Leucemia linfoblástica aguda de línea T
Anuncio

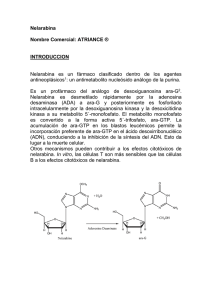
Documento descargado de http://www.elsevier.es el 18/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Leucemia linfoblástica aguda de línea T* Blanca Xicoya, Carlos Grandeb y Josep-Maria Riberaa a Servicio de Hematología Clínica. Institut Català d’Oncologia-Hospital Universitari Germans Trias i Pujol. Badalona. Universidad Autònoma de Barcelona. b Servicio de Hematología. Hospital Doce de Octubre. Madrid. España. La leucemia linfoblástica aguda fenotipo T (LLA-T) es una enfermedad con una notable heterogeneidad clínica y biológica. El primer paso para el éxito del tratamiento de la LLA-T es la identificación de las características inmunofenotípicas, citogenéticas y moleculares de la enfermedad, que pueden tener una implicación pronóstica. Para ello, son necesarias las técnicas convencionales asociadas a otras, como la reacción en cadena de la polimerasa y los recientes estudios de perfiles de expresión génica. Con estos últimos se ha profundizado en la compleja fisiopatología de la enfermedad, resultado de translocaciones cromosómicas y activación de protooncogenes, cuyos productos alteran vías de transducción de señales relacionadas entre sí. El mejor conocimiento de las alteraciones del metabolismo del linfocito T y el estudio sistemático de los perfiles genómicos de los pacientes y las vías implicadas ha permitido explorar el uso de fármacos más específicos para la LLA-T, aislados o asociados a la quimioterapia convencional. Es probable que, en un futuro próximo, la expresión génica, más que el subtipo de LLA-T, sea la que constituya la base para seleccionar el tratamiento de las LLA-T y permita mejorar su pronóstico. Palabras clave: Leucemia linfoblástica aguda T. Mecanismos genéticos. Nuevos tratamientos. La leucemia linfoblástica aguda (LLA) tiene un fenotipo T (LLA-T) en un 15% de los niños y en el 25% de los adultos. La LLA-T se presenta con frecuencia en adultos jóvenes, por lo general varones, con una cifra de leucocitos elevada, masa mediastínica y posible invasión del sistema nervioso central (SNC). La LLA-T es una enfermedad heterogénea que incluye formas de fenotipo inmaduro, otras de fenotipo intermedio y otras de fenotipo maduro. Según algunos estudios, los subtipos más inmaduros y los más maduros tienen peor pronóstico en comparación con los de madurez intermedia1-3. El mejor conocimiento de los mecanismos genéticos y moleculares implicados en la patogenia de la enfermedad ha permitido explicar mejor la heterogeneidad clínica y pronóstica de la LLA-T. Clasificación de la leucemia linfoblástica aguda de fenotipo T Clasificación morfológica y citoquímica T-cell acute lymphoblastic leukemia T-cell acute lymphoblastic leukemia (T-ALL) is a disease with marked clinical and biological heterogeneity. The first step for the successful treatment of T-ALL is to identify the immunophenotypic, cytogenic and molecular characteristics of the disease, which can have prognostic implications. To do this, conventional techniques associated with other methods, such as polymerase chain reaction and gene expression profiling, are required. These latter methods have deepened understanding of the complex physiopathology of the disease, which is a result of chromosomal translocations and proto-oncogene activation, the products of which alter interrelated signal transduction pathways. Greater knowledge of alterations of T lymphocyte metabolism and systematic study of patients’ genomic profiles and the pathways involved has allowed the use of more specific drugs for T-ALL to be explored, either alone or in combination with conventional chemotherapy. In the near future, gene expression, rather than T-ALL subtype, will probably form the basis for treatment selection in T-ALL and will improve the prognosis of this disease. Key words: T-cell acute lymphoblastic leukemia. Genetic mechanisms. New treatments. *Trabajo financiado en parte con la beca PEF/06 de la Fundación Internacional José Carreras para la Lucha contra la Leucemia. Correspondencia: Dra. B. Xicoy. Servicio de Hematología Clínica. Institut Català d’Oncologia. Hospital Universitari Germans Trias i Pujol. C. Canyet, s/n. 08916 Badalona. Barcelona. España. Correo electronico: bxicoy@comb.es Según los criterios del grupo cooperativo franco-americanobritánico (FAB), la LLA de estirpe B o T se clasifica en 3 variedades4: LLA1, LLA2 y LLA3. Como es característico, en todas las LLA existe negatividad para la reacción citoquímica de las peroxidasas y, en la LLA-T en particular, positividad centrosómica para la reacción de la fosfatasa ácida. Esta clasificación puramente morfológica ha caído en desuso. Clasificación inmunofenotípica La LLA-T se define por la expresión citoplasmática o en membrana de CD3. La reactividad para CD2 y/o CD7 no es suficiente para adscribir un caso como de línea T, aunque el CD7 se exprese en la práctica totalidad de las LLA-T. Es de interés recordar que, en las LLA-T, la expresión de CD2 se ha asociado a un buen pronóstico, al menos en niños. Por otra parte, es de resaltar que una característica de las neoplasias T, en general, y de las LLA-T, en particular, es la pérdida de antígenos de línea y la expresión aberrante de antígenos de otras líneas, lo que puede dificultar su clasificación. Asimismo, merece destacarse la baja frecuencia de positividad para los antígenos CD34 y HLA-DR, particularmente en niños, en quienes se asocia a un peor pronóstico. Según la clasificación del European Group for Immunological Characterization of Leukemias (EGIL) (tabla 1), entre las LLA-T se reconocen 4 subgrupos de menor a mayor diferenciación: pro-T, pre-T, tímica cortical y tímica madura5. El fenotipo pro-T, que para algunos es el de peor pronóstico dentro de las LLA de línea T, suele presentar únicamente positividad para CD3 citoplasmático (CD3c) y CD7, y es negativo para CD3 de superficie (CD3s), CD4, CD8 y otros antígenos de línea T. La LLA pre-T es CD2+, habitualmente CD3s–, y presenta positividad variable para CD5, CD7, y positividad intensa para la desoxinucleotidiltransferasa terminal (TdT). La LLA-T tímica o cortical es el Med Clin (Barc). 2007;129(Supl 1):45-9 45 Documento descargado de http://www.elsevier.es el 18/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. XICOY B ET AL. LEUCEMIA LINFOBLÁSTICA AGUDA DE LÍNEA T TABLA 1 Clasificación del European Group for Immunological Classification of Leukemia (EGIL) para las leucemias linfoblásticas agudas T Pro-T (EGLIL-I) Pre-T (EGIL-II) Tímica (EGIL-III) Madura (EGIL-IV) cCD3 sCD3 CD7 CD1a TdT CD2 CD5 + + + + + + – +/– + + + + + – – – +/– +/– +/– +/– + + – + + + CD4/CD8 – + +/– +/– –/– –/+ Tdt: desoxinucleotifiltransferasa terminal. subtipo inmunológico más frecuente. Su fenotipo se define por la positividad para CD1a con independencia de la positividad para otros marcadores incluyendo CD3s, CD2, CD5 e incluso CD7; algunos casos pueden presentar positividad simultánea para CD4 y CD8, y positividad débil para CD3s; la TdT con frecuencia es positiva. Por último, el fenotipo de la LLA-T madura es negativo para CD1a, y presenta positividad para CD3s y expresión variable para CD2, CD5, CD7; en algunos casos, puede haber positividad para TdT (tabla 1). Al igual que las LLA de línea B, hasta el 25% de las LLA-T pueden expresar algún marcador de línea mieloide. Clasificación citogenética y molecular En la LLA-T, los trastornos genéticos y moleculares están peor caracterizados que en las LLA de estirpe B. En la mitad de los pacientes con LLA-T, hay trastornos estructurales identificables por citogenética convencional. Sin embargo, un porcentaje más elevado de anomalías citogenéticas crípticas y deleciones son detectadas por técnicas de hibridación in situ fluorescente (FISH) o por la reacción en cadena de polimerasa (PCR). Los trastornos genéticos incluyen reordenamientos que afectan a los genes del receptor de células T (TCR), situados en la región 14q11 [t(1;14), t(10;14), t(8;14), t(5;14), t(11;14)], donde se hallan los locus α y δ, o en 7q34 [t(1;7), t(7;9), t(7;11), t(7;19)], donde se halla el locus β del receptor T, o 7p15, donde se halla el locus γ. Un segundo grupo de alteraciones comportan la formación de genes de fusión (p. ej., SIL-TAL1, PICALM-MLLT10, NUP214-ABL1 y MLL-ENL, entre otros). El tercer grupo de lesiones incluye mutaciones de genes (NOTCH1, FLT3 y n-RAS, entre otros). La mayoría de los trastornos genéticos implican la activación de protooncogenes que provocan la expresión aberrante de factores de transcripción e interfieren en, al menos, 4 vías de señal interconectadas entre sí que, respectivamente, confieren a la célula una ventaja proliferativa y una mayor supervivencia; provocan una desdiferenciación y muerte celular precoz; interfieren en el ciclo celular, o confieren a la célula una capacidad de autorrenovación. La primera vía se debe fundamentalmente a la activación de tirosincinasas y en ella están implicados genes como NUP214-ABL1 —resultado de la traslocación t(9;9)(q34;q34) y presente en un 6% de pacientes con LLA-T— y otros como JAK2, LCK, FLT3 y n-RAS. En la segunda vía son claves los factores de transcripción, y en ella están implicados genes como TAL1 (sobreexpresado en el 40% de las LLA-T), así como TAL2, LYL1, bHLHB1, MYC, LMO1 y LMO2, TLX1 (HOX11) y TLX3 (HOX11L2) y miembros del clúster HOXA. La activación de HOX-11 se halla en el 33% de los adultos y el 3% de los niños con LLA-T y la de HOX11L2 en el 20% de los casos de LLA-T infantil y entre el 5 y el 10% de los casos de LLA-T en la edad adulta. Los loci CDKN2A y CDKN2B en 9p21 contienen genes involucrados en la regulación del ciclo celular; 46 Med Clin (Barc). 2007;129(Supl 1):45-9 así, la tercera vía suele ser consecuencia de la deleción de CDKN2A, a menudo junto con CDKN2B. Por último, la activación de NOTCH1, cuando es resultado, en ocasiones, de la traslocación t(7;9)(q34;q34.3), está presente en más del 50% de las LLA-T y es la causa de que la célula adquiera una capacidad de autorrenovación. NOTCH1 es una proteína transmembrana que, en estado activo, es anclada por la enzima gammasecretasa, que transloca la porción intracelular de la proteína al núcleo, donde actúa en consonancia con un número de factores de transcripción activadores. El perfil de expresión génica ha permitido correlacionar la expresión de algunos genes con los diferentes estadios del desarrollo del timocito. Así, LYL1 representa el estadio de timocito inmaduro (pro-T); TLX1 (HOX11), el timocito cortical precoz, y TAL1 se correlaciona con el estadio de timocito cortical tardío. Los estudios sobre la interrelación entre estos genes y las vías implicadas han identificado c-MYC como la diana directa de NOTCH1, a NUP214-ABL1 asociado a la sobreexpresisón de HOX11 y la deleción de CDKN2A, y a LMO1 y LMO2 a desregulación de LYL1 o TAL1 (tabla 2)6-8. Cuadro clínico La LLA-T afecta con mayor frecuencia a adultos jóvenes, en general adolescentes, cursa con hiperleucocitosis, puede presentarse con infiltración del SNC (5-10%) y en un tercio de los casos se detecta una masa mediastínica en una radiografía de tórax o una tomografía computarizada torácica con posible derrame pleural asociado, por lo que con frecuencia el paciente refiere dolor torácico. La clínica y la morfología son indistinguibles del linfoma linfoblástico (LLB). Pronóstico La LLA-T en niños tiene un pronóstico mejor que en adultos. En estos últimos la tasa de supervivencia es del 3545% a los 5 años y la supervivencia libre de enfermedad (SLE) estimada a los 3 años es del 40-45%. La mejoría en los resultados del tratamiento ha sido más evidente en la LLA-T que en la LLA de estirpe B, y se ha atribuido, por un lado, a la introducción de citarabina, ciclofosfamida y/o tenipósido, en las consolidaciones, y a la aplicación del trasplante de progenitores hematopoyéticos (TPH) alogénico en primera remisión completa (RC) a los pacientes con los subtipos inmunológicos pro-T y maduro, a aquellos con factores de alto riesgo en el diagnóstico o respuesta subóptima al tratamiento. Los rasgos clínicos y citogenéticos, salvo la edad y la hiperleucocitosis, no son predictivos de la respuesta al tratamiento en la LLA-T. De hecho, la edad adulta y la presencia de una cifra de leucocitos superior a 100 × 109/l en el momento del diagnóstico se consideran factores de alto riesgo. En la infancia, los varones tienen un pronóstico peor en la ma- Documento descargado de http://www.elsevier.es el 18/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. XICOY B ET AL. LEUCEMIA LINFOBLÁSTICA AGUDA DE LÍNEA T TABLA 2 Alteraciones cromosómicas y moleculares en la leucemia aguda linfoblástica T Vía molecular Ateraciones cromomosómicas Cambios moleculares tTCR GF tTCR tTCR tTCR tTCR tTCR tTCR tTCR tBCL11B TLX1(HOX11)* t(1;14)(p32;q11) o t(1;7)(p32q34) del(1)(p32) t(7;9)(q34;q32) t(7;19)(q34;p13) t(14;21)(q11;q22) t(7;12)(q34p13.3) o t(12;14)(p13;q11) t(8;14)(q24;q11) o t(7;8)(q34;q24) t(11;14)(p15;q11) o t(7;11)(q34;p15) t(11;14)(p13;q32) o t(7;11)(q34;p13) t(11;14)(p13q32) del(11)(p13p13) inv(7)(p15q34) o t(7;7)(p15;q34) o t(7;14)(p15;q32) t(7;14)(p15;q32) t(10;14)(q24;q11) o t(7;10)(q34 ;q24) tTCR tBCL11B tTCR 33 3 TLX3 (HOX11L2) t(5;14)(q35;q32) tBCL11B 5-10 20 PICALM-MLLT10* MLL-ENL t(5;7)(q35;q21) t(2 ;5)(p21;q35) t(10 ;11)(p13;q14) t(11;19)(q23;p13.3) CDK6 ? GF GF 10 8 10 8 Proliferación y supervivencia BCR/ABL1 NUP214-ABL1 t(9;22)(q34;q11) t(9;9)(q34;q34) GF GF 4 <6 ETV6-ABL1 EML1-ABL1 ETV6-JAK2 LCK FLT3 N-RAS t(9;14)(q34;q32) t9;14)(q34;q32) t(9;12)(p24;p13) t(1;7)(p34;q34) Mutaciones Mutaciones GF GF GF tTCR 6 < 10 6 < 10 Defectos del ciclo celular CDKN2A/CDKN2B del(9)(p21) Desdiferenciación TAL1* SIL-TAL1 TAL2* LYL1* HLHB1 CCND2 MYC LM01* LM02 HOXA Capacidad de autorrenovación NOTCH1 Mutaciones t(7;9)(q34;q34.3) Frecuencia (%) Adultos Niños 30 <5 <1 3 9-30 <1 Comentarios Timocito cortical tardío Timocito inmaduro. Peor pronóstico Desregulación de TAL1 Desregulación de LYL1 o TAL1 Timocito cortical precoz. Buen pronóstico Peor pronóstico que TLX1 (HOX11) Buen pronóstico Sobreexpresión HOX11 y del CDKN2A A menudo asociada a CDKN2B Homozigotas 65%, heterozigotas 15% GF <1 50-60 <1 50-60 Diana: c-MYC Buen pronóstico en niños *Genes sobreexpresados en ausencia de anormalidades visibles. ?: gen implicado desconocido; CDK6: gen sobreexpresado por el promotor CDK6; GF: genes de fusión resultado de traslocaciones; tBCL11B (Krüppel-like zinc-finger gene): traslocaciones que afectan al gen BCL11B; tTCR: traslocaciones que afectan al receptor de células T, un 35% detectables por hibridación in situ (crípticas por estudio citogenético). yoría de los estudios, quizá por la presencia de recaídas en la zona testicular. Las recidivas aisladas en el SNC son poco frecuentes. La respuesta al tratamiento puede evaluarse por la tasa de aclaramiento de los blastos leucémicos en sangre periférica tras una semana de tratamiento con prednisona o bien en el día 8 del tratamiento de inducción, por la desaparición de los blastos en médula ósea el día 14 del tratamiento de inducción, por el tiempo requerido para la obtención de la RC o bien por el seguimiento de la enfermedad residual mínima (ERM), mediante citofluorometría o reacción en cadena de la polimerasa (PCR). Todo ello refleja la rapidez y la intensidad de desaparición de los blastos. Las recidivas más allá de los 5 años de la RC son poco frecuentes en la LLA-T. En la mayoría de los estudios, las LLA-T tienen un pronóstico mejor que las LLA de línea B «inmadura» en adultos9,10. El grupo cooperativo alemán GMALL ha demostrado que las que tienen un fenotipo muy inmaduro (pro-T o EGIL-I) o muy maduro (EGIL-IV) tienen un pronóstico peor que las de fenotipo intermedio (pre-T o EGIL-II y tímica cortical o EGIL-III). Así, un 60% de los pacientes con LLA-T tímica o cortical permanecieron en RC continuada frente a sólo un 20-30% de los pacientes con LLA pro-T/pre-T y T madura11. Esta di- ferencia en el pronóstico en función de la diferenciación linfoide T no se demostró en la serie de LLA-T del grupo PETHEMA (Programa Español para el Tratamiento en Hematología), al menos en las LLA-T maduras. Aunque los pacientes con el subtipo T-maduro tuvieron una respuesta más lenta al tratamiento, esto no se tradujo en una diferente tasa de RC o unas probabilidades de SLE y supervivencia global (SG) significativamente menores. La expresión de marcadores mieloides no tiene un valor pronóstico en la LLA-T12. La correlación de la expresión de oncogenes con diferentes estadios del desarrollo de los linfocitos T podría explicar en parte el pronóstico distinto de los subtipos diferentes de LLA-T. Así, la sobreexpresión de HOX11, que caracteriza a la LLA-T tímica o cortical, se asocia a un mejor pronóstico en la mayoría de los estudios, mientras que es peor el de los pacientes que expresan HOX11L213,14. Los estudios de perfil de expresión génica mediante micromatrices (microarrays) han permitido identificar un limitado número de genes que son predictivos de la respuesta al tratamiento de inducción y de la duración de la RC en pacientes con LLA-T15. Así, en adultos, el gen de la interleucina 8 se expresa con frecuencia en las LLA-T refractarias en comparación con las LLA-T que responden Med Clin (Barc). 2007;129(Supl 1):45-9 47 Documento descargado de http://www.elsevier.es el 18/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. XICOY B ET AL. LEUCEMIA LINFOBLÁSTICA AGUDA DE LÍNEA T al tratamiento de inducción. Recientemente, el modelo de patrón de expresión de 3 genes, como AHNAK, CD2 y TTK, fue predictivo de la duración de la remisión en un estudio y el patrón de expresión de los genes FLAR, NOTCH2 y BTG3 en niños se correlacionó con la duración de la RC y la supervivencia en otro16. Se ha descrito que la presencia del gen de fusión NUP214/ABL1 se asocia a un curso más agresivo de la enfermedad, pero ello no se ha confirmado; es incierto si la presencia de este gen de fusión puede conducir a la amplificación del gen ABL1 en la LLA-T y, por tanto, ser potencialmente susceptible a inhibidores de tirosincinasa de ABL, como imatinib17. También, la sobreexpresión de ERG, un factor de transcripción ETG, modulador de la maduración de células linfoides en la LLA-T tímica o cortical, puede causar un curso más agresivo de la enfermedad y conferir un curso desfavorable a este subgrupo de LLA-T de relativo buen pronóstico. En este sentido, la baja expresión de este factor de transcripción junto con la de BAALC (brain and acute leucemia, cytoplasmic) identifica a pacientes con LLA-T de curso favorable18,19. Las mutaciones NOTCH1 en niños y la proteína de fusión MLL-ENL, resultado de la traslocación t(11;19)(q23;p13.3), frecuente en adolescentes, se han asociado a buen pronóstico20,21. Por último, la expresión de CD52, diana para alemtuzumab, un anticuerpo monoclonal, no parece tener implicaciones pronósticas. provoca la acumulación de dGTP con inhibición de la ribonucleótido reductasa y de la síntesis de ADN, lo que provoca la muerte celular. Se ha observado un 35% de RC en pacientes con LLA-T sin invasión del SNC y de un 49% en pacientes en primera recaída. La nelarabina ha sido aprobada por la Food and Drug Administration (FDA) para el tratamiento de pacientes con LLA-B o LLA-T en recidiva o refractarios a 2 líneas de tratamiento o más. La dosis recomendada en adultos es de 1500 mg/m2 i.v. en infusión de 2 h los días 1, 3, y 5 de un ciclo de 21 días. La dosis está a menudo limitada por la neurotoxicidad y la tolerancia en adultos es peor que en niños, por lo que a menudo debe disminuirse la dosis. La mielodepresión es menos frecuente. Se ha demostrado su actividad tanto aislada como asociada a la quimioterapia24-28. Tratamiento Clofarabina En el momento actual, el tratamiento de las LLA se adapta al riesgo y es independiente del subtipo inmunológico, aunque algunos grupos tratan a los subtipos LLA-T proT/pre-T y tímica madura con protocolos más intensivos, que incluyen el TPH en primera RC, con resultados preliminares favorables2. Por su parte, los perfiles de expresión génica pueden identificar genes asociados a la resistencia a fármacos empleados en el tratamiento como los glucocorticoides, asparaginasa, vincristina y daunorrubicina. Recientemente han aparecido nuevos tratamientos más específicos para las LLA-T. Se está investigando, en ensayos clínicos, el papel de nuevos fármacos con actividad frente a la LLA-T, como clofarabina, nelarabina y forodesina, análogos de las purinas, de forma aislada o asociados a quimioterapia convencional, o con anticuerpos monoclonales con actividad potencial frente a la LLA-T, como el anti-CD52 (alemtuzumab). También se está investigando la posible eficacia de los inhibidores de las tirosincinasas, como imatibib, nilotinib y dasatinib, en las LLA-T con el gen de fusión NUP214/ABL1 y los inhibidores de gammasecretasa, que pueden ser activos frente a las LLA-T con activación de NOTCH1. La clofarabina es otro análogo de dGuo. Con este fármaco se ha observado un 20% de RC en pacientes con LLA refractaria como paso previo a la posterior realización de un TPH alogénico. La clofarabina ha sido aprobada por la FDA para el tratamiento de los pacientes entre 1 y 21 años en recidiva o refractarios a 2 o más líneas de tratamiento. La dosis en niños es de 52 mg/m2 i.v., durante 5 días (en adultos se recomienda una dosis inferior —40 mg/m2 i.v. durante 5 días—). Se está estudiando la posible sinergia con quimioterapia, como la ciclofosfamida31-33. Análogos de las purinas La vía de las purinas desempeña un papel importante en el metabolismo del linfocito T y en el desarrollo de la LLA-T. La purina-nucleósido fosforilasa (PNP) es una enzima presente en todos los tejidos, especialmente en linfocitos, y es clave en la vía de metabolismo de las purinas. La inhibición de esta enzima provoca una acumulación de desoxiguanosina (dGuo) en plasma e intracelular de desoxiguanosina trifosfato (dGTP), que inhibe la síntesis del ADN e induce la muerte celular por apoptosis22,23. Nelarabina La nelarabina (2-amino-9-β-D-arabinofuranosil-6-metoxi-9H -purina) es un profármaco análogo de deoxiguanosina que 48 Med Clin (Barc). 2007;129(Supl 1):45-9 Forodesina La forodesina es un análogo de PNP y un inhibidor potente y selectivo de esta enzima. Se ha demostrado su actividad selectiva como fármaco único en neoplasias malignas, especialmente en la LLA-T. La respuesta al tratamiento se correlaciona con la acumulación intracelular de dGTP. Se ha descrito una tasa de RC del 23% en 13 pacientes multitratados con LLA-T. La dosis es de 40 mg/m2 i.v., y se dispone de una formulación oral. El perfil de toxicidad es aceptable, incluso en un tratamiento prolongado, y no es neurotóxica29,30. Fármacos con acción en las alteraciones 9q34 El estudio de las alteraciones 9q34 en la LLA-T ha identificado 2 nuevos subtipos moleculares susceptibles de dianas terapéuticas. – NOTCH1 es el resultado de la rara traslocación t(7;9)(q34;q34.3). Estudios en marcha están investigando la eficacia de los inhibidores gammasecretasa de NOTCH1, como LY-450139 o MK-0752, en pacientes previamente tratados con LLA-T y LLA-B. La presencia de la mutación FBW7 confiere resistencia a los inhibidores de gammasecretasa34. – La segunda alteración 9q34 es el gen de fusión NUP214ABL1. Estudios in vitro indican que las células con este gen de fusión son sensibles a imatinib o dasatinib. Se están planeando ensayos clínicos con regímenes similares a los usados en la LLA con cromosoma Filadelfia35. El mejor conocimiento de los mecanismos moleculares implicados en el desarrollo de la LLA-T y el uso de terapias específicas contra diversas alteraciones moleculares, junto con un tratamiento individualizado, puede mejorar el pronóstico de los pacientes. Estas estrategias incluirían un tratamiento en función de la ERM o el uso del perfil de expresión génica para establecer los factores pronósticos y seleccionar el tratamiento. Documento descargado de http://www.elsevier.es el 18/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. XICOY B ET AL. LEUCEMIA LINFOBLÁSTICA AGUDA DE LÍNEA T REFERENCIAS BIBLIOGRÁFICAS 1. Vitale A, Guarini A, Ariola C, Mancini M, Mecucci C, Cuneo A, et al. Adult T-cell lymphoblastic leukemia: biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LLA 0496 protocol. Blood. 2006;107:473-9. 2. Göekbuget N, Thiel E, Beck J, Griesinger F, Hartmann, Jentsch-Ullrich K, et al. Improved outcome in adult T-ALL by risk-adapted treatment strategies. Blood. 2004;104:682a. 3. Crist WM, Shuster JJ, Falleta J, Pullen DJ, Berard CW, Vietti TJ, et al. Clinical features and outcome in childhood T-cell leukemia-lymphoma according to stage thymocyte differentiation: a Pediatric Oncology Group Study. Blood. 1998;72:1783-6. 4. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of acute leukaemias. FrenchAmerican-British (FAB) Co-operative Group. Br J Haematol. 1976;33:451-8. 5. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995;9:1783-6. 6. Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006;20:1496-510. 7. Harrison CJ. Genetics of T-cell lymphoblastic leukemia. Hematology (EHA Education Program). 2007;1:154-60. 8. Armstrong SA, Look T. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23:6306-15. 9. Rowe JM, Buck G, Burnett AK, Chopra R, Wiernik PH, Richards SM, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106:3760-7. 10. Thomas X, Boiron JM, Huguet F, Dombret H, Bradstock K, Vey N, et al. Outcome of treatment in adults with acute lymphoblastic leukemia: analysis of the LLAA-94 trial. J Clin Oncol. 2004;22:4075-86. 11. Hoelzer D, Gökbuget N, Ottmann O, Ching-Hon P, Relling MV, Appelbaum FR, et al. Acute lymphoblastic leukemia. Hematology (Am Soc Hematol Educ Program). 2002;162-92. 12. Xicoy B, Ribera JM, Oriol A, Sanz MA, Abella E, Tormo M, et al. Significado pronóstico de los subtipos inmunológicos de la leucemia aguda linfoblástica T del adulto. Estudio de 81 pacientes. Med Clin (Barc). 2006;126:41-6. 13. Ferrando AA, Neuberg DS, Dodge RK, Paietta E, Larson RA, Wiern PH, et al. Prognostic importance of TLX1(HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukemia. Lancet. 2004;363:5356. 14. Berger R, Dastungue N, Busson M, Van den Akker J, Pérot C, Ballerini P, et al. t(5;14)/HOX11L2-positive T-cell acute lymphoblastic leukemia. A collaborative study of the Groupe Français de Cytogénétique Hématologique (GFCH). Leukemia. 2003;17:1851-7. 15. Chiaretti S, Li X, Gentleman R, Vitale A, Vignetti M, Mandelli F, et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood. 2004;103:2771-8. 16. Gottardo NG, Hoffmann K, Beesley AH, Freitas JR, Firth MJ, Perera KU, et al. Identification of novel moleculaar prognostic markers for paediatric T-cell acute lymphoblastic leukaemia. Br J Hematol. 2007;137:319-28. 17. Burmeister T, Gökbuget N, Reinhardt R, Rieder H, Hoelzer D, Schwartz S, et al. NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood. 2006;108:3556-9. 18. Baldus CD, Burmeister T, Martus P, Schwarz S, Gökbuget N, Bloomfield CD, et al. High expression of the ETS Transcription factor ERG predicts adverse outcome in acute T-lymphoblastic leukemia in adults. J Clin Oncol. 2006;24:4714-20. 19. Baldus CD, Martus P, Burmeister T, Schwartz S, Gökbuget N, Bloomfield CD, et al. Low ERG and BAALC expression identifies a new sub- 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. group of adult T-lymphoblastic leukemia with highly favorable outcome. J Clin Oncol. 2007;25:3739-45. Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151-7. Rubnitz JE, Camitta BM, Mahmoud H, Raimondi SC, Carroll AJ, Borowitz MJ, et al. Childhood acute lymphoblastic leukemia with the MLLENL fusion t(11;19)(q23;p13.3) traslocation. J Clin Oncol. 1999;17:191-6. Ravandi F, Gandhi V. Novel purine nucleoside analogues for T-cell-lineage acute lymphoblastic leukaemia and lymphoma. Expert Opin Investig Drugs. 2006;15:1601-13. Beesley AH, Palmer ML, Ford J, Weller RE, Cummings AJ, Freitas JR, et al. In vitro cytotoxicity of nelarabine, clofarabine and flavopiridol in paediatric acute lymphoblastic leukemia. Br J Haematol. 2007;137:109-16. Kurtzberg J, Ernst TJ, keating MK, Gandgi V, Hodge JP, Kisor DF, et al. Phase I study 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol. 2005;23:3396-403. Berg SL, Blaney SM, Devidas M, Lampkin TA, Murgo A, Bernstein M, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children’s Oncology Group. J Clin Oncol. 2005;23:3376-82. DeAngelo D, Yu D, Johnson JL, Coutre SE, Stone RM, Stopeck AT, et al. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group study 19801. Blood. 2007;109:5136-42. Göekbuget N, Arnold R, Atta J, Brück P, Hermann S, Horst H, et al. Compound GW506U78 has high single-drug activity and good feasibility in heavily pretreated relapsed T-lymphoblastic leukemia (T-ALL) and Tlymphoblastic lymphoma (T-LBL) and offers the option for cure with stem cell transplantation (SCT). Blood. 2005;104:150a. Dunsmore K, Devidas M, Borowitz MJ, Winick N, Hunger S, Carroll WL, et al. Nelarabine can be safely incorporated into an Intensive, multiagent chemotherapy regimen for the treatment of T-Cell acute lymphocytic leukemia (ALL) in children: a report of the Children’s Oncology Group (COG) AALL00P2 protocol for T-cell leukemia. Blood. 2006;108:1864a. Furman RR, Gore L, Ravandi F, Hoelzer D. Forodesine (bcx-1777) is clinically active in relapsed/refractory T-cell leukemia: results of a phase II study (interim report). Blood. 2006;108:1851a. Stelljes M, Kienast J, Berning B, Gokbuget N, Hoelzer D, Silling G, et al. Forodesine in patients with refractory/relapsed T-ALL can induce prolonged stable remission with minimal toxicity before and after allogeneic stem cell transplantation. Kearns P, Michel G, Nelken B, Joel S, AL-Ghazaly E, Beishuizen A, et al. BIOV-111 a European Phase II trial of Clofarabine (Evoltra®) in refractory and relapsed childhood acute lymphoblastic leukemia. Blood. 2006;108:1859a. Karp JE, Ricklis RM, Balakrishnan K, Briel J, Greer J, Gore SD, et al. A phase I clinical-laboratory study of clofarabine followed by cyclophosphamide for adults with refractory acute leukemias. Blood. 2007;110:1762-9. Jeha S, Kantarjian H. Clofarabine for the treatment of acute lymphoblastic leukemia. Expert Rev Anticancer Ther. 2007;7:113-8. Quintas-Cardama A, Tong W, Manshouri T, Cools J, Gilliland DG, Kantarjian H, et al. Frequency of NUP214-ABL1 oncogene in patients with Tcell acute lymphoblastic leukaemia (T-ALL) and analysis of the activity of imatinib and nilotinib in NUP214-ABL1-expressing T-ALL cell lines. Blood. 2006;108:710a. O’Neil J, Grim J, Strack P, Rao S, Tibbits D, Winter C, et al. FBW7 mutacions in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813-24. Med Clin (Barc). 2007;129(Supl 1):45-9 49