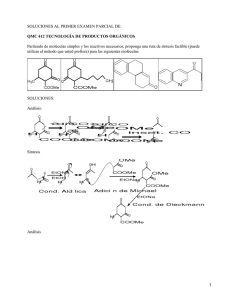
James Clerk Maxwell Ludwig Boltzmann Germán
Anuncio

TERMODINÁMICA ESTADÍSTICA TEORÍA CINÉTICA DE LOS GASES FENÓMENOS DE TRANSPORTE CINÉTICA QUÍMICA CINÉTICA MOLECULAR FENÓMENOS DE SUPERFICIE Ludwig Boltzmann James Clerk Maxwell Germán Fernández 31-01-2016 Índice general 1. MECÁNICA ESTADÍSTICA 3 1.1. Macroestado de un sistema . . . . . . . . . . . . . . . . . . . . . . 3 1.2. Microestado de un sistema . . . . . . . . . . . . . . . . . . . . . . 4 1.3. Relación entre macroestado y microestado . . . . . . . . . . . . . . 5 1.4. El colectivo canónico . . . . . . . . . . . . . . . . . . . . . . . . . 6 1.5. Evaluación de la entropía . . . . . . . . . . . . . . . . . . . . . . . 7 1.6. Probabilidad de un microestado de un colectivo canónico . . . . . . 9 1.7. Funciones termodinámicas en el Colectivo canónico . . . . . . . . . 10 1.7.1. Evaluación de U (Energía interna) . . . . . . . . . . . . . . 10 1.7.2. Evaluación de P (Presión) . . . . . . . . . . . . . . . . . . 11 1.7.3. Evaluación de S (Entropía) . . . . . . . . . . . . . . . . . . 12 1.7.4. Evaluación de A (Energía libre de Helmholtz) . . . . . . . . 12 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.7.5. Evaluación de µ (Potencial químico) . . . . . . . . . . . . . 13 1.8. Resumen de ecuaciones . . . . . . . . . . . . . . . . . . . . . . . . 13 1.9. Propiedades de la función de partición canónica . . . . . . . . . . . 14 1.10. Función de partición canónica de un sistema de partículas que no interaccionan . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 1.11. Función de partición canónica de un gas ideal puro . . . . . . . . . 18 1.12. Cálculo de la energía interna . . . . . . . . . . . . . . . . . . . . . 19 1.13. Función de partición traslacional . . . . . . . . . . . . . . . . . . . 20 1.14. Función de partición rotacional . . . . . . . . . . . . . . . . . . . . 23 1.15. Función de partición vibracional . . . . . . . . . . . . . . . . . . . 24 1.16. Función de partición electrónica . . . . . . . . . . . . . . . . . . . 25 1.17. Ecuación de Estado . . . . . . . . . . . . . . . . . . . . . . . . . . 25 1.18. Energía Interna . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 1.19. Capacidad calorífica . . . . . . . . . . . . . . . . . . . . . . . . . . 28 1.20. Entropía . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 1.21. PROBLEMAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 2. TEORÍA CINÉTICA DE LOS GASES 41 2.1. Introducción . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 2.2. Fundamentos de la teoría cinético-molecular de los gases . . . . . . 42 2.3. Funciones de distribución de velocidad . . . . . . . . . . . . . . . . 42 2.4. Funciones de distribución de las componentes de la velocidad g(vx ), g(vy ) y g(vz ) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 43 2.5. Obtención de la función de distribución para vx g(vx ) . . . . . . . . 43 2.6. Función de distribución φ(~v ) = g(vx )g(vy )g(vz ) . . . . . . . . . . 46 2.7. Obtención de la función de distribución del módulo de la velocidad G(v) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47 2.8. Cálculo de la velocidad media < v > . . . . . . . . . . . . . . . . 50 2.9. Velocidad más probable . . . . . . . . . . . . . . . . . . . . . . . . 50 2.10. G(v) en función de energías traslacionales . . . . . . . . . . . . . . 50 2.11. Colisiones con una pared y efusión . . . . . . . . . . . . . . . . . . 51 2.12. Ley de Graham: Efusión . . . . . . . . . . . . . . . . . . . . . . . 54 2.13. Método de Knudsen para determinar presiones de vapor de líquidos 54 2.14. Colisiones moleculares . . . . . . . . . . . . . . . . . . . . . . . . 55 2.15. El recorrido medio libre: λ . . . . . . . . . . . . . . . . . . . . . . 59 2.16. Problemas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 3. FENOMENOS DE TRANSPORTE 69 3.1. Introducción . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69 3.2. Tipos de fenómenos de transporte . . . . . . . . . . . . . . . . . . 70 3.3. Conductividad térmica . . . . . . . . . . . . . . . . . . . . . . . . 71 3.4. Ejemplo de aplicación de la Ley de Fourier . . . . . . . . . . . . . 73 3.5. Evaluación de k mediante la teoría cinética de los gases . . . . . . . 74 3.6. Ejemplo de evaluación de la conductividad térmica (k) . . . . . . . 78 3.7. Ley de Newton de la viscosidad . . . . . . . . . . . . . . . . . . . 79 3.8. Ley de Newton en función del momento lineal . . . . . . . . . . . . 80 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 4 3.9. Viscosidad en gases y líquidos . . . . . . . . . . . . . . . . . . . . 81 3.10. La ecuación de Poiseuille . . . . . . . . . . . . . . . . . . . . . . . 82 3.11. Teoría cinética aplicada a la viscosidad de los gases . . . . . . . . . 86 3.12. Problemas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88 3.13. Difusión: Primera Ley de Fick . . . . . . . . . . . . . . . . . . . . 88 3.14. Teoría cinética de la difusión en los gases . . . . . . . . . . . . . . 90 3.15. Problemas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com Capı́tulo 1 MECÁNICA ESTADÍSTICA La mecánica estadística es la parte de la química física que permite la predicción de propiedades termodinámicas a partir de los datos proporcionados por la mecánica cuántica. Así, a partir de propiedades microscópicas de la materia (cuánticas) se pueden obtener propiedades macroscópicas como entropía, capacidades caloríficas, energía interna , tensión superficial, viscosidad,.... 1.1. Macroestado de un sistema La mecánica estadística es el nexo de unión entre el mundo microscópico estudiado por la mecánica cuántica, y el mundo macroscópico estudiado por la termodinámica clásica. El estado de un sistema macroscópico, formado por un gran número de partículas, _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 6 CAPÍTULO 1. MECÁNICA ESTADÍSTICA viene definido por unas variables macroscópicas denominadas funciones de estado, dependientes sólo de las condiciones iniciales y finales pero no del camino seguido. En una sustancia pura en equilibrio el estado del sistema (macroestado) queda definido por tres variables, presión, temperatura y número de moles. No es necesario especificar el volumen porque existe una ecuación de estado que relacina las cuatro variables termodinámicas. Por ejemplo, en el caso de un gas ideal PV=nRT. En este caso el macroestado del sistema queda especificado por tres variables de estado, ya que cualquier otra magnitud termodinámica se puede obtener a partir de ellas. Así, U=U(P,T,n)=U(V,T,n)=.... Dar el macroestado de un sistema consiste, por tanto, en especificar el menor número posible de variables independientes que determinan su estado termodinámico. 1.2. Microestado de un sistema En mecánica clásica el estado microscópico de un sistema o microestado se obtiene especificando las coordenadas y velocidades de todas las partículas que lo componen en un instante de tiempo dado. Para un sistema clásico de N partículas (1,2,3,.....N), son necesarias 3N coordenadas para especificar la posición de todas las partículas: x1 , y1 , z1 , x2 , y2 , z2 ......xN , yN , zN . Además son necesarias otras 3N coordenadas para especificar sus velocidades, vx1 , vy1 , vz1 Por tanto, son necesarias 6N coordenadas para describir el sistema en mecánica clá_________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.3 Relación entre macroestado y microestado 7 sica y calcular sus propiedades. Por ejemplo, la energía total del sistema es suma de su energía cinética, que depende de las velocidades, y de la energía potencial, dependiente de la posición de las partículas. Puede demostrarse que la energía del sistema depende del número de partículas y del volumen disponible para el movimiento E=E(N,V). En sistemas cuánticos el estado del sistema viene dado por la función de onda, dependiente de los números cuánticos n,l,m y ms , que deben especificarse para cada partícula, lo que supone 4N números cuánticos para un sistema de N partículas. La energía del sistema se calcula a partir de la ecuación de Schrödinger una vez conocida la función de onda. Igual que en el caso clásico la energía depende de N y V. 1.3. Relación entre macroestado y microestado Consideremos un sistema termodinámico formado por una gas encerrado en un recipiente, sumergido en un baño termostático, de paredes rígidas y conductoras. Este sistema se encuentra en un macroestado que viene definido por las variables termodinámicas: número de partículas, temperatura y volumen (por ejemplo, 1 mol, 298 K y 1L). Si medimos estas magnitudes en distintos instantes veremos que no cambian, es decir su estado macroscópico (macroestado) de este sistema en equilibrio no varía con el tiempo. Sin embargo, a nivel molecular se producen fluctuaciones en las velocidades y posiciones de las moléculas que componen dicho gas. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 8 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Tenemos un único macroestado pero muchos microestados compatibles con él. Durante la medida de una propiedad macroscópica el sistema pasará por un gran número de microestados y el valor obtenido corresponde al promedio temporal sobre todos esos microestados. El cálculo de este promedio temporal es complejo ya que implica saber la energía de los diferentes microestados y qué microestados visita el sistema durante la medida y durante cuánto tiempo. 1.4. El colectivo canónico Para simplificar el cálculo de este promedio vamos a considerar un número grande de sistemas idénticos, todos ellos en el mismo macroestado, pero congelados en distintos microestados. Este conjuto recibe el nombre de colectivo canónico. Las propiedades termodinámicas se calculan multiplicando la probabilidad de que el sistema se encuentre en un microestado por el valor de la propiedad en dicho microestado y sumando sobre todos los posibles microestados del colectivo. Por ejemplo, la energía interna de un gas es igual al promedio de las energías de P todos los microestados que forman el colectivo canónico. U = pi Ei , donde pi es la probabilidad de que el sistema se encuentre en el microestado i y Ei la energía de dicho microestado. Si el colectivo está formado por A sistemas y hay ai sistemas en el micrestado i, la probabilidad de ese microestado viene dada por pi = _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com ai A. 1.5 Evaluación de la entropía 9 Podemos calcular la presión ejercida por un gas mediante la ecuación P = P p i Pi , siendo pi la probabilidad del microestado i y Pi su presión. Existen magnitudes termodinámicas, como la entropía, que no están definidas a nivel microscópico y no pueden ser calculadas de esta forma. 1.5. Evaluación de la entropía La entropía en colectivo está relacionada con el grado de desorden del mismo. Este desorden podemos evaluarlo calculando el número de maneras de repartir los A sistemas que forman el colectivo de manera que haya a1 en el microestado 1, a2 en el microestado 2....., aN en el microestado N. El número de formas de repartir los A sistemas viene dado por: A! W =Q j aj ! (1.1) La relación entre el desorden (W) y la entropía viene dada por la ecuación de Boltzmann, S = klnW , siendo k la constante de Boltzmann. Tomando logaritmos neperianos en W: lnW = lnA! − X lnaj ! (1.2) j Utilizando la aproximación de Stirling lnN ! = N lnN − N . Sustituyendo en la ecuación de Boltzman, obtenemos la entropía de nuestro colec_________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 10 CAPÍTULO 1. MECÁNICA ESTADÍSTICA tivo de sistemas Scol = k(AlnA − A − X aj lnaj − aj ) = j − k kAlnaA − kA X aj lnaj + k j X X j j aj = kAlnA − k aj lnaj (1.3) Por tanto, Scol = kAlnA − k X aj lnaj (1.4) j Como todos los sistemas del colectivo son iguales desde el punto de vista macroscópico, tendrán la misma entropía. Dividiendo por el número de sistemas obtenemos la entropía de cada uno. S= X aj Scol = klnA − k lnaj A A (1.5) j Dado que la probabilidad de un microestado viene dada por: pj = aj A, la ecuación anterior puede escribirse como: S = klnA − k X pj lnApj (1.6) j Aplicando la propiedad del neperiano de un producto: − k S = klnA X p j lnA − k j X pj lnpj = −k j _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com X j pj lnpj (1.7) 1.6 Probabilidad de un microestado de un colectivo canónico 1.6. 11 Probabilidad de un microestado de un colectivo canónico Las ecuaciones obtenidas anteriormente conectan la mundo microscópico con el macroscópico, permitiendo el cálculo de propiedades termodinámicas, energía interna y entropía, a partir de propiedades microscópicas calculadas mediante la mecánica cuántica. U= X pj Ej (1.8) j S = −k X pj lnpj (1.9) j Siendo pj la probabilidad del microestado j del colectivo canónico y Ej su energía. La probabilidad, pj , de un microestado del colectivo tiene la siguiente forma matemática: pj = Ce−βEj (1.10) Siendo, C y β parámetros a determinar. La constante C se puede determinar con la P condición de normalización j pj = 1 X 1=C e−βEj (1.11) j Despejando C: 1 −βEj e j C=P (1.12) Sustituyendo en la probabilidad del microestado j: e−βEj pj = P −βE j je _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.13) 12 CAPÍTULO 1. MECÁNICA ESTADÍSTICA El denominador de esta expresión recibe el nombre de función de partición canónica, Q, función de N, V y T. Q= X e−βEj (1.14) j Obsérvese que la función de partición canónica es una suma a lo largo de todos los microestados del colectivo. Sustituyendo el parámetro β por su valor, β = Q= X 1 kT . e−Ej /kT (1.15) j 1.7. Funciones termodinámicas en el Colectivo canónico Una vez determinada la probabilidad de cada microestado pasamos a calcular las funciones termodinámicas del colectivo: U, P, S, G, A, µ. 1.7.1. Evaluación de U (Energía interna) U= X P j pj Ej = P Ej e−βEj k j e−βEk P = j Ej e−Ej /kT Q (1.16) Derivando (1.15) respecto de T a volumen y composición constantes: ∂Q ∂T = V,N 1 X Ej e−Ej /kT kT 2 j _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.17) 1.7 Funciones termodinámicas en el Colectivo canónico 13 Despejando el sumatorio de la ecuación (1.17): X 2 ∂Q kT = Ej e−Ej /kT ∂T V,N (1.18) j Sustituyendo (1.18) en (1.16) U = kT 2 1.7.2. ∂lnQ ∂T (1.19) V,N Evaluación de P (Presión) Supongamos que Pj es la presión del microestado j con probabilidad pj . P = X p j Pj (1.20) j Como pj = e−βEj Q y además, dE = P dV a NB = cte nos da Pj = − ∂Ej ∂V N sustituyendo ambas ecuaciones en (1.20) 1 X −βEj e P =− Q j Derivando la función de partición Q = ∂Q ∂V = T,N P X j ∂Ej ∂V (1.21) N e−βEj respecto al volumen: −β j ∂Ej ∂V e−βEj (1.22) e−βEj (1.23) N Despejando de (1.22): 1 β ∂Q ∂V =− T,N X ∂Ej j ∂V _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com N 14 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Sustituyendo (1.23) en (1.21): 1 P = Qβ Sustituyendo β por 1.7.3. 1 KT ∂Q ∂V T,N 1 = β ∂lnQ ∂V (1.24) T,N en (1.24) nos queda: ∂lnQ P = kT ∂V T,N (1.25) Evaluación de S (Entropía) Partimos del primer principio de la termodinámica: dU = T dS − P dV (1.26) Despejando dS en (1.26) y dividiendo por T dS = dU P + dV T T (1.27) Sustituyendo P en (1.27) por (1.25): dU dS = +k T ∂lnQ ∂V dV (1.28) T,N Integrando (1.28) S= 1.7.4. U + klnQ T (1.29) Evaluación de A (Energía libre de Helmholtz) Partimos de la expresión termodinámica para la energía libre de Helmholtz: A = U − TS _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.30) 1.8 Resumen de ecuaciones 15 Sustituyendo S en (1.30) por (1.29) U A=U −T + klnQ = −kT lnQ T (1.31) Así, la ecuación de Helmholtz en mecánica estadística nos queda: A = −kT lnQ 1.7.5. (1.32) Evaluación de µ (Potencial químico) A partir de las ecuaciones G = H − T S, A = U − T S y H = U + P V se llega a G = A + PV ∂G ∂A ∂lnQ µA = = = −kT ∂nA T,V,ni6=A ∂nA T,V,ni6=A ∂nA T,V,ni6=A (1.33) Por tanto, el potencial químico del componente B en una mezcla binaria AB, es: ∂lnQ µA = −kT (1.34) ∂nA T,V,ni6=A 1.8. Resumen de ecuaciones Q= X e −Ek kT (1.35) k ∂lnQ ∂T V,N ∂lnQ P = kT ∂V T,N U = kT 2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.36) (1.37) 16 CAPÍTULO 1. MECÁNICA ESTADÍSTICA S= U + klnQ T A = −kT lnQ ∂lnQ ∂lnQ H = U + P V = kT 2 + kT V ∂T N,V ∂V N,T ∂Q G = H − T S = A + P V = −kT lnQ + kT V ∂V N,T ∂lnQ µA = −kT ∂nA T,V,ni6=A 1.9. (1.38) (1.39) (1.40) (1.41) (1.42) Propiedades de la función de partición canónica La función de partición canónica depende de (N,V,T) y viene dada por: Q(N, V, T ) = P j e−Ej /kT . Supongamos un sistema con niveles de energía no degenerados (gi = 1), con energías 0,1,2,3... La función de partición se desarrolla según la siguiente expresión: Q= X e−Ej /kT = 1 + e−1/kT + e−2/kT + ..... (1.43) j Ahora evaluamos los valores de Q en los límites de alta y baja temperatura. Si T → 0, Q=1. Nos indica que a baja temperatura sólo el primer estado cuántico es accesible. Si T → ∞, Q=1+1+1+1.... Nos da el número total de estados cuánticos (microestados) accesibles por el sistema. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.9 Propiedades de la función de partición canónica 17 A medida que la temperatura sube aumentan el número de estados accesibles por el sistema. Como podemos ver en la gráfica la probabilidad de ocupación de un microestado disminuye con la temperatura. El microestado más probable es el de menor energía, disminuyendo la probabilidad de ocupación a medida que aumenta la energía. Consideremos ahora sistemas con degeneración, es decir, cada nivel de energía posee más de un estado cuántico. En esta situación podemos seguir sumando sobre niveles de energía introduciendo _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 18 CAPÍTULO 1. MECÁNICA ESTADÍSTICA la degeneración en la función de partición. Q= X gj e−Ej /kT = g0 + g1 e−E1 /kT + g2 eE2 /kT + .... (1.44) j Evaluando los límites de Q a altas y bajas temperaturas: Si T → 0, Q = g0 . Es decir, a cero kelvin sólo el nivel fundamental es accesible. Si T → ∞, Q = g0 + g1 + g2 + ...... En este caso son accesibles todos los niveles del sistema. Cuando un sistema presenta degeneración la probabilidad de ocupación de un nivel j viene dada por: pj = gj e−Ej /kT Q (1.45) En esta ecuación se observa que la probabilidad de un nivel depende de su degeneración y puede darse el caso de que el nivel fundamental tenga menor ocupación que un nivel excitado, siempre que éste último sea más degenerado. 1.10. Función de partición canónica de un sistema de partículas que no interaccionan Una vez calculada la función de partición canónica Q de un sistema sumando e−Ej /kT sobre todos los estados posibles, pueden calcularse todos las propiedades termodinámicas del sistema (P, U, S, A, µA ). La existencia de interacciones entre _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.10 Función de partición canónica de un sistema de partículas que no interaccionan 19 partículas hace difícil el cálculo de Q. Supongamos un sistema sin interacciones, el hamiltoniano del sistema puede separarse en suma de hamiltonianos para cada partícula Ĥ = Ĥ1 + Ĥ2 + ....... + ĤN y la energía es suma de energías de las moléculas individuales Ej = 1,r + 2,s + ...... + N,w donde es la energía de una molécula y r,s....,w indica el estado cuántico en que se encuentran las respectivas moléculas. Sustituyendo en la función de partición: Q= X −β1,r X e−βEj = j X r e X e−β (1,r +2,s +.....+N,w ) = j e −β2,s s ........ X e−βN,w = q1 q2 ......qN (1.46) w q1 , q2 , ...., qN son las funciones de partición moleculares. Si todas las moléculas son de la misma clase q1 = q2 = ..... = qN . Por tanto: Q = (q)N (1.47) Si las moléculas no son todas iguales, sino que hay dos tipos, NB moléculas de B y NC moléculas de C, la función de partición se transforma en: Q = (qB )NB (qC )NC (1.48) donde: qB = X e−βB,r y qC = X r e−βC,s (1.49) s La mecánica cuántica impide distinguir entre partículas de un mismo gas. Para una mezcla de gases ideales con NB moléculas de B y NC moléculas de C, se pueden _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 20 CAPÍTULO 1. MECÁNICA ESTADÍSTICA distinguir las moléculas de B de las moléculas de C, pero no podemos distinguir las moléculas de B entre si, ni tampoco las de C. La ecuación (1.47) distingue entre moléculas del propio gas y por tanto debemos corregirla dividiendo por N !. Q= qN N! (1.50) Para dos gases la ecuación (1.48)se transforma en: Q= 1.11. (qB )NB (qB )NC NB !NC ! (1.51) Función de partición canónica de un gas ideal puro La relación entre la función de partición canónica y molecular de un gas puro puede escribirse como: Q= qN N! (1.52) Siendo Q la función de partición canónica y q la función de partición molecular. Tomando neperianos en (1.52): lnQ = N lnq − lnN ! (1.53) Utilizando la aproximación de Stirling lnN ! = N lnN − N en (1.53): lnQ = N lnq − N lnN + N Consideremos ahora la función de partición molecular q = (1.54) P j e−βj . La energía molecular es suma de energía traslacional, rotacional, vibracional y electrónica. j = tr,n + vib,v + rot,J + ele,u _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.55) 1.12 Cálculo de la energía interna 21 Sustituyendo en la función de partición molecular q q= XX XX X −βtr,n n v e n e−β (tr,n +vib,v +rot,J +ele,u ) = u J X e−βvib,v X v e−βrot,J X e−βele,u (1.56) u J q = qtr · qvib · qrot · qele (1.57) Tomando neperianos en (1.57) lnq = lnqtr + lnqrot + lnqvib + lnqele (1.58) Sustituyendo en (1.54) lnQ = N lnqtr + N lnqvib + N lnqrot + N lnqele − N (lnN − 1) 1.12. (1.59) Cálculo de la energía interna U = kT 2 ∂lnQ ∂T (1.60) V,N Sustituyendo (1.59)en (1.60) dlnqvib dlnqrot dlnqele ∂lnqtr 2 U = N kT + + + ∂T dT dT dT V tr vib donde Utr = N kT 2 ∂lnq Uvib = N kT 2 dlnq ∂T dT , ......... (1.61) V U = Utr + Uvib + Urot + Uele _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.62) 22 CAPÍTULO 1. MECÁNICA ESTADÍSTICA 1.13. Función de partición traslacional Los niveles de energía traslacionales vienen dados por el modelo de la partícula en una caja tridimensional. h2 tr = 8m n2x n2y n2z + + a2 a2 a2 ! (1.63) Separando esta energía en tres sumandos, tr = h2 n2y h2 n2z h2 n2x + + = tr,x + tr,y + tr,z 8ma2 8mb2 8mc2 (1.64) Sustituyendo esta energía en la función de partición traslacional, qtr = X e −βtr,i = ∞ X ∞ X ∞ X e−β(tr,x +tr,y +tr,z ) (1.65) nx =1 ny =1 nz =1 i Separando la exponencial en producto de exponenciales: qtr = ∞ X nx =1 ∞ X −βtr,x e ny =1 −βtr,y e ∞ X e−βtr,z = qtr,x qtr,y qtr,z (1.66) nz =1 Como podemos ver en la ecuaciones anteriores la energía traslacional es suma de energías en cada una de las direcciones del movimiento, mientras que la función de partición es producto de las tres funciones de partición. Dado que las tres funciones de partición son idénticas sólo evaluaremos la qtr,x . Para simplificar los cálculos trasladaremos todos los niveles de energía en una cantidad h2 /8ma2 para que el nivel fundamental tenga energía cero. tr,x = n2x h2 h2 h2 − = (n2 − 1) 2 2 8ma 8ma 8ma2 x _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.67) 1.13 Función de partición traslacional 23 Con lo que la función de partición traslacional en dirección x nos queda: ∞ X qtr,x = h2 kT 2 e− 8ma2 (nx −1) (1.68) nx =1 Definimos la temperatura traslacional característica en dirección x como: θtr,x = h2 /8ma2 k, que llevada a la función de partición nos da: qtr,x = ∞ X e− θtr,x T (n2x − 1) (1.69) nx =1 A T=0 K, la función de partición vale 1, es decir sólo está accesible para las moléculas el estado fundamental. A T → ∞, la función de partición también tiende al infinito, lo que nos indica que todos los estados son accesibles. Separando en la ecuación anterior en dos términos y sacando fuera del sumatorio el independiente de nx nos queda: qtr,x = e θtr,x T ∞ X e− θtr,x 2 nx T (1.70) nx =1 La temperatura traslacional característica depende del tipo de molécula, pero en general toma valores muy pequeños 10−14 K. Salvo en las proximidades a 0 K se puede realizar la aproximación θtr,x << T , que equivale a θtr,x /T << 1 y eθtr,x /T ≈ 1, lo que nos deja la función de partición como sigue: qtr,x = ∞ X e− θtr,x 2 nx T (1.71) nx =1 Otra aproximación que podemos realizar es el cambio del sumatorio por integral aprovechando que los términos del sumatorio son muy próximos entre sí. Z ∞ θ tr,x 2 qtr,x = e− T nx dnx 0 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.72) 24 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Dado que la suma tiene un gran número de términos cambiamos el límite inferior de la integral sin que se cometa un error importante. Para encontrar la solución de 1/2 R∞ 2 la integral la comparamos con: 0 e−bx dx = 21 πb ∞ Z qtr,x = e− θtr,x 2 nx T dnx = 0 π 1/2 1 2 (θtr,x /T )1/2 (1.73) Sustituyendo θtr,x por su valor nos queda: qtr,x = 2πmkT h2 1/2 a (1.74) De forma análoga obtenemos los valores para qtr,y y qtr,z qtr,y = qtr,z = 2πmkT h2 1/2 2πmkT h2 1/2 b (1.75) c (1.76) La función traslacional total nos queda: qtr = qtr,x qtr,y qtr,z = 2πmkT h2 3/2 V (1.77) Esta ecuación se cumple siempre que la temperatura sea mucho mayor que la temperatura característica de la molécula, condición que se cumple incluso a temperaturas muy bajas. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.14 Función de partición rotacional 1.14. 25 Función de partición rotacional La energía de los diferentes niveles rotacionales vienen dados por el modelo del rotor rígido: rot,J = ~2 J(J+1) , 2I ∞ X qrot = la degeneración de los niveles es gJ = 2J + 1 gJ erot,J /kT = J=0 ∞ X (2J + 1)e− ~2 J(J+1) 2IkT (1.78) J=0 Llamamos temperatura rotacional θrot a: θrot = ~2 2Ik , tiene unidades de temperatura (K) pero no es temperatura en sentido físico. Por tanto: ∞ X qrot = (2J + 1)e− θrot J(J+1) T (1.79) J=0 Si θrot T es pequeño, la separación entre niveles rotacionales es pequeña comparada co n kT y podemos aproximar la suma mediante una integral. Z ∞ qrot = (2J + 1)e− θrot J(J+1) T dJ (1.80) 0 Haciendo el cambio de variable w = J(J + 1) ⇒ dw = (2J + 1)dJ ⇒ dJ = Z qrot = ∞ e 0 − θrot w T T − θrot w ∞ T e T dw = − = θrot θrot 0 dw 2J+1 (1.81) La función de partición rotacional debe modificarse introduciendo el número de simetría σ que toma valor 1 para moléculas homonucleares y 2 para heteronucleares. qrot = T σθrot _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.82) 26 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Para T > θrot la ecuación anterior comete un error imprortante y debe utilizarse el siguiente desarrollo: qrot " # T 1 θrot 1 θrot 2 1 θrot 3 = 1+ + + + .... σθrot 3 T 15 T 315 T (1.83) Para T < θrot debe evaluarse la función de partición mediante el sumatorio: qrot = θ P∞ − rot J(J+1) T J=0 (2J + 1)e Para moléculas poliatómicas lineales sigue siendo válida la expresión anterior. En el caso de poliatómicas no lineales, la función de partición cuando T >> θrot , es: √ qrot (T ) = π T 3/2 √ σ θa θ b θc (1.84) Siendo θa , θb y θc las temperaturas características correspondientes a las rotaciones en torno a los ejes principales de inercia (a,b,c). El número de simetría, σ, coincide con el número de configuraciones indistinguibles obtenidas por rotación de la molécula. Para el agua debido a su eje C2 , sigma vale 2. En el amoniaco sigma toma valor 3. 1.15. Función de partición vibracional La aproximación del oscilador armónico da vib = v + 1 2 hν donde v es el número cuantico vibracional que oscila entre [0, ∞] y no hay degeneración. Es usual en mecánica estadística tomar el nivel más bajo de energía como cero vib = v + 21 hν − 21 hν = vhν _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.16 Función de partición electrónica qvib = X e −βvib,v v Donde θvib = = ∞ X −βvhν e 27 = ∞ X v=0 hν − kT v e = 0 ∞ X e− vθvib T (1.85) 0 hν k llamada temperatura vibracional cararcterística. P n 2 3 Utilizando el desarrollo en serie ∞ n=0 x = 1 + x + x + x + ...... = 1 1−x , la función de partición vibracional se puede escribir como sigue: qvib = 1 (1.86) 1 − e−θvib /T Esta ecuación es válida siempre que la temperatura no sea muy elevada. 1.16. Función de partición electrónica Calculamos qele como una suma extendida a los niveles de energía electrónicos, por lo que incluiremos la degeneración gele de cada nivel. qele = X gele,i e− ele,i kT = gele,0 + gele,1 e− ele,1 kT + gele,2 e− ele,2 kT + .... (1.87) i En esta última ecuación hemos tomado como cero la energía electrónica del nivel fundamental 1.17. Ecuación de Estado A partir de la función de partición traslacional se puede obtener la presión de un gas ideal. P = N KT ∂lnqtr ∂V _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.88) T 28 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Escribimos la función de partición traslacional: 2πmkT 3/2 qtr = V h2 (1.89) Tomando logaritmos neperiano lnqtr = ln 2πmkT h2 3/2 + lnV (1.90) Derivando dlnqtr 1 = dV V (1.91) Sustituyendo en la fórmula de la presión y teniendo en cuenta que N k = nR P = 1.18. N kT nRT = V V (1.92) Energía Interna E = Etr + Erot + Evib + Eele (1.93) Cálculo de la energía interna traslacional Etr = N kT qtr = 2πmkT h2 3/2 2 ∂lnqtr ∂T (1.94) V 3 V ⇒ lnqtr = lnT + ln 2 2πmk h2 (1.95) V Derivando: ∂qtr ∂T = 31 2T V _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (1.96) 1.18 Energía Interna 29 Etr = N kT 2 · 3 3 3 = N kT = nRT 2T 2 2 (1.97) Cálculo de la energía interna rotacional Erot = N kT qrot = 2 dlnqrot dT (1.98) T ⇒ lnqrot = lnT − lnσθrot σθrot (1.99) Derivando: dlnqrot 1 = dT T (1.100) Sustituyendo la derivada en la expresión de la energía Erot = N kT 2 · 1 = N kT = nRT T (1.101) Cálculo de la energía interna vibracional Evib = N kT qvib = 1 1− e−θvib /T 2 dlnqvib dT ⇒ lnqvib = −ln(1 − e−θvib /T ) (1.102) (1.103) Derivando: dlnqvib θvib /T 2 e−θvib /T θvib /T 2 = = dT 1 − e−θvib /T eθvib /T − 1 (1.104) Sustituyendo esta derivada en la ecuación de la energía Evib = nRθvib 1 eθvib /T _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com −1 (1.105) 30 CAPÍTULO 1. MECÁNICA ESTADÍSTICA 1.19. Capacidad calorífica La capacidad calorífica a volumen constante se define como la variación de la energía interna con la temperatura. Cv = ∂U ∂T (1.106) N,V De igual modo que hicimos con la energía intena, podemos calcularla como suma de sus contribuciones: traslacional, rotacional, vibracional y electrónica. Comenzamos por la capacidad calorífica traslacional. Cv,tr = ∂Utr ∂T (1.107) N,V Dado que Utr = 32 nRT Cv,tr = ∂Utr ∂T N,V 3 = nR 2 (1.108) Derivando respecto de T la Urot , obtenemos la Cv,rot Cv,rot = ∂Urot ∂T = nR (1.109) N,V Por último derivamos la Uvib para obtener la Cv,vib Cv,vib = ∂Uvib ∂T = nR N,V θvib T 2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com eθvib /T (eθvib /T − 1)2 (1.110) 1.20 Entropía 1.20. 31 Entropía La entropía de un sistema termodinámico se puede obtener como suma de sus cuatro contribuciones: entropía traslacional, rotacional, vibracional y electrónica. S = Str + Srot + Svib + Sele (1.111) Comenzamos con el cálculo de la entropía traslacional. Str = Utr + N klnqtr − N k(lnN − 1) T (1.112) El término N k(lnN − 1) es necesario para corregir la distinguibilidad de las partículas. 3 Str = nR + nRln 2 2πmkT h2 3/2 V − nRlnN + nR (1.113) Agrupando términos: 5 Str = nR + nRln 2 " 2πmkT h2 3/2 1 V N # (1.114) Reemplazamos el volumen por la ecuación de estado del gas ideal V = N kT /P , recordando que N k = nR. 5 Str = nR + nRln 2 " 5 Str = nR + nRln 2 2πmkT h2 " 3/2 2πmkT h2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com kT 1 N P N 3/2 kT P # (1.115) # (1.116) 32 CAPÍTULO 1. MECÁNICA ESTADÍSTICA Cálculo de la entropía rotacional: Srot = Svib = 1.21. Urot T + N klnqrot = nR + nRln T σθrot (1.117) Uvib θvib 1 + N klnqvib = nR − nRln(1 − e−θvib /T ) (1.118) θ /T T T e vib − 1 PROBLEMAS 1. Considere todas las posibles distribuciones de 6 elementos distinguibles en tres categorías y calcular el peso de cada una de ellas. Si todas las formas de repartir los elementos fuesen igualmente probables, ¿qué distribución resultaría más probable?. Respuesta: (2,2,2) 2. Se tiene un sistema formado por N=4 espines (s=1/2) independientes y distinguibles, teniendo cada uno de ellos dos posibles estados degenerados (ms = ±1/2). a) Representa todos los microestados posibles del sistema, agrupándolos en función del valor de Ms . ¿Cuál es la probabilidad de cada microestado? ¿y la de cada valor de Ms ?. b) Dibuja una curva de probabilidad (casos favorables/casos totales) frente a Ms . c) Repite la curva anterior para N=6. Dibújala también para N=100. d) ¿Cuál sería la entropía de un sistema formado por 1 mol de espines de este _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.21 PROBLEMAS 33 tipo a T=0K? 3. Considere un sistema con niveles energéticos dados por un número cuántico que toma valores enteros positivos desde cero hasta infinito. La energía de los niveles puede expresarse de la forma EJ = J(J + 1)a, donde a es una constante con unidades de energía. La degeneración de los niveles es 2J+1. a) Calcular la función de partición a dos temperaturas, tales que kT/a=10 y kT/a=20. b) Represente la probabilidad de ocupación de los niveles energéticos en función de J. ¿Qué nivel es el más probable para cada una de las temperaturas dadas?. Respuesta: a) 10.34 y 10.24; b) J=2 y J=3. 4. Suponer un sistema con microestados no degenerados y con energías equidistantes: En = n∆E, siendo ∆E > 0 y n cualquier número entero comprendido entre cero e infinito. a) Obtener una expresión para la función de partición. (Nota: la suma de infinitos términos de una progresión geométrica es 1/(1-r) siendo r la razón de la progresión). b) Representar la función de partición frente a kT para diferentes valores de ∆E. c) ¿Cuál sería la función de partición si kT >> ∆E?. d) Para el caso del apartado c) deducir una expresión para la energía interna _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 34 CAPÍTULO 1. MECÁNICA ESTADÍSTICA del sistema. 5. Considere un sistema formado por N=10000 partículas distinguibles, cada una de las cuales es capaz de acceder a tres estados de energías 0, y 2, siendo la energía total diponible 5000. Determine cuántas partículas habrá en cada uno de los estados para la distribución más probable. Compruebe si la distribución resultante cumple con la distribución de Boltzmann. 6. Un sistema formado por N partículas idénticas e independientes, cuyos niveles de energía dependen del número cuántico n de la forma = b(n − 1), donde b es una constante positiva con dimensiones de energía. Sabiendo que n toma valores positivos (1,2,3...) y que la degeneración de los niveles es 2n: a) Obtener una función de partición para las partículas. ¿Cuál es el valor a T=0K?. Interpretar el resultado. b) A partir del resultado anterior, obtener una expresión aproximada de la función de partición para temperaturas altas. ¿Cuál es el valor si kT=1000b?. c) Obtener una expresión para la energía interna molar del sistema en el límite de altas temperaturas. 7. a) Demostrar que si se usa la aproximación del oscilador armónico, la población relativa de un nivel vibracional v de una molécula diatómica es: vθvib θvib Nv =e T 1 − e− T (1.119) N b) Calcular para un mol de CO el número de moléculas en el nivel v=0 y v=1 a 25o C y a 1000o C. Para el CO ν¯e = 2143 cm−1 . _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.21 PROBLEMAS 35 c) Para el N2 (g) con θvib = 3352 K, representar la población relativa del nivel vibracional v=1 frente a T, desde 0 a 15000 K (0, 2000, 4000, 6000, 8000, 10000, 12000, 15000), utilizando la aproximación del oscilador armónico. Explicar qué parte de la curva no es exacta. 8. Los átomos de sodio tienen términos electrónicos fundamentales doblete. Calcular la función de partición molecular molar estandar (es decir, usando un volumen molar correspondiente a 1 bar) para los átomos de sodio a T=1000 K. ¿Cuánto valdría la función de partición a T=0K? 9. Para el HF, θrot = 29,577K. Calcular qrot para el HF(g) a 20K, 30K y 40K: a) con la aproximación de altas temperaturas. b) de forma exacta. c) comparar resultados. 10. Un átomo de argón se encuentra atrapado en una caja cúbica de volumen V. ¿Cuál es su función de partición traslacional a: (a) 100 K; (b) 298 K; (c) 10000 K; (d) 0K, si el lado de la caja es 1cm? 11. La forma de la función de partición traslacional, tal como se utiliza normalmente, es válida cuando son accesibles un gran número de niveles de energía: (a) ¿Cuándo resulta incorrecta la expresión normal y hemos de utilizar el sumatorio de forma explícita?; (b) Calcula la temperatura del argón en el problema anterior para que la función de partición disminuya hata 10; (c) ¿Cuál es el valor exacto de la función de partición a esa temperatura? _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 36 CAPÍTULO 1. MECÁNICA ESTADÍSTICA 12. La mayoría de las moléculas tienen estados electrónicos que están muy altos en energía respecto del fundamental, de manera que sólo hay que considerar este último para calcular sus propiedades termodinámicas. Hay diversas excepciones. Un caso interesante es la molécula de NO que tiene un estado electrónico excitado sólo 121.1 cm−1 por encima del estado fundamental, siendo este estado excitado y el fundamental doblemente degenerados. Calcula y dibuja la función de partición electrónica desde cero a 1000 K. ¿Cuál es la distribución de poblaciones a temperatura ambiente? 13. Calcula la energía interna electrónica del NO a 298 K. Obtén primero una expresión para U a una temperatura cualquiera, T, (utiliza la función de partición anterior) y sustituye entonces el valor T=298K. 14. Calcula la contribución electrónica a la entropía molar de la molécula de NO a 298 K y 500 K. Haz servir los datos del problema 15. La molécula de NO tiene un estado fundamental doblemente degenerado, y a sólo 121,1 cm−1 se encuentra un estado excitado doblemente degenerado. Calcula la contribución electrónica a la capacidad calorífica molar de esta molécula a 50, 298, 500 K. 16. Los niveles de energía vibracional de la molécula de iodo respecto al estado fundamental tienen los siguientes números de ondas: 213.30, 425.39, 636.27, 845.39, 1054.38 cm−1 . Obtén la función de partición vibracional calculando explicitamente la suma en la expresión de la función de partición molecular q, _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.21 PROBLEMAS 37 a 100 K y 298 K. Compárala con la función de partición obtenida, suponiendo que la vibración de la molécula de iodo se puede considerar armónica, siendo ν = 214,6 cm−1 . ¿Qué proporción de moléculas de iodo se encuentra en el estado fundamental y en los dos estados excitados vibracionales inferiores a la temperatura de a) 100 K y b) 298 K?. 17. Una muestra de cinco moleculas tiene una energia total E = 5(0 + ). Cada molecula es capaz de ocupar estados de energia con un valor 0 + j, siendo j un numero entero (0,1,2.....). Dibuja una tabla con columnas encabezadas por la energia de una molecula (desde 0 a 0 + 5) y escribe debajo de estas todas las configuraciones del sistema compatibles con una energia total de 5(0 +). Encuentra el peso (W) de cada configuracion. .Cual es la configuracion mas probable?. 18. Se considera un sistema compuesto por un gran numero de moleculas, N, cada una de las cuales tiene acceso a dos niveles de energia 0 y 1 con la misma degeneracion, g, (0 < 1 ), = 1 −0 . Este sistema obedece la estadistica de Maxwell-Boltzmann y esta en equilibrio con un termostato a la temperatura T. 1) Deduce una expresion que nos permita calcular la funcion de particion molecular. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 38 CAPÍTULO 1. MECÁNICA ESTADÍSTICA 2) Escribe las expresiones de los numeros de particulas, N0 y N1 , que en promedio tienen las energias 0 y 1 , respectivamente. Calcula la probabilidad de encontrar una molecula en los niveles 0 y 1 , respectivamente. 3) Calcula < N0 > − < N1 >. ¿Cuales son los limites de < N0 >, < N1 > y de < N0 > − < N1 >, en los dos casos siguientes: i) T bajas ( >> kT ) ii)T altas ( << kT ) Explica los resultados anteriores con argumentos físicos simples. 4) Obten una expresion para la energia interna U del sistema considerado. 19. Calcula el valor de la entropia que proporciona la ecuacion Sekur-Tetrode para un mol de atomos de neon a 200 K y 1 atmosfera de presion. R: 137,92 J/Kmol 20. Calcula Ū − Ū 0 , Ḡ − Ḡ0 y S̄ a 300 K para la molecula de CO2 . Los modos normales de vibracion tienen los siguientes numeros de onda: 1340 (1), 667 (2) y 2349 (1) cm-1. La constante rotacional es B = 0,390 cm−1 . R: 6,94x103 J/mol ; -54,77x103J/mol; 214,01 J/Kmol 21. Calcula el valor termodinamico estadistico de la constante de equilibrio Kp de la reaccion I2 2I a 1000 K. Los datos espectroscopicos para la molecula de I2 son los siguientes: B = 0,0373 cm−1 , ν̄ = 214,56 cm−1 , D0 = 1,5417 eV. Los atomos de iodo tienen un estado fundamental 2 P3/2 . R: 3,22x10-3 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.21 PROBLEMAS 39 22. Sea un sistema formado por 3 moléculas distinguibles e independientes cuya energía total es 3. Cada molécula puede encontrarse en 4 estados diferentes con energías 0 = 0, 1 = , 2 = 2 y 3 = 3. (a) Escribir todos los microestados posibles del sistema. (b) ¿Cuántas y cuáles son las posibles distribuciones del sistema? (c) Hallar la probabilidad de cada una de las distribuciones. 23. Sea un sistema formado por 4 partículas distinguibles e independientes cuya energía total es 2. Cada partícula puede hallarse en 4 niveles diferentes con energías 0 = 0, 1 = , 2 = 2 y 3 = 3. (a) Escribir todos los microestados posibles del sistema. ¿Cuántas y cuales son las posibles distribuciones del sistema? Suponer que los niveles energéticos no son degenerados. (b) ¿Cómo se modifica el número de microestados si el nivel 0 está dos veces degenerado y el nivel 1 está tres veces degenerado? 24. Sean 1 , 2 , 3 .... los niveles de energía moleculares de un cierto gas de moléculas que no interaccionan entre sí. En el equilibrio, ¿es posible tener más moléculas en el nivel 2 que en el nivel 1 si 2 > 1 ?. Explícalo. 25. Un cierto sistema se compone de 1 mol de moléculas idénticas que no interaccionan entre sí. Cada molécula tiene disponibles únicamente 3 niveles de energía cuyos valores y degeneraciones son: 1 = 0, g1 = 1; 2 /k = 100K, g2 = 3; 3 /k = 300K, g3 = 5 (k es la constante de Boltzmann). (a) Calcular la función de partición a 200K. (b) Calcular el número de moléculas _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 40 CAPÍTULO 1. MECÁNICA ESTADÍSTICA en cada nivel a 200K. (c) Calcular el número de moléculas en cada nivel en el límite T → ∞. 26. Considerar un sistema de N partículas distinguibles e independientes, cada una de las cuales tiene sólo dos estados accesibles: un estado fundamental de energía 0 y un estado excitado de energía . (a) Obtener la función de partición molecular, q, a la temperatura T. (b) Obtener el número de partículas en cada estado molecular, N0 y N , a la temperatura T. (c) Obtener la energía del sistema E a la temperatura T. (d) ¿A qué valores tienden N0 , N y E cuando T → 0? ¿y cuando T → ∞? Comenta los resultados. (e) A la temperatura T ∗ , la población del estado fundamental es doble de la del estado de energía . Calcular en función de T ∗ y la constante de Boltzmann k. (f) Repetir los apartados a, b y c considerando que las energías de los dos estados aumentan en una cantidad 0 27. La función de Helmoltz A(N, V, T ) = −kB T lnZ(N, V, T ) es el potencial termodinámico fundamental del colectivo canónico, del que podemos deducir todas las relaciones termodinámicas. Utilizando su definición A = E − T S y su forma diferencial dA = −SdT − P dV + µdN , encuentra expresiones termodinámico-estadísticas (en términos de la función de partición canónica) para S, P, µ y E, y comprueba que coinciden con las obtenidas explícitamente a partir de Z. 28. En un sistema formado por N partículas distinguibles e independientes, cada _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 1.21 PROBLEMAS 41 una de las partículas puede hallarse en 3 niveles de energía no degenerados con valores 0, 1 , 2 . A una temperatura de 300K la mitad de las partículas se hallan en el nivel fundamental, la tercera parte en el nivel 1 y la sexta parte en el nivel molecular 2 . (a) Determinar los valores de 1 /k y 2 /k. (b) Determinar la función de partición molecular q, 10, 300 y 1000K e interpretar los resultados obtenidos. (c) Demostrar que a cualquier temperatura la fracción de partículas suma de las de los dos niveles excitados es 1 − 1q . (d) Demostar que la temperatura a la que es máxima la población del nivel de energía 1 vie i h 1 . (e) Calcular la fracción de partículas ne dada por: Tmax = k2 ln 2− 1 del nivel fundamental a Tmax 29. Considera un sistema de 3 partículas (a,b,c) con dos niveles de energía 1 y 2 y degeneraciones g1 = 1 y g2 = 2. Hay por tanto tres estados disponibles sin restricciones en cuanto al número de partículas en cada uno de ellos. Escribe todos los microestados asociados al macroestado en el que en el primer nivel hay 1 partícula y en el segundo nivel hay 2 partículas (N1 = 1, N2 = 2) _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 42 CAPÍTULO 1. MECÁNICA ESTADÍSTICA _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com Capı́tulo 2 TEORÍA CINÉTICA DE LOS GASES 2.1. Introducción La teoría cinética de los gases permite deducir las propiedades del gas ideal empleando un modelo en el que las moléculas del gas son esferas que cumplen las leyes de la mecánica clásica. Las propiedades calculables mediante este modelo son: presión del gas, distribución de velocidades moleculares, velocidad molecular media, velocidad de colisión y distancia media entre colisiones. Estas propiedades permiten el estudio de la cinética de reacciones en fase gaseosa así como el flujo de fluidos y la transmisión de calor. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 44 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES 2.2. Fundamentos de la teoría cinético-molecular de los gases La teoría cinética puede considerarse como una rama de la termodinámica estadística ya que deduce propiedades macroscópicas de la materia a partir de propiedades moleculares. Los principios en los que se fundamenta son los siguientes: Un gas está formado por un gran número de partículas esféricas cuyo tamaño es despreciable comparado con la distancia entre las partículas. Las moléculas se mueven en línea recta a gran velocidad y sólo interaccionan cuando colisionan. Los choques entre partículas y con las paredes del recipiente se consideran perfectamente elásticos, conservándose la energía cinética traslacional. La teoría cinética supone que las partículas obedecen las leyes de Newton. Esta suposición es incorrecta (las moléculas cumplen las leyes de la mecánica cuántica) y conduce a resultados incorrectos en la predicción de las capacidades caloríficas del gas, aunque da resultados aceptables en propiedades como presión o difusión. 2.3. Funciones de distribución de velocidad Las funciones de distribución de velocidades nos van a permitir conocer la fracción de moléculas con velocidades comprendidas entre dos valores dados. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.4 Funciones de distribución de las componentes de la velocidad g(vx ), g(vy ) y g(vz ) 45 Vamos a utilizar 3 funciones de distribución : Para los componentes de la velocidad g(vx ), g(vy ) y g(vz ). Para el vector velocidad φ(~v ) para el módulo de la velocidad G(v). 2.4. Funciones de distribución de las componentes de la velocidad g(vx ), g(vy ) y g(vz ) La función de distribución g(vx ) permite conocer la fracción de moléculas con velocidades comprendidas entre vx y vx + dvx . dNvx /N = g(vx )dvx (2.1) Pueden escribirse ecuaciones análogas para las funciones de distribución en dirección y,z 2.5. dNvy /N = g(vy )dvy (2.2) dNvz /N = g(vz )dvz (2.3) Obtención de la función de distribución para vx g(vx ) La probabilidad de encontrar una molécula en un determinado nivel energético, j, viene dada por: e−j /kT pj = P − /kT e i _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.4) 46 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Desde el punto de vista clásico la energía cinética de una molécula varía de forma continua y viene dada por la expresión x = 12 mvx2 . Llevando este valor de energía a la expresión de la probabilidad se obtiene: 2 mvx dNvx e− 2kT dvx dp(vx ) = =R 2 N ∞ − mvx 2kT dvx e −∞ (2.5) Donde, dp(vx ) representa la fracción de moléculas con velocidad comprendida entre vx y vx + dvx . La integral del denominador representa la suma a lo largo de todos R los posibles valores de vx y tiene la forma e−bx dx = (π/b)1/2 2 mvx e− 2kT dvx dNvx = dp(vx ) = 2πkT 1/2 N (2.6) m Teniendo en cuenta que, dNvx N = g(vx )dvx , la función de distribución para vx viene dada por: g(vx ) = m 1/2 2 e−mvx /2kT 2πkT (2.7) Donde m es la masa de una molécula del gas, k es la constante de Boltzmann y T la temperatura. Las expresiones para g(vy ) y g(vz ) son análogas dada la equivalencia entre las direcciones espaciales. g(vx ) = m 1/2 2 e−mvy /2kT 2πkT (2.8) g(vx ) = m 1/2 2 e−mvz /2kT 2πkT (2.9) _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.5 Obtención de la función de distribución para vx g(vx ) 47 Las unidades de la función de distribución son inversas a las de la velocidad, para que la probabilidad obtenida al multiplicarla por dvx sea adimensional. La función g(vx ) es gaussiana, centrada en vx = 0 y simétrica respecto al eje de ordeandas (y). Esta simetría hace que la densidad de probabilidad de encontrar moléculas en vx = a sea igual a la probabilidad de encontrarlas en vx = −a. La densidad de probabilidad alcanza su máximo en cero, tendiendo hacia cero a medida que vx se va hacia ±∞. Un aumento de la temperatura del gas produce un aumento en la densidad de probabilidad para valores altos, tanto positivos como negativos, de la velocidad. Un efecto similar al de la temperatura podemos observarlo al variar la masa del gas. En la siguiente gráfica se aprecia como al disminuir la masa del gas la función de distribución se ensancha. Por tanto, la masa y la temperatura tienen efectos contrarios en la función de distribución, una disminución de masa o aumento de la temperatura producen el ensanchamiento de la curva. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 48 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES 2.6. Función de distribución φ(~v ) = g(vx )g(vy )g(vz ) La probabilidad de que una molécula tenga su componente de velocidad en dirección x comprendida entre vx y vx+dx , en dirección y entre vy y vy+dy y en dirección z entre vz y vz+dz vendrá dada por: dNvx ,vy ,vz /N = g(vx )g(vy )g(vz )dvx dvy dvz (2.10) dNvx ,vy ,vz /N representa la probabilidad de que una molécula (fracción de moléculas) tenga el extremo de su vector velocidad en el interior de una caja de lados dvx , dvy , dvz Sustituyendo las expresiones calculadas anteriormente para las funciones de distribución en cada dirección espacial: φ(v̄) = g(vx )g(vy )g(vz ) = m 1/2 2 e−mv /2kT 2πkT _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.11) 2.7 Obtención de la función de distribución del módulo de la velocidad G(v)49 Siendo, v 2 = vx2 + vy2 + vz2 , el cuadrado del módulo de la velocidad. Esta nueva función de distribución no depende de la orientación del vector, sino exclusivamente de su módulo. 2.7. Obtención de la función de distribución del módulo de la velocidad G(v) En esta sección vamos a deducir la ecuación que nos permite conocer la fracción de moléculas que tienen el módulo de su velocidad comprendida entre dos valores determinados. Llamamos dNv /N a la fracción de moléculas con velocidad comprendida entre v y v+dv. Donde N representa el número total de moléculas del gas. Nv = G(v)dv N (2.12) G(v) es la función de distribución de velocidades o densidad de probabilidad. Al multiplicar esta función por dv se obtiene la fracción de moléculas con velocidad comprendida entre v y v+dv. Si nos interesa calcular la fracción de moléculas con velocidad comprendida entre v1 y v2 , integramos la función de distribución de velocidades en ese intervalo. (Es equivalente hablar de fracción de moléculas que tienen su velocidad en un intervalo dado y probabilidad de que una molécula tenga su velocidad comprendida en dicho _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 50 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES intervalo) Z v2 P [v1 , v2 ] = G(v)dv (2.13) v1 La probabilidad de que una molécula tenga su velocidad comprendida en el intervalo [0, ∞] es 1 Z ∞ G(v)dv = 1 (2.14) 0 Para obtener G(v) debemos integrar la función φ(~v ) = g(vx )g(vy )g(vz ) para un valor v del módulo de velocidad a lo largo del espacio. Dado que esta integral se extiende sobre un casquete esférico es conveniente el uso de coordenadas esféricas (v, θ, ϕ). g(vx )g(vy )g(vz )dvx dvy dvz = m 3/2 2 e−mv /2kT dvx dvy dvz 2πkT (2.15) Haciendo el cambio a esféricas, dvx dvy dvz = v 2 senθdvdθdϕ, e integrando: dNv = G(v)dv = N Z 0 π Z 2π 0 m 3/2 2 e−mv /2kT v 2 senθdvdθdϕ 2πkT (2.16) Dado que v y su diferencial son constantes podemos sacarlas fuera de la integral. Z π Z 2π m 3/2 dNv 2 = G(v)dv = e−mv /2kT v 2 dv senθdθdϕ (2.17) N 2πkT 0 0 m 3/2 dNv 2 e−mv /2kT v 2 dv4π = G(v)dv = N 2πkT (2.18) Donde la función de distribución del módulo de la velocidad G(v) es: G(v) = m 3/2 2 e−mv /2kT 4πv 2 2πkT _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.19) 2.7 Obtención de la función de distribución del módulo de la velocidad G(v)51 A valores pequeños del módulo de la velocidad predomina el término parabólico (v 2 ) y la función crece cuadráticamente. A valores elevados de v comienza a domi2 nar la exponencial negativa e−v haciendo que la función G(v) disminuya su valor, anulándose en el infinito. El incremento de la temperatura produce un ensanchamiento en la función de distribución y un desplazamiento de su máximo a velocidades mayores. Un incremento en la masa molecular, sin embargo, produce el efecto contrario, estrechamiento de la función y desplazamiento de su máximo a velocidades menores. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 52 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES 2.8. Cálculo de la velocidad media < v > Z ∞ < v >= vG(v)dv = 4π 0 Utilizando la integral R∞ 0 m 3/2 Z ∞ 2 e−mv /2kT v 3 dv 2πkT 0 2 x2n+1 e−ax dx = (2.20) n! 2an+1 m 3/2 1 8kT 1/2 = < v >= 4π 2πkT 2(m/2kT )2 πm (2.21) Multiplicando y dividiendo por el número de avogadro y teniendo en cuenta que kNA = R y NA m = M < v >= 2.9. 8RT πM 1/2 (2.22) Velocidad más probable La velocidad más probable es la velocidad a la que es máxima la función G(v) y se obtiene igualando a cero la derivada de G(v) respecto de v. 2RT 1/2 dG(v) = 0 → vmp = dv M 2.10. (2.23) G(v) en función de energías traslacionales La distribución de Maxwell puede escribirse en función de energías traslaciona1/2 les tr = 12 mv 2 , despejando v, v = 2mtr , derivando: 1/2 2 1 −1/2 1 1/2 dtr dv = dtr = (2.24) 1/2 m 2 tr 2m tr _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.11 Colisiones con una pared y efusión Sustituyendo v y dv en dNv N = 53 3/2 −mv2 m e 2kT 4πv 2 dv 2πmkT m 3/2 −m(2tr /m) 2 1 1/2 d dNtr tr tr = e 2kT 4π 1/2 N 2πmkT m 2m tr (2.25) Simplificando: dNtr = 2π N 1 πkT 3/2 e −tr 1/2 kT tr dtr (2.26) dNtr representa el número de moléculas cuya energía traslacional está comprendida entre tr y tr + dtr La energía traslacional media es la misma para todos los gases a la misma temperatura. 2.11. Colisiones con una pared y efusión Vamos a calcular la frecuencia de colisión de las moléculas de un gas con la pared de un recipiente en un dt. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 54 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Si tomamos la pared perpendicular al eje y, sólo las moléculas con componente y de velocidad chocarán contra dicha pared. La fracción de moléculas con componente y de velocidad comprendida entre vy y vy + dvy viene dada por: dNvy = g(vy )dvy N (2.27) Solamente las moléculas que se encuentran a una distancia vy dt de la pared podrán chocar contra ella en el dt. El volumen de la caja en el que se encuentran las moléculas que pueden chocar contra la pared sera: V 0 = Avy dt. Dividiendo este voluen entre el total V 0 /V se obtiene la fracción de moléculas que por su proximidad a la pared pueden chocar. Multiplicando el número de moléculas que tienen la velocidad comprendida entre vy y vy + dvy por la fracción de moléculas que pueden llegar a la pared en el dt obtenemos: dNp,vy = Avy dt V0 dNvy = N g(vy )dvy V V (2.28) Integrando en el intervalo vy [0, ∞], obtenemos las colisiones totales: Z dNp = 0 ∞ Avy dt AN dt N g(vy )dvy = V V Z ∞ vy g(vy )dvy (2.29) 0 Evaluando el promedio de vy Z ∞ Z ∞ m 1/2 −mvy2 e 2kT vy dvy = 2πkT g(vy )vy dvy = 0 0 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com kT 2πm 1/2 (2.30) 2.11 Colisiones con una pared y efusión 55 Llevando este resultado a la ecuación de colisiones por unidad de tiempo. dNp N Adt A = V kT 2πm 1/2 (2.31) Esta última ecuación representa el número de moléculas que colisionan contra la pared de área A en un dt. dN Definimos la frecuencia de colisión por unidad de área y tiempo como:Zp = A1 dtp N kT 1/2 1 8kT 1/2 N 1 N Zp = = = <v> (2.32) V 2πm 4 2πm V 4 V Siendo < v > el módulo de la velocidad media. Zp = 1 N <v> 4 V (2.33) La frecuencia de colisión depende de la densidad del gas (N/V ) y de la velocidad media de las moléculas. A mayor densidad y velocidad media habrá mayor frecuencia de colisiones. De la ecuación de estado del gas ideal, P V = k = R/NA , se obtiene, N V = P kT . N NA RT y considerando que, Con este resultado podemos simplificar la ecuació anterior. 1 N 1 Zp = < v > = 4 V 4 8kT πm 1/2 P kT (2.34) Agrupando términos nos queda: Zp = P (2πmkT )1/2 El cálculo de Zp para el oxígeno a 298K y 1atm da: 2,7x1023 col/cm2 s _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.35) 56 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES 2.12. Ley de Graham: Efusión Si un recipiente que contiene un gas tiene un pequeño agujero de área Aor y en el exterior del recipiente hay vacío, las moléculas que chocan con el agujero escapan del recipiente a una velocidad que viene dada por: dN −Aor P = dt (2πmkT )1/2 (2.36) El signo menos indica que las moléculas salen del recipiente. A este fenómeno se le denomina efusión, siendo las velocidades de efusión inversamente proporcionales a las raices cuadradas de las masas moleculares. Para que se cumpla la Ley de Graham es necesario que el orificio sea muy pequeño, para que el gas se escape lentamente y no rompa la ditribución Maxweliana de velocidades. El diámetro del agujero debe ser sustancialmente menor que la distancia que las moléculas del gas recorren entre dos colisiones (recorrido medio libre), para evitar que choquen unas con otras en las proximidades del orificio, lo que daría lugar a un flujo colectivo que rompe la distribución de Maxwell 2.13. Método de Knudsen para determinar presiones de vapor de líquidos Se mide la presión de vapor de un líquido determinando la velocidad de efusión del gas en equilibrio con él. Para ello se mide la pérdida de peso w en el interior _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.14 Colisiones moleculares 57 debido a las moléculas que escapan. m 1/2 dw dN Aor Pv m 1/2 =m =− = −Pv Aor dt dt (2πmkT ) 2πkT (2.37) En un intervalo de tiempo finito ∆t, se producirá la siguiente pérdida de peso: m 1/2 ∆w = −Pv Aor ∆t 2πkT (2.38) Pesando el recipiente al principio y al final del experimento determinamos ∆w y utilizando la ecuación anterior la presión de vapor. 2πkT 1/2 ∆w Pv = Aor ∆t m 2.14. (2.39) Colisiones moleculares La teoría cinética permite calcular la frecuencia de colisiones entre moléculas. Suponemos que las moléculas son esferas rígidas de diámetro d y despreciamos las fuerzas intermoleculares excepto en el momento de la colisión. A presiones elevadas las fuerzas intermoleculares no pueden ser despreciadas y las ecuaciones obtenidas no son aplicables. Utilizaremos la siguiente notación: z12 : número de colisiones por unidad de tiempo (frecuencia de colisiones) que una molécula del gas 1 experimenta con las moléculas del gas 2. z11 : frecuencia de colisiones que una molécula del gas 1 experimenta con otras moléculas del gas 1. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 58 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Z12 : frecuencia total de colisiones entre moléculas del gas 1 con las del gas 2 por unidad de volumen. Sean N1 moléculas del gas 1 con diámetro d1 y N2 moléculas del gas 2 con diámetro d2 . Las moléculas se mueven a velocidades medias v1 > y < v2 >. Comenzamos calculando las colisiones de las moléculas tipo 1 con las tipo 2 (z12 ). Consideremos las moléculas de tipo 2 en reposo y las de tipo 1 moviéndose a velocidad < v12 >, velocidad media relativa de las moléculas tipo 1 respecto a las tipo 2. En un dt la molécula 1 reacorrerá una distancia < v12 > dt y chocará contra todas las moléculas 2 que se encuentren en el interior de un cilindro de diámetro d1 + d2 . El volumen del ciclindro es: N2 d1 + d2 2 N2 N2 Vcil =π < v12 > dt = πd212 < v12 > dt V 2 V V _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.40) 2.14 Colisiones moleculares 59 Siendo d12 el diámetro de colisión. También podemos definir una sección eficaz, σ12 = πd212 . Las colisiones que sufre una partícula de tipo 1 con las de tipo 2 por unidad de tiempo, llamada frecuencia de colisión z12 nos queda: z12 = σ12 < v12 > N2 N (2.41) Tan solo nos falta obtener el módulo de la velocidad relativa media, < v12 >. considerando colisiones a 90o (media entre 0o y 180o ) < v12 >2 =< v1 >2 + < v2 >2 . q Dado que < v >= 8RT πM < v12 >2 = 8RT 8RT + πM1 πM2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (2.42) 60 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Sustituyendo en z12 z12 = σ12 8RT π 1 1 + M1 M2 1/2 N2 V (2.43) Definimos la frecuencia total de colisiones de las moléculas tipo 1 con las tipo 2 como: Z12 = z12 · N1 . Dado que es un número muy grande se suele tomar por unidad de volumen: Z12 = z12 N1 V (2.44) Sustituyendo z12 Z12 = σ12 8RT π 1 1 + M1 M2 1/2 N1 N1 V V (2.45) En general z12 6= z21 pero Z12 = Z21 Las colisiones entre moléculas de tipo 1 son: z11 = πd211 < v11 > N1 V (2.46) donde d11 = d1 + d1 = d1 2 (2.47) < v11 >2 =< v1 >2 + < v1 >2 = 2 < v1 >2 = 2 8kT πm (2.48) Sustituyendo z11 = √ 2πd21 8kT πm 1/2 N1 V (2.49) Las colisiones totales entre moléculas de tipo 1 por unidad de tiempo y de volumen, vendrán dadas por: Z11 1 =√ 2πd21 8kT πm 1/2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com N1 V (2.50) 2.15 El recorrido medio libre: λ 61 Obsérvese que es necesario dividir la ecuación entre 2 para no contar dos veces la misma colisión. 2.15. El recorrido medio libre: λ Se define como la distancia que una molécula avanza entre dos colisiones sucesivas. Sea < v1 > la velocidad media de las moléculas tipo 1. En un tiempo t recorre una distancia < v1 > t siendo el número de colisiones [z11 + z12 ]t. Por tanto, la distancia media recorrida por una molécula 1 entre dos colisiones es: < v1 > z11 + z12 (2.51) 1 V < v1 > =√ 2 z11 N 2πd1 1 (2.52) λ= Si el gas es puro: λ= Utilizando la ecuación PV=NkT, V/N=kT/P. λ= √ 2.16. 1 kT 2πd21 P (2.53) Problemas 1. La función de distribución o densidad de probabilidad, f (vx ), de la componente vx de la velocidad molecular de un gas en equilibrio térmico viene dada, en la teoría cinética de los gases, por la expresión f (vx )dvx = _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 62 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES 2 1/2 −mvx m e 2kT dvx , 2πkT donde m es la masa moleuclar, k la constante de Boltz- mann y T la temperatura absoluta. a) ¿Qué dimensiones tienen f (vx ) y f (vx )dvx ? b) Calcula el valor más probable de vx , vxmp , o valor de vx para el que la función de distribución es máxima, y obtén f (vxmp ) c) Discute los efectos de la masa molecular y la temperatura en f (vxmp ) comparando los valores para el He y el Xe a 300 y a 1000 K. d) Calcula la velocidad cuadrática media, < vx2 >, y observa que el producto f (vxmp ) < vx2 > es una magnitud independiente de la masa del gas y de la temperatura. e) Calcula el promedio estadístico, o promedio sobre la distribución, de vx , < vx >, y comprueba que la disviación típica, σ(vx ), coincide con la raíz de la velocidad cuadrática media, vxrcm . f) Obtén la función de distribución para el módulo de la velocidad integrando sobre los ángulos el producto de las distribuciones f (vx )f (vy )f (vz )dvx dvy dvz . Compara el significado de f (vx )dvx con el de este producto y con el de f (v)dv. g) Calcula la velocidad más probable v mp , la velocidad media, < v > y la raíz de la velocidad cuadrática media, v rcm , y comprueba que estas tres magnitudes estan en la relación v mp :< v >: rrcm = 1,4142 : 15958 : 17321 h) Deduce la función de distribución de la energía traslacional f ()d a partir de la distribución molecular (f). Observa que esta distribución no es gausiana _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.16 Problemas 63 sino exponencial y que es función exclusiva de la temperatura, es decir, no depende de la masa del gas. i) La función de distribución de energía anterior (distribución de Maxwell) es el equivalente continuo de la distribución de energías discretas dentro del colectivo canónico, o de la distribución de energía molecular de Boltzmann. ¿Cómo interpretarías los tres factores que aparecen en la distribución del aparatado anterior (constante, exponencial y término 4πv 2 2. Para 1 mol de oxígeno a 300K y 1 atm calcular: a) El número de moléculas cuyas velocidades estén comprendidas entre 500,000 y 500,001 m/s (puesto que este intervalo de velocidades es pequeño, la función de distribución prácticamente no varía, y podemos considerarlo infinitesimal). b) El número de moléculas con vz comprendido entre 150000 y 150001 m/s. c) El número de moléculas que simultáneamente tienen vz y vx entre 150,000 y 150,001 m/s. Solución a) dNv = G(v)dv → Nv = N G(v)∆v N m 3/2 −mv2 G(v) = e 2kT 4πv 2 2πkT (2.54) (2.55) Sustituyendo, v = 500 m/s; k = 1, 38x10−23 J/s; T = 300K; m = 0,032 = 5,33x10−26 kg, se obtiene: G(v) 6x1023 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com = 1,85x10−3 s/m Por tanto, 64 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Nv = 6x1023 · (1,85x10−3 s/m) · (0,001 m/s) = 1,1x1018 moleculas b) dNvz = G(vz )dvz → Nvz = N G(vz )∆vz = 7,75x1017 moleculas N (2.56) m 1/2 −mvz2 g(vz ) = e 2kT (2.57) 2πkT Se resuelve de forma análoga al apartado a) c) Nvx vy vz = N g(vx )g(vz )∆vx ∆vz = 9, 22x10−11 moleculas (2.58) 3. Calcular la densidad de probabilidad para la componente x de la velocidad de una muestra de moléculas de oxígeno a 300K en el intervalo 0 < |vx | < 1000m/s. Representar la función resultante. Solución: −6 m−2 s2 )v 2 x g(vx ) = (1,04289x10−3 s/m)e(−6,4145x10 (2.59) 4. Para el metano gas a 300K y 1 bar, calcular la probabilidad de que una molécula elegida al azar tenga una velocidad comprendida entre 400,000 m/s y 400,001 m/s. Este intervalo es lo suficientemente pequeño para considerarse infinitesimal. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.16 Problemas 65 Solución: Nv = G(v)∆v = 1,24x10−6 N (2.60) 5. a) Para el oxígeno a 25o C y 1 atm, calcular el cociente de probabilidad de que una molécula tenga su velocidad en un intervalo infinitesimal dv localizado a 500 m/s y la probabilidad de que su velocidad esté en un intervalo infinitesimal dv centrado en 1500 m/s. b) ¿A qué temperatura tendrá el oxígeno su función de distribución del módulo de velocidad de 500 m/s igual a la del módulo de 1500 m/s? Solución: a) La probabilidad de que una molécula tenga su velocidad comprendida entre v y v+dv viene dada por: ambas velocidades, Nv1 N Nv N = G(v)dv. Escribiendo esta expresión para = G(v1 )dv y Nv2 N = G(v2 )dv. Dividiendo ambas expresiones: Nv1 N Nv2 N = 2 −v 2 ) 2 −m(v2 G(v1 ) 1 v1 = e 2kT = 70219 Gv2 v22 (2.61) Por cada molécula con velocidad comprendida entre 1500 y 1500+dv hay 70219 moléculas con velocidad entre 500 y 500+dv b) Igualando a 1 el cociente de ambas probabilidades: Nv1 2 −v 2 ) −m(v2 1 N 2kT = 1 = e Nv2 N _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com v12 v22 (2.62) 66 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES Despejando la temperatura, T=1751.6 K 6. Calcula para el dióxido de carbono a 500 K: (a) vrms ; (b) <v>; (c) vp Solución: (a) < vrms >= 3kT m 1/2 8RT πM = 3RT M = 532 m/s (2.63) (b) < v >= 1/2 = 490 m/s (2.64) (c) vp = 2RT M 1/2 = 435 m/s (2.65) Donde M = 0,044 kg/mol y R = 8,314 J/molK 7. Calcula < v 3 > para las moléculas de un gas ideal. ¿Es < v 3 > igual a < v >< v 2 > Solución: El promedio de una suma puede expresarse como suma de promedios, no cumpliéndose esta propiedad para el producto. Z ∞ Z ∞ 2 m 3/2 −mv e 2kT 4πv 2 v 3 dv (2.66) < v 3 >= G(v)v 3 dv = 2mkT 0 0 R∞ 2 Escribimos la solución de la integral comparándola con: 0 x2n+1 e−αx dx = n! 2αn+1 27/2 < v >= √ π 3 kT m _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3/2 (2.67) 2.16 Problemas 67 8. Obtener la energía de traslación molecular media a partir de la distribución de Maxwell expresada como distribución de energías. 9. Calcular la energía traslacional más probable para las moléculas de un gas ideal. Compararla con la energía traslacional media de la moléculas del gas ideal. 10. Deducir la fórmula de un hidrocarburo gaseoso que efluye 0.872 veces más rápido que el oxígeno a través de un pequeño orificio, siendo iguales la presión y la temperatura. 11. Un recipiente que contiene escandio sólido en equilibrio con su vapor a 1690 K, experimenta una pérdida de peso de 10.5 mg en 49.5 minutos, a través de un agujero circular de 0.1763 cm de diámetro. Calcular la presión de vapor del Sc a 1690 K en torr. 12. Un vehículo espacial con un volumen interno de 3 m3 choca con un meteorito originándose un orificio de 0.1 mm de radio. Si la presión del oxígeno dentro del vehículo es inicialmente de 0.8 atm y su temperatura de 298 K. ¿Cuánto tiempo tardará la presión en reducirse a 0.7 atm?. 13. Para el Octoil C6 H4 (COOHC8 H17 )2 , la presión de vapor es 0.01 torr a 293 K. (a) Calcular el número de moléculas que se evaporan en vacío a partir de 1,00 cm3 de superficie de líquido a 393 K en 1.0 s. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 68 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES (b) Calcular la masa que se evapora a esa temperatura durante 1.0 s. 14. Para el N2 (g) con un diámetro de colisión de 3.7 Anstrong y a 300 K, calcular el recorrido libre medio a: (a) 1 bar; (b) 1.0 torr; (c) 1,0x10−6 torr. 15. Calcular el número total de colisiones por segundo y por cm3 en la atmósfera terrestre a 25o C y 1 atm entre: (a) Moléculas de oxígeno (b) Moléculas de nitrógeno. (c) Moléculas de oxígeno y de nitrógeno. Utilizar los siguientes radios moleculares r(O2 ) = 178 pm y r(N2 ) = 185 pm 16. Sea un gas ideal encerrado en un recinto a volumen constante. Al disminuir la temperatura: a) La densidad de frecuencias de colisiones mutuas Z11 , no varía b) La densidad de frecuencias de colisiones mutuas Z11 , disminuye c) La densidad de frecuencias de colisiones mutuas Z11 , aumenta. 17. Cuando disminuye el volumen permitido a un gas ideal permaneciendo constante la temperatura, disminuye: a) La velocidad media de traslación. b) el recorrido libre medio. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 2.16 Problemas 69 18. El recorrido libre medio de las moléculas de un gas a temperatura ambiente y presión atmosférica es del orden de: a) 10−7 m b) 10−12 m c) 103 m 19. Para un gas ideal contenido en un recipiente de volumen constante, al aumentar la temperatura también aumenta: a) el recorrido libre medio b) la sección eficaz de colisión de las moléculas. c) número de colisiones por unidad de área y unidad de tiempo. 20. a) Indicar el rango de v. b) Indicar el rango de vx c) Sin hacer ningún cálculo, indicar el valor de < vz > para el O2(g) a 298 K y 1 bar. d) Para la fórmula dNv /N = G(v)dv, explicar lo que significa dNv . 21. ¿Verdader o falso? a) A 300K y 1 bar , vx,p es la misma para el He(g) que para el N2 (g). b) A 300 K y 1 bar, vp es la misma para el He(g) que para el N2 (g). c) A 300 K y 1 bar < tr,p > es la misma para el He(g) que para el N2 (g). d) La función de distribución G(v) es adimensional. 22. ¿A qué temperatura deben enfriarse los átomos de He para que tengan la misma vrms que el O2 (g) a 25o C?. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 70 CAPÍTULO 2. TEORÍA CINÉTICA DE LOS GASES _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com Capı́tulo 3 FENOMENOS DE TRANSPORTE 3.1. Introducción La termodinámica clásica estudia las propiedades de equilibrio de los sistemas, siendo los procesos reversibles. Ahora vamos a estudiar sistemas que estan fuera del equilibrio, en los cuales ocurren procesos irreversibles. Un sistema se encuentra fuera del equilibrio porque materia o energía son transportadas entre el sistema y los alrededores, o bien, entre diferentes partes del sistema. Estos sistemas fuera del equilibrio evolucionan, con el paso del tiempo, siguiendo procesos irreversibles hacia estados de equilibrio. Dos ejemplos de procesos irrevesibles son la disolución de una sal y la transferencia de calor entre dos focos térmicos a diferente temperatura. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 72 CAPÍTULO 3. FENOMENOS DE TRANSPORTE En el esquema (a) tenemos un sistema formado por dos fases (sulfato de cobre y agua), que evoluciona de modo irreversible hasta la disolución total de la sal o saturación de la disolución En el esquema (b) dos focos caloríficos en contacto evolucionan hasta alcanzar el equilibrio térmico. 3.2. Tipos de fenómenos de transporte Flujo de fluidos: Existen fuerzas no equilibradas en el sistema, no hay equilibrio mecánico y partes del sistema se mueven. La Ley de Newton de la viscosidad relaciona la fuerza de rozamiento entre las láminas del fluido y el gradiente de velocidad. En esta Ley a parece una constante de porporcionalidad llamada viscosidad. Fx = −ηA dvx dz (3.1) Conducción electrica: Al aplicar un campo eléctrico las partículas con carga (electrones, iones) se mueven produciendo una corriente electrica. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.3 Conductividad térmica 73 Conducción térmica: Existen diferencias de temperatura entre sistema y alrededores o dentro del sistema, produciéndose un flujo de energía calorífica hasta igualar las temperaturas. El flujo de calor entre dos focos viene descrito por la Ley de Fourier y es proporcional al gradiente de temperatura. La constante de porporcionalidad se denomina conductividad térmica. dT dq = −kA dt dz (3.2) Transporte de materia: Existen diferencias de concentraciones de sustancias entre diferentes regiones de la disolución. Se produce un flujo de materia hasta que se igualan las concentraciones. El flujo de materia viene dado por la Ley de Fick y es proporcional al gradiente de concentración. La constante de proporcionalidad se donomina coeficiente de difusión. dc dn = −DA dt dz 3.3. (3.3) Conductividad térmica La sustancia en contacto con los depósitos a distintas temperaturas alcanzará al final un estado estacionario en el que habrá un gradiente uniforme de temperatura dT/dz. El flujo de calor dq/dt (medido en J/s) a través de cualquier plano perpendicular a z es proporcional al área de la sección transversal y al gradiente de temperatura. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 74 CAPÍTULO 3. FENOMENOS DE TRANSPORTE La expresión matemática que relaciona dichas magnitudes físicas se conoce como Ley de Fourier de la coducción térmica. dq dT = −kA dt dz (3.4) Siendo k una constante de proporcionalidad, llamada conductividad térmica de la sustancia, cuyas unidades son: J/K cm s. Los buenos conductores térmicos tienen constantes elevadas k(Cu(s)) = 10J/Kcms. Los malos conductores térmicos poseen costantes pequeñas k(CO2 (g)) = 10−4 J/Kcms La constante k depende de temperatura y presión para sustancias puras, en el caso de mezclas también depende de la composición. En los gases aumenta con la temperatura. La Ley de Fourier también puede escribirse en función del flujo de calor por unidad _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.4 Ejemplo de aplicación de la Ley de Fourier 75 de tiempo y área: J. Denominado densidad de flujo de calor, cuyas unidades son J/m2 s J= 1 dq dT = −k A dt dx (3.5) La transferencia de calor en los gases tiene lugar por colisiones moleculares. Las moléculas de temperatura más alta tienen mayor energía y en las colisiones ceden parte de esta energía a las moléculas de menor temperatura. Dado que las moléculas de gas tienen gran libertad de movimiento la transmisión de calor suele producirse también por convección, debido al movimiento del fluido. Otra forma de transmisión de calor es la radiación, debida a la emisión de ondas electromagnéticas por parte de los cuerpos calientes que son absorbidas por los fríos. Para que la Ley de Fourier sea aplicable deben cumplirse tres condiciones: Sistema isótropo (todas las direcciones son iguales) Gradiente de temperatura pequeño. No hay transferencia de calor por conducción ni radiación. 3.4. Ejemplo de aplicación de la Ley de Fourier Dos depósitos de calor con temperaturas respectivas de 325 y 275 K se ponen en contacto mediante una varilla de hierro de 200 cm de longitud y 24cm2 de sección transversal. Calcular el flujo de calor entre los depósitos cuando el sistema alcanza su estado estacionario. La conductividad térmica del hierro a 25o C es 0.804 J/Kcms. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 76 CAPÍTULO 3. FENOMENOS DE TRANSPORTE El sistema se encuentra en estado estacionario y tiene un gradiente de temperatura constante que viene dado por: dT ∆T 275 − 325 = = = −0,25K/cm dz ∆z 200 (3.6) El flujo de calor que atraviesa una sección transversal del conductor viene dada por la Ley de Fourier: dq dT = −kA = −0804 J/Kcms · 24 cm2 · (−0,25K/cm) = 4,824 J/s (3.7) dt dz 3.5. Evaluación de k mediante la teoría cinética de los gases La teoría cinética de los gases da expresiones teóricas para la conductividad térmica con resultados acordes con la experiencia. Partimos de las siguientes aproximaciones: Las moléculas son esferas rígidas de diámetro d. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.5 Evaluación de k mediante la teoría cinética de los gases Las moléculas se mueven al velocidad media < v >= 77 8RT 1/2 πM La distancia media recorrida por una molécula entre dos colisiones es: λ = kT √ 1 2πd2 P En cada colisión las propiedades moleculares se ajustan a las de esa posición. Para calcula k, debemos obtener el flujo neto de calor por unidad de tiempo y área a través del plano x0 Jz = J↑ − J↓ = ↑ dN↑ − ↓ dN↓ (3.8) Donde, dN↑ , representa el número de moléculas que atraviesan z0 desde arriba por unidad de área en un dt. Donde, dN↓ , representa el número de moléculas que atraviesan z0 desde abajo por unidad de área en un dt. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 78 CAPÍTULO 3. FENOMENOS DE TRANSPORTE ↑ y ↓ representan la energía media de cada una de esas moléculas. Al no existir convección dN↑ = dN↓ . Calculamos dN↑ considerando el plano z0 como una pared de área A sobre la que chocan las moléculas. El número de colisiones en un dt vendrá dado por: dN↑ = dN↓ = 1 N <v> 4 V (3.9) Sustituyendo en la expresión del flujo de calor Jz = 1 N < v > (↑ − ↓ ) 4 V (3.10) La energía de las moléculas que cruzan z0 es la que adquieren durante la última colisión, que de media se produce en z0 ± 2/3λ. ↑ : es la energía que posee la molécula en z0 − 2/3λ ↓ : es la energía que posee la molécula en z0 + 2/3λ Suponiendo una variación lineal de la energía con z, en las proximidades de z0 , podemos escribir: ↑ = (z0 − 2/3λ) = 0 − ↓ = (z0 + 2/3λ) = 0 + ∂ ∂z ∂ ∂z · 2/3λ (3.11) · 2/3λ (3.12) 0 0 Sustituyendo las energías medias de las moléculas que cruzan z0 en el flujo neto de calor: 1 N ∂ ∂ Jz = < v > 0 − 2/3λ − 0 − 2/3λ = 4 V ∂z 0 ∂z 0 1N −4 ∂ <v> λ 4V 3 ∂z 0 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (3.13) 3.5 Evaluación de k mediante la teoría cinética de los gases 79 Simplificando nos queda: 1N Jz = − <v>λ 3V ∂ ∂z (3.14) 0 La energía, de una molécula depende de la temperatura y ésta a su vez depende de z. Aplicando la regla de la cadena: ∂ dT ∂Um /NA dT Cvm dT ∂ = = · = ∂z ∂T dz ∂T dz NA dz (3.15) Donde se ha tenido en cuenta que: Cvm = dUm /dT Sustituyendo en la densidad de flujo de calor Jz : Jz = − 1N Cvm dT <v>λ 3V NA dz (3.16) Obteniéndose para la conductividad térmica el valor: k= 1N Cvm <v>λ 3V NA (3.17) Ecuación en la que habitalmente se reempla el cociente N/V por ρ (densidad de moléculas por unidad de volumen) 1 Cvm k= ρ<v>λ 3 NA (3.18) Existen desarrollos más rigurosos, suponiendo que las moléculas siguen la distribución de velocidades de Maxwell en vez de la velocidad media: k= 25π Cvm λ<v>ρ 64 NA _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (3.19) 80 CAPÍTULO 3. FENOMENOS DE TRANSPORTE Ecuación que resulta más manejable si hacemos las siguientes sustituciones: λ = P 8kT 1/2 kT √ 1 ;ρ= N : V = RT y < v >= πm 2πd2 P k= 3.6. 25 32 RT πM 1/2 1 Cvm NA d2 (3.20) Ejemplo de evaluación de la conductividad térmica (k) Calcula la conductividad térmica del He a 1 atm y 0o C y a 10 atm y 100o C. Utilizar el valor del diámetro molecular que se obtiene a partir de medidas de viscosidad a 1 atm y 0o C, d=2,2 Anstrong. El valor experimental a 0o C y 1 atm es 1, 4x10−3 J/Kcms Utilizaremos la ecuación más exacta: 25 k= 32 RT πM 1/2 1 Cvm NA d2 (3.21) Hacemos las sutituciones: P=1atm; T=273K; d = 2,2x10−10 m; Cvm = 3/2R con R=8.314 J/molK; M = 4x10−3 Kg/mol El resultado es k=0.142 J/Kms _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.7 Ley de Newton de la viscosidad 3.7. 81 Ley de Newton de la viscosidad La viscosidad es la propiedad que caracteriza la resistencia de un fluido a fluir. Los fluidos que fluyen fácilmente son poco viscosos. La viscosidad se representa por η y sus unidades son N s/m2 Consideremos un fluido que fluye entre dos láminas grandes, planas y paralelas. La experiencia nos dice que la velocidad vx es máxima en el centro y cero sobre las láminas. Las capas horizontales de fluido se deslizan unas sobre otras, ejerciendo una fuerza de fricción que opone resistencia al desplazamiento. La fuerza Fx ejercida por el fluido de movimiento más lento (1) es proporcional al área de la superficie de contacto, A, y al gradiente de la velocidad dvx dz . La constante de porporcionalidad es la viscosidad del fluido η Fy = −ηA dvy dx (3.22) Esta ecuación es conocida como la Ley de Newton de la viscosidad. El signo menos indica que la fuerza de viscosidad sobre el fluido que se mueve más rápido es opuesta a la dirección de su movimiento. Por la tercera Ley de Newton, el fluido que se mueve más rápido ejerce una fuerza dv ηA dxy en dirección z positiva sobre el fluido que se mueve más lento. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 82 CAPÍTULO 3. FENOMENOS DE TRANSPORTE La Ley de Newton se ajusta bien a los gases y líquidos, siempre que la velocidad no sea demasiado alta. Cuando se cumple la Ley de Newton tenemos un flujo laminar. A velocidades muy altas el flujo se vuelve turbulento y la ecuación no es válida. Se llama fluido Newtoniano al fluido en el que η es independiente de dvx /dz. En los fluidos no Newtonianos η varía a medida que lo hace dvx /dz. La mayoría de los gases son Newtonianos, mientras que las soluciones de polímeros, suspensiones coloidales generalmente no lo son, de manera que un incremento en la velocidad de flujo puede cambiar la forma de las moléculas de polímero (flexibles) facilitando el flujo por reducción de la viscosidad. 3.8. Ley de Newton en función del momento lineal Vamos a expresar la Ley de Newton en función del momento lineal. Escribimos x la segunda Ley de Newton, Fx = max = m dv dt = d(mvx ) dt = dpx dt Sustituyendo en la Lewy de Newton de la viscosidad dpx dvx = −ηA dt dz _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (3.23) 3.9 Viscosidad en gases y líquidos 83 dpx /dt (flujo de cantidad de movimiento), representa la variación de la componente y del momento lineal de una capa situada a un lado en el interior del fluido, debida a su interacción con el fluido que está del otro lado. La explicación molecular de la viscosidad supone el transporte de momento lineal a través de los planos perpendiculares al eje z. Se define la densidad de flujo de cantidad de movimiento por unidad de área y tiempo: Jz = 3.9. 1 dpx dvx = −η A dt dz (3.24) Viscosidad en gases y líquidos El coeficiente de viscosidad (η) depende de temperatura, presión y tipo de sustancia. En los líquidos la viscosidad disminuye de forma importante al aumentar la temperatura debido al debilitamiento de las fuerzas intermoleculares. En los gases la viscosidad tiende a aumentar con el incremento de la temperatura. Dado que la unidad de la viscosidad N s/m2 es excesivamente grande se definen poise y centipoise: 1 poise = 0,1 N s/m2 ; 100 cp = 1 poise Viscosidades de algunos fluidos: CH4 η = 10−5 N s/m2 ; H2 O η = 10−3 N s/m2 ; aceite η = 0,1N s/m2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 84 CAPÍTULO 3. FENOMENOS DE TRANSPORTE 3.10. La ecuación de Poiseuille Permite determinar el flujo laminar estacionario (dV/dt) para un fluido incompresible, de viscosidad constante, a través de un tubo cilíndrico de radio r. Consideremos un fluido que circula por una conducción cilíndrica de radio r. Tomemos un elemento infinitesimal de longitud z y radio interno s, con una presión p1 a la entrada y p2 a la salida. Vamos a escribir las fuerzas que actúan sobre la capa de fluido situada entre s y s+ds. 1. Fuerzas hidrostáticas de presión (F = P S) Fuerza debida a la presión de entrada: F1 = 2πSdS · P1 Fuerza debida a la presión de salida: F2 = −2πSdS · P2 2. Fuerzas de rozamiento. Rozamiento con la parte interior FRI = −η · 2πSdz(dV /dS)s . Como el gradiente de velocidad es negativo la fuerza de rozamiento es positiva _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.10 La ecuación de Poiseuille 85 ya que las capas internas van más rápidas y aceleran la capa de fluido considerada. Rozamiento con la pared exterior FRE = η·2π(S+dS)dz(dV /dS)S+dS . Esta fuerza de rozamiento es negativa ya que se opone al movimiento de la capa considerada. Una vez alcanzado el régimen estacionario: P~ Fi = 0 2πSdS ·P1 −2πSdS ·P2 −η2πSdz(dv/dS)S +η2π(S +dS)dz(dV /dS)S+dS = 0 (3.25) Dado que P2 = P1 + dP 2πSdS·P1 −2πSdS(P1 +dP )−η2πSdz(dv/dS)S +η2π(S+dS)dz(dV /dS)S+dS = 0 (3.26) Simplificando las expresión anterior − 2πSdSdP + 2πηdz [(S + dS)(dv/dS)S+dS − S(dv/dS)s ] = 0 (3.27) Teniendo en cuenta la definición de derivada: df (x) = f (x + dx) − f (x), podemos escribir la expresión anterior de la siguiente forma dv =0 − SdSdP + ηdzd s dS (3.28) dP dv SdS = ηd s dz dS (3.29) _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 86 CAPÍTULO 3. FENOMENOS DE TRANSPORTE Integrando con el gradiente de presión constante dP S 2 dv = ηS +C dz 2 dS (3.30) Dado que dv/ds no puede ser infinito, si haciendo S=0 0=0+C → C =0 (3.31) Despejando dv dv = 1 dP SdS 2η dz (3.32) 1 dP s2 +C 2η dz 2 (3.33) Integrando de nuevo v(S) = Calculamos la nueva constante de integración, C, sabiendo que en las paredes de la conducción el flujo se anula, v(S = r) = 0 0= 1 dP r2 1 dP 2 +C →C =− r 2η dz 2 4η dz (3.34) Reemplazando la constante de integración en la ecuación de la velocidad v(s) v(s) = − 1 dP 2 (r − s2 ) 4η dz (3.35) Ecuación que nos da un perfil parabólico para la velocidad de un fluido en estado estacionario. _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.10 La ecuación de Poiseuille 87 En un dt el volumen de fluido que se ha desplazado por la corona de espesor ds es: dV = 2πSdS ·v(s)dt. El volumen total que circula por la conducción lo obtenemos integrando desde S=0 hasta S=r. dV = dt Z r 2πS · v(S)dS (3.36) 0 Sustituyendo v(S) por su valor: dV = 2π dt Z r −1 2 dP −π dP S (r − s2 ) = 4η dz 2η dz 0 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com r4 r4 − 2 4 (3.37) 88 CAPÍTULO 3. FENOMENOS DE TRANSPORTE Haciendo la diferencia y simplificando se obtiene la ecuación diferencial de Poiseuille. dV −πr4 dP = dt 8η dz (3.38) En el caso de los líquidos podemos integrar ya que son poco compresibles y el volumen es independiente de la presión. ∆V −πr4 ∆P = ∆t 8η ∆z 3.11. (3.39) Teoría cinética aplicada a la viscosidad de los gases Vamos a deducir una expresión para la viscosidad empleando la teoría cinética de los gases. la deducción es análoga a la realizada para la conductividad térmica, con la excepción de que en vez de transportar calor se transporta cantidad de movimiento. Jz = J↑ − J↓ = dN↑ p↑ − dN↓ p↓ Dado que dN↑ = dN ↓ = 1N 4V (3.40) <v> Jz = 1N < v > (p↑ − p↓ ) 4V (3.41) Donde, p↑ es la cantidad de movimiento de una molécula en el plano x0 − 2/3λ y p↓ el momento lineal correspondiente al plano x0 + 2/3λ. Jz , representa el flujo neto de momento lineal por unidad de área y tiempo. p↑ = m vx0 − ∂vx ∂z 2 · λ 0 3 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (3.42) 3.11 Teoría cinética aplicada a la viscosidad de los gases p↓ = m vx0 + ∂vx ∂z 2 · λ 0 3 89 (3.43) Sustituyendo en Jz Jz = ∂vx 2 ∂vx 2 1N m vx0 − · λ − vx0 + · λ 4V ∂z 0 3 ∂z 0 3 (3.44) Simplificando 1N Jz = < v > mλ 3V ∂vx ∂z (3.45) 0 x Comparando esta última ecuación con Jz = −η dv dz , obtenemos el valor de η η= 1N < v > mλ 3V (3.46) Llamando ρ = N/V y m = M/NA , nos queda η= 1 M < v > λρ 3 NA (3.47) Un tratamiento más riguroso nos lleva a la ecuación: η= 5π M < v > λρ 32 NA (3.48) Ecuación que resulta más manejable si hacemos las siguientes sustituciones: λ = kT N P 8kT 1/2 √ 1 : ; ρ = = y < v >= 2 P V RT πm 2πd η= 5 M RT 1/2 √ 16 π NA d2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com (3.49) 90 CAPÍTULO 3. FENOMENOS DE TRANSPORTE 3.12. Problemas 1. Las viscosidades del dióxido de carbono a 1 atm y 0, 490, y 850o C son 139, 330 y 436 mP, respectivamente. Calcule el diámetro de esfera rígida aparente del dióxido de carbono a cada una de estas temperaturas. Aplicamos la ecuación η= 5 (M RT )1/2 16π NA d2 (3.50) d2 = 5 (M RT )1/2 16π NA η (3.51) Pasamos η a las unidades del sistema internacional 139 mp 1p 1N s/m2 = 139x10−4 N S/m2 1000 mp 10p (3.52) M=0.044 kg/mol; R=8.314 J/molK T=273 K; NA = 6,023x1023 mol−1 . Sustituyendo en la ecuación se obtiene un diámetro para el dióxido de carbono de 4,59x10−10 m 3.13. Difusión: Primera Ley de Fick Si cj,1 6= cj,2 y ck,1 6= ck,2 al quitar el tabique se produce un movimiento de las espacies j,k que reducirá o eliminará la diferencia de concentraciones entre ambos recipientes. La difusión es un movimiento macroscópico de los componentes de un sistema debido a diferencias de concentración. Si cj,1 < cj,2 , hay un flujo neto de j desde 2 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.13 Difusión: Primera Ley de Fick 91 hacia 1 hasta que se igualen concentraciones y potenciales químicos de j y k en toda la celda. En la difusión todas las moléculas tienen velocidades al azar. Sin embargo, dado que la concentración en la parte derecha de un plano perpendicular es mayor que en la parte izquierda, es más probable que las moléculas de j crucen de derecha a izquierda el plano que de izquierda a derecha, produciéndose un flujo neto de j hacia la izquierda. La primera Ley de Fick nos da la velocidad de difusión. dnj dt , dcj dnj = Djk A dt dz (3.53) dnk dck = Dkj A dt dz (3.54) representa la velocidad de flujo de j (moles/s) a través del plano P de área A, perpendicular al eje z, siendo cj dz el gradiente de concentración en dicho plano. Djk , es una constante de proporcionalidad denominada coeficiente de difución. Djk , depende de T,P y de la composición local, variando de forma importante con _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 92 CAPÍTULO 3. FENOMENOS DE TRANSPORTE la distancia y con el tiempo a lo largo de la celda de difusión. 3.14. Teoría cinética de la difusión en los gases La deducción es análoga a la conductividad térmica o viscosidad con al diferencia de que se transporta materia. El flujo neto de materia (en moles) que atraviesa el plano z0 viene dado por: Jz = J↑ − J↓ = dN↑ dN↓ − NA NA (3.55) El número de moléculas que atraviesan z0 desde abajo viene dado por: dN↑ = c↑ N↑ NA 1 1 <v> = < v > NA 4 V NA 4 V Sustituyendo c↑ por la concentración de moléculas en el plano z0 − 2/3λ dcj 1 2 dN↑ = < v > NA cj0 − λ 4 3 dz 0 (3.56) (3.57) De forma análoga escribimos el número de moléculas que cruzan z0 desde arriba. dcj 1 2 dN↓ = < v > NA cj0 + λ (3.58) 4 3 dz 0 _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 3.15 Problemas 93 Sustituyendo en Jz Jz = dcj 1 −4 dcj −1 <v> λ <v>λ 4 3 dz 3 dz (3.59) De donde se deduce que el coeficiente de difusión viene dado por: D= 1 <v>λ 3 (3.60) D= 3π λ<v> 16 (3.61) Un tratamiento riguroso nos da: Reemplazando λ = kT √ 1 2πd2 P y < v >= 8 D= 2 8d 3.15. 8kT 1/2 , πm kT πm 1/2 nos queda: kT P (3.62) Problemas 1. El hidrógeno gaseoso se difunde a través de una lámina de paladio de 0.0050 cm de espesor. Del lado izquierdo de la lámina, el hidrógeno se mantiene a 25.0o C y una presión de 750 mm de Hg, mientras que del lado derecho se mantiene un buen vacío. Después de 24 horas, el volumen de hidrógeno en el compartimento de la izquierda disminuye en 14,1 cm3 . Si el área de la lámina a través de la cual ocurre la difusión es 0.743 cm2 . ¿Cuál es el cociente de difusión del hidrógeno en el paladio? _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com 94 CAPÍTULO 3. FENOMENOS DE TRANSPORTE 2. El diámetro molecular que se obtiene para el oxígeno a partir de medidas de viscosidad a 0o C y 1 atm es 3.6 Anstrong. Calcular el coeficiente de autodifusión del oxígeno a 0o C y presiones de 1 atm y 10 atm. El valor experimental a 0o C y 1 atm es 0.19 cm2 /s _________________________________________________ Germán Fernández www.quimicafisica.com www.academiaminas.com