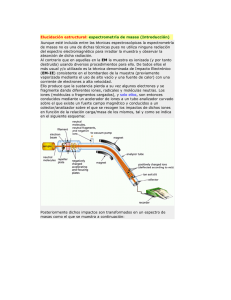
el espectrómetro de masas - Instituto de Biotecnología
Anuncio

Espectrometría de Masas Instituto de Biotecnología UNIVERSIDAD MAESTRÍA NACIONAL EN AUTÓNOMA CIENCIAS DE MÉXICO BIOQUÍMICAS CURSO DE MÉTODOS ESPECTROMETRÍA DE MASAS PRESENTA: GERMÁN PLASCENCIA VILLA JUNIO DE 2003 CUERNAVACA, MORELOS 1 Espectrometría de Masas Espectrometría de Masas INDICE INDICE INTRODUCCIÓN HISTORIA DE LA ESPECTROMETRIA DE MASAS EL ESPECTRÓMETRO DE MASAS ENTRADA DE LA MUESTRA MÉTODOS DE IONIZACIÓN Impacto Electrónico (Electron Impact EI) Ionización Química (Chemical Ionization CI) Ionización de Campo (Field Ionization FI) Desorción de Campo (Field Desorption FD) Bombardeo con Átomos Rápidos (Fast Atom Bombardment FAB) Ionización por Electrospray (Electrospray Ionization ESI) Matrix-Assited Laser Desorption/Ionization (MALDI) ANALIZADORES DE MASAS Efecto de campos magnéticos sobre los iones Principios de los analizadores de masas Espectrómetro de masas de sector magnético (Magnetic Sector Mass Spectrometer) Espectrómetros de masas de cuadrupolo (quadrupole) Analizador de Tiempo de Vuelo (Time-of-flight TOF) Analizadores de trampa de iones (Ion-Trap) Analizador FT-ICR (Fourier Transform Ion Cyclotron Resonance) DETECTORES DE IONES Copa Faraday Multiplicador de Electrones (Electron Multiplier) Detector Daly Detector de Microcanales ADQUISICIÓN Y PROCESAMIENTO DE DATOS Procesamiento de Datos APLICACIONES Análisis de estructura y masa Sistemas acoplados Análisis cuantitativos Análisis de péptidos Iones de péptidos Secuenciación de péptidos Identificación de proteínas HERRAMIENTAS PARA ANÁLISIS DE PROTEÍNAS POR ESPECTROMETRÍA DE MASAS DEFINICIÓN DE TÉRMINOS REFERENCIAS 2 2 3 4 6 8 9 9 10 11 11 13 14 16 19 19 19 20 21 22 24 25 27 27 27 28 29 30 31 32 32 33 34 35 36 36 37 38 39 40 Espectrometría de Masas INTRODUCCIÓN La Espectrometría de Masas es una poderosa técnica microanalítica usada para identificar compuestos desconocidos, para cuantificar compuestos conocidos, y para elucidar la estructura y propiedades químicas de moléculas. La detección de compuestos puede ser llevada a cabo con cantidades realmente pequeñas (algunos pmoles) de muestra y obtener información característica como el peso y algunas veces la estructura del analito. En todos los casos, alguna forma de energía es transferida a las moléculas a analizar para afectar la ionización. En la técnica clásica de impacto electrónico (electron ionization EI), algunas de las moléculas ionizadas del analito “explotan” en una variedad de fragmentos ionizados, el patrón de fragmentación resultante así como los iones residuales constituyen el espectro de masas. En principio, el espectro de masas de cada compuesto es único y puede ser usado como se “huella química” para caracterizar el analito. El proceso de análisis por espectrometría de masas comienza en llevar el compuesto a analizar a fase gaseosa, -2 la muestra debe tener una presión de vapor de que 10 mmHg, debido a que las moléculas deben migrar por difusión desde el sistema de entrada hacia la cámara de ionización. Las muestras pueden ser introducidas al espectrómetro de masas usando una sonda directa o por entrada en lote (batch) para sólidos puros o líquidos volátiles. Analitos purificados por diferentes técnicas de separación (cromatografía de gases, cromatografía de líquidos, electroforesis capilar, etc.) pueden entrar al espectrómetro de masas tan pronto como vayan saliendo. Ya que las moléculas neutras difunden en forma aleatoria por la fuente de ionización, solo una porción es ionizada. El proceso de ionización más común en análisis en fase gaseosa es el de ionización electrónica (EI), en el cual se transfiere energía a la molécula neutra en estado de vapor, dándole suficiente energía para expulsar uno de sus electrones y de ese modo tener una carga residual positiva. Este proceso produce un ion con carga positiva y un electrón suelto. La molécula ionizada puede tener energía excesiva que puede ser disipada a través de la fragmentación de ciertos enlaces químicos. El rompimiento de varios enlaces químicos permite la producción de fragmentos de ion cuya masa es igual a la suma de las masas atómicas de un grupo de átomos que retienen la carga positiva durante el proceso de fragmentación. Para compuestos no volátiles, iones de la molécula intacta son producidos al pasar la solución por un campo eléctrico (electrospray ionization) o por bombardeo de partículas (fast atom bombardment) o por interacción con especies fotoexcitadas (matrix-assisted laser desorption). Después de producir los iones, el siguiente paso es su análisis en el analizador de iones de acuerdo a su relación masa/carga (m/z). Los iones tienen una carga eléctrica que les permite ser controlados por campos eléctricos; son separados por su valor m/z en el analizador de masas. Existen diferentes tipos de analizadores de masas: magnéticos, de cuadrupolo, trampa de iones cuadrupolo, trampa de iones magnética, o tiempo de vuelo (time-of-flight). Los iones son analizados de acuerdo a su abundancia a lo largo de la escala m/z. Durante el proceso de adquisición de datos provenientes del detector, los datos pueden ser organizados en forma tabular o en formato de gráfica de barras para dar finalmente el espectro de masas de la muestra analizada. 3 Espectrometría de Masas HISTORIA DE LA ESPECTROMETRÍA DE MASAS La espectrometría de masas tiene su origen en los experimentos desarrollados en el Cavendish Laboratory de la Universidad de Cambridge en Inglaterra en 1897 por Joseph John Thomson, él descubrió que descargas eléctricas en gases producían iones y que estos rayos de iones podían adoptar diferentes trayectorias parabólicas de acuerdo a su masa cuando pasaban a través de campos electromagnéticos. Thomson realizó la construcción del primer espectrómetro de masas, llamado parábola espectrográfica (Figura 1), para la determinación de la relación masa/carga de iones. Figura 1. Parábola espectrográfica construida por J.J. Thomson Uno de los estudiantes de Thomson, Francis William Aston fue quien diseño varios espectrómetros de masas en los cuales los iones eran dispersados por sus masas y enfocados de acuerdo a su velocidad. Esto permitió mejorar el poder de resolución de masas y el posterior descubrimiento de isótopos de diferentes elementos que están presentes en la naturaleza. Thomson recibió el Premio Nobel de Física en 1906 y Aston el Premio Nobel de Química en 1922. En la década de los 20´s, Arthur J. Dempster de la Universidad de Chicago, desarrolló un analizador magnético que enfocaba iones formados por impacto electrónico (electron impact) sobre un colector eléctrico. Este diseño fue adaptado por Josef Mattauch, Richard Herzog, Kenneth Bainbridge, Alfred Nier y otros, permitiendo importantes descubrimientos en física atómica y nuclear en los 30´s. Pero fue hasta la década de los 40´s que los primeros espectrómetros de masas comerciales aparecieron. Durante los 30´s y 40´s, Nier y algunos otros incorporaron las tecnologías de vacío y electrónica para mejorar el desempeño de los primeros diseños de espectrómetros. Instrumentos de doble enfoque, que combinan analizadores magnéticos y electrostáticos fueron introducidos permitiendo incrementar la precisión. Estos instrumentos fueron originalmente desarrollados con el propósito de determinar en forma precisa los pesos atómicos de los elementos y sus isótopos. En la década de los 40´s se construyeron los primeros espectrómetros de masas disponibles comercialmente por medio de diferentes compañías de Europa y Estados Unidos. En esta década también se desarrolló el espectrómetro de masas de tiempo de vuelo (time-of-flight, TOF), un concepto propuesto como un analizador mas barato y simple en el cual los iones son separados en base a sus diferencias en velocidad cuando son acelerados en un tubo de vuelo lineal. A principios de los 50´s, los patrones de fragmentación de los iones comenzaron a ser entendidos, aunque los aparatos disponibles estaban limitados en el rango de masas. Otro tipo de instrumento desarrollado para el propósito de acoplar el espectrómetro de masas a un cromatógrafo de gases fue el filtro de masas cuadrupolo, el cual usa un campo eléctrico cuadrupolar conteniendo componentes de radiofrecuencia y corriente directa para separar iones. En 1953 el analizador de masas cuadrupolo fue patentado. Este analizador fue reportado por Wolfgang Paul de la Universidad de Bonn, quien después ganó el Premio Nobel de Física en 1989 por su trabajo de trampa de iones (ion trapping). En 1956 las primeras muestras biológicas (esteroides) fueron exitosamente analizadas. En los 60´s la espectrometría de masas se estableció como una técnica para caracterizar compuestos orgánicos. Este fue asistido con la introducción de una fuente de ionización química. Otros métodos de ionización surgieron incluyendo desorción de campo, ionización secundaria (secondary ionization MS o SIMS), desorción de plasma y desorción láser. Aparecieron las primeras publicaciones científicas especializadas incluyendo Organic Mass Spectrometry. La espectrometría de masas en tándem (MS/MS) surgió con el desarrollo de procedimientos de disociación por inducción de colisión, permitiendo obtener información estructural de componente de una mezcla usando dos etapas de análisis de masas. 4 Espectrometría de Masas En 1974 se desarrollo la espectrometría de masas Fourier ICR (FT-ICR MS). Una importante ventaja de este sistema es que permite detectar simultáneamente varios iones y estos espectrómetros de masas también logran tener muy alta resolución de masas. En los 80´s se desarrollaron las técnicas de ionización capaces de ionizar eficientemente moléculas biológicas. Michael Barber en Manchester desarrollo la técnica FAB (Fast Atom Bombardment) la cual utiliza una fuente de átomos pesados neutrales para ionizar compuestos de una superficie de una matriz líquida. John Fenn de la Universidad Yale refinó la fuente de iones originalmente reportada por Malcolm Dole de la Universidad Northwertern casi dos décadas antes de la técnica de ionización de electrospray (ESI). Matrix-assisted laser desorption ionization (MALDI) fue también desarrollada en los 80´s por Franz Hillenkamp y Michael Karas. En esta técnica, las moléculas son desorbidas por un láser de una superficie sólida o líquida conteniendo una matriz de compuesto orgánico. Las técnicas ESI y MALDI han permitido la introducción de moléculas biológicas de más de 1 millón de Daltons en los espectrómetros de masas como iones estables en fase gaseosa. Actualmente, los espectrómetros de masas son utilizados para el estudio de un amplio rango de sustancias y materiales con alta precisión y sensibilidad. Los campos de aplicación son desde estudios astronómicos del sistema solar, geofísica y geoquímica, hasta análisis químicos y en forma fundamental en investigaciones de química, toxicología, ciencias ambientales, y en forma creciente en ciencias biológicas y biomédicas. 5 Espectrometría de Masas EL ESPECTRÓMETRO DE MASAS Un Espectrómetro de Masas es un instrumento que mide las masas de moléculas individuales que han sido convertidas en iones. Un espectrómetro de masas no mide la masa molecular directamente, pero mide la relación masa/carga de los iones formados de las moléculas. Los espectrómetros de masas tienen siete componentes mayores: un sistema de entrada, una fuente de iones, un analizador de masas, un detector, un sistema de vacío, un sistema de control y un sistema de datos. El sistema de entrada, junto con la fuente de iones y el tipo de analizador de masas definen el tipo de espectrómetro y las capacidades del sistema. Figura 2. Componentes básicos de un espectrómetro de masas Los instrumentos usados están compuestos de una combinación de sistemas de entrada, fuentes de iones, y analizadores de masas (Tabla 1). Tabla 1. Variantes de componentes del sistema de espectrometría de masas COMPONENTE VARIANTES Sistema de entrada 1. Directo. 2. Columna capilar cromatografía líquida. Fuente de iones 1. Impacto electrónico. 2. Ionización Química. 3. Ionización de campo. 4. Deserción de campo. 5. Bombardeo con Átomos Rápidos. 6. Electrospray, incluyendo nanospray y microspray. 7. Matrix-assisted laser desorption (MALDI) Analizador de masas 1. Cuadrupolo. 2. Trampa de iones (Ion-trap). 3. Tiempo de vuelo (Time-of-flight TOF). 4. Analizador FT-ICR Los procesos básicos asociados con la espectrometría de masas abarcan desde la generación de iones en fase gaseosa de un analito, y la medición de las relaciones masa/carga (m/z) de estos iones. La formación de iones de la muestra en fase gaseosa es un prerequisito esencial en el proceso del espectrómetro de masas en el proceso de verificación de masas moleculares o análisis. Aunque los primeros espectrómetros de masas requerían que la muestra estuviera en forma gaseosa, nuevos desarrollos han permitido incluir muestras en soluciones líquidas o en una matriz sólida. Dependiendo del tipo de entrada y técnica de ionización usada, la muestra puede ya estar en forma de iones en solución, o puede ser ionizada en conjunto con su volatilización por otros métodos en la fuente de iones. El estudiar el analito como un ion en fase gaseosa le dan dos características fundamentales. La primera característica fundamental es que el movimiento de los iones en fase gaseosa puede ser controlado en forma precisa en campos electromagnéticos que contienen los iones a estudiar y pueden ser manipulados para probar el movimiento del ion en el campo. Debido a que el movimiento de los iones es proporcional a la relación m/z del ion, esto justifica la medición de la m/z y por lo tanto la masa del analito. Los detalles concernientes a diferentes tipos de técnicas de análisis de masas y técnicas de ionización utilizadas determinan factores tales que la precisión y exactitud de la medición de m/z, la resolución, y el rango de m/z del analizador. La segunda 6 Espectrometría de Masas característica fundamental es que el uso de iones en fase gaseosa incrementa la sensibilidad del espectrómetro de masas. El movimiento preciso de iones en campos electromagnéticos que permiten medir m/z también les provee contención y enfoque. Durante el proceso de medida de m/z, los iones son transmitidos con gran eficiencia a los detectores de partículas que registran la llegada de estos iones. Estas llegadas son detectadas con gran sensibilidad debido a una combinación de señales de bajo fondo y la eficiente generación de electrones secundarios que pueden 5 subsecuentemente ser multiplicados por factores de 10 o mayores. El mantenimiento de la precisión y exactitud de estos movimientos y por lo tanto de la sensibilidad del análisis requiere operación experta del sistema de espectrometría de masas. Algunas desventajas del uso de iones en fase gaseosa es la dificultad de generación de los iones y colocarlos en la fase gaseosa, además de la complejidad de los instrumentos utilizados en este tipo de experimentos. El desarrollo de la ionización por electrospray y MALDI han aportado dos importantes métodos para la producción de iones de péptidos y proteínas en fase gaseosa. Otra consecuencia de la complejidad de los instrumentos es el alto precio de tales instrumentos en términos su adquisición y costos de mantenimiento. Sin embargo, en general y considerando la sofisticación, productividad y reproducibilidad de los experimentos desarrollados, se puede justificar que el costo de la espectrometría de masas es proporcional con los costos asociados con otros componentes en los proyectos de investigación que requieren este tipo de análisis. 7 Espectrometría de Masas ENTRADA DE LA MUESTRA Para sólidos razonablemente puros, la muestra es colocada sobre la punta de una barra que es introducida dentro del espectrómetro a través de un sistema de vacío. La muestra es entonces evaporada o sublimada en una fase gaseosa, usualmente por medio de calor. Gases y líquidos pueden ser introducidos a través de sistemas de entrada diseñados especialmente con un control de flujo, llamados sondas directas (direct probes), estos dispositivos transfieren la muestra desde el exterior a través de un sistema de vacío hacia la fuente de ionización que esta a alta presión (Figura 3). Estas sondas pueden ser diseñadas para soportar muestras para ser ionizadas por distintos métodos. Una vez que la muestra está es fase gaseosa es ionizada y frecuentemente acompañado de fragmentación para proceder al análisis de masas. En algunas técnicas especializadas, la volatilización y la ionización de la muestra ocurren al mismo tiempo. Figura 3. Sondas directas (direct probes) Para obtener el espectro de masas de un compuesto simple de una mezcla, los componentes individuales deben ser separados antes del análisis de espectrometría de masas. La separación de los compuestos es necesaria para tener certeza de la identificación, ya que dos o más compuestos presentes en la muestra pueden crear espectros que se sobrelapen o mezclen ya que los iones generan fragmentos simples que dificultarían el análisis de los datos obtenidos. Desde que en la década de los 60´s se acopló la cromatografía de gases (GC) a la espectrometría de masas, esta conexión permite separar compuestos ya en fase gaseosa para su entrada al espectrómetro al entrar las diferentes fracciones en diferentes tiempos en forma continua permite un análisis secuencial de las muestras. Recientemente, equipos de cromatografía de líquidos (LC), cromatografía de fluidos supercríticos, HPLC y electroforesis capilar, utilizados para separar compuestos, se pueden acoplar a espectrómetros de masas para análisis de muestras complejas. 8 Espectrometría de Masas MÉTODOS DE IONIZACIÓN Un reto fundamental en la espectrometría de masas de cualquier tipo de analitos es la producción de iones en fase gaseosa de estas especies, y dificultades en la producción de estos iones en fase gaseosa pueden impedir el análisis por espectrometría de masas de ciertas clases de moléculas. Esta situación fue el caso de proteínas y péptidos. Las primeras técnicas que fueron aplicadas, ionización electrónica (electron ionization EI) e ionización química (chemical ionization CI), son procesos de dos etapas en los cuales los analitos son vaporizados con calor y la ionización ocurre una vez que el analito esta en fase gaseosa. Esta etapa de vaporización limitó los experimentos de análisis por espectrometría de pequeños péptidos, usualmente a máximo 4 o 5 aminoácidos. Adicionalmente, estos péptidos tenían que ser derivatizados para minimizar su polaridad y darles suficiente volatilidad. El análisis de proteínas no era simplemente posible, y problemas similares se encontraban en otras clases de moléculas polares. La ionización por bombardeo con átomos rápidos (fast atom bombardment FAB) fue el primer método de ionización que hizo posible analizar rutinariamente por espectrometría de masas a moléculas polares, también fue fundamentalmente diferente de otros métodos de ionización tal como ionización electrónica (IE) debido a que los iones en fase gaseosa fueron generados directamente de una solución del analito en fase líquida. Como resultado, las diferentes formas de vaporización y de ionización del analito se convirtieron en una etapa estrechamente ligada en el proceso total de análisis por espectrometría de masas. Impacto Electrónico (Electron Impact EI) El Impacto Electrónico (Electron Impact EI) o comúnmente llamado Ionización Electrónica (Electron Ionization EI) es un método clásico de generación de iones en espectrometría de masas utilizado aún en muchas -6 aplicaciones. El analito es introducido dentro de la fuente de ionización que esta al vacío (<10 mbar) y sebsecuentemente ionizado por colisiones con un flujo de electrones (a 70 eV). - +. M+e _M +2e - Los electrones para este proceso son generados por emisión térmica de un filamento caliente. El filamento esta típicamente hecho de tungsteno o de renio. Los electrones son subsecuentemente acelerados al aplicar un potencial entre el filamento y la muestra. El rayo de electrones cruza la fuente de iones, afectando las moléculas -4 neutras que provienen de la muestra (Figura 4). La corriente típica del filamento es de 1x10 Amp. Figura 4. Ionización por Impacto Electrónico (EI) La muestra es introducida por medio de una sonda directa (direct probe) operada de 20 a 500°C o por medio de un cromatógrafo de gases. Las muestras deben ser de polaridad baja o media y con cierta estabilidad térmica, además de que deben ser evaporadas antes de su ionización. El proceso de EI causa mucha fragmentación de las moléculas lo cual puede tener ventajas en la deducción de su estructura. El mecanismo de formación de iones para la especie AB es el siguiente: -* + 1. AB + e _ A + B + e -* + ° 2. AB + e _ A + B + 2e -* +°* +°* +° 3. AB + e _ [AB ] + 2e seguido de [AB ] _ AB -* 2+* 2+* + + 4. AB + e _ [AB ]" + 3e seguido de [AB ]" _ A + B - (muy baja abundancia) -* + 5. AH + e _ AH* + e seguido de AH* + AH _ [AH+H] + A 9 Espectrometría de Masas Donde: * = Especies de alta energía. ° = Radicales. " = Intermediarios de vida corta. En 1 y 2 se indican las especias de mayor abundancia las cuales sufren de fragmentación instantánea. En 3 indica especias de abundancia media. En 4 indica especies de abundancia muy baja. En 5 son especies que pueden formarse a altas presiones Ionización Química (Chemical Ionization CI) La Ionización Química (Chemical Ionization CI) es un método de ionización relativamente suave, y es el primer método de ionización suave introducido a la espectrometría de masas. La ionización es afectada por reacciones sobre la molécula con iones de un reactivo gaseoso generados por medio de ionización de impacto electrónico (EI) con moléculas del analito neutras. 3 4 El reactivo gaseoso es empleado con un exceso de 10 -10 veces la molaridad del. Esto puede ser logrado al usar volumen de iones bastante pequeño en comparación a las condiciones de EI. El volumen de reactivo gaseoso se controla por medio de una válvula. Figura 5. Ionización Química (CI) + Entre los agentes gaseosos utilizados se incluyen el metano, el isobutano y al amoniaco, formando iones CH5 , + + C4H9 , y NH4 respectivamente como especies predominantes. En caso del metano, el siguiente proceso es el que permite la formación de iones reactivos: - +. 1. CH4 + e _ CH4 Y fragmentos tales como CH3 +. + . 2. CH4 + CH4 _ CH5 + CH3 (autoprotonación) + + 3. CH3 + CH4 _ C2H5 + H2 + La protonación de un analito es dependiente de la diferencia de afinidades por protones en las bases + + correspondientes. Por ejemplo, el CH5 protona la mayoría de los analitos, mientras que el NH4 es más selectivo debido a que necesita un sitio ligeramente básico en las moléculas del analito. + + M + CH5 _ [M+H] + CH4 + Los iones [M+H] son llamados iones cuasi-molecular. La muestra es introducida por medio de una sonda directa (direct probe) a una temperatura de operación de 20 a 500°C (para sólidos y líquidos de baja volatilidad, así como líquidos y gases) o por medio de un cromatógrafo de gases (mezclas). Los analitos deben tener polaridad baja o media, así como estabilidad térmica. La ionización química genera iones de energía interna relativamente baja, exhibiendo un bajo nivel de fragmentación, la evaporación del analito antes de su ionización es el paso crítico. 10 Espectrometría de Masas Tipo de analito Polaridad baja Polaridad media Polaridad alta + Iones K A Tabla 2. Iones producidos por CI Iones positivos Iones negativos dominante // raramente detectado dominante // raramente detectado + -. [M+H] M // [M-H] + + + -. [M+H] // [M+R] [2M+H] M [M-H] + + + -. [M+H] [M+R] // [2M+H] [M-H] // [M+R] M descomposición descomposición Ionización de Campo (Field Ionization FI) La Ionización de Campo (Field Ionization FI) es un método de ionización suave. La muestra es introducida por las mismas técnicas que en EI y CI. La ionización de campo lleva el riesgo de descomposición térmica de la muestra. Las moléculas son ionizadas por medio de ionización de campo (FI) en los bordes del campo del ánodo emisor. El emisor consiste de un alambre de tungsteno de 10-13mm de diámetro que es activado por un procedimiento especial creando miles de microagujas en su superficie. Un voltaje alto de 10-12 kV es aplicado al emisor causando polarización y finalmente ionización de las moléculas que están cerca de las puntas de las microagujas donde el campo eléctrico alcanza una gran fuerza. Este proceso tiene una eficiencia de ionización +. + muy baja y por lo tanto, FI produce una corriente de iones muy baja. FI produce iones del tipo M y/o [M+H] , dependiendo del analito. Aunque los iones generados pueden estar casi libres de fragmentación, se prefiere la ionización CI en lugar de FI no puede ser utilizada debido a la volatilidad del analito. La muestra es introducida por medio de una sonda directa (direct probe) a una temperatura de operación de 20 a 500°C (para sólidos y líquidos de baja volatilidad, así como líquidos y gases), el uso de un cromatógrafo de gases acoplado al espectrómetro de masas es complicado ya que las condiciones son difíciles de estabilizar en las corridas de las muestras. Mientras que F I genera iones con muy baja energía interna y casi nula fragmentación, la evaporación del analito antes de su ionización es la etapa crítica. Tipo de analito Polaridad baja Polaridad media Polaridad alta Tabla 3. Iones producidos por FI Iones positivos Iones negativos dominante // raramente detectado Dominante // raramente detectado +. + M // [M+H] +. + M [M+H] + +. [M+H] // M - Desorción de Campo (Field Desorption FD) La Desorción de Campo (Field Desorption FD) es un método de ionización muy suave utilizado en espectrometría de masas. Este método esta basado en el de Ionización de Campo (Field Ionization FI), FD ha sido desarrollado al combinar la desorción y la ionización del analito, pero no hay necesidad de evaporación antes de la ionización, generando iones con muy baja energía interna y casi nula fragmentación. En la Figura 6 se muestra un equipo de espectrometría de masas con una fuente de FD. Figura 6. Fuente de FD 11 Espectrometría de Masas Moléculas neutras son ionizadas principalmente por FI en el borde del campo del ánodo emisor, los cationes son desorbidos tal cual. Un alto voltaje de 10-12 kV es aplicado al emisor causando polarización y finalmente ionización de las moléculas que están cerca de las puntas de las microagujas donde el campo eléctrico alcanza una enorme fuerza (Figura 7). Un ligero calentamiento de la muestra ayuda a mantener el equipo limpio al evitar su acumulación. Figura 7. Esquema general de ionización por FD Usualmente, se pasa una corriente de 50 mA sobre el emisor (un alambre de 10mm) o incluso hasta 80-100 mA en emisores de 13mm. Esto incrementa la movilidad de las moléculas sobre la superficie del emisor, ayudando a la migración hacia las puntas de las agujas. El emisor puede resistir hasta 800°C. La muestra es introducida dentro de la fuente de iones por medio de una sonda directa que lleva el emisor de campo en su punta (Figura 8), el alambre activado entre las dos puntas de acero inoxidable es de 5mm de longitud. Este par de alfileres de acero están aislados eléctricamente mutuamente por medio de una pieza de cerámica. La sonda transfiere el emisor F D dentro de la muestra y se suministra un alto voltaje para la desorción/ionización y ocurre el calentamiento del emisor. Figura 8. Sonda y emisor de FD El emisor FD consiste de un alambre central de tungsteno de 10-13mm de diámetro. La activación a alta temperatura esta basada en una descomposición térmica de la superficie del alambre de tungsteno caliente al vacío. La activación crea miles de microagujas en la superficie del emisor (Figura 9). Figura 9. Supericie del emisor (80x80mm) La muestra es introducida como una solución (1 a 2 ml a 0.1-1.0 mg/ml en solventes volátiles) hacia el emisor. Durante este proceso una solo gota colgante en la punta de una jeringa de un microlitro puede tocar la superficie del emisor, de otra manera se rompería el emisor (Figura 10). 12 Espectrometría de Masas Figura 10. Proceso de montaje de una gota de 1.5ml sobre el emisor activado. La gota permanece en el alambre donde el solvente se avaporará para permitir tener solo una delgada capa de muestra. Es preferible que la muestra sea soluble en solventes volátiles, se pueden analizar compuestos de baja o alta polaridad, inclusive es posible analizar mezclas de compuestos, aunque solo se pueden cuantificar compuestos puros. Si la muestra esta contaminada con sales inorgánicas puede ocurrir daño en los componentes del sistema. Se pueden analizar muestras de 1-5000 amu. Tabla 4. Iones producidos por FD Iones positivos Iones negativos Tipo de analito dominante // raramente detectado dominante // raramente detectado +. + 2+ +. Polaridad baja M // [M+H] M [2M] +. + + 2+ + + Polaridad media M [M+H] // [M+Cat] M [2M+H] [2M+Cat] + + + + +. 2+ Polaridad alta [M+H] [M+Cat] [2M+H] [2M+Cat] // M M [M-H] // [M+An] + + + +. Iones K A K [Kn+An-1] // [KA] A [An+Kn-1] + + + Cat: cationización por Li , Na , K , y otros iones metálicos. - An: anionización por Cl , Br , I , HSO4-, y otros aniones. Bombardeo con Átomos Rápidos (Fast-Atom Bombardment FAB) Los métodos de desorción de partículas fueron desarrollados en los 70´s y 80´s, y a diferencia del método de desorción de campo (field desorption), evito complicaciones en diseño de equipo y preparación de la muestra. FAB es un método de ionización suave, resulta de la combinación de desorción del analito desde una fase condensada (mezcla de analito sólido+matriz) con su subsecuente ionización. La matriz esta formada de moléculas orgánicas (glicerol o 3-nitrobencil alcohol) que mantienen una superficie homogénea para el + bombardeo. Ocurre el bombardeo de la muestra con átomos rápidos (Ar o Xe de 3-10 keV) o iones rápidos (Cs arriba de 35 keV) (Figura 11). Si se utilizan iones rápidos la técnica se conoce también como liquid secondary ion mass spectrometry (LSIMS o liquidSIMS). En la práctica, no hay una diferencia significativa entre los espectros obtenidos por FAB o LSIMS. Figura 11. Sistema general de FAB El rayo primario de átomos rápidos es librado por una pistola de átomos rápidos (FAB gun) (Figura 12) o por + + una pistola de iones Cs que utiliza un horno conteniendo CsI como fuente de Cs . La pistola FAB esta localizada fuera de la fuente de iones, a través de ella pasa un flujo de Xe neutro, los iones Xe son generados 13 Espectrometría de Masas por EI. Los iones son acelerados, enfocados y neutralizados por intercambio de cargas con Xe neutro al haber colisión. El rayo de átomos pega con una gota de muestra y debido a colisiones y disrupciones se forman iones secundarios que son expulsados de la superficie de la muestra, los iones generados son acelerados y enfocados al analizador. Figura 12. FAB gun La muestra es introducida por medio de una sonda directa. Básicamente la sonda FAB consiste de una simple barra con un contenedor para la muestra donde se dirigen los átomos rápidos que soporta la gota de la solución de analito contenida en una matriz. Este contenedor de la muestra esta aislado eléctricamente. La función de la matriz es absorber energía de las partículas primarias que la impactan, actuar como solvente para el analito, refrescar la superficie de la gota y ayudar en el proceso de ionización, ya que puede ser aceptor/donador de protones o electrones. Para que una matriz lleve a cabo todas sus funciones es necesario que la muestra sea 100% soluble en ella, solo aquellos solventes con baja presión de vapor pueden ser usados fácilmente, el solvente debe tener una viscosidad suficientemente baja que permita la difusión de los solutos a la superficie, ser químicamente inerte. Las muestra que pueden ser analizadas por FAB deben ser altamente solubles, e incluso altamente polares (péptidos, oligosacáridos), ionicos (sales), o apolares (compuestos neutros, oligomeros sintéticos). FAB puede ser utilizado para detección directa de iones positivos o negativos. El rango de masa es desde 300 amu hasta 5000 amu, en compuestos de menor masa resultan algunas complicaciones en la detección de los picos del espectro y en moléculas mayores se presentan problemas en la desorción. Los iones secundarios generados pueden permanecer estables por varios minutos, ya que poseen baja energía interna y exhiben un bajo nivel de fragmentación. Tabla 5. Iones producidos por FAB Iones positivos Iones negativos Tipo de analito dominante // raramente detectado dominante // raramente detectado +. + -. Polaridad baja M // [M+H] M // [M-H] +. + + -. Polaridad media M [M+H] // [M+Cat] M [M-H] + + +. -. Polaridad alta [M+H] [M+Cat] // M [M-H] // [M+An] M + + + +. -. Iones K A K [Kn+An-1] // [KA] A [An+Kn-1] // [KA] + + + Cat: cationización por Li , Na , K , y otros iones metálicos. - An: anionización por Cl , Br , I , HSO4-, y otros aniones. Ionización por Electrospray (Electrospray Ionization ESI). La Ionización por Electrospray (ESI) es uno de los métodos de ionización mas recientemente desarrollados en espectrometría de masas. El diseño y operación de fuentes de ionización por electrospray usadas comúnmente en los espectrómetros de masas están basados en diseños descritos por Fenn y colaboradores en 1985. Este método es llevado a cabo a presión atmosférica a diferencia de otros métodos, por lo que se le conoce también como un método de ionización a temperatura ambiente (atmospheric pressure ionization API). ESI es 14 Espectrometría de Masas ampliamente utilizado en aplicaciones de ciencias bioquímicas y biomédicas debido a su capacidad de analizar moléculas altamente polares tales como péptidos, oligonucleótidos y oligosacáridos. En el proceso general de electrospray, que ocurre en la punta del emisor (capilar o aguja), una solución acuosa -4 -5 ácida o básica (dependiendo del la muestra) diluida del analito (10 -10 molar) es rociada desde la punta del emisor en el cual se aplica un potencial de 3-4 kV, la solución debe proveer conductividad eléctrica que puede ser obtenida por el uso de analitos iónicos o aditivos iónicos tales como buffers o por algún grado de disociación electrolítica del solvente. El líquido comienza a salir de la aguja, incrementa su carga y asume una forma cónica, llamada cono de Taylor, en honor a G.I. Taylor quien describió este fenómeno en 1964 (Figura 13). El líquido asume esta forma cuando incrementa su carga ya que una forma cilíndrica puede retener mas carga que una esfera. En la punta del cono, el líquido cambia de forma a una línea fina, que se vuelve inestable ya que es forzado a retener mas y más carga, y finalmente llega a un punto crítico donde no puede soportar mas carga eléctrica y la solución entonces se dispersa en forma de niebla de pequeñas gotas (de menos de 10 mm de diámetro) altamente cargadas que vuelan buscando una superficie de carga opuesta. Debido a que las gotas están altamente cargadas con la misma carga eléctrica se repelen fuertemente, las gotas vuelan y se dispersan cubriendo un área cada vez mayor y se van reduciendo de tamaño ya que las moléculas de solvente se evaporan en su superficie, y la distancia entre las moléculas cargadas disminuye dramáticamente. Si la gota no encuentra donde disipar su carga, las cargas eléctricas llegan a un estado crítico y la gota explota violentamente (Figura 14). Este proceso fue originalmente observado por el físico John Zelany en 1914. Figura 14. Reducción de tamaño de gota en ESI Figura 13. Cono de Taylor Aún no existe una explicación 100% aceptada de lo que le sucede a las gotas, pero algunas de las teorías son: 1. Modelo de Residuo Cargado (charged residue model) de Dole. Las gotas sufren repentinamente de explosiones coulombicas produciendo gotas más pequeñas y más pequeñas que finalmente contendrán solo una molécula cargada y quizá algunas moléculas de solvente. 2. Modelo de Evaporación Iónica (ion evaporation model) de Iribarne y Thomson. Ocurre la expulsión de moléculas cargadas para reducir la densidad de carga de la superficie. 3. La gota original sobrevive después de haber expulsado algunas micro gotas cargadas. De cualquier forma, el proceso termina con moléculas cargadas que pueden todavía llevar moléculas de solvente. El proceso de evaporación y rompimiento de gotas se repite hasta que el tamaño y carga de las gotas desorba moléculas protonadas dentro de la fase gaseosa, donde pueden ser dirigidas en el espectrómetro de masas por medio de campos eléctricos apropiados (Figura 15). El proceso de evaporación puede ser suplementado con un flujo de gas (típicamente nitrógeno) y calor. Figura 15. Proceso general de Ionización por Electrospray (ESI) Una característica de ESI es que debido a las condiciones ácidas usadas para producir las gotas cargadas positivamente se tienden a protonar todos los sitios básicos en las moléculas de analito. Una segunda característica de ESI es la eficiencia del proceso de ionización y, como resultado, la sensibilidad de los experimentos basados en esta forma de ionización, la eficiencia en la protonación de estos sitios básicos en 15 Espectrometría de Masas ambientes ácidos parece contribuir a la sensibilidad. Una tercera característica es su compatibilidad con los solventes de HPLC de fase reversa (reverse-phase high-performance liquid chromatography), ya que las mezclas agua/solvente tienen excelentes propiedades compatibles con ESI. Figura 17. Rociador (sprayer) de ESI y el orificio del capilar (0.4mm de ancho). La señal observada en el proceso ESI es proporcional a la concentración del analito en la solución que esta siendo rociada pero no a su velocidad de flujo. Esto es debido a que un incremento en la velocidad de flujo simplemente provoca tener que eliminar un mayor porcentaje de solvente, debido a que en muchas ocasiones se tienen muestras biológicas en un volumen pequeño y en poca cantidad, es preferible poder rociar un volumen extremadamente pequeño, por lo que a partir de ESI que maneja flujos de 5-1000ml/min se han derivado el micro ESI que maneja una velocidad de flujo de 0.5-5ml/min y el nano ESI 0.02-0.5ml/min (Figura 17). Figura 17. ESI, microESI y nanoESI En dirección contraria, se han diseñado ESI asistidos neumáticamente, llamados Ion Spray (ISP) donde una corriente de gas inerte es aplicada en conjunto con un alto voltaje para ayudar al proceso de rociado de volúmenes grandes. Matrix-Assisted Laser Desorption/Ionization (MALDI) Esta técnica de ionización, comúnmente llamada MALDI, fue descrita por Karas y Hillenkamp en 1988. En este método de ionización los analitos son disueltos en una solución de un compuesto que absorbe UV, llamado matriz (matrix), y colocado dentro del espectrómetro de masas. Debido a que los solventes se van secando, los compuestos de la matriz cristalizan y los analitos quedan contenidos dentro de la matriz de cristales (Figura 18). Figura 18. Proceso de Matrix-Assisted Laser Desorption/Ionization (MALDI) 16 Espectrometría de Masas 7 2 Pulsos de láser de luz UV (3-5 ns, 337 nm, 10 W/cm ) son utilizados para vaporizar pequeñas cantidades de la matriz donde se incluyen los iones del analito y llevados a fase gaseosa en el proceso (Figura 19). La ionización ocurre por protonación en el ambiente ácido producido por los compuestos de la matriz y por la adición de ácido diluido en la muestra. La protonación de péptidos es particularmente eficiente en ambientes ácidos, haciendo de MALDI, un método efectivo para producir péptidos protonados. Debido a que con la desorción por láser la producción de iones es discreta y es pequeños paquetes, el MALDI esta comúnmente combinado con un analizador de tipo time-of-flight (TOF) dando un sistema de alta sensibilidad. Figura 19. Laser UV Mientras que ESI es afectado por especies contaminantes, MALDI puede tolerar niveles variables de algunos contaminantes. Cada pulso láser genera una pequeña cantidad de plasma que se expande en vacío, del cual se extraen los iones (positivos o negativos) del analito que son acelerados por un potencial de 10-30 kV y controlados por medio de campos electromagnéticos. Debido a diferencias de velocidades que resultan de sus diferencias en masas los iones llegan al detector en diferentes tiempos. Los tiempos de vuelo son del rango de 10-100 ms. Una muestra de 1 ml de la solución del analito o de la solución analito/matriz es aplicada en un contenedor y se seca, formando una capa delgada y cristalina. Los contenedores son de acero pulido, plata o cromo plateado (Figuras 20 y 21). Figura 20. Contenedor de plata para 10 muestras, cada pozo mide 23 mm de diámetro. Figura 21. Detalle del pozo 10 del contenedor. El rectángulo muestra el lugar donde impactará el láser. La línea blanca es un rasguño. La muestra aplicada es una solución en acetona que formo una capa cristalina. Se necesitan entre 10-100 disparos del láser para evaporar la capa completamente. La función de la matriz es en primer lugar absorber la energía del láser. Por lo que las matrices para MALDI están formadas de compuestos aromáticos (Figura). La ionización del analito puede ser afectada por deprotonación, al aceptar o ceder un electrón, por unión de cationes o fotoionización, por lo que los iones generados por MALDI son de una amplia variedad de moléculas, ya que dependiendo de la combinación entre el analito y la matriz será el tipo de iones predominantes en el espectro obtenido. 17 Espectrometría de Masas Figura 22. Compuestos utilizados en las matrices MALDI Tabla 6. Iones producidos por MALDI Iones positivos Iones negativos Tipo de analito dominante // raramente detectado dominante // raramente detectado +. + -. Polaridad baja M // [M+H] M // [M-H] +. + + + -. M [M+H] // [M+Cat] [2M+H] M [M-H] // [2M-H] Polaridad media n+ nMW > 3000: [M+nH] , n = 2, 3 MW > 3000: [M-nH] , n = 2, 3 + + + + -. [M+H] [M+Cat] // [2M+H] [2M+Cat] [M-H] // M [2M-H] Polaridad alta n+ nMW > 3000: [M+nH] , n = 2, 3 MW > 3000: [M-nH] , n = 2, 3 + + + +. -. Iones K A K // [Kn+An-1] [KA] A // [An+Kn-1] [KA] + + + Cat: cationización por Li , Na , K , y otros iones metálicos. - An: anionización por Cl , Br , I , HSO4-, y otros aniones. + Residuos de la matriz pueden formarse con todos los analitos dando: [M+H+matriz] . 18 Espectrometría de Masas ANALIZADORES DE MASAS Todos los espectrómetros de masas combinan la formación de iones, el análisis de masas, y la detección de iones. Los analizadores de masas llevan a cabo la separación de los iones producidos por diferentes métodos en una fuente de iones, de acuerdo a su relación masa/carga (m/z) para que puedan llegar al detector. La unidad para m/z es el Thomson (Th). En los experimentos de espectrometría de masas aparecen dos clases principales de iones, el ion molecular, que es la molécula completa del analito, y fragmentos iónicos, los cuales contienen solo una parte de la estructura. El peso molecular de una molécula puede ser calculado de la m/z del ion molecular, si la carga (z) es conocida, la información estructural se obtiene a partir de las m/z de los fragmentos del ion. Una variedad de analizadores de masas están disponibles para realizar estas mediciones, incluyendo cuadrupolo (quadrupole), trampa de iones (ion-trap), tiempo de vuelo (time-of-flight), sector magnético (magnetic sector), ciclotrón (ion cyclotron resonance) y otros. Cada tipo de analizador posee características y aplicaciones especiales, así como beneficios y limitaciones. La selección del tipo de analizador a utilizar es en base a que aplicación se le dará, el costo y el rendimiento deseado, ya que el analizador determina varias características del resultado del experimento, entre estas la resolución y el rango de m/z de los iones que pueden ser medidos. Efecto de campos magnéticos sobre los iones Todos los analizadores de masas comúnmente usados utilizan campos eléctricos y magnéticos para aplicar una fuerza a partículas cargadas (iones). La relación entre fuerza, masa y los campos aplicados puede verse a través de la segunda ley de Newton y la ley de Lorentz. Segunda ley de Newton Ley de Lorentz F=ma F=e(E+vB) Donde, F= fuerza aplicada al ion, M= masa del ion, a= aceleración, e= carga iónica, E= campo eléctrico, vB= producto de la velocidad del ion y el campo magnético aplicado. De la segunda ley de Newton se observa que la fuerza aplicada causa una aceleración que es dependiente de la masa del ion, y la Ley de Lorentz indica que la fuerza aplicada es también dependiente de la carga del ion. Por lo tanto, los espectrómetros de masas separan los iones de acuerdo a su relación masa/carga (m/z) y no solo a partir de la masa únicamente. Principios de los analizadores de masas Un analizador de masas es análogo al equipo utilizado en espectroscopia óptica para el análisis del contenido de color de luz visible. En espectroscopia óptica, se inicia con luz visible que esta compuesta de colores individuales (a diferentes longitudes de onda) que están presentes en diferentes intensidades. Un prisma separa la luz es sus diferentes longitudes de onda, y pequeña salida es usada para seleccionar cual longitud alcanza el detector. Las diferentes longitudes de onda son barridas o escaneadas al otro lado de la salida del detector y la intensidad de luz es registrada como una función del tiempo (longitud de onda) (Figura 23). En espectrometría de masas, se inicia con una mezcla de iones que tienen diferentes relaciones masa/carga y diferentes abundancias relativas. Campos electromagnéticos separan los iones de acuerdo a sus relaciones masa/carga, y una hendidura es usada para seleccionar cuales iones llegan al detector. Las diferentes relaciones masa/carga son entonces analizadas en la salida del detector y las corrientes de iones son registradas como una función del tiempo (figura 23). 19 Espectrometría de Masas Figura 23. Comparación entre espectrofotómetro y espectrómetro de masas Espectrómetro de masas de sector magnético (Magnetic Sector Mass Spectrometer) En un espectrómetro de masas de deflección magnética, los iones que llegan de la fuente de iones son acelerados a una gran velocidad. Los iones entonces pasan a través de un sector magnético (magnetic sector) en el cual se aplica un campo magnético en una dirección perpendicular al movimiento de los iones. Cuando se aplica aceleración perpendicular al movimiento de los iones, su velocidad permanece constante, pero se mueven en dirección circular, es por esto que el sector magnético tiene forma de arco, el radio y el ángulo del arco varían de acuerdo a diferentes diseños. Un sector magnético por si mismo es capaz de separar iones de acuerdo a su relación masa/carga, pero su resolución se ve limitada ya que no todos los iones que llegan de la fuente de iones tienen la misma energía ni la misma velocidad. Para tener una mejor resolución se añade un sector eléctrico (electric sector) que enfoca los iones de acuerdo a su energía cinética, esto se realiza aplicando una fuerza perpendicular a la dirección del movimiento de los iones, también en forma de arco. Figura 24. Espectrómetro de masas de sector magnético. La configuración de este aparato es también llamada de geometría reversa, ya que el sector magnético esta antes del sector eléctrico. La operación del espectrómetro de masas se logra manteniendo constantes el potencial de aceleración y el potencial del sector eléctrico y variando su campo magnético. Los iones que tienen una energía cinética constante, pero una relación m/z diferente son captados por el detector a diferentes fuerzas de campo magnético. La dependencia de la relación m/z sobre los campos eléctrico y magnético es deducida a partir de que todos los iones formados en la fuente de iones son acelerados a una energía cinética (T) de acuerdo a: 20 Espectrometría de Masas Despejando velocidad (v) tenemos: 2 De la Ley de Lorentz, el campo magnético aplicado a una fuerza evB debe ser igual a la fuerza centrípeta mv /r ya que los iones se mueven en un arco a través del sector: Substituyendo velocidad (v), se llega a la ecuación de trabajo aplicable al espectrómetro de masas de sector magnético: El sector magnético se mantiene constante usualmente a un valor en el cual pasen solo los iones que tengan una energía cinética específica, por lo que el parámetro que es variable es B, la fuerza del campo magnético. El campo magnético es generalmente analizado exponencialmente o linealmente para obtener el espectro de masas. El análisis de campo magnético puede ser usado para cubrir un amplio rango de m/z con una sensibilidad que es independiente de la relación masa/carga. Una alternativa es mantener B constante y analizar V. El potencial del sector eléctrico rastrea el voltaje, esto tiene la ventaja de que la relación m/z y el voltaje tienen una relación linear, pero una desventaja al analizar el voltaje es que la sensibilidad es proporcional a la relación m/z. El analizador de masas de sector magnético es un modelo clásico que posee varias ventajas: alta reproducibilidad, alto desempeño en análisis cuantitativos, alta resolución, sensibilidad y rango dinámico. Algunas limitaciones son que no pueden adaptarse a algunos métodos de ionización (por ejemplo: MALDI), costos mayores que otros analizadores. Son ampliamente usados en análisis de compuestos orgánicos, realizan mediciones exactas de masas, en cuantificación y en mediciones de isótopos. Espectrómetros de masas de cuadrupolo (quadrupole) El analizador de masas de cuadrupolo (quadrupole) es probablemente el analizador de masas mas utilizado, ya que además de su alto desempeño analítico combina facilidad de uso. Este tipo de analizador esta compuesto de cuatro barras organizadas paralelamente como dos grupos de dos barras conectadas eléctricamente (Figura 25). Una combinación de voltajes de radiofrecuencia (rf) y corriente directa (dc) son aplicados a cada par de barras, este arreglo simétrico permite producir campos hiperbólicos. Superficies diagonalmente opuestas están conectadas juntas a fuentes de voltajes rf y dc. Los iones se extraen de la fuente de iones y son acelerados (515 V) dentro del espacio central formado por el cuadrupolo a lo largo del eje longitudinal hacia el detector. Miller y Denton describieron el concepto de filtro de masas cuadrupolo como la combinación o superposición de filtros de pase bajo y pase alto. Al menos para iones relativamente ligeros (<300 Da), el análisis m/z no es afectado por la estructura del ion. Figura 25. Analizador cuadrupolo (quadrupole) El filtro cuadrupolo m/z es examinado al modificar la magnitud de la amplitud de voltaje de rf y manteniendo dc a una proporción fija. El poder de resolución es establecido por la relación de los voltajes rf y d c . Las trayectorias de los iones a través del espacio central de las barras es complicado, y para cada par de voltajes dc y rf solo iones de un valor m/z específico evitaran colisionar con las barras y pasaran el filtro cuadrupolo a través del eje Z hasta alcanzar el detector, todos los demás iones chocarán con las superficies del cuadrupolo a 21 Espectrometría de Masas estos valores de rf y dc. El espectro de masas completo puede ser examinado ya que se hace un barrido desde un valor de voltaje mínimo preestablecido hasta un máximo, pero manteniendo la relación dc/rf constante. Una exploración rigurosa de la trayectoria de los iones a través del filtro cuadrupolo incluye un gran número de variables físicas que afectan los campos eléctricos instantáneos que afectan los iones. Para un filtro de masas hiperbólico clásico, el potencial de distribución (F) a cualquier tiempo (t) es descrito por la siguiente ecuación: 2 F = [U+Vcos(wt)] x - y 2 2r0 2 2 donde x y y son las distancias a lo largo de ejes de coordenadas, r0 es la distancia desde el eje z a cualquiera de las superficies del cuadrupolo, w es la frecuencia angular (2p¶) de la corriente alterna aplicada (AC), V es la magnitud de la señal rf aplicada, y U es la magnitud del potencial dc aplicado a las superficies. El campo eléctrico instantáneo a lo largo de cualquiera de los tres ejes (x-y-z) puede ser calculado al derivar parcialmente la ecuación anterior en función de un eje particular, al realizar esto, se observa que los potenciales dc y AC aplicados a las superficies no afecta la aceleración de los iones en la dirección Z, es decir a través del eje entre la fuente de iones y el detector, ya que la formula no tiene dependencia de Z. Desde un punto de inicio del espectrómetro de masas de cuadrupolo, una solución frontera para la ecuación de Mathieu corresponde a un desplazamiento finito de los iones a lo largo de los ejes x y y, esto es que los iones siguen una trayectoria para evitar colisiones con las superficies y llegar al detector. Otro tipo de solución describe el desplazamiento radial de los iones que chocan con las superficies del cuadrupolo para excluir transmisión al detector. Definición de los términos a y q de la ecuación de Mathieu, en la cual a se relaciona con dc y q con rf. 2 2 2 2 ax = -ay = 4zeU/m r0 qx = -qy = 2zeV/m r0 Los valores a (proporcional a U/m) y q (proporcional a V/m) corresponden a trayectorias estables de los iones en el filtro cuadrupolar. Estas trayectorias le permiten a los iones llegar a la superficie del detector sin chocar o colisionar con las superficies del cuadrupolo. Estas transformaciones matemáticas permiten crear un diagrama a-q que simplifica los parámetros que afectan el movimiento de los iones en el cuadrupolo, estos diagramas son bidireccionales (en a y en q) pero incluyen seis variables (e, w , r 0, m, U, y V), este diagrama de estabilidad (Figura 26) relaciona el potencial dc aplicado (U) y el potencial rf aplicado (V). Figura 26. Diagrama de estabilidad a-q El analizador cuadrupolo ofrece alta reproducibilidad, además de costos relativamente bajos de manejo y mantenimiento del sistema, pero puede llegar a tener una resolución limitada, puede no adaptarse a algunos métodos de ionización. Este analizador esta frecuentemente acoplado a sistemas de cromatografía de gases/espectrómetro de masas (GC/MS) y cromatografía líquida/espectrómetro de masas (LC/MS). Analizador de Tiempo de Vuelo (Time-of-flight TOF) El principio de operación del analizador de tiempo de vuelo (time-of-flight TOF) involucra la medición del tiempo requerido por un ion para viajar desde la fuente de iones hasta el detector localizado a 1-2m de la fuente. Todos los iones reciben la misma energía cinética durante la aceleración instantánea (3000 eV), pero debido a que 22 Espectrometría de Masas tienen diferentes valores de m/z, se separan en grupos de acuerdo a su velocidad (por lo tanto m/z) como van recorriendo la región libre de campo entre la fuente de iones y el detector. Los iones chocan secuencialmente en el detector en forma de un incremento de m/z. Los iones de baja m/z llegan al detector antes que aquellos con m/z alta debido a que entre mas m/z tengan los iones tendrán una velocidad menor (Figura 27). Figura 27. Analizador TOF La energía cinética de un ion se define por: T = eV = mv 2 2 La velocidad del ion: v = 2zeV m 1/2 Debido a que es impráctico medir directamente la velocidad del ion, el tiempo de vuelo (time-of-flight tof) desde la fuente de iones hasta el detector (separados por una distancia L) provee la información necesaria: [ ] tof = L = m v 2zeV _ El valor de m/z para un ion es determinado por su tof al detector calibrado con los tof de al menos dos iones de m/z conocida. El tiempo de tránsito de un ion de 3000 eV de 800 m/z es aproximadamente 70 mseg a través de un tubo de 1m de vuelo, el ciclo de producción de iones, aceleración y detección es completado en un periodo de aproximadamente 100mseg para un rango mayor de 1000 m/z. Los iones que parten de la fuente de iones en un espectrómetro de masas TOF no tienen los mismos tiempos de partida ni exactamente la misma energía cinética, por lo que estos aparatos han sido diseñados para compensar estas diferencias. Un reflectron es un aparato óptico en el cual los iones en un espectrómetro TOF pasan a través de un espejo o reflectron y su vuelo es invertido (Figura 28). Un campo de reflección linear permite a los iones con mayores energías cinéticas penetrar más profundamente dentro del reflectron en comparación con iones de energías cinéticas menores. Los iones que estén más dentro del reflectron tardarán más en llegar al detector. Si un paquete de iones de una m/z dada contiene iones con diferentes energías cinéticas, el reflectron disminuirá la dispersión de los tiempos de vuelo de los iones, mejorando la resolución del espectrómetro. Figura 28. Reflectron El analizador TOF es el analizador de masas más rápido, puede adaptarse a los métodos de ionización por pulsos (MALDI), tiene alta transmisión de iones y el rango de masas más grande de todos los analizadores de masas. Algunas limitantes son que en muchas ocasiones requieren exclusivamente de un método de ionización por pulsos y que la selectividad de los iones puede limitarse en algunos experimentos. Casi todos los sistemas 23 Espectrometría de Masas MALDI están en conjunto con un analizador de TOF, los sistemas GC/MS con TOF poseen una alta velocidad de análisis. Analizador de trampa de iones (Ion-Trap) El espectrómetro de masas de trampa de iones cuadrupolo (quadrupole ion-trap), así como el filtro cuadrupolo, utilizan campos eléctricos oscilantes para atrapar los iones en forma controlada. En la trampa de iones cuadrupolo, los iones son atrapados en un espacio tridimensional, mientras que en el filtro de masas cuadrupolo solo es en un espacio bidimensional. La trampa de iones cuadrupolo atrapa los iones en un pequeño volumen entre tres electrodos, uno circular (ring electrode) y dos electrodos hiperbólicos en los extremos (end-cap electrodes), por medio de campos eléctricos oscilantes (Figura 29). Figura 29. Analizador de trampa de iones (ion-trap) La trampa de iones es operada al aplicar un potencial sinusoidal (rf a una frecuencia determinada) al electrodo circular, mientras que los de los extremos se mantienen constantes (frecuentemente en cero), o mantenidos a un potencial oscilante. La variedad de potenciales posibles que pueden aplicarse a los electrodos de los extremos permite atrapar los iones dentro de un rango m/z específico, es decir atrapar iones por encima de un valor m/z específico, atrapar iones solo de un valor m/z seleccionado, o expulsar iones de un valor m/z específico. Como fue originalmente concebido, la trampa de iones puede ser ajustada para almacenar iones de un valor m/z dado. Al cambiar las condiciones de operación, iones de un valor m/z especificado podrán salir o ser expulsados de la trampa. Stafford y colaboradores establecieron el modo de operación de la trampa de iones, en el cual los iones los iones que tienen un valor m/z por encima de un valor predeterminado son almacenados, pero eventualmente son expulsados de acuerdo a valores secuenciales de m/z al cambiar los parámetros de operación para permitir un examen analítico. El movimiento dentro de la trampa de iones, al igual que en el filtro cuadrupolo, puede ser descrito por las ecuaciones de Mathieu. Debido a que potenciales dc pueden ser aplicados a la trampa, puedes usarse diagramas de estabilidad basados en a y q para indicar cuales iones de acuerdo a su valor m/z permanecen estables dentro de la trampa de iones y cuales serán expulsados bajo unas condiciones específicas. Las condiciones para estabilidad de iones dentro de la trampa de iones pueden ser graficadas en función de a-q de Mathieu. Los valores de a y q de la ecuación de Mathieu dependen de las dimensiones de la trampa de acuerdo a las ecuaciones: az = -2ar = -16 eU 2 2 mr0 w RF . qz = -2qr = -8 eV 2 2 mr0 w RF . Los subíndices z y r representan el movimiento axial y radial entre los electrodos de los extremos, U es el potencial dc aplicado a cualquiera de los electrodos de los extremos, V es potencial rf aplicado al electrodo 24 Espectrometría de Masas 2 central, r0 es el radio del electrodo central, y w RF es la frecuencia angular. Iones cuyos valores a y q caen dentro de la gráfica tendrán trayectorias estables en la trampa de iones (Figura 30). Figura 30. Diagrama de estabilidad a-q Durante la operación del espectrómetro de trampa de iones, las oscilaciones de los iones atrapados son -3 reducidas por colisiones con helio a alta presión (10 torr). Durante el proceso de detección, los potenciales de los electrodos son alterados para producir una rampa de amplitud de radio frecuencia en las trayectorias de los iones y así expulsar los iones en dirección axial. Los iones son expulsados en orden de decrementos de m/z, enfocados a la salida de la trampa y detectados por el sistema de detección de iones. Algunos investigadores han demostrado gran resolución y rango de masas en sistemas de trampas de iones diseñadas especialmente. Sin embargo, estas características no son aplicables a todos los sistemas de trampa de iones disponibles comercialmente. Este sistema posee alta sensibilidad además de ser equipos relativamente compactos, pero ofrecen poca capacidad en análisis cuantitativos, pueden sufrir de efectos por cargas y reacciones de los iones, muchos parámetros influyen la calidad del espectro obtenido por este método (excitación, atrapado y detección de los iones) por lo que debe contarse con sistemas de control sumamente precisos. Puede acoplarse a sistemas cromatográficos. Analizador FT-ICR (Fourier Transform Ion Cyclotron Resonance) El analizador FT-ICR (Fourier-Transform Ion Cyclotron Resonance) es probablemente el método más complejo de análisis de masas. Esta técnica fue desarrollada en los 50´s, es altamente sensible, pero tiene limitaciones de resolución de masas (>106 Da). Este tipo de espectrómetros consisten principalmente de tres secciones, la primera es la fuente de iones (ESI y MALDI son las más comunes), después la región de transferencia de iones donde los iones producidos son captados, enfocados y transferidos a una región de alto vacío antes de entrar al analizador, y finalmente el analizador. El arreglo estándar para el analizador FT-ICR, es una trampa de iones localizada dentro de un campo magnético especialmente uniforma de una fuerza B0. El efecto del campo magnético es obligar a los iones incidentes a circular en una órbita (Figura 31), la frecuencia es determinada por la masa (m), la carga (z) y velocidad (v) del ion, por acción de la ley de Lorentz, ecuación. El ion es llevado a una trayectoria circular en un plano perpendicular al campo magnético. La frecuencia angular de los iones (wc) se define por la ecuación 2. El sentido contrario de rotación es experimentado por iones de carga opuesta. Ecuación 1. Ecuación 2. F = zivB0 wc = ziB0 2pmi Ecuación 3. mi = B0 . 2zipwc 25 Espectrometría de Masas Donde, F = fuerza de lorentz B0 = campo magnético mi = masa del ion zi = carga del ion v = velocidad incidente del ion wc = frecuencia rotacional inducida (ciclotrón) Figura 31. Analizador FT-ICR La presencia de iones entre el par de electrodos (en trampa) no producirá ninguna señal medible, por lo que es necesario excitar los iones de una m/z dada como un paquete a un radio orbital mayor al aplicar barrido de potencial rf de milisegundos. Una frecuencia excitará una m/z particular (la transformación de Fourier permite medir simultáneamente todas las frecuencias). La medición de la frecuencia angular (wc) lleva a valores de m/z y al espectro de masas. Debido a que la frecuencia puede ser medida más exactamente que otras propiedades físicas, esta técnica de análisis tiene una gran resolución. Después de la excitación, los iones regresan a su estado normal. Este tipo de analizadores poseen la resolución de masas mas alta, alta capacidad, y acoplamiento a métodos de ionización por pulsos (MALDI), es un método no destructivo, pero posee un rango de masa limitado, puede sufrir de efectos por cargas y por reacciones entre los iones, se deben cuidar muchos parámetros para tener buenas mediciones. Se acopla bien a MALDI y ESI. 26 Espectrometría de Masas DETECTORES DE IONES Sensibilidad, precisión, y tiempo de respuesta son criterios importantes que distinguen los diferentes sistemas de detección de iones. Alta precisión y rápido tiempo de respuesta son características mutuamente excluyentes. Idealmente, la sensibilidad debe definir explícitamente la corriente de ion recibida por el detector. La definición debe incluir: (1) el nombre del compuesto de calibración, (2) el valor m/z del ion medido, (3) el poder de resolución del instrumento, (4) la cantidad de compuesto de calibración consumido por minuto, (5) la intensidad de la corriente de electrones y presión de la fuente de iones (para CI), (6) la corriente del ion que llego al detector. Estas unidades de sensibilidad permiten comparar instrumentos, que en ocasiones es difícil ya que las diferentes compañías usan diferentes criterios. Copa Faraday Este es un detector eléctrico convencional, iones positivos que impactan sobre el colector son neutralizados por electrones después de pasar a través de una resistencia. La caída de voltaje en la resistencia de acuerdo a un estándar es la medición de la corriente del ion. La señal del voltaje de la resistencia es entonces amplificada por un amplificador DC o por unn electrómetro. El pequeño electrodo de metal que intercepta los iones positivos esta montado en una jaula de Faraday (Figura 32). La corriente del ion medida y amplificada es directamente proporcional al número de iones y al número de cargas de los iones. La respuesta de la copa faraday es independiente de la energía, la masa, o la naturaleza química de los iones. Este tipo de detector es simple, barato, resistente y fiable. Posee alta precisión, sensibilidad y produce poco ruido de salida. La principal desventaja es que tiene un gran retraso inherente a la simplificación del sistema. Por esto es que se utiliza para mediciones precisas o de corrientes de iones poco cargados y no debe ser usado para realizar examinaciones rápidas (por ejemplo en sistemas GC/MS). Figura 32. Copa Faraday Multiplicador de Electrones (Electron Multiplier) El multiplicador de electrones (electron multiplier) utiliza el principio de emisión secundaria de electrones para afectar la amplificación. En el dinodo discreto (discrete-dynode) los iones provenientes del analizador de masas son enfocados sobre un dinodo que emite electrones en proporción directa al número de iones bombardeados hacia él. Los electrones secundarios del dinodo (iones en electrones) son acelerados y enfocados hacia un segundo dinodo, el cual también emite electrones secundarios (electrones en electrones). De este modo, la amplificación es llevada a cabo a través de un “efecto cascada” de electrones secundarios de dinodo a dinodo, ya que el número de electrones que llegan a un dinodo es menos al que sale del mismo (Figura 33). Cada etapa es un dinodo de cobre-berilio conectado a un potencial sucesivamente mayor por un divisor de voltaje. 7 Pueden llegar a tener de 10 a 20 etapas amplificando la señal en un orden de hasta 10 . 27 Espectrometría de Masas Figura 33. Multiplicador de electrones de dinodo discreto (discrete-dynodo) Este tipo de detectores son de amplio uso, ya que además de ser sistemas confiables, baratos y que pueden captar casi todos los electrones poseen sistemas electrónicos y mecánicos simples, aunque pueden no ser tan sensibles (al no tener amplificación de señal) y un tiempo de respuesta lento debido a alta impedancia en el amplificador. Un diseño alternativo de multiplicador de electrones utiliza un dinodo continuo en forma de cuerno hecho de vidrio cubierto con plomo. Este vidrio cubierto es eléctricamente resistente y provee un gradiente eléctrico que esta conectado a una fuente de poder. Hacia el final del cuerno se ajusta el ángulo para poder captar todos los iones y convertirlos en electrones secundarios. Estos electrones secundarios son atraídos a lo largo de un gradiente eléctrico positivo en el cuerno, estos electrones chocan con la pared y más electrones secundarios son expulsados, provocando amplificación (Figura 34). La forma de dinodo continuo (frecuentemente llamado channeltron) es curva para prevenir que iones positivos causen señales de ruido debido a ionización secundaria de moléculas residuales de gas. Figura 34. Principio general y entrada de muestra de un dinodo continuo (channeltron) 6 La rápida respuesta y alta sensibilidad, con una ganancia de 10 hacen que el detector multiplicador de electrones sea indispensable en sistemas como el GC/MS. Detector Daly Una corriente de iones positivos pasan por la hendidura del detector y son atraídos hacia un cátodo de aluminio (el botón Daly) teniendo un gran potencial negativo (-15000 V). Ocurre el impacto con el botón Daly, el cual esencialmente sirve como un dinodo de conversion que produce más de ocho veces electrones secundarios que son atraídos hacia un aparato centelleante, los electrones secundarios generan fotones que son amplificados en un fotomultiplicador (Figura 35). Figura 35. Detector Daly 28 Espectrometría de Masas El detector Daly tiene la ventaja de que no opera bajo vacío, puede detectar iones de masas grandes, y puede detectar iones estables y metaestables, pero solo es capaz de detectar iones de alguna m/z específica ya es un detector de canal simple. Detector de Microcanales Un arreglo de capilares de vidrio huecos (de 10-25mm de diámetro), unidos a un material emisor de electrones, formando un microcanales. Manteniendo estos microcanales a un alto voltaje permite a los iones golpear el material y expulsar electrones, esto causa una avalancha, creando electrones secundarios con una ganancia de 3 4 10 -10 (Figura 36). Estos detectores son útiles en señales de baja intensidad. Figura 36. Principio del detector de microcanales Los microcanales pueden también emitir electrones a través de pantallas fosforescentes. Un detector de arreglo de fotodiodo es usado para convertir los fotones liberados en señales eléctricas (Figura). Figura 37. Detector de arreglo de fotodiodo. 29 Espectrometría de Masas ADQUISICIÓN Y PROCESAMIENTO DE DATOS En los 60´s y 70´s el oscilógrafo de rayo de luz fue el primer aparato de registro con suficiente respuesta de alta frecuencia para registrar en forma precisa los espectros de masas. Hasta el desarrollo de la minicomputadora fue posible hacer procesamiento de datos al tener algoritmos para su proceso e interpretación. Figura 37. Componentes esenciales de un sistema de adquisición de datos La salida análoga del detector es típicamente una señal de voltaje que varía en función del tiempo, esta es convertida a una señal digital por medio de un convertidor análogo-digital. Los datos digitales son procesados y almacenados en el CPU de una computadora. El sistema de datos almacena solo la información de los picos del espectro de masas (valor de m/z y la intensidad) para cada análisis. Estos datos son entonces procesados para producir diferentes representaciones al interpretar y procesar los datos por medio de la computadora. Los datos procesados son entonces desplegados ya sea en el monitor o impresos en formato de gráfica de barras. Figura 38. Diagrama de procesamiento de datos. ARRIBA: Señal analoga. EN MEDIO: Señal procesada. ABAJO: Datos procesados mediante software y produciendo el espectro de masas. La calibración del espectrómetro de masas, en el eje de masa es llevado a cabo al introducir una muestra de un compuesto cuyo espectro de masas es bien conocido. Dependiendo del tipo de algoritmo utilizado, el operador puede ingresar los valores conocidos de m/z a los picos en el espectro de masas del compuesto de calibración, la computadora realizará los ajustes necesarios en la escala de masa. En otros casos, el operador puede ajustar los parámetros de la escala de masa hasta que en la computadora se indiquen los valores apropiados de m/z de los picos conocidos en el espectro de masas del compuesto de calibración. 30 Espectrometría de Masas Procesamiento de datos Debido a las grandes cantidades de información que son generadas y la gran cantidad de parámetros que cambian en un periodo de tiempo, las computadoras son una herramienta esencial en espectrometría de masas, tanto para control del equipo, adquisición, almacenamiento y presentación de datos. Los sistemas de procesamiento de datos generalmente incluyen programas útiles en cuantificación, interpretación de los espectros de masas y comparación de compuestos a través de consulta de bases de datos vía internet. La estrategia es utilizar un sistema de procesamiento datos que permita adquirir datos constantemente, y obtener los datos tan rápido como sea posible teniendo una buena relación señal-ruido sin abusar de los principios de operación del espectrómetro de masas. Esto usualmente es completado en un ciclo de examen del orden de 1 segundo, adquiriendo un nuevo espectro de masas cada segundo, esto tiene la ventaja de no perder datos importantes pero tiene la desventaja de que se deben adquirir grandes cantidades de datos. El análisis será realizado con el problema de tener que determinar cuales datos son útiles y necesarios para tener el resultado final. Los sistemas de procesamiento de datos emplean procedimientos de búsqueda hacia adelante o de búsqueda reversa. En la búsqueda hacia adelante, un espectro no definido es comparado con cada miembro de una biblioteca de espectros de referencia. Un resultado siempre será alcanzado, al calificar son cierta clase de parecido respecto a las referencias, esto tiene la ventaja de sugerir el tipo de compuestos presentes en la muestra si el grado de parecido no es aceptable o suficiente. En la búsqueda reversa, un banco de datos es comparado con el espectro no definido para determinar si algunos picos de referencia están presentes en el espectro analizado. Un resultado puede lograrse si el espectro no definido tiene algunos picos “extras” que pueden ser comparables a los espectros de referencia. No se podrá identificar un compuesto si no se tiene un espectro de referencia que corresponda en la base de datos. Las computadoras pueden realizar búsquedas en bases de datos de espectros de masas, para ello se basan en algoritmos de probabilidad, llamados algoritmos PBM (probability-based-matching) y variantes del algoritmo INCOS, el cual busca parecido entre los picos más intensos del espectro (utiliza los 16 picos más intensos) contra una base de datos. 31 Espectrometría de Masas APLICACIONES Análisis de estructura y masa En el análisis de estructura por medio de espectrometría de masas de la acetona (C3H6O) se observan diferentes fragmentos de iones y el ion molecular en m/z=58 (Figura 39). Figura 39. Espectro de masas de la acetona (C3H6O) Los iones más intensos han sido marcados con su m/z. Los fragmentos de ion son usados por los espectrometristas de masas para deducir las estructuras moleculares. Algunas veces los símbolos [+·] y [-·] son usados para indicar los iones radicales, esta información es útil para determinar los patrones de fragmentación. Se observa que la pérdida de 15 Da del ion molecular de la acetona da un ion de m/z=43 indicando la presencia de un grupo metil (CH3) en la molécula original, la pérdida de 28 Da da un ion con m/z= 15 sugiriendo la presencia de CO. Al analizar estas pérdidas y organizando las posibles estructuras a partir de los iones obtenidos, se puede deducir la estructura del compuesto original. Algunas pérdidas comúnmente observadas son de agua (H2O 18 Da), amonio (NH3 17 Da) y grupo fenil (C6H5 77 Da). Otra aplicación de la espectrometría de masas es la determinación de masas. Cada isótopo de cada elemento (excepto para carbono al cual se le asigno un valor de 12.00000 Da) tiene una masa no entera única. La determinación de mediciones de masa permite determinar composición química. En el espectro de masas (Figura 40) es posible distinguir entre monóxido de carbono (CO, m/z=27.995) y nitrógeno (N2 m/z=28.006) por mediciones exactas de sus masas Figura 40. Espectro de masas CO y N2. 32 Espectrometría de Masas Sistemas Acoplados La espectrometría de masas es un sistema de detección útil en técnicas de separación tales como cromatografía de gases (G C), cromatografía líquida (LC), electroforesis capilar y otras, debido a que es altamente sensible y es capaz de identificar compuestos químicos en forma precisa. El reto al acoplar el espectrómetro de masas a un sistema de separación es el mantener el vacío necesario al introducir la muestra desde el cromatógrafo. Interfaces que restringen o reducen el flujo de gas de entrada al espectrómetro permiten acoplar el cromatógrafo de masas y el espectrómetro de masas (GC/MS) (Figura 41). Figura 41. Sistema GC/MS Al vaporizar el solvente de una cromatografía líquida este representaría un volumen de 100-100 veces más grande que el gas usado en cromatografía de gases. Se han desarrollado interfaces que resuelven este problema al eliminar este exceso de gas usando calor y vacío, en algunos casos con ayuda de una corriente de gas que ayuda a secar. Los sistemas acoplados LC/MS utilizan ionización química (CI) o electrospray (ESI) como métodos de ionización. Tabla 7. Compuestos aptos para análisis LC/MS Bajo peso molecular Peso molecular medio Peso molecular alto < 1 kDa 1 -10 kDa > 10 kDa Drogas, compuestos endógenos, vitaminas, pesticidas, toxinas, Polipéptidos sintéticos y Polipéptidos, proteínas, conjugados (glucoronidos, SO4) polisacáridos. oligonucleótidos, polisacáridos. de compuestos con m/z > 50. Figura 42. Sistema LC/MS En GC/MS y LC/MS, los datos obtenidos consisten en una serie de espectros de masas que son adquiridos secuencialmente en tiempo. Para generar esta información, el espectrómetro de masas examina un rango de masas (por ejemplo m/z de 30 a 500) repetitivamente durante la corrida cromatográfica. Si se realiza un examen cada segundo y se hace una corrida de 30 minutos, se obtendrán 1800 espectros. La información obtenida puede ser analizada en diferentes formas. En primer lugar, las intensidades de todos los iones en cada espectro son sumadas, y esta suma graficada como función del tiempo de retención cromatográfico (TIC) cuya apariencia es similar a los datos de salida de un detector cromatográfico convencional (Figura 43 parte superior). Otra forma es desplegar cualquiera de los espectros obtenidos (Figura 43 parte central), cada pico en TIC representa un compuesto eluido que puede ser identificado al analizar su espectro de masas obtenido es ese punto. La intensidad a una m/z específica en el transcurso de la corrida 33 Espectrometría de Masas cromatográfica puede ser desplegada para producir un perfil de corrientes de iones seleccionados o un cromatograma de masas (Figura 43 parte inferior). Esta técnica puede ser usada para encontrar componentes en una mezcla compleja sin tener que examinar cada espectro en forma individual. Figura 43. Análisis de espectros en sistemas acoplados. Acoplando dos etapas de análisis de masas (MS/MS) puede ser muy útil en la identificación de compuestos en mezclas complejas y en la determinación de sustancias no conocidas. El examen de iones producidos es el modo MS/MS mas frecuentemente usado, se generan los espectros de masas de iones producidos de un valor m/z específico. De una mezcla de iones generados o colectados, se seleccionan aquellos iones que tengan un valor m/z particular, esta es la primera parte del análisis. Estos iones son fragmentados y los productos de la fragmentación de estos iones son analizados es una segunda etapa del análisis de masas (Figura). Si la muestra es un compuesto puro y se realiza fragmentación de los iones producidos, los espectros producidos de esta fragmentación proveerán información adicional respecto la estructura del analito. Si la muestra original es una mezcla y se usa ionización suave, la segunda etapa de MS puede usarse para obtener un espectro de masas para cada componente en la mezcla. MS/MS puede también usarse para eliminar interferencias en experimentos SIM donde se producen varias señales de iones de una m/z específica. Análisis cuantitativos Cuando ya se conoce el espectro de masas de un compuesto específico, se puede confirmar la presencia de sustancias específicas o medir la cantidad presente en la muestra. Este tipo de análisis son rutinarios en mediciones de contaminantes ambientales y estudios farmocinéticos donde se requiere cuantificar analitos en concentraciones muy bajas y además en mezclas complejas. El espectrómetro de masas se ajusta para monitorear solo valores específicos de m/z representativos de las moléculas a analizar. El tipo de ionización utilizado puede favorecer la producción de ciertos tipos de iones, incrementando la sensibilidad y manteniendo la señal de ion en su valor de m/z. Figura 44. Sistema MS/MS En la Figura 45, se muestra un espectro en el cual se monitorearon solo los valores de m/z=158 y m/z=160 durante una corrida cromatográfica. Este proceso se conoce como monitoreo selectivo de iones (SIM). 34 Espectrometría de Masas Figura 45. Monitoreo selectivo de iones (SIM) Este espectro es un análisis cuantitativo de un derivado de 5-fluorouracil en plasma, la respuesta de la sustancia de interés es medida relativamente en comparación con un estándar interno añadido a la muestra. En la parte superior de la Figura se muestra el perfil SIM del ion a una m/z=158, el compuesto de interés produce el pico central, dos pequeños picos aparecen y corresponden a compuestos que también producen iones a m/z=158. En la parte inferior de la Figura es el perfil SIM de la misma molécula teniendo el N15 sustituido por N14 normal en dos posiciones, debido a que ambos nitrógenos están sustituidos, la señal de este ion es a m/z=160, 2 Da mas pesado. Aunque la muestra y es estándar tienen el mismo tiempo de retención, son detectados en forma separada ya que tienen masas diferentes. Los estándares pueden ser sustancias altamente relacionadas o químicamente idénticas pero sintetizadas sustituyendo un isótopo de uno de los elementos como en este caso. La medición de isótopos es útil en diferentes áreas tales como estudios metabólicos que utilizan sustancias marcadas isotópicamente, en estudios climáticos al usar isótopos de oxígeno y carbono que son temperatura dependientes, en cálculo de edades de materiales (por ejemplo rocas) al medir plomo, neodimio o estroncio. La relación de isótopos de un elemento puede ser determinada en forma precisa y exacta por medio de espectrometría de masas, pero es necesario reducir los compuestos a moléculas simples antes de la medición, los compuestos orgánicos son reducidos a CO2, H2O y N2. Análisis de péptidos Para realizar análisis de péptidos, una vez obtenida la proteína o extracto de proteínas, se realiza una digestión proteolítica para obtener péptidos, esta mezcla de péptidos son separados y purificados por medio de cromatografía de gases (GC), HPLC u otras técnicas de separación (Figura 45). Los péptidos separados y purificados son sometidos a espectrometría de masas para obtener los espectros de las diferentes fracciones. Se analizan los espectros obtenidos, la relación m/z y la intensidad de los iones obtenidos en los diferentes espectros son usados para identificar los péptidos de las muestras. Figura 45. Análisis de péptidos por medio de espectrometría de masas. 35 Espectrometría de Masas Iones de péptidos Cuando un péptido es fragmentado por colisión con moléculas un de gas neutro, la fragmentación es inducida por transferencia de energía cinética de las moléculas del gas al péptido. Cuando un péptido es fragmentado en un enlace peptídico particular entre carbonilo (-CO-) y nitrógeno (-NH-), se forman dos fragmentos, un fragmento del péptido retiene la carga positiva en el C terminal del péptido ionizado y es llamado ión-y, el otro fragmento retiene la carga positiva en el N terminal y es llamado ión-b. Un ión-b generalmente tiene un ión-y complementario. Cuando un péptido que tiene una sola carga es fragmentado, la carga se mantiene solo en un fragmento y solo aquel que tenga la carga podrá ser detectado mientras que el otro permanecerá como fragmento neutro no detectable. Péptidos doblemente cargados producirán dos iones cargados. Figura 46. Iones formados durante fragmentación de péptidos. Durante la fragmentación de péptidos pueden formarse otros tipos de iones además de los iones-y y los ionesb. Los iones-a y los iones-x, los iones-c y los iones-z, siendo complementarios entre a-x y c-z. Los iones a-x se forman por rompimiento entre el péptido y el carbonilo en el extremo N terminal. Los iones c-z se forman al haber un rompimiento entre el nitrógeno (-NH-) y el resto del péptido. Los iones a, x, c y z se forman en condiciones de alta energía y los b-y en baja energía. Secuenciación de péptidos La secuenciación de novo de péptidos es llevada a cabo al determinar la secuencia directamente del espectro de masas sin consulta de bases de datos. Las diferencias entre m/z sucesivas se usan para determinar la secuencia aminoacídica del péptido. Se asume que solo ocurre formación de iones b-y durante el proceso de fragmentación del péptido, se forman iones-b ascendentes desde el extremo N terminal mientras que se forman iones-y en forma descendente desde el C terminal. Se determinan las m/z de los fragmentos aminoacídicos. Al comparar las diferencias entre masas sucesivas de cada serie y comparándolos con las masas conocidas de residuos de aminoácidos, se puede determinar la secuencia de un péptido. Tanto los iones de la serie b como de la serie y indican la misma secuencia. Figura 47. Secuenciación de novo de un péptido AVAGCAGAR La precisión de la secuencia deducida es limitada por las características del equipo. La calidad de la reacción de fragmentación, ya que en ocasiones no ocurre el corte y los fragmentos obtenidos diferirán en algún aminoácido que no podrá deducirse, por ejemplo, en presencia de prolina la fragmentación puede no ocurrir y causar errores en la secuencia. 36 Espectrometría de Masas Identificación de proteínas Para identificar proteínas se utilizan las huellas de masas de péptidos (peptide mass fingerprinting) o huellas peptídicas, este método utiliza los péptidos obtenidos de la digestión enzimática de la proteína para obtener sus espectros de masas. Los péptidos son identificados por comparación contra una base de datos, ya que las masas de los péptidos forman en conjunto la huella peptídica de la proteína, es posible identificar la proteína de interés. Factores importantes que deben ser considerados en este procedimiento es la pureza de la proteína y la precisión de los datos obtenidos, la precisión de los datos disponibles en las bases de datos y el tipo de algoritmos usados al comparar las masas de los péptidos (Figura 48). Figura 48. Proceso de identificación de péptidos por medio de huellas peptídicas. El resultado de este análisis puede sufrir algunos problemas al tener presencia de señales provenientes de péptidos que no están presentes en la proteína de interés debido a que la purificación dejo algunas proteínas que contaminaron la muestra o presencia de péptidos contaminantes. Es posible además obtener resultados falsos positivos al comparar los espectros obtenidos contra las bases de datos, o que la proteína de estudio tenga modificaciones postraduccionales desconocidas que podrían alterar el análisis, algunas veces es difícil distinguir entre proteínas que son muy similares o que podrían tener las mismas huellas peptídicas. Se dispone de tres tipos de herramientas para al análisis de huellas peptídicas: 1. Programas que asignan puntuaciones a las masas de los péptidos obtenidos que coinciden con las masas de péptidos de las bases de datos. Por ejemplo: PepSea: http://www.unb.br/cbsp/paginiciais/pepseafingerprint.htm Peptldent: http://us.expasy.org/tools/peptident.html 2. Programas que asignan puntuaciones basados en la longitud del péptido y la proteína. Por ejemplo: MOWSE: http://srs.hgmp.mrc.ac.uk/cgi-bin/mowse MS-Fit: http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm 3. Programas que asignan puntuaciones probabilisticas para determinar la significancia de los resultados obtenidos entre las muestras y las bases de datos. Esto es para evitar resultados no significativos. Por ejemplo: PROWL: http://prowl.rockefeller.edu/ MASCOT: http://www.matrixscience.com/cgi/index.pl?page=/search_form_select.html 37 Espectrometría de Masas HERRAMIENTAS PARA ANÁLISIS DE PROTEÍNAS POR ESPECTROMETRÍA DE MASAS Protein Prospector Colección de herramientas de la UCSF Mass Spectrometric Facility usadas para identificar proteínas usando datos de espectrometría de masas y bases de datos. http://prospector.ucsf.edu/ ExPASy Proteomics Tools Programas del Expert Protein Analysis System (ExPASy) del Swiss Institute of Bioinformatics. Para identificación y caracterización de proteínas, predicción de modificaciones postraduccionales, topología y estructura de proteínas. http://us.expasy.org/tools/ MASCOT Identificación de proteínas basado en bases de datos de pesos de péptidos, usa métodos probabilisticos para calcular significancia entre datos. http://www.matrixscience.com/cgi/index.pl?page=/search_form_select.html PeptideSearch Identificación de proteínas en base a sus espectros de masas. http://www.mann.embl-heidelberg.de/GroupPages/PageLink/peptidesearchpage.html MOWSE MOWSE (MOlecular Weight Search). Utiliza pesos moleculares obtenidos por digestión de proteínas con una proteasa conocida y busca en bases de datos de péptidos para identificar proteínas, de 3 a 4 péptidos son necesarios para identificar una proteínas entre una base de datos de 50000 proteínas. http://srs.hgmp.mrc.ac.uk/cgi-bin/mowse Mass Spec Tools Herramientas de Scientific Information Services usadas para generar y graficar espectros de masas y calcular distribución de isótopos. http://www.chemsw.com/13057.htm 38 Espectrometría de Masas DEFINICIÓN DE TÉRMINOS Relación masa/carga Esta expresión, abreviada m/z, es la relación del número de masa (m) de una partícula dada entre el número (z) de unidades cargadas electrostáticamente (e) que posee la partícula. Así, m/z es una relación adimensional que es el parámetro medido por el analizador de masas. La masa de una partícula dada es igual a la suma de sus masas atómicas (en Daltons) de todos los elementos que componen la partícula. El símbolo para las unidad 12 de masa es u, y corresponde a 1/12 de la masa del C, al que se la ha asignado un valor de 12.000000 por convención. Iones doblemente cargados Es posible para una molécula perder dos electrones durante el proceso de ionización. Estos iones que están doblemente cargados producirán un pico en el espectro de masas en un valor m/z numéricamente igual a la mitad de la masa molecular del ion. Los iones doblemente cargados son insignificantes en el espectro de masa para la mayoría de los compuestos. Sin embargo, para aquellas moléculas que los produzcan en forma estable, los picos correspondientes en el espectro de masas pueden ser usados en la interpretación de datos. Ion molecular El ion molecular resulta de la ionización de la molécula a analizar. Este ion representa la molécula intacta y es el último precursor de todos los iones fragmentados que componen el espectro de masas. El pico del ion molecular aparece a un valor m/z numéricamente igual al peso molecular del compuesto. Pico base Es el pico más intenso en el espectro de masas. Es usado como base para normalizar las intensidades de los otros picos. Al pico base se le asigna una intensidad relativa de 100%. Intensidad relativa La intensidad relativa de un pico representa su intensidad en comparación con el pico base. Se representa como % respecto al pico base que es 100%. Por convención, este dato se indica en la izquierda del espectro de masas. Porcentaje total de ionización Este término expresa la abundancia de un ion individual comparado con la suma de las abundancias de todos los iones en un rango de masa especifico. La escala de porcentaje total de ionización esta representada a la derecha del espectro de masas con el símbolo (%Sn), este dato puede ser utilizado para comparar diferentes espectros de masas de diferentes compuestos. Espectro de masas Un espectro de masas es una gráfica de intensidad relativa del ion como función de la relación masa/carga (m/z). El espectro de masas es frecuentemente representado como un histograma simple. Esta forma de registro de iones y sus intensidades sirven para establecer el peso molecular y estructura del compuesto a ser analizado. Debido a que ocurre la fragmentación del analito, las fracciones del ion aparecen en el espectro a valores m/z menores que la molécula completa ionizada (ion molecular) a partir de es tos datos se deduce la estructura y peso molecular de la molécula completa. 39 Espectrometría de Masas REFERENCIAS Bafna V and Edwards N. SCOPE: A probabilistic model for scoring tandem mass spectra against a peptide database. Bioinformatics. 2001;17 Suppl 1:S13-21 Clauser KR, Baker P, Burlingame AL. Role of Accurate Mass Measurement (±10 ppm) in Protein Identification Strategies Employing MS or MS/MS and Database Searching. Anal Chem 1999 Jul 15;71(14):2871-82. Coligan (Editor), Ben M. Dunn (Editor), Hidde L. Ploegh (Editor), David W. Speicher (Editor), Paul T. Wingfield (Editor). Current Protocols in Protein Science. June 1995 Wiley Eng J, McCormack A and Yates J. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society of Mass Spectrometry, 1994, 5, 976-989 Kinter, M.; Sherman, N. E.. Protein Sequencing and Identification Using Tandem Mass Spectrometry. Wiley Interscience. 2000. New York, USA. Pappin DJC, Hojrup P, Bleasby AJ. Rapid Identification of proteins by peptide-mass fingerprinting. Current Biology (1993), vol 3, 327-332 Perkins, DN, Pappin, DJ, Creasy, DM and Cottrell, JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999; 20(18):3551-3567. Pevzner PA, Mulyukov Z, Dancik V, Tang CL. Efficiency of database search for identification of mutated and modified proteins via mass spectrometry. Genome Res 2001 Feb;11(2):290-9 Watson, J.T.. Introduction to Mass Spectrometry. Lippincott-Raven Publishers. Third Edition. 1997. Philadelphia, PA, USA. Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999;112:531-52. 40