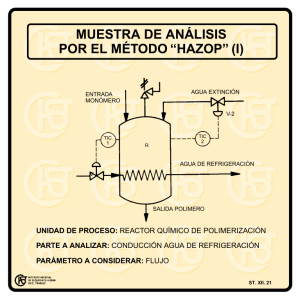
Sintesis de Polimeros
Anuncio

Asignatura: Materiales Poliméricos y Compuestos. Tema 2.- Síntesis de Polímeros. Técnicas de Polimerización. ÍNDICE 1.- Introducción. 2.- Polimerización por adición. 2.1.- Introducción 2.2.- Polimerización por radicales libres o radicalaria. 2.2.1.- Introducción. Proceso de polimerización. 2.2.2.- Cinética de la polimerización por radicales libres. 2.2.3.- Efecto Norris – Trommsdorff 2.2.4.- Características de la polimerización por radicales libres. 2.3.- Polimerización iónica 2.3.1.- Introducción. 2.3.2.- Polimerización catiónica. 2.3.3.- Polimerización aniónica. 2.4.- Polimerización con estereoquímica controlada. 2.3.2.- Polimerización de Ziegler-Natta 2.3.3.- Polimerización catalizada con metalocenos 3.- Polimerización por condensación o escalonada (por etapas). 3.1.- Introducción. 3.2.- Cinética de la polimerización por etapas. Poliesterificación. 3.3.- Polimerizaciones escalonadas con reticulación 3.4.- Estequiometría y control del peso molecular 3.5.- Práctica de las polimerizaciones escalonadas. 3.6.- Síntesis de poliesteres 3.7.- Formación de poliamida. 3.6.1.- Síntesis Nylon 6.6 3.6.2.- Síntesis de Nylon-6 3.8.- Síntesis de poliuretanos 3.9.- Síntesis de policarbonatos. 3.10.- Síntesis de resinas epoxy. 3.11.- Síntesis de siliconas. 3.12.- Polimerización metatésica de olefinas. 4.- Copolimerización. 4.1.-Introducción. 4.2.- Ecuación de composición del copolímero. 4.3.- Composición instantánea de monómeros y de copolímero 4.4.- Cinética de copolimerización. 5.- Síntesis de caucho 6.- Técnicas de polimerización. 6.1.- Introducción. 6.2.- Polimerización en masa o en bloque. 6.3.- Polimerización en disolución. 6.4.- Polimerización en suspensión en fase acuosa. 6.5.- Polimerización en emulsión. 1.- Introducción. La polimerización es el proceso químico (Reacción química) por el cual, mediante el calor, la luz o un catalizador, se unen varias moléculas de un compuesto para formar una cadena de múltiples eslabones de estas y obtener una macromolécula (Polímero). Existen muchas de estas reacciones y son de distintas clases. Pero todas las polimerizaciones tienen un detalle en común: comienzan con moléculas pequeñas, que luego se van uniendo entre sí para formar moléculas gigantes. Llamamos monómeros a esas moléculas pequeñas. Así, los procesos de polimerización persiguen la obtención de estructuras de alto peso molecular partiendo de materiales de bajo peso molecular. El proceso de polimerización puede entenderse como un proceso de ensamblaje de unidades monoméricas que se repiten para formar estructuras de mayores dimensiones. Los procesos de polimerización permiten obtener estructuras poliméricas de tipo lineal o reticular: Las reacciones de polimerización se pueden clasificar del modo siguiente: Sistema de Adición-Condensación. En este sistema las reacciones de polimerización se pueden clasificar según la forma en que se lleva a cabo el proceso de polimerización y la naturaleza de las reacciones que tienen lugar. Así, este sistema divide las polimerizaciones en dos tipos que son: (i).- Polimerización por Adición (Poliadiciones) Formación de cadenas a través de la adición de monómeros activados. Da lugar a polímeros lineales. (ii).- Polimerización por Condensación (Policondensaciones) Formación de cadenas por condensación mediante reacciones de grupos funcionales. Dos compuestos orgánicos reaccionan químicamente para formar uno de mayor peso molecular. Puede dar lugar a polímeros lineales cuando la funcionalidad es 2 (f = 2) o polímeros reticulares cuando la funcionalidad es superior a 2 (f > 2). Decimos que una polimerización es por adición, si la molécula entera de monómero pasa a formar parte del polímero, por lo que no se forman subproductos (Figura 1.1). Figura 1.1.- Polimerización por adición. En la tabla 1.1 se muestran algunos ejemplos de polímeros típicos que se obtienen por polimerización de adición. Tabla 1.1.- Algunos ejemplos de polímeros típicos que se obtienen por polimerización de adición. Por otro lado, llamamos polimerización por condensación, si parte de la molécula de monómero se pierde cuando el monómero pasa a formar parte del polímero. Se unen dos tipos diferentes de grupos funcionales con eliminación de una pequeña molécula estable. Aparecen subproductos de la reacción en forma de moléculas sencillas (H2O, NH3, HCl, CH3OH, …) que deben de eliminarse del medio de reacción para que las reacciones de desarrollen de forma adecuada (Figura 1.2). Los procesos de policondensación, de forma general, requieren dos componentes para llevar a cabo la reacción, pero en algunos casos es posible formar una estructura de condensación a partir de un solo componente (autocondensación), como es el caso de la Poliamida 6. Figura 1.2.- Polimerización por condensación (Policondensación). Formación del fenol - formaldehido Figura 1.2.- Polimerización por condensación (Policondensación). Formación del monómero (PET) Los polímeros resultantes de la polimerización por condensación se denominaron policondensados (por ejemplo, poliésteres, poliamidas, resinas fenólicas, etc.) y los de la polimerización por adición poliaductos (por ejemplo, polietilenos, polibutadieno, poliuretanos, etc.). En base a esta clasificación se estudiaron las cinéticas de las reacciones y los mecanismos de crecimiento de las moléculas a partir de los monómeros. En la tabla 1.2 se muestran algunos ejemplos de polímeros típicos que se obtienen por polimerización de condensación. Tabla 1.2.- Algunos ejemplos de polímeros típicos que se obtienen por polimerización de condensación. Cuando se polimeriza el etileno para obtener polietileno, cada átomo de la molécula de etileno se transforma en parte del polímero. El monómero es adicionado al polímero en su totalidad. Podría decirse que un polímero de adición acepta todo. Pero en un polímero de condensación, algunos átomos del monómero no pasan a formar parte del polímero. En la obtención del nylon 6,6 a partir de cloruro de adipoilo y hexametilén diamina, cada átomo de cloro del cloruro de adipoilo juntamente con uno de los átomos de hidrógeno de la amina, son expulsados como HCl gaseoso. Debido a que ahora hay menos masa en el polímero que en los monómeros originales, decimos que el polímero está condensado con respecto a los monómeros. El subproducto, ya sea HCl gaseoso, agua o lo que fuere, se denomina condensado. Sistema Crecimiento de Cadena-Crecimiento en Etapas. Este sistema de clasificación de las polimerizaciones divide las reacciones de polimerización en dos categorías, que son: (i).- Polimerizaciones por crecimiento de cadena Un iniciador reacciona con una molécula del monómero para dar un producto intermedio que vuelve a reaccionar sucesivamente con moléculas del monómero para dar nuevos productos intermedios. Las cadenas crecen (no se unen) (ii).- Polimerizaciones por crecimiento en etapas Las unidades del monómero tienen grupos funcionales que pueden reaccionar entre sí, las reacciones son más lentas y el crecimiento es a saltos en lugar de unidad a unidad. En la polimerización por crecimiento en etapas (o pasos) es posible que un oligómero reaccione con otros, por ejemplo un dímero con un trímero, un tetrámero con un dímero, etc., de forma que la cadena se incrementa en más de un monómero. En la polimerización por crecimiento en etapas, las cadenas en crecimiento pueden reaccionar entre sí para formar cadenas aún más largas. Esto es aplicable a cadenas de todos los tamaños. En una polimerización por crecimiento de cadena sólo los monómeros pueden reaccionar con cadenas en crecimiento. Las diferencias entre polimerización por crecimiento de cadena y polimerización por crecimiento en etapas son un poco más complicadas que las diferencias entre polimerización por adición y polimerización por condensación. En una polimerización por crecimiento de cadena, los monómeros pasan a formar parte del polímero de uno en uno cada vez. Primero se forman dímeros, después trímeros, a continuación tetrámeros, etc. La cadena se incrementa monómero a monómero. En la figura 1.3 se detalla una polimerización por crecimiento de cadena, que corresponde a la polimerización aniónica del estireno, para obtener poliestireno. Figura 1.3.- Polimerización por crecimiento de cadena. Pero en una polimerización por crecimiento en etapas, las cosas son más complicadas. Veamos un ejemplo de reacción entre dos monómeros, el cloruro de tereftoilo y el etilenglicol, para formar un poliéster llamado poli(etilen tereftalato). Lo primero que sucede, es la reacción de los dos monómeros para formar un dímero. En este punto de un sistema de crecimiento de cadena, sólo podría suceder una cosa: que se adicione un tercer monómero al dímero para dar lugar a un trímero, luego un cuarto para formar un tetrámero y así sucesivamente. Pero en la polimerización por crecimiento en etapas, ese dímero puede hacer un montón de cosas diferentes. Obviamente, puede reaccionar con uno de los monómeros para formar un trímero: o bien puede reaccionar con una molécula de etilenglicol: Pero también puede hacer otras cosas, como es reaccionar con otro dímero para formar un tetrámero: o puede reaccionar con un trímero para formar un pentámero. Haciendo las cosas aún más complicadas, esos tetrámeros y pentámeros pueden reaccionar para formar oligómeros aún más grandes. Y así crecer y crecer hasta que los oligómeros sean lo suficientemente grandes como para transformarse en polímeros. En conclusión, la principal diferencia es ésta: en una reacción por crecimiento en etapas, las cadenas en crecimiento pueden reaccionar entre sí para formar cadenas aún más largas. Esto es aplicable a cadenas de todos los tamaños. El monómero o dímero puede reaccionar del mismo modo que una cadena de cientos de unidades monoméricas. Pero en una polimerización por crecimiento de cadena, sólo los monómeros pueden reaccionar con cadenas en crecimiento. Dos cadenas en crecimiento no pueden unirse como si se tratara de una polimerización por crecimiento en etapas. La polimerización por crecimiento en etapas para formar un poliéster, generó un subproducto: HCl gaseoso. Esto, hará que se trate tanto de una polimerización por condensación como de una polimerización por crecimiento en etapas. Por otro lado, la polimerización por crecimiento de cadena del estireno, no produjo ningún subproducto. Por lo tanto, una polimerización por adición también puede involucrar una polimerización por crecimiento de cadena. Resulta sencillo concluir aquí, que polimerización por crecimiento en etapas y polimerización por condensación son la misma cosa, y por otra parte, que polimerización por crecimiento de cadena y polimerización por adición también son la misma cosa. Dicha conclusión no es verdad, ya que existen polimerizaciones por adición que son polimerizaciones por crecimiento en etapas. Un ejemplo es la polimerización que produce los poliuretanos. También existen polimerizaciones por condensación que son polimerizaciones por crecimiento de cadena. Lo importante es saber que las polimerizaciones pueden producirse por crecimiento en etapas o crecimiento de cadena y que pueden ser por condensación o por adición. La característica decisiva es la presencia en los monómeros, por una parte, de dobles enlaces entre átomos de carbono, que podían ser activados por iniciadores de tipo iónicos o de radicales, reaccionando en cadena y dando lugar a macromoléculas cuyo crecimiento se interrumpe cuando sus centros activos quedan neutralizados, o bien, por otra parte, de varios grupos reactivos (alcohol, amina, isocianato, ácido) que reaccionando entre sí (con separación de una molécula simple, en algunos casos, o por simple adición) dan origen a moléculas cada vez mayores, sin que se llegue a interrumpir la reacción nada más que por ausencia de un reactivo o por dificultad de que los grupos reactivos presentes en la masa reaccionante establezcan contacto mutuo. Los grupos funcionales son estructuras submoleculares, caracterizadas por una conectividad y composición elemental específica que confiere reactividad a la molécula que los contiene. Estas estructuras reemplazan a los átomos de hidrógeno perdidos por las cadenas hidrocarbonadas saturadas. Los grupos alifáticos, o de cadena abierta, suelen ser representados genéricamente por R (radicales alquílicos), mientras que los aromáticos, o derivados del benceno, son representados por Ar (radicales arílicos). Los grupos funcionales confieren una reactividad química específica a las moléculas en las que están presentes. En la tabla 1.1 se dan los grupos funcionales más comunes. Una serie homóloga es un conjunto de compuestos que comparten el mismo grupo funcional y, por ello, poseen propiedades similares. Por ejemplo: la serie homóloga de los alcoholes primarios poseen un grupo OH (hidroxilo) en un carbono terminal o primario. Las series homólogas y grupos funcionales listados a continuación son los más comunes. En la tabla, los símbolos R, R', o similares, pueden referirse a una cadena hidrocarbonada, a un átomo de hidrógeno, o incluso a cualquier conjunto de átomos Tabla 1.1.- Grupos funcionales más comunes Funciones oxigenadas. Presencia de algún enlace carbono-oxígeno: sencillo (C-O) o doble (C=O) Grupo funcional Serie homóloga Fórmula Grupo hidroxilo Alcohol Grupo alcoxi (o ariloxi) Estructura Prefijo Sufijo R-OH hidroxi- -ol Éter R-O-R' -oxi- R-il R'-il éter Aldehído R-C(=O)H oxo- -al -carbaldehído Cetona R-C(=O)-R' oxo- -ona Grupo carboxilo Ácido carboxílico R-COOH carboxi- Ácido -ico Grupo acilo Éster R-COO-R' Grupo carbonilo -iloxicarbonil- R-ato de R'-ilo Ejemplo Tabla 1.1.- Grupos funcionales más comunes (Continuación) Funciones nitrogenadas. Amidas, aminas, nitrocompuestos . Enlaces carbono-nitrógeno: C-N, C=N ó C≡N Grupo Tipo de Fórmula Estructura Prefijo Sufijo funcional compuesto Amina R-NR2 amino- -amina Imina R-NCH2 _ _ Grupos amino y carbonilo Amida R-C(=O)N(-R')R" _ _ Grupo nitro Nitrocompuesto R-NO2 nitro- _ Nitrilo o cianuro R-CN ciano- -nitrilo Isocianuro R-NC alquil isocianuro _ Isocianato R-NCO alquil isocianato _ Isotiocianato R-NCS alquil isotiocianato Azoderivado R-N=N-R' azo- -diazeno Diazoderivado R=N=N diazo- _ Azida R-N3 azido- -azida Sal de diazonio X- R-N+≡N _ ...uro de ...diazonio _ Hidrazina R1R2N-NR3R4 _ -hidrazina _ Hidroxilamina -NOH _ -hidroxilamina Grupo amino Grupo nitrilo Grupo azo Ejemplo Cuando en los monómeros existen dobles enlaces entre átomos de carbono de las moléculas de los monómeros, la polimerización en cadena es siempre por adición, distinguiéndose tres reacciones diferentes, aunque simultáneas: iniciación, propagación y terminación. Cuando en los monómeros existen varios grupos reactivos, la reacción entre los grupos funcionales reactivos de dos moléculas (poliadiciones y policondensaciones) es casi siempre por un mecanismo en etapas con una única reacción, con una única cinética y mecanismo, prácticamente independiente del tamaño de la molécula en que se encuentren. Se suelen denominar impropiamente policondensaciones, pero su designación correcta es la de polimerizaciones escalonadas o en etapas. En la tabla 1.2 se muestran las características diferentes de las polimerizaciones en cadena y en etapas Tabla 1.2 – Diferencias entre las polimerizaciones en cadena y en etapas POLIMERIZACIÓN EN CADENA POLIMERIZACIÓN EN ETAPAS Apenas el monómero y las especies propagantes pueden reaccionar entre si. La polimerización envuelve al mínimo dos PROCESOS CINÉTICOS. La concentración del monómero disminuye gradualmente durante la reacción. La velocidad de reacción aumenta con el tiempo hasta alcanzar un valor máximo, en el que permanece. Polímeros con alto peso molecular se forman desde el inicio de la reacción, y este no se altera con el tiempo. La composición química porcentual del polímero es igual que la del monómero que lo origina. Cualesquiera de las especies moleculares presentes en el sistema pueden reaccionar entre si. La polimerización solo tiene un proceso cinético. El monómero se consume totalmente ya en el comienzo de la reacción, restando menos de 1% al final. La velocidad de reacción es máxima en el comienzo y disminuye con el tiempo. Mucho tiempo de reacción es esencial para obtener un polímero con elevado peso molecular, el cual aumenta durante la reacción. La composición química porcentual del polímero es diferente de aquella del monómero que lo origina. Con estas clasificaciones, polímeros que antes eran incorrectamente considerados como productos de poliadición, como los poliuretanos (que no liberan moléculas de bajo peso molecular, mas bien son característicamente obtenidos por una reacción de condensación), pasan a recibir una clasificación más precisa al considerarlos provenientes de una polimerización en etapas. 2.- Polimerización por adición. 2.1.- Introducción. En la formación de polímeros por adición, el mecanismo de reacción es en cadena con tres etapas bien diferenciadas: (i).- Iniciación (ii).- Propagación (iii).- Terminación En este tipo de polimerización se genera una especie reactiva a partir del monómero, la cual participa en una reacción que la consume y que a su vez genera otra especie similar, de modo que cada reacción depende de la formación de una especie reactiva en la reacción anterior, por lo cual esta reacción también se denomina reacción en cadena. Las especies reactivas pueden ser radicales, cationes o aniones. Los polímeros de adición así formados tienen pesos moleculares superiores a 100000. La polimerización en cadena es generalmente el resultado de la apertura del doble enlace existente en muchos compuestos monoméricos como los derivados vinílicos o acrilicos. Propileno Poli (metacrilato de metilo) Poli (cloruro de vinilo) Poliestireno La ruptura, además de por la propia estructura del monómero, está condicionada por las condiciones de la reacción y, sobre todo, por la acción de un iniciador que activa la densidad electrónica del monómero de forma y manera que rompe el doble enlace bien en una rotura homolítica (se produce cuando cada átomo que se separa retiene un electrón de los dos que constituyen el enlace, formando radicales libres) o heterolítica (uno de los átomos separados se lleva los dos electrones que constituían el enlace, formándose un anión o un catión) . En el primer caso, se generan radicales. En el segundo caso, dependiendo del carácter electrófilo (reactivos aceptores de electrones) o nucleófilo (reactivos dadores de electrones) del iniciador, se genera una especie catiónica o aniónica que condiciona el posterior desarrollo de la cadena en crecimiento. De todos esos procesos, la polimerización radical es la más importante. Casi el 50% de todos los polímeros sintetizados en el mundo se producen por vía radical ya que muchos de los monómeros de tipo vinílico y acrílico habituales polimerizan por vía radical. Las razones fundamentales de este éxito descansan en la posibilidad de realizarlas a temperaturas bajas (entre ambiente y 100 °C) y los menores requerimientos de pureza que son necesarios, tanto en lo referente a los monómeros como a los iniciadores, para que la reacción se desarrolle eficientemente. Es también importante recalcar que la polimerización radical es el método más versátil para preparar copolímeros, al poner más de un monómero a polimerizar en el mismo medio. Hasta hace poco tiempo, sin embargo, la polimerización radical adolecía de una serie de limitaciones que le impedían competir con las polimerizaciones aniónica o catiónica en la preparación de determinados materiales. Y así, las polimerizaciones aniónicas, y más recientemente las catiónicas, permitían preparar polímeros y copolímeros de estructuras bien definidas y con pesos moleculares de una muy baja polidispersidad. Permitían, por ejemplo, preparar copolímeros de bloque e injerto, con separación de microfases y que podían dar lugar a estructuras utilizables como elastómeros termoplásticos, surfactantes poliméricos, compatibilizantes de mezclas de polímeros, etc. Estos materiales se preparaban (y se preparan) gracias a las llamadas polimerizaciones iónicas sin terminación que, tras una rápida iniciación, propagan las cadenas rápidamente sin ningún otro proceso paralelo que impida ese crecimiento. Desafortunadamente, llevar a cabo esas reacciones implica trabajar a temperaturas muy bajas (fusión del CO2) y con monómeros y disolventes de extraordinaria pureza. Las reacciones de polimerización radical convencionales no son capaces de generar esos materiales debido a una serie de reacciones que compiten con el propio crecimiento de la cadena y que más adelante se denominaran de terminación o transferencia de cadena. Sin embargo, en los últimos años, se han descubierto una serie de procesos por vía radical (denominados bajo el término polimerización radical controlada) que permiten competir con los productos derivados de las polimerizaciones aniónicas mediante procesos mucho menos complicados. Como ya se ha comentado, las polimerizaciones en cadena presentan reacciones de iniciación, propagación y terminación distintas y bien definidas. La iniciación de una polimerización en cadena puede ser inducida por calor, por agentes químicos (INICIADORES) o por radiación (ULTRAVIOLETA y RAYOS γ). La iniciación por calor o radiación proporciona una HOMÓLISIS (rompimiento de un enlace químico resultando en la formación de dos radicales libres) del doble enlace del monómero, resultando en un mecanismo de reacción vía RADICALES LIBRES (átomo u grupo de átomos que poseen un electrón sin pareja, o sea, libre), mientras que la iniciación química (la que se emplea en la mayoría de las industrias), se consigue con iniciadores, sustancias que pueden provocar tanto la homólisis como la HETERÓLISIS (rompimiento de enlace químico resultando en dos iones de cargas opuestas) del doble enlace. Por tanto, la polimerización puede transcurrir a través de radicales libres, por vía CATIÓNICA o por vía ANIÓNICA, o todavía, por COORDINACIÓN. En el caso la polimerización sea iniciada por un iniciador radicalario se llama polimerización radicalaria o de radicales libres, si el iniciador sea un catión se denomina catiónica, si el iniciador es un anión la polimerización se dice aniónica En el caso de la polimerización por coordinación los iniciadores son también CATALIZADORES. Si se utilizan complejos constituidos por COMPUESTOS DE TRANSICIÓN (compuesto formado por elementos de los llamados de transición en la tabla periódica.) y ORGANOMETÁLICOS (sustancias orgánicas en que uno de los átomos de carbono de la molécula está directamente ligado a un átomo metálico), como los de Ziegler-Natta o los metalícenos, la polimerización se denomina de Ziegler-Natta o catalizada por metalícenos, respectivamente. Este tipo de catálisis se aplica solamente a monómeros apolares, y tiene como ventaja, la obtención de polímeros altamente estereorregulares. Cuando las moléculas tienen dobles enlaces C=C, la polimerización tiene lugar por adición, debido a la alta reactividad de las uniones de estos enlaces, que pueden ser activados por radicales libres o iónicos, denominados iniciadores: I I I I C C I I I I C C I I I I C C La presencia de sustituyentes determinados en los carbonos del doble e n l a c e altera la negatividad de los electrones π, aumentando o disminuyendo la densidad electrónica en el doble enlace. En el caso de derivados vinílicos, esta alteración puede representarse: C H 2 CH R 1 C H 2 CH R 1 Entre los sustituyentes del tipo R1 , (electrófilos) están los grupos –CH3 , -COOR, CONH2, que requieren iniciadores aniónicos para estabilizar el exceso de carga positiva del doble enlace. Por ejemplo: KNH2 CH2 CH R1 K H2N CH2 CH R1 Contrariamente, los sustituyentes R2 (nucleófilos), tales como –O-R requieren iniciadores catiónicos para estabilizar el exceso de carga negativa: HCl 4 Al CH2 CH R2 CH3 C H R2 Cl 4 Al En el primer caso, las polimerizaciones así iniciadas se denominan aniónicas y, en el segundo, catiónicas. La presencia de dobles enlaces conjugados y de sustituyentes de anillos bencénicos origina una resonancia que posibilita la polimerización aniónica o catiónica indistintamente. Debido a la neutralidad eléctrica, los iniciadores del tipo de radical libre son compatibles con la mayor parte de los sustituyentes electrófilos y nucleófilos, sobre todo cuando pueden estabilizarse las cargas eléctricas mediante resonancia, lo que les da un carácter no selectivo y de amplio uso. En la tabla 2.1.1 se dan unos ejemplos de las más frecuentes aplicaciones de los distintos tipos de iniciadores. Tabla 2.1.1.- Tipos de iniciadores utilizados en las polimerizaciones en cadena 2.2.- Polimerización por radicales libres o radicalaria. 2.2.1.- Introducción. Proceso de polimerización. Una de las reacciones más comunes y útiles para la obtención de polímeros, es la polimerización por radicales libres. Puesto que la mayoría de los plásticos, los elastómeros y algunas fibras se fabrican por polimerización de radicales libres, este método es el de mayor importancia desde el punto de vista comercial. Si a esto le añadimos los recientes avances que se han logrado en esta área como, por ejemplo, la puesta a punto de la denominada polimerización radical controlada, que permite preparar copolímeros de bloque e injerto (compitiendo con la polimerización iónica), cuyas aplicaciones industriales son muy importantes, ha hecho que este tipo de sintésis refuerce su predominio en la producción de los materiales polímeros. Casi todos los compuestos conteniendo un doble enlace pueden ser polimerizados o copolimerizados por vía radical. Las únicas condiciones que deben cumplir son la capacidad termodinámica de polimerizar y la posibilidad cinética de hacerlo en parámetros que hagan rentable el proceso. La primera condición suele cumplirse en casi todos los casos, al ser la apertura del doble enlace y posterior concatenación de unidades un proceso espontaneo. La condición cinética, sin embargo, es algo más restrictiva y elimina ciertos monómeros de las vías comerciales de polimerización radical. Se emplea para sintetizar polímeros a partir de monómeros vinílicos, es decir, pequeñas moléculas conteniendo dobles enlaces carbono-carbono (C=C). Entre los polímeros obtenidos por polimerización por radicales libres tenemos el poliestireno, el poli(metacrilato de metilo, el poli(acetato de vinilo) y el polietileno ramificado. En la tabla 2.2.1.1 se muestran algunos ejemplos de polímeros típicos que se obtienen por polimerización de adición por radicales libres. Tabla 2.2.1.1.- Algunos ejemplos de polímeros típicos que se obtienen por polimerización de adición por radicales libres. Como ocurre en otras reacciones en cadena, la polimerización por radicales libres es una reacción rápida que consta de las etapas de reacción en cadena características: iniciación, propagación y terminación. En general, las reacciones de polimerización radical son poco estereoselectivas, generándose polímeros atácticos. En la etapa inicial o iniciación se necesita el concurso de alguna molécula capaz de generar radicales. Muchos iniciadores de la polimerización radical son compuestos que tienen algún enlace fácil de romper homolíticamente por la acción de la luz o el calor. Este tipo de polimerización tiene lugar, generalmente, por apertura del doble enlace, lo que origina la formación de un radical libre, generalmente, por combinación con un radical libre previamente preformado (R˙), que proviene de la descomposición de un iniciador. La cadena en crecimiento es, por lo tanto, un radical libre. La activación del doble enlace en la polimerización por radicales libres o radicalaria puede realizarse no sólo mediante la adición de determinadas moléculas que actúan como iniciadores químicos, es decir, de una molécula que genere radicales en condiciones suaves (temperatura < 100 ºC, luz ultravioleta) ,que es el caso más frecuente en la práctica, sino también por acción del calor (activación térmica) necesitándose temperaturas elevadas o de determinadas radiaciones (fotoquímica o radioquímica). Los iniciadores pueden ser orgánicos o inorgánicos. Entre los primeros destacan los peróxidos y los diazocompuestos. Todos ellos se caracterizan por generar radicales libres muy estables. A veces se descomponen generando moléculas sencillas. Iniciación con peróxidos. De todos ellos, el más empleado es el peróxido de benzoilo (BPO),que se descompone térmicamente a suficiente velocidad en dos radicales libres a temperaturas próximas a 70 ºC. Los radicales resultantes pueden adicionarse al monómero (por ejemplo, estireno), pueden participar en una sustitución aromática o pueden descomponerse por medio de una escisión en β y formar radicales fenilos reactivos: Otros iniciadores habituales de este tipo son el peróxido o el hidroperóxido de terbutilo. Iniciación con azocompuestos. De estructura muy diferente e igualmente muy usado por su intervalo de temperatura y relativa facilidad de manejo es el azo-bis-isobutironitrilo (AIBN): Lo que hace especiales a estas moléculas que actúan como iniciadores, es que poseen la inexplicable habilidad de escindirse de un modo bastante inusual. Cuando lo hacen, el par de electrones del enlace que se rompe, se separa, lo que es extraño, dado que siempre que sea posible, los electrones tienden a estar apareados. Cuando ocurre esta escisión, nos quedamos con dos fragmentos llamados fragmentos de iniciador, provenientes de la molécula original, cada uno con un electrón desapareado. Las moléculas como éstas, con electrones desapareados reciben el nombre de radicales libres. Para el caso del peróxido de benzoilo (BPO), se tiene: y para el caso del azo-bis-isobutironitrilo (AIBN): La velocidad de descomposición de los peróxidos tales como el peróxido de benzoilo, puede aumentarse añadiendo pequeñas cantidades de aminas terciarias como la N,N-dimetilanilina. La velocidad de descomposición de los iniciadores puede incrementarse por exposición a la radiación ultravioleta (UV). Por ejemplo, el azo-bis-isobutironitrilo (AIBN) puede descomponerse a bajas temperaturas mediante radiación ultravioleta de 360 nm de longitud de onda. Los persulfatos son los iniciadores inorgánicos más usados en las polimerizaciones en emulsión, descomponiéndose en la fase acuosa: S2O8 2SO4 y estos radicales se difunden hasta la fase orgánica. También se emplean sistemas redox, como sales ferrosas en presencia de agua oxigenada o cuprosas en presencia de ácidos orgánicos: En términos generales la iniciación por descomposición de un iniciador I puede representarse por la siguiente reacción: I 2R In2 2In siendo: R˙ = Radical libre de bajo peso molecular (radical primario). La velocidad de descomposición de iniciadores normalmente sigue una cinética de primer orden y depende del disolvente y de la temperatura de polimerización. En la tabla 2.2.1.2 se dan las constantes de equilibrio de varios iniciadores de uso común. Debe notarse que debido a la recombinación, la cual depende del disolvente, y otras reac ciones secundarias de los radicales libres R˙, la eficiencia del iniciador raramente alcanza el 100 %. Por tanto, se emplea el factor de rendimiento f para mostrar el porcentaje de radicales libres efectivos producidos. Tabla 2.2.1.2.- Constantes de equilibrio de iniciadores comunes en varios disolventes (a) . Propagación. Sin embargo, los electrones desapareados no se sentirán cómodos estando aislados y tratarán de aparearse. Si son capaces de encontrar cualquier electrón con cual aparearse, lo harán. El doble enlace carbono-carbono de un monómero vinílico como el etileno, tiene un par electrónico susceptible de ser fácilmente atacado por un radical libre. Así, el electrón desapareado, cuando se acerca al par de electrones, toma uno de ellos para aparearse. Este nuevo par electrónico establece un nuevo enlace químico entre el fragmento de iniciador y uno de los carbonos del doble enlace de la molécula de monómero. El otro electrón, sin tener dónde ir, se asocia al átomo de carbono que no está unido al fragmento de iniciador. Esto nos conduce a la misma situación con la que comenzamos, ya que ahora tendremos un nuevo radical libre cuando este electrón desapareado venga a colocarse sobre ese átomo de carbono. El proceso completo, desde la ruptura de la molécula de iniciador para generar radicales hasta la reacción del radical con una molécula de monómero, recibe el nombre de etapa de iniciación de la polimerización. Para el caso del peróxido de benzoilo (BPO), se tiene: Este nuevo radical reacciona con otra molécula de etileno, del mismo modo que lo hizo el fragmento de iniciador. De hecho, esto no nos lleva a ninguna parte en cuanto al apareamiento de los electrones, ya que cuando esta reacción tiene lugar una y otra vez, siempre formamos otro radical. El proceso de adicionar más y más moléculas monoméricas a las cadenas en crecimiento, se denomina reacción propagación, que supone la verdadera formación del polímero. La mayor parte de los procesos de propagación implican uniones cabeza-cola (casi el 99%): En algunos polímeros como el poli (fluoruro de vinilo), las uniones cabeza-cabeza pueden suponer porcentajes del orden del 13% Puesto que seguimos regenerando el radical, podemos continuar con el agregado de más y más moléculas de etileno y constituir una larga cadena del mismo. Las reacciones como éstas que se auto-perpetúan, son denominadas reacciones en cadena. Por lo tanto, mientras la cadena sigue creciendo van quedando unos pocos electrones desapareados. En el caso del peróxido de benzoilo (BPO), se tiene: De una forma general, los radicales libres activan rápidamente el doble enlace C=C de los monómeros creando nuevos radicales de iniciación de la cadena, con una determinada velocidad. R RADICAL LIBRE M k i MONOMERO RM1 NUEVO RADICAL LIBRE que a su vez activan los dobles enlaces de otras moléculas de monómero, y así sucesivamente: k p1 RM1 M RM 2 …………………………... k pi RM i M RM i 1 ……………………………. constituyendo las reacciones de propagación. La propagación es una reacción bimolecular, que se produce mediante la adición de un radical libre nuevo (RM ˙) a otra molécula del monómero (M), y la repetición sucesiva de esta operación, Aunque es posible que se produzcan pequeñas variaciones de la constante de equilibrio de propagación (kp ) en las primeras etapas, la constante de equilibrio se considera normalmente independiente de la longitud de cadena. Así, los símbolos M ˙, RM ˙ y RM n M ˙ pueden considerarse equivalentes en las ecuaciones de polimerización en cadena de radicales libres. En monómeros de tipo vinílicos asimétricos (RCH=CH2), el iniciador se une al carbono menos sustituido del alqueno para dar el radical más sustituido (el más estable): A continuación se producen una serie de reacciones de adiciones cabeza-cola: Los electrones desapareados dan lugar a radicales inestables y finalmente van a encontrar una forma de aparearse sin generar un nuevo radical. Entonces nuestra pequeña reacción en cadena comenzará a detenerse y la propagación finalizará. Los radicales terminan con su actividad en las llamadas reacciones de terminación y transferencia de cadena, de ese modo la propagación concluye cuando: (a).- Se encuentren dos cadenas en crecimiento. Dos radicales en crecimiento se encuentran en la masa reaccionante dando lugar a una o dos cadenas que no presentan radicales en sus extremos. En el primer caso, los dos radicales forman un enlace covalente entre las dos cadenas en crecimiento, generando una única nueva cadena. Este tipo de terminación se denomina por combinación. Los dos electrones desapareados se unirán para formar un par y se establecerá un nuevo enlace químico que unirá las respectivas cadenas. Dos macrorradicales de cadena se combinan o acoplan entre sí, DP denominándose acoplamiento. Así, la longitud de cadena cinética (V) es igual a , donde DP es el 2 grado de polimerización medio. RM i RM j R M i j R Debe resaltarse que existe una configuración de cabeza a cabeza en la unión de los dos macrorradicales del polímero muerto. (b).- Otra forma en la que nuestro par electrónico puede concluir la polimerización es por desproporción de dos radicales (dos radicales en crecimiento pueden dar lugar a dos cadenas diferenciadas). Esta es una manera bastante complicada en la cual dos cadenas poliméricas en crecimiento resuelven el problema de sus electrones desapareados. En la desproporción, cuando los extremos de dos cadenas en crecimiento se acercan, el electrón desapareado de una de ellas hace algo extraño. En lugar de unirse al electrón desapareado de la otra cadena, busca un compañero en cualquier parte. Encuentra uno en el enlace carbono-hidrógeno del átomo de carbono vecino al otro carbono radical. De modo que nuestro electrón desapareado no sólo toma uno de los electrones de este enlace, sino también el átomo de hidrógeno. Ahora, nuestra primera cadena no tiene electrones desapareados y el carbono terminal comparte ocho electrones. Tenemos pues, que este proceso de terminación incluye la transferencia de un átomo de hidrógeno del extremo de una cadena al radical libre del extremo de otra cadena en crecimiento, quedando uno de los polímeros "muerto" con un extremo de la cadena insaturado. De esta forma, el tipo y/o la proporción de cada tipo de proceso de terminación de cadena se puede determinar comprobando la proporción de configuraciones de cabeza a cabeza y la de grupos terminales insaturados. RM i RM j RMi RM j M j insaturado La cadena polimérica pierde su átomo de hidrógeno y ahora no sólo posee un átomo de carbono con un electrón desapareado, sino dos. Pero aunque parece un gran problema, en realidad no lo es tanto. Los dos carbonos radicales, siendo vecinos, pueden unir fácilmente sus electrones desapareados para formar un par y por lo tanto un enlace químico entre ambos átomos de carbono. Como éstos ya compartían un par electrónico, el segundo par creará un enlace doble en un extremo de la cadena polimérica. En general, predomina la terminación por combinación. sustituyentes, R, son voluminosos, aumenta la desproporción. Cuando los grupos Los tipos de terminaciones (a) y (b) están muy condicionados por las posibilidades de difusión de las cadenas en crecimiento en el medio de alta viscosidad en el que se va convirtiendo la masa reaccionante. La probabilidad de que se encuentren es también baja debido a la limitada concentración de cadenas en crecimiento, condicionada por la baja concentración de iniciador (c).- Por transferencia de cadena al polímero: Es la reacción de un radical polimérico con una molécula (monómero, solvente (por ejemplo, Cl4C), un agente de transferencia) o con otro radical polimérico por transferencia de un átomo de hidrógeno. A veces, el electrón desapareado en el extremo de la cadena se encuentra tan incómodo, que se aparea con un electrón de un enlace carbono-hidrógeno de otra cadena polimérica. Esto deja un electrón desapareado en el medio de la cadena que no puede formar un doble enlace terminal, pero sí puede y de hecho lo hace, reaccionar con una molécula de monómero, del mismo modo que lo hace el fragmento de iniciador. Eso origina una nueva cadena creciente en la mitad de la primera cadena, dando lugar a un polímero ramificado. Esta reacción constituye un problema en el polietileno, tan grave que es imposible obtener polietileno lineal no ramificado por polimerización por radicales libres. Estas ramificaciones ejercen un notable efecto en el comportamiento del polietileno. Por ejemplo, el polietileno de baja densidad (0.92 g/cm3) obtenido a alta presión (200 atmósferas) y temperaturas cercanas a los 200 ºC, utilizando peróxidos como iniciadores, posee una estructura ramificada, poco cristalina. Se estima que presenta un 43 % de cristalinidad. Sus cadenas poliméricas poseen alrededor de 25000 unidades y debido a su elasticidad, tenacidad y gran resistencia al impacto se utiliza para fabricar bolsas de plástico y como material de empaque. El polietileno de alta densidad (0,97 g/cm3) presenta un 76 % de cristalinidad y un punto de fusión más elevado (130 ºC) que el de baja densidad, lo que permite su esterilización térmica. Las reacciones de transferencia de cadena se pueden utilizar para controlar el peso molecular del polímero, por ejemplo, utilizando tioles: Otro ejemplo de polimerización por radicales libres es la del estireno (Figura 2.2.1.1). Este monómero se polimeriza fácilmente utilizando peróxido de benzoílo como iniciador. El producto, poliestireno, tiene un peso molecular promedio de 1 a 3 millones y es un polímero amorfo y termoplástico. (i).- Iniciación (ii).- Propagación. (iii).- Terminación por acoplamiento. Figura 2.2.1.1.- Etapas de la polimerización radicalaria del estireno. El grado de polimerización es proporcional a la concentración de monómero e inversamente proporcional a la raíz cuadrada de la concentración del iniciador, además de depender de la temperatura, a través de las constantes cinéticas. Sin embargo, esta hipótesis no es siempre correcta debido a la posible transferencia de cadena, que desactiva los radicales de cadena, creando nuevos radicales en moléculas de monómero, de iniciador, del propio disolvente y/o de las impurezas existentes: RM XY RMX Y Los nuevos radicales Y˙ así creados, pueden tener una baja actividad, recibiendo el nombre de retardadores (es el caso del nitrobenceno en la polimerización del estireno) o inhibidores, cuando se estabilizan fácilmente. Si, por el contrario, su actividad es muy alta, aceleran el proceso de la polimerización (acelerantes), aunque, en ambos casos, originan una disminución del grado de polimerización de las macromoléculas resultantes. Los agentes de transferencia tienen como finalidad regular el peso molecular en una determinada polimerización. La constante de transferencia es mayor que la constante de propagación o crecimiento, de ésta manera se impide el crecimiento de largas cadenas con pequeñísimas cantidades. Dichos agentes transfieren un átomo o grupo a la cadena en crecimiento acabando con su actividad radicalaria. El átomo transferido más habitualmente es el átomo de hidrógeno, lo que se consigue desde moléculas que contengan grupos como S-H, P-H, Sn-H, etc., o desde moléculas con hidrógenos activos en posición β a grupos carbonilo (acetona), fenilo (tolueno, cumeno), o grupos vinílicos (derivados de alilo): La adición de modificadores no tiene efectos sobre la velocidad de polimerización. Los agentes de transferencia de cadena pueden disminuir el promedio de la longitud de la cadena y en casos extremos, cuando se usan en grandes proporciones pueden conducir a la formación de telómeros. Algunos agentes de transferencia de cadena producen radicales con baja actividad y si la reacción de reiniciación es lenta la velocidad de polimerización decrece porque hay un incremento de concentración de radicales que aumenta la terminación por acoplamiento. Cuando esto sucede, se dice que la sustancia responsable es un RETARDADOR (por ejemplo nitrobenceno en el caso del estireno). En casos extremos el reactivo agregado puede impedir totalmente la polimerización, reaccionando con la especie radical iniciadora convirtiéndola en especie no reactiva. En este caso es un INHIBIDOR 2.2.2.- Cinética de la polimerización radicalaria. La velocidad total de polimerización, como también la longitud de las cadenas poliméricas formadas en la polimerización de adición, están determinadas por las velocidades de los procesos individuales de iniciación, propagación y término. El mecanismo general para la conversión de monómero o polímeros, usando un iniciador típico de radicales I, puede describirse con el siguiente conjunto de ecuaciones: (1).- Iniciación: v i k i d 2R I ii k i RM R M 1 vd d I dt vi v d kd I d RM dt velocidad de iniciación velocidad de descomposición k i M M I = Iniciador, peróxido orgánico o AIBN M = Monómero R˙ = Radical de bajo peso molecular (radical primario) M1˙ = Radical polimérico con una unidad de monómero (M) en la cadena. (2).- Propagación de la cadena: iii R M RM1 RM M RM 1 2 ------------------------kp RM i M RM i1 --------------------------------Mi˙ = Radical polimérico con i unidades de monómero (M) en la cadena. La velocidad de desaparición del monómero se expresa como: d M dt k p M M k i R M Para las cadenas largas, la cantidad de monómero consumido en la iniciación es pequeña comparad a con la que se consume en la propagación, lo que permite escribir la ecuación anterior de la siguiente manera: d M dt k p M M (3).- Terminación de la cadena. (a).- Por combinación. k t ,c RM i RM j R Mi j R v tc , d M dt 2ktc M M 2ktc M (b).- Por desproporción: k t ,d RM i RM j RM i RM j M ...insaturado , j v td d M dt 2ktd M 2 2 Si se asume que ambas reacciones son cinéticamente equivalentes, se puede escribir una ecuación de velocidad generalizada para el proceso de terminación. iv k t P , k k k RM i RM j t t ,c t ,d Con algunas suposiciones válidas, se pueden obtener expresiones simples para la velocidad de polimerización (vp) y grado de polimerización (GP). 1.- La velocidad de formación de los radicales libres (R˙) es igual a la velocidad de consumo de radicales. 2.- En la etapa de propagación, la actividad radical es independiente de la longitud de cadena, por tanto las constantes específicas de cada paso de la propagación se consideran iguales, permitiendo que todos los pasos de la propagación puedan representarse mediante una única constante de equilibrio específica kp) 3.- Conforme a la aproximación de Bodestein, la velocidad de iniciación y la de terminación tienden a igualarse y la concentración de radicales de cadena permanece prácticamente constante, con lo que el sistema queda en estado estacionario durante casi todo el proceso de polimerización. La velocidad de producción de cadenas radicales es igual a la velocidad de término de cadenas radicales (etapa determinante). d RM n (equilibrio dinámico Vi=Vt) 0 dt 4.- La velocidad de polimerización es igual a la velocidad de propagación (el monómero consumido mediante la ecuación (ii) es insignificante comparada con la consumida en la ecuación (iii)) La expresión de velocidad para las cuatro etapas anteriores puede obtenerse de las ecuaciones (i) – (iv) como sigue: v d 2kd f I donde f es un factor de eficacia, con el que se tienen en cuenta pérdidas de radicales por actuación sobre el propio iniciador, sobre el disolvente o sobre impurezas presentes en el monómero. v i k i R M v p k pC M v t 2ktC 2 siendo: C RM i i 1 De acuerdo con la suposiciones (1) y (3) respectivamente, luego: Vd debe ser igual a Vi y 2kd f I 2ktC2 de donde: C kd f I kt esta última igual a Vt, Considerando que la mayor parte del monómero se consume en las reac ciones de propagación, la velocidad que definirá el progreso de la polimerización será, en términos globales (suposiciones (2) y (4): v p k pC M k p kd f kt I M k1 I M donde: k1 k p kd f kt Esta ecuación predice que la velocidad de formación de polímero debería ser proporcional a la raíz cuadrada de la concentración del iniciador y a la primera potencia de la concentración de monómero. En la práctica, sin embargo, se han comprobado expresiones cinéticas del tipo: v p k1 I M con valores de α entre 0,5 y 1 y de β entre 1 y 1,5. Longitud de cadena cinética. La longitud de cadena cinética V, es el número de moléculas de monómero consumidas por cada radical primario, siendo igual a la velocidad de propagación dividida por la velocidad de iniciación: V Velocidad de propagación v p v p Velocidad de iniciación vi vt l uego: V y com o, C k pC M kd f I , resul t a: kt V k p M 2kt C 2kt C 2 k p M 2 kt kd f I De esta expresión, se desprende que V es inversamente proporcional a la velocidad de iniciación (o velocidad de descomposición del iniciador). Probado experimentalmente en muchas reacciones de polimerización vinílicas. Un incremento en la temperatura produce un incremento en Vi y un correspondiente decrecimiento en la longitud de la cadena cinética. La longitud de cadena cinética es independiente del tipo de proceso de terminación, mientras que el grado de polimerización real o longitud de cadena depende del modo de terminación. Para la terminación por acoplamiento, GP 2V , dado que el acoplamiento hace que se doble la longitud de cadena real. En la terminación por desproporción, GP V . Teóricamente el grado de polimerización resultará tanto mayor, cuanto mayor resulte la velocidad de propagación rp , y menor la de terminación rt .Si la terminación es por combinación, es válida la siguiente expresión: k p M RM vp k M GP p 2 vt kt RM kt RM 2 k p M GP kt kd f kt I k p M kd kt f I k' M I si endo: k ' kp kd kt f mientras que, si la terminación es por neutralización (o desproporción) con creación de un doble enlace, resulta: GP s i endo: k '' rp rt k p M 2kt kd f kt I k p M 2 kd kt f I k '' M I kp 2 kd kt f Por tanto, se pueden sacar las siguientes conclusiones sobre la polimerización en cadena de radicales libres utilizando un iniciador químico: 1.- La velocidad de propagación es proporcional a la concentración de monómero y a la raíz cuadrada de la concentración del iniciador. 2.-La velocidad de terminación es proporcional a la concentración del iniciador. 3.- El peso molecular medio es proporcional a la concentración del monómero e inversamente proporcional a la raíz cuadrada de la concentración del iniciador. 4.- La primera cadena iniciada produce rápidamente un polímero de alto peso molecular. 5.- La concentración del monómero decrece constantemente durante la reacción y tiende a cero al final. 6.- Los aumentos de las velocidades de iniciación, propagación y terminación con la temperatura siguen la ley de Arrhenius. Las energías de activación de la iniciación, de la propagación y de la terminación son aproximadamente 146, 21, y 12.5 kJ/mol, respectivamente. En las tablas 2.2.2.1 y 2.2.2.2 se encuentran datos sobre energías de activación típicas. 7.-Al aumentar la temperatura aumenta la concentración de radicales libres, y por tanto la velocidad de las reacciones, pero disminuye el peso molecular medio. 8.- Si la temperatura excede un valor límite (Tc), el polímero se descompone sin que se produzca reacción de propagación a temperaturas por encima de Tc. La temperatura de techo (límite) para el estireno es 310 °C y sólo 61 °C para el α-metilestireno. Tabla 2.2.2.1.- Energías de activación para las reacciones de propagación (Ep) y de terminación (Et ) en la polimerización en cadena de radicales libres. Tabla 2.2.2.2.- Valores cinéticos de radicales libres típicos. Existe una cierta tendencia a la formación de segmentos estereorregulares, particularmente a temperaturas bajas, aunque se prefiere el uso de catalizadores iónicos y de coordinación para la obtención de polímeros estereorregulares. Si no ocurren otras reacciones que las mencionadas, la longitud de la cadena cinética V debería estar relacionada con el grado de polimerización - Para terminación por combinación: GP , así: GP 2V - Para terminación por desproporción: GP V Esto es cierto para algunos sistemas, pero para otros ocurren amplias desviaciones en que hay más moléculas de polímeros que centros activos. Estas son como resultado de las reacciones de transferencia de cadena. Cada uno de los procesos de transferencia de cadena produce la terminación de un macrorradical y da lugar a otro macrorradical. En los dos casos, el electrón no apareado no se hallará ya en el carbono terminal. Las nuevas posiciones del radical sirven de puntos de ramificación para la extensión de la cadena o su ramificación. La transferencia de cadena puede producirse también con el monómero, el ini ciador, el disolvente o cualquier otro de los aditivos presentes en el sistema de polimerización, siendo de mayor interés la transferencia de cadena con los disolventes y con otros aditivos. La transferencia de cadena a otras moléculas exceptuando el disolvente o los aditivos es normalmente despreciable. La reacción de transferencia de cadena disminuye la longitud de cadena media de acuerdo con la concentración del agente de transferencia de cadena S. El grado de polimerización GP resultante es igual al que se habría obtenido sin disolvente ni aditivos más un factor relacionado con el producto del cociente de la velocidad de propagación (vp) y la velocidad de transferencia de cadena (vtr) y el cociente de la concentración de monómero [M] y la concentración del agente de transferencia de cadena [XP]. La ecuación de Mayo, que proporciona pendientes positivas cuando se representan los valores, es la relación inversa que se deriva de la expresión citada anteriormente. El cociente de la velocidad de cese o terminación por transferencia y la velocidad de propagación se llama constante de transferencia de cadena (C s). Esta última está relacionada con fuerzas de enlace relativas en la molécula del disolvente) del aditivo y la estabilidad del nuevo radical producido. A continuación se muestra la ecuación de Mayo. k tr M X P M XP n n ' kp P M PM donde: P = Monómero, iniciador, solvente, modificador. A menos que k’p » kp (en cuyo caso el agente podría actuar también como inhibidor), el agente de transferencia de cadena tiene efecto despreciable sobre la velocidad de polimerización. Por su parte, la expresión para la longitud de la cadena cinética queda: V Sustituyendo C e invirtiendo: v i ktr XR C 1 1 ktr XP (Ecuación de Mayo), GP GP 0 k p M Donde vp 2kt 1 GP 0 k p M GP y GP0 son los grados de polimerización con y sin agente de transferencia, en el caso que XP Solvente . S 1 1 Cs GP GP 0 M , Cs ktr Cons tan te de transferencia de cadena kp La constante de transferencia de cadena está relacionada con fuerzas de enlace relativas en la molécula del disolvente o del aditivo y la estabilidad del nuevo radical producido. En la figura 2.2.2.1 se muestra la representación gráfica de la ecuación de Mayo, que n proporciona pendientes positivas cuando se representan los valores. Figura 2.2.2.1.- Representación gráfica de la ecuación de Mayo Como se puede ver en la figura 2.2.2.2, el peso molecular del poliestireno disminuye cuando se polimeriza con disolventes, y el incremento o la disminución de la pendiente están relacionados con la eficacia de transferencia de cadena del disolvente. Las pendientes de dicha gráfica son iguales a C s. Figura 2.2.2.2.- Peso molecular del poliestireno en función del disolvente y de su concentración; A = benceno, B = tolueno, C = n-heptano, D = cloroformo, E = etilbenceno, F = cumeno, G = secbutilbenceno, H = fenol, I = m-cresol, J = p-cresol, K = o-cresol, L = tetracloruro de carbono, M = tetrabromuro de carbono, N = n-butilmercaptano, [S] = concentración de agente de transferencia de cadena, [M] = tetrabromuro de carbono, N = n-butilmercaptano, [S] = concentración de agente de transferencia de cadena, [M] = concentración de monómero de estireno. Los agentes de transferencia de cadena también han sido llamados reguladores (del peso molecular). Cuando se utilizan en grandes proporciones, se denominan telógenos, dado que producen polímeros de bajo peso molecular (telómeros) en reacciones de telomerización. Evaluación de vi y vp. Puesto que: k d 2R I k i RM R Se tiene: v d k d I k i I y v p k p M kd kt I elevando al cuadrado: v p2 2 kp kt 2 M 2 kp kd I de donde: kt v p2 2 M k d I v p2 2 M vi o también: kp kt 1 2 v p M v i 1 2 constantes separables solo por métodos estacionarios. La ecuación de Arrhenius, puede expresarse de la siguiente forma: Constante de velocidad: k p Ap E A RT e Donde: Ap = Factor de frecuencia o choque, que es próximo a la constante de velocidad de la reacción, si todos los choques o colisiones conducen a reacción. Ep = Energía de activación. Aplicando la ecuación de Arrhenius a la razón de velocidad: kp kt 1 2 A p 1 At 2 e 1 E p Et / RT 2 Ln (logaritmos neperianos) a ambos lados de la expresión anterior y representando k A E p 1 Et 1 p 2 p gráficamente Ln 1 Ln 1 en función de 1/T es posible determinar R T 2 2 kt At Tomando E: energía de activación de la reacción total de polimerización. Para la mayoría de las reacciones de E kJ polimerización: E p t 21 25 . 2 mol Relación entre grado de polimerización y T. De la expresión de longitud de cadena cinética. k p M V kt ki I Tomando Ln de ambos lados y reemplazando las constantes de velocidad por las correspondientes expresiones de Arrhenius, se tiene: 1 LnV Ln k p M Ln kt ki I 2 E p 1 Et 1 Ei LnV Ln K Ln K ' RT 2 RT 2 RT y derivando: d LnV 1 1 1 E p Et Ei dT 2 2 RT 2 donde: Ep Et kJ 21 25 2 mol , Ei kJ 125 2 mol con lo que el coeficiente de 1/RT2 es negativo. El decrecimiento en el grado de polimerización con el incremento de la temperatura resulta del hecho que la energía de activación de la reacción de iniciación es relativamente alta comparada con la energía de activación de la reacción de crecimiento. Aunque la velocidad de crecimiento de la cadena (propagación) también aumenta con el incremento de temperatura (el cual por sí mismo podría conducir a un incremento en el DP ), el incremento en la velocidad de iniciación es el factor que contrapesa y de acuerdo a la ecuación: V el grado de polimerización decrece. La fotopolimerización es un caso aparte: vp vi vt d LnGP dT A p 1 1 At 0 2 RT 2 2.2.3.- Efecto Norrish-Trommsdorff (Autoaceleración). La velocidad de polimerización de los monómeros líquidos tales como el metilmetacrilato puede seguirse controlando el cambio de volumen por dilatometría o el incremento de la viscosidad. La viscosidad tiene muy poca influencia en la velocidad de polimerización o en el peso molecular a menos que sea relativamente alta. Cuando la viscosidad es alta, la reacción de terminación se ve obstaculizada dado que los macrorradicales no gozan de una difusión rápida en el medio viscoso. Por otro lado, el monómero tiene una difusión bastante rápida lo que ocasiona la formación de macrorradicales de alto peso molecular como resultado de una propagación sin terminación. Esta autoaceleración, llamada efecto de Norris-Trommsdorff o de gel es la disminución de la velocidad de terminación en un medio viscoso (Viscosidades altas), lo que ocasiona la formación de polímeros de peso molecular anormalmente alto. El efecto observado es la disminución de la constante kt, que corresponde al numero de moles por segundo de cadenas radicales que desaparecen a través de terminación cuando [RnMi] = 1 mol. Por lo tanto, si kt disminuye a viscosidad (η) alta la concentración de radicales aumenta sobre la correspondiente en el equilibrio del estado estacionario, entonces se tiene: v i vt k i I kt R M i k R M i i I k t 2 R M i k i I 1 1 Cte kt kt 1 2 Pero: v t kt R M i 2 y sustituyendo en la expresión anterior la (2.2.3.1), se tiene: v t kt Cte 1 Cons tan te kt La disminución de kt → Aumenta [RMi˙], lo que lleva a que: 1 2 (2.2.3.1) (a).- La velocidad de polimerización aumenta rápidamente con el incremento de viscosidad: v p k p M R M i 2 (b).- El largo de la cadena cinética aumenta con el incremento de la velocidad de polimerización y velocidad de término constante V Vp Vi El calor desprendido en la polimerización puede disiparse en el caso de tubos de ensayo pequeños, pero para las polimerizaciones en bloque (en masa o bloque) a gran escala serán necesarios equipos especialmente diseñados. Afortunadamente, los monómeros como el metacrilato de metilo pueden polimerizarse sin dificultad en hojas de hasta varios centímetros de grosor en sistemas continuos o estáticos. El producto de peso molecular muy alto obtenido, a causa del incremento de viscosidad que ocasiona la autoacelración, es de utilidad como plástico de fundición pero no para el moldeo o la extrusión. Efecto de la temperatura. La influencia de la temperatura en el curso de la polimerización depende de la eficiencia del iniciador y velocidad de descomposición, transferencia de cadena y propagación de la cadena. La energía total de activación es: Ea 1 1 Ed E p Et 2 2 Ep –(1/2)Et es una medida de la energía necesaria para polimerizar un monómero particular: Estireno : Vinilacetato : 27.2 kJ/mol 19.7 kJ/mol Los iniciadores tienen una energía de activación, Ed en el rango de 125 a 170 kJ/mol. Este es el papel controlante en la etapa de iniciación en la polimerización de radicales libres. Consecuentemente, la energía total de activación, Ea, en las polimerizaciones de radicales libres está en el rango de 85 – 150 kJ/mol. 2.2.4.- Características de las polimerizaciones radicalarias. En resumen y en términos generales, las polimerizaciones por radicales son fuertemente exotérmicas, requiriendo altos niveles de activación. Las necesi dades de refrigeración son importantes, debiendo mantenerse la temperatura de reacción lo más constante y uniforme posible, para evitar grandes dispersiones de peso molecular. La velocidad de reacción aumenta con la temperatura, pero el peso molecular y, en consecuencia, el grado de polimerización promedio disminuye, mientras que el tiempo que dura la reacción no afecta a la longitud de las macromoléculas, si las concentraciones no se modifican apreciablemente. La viscosidad aumenta con el grado de conversión, dificultando la transferencia de calor y en la etapa final de la reacción se puede originar un desequilibrio entre las velocidades de iniciación y de terminación, produciendo una aceleración de la reacción (autoaceleración) e, incluso, una explosión, en particular en las polimerizaciones en soluciones concentradas y en masa. 2.3.- Polimerización iónica. 2.3.1.- Introducción. La reacción de polimerización en cadena puede proceder con iones en lugar de radicales libres como partículas propagadoras de la cadena polimérica. Estas pueden ser cationes o aniones, dependiendo del iniciador que se emplee. Las polimerizaciones iónicas son muy sensibles a la presencia de agua y sustancias extrañas en el medio de reacción. Requieren reactivos muy puros y se llevan a cabo en masa o en presencia de un disolvente. Las polimerizaciones iónicas se basan en una ruptura heterolítica de los dobles enlaces de los monómeros olefínicos merced al papel de un iniciador que ataca de forma adecuada la densidad electrónica del monómero. Y así, en una típica polimerización aniónica, un anión fuertemente básico A- puede atacar a una olefina: A- + → A-C-C- El producto de esta adición es otro anión que puede adicionar nuevos monómeros en un clásico proceso de propagación. No todas las olefinas, sin embargo, son susceptibles de una polimerización aniónica. Para hacer posible la reacción, el monómero debe tener una estructura en la que la densidad electrónica del doble enlace se mueva con facilidad o que dicho enlace este ya parcialmente polarizado. Monómeros como el estireno y sus derivados, las piridinas o los dienos están entre los monómeros que cumplen la primera condición. Otros, como los acrilatos, metacrilatos o acrilonitrilo tienen sustituyentes próximos al doble enlace que provocan la adecuada polarización. Sin embargo, la consecución de altos polímeros en polimerizaciones iónicas (tanto aniónicas como catiónicas) necesitan que esas especies iónicas presentes en el medio tengan una adecuada estabilidad o, en otros términos, un adecuado tiempo de vida medio que les permita continuar el proceso de propagación 2.3.2.- Polimerización catiónica. La polimerización vinílica catiónica es una forma de obtener polímeros a partir de moléculas pequeñas, o monómeros, que contengan dobles enlaces carbono-carbono. Su empleo comercial principal es para sintetizar poliisobutileno. Como iniciadores (o catalizadores) catiónicos pueden usarse los ácidos sul fúrico, fosfórico, perclórico, y los denominados ácidos de Lewis, es decir, aquellos compuestos que, debido a su estructura molecular, forman compuestos iónicos de coordinación en presencia de HCl, H2O, etc. (sustancia denominada cocatalizador) y protones libres, que se adicionan al doble enlace. Tal es el caso del cloruro de aluminio con HCl, el trifluoruro de boro y el tetracloruro de titanio con H2O, etc. En la polimerización vinílica catiónica, el iniciador es un catión, es decir, un ión con carga eléctrica positiva, A+, que se une a una olefina, a un ciclo o a otro monómero, compensando la carga con un contraión colocado en sus proximidades. El catión resultante adiciona más monómero hasta que la reacción se termina. El mecanismo de terminación en la catiónica tiene lugar mediante una reacción entre el contraión y la cadena en crecimiento que, de esta forma, termina su vida activa. El proceso regenera el iniciador que puede iniciar una nueva cadena. Por lo tanto, el conjunto de secuencias es similar a lo que en las radicales hemos denominado una transferencia de cadena. Tal proceso de terminación es el factor dominante de una polimerización catiónica. Debe ser cuidadosamente controlado para impedir que agua u otras impurezas generen procesos de terminación no deseados. Las reacciones, además, solo transcurren bien a temperaturas bajas (-70 °C). El catión atraerá un par de electrones, con carga negativa, del doble enlace carbono-carbono y por lo tanto se formará un enlace simple con el iniciador, tal como se muestra en la figura 2.3.2.1. Esto deja deficiente de electrones a uno de los carbonos que estaban comprometidos en el doble enlace, portando una carga positiva. Este nuevo catión va a reaccionar con una segunda molécula de monómero, del mismo modo que hizo el iniciador cuando reaccionó con el primer monómero. El proceso ocurre una y otra vez, hasta que se alcanza un alto peso molecular, lo suficiente como para que el polímero pueda prestarnos alguna utilidad. Figura 2.3.2.1.- Polimerización catiónica. Muchas veces, sin embargo, esto comienza de una manera más complicada. Normalmente, el iniciador empleado es el tricloruro de aluminio, AlCl3 (ácido de Lewis con un orbital sp3 disponible). Según la regla del octeto todos los átomos del segundo período de la tabla periódica buscan tener ocho electrones en su capa o nivel externos. El átomo de aluminio en el AlCl3 comparte pares de electrones con otros tres átomos, por lo que queda con apenas seis electrones, dos menos del octeto. No obstante, tiene un orbital completo (es decir, un lugar vacante donde debería haber electrones) que está vacío y listo para ser ocupado. Y justamente lo que ocurre, para suerte de ese átomo de aluminio, es que por lo general, se encuentra presente en el sistema una pequeña cantidad de agua. Entonces el átomo de oxígeno del agua, que tiene dos pares electrónicos no compartidos, dona uno de ellos al átomo de aluminio, formando un complejo entre el AlCl3 y el H2O. Al ser extremadamente electronegativo, el oxígeno tratará de atraer hacia sí mismo los electrones que comparte con los átomos de hidrógeno, dejando a éstos con una ligera carga positiva. Por lo tanto, se crea una situación propicia para el ataque de un par de electrones proveniente del doble enlace de la molécula monomérica. Así, el monómero podrá tomar un hidrógeno, convertirse en catión y el complejo AlCl3/H2O se transforma en su anión complementario, AlCl3OH-. Este proceso, por el cual se forma el complejo AlCl3/H2O y reacciona con la primera molécula de monómero, se denomina iniciación. Ahora tenemos un nuevo catión. Más que un catión, es un carbocatión. Así lo llamamos por poseer la carga positiva sobre un átomo de carbono. Los carbocationes son sumamente inestables, porque el átomo de carbono sólo posee seis electrones en su capa externa. O sea dos electrones menos de los que todo átomo de carbono desea poseer en su capa externa. Por lo tanto un carbocatión hará todo lo posible para ganarse esos dos electrones que le faltan y alcanzar el octeto. De modo que el carbocatión mira a su alrededor y encuentra un par de electrones en el doble enlace de la molécula monomérica más cercana. (En un doble enlace, existen dos pares de electrones). Entonces el carbocatión toma esos electrones y al hacerlo, establece un enlace simple con la molécula de monómero. Esto también genera otro carbocatión, que a su vez puede reaccionar con otro monómero, luego con otro y así sucesivamente. Finalmente, obtenemos una larga cadena polimérica. De igual modo, el tricloruro de aluminio, con el orbital sp3 disponible, también puede formar un complejo con un alcohol: donde el protón está unido al oxígeno en un sistema de tres enlaces similar al ión H3O+. Además los átomos de halógeno ejercen una acción desplazante sobre los electrones del oxígeno, como consecuencia de lo cual el enlace con el hidrógeno se encuentra en una situación extremadamente debilitada, siendo un poderoso ácido que puede actuar como iniciador de una polimerización catiónica. Además el anión resultante de la perdida de ese protón, AlC13(OR)- puede actuar como contraión en una polimerización catiónica. Dependiendo del grupo sustituyente, su organización estérica y la afinidad del protón pueden dar lugar a toda una escala de iniciadores catiónicos de diferentes potencialidades. Y así, algunos halógenos pueden reemplazarse por otros alcóxidos o alquilos, se pueden usar diferentes coiniciadores, etc. El proceso por el cual se agrega un monómero atrás de otro dando origen a un polímero, se denomina propagación. Las especies en propagación, al igual que ocurre en la polimerización radical pero no en la aniónica, son proclives a procesos de terminación y de transferencia de cadena. La verdadera terminación tiene lugar cuando el catión se combina bien con una impureza o bien con su propio contraión. Esta reacción con el contraión puede consumir el coiniciador El modo más común de terminación, por el que se detiene el ciclo de agregar más moléculas de monómero al polímero en crecimiento es algo como esto. Imaginemos la cadena en crecimiento de poliisobutileno. Los grupos metilo unidos al átomo de carbono catiónico tienen un pequeño problema: sus hidrógenos. Esos hidrógenos no tendrán inconvenientes en separarse y unirse a otras moléculas. Esto es justamente lo que puede suceder cuando se acercan a una molécula de monómero isobutileno. Ávidos de electrones como están por ser parte de un catión, estos hidrógenos son fácilmente atacados por un par de electrones proveniente del doble enlace de la molécula de isobutileno. Cuando todos los electrones se hayan reordenado, como muestran las flechas, nos quedaremos con una cadena polimérica neutra que contiene un doble enlace terminal y un nuevo catión, formado a partir de la molécula de isobutileno. El extremo de la cadena ahora será neutro y ya no podrá reaccionar para seguir creciendo. Pero el nuevo catión puede comenzar una nueva cadena en crecimiento, del mismo modo en el que lo hizo nuestro iniciador. Este proceso se denomina transferencia de cadena. Ocurre además en una polimerización por radicales libres, como también en otras clases de polimerización. Ese tipo particular de transferencia de cadena se llama transferencia de cadena al monómero. Pero existe otra clase de transferencia. Para comprenderlo, debemos recordar que para cada catión, en el mismo recipiente (vaso de precipitados) existe un anión acechando en alguna parte, como puede ser el, AlCl3OH. Tal como todos sabemos, cationes y aniones tienen tendencia a reaccionar entre sí, lo cual puede ser problemático cuando deseamos que nuestro catión reaccione con alguna otra cosa, como por ejemplo una molécula de monómero. Veamos cómo ocurre. Cuando reacciona el iniciador catiónico y forma una cadena catiónica en crecimiento, el anión del mismo pasa a ser el anión del catión polimérico en crecimiento. Hay que tener presente, que los aniones tienden a poseer pares de electrones flotando a su alrededor sin nada que hacer y eso puede ser un inconveniente. Esto es justamente lo que ocurre aquí. Los electrones del anión, de vez en cuando, atacarán los átomos de hidrógeno de los grupos metilo adyacentes al carbono catiónico. Estos átomos de hidrógeno son los que estaban tan ansiosos de ser secuestrados por una molécula monomérica cercana. No a menudo, pero sí a veces, estos hidrógenos reaccionan muy fácilmente y de la misma manera, con los electrones no compartidos del átomo de oxígeno del anión, dejando a la moribunda cadena polimérica con un doble enlace terminal. Pero por otro lado, el complejo AlCl3/H2O es regenerado y puede comenzar a producir nuevos polímeros crecientes, tal como lo había hecho antes. Y es otro caso de transferencia de cadena. Claro que no todo sale siempre tan bien. A veces, el que reacciona es el par de electrones no compartidos del átomo de cloro, y no con uno de los átomos de hidrógeno, sino con el mismo átomo de carbono catiónico. Este abandona el contraión y se une al polímero. Y entonces nos quedamos con otra clase distinta de polímero inactivo, que termina con un átomo de cloro y con, AlCl2OH. , que no va a iniciar ninguna nueva cadena en crecimiento. Este proceso se denomina terminación, porque no se inicia ninguna cadena. Es la última de las tres etapas principales en cualquier polimerización por crecimiento de cadena, en la cual las dos primeras son, obviamente, la iniciación y la propagación. Cuando la terminación toma lugar, la polimerización se da por concluida. De un modo general y designando con A al catalizador y con RH al cocatalizador, la polimerización catiónica se puede desarrollar así: K H AR A RH k i H AR M HM AR k p HM x1 AR HM AR M x k t HM AR M x H AR x considerando que la primera reacción es más rápida que la segunda, la veloci dad de iniciación será: ri Kk i ARH M La reacción de terminación es de primer orden y su velocidad es: rt kt HM x AR Admitiendo la hipótesis de un estado estacionario (difícil de comprobar por que, en general, las polimerizaciones catiónicas proceden muy rápidamente) resulta (rt = ri ) : Kk i ARH M kt HM x AR Kk HM x AR i ARH M kt kk 2 rp K p i ARH M kt r k dp N p p M rt kt En estas reacciones la energía de activación de la etapa de propagación es baja y la de la etapa de terminación es más alta, es decir Et >Ep . El valor de E=E i +Ep -Et , está normalmente en el intervalo de -167 a 251 kJ/mol y cuando es negativo se tiene un aumento de rp al disminuir la temperatura, con un incremento del grado de polimerización. Esta situación es la inversa de la que se presenta en las polimerizaciones por radicales libres. Si el valor de E es positivo, la velocidad aumenta con la temperatura, pudiendo producirse una explosión. En la práctica, las polimerizaciones catiónicas se realizan a bajas temperaturas. La velocidad de reacción depende también de la constante dieléctrica del medio en que se verifica la reacción y de la presencia de sustancias extrañas, creciendo con la concentración del monómero y del catalizador, mientras que el peso molecular medio aumenta al aumentar la concentración del monómero y es independiente de la concentración del iniciador. La polimerización catiónica de eteres cíclicos tiene lugar mediante la formación de un ion oxonio cíclico, que sirve como especie activa. El caso más simple, el polióxido de etileno. El polióxido de etileno así obtenido es de bajo peso molecular, prefiriéndose la polimerización aniónica para alcanzar pesos moleculares más altos. El óxido de propileno o la epiclorhidrina no funcionan bien con iniciadores catiónicos. Otra polimerización catiónica de interés industrial es la del trioxano (el trímero cíclico del formaldehido) para dar lugar a polióxido de metileno. La reacción puede iniciarse casi con cualquier ácido de Lewis, dando lugar a un polímero altamente cristalino y de alto peso molecular. Incidentalmente, debe mencionarse que esta reacción tiene un importante periodo de inducción que puede eliminarse mediante la adición de trazas de formaldehido. 2.3.3.- Polimerización aniónica. La polimerización vinílica aniónica es un método por el cual se obtienen polímeros a partir de pequeñas moléculas que contengan dobles enlaces carbono-carbono. Es un tipo de polimerización vinílica. La polimerización aniónica se inicia por adición de un anión al doble enlace del monómero. Como aniones iniciadores se utilizan OH-, NH2-, y carbaniones de compuestos organometálicos como el butil-litio (BuLi): El más usado es el butil litio. El use comercial de iniciadores como el butil litio se debe fundamentalmente a su solubilidad en disolventes hidrocarbonados, condición que no satisfacen derivados de alquilo o arilo de otros metales alcalinos. En disolventes más polares reaccionan con ellos debido a la reactividad que les confiere el enlace metal-carbono. Una ínfima porción del butil litio siempre se encuentra escindida. No mucho, sino una parte. Se escinde para formar un catión litio y un anión butilo. Un anión como éste, donde la carga negativa se localiza sobre un átomo de carbono, se denomina carbanión. Por lo tanto el anión butilo donará un par de electrones a uno de los átomos de carbono del monómero involucrados en el doble enlace. Ahora este átomo de carbono ya posee ocho electrones en su capa externa, que comparte con los átomos al cual está unido, de modo que un par de estos electrones, específicamente el par del doble enlace carbono-carbono, abandonará el átomo de carbono y se establecerá sobre el otro átomo de carbono del doble enlace. El proceso por el cual el butil-litio se escinde y el anión butilo reacciona con la molécula del monómero, se denomina iniciación. El carbanión (extremo aniónico de la cadena) reacciona ahora con otra molécula de monómero exactamente de la misma manera en que el iniciador reaccionó con la primera molécula monomérica, por lo tanto se genera otro carbanión. Este proceso se sucede en el tiempo y cada vez que se agrega otro monómero a la cadena en crecimiento, se genera un nuevo anión, permitiendo la incorporación de otro monómero. Así es como crece la cadena polimérica. La adición sucesiva de monómeros se denomina propagación. Pero lo anterior, no puede continuar indefinidamente y algo debe poner fin al proceso. En la mayoría de los casos, lo único que impide que se sigan agregando más monómeros a la cadena en crecimiento, es que al final se agoten las moléculas de monómero que teníamos en nuestro recipiente (vaso de precipitados). Así, en ausencia de impurezas las cadenas siguen creciendo hasta que se termina el monómero. El extremo aniónico es perfectamente estable y el tamaño de las cadenas aumentará por adición de más monómero. Por esta razón estos materiales fueron llamados polímeros vivientes. Impurezas dadoras de protón (agua o ácidos) terminan la cadena: Es conocida la polimerización del butadieno con sodio metálico, que justi fica el antiguo nombre del polibutadieno (goma BUNA, es decir Butadieno - Sodio) cuya iniciación se verifica como se indica seguidamente: Na CH 2 CH CH CH 2 Na C H 2 CH CH C H 2 También se polimeriza el acrilonitrilo con sodio y potasio metálico en amoníaco líquido (formándose previamente la amida correspondiente): 2Na 2NH3 2NaNH2 H2 CN CN CN I I I 2NaNH 2 CH 2 CH CN NH 2 CH 2 C H Na NH 2 CH 2 C H CH 2 C H Na La desactivación se debe a la presencia de sustancias extrañas que desactivan el macroión. Si se utilizan reactivos muy puros, la reacción continúa hasta que se consume el monómero, quedando macroiones activados, que mantienen su actividad durante mucho tiempo, de forma que si, posteriormente, se adiciona monómero, la reacción continúa; por este motivo se dice que estos polímeros están «vivos». Así, si dentro de un rato se agregara más monómero en el recipiente (vaso de precipitados), éste se adicionaría a la cadena haciendo que creciera más. Se sabe que algunas cadenas de poliestireno se han mantenido así de activas durante años. Para detenerlas, se debe agregar algo que reaccione con los carbaniones, como por ejemplo agua. Este tipo de sistemas se denominan polimerizaciones aniónicas vivientes. Este recipiente (vaso de precipitados) repleto de polímero, que se mantiene activo durante años, si agrega más monómero, éste se adicionará a las cadenas poliméricas vivientes. En lugar de agregar el mismo monómero a la solución de polímero viviente, ¿por qué no probar con un monómero diferente? El resultado fue un polímero cuyas cadenas consistían en un largo segmento de un tipo de polímero y un segundo largo segmento de otro polímero. Los polímeros de este tipo de llaman copolímeros en bloque. Por ejemplo, una solución de cadenas vivientes de poliestireno reacciona con butadieno para dar un copolímero en bloque estireno-butadieno. También se puede obtener de ese modo el copolímero en tribloque estireno-butadieno-estireno. La polimerización aniónica tiene consecuencias prácticas importantes: Copolimerización en bloque: si se introduce un segundo monómero después de que se haya terminado la carga inicial del primero, las cadenas aniónicas continúan la polimerización con el segundo, dando un copolímero en bloque. Flexibilidad sintética: permite la introducción de una amplia variedad de grupos terminales. Por ejemplo, burbujeando CO2 a través de un conjunto de cadenas aniónicas y por posterior exposición al H2O se obtiene un polímero terminado en grupos carboxilo: En forma similar, si se agrega óxido de etileno, se obtienen cadenas terminadas en grupos hidroxilo: Resumiendo, la característica más importante de muchas polimerizaciones aniónicas es la falta de un mecanismo inherente de terminación. Las especies activas generadas en los primeros instantes del proceso por reacción del iniciador con el monómero mantienen esa actividad hasta que el monómero desaparece, tras lo cual es incluso posible mantener esa actividad durante un cierto tiempo, por lo que si se adiciona nuevo monómero desde el exterior la polimerización se reinicia. Dado que las especies activas se generan al mismo tiempo y todas acaban de crecer cuando el monómero desaparece, la longitud (el peso molecular) de las cadenas es muy homogénea, lo que conduce a distribuciones estrechas de peso molecular o índices de polidispersidad muy bajos. Por otro lado, la concentración de iniciador condiciona la concentración de especies activas y ésta y la concentración de monómero el tamaño medio de las cadenas (o peso molecular). El proceso de terminación se puede conseguir desde fuera adicionando sustancias que acaben con las especies activas. Por ejemplo, la adición de alcoholes conduce a alcóxidos, que no pueden iniciar nuevas reacciones aunque quedara algo de monómero. Cuando un segundo monómero se adiciona a la masa reaccionante tras el consumo total del primero y en un intervalo de tiempo tal que las especies activas siguen todavía "vivas", es posible obtener copolímeros de bloque o con estructuras más o menos complejas si se van sucediendo las adiciones de monómero de uno y otro tipo (dibloque, tribloque, etc.). Este tipo de copolímeros han encontrado un acomodo notable en el ámbito de las mezclas de polímeros. Muchos polímeros son inmiscibles pero pueden obtenerse mezclas estables con propiedades aprovechables si las fases separadas de ambos componentes se estabilizan mediante la adición de copolímeros que contienen unidades de uno y otro tipo. Cada bloque se disuelve en la fase semejante, rebaja la tensión interfacial y juega un papel parecido a los surfactantes en las mezclas entre agua y compuestos hidrocarbonados. Las olefinas no son los únicos monómeros susceptibles de polimerizar aniónicamente, ya que muchos heterociclos saturados pueden ser polimerizados aniónicamente mediante un mecanismo que implica la apertura de anillo. Tal es el caso del óxido de etileno, que se puede polimerizar mediante el concurso de una iniciación debida, por ejemplo, al fenolato sódico: reacción que, sin embargo, no funciona si no existe fenol libre en la mezcla reaccionante. 2.3.4.- Polimerización con estereoquímica controlada o estereoespecífica. 2.3.4.1.- Introducción. Las propiedades de los materiales polímeros dependen fundamentalmente de su estructura química, del peso molecular y de la conformación macromolecular. En el caso de α-olefinas, debido a la formación de centros estereogénicos, cada carbono asimétrico puede adoptar dos posibles configuraciones, y estos materiales tienen propiedades físicas muy diferentes en función del orden configuracional que presenten. Los métodos de síntesis de poliolefinas permiten obtener materiales con diferente esterorregularidad. Así cadenas de polímero formadas por la sucesión regular de átomos de carbono con la misma configuración constituyen un polímero isotáctico, mientras que si existe alternancia en la configuración de los átomos de carbono asimétricos, el polímero es sindiotáctico. Cuando no existe orden configuracional el polímero es atáctico. El desarrollo de sistemas catalíticos con alta especificidad ha permitido la obtención de poliolefinas con elevada estereorregularidad y con microestructuras diferentes, siendo posible también la síntesis de polímeros hemiisotácticos y polímeros estereobloque (Figura 2.3.4.1.1). Figura 2.3.4.1.1.- Diferente esterorregularidades Factores que afectan a la estereorregularidad en la polimerización de olefinas. Los métodos empleados habitualmente en la síntesis de poliolefinas son la polimerización radical, polimerización iónica y polimerización de coordinación y, dependiendo de la técnica empleada, los factores que controlan el curso estereoquímico de la polimerización son diferentes. El control estereoquímico depende de la temperatura, como en el caso de poliolefinas obtenidas por polimerización radical o mediante polimerización iónica en disolventes con alta capacidad de solvatación. O también puede estar ejercido por la coordinación entre el iniciador, el monómero y la cadena en crecimiento, como ocurre en polimerizaciones iónicas en disolventes con baja capacidad de solvatación, o en polimerización de coordinación utilizando catalizadores metálicos. En estos casos, es la coordinación la que dirige la aproximación de la olefina a través de una de las caras enantiomórficas, conduciendo a una polimerización estereoespecífica. En estos sistemas el iniciador (catalizador) no sólo es la especie que inicia la polimerización, sino que también promueve la orientación del monómero (Figura 2.3.4.1.2). Figura 2.3.4.1.2.- Iniciador (catalizador) iniciando y promoviendo la orientación del monómero 2.3.4.2.- Polimerización de Ziegler-Natta. El descubrimiento por Carl Ziegler en Alemania en 1955, de los catalizadores basados en metales de transición para la polimerización de alquenos ha sido de vital trascendencia en la producción industrial de poliolefinas. Así, especial interés tienen los catalizadores estereoespecíficos de Ziegler-Natta usados en algunas polimerizaciones industriales, como la del propileno. Carl Ziegler estudió la polimerización del etileno y Giulio Natta en Italia la del propileno. Ambos obtuvieron el premio Nobel en el año 1963. La polimerización de Ziegler-Natta es un método utilizado en la polimerización vinílica. Es importante porque permite obtener polímeros con una tacticidad específica. Es sobre todo útil, porque permite hacer polímeros que no pueden ser hechos por ningún otro camino, como el polietileno lineal no ramificado y el polipropileno isotáctico. La polimerización vinílica de radicales libres sólo puede dar polietileno ramificado y el propileno no polimeriza del todo por polimerización de radicales libres. Pero como los centros activos encuentran en entornos distintos, la catálisis dar lugar a velocidades diferentes de polimerización (propagación y terminación). Entonces se produce polímeros con una gran variedad de pesos moleculares. El catalizador de Ziegler-Natta por lo general TiCl3 o TiCl4, como puede observarse implica la presencia de un metal de transición. También están implicados cocatalizadores y estos, por lo general, están basados en los elementos del grupo III, que son metales como el aluminio. Así, en general, nuestro par (Catalizador/No-catalizador) será TiCl3 y Al(C2H5)2Cl, o TiCl4 con Al(C2H5)3. La presencia del cloruro de titanio (u otro metal de transición, como el vanadio o el cobalto), junto con el aluminio trietilo (u otro compuesto órgano-metálico del segundo o tercer grupo) orienta la posición de los sustituyentes de las moléculas del monómero de una manera ordenada, debido a la formación de un complejo de coordinación. El propileno es activado por este complejo, intercalándose entre el enlace titanio-alquilo, pero, debido al tamaño relativamente grande de los átomos de cloro, sólo puede hacerlo en una determinada posición. El crecimiento del grupo alquilo se hace, por consiguiente, a partir del titanio, de forma estéreo-específica. A este tipo de polimerizaciones se les denomina de coordinación. La polimerización resulta ser así una inserción de moléculas de monómero en el enlace entre el metal y la cadena polimérica en crecimiento. A diferencia de las polimerizaciones radicalaria e iónica (catiónica y aniónica) en las cuales el iniciador transfiere su función (radical o especie cargada) a la cadena en crecimiento, en la polimerización por coordinación cada paso de crecimiento regenera la capacidad de coordinación del complejo del metal. La polimerización termina por agregado de sustancias como agua, metanol, aminas, ácidos, las cuales destruyen la unión de la cadena polimérica con el metal. Ventajas. - Las condiciones de reacción son muy suaves, a presión atmosférica y a bajas temperaturas (-70 ºC). - Origina moléculas lineales. Por ejemplo, polietileno de alta densidad, el cual tiene un alto grado de cristalinidad, lo cual resulta en un polietileno de mayor punto de fusión y con una resistencia mecánica mucho mayor. - Permite un control esteroquímico de la reacción. Por ejemplo, en la obtención de polipropileno isotáctico, altamente cristalino. Se usa en decoración de interiores, cajas de baterías de autos, empaques, pasto sintético, sillas. Estudiaremos el sistema formado por el TiCl3 como catalizador y el Al(CH2-CH3)2Cl como cocatalizador, viendo como trabaja el sistema para hacer polímeros. El TiCl3 puede disponerse en diversas estructuras cristalinas, estando interesados en la α-TiCl3, que se puede representar como (En la superficie hay átomos de Ti con un orbital vacio): Estructura cristalina del α-TiCl3 Como puede verse, cada átomo de titanio esta coordinado con seis átomos de cloro, con la geometría octaedral. Esta disposición es como el titanio se encuentra más cómodo, es decir cuando esto esta coordinado a otros seis átomos. Esto presenta un problema para los átomos de titanio en la superficie del cristal. En el interior del cristal, cada titanio es rodeado por seis cloros, pero sobre la superficie, un átomo de titanio esta rodeado sobre un lado por cinco átomos de cloro, pero del otro lado por el espacio vacío. El titanio es un metal de transición que tiene seis orbitales vacíos siendo el resultado de un orbital 4s y cinco orbitales 3d) en su capa más exterior de electrones (Figura 2.3.4.2.1). Figura 2.3.4.2.1.- Orbital 4s y cinco orbitales 3d. El titanio tiene que estar coordinado con bastantes átomos para poner dos electrones en cada uno de sus orbitales. El átomo de titanio sobre la superficie del cristal tiene bastantes átomos vecinos para llenar cinco de los seis orbitales. El sexto orbital esta vacío, y se muestra como un cuadrado vacío en la imagen. Pero el titanio quiere llenar su orbital. Entonces entre en juego el co-catalizador, Al(C2H5)2Cl y dona uno de sus grupos de etilo al titanio empobrecido, pero en dicho proceso echa fuera uno de los cloros. Todavía tenemos un orbital vacío. Como puede verse, el aluminio primeramente se queda coordinado, aunque no enlazado covalentemente, al átomo de carbono del grupo de etilo CH2, que justamente se ha donado al titanio. No sólo eso, también se coordina a uno de los átomos de cloro adyacentes al titanio. Pero el titanio todavía tiene un orbital vacío que puede llenarse. Entonces un monómero vinílico, como por ejemplo el propileno, aparece. Hay dos electrones en el sistema-p de un doble enlace de carbono – carbono, que pueden ser usados para llenar el orbital vacío del titanio. Se dice que el propileno y el titanio forman un complejo, y se dibuja como sigue: Figura 2.3.4.2.2.- Formación de un complejo entre el propileno y el titanio Pero el proceso de formación de complejos es un proceso más bien complicado, no tan simple como nos indica la figura 2.3.4.2.2. Complejos alqueno-metal. Supongamos que aparece un monómero vinílico, como por ejemplo, una molécula de propileno. El zirconio va a disfrutar de esta situación. Para entenderlo mejor, observemos el monómero vinílico, concretamente, su doble enlace. Un enlace doble carbono-carbono está constituido por un enlace σ y un enlace π. El enlace π consiste en dos orbítales π. Uno es el orbital π enlazante (esquematizado en azul) y el otro es el orbital π antienlazante (mostrado en rojo). El orbital π enlazante posee dos lóbulos situados entre los átomos de carbono, mientras que el orbital π antienlazante tiene cuatro lóbulos que se asoman desde los dos átomos de carbono. Por lo general el par de electrones permanece en el orbital π enlazante. El orbital π antienlazante tiene demasiada energía, por lo que bajo circunstancias normales, se encuentra vacío. Veamos el zirconio (1s22s22p63s23p63d104s24p64d25s2). La figura 2.3.4.2.3 nos muestra el zirconio y dos de sus orbitales d. En realidad, el zirconio posee cinco orbitales d, pero para mayor claridad, sólo se muestran dos. Figura 2.3.4.2.3.- Zirconio y dos de sus orbitales d Uno de los orbitales d que se muestran, es el orbital vacío. Está representado por lóbulos verdes. Los lóbulos rosa constituyen uno de los orbitales d completos. Ese orbital d vacío va a buscar un par de electrones y sabe que el orbital π enlazante del alqueno posee un par para compartir. De modo que el orbital π enlazante del alqueno y el orbital d del zirconio se combinan y comparten un par de electrones. Pero una vez que se combinan, el otro orbital d se acerca extremadamente al orbital π antienlazante vacío. De modo que finalmente el orbital d y el orbital π antienlazante también comparten un par de electrones. Esta coparticipación adicional de electrones hace que el complejo sea más poderoso. Este acomplejamiento entre el alqueno y el zirconio establece las cosas para la polimerización. Polimerización. Polimerización isotáctica. La naturaleza precisa del complejo que se forma entre el titanio y el propileno es complicada. Con el fin de hacer las cosas simples solamente vamos a dibujarlo como se hizo antes, resultando: El complejo anterior soluciona de forma adecuada el problema que tenía el titanio con sus orbitales-d que no tenían bastantes electrones. Pero eso no puede continuar así. El complejo no va a quedarse así siempre y debe tener lugar algún reordenamiento de los electrones. Varios pares de electrones van a cambiar posiciones, como se muestra a continuación: No se sabe exactamente que pares cambian de sitio en primer lugar, pero se piensa que el primero en moverse es el par del enlace- π del carbono-carbono, que esta complejado con el titanio .Dicho par electrónica va a cambiar para formar el enlace simple carbono-titanio. Entonces los electrones del enlace entre el titanio y el carbono del grupo de etilo que titanio conseguido de Al(C2H5)2Cl. Este par de electrones va a cambiar de sitio para formar un enlace entre el grupo etilo y el carbono substituido del grupo metilo del monómero propileno. Se termina con la estructura que se ve en el lado derecho del esquema anterior. Que pasa después es lo que llamamos una migración. No se sabe porque ocurre, solamente sabemos que pasa. Los átomos se reorganizan para formar una estructura ligeramente diferente, como esta: El aluminio esta ahora complejado con uno de los átomos de carbono del monómero propileno. Como puede verse, el titanio esta como al comienzo, con un orbital vacío, necesitando electrones para llenarlo. Cuando viene otra molécula de propileno, el proceso entero comienza por todas partes, y el resultado final es algo como esto: y desde luego, cada vez más y más moléculas de propileno reaccionan, y nuestra cadena de polímero crece y crece. Se puede ver en la figura que todos los grupos metilo en el polímero creciente están en el mismo lado de la cadena. Con este mecanismo se obtiene el polipropileno isotáctico. El truco está en que cada molécula de propileno que viene se coordinará exactamente del mismo modo que el anterior. Esto es debido a la simetría del sitio activo, que a su turno esta determinado según la disposición de la cadena del polímero creciente. Polimerización sindiotáctica. El sistema descrito anteriormente solamente da polímeros isotácticos. Pero otros sistemas pueden dar lugar a polímeros sindiotácticos, como el basado en el vanadio, en vez del titanio. El sistema es VCl4/Al(C2H5)2Cl, que no es demasiado diferente del sistema del titanio. La estructura del complejo es análoga, pero no se da la migración. El siguiente se adiciona en el otro orbital. Este complejo actuara del mismo modo que el sistema del titanio cuando una molécula propileno viene en su camino. Primeramente, el propileno forma un complejo con el vanadio y entonces se produce un movimiento de electrones justamente como antes, y el propileno es insertado entre el metal y el grupo de etilo, igual que antes. Todo eso se muestra esto en la imagen siguiente: : pero también puede verse una diferencia importante en la imagen. En el sistema del titanio, el polímero creciente encadena posiciones de cambios sobre el átomo de titanio. Esto no pasa aquí. La cadena de polímero creciente permanece en su nueva posición. Es decir, hasta que venga otra molécula de propileno. Este segundo propileno reacciona mientras que la cadena creciente esta todavía en su nueva posición, justo como se ve a continuación: Pero puede observarse que cuando el segundo propileno se añade a la cadena, esta cambia de posición otra vez. Esta por detrás de la posición donde comenzó. Se pueden observar los grupos metilo del primer monómero, en azul, y el segundo monómero, en rojo, que están en lados opuestos de la cadena de polímero. Cuando la cadena de polímero creciente está en una posición el monómero propileno sólo puede añadirse de modo que el grupo de metilo esté en un lado de la cadena. Cuando la cadena está en otra posición, el propileno sólo puede añadirse de modo que el grupo metilo cuelgue del otro lado. Debería ser obvio por qué las inserciones sucesivas de propileno son diferentes, ya que los sitios catalíticos tienen simetrías diferentes. En resumen, se tiene la siguiente secuencia: Limitaciones. La polimerización de Ziegler-Natta es un procedimiento de hacer polímeros a partir de monómeros hidrocarbonados, como por ejemplo el etileno y el propileno. Pero no trabaja para algunas otras clases de monómeros. Por ejemplo, no se puede hacer el cloruro de polivinilo (PVC) mediante la polimerización de Ziegler-Natta. Cuando el catalizador y el cocatalizador vienen juntos para formar el complejo de iniciación, los radicales se producen durante los pasos intermedios de la reacción. Estos pueden iniciar la polimerización de radicales libres del monómero cloruro de vinilo. Los acrilatos también están fuera, porque los catalizadores de Ziegler-Natta a menudo inician la polimerización aniónica de vinilo en dichos monómeros. Durante un largo periodo de tiempo, la polimerización de Ziegler-Natta era la reacción más útil y versátil para producir los polímeros de tacticidad específico deseada. Pero recientemente un nuevo tipo de polimerización, usando también complejos metálicos como iniciadores, ha sido desarrollada, y se denomina polimerización catalizada con metalocenos. 2.3.4.2.- Polimerización catalizada con metalocenos. La polimerización catalizada por metalocenos resulta ser la más indicada para competir con los polímeros vinílicos desde que se inventó la polimerización Ziegler-Natta. La razón es que la polimerización catalizada por metalocenos permite producir polietileno capaz de detener las balas. Este nuevo polietileno es mejor que el Kevlar para la fabricación de chalecos a prueba de balas. Y puede lograrlo porque tiene un peso molecular mucho más alto (Hasta seis o siete millones) que el polietileno sintetizado por medio del procedimiento de Ziegler-Natta. Pero hay más que elevados pesos moleculares. Así, se pueden enumerar las siguientes características generales de los catalizadores metalocenos 1.-Pueden polimerizar casi cualquier monómero 2.-Producen polímeros extremadamente uniformes. 3.- Permite hacer polímeros con tacticidades muy específicas. Dependiendo de las necesidades, puede ponerse a punto para hacer polímeros isotácticos y sindiotácticos. Polimerizan α-olefinas con una alta estereoregularidad para dar polímeros isotácticos o sindiotácticos Los metalocenos son iones metálicos con carga positiva, entre medio de dos aniones ciclopentadienilo, con carga negativa. Un anión ciclopentadienilo es un pequeño ión formado a partir de una molécula llamada ciclopentadieno. Se puede observar que existe un átomo de carbono con dos hidrógenos, mientras que el resto tiene sólo uno. Estos dos hidrógenos son ácidos, es decir, pueden desprenderse con facilidad. Cuando uno de ellos se va, abandona los electrones del enlace. De modo que el carbono que queda, tiene un par electrónico extra. Además el ciclopentadieno tiene dos enlaces dobles en la molécula y cada uno de ellos tiene dos electrones, de modo que en total suman cuatro. Si se le suman esos dos electrones de más sobre el carbono que perdió un hidrógeno tendremos seis. Esto es importante, ya que un anillo con seis electrones, se volverá aromático y en esa forma aniónica será sumamente estable. Ciclopentadieno Ciclopentadienilo Los iones ciclopentadienilo tienen carga -1, de modo que cuando aparece un catión como el Fe con carga +2, dos de los aniones formarán un "sandwich" con el hierro. Este "ferro-sandwich" se denomina ferroceno [(C5H5)2Fe]. A veces se encuentra involucrado un catión de carga mayor, como el zirconio con carga +4. Para balancear la carga, el zirconio se unirá a dos iones cloruro, cada uno con carga -1, para dar un compuesto neutro. Bis- Clorozirconoceno Los zirconocenos son un poco diferentes de los ferrocenos. Esos ligandos extras, los cloruros, ocupan un espacio. Resulta difícil para ellos deslizarse entre los anillos ciclopentadienilo. Por lo tanto, para hacerles lugar a los cloruros, los anillos se inclinan entre sí, abriéndose como el caparazón de una almeja. En los metalocenos con más de dos ligandos los anillos pueden no estar paralelos sino en ángulo lo que permite la movilidad de los otros ligandos. Como puede verse, los anillos ciclopentadienilo, representados con líneas oscuras gruesas, son paralelos entre sí en el ferroceno, pero forman un ángulo en el zirconoceno. Esta inclinación ocurre siempre y cuando un metaloceno tenga más ligandos que sólo los dos anillos ciclopentadienilo. Podemos emplear algunos derivados del bis-clorozirconoceno para obtener polímeros. Por ejemplo, éste: Anillos indenilo Se diferencia del bis-clorozirconoceno en el sentido que cada anillo ciclopentadienilo tiene fusionado un anillo aromático de seis carbonos, mostrado en rojo. Este sistema de dos anillos formado a partir de un anillo ciclopentadienilo fusionado con un anillo fenilo, se denomina ligando indenilo. Por otra parte, hay un puente etileno, representado en azul, que une los anillos ciclopentadienilo, el superior y el inferior. Estas dos características hacen de este compuesto, un excelente catalizador para la obtención de polímeros isotácticos. Los voluminosos ligandos indenilo, dispuestos en posiciones opuestas como están, dirigen los monómeros entrantes, así sólo pueden reaccionar cuando se encuentren en la dirección correcta, para dar polímeros isotácticos. Ese puente etileno mantiene en su lugar a los dos anillos indenilo. Sin el puente, girarían y podrían no situarse en el lugar correcto para dirigir la polimerización isotáctica. Hemos hablado sobre lo que son los metalocenos y que son capaces de hacer polímeros con una tacticidad específica. Pero no hemos dicho nada acerca de cómo ocurre realmente la polimerización. Para lograr que nuestro complejo zirconoceno catalice una polimerización, lo primero que debemos hacer es agregar algo de MAO, que es la abreviatura de metil alumoxano, el cual es un polímero, cuya estructura es la siguiente: Es un polímero extraño porque posee átomos metálicos en su cadena principal. Pero estamos más interesados en lo que hace que en lo que es. Coloquemos una pequeña porción de este compuesto (de un centésimo a un décimo del tanto por ciento total del catalizador) y hará algo con los cloruros del zirconoceno, que son lo que llamamos lábiles. Esto quiere decir que pueden desprenderse fácilmente del zirconoceno. Por lo tanto el metil alumoxano puede reemplazarlos con algunos de sus grupos metilo (Por reacción con metil alumoxano (MAO) se pueden sustituir los cloros por grupos metilo) y nos queda un catalizador similar a éste: Sin embargo, los grupos metilo también pueden desprenderse fácilmente (Puede perder un grupo metilo para dar un catión). Cuando uno de ellos lo hace, obtenemos un complejo como éste : Se puede observar que el zirconio cargado positivamente es estabilizado, ya que los electrones del enlace carbono-hidrógeno son compartidos con el zirconio. Esto se denomina α-asociación agóstica (El catión se estabiliza por cesión de densidad electrónica del enlace carbono-hidrógeno). Pero aún así, al zirconio le faltan electrones. Necesita más de una asociación agóstica para ser satisfecho. Ahí es cuando entra en juego nuestro monómero olefínico, por ejemplo un alqueno como el propileno, que puede estabilizar la carga positiva formando un complejo. La estereoquímica de este complejo controla la del producto final. Su doble enlace carbono-carbono está repleto de electrones para compartir. De modo que comparte un par con el zirconio. Figura 2.3.4.2.1.- Monómero olefínico (propileno) estabilizando la carga positiva formando un complejo. Pero el acomplejamiento es un proceso más bien complicado, no tan simple como sugiere la figura 2.3.4.2.1 anterior, y que ya se ha visto al estudiar la polimerización de Ziegler-Natta. Pero ahora los complejos son algo diferentes de los existentes en la polimerización de Ziegler-Natta, donde el otro orbital-d está sumamente próximo al orbital π-antienlazante vacío. Por tanto, el orbital-d y el orbital π-antienlazante comparten un par de electrones, pero en el caso del metalocenos esto no es posible. El metal en los complejos de metalocenos es d0 (a diferencia de d2 en los sistemas de ZieglerNatta) y simplemente no hay bastantes electrones alrededor para que ocurra el ―enlace por la parte de atrás. El complejo esta estabilizado por los orbitales π-antienlazantes de la olefina, que interaccionan con los orbitales-d vacíos del metal de transición. Esta formación de complejos entre el alqueno y el circonio establece las cosas para el siguiente paso de la polimerización. Polimerización. La naturaleza exacta del complejo entre el zirconio y el propileno es complicada. Para simplificar, de ahora en adelante vamos a representarlo tal como lo hicimos antes, es decir, así: Este acomplejamiento estabiliza el zirconio, pero no por mucho tiempo. Es decir, cuando este complejo se forma, puede reordenarse en una nueva forma. Los electrones comienzan a moverse, como se puede observar en la figura 2.3.4.2.2. Los electrones del enlace zirconio-carbono metílico se desplazan para formar un enlace entre el carbono metílico y uno de los carbonos del propileno. Entre tanto, el par electrónico que había participado del enlace alqueno-complejo metálico se desplaza para formar un enlace entre el zirconio y uno de los carbonos del propileno. Figura 2.3.4.2.2.-. Estado de transición cíclico de cuatro miembros a través del cual se da la primera adición. Como se puede observar, esto ocurre a través de un estado de transición de cuatro miembros se da la primera adición. Además, el zirconio termina tal como empezó, perdiendo un ligando pero con una asociación agóstica con un enlace C-H del monómero propileno. Volviendo al comienzo, otro monómero propileno puede aparecer y reaccionar del mismo modo que lo hizo el primero. El propileno se coordina con el zirconio y luego los electrones cambian de posición: Así, un segundo monómero propileno se ha agregado a la cadena y se observa que terminamos con un polímero isotáctico. Los grupos metilo se encuentran siempre del mismo lado de la cadena polimérica. El próximo monómero que aparezca se coordinará con el zirconio en el mismo lado que el primero. La dirección en que se aproximan, cambia con cada monómero agregado. Pero entonces, ¿por qué se obtiene un polímero isotáctico? Fijémonos por un momento en el catalizador y en un monómero propileno entrante. Como puede apreciarse, el monómero propileno siempre se acerca al catalizador con su grupo metilo dirigido en el sentido opuesto del ligando indenilo. Si el grupo metilo se dirigiera hacia el ligando indenilo, ambos se encontrarían, evitando que el propileno se acercara lo suficiente al zirconio para formar el complejo. De modo que sólo cuando el metilo apunta en sentido contrario respecto al ligando indenilo, el propileno puede acomplejarse con el zirconio. Cuando se agrega el segundo monómero, éste debe aproximarse desde el otro lado y también sus grupos metilo deben apuntar en sentido contrario a los anillos indenilo. Pero se puede observar que esto significa que el grupo metilo apunte hacia arriba en lugar de hacerlo hacia abajo. Dado que el segundo propileno se adiciona desde el lado opuesto al primero, debe dirigirse en dirección opuesta para que los grupos metilo terminen del mismo lado de la cadena polimérica. 30 31 32 33 34 35 36 37 38 39 40 41 42 43 Sabiendo que el catalizador anterior nos conduce a polímeros isotácticos, ¿qué tipo de catalizador nos dará polipropileno sindiotáctico? Es un catalizador como éste, que fue investigado por Ewen y Asanuma. Es fácil comprender porqué se obtiene polimerización sindiotáctica con este catalizador. Los monómeros se aproximan sucesivamente desde los lados opuestos del catalizador, pero siempre dirigen sus grupos metilo hacia arriba. De esta forma, los grupos metilo terminan en lados alternados de la cadena polimérica. Sin embargo los catalizadores metalocénicos pueden hacer cosas más extrañas que esas. Consideremos el dicloruro de bis(2-fenilindenil)zirconio. Este metaloceno, como puede apreciarse debajo, no tiene un puente entre los dos anillos indenilo. Esto quiere decir que los dos anillos pueden girar libremente. A veces pueden estar apuntando en direcciones opuestas. Llamamos a ésta, forma rac. Otras veces pueden apuntar en la misma dirección. A esta situación le decimos forma meso. El compuesto pasa un tiempo en la forma rac y luego se da vuelta, pasando a la forma meso. Al cabo de un instante, rotará nuevamente. Esto sucede una y otra vez. Con respecto a la polimerización, lo que ocurres es que cuando el zirconoceno está en la forma rac, el monómero polipropileno sólo puede acercarse en una orientación que generará polipropileno isotáctico. Pero cuando el zirconoceno gira y adopta su forma meso, el monómero propileno puede aproximarse con cualquier orientación. Esto originará un polipropileno atáctico. Por tanto, el zirconoceno se encuentra alternando constantemente entre las formas. Lo hace aún cuando se está produciendo la polimerización. Esto quiere decir que la misma cadena polimérica tendrá bloques atácticos y bloques isotácticos, como éstos: Esta clase de polipropileno es denominada polipropileno elastomérico porque es realmente eso, un tipo especial de elastómero llamado elastómero termoplástico. 3.- Polimerización por condensación o escalonadas. 3.1.- Introducción. En este apartado se aborda la otra gran familia de polimerizaciones, las polimerizaciones denominadas por etapas y sus aspectos cinéticos. Tales polimerizaciones son la vía de obtención de termoplásticos tan importantes como las poliamidas o los poliésteres que, además de su uso como termoplásticos convencionales (por ejemplo, el polietilentereftalato, PET), tienen también una amplia implantación en el ámbito de las fibras. Por otro lado, son igualmente procesos por etapas los que proporcionan la mayor parte de los polímeros reticulados o termoestables. Los aspectos cinéticos de este tipo de polimerización son bastante diferentes de los que se dan en las polimerizaciones en cadena. La característica diferencial de las polimerizaciones par etapas es la estrecha relación existente entre el grado de conversión y el peso molecular finalmente obtenido. Por otro lado, y en lo que se refiere a los polímeros termoestables, son aspectos ligados a la cinética de polimerización los que permiten simular el grado de conversión critico necesario para que se alcance la reticulación necesaria y que el material se comporte como tal termoestable. Una parte importante de los materiales sintéticos más utilizados con relevante incidencia en aplicaciones muy concretas como fibras, adhesivos, resinas, elastómeros, etc., son los llamados polímeros de condensación, nombre que surge del tipo de proceso de polimerización que los genera. Desde el punto de vista del mecanismo de crecimiento del polímero, la polimerización de condensación procede mediante una sucesión de diferentes etapas que implican una serie de reacciones elementales entre centros reactivos que, generalmente, son grupos funcionales en el sentido clásico de la química orgánica. Por esta razón, este tipo de polimerizaciones se conocen también bajo el nombre de polimerización por etapas. La química de la polimerización por etapas es básicamente la misma que la de las reacciones de condensación clásicas como, por ejemplo, las que conducen a la formación de ésteres no poliméricos como el acetato de etilo. La obtención de polímeros de alto peso molecular exige el requerimiento adicional de emplear reactivos (monómeros) que tengan en su molécula dos o más grupos funcionales, iguales o distintos, con objeto de que la reacción progrese. Mientras que los polímeros de condensación sólo constituyen un pequeño porcentaje de los polímeros sintéticos, la mayoría de los polímeros naturales son de este tipo. La baquelita, que fue el primer polímero completamente sintético, se obtuvo por condensación escalonada de fenol y de formaldehído. La reacción tiene lugar simultáneamente entre moléculas de monómero entre sí, que producen dímeros, trímeros, tetrámeros, etc. y de éstas con otras, dando origen a macromoléculas de mayor tamaño, sin que se encuentren diferencias en cinéticas, ni de mecanismos, cualquiera que sea el tamaño de las moléculas reaccionantes. Los grupos reactivos pueden estar en la misma molécula (aminoácidos como la ε-caprolactama, por ejemplo, como en el caso del «nylon» 6 perteneciente al grupo de las poliamidas) y entonces sólo existe un único monómero que reacciona y que contiene ambos grupos funcionales: 3nA B A B A B A B n En el caso del nylon 6 se tiene: H 2 N-CH 2 -CH 2 -CH 2 -CH 2 -CH 2 - COOH - → Poliamida(Nylon 6)+ H2O Los grupos reactivos pueden estar en moléculas distintas: 3nA A 3nB B A A B B A A B B A A B B n como en el caso del polietilentereftalato, que es un polímero que se obtiene mediante una reacción de policondensación entre el ácido tereftálico y el etilenglicol. Pertenece al grupo de materiales sintéticos denominados poliésteres. Es un polímero termoplástico lineal, con un alto grado de cristalinidad. O también como en el caso del Nylon 6.6, que pertenece al grupo de materiales sintéticos denominados poliésteres y que se genera formalmente por policondensación de un diácido (Ácido adípico) con una diamina (Hexametilenediamina), mediante el proceso siguiente: Este tipo de reacciones puede también llevarse a cabo con monómeros que posean más de dos grupos funcionales (multifuncionales), obteniéndose en esos casos cadenas que presentan estructuras no lineales debido a la presencia de ramificaciones. Además, bajo adecuadas combinaciones de monómeros bi- o multifuncionales, las cadenas en crecimiento pueden unirse entre si, dando lugar a estructuras en forma de red (reticuladas). Hay otros hechos relevantes a recordar, por las implicaciones que ello tiene sobre la cinética del proceso. Uno de ellos se refiere a la obtención en cada etapa de las polimerizaciones por etapas (aunque no en todos los casos) de una molécula pequeña. Esa molécula suele ser agua en muchos casos aunque también puede obtenerse ácido clorhídrico u otras moléculas. Un segundo hecho relevante es la posible reversibilidad de cada una de esas etapas. La única manera de obtener altos grados de conversión y altos grados de polimerización es desplazar ese equilibrio en el sentido de la formación de polímero, lo que puede llevarse a cabo eliminando los productos secundarios antes mencionados (agua, etc.) mediante métodos físicos (aplicación de vacío, altas temperaturas) o químicos (neutralización de ácidos con álcalis). Sin embargo, la utilización de éstos u otros métodos debe hacerse de forma que no afecten a la estequiometria de los reactantes pues ello puede afectar a distintas características del policondensado El grado de polimerización medio numérico (dp) N definido como la relación entre el peso molecular medio numérico del polímero M N y el del residuo monomérico M 0 puede expresarse también como la relación entre el número de moléculas iniciales de monómero N0 y el número final de moléculas presentes en la masa después de la polimerización N: dp N N0 N El grado de conversión X necesario para conseguir un grado de polimerización determinado (dp) N queda definido por las ecuaciones de Carothers: X N0 N , N0 dp N 1 1 X Para polimerizaciones de monómeros AB, o de AA + BB en la relación 1/1, el grado de conversión de las moléculas es igual al grado de conversión de los grupos funcionales A y B, es decir: X X A XB 0 N AA En términos generales cuando 0 r , la relación entre los grados de conversión de los grupos NBB funcionales A y B es: X B rX A por lo que el grado de polimerización medio numérico definido en (1) puede cal cularse mediante la siguiente expresión: 0 0 N AA NBB 0 0 N AA 1 X A NBB 1 rX B dp N y teniendo en cuenta que rNBB N AA resulta: 0 0 dp N 1 r r 1 2rX A Esta expresión permite analizarla influencia de la concentración de los reactivos y del grado de conversión sobre el grado de polimerización. Así, por ejemplo, para obtener un polietilentereftalato de grado de polimerización 20, con relación de monómero 1/1 (r = 1), el grado de conversión necesario sería: 20 1 de donde: 1 X A X A 95 % y para (dp) N = 40 , sería: 40 1 de donde: 1 X A X A 97.5 % Si la reacción se realiza con un exceso del 5 % de glicol sobre el estequiométrico 0 N AA 1 0.952 se tienen las siguientes relaciones: 0 NBB 1.05 1.952 13.6 X A 95 %, dp N 1.952 1.8088 X A 97.5 %, dp N 1.952 20.4 1.952 1.8564 Queda así puesta de manifiesto una posible manera de limitar el grado de polimerización (y del peso molecular, por lo tanto), mediante uso de un reactivo en exceso. Si la polimerización finaliza sin que se agote ninguno de los grupos funcio nales, el grado de polimerización podría incrementarse cuando el material se calienta y plastifica para su transformación mientras que, si la reacción se detiene porque se ha consumido totalmente uno de los grupos funcionales (XA → 1) , no puede continuar durante el proceso de transformación. Otra posibilidad es la adición controlada de un reactivo monofuncional (etanol, en el ejemplo utilizado), siendo entonces: 0 N AA r 0 NBB NB0 0 N AA 0 0 Sea el caso en que, con una relación 0 1 , se añade un 2.5 % de N B con respecto a NBB : NBB r 1 0.952 1.05 con lo que se obtendrían los mismos resultados que añadiendo un 5 % de exceso de BB. En la mayoría de los casos, esta alternativa resulta más económica. Características de las polimerizaciones escalonadas. En general las reacciones de este tipo no son excesivamente exotérmicas y sus energías de activación son moderadas. Las velocidades de reacción son reducidas a temperatura ambiente, requiriéndose mucho tiempo para alcanzar un grado de polimerización elevado (y en consecuencia un grado de conversión próximo al 100 %). Siempre que se condensen moléculas sencillas de bajo punto de ebullición, hay que retirarlas del reactor, mediante un condensador de reflujo. A medida que aumenta el grado de conversión la viscosidad de la masa reaccionante aumenta y es necesario mantener un alto nivel de agitación para que no disminuya la velocidad de reacción. Aumentando la temperatura, además de aumentar la velocidad de reacción, la viscosidad de la masa reaccionante disminuye y se mejoran las condiciones de transferencia de calor. Generalmente se trabaja entre 150 y 250 °C. El grado de polimerización depende fundamentalmente del grado de conversión obtenido y de la relación de los reactivos, según indican las ecuaciones de Carothers. La temperatura tiene muy poco efecto en la longitud media de las cadenas moleculares del producto final. 3.2.- Cinética de la polimerización por etapas. Poliesterificación. La polimerización por etapas es un proceso que implica el concurso de uno o más monómeros que contengan, por lo menos, dos grupos funcionales iguales o distintos. Estos monómeros, al reaccionar entre ellos (con o sin eliminación de moléculas pequeñas), forman un primer producto con un grupo funcional diferente. Por ejemplo, diácidos y dioles forman una primera molécula con un grupo éster en el medio y un grupo ácido y un grupo alcohol en los extremos. Este primer resultado de la polimerización puede reaccionar con los monómeros iniciales o con una estructura similar así misma. A su vez, estos nuevos productos pueden reaccionar entre sí, con los monómeros iniciales o con el primer producto de su reacción mutua. De esta formas, los monómeros iniciales desaparecen rápidamente en una fase temprana de la polimerización, generándose oligómeros de diversa longitud que van creciendo a medida que progresa la reacción entre las diversas cadenas presentes en el medio o, lo que es lo mismo, a medida que van desapareciendo del medio los grupos funcionales capaces de generar la unidad representativa del polímero que se está formando. Deben recordarse las diferencias o entre este tipo de procesos y los denominados en cadena, donde una vez que se ha generado una especie activa, ésta adiciona rápidamente monómero hasta completar una cadena. De esta forma, transcurridos tiempos importantes de reacción, la mezcla reaccionante contiene muchas cadenas que ya no crecerán más y cantidades importantes del monómero original. Ambas peculiaridades no se dan nunca en una polimerización por etapas. La polimerización por etapas procede, por tanto, mediante un conjunto enorme de reacciones separadas y consecutivas. El sistema, en cualquier instante, contiene una mezcla de moléculas de distinto tamaño, lo que de cara a su consideración cinética obligaría a introducir un número elevado de constantes de reacción (una para cada etapa), ya que la velocidad de polimerización es la suma de todas las velocidades de reacción entre moléculas de distintos tamaños. Este hecho, que dificulta el análisis cinético del proceso, puede simplificarse adecuadamente si se introduce el principio de igual reactividad de los grupos funcionales. Este principio supone considerar tres aspectos diferentes: por un lado, el que las reactividades de los diferentes grupos funcionales de un monómero son iguales; segundo, que la reactividad de un grupo funcional de un monómero es la misma independientemente del hecho de que el otro u otros grupos funcionales hayan reaccionado o no y tercero, y más importante por el número de etapas implicadas, la reactividad del grupo funcional no depende del tamaño de la molécula al que este unido. Conviene matizar, sin embargo, que este principio de igual reactividad no se cumple para todos los sistemas, sobre todo cuando se utilizan monómeros aromáticos y monómeros multifuncionales asimétricos. Otro factor importante que puede afectar a la reactividad es el hecho de que a medida que progresa la reacción, con el consiguiente aumento del peso molecular, la viscosidad del medio puede aumentar lo suficiente como para que las velocidades de reacción estén gobernadas por la difusión, anulando o matizando la hipótesis de igual reactividad. El efecto de la viscosidad en estas reacciones no es, sin embargo, tan importante como en las polimerizaciones en cadena. Aunque se pueden presentar situaciones más complejas, sólo consideraremos la cinética de la poliesterificación simple. La cinética de la mayoría las condensaciones habituales sigue mecanismos análogos. La cinética de la poliesterificación de un diácido (actúa como catalizador) con un glicol (diol), en relación equimolecular, con una concentración inicial C0 de los grupos funcionales, ácido y alcohol, idéntica para ambos. A medida que progresa la reacción, la concentración disminuirá a C, manteniéndose también idéntica, pues en la reacción se eliminan dichos grupos funcionales uno a uno. Se supone que el H2O totalmente a medida que se va formando, por lo que el grado de conversión será: X se elimina C0 C C0 y por consiguiente: C C0 (1 X ) La esterificación puede ser autocatalizada por el ácido, en cuyo caso la ecuación cinética será: Velocidad de policondensación r donde d A dt k A D 2 A Concentración de diácido D Concentración de diol Si A D C se puede escribir: r dC 2 k COOH OH kC 3 dt de donde: dC kdt C3 considerando que la velocidad de reacción es independiente de la longitud de la cadena molecular que separa a los grupos funcionales reactivos, es decir k = Cte , por integración se obtiene: 2kt 1 1 2 2 C C0 o bien: 2ktC02 1 1 X 2 1 En la síntesis de los poliésteres, serán necesarios tiempos de reacción mucho más altos para conseguir la formación de polímeros grandes en esterificaciones sin catalizadores que en los sistemas con catalizadores ácidos o básicos. Puesto que el ácido o la base añadidos se comportan como un catalizador, sus concentraciones aparentes no cambiarán con el tiempo, y por tanto no necesitan ser incluidas en la expresión de la velocidad de reacción. En el caso de añadirse un ácido fuerte como catalizador, la reacción se acelera y la ecuación cinética puede escribirse r dC k 'COOH OH k 'C 2 dt siendo k' una nueva constante cinética que incluye la constante de velocidad k y la concentración del catalizador, que permanece constante. Integrando: k 't o bien: k ' tC0 1 1 1 X 1 1 C C0 Ambas ecuaciones fueron verificadas experimentalmente por Flory en 1939, a diferentes temperaturas para la polimerización del ácido adípico con etilenglicol. El poliéster obtenido, no obstante, no tiene interés industrial. 3.3.- Polimerizaciones escalonadas con reticulación Cuando los reaccionantes son difuncionales (diol y diácido, por ejemplo), el grado de polimerización depende, no sólo del grado de conversión, sino también de la proporción inicial de los monómeros. Cuando parte de un reactivo difuncional se sustituye por otro trifuncional del mismo grupo (glicerina, en lugar de etilenglicol, en el ejemplo considerado), la ecuación de Carothers se modifica tomando la forma: dp N 2 2 Xfav de donde resulta X = grado de conversión de grupos funcionales 2 2 fav dp N fav Siendo fav, la funcionalidad media de los reactivos, definida en función de las relaciones molares en que intervienen los tres reactivos, por ejemplo: triol: fav N A0 moles de diol, N B0 moles de diácido y NC0 moles de 12r 2 3 2r 3r 1 siendo: 3NC0 0 2N A 3NC0 2N A0 3NC0 r 1 , 2N 0 Con lo que para el mismo grado de conversión el grado de polimerización resulta mucho mayor. Así, se puede comprobar que para una mezcla equimolar de diol y diácido (fav = 2), con conversiones del 90 y 95 %, los grados de polimerización son 10 y 20, respectivamente, mientras que, si la mezcla es de 0.7 moles de diol, 1 mol de diácido y 0.2 moles de triol se verifica que: r 2x 0.7 3x 0.2 1 , 2 x1 fav 3x 0.2 0.3 2x 0.7 3x 0.2 12x1 2.105 3 2x1x 0.3 3x1x 0.7 y con conversiones de 90 y 95 % se tienen grados de polimerización de 18.95, en el primer caso, y de 8000 en el segundo. La polimerización tridimensional alcanza un punto en el que se produce la gelación o formación de un reticulado de cadenas de polímero esencialmente de tamaño infinito en la mezcla en reacción (punto de gel). El súbito comienzo de la gelación señala la división de la mezcla en dos partes: el gel, que es insoluble en todos los disolventes no degradantes, y el sol, que sigue siendo soluble y puede extraerse del gel. Al proseguir la polimerización más allá del punto de gel, la cantidad de gel aumenta a expensas del sol y la mezcla se transforma rápidamente de un líquido viscoso en un material elás tico de viscosidad infinita. Una característica importante del comienzo de la gelación es el relativamente bajo peso molecular medio en número de la mezcla en ese punto, en el que su peso molecular medio en peso o ponderal se hace infinito. La relación entre el peso molecular medio en peso y el grado de conversión requiere conocer la función de distribución de los pesos moleculares en la mezcla reaccionante, sin embargo, puede determinarse un valor crítico del grado de conversión a partir del cual ya se puede afirmar que se ha producido la gelificación. Este valor crítico XG, será el que haga infinito el peso molecular medio numérico. Por lo tanto, XG 2 fav . La obtención experimental del punto gel en base a determinaciones de viscosidad dará un valor más bajo del grado de conversión que XG (debido a la relación f Mw ). Las variaciones de viscosidad se acentuarán en la vecindad del punto gel debido al incremento en la dispersión de pesos moleculares que se produce en este punto. La figura 3.3.1 muestra las variaciones del grado de conversión X, del peso molecular medio numérico (dp) N y de la viscosidad, en función del tiempo de reacción. Tiempo de reacción Figura 3.3.1.- Variaciones del grado de conversión X, del peso molecular medio numérico (dp) N y de la viscosidad, en función del tiempo de reacción. En el ejemplo que se consideraba más arriba, el punto gel se encontraría antes de XG = 0,9502. 3.4.- Estequiometría y control del peso molecular Se denomina relación estequiométrica r al cociente entre los números iniciales de moléculas de los monómeros A-A y B-B: r N0,b (3.4.1) N0,a definida siempre de forma que r ≤ 1 , es decir, en función del reactante cuyos grupos funcionales no están en exceso. Cuando r es diferente de uno se suele hablar de desajuste estequiométrico Los grados de conversión de la reacción se definen no con respecto al número de moléculas de los monómeros sino a los números de grupos funcionales A y B, que son los que desaparecen para formar la entidad que caracteriza al polímero: pa 2N F Número de grupos A reaccionados 0,a a Número inicial de grupos A 2N0,a (3.4.2) pb 2N F Número de grupos B reaccionados 0,b b Número inicial de grupos B 2N0,b (3.4.3) siendo Fa y Fb el número de grupos funcionales A y B presentes en un instante determinado. Como en cada instante, el número de grupos A que han reaccionado debe ser igual al número de grupos B que han reaccionado (las polimerizaciones por etapas como las consideradas implican reacciones uno a uno entre los grupos funcionales implicados), ello permite escribir que: r p b p a (3.4.4) La dependencia del peso molecular con el grado de conversión o, lo que es lo mismo, con el tiempo de reacción, permite suponer que si detenemos la polimerización por algún método en un instante determinado, podemos obtener un polímero con un peso molecular concreto. Sin embargo, esta situación debe matizarse ya que si las moléculas de polímero así obtenidas contienen grupos funcionales en sus extremos, éstos podrían reaccionar con posterioridad si se dan las adecuadas condiciones, originando una variación del peso molecular que puede no ser interesante en el tipo de aplicación en la que pretenda emplearse el policondensado. Esta situación puede evitarse polimerizando de forma que exista un exceso de alguno de los monómeros. Este grupo funcional en exceso asegura que todas las moléculas de polímero tendrán ese mismo grupo funcional en los extremos, una vez que el monómero en defecto haya desaparecido completamente. Con esa estrategia se obtiene un polímero estable en tanto que incapaz de progresar o reaccionar ulteriormente. Esta diferencia estequiométrica puede provocarse directamente, poniendo en la alimentación inicial un exceso de alguno de los mono-meros (por ejemplo, un exceso de diamina en la polimerización por etapas de una poliamida). También puede alterarse la estequiometria mediante la adición de un monómero mono-funcional (por ejemplo, ácido laúrico, benzoico, etc.). El efecto cuantitativo de estos desajustes estequiométricos sobre el peso molecular puede llegar a ser significativo. Se comenzará con polimerizaciones por etapas de monómeros bifuncionales del tipo A-A con monómeros de tipo B-B, donde se considerará que el monómero B-B está en exceso. La relación o desajuste estequiométrico, ecuación (3.4.1) ha sido ya definida de forma que su valor sea menor que la unidad, por tanto, en este caso. r= (N0a/N0b). El grado de polimerización promedio en número se expresa, de nuevo, como el cociente entre el número total de moléculas iniciales N0 y el número total de moléculas de polímero N. El número total de moléculas de monómero inicialmente presentes es ahora: N0 N0,a N0,b 2 1 N0,a 1 r 2 (3.4.5) El número total de moléculas de polímero es equivalente a la mitad del número total de grupos A y B que no han reaccionado, ya que cada cadena tiene dos extremos pero con un grupo funcional diferente en cada uno de ellos. Este número total de moléculas de polímero se puede relacionar con la conversión p, definida como la fracción de grupos A que han reaccionado en un tiempo determinado. Si p es la fracción de grupos A que han reaccionado, la fracción de grupos A no reaccionados será (1 — p). En el presente caso, la fracción de grupos B reaccionados es distinta, debido al desajuste estequiométrico, pero puede escribirse como rp, con lo que la fracción de grupos B que no han reaccionado será (1- rp). Por tanto, el número total de grupos A y B no reaccionados es N0,a(1 — p) y N0,b(1- rp), respectivamente, con lo que el número total de moléculas de polímero será la mitad de la suma de ambos. Si ahora se llevan los valores de N0 y N a la definición del grado de polimerización promedio en número, se obtiene la siguiente expresión: xn N0 N N0,a 1 1 r 1 r 2 N0,a 1 p N0,b 1 rp 1 r 2rp (3.4.6) 2 La cual refleja la dependencia del grado de polimerización con el desajuste estequiométrico y la conversión y donde pueden significarse dos casos extremos. Si r = 1, se obtiene la ecuación: xn N0 N M 0 M 0 M M 0 1 p 1 1 p (3.4.7) mientras que si el grado de conversión (p) lo igualamos a la unidad, se obtiene: xn 1 r 1 r (3.4.8) que refleja el máximo valor del grado de polimerización que se puede obtener cuando se polimeriza en condiciones no estequiométricas al 100% de conversión. Este valor es siempre más pequeño que el que se obtiene en condiciones estequiométricas. En la figura 3.4.1 se observa la dependencia de x n con respecto a la relación estequiométrica para diversas conversiones según la ecuación (3.4.7). La constatación más evidente es que la obtención de un determinado peso molecular requiere un perfecto control de la estequiometria y de la conversión. Pequeñas variaciones en la estequiometria suponen reducciones considerables del grado de polimerización. Y así, una polimerización realizada con un desajuste estequiométrico r = 0,99 hasta una conversión p = 0,99 da lugar a que x n se reduzca de 100 a 67 al compararlo con el que se obtiene en condiciones estrictamente equimolares. Asimismo, para la obtención de relativos altos pesos moleculares, es necesario alcanzar conversiones mayores que el 98%. Una cuidadosa consideración de la figura 3.4.1 permite vislumbrar la importancia que pueden tener la existencia de impurezas en los monómeros, que alteran la estequiometria, así como la pérdida de alguno de los monómeros en los procesos de eliminación de los subproductos (evaporación, arrastre, etc.). Figura 3.4.1.-Influencia de la relación estequiométrica en el grado de polimerización. Los números indican el grado de conversión (p). En un segundo caso, se va a considerar la polimerización por etapas (en condiciones igualmente equimolares) de monómeros del tipo A-A con monómeros B-B, acompañados ambos por la presencia de un monómero monofuncional que se va a suponer es del tipo B. Las expresiones que se obtienen son las mismas que anteriormente, con la diferencia de que la relación estequiométrica viene dada ahora por la expresión: r N0,a N0,b 2N0,b´ (3.4.9) siendo N0,b´ el número de moléculas del monómero monofuncional B y N0a= N0,b´. Debido a que una molécula de monómero monofuncional B tiene el mismo efecto que una molécula B-B para terminar el crecimiento de una cadena de polímero se introduce el factor 2. Otro caso posible son las polimerizaciones por etapas con monómeros del tipo A-B (que aportan implícitamente condiciones estequiométricas) cuando se llevan a cabo en presencia de un monómero monofuncional con objeto de estabilizar el polímero resultante. En este caso la relación estequiométrica es la misma que la expresada en la ecuación (3.4.9). Las consecuencias que se han inferido al considerar la figura 3.4.1 son perfectamente aplicables a las polimerizaciones por etapas desarrolladas en estos dos últimos casos. 3.5.- Práctica de la polimerización escalonada. Este tipo de polimerizaciones se realiza habitualmente de forma discontinua en autoclaves provistos de agitador (de ancla o de hélice, según la viscosidad de la mezcla en la etapa final de la reacción) con serpentines internos y/o camisa externa, por los que, inicialmente, se introduce vapor para activar la reacción y, posteriormente, agua de refrigeración para mantener la temperatura por debajo del límite de estabilidad química de los productos. A través del refrigerante de reflujo se condensa el agua (que se separa) y los monómeros y el disolvente evaporados (que se retornan al reactor). Una vez finalizada la reacción, el polímero obtenido se descarga en disolución o fundido. 3.6.- Síntesis de poliésteres. Para hacer un poliéster, se parte de dos compuestos que son el etilenglicol y el dimetil tereftalato, este último es un éster y el etilenglicol es un alcohol. Cuando alcoholes y ésteres se unen, ejecutan un pequeño baile en una reacción que se llama transesterificación, donde hay muchos cambios de pareja. Se debe fijar la atención en uno de los grupos metoxi (en rojo) del dimetil tereftalato y en el grupo hidroxietoxi (en azul) del etilenglicol. Estos grupos van a canjear posiciones. El grupo hidroxietoxi va a terminar en el éster, donde había estado el grupo metoxi. Y el grupo metoxi se unirá a un hidrógeno, donde antes estaba el grupo hidroxietoxi. Esto es lo que ocurre en la primera etapa de la síntesis de un poliéster. Veamos cómo ocurre. El lugar indicado para fijarnos ahora es el éster, el dimetil tereftalato, especialmente en el carbono del carbonilo. Puede observarse que está unido al oxígeno por un enlace doble. El oxígeno es mucho más electronegativo que el carbono, por lo tanto atrae hacia sí los electrones. Esto deja al carbono con una carga parcial positiva, lo cual significa que puede ser fácilmente atacado por un par de electrones que provenga de cualquier parte. Cuando hay un alcohol cerca, como el etilenglicol, los pares electrónicos no compartidos del oxígeno del alcohol lo harán con todo gusto. Y atacan tal como se ve en la figura siguiente: Por lo tanto, un par de electrones del doble enlace carbono-oxígeno se desplaza hacia el oxígeno del carbonilo, creándole una carga negativa. Entre tanto, el oxígeno del alcohol adquiere una carga positiva. Esta etapa intermedia realiza un reordenamiento de electrones, mostrado con las flechas. El resultado final de todo este intercambio, es que el grupo hidroxietoxi del etilenglicol termina unido al éster y el grupo metoxi del éster es expulsado para formar una molécula de metanol. Y también sucede que el grupo metoxi y el grupo hidroxietoxi terminan canjeando sus posiciones. Ese metanol es un subproducto. No lo queremos en el medio de reacción. Si efectuamos dicha reacción a temperatura lo suficientemente alta, el metanol entrará en ebullición. Esto es conveniente. No sólo nos liberamos del metanol, sino que el hecho de eliminarlo del medio de reacción, conduce la reacción de transesterificación a altas conversiones, según nos dice el principio de Le Chatelier. La misma reacción ocurrirá en el otro extremo del tereftalato, de modo que obtendremos el bis-(2hidroxietil) tereftalato. Una vez que se obtiene el bis-(2-hidroxietil) tereftalatos, éste sigue reaccionando. Sin embargo estas reacciones son fáciles de seguir, porque son todas transesterificaciones similares a la que ya se vio. Esta molécula es un alcohol y un éster. Por lo tanto, puede transesterificar consigo misma, como muestra la figura siguiente: Esta forma un producto intermedio, parecido al que se vio antes, realiza un reordenamiento similar de electrones, para obtener el producto que se ve en la figura siguiente: Este producto puede reaccionar nuevamente, tal como hizo el bis-(2-hidroxietil)tereftalato. A medida que esto sucede, las moléculas formadas se hacen más y más grandes, hasta que se obtiene un PET de alto peso molecular. Las propiedades más importantes que hacen que el PET sea un polímero tan extendido en su uso son: 1.- Químicamente estable. 2.- Por su excepcional resistencia química, no es posible aplicar sobre él, adhesivos con disolventes. 3.- Actúa como barrera de gases. 4.- Excelente resistencia al fuego, no transmite la llama. 5.- Excelente transparencia y brillo. 6.- Excelente moldeabilidad. 7.- Buena maquinabilidad. 8.- En general reciclable. 9.- Respetuoso con el medioambiente; su combustión no genera sustancias contaminantes ( a no ser que se encuentren con cloro y formen dioxinas ). 10.- No genera en su producción gases perjudiciales para la capa de ozono. 11.- Reducen la transmisión del ruido. 12.- No presenta riesgos de impactos severos, no se le considera tóxico aunque en la síntesis del PET, se utilizan sustancias que producen irritación de ojos y vías respiratorias, también su fabricación se asocia a un pequeño aumento de incidencia de cáncer. Los metales pesados se pueden emplear como catalizadores durante el proceso de producción y finalmente estos acaban en el medio ambiente, siendo contaminantes. 13.- Apropiado para aplicaciones de contacto directo con alimentos al ser inodoro e insípido y por sus propiedades autoadherentes. En el caso del film, en el proceso de manipulación o en el mismo acto de desenrollarlo, se inducen cargas negativas sobre el plástico. Entonces, al acercarla a otros cuerpos, genera por inducción cargas positivas, y esto hace que ésta la atraigan hacia sí. Por otra parte, el material plástico es un aislante y mantiene durante mucho tiempo, su estado de carga, a menos que el ambiente esté muy húmedo. 14.- En cuanto a la estabilidad frente al calor, son termoplásticos; los artículos fabricados con este producto no deben exponerse a un uso continuado a temperaturas superiores a 65/70 °C. En definitiva, se ablanda por acción del calor. 15.- El componente ultravioleta de los rayos solares ocasiona degradación, ésta depende de las condiciones de exposición, es decir, de la duración real de la exposición a la luz solar, de la temperatura, humedad y de la intensidad de los rayos. La degradación se pone de manifiesto por un progresivo amarilleamiento, una disminución en la transmisión de luz y una pérdida de las propiedades mecánicas. El propio PET posee una cierta resistencia a la intemperie por lo que puede utilizarse en aplicaciones exteriores en lugares en los que la acción de los rayos solares no incida de forma permanente sobre la placa y sea de baja intensidad. 16. En general resiste a la mayoría de ácidos, alcoholes y sales, así como a los plastificantes. También es resistente a hidrocarburos como el xileno, los aceites minerales y el petróleo. La resistencia a los hidrocarburos alifáticos es limitada. También resiste el ataque químico de la lluvia ácida, humos de escape de motores diesel y aire con cierta salinidad. 3.6.- Síntesis de poliamidas. 3.6.1.- Síntesis de nylon 6.6. Se trata de cómo obtener nylon. Para empezar, el nylon se obtiene por medio de una reacción de polimerización por crecimiento en etapas y por una polimerización por condensación. Los nylons se sintetizan a partir de diácidos y diaminas. Para hacer nylon 6.6 no se necesitan catalizadores, ya que los ácidos catalizan la reacción y uno de los monómeros es precisamente un ácido. Entre dos moléculas de ácido adípico ocurre una pequeña reacción. Una le dona un protón al oxígeno del carbonilo de la otra. Cuando el oxígeno del carbonilo es protonado, se vuelve mucho más vulnerable al ataque del nitrógeno de la diamina. Esto ocurre porque el oxígeno protonado porta una carga positiva. Al oxígeno no le gusta tener una carga positiva. Entonces atrae hacia sí mismo los electrones que comparte con el carbonilo. Esto deja al carbono del carbonilo deficiente de electrones y listo para que el nitrógeno de la amina le done un par: Entonces los electrones hacen su papel. Uno de los pares del doble enlace del carbonilo se desplaza totalmente hacia el oxígeno, haciéndose cargo del problema de la carga positiva sobre ese átomo, pero ahora el nitrógeno queda con una carga positiva. Por lo tanto, obtenemos una distribución electrónica aún más elaborada. Los electrones provenientes del enlace oxígeno - hidrógeno, vuelven al oxígeno liberando el protón y regenerando el catalizador ácido. Entonces el oxígeno del carbonilo comparte sus nuevos electrones con el átomo de carbono, regenerando el doble enlace del carbonilo. De hecho, no es suficiente. El oxígeno del grupo hidroxilo decide por su cuenta, hacer un pequeño reordenamiento de electrones. Toma el par que comparte con el carbono y lo acapara para sí mismo, rompiendo el enlace entre él y el carbono. Luego dona un par de electrones al hidrógeno unido al nitrógeno. Esto hace que el hidrógeno, que ahora comparte un par electrónico con el oxígeno, no necesite mantener el par que comparte con el nitrógeno, de modo que deja escapar dicho par, dándoselo al nitrógeno. Este desplazamiento de electrones rompe el enlace entre el hidrógeno y el nitrógeno y elimina la carga positiva sobre ese nitrógeno. Se libera H2O y se genera un dímero conteniendo un enlace amida. Ese dímero tiene un grupo ácido en un extremo y un grupo amino en el otro. Esto significa que puede reaccionar con una molécula del diácido o una molécula de la diamina. Sea como fuere, se obtiene un trímero. El dímero, si lo desea, también puede reaccionar con otros dímeros para formar un tetrámero. O puede reaccionar con un trímero para formar un pentámero y a su vez reaccionar con oligómeros más grandes. Finalmente, cuando esto sucede, los dímeros se transforman en trímeros, tetrámeros y oligómeros más grandes y estos oligómeros reaccionan entre sí para formar oligómeros aún más grandes. Esto sigue así hasta que se hacen lo suficientemente grandes como para ser considerados polímeros. Para que las moléculas crezcan lo suficiente como para ser consideradas polímeros, tenemos que hacer esta reacción bajo vacío. En este caso, todo el subproducto agua se evaporará y será eliminado del medio de reacción. Debemos deshacernos del agua debido a una pequeña regla llamada Principio de Le Chatelier. Hay que recordar que la reacción no necesita un catalizador ácido para llevarse a cabo. La razón es que cuando nos acercamos al final de la polimerización, donde no hay muchos grupos ácidos remanentes para comportarse como catalizadores, la reacción aún prosigue. Es decir, la amina puede reaccionar con los ácidos carboxílicos no protonados. Si no fuera así, no se podría obtener nylon 6.6 de alto peso molecular sin un catalizador externo, ya que la reacción se detendría a conversiones más altas, cuando no haya suficientes grupos ácidos para actuar como catalizadores. Los nylons también pueden obtenerse a partir de una diamina y un dicloruro de ácido: Esta reacción sigue el mismo mecanismo, pero aquí sí se necesita agregar trazas de ácido que actúen como catalizador. (Cuando se obtiene nylon de la otra forma, el ácido adípico actúa como catalizador). Además, se produce HCl gaseoso como subproducto, en lugar de agua. Si desea ver una animación de este método, haga clic aquí. En el nylon 6,6 los oxígenos del carbonilo y los hidrógenos de la amida pueden unirse mediante un enlace de puente de hidrógeno. Esto permite que las cadenas puedan alinearse ordenadamente para formar fibras, como puede apreciarse en la figura 3.6.1.1. Figura 3.6.1.1.- Enlace de puente de hidrógeno entre los oxígenos del carbonilo y los hidrógenos de la amida 3.6.2.- Síntesis de Nylon-6 El nylon 6 es muy parecido al nylon 6.6, como se puede observar en la figura siguiente: Pero sintetizar nylon 6 es totalmente diferente que sintetizar nylon 6.6. En primer lugar, el nylon 6 se obtiene a partir de una sola clase de monómero, llamado caprolactama. En cambio el nylon 6,6 se sintetiza a partir de dos monómeros, el cloruro de adipoilo y la hexametileno diamina. El nylon 6 se obtiene calentando caprolactama a unos 250 oC en presencia de aproximadamente 5-10 % de agua. ¿Qué ocurre con la caprolactama cuando hay agua a su alrededor?. El oxígeno del carbonilo toma uno de los átomos de hidrógeno del agua. Y como sucede a menudo, una insignificancia como ésta, puede desencadenar algo mucho más grande. El oxígeno del carbonilo dona un par de electrones al átomo de hidrógeno del agua, robándole ese hidrógeno al agua. Esto nos conduce a un carbonilo protonado y a un grupo hidroxilo libre, el cual volverá para acosar a la codiciosa caprolactama. Pero primero, tengamos en cuenta que el oxígeno del carbonilo ahora tiene una carga positiva. Al oxígeno no le gusta esto, de modo que toma un par de electrones del doble enlace del carbonilo, dejando la carga positiva sobre el átomo de carbono del carbonilo. Pero colocar un carbocatión en una molécula es suplicar por algún nucleófilo que venga y lo ataque. Pero se había formado un ión hidróxido cuando la caprolactama le robó un protón a una molécula de agua. Este pequeño hidróxido le había cedido su protón a la caprolactama. Así, que aún demostrando una gran hostilidad, ataca el carbocatión. La molécula que se forma ahora es un gem diol inestable. A continuación se produce un loco reordenamiento de electrones. El átomo de nitrógeno dona un par de electrones a un átomo de hidrógeno de uno de los grupos hidroxilo tomándolo para sí. Los electrones que compartía el hidrógeno con su oxígeno, pasan a formar un doble enlace entre el oxígeno y el átomo de carbono. Y por último, los electrones compartidos por el carbono y el nitrógeno se mudan hacia el nitrógeno, rompiendo el enlace carbono-nitrógeno. El anillo se rompió y no hay más caprolactama, pagando caro su codicia, ya que ahora nos quedamos con un aminoácido lineal, el cual puede reaccionar con otra molécula de caprolactama, de forma muy parecida a como hizo el agua. Las moléculas de caprolactama no son muy brillantes. El hecho de ver que una de ellas haya sido destruida por su codicia, no las hace menos codiciosas. Entonces tratan de robar lo que puedan de su pariente desvalida, y así la molécula de caprolactama se apoderará del hidrógeno ácido del aminoácido lineal. El oxígeno del carbonilo dona un par de electrones a ese hidrógeno, separándolo del aminoácido. Tal como se esperaba, los electrones se reordenan para formar el carbocatión, igual que antes: Este carbocatión es todavía una invitación abierta a cualquier nucleófilo que ande cerca, pero esta vez, existe un nuevo nucleófilo. Es el aminoácido que acaba de perder su hidrógeno ácido y realiza el mismo papel que hacía antes el ión hidróxido. Esto nos da un derivado de amonio y éste en particular es sumamente inestable. Por lo tanto los electrones hacen su juego. El nitrógeno del anillo toma un hidrógeno del nitrógeno del amonio. Además, el enlace entre el carbono y el nitrógeno se rompe, abriendo el anillo. Y así se termina otra codiciosa molécula de caprolactama. Ese grupo carboxilato en el extremo de la molécula va a merodear por ahí y se va a robar el hidrógeno del alcohol. Esto origina un nuevo grupo carbonilo en la mitad de la molécula y regenera el ácido carboxílico. Nadie sabe realmente el orden de los dos últimos pasos. Podrían ocurrir en sentido inverso. Sólo se sabe que los dos ocurren antes de que todo termine. Ahora que tenemos otra vez el ácido, es seguro que reaccionará con otra avarienta molécula de caprolactama, y luego con otra y otra, hasta que obtengamos largas cadenas de nylon 6. 3.7.- Síntesis de poliuretanos. Para comenzar, los poliuretamos se obtienen a partir de dos monómeros, un diol y un diisocianato. Con la ayuda de una pequeña molécula llamada diazobiciclo[2.2.2]octano, o DABCO, se puede lograr que ambos polimericen. Cuando mezclamos los dos monómeros en presencia de DABCO, ocurre algo interesante. El DABCO es un nucleófilo muy bueno, es decir, tiene un par de electrones no compartidos a quienes les encantaría atacar algún núcleo vulnerable. Los electrones tienen cargas negativas y los núcleos de los átomos tienen cargas positivas y ambas cargas se atraen. Por lo tanto, los electrones del DABCO miran a su alrededor y encuentran un núcleo en los hidrógenos alcohólicos del diol. Estos hidrógenos son vulnerables porque se encuentran unidos a átomos de oxígeno. El oxígeno es electronegativo. O sea que atrae hacia sí los electrones de otros átomos. Esto deja no balanceada la carga positiva de sus núcleos. Los electrones que podrían balancear esa carga positiva con sus propias cargas negativas, fueron atraídos por el oxígeno. Por lo tanto, quedó una ligera carga positiva sobre el hidrógeno. De modo que los electrones del DABCO observan esto y forman un enlace por puente de hidrógeno entre el hidrógeno y el nitrógeno del DABCO. Este enlace por puente de hidrógeno deja una carga positiva parcial sobre el nitrógeno, y más importante aún, una carga parcial negativa sobre el oxígeno. Esta carga parcial negativa activa mucho más al oxígeno. Y siendo así, quiere reaccionar con cualquier cosa. El oxígeno tiene un exceso de electrones, de modo que reaccionará con algo que se encuentre deficiente de electrones. Si se observa el isocianato, se verá que el carbono del grupo isocianato se sitúa justo en el medio de dos elementos electronegativos como el oxígeno y el nitrógeno. Esto quiere decir que dicho carbono se verá muy pobre en electrones. Por eso, el activo oxígeno reacciona con él. Arroja un par de electrones sobre ese carbono y se establece un enlace. De hecho, esto desplaza un par electrónico del doble enlace carbono-nitrógeno. Este par se sitúa sobre el nitrógeno, confiriéndole una carga negativa. Mientras tanto el oxígeno, que donó un par electrónico, quedará con una carga positiva. No hay cosa peor para un átomo de nitrógeno, que portar una carga negativa. Por eso va a tratar de liberarse de ella en cuanto pueda. La manera más sencilla es donarle ese par al átomo de hidrógeno del alcohol. Esto forma un enlace entre ese hidrógeno y el nitrógeno. Los electrones que el hidrógeno había compartido con el oxígeno ahora pertenecen sólo al oxígeno. Esto elimina esa carga positiva que portaba el oxígeno. Cuando todo esto termina, se obtiene una nueva clase de dímero de uretano. Este dímero de uretano tiene un grupo alcohol en un extremo y un grupo isocianato en el otro, de modo que puede reaccionar ya sea con un diol o con un diisocianato para formar un trímero. O puede reaccionar con otro dímero, o un trímero, o aún oligómeros más grandes. De esta forma, monómeros y oligómeros se combinan y combinan hasta que obtenemos un poliuretano de alto peso molecular. Se puede observar, que no sólo reaccionan los monómeros, sino también los dímeros, trímeros y así sucesivamente. Esto nos habla de una polimerización por crecimiento en etapas. Además, debido a que no se producen pequeñas moléculas como subproductos, decimos que se trata de una polimerización por adición. Polímeros dentro de polímeros. A veces, en lugar de emplear una molécula pequeña de un diol como el etilenglicol, usamos un poliglicol de un peso molecular de alrededor de 2.000. Esto nos conduce a un polímero dentro de otro polímero, por así decirlo, y obtenemos un poliuretano parecido a éste: 3.8.- Síntesis de policarbonatos. Para hacer policarbonatos los dos compuestos principales serán las moléculas de bisfenol A, que se obtiene a partir de fenol y acetona, y fosgeno. Por el momento, vamos a ocuparnos del bisfenol A y luego hablaremos del fosgeno. El primer paso para obtener un policarbonato es tratar el bisfenol A con hidróxido de sodio, NaOH. El grupo hidroxilo va a cumplir la función que cumplen los álcalis, tomando un protón del bisfenol A. Cuando esto sucede, el grupo hidroxilo se transforma en una molécula de agua y el bisfenol A, que es un alcohol, se encontrará en su forma de sal sódica. Luego, sobre el grupo alcohol del bisfenol A, ocurre la misma reacción otra vez. Ahora que el bisfenol A es una sal, puede actuar sobre el fosgeno. Puede verse que el oxígeno de la sal de bisfenol A tiene ahora una carga negativa. Esto quiere decir que puede donar un par de electrones al átomo de carbono del fosgeno. Hay que tener en cuenta que ese carbono se encuentra deficiente de electrones, porque es vecino del oxígeno electronegativo. Cuando ese átomo de carbono gana un nuevo par electrónico proveniente de la sal de bisfenol A, deja escapar uno de los pares que estaba compartiendo en forma no equitativa con el oxígeno del carbonilo. Este par quedará sobre ese oxígeno, dándole una carga negativa. Los electrones de ese oxígeno volverán hacia el carbono, restituyendo el doble enlace carbono-oxígeno. De hecho, se sabe que el carbono no puede compartir diez electrones, de modo que tiene que deshacerse de dos. Y los dos electrones que se van a embarcar, son el par que el carbono había estado compartiendo con uno de los átomos de cloro. Así, el cloro y sus electrones serán expulsados de la molécula. La molécula que se forma ahora se llama cloroformato. El ión cloruro que fue expulsado, se unirá con ese ión sodio para formar NaCl. El cloroformato puede ser atacado por otra molécula de bisfenol A, tal como lo hizo el fosgeno. Y una segunda molécula de bisfenol A puede atacar tal como lo hizo la primera. Y lo hace a través de un intermediario similar y un juego electrónico similar al que vimos, para obtener el carbonato constituido por las especies mostradas. Después de esto, los grupos salinos de la gran molécula pueden reaccionar con más fosgeno y de ese modo, la molécula crece hasta que obtenemos el policarbonato. 3.9.- Síntesis de resinas epoxi. Existen dos pasos para obtener resinas epoxi. Primeramente debemos sintetizar un diepoxi y luego tenemos que entrecruzarlo con una diamina. Por lo tanto, vamos a tratar ambos pasos por separado. Sintetizando el diepoxi. Este paso es un tipo de polimerización por crecimiento en etapas en el mejor sentido de la palabra. Obtenemos el prepolímero por medio de bisfenol A y epiclorhidrina. La reacción es la siguiente: Lo primero que sucede, es que el NaOH hace un pequeño canje con el bisfenol A, para dar la sal sódica de bisfenol A: Como se puede apreciar, la sal tiene un oxígeno con tres pares de electrones sin compartir. Por lo tanto, este oxígeno especial quiere compartir sus electrones con otros átomos. De modo que en la epiclorhidrina más cercana, encuentra un átomo de carbono que podría usar algunos electrones. Ese átomo es el carbono vecino al cloro. Se supone que debería ser el cloro el que compartiera un par de electrones con ese carbono, pero siendo tan electronegativo como es, el cloro tiende a acaparar ese par. Por lo tanto, el oxígeno dona un par de sus electrones al carbono. Claro que el carbono sólo puede compartir cuatro pares de electrones por vez, por lo que si quiere tomar el par del oxígeno, otro par deberá irse. De modo que se deshace del par que compartía con el cloro y libera éste fuera de la molécula. Terminamos con una molécula similar al bisfenol A, sólo que posee un único grupo epoxi. Y también obtenemos NaCl. A continuación pueden suceder distintas cosas. Hay que tener en cuenta que estos prepolímeros pueden existir en distintos pesos moleculares. A veces el grado de polimerización puede llegar hasta 25. Pero otras, puede ser tan pequeño como esto: El tamaño del prepolímero depende de la relación epiclorhidrina - bisfenol A en la mezcla de reacción. Supongamos que fijamos dicha relación en dos moléculas de epiclorhidrina por cada molécula de bisfenol A. Veamos lo qué sucede en ese caso: Es decir, también se obtiene un grupo epoxi en el otro extremo. La reacción entonces se detiene, dado que no queda más sal sódica de bisfenol A para reaccionar. Pero si hay menos de dos moléculas de epiclorhidrina por cada molécula de bisfenol A, no toda la sal sódica de bisfenol A podrá reaccionar con la epiclorhidrina. Supongamos que nuestra relación es ahora tres moléculas de epiclorhidrina por cada dos moléculas de bisfenol A. Cuando todas las moléculas de epiclorhidrina hayan reaccionado, tendremos una mezcla 50:50 de estas dos moléculas: Estas dos moléculas pueden reaccionar entre sí para dar lugar a esta otra: Ahora tenemos un dímero, que resulta ser una sal sódica. Hay que tener en cuenta la carga negativa sobre el átomo de oxígeno. Cuando aparece una molécula de agua (se forman muchas moléculas de agua cuando se hace la sal sódica de bisfenol A) un par electrónico del oxígeno atacará uno de los hidrógenos del agua, robándolo de ésta. El oxígeno forma un grupo alcohol y otra vez obtenemos nuestro NaOH. Cuanta más epiclorhidrina se tenga con respecto a la sal de bisfenol A, mayor será el oligómero que obtendremos. Curando el diepoxi con una diamina Una vez que se obtienen los prepolímeros diepoxi hay que unirlos. Se hace mediante la adición de una diamina, la cual hace su papel cuando ve esos grupos epoxi en los extremos del prepolímero. Los pares de electrones de los grupos amino van a observar esos grupos epoxi y se van a dar cuenta que el oxígeno del epoxi, siendo tan electronegativo como es, está atrayendo todos los electrones de los átomos de carbono vecinos. Por lo tanto, los grupos amino observan esos átomos de carbono y ven que pueden donarle fácilmente sus electrones al átomo de carbono que está en el extremo de la molécula. Cuando lo hacen, el carbono abandona los electrones que compartía en forma no equitativa con el oxígeno. El enlace entre el carbono y el oxígeno se rompe y se forma uno nuevo entre el carbono y el nitrógeno de la amina. Por lo tanto, nos queda una carga negativa sobre el oxígeno y una carga positiva sobre el nitrógeno. Al oxígeno le gustan los electrones, pero tiene tres pares que no comparte con ningún otro átomo. Por lo tanto, uno de esos pares de electrones sin compartir busca algo con qué unirse y encuentra el hidrógeno unido al nitrógeno positivo. Estos electrones entonces atacan a ese hidrógeno. Cuando atacan, forman un enlace con el hidrógeno y éste se separa del nitrógeno, dejando su electrón. Este se encarga de esa carga positiva, dejando al nitrógeno neutro. Y, el oxígeno también queda neutro, ya que ganó un protón y ahora forma un grupo alcohol. El grupo amino aún tiene un hidrógeno de más y puede reaccionar con otro grupo epoxi, exactamente de la misma manera. De acuerdo con cuántos hidrógenos tenga la amina, el mismo número de grupos epoxi podrán reaccionar con éstos. Esto es lo que se obtiene finalmente: Hay que hacer notar que se esta usando una diamina, por lo que los grupos amino del otro extremo de la diamina pueden también reaccionar con dos grupos epoxi. En definitiva, se obtiene finalmente cuatro prepolímeros epoxi unidos a una sola molécula de diamina. Y también hay que señalar, que los otros extremos de los prepolímeros diepoxi están unidos a otras moléculas de diamina. De este modo, todas las moléculas de diamina y todas las moléculas de diepoxi se unen formando una sola molécula gigante, o una red entrecruzada. Esa red es algo parecido a esto: 3.10.- Síntesis de siliconas. Se trata de hacer polisiloxanos. Por lo general, estos polímeros se obtienen a partir de monómeros como el octametil ciclotetrasiloxano, que se puede representar así. Esta molécula hace algo curioso en presencia de bases como el NaOH. Un grupo hidroxilo dona un par de electrones a uno de los átomos de silicio del anillo, el cual lo acepta. El único problema es que el silicio ya tiene ocho electrones compartidos. No puede tener diez. Por lo tanto, debe deshacerse de un par, que es el que forma el enlace silicio-oxígeno. Así, el par se desplaza totalmente hacia el oxígeno. Esto rompe el enlace entre el oxígeno y el silicio y el anillo ya deja de ser tal. Se abre. Y además, ese oxígeno que ganó ese par de electrones, ahora posee una carga negativa. Puede atacar a una segunda molécula de monómero, exactamente como el hidróxido atacó a la primera. Lo anterior conduce a que se agrega más y más monómero y finalmente, se obtiene una nueva cadena de polisiloxano. Debido a que se abren los anillos del monómero para hacer el polímero, ésta es por supuesto, una polimerización por apertura de anillo. 3.11.- Polimerización metatésica de olefinas Los polímeros vinílicos, obtenidos a partir de monómeros vinílicos es decir, pequeñas moléculas conteniendo dobles enlaces carbono-carbono, no poseen dobles enlaces en su cadena principal. Poli (metacrilato de metilo) Ahora vamos a tratar sobre cómo partiendo de monómeros con dobles enlaces se pueden obtener polímeros con dobles enlaces en su cadena principal. A estos polímeros los llamamos polialquenámeros. Una forma de obtener polialquenámeros es por medio de una ingeniosa reacción denominada metátesis olefínica. Una olefina es lo mismo que un alqueno, es decir, una molécula, que presenta al menos un doble enlace carbono-carbono. La metátesis olefínica es obviamente una reacción que involucra olefinas. Para ser exactos, dos olefinas. Y transcurre así: los carbonos unidos por el doble enlace se intercambian para formar dos nuevas olefinas, tal como lo describe la siguiente figura. Pero no resulta obvio que esta reacción pueda ser usada para hacer polímeros. De hecho, esta reacción fue descubierta en los años 20 y cincuenta años después los científicos idearon una manera de utilizarla en la obtención de polímeros. Pero tuvieron que pasar otros veinte años hasta que alguien hiciera algo útil con esta reacción. Y aunque el proceso demandó mucho estudio por parte de algunos químicos trabajando en polímeros, finalmente tuvo éxito. Los químicos idearon dos métodos para emplear la metátesis olefínica en la obtención de polímeros. Esas dos polimerizaciones se denominan polimerización metatésica por apertura de anillo (del inglés, ROMP) y polimerización metatésica de dienos acíclicos (del inglés, ADMET). Se estudiara primero la ADMET porque es más sencilla, si bien la ROMP ha sido más extensamente investigada. Polimerización metatésica de dienos acíclicos (ADMET). La polimerización metatésica de dienos acíclicos (ADMET), es una polimerización por crecimiento en etapas y por condensación, ya que se obtiene gas etileno como subproducto. En la ADMET partimos de un dieno acíclico como el 1,5-hexadieno y terminamos con un polímero con un doble enlace en la cadena principal, además de gas etileno como subproducto. Usando el ejemplo de dos moléculas de 1,5-hexadieno que reaccionan entre sí, se puede mostrar como la metátesis hace eso posible Los dobles enlaces de los extremos de la nueva molécula pueden seguir reaccionando por metátesis, de modo que el polímero crece. Hemos mostrado el isómero cis del producto formado, pero por lo general se pueden formar ambos, el cis y el trans, y en el polímero final hay suficiente lugar para los dos. Polimerización Metatésica por Apertura de Anillo (ROMP). También podemos obtener polímeros a partir de olefinas cíclicas. Observando la figura de abajo se puede ver que una olefina cíclica, en este caso ciclopenteno, es empleada para sintetizar un polímero que no posee estructuras cíclicas en su cadena principal. Por esta razón es que hablamos de una "polimerización metatésica por apertura de anillo". La ROMP se emplea para obtener algunos productos muy útiles. Por ejemplo, una formidable pequeña molécula como el norborneno, es polimerizada mediante la ROMP para obtener polinorborneno. Es una clase de caucho usada para hacer, entre otras cosas, componentes de automóviles, esos componentes de la carrocería que evitan la vibración. También, mediante la ROMP, podemos obtener otro polímero a partir de una molécula similar: el endodiciclopentadieno. Cuando lo polimerizamos, obtenemos un polímero con una olefina cíclica como grupo pendiente. Este polímero es por supuesto, el polidiciclopentadieno. Esta olefina pendiente puede hacerse reaccionar para entrecruzar el polímero, posiblemente por la tradicional polimerización vinílica u otra. El polímero es un plástico rígido que se emplea para fabricar platos de antenas de satélites, componentes de carrocería para vehículos aptos para la nieve y otros objetos que por lo general se empleen en exteriores. No se usa en interiores, porque siempre hay una pequeña porción de endo-diciclopentadieno sin reaccionar que queda ocluida en el polímero. Se trata de un monómero maloliente que en poco tiempo es capaz de impregnar una habitación. Comienza con una etapa de iniciación. En este caso, el iniciador es generalmente un átomo metálico como el tungsteno o el molibdeno. El iniciador reacciona con una molécula de ciclopenteno, por medio de una reacción metatésica como las que hemos estado viendo, para dar una molécula con un doble enlace carbono-carbono en un extremo y un doble enlace carbono-metal en el otro. Ese doble enlace carbono-metal puede reaccionar con otra molécula de ciclopenteno exactamente de la misma forma: Y así el polímero crece, hasta que obtenemos esto: 4.- Copolimerización. 4.1.-Introducción. Cuando una polimerización en cadena se lleva a cabo en presencia de dos o más monómeros, el producto que se obtiene es una cadena formada por la sucesión de esas diversas unidades estructurales, combinadas en formas muy diversas dependiendo de la concentración de los monómeros y sus reactividades relativas. Estos procesos reciben el nombre de copolimerización si se utilizan solo dos monórneros distintos. La utilización de tres o más monómeros recibe el nombre genérico de copolimerización multicomponente, reservándose el nombre de terpolimerización al caso particular en el que se empleen tres monómeros. Ello no quiere decir que no se puedan obtener copolímeros mediante reacciones de condensación. Por ejemplo, la reacción entre un diol y dos diacidos distintos puede conducir a la formación de un copoliester. Sin embargo, son los copolímeros obtenidos mediante reacciones en cadena los más introducidos en el mercado. Se pondrá énfasis en aspectos los cineticos que permiten regular la composición de los mismos y, por tanto, sus propiedades. La copolimerización consiste en la formación de macromoléculas (polímeros) a partir de dos o más monómeros con estructuras moleculares diferentes. Esto conduce a la obtención de una extensa gama de productos cuya naturaleza va a depender de: ( i).- La naturaleza de los monómeros (ii).- De las concentraciones relativas de los monómeros presentes en la reacción (mezclas apropiadas de monómeros) ( iii).- De la distribución de las secuencias, es decir de la forma en la que se sitúen los monómeros a lo largo de la cadena macromolecular durante el proceso de polimerización. Por estos motivos, la técnica de la copolimerización constituye el método ideal para obtener una gran variedad de polímeros con estructuras y propiedades diferentes, que presentan determinadas características quimico-fisicas útiles para aplicaciones específicas. Las unidades de repetición en los copolímeros pueden distribuirse en la cadena de forma alternante, cuando hay una ordenación regular de M1 y M2 en la cadena, es decir, (M1M2)n; de manera aleatoria, si las secuencias de M1 y M2 se ordenan de forma arbitraria, es decir, M1M1M2M1M2M2...; de bloque, si se encuentran secuencias largas con la misma unidad d repetición en la cadena, es decir, (M1)m(M2)n; de injerto, si las extensiones de cadena con el segundo monómero son ramificaciones, es decir, Los copolímeros al azar (o estadísticos) y alternantes pueden obtenerse mediante polimerizaciones en cadena y policondensaciones, los demás tipo de copolímeros requieren estrategias específicas para su producción, como puede ser la polimerización radical mediante transferencia de átomo (ATRP). Los copolímeros estadísticos y alternantes pueden obtenerse mediante polimerizaciones en cadena y policondensaciones. En este último caso, el copolímero suele tener una composición global muy parecida a la de la mezcla de monómeros, ya que estos procesos además de llevarse a cabo hasta conversiones próximas al 100%, con objeto de obtener copolímeros de alto peso molecular, tienen la peculiaridad de emplear monómeros (por ejemplo, diacidos) de reactividades muy parecidas ante el segundo grupo funcional necesario (por ejemplo, dioles), así como la necesidad de estequiometrias muy ajustadas si no se quiere malgastar alguno de los monómeros. Sin embargo, en los procesos de polimerización en cadena, y salvo en ciertas condiciones muy particulares, la composición depende mucho más de las diferentes reactividades de los monómeros y de sus proporciones en la mezcla, que pueden, en principio, tomar cualquier valor. Aunque el mecanismo de copolimerización se parece al de la polimerización con un único reactivo (homopolimerización), las reactividades de los monómeros pueden diferir cuando se usa más de uno. Los copolímeros se pueden obtener por reacciones escalonadas o por polimerización en cadena de radicales. Es importante resaltar que si las especies reactantes son M1 y M2, la composición del copolímero no es una mezcla de [M1] n +[ M2] n . La mayoría de los polímeros naturales son homopolímeros en gran parte, aun que tanto las proteínas como los ácidos nucleicos son copolímeros. Aunque muchos de los polímeros sintéticos son homopolímeros, el SBR, el caucho sintético de uso más extendido, es un copolímero de estireno (S) y butadieno (B). El ABS, que es un plástico también de muy amplia utilización, es un copolímero o mezcla de polímeros de acrilonitrilo, butadieno y estireno. El Spancex, que es una fibra especial, es un copolímero de bloque de un poliuretano rígido y de un poliéster flexible. Los copolímeros más importantes son el SBR, el caucho de butilo, el poli(cloruro de vinilo-co-acetato de vinilo), el Saran, los ionómeros y las fibras de Acrilan. Los copolímeros injertados y de bloque se diferencian de las mezclas, pero tienen propiedades de cada uno de los componentes. Así, los copolímeros de bloque pueden utilizarse como elastómeros termoplásticos y los copolímeros injertados con una cadena principal flexible, pueden utilizarse como plásticos de alto impacto. Los copolímeros de bloque e injertados pueden obtenerse por polimerización de reacción en cadena y por polimerización escalonada. Los principales copolímeros de bloque son los elastómeros termoplásticos y las fibras elásticas. Los principales copolímeros de inserción son el ABS, el HIP, los plásticos de poliéster y el almidón o la celulosa injertados. Un modelo que describa adecuadamente el proceso de copolimerización debe ser capaz de predecir la composición global del copolímero, un aspecto complicado ya que la composición de las cadenas que se van obteniendo a lo largo del proceso puede ir cambiando en el tiempo. Debe también ser capaz de predecir la velocidad global del proceso y la distribución de secuencias de los comonómeros (microestructura), ya que una misma composición global puede ser el resultado de muchas posibles combinaciones de los monómeros a lo largo de las grandes cadenas que se forman. Velocidad y microestructura pueden también ir cambiando a lo largo del proceso de polimerización, al ir variando la relación o proporción entre las cantidades de los monómeros que quedan por reaccionar (alimentación), debido a la diferente reactividad de los mismos. La complejidad de estas dependencias ha obligado a la introducción de numerosas simplificaciones a la hora de dar lugar a diversos modelos que expliquen el proceso. Durante más de 50 años, el modelo más aceptado ha sido el llamado modelo terminal, basado en que la reactividad de las especies propagantes solo depende de la última unidad adicionada a la cadena, admitiéndose también la ausencia de reacciones secundarias. Este modelo sencillo es capaz de ajustar aceptablemente datos experimentales correspondientes a la composición global del copolímero usando únicamente dos parámetros, denominados relaciones de reactividad. 4.2.- Ecuación de composición del copolímero. El modelo terminal se basa en tres premisas bien definidas: en primer lugar, se supone que la reactividad de las cadenas propagantes solo depende de la naturaleza de la última unidad de la cadena, siendo por tanto independiente de la composici6n previa de la misma. En segundo lugar, se supone que las cadenas de copolímero formadas son lo suficientemente largas como Para desechar la influencia de los procesos de iniciación y de terminación, dando solo importancia a las reacciones de propagación. Finalmente, y aunque en general suele ser una suposici6n bien asentada, las reacciones de copolimerización se consideran irreversibles. La polimerización simultánea y conjunta de dos monómeros, puede dar lugar a macromoléculas mixtas, en las que aparecen mezclados los dos monómeros, constituyendo lo que se denomina un copolímero. La incorporación de los monómeros al copolímero que se está formando depende no solo de la composición de la mezcla de los monómeros sino también de sus respectivas reactividades. Entonces como la diferencia de reactividad de los monómeros, expresada como relaciones de reactividad (r), la composición del copolímero (n) puede ser diferente de la de la mezcla de reactivos utilizada como materia prima (x). Cuando x es igual a n, el producto es un copolímero azeotrópico. Vamos a deducir una expresión que permita determinar la composición instantanea del copolímero en función de las concentraciones de los monómeros y de sus reactividades. En la polimerización por adición la etapa de la propagación es la más interesante. Sin embargo, en el caso de la copolimerización la propagación es la reacción decisiva y adquiere una importancia fundamental, ya que al haber dos monómeros diferentes, se pueden dar cuatro tipos distintos de poliadición a la cadena en crecimiento. Entonces, el problema inicial es determinar que proporción de un determinado monómero entra en la composición del copolímero y cuál es su estructura. Wall, Dostal, Lewis, Alfrey, Simha y Mayo fueron los investigadores que, en la década de los treinta, dedujeron una ecuación diferencial capaz de darnos la composición del copolímero formado. • • Llamemos M1 y M2 a los dos monómeros presentes al comienzo de la copolimerización y R-M1 y R-M2 a los radicales polímeros que poseen las unidades estructurales, M1 y M2, respectivamente, en el extremo radical. Pueden considerarse cuatro reacciones de propagación alternativas: k 11 R M1 M1 R M1 (4.2.1) k 12 R M1 M 2 R M 2 (4.2.2) k 21 R M 2 M1 R M1 (4.2.3) k 22 R M2 M 2 R M 2 (4.2.4) en las que k11, k12 k21 y k22 son las correspondientes constantes cinéticas y el círculo negro tiene el significado de una especie reactiva o propagante, que puede ser un radical o un ión. Los subindices en las constantes de velocidad de reacción se refieren el primero al radical y el segundo al monómero que se incorpora a la cadena en crecimiento. en las que k11, k12 k21 y k 22 son las correspondientes constantes cinéticas. Dos de las reacciones son homopolimerizaciones o etapas autopropagadas, y las otras dos son heteropolimerizaciones o etapas de propagación cruzada. Empíricamente se ha descubierto que las constantes de velocidad específicas de las reacciones anteriores son básicamente independientes de la longitud de cadena, siendo la velocidad de adición del monómero principalmente dependiente de la unidad de monómero que se suma al final de cadena en crecimiento. Así, las cuatro copolimerizaciones entre dos comonómeros se pueden representar con sólo cuatro ecuaciones. Al igual que en la homopolimerización, en la copolimerización se alcanza un estado estacionario. La velocidad de formación • de los radicales libres R-M2 es igual a su velocidad de desaparición. Análogo razonamiento se puede hacer con respecto a • los radicales libres R-M1 . Por otra parte, la velocidad de desaparición del monómero M1, por incorporación en el copolímero, viene dada por la ecuación diferencial: d M 1 k11 M1 M1 k21 M 2 M1 dt (4.2.5) d M 2 k22 M 2 M 2 k12 M1 M 2 dt (4.2.6) y la del monómero M2: Por tanto, la relación entre ambas velocidades de desaparición expresa la composición del copolímero que puede escribirse como: n d M1 d M 2 k11 M1 M1 k21 M 2 M1 k22 M 2 M 2 k12 M1 M 2 (4.2.7) Es interesante recalcar que esa es la composición del copolímero que se forma en un instante dado, instante en el que las composiciones de los monómeros en la alimentación son las que se introducen en la ecuación (4.2.7). En un instante siguiente la composición de los monómeros en la alimentación puede cambiar y, por tanto, cambia también la composición del copolímero que se va a formar. Con objeto de eliminar la concentración de especies propagantes se aplicará la hipótesis de estado estacionario a cada una de las especies propagantes. Considerando las cuatro reacciones posibles durante la copolimerización mencionadas, la primera y la cuarta no suponen variación en la concentración de las especies activas terminadas en M 1 y M 2, mientras que si lo hacen los procesos de interconversión (reacciones segunda y tercera). Por tanto, la velocidad de estos dos procesos deben ser iguales para que las concentraciones de las especies reactivas permanezcan constantes, pudiendo escribirse entonces que: k21 R M2 M1 k12 M1 M2 (4.2.8) La misma ecuación se obtiene desde la óptica de M 1 como de M2. • • Despejando de la ecuación (4.2.8) la concentración de [R-M1 ] o de [R-M2 ] se obtiene: k 21 R M 2 M1 k12 M 2 k R M M 1 2 R M 2 12 k 21 M1 R M1 y sustituyendo en la ecuación (4.2.7) se obtiene finalmente la expresión que nos da desaparición de los monómeros (n = M1/ M2): (4.2.9) el cociente de 1 k11 M1 d M k12 M 2 M1 n 1 d M 2 k 22 M 2 M 2 1 k 21 M1 r1 M1 M 2 (4.2.10) M1 r2 M 2 o bien: d M n 1 d M 2 M 1 r1 1 M 2 M 1 r2 2 M1 (4.2.10’) en donde: r1 = (k11/k12) y r2 = (k22/k21) (4.2.11) que son las llamadas relaciones de reactividad de los monómeros y expresan la reactividad relativa de cada tipo de radical con su propio monómero respecto del otro, es decir reflejan la tendencia de cada uno de los monómeros a copolimerizar consigo mismo o con el comonómero. Un valor de r1 > 1 significa que la especie • propagante M1 prefiere añadir el monómero M1 antes que el M2. . Un valor de r1 < 1 significa que añade • preferentemente M2. . Un valor de r1 = 0 significa que la especie propagante M1 siempre añade el monómero M2. Solo cuando ambas relaciones de reactividad sean iguales a 1, a cada extremo propagante le da lo mismo reaccionar consigo mismo o con el otro, pero que en los demás casos hay una tendencia definida que puede ir alterando la composición inicial de la mezcla de monómeros a lo largo del tiempo. La expresión (4.2.10) es la denominada ecuación de composición instantánea del copolímero o de Lewis-Mayo porque expresa la composición instantánea del copolímero en función de la concentración de los monómeros 1 • • y 2 y de la concentración de los centros activos y R-M1 y R-M2 . La ecuación (4.2.10) tiene validez a cualquier grado de conversión de la mezcla de monómeros, ya que tiene en cuenta solo la composición del copolímero que se esté formando instantaneamente en el tiempo t, con la composición instantánea de la mezcla de monómeros en el mismo momento. • Si r1 es mayor que la unidad, esto quiere decir que el radical M1 reacciona más rapidamente con el monómero de su propia estructura que con el otro monómero, mientras que si r1 es menor que la unidad, entonces reacciona más velozmente con el monómero de distinta estructura que con su propio monómero. Asi, por ejemplo, en la copolimerización del estireno con el metacrilato de metilo por radicales libres, se ha obtenido que r1 = 0,52 y r2 = 0,46, es decir, que cada radical libre reacciona unas dos veces más de prisa con el monómero opuesto. La ecuación del copolímero también puede ponerse en la forma: n r1x 1 r 1 2 x donde n y x son las relaciones molares de los monómeros en el copolímero y en la mezcla de reactivos, respectivamente, y r1 y r2 son los cocientes de reactividad. La ecuación del copolímero puede utilizarse para mostrar el efecto de la composición de la mezcla de reactivos sobre la composición del copolímero (n). Mientras que los valores de estos dos parámetros son iguales en las copolimerizaciones azeotrópicas, serán distintos en la mayoría de las otras copolimerizaciones. Por tanto, se acostumbra a compensar la diferencia de cocientes de reactividad añadiendo monómeros de manera continua a la mezcla para producir así copolímeros de composición uniforme.En la tabla 4.2.1 se dan algunos valores de reactividades. Tabla 4.2.1.- Reactividades de los monómeros que intervienen en algunas copolimerizaciones por radicales libres. Existen otros modelos, como el de la penúltima unidad que permite obtener también una expresión para evaluar la composición del copolímero. Este modelo se basa en la hipótesis de que la reactividad de las especies propagantes está afectada por la naturaleza química de la penúltima unidad. Si tenemos en cuenta esta hipótesis entonces tendremos las siguientes ocho reacciones de propagación: k 111 R M1 M1 M1 R M1 M1 k 111 R M 2 M 2 M 2 R M 2 M 2 k 221 R M1 M1 M 2 R M 2 M1 k 222 R M 2 M 2 M1 R M 2 M 2 k 211 R M 2 M1 M1 R M1 M1 k 121 R M1 M 2 M1 R M 2 M1 k 212 R M 2 M1 M 2 R M1 M 2 k 122 R M1 M 2 M 2 R M 2 M 2 Si utilizamos razonamientos similares a los empleados en el modelo anterior, es posible obtener la siguiente ecuación que nos permite calcular la composición instantánea del copolímero: d M1 d M 2 1 r21 M1 / M 2 r11 M1 / M 2 1 r21 M1 / M 2 1 1 M / M r M / M r21 r22 M1 / M 2 1 2 12 1 2 En donde las relaciones de reactividad r 12 , r21, r11 y r22 valen: r11 k111 k112 r12 k122 k121 r22 k222 k221 r21 k211 k212 Si el efecto de la penúltima unidad sobre la reactividad del radical que se propaga no es importante, entonces se cumple que r21 = r11 y r12 = r22, con lo que ambos modelos se hacen iguales. Como puede apreciarse, en la ecuaciones que nos da la composición del copolímero, en ambos modelos, no aparecen constantes de velocidad de reacción ni para la iniciación ni la terminación, lo que quiere decir que la composición del copolímero no depende de las velocidades totales de polimerización ni del proceso de iniciación, cualquiera que sea su naturaleza, como se ha comprobado experimentalmente. Se ha observado que el cambio del medio de reacción no ejerce una influencia apreciable en las relaciones de reactividad r1 y r2, debido a que las reacciones en cadena a través de radicales libres no suelen ser sensibles al cambio del disolvente. La observación de la ecuación de composición permite la predicción de una serie de comportamientos en función de que las relaciones de reactividad adopten una serie de valores determinados. Los tipos de copolimerización límites, teóricamente posibles, son tres: (1).- Copolimerización ideal. Es el tipo más sencillo, pero el más excepcional. Aquí las reactividades relativas de ambos radicales muestran la misma preferencia por un tipo de monómero respecto del otro, es decir: k11 k12 k 21 k 22 es decir: r1 1 r2 En estas condiciones, las velocidades de incorporación de ambos monómeros son independientes de la naturaleza de la última unidad de las especies propagantes o, lo que es lo mismo, ambas especies tienen la misma preferencia para añadir uno u otro monómero. El grupo final no ejerce influencia en la velocidad de adición y los dos tipos de unidades se distribuyen al azar a lo largo de la cadena y en cantidades relativas determinadas por la composición de monómeros en la mezcla y por los valores de r1 y r2. La ecuación de composición del copolímero en este caso vendrá dada por d M1 M r1 1 M 2 d M 2 o bien en función de las fracciones molares: F1 r1f1 r1f1 f2 y las unidades monoméricas se disponen al azar a lo largo de la cadena: M1-M1-M2-M1-M2-M2-M1-M1 Se dice que estos copolímeros, presentan un comportamiento Bernaulliano (Una distribución bernoulliana es aquella en la que un determinado evento no está condicionado por eventos previos). La mayoría de las copolimerizaciones aniónicas y catiónicas suelen ser de este tipo. En el caso particular de que r1 = r2 = 1, ambos monómeros tienen igual preferencia hacia las especies propagantes, colocándose de una manera completamente al azar en la cadena, obteniéndose un copolímero cuya composición global es idéntica a la de la alimentación. En la figura 4.2.1 se observa la dependencia de la composición instantánea del copolímero con la composición de la alimentación para una serie de diversos valores de r1 y r2 que cumplen la condición r1r2 = 1. Se observa que a medida que la diferencia entre las relaciones de reactividad aumenta (r1 = 10, r2 = 0,1) se obtienen copolímeros que son más ricos en el comonómero que es más reactivo, como en el caso de las copolimerizaciones iónicas. Estas copolimerizaciones presentan poco interés, debido a la limitada variación de composiciones del copolímero que se puede obtener. Figura 4.2.1.- Relación entre las composiciones instantáneas del copolímero y la alimentación en diversas copolimerizaciones ideales (r1r2 = 1). El valor indicado en la gráfica corresponde a r1. (2).- Copolimerización alternante. Aquí cada radical prefiere reaccionar exclusivamente con el monómero de tipo opuesto. Entonces, k11 k12 k21 k22 0 es decir: r1 r2 0 (r1r2 0) Ello es tanto como decir que ambas especies propagantes prefieren añadir el otro monómero antes que el propio. Como consecuencia de ello, los monómeros entran en el copolímero de manera perfectamente alternada (copolimerización alternante) y, por tanto, en cantidades equimolares. La ecuación de composición del copolímero vendrá dada por d M1 d M 2 1 y las unidades monoméricas se dispondrán, por lo tanto, de modo alternado y regular a lo largo de la cadena: M1-M2-M1-M2-M1-M2-M1-M2-M1-M2 Luego cuando las reactividades son muy bajas, las reacciones primera y cuarta son muy lentas y se produce un copolímero alternado. Es interesante observar que los monómeros que cumplen esta condición no suelen formar homopolímeros. En este caso, la composición inicial de monómeros más adecuada es la equimolar, ya que ello permite llegar hasta el final de la conversion manteniendo la misma concentración en la alimentación. Si la composición inicial fuera, por ejemplo, 70/30 en moles, llegaría un momento en el que la producción de copolímero alternante cesaría, al haberse consumido el monómero minoritario. A partir de ahí, la prolongación del proceso solo daría lugar a homopolímero del componente mayoritario, obteniéndose por tanto al final una mezcla de copolímero alternante y homopolímero que, en general, suele ser una mezcla inmiscible de deficientes propiedades. (3).- Caso de que r1 > 1 y r2 < 1. En este caso las reacciones (4.2.1) y (4.2.3) son preferidas a las (4.2.2) y (4.2.4), lo que significa que el monómero M1, entra preferentemente a formar parte del copolímero. Si además r1 y r2 difieren apreciablemente entre sí, el copolímero formado estará compuesto preferentemente por el monómero M1. Para controlar la composición del copolímero formado, de forma que este contenga una cantidad apreciable del monómero M2 en su estructura es necesario mantener la alimentación de forma que la relación [M1]/[M2] sea siempre menor que la unidad. En estas circunstancias se obtiene un copolímero polidisperso en composición y en pesos moleculares. (4).- Caso de que r1 < 1 y r2 > 1. En este caso se obtiene el resultado opuesto al caso anterior. (5).- Copolimerización normal. Este es el tipo intermedio entre los dos anteriores; aquí se cumple que: 0 < r1.r2 < 1 y la tendencia alternante se ve parcialmente compensada por la disposición al azar. Existe también la posibilidad de que cada radical prefiera reaccionar con el monómero de su propia estructura, o lo que es lo mismo r1 > 1 y r2 > 1 aunque se conocen muy pocos casos en los que r1.r2 >1, por ejemplo, para el sistema benzoato de alilo/cloruro de alilo, r1 = 2,5 y r2 = 1,25. En este caso la estructura que se obtiene es: M1-M1-M1-M1-M1-M2-M2-M2-M2-M2 Por tanto, si las reactividades son muy grandes se obtiene una mezcla de homopolímeros, más que un verdadero copolímero. El comportamiento descrito anteriormente comienza a no cumplirse a medida que los valores de r1 y r2 van siendo cada vez más diferentes. Este tipo de copolimerizaciones no suele ser corriente en la realidad, por lo que la obtención de copolímeros de bloque se ha realizado mediante diferentes estrategias (polimerización aniónica sin terminacion, polimerizaciones controladas, iniciadores bifuncionales, etc). A modo de resumen, en la tabla 4.2.1 se dan los posibles casos de copolímeros asi como las condiciones que deben cumplir las relaciones de reactividad de ambos monómeros. Tabla 4.2.1.- Casos especiales en la copolimerización de dos monómeros por radicales libres. 4.3.- Composición instantánea de monómeros y de copolímero. Las composiciones de la mezcla monomérica (alimentación) y del copolímero que se forma, no van a ser constantes en el tiempo, sino que irán variando durante el proceso de copolimerización. El primer copolímero que se forma no va a tener la misma composición que la mezcla monomérica, excepto en casos especiales. Por esta razón, la composición de la mezcla de monómeros variará, y como se deduce de la ecuación del copolímero, variará a su vez la composición del copolímero formado. Por lo tanto, la mayoría de los copolímeros son heterogéneos en composición, aunque como es lógico a conversion del 100%, la composición media global del copolímero deberá ser igual a la de la mezcla inicial de monómeros. La ecuación de composición del copolímero se suele expresar también en función de la fracción molar de la alimentación (mezcla de los monómeros) en vez de su concentración molar y de la composición del copolímero. Para ello basta con definir Fi y fi como la fracción molar del componente i en el copolímero y en la mezcla de monómeros, respectivamente, entonces: F1 1 F2 d M1 (4.3.1) d M1 d M 2 y f1 1 f2 M1 M1 M 2 (4.3.2) Sustituyendo estos valores en las ecuaciones (4.2.10) y (4.2.10´) se obtiene que: r1f12 f1f2 F1 2 r1f1 2f1f2 r2f22 (4.3.3) o bien: F1 f1 r1f1 f2 F2 f2 r2f2 f1 (4.3.3´) Estas ecuaciones relacionan la composición del copolímero formado en un instante dado con los parámetros r1 y r2. La figura 4.3.1 muestra la representación de la fracción molar de monómero 1 en el copolímero, F1, en función de la fracción molar del monómero 1 en la alimentación, f1, para algún valor de las relaciones de reactividad r1 y r2. Figura 4.3.1.- Representación de la fracción molar del componente 1 en el copolímero, F1, en función de la fracción molar del componente 1 en la alimentación de monómeros, f1, para algunos valores de los parámetros r1 y r2. Como puede verse en la figura 4.3.1, cuando r1 = r2 = 1, tanto el copolímero como la alimentación de monómeros tienen siempre la misma composición. Este caso es muy difícil de encontrar en la práctica. En este caso la ecuación de composición del copolímero se reduce a: F1 f1 f1 f2 f1 f2 2 f1 f f1 f2 1 (4.3.4) ya que f1 + f2 = 1. Si se cumple que r1 = r2 = ri, el copolímero y la alimentación tienen la misma composición cuando fi = 0,5. En este caso la ecuación (4.3.3) puede escribirse: F1 ri 1 0,5 2 ri 1 (4.3.5) Por otra parte, si r1 = r2 pero de forma que ambos valores de r sean menores que la unidad, el copolímero que se forma es más rico en monómero 1 para valores de la alimentación f1 < 0,5 y más rico en monómero 2 a partir de f1 > 0,5. Existen casos especiales en los que la composición del copolímero no varía y es igual a la de la alimentación. Esta situación se denomina copolimerización azeotrópica y da lugar a copolímeros de composición homogénea. En la copolimerización azeotrópica, puesto que la composición del copolímero es igual a la de la mezcla monomérica, se verificará que: F1 f1 d M1 M1 d M 2 M 2 (4.3.6) sustituyendo estas condiciones en la ecuación de la composición del copolímero se obtiene que: r1 M1 M 2 r2 M 2 M1 1 (4.3.7) de donde: M1 1 r2 M 2 1 r1 (4.3.8) De esta última ecuación se deduce que solo existe azeótropo cuando se cumple que: (a).- r1<1 y r2<1 (b).- r2>1 y r2>1 El primer caso solo se encuentra en las copolimerizaciones radicales. La composición a la que tanto la alimentación como el copolímero formado tienen la misma composición se denomina composición azeotrópica y se puede calcular a partir de la ecuación (4.3.8). Si sustituimos en esta ecuación la condición dada en la ecuación (4.3.2) se obtiene que: f1crítica 1 r2 2 r1 r2 (4.3.9) Esta composición azeotrópica origina un copolímero monodisperso en composición. A cualquier otra composición se obtiene polidispersividad química. En la figura 4.3.2 se muestra la dependencia de F1 con f1 para distintos valores de las relaciones de reactividad (r1/r2). La ecuación de composición del copolímero ha sido comprobada experimentalmente en numerosos sistemas siendo aplicable a copolimerizaciones radicales, aniónicas y catiónicas. Sin embargo, hay que tener en cuenta que los valores de las relaciones de reactividad r1 y r2 pueden diferir drásticamente. Así, por ejemplo, en el caso de la copolimerización del estireno con el metacrilato de metilo se obtiene que r1 = 0,52 y r2 = 0,46, si el mecanismo de copolimerización es por radicales libres, mientras que r1 = 10 y r2 = 0,1 si la reacción de copolimerización procede por vía catiónica y, finalmente, r1 = 0,1 y r2 = 6 si se copolimeriza aniónicamente. En la figura 4.3.3 se muestra como varia la composición del copolímero en función de la alimentación dependiendo del tipo de polimerización que se emplee, para el caso del par estireno/ metacrilato de metilo. La diferencia de valores, cuando se comparan sobre todo las relaciones de reactividad de las copolimerizaciones iónicas, refleja la gran selectividad de las mismas. El estireno presenta una mayor reactividad en la polimerización catiónica que en la aniónica, debido a que el anillo aromático favorece la formación y estabilización de un carbocatión. Por el contrario, el grupo éster del metacrilato de metilo favorece la formación de un carbanión. El resultado de esta selectividad es que ambos procesos iónicos producen copolímeros que son ricos en el comonómero más reactivo, sea cual fuere la alimentación. Sin embargo, esta selectividad no se produce en la copolimerización radical; los radicales propagantes quedan perfectamente estabilizados por sus sustituyentes y presentan relaciones de reactividad semejantes lo que origina copolfímeros de muy diversas composiciones con el simple hecho de variar la alimentación. En general, la copolimerización iónica es mucho más selectiva que la radical, lo que limita el uso práctico de la primera. Figura 4.3.2.- Dependencia de F1 con f1 para distintos valores de las relaciones de reactividad (r1/r2). Figura 4.3.3.- Composición instantánea del copolímero, F1, en función de la composición de la alimentación, f1, para el par estireno/metacrilato de metilo en el caso de copolimerizacian catiónica, radical y aniónica, respectivamente Así mismo, con los datos de la tabla 4.2.1, los cocientes de reactividad del butadieno y del estireno son r1 = 1,39 y r2 = 0,78, respectivamente. Puesto que la velocidad de desaparición del butadieno (M1 ) es más rápida que la del estireno (M 2 ), la composición de la mezcla de reactivos (x) cambiaría muy rápidamente si no se añadiera butadieno para evitar cambios de la composición, es decir, se produciría deriva de la composición. Como se ve en la ecuación siguiente, una relación equimolar de butadieno y estireno produciría un copolímero con mucho butadieno en el cual habrá cuatro moléculas de butadieno por cada tres de estireno en la cadena del polímero. n M1 1.39M1 M2 1.39 1 2.39 4 1.34 3 M2 M1 0.78M2 1 0.78 1.78 A partir de f1, la fracción molar del monómero M1, en la mezcla reaccionante (f2 la del monómero M2) y F1 la fracción molar del monómero M1, en el copolímero resultante (F1 la del monómero M2), haciendo un balance de masas, en un determinado instante se tiene: r1f12 f1f2 F1 2 r1f1 2f1f2 r2f22 Siendo: f2 1 f1 , F2 1 F1 En función de los valores de r1 y r2 pueden dibujarse gráficos (Figura 4.3.4) que proporcionan los valores de F1 en función de f1. Para r1 = r2 = 1 resulta la bisectriz F1 = f1 (a) y para r1 = r2 = 0 resulta F1 = 0,5 (b) Figura 4.3.4.- Valores de F1 en función de f1 . Es interesante considerar la forma de estas curvas en función de los valores del producto r1 r2 . Cuando r1r2 =1 se obtienen curvas simétricas como las representadas en la figura central, para r1 = 0,1-10. Cuando tanto r1 como r2 son aproximadamente iguales a 1, se dice que las condiciones son ideales, y se produce un copolímero aleatorio (no alternante),. Así, al copolimerizar clorotrifluoroetileno con tetrafluoroetileno se obtendrá un copolímero perfectamente aleatorio (copolímero ideal). Cuando r1 y r2 son aproximadamente iguales a cero, como es el caso de la copolimerización de anhídrido maleico con estireno, se obtiene un copolímero alternante. En general, existirá una tendencia a la alternancia cuando el producto r1 r2 se acerque a cero. Por el contrario, si los valores de r1 y r2 se parecen, y el producto r1 r2 se acerca a 1, la tendencia será producir copolímeros aleatorios. El valor de r1 r2 para la mayoría de las copolimerizaciones se halla entre 0 y 1, pudiéndose utilizar dicho valor para estimar el grado de aleatoriedad en un copolímero. Para r1 r2 < 1 (caso más frecuente) se obtienen curvas entre la «b» y la «a», es decir, con una cierta tendencia a la alternancia. Si r1 y r2 son ambas menor (o mayor) que la unidad se obtienen curvas del tipo «c» (figura de la derecha) que ofrecen una intersección P, con la bisectriz. En este punto la composición de los monómeros reaccionantes f 1c coincide con la composición del polímero F1c y, por lo tanto, permanecen ambas constantes a lo largo de la polimerización. Es tas composiciones se denominan azeotrópicas: f1c 1 r2 2 r1 r2 Cuando r1 y r2 son mayores que 1 se favorece la formación de largas secuencias o bloques de cada monómero y, en casos extremos, podría predominar la formación de homopolímeros. Este caso es poco frecuente. Cuando interesa producir copolímeros con estructura en bloque, del tipo: M1 M1 M1 M1 M2 M2 M2 M2 M2 M1 M1 M1 o bien: M2 M2 M2 M2 I M1 M1 M1 M1 M 1 M1 M1 M1 M1 M1 M1 M1 I M2 M2 M2 M2 M2 puede recurrirse a la preparación de prepolímeros de bajo grado de polimerización, que luego se copolimerizan entre sí. Esta forma de operar se utiliza con frecuencia para los policondensados. En el caso de monómeros que polimerizan por un mecanismo en cadena, la polimerización aniónica proporciona polímeros «vivos» que pueden formar copolímeros de bloque por reacción posterior con un segundo monómero. También pueden obtenerse copolímeros por masticación, pues, mediante acciones mecánicas (de cortadura), se rompen los enlaces covalentes de las macromoléculas, creándose radicales libres que reaccionan entre sí, dando lugar a copolímeros tipo bloque, altamente desordenados. La extensión del análisis efectuado a la copolimerización catiónica o aniónica es directa pero el orden de reactividad de los monómeros es diferente. En la copolimerización mediante reacción por etapas, dado que las reactividades de todos los grupos funcionales son muy similares, independientemente de la longitud de la molécula, los comonómeros se distribuyen aleatoriamente a lo largo de la cadena en cantidades proporcionales a sus concentraciones en la mezcla. Ocasionalmente se encuentran algunas desviaciones a esta generalización. 4.4.-Cinética de la copolimerización. La ecuación de composición del copolímero obtenida anteriormente no nos dice nada acerca del proceso cinético. En este caso es más complicado que en el caso de la homopolimerización, pero a pesar de ello, tiene solución teórica en los casos más sencillos de mezclas binarias. Las magnitudes directamente observables son cinco, a saber: (1).- Velocidad de iniciación (2).- Velocidad total de copolimerización (3).- Composición del copolímero (4).- Peso molecular (5).- Tiempo de vida de los radicales libres. La velocidad de iniciación total será la suma de las velocidades de iniciación de los dos monómeros. El esquema cinético global será: M1 M1 Iniciación M 2 M 2 k 11 R M1 M1 R M1 k 12 R M1 M 2 R M 2 Pr opagación k 21 R M 2 M1 R M1 k 22 R M 2 M 2 R M 2 k 11 R M1 R M1 R M1 M 1 R k 12 R M1 R M 2 R M1 M 2 R Ter min ación k 21 R M 2 R M1 R M 2 M1 R k 22 R M 2 R M 2 R M 2 M 2 R Las condiciones del estado estacionario son dos: una se aplica a la concentración total de radicales libres y la otra a los radicales libre [M1• y [M2•] por separado, es decir: k21 M2 M1 k12 M1 M2 (4.4.1) y como: Vi = vt 2 v i v1 v 2 kt11 M1 2kt12 M1 M2 kt 22 M2 2 (4.4.2) despejando de la ecuación (4.4.1) la concentración de radicales libres [M2•] y sustituyéndola en la ecuación (4.4.2) se obtiene: v 0,5 k k 2 M k k M 2 M1 1 t 122 12 2 t 22 12 2 2 2kt 12 k 21 M1 k21 M1 0,5 (4.4.3) Las velocidades de desaparición de los monómeros serán: d M 1 k11 M1 M1 k21 M 2 M1 dt (4.4.4) d M 2 k22 M 2 M 2 k12 M1 M 2 dt (4.4.5) La velocidad total de polimerización será: d M1 M 2 dt 2 2 r1 M1 2 M1 M 2 r2 M 2 v10,5 / y xz z r1 M1 2 r1r2 M1 M 2 r2 M 2 y y 2 en donde se ha hecho: x kt 12 kt 11kt 22 y kt 11 2 k11 y kt 22 2 k 22 (4.4.6) 5.- Síntesis de caucho Se trata de hacer caucho SBS. Es algo así: Comenzamos empleando una técnica llamada polimerización aniónica viviente. Una polimerización viviente es una que tiene lugar sin reacciones de terminación. Esto quiere decir que una vez que el monómero en su recipiente (Vaso de precipitados) ha sido agotado y se ha transformado en polímero, las cadenas poliméricas aún se encuentran activas. Si se colocara más monómero dentro del recipiente, se adicionaría al polímero, haciéndolo más grande. Lo primero que tenemos que hacer es obtener una cadena de poliestireno viviente. Esto se logra polimerizando el monómero estireno con un iniciador aniónico como el butil-litio. Esta cadena de poliestireno es viviente, por lo que si se le agrega un segundo monómero, éste se adicionará al polímero. Entonces se adicionará monómero butadieno. Esto nos dará un copolímero en bloque estireno-butadieno viviente. El próximo paso es adicionar más monómero estireno y obtendremos un copolímero en tribloque estireno-butadieno-estireno. Aunque el monómero butadieno se adicionará al anión del extremo de la cadena de poliestireno, el monómero estireno no se agregará al anión del extremo de la cadena de polibutadieno. Esto resulta muy inconveniente. Para solucionarlo, se le hace reaccionar con un compuesto llamado diclorodimetilsilano. La cadena aniónica viviente expulsa un átomo de cloro del silano y se obtiene un polímero que termina en un clorosilano, y nuestro polímero ya no es más viviente. Y sirve porque se puede hacer algo con este polímero terminado en clorosilano. Es decir, si se toma el homopolímero poliestireno viviente, éste reaccionará con el polímero terminado en clorosilano, de la misma forma que lo hizo el copolímero estireno-butadieno con el diclorodimetilsilano. Esto nos da el copolímero en tribloque que queríamos. 6.- Técnicas de polimerización. 6.1.- Introducción. Muchos monómeros, como el estireno, el acrilonitrilo y el cloruro de vinilo son tóxicos, además de dar lugar a reacciones de polimerización muy exotérmicas. Por tanto, deberán tomarse las precauciones necesarias para minimizar el contacto con estos compuestos y para controlar la temperatura de la reacción de polimerización. Existen cuatro técnicas industriales empleadas en la polimerización de un monómero, la polimerización en masa, en solución, en suspensión y en emulsión (Solución coloidal en que pequeñas partículas de un líquido están dispersas en otro líquido) (Tabla 6.1.1). Cada una de estas técnicas tiene condiciones específicas y dan origen a polímeros con características diferentes. Cada uno de esos métodos tiene sus ventajas y sus inconvenientes (Tabla 6.1.2). Tabla 6.1.1.- Tipos de sistemas de polimerización. Tabla 6.1.2.- Comparación de los distintos sistemas de polimerización. 6.2.- Polimerización en masa o en bloque. Este tipo de polimerización es quizás el más obvio de todos y es el empleado más frecuentemente para la obtención de polímeros de condensación, en los que las reacciones son ligeramente exotérmicas, por lo que el control del calor que se produce es relativamente fácil. En cambio en el caso de monómeros vinílicos este control es más difícil, debido a que las reacciones son muy exotérmicas. Este incremento de temperatura, por otra parte, provoca una mayor velocidad de descomposición del iniciador, lo que origina una aceleración de la velocidad de polimerización y una disminución del peso molecular del polímero obtenido. Además, y como consecuencia del incremento de viscosidad de la mezcla, se producen puntos calientes localizados que originan la formación de burbujas, estropeándose la apariencia física del material. Por estas razones, excepto en la preparación de piezas coladas de polimetacrilato de metilo, poliestireno y policloruro de vinilo, este método de polimerización en masa se utiliza raramente. En la figura 6.2.1 puede verse el esquema de un reactor de polimerización en continuo para la obtención de poliestireno a escala industrial. Figura 6.2.1.- Esquema de un reactor de polimerización continua empleado para la obtención de poliestireno. La polimerización en masa es una técnica simple, homogénea, donde solo el monómero y el iniciador están presentes en el sistema. Se realiza añadiendo directamente el iniciador al monómero líquido. En el caso de que la polimerización sea iniciada térmicamente o por radiación, solo habrá monómero en el medio reaccionante. Por consiguiente, esta técnica es económica, además de producir polímeros con un alto grado de pureza, obteniéndose productos libres de contaminantes y de alta claridad óptica. Esta polimerización es altamente exotérmica, presentándose dificultades en el control de la temperatura y de la agitación del medio reaccionante, que rápidamente se vuelve viscoso desde el inicio de la polimerización, dificultándose la evacuación del calor desarrollado, por lo que se crean zonas calientes en las que puede tener lugar la autoaceleración. La agitación durante la polimerización debe ser vigorosa para que haya la dispersión del calor de formación del polímero, evitándose puntos sobrecalentados, que dan un color amarillento al producto. Este inconveniente puede ser evitado usándose inicialmente un prepolímero (mezcla de polímero y monómero), que es producido a una temperatura más baja, con una baja conversión del monómero y condiciones moderadas. El monómero residual generalmente debe ser eliminado del polímero mediante arrastre con vapor (« stripping»). En algunos casos, la polimerización se lleva a cabo en moldes sin agitación, como, por ejemplo, para producir placas de polimetilmetacrilato altamente transparentes. A camino del molde, se calienta el prepolímero completándose la polimerización. La polimerización en masa es muy usada en la fabricación de lentes plásticas amorfas, debido a las excelentes cualidades ópticas conseguidas en las piezas moldeadas, sin presión, como en el caso del poli (metacrilato de metilo.)Se usa para la polimerización del estireno, VCM y metacrilato de metilo. En la figura 6.2.2 se representa un proceso para obtener ABS por polimerización en masa. El proceso de masa ABS fue originalmente adaptado del proceso para obtener poliestireno. Este proceso tiene dos ventajas inherentes sobre la polimerización por suspensión y por emulsión. Una es que el agua residual de tratamiento es mínima y otra es el ahorro de energía por evitar la etapa de separación y secado de la resina del agua de proceso. Otra ventaja es que produce ABS poco pigmentado, incluso algo traslucido, lo que reduce la concentración de colorantes necesarios. Generalmente es más eficiente a modificaciones por impacto que el realizado por emulsión, sin embargo, la cantidad de caucho que se puede incorporar está limitada por limitaciones del proceso respecto a la viscosidad. El brillo superficial es menor debido a que las partículas de caucho son mayores. Figura 6.2.2.- Proceso para obtener ABS por polimerización en masa 6.3.- Polimerización en disolución. En la polimerización en disolución, además del monómero y del iniciador, se emplea un disolvente, que debe disolverlos, formando un sistema homogéneo. El disolvente ideal debe ser barato, de bajo punto de ebullición y de fácil separación posterior del polímero. Al final de esta polimerización, el polímero formado puede ser soluble o no en el disolvente usado. En el caso de que el polímero sea insoluble, se obtiene un lodo, que es fácilmente separable del medio reaccionante por filtración. Si el polímero fuese soluble, se utiliza un no-disolvente para precipitarlo en forma de fibras o polvo. En la polimerización en disolución con la presencia de un disolvente se facilita la transmisión del calor y se reduce la viscosidad del medio, con lo que tiene como ventaja una temperatura homogénea debido a la fácil agitación del sistema, que evita el problema del sobrecalentamiento. Sin embargo, se puede producir transferencia de cadena a las moléculas de este nuevo componente, que se traduce en una reducción del peso molecular. Por ello se deben seleccionar con cuidado los disolventes a emplear. La polimerización en solución se utiliza principalmente cuando se desea aplicar la propia solución polimérica, y se emplea bastante en policondensación. 6.4.- Polimerización en suspensión en fase acuosa. Es uno de los procesos de polimerización de mayor importancia industrial. El monómero se dispersa en un medio acuoso debido a que es insoluble en el agua y el iniciador es soluble en la fase orgánica. En general se añade un estabilizador que asegure que los glóbulos (tamaños de 50 a 500 μm) permanezcan dispersos sin coalescer en el transcurso de la polimerización. El alcohol polivinilico disuelto en la fase acuosa es un agente tipico de suspensión. El polímero final queda en forma de pequeñas perlas. Las caracteristicas de la polimerización en suspension son las mismas de la polimerización en masa, con la única diferencia que el monómero forma unidades discretas, sin embargo, dentro de cada perla la polimerización transcurre igual que con el monómero puro. El tamaño medio de las perlas puede variar entre amplios límites, dependiendo de la velocidad de agitación, la relación (monomero/agua) y el tipo de estabilizador. La distribución de tamaños de las perlas suele ser muy estrecha si la reacci6n se ha llevado a cabo en condiciones uniformes. La polimerización en suspensión (Mezcla en que pequeñas partículas de un sólido o líquido se mantienen suspensas en un líquido o un gas), también conocida como polimerización en perlas, por la forma como se obtienen los polímeros, es una polimerización heterogénea donde el monómero y el iniciador son insolubles en el medio dispersante, en general el agua. En la polimerización en suspensión en fase acuosa mediante agitación se mantiene el monómero (insoluble en agua) en forma de gotas en las que tiene lugar la reacción. Esta modalidad se distingue de la aparentemente similar polimerización en emulsión por la localización del iniciador y la cinética a que obedece. El iniciador está disuelto en el monómero y la cinética es la misma que la de la polimerización en masa. El monómero es la fase dispersa y da lugar a un polímero en fase sólida también dispersa. La dispersión se mantiene por una combinación de agitación y el empleo de estabilizadores solubles en agua (por ejemplo, metilcelulosa o gelatina). La polimerización tiene lugar dentro de las partículas en suspensión, las cuales tienen tamaño medio entre 2 a 10 mm, y donde se encuentran el monómero y el iniciador. La agitación del sistema es un factor muy importante en esta técnica, pues según la velocidad de agitación empleada, varía el tamaño de las partículas. Además del monómero el iniciador y el solvente, también se adicionan agentes tensoactivos (Sustancia, como un detergente, que al adicionarla a un líquido aumenta la capacidad de este desparramarse y humedecer, debido a la disminución de su tensión superficial), que auxilian en la suspensión del polímero formado, evitando la adhesión entre las partículas y, como consecuencia, la precipitación del polímero sin la formación de las perlas. La precipitación del polímero también puede ser evitada por la adición al medio reaccionante de un polímero hidrosoluble, de elevado peso molecular, que aumente la viscosidad del medio. No obstante, la incorporación de estos aditivos al sistema dificulta la purificación del polímero resultante. El problema de este tipo de polimerización es mantener la suspension durante el period() critic() que va del 20 al 30% de conversion, ya que en este periodo los glóbulos formados son muy viscosos y tienden a agregarse. Cuando se pasa a conversiones superiores los glóbulos son bastante duros para no agregarse. 6.5.- Polimerización en emulsión. La definición de emulsión es: "Una suspensión coloidal estable como por ejemplo la leche, constituida por un líquido inmiscible disperso y retenido en otro líquido por una sustancia llamada emulsificante". La polimerización en emulsión es una polimerización heterogénea en medio líquido, que requiere una serie de aditivos con funciones específicas, como emulgente, que es un producto que, en pequeñas cantidades, ayuda a formar o estabilizar una emulsión (generalmente un detergente), soluciones tampón de pH (Mezcla de compuestos utilizada para mantener el pH constante en una disolución) , coloides, protectores, reguladores de tensión superficial, reguladores de polimerización (modificadores) y activadores (agentes de reducción). Tiene una alta velocidad de reacción y conversión, siendo de fácil control de agitación y temperatura. Los polímeros obtenidos con esta técnica presentan altos pesos moleculares, son más difíciles de purificar por la gran cantidad de aditivos adicionados. Sin embargo esta técnica tiene gran importancia industrial y es muy empleada en poliadiciones, principalmente cuando se aplica directamente el látex resultante. La polimerización en emulsión, es similar a la anterior, con la diferencia que, mediante un agente emulsionante, se logra que el diámetro de las gotas de la f ase orgánica sea mucho menor; además, el iniciador debe ser soluble en la fase acuosa (persulfatos, agua oxigenada, sistemas redox, etc.). Como agentes emulsionantes se usan, además de los detergentes clásicos, otros surfactantes, como alcoholes sulfonados, aminas alifáticas, alcohol polivinílico, etc. Es una modalidad ampliamente utilizada en la obtención del PVC, poli(acetato de vinilo), el policloropreno, los polimetacrilatos, el poli(cloruro de vinilo), la poliacrilamida y los copolímeros de poliestireno, polibutadieno y poliacrilonitrilo. La cinética es distinta de la cinética de la polimerización en suspensión. La velocidad de polimerización y el peso molecular que pueden conseguirse son mayores que los de la polimerización en suspensión o en masa. En esta polimerización, el iniciador es soluble en agua, mientras que el monómero es apenas parcialmente soluble. El agente emulsionante juega un papel importante en este tipo de polimerizaciones y tiene como objetivo formar micelas (Agregados de 50 a 100 moléculas de un coloide con sus extremos hidrofílicos orientados hacia la fase acuosa, creando en su interior un medio hidrocarbonado), de tamaño entre 1 nm y 1 mm, donde queda contenido el monómero. Algunas micelas son activas, o sea, la reacción de polimerización se procesa dentro de ellas, mientras que otras son inactivas (gotas de monómeros), constituyendo apenas una fuente de monómero. A medida que la reacción ocurre, las micelas inactivas suplen a las activas con monómero, que crecen hasta formar gotas de polímero, originando posteriormente el polímero sólido. La figura 6.5.1 representa el esquema de un sistema de polimerización en emulsión. La mayor parte del monómero está en forma de gotas pero algo se introduce en las micelas y las hincha. El polímero se forma dentro de estas micelas (más abundantes que las gotas de monómero y con una relación (Superficie/Volumen) mayor) hacia donde se dirigen los radicales libres generados en la fase acuosa. Las gotitas actúan como depósitos de monómero y van disminuyendo de tamaño, ya que hay una difusión del monómero hacia las micelas. Figura 6.5.1.- Representación esquemática de un sistema de polimerización en emulsión En la polimerización en masa, la velocidad puede incrementarse solamente aumentando la velocidad de iniciación (dado que no puede regularse la concentración del monómero), pero esto causa una disminución en el grado de polimerización. En la polimerización en emulsión puede aumentarse la velocidad de polimerización aumentando el número de partículas de polímero en crecimiento. Si la velocidad de iniciación se mantiene constante, el grado de polimeriza ción aumenta en lugar de disminuir como en la polimerización en masa o en suspensión. El número de partículas de polímero está determinado por el número de micelas del agente emulsionante inicialmente presente, que se puede incrementar aumentando su concentración. Para comprender la polimerización por emulsión y los emulsificantes, primero debemos entender cómo actúa un jabón o surfactante, como se le dice en las polimerizaciones por emulsión, el cual posee dos extremos de distinta solubilidad. Un extremo, denominado cola, consiste en una larga cadena hidrocarbonada soluble en compuestos orgánicos, no polares. El otro, llamado cabeza, a menudo es una sal de sodio o potasio, soluble en agua. La sal soluble en agua puede ser la sal de un ácido carboxílico o ácido sulfónico. El término técnico para la exhibición química de "personalidades duales" es anfipático. Cuando se tienen una gran cantidad de moléculas de jabón y a una concentración dada de agua, las moléculas de jabón se aglomeran para formar micelas, las cuales se ordenan con sus colas hacia adentro. Cualquier suciedad, por ejemplo, grasa que se pueda tener en sus manos, es principalmente de origen orgánico y puede representarse así: Cuando se lavan las manos con agua jabonosa, la partícula de suciedad, salta justo hacia el medio, de modo que permanece disuelta en las colas orgánicas de la micela. Por lo tanto, la suciedad queda disuelta en la micela, la micela queda disuelta en agua. En una polimerización por emulsión, el jabón o surfactante es disuelto en agua hasta alcanzar la concentración micelar crítica (CMC). El interior de la micela provee el lugar necesario para la polimerización. Luego se agregan un monómero (como el estireno o el metacrilato de metilo) y un iniciador radicalario soluble en agua y toda la mezcla se sacude o se agita. Las polimerizaciones en emulsión siempre se realizan por medio de radicales libres. Los extremos aniónicos y catiónicos de las cadenas serán rápidamente saciados por el agua. El producto de una polimerización en emulsión recibe el nombre de látex. Una vez que se pone todo en el reactor, el monómero puede encontrarse en tres lugares distintos. Para empezar, puede estar formando grandes gotas que flotan sin rumbo en el agua. En segundo lugar, alguna porción podría estar disuelta en agua, pero es poco probable, ya que los monómeros orgánicos como el estireno y el metacrilato de metilo son hidrofóbicos. Por último, el monómero puede encontrarse en las micelas, que es exactamente lo que se quiere. El líquido inmiscible es el monómero hidrofóbico, el líquido madre es el agua y el emulsificante es el jabón. La iniciación tiene lugar cuando un fragmento de iniciador migra hacia una micela y reacciona con una molécula de monómero. Comúnmente se emplean iniciadores solubles en agua como los peróxidos y persulfatos (lo cual también impide la polimerización en las grandes gotas de monómero). Una vez que comienza la polimerización, la micela es considerada como una partícula. Las partículas poliméricas pueden crecer hasta pesos moleculares extremadamente elevados, especialmente si la concentración de iniciador es baja. Eso hace que la concentración de radicales y la velocidad de terminación también sean bajas. A veces se adiciona a la mezcla un agente de transferencia de cadena para impedir que el peso molecular sea demasiado alto. Prosiguiendo con la polimerización, el monómero migra desde las grandes gotas que forma hacia las micelas. En promedio, existe un radical por micela. Debido a esto, no existe mucha competencia por el monómero entre las cadenas en crecimiento de las partículas, por lo tanto éstas crecen a pesos moleculares casi idénticos y la polidispersidad es muy cercana a uno. En las polimerizaciones por emulsión, prácticamente todo el monómero se consume, lo cual quiere decir que el látex puede ser usado sin purificación. Esto es importante para pinturas y revestimientos. Sólo hay que agregarle color al látex, envasarlo en una lata y ya estará listo para ser usado. Este es el aspecto ingenioso de la polimerización por emulsión: cada micela puede considerarse como una polimerización en mini-masa. A diferencia de las tradicionales polimerizaciones en masa, no existe monómero residual sin reaccionar y no se forman "puntos calientes". En las polimerizaciones en masa (es decir, sólo el monómero y el iniciador, sin solvente) los puntos calientes causan degradación y decoloración y la transferencia de cadena ensancha la distribución de pesos moleculares. A veces un incremento de la temperatura puede hacer que la velocidad de polimerización se incremente en forma explosiva. En cambio el agua actúa aquí como un dispersante del calor para todos estos mini-reactores impidiendo que exploten. La velocidad de polimerización es la misma que la velocidad de desaparición del monómero, el cual desaparece más rápidamente cuando existen más partículas. Para que existan más partículas debe haber más micelas. Si se incrementa la concentración de jabón, obtendremos más micelas. Pero supongamos que la concentración de iniciador no se modifica. En ese caso, tendremos más partículas y menos radicales. Lo cual significa que el número de radicales por micela cae por debajo de uno. En otras palabras, la velocidad de terminación será baja, ya que hay menos radicales. Y el resultado final es que disminuyendo la concentración de iniciador se incrementa el peso molecular y la velocidad de polimerización. Esto es exactamente lo contrario a las polimerizaciones en masa y en solución. Para incrementar la velocidad de polimerización en estos casos, debemos aumentar la temperatura o la concentración de iniciador, con lo que se logra un incremento en la velocidad de terminación y una disminución en el peso molecular. En la tabla 6.5.1 se comparan las características de las polimerizaciones en masa, solución, suspensión y emulsión. Tabla 6.5.1.- Comparación de los sistemas de polimerización. TIPO VENTAJAS Masa # Alto grado de pureza # Requiere equipos sencillos # Altos pesos moleculares Solución Emulsión Suspensión DESVENTAJAS # Control de temperatura difícil (Puntos calientes) # Distribución de peso molecular ancha # Presencia de monómero residual sin reaccionar #Alta viscosidad # Control de temperatura fácil # El disolvente causa reducción en el peso # La disolución polimérica formada molecular y en la velocidad de reacción puede ser utilizada directamente # Transferencia de cadena al solvente # Control del peso molecular # Dificultades en la extracción del disolvente # Polimerización rápida # Contaminación del polímero con agentes # Obtención de polímeros con alto emulsionantes y agua peso molecular # Fácil control de la temperatura # Reacciona todo el monómero # Contaminación del polímero con agentes # Control de temperatura fácil estabilizadores y agua # Obtención del polímero en forma # La presencia del surfactante puede causar de perlas sensibilidad al agua # Se pueden obtener materiales con # Requiere agitación continua baja Tg, Baja viscosidad Además de las técnicas de polimerización anteriores, algunos polímeros se pueden producir por la técnica de polimerización interfacial. En esta, la polimerización ocurre en la intercara entre dos solventes inmiscibles, en la que cada uno de los monómeros está en una de las fases. El polímero se forma en esa intercara, luego se remueve a fin de permitir la continuidad de la polimerización. Este método es limitado a un pequeño número de polimerizaciones en etapas, debido a las condiciones de reacción necesarias. En la figura 6.5.2 se representa un proceso para obtener ABS en el cual el látex de caucho se forma a partir de polibutadieno. Figura 6.5.2.- Proceso para obtener ABS, en el cual el látex de caucho se forma a partir de polibutadieno.