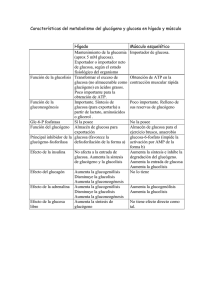
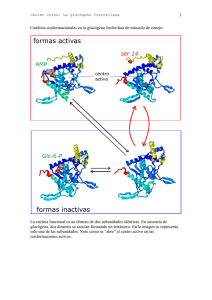
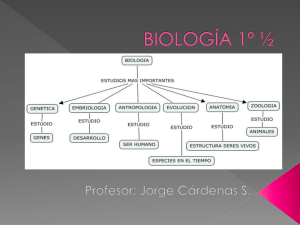
TEMA 10-Glucogenosis - Blog de Química Biológica Patológica
Anuncio

Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 1. INTRODUCCIÓN Las glucogénesis son un grupo de enfermedades hereditarias con una característica bioquímica común: una alteración del depósito de glucógeno en los tejidos afectados en los que puede estar aumentado o tener una estructura anómala. Se producen cuando existe deficiencia genética de la actividad de alguna de las enzimas que lo degradan o lo sintetizan. De aquí, que los dos tejidos más afectados sean aquellos en los que el metabolismo del glucógeno es más importante: el hígado y el músculo. En la mayoría de las glucogenosis las manifestaciones clínicas se consideran, esencialmente, en la expresión de la dificultad que existe en estos tejidos para movilizar sus depósitos de glucógeno. Así, si el hígado es el afectado, se produce hepatomegalia, alteración en la regulación de la glucemia en el periodo postabsortivo y disminución en el crecimiento. Cuando es el músculo, puede aparecer debilidad muscular, fatiga precoz al ejercicio e incluso, en algunos tipos, dolor muscular y contracturas cuando el ejercicio es rápido e intenso. También existen otras glucogenosis cuyas manifestaciones clínicas no están relacionadas con la existencia de un defecto en la degradación fosfolítica del glucógeno como ocurre en la deficiencia de α-glucosidasa ácida y en la deficiencia de la enzima ramificante. En el primer caso, el glucógeno se acumula en una inusual localización subcelular y en el segundo posee una estructura anómala. En general, se pueden distinguir tres tipos de glucogenosis atendiendo a la expresión clínica y hallazgos histopatológicos: glucogenosis hepática, glucogénesis muscular y glucogenosis generalizada (con manifestaciones hepáticas, musculares y cardíacas). A partir que Carl y Gerty Cori descubrieran en 1952 la deficiencia específica de actividad Glucosa-6-fosfatasa, se fueron identificando otras deficiencias enzimáticas como causa de diferentes glucogenosis. Atendiendo a la actividad enzimática deficiente y distinguiendo entre las isoenzimas de uno u otro tejido, Cori sugirió una clasificación numérica, según un orden cronológico, que fue generalmente aceptada. Se llegaron a establecer hasta siete tipos bien definidos. Posteriormente, se han caracterizado nuevas deficiencias enzimáticas y en los últimos años se están empezando a conocer las mutaciones que las producen. En la actualidad se tiende a designar las glucogenosis según la naturaleza del déficit enzimático, y en algún caso, con subtipos. Aquí se utiliza la clasificación de Cori y el número del catalogo de MIM (Mendelian Inheritance in Man), (Tabla Nº1). Área Química Biológica Dra. Silvia M. Varas 1 Química Biológica Patológica Tipo y Nombre común Ia; Enfermedad de von Gierke Ib Ic II; Enfermedad de Pompe IIIa; Enfermedad de Cori o de Forbes IIIb Tema 10: Glucogenosis Tabla Nº1: Clasificación de la glucogenosis Principal Estructura tejido Glucógeno afectado Glucosa-6-fosfatasa Hígado, riñón Normal Déficit enzimático Glucosa-6-fosfatasa translocasa Transportador de Pi α- Glucosidasa ácida lisosomal Enzima desramificante Hígado, leucocitos Hígado “ “ Músculo esquelético y cardíaco Normal Hígado, músculo Ausencia de cadenas externas o muy cortas “ Enzima desramificante Enzima ramificante Hígado (Músculo: N) Hígado V; Enfermedad de McArdle VI; Enfermedad de Hers VII; Enfermedad de Tarui VIII o IX Fosforilasa muscular Fosforilasa hepática Fosfofructoquinasa Músculo esquelético Hígado Fosforilasa quinasa 0 Glucógeno sintasa IV; Enfermedad de Andersen Año: 2013 Cadenas no ramificantes muy largas Normal Síntomas Hepatomegalia, fallo renal Como en Ia. Susceptibilidad elevada a infecciones bacterianas. Neutropenia Como en la Ia Forma infantil: muere a la edad de 2 años; forma juvenil: defectos musculares (miopatía); forma adulta: como en la distrofia muscular. Hepatomegalia en los recién nácidos; miopatia Hepatomegalia en los recién nácidos Hepato y esplenomegalia, mioglobina en la orina. Normal Calambres inducidos por el ejercicio y dolor; mioglobina en orina Hepatomegalia Músculo eritrocitos Normal Como en V; también anemia hemolítica Hígado, leucocitos, músculo Hígado “ Hepatomegalia Normal, deficiente en cantidad Hipoglucemia, hipercetonemia, muerte temprana 2. EL GLUCÓGENO: ESTRUCTURA, METABOLISMO Y FUNCIÓN Las células animales almacenan glucosa en su citosol en forma de glucógeno, polímero muy ramificado y fácilmente movilizable. Su masa molecular es variable, dependiendo de las unidades glucosídicas que lo formen, aunque posee una estructura definida. Las unidades de glucosa, en numero de 20.000 a 30.000, están unidas por enlaces glucosídicos α-1-4 (amilosa), y α(1-6) (amilopectina). Aproximadamente el 90% de los enlaces glucosídicos son del tipo α (1-4) y el 10% del tipo α (1-6), formando estos los puntos de ramificación. Las cadenas internas entre dos puntos de ramificación están formadas por 3 o 4 unidades glucosídicas y las externas por 8-10 unidades (Fig. 1). Esta estructura le proporciona varias ventajas: una buena solubilidad para su tamaño, muchos puntos de acceso a las enzimas glucógenolíticas y la posibilidad de almacenar muchos residuos glucosilo sin modificar apreciablemente la presión osmótica intracelular. El músculo y el hígado son los tejidos donde se almacena la mayor parte del glucógeno del organismo. Área Química Biológica Dra. Silvia M. Varas 2 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 El músculo esquelético contiene alrededor de los 2/3 del glucógeno total en una concentración de hasta el 1 g por 100 g de tejido fresco. El hígado posee la concentración de glucógeno más elevada: en periodo postprandial, puede llegar hasta 5-7 g por 100 g de tejido fresco. En el músculo, como en otros tejidos, el glucógeno se utiliza como combustible glucolítico de la propia célula, aunque, en este caso, acoplado a las necesidades de su específica función contráctil. Su papel es muy diferente en el hígado; la glucosa producida en la glucógenolisis y liberada al líquido extracelular ayuda a mantener la glucemia, principalmente durante el ayuno temprano, y de esa forma será utilizada por todos los tejidos. Figura Nº1: Estructura del glucógeno En todos los tejidos, la síntesis y degradación del glucógeno se produce por vías metabólicas diferentes, en las que, en último término, dos enzimas: glucógeno sintasa y glucógeno fosforilasa, actúan directamente en cada una de ellas. Sus actividades son reguladas, de una forma coordinada, en una cascada multicíclica por una serie de mecanismos en los que se incluyen la fosforilación y la modulación por efectores alostéricos. 2.1- Las enzimas involucradas en el metabolismo del glucógeno Las enzimas involucradas en el metabolismo del glucógeno. Los alumnos repasaran la intervención de cada una de las siguientes enzimas: Área Química Biológica Dra. Silvia M. Varas 3 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 a) Las enzimas que sintetizan y descomponen el glucógeno: - Uridina difosfo(UDP)-glucosa pirofosforilasa - Glucógeno sintasa - La ramificante: transfiere un mínimo de 6 unidades glucosilos unidos (α-1→4) de un extremo externo a una cadena de glicógeno en una posición (α-1→6) sobre la misma cadena o una nueva. Figura Nº1. - La desramificante : tiene 2 actividades catalíticas independientes que ocurren en distintos sitios en una única cadena polipeptídica: tiene una actividad de transferasa y otra hidrolasa. La fosforilasa degrada las cadenas de glicógeno hasta 4 unidades glucosilos antes del punto (α-1→6) de ramificación. Luego la desramificante usando su actividad de transferasa (1,4-α-D-glucan, 1,4-α-D-glucan, 4-α-D- glicosiltransferasa) transfiere esos 3 residuos al extremo de otra cadena. Luego usando su actividad de hidrolasa (amilo-1,6-glucosidasa) escinde la glucosa unida en el punto de ramificación (α-1→6). Ver figura Nº 2. - Fosforilasa: cataliza el clivaje secuencial de unidades glucosilos de cadenas unidas (α-1→4) del glucógeno y libera Glu-1-P. Tres isoenzimas fosforilasas humanas han sido identificadas y clonadas: en hígado, músculo y cerebro. Se regula por interacción alostérica y modificaciones covalentes (fosforilación y defosforilación). Esta codificada por distintos genes. Es altamente regulada: es activa por fosforilacion en la Ser 14 en respuesta a epinefrina o glucagón y son inhibidas por desfoforilacion por la fosfoproteína fosfatasa-1. - α-glucosidasa ácida: tiene la capacidad de hidrolizar ambos tipos de uniones y lleva a cabo la completa degradación del glucógeno en el lisosoma. Su déficit produce la enfermedad de Pompe. Área Química Biológica Dra. Silvia M. Varas 4 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº2: Acción de la enzima desramificante. b) Enzimas que regulan la glucógeno fosforilasa y glucógeno sintasa: La glucógeno sintasa y la fosforilasa y sus actividades son a la vez reguladas por hormonas, a través de una variedad de mecanismos de fosforilación y desfosforilación. Ver figura Nº3. - Fosforilasa quinasa: es una proteína que fosforila y activa fosforilasa; constituída por múltiples unidades α, β, γ y δ. La unidad estructural es (αβγδ)4. Es activada por Ca2+ y por fosforilación de proteína quinasa dependiente de AMPc (PKA). Las subunidades α y β son subunidades regulatorias (reguladas por fosforilación), γ es la subunidad catalítica y la subunidad δ es la proteína calmodulina que es regulada por Ca2+. Calmodulina tiene dos sitios de unión al Ca2+, estos sitios de unión están formados por motivos hélice-bucle-hélice, casi superponibles, conocidos como manos EF. El gen PRKAG2 sobre el cromosoma 7 codifica la subunidad γ2 de proteína quinasa activada por AMP (AMPK), una subunidad regulatoria y mutaciones en PRKAG2 que da lugar a glicogenosis cardíaca asociada con deficiencia de proteína quinasa. Área Química Biológica Dra. Silvia M. Varas 5 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº3: Sistema interconvertible de la glucógeno fosforilasa - Proteína quinasa activadas por AMPc (AMPK): el complejo AMPK es un heterotrímero compuesto por subunidades α, β y γ que median la respuesta celular a cambios en la producción o consumo de ATP. - Proteína quinasa dependiente de AMPc (PKA): fosforila y activa la fosforilasa quinasa. La unión del AMPc a las subunidades regulatorias marca la disociación del tetrámero y liberación de las subunidades catalíticas las cuales son capaces de fosforilar proteínas target. - Fosfoproteína Fosfatasa-1: Hay 4 tipos de fosfatasas (serina/treonina) en humanos pero solo la Fosfatasa 1 y la Fosfatasa 2A son las únicas en músculo con actividad significativa en el metabolismo del glucógeno. La fosfoproteína fosfatasa elimina grupos fosforilos de la glucógeno fosforilasa α y de las subunidades α y β de la fosforilasa quinasa. La fosfoproteína fosfatasa-1 se controla diferente en músculo e hígado. En el músculo, la subunidad catalítica llamada PP1c es activa sólo cuando está unida al glucógeno a través de su subunidad GM unida al glucógeno. Es regulada por la fosforilación en dos sitios separados de GM (sitio 1 y sitio 2). La fosforilación del sitio 1 activa PP1c, mientras que la fosforilación del sitio 2 (por PKA) hace que PP1c se libere dentro del Área Química Biológica Dra. Silvia M. Varas 6 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 citoplasma inactivándola. En el hígado la fosfoproteína fosfatasa-1 también se une al glucógeno por la subunidad unida al glucógeno llamada GL, y no esta sujeta a fosforilación. c) Las enzimas que catalizan pasos reversibles. Las enzimas que catalizan los pasos reversibles no son reguladas. - Fosfoglucomutasa - Fosfohexosa isomerasa - La interconversión de glucosa y la glucosa 6-fosfato - Hexoquinasa - Glucoquinasa d) El sistema de la glucosa-6-fosfatasa La glucosa 6-fosfatasa cataliza el paso final de tanto la gluconeogénesis y la degradación del glucógeno (ver figura Nº3). Cataliza la hidrólisis de la glucosa 6fosfato a la glucosa y fosfato y es la única enzima que es capaz de producir cantidades significativas de glucosa en el cuerpo; la enzima desramificante y la α-glucosidasa ácida puede hacer pequeñas cantidades de glucosa. La glucosa 6-fosfatasa se encuentra en niveles tan altos sólo en el hígado, los riñones, y las células β de los islotes del páncreas. El sistema de la glu 6-fosfatasa (G6P) esta compuesto por: - La subunidad catalítica de la glucosa 6-fosfatasa La subunidad 1 de glucosa 6-fosfatasa humana (G6PC1, también conocida como la glucosa 6-fosfatasa-α) es un gen simple copia (GenBank 401120), ubicado en el cromosoma 17q21. Una segunda subunidad catalítica de la glucosa-6-fosfatasa (G6PC2) conocida como la proteína relacionada a glucosa 6-fosfatasa específica de los islotes (IGRP) se expresa selectivamente en las células β de los islotes del páncreas. La tercera subunidad catalítica de la glucosa-6-fosfatasa (G6PC3), también conocida como conocida como Glu- 6-fosfatasa-β, proteína relacionada a la subunidad catalítica de glucosa 6-fosfatasa expresada ubicuamente. Se expresa ampliamente entre los tejidos y en paralelo a la glucosa 6-fosfatasa-α. Área Química Biológica Dra. Silvia M. Varas 7 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 - Transporte de Glucosa 6-fosfato microsomal La actividad de glucosa 6-fosfatasa está latente. Es decir, hay más actividad enzimática en vesículas microsomales rotas que en vesículas intactas. Para explicar este fenómeno, se propuso que había una proteína de transporte de glu-6-P microsomal que limita la velocidad, denominado T1, translocasa 1 (G6P T1) (EMBLY 15409). Ahora se sabe que codificada por el gen SLC 37 A4 (solute carrier family 37 (glucose-6-phosphate transporter), member 4). - Transporte de fosfato microsomal El fosfato es uno de los productos de la hidrólisis de glucosa 6-fosfato. El fosfato es un inhibidor de la enzima glucosa-6-fosfatasa, y si se limita a la luz del retículo endoplásmico, se inhibiría la enzima. Se ha informado una proteína que transporta fosfato microsomal de 37kDa, lo transportaría hacia el citosol. Esta proteína también transporta pirofosfato y carbamil fosfato. - El transporte de glucosa microsomal Se han descriptos un total de 13 miembros de una familia de proteínas de facilitan el transporte de glucosa (GLUT). El hígado y las células β de los islotes pancreáticos expresan predominantemente el transportador de glucosa 2 (GLUT2). Las células musculares expresan predominantemente GLUT4 y GLUT1. La principal diferencia entre GLUT2 y los demás GLUT (GLUT1, GLUT3, GLUT4, y GLUT5) es que GLUT2 tiene un Km mucho mayor para la glucosa. e) Las proteínas glicolíticas - Fosfofructoquinasa (fosfofructoquinasa cataliza la conversión de la fructosa 6fosfato en fructosa 1,6-bisfosfato). Área Química Biológica Dra. Silvia M. Varas 8 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº4: Sistema de la glucosa-6-fosfatasa en el retículo endoplásmico (RE) f) Regulación del metabolismo del glucógeno En general, se asume que la glucógeno sintasa controla la velocidad del metabolismo del glucógeno. La síntesis está indirectamente regulado a través de la expresión del receptor de esteroides coactivador 2 (SRC-2). SRC-2 ha sido implicado en la regulación de las respuestas endocrinas múltiples, incluyendo la reproducción y el metabolismo energético, y ratones KO (por la técnica de knock out, se anula la expresión del gen en estudio) para SRC-2 mostraron una fenocopia (fenotipo idéntico) para la enfermedad por almacenamiento de glucógeno tipo Ia. Área Química Biológica Dra. Silvia M. Varas 9 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº5: Enzimas involucradas en la síntesis y degradación del glucógeno DEGRADACION DEL GLUCÓGENO La desintegración del glucógeno o glucogenólisis (ver figura Nº5) requiere de 3 enzimas: 1-La glucógeno fosforilasa (o simplemente fosforilasa), cataliza la fosforolisis de glucógeno para obtener Glu-1-P. 2- La enzima desramificante, que elimina las ramificaciones del glucógeno, haciendo accesibles a la acción de la fosforilasa y 3- La fosfoglumutasa que convierte Glu-1-P en Glu-6-P que tienen distintos destinos metabólicos. SINTESIS DEL GLUCÓGENO La síntesis del glucógeno o glucogenogenesis requiere 3 enzimas: 1- UDP-glucosa pirofosforilasa, que a partir de Glu-1-P + UTP→ UDP-Glu + 2PPi 2- Glucógeno sintasa: transfiere la unidad UDP-glu a un extremo no reductor del glucógeno para formar un enlace α (1→4). Solo genera enlaces α (1→4), para producir α-amilosa. Área Química Biológica Dra. Silvia M. Varas 10 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 3- La enzima ramificante transfiere segmentos de 7 residuos de una cadena con enlaces α(1→4) hasta el grupo C6OH del residuo glucosa en la misma cadena o en otra. Cada punto de ramificación debe estar alejado al menos por 4 residuos. REGULACION DEL METABOLISMO DEL GLUCÓGENO (A) Control alostérico directo de la glucógeno fosforilasa y la glucógeno sintasa La glucógeno fosforilasa y la glucógeno sintasa están bajo el control alostérico por efectores (+ y -). La glucógeno fosforilasa se activa por AMPc y se inhibe por ATP y Glu-6-P. Y la glucógeno sintasa se activa con altas concentraciones de ATP y Glu-6-P. (B) Modificaciones covalentes de la glucógeno fosforilasa y la glucógeno sintasa La glucógeno fosforilasa es activa cuando esta fosforilada. Involucran tres enzimas: la fosforilasa quinasa, PKA y la Fosfoproteína fosfatasa-1. La glucógeno sintasa puede ser fosforilada en al menos 9 residuos Ser por distintas quinasas (fosforilasa quinasa, PKA) y ser inactivada (C) Efectos hormonales sobre el metabolismo del glucógeno El metabolismo del glucógeno en el hígado esta controlado en gran parte por las hormonas polipeptídicas insulina y glucagón. En músculo y otros tejidos el control se ejerce por insulina y adrenalina y noradrenalina. Ver figura Nº 6. Figura Nº6: Cascada desencadenada por glucagón en hígado Área Química Biológica Dra. Silvia M. Varas 11 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 3. DIAGNÓSTICO DE LAS GLUCOGENOSIS HEPÁTICAS El diagnóstico de las glucogenosis y la posterior identificación de la deficiencia enzimática concreta se debe efectuar mediante el siguiente procedimiento: 3.1. Historia clínica - Anamnesis. - Examen físico: - Antropometría. - Fenotipo. - Hepatomegalia. 3.2. Técnicas de imagen a. Ultrasonografía: siempre indicada en la evaluación de la hepatomegalia y/o de la disfunción hepática mantenida. En el caso de las glucogenosis útil para: - Valoración de la alteración de la ecoestructura (sugerente de enfermedad de depósito). - Detección y seguimiento de las lesiones ocupantes de espacio: adenomas. b. Tomografia axial computarizada. c. Resonancia magnética. 3.3. Diagnóstico Bioquímico: Determinaciones básales, en ayuno: - Glucemia, ácido láctico. - Pruebas de función hepática. - Enzimas musculares: CPK. - Ácido úrico. - Metabolismo lipídico: colesterol y triglicéridos. - Hemograma y estudio de coagulación con plaquetas. - Cuerpos cetónicos en sangre/orina. Tabla Nº2: Características bioquímicas de la glucogenosis con afectación hepática Glucogenosis Hipoglucemia I Si <45 mg/dl III IV VI y IX Si No No No No No ± (ayunas) Ácido Láctico Hipelipidemia > 45,0045 22,50-45.00 mg/dl mg/dl Si Si Colesterol Triglicéridos Área Química Biológica Normal Dra. Silvia M. Varas - Normal 12 Química Biológica Patológica Ácido Úrico Tema 10: Glucogenosis Año: 2013 Normal Normal Normal Cetosis No Si No ±/Si Cetonuria No Si No ±/Si GOT, GPT CPK Afectación Normal Normal o Normal No Si No Si/No Afectación Renal Si No No No Afectación No Si Si/No No muscular cardíaca Pruebas de sobrecarga: Test dinámicos: Los detalles técnicos de cada prueba serán incluidos en el diagnóstico bioquímico (Pagina 42). - Curva de sobrecarga oral de glucosa: Se administran de 1,75 – 2 g/Kg de peso de glucosa oral, con determinaciones de glucosa a los tiempos: 0, 30, 60, 90 y 120 minutos. Respuesta normal: glucemia menor 140 a los 0 y 120 minutos. - Curva de glucagón: Valora la capacidad del hígado para liberar glucosa. Se administra 30µg/Kg im (máximo, 1 mg) de glucagón y se determina glucemia a los 0, 15, 30, 45, 60 90 y 120 minutos. Respuesta normal: elevación de la glucemia >25% entre los 15 y 45, volviendo después a la normalidad. En la glucogenosis de tipo I hay ausencia de respuesta pero con aumento de lactato mientras que en el déficit de la enzima desramificante (tipo III) existe una elevación cuando se realiza la prueba en etapa postprandial (ya que entonces puede movilizar los residuos de glucosa en posición α(1→4). - Curva de sobrecarga oral de galactosa: Se administran 2 g de galactosa al 10 % por Kg de peso. Se realizan extracciones para la determinación de glucosa y lactato cada 15 minutos durante 2 horas. - Test de ejercicio isquémico (prueba del lazo): se insufla el manguito de un manómetro por encima de la Presión arterial sistólica; se le dice al paciente que apriete una bola de goma con la mano del brazo isquémico de forma repetida. Se extrae al finalizar la prueba sangre para medir lactato plasmático del brazo isquémico. En algunas glucogenosis de afectación muscular se produce una respuesta tetánica de la extremidad sometida a isquemia, sin producirse un aumento de lactato. Área Química Biológica Dra. Silvia M. Varas 13 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Tabla Nº3: Respuesta a pruebas funcionales: sobrecarga de glucosa y test de glucagón Con hipoglucemia Respuesta a Respuesta al Respuesta al glucosa oral glucagón glucagón (Ayunas) (Postprandial) Tipo TG Ácido Lacta- Glucos Lactat Úrico to a o Glucosa Lactat Glucosa o I 0 III VI 0- N N N N Lactato 0- 0 0 0 0 0 0- 0 IX 3.4- Biopsia hepática 3.4.1. Morfología. Estudio histológico (MO): a. Confirmación del aumento de glucógeno hepático (intrahepatocitario). b. Valoración de la presencia de grasa (esteatosis). c. Identificación de fibrosis (hepatopatía progresiva) o cirrosis. d. Histoquímica. 3.4.2. Estudio ultraestructural y cuantificación del glucógeno acumulado. 3.5-. Diagnóstico Molecular Área Química Biológica Dra. Silvia M. Varas 14 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 ALGORITMO DIAGNÓSTICO PARA LAS GLUCOGENOSIS: Figura Nº5a: Diagnóstico de sospecha de glucógenolisis hepáticas: clínica y pruebas básicas Figura Nº5b: Diagnóstico de sospecha de glucógenolisis hepáticas: pruebas básicas y funcionales En la Figura Nº5 a y b se muestra un esquema diagnóstico de sospecha de glucogenosis hepática a partir de la clínica y unas determinaciones analíticas basales. Área Química Biológica Dra. Silvia M. Varas 15 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 4. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO I (DEFICIENCIA DE GLUCOSA-6-FOSFATASA, ENFERMEDAD DE VON GIERKE) La enfermedad por almacenamiento de glucógeno tipo I fue descrita por primera vez como "hepatonefromegalia glicogenica" de von Gierke en 1929, y sigue siendo ampliamente conocido como enfermedad de Von Gierke. Las glucogenosis tipo I son un grupo de enfermedades metabólicas hereditarias producidas por un defecto genético de algunos de los componentes del sistema enzimático de la glucosa-6-fosfatasa. Mediante ensayos enzimáticos en microsomas intactos o rotos, se identificaron cuatro subtipos: Glucogenosis tipo Ia, por deficiencia de glucosa-6-fosfato hidrolasa, Glucogenosis tipo Ib, por deficiencia del transportador de glucosa-6- fosfato, Glucogenosis tipo Ic y Glucogenosis tipo Id, posiblemente por deficiencia de los transportadores de la glucosa o del Pi/PP, respectivamente. Es una de las glucogenosis mas frecuentes, entre 1:100.000 y 1:300.000 recién nácidos. En la práctica parece que solo hay dos tipos de glucogenosis I (Ia e Ib), que se diferencian por medio de la determinación de la actividad enzimática y por el estudio de las mutaciones genéticas. El fenotipo clínico es similar en todas ellas, aunque los pacientes con el tipo Ib asocian neutropenia constante o cíclica, cuya gravedad oscila desde leve hasta la agranulocitosis y alteración de la función de los neutrófilos que condicionan infecciones bacterianas recurrentes y ulceras bucales e intestinales. Hallazgos clínicos y de laboratorio Los pacientes con enfermedad de tipo I de almacenamiento de glucógeno pueden presentar en el período neonatal con la hipoglucemia y ácidosis láctica, sin embargo, se presenta más comúnmente a los 3-4 meses de edad, con hepatomegalia y / o convulsiones hipoglucémicas. Estos niños a menudo tienen caras regordetas como muñecas con exceso de tejido adiposo en las mejillas, extremidades relativamente delgadas, baja estatura, y un abdomen prominente que es debido a la masiva hepatomegalia sin esplenomegalia; es habitual la nefromegalia. El corazón es de tamaño normal. Área Química Biológica Dra. Silvia M. Varas 16 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº7 : Paciente de 9 meses de edad con glucogenosis tipo I. El contorno del hígado ha sido demarcado con un lápiz marcador. Las características de la enfermedad son la hipoglucemia, ácidosis láctica, la hiperuricemia y la hiperlipidemia. La hipoglucemia y ácidosis láctica pueden ocurrir después de un ayuno corto. En el pasado, muchos pacientes con la enfermedad por almacenamiento de glucógeno no reconocido y sin ser tratados murieron en la infancia temprana con hipoglucemia y ácidosis profunda. La hiperuricemia está presente en los niños pequeños, pero rara vez se desarrolla la gota antes de la pubertad. A pesar de la marcada hepatomegalia, las transaminasas hepáticas suelen ser normales o sólo ligeramente elevadas. Recientes estudios han demostrado que aumenta la actividad de biotinidasa en plasma en los pacientes con glucogenosis tipo Ia y Ib, tipo III, VI y IX (Paesold-Burda et al., 2007). Ocasionalmente los pacientes con glucogenosis tipo I refieren diarrea y en la tipo Ib se ha descripto una enfermedad inflamatoria intestinal, similar a la enfermedad de Crohn y que responde al factor estimulante de colonias de granulocitos (Melis y cols., 2003). Son comunes las menorragia, contusiones y epistaxis y se asocian con un tiempo de sangría prolongado como resultado de la agregación plaquetaria y deterioro de la adherencia. Área Química Biológica Dra. Silvia M. Varas 17 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº8: Fisiopatología de la glucogenosis tipo I El plasma puede ser "lechoso" en apariencia, como resultado de una elevación notable de los triglicéridos. Colesterol y fosfolípidos estan también elevados, pero en menor proporción. Las cifras de triglicéridos pueden alcanzar los 4.000 a 6.000 mg/dl y de colesterol de 400 a 600 mg/dl. La histología del hígado se caracteriza por una distensión universal de los hepatocitos por acúmulo de glucógeno y grasa. Las vacuolas de lípidos son particularmente grandes y prominentes. Complicaciones a largo plazo Sin embargo, a pesar de un buen control metabólico, algunas de las complicaciones a largo plazo, son el desarrollo de adenomas hepáticos con la progresión a carcinoma hepatocelular y enfermedad renal progresiva Fisiopatología Los hallazgos bioquímicos más importantes en la enfermedad por almacenamiento de glucógeno de tipo I son la hipoglucemia, ácidosis láctica, la hiperuricemia y la hiperlipidemia (Figura Nº8). Además, el tipo de enfermedad por almacenamiento de Área Química Biológica Dra. Silvia M. Varas 18 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 glucógeno Ib presenta una anormalidad celular, clínicamente significativa la neutropenia. La fisiopatología de estos hallazgos clínicos son: - La hipoglucemia La deficiencia de glucosa 6-fosfatasa en el hígado bloquea los últimos pasos de las vías glucogenolíticas y la gluconeogénesis. También interrumpe el reciclaje constante de glucosa en el hígado a través de la glucosa a glucosa 6-fosfato al sistema de la glucosa. Como era de esperar, estos pacientes no son capaces de mantener niveles normales de glucosa en sangre en ayunas. - La ácidosis láctica Los altos niveles de fructosa-6-fosfato (en equilibrio con la glucosa 6-fosfato) conduce a un aumento de la fructosa 2,6-bisfosfato (F26BP) y por lo tanto la activación de la fosfofructoquinasa y de la vía glicolítica; con aumento de piruvato y finalmente de ácido lactico. - La hiperuricemia La hiperuricemia es causada por la combinación de una disminución del aclaramiento renal asociado a un aumento de la producción endógena. La 1º causa, lactato compite con el ácido úrico para la excreción por riñón. Para la 2º causa, la acumulación de ésteres de fosfato resulta en disminución del fosfato inorgánico intrahepático. La disminución de la concentración de ATP y Pi estimula la actividad de AMP-desaminasa hepática, así que aumenta la degradación de los nucleótidos de adenina y se produce ácido úrico. Por otro lado, se ha visto que la administración de glucagón produce una depleción de ATP y fosfato (por generación de AMPc y PPi), (este de AMP refuerza la activación de AMP deaminasa), que conduce a una marcada hiperuricemia. Además la síntesis de la PP-ribosa-P puede ser estimulada por el incremento de las vía de las pentosas y estimula la síntesis de novo de ácido úrico. - Hiperlipidemia La hiperlipidemia es un resultado tanto del aumento de la síntesis de triglicéridos como de colesterol. El estímulo de la vía glicolítica en forma exagerada produce una generosa cantidad de NADH y NADPH y abundante restos de acetil-CoA y glicerol, por lo que ambos sustratos y cofactores se encuentran disponibles para la síntesis hepática de triglicéridos. El aumento de piruvato y la estimulación de la piruvato carboxilasa por el glucagón lleva a un aumento de oxalacetato, lo que, con acetil-CoA, forma citrato. Citrato proporciona una fuente de Acetil CoA para la enzima AcetilÁrea Química Biológica Dra. Silvia M. Varas 19 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 CoA carboxilasa, citosólica, que la carboxila a malonil-CoA. Malonil-CoA por un lado sigue la biosíntesis de los ácidos grasos y por otro lado, inhibe la carnitina palmitoil transferasa I, evitando la transferencia de los ácidos grasos en la mitocondria para su degradación por β-oxidación. No hay aumento de la síntesis de cuerpos cetónicos. - Neutropenia: Solo el tipo Ib se asocia con neutropenia. La médula ósea aparece normal a pesar que los neutrófilos en la sangre están muy reducidos. Los neutrófilos tienen una estallido oxidativo reducido cuando se estimulan y una defectuosa quimiotaxis. El tratamiento con G-CSF (Granulocyte colony-stimulating factor) es eficaz para corregir la neutropenia y la mejora de la enfermedad inflamatoria del intestino, pero se ha asociado con una evolución a leucemia mielógena aguda en los pacientes. Los neutrófilos de ratones KO para el gen de glucosa 6-fosfato translocasa presentan un incremento del estrés en RE, evidenciado por aumento de la expresión de chaperonas en el RE y estrés oxidativo. Además existe un incremento de activación de caspasa 3 y Bax (proteína pro-apoptótica) con muerte por apoptosis. Bioquímica y base molecular La enfermedad por almacenamiento de glucógeno tipo Ia es la deficiencia de la subunidad catalítica de la glucosa 6-fosfatasa (G6PC1, también conocida como la glucosa 6-fosfatasa-alfa). La enfermedad por almacenamiento de glucógeno tipo Ib es la deficiencia de la translocasa de la glucosa 6-fosfato hepática microsomal. In vivo, se traduce en anormalmente baja o ausente actividad de glucosa 6-fosfatasa hepática en microsomas intactos, dependiendo si el defecto es completo o parcial. Se aisló un cDNA que codifica la translocasa de glucosa 6-fosfato y caracterizada (EMBL Y15409). El gen del transportador de glucosa-6-fosfato ( SLC37 A4), antes llamado de G6P-translocasa (G6PT1) está localizado en el cromosoma 11q23, mutaciones en el gen del transportador es el responsable del trastorno tipo Ib. Dos mutaciones, G339C y 1211delCT, parecen ser las mutaciones mas frecuentes en pacientes de raza blanca. Diagnóstico El método mas seguro de diagnóstico lo constituye la determinación del nivel de actividad de la glucosa-6- fosfatasa en hígado. Las determinaciones de glucemia y lactato en ayunas y las respuestas a la sobrecarga oral de glucosa y al test del glucagón (escaso o nulo aumento de la glucemia y marcado incremento del nivel de lactato, ya Área Química Biológica Dra. Silvia M. Varas 20 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 basalmente elevado), o las respuestas a la administración de galactosa o fructosa apoyan la sospecha diagnóstica, pero no permiten un diagnóstico definitivo. Ver Tabla Nº4. El estudio de dos casos con glucogenosis tipo Ib (E. Pallarés Querol y col., 2002) demostraron que la sobrecarga con glucosa generaba que los valores de lactato plasmático disminuyeron con el tiempo en los pacientes y permanecía sin cambios en los controles. Ver figura Nº 9. Figura Nº 9: Sobrecarga de Glucosa en pacientes controles y con tipo Ib: niveles de lactato. Cuando se le hizo una sobrecarga de galactosa pone de manifiesto que la galactosa no se puede convertir en glucosa, pero sí en lactato. Tabla Nº 4. Glucosa (mmol/L) Lactato (mmol/L) Basal 5,8 4,9 Tabla Nº 4 : Sobrecarga de Galactosa 20 min 40 min 60 min 80 min 4,3 3,5 3,3 2,7 7,1 7,2 8,0 100 min 2,4 120 min 2,1 8,5 9,6 7,8 Y en la sobrecarga con glucagón (Tabla Nº 5) se vio cómo la glucosa sérica no respondió al glucagón pero hubo aumento de lactato (de 5 a 11 mmol/L) con 60 minutos de ayuno. Se confirmó así una hipoglucemia normocetósica con hiperlacticoacidemia. Basal (ayuno) Glucosa (mmol/L) Caso 1 Caso 2 1 1,2 Área Química Biológica Tabla Nº 5: Sobrecarga de Glucagón 10 min. 20 min. 40 min. 60 min. 0,7 1,1 0,6 1,0 0,5 -- Dra. Silvia M. Varas 0,4 -- Observaciones Asintomático Sintomático 21 Química Biológica Patológica Lactato (mmol/L) Caso 1 Caso 2 5 9,3 Tema 10: Glucogenosis -13,5 Año: 2013 11 -- Genética El diagnóstico de la enfermedad por almacenamiento de glucógeno tipo I se hereda como un rasgo autosómico recesivo. El gen de la glucosa-6-fosfatasa α esta el cromosoma 17q21 y la translocasa se encuentra en el cromosoma 11q23. El diagnóstico prenatal de la enfermedad de tipo Ia se ha logrado a través de biopsia de hígado fetal. En este momento, el análisis de la mutación del ADN en las vellosidades coriónicas o amniocitos en tanto en la enfermedad del tipo Ia como Ib evita la necesidad de una biopsia de hígado fetal. Se han identificado 56 mutaciones, en 600 alelos de 300 pacientes de familias no emparentadas. Tres mutaciones: R83C, Q347X y 727G—> T, constituyen el 60% de todos los alelos mutados (32,5%, 14,3% y 11,3% respectivamente); mientras que las restantes ninguna llegan al 5% e incluso 28 mutaciones se encuentra en un solo alelo. Por tanto, cuando se plantee el diagnóstico de una Glucogenosis tipo I, deficiencia en el complejo multienzimático de la glucosa-6- fosfatasa, será necesario realizar el ensayo en tejido hepático sin congelar (mantenido en hielo), y recién extraído; para poder medir la actividad glucosa-6-fosfatasa “total” así como la actividad “latente” y poder distinguir entre los dos subtipos, actualmente establecidos, de esta glucogenosis: Ia, Ib. En esta deficiencia no se puede realizar el diagnóstico enzimático utilizando células sanguíneas. Evolución Clínica y complicaciones Es fundamental un adecuado tratamiento dietético de forma mantenida para prevenir o reducir la mayoría de las complicaciones. Puede existir una talla baja además de un retraso puberal. La fertilidad posterior no parece estar afectada. Los enfermos no tratados presentan alrededor de la pubertad las siguientes complicaciones: - Hiperlipidemia. - Pancreatitis. - Mayor susceptibilidad para el desarrollo de arteriosclerosis. Área Química Biológica Dra. Silvia M. Varas 22 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 - Alteración de la agregabilidad plaquetaria en el tipo Ib, que puede ser un factor protector frente a la arteriosclerosis. - Pueden aparecer adenomas hepáticos en la 2-3ª década de la vida, que pueden malignizarse ocasionalmente. - Afectación renal: nefromegalia bilateral y, de forma progresiva, proteinuria, hipertensión arterial, litiasis renal, hipofosfatemia, nefrocalcinosis, glomeruloesclerosis y fibrosis intersticial progresiva; todo ello puede ocasionar una insuficiencia renal, susceptible de diálisis e incluso trasplante. - Hiperuricemia, con episodios de gota en el adulto. - Con el embarazo pueden exacerbarse los síntomas. - Excepcionalmente puede presentarse hipertensión pulmonar con insuficiencia cardiaca secundaria. - Osteoporosis. - Ocasionalmente puede evolucionar a una cirrosis con hipertensión portal. - La presencia de episodios recurrentes de ácidosis láctica constituirá un signo de mal pronóstico. Si se interrumpe el tratamiento dietético prescrito en un paciente adolescente o adulto puede presentarse una cierta tolerancia a la hipoglucemia. A medida que pasan los años, los problemas metabólicos se vuelven menos intensos y más fáciles de tratar. 5. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO III (ENFERMEDAD POR DEFICIENCIA DE LA ENZIMA DESRAMIFICANTE, DEXTRINOSIS LÍMITE, ENFERMEDAD DE CORI O DE FORBES) En 1952, Illingworth y Cori, reconoce la presencia de excesivas cantidades de estructura anormal de glicógeno en hígado y músculo de un paciente que fue estudiado clínicamente por Forbes. El glucógeno tenía las cadenas externas muy cortas como una dextrina limite. La enfermedad es también llamada dextrinosis límite o enfermedad de Cori o Forbes. Es causada por la ausencia de la amilo-1-6-glucosidasa o enzima desramificante, lo que condiciona la acumulación de dextrinas límites, disminuyendo la liberación de glucosa, aunque los pacientes conservan intacto el mecanismo de la gluconeogenesis. En la Glucogenosis tipo III, la actividad amilo-1,6- glucosidasa puede ser deficiente Área Química Biológica Dra. Silvia M. Varas 23 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 en el hígado y en el músculo, tipo IIIa, el mas frecuente (85% de todos los pacientes), o solo en el hígado, tipo IIIb. Historia y descripción general La mayoría de los pacientes con enfermedad por almacenamiento de glucógeno tipo III sobreviven a una larga vida, tal como, por ejemplo, los dos pacientes que originalmente fueron informados por Snappes y Van Creveld en 1928, quienes presentaban hepatomegalia y una incapacidad para movilizar el glucógeno del hígado. Estos pacientes fueron estudiados luego por Van Creveld y en Huijing en 1964, y demostraron que eran deficientes en la actividad de la enzima desramificante. Ambos pacientes, un hombre y una mujer, habían mejorado clínicamente y mostraron una menor hepatomegalia después de la pubertad, un hallazgo relativamente común en los individuos afectados. Se ha afirmado desde hace tiempo que los pacientes adultos la mayoría son asintomáticos. En las últimas tres décadas, sin embargo, varios informes han documentado la presencia de una miopatía esquelética progresiva, cardiomiopatía, fibrosis miocárdica, cirrosis hepática y / o carcinoma hepatocelular en estos pacientes. En los dos tejidos se acumula un polisacárido que, cuando se caracteriza por el espectro del complejo iodado, tiene una estructura parecida a la dextrina limite que produce la fosforilasa. La variabilidad fenotípica clínica y enzimática en las Glucogenosis tipo III parece que también esta relacionada con la complejidad de las características del gen de la enzima desramificante. Hallazgos clínicos y de laboratorio La clínica es variable dependiendo de la diferente expresión tisular de la enzima deficiente. Los pacientes afectados tienen un cuadro clínico similar, pero más leve que el tipo I y son capaces de tolerar períodos de ayuno más prolongados, sobre todo al aumentar la edad. Sin embargo, en el lactante y en el niño pequeño el cuadro clínico de ambas glucogenosis es indistinguible: hepatomegalia, hipoglucemia en ayunas con cetosis, hiperlipidemia y retraso del crecimiento. Las transaminasas están discretamente elevadas a no ser que la enfermedad hepática este muy avanzada. La función hepática tiende a normalizarse con la edad al tiempo que disminuye el tamaño hepático. La hepatomegalia depende del depósito de glucógeno y la infiltración grasa es mínima o nula. La fibrosis periportal o septal esta siempre presente Área Química Biológica aunque solo excepcionalmente Dra. Silvia M. Varas progrese a cirrosis. 24 La Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 esplenomegalia solo aparece en caso de hepatopatía fibrosante progresiva. En el 25% de los pacientes se ha descrito la aparición de adenomas hepáticos, ninguno de los cuales ha sufrido malignización. Los pacientes con glucogenosis III pueden tener afectación muscular que se manifiesta en la tercera o cuarta década de la vida como debilidad muscular que empeora con el ejercicio y atrofia de la masa muscular (glucogenosis IIIa). Los niveles plasmáticos de CPK están elevados. En este tipo de glucogenosis no hay afectación renal ni nefromegalia, pero si afectación cardíaca con cardiomegalia por cardiomiopatia hipertrófica; el aumento concéntrico del grosor de la pared del ventrículo izquierdo se hace mas evidente con la pubertad. En los pacientes con afectación muscular (tipo IIIa), la debilidad muscular suele ser mínima durante la infancia, pero puede llegar a ser severa después de la tercera o cuarta década de la vida, evidenciada por una perdida lenta y progresiva del tejido muscular. La hipoglucemia y la hiperlipidemia son hallazgos comunes. En contraste con la enfermedad por almacenamiento de glucógeno, tipo I, la elevación de las transaminasas hepáticas y cetosis en ayunas son prominentes, y el lactato sanguíneo y las concentraciones de ácido úrico son usualmente normales, Tabla Nº3. La administración de glucagón 2 horas después de comer hidratos de carbono provoca un aumento normal de la glucosa en sangre, pero después de un ayuno nocturno, el glucagón no provoca ningún cambio en la glucosa en sangre. Tabla Nº4. La histología del hígado se caracteriza por una distensión universal de hepatocitos por acúmulo de glucógeno y la presencia de septos fibrosos. La fibrosis y la escasez de grasa permite distinguir la glucogenosis tipo III de tipo I. La fibrosis, que oscila entre una fibrosis periportal mínima a la cirrosis micronodular, aparece en la mayoría de los casos por no ser progresiva. Sin embargo, esto debe ser seguido muy de cerca porque se ha informado de que los pacientes desarrollan cirrosis hepática manifiesta en la edad adulta Genética La glucogenosis tipo III se hereda de forma autosómica recesiva. La enfermedad ha sido reportada en los caucásicos, africanos, hispanos y asiáticos. La frecuencia de la enfermedad es relativamente alta en los judios Sefaradíes de ascendencia del norte de Área Química Biológica Dra. Silvia M. Varas 25 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Africa (la prevalencia de 1:5400). El diagnóstico prenatal y la detección de portadores son técnicamente difíciles con los ensayos enzimáticos, pero son posibles utilizando técnicas de biología molecular con análisis de la mutación o por estudio de ligamiento. Para el diagnóstico se pueden utilizar biopsias de ambos tejidos y también los eritrocitos. Si al medir la actividad en estas células, no es deficiente y el cuadro clínico así lo sugiere no se excluye que el déficit se exprese solo en hígado o músculo. Este gen tiene un tamaño de 85 kilobases comprende 35 exones. Actualmente se han identificado 24 mutaciones en pacientes con Glucogenosis tipo III; se destaca el hecho de que no hay ninguna predominante como ocurre en otras enfermedades metabólicas hereditarias. En el tipo IIIa y en pacientes caucásicos y afroamericanos las más frecuentes son: R864X (10,3%), 3964delT (6,7%), IVS32-12A > G (5,5%) y R1228X (5,2%). En los pacientes con el subtipo III b, se ha demostrado que dos mutaciones en el exón 3 se asocian de manera exclusiva: 17delAG y Q6X. Diagnóstico Bioquímico: La hipoglucemia es menos intensa que en el tipo I y los pacientes toleran periodos de ayuno más prolongados. El ayuno se acompaña de una importante cetonemia. Tabla Nº3. Tras el ayuno nocturno no hay aumento de la glucemia ni de lactato tras administración de glucagón. Ver Tabla Nº4. La glucemia sí aumenta cuando la curva se repite tras una comida rica en carbohidratos. Las curvas de galactosa y fructosa son normales. El lactato y el ácido úrico son normales o están moderadamente elevados. La hiperlactacidemia tras la ingesta sugiere una glucogenosis tipo III, sobre todo si el cuadro se acompaña de hepatomegalia. La hiperlipidemia no es tan pronunciada como en el tipo I. Los niveles séricos de creatina quinasa a veces pueden ser útiles para identificar pacientes con afectación muscular, pero los niveles normales no descartan la deficiencia de enzimas musculares. El diagnóstico definitivo se hace por medio de la determinación de la actividad enzimática en hígado o músculo. Área Química Biológica Dra. Silvia M. Varas 26 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Evolución Clínica y complicaciones En estos pacientes el tamaño renal es normal; la hipoglucemia es poco frecuente y no representa un problema grave. Generalmente no evoluciona a cirrosis, ya que la fibrosis objetivada permanece estable. 6. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO IV (DEFICIENCIA DE LA ENZIMA RAMIFICANTE, AMILOPECTINOSIS, O ENFERMEDAD DE ANDERSEN) En la enfermedad del almacenamiento de glucógeno, tipo IV es también conocida como enfermedad de Andersen o amilopectinosis en reconocimiento a Dorothy Hansine Andersen quien en 1956 realiza la descripción clínica de un paciente con una progresiva hepatoesplenomegalia que había acumulaba un polisacárido anormal con pobre solubilidad en el agua. En la Glucogenosis tipo IV el diagnóstico se realiza en una biopsia de hígado, demostrando que la actividad de la enzima ramificante es deficiente y que la estructura del glucógeno es similar a la de una amilopeptina, cuando se caracteriza por el espectro del complejo iodado. La cantidad de polisacárido que se acumula en el hígado (< 5 g/100 g) no esta aumentada, ni tampoco en los demás tejidos. La patogénesis de la lesión en el hígado no está clara. El material almacenado con estructura de glucógeno anormal y / o insolubilidad es más probable que sea la causa de la lesión celular, como en la enfermedad de tipo de almacenamiento de glucógeno tipo III. En los hepatocitos se acumula glucógeno y amilopectina. El comienzo de los síntomas suele ocurrir entre los 3 y los 15 meses. Los síntomas mas frecuentes suelen ser fallo de medro, hepatomegalia, distensión abdominal y otros síntomas digestivos. Al progresar la enfermedad hay evidentes signos y síntomas de hepatopatía crónica (cirrosis). En la mitad de los pacientes hay datos de afectación neuromuscular como hipotonía, atrofia muscular e hiporreflexia. Diagnóstico El cuadro clínico y bioquímico de la enfermedad no se puede distinguir de otras causas de cirrosis en la infancia. El diagnóstico del tipo IV de la enfermedad requiere una biopsia para la demostración de glucógeno anormal (largas cadenas externas y Área Química Biológica Dra. Silvia M. Varas 27 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 un polisacárido similar a la amilopectina) y una deficiencia de la enzima de ramificación en el hígado, músculo, leucocitos, eritrocitos, o fibroblastos Genética Enfermedad por almacenamiento de glucógeno tipo IV se hereda como un rasgo autosómico recesivo. La detección de portadores es posible mediante análisis de ADN. La deficiencia enzimática parcial se ha observado en portadores obligados por medición de la actividad enzimática en los leucocitos, eritrocitos, o fibroblastos de la piel. El diagnóstico prenatal por el ensayo de la enzima está disponible para la forma mortal de glucogenosis mediante el uso de cultivo de amniocitos o vellosidades coriónicas. La caracterización de las mutaciones en curso permitirá un diagnóstico basado en el ADN. Diagnóstico bioquímico La hipoglucemia es infrecuente. Las respuestas a las sobrecargas de galactosa y fructosa son normales. El lactato y piruvato son normales. Las transaminasas están moderadamente elevadas. La cuantificación de la actividad del enzima en tejido hepático confirma el diagnóstico. La hepatopatía suele progresar a una cirrosis macronodular con abundantes depósitos PAS positivos en los hepatocitos. Si la enfermedad no es tratada muchos pacientes mueren por falla hepática en los tres primeros años de vida. Evolución Clínica y complicaciones Existe hepato y esplenomegalia manifiesta y progresiva, que lleva a una insuficiencia hepática con fallecimiento en la infancia. Ocasionalmente el tratamiento con esteroides puede inducir una remisión temporal. 7. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO V (DEFICIENCIA ENFERMEDAD DE DE FOSFORILASA MCARDLE, MUSCULAR, DEFICIENCIA DE MIOFOSFORILASA) La enfermedad por almacenamiento de glucógeno, tipo V también se conoce como la enfermedad de McArdle, en reconocimiento de la descripción clínica del Dr. Brian Área Química Biológica Dra. Silvia M. Varas 28 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 McArdle en 1951 de un paciente de 30 años de edad que sufría de dolor muscular, debilidad y rigidez después de un ligero ejercicio. El lactato de la sangre en este paciente cayó abruptamente durante el ejercicio en lugar de subir como ocurre en la normalidad, lo que sugiere que el paciente no pudo convertir el glucógeno muscular en lactato. El inicio de los síntomas se suele producir en la infancia, pero el diagnóstico se realiza en la segunda o tercera década de vida ya que manifestaciones clínicas tales como calambres y mioglobinuria, no se suelen producir antes de los 10 años de edad. En los enfermos de McArdle presentan un fenómeno patognomónico de la enfermedad denominado “segundo aliento”. Tras la aparición de los primeros síntomas (intolerancia al ejercicio con cansancio prematuro, mialgias, rigidez, debilidad y contracturas musculares) y en un periodo variable de 5-10 minutos, pueden experimentar una mejoría por la mayor disponibilidad de glucosa sanguínea para los músculos, fenómeno conocido como “segundo aliento” (del inglés second wind). Y que supone la utilización de otras rutas metabólicas alternativas para la obtención de ATP, de origen no muscular, tales como utilización de ácidos grasos y la oxidación de aminoácidos. Fisiopatología: - Existen dos mecanismos que permiten explicar la intolerancia al ejercicio asociada a esta enfermedad. Por un lado el déficit de miofosforilasa priva al organismo del sustrato para la glucólisis anaeróbica, clave para la fase inicial de la contracción muscular, y por otro, la imposibilidad de metabolizar aeróbicamente el glucógeno provoca su depósito intracelular en las fibras de músculo estriado. - La mioglobinuria es causada por la necrosis de las fibras musculares, producida por los mismos mecanismos que producen la fatiga y las contracturas, aunque sostenidos en el tiempo y/o de mayor intensidad. - El ADP, es convertido en AMP, IMP, amonio y productos de degradación de adenina, incluyendo inosina, hipoxantina y ácido úrico (“hiperuricemia miogénica”) - La debilidad persistente se relaciona con el daño muscular permanente y la consecuente pérdida de fibras musculares. Durante el ejercicio intenso, se produce una disminución en los niveles de ATP, y un aumento en los de IMP. El ATP es degradado a ADP, y la adenilato quinasa transfiere una molécula de fosfato de un ADP a otro, produciendo una molécula de ATP y otra de AMP, el AMP es desaminado Área Química Biológica Dra. Silvia M. Varas 29 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 a IMP y amonio (Figura Nº8). La reacción catalizada por la MADA (mioadenilato desaminasa), permite desplazar la reacción de la adenilato quinasa hacia la formación de ATP, al degradarse rápidamente el AMP formado. Sin embargo durante el ejercicio intenso y prolongado se consume más ATP del que se produce, apareciendo la fatiga muscular. Figura Nº8: La adenilato quinasa convierte dos moléculas de ADP en una molécula de ATP y una molécula de AMP. Esta reacción se encuentra acoplada a la catalizada por la AMP desaminasa (o mioadenilato desaminasa o MADA), que convierte el AMP formado en IMP. Bioquímica / base molecular de la enfermedad La enzima deficiente es la glucógeno fosforilasa muscular (GPM), que cataliza la fosforolisis de los enlaces α-1,4-glucosil del glucógeno para formar D-glucosa-1fosfato, según la reacción: Glucógeno (n+1) + Pi Glucógeno (n) + Glucosa-1-P En humanos existen 3 isoformas; forma hepática (GPL, codificada por el cromosoma 14), cerebral (también llamada fetal, GPB, codificada por el cromosoma 10) y muscular (GPM, codificada por el cromosoma 11). El cerebro y el corazón, expresan la GPB y la GPM, mientras que el hígado expresa exclusivamente la GPL. En el músculo esquelético inmaduro, se expresan la GPB y la GPM, en el proceso de maduración muscular la isoforma fetal va siendo reemplazada por la isoforma muscular, presente exclusivamente en las fibras musculares adultas. La forma muscular es la única isoenzima presente en el músculo maduro; representa aproximadamente la mitad del total de enzima en el corazón y un 20-30 por ciento en el cerebro, y ninguna en el hígado. Esto probablemente explica por qué, clínicamente, la deficiencia de fosforilasa muscular no afecta a otros órganos además de los músculos. La GPM, existe como un homodímero (M2) que se compone de dos subunidades idénticas de 97.440 Da cada una (842 aminoácidos), el centro activo de GPM contiene piridoxal fosfato. Área Química Biológica Dra. Silvia M. Varas 30 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Bioquímicamente, la actividad de la enzima es muy baja o indetectable en el músculo. La concentración de glucógeno esta incrementada; el glucógeno acumulado es de estructura normal. El gen de la fosforilasa muscular ha sido clonado y localizado en el cromosoma 11q13 (GenBank M16013). A nivel de los genes, existe una gran heterogeneidad genética, con muchas mutaciones diferentes que causa la enfermedad de McArdle identificados a la fecha. Una mutación sin sentido cambiando arginina a un stop en el codón 49 (R49X) y una deleción de un solo codón en el exón 17 son prevalentes en caucásicos. Genética La enfermedad por almacenamiento de glucógeno, tipo V se hereda como un rasgo autosómico recesivo. El gen PGYM (MIM # 608455) ha sido clonado, secuenciado y asignado al locus 13 del brazo largo del cromosoma 11, y fue identificado como causante de la enfermedad en 1993. En la mayoría de los casos, los portadores heterocigotos no están afectados clínicamente, sin embargo, hay informes de heterocigotos que cursan con esta enfermedad. La mutación sin sentido, p.R50X es la más frecuente en los diferentes grupos de poblaciones donde se ha observado, ver tabla 5a. Tabla Nº 5a: Frecuencia de las mutaciones en las distintas poblaciones Grupos de Frecuencia Frecuencia Frecuencia poblaciones Mutación Mutación Mutación W798R R50X G205S Reino Unido 81 % Estados Unidos 64 % 10% Alemania 56 % Italia 43 % Holanda 31 % Francia 72 % 3,2% España 47-55 % (53,8) 9% (10,2) 16,5% La segunda mutación más frecuente descrita en el gen PYGM es la mutación con cambio de aminoácido p.G205S que representa el 10% de alelos en pacientes americanos y 9% de alelos en pacientes españoles. La mutación con cambio de aminoácido p.W798R se ha encontrado en el 16,5% de los alelos de los pacientes españoles estudiados. Esta alteración particular de población es además la segunda más frecuente en esta población. Área Química Biológica Dra. Silvia M. Varas 31 Química Biológica Patológica Exón Codón 1 5 20 50 205 798 Tema 10: Glucogenosis Tabla Nº5b : Características de las mutaciones más frecuentes Secuencia Cambio nt Cambio Tipo de mutación aminoacídico c.148C>T CGA→TGA p.R50X Arg →Stop c.613G>A GGC→AGC p.G205S Gly→Ser p.W798R Tryp→Arg c.2392T>C TGG→CGG Año: 2013 PCR-RFLP? Si, Nla III (+) Si, Hae III (-) Si, BsrBI (+) La mutación terminadora p.R50X, es decir una Arginina es sustituida por un codón de terminación y se obtiene una proteína truncada susceptible a la degradación. La mutación genera un sitio de corte para la enzima NlaIII. Se la puede detectar por amplificación por PCR y digestión con Nla III. La mutación con cambio de sentido (missense) p.G205S, es decir hay un cambio de Glicina por Serina. La mutación desaparece un sitio de corte para la enzima HaeIII. Se la puede detectar por amplificación por PCR y digestión con HaeIII. La mutación con cambio de sentido (missense) p. W798R, es decir hay un cambio de Triptofano por Arginina. La mutación genera un sitio de corte para la enzima BsrBI. Se la puede detectar por amplificación por PCR y digestión con BsrBI. Área Química Biológica Dra. Silvia M. Varas 32 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Figura Nº9a: Gel de poliacrialmida al 12%, donde se analiza las mutaciones R50X, G205S y K543T (Tomado de Tsujino y col. 1993). Figura Nº9b: Gel de agarosa al 2%, donde se analiza la mutación W797R. Muestra la digestión con BsrBI en paciente normal (línea 6), en dos pacientes homocigotas para W797R (línea 3 y 5) y en uno heterocigota (linea4). Linea2: producto no digerido (903pb). La mutación genera 2 fragmentos: 664 y 239 pb (Tomado de Rubio JC y col., 2006). Diagnóstico a-Determinación de la creatina quinasa sérica (CK) Los niveles séricos de CK, aportan una información importante, dado que los pacientes con enfermedad de McArdle presentan niveles aumentados en suero en Área Química Biológica Dra. Silvia M. Varas 33 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 forma persistente, con cifras de CK en torno a 1000 U/L (valor de referencia < 170 U/l). b- Prueba de ejercicio en isquemia Desde la primera descripción de la enfermedad por el Dr. Brian McArdle, en la que se indicaba la incapacidad de los músculos esqueléticos de los pacientes para producir lactato durante el ejercicio físico se introdujo una prueba funcional de ejercicio en isquemia para diagnosticar a los pacientes. El fundamento de esta prueba se basa en la estimulación al máximo de la glucólisis muscular durante el ejercicio isquémico y la determinación de lactato y amonio (sirve como control de la prueba) producido. Para ello se coloca un catéter en la vena antecubital, y se realiza una primera toma de sangre en condiciones basales, a continuación se sitúa un esfingomanómetro en el antebrazo del paciente inflándolo por encima (unos 50 mmHg) de su presión arterial, después el paciente comienza a realizar ejercicio abriendo y cerrando la mano durante 1 minuto, tras finalizar el ejercicio se toman muestras sanguíneas para medir lactato a los minutos 1, 3, 5 y 10. Interpretación: En los individuos sanos se produce un aumento de lactato entre 3 y 5 veces respecto al valor basal. En los pacientes con enfermedad de McArdle no se observa dicho aumento y se obtiene una curva plana respecto al tiempo, mientras que la curva de amonio será normal o con respuesta exagerada y sirve para detectar falsos positivos. Figura Nº10 : Prueba de ejercicio en isquemia. A: curva de respuesta normal; B: curva plana de pacientes con glucogenosis tipo V. Área Química Biológica Dra. Silvia M. Varas 34 Química Biológica Patológica Tema 10: Glucogenosis Parámetro medido Año: 2013 Paciente Homocigota Paciente Heterocigota (n=18) (n=5) Glucosa (mg/dl) (antes ingestión de sacarosa) 5,o±0,3 4,8 ±0,4 Glucosa (mg/dl) (despues ingestión de sacarosa) 7,3±0,3 7,1±0,3 Lactato (mM/L) (antes ingestión de sacarosa) 0,6±0,1 0,6±0,1 95,7± ±17,4 37,6± ±6,3 2889±588 3118±1520 1,7±0,2 1,7±0,5 173,8±22 141,2±39,2 3157±718 3229±1595 Variables séricos a nivel basal NH3+ (umol/L) CK (UI/L) Variables séricos a luego sobrecarga Lactato (mM/L) (al final del test) NH3+ (umol/L) (al final del test) CK (UI/L) después de la sesión de ejercicio CK: actividad sérica de creatin quinasa *- Tabla tomada de Rubio y col. (2007); donde los pacientes son homocigota o heterocigota para la mutación C34T. Con negrita se denota las diferencias estadísticamente significativas. OJO FALTA LOS VALORES DE LOS PACIENTES WT! Si bien es una prueba con aplicabilidad diagnóstica, presenta algunos problemas; i) depende en gran medida, de la capacidad y colaboración del paciente para realizar el test, ii) existe variabilidad preanalítica en la medida del lactato sérico, iii) existe riesgo de que se produzca un síndrome compartimental (causado por la inflamación muscular resultante de la necrosis y de la disminución del flujo sanguíneo local), como consecuencia de la prueba. Se ha indicado la posibilidad de realizar esta prueba diagnóstica en ausencia de isquemia, siendo esta metodología muy útil para el diagnóstico de alteraciones en la glucólisis, sin producir las complicaciones que genera el test isquémico. c- Prueba en cicloergómetro Es una prueba fisiológica, en la que se monitoriza el ritmo cardíaco durante el pedaleo en un cicloergómetro cuando se ejerce una resistencia constante al mismo, con el fin de detectar la aparición del fenómeno del “segundo aliento” que es patognomónico en esta enfermedad. La prueba realizada de manera controlada, a un ritmo moderado con una carga constante de trabajo. Es un método específico, sensible y simple para diagnosticar pacientes con GSD V, (para mas detalles de su realización ver al final en Rubio JC y col., 2007). Estrategia de diagnóstico Área Química Biológica Dra. Silvia M. Varas 35 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 El protocolo propuesto por JC Rubio y col. (2009) comienza con criterios de inclusión clínicos: intolerancia al ejercicio, elevación persistente de CK, curva plana de ácido láctico en la prueba de ejercicio en isquemia, fenómeno del segundo aliento y mioglobinuria. A partir de DNA obtenido de muestra sanguínea, la estrategia propuesta consta de las siguientes etapas: i) cribado secuencial mediante métodos sencillos de PCR-RFLP en función de la frecuencia de las tres mutaciones más comunes en la población española, p.R50X, p.W798R y p.G205S, lo que permitiría caracterizar definitivamente cerca del 60% de los pacientes, ii) secuenciación completa de los exones, 1, 14, 17 y 18, que presentan una mayor incidencia de mutaciones, lo que aumentaría la eficiencia de la estrategia llegando a una identificación de los dos alelos mutados en el 75% de los pacientes. Siguiendo este protocolo, en tres de cada cuatro pacientes (75%), no sería necesario practicar una intervención invasiva como es la biopsia muscular para diagnosticar la enfermedad. 8. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO VI (DEFICIENCIA DE FOSFORILASA HEPÁTICA, ENFERMEDAD DE HERS) El diagnóstico de la glucogenosis hepática por deficiencia de la actividad de glucógeno fosforilasa, solo se puede realizar en una biopsia de hígado; ya que es una isoforma que solo se expresa en este tejido. La actividad glucógeno fosforilasa en el hígado puede estar disminuida, tanto en la deficiencia primaria de esta enzima o como consecuencia del déficit de la glucógeno fosforilasa quinasa. Por lo tanto, para poder llegar al diagnóstico fiable de uno u otro déficit habrá que medir ambas enzimas. Historia y descripción general Entre los años 1950 y 1960, una gran proporción de pacientes con enfermedad por almacenamiento de glucógeno no pudo ser clasificada como perteneciente a cualquiera de los tipos que habían sido descritas anteriormente. En 1959, Hers describe 3 pacientes, estos pacientes tenían hepatomegalia, hipoglucemia leve, y marcado incremento del contenido de glucógeno en el hígado con actividad normal de glu-6-fosfatasa. Área Química Biológica Dra. Silvia M. Varas 36 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 La clínica de estos procesos, habitualmente mas leve que en otras glucogenosis, suele ser hepatomegalia, distensión abdominal y retraso del desarrollo, manifestándose en la lactancia o infancia. La hipoglucemia sintomática es excepcional, excepto en los períodos de ayuno muy prolongados, los cuales provocan cetonemia, aunque menor que en la glucogenosis III. No suele haber ácidosis metabólica y los niveles de ácido láctico y ácido úrico son normales. La hiperlipidemia es moderada con aumento de los triglicéridos y del colesterol. Las transaminasas pueden estar moderadamente elevadas. El curso clínico es benigno y las alteraciones clínicas y bioquímicas así como la hepatomegalia disminuyen con la edad; muchos adultos están asintomáticos. Algunas mutaciones del gen de la fosforilasa quinasa se han asociado con fenotipos clínicos más graves, describiéndose algún caso con progresión a cirrosis en la infancia. Puede haber retraso motor por hipotonía muscular en la forma autosómica recesiva con afectación muscular y hepática. Luego del ayuno nocturno la curva de glucagón se caracteriza por aumento de la glucemia sin aumento del lactato. Esta curva no diferencia entre el déficit de fosforilasa quinasa y el de fosforilasa. El diagnóstico exacto exige la determinación de las actividades enzimáticas en diferentes muestras de tejido y células hemáticas. Genética Tres isoformas de fosforilasa son conocidos: las isoformas de músculo (M), hígado (L), y el cerebro (B). Ellos son codificadas por los genes por separado, PGYM:es el gen para la isoforma muscular de la fosforilasa; PGYL: es el gen para la isoforma hepática de la fosforilasa y PGYB: es el gen para la isoforma cerebro de la fosforilasa, que han sido asignadas a los cromosomas 11, 14 , y 20, respectivamente. Evolución Clínica y complicaciones En los pacientes con glucogenosis VI la hepatomegalia puede ser masiva, aunque los pacientes pueden estar libres de síntomas y llevar una vida normal. Suelen presentar, sin embargo, cierta elevación de las transaminasas y de los lípidos en el suero. La hepatomegalia puede remitir a medida que el niño crece. Área Química Biológica Dra. Silvia M. Varas 37 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 9. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO VII (DEFICIENCIA DE FOSFOFRUCTOQUINASA MUSCULAR, ENFERMEDAD DE TARUI) La enfermedad por almacenamiento de glucógeno es conocido como enfermedad de Tarui en reconocimiento de la descripción enzimática y clínica en 1965 de una familia con 3 hijos afectados (una mujer y dos varones) en su segunda década de vida. Ellos presentaban fatiga e intolerancia al ejercicio como en el tipo V. El glicógeno muscular estaba aumentado con actividad de fosforilasa normal y con actividad no detectable de fosfofructoquinasa. Hallazgos clínicos Las características clínicas son muy similares a la enfermedad por almacenamiento de glucógeno tipo V. Los pacientes experimentan la aparición temprana de la fatiga y el dolor con el ejercicio. El ejercicio vigoroso produce calambres musculares severos y mioglobinuria. Hay, sin embargo, varias características que diferencian el tipo VII de la enfermedad de tipo V. En la enfermedad de tipo VII, (1) para la mayoría de los pacientes, la intolerancia al ejercicio es evidente en la infancia, los síntomas son más severos que los observados en el tipo de enfermedad por almacenamiento de glucógeno V, y el ataque puede estar asociado con náuseas y vómitos; (2) se produce una anemia hemolítica compensada, como lo demuestra el aumento del nivel de la bilirrubina sérica y recuento de reticulocitos y 2,3-bisfosfoglicerato de eritrocitos que también disminuye, en forma secundaria al bloqueo de la glucólisis en el paso de la fosfofructoquinasa, (3) hay hiperuricemia siempre presente y exagerado aún más por el ejercicio muscular a un grado más grave que la observada en la enfermedad de almacenamiento de glucógeno tipo V o tipo III; (4) un polisacárido anormal está presente en las fibras musculares y ácido peryódico de Schiff-positivo, pero resistentes a la digestión de diastasa y (5) intolerancia grave al ejercicio particularmente aguda después de las comidas especialmente las que son ricas en hidratos de carbono. Bioquímica / base molecular de la enfermedad Fosfofructoquinasa es una enzima tetramérica derivada de tres loci genéticos que codifican para las subunidades de músculo (M), hígado (L), y plaquetas (P). La enzima de plaquetas es del mismo tipo que el de los fibroblastos. Los genes para el músculo, el hígado y los tipos de las plaquetas se asignan a los cromosomas 12, 21 y Área Química Biológica Dra. Silvia M. Varas 38 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 10, respectivamente. Estas subunidades se expresa de forma variable en diferentes tejidos. El músculo maduro sólo expresa la subunidad M y contiene únicamente el homotetrámero M4. Los eritrocitos expresan tanto la subunidad M como la L, y la formación de tetrámero aleatoria produce cinco isoenzimas, M4 y L4 y tres formas híbridas (M3L, L2M2, y ML3). En la deficiencia clásica de fosfofructoquinasa de músculo, un defecto genético de la subunidad M (GenBank U24183) provoca una falta total de la actividad de la enzima en el músculo, mientras que los eritrocitos carecen de las isoenzimas M4 e híbridos, pero todavía expresa L4, que representa alrededor del 50 por ciento de la actividad de fosfofructoquinasa de los eritrocitos normales. En contraste, los pacientes con la forma miopática de aparición tardía de la deficiencia de fosfofructoquinasa tiene tanto las isoenzimas híbridas que contienen M y L4 en sus eritrocitos. La subunidad M residual puede explicar los síntomas leves y la aparición tardía de esta variante de la deficiencia fosfofructoquinasa. La relación entre el nivel de actividad residual fosfofructoquinasa en el músculo esquelético y la expresión fenotípica de la enfermedad no está todavía clara. El mecanismo para la anemia hemolítica compensada se ha atribuido a una disminución de recambio de ATP, el aumento de los iones de calcio intracelular, y la disminución de la deformabilidad en los eritrocitos. Diagnóstico El diagnóstico molecular es útil sólo en poblaciones de pacientes limitados o en familias con mutaciones conocidas. Para la mayoría de los pacientes, el diagnóstico requiere la demostración bioquímica o histoquímica del defecto enzimático en el músculo. La ausencia de la isoenzima M de fosfofructoquinasa también puede demostrarse en las células sanguíneas y fibroblastos. Tratamiento No existe un tratamiento específico para esta condición. Evitar el ejercicio intenso es recomendable para prevenir los ataques agudos de calambres musculares y mioglobinuria. El beneficio clínico de una dieta cetogénica ha sido reportada en un niño con deficiencia de fosfofructoquinasa infantil con artrogriposis. Como por otras enfermedades musculares de almacenamiento de glucógeno se debe evitar las Área Química Biológica Dra. Silvia M. Varas 39 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 estatinas y las precauciones de la hipertermia maligna en los pacientes sometidos a anestesia. Genética La enfermedad de almacenamiento de glucógeno, tipo VII se hereda como un rasgo autosómico recesivo. Se han sido identificadas las mutaciones múltiples, incluyendo los defectos de empalme, marco de lectura, y mutaciones missense. No hay ninguna correlación genotipo-fenotipo. 10. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO IX, (DEFICIENCIA FOSFORILASA QUINASA) Historia y descripción general McKusick, en OMIM (Online Mendelian Inheritance in Man) clasificó a la deficiencia de fosforilasa quinasa hepática como una enfermedad ligada al cromosoma X, como del tipo VIII. La clasificación numérica confusa fue debida en parte a una falta de conocimiento del complicado sistema que activa a la fosforilasa. La complejidad genética es ahora más clara, sabiendo que la fosforilasa quinasa se compone de cuatro subunidades codificadas por diferentes genes en diferentes cromosomas (tanto el cromosoma X como distintos autosomas) y expresados diferencialmente en los distintos tejidos. En este capítulo nos referimos a todas las deficiencias de la fosforilasa quinasa como el tipo de enfermedad por almacenamiento de glucógeno tipo IX. Basado sobre el gen / subunidad implicados, los tejidos que son principalmente afectados, y el modo de herencia, la deficiencia de la fosforilasa quinasa en el ser humano se divide en seis subtipos (tabla Nº 6). Dos modelos de roedores, que se diferencian en los tejidos afectados y los modos de herencia, se incluyen para su comparación. Diagnóstico Establecer el diagnóstico del tipo de enfermedad por almacenamiento de glucógeno IX es un reto debido a la heterogeneidad genética que subyace y expresividad variable. La deficiencia de la fosforilasa quinasa no es siempre detectable por el ensayo de los eritrocitos y la biopsia de hígado, de músculo o biopsia del corazón son necesarios para confirmar algunos casos bioquímicamente. El ensayo de la enzima Área Química Biológica Dra. Silvia M. Varas 40 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 también es incapaz de diferenciar entre la deficiencia de la subunidad α, con la consiguiente herencia ligada al X, y la deficiencia de las subunidades β y γ , con un patrón de herencia autosómico recesivo. Así, en muchos casos, el análisis de mutación se necesita para la subtipificación de la enfermedad. Tabla Nº 6: Subtipos causantes de Enfermedad por almacenamiento de glucógeno tipo IX Subtipos Especies Tejidos Herencia Gen mutante Subunidad Ligada al X PHK A2 αL afectados IX-a1 humano Hígado, células sangre IX-a2 humano hígado Ligada al X PHK A2 αL IXb humano Hígado, células Autosómica PHK B β Autosómica PHK G2 γTL αM de la sangre, músculo IXc humano Hígado, células sangre IXd humano Músculo Ligada al X PHK A1 IXe humano músculo Autosómica ? IXf humano corazón Autosómica ? ? I-ratón músculo Ligada al X PHKA1 αM gsd-rata hígado Autosómica PHKG2 γTL La detección de mutaciones de tipo IX requiere secuenciar los genes para tres subunidades del hígado. El gen que codifica la subunidad PHKA2 α ha estado implicado más comúnmente, seguido por el gen que codifica la subunidad PHKB β, independientemente de la presencia de la deficiencia en los eritrocitos (Beauchamp y col., 2007). Una deleción única grande del gen PHKA2 se atribuyó a la recombinación homóloga desigual entre las repeticiones Alu en los intrones 19 y 26 en un niño japonés (Fukao et al., 2007). Las mutaciones la subunidad γ del gen PHKG2 esta asociado con afectación hepática grave, con la hipoglucemia recurrente y la fibrosis hepática (Beauchamp et al, 2007; Burwinkel et al, 2003.). La elevación de tetrasacárido urinario se confirmó en cuatro de siete pacientes con deficiencia de la fosforilasa quinasa en los eritrocitos, proporcionando una prueba de detección menos invasiva y potencialmente útil (Morava et al., 2005). Área Química Biológica Dra. Silvia M. Varas 41 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Tratamiento El tratamiento para la deficiencia de la fosforilasa quinasa de hígado es sintomático. Una dieta alta en carbohidratos y comidas frecuentes son eficaces para prevenir la hipoglucemia, pero la mayoría de los pacientes no requieren tratamiento específico. El pronóstico es generalmente bueno. Evolución Clínica y complicaciones En la tipo IX puede existir hepatomegalia desde estadíos tempranos de la vida, pero remite a medida que el niño se hace mayor. La elevación de las transaminasas suele ser mínima. En general, salvo en formas graves, no precisan tratamiento. La monitorización del seguimiento de pacientes con glucogenosis se detalla en la tabla Nº8. 11. ENFERMEDAD POR ALMACENAMIENTO DE GLUCÓGENO, TIPO 0 (DEFICIENCIA GLUCÓGENO SINTASA HEPÁTICA) La glucógeno sintasa cataliza la formación de puentes α-1,4 que alargan las cadenas de moléculas de glucosa para formar glucógeno. En la enfermedad por almacenamiento de glucógeno tipo 0, sólo se deteriora la síntesis de glucógeno en el hígado. La deficiencia genética de la actividad de sintasa de glucógeno hepático (GYS2) en los seres humanos parece ser muy raro. 12. DIAGNÓSTICO BIOQUÍMICO Pruebas funcionales: dentro de estas pruebas se encuentra la sobrecarga oral de glucosa y determinación de glucosa posterior la sobrecarga IV de glucagón. Prueba de sobrecarga con glucagón: Procedimiento: Tener una cánula en la fosa antecubital con un buen acceso para la administración de glucosa, si es necesario. tiempo 0 min. tomar 2 ml de sangre para la glucosa y el lactato. Inmediatamente después administrar glucagón 20 microgramos / kg im (los niños). Para los adultos se debe administrar 1 mg im. tiempo de 30 ‘ tomar 2 ml de sangre para la glucosa y el lactato tiempo de 60 ‘ tomar 2 ml de sangre para la glucosa y el lactato Área Química Biológica Dra. Silvia M. Varas 42 Química Biológica Patológica Tema 10: Glucogenosis tiempo de 90’ tomar 2 ml de sangre para la glucosa y el lactato tiempo de 120 ‘ tomar 2 ml de sangre para la glucosa y el lactato Año: 2013 Interpretación Adultos: en sujetos normales, hay un aumento en la glucosa plasmática > 4 mmol / L (>72mg/dl). La mayor tolerancia en adultos puede ser explicado por una severidad menor de la enfermedad en los adultos. Niños: en sujetos normales, hay un aumento en la glucosa plasmática > 2 mmol /L (32mg/dl) y una concentración de lactato en plasma en pacientes ayunados o estimulados < 2,4 mmol / L (21,60 mg/dl). Figura Nº 11: Prueba de tolerancia a glucagón. A: curva de respuesta normal; B: curva plana. 12.1- Diagnóstico bioquímico-genético de las glucogenosis hepáticas El diagnóstico bioquímico de las glucogenosis hepáticas se basa en la detección de la actividad enzimática deficiente, mediante su medida en una biopsia de hígado. Se completa con la cuantificación de la concentración del glucógeno y con la determinación de su estructura. En algunos tipos de glucogenosis la actividad enzimática deficiente en el hígado también lo es en el músculo. En otros casos, la deficiencia de la enzima hepática se expresa parcialmente en células sanguíneas (eritrocitos o leucocitos). Por tanto, la indicación de que enzima medir y en que tejido dependerá de las características clínicas y biológicas de cada paciente (Tabla Nº7). Área Química Biológica Dra. Silvia M. Varas 43 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Tabla Nº7: Diagnóstico bioquímica-genérico. Obtención de muestras Muestra a tomar Diagnóstico de Músculo Hígado Eritrocitos Sangre Total * presunción Glucogenosis Ia + + Glucogenosis Ib (1) + + Glucogenosis III (2) (4) + + Glucogenosis IV (3) + + + + + Glucogenosis VI Glucogenosis IX (4) + + + + + + * Para el diagnóstico genético-molecular (1) El diagnóstico enzimático solo se puede realizar en una biopsia de hígado no congelado. (2) En este tipo, de ser posible, se enviará biopsia de hígado y de músculo. (3) El diagnóstico enzimático se puede realizar en una biopsia de hígado y de músculo (4) La actividad normal en los eritrocitos no descarta que exista una deficiencia que solo se expresa en el hígado o en el músculo; en estos casos el diagnóstico solo se puede hacer en estos tejidos. 13. CONDICIONES PARA LA OBTENCIÓN Y ENVÍO DE LAS MUESTRAS 13.1- Tipo de muestra El tipo de muestra a enviar dependerá del diagnóstico de presunción, como se indica en la tabla 7. En todos los casos se enviara, además, una muestra de sangre total en EDTA para el diagnóstico genético- molecular. 13.2- Obtención de biopsias La biopsia hepática o muscular se puede tomar por punción con aguja o quirúrgicamente. El material obtenido por punción, por su escasez, solo permite estudios enzimáticos muy limitados. Por ello, se recomienda la biopsia quirúrgica. Si además del hígado, se quiere estudiar el tejido muscular, en el mismo acto quirúrgico se puede tomar una biopsia de los músculos de la pared abdominal. Inmediatamente después de la toma, cada biopsia se colocara por separado, en un tubo con tapón, resistente a la congelación, pequeño (si es posible un criotubo de 2-5 ml) y vacío, claramente etiquetado (nombre y apellido del enfermo, naturaleza de la muestra) que Área Química Biológica Dra. Silvia M. Varas 44 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 se cerrara y se congelara (-60°C o a temperatura inferior). La cantidad mínima recomendada para hígado y músculo es de unos 25 mg (el tamaño de un garbanzo). 13.3- Obtención de muestras de sangre y eritrocitos Como se indica en la tabla 7, en algunos casos, se pueden necesitar muestras de sangre tratada de distinta forma: a) Sangre total: tres muestras de 2 ml de sangre, extraída en los tubos con EDTA. Se agitarán y congelarán, inmediatamente, a – 60ºC. b) Eritrocitos separados de la siguiente forma: - Centrifugar al menos 2 ml de sangre heparinizada a 1.500 rpm., 10 min. - Eliminar el plasma. Añadir doble volumen de ClNa 0,9% al sedimento de eritrocitos, mezclar cuidadosamente y centrifugar 1.500 rpm., 10 min. - Eliminar el sobrenadante y repetir la operación anterior por tres veces. Congelar inmediatamente el último precipitado de eritrocitos (-60° C) y guardar a esta temperatura hasta su envío. 13.4- Transporte Todas las muestras, biopsias, sangre o eritrocitos, deben permanecer congeladas durante toda la duración del transporte. Se colocaran en una caja de telgopor con hielo seco. La cantidad de hielo seco se adaptara en función del tiempo de transporte (1 Kg. dura 24 horas, aproximadamente). 14. TRATAMIENTO EN LA GLUCOGENOSIS TIPO I: 1. Tratamiento dietético-nutricional El objetivo del tratamiento nutricional es prevenir la hipoglucemia (mantener los niveles plasmáticos de glucosa > 3,89 mmol/L) y sus consecuencias metabólicas. Para ello, se utilizan básicamente dos estrategias: la realización de comidas frecuentes (cada 2 a 4 horas) ricas en hidratos de carbono durante el día junto a una infusión nocturna de glucosa a través de una sonda nasogástrica o bien la administración de almidón crudo de maíz. Los resultados obtenidos con ambos métodos son similares. a) Modificaciones en la dieta oral • Uso de comidas ricas en hidratos de carbonos de absorción lenta o semilenta: arroz, pasta, pan, legumbres, etc. Área Química Biológica Dra. Silvia M. Varas 45 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 Restringir los carbohidratos de absorción rápida: sacarosa y fructosa. Limitar la ingesta de leche a 0,5 litros al día. Tabla Nº8: Monitorización adecuado de la dieta (objetivos) Parámetro Sangre Medida - Glucemia ≥ 3,9mmol/L (70 mg/dL) - Lactato (antes de la comida) 2,0 – 5,0 mmol/L (18-45 mg/dL) Orina - Lactato (orina de 12 hs o ≥ 0,6 mmol/L ( 5,400 mg/dL) ≥ 0,12 muestras) - Cociente lactato/creatinina Tabla Nº 9: Seguimiento en pacientes con glucogenosis I Laboratorio - Determinación de glucemia capilar a lo largo del día - Estudio basal - Estudio lipídico completo (Ct, Tg, HDLc LDLc y VLDL) Ácido Úrico Equilibrio ácido-base Función hepático y renal Hemograma y recuento diferencial - Orina de 24 hs: Albúmina Aclaramiento Excreción de lactato Frecuencia 3-6 meses Algunos autores recomiendan restringir las purinas y la grasa. Se sugiere que el contenido de la dieta siga la siguiente distribución: Hidratos de carbono 60-65%; proteínas 10-15% y lípidos 20-30% del aporte calórico total. No esta claro si estos pacientes tienen requerimientos energéticos más elevados. b) Nutrición enteral nocturna Administración de una infusión de glucosa o de polímeros de glucosa o de una formula enteral sin lactosa ni fructosa a lo largo de 10- 12 horas. La infusión no debe comenzar después de 1 hora de la última comida ni el desayuno mas tarde de 15 minutos después de suspender la infusión. Los requerimientos de glucosa se basan en la tasa estimada de producción endógena de glucosa, que disminuyen con la edad. Se recomienda comenzar con una cantidad de glucosa de 7 mg/kg/minuto y ajustar Área Química Biológica Dra. Silvia M. Varas 46 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 posteriormente a la baja (4-6 mg/kg/minuto) para mantener glucemias entre 3,9 y 5,3 mmol/L. Como la necesidad de la nutrición enteral nocturna es prolongada, sugerimos la realización de una gastrostomia para alimentación. Con el fin de evitar complicaciones se realiza mediante control ecográfico. Asimismo se utiliza profilaxis antibiótica quirúrgica en todos los casos (cefalosporina de 3º generación). c) Almidón crudo de maíz Su efecto se basa en el hecho de que la glucosa procedente del almidón de maíz no cocinado se libera y absorbe mas lentamente, permitiendo mantener la glucemia durante 6 a 8 horas en vez de las 3 horas de una toma equivalente de glucosa. Este régimen puede usarse con seguridad por encima de los 2 años de edad con dosis de almidón entre 1,6 g/kg y 2,5 g/kg, cada 4 a 6 horas. No existe tanta unanimidad en lo referido a lactantes. El almidón de maíz (Maicena) se prepara en una suspensión de agua a temperatura ambiente con una relación peso: volumen de 1:2, utilizando una cantidad de almidón similar a la tasa estimada de producción endógena de glucosa durante el ayuno. No debe administrarse con azucares de absorción rápida. El uso de otros almidones (arroz, tapioca, trigo) obtiene resultados discretamente inferiores a los del almidón de maíz. Este aporte nocturno de glucosa debe mantenerse incluso una vez terminado el crecimiento y el desarrollo. d) Suplementos vitamínicos y de minerales Es necesario suplementar con vitaminas y minerales, especialmente con calcio, para cubrir requerimientos. e) Planificación y monitorización de la dieta f) Otros tratamientos para evitar la hipoglucemia (no contrastados) - Inhibidores de la amilasa intestinal: Voglibose, 0,1-0,2 mg antes de cada comida. - Diazoxido a dosis bajas: 3-5 mg/kg/día. 2. Tratamiento de las complicaciones a- Complicaciones renales - No existe tratamiento preventivo. Persiste hiperfiltracion glomerular a pesar de un tratamiento dietética adecuada precoz. - Moderada restricción proteica. - Inhibidores de la enzima de conversión de angiotensina: captopril. - Tratamiento de la insuficiencia renal crónica. Área Química Biológica Dra. Silvia M. Varas 47 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 b- Osteoporosis - Valoración de la dieta. - Ejercicio físico regular. - Medidas preventivas: 400-800 UI de vitamina D3 + 0,5-1,0 g de calcio al día. c- Hiperuricemia - Restricción de purinas. - Alopurinol 10-15 mg/kg/dia. - Opcional: alcalinizar la orina con bicarbonato sodico 1-2 mmol/kg/dia (80-170 mg). TIPO III El tratamiento dietético es menos exigente que en glucogenosis I: no precisa restricción de fructosa, sacarosa ni lactosa. La distribución calórica de la dieta: hidratos de carbono 55-60%; proteínas 15-20% y lípidos 20-30%. Es bastante discutida la necesidad de hacer una dieta hiperproteica. Para unos autores, en presencia de hipoglucemia el manejo ha de ser similar al de una glucogenosis I (tomas frecuentes durante el día ricas en hidratos de carbono de absorción lenta junto a una enteral nocturna o suplementos de almidón crudo de maíz); mientras que para otros el aspecto basal de la dieta es un aumento de las proteínas y una limitación de los azucares. La nutrición enteral nocturna quedaría reservada para lactantes y niños pequeños con formas graves de glucogenosis III (hipoglucemia precoz, miopatía o disfunción hepática importante). TIPO IV El almidón crudo de maíz y la nutrición enteral continua pueden mejorar temporalmente la situación clínica, pero el único tratamiento efectivo es el trasplante hepático. TIPO VI No esta claro el papel del tratamiento dietético excepto en lactantes y niños pequeños. Se recomienda evitar los períodos prolongados de ayuno y las tomas nocturnas adicionales en los episodios infecciosos. Área Química Biológica Dra. Silvia M. Varas 48 Química Biológica Patológica Tema 10: Glucogenosis Año: 2013 BIBLIOGRAFÍA: Priya S. Kishnani, Dwight Koeberl, Yuan-Tsong Chen. PART 7: CARBOHYDRATES. Chapter 71: Glycogen Storage Diseases. .The Online Metabolic & Molecular Bases of Inherited Disease. Editors: Valle D, Beaudet AL, Vogelstein, Kinzler, Antonarakis & Ballabio. Editors Emeritus: Scriver CR, Childs & Sly WS Scriver CR, Beaudet AL, Sly WS & Valle D. The metabolic and molecular basis of inherited disease. 7º Edition. 1995. New York Mc Graw-Hill. Roseline Froissart, Monique Piraud, Alix Mollet Boudjemline, Christine Vianey-Saban1, François Petit, Aurélie Hubert-Buron, Pascale Trioche Eberschweiler, Vincent Gajdos and Philippe Labrune: Glucose-6phosphatase deficiency. Orphanet Journal of Rare Diseases 2011, 6:27 E. Pallarés Querol, M. Migueláñez Díaz, J. Rubí Cervino, E. Ripoll Sevillano y M. Martínez Pardo Ácidosis láctica en pediatría. Química Clínica 2002; 21 (4) 280-284 Rochelle Hirschhorn, Arnold J.J. Reuser. PART 4: LYSOSOMAL DISORDERS- Chapter 135: Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. The Online Metabolic & Molecular Bases of Inherited Disease. Editors: Valle D, Beaudet AL, Vogelstein, Kinzler, Antonarakis & Ballabio. Editors Emeritus: Scriver CR, Childs & Sly WS J H Barth, G E Butler & P J Hammond: Biochemical Investigations in Laboratory Medicine. Clinical Biochemistry, Paediatric Endocrinology & General Medicine, Leeds General Infirmary & Harrogate General Hospital. http://www.pathology.leedsth.nhs.uk (Pruebas de laboratorio e interpretación). J. C. Rubio, M. Pérez , J. L. Maté-Muñoz , I. García-Consuegra , C. Chamorro-Viña, M. Fernández del Valle, A. L. Andreu, M. A. Martín1, J. Arenas1, A. Lucia. AMPD1 Genotypes and Exercise Capacity in McArdle Patients. Int J Sports Med 2007; 28: 1–5 Juan C. Rubio, Ines Garcia-Consuegra, Gisela Nogales-Gadea, Alberto Blazquez , Ana Cabello, Alejandro Lucia, Antoni L. Andreu, Joaquin Arenas, and Miguel A. Martin: A proposed molecular diagnostic flowchart for myophosphorylase deficiency (McArdle disease) in blood samples from Spanish patients. Hum Mutat. 2007 Feb;28(2):203-4. Tsujino S, Shanske S, DiMauro S. 1993. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle's disease). N Engl J Med 329(4):241-5. Juan C. Rubio; Alejandro Lucia; Israel Fernández-Cadenas; Ana Cabello; Alberto Blázquez; Josep Gamez; Antoni L. Andreu; Miguel A. Martín; Joaquin Arenas: Novel Mutation in the PYGM Gene Resulting in McArdle Disease. Arch Neurol. 2006;63:1782-1784 Área Química Biológica Dra. Silvia M. Varas 49