con las vacunas contra el cólera, antitifoídicas y antiparatifoídicas A y B inyectables está contraindicada, recomendándose un intervalo de 3 semanas. POSOLOGÍA: La primovacunación consiste en una inyección subcutánea o intramuscular de una dosis de 0,5 ml de vacuna reconstituída. Revacunaciones: las normativas internacionales de salud exigen una inyección de revacunación de 0,5 ml cada 10 años. MODO DE EMPLEO: Reconstituir cuidadosamente la vacuna inyectando el diluyente contenido en la jeringuilla en la ampolla de liofilizado (vacuna). Después de la completa disolución, reaspirar la vacuna con la jeringuilla e inyectar. La vacuna se inyecta por vía subcutánea. La vacuna reconstituída deberá protegerse de la luz y se utilizará dentro de la hora siguiente a su reconstitución. EFECTOS SECUNDARIOS: Entre el cuarto y el séptimo día, pueden aparecer picks febriles, cansancio y dolores de cabeza. Se señalará al médico cualquier efecto no deseado molesto que no se haya mecionado en este texto. CONSERVACIÓN: No sobrepasar la fecha límite de utilización que figura en el envase. PRECAUCIONES ESPECIALES DE CONSERVACIÓN: Conservar entre +2°C y+8°C (en cámara fría). No congelar. Utilícese inmediatamente después de la reconstitución. 69 Bacteria Tétanos “Crear vacunas, es proteger la vida” 70 TETAVAX Vacuna Adsorbida Suspensión Inyectable40 U.I./Dosis 1. NOMBRE DEL MEDICAMENTO. TETAVAX, suspensión inyectable. Vacuna antitetánica adsorbida 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA. Una dosis (0.5 ml) contiene: Toxoide tetánico. Adsorbido en Hidróxido de Aluminio hidratado Para lista completa de excipientes, ver sección 6.1 ≥ 40 U.I. 0.6 mg Al 3. FORMA FARMACEUTICA. Suspensión inyectable. 4. DATOS CLINICOS 4.1 Indicaciones terapéuticas. Prevención del tétanos Usos - Profilaxis del Tétanos post exposición en caso de heridas recientes que pueden estar contaminadas por esporas tetánicas en sujetos que no han recibido inmunización primaria o en los cuales, ésta es incompleta o se desconoce. - Profilaxis del Tétanos neonatal en las mujeres no inmunizadas contra el Tétanos en edad fértil o embarazadas, en los países en los que el tétanos neonatal es frecuente. - Vacunación Primaria. - Vacunación de refuerzo. 4.2 Posología y forma de Administración Posología. Profilaxis del tétanos post exposición En caso de una herida menor, el médico debe evaluar previamente el riesgo de contaminación de ésta por Clostridium tetani al nivel del sitio de la herida. Además de la desinfección de la herida, su desbridamiento y la administración de la vacuna, en algunos casos debe administrarse simultáneamente en otro lugar del cuerpo una inmunización pasiva con una inmunoglobulina humana antitetánica (ver tabla a continuación). 71 Es necesario iniciar una vacunación primaria en los sujetos que hayan padecido tétanos, porque la enfermedad clínica no induce la producción fiable de anticuerpos. Profilaxis del Tétanos neonatal: Las mujeres en edad fértil y las embarazadas aún no inmunizadas deben recibir dos dosis sucesivas de esta vacuna con un intervalo mínimo de 4 semanas. De ser posible, la primera inyección debe administrase preferentemente al menos 90 días antes del parto. Vacunación primaria: Si es necesario administrar una vacunación primaria en un adulto, el esquema vacunal consiste en 2 dosis sucesivas con 1-2 semanas de intervalo, seguidas de un refuerzo administrado entre 6 y 12 meses después de la segunda inyección. Vacunación de refuerzo: Una dosis de 0.5 ml 10 años después de la vacunación primaria y luego cada 10 años. FORMA DE ADMINISTRACIÓN Debido a que esta vacuna se adsorbe, es preferible administrarla por vía intramuscular con el fin de disminuir al máximo las reacciones locales. Los sitios recomendados son la cara anterolateral del muslo o del brazo. La vía subcutánea profunda también puede emplearse. La vía intradérmica no debe utilizarse. 4.3 Contraindicaciones El riesgo letal asociado al tétanos en la profilaxis post exposición en caso de herida excluye toda contraindicación potencial. En otros casos: -Hipersensibilidad a uno de los componentes de la vacuna -Contraindicaciones habituales para todas las vacunaciones: es preferible aplazar la vacunación en caso de fiebre, enfermedad aguda o enfermedad crónica evolutiva. -Reacción de hipersensibilidad o trastorno neurológico aparecidos después de una inyección anterior de esta vacuna. 4.4 Advertencias y precauciones especiales de empleo En aquellos sujetos que presentan un síndrome de Guillain Barré o una neuropatía del plexo braquial tras la administración anterior de una vacuna que contenga toxoide tetánico, la decisión de vacunar con una vacuna que contenga toxoide tetánico deberá basarse en la evaluación cuidadosa de los posibles beneficios y riesgos. La vacunación normalmente está justificada cuando no está completado el programa de primovacunación ( es decir con menos de tres dosis administradas). No inyectar por vía intravascular: asegurarse de que la aguja no penetre en un vaso sanguínea. Al igual que todas las vacunas inyectables, debe disponerse de un tratamiento médico apropiado y vigilar al sujeto en el caso raro de una reacción anafiláctica después de la administración de la vacuna. La inmunogenicidad de la vacuna se podría alterar con un tratamiento inmunosupresor o en un estado de inmunodeficiencia. En estos casos, se recomienda esperar el final del tratamiento para vacunar o asegurarse de la buena protección del sujeto. No obstante, se recomienda la vacunación de individuos con inmunodeficiencia crónica, como la infección del VIH, si la patología presente permite la inducción de una respuesta de anticuerpos aunque sea limitada. Con el fin de prevenir reacciones de hipersensibilidad, evitar la administración a personas que han recibido una inmunización primaria completa o una dosis de refuerzo durante los 5 años anteriores. Debe tenerse muy en cuenta el riesgo potencial de apnea y la necesidad de vigilancia respiratoria durante 48-72 horas cuando se adminístren las dosis de primovacunación en los muy prematuros (recién nacidos < 28 semanas de embarazo) y particularmente aquellos que tienen antecedentes de inmadurez respiratoria. Debido al beneficio considerable de la vacunación en estos lactantes, la administración no se debe suspender ni aplazarse. 4.5 Interacción con otros medicamentos y otras formas de interacción No hay inconveniente conocido a la administración de esta vacuna en el curso de una misma sesión de vacunación con otras vacunas usuales inyectadas en lugares separados. 4.6 Embarazo y lactancia Los datos experimentales y clínicos permiten prescribir esta vacuna durante el embarazo en caso necesario, y en cualquier momento del mismo. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas No se aplica 4.8 Reacciones adversas Según los datos, de las declaraciones espontáneas, los siguientes acontecimientos se informaron después de la comercialización de TETAVAX. Estos acontecimientos se informaron muy raramente (< 0.01 %). Sin embargo, su incidencia exacta no puede ser calculada de manera precisa. - Trastornos de la sangre y del sistema linfático - Linfadenopatía - Trastornos del Sistema Inmunitario - Reacciones de hipersensibilidad tipo I - Trastornos del Sistema Nervioso - Cefalea, vértigo - Trastornos vasculares 72 - Hipotensión (en un contexto de reacción de hipersensibilidad de tipo I) - Trastornos de la piel y del tejido subcutáneo - Síntomas de tipo alérgico como una urticaria, prurito generalizado o eritema - Trastornos musculo-esqueléticos y del tejido conjuntivo - Mialgia, artralgia - Trastornos generales y anomalías en el lugar de administración Pueden aparecer en el lugar de la inyección como por ejemplo dolor, rash, endurecimiento o edema durante las 48 horas siguientes y persistir durante uno o dos días. La formación de un nódulo subcutáneo puede, a veces, acompañar estas reacciones. Se han informado raramente casos de abscesos asépticos. La incidencia y la gravedad de estos fenómenos locales pueden verse influenciadas por el lugar, la vía y el método de administración al igual que por el número de dosis anteriores recibidos. Fiebre temporal. Malestar. Todas estas reacciones se han observado la mayoría de las veces, en personas hiperinmunizadas, en particular en aquellas que han recibido refuerzos demasiado frecuentes. Reacciones adversas potenciales (es decir, que no han sido informados directamente con TETAVAX, sino con otras vacunas que contenían uno o varios de los elementos antígenos de TETAVAX). Síndrome de Guillain Barré y neuropatía del plexo braquial tras la administración de una vacuna que contenga el toxoide tetánico. Apnea en los muy prematuros (nacidos < 28 semanas de embarazo) ver sección 4.4 4.9 Sobredosis No documentado 5 PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas VACUNA CONTRA EL TÉTANOS (JO7 AM01: Toxoide tetánico). La vacuna se prepara a partir de la toxina tetánica destoxificada por el formaldehido y purificada. La inmunidad aparece a partir de la segunda inyección. Se refuerza con la tercera inyección y permanece 5 a 10 años tras la cuarta inyección. 5.2 Propiedades farmacocinéticas No se aplica. 5.3 Datos Preclínicos sobre seguridad No se aplica 6 DATOS FARMACEUTICOS 6.1 Lista de excipientes Incluir listado cualitativo de acuerdo a la última fórmula autorizada en el registro sanitario. 6.2 Incompatibilidades Debido a la ausencia de estudio de compatibilidad, este medicamento no debe estar mezclado con otros medicamentos. 6.3 Período de validez 3 años. 6.4 Precauciones especiales de conservación Conservar en el refrigerador (entre 2° C y 8°C) 6.5 Naturaleza y contenido del envase Indicar de acuerdo a lo autorizado en el registro sanitario. 6.6 Precauciones especiales de eliminación y otras manipulaciones Agitar antes de la inyección, hasta obtener una suspensión homogénea. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN SANOFI PASTEUR 2 AVENUE PONT PASTEUR 69007 LYON FRANCIA 73 Bacteria Fiebre Tifoidea “Crear vacunas, es proteger la vida” 74 TYPHIM VI Vacuna Antitifoídea Poliosídica 1. PRESENTACIONES: TYPHIM Vi, solución inyectable en jeringa prellenada TYPHIM Vi, solución inyectable en multidosis 2. COMPOSICIÓN CUALITATIVA y CUANTITATIVA Una dosis de 0,5 mL de vacuna contiene: Poliósido capsular purificado de Salmonella typhi (cepa Ty2) 25 µg Lista de excipientes, ver epígrafe 6.1. 3. FORMA FARMACÉUTICA Solución inyectable 4. DATOS CLÍNICOS 4.1. Indicaciones terapéuticas Este medicamento es una vacuna recomendada para la prevención de la fiebre tifoidea en los adultos y los niños mayores de 2 años. TYPHIM Vi está destinada a los viajeros que se dirigen a zonas de endemia, a los emigrantes y al personal de salud. 4.2. Posología y forma de administración Posología RESERVADO AL ADULTO Y AL NIÑO MAYOR DE 2 AÑOS. Una sola inyección asegura la protección. Se practicará una revacunación cada 3 años si se mantiene la exposición al riesgo. El esquema de vacunación es el mismo en el niño y en el adulto. Forma de administración Vía intramuscular o subcutánea. 4.3. Contraindicaciones Hipersensibilidad conocida a cualquiera de los componentes de la vacuna. 4.4. Advertencias y precauciones especiales de empleo No inyectar por vía intravascular: verificar que la aguja no penetre en un vaso sanguíneo. Esta vacuna protege contra el riesgo infeccioso vinculado a la Salmonella typhi pero no ofrece protección contra las bacterias Salmonella paratyphi A o B. En los niños menores de 2 años, no se indica esta vacuna debido a un riesgo de respuesta insuficiente en anticuerpos. Es preferible diferir la vacunación en caso de fiebre, enfermedad aguda o enfermedad crónica evolutiva. 4.5. Interacción con otros medicamentos y otras formas de interacción Esta vacuna puede asociarse a otras vacunas usuales durante una misma sesión de vacunación (hepatitis A, fiebre amarilla, difteria, tétanos, poliomielitis, rabia, meningitis A + C y hepatitis B). 4.6. Embarazo y lactancia Embarazo No se dispone de información confiable de teratogénesis en el animal. En clínica, no existen en la actualidad datos suficientes que permitan evaluar un posible efecto malformativo o fetotóxico de la vacuna cuando se administra durante el embarazo. Debido a la gravedad de la enfermedad y en caso de alto riesgo de exposición a la fiebre tifoidea en el embarazo se debe hacer necesariamente una evaluación riesgo-beneficio para decidir la administración o no de Typhim Vi Lactancia No se conoce si la vacuna se excreta por la leche materna. Se deben tener precaución en mujeres que están amamantando. 4.7. Efectos sobre la capacidad para conducir vehículos y utilizar máquinas No se aplica. 75 4.8. Reacciones adversas Las reacciones observadas después de la vacunación son generalmente moderadas y de corta duración. Frecuentes: reacciones locales en el lugar de inyección (dolor, edema, enrojecimiento). Raras: reacciones generales: fiebre, astenia, cefalea, malestar, artralgias, mialgias, náuseas, dolores abdominales. Muy raras: reacciones de tipo alérgico (prurito, erupción cutánea, urticaria). Han sido observados casos aislados de enfermedad del suero y de reacciones anafilactoides. 4.9. Sobredosis No se aplica. 5. PROPIEDADES FARMACOLÓGICAS 5.1. Propiedades farmacodinámicas VACUNA ANTI-TIFOÍDICA (Código ATC: J07AP) Vacuna preparada a partir del polisacárido Vi purificado de Salmonella typhi. La inmunidad aparece dentro de 15 días a 3 semanas después de la inyección. La duración de la protección es de por lo menos 3 años. En el curso de estudios realizados en zonas de fuerte endemia, la tasa de protección (respecto a la fiebre tifoidea) conferida por una inyección de vacuna es del 77% en Nepal y del 55% en Sudáfrica. En los países industrializados, la seroconversión se nota en más del 90% de los sujetos después de una sola inyección. 5.2. Propiedades farmacocinéticas No se aplica. 5.3. Datos preclínicos sobre seguridad No se aplica. 6. DATOS FARMACÉUTICOS 6.1. Lista de excipientes Fenol y solución tampón que contiene cloruro de sodio, fosfato disódico dihidratado, fosfato monosódico dihidratado y agua para preparados inyectables. 6.2. Incompatibilidades En ausencia de estudios de compatibilidad esta vacuna no debe mezclarse con otros medicamentos. 6.3. Periodo de validez 3 años. 6.4. Precauciones especiales de conservación Conservar en refrigerador (entre +2°C y +8°C). No congelar. 6.5. Naturaleza y contenido del recipiente - 0,5 mL de solución en jeringa prellenada (vidrio de tipo I) con un tapón-émbolo (clorobromobutilo) - caja de 1. - 10 x 0,5 mL de solución en frasco (vidrio) con un tapón (clorobutilo) - caja de 1. - 20 x 0,5 mL de solución en frasco (vidrio) con un tapón (clorobutilo) - caja de 1. 6.6. Instrucciones de uso, y manipulación La vacuna debe quedarse algunos minutos a temperatura ambiente antes de utilizarla. La eliminación de los productos no utilizados o de los desechos se establecerá de acuerdo con las exigencias locales. 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN SANOFI PASTEUR S.A. - 2, avenue Pont Pasteur 69007 Lyon, Francia 8. PRESENTACIÓN Y NÚMERO DE IDENTIFICACIÓN ADMINISTRATIVA - 339 841-1: 0,5 mL de solución en jeringa prellenada (vidrio de tipo I) con un tapón-émbolo (clorobromobutilo) - caja de 1. - 332 206-9: 10 x 0,5 mL de solución en frasco (vidrio) con un tapón (clorobutilo) - caja de 1. - 332 206-9: 20 x 0,5 mL de solución en frasco (vidrio) con un tapón (clorobutilo) - caja de 1. 76 Virus Influenza “Crear vacunas, es proteger la vida” 77 VAXIGRIP Vacuna anti-influenza inactivada suspensión inyectable 1. NOMBRE DEL MEDICAMENTO VAXIGRIP, suspensión inyectable en jeringa precargada Vacuna antiinfluenza de virus fraccionados, inactivada. 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Virus de la influenza*, fraccionado, inactivado, conteniendo 15 microgramos de HA** de cada uno de los tres antígenos análogos o derivados de las cepas recomendadas por la OMS para el Hemisferio Sur, para una dosis de 0.5 ml. *Cultivados en huevos fertilizados de gallinas provenientes de lotes sanos. **Antígenos de Hemaglutinina. La vacuna cumple con las recomendaciones de la OMS (en el hemisferio sur) para cada temporada. 3. FORMA FARMACÉUTICA Suspensión inyectable en jeringa precargada La vacuna después de agitar suavemente, es un líquido blanquecino y opalescente. 4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas. Prevención de la influenza, particularmente en sujetos que presentan un elevado riesgo de complicaciones asociadas. Vaxigrip está indicada para adultos y niños a partir de los 6 meses de edad. Vaxigrip debe ser usado siguiendo recomendaciones oficiales. 4.2 Posología y método de administración Posología. Adultos 0,5ml. Población pediátrica. Niños a partir de los 36 meses de edad: 0.5 ml. Niños a partir de los 6 a 35 meses: los datos clínicos son limitados. Se puede administrar una dosis de 0,25 ml. La dosis administrada debe cumplir con las recomendaciones nacionales en vigor. Los niños que no han sido vacunados anteriormente, deberán recibir una segunda dosis, después de un intervalo de al menos 4 semanas. Forma de administración Administrar por vía intramuscular o subcutánea profunda. Precauciones que deben tomarse antes de manipular o administrar este medicamento Para consultar las instrucciones de preparación del medicamento antes de la administración, ver sección 6.6. 4.3 Contraindicaciones. Hipersensibilidad a los principios activos, a alguno de los excipientes o a cualquier compuesto que pudiera estar presente como traza, tales como huevo (ovoalbúmina, proteínas de pollo), neomicina, formaldehído y octoxinol 9. Se debe posponer la vacunación en caso de enfermedad febril o infección aguda. 4.4 Advertencias y precauciones especiales de uso. Al igual que con todas las vacunas inyectables, se recomienda disponer de un tratamiento médico apropiado y vigilar al sujeto en el caso raro de una reacción anafiláctica después de la administración de la vacuna. VAXIGRIP no debe administrarse en ningún caso por vía intravascular. La respuesta de anticuerpos en los pacientes que presentan una inmunosupresión congénita o adquirida puede ser insuficiente. 78 Interferencia con análisis serológicos: Ver sección 4.5. 4.5 Interacciones con otros medicamentos y otras formas de interacción. VAXIGRIP puede ser administrado al mismo tiempo que con otras vacunas. Sin embargo, las inyecciones deben administrarse en dos miembros diferentes. Debe tenerse en cuenta que pueden intensificarse los efectos adversos. La respuesta inmunitaria puede verse reducida si el paciente está recibiendo un tratamiento inmunosupresor. Tras la vacunación contra la influenza se han observado respuestas falsamente positivas a las pruebas serológicas que utilizan el método ELISA para detectar los anticuerpos contra la HIV1, hepatitis C y sobre todo HTLV1. Descartadas por el Western Blot, estas reacciones transitorias falsamente positivas se deberían a la respuesta IgM inducida por la vacunación. 4.6 Fertilidad, embarazo y lactancia. Embarazo Las vacunas antiinfluenza inactivadas se pueden utilizar en todas las fases del embarazo. Los datos de seguridad disponibles son más amplios para el segundo y tercer trimestres que para el primero. Sin embargo los datos sobre el uso de vacunas antigripales inactivadas a nivel mundial no indican que tengan ningún efecto anormal para el feto y la madre atribuible a la vacuna. Lactancia. La vacuna puede administrarse durante la lactancia. Fertilidad. No se dispone de datos de fertilidad. 4.7 Efectos sobre la capacidad para conducir y el uso de máquinas. La influencia de VAXIGRIP sobre la capacidad de conducir y utilizar máquinas es nula o insignificante. 4.8 Reacciones adversas Efectos adversos observados durante los ensayos clínicos: La tolerancia de las vacunas antiinfluenza trivalentes inactivadas se evalúa durante ensayos clínicos abiertos, no controlados, realizados anualmente según las exigencias reglamentarias. Estos ensayos incluyen al menos a 50 adultos de entre 18 - 60 años y al menos 50 personas de 61 años o mayores. La evaluación de la tolerancia se realiza durante los primeros 3 días siguientes a la vacunación. Se han observado los efectos adversos siguientes durante los ensayos clínicos con las frecuencias siguientes: Muy frecuentes (≥ 1/10); frecuentes (≥1/100, <1/10); poco frecuentes (≥ /1000, <1/100). Eventos adversos informados durante la vigilancia posterior a la comercialización Los eventos adversos informados durante la vigilancia posterior a la comercialización, junto con los ya observados durante los ensayos clínicos, son los siguientes: Trastornos de la sangre y del sistema linfático: Trombocitopenia transitoria, linfadenopatía transitoria. Trastornos del sistema inmunológico: Reacciones alérgicas, que provocan shock en casos raros, angioedema. Trastornos del sistema nervioso: Neuralgia, parestesia, convulsiones febriles, trastornos neurológicos tales como encefalomielitis, neuritis y síndrome de Guillain-Barré. Trastornos vasculares: Vasculitis asociada en casos muy raros a problemas renales transitorios. Trastornos de la piel y del tejido subcutáneo: Reacciones cutáneas generalizadas que incluyen prurito, urticaria, rash no específico. 4.9 Sobredosificación. Es improbable que una sobredosis produzca algún efecto adverso. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas. Grupo farmacoterapéutico: Vacuna antiinfluenza La seroprotección se obtiene generalmente a las 2 ó 3 semanas. La duración de la inmunidad posterior a la vacunación contra las cepas homólogas o cepas estrechamente relacionadas con las cepas de la vacuna varía, pero generalmente es de 6 a 12 meses. 79 5.2 Propiedades farmacocinéticas. No se aplica 5.3 Datos preclínicos sobre seguridad. No se aplica 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes. Cloruro de sodio, cloruro de potasio, fosfato disódico dihidratado, fosfato monopotásico y agua para inyectables. 6.2 Incompatibilidades. En ausencia de estudios de compatibilidad, esta vacuna no debe mezclarse con otros medicamentos. 6.3 Período de validez. 1 año 6.4 Precauciones especiales de conservación. Conservar en refrigerador (entre 2ºC a 8º C). No congelar. Conservar la jeringa en el embalaje exterior para protegerla de la luz. 6.5 Naturaleza y contenido del envase. 0,5 ml de suspensión en jeringa precargada (vidrio tipo I) con aguja unida equipada de un tapón émbolo (elastómero clorobromobutilo o clorobutilo o bromobutilo). 6.6 Instrucciones de uso, manipulación y eliminación. La vacuna debe estar a temperatura ambiente antes de usar. Agitar antes de usar. Antes de administrar, verifique el contenido visualmente. La vacuna no debe usarse si hay partículas extrañas en la suspensión. Cuando se indica una dosis de 0,25 ml en niños, empuje el tapón émbolo exactamente hasta el límite de la marca en la jeringa para que se elimine la mitad del volumen. Debe inyectarse el volumen restante. Ver también la rúbrica 4.2. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN SANOFI PASTEUR S.A. 2, avenue Pont Pasteur 69007 LYON - FRANCIA 80 Virus Rabia “Crear vacunas, es proteger la vida” 81 VERORAB Vacuna Antirrábica Celular Inactivada Polvo Liofilizado para Inyectable 2,5 U.I. con Solvente 1.DENOMINACIÓN DEL MEDICAMENTO VERORAB, polvo y disolvente para suspensión inyectable en jeringa precargada, Vacuna antirrábica para uso humano preparada en cultivos celulares (inactivada). 2.COMPOSICIÓN CUALITATIVA y CUANTITATIVA Después de la reconstitución, 1 dosis (0,5 mL) contiene: Virus de la rabia*; cepa Wistar Rabies PM/WI38 1503-3M (inactivada)…> 2,5 UI * Producido en células VERO Para la lista completa de excipientes, ver sección 6.1. 3.FORMA FARMACÉUTICA Polvo y disolvente para suspensión inyectable en jeringa precargada. Polvo de color blanco uniforme. 4.DATOS CLÍNICOS 4.1Indicaciones terapéuticas VERORAB está indicado para la prevención de la rabia. Prevención de la rabia antes de la exposición (vacunación en preexposición) La vacunación antes de la exposición debe recomendarse a los sujetos que presenten un riesgo elevado de contaminación por el virus de la rabia. Debe ser vacunada toda persona con riesgo permanente, como el personal de laboratorio de diagnóstico, de investigación o producción que trabaje con el virus de la rabia. Se aconseja realizar un control serológico cada 6 meses (ver sección 4.4). La vacunación anterior a la exposición debe recomendarse igualmente a los sujetos con riesgo frecuente de exposición al virus de la rabia. - Los veterinarios y sus ayudantes, los cuidadores de animales. - Las personas que, debido a su actividad profesional o durante su tiempo libre, entren en contacto con especies como el perro, el gato, la mofeta, el mapache o el murciélago u otras especies que puedan tener la rabia. Por ejemplo, Ios guardias de caza, los cazadores, los trabajadores forestales y espeleólogos, los taxidermistas. - Adultos y niños que residan o pasen temporadas en zonas de enzootia. Pueden realizarse controles serológicos cada 2 ó 3 años en todos los individuos sometidos a exposición discontinua. En las zonas de baja enzootia rábica, se considera que los veterinarios y sus asistentes (incluidos los estudiantes), los cuidadores de animales y el personal de las reservas naturales (guardas de caza) corren un riesgo ocasional de exposición y deben recibir una primovacunación antirrábica. Deben realizarse controles serológicos de anticuerpos contra la rabia a intervalos regulares en función del riesgo presentado por cada sujeto. Se administrarán inyecciones de refuerzo sistemáticamente en función del riesgo que corra el sujeto. La frecuencia de administración de las inyecciones de refuerzo se describe en la sección 4.2. Prevención de la rabia después de la exposición (vacunación en postexposición): 82 Al menor riesgo de contaminación rábica, la vacunación postexposición debe realizarse lo antes posible. En algunos países, la vacunación debe realizarse en un centro antirrábico especializado. El tratamiento posterior a Ia exposición incluye el tratamiento local no específico de la herida, una inmunización pasiva con inmunoglobulinas antirrábicas (IGR) y la vacunación en función de la naturaleza de la herida y el estado del animal (ver tablas 1 y 2). Tabla 1: Instrucciones a seguir dependiendo del estado del animal Tabla 2: Guía de la OMS para el tratamiento posterior a la exposición en función de la gravedad de la herida 83 4.2 Posología y forma de administración Posología VERORAB puede administrarse a los adultos y a los niños utilizando la misma posología. EI esquema de vacunación debe adaptarse a las circunstancias de la vacunación y al estado de inmunidad antirrábica del sujeto. 4.2.1 Vacunación y preexposición Deben administrarse tres dosis de VERORAB (0,5 mL) los días D0, D7 y D28 o D21. Refuerzo después de la vacunación de preexposición Se administrará una inyección de refuerzo de VERORAB (0,5 mL) un año después de la primovacunación, y después una inyección de refuerzo cada cinco años (tabla 3). Tabla 3: Recomendaciones relativas a la primovacunación y los refuerzos * La inyección del D28 puede administrarse el D21 VERORAB puede administrarse como inyección de refuerzo después de una primovacunación con una vacuna antirrábica de cultivo celular (vacuna antirrábica preparada en células VERO o preparada en células diploides humanas (HDCV)). 4.2.2 Vacunación de post exposición: Primeros auxilios: tratamiento local de la herida. Todas las mordeduras y arañazos deben lavarse inmediatamente con agua y jabón o detergente. Esto puede permitir eliminar de manera eficaz el virus de la rabia en el lugar de la infección. Podrá aplicarse a continuación una solución de alcohol al 70%, tintura (o una solución) de yodo, o una solución al 0,1% de amonio cuaternario (siempre que no existan restos de jabón, pues ambas sustancias se neutralizan mutuamente). En función de la gravedad de las heridas, si las inmunoglobulinas antirrábicas (IGR) deben administrarse en asociación con la vacuna, consultar las condiciones de utilización de las IGR que aparecen en el prospecto. Si es necesario, el tratamiento deberá completarse mediante la administración de una profilaxis antitetánica y/o una antibioticoterapia. Sujetos completamente inmunizados Deben administrarse dos dosis de refuerzo de VERORAB (0,5 mL) el D0 y el D3. En caso de duda o si el refuerzo se remonta a más de 5 años o si la vacunación es incompleta, el paciente no debe considerarse como completamente inmunizado, y debe establecerse el tratamiento completo de post exposición. Tabla 4: Recomendaciones relativas a la vacunación antirrábica post exposición según las inyecciones anteriores Sujetos no inmunizados: Deben administrarse cinco dosis de VERORAB (0,5 mL) los D0, D3, D7, D14 y D28. En caso de herida grave, deben administrarse inmunoglobulinas antirrábicas (IGR) al mismo tiempo que la primera inyección (categoría III según la clasificación OMS de riesgo rábico). Pueden utilizarse inmunoglobulinas equinas y humanas con VERORAB. La posología de las IGR reconocida a nivel internacional es la siguiente: Inmunoglobulinas humanas antirrábicas: 20 UI/kg de peso corporal. lnmunoglobulinas equinas antirrábicas: 40 UI/kg de peso corporal. Puesto que los IGR son capaces de inhibir parcialmente la producción activa de anticuerpos, la dosis administrada no debe superar la dosis recomendada. La vacuna se inyectará en la zona contralateral a los lugares de administración de las IGR. En las zonas geográficas con rabia endémica, la administración de dos inyecciones de vacuna el D0 puede justificarse, por ejemplo, cuando las lesiones son muy graves o se encuentran cerca del sistema nervioso, en caso de inmunodeficiencia del sujeto, o cuando se produce un retraso entre la exposición y la consulta médica. Forma de administración. VERORAB se administra por vía intramuscular solamente, en el deltoides en los adultos o en la región anterolateral del muslo en los niños pequeños (ver igualmente secciones 4.4 y 6.6). 84 4.3 Contraindicaciones 4.3.1 Preexposición Contraindicaciones habituales de cualquier vacunación: la vacunación debe diferirse en caso de fiebre o enfermedad aguda. Hipersensibilidad conocida al principio activo a uno de los excipientes, a la polimixina B, a la estreptomicina o a la neomicina. En todos los casos, debe valorarse la relación riesgo-beneficio. 4.3.2 Post exposición Debido a la evolución siempre fatal de la infección rábica, no existe ninguna contraindicación de la vacunación post exposición. 4.4 Advertencias y precauciones especiales de empleo Las recomendaciones relativas al calendario de inyección deben respetarse escrupulosamente. Especialmente en los casos de post exposición, se debe administrar el tratamiento en función del estado del animal, de las circunstancias del contacto así como de la naturaleza de la herida (ver sección 4.2). Al igual que con todas las vacunas inyectables, se recomienda disponer de un tratamiento médico apropiado para hacer frente a una posible reacción anafiláctica inmediata después de la inyección de la vacuna, en especial en caso de post exposición en los sujetos que padezcan una hipersensibilidad conocida a la polimixina B, Ia estreptomicina o la neomicina. No inyectar por vía intravascular. Comprobar que la aguja no penetre en un vaso sanguíneo antes de la inyección de la vacuna. VERORAB no debe administrarse por vía subcutánea. VERORAB no debe inyectarse en la región del glúteo ya que se han observado los títulos de anticuerpos neutralizantes más débiles en esta región. Son necesarios controles serológicos regulares. Estos controles serológicos se realizan mediante la verificación de la neutralización completa de un virus de prueba por el método RFFIT (Prueba rápida de inhibición de foco fluorescente). Esta prueba debe efectuarse cada 6 meses en personas expuestas a un riesgo permanente, y cada 2 ó 3 años después de cada refuerzo en sujetos sometidos a una exposición discontinua. Si el índice de anticuerpos está por debajo de un título considerado como protector, 0,5 UI/mL (RFFIT), debe administrarse una inyección de refuerzo. Cuando la vacuna se administra a sujetos que presentan una inmunodeficiencia conocida, debido a una enfermedad inmunosupresora o a un tratamiento inmunosupresor concomitante (como por ejemplo, corticoides), debe realizarse un control serológico de sus índices de anticuerpos entre 2 y 4 semanas después de la vacunación. Si el índice de anticuerpos está por debajo del título considerado como protector, 0,5 UI/mL (RFFIT), debe administrarse una inyección suplementaria. Debe tenerse muy en cuenta el riesgo potencial de apnea y la necesidad de vigilancia respiratoria durante 48-72 horas cuando se administren las dosis de primovacunación en los muy prematuros (nacidos < 28 semanas de embarazo) y particularmente aquéllos que tienen antecedentes de inmadurez respiratoria. Debido al beneficio considerable de la vacunación en estos lactantes, la administración no debe suspenderse ni aplazarse. 4.5 Interacción con otros medicamentos y otras formas de interacción Los corticoides así como los otros tratamientos inmunosupresores pueden interferir en la producción de anticuerpos y hacer que la vacuna no surta efecto, ver sección 4.4. Las inmunoglobulinas deben administrarse en un lugar diferente al de la vacuna (lado contralateral), ver sección 6.2. 4.6 Embarazo y lactancia Embarazo: No se dispone de información fiable de teratogénesis en el animal. En cuanto al aspecto clínico, el uso de la vacuna en un número limitado de embarazos no ha demostrado, al parecer, ningún efecto tóxico o fetotóxico particular hasta ahora. De cualquier modo, son necesarios estudios complementarios para evaluar las consecuencias de una exposición durante el embarazo. Debido a la gravedad de la enfermedad, no debe modificarse la aplicación de la vacunación durante el embarazo. Lactancia: Se puede administrar la vacuna durante la lactancia. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas Se ha informado con frecuencia la presencia de mareos después de la vacunación. Esto puede tener efectos temporales sobre la capacidad para conducir vehículos o utilizar máquinas. 4.8 Reacciones adversas Reacciones locales y benignas: dolor, eritema y edemas, prurito e induración en el lugar de la inyección. Reacciones generales: fiebre moderada, escalofríos, malestar general, astenia, cefaleas, mareos, artralgia, mialgias, trastornos gastrointestinales (náuseas, dolores abdominales). Excepcionalmente: reacciones anafilactoides, urticaria, erupción cutánea. Apnea en los muy prematuros (nacidos < 28 semanas de embarazo) (ver sección 4.4). 4.9 Sobredosis No se han descrito casos de sobredosis. 5 PROPIEDADES FARMACOLÓGICAS Grupo farmacoterapéutico: VACUNAS ANTIRRÁBICAS Código ATC: J07B G 5.1 Propiedades farmacodinámicas La inyección de VERORAB provoca una respuesta inmunitaria específica. La neutralización del virus de la rabia mediante los anticuerpos antirrábicos juega un papel fundamental en la protección. Debido a la evolución fatal de la enfermedad, no puede realizarse un estudio controlado sobre la eficacia. De cualquier modo, 85 un título de anticuerpos antirrábicos > 0,5 UI/mL (medido por RFFIT) se considera por la OMS como un elemento indicativo de protección. 5.2 Propiedades farmacocinéticas No aplicable. 5.3 Datos preclínicos sobre seguridad No aplicable. 6 DATOS FARMACÉUTICOS 6.1 Lista de excipientes Polvo: maltosa, albúmina humana. Disolvente: cloruro de sodio, agua para inyectables. 6.2 Incompatibilidades Las inmunoglobulinas antirrábicas y las vacuna antirrábicas nunca deben asociarse en la misma jeringa ni administrarse en el mismo sitio anatómico. 6.3 Periodo de validez 3 años. La vacuna debe utilizarse de inmediato después de su reconstitución. 6.4 Precauciones especiales de conservación Conservar en nevera (entre 2°C y 8°C). No congelar. 6.5 Naturaleza y contenido del recipiente Polvo en frasco (vidrio de tipo I) provisto de un tapón (clorobutilo) y de una cápsula + 0,5 mL de disolvente en jeringa precargada (vidrio de tipo I) provista de un tapón-émbolo (clorobromobutilo) - Caja de 1 6.6 Instrucciones de uso y manipulación Para reconstituir la vacuna: - Retirar la tapa del frasco de vacuna. - Inyectar el contenido de la jeringa precargada en el frasco de polvo. - Agitar suavemente hasta obtener una suspensión homogénea de vacuna. La vacuna reconstituida se presenta en forma de líquido límpido. - Aspirar inmediatamente 0,5 mL de suspensión. - Inyectar 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN SANOFIPASTEUR 2 avenue Pont Pasteur 69007 LYON 86 87 88 89 Vademécum Vacunas SCLSPA140701 Sanofi Pasteur 90