Tema 1 – INTRODUCCIÓN A LA MECÁNICA ESTADÍSTICA Y
Anuncio

Tema 1 – INTRODUCCIÓN A LA MECÁNICA ESTADÍSTICA Y
REPASO DE TERMODINÁMICA
Introducción: conceptos básicos de la termodinámica y la
mecánica estadística.
La distrbución canónica.
Postulados de la mecánica estadística. El espacio de las fases
molecular. Distribución de velocidades moleculares de MaxwellBoltzmann.
Las tres leyes de la termodinámica. Los potenciales
termodinámicos y las relaciones de Maxwell.
La ecuación de Gibbs-Duhem. Criterios de estabilidad
termodinámica.
[HUA-1; CHA-1,2; CAL; AGU]
1
Introducción: conceptos básicos de la termodinámica y la mecánica estadística.
2
Objetivo de la Mecánica Estadística:
Deducir las propiedades de un sistema macroscópico a partir
del conocimiento de sus constituyentes microscópicos.
La Mecánica Estadística: una disciplina que proporciona una
explicación microscópica de los conceptos y problemas que
previamente se han visto en Termodinámica.
3
Ejemplo: el gas ideal en equilibrio
Termodinámica
Presión
Volumen
Temperatura
Número de moles
Ecuación de estado:
PV=nRT
Física Estadística
N moléculas (~1023)
Se mueven y chocan
entre sí y contra las
paredes
¿Cómo obtener la ecuación de estado?
4
Mecánica Estadística El gas ideal en equilibrio
N moléculas (~1023)
¿Cómo describimos el estado del gas?
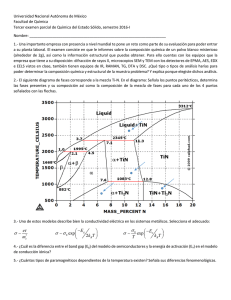
¿Cómo describimos la situación de equilibrio?
Mismo número de moléculas en cada celdilla
Pero,... ¿es exactamente el mismo número?
La Física Estadística permite tratar algo nuevo: fluctuaciones
(Ejemplo: fluctuaciones nº partículas sim-DM)
¿Cómo?
Con el cálculo de probabilidades (estadística)
5
Definiciones:
Estado microscópico o microestado:
Especifica con detalle toda la información sobre las moléculas del gas, y permite
describir con detalle el gas
Estado macroscópico o macroestado:
Se puede describir perfectamente el estado del gas diciendo cuántas moléculas hay
en parte del recipiente (en cada celdilla)
Puede haber varios microestados diferentes correspondientes a un mismo macroestado.
Si el macroestado del sistema tiende a no variar en el tiempo, decimos que el sistema
está en equilibrio.
El estado de equilibrio es el más aleatorio, el que tiene más microestados.
Si un sistema aislado está en una situación poco aleatoria, variará en el tiempo,
aproximándose finalmente a su situación de mayor azar o equilibrio.
Se dice que un proceso es irreversible si, invertido en el tiempo, es tal que casi nunca
ocurre en la realidad
Sólo se ve un sentido preferente del tiempo si se parte de una situación de falta de
azar en un instante determinado.
6
Presión de un gas ideal
F
p =
A
mv
dA
F ∆t = m ∆v
Presión
media
=
Momento medio
adquirido por la
pared tras el choque
x
Número medio de
choques por unidad de
tiempo y de unidad de
área de la pared
7
Presión de un gas ideal
v dt
Presión
media
p
=
=
Momento medio
adquirido por la
pared tras el choque
2mv
x
1
2
2
p ≈ n m v = nε c
3
3
x
dA
Número medio de
choques por unidad de
tiempo y de unidad de
área de la pared
1
n
6
(dA v dt )
1
2
εc = mv
2
mv
1
dA dt
Densidad de flujo molecular:
1
dΦ = n (dA v dt )
6
8
Recorrido libre medio
Distancia media entre colisiones
mv
Recorrido libre medio = tiempo medio entre colisiones × velocidad media
Volumen barrido por una molécula
hasta que se encuentra con otra:
Sección eficaz de dispersión:
Recorrido libre medio :
πD
2
1
=
n
σ = π D2
≈
1
1
=
nπ D 2 σ n
9
Estimaciones numéricas:
N A = 6.0225 10 23 moléculas / mol
1 litro de nitrógeno a presión y temperatura
ambiente
p = 10 6 dinas / cm 2 , T = 300 K ,
V = 1l = 10 3 cm 3
28 g de N 2
n = p / kT =
m = 1.15 g ,
PM = 2 × 14 = 28
en 1 litro : N = N A
N A moléculas
m
= 2.47 10 22 moléculas
PM
N
≈ 2.5 1019 moléculas / cm 3
V
3 p
≈ 6.0 10 −14 ergios
2n
m1 molec = 28 / N A = 4.65 10 − 23 g
E=
2
2E
v ≈
v ≈ 5.1 10 4 cm / s
≈ 2.610 9
m
radio molecular : a ≈ 10 −8 cm
2
σ = 4π a 2 ≈ 12 10 −16 cm 2 →
≈
1
≈ 3 10 −5 cm >> a
nσ
10
Vamos a tratar sobre la interacción térmica entre sistemas macroscópicos
Situación:
Los parámetros externos de los sistemas (y los niveles de energía)
permanecen fijos.
Objetivo:
¿Cómo calcular las propiedades de un sistema macroscópico en
equilibrio térmico?
11
La distribución canónica.
12
Distribución de energía entre sistemas macroscópicos.
Dos sistemas A y A’, con energías E y E’
A
A’
E
E’
Ω(E)
Ω’(E’)
(intervalos δE)
A*= A+A’
E* = E+E’
Una vez puestos en contacto, y alcanzado el equilibrio,
¿cuál es la probabilidad de que A tenga una determinada energía?
¿de todos los estados accesibles al sistema A*,
cuántos estados de A* se tienen con energía E para el sistema A?
Ω* (E)
P( E ) = *
Ω total
¿cómo varía P(E) con la energía?
Ejemplo: 1 sólido de Einstein
13
Ω* (E)
P( E ) = *
Ω total
¿cómo varía P(E) con la energía?
P ( E ) = Ω * ( E ) × cte
Ω * ( E ) = Ω( E ) ⋅ Ω' ( E ' ) = Ω( E ) ⋅ Ω' ( E * − E )
E' = E * − E
Estudiaremos Ln P(E)
En equilibrio, el sistema estará en el estado más probable: maximizar Ln P(E)
ln P ( E ) = ln Ω * ( E ) + ln cte = ln cte + ln Ω( E ) + ln Ω' ( E ' )
Maximizar: ∂ ln P = 0 = 1 ∂P
∂E
P ∂E
Y la condición de equilibrio es:
∂ ln Ω( E ) ∂ ln Ω( E ' )
=
∂E '
∂E
∂ ln Ω( E ) ∂ ln Ω( E ' ) ∂E '
+
=0
∂E '
∂E
∂E
Podemos definir: β ( E ) =
∂ ln Ω( E ) 1 ∂Ω
=
∂E
Ω ∂E
β (E) = β ' (E' )
14
Definiciones:
Vemos que β y lnΩ son magnitudes importantes en la interacción térmica.
β tiene unidades de inverso de energía:
1
β
≡ kT
k : constante de Boltzmann
β (E) =
∂ ln Ω( E ) 1 ∂Ω
=
∂E
Ω ∂E
k : cte con dimensiones de energía
T : parámetro que da una medida de
la energía en unidades de k
k = 1.381×10−23 J/K = 8.617×10−5 eV/K
T : temperatura absoluta
β (E) =
∂ ln Ω
∂ ln Ω
1
→
=
∂E
∂E
kT
S = k ln Ω
1 k ∂ ln Ω
1 ∂S
=
→ =
∂E
T
T ∂E
Entropía, es una medida cuantitativa del grado de
aleatoriedad del sistema
15
La condición de P(E) máxima equivale a S total máxima:
P ( E ) max
S * = S + S ' max
β = β'
T = T'
Por tanto,, los dos sistemas están a la misma temperatura.
En equilibrio térmico el sistema total está distribuido entre el mayor número
de estados posibles, es decir, está en su macroestado más aleatorio.
Ejemplo: sólido de Einstein
2 sistemas en contacto
16
Sistema en contacto con un foco térmico
Sistema A en contacto con un foco A’ (mucho más grande)
A
A’
Ω’(E’)
A*= A+A’
Ω(E)
E’
E* = Er +E’
¿Cuál es la probabilidad, Pr, de que A esté en un estado cualquiera r
con energía Er?
Es igual a la probabilidad de que A’ esté en un estado con energía E’
¿Cómo introducimos la temperatura en la probabilidad?
Sistema total aislado: A*= A+A’
E total constante:
E* = Er +E’, y
Er<< E*
Pr = a ⋅ Ω' ( E * − E r )
17
Pr = a ⋅ Ω' ( E * − E r )
Haremos lnPr, y desarrollaremos en serie
alrededor de E’=E*
ln Ω' ( E * − E r ) = ln Ω' ( E * ) −
∂ ln Ω'
E r = ln Ω' ( E * ) − β E r
∂E '
β =(kT)-1 es el parámetro asociado a la temperatura constante del foco térmico A’
Por tanto tendremos,
Ω' ( E * − E r ) = Ω' ( E * ) exp (− β E r ),
Pr = C ⋅ exp (− β E r )
Ω' ( E * ) = cte
exp (− β E r ) ≡ Factor de Boltzmann
Esta función de distribución se llama distribución canónica
Ejemplo: sólido de Einstein
2 sistemas en contacto: A<<A’
18
Distribución canónica:
Pr = C ⋅ exp (− β E r )
Conjunto canónico:
Conjunto de sistemas en el que todos ellos están en
contacto con un foco térmico a temperatura T
¿Cómo hallamos C ?
Normalización de la probabilidad
exp (− β E r ) = 1
C⋅
Pr = 1
r
r
exp (− β E r )
Pr =
exp (− β E r )
r
Y ya podemos calcular los valores medios de las distintas magnitudes:
y=
Pr y r =
y r exp (− β E r )
r
exp (− β E r )
r
r
19
Gas ideal: Partículas en una caja 3D
N partículas en una caja de lados Lx, Ly, Lz, V= Lx × Ly × Lz
Gas ideal, gas diluído:
- interacciones moleculares despreciables
- gas no degenerado (muchos más estados accesibles que moléculas)
Esto permite fijarnos en una sola molécula.
Gas en equilbrio a temperatura T:
(haremos el tratamiento habitual)
Sistema A (1 molécula) en contacto con un sistema A’ mucho
mayor (foco térmico, el resto del gas) a temperatura T.
Objetivo:
¿Cuál es la probabilidad, Pr, de hallar la molécula en uno de sus
estados cuánticos ( r, εr ) ?
exp (− β ε r )
Pr =
exp (− β ε r )
r
εr =
π
2
2m
2
nx
2
Lx
2
+
ny
2
Ly
2
+
nz
2
Lz
2
20
Calcularemos la energía media
de la molécula:
ε=
Pr ε r =
ε r exp (− β ε r )
r
exp (− β ε r )
r
r
Si operamos...
ε r exp (− β ε r ) = −
r
r
∂
∂
exp (− β ε r ) = −
∂β
∂β
exp (− β ε r )
r
Y podemos escribir,
ε=
1 ∂Z
− ∂Z / ∂β
∂ ln Z
=−
=−
∂β
Z
Z ∂β
exp (− β ε r )
Z≡
r
Z : Función de partición
Esta es la forma general de calcular la energía media de un sistema.
Hay que evaluar la función de partición.
21
Calculemos la energía media del gas, y la función de partición.
∂ ln Z
ε =−
∂β
exp − β
nx
εr =
r
Z=
¿cómo?
exp (− β ε r )
Z≡
ny
nz
π
2
2m
2
nx
2
Lx
2
+
ny
2
Ly
2
+
π2
2
2m
nz
2
Lz
2
nx
2
Lx
2
+
ny
2
Ly
2
+
nz
2
Lz
2
Z es separable: Z = Z x ⋅ Z y ⋅ Z z
Si la energía es grande comparada con ∆εi ,
Calcularemos
Zx
y
→
Zx = Zy = Zz
22
u≡
Zx =
∞
0
exp
−β
π2 2
2m
nx 2
dn x = 2m
2m
1/ 2
β
Lx 2
1/ 2
β
π
Lx
Lx
π
∞
0
nx
exp(−u 2 ) du
= cte
Por tanto,
Z x = b L x β −1 / 2
Z = Zx ⋅Zy ⋅Zz
Y la energía media por molécula...
∂ ln Z
ε =−
∂β
Z = b 3 V β −3 / 2
(nos interesa la dependencia de la
energía con la temperatura y el volumen)
3
ln Z = 3 ln b + ln V − ln β
2
3
31
3
ε =− −
, ε = kT
=
2
2β
2β
La energía media por molécula es
independiente del volumen y
proporcional a la temperatura
Para todo el gas: E = N ε =
3
N kT
2
23
¿cómo calculamos la presión media del gas?
F
p =
dA
mv
dA
- la molécula está en uno de sus estados cuánticos, con energía εr
- la molécula hará una fuerza Fr sobre la pared,
- y producirá un “desplazamiento” dLx de la pared,
- la molécula habrá ejercido un trabajo (igual a la variación de su energía):
Fr dL x = −dε r
Fr = −
∂ε r
∂L x
Por tanto, debemos hallar la fuerza media,
exp (− β ε r ) −
F=
Pr Fr =
r
∂ε r
∂L x
exp (− β ε r )
r
r
24
exp (− β ε r ) −
F=
Procedemos como antes...
exp (− β ε r ) −
r
Pr Fr =
r
∂ε r
∂L x
exp (− β ε r )
r
r
∂ε r
=−
∂L x
−
r
1 ∂
∂
exp (− β ε r ) =
β ∂L x
β ∂L x
1
Y la fuerza ejercida por una molécula es:
F=
exp (− β ε r )
r
β −1 ∂Z / ∂L x
Z
=
1 ∂ ln Z
β ∂L x
Z = b 3 V β −3 / 2
3
ln Z = 3 ln b + ln V − ln β
2
V= Lx × Ly × Lz
F=
kT
1 ∂ ln V
1
=
=
β ∂L x
β Lx Lx
Finalmente, la presión:
mv
dA
Ly
p=
Lz
FN
kT N
=
L y Lz
V
pV = N k T
Ecuación de estado del gas ideal
25
Postulados de la mecánica estadística.
El espacio de las fases molecular.
Distribución de velocidades moleculares de Maxwell-Boltzmann.
26
¿Cómo se especifica el estado microscópico de un sistema descrito en
términos de la mecánica clásica?
Especificar las posiciones y
momentos de cada partícula
Puntos en el espacio de fases
El estado de un sistema, en mecánica clásica,
puede describirse especificando la celdilla del
espacio de fases en el que se encuentran las
posiciones y momentos de las partículas del
sistema.
Sistema de N partículas:
El espacio de fases tiene N x 6 dimensiones
Definición de equilibrio:
Un sistema aislado está en equilibrio si se halla con igual probabilidad en
cada uno de sus estados accesibles, es decir, en cada una de las celdillas
accesibles del espacio de fases
27
Definición:
Un sistema aislado está en equilibrio si la probabilidad de hallarlo en cada
uno de sus estados accesibles es independiente del tiempo.
Y viceversa...
Si un sistema aislado se halla con la misma probabilidad en
cada uno de sus estados accesibles, estará en equilibrio
Si no es así, variará en el tiempo hasta llegar al equilibrio.
Hipótesis de trabajo. Postulados de la mecánica estadística.
•
“Hipótesis ergódica”. El promedio de una propiedad mecánica entre
los estados accesibles es igual al promedio temporal de la variable
termodinámica correspondiente.
•
“Principio de igual probabilidad a priori”. Todos los microestados
accesibles para el sistema en un macroestado de equilibrio son
igualmente probables.
28
Volvemos a la distribución canónica, ahora en términos de posiciones y
momentos de las partículas
2
Antes:
εr =
Pr ∝ exp (− β E r )
π2
2m
2
nx
Lx
2
+
ny
2
Ly
2
+
E r = E {n1x , n1 y , n1z , ....., n Nx , n Ny , n Nz }
Ahora:
E r = E {r1x , r1 y , r1z , ....., rNx , rNy , rNz ; p1x , p1 y , p1z , ....., p Nx , p Ny , p Nz }
y usaremos una densidad de probabilidad:
(r , p ) dr dp ∝ exp( − β E (r , p )) ⋅
dr dp
h03 N
h0 = dr dp
(r , p ) dr dp = C ⋅ exp(− β E (r , p )) dr dp
C
( r , p ) dr dp = 1
29
nz
2
Lz
2
Distribución de velocidades de Maxwell
Gas ideal, N moléculas en un volumen V, en equilbrio a temperatura T.
Tratamos una molécula (clásicamente):
1
p2
2
ε = mv =
2
2m
Buscamos la probabilidad de hallar la molécula en una celdilla del espacio de fases:
(r , p ) dr dp ∝ exp[− β ( p 2 / 2m)] dr dp
En el equilibrio, el gas está uniformemente distribuido en V :
Buscaremos el nº medio de moléculas, por unidad de volumen,
con velocidad entre v y v + dv
1
− β mv 2
N ( r , v ) dr dv
= C ⋅e 2
dv
f ( v ) dv =
dr
Función de distribución de
velocidades de Maxwell
30
¿Cómo hallamos C ?
1
Si integramos a todas las velocidades
tendremos el número de moléculas por
unidad de volumen, n
C⋅ e
− 1 β mv 2
2
− β mv 2
N (r , v ) dr dv
= C ⋅e 2
dv
f ( v ) dv =
dr
2
2
v 2 = vx + v y + vz
dv = n
2
es separable
Para vx
π
1
βm
2
1
2
exp − β mv x dv x =
−∞
2
∞
βm
f (v )dv = n
2π
3/ 2
e
1/ 2
− 1 β mv 2
2
βm
C=n
2π
dv
3/ 2
Función de distribución de
velocidades de Maxwell
31
Distribución de una componente de la velocidad
βm
f ( v ) dv = n
2π
3/ 2
e
− 1 β mv 2
Buscaremos el nº medio de moléculas, por unidad de volumen,
con la componente x de la velocidad entre vx y vx+dvx
g (v x ) dv x =
f (v )dv =
v y vz
1
2
= C ⋅ exp − β mv x dv x
2
1
2
= C ' ⋅ exp − β mv x dv x
2
∞
¿cómo hallamos C’ ?
⋅
1
exp − β m (v y2 + v z2 ) dv y dv z =
2
− ∞− ∞
⋅
g (v x ) dv x = n
−∞
g (v x ) dv x = n
∞ ∞
βm
2π
1/ 2
C' = n
βm
2π
1
2
⋅ exp − β mv x dv x
2
1/ 2
⋅
32
2
dv
¿Cuál es el valor medio de vx ? ¿se desplaza el gas?
βm
g (v x ) dv x = n
2π
∞
1
vx =
g (v x ) v x dv x = 0
n −∞
1/ 2
1
2
⋅ exp − β mv x dv x
2
Anchura cuadrática media:
g (v x )
nβ m / 2 π
1.0
∞
1
kT
v =
g (v x ) v x2 dv x =
m
n −∞
0.8
2
x
0.6
0.4
(v )
2 1/ 2
x
0.2
=
kT
m
0.0
-4
-2
0
2
vx
4
⋅
βm
33
Distribución de los módulos de las velocidades moleculares
Buscaremos el nº medio de moléculas, por unidad de volumen,
con el módulo de la velocidad entre v y v+dv
F (v) dv =
vy
A : zona de velocidades
f (v ) dv
cuyo módulo está entre
v y v+dv
A
Volumen de la corteza:
v
(4π v )dv
vx
2
vz
F (v) dv = 4 π f (v ) v dv
2
F (v) dv = 4 π C e
− 1 β mv 2
2
βm
2
v dv = 4 π n
2π
3/ 2
e
v+δv
− 1 β mv 2
2
v 2 dv
34
βm
Propiedades de la función de distribución de
F (v) dv = 4 π n
los módulos de las velocidades moleculares
2π
F (v ) dv = n
n
π
m
kT
π
m
kT
3/ 2
e
− mv 2 / 2 kT
e
− 1 β mv 2
2
v 2 dv
v 2 dv
1.0
F (v )
2
2
3/ 2
1/ 2
0.8
¿cuál es la velocidad media?
0.6
¿cuál es la velocidad más probable?
0.4
¿cuál es la velocidad raiz cuadrática
media?
0.2
¿cuál es la energía media por molécula?
0.0
0
1
2
3
4
5
v
kT / m
35
F (v ) dv = n
2
π
m
kT
3/ 2
e
∞
velocidad media:
m
kT
1
2
v=
F (v ) v dv =
n0
π
velocidad más probable, v*: máximo de F(v)
velocidad raiz cuadrática media:
− mv 2 / 2 kT
3/ 2
e
v 2 dv
− mv 2 / 2 kT
d F (v )
=0
dv
( )
v rcm ≡ v 2
1/ 2
∞
1
2
v2 =
F (v ) v 2 dv =
π
n0
energía media por molécula:
v 3 dv
mv 2
,
ε=
2
m
kT
3/ 2
e
− mv 2 / 2 kT
v 4 dv
( )
v rcm ≡ v 2
∞
1/ 2
1
m1
F (v) v 2 dv =
ε = mv 2 =
2
2 n0
m
π
m
kT
3/ 2
e
− mv 2 / 2 kT
36
v 4 dv
F (v ) dv = n
2
π
m
kT
3/ 2
e
− mv 2 / 2 kT
v 2 dv
v rcm = 3
F (v )
n
2
π
m
kT
8
0.8
v=
0.6
v* = 2
0.4
ε=
1/ 2
π
kT
m
kT
m
kT
m
3
kT
2
0.2
0.0
0
1
v*
2
v
3
4
5
v
kT / m
v rcm
Ejemplo: Nitrógeno, 1 atmósfera, 300K:
v * ≈ 500 m/s
37
F (v ) dv = n
2
π
m
kT
3/ 2
e
− mv 2 / 2 kT
v 2 dv
N2
H2
Efecto de la temperatura
Efecto de la masa
¿Por qué en la atmósfera terrestre hay mucho más nitrógeno y oxígeno que hidrógeno?
¿Por qué hay hidrógeno en la de Júpiter?
38
Validez del estudio clásico:
Gas: las distancias y velocidades típicas
asociadas a las moléculas deben cumplir:
r0 >>
p0
, r0 >> λ0 , longitud de onda de de Broglie
p0 ≈ m v * = 2 m k T ,
λ0 =
r0 ⋅ p 0 >>
h
≈
2π p 0
3
r0 ≡ volumen por molécula =
h
2mkT
Límite clásico:
V
N
r0 = n −1 / 3
λ0
h n1 / 3
≈
<< 1
r0
mkT
Gas diluido, masa no muy
pequeña, alta temperatura
Ejemplo: Helio, 1 atmósfera, 300K:
p ≈ 10 5 Pa, kT ≈ 4 10 −19 J , n = p / kT ≈ 2.5 10 25 molecs / m 3
λ0 ≈ 0.01 nm, r0 ≈ 3 nm
39
Repaso de Termodinámica
Las tres leyes de la termodinámica.
Los potenciales termodinámicos y las relaciones de Maxwell.
40
Descripción macroscópica de sistemas termodinámicos
Un sistema termodinámico es cualquier cantidad de materia o radiación lo
suficientemente grande como para ser descrito por parámetros macroscópicos, sin
ninguna referencia a sus componentes individuales (microscópicos).
Para una descripción completa del sistema también se necesita un descripción del
contorno (los límites), y de las interacciones que este permite con el entorno. Los
contornos pueden permitir el paso de materia y energía.
Sistema aislado: no intercambia energía ni masa con su entorno.
Sistema cerrado: sólo puede intercambiar energía.
Sistema abierto: puede intercambiar materia y energía.
Sistema móvil / rígido: las paredes permiten (o no) transferir energía en forma de
trabajo mecánico.
Sistema diatérmico: transferencia de calor sin trabajo.
Sistema adiabático: no hay transferencia de calor por las paredes.
Sistemas en contacto térmico, permeables, en contacto difusivo, etc
Parámetros termodinámicos: variables termodinámicas que describen el
macroestado del sistema.
Los macroestados se pueden describir en términos de un pequeño número de
variables de estado. (Ej: macroestado de un gas: masa, presión y volumen lo
describen totalmente)
41
Variables intensivas: independientes de la masa (ej: temperatura)
Variables extensivas: proporcionales a la masa (ej: energía interna)
Cantidades específicas: expresadas por unidad de masa.
Cantidades molares: expresadas por mol.
(EJ: Capacidad calorífica específica y molar)
Un sistema está en equilibrio termodinámico cuando sus variables de estado son
constantes a escala macroscópica.
No se requiere que los parámetros termodinámicos sean estrictamente
independientes del tiempo.
Los parámetros termodinámicos son promedios macroscópicos del movimiento
microscópico, por tanto habrá fluctuaciones. El valor relativo de estas fluctuaciones
es casi despreciable en sistemas macroscópicos, excepto cerca de las transiciones
de fase.
Sistema homogéneo: los parámetros intensivos son los mismos en todo el sistema.
Sistema inhomogéneo: uno o mas de los parámetros intensivos presenta variaciones
espaciales.
Un sistema inhomogéneo puede estar formado por distintas fases, separadas por
contornos de fase, de forma que cada fase sea homogénea. (EJ: En el punto triple del
agua coexisten hielo, agua y vapor de agua.)
Los contornos pueden ser arbitrarios, pero la termodinámica sigue siendo válida.
42
Ecuación de estado: es una relación funcional entre los parámetros de un sistema en
equilibrio.
Un estado de un sistema descrito por los parámetros p,V y T tendrá una ecuación de
estado f[p,V,T]=0, y por tanto reduce en uno el número de variables independientes.
Esta ecuación describe una superficie, la superficie de equilibrio.
Un estado de equilibrio puede ser representado por un punto en esta superficie. Un punto
fuera de ella es un estado de no-equilibrio.
Diagrama de estado: una proyección de una curva en la superficie de estado. (ej:
diagrama P-V)
Superficie de equilibrio para un
gas ideal con número fijo de
partículas.
U
3
2
pV
43
Leyes de la Termodinámica
Ley Cero (o principio cero) de la Termodinámica
Si dos sistemas están por separado en equilibrio con un tercero, entonces también deben
estar en equilibrio entre ellos.
Si tres o mas sistemas están en contacto térmico y todos juntos en equilibrio, entonces
cualquier par está en equilibrio por separado.
El concepto de temperatura se basa en este principio cero.
44
Primera Ley de la Termodinámica
Es una adapación para la termodinámica de la ley de conservación de la energía.
Se define la energía interna del sistema, E, como su energía respecto del SR del centro de masa.
El trabajo necesario para cambiar el estado de un sistema aislado depende unicamente de los estados
inicial y final, y es independiente del método usado para realizar el cambio.
Por tanto, existe una función de estado que identificamos como la energía interna. El trabajo
realizado sobre el sistema es W. Por tanto, el cambio de la energía interna durante una
transformación adiabática es ∆ E = W.
El sistema también puede variar su energía sin realizar trabajo mecánico, se transfiere de otra forma,
como calor.
Definición de calor: La cantidad de calor Q absorbido por un sistema es el cambio en su energía interna
que no se debe al trabajo.
La conservación de energía será:
∆ E = Q + W.
Si realizamos variaciones cuasiestáticas (p.ej., de volumen) escribiremos: δ W = - p dV.
Si movemos el pistón muy rápido el gas no hará trabajo sobre el pistón y δ W → 0 aunque varíe el
volumen.
Usamos d para una diferencial (propia) que depende sólo del cambio de estado. Usamos δ para indicar una diferencial
(impropia) que también depende del proceso usado para cambiar el estado.
Por tanto se escribe:
d E = δ Q + δ W.
45
Segunda Ley de la Termodinámica
La base de esta ley es el hecho de que si mezclamos partes iguales de dos gases nunca los
encontraremos separados de forma espontánea en un instante posterior.
Enunciado de Clausius: No hay ninguna transformación termodinámica cuyo único efecto sea transferir
calor de un foco frío a otro caliente.
Enunciado de Kelvin: No hay ninguna transformación termodinámica cuyo único efecto sea extraer calor
de un foco y convertirlo totalmente en trabajo.
La segunda ley proporciona la base para el concepto termodinámico de entropía.
Principio de máxima entropía: Existe una función de estado de los parámetros extensivos de cualquier
sistema termodinámico, llamada entropía S, con las siguientes propiedades:
1. los valores que toman las variables extensivas son los que maximizan S consistentes con
los parámetros externos,
2. la entropía de un sistema compuesto es la suma de las entropías de sus subsistemas.
(2º y 3º postulados de Callen)
Introducción de la definición de temperatura. Sistema hidrostático: E=E(S,V,ni)
dE =
∂E
∂E
dS +
dV +
∂S
∂V
∂S
>0
∂E
i
∂E
dni = T dS − pdV +
∂ni
1
>0
T
µ i dni
i
T >0
46
Teorema de Clausius:
Para cualquier ciclo cerrado
Q
T
0
donde δ Q es el calor absorbido por el sistema de un foco a temperatura T.
Si el ciclo es reversible
Q
0
T
Qrev
T S
es el calor absorbido durante un proceso reversible el que
la temperatura cambia en δ T
Podemos definir la capacidad calorífica:
Cx
Qrev
T
x
CX = T
Relacionamos capacidad calorífica y entropía:
∂S
∂T
dE = δQ + dW = T dS − pdV
Capacidad calorífica a volumen constante: dV = 0
dE = TdS
X
CV = T
Capacidad calorífica a presión constante:
(entalpía)
H = E + pV
dp = 0
∂S
∂T
=
V
∂E
∂T
V
dH = TdS + Vdp
dH = TdS
CP = T
∂S
∂T
=
P
∂H
∂T
P
47
Tercera Ley de la Termodinámica
Terorema de Nerst: Una reacción química entre fases puras cristalinas que ocurre en el cero absoluto
no produce ningún cambio de entropía.
Enunciado de Nerst-Simon: El cambio de entropía que resulta de cualquier transformación isoterma
reversible de un sistema tiende a cero según la temperatura se aproxima a cero.
lim
T 0
ST
0
Enunciado de Planck: Para T→0, la entropía de cualquier sistema en equilibrio se aproxima a una
constante que es independiente de las demás variables termodinámicas.
Teorema de la inaccesibilidad del cero absoluto: No existe ningún proceso capaz de reducir la
temperatura de un sistema al cero absluto en un número finito de pasos.
4º Postulado de Callen: La entropía de cualquier sistema se anula en el estado para el cual
∂E
∂S
=0
V , ni
48
Potenciales termodinámicos
Necesitamos relaciones entre las funciones termodinámicas, definir diferentes
potenciales termodinámicos dependiendo de qué se mantiene constante en el
sistema.
Partimos de:
µ i dni ,
dE = T dS − pdV +
E = E ( S , V , ni )
i
µ i dni )
Definimos los potenciales termodinámicos: (todos con +
i
Energía interna:
E = E (S ,V )
Entalpía:
H = E − PV
Energía libre de Helmholtz:
dE = T dS − pdV
H (S , P)
F = E − TS
Energía libre de Gibbs (entalpía libre):
dH = T dS + Vdp
F (T , V )
G = H − TS
dF = − SdT − pdV
G (T , p )
dG = − SdT + Vdp
Esto se obtiene usando las transformaciones de Legendre:
Sea:
f ( x, y ) → df = u dx + v dy
Si definimos:
g ≡ f −ux
Entonces:
g (u , y ) → dg = − x du + v dy
49
Principio de energía mínima: (es un corolario del de S máxima):
El valor de equilibrio de cualquier parámetro interno sin ligadura es tal que hace mínima la
energía interna para el valor dado de la entropía.
Principios extremales:
Los valores de equilibrio de los parámetros internos sin ligadura minimizan los potenciales
termodinámicos correspondientes:
F: sistema en contacto con foco térmico, T cte.
H: sistema en contacto con fuente de presión, P cte.
G: sistema en contacto con ambps focos, T y P ctes.
50
Relaciones de Maxwell
Se obtienen usando las transformaciones de Legendre, más la diferencial exacta:
f ( x, y ) → df = u dx + v dy
∂2 f
∂2 f
=
⇔
∂x∂y ∂y∂x
E = E (S ,V )
H = E − PV
F = E − TS
G = H − TS
∂u
∂y
=
x
∂v
∂x
y
dE = T dS − pdV →
dH = T dS + Vdp →
dF = − SdT − pdV →
dG = − SdT + Vdp →
∂T
∂V
∂T
∂P
∂S
∂V
∂S
∂P
=−
S
=
S
=
T
=
T
∂P
∂S
∂V
∂S
∂P
∂T
∂V
∂T
V
P
V
P
51
Aplicaciones:
Relaciones entre capacidades caloríficas, y coeficientes de dilatación y de compresibilidad.
dS =
dS =
∂S
∂S
∂P
dV → TdS = CV dT + T
dT +
∂T
∂V
∂T
∂V
∂S
∂S
dP → TdS = C P dT − T
dT +
∂T
∂P
∂T
∂V
C P − CV = T
∂T
α≡
1 ∂V
V ∂T
P
∂P
∂T
κT oS ≡
P
α2
,
C P − CV = T V
κT
V
∂P
= −T
∂V
1 ∂V
V ∂P
T
∂V
∂T
dV
V
dP
P
2
P
T oS
Constante adiabática : γ =
CP κT
=
CV κ S
52
Ecuación de Gibbs-Duhem
Si comparamos la expresión diferencial para E con la relación fundamental:
µ i dni ,
dE = T dS − pdV +
i
µ i dni +
dE = T dS + S dT − Vdp − pdV +
i
n i dµ i ,
i
... vemos que los parámetros intensivos no son independientes, sino que cumplen la
relación de Gibss-Duhem:
ni dµ i = 0
S dT − Vdp +
i
Y para la energía libre de Gibbs tenemos:
E = TS − pV + µN ,
G = E − TS + pV = µN
Es decir, la energía libre de Gibbs molar es igual al potencial químico.
53
Estabilidad termodinámica.
Principio de Le Chatelier: Si un sistema está en eauilibrio estable, cualquier perturbación
produce procesos que tienden a devolver al sistema a su estado original de equilibrio.
Cómo responde el sistema a fluctuaciones locales de los distintos parámetros.
El principio de máxima entropía requiere:
S 0
2
La estabilidad térmica requiere: Cp
S
CV
0
Partiendo de fluctuaciones de presión:
Ti
Si
0
0
Vi
Vi
Vi
T
Vi
S
T,i
S,i
0
pi
pi
0
2
S
2
S
1
Vi
2
1
Ti
Vi S,i
2
i
T,i
Ti
i
S
pi
2
pi
2
1
V
V
p
T
S
0
0
S
54