CAPITULO 10 CATÁLISIS HETEROGENEA 1. INTRODUCCIÓN
Anuncio

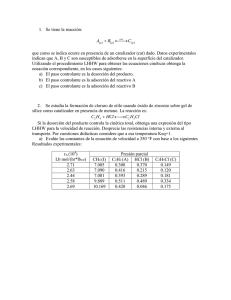
Reactores Químicos y Biológicos Catálisis Heterogénea CAPITULO 10 CATÁLISIS HETEROGENEA 1. INTRODUCCIÓN Para que una reacción química tenga lugar se debe superar el valor de la energía de activación. Una vez vencida esa barrera el sistema evoluciona de forma tal que llegará al estado final de la reacción. La velocidad de reacción podría incrementarse de dos maneras: aumentando la concentración del "complejo activado" o eventualmente disminuyendo la energía de activación. Este último mecanismo es el que se pone de manifiesto cuando se emplea determinadas sustancias llamadas catalizadores. Estas sustancias aceleran las reacciones químicas disminuyendo la energía libre de activación, se combinan con los reactivos para producir un estrato de transición de menor energía que el estado de transición de la reacción no catalizada. Por ejemplo el hidrógeno y el oxígeno gaseoso son inertes a temperatura ambiente pero rápidamente reaccionan cuando están expuestos con platino. Cuando los productos de la reacción se forman, se regenera el catalizador al estado libre. El catalizador cambia una velocidad de reacción promoviendo un mecanismo molecular diferente para la reacción pero no modifica el equilibrio termodinámico. Figura 1 Hay principalmente dos tipos de catálisis: Catálisis homogenea: tiene lugar cuando los reactivos y el catalizador se encuentran en la misma fase, sea líquida o gaseosa. En la catálisis homogénea se tiene un acceso más fácil al mecanismo de reacción y por consecuencia se puede dominar mejor el proceso catalítico correspondiente. Otra ventaja no menos despreciable de este tipo de catálisis es la ausencia de efectos de envenenamiento tan frecuentes en 1 Reactores Químicos y Biológicos Catálisis Heterogénea el caso de la catálisis heterogénea, y que obliga a tratamientos costosos de eliminación de impurezas. Algunos de los procesos más importantes: - Polimerización de olefinas: polietileno - Producción de biodiesel - Adición de olefinas: polibutadieno - Oxidación de olefinas: óxido de propileno - Polimerización-condensación: fibra de poliéster. Uno de los inconvenientes de la catálisis homogénea es la dificultad de separar el catalizador del medio reaccionante, lo que presenta un mayor costo que el de los procesos heterogéneos convencionales. Catálisis heterogénea: el catalizador esta presente en la reacción en una fase diferente a la de los reactivos. Generalmente el catalizador es un sólido y los reactivos son líquidos o gases. La separación más simple y completa del catalizador del producto provoca que la catálisis heterogénea sea mas atractiva económicamente. Uno de los inconvenientes que presenta los catalizadores heterogéneos es la desactivación, esta puede originarse por sinterizado de la superficie, envenenamiento irreversible provocado por alguna sustancia o ensuciamiento provocado por la deposición de carbón u otras sustancias. Otras posibles clasificaciones de los catalizadores presentes en la literatura se basan en su estado físico, es así que el catalizador puede ser gaseoso, líquido o sólido. Dependiendo de la sustancia sobre la cual se ha manufacturado, el catalizador puede ser orgánico (enzimas o ácidos orgánicos) o inorgánicos (metales, óxidos metálicos, etc.). Finalmente, basado en la acción la catálisis puede clasificarse en enzimática, acido-base, fotocatalítica, etc. 2. CATALISIS HETEROGENERA Tipos de catalizadores Una reacción heterogénea ocurre en una la interfase sólido-fluido por lo que es esencial disponer de una área interfacial muy grande con el fin de conseguir altas velocidades de reacción. Esta área puede proveerse mediante un estructura porosa interna de pequeños poros. Los catalizadores porosos pueden tener áreas intersticiales muy grandes. Un catalizador de silice-alumina para cracking catalítico puede tener un área superficial de 300 m2/seg (una casa muy grande o una cancha de futbol en un gramo). 2 Reactores Químicos y Biológicos Catálisis Heterogénea En determinados materiales los tamaños de poros son tan pequeños que solo moléculas pequeñas pueden acceder por lo que pueden proveer altas selectividades. Estos materiales se denominan tamices moleculares y comprenden las zeolitas y ciertas arcillas. Las zeolitas son alumino silicatos cristalinos microporosos con tamaños de poro muy uniforme. Son muy utilizados en el cracking del petróleo. En algunos casos el catalizador consiste de pequeñas partículas de un material activo sobre un material llamado inerte llamado soporte. El material activo frecuentemente es un metal o una aleación metálica. Tales catalizadores se denominan catalizadores soportados. Catalizadores estructurados: son estructuras rígidas con grandes poros o canales que aseguran una baja pérdida de carga y que exponen una elevada área superficial lateral sobre la que se puede pegar una delgada película de catalizador. Algunos tipos de catalizadores estructurados son los monolitos, espumas y mallas. Durante los años 60 se desarrollaron diferentes tipos de catalizadores “de flujo paralelo”, formados por placas o tubos y finalmente aparecieron los catalizadores que se denominaron de “panal de abeja” (honeycomb), por su parecido estructural a dichos elementos. Actualmente, a este tipo de catalizadores formados por “estructuras unitarias atravesadas longitudinalmente por canales paralelos” se les da el nombre de “monolitos” (ver figura 2). 3 Reactores Químicos y Biológicos Catálisis Heterogénea Figura 2. Catalizadores estructurados Reacciones gas-sólido Consideremos las reacciones en fase gas catalizada por superficies sólidas. Para que una reacción catalítica ocurra, al menos uno de los reactivos debe estar unido a la superficie. Esta unión se denomina adsorción y puede tener lugar por dos procesos: adsorción física y quimisorción. Adsorción física o fisisorción: las moléculas del gas se mantienen unidas a la superficie del sólido por medio de fuerzas de Van der Waals. Este hecho define todas las características propias de la fisisorción: i) es una interacción débil. ii) Es un proceso exotérmico (las fuerzas de van der Waals son atractivas) en el que los calores liberados, ΔHads (aprox. 20-40 kJ/mol) son semejantes a las entalpías de condensación de la sustancia adsorbida. iii) La molécula fisisorbida mantiene su identidad ya que la energía es insuficiente para romper el enlace aunque su geometría puede estar distorsionada. iv) La adsorción física es un proceso no especifico ya que las fuerzas que intervienen no lo son y no existe una selectividad marcada entre adsorbato y adsorbente. 4 Reactores Químicos y Biológicos Catálisis Heterogénea v) La fisisorción se produce en multicapas. Sobre una capa de gas fisisorbida puede adsorberse otra. Adsorción química o quimisorción: fue propuesta por Langmuir en 1916. En este caso las moléculas de gas se mantienen unidas a la superficie formando un enlace químico fuerte. Este hecho define las características propias de la quimisorción: i) se trata de una interacción más fuerte que la fisisorción. ii) las entalpías de quimisorción son mucho mayores que las de fisisorción y del orden de las que se liberan en la formación de enlaces químicos, ΔH°ads = - (100500) kJ/mol. iii) La quimisorción es específica. Por ejemplo el N2 es quimiadsorbido a temperatura ambiente sobre Fe, W, Ca y Ti, pero no sobre Ni, Zn, Ag, Cu o Pb. iv) Dado que implica la formación de un enlace entre adsorbato y el adsorbente, el proceso se detiene tras la formación de una monocapa sobre la superficie. v) En general, la quimisorción implica la rotura y formación de enlaces, por lo que la molécula quimisorbida no mantiene la misma estructura electrónica (enlaces) que en fase gaseosa El tipo de adsorción que afecta a la velocidad de reacción química es la quimisorción. Los sitios activos sobre la superficie son puntos sobre la superficie catalítica que pueden formar enlaces químicos fuertes con un átomo o molécula adsorbido. 3. ETAPAS EN UNA REACCIÓN QUIMICA Un esquema de la magnitud de los tamaños de un reactor empacado y de la partícula catalítica se muestra en la Fig.3. Figura 3 El proceso global de una reacción heterogenea procede a través de una serie de etapas representadas en la Figura 4: 5 Reactores Químicos y Biológicos Catálisis Heterogénea 1. Transporte de reactivos hasta desde el fluido a la superficie externa del catalizador 2. Difusión de los reactivos desde la boca del poro a la vecindad de la superficie catalítica 3. Adsorción de los reactivos sobre el catalizador 4. Reacción sobre la superficie catalítica 5. Desorción de los productos de reacción 6. Difusión de los productos desde el interior de los poros a la superficie externa. 7. Transporte de los productos hacia el seno del gas En este capítulo las etapas de adsorción, reacción superficial y la desorción serán consideradas para determinar la ecuación de velocidad de la reacción catalítica. Para ello será necesario disponer de información experimental obtenida en condiciones tales que las etapas de difusión interna y externa sean lo suficientemente altas como para que puedan ser despreciadas. Figura 4. Etapas en una reacción heterogénea Adsorción Hemos visto que el fenómeno catalítico heterogéneo requiere de la adsorción química en la superficie del catalizador de al menos uno de los reactivos. Dado que la reacción se lleva a cabo en la superficie del catalizador, el conocimiento de la cantidad de moléculas adsorbidas en esta superficie reviste gran importancia. La adsorción de un reactivo A sobre un sitio S puede representarse: A+S AS Donde AS representa una nueva especie “A adsorbida” A 6 Reactores Químicos y Biológicos Catálisis Heterogénea La concentración de reactivo adsorbido se relaciona por lo tanto con la concentración (presión) del reactivo en la fase gas (fluido). Para encontrar esta relación supongamos un sólido al cual se le suministra una cierta cantidad de gas (por ejemplo hidrógeno). Parte del gas se adsorberá en la superficie del sólido y parte quedará en la fase gas. Cuando la adsorción se ha completado y se alcanza el equilibrio, la relación entre la concentración de gas adsorbido y la presión del gas con la que está en equilibrio a temperatura constante se denomina isoterma de adsorción (Figura 5). Figura 5. Isoterma de adsorción de Langmuir. La forma de expresión cinética de la adsorción estará conectada al mecanismo adoptado. En una primera etapa se propone un modelo y la isoterma obtenida se compara con los resultados experimentales. Si los resultados del modelo son correctos, el modelo puede describir en forma razonable lo que ocurre en la realidad. Consideremos el ejemplo de la adsorción de CO sobre una superficie metálica. En la literatura se postulan dos modelos para la adsorción del CO: 1) La adsorción molecular CO + S CO S (1) 2) Adsorción disociativa CO + 2 S CS+OS (2) La forma en que se adsorba el CO dependerá de la superficie Vamos a considerar el primer caso, esto es, la adsorción molecular del CO. Con el fin de encontrar la ley de velocidad la reacción (1) puede tratarse como una reacción elemental. La velocidad será proporcional al número de colisiones de la molécula sobre la superficie la cual será función de la presión parcial de monóxido de carbono. Además las moléculas de CO sólo se adsorberán en los sitios vacantes. Por lo tanto la podemos expresar la velocidad neta de adsorción: 7 Reactores Químicos y Biológicos Catálisis Heterogénea rads = k A PCOCv − k − ACCOS (3) La relación KA= kA/k-A es la constante de equilibrio de adsorción. La ecuación puede expresarse por lo tanto como: ⎛ C rads = k A ⎜⎜ PCOCv − COS KA ⎝ ⎞ ⎟⎟ ⎠ (4) Dado que es solo el CO la molécula adsorbida, el balance de sitios totales resulta: Ct = Cv + CCOS (5) En el equilibrio, la velocidad neta de adsorción es cero. Por lo tanto igualando a cero la ecuación (3) y relacionándola con la ecuación (5), resulta: CCOS = K A PCOCt 1 + K A PCO (6) Esta ecuación cuantifica la concentración de CO adsorbido sobre la superficie del sólido y representa la ecuación para la isoterma de adsorción. Este tipo particular de ecuación representa la isoterma de Langmuir. La figura 6a presenta un grafico de la cantidad adsorbida por unidad de masa de catalizador en función de la presión parcial de CO. CCOS (mol/gcat.) CCOS (mol/gcat.) Parabólica Lineal PCo (a) PCo (b) Figura 6. Isoterma de Langmuir para la adsorción molecular Un método para chequear que un modelo de adsorción predice el comportamiento experimental observado es linealizar la ecuación obtenida, en este caso la ecuación (6): PCO 1 P = + CO CCOS K ACt Ct (7) 8 Reactores Químicos y Biológicos Catálisis Heterogénea A partir del grafico (fig 7) se obtienen los parámetros de la isoterma PCO CCOS 1 Ct 1 KACt PCo Figura 7 Si se plantea el modelo de adsorción disociativa (ec 2), se requerirán dos sitios activos vacantes, por lo tanto la velocidad neta de adsorción será: ⎛ C C rads = k A ⎜⎜ PCO C v2 − CS OS KA ⎝ ⎞ ⎟⎟ ⎠ (8) Planteando la condición de equilibrio y considerando el balance de sitios: Ct = Cv + COS + CCS (9) Asumiendo CCS=COS, se obtiene la siguiente para la isoterma presentada en la Fig. 6b COS = ( K A PCO )1 / 2 Ct 1 + 2( K A PCO )1 / 2 (10) Linealizando la expresión: ( PCO )1 / 2 1 2( PCO )1 / 2 = 1/ 2 + COS K A Ct Ct Si la adsorción disociativa se verifica, un grafico de (11) ( PCO )1 / 2 versus ( PCO )1 / 2 debería COS ser lineal. Cuando mas de una sustancia está presente la ecuación de la isoterma es mas compleja. Los principios son los mismos y la derivación de las expresiones es sencilla. Demuestre que la ecuación de la isoterma de A cuando hay un adsorbato B esta dado por la siguiente relación: 9 Reactores Químicos y Biológicos C AS = Catálisis Heterogénea K A PACt 1 + K A PA + K B PB (12) Es importante aclarar que para las derivaciones de las ecuaciones de las isotermas de Langmuir se han realizado algunas suposiciones: -Se asume una superficie uniforme, esto es la superficie proporciona un cierto número de posiciones para la adsorción y todas son equivalentes - sólo se adsorbe una molécula sobre cada posición - su adsorción es independiente de la ocupación de las posiciones vecinas (las moléculas adsorbidas no interaccionan entre si). Las isotermas de adsorción se utilizan para determinar áreas superficiales de sólidos y catalizadores al igual que para determinar constantes de equilibrio de adsorcióndesorción. Reacción superficial Después de que un reactivo se adsorbe sobre la superficie, la reacción hacia los productos puede darse de diferentes maneras. 1. Sitio único: solo un sitio activo está involucrado en la reacción: B A AS BS Dado que cada etapa del mecanismo de reacción es elemental, la velocidad de reacción está dada por: ⎛ C rs = k s ⎜⎜ C AS − BS KS ⎝ ⎞ ⎟⎟ ⎠ (13) Donde KS es la constante de equilibrio de la reacción superficial. 2. Sitio dual: en este caso el reactivo interactúa con otro sitio para formar el producto: AS+S BS+S A B La correspondiente expresión de la velocidad de reacción es: ⎛ C C rs = k s ⎜⎜ C AS C v − BS V KS ⎝ ⎞ ⎟⎟ ⎠ Un ejemplo de mecanismo dual es la reacción entre dos especies adsorbidas, tales como el CO y el O. La forma genérica en este caso es: 10 Reactores Químicos y Biológicos AS+BS Catálisis Heterogénea CS+DS A C B D y la velocidad de reacción: ⎛ C C rs = k s ⎜⎜ C AS C BS − CS DS KS ⎝ ⎞ ⎟⎟ ⎠ (14) Un tercer mecanismo dual se presenta cuando dos especies se adsorbe sobre diferentes sitios S y S’. La reacción genérica: A S + B S’ C S’ + D S ⎛ C C rs = k s ⎜⎜ C AS C BS' − CS' DS KS ⎝ A B ⎞ ⎟⎟ ⎠ C D (15) Las reacciones que involucran un sitio único o sitios duales se refieren como cinéticas tipo Langmuir -Hinshelwood. 3. Eley-Rideal. Un tercer mecanismo posible para la reacción surge de la interacción entre una molécula adsorbida y una molécula en fase gas., tal como la reacción entre el propileno y el benceno. En forma genérica: AS+B CS ⎛ C ⎞ rs = k s ⎜⎜ C AS PB − CS ⎟⎟ KS ⎠ ⎝ (16) Este tipo de mecanismo se denomina “mecanismo de Eley Rideal” Desorción Los productos adsorbidos generados en la reacción superficial deben desorberse a la fase gaseosa: CS C+S La velocidad de desorción es: ⎛ PC ⎞ rDC = k D ⎜⎜ CCS − C v ⎟⎟ K DC ⎠ ⎝ (17) donde KDC es la constante de desorción. La etapa de desorcion de C es la reversa de la etapa de adsorción de C y solo difiere en el signo: rDC = − rAC Y la constante de desorción es la inversa de la cte de equilibrio de la adsorción: 11 Reactores Químicos y Biológicos K DC = Catálisis Heterogénea 1 KC (18) Concepto de etapa controlante Cuando se lleva a cabo una reacción heterogénea, las velocidades de cada una de las etapas es serie (adsorción, reacción superficial y desorción) son iguales: − rA' = rAD = rS = rD Sin embargo una etapa en particular es general la etapa controlante o limitante. Una metodología generalmente adoptada para determinar la cinética de reacciones heterogéneas es la aproximación de Langmuir-Hinshelwood dado que se basa en ideas de Hinshelwood basadas en los principios de adsorción de Langmuir. Se propone la siguiente metodología - Se asume una serie de etapas en la reacción. - Deducción de las expresiones de las velocidades de cada etapa asumiendo que todas las etapas son reversibles. - Se postula una etapa como la controlante y las demás se usan para eliminar todos los términos de cubrimientos. - Comparación de las predicciones del modelo con los datos experimentales. El procedimiento descripto será discutido con un ejemplo particular de una reacción catalítica; la descomposición de cumeno para formar benceno y propileno. La reacción global es: C6H5CH(CH3)2 C6H6 + C3H6 La secuencia propuesta para la descomposición es la siguiente: C+S kA CS k-A CS ks BS +P k-s BS kD k-D B+S Adsorción del cumeno rAD = k A PC C v −k − ACCS 12 Reactores Químicos y Biológicos ⎛ C rAD = k A ⎜⎜ PC C v − CS KC ⎝ Catálisis Heterogénea ⎞ ⎟⎟ ⎠ (19) Reacción superficial: rS = k S CCS − k − S C BS PP ⎛ C P ⎞ rS = k S ⎜⎜ CCS − BS P ⎟⎟ KS ⎠ ⎝ (20) Desorción del benceno rD = k D C BS − k − D C s PB ⎛ C P ⎞ rD = k D ⎜⎜ C BS − v B ⎟⎟ K DB ⎠ ⎝ (21) Dado que no hay acumulación de las especies reactivas sobre la superficie, las velocidades de cada etapa son iguales: − rC = rAD = rS = rD Etapa controlante la adsorción de cumeno? Si asumimos que la etapa controlante es la adsorción de cumeno, la constante de velocidad de reacción de esta etapa (kA) es pequeña respecto a las constantes de las otras etapas (ks y kD). Por lo tanto: ⎛ C − rC = r AD = k A ⎜⎜ PC C v − CS KC ⎝ ⎞ ⎟⎟ ⎠ (22) Dado que Cv y CCS no son variables medibles, deben ser reemplazadas por cantidades medibles para cuantificar la velocidad de reacción: Asumiendo que la etapa controlante es la adsorción, se cumple que las ecuaciones (19) y (20): rs r ≅0 , D ≅0 ks kD Despejando de estas ecuaciones las concentraciones de las especies adsorbidas intermedias: C CS = C BS PP KS C BS = K B PB Cv (23) (24) Reemplazando la ecuación (24) en la (23), resulta: C CS = K B PB PP Cv KS (25) 13 Reactores Químicos y Biológicos Catálisis Heterogénea Reemplazando en la ecuación de la velocidad de reacción ⎛ PP ⎞ − rC = r AD = k ACv ⎜⎜ PC − B P ⎟⎟ KP ⎠ ⎝ Donde KP=KSKC/KB , es la constante de equilibrio expresada en presiones parciales de la reacción estudiada C B+P La concentración de sitios vacantes Cv, puede eliminarse utilizando el balance de sitios: C t = Cv + CCS + C BS (26) Sustituyendo las ecuaciones (22) y (23) en la (26) resulta C t = Cv + K B PB PP Cv + K B PB Cv KS Resolviendo para Cv Cv = Ct PP 1 + + K B B P + K B PB KS Finalmente la expresión de la ecuación de velocidad de reacción de la descomposición de cumeno que resulta de asumir la adsorción del cumeno como etapa controlante es: − rC = r AD = Ct k A (PC − PB PP / K P ) PP 1 + K B B P + K B PB KS (27) La velocidad de reacción inicial y considerando que no hay productos presentes resulta: − rCo = Ct k A PCo = kPCo (28) Etapa controlante la reacción superficial? La expresión de la velocidad de reacción para la etapa de reacción superficial: ⎛ C P ⎞ rS = k S ⎜⎜ CCS − BS P ⎟⎟ KS ⎠ ⎝ Asumiendo rA r ≅0 , D ≅0 kA kD Y operando del mismo modo se evalúan las concentraciones de CBS y CCS CCS = K C Cv PC C BS = K B Cv PB 14 Reactores Químicos y Biológicos Catálisis Heterogénea El balance de sitios queda expresado en este caso: Ct = Cv + CCS + C BS = Cv + K C Cv PC + K B Cv PB Resolviendo esta ecuación para Cv y reemplazando en la ecuación de velocidad junto con CBS y CCS se obtiene la velocidad de reacción para la descomposición de cumeno cuando se asume como etapa controlante la reacción superficial: k 6 47 4 8 Ct k A K C (PC − PB PP / K P ) − rC = r S = 1 + K B PB + K C PC (29) La velocidad inicial queda expresada de la siguiente manera: − rCo = kPCo 1 + K C PCo En estas condiciones pueden analizarse dos situaciones límites: - A bajas velocidades presiones parciales de cumeno se obtiene una relación lineal: − rCo = kPCo - Mientras que a altas presiones parciales, la velocidad es de orden cero − rCo = kPCo k = K C PCo K C Etapa controlante la desorción del benceno? La velocidad de la etapa de desorción del producto queda expresada como: ⎛ CP ⎞ rD = k D ⎜⎜ C BS − v B ⎟⎟ K DB ⎠ ⎝ En este caso se asume que: rS r ≅0 , D ≅0 kS kD Despejando de las expresiones de velocidad de las otras etapas CBS y CCS y planteando el balance de sitios totales, se obtiene la expresión de la velocidad de descomposición de cumeno asumiendo como etapa controlante la desorcion del benceno: k 647 48 Ct k D K S K C (PC − PB PP / K P ) − rC = r D = PP + K C K S PC + K C PC PP (30) 15 Reactores Químicos y Biológicos Catálisis Heterogénea La velocidad inicial es independiente de la presión parcial de cumeno: − rCo = k D C t Graficando la velocidad inicial vs. la presión inicial del reactivo, es posible analizar cuál de los modelos propuestos es consistente con los datos experimentales. Las predicciones de los modelos propuestos en las condiciones iniciales se resumen en la siguiente tabla: Etapa controlante Ecuación de velocidad inicial -rCo Adsorción del cumeno − rCo = kPCo PCo -rCo − rCo = kPCo Reacción superficial − rCo = k KC PCo -rCo Desorción del benceno − rCo = k D C t PCo Los datos experimentales de –rCO vs PCo se muestran en la figura 8 16 Reactores Químicos y Biológicos Catálisis Heterogénea -rCo PCo Por lo tanto puede concluirse que la etapa controlante del esquema cinético propuesto corresponde a la reacción superficial. En la siguiente tabla se resume las expresiones de velocidad para diferentes mecanismos que son reversibles y en donde la reacción superficial es la etapa controlante. Sitio único AS BS Dos sitios AS+S BS+S AS+BS CS+S Eley Ridel A S + B (g) CS − rA = kPA 1 + K A PA + K B PB − rA = kPA (1 + K A PA + K B PB )2 − rA = kPA (1 + K A PA + K B PB )2 − rA = kPA 1 + K A PA + K C PC Un importante porcentaje de las reacciones catalíticas de interés se verifica que la reacción superficial es la etapa controlante en el mecanismo de reacción la reacción superficial. En esta situación se puede generalizar la expresión de la velocidad de reacción de la siguiente manera: velocidad = factor cinético ( fuerza impulsora) (ter min o de adsorcion )n Ecuaciones de velocidad derivadas de la hipótesis del pseudo estado estacionario Cuando más de una etapa del mecanismo de reacción puede ser el limitante, un alternativa para derivar la ecuación de velocidad es adoptar la hipótesis de pseudo estado estacionario. De acuerdo con esta hipótesis cada una de las especies 17 Reactores Químicos y Biológicos Catálisis Heterogénea adsorbidas sobre la superficie es un intermediario y las velocidades netas de formación de todas las especies adsorbidas serán cero. Como ejemplo consideremos la siguiente reacción enzimática: La velocidad global desaparición del sustrato es: − rS = k1 [E ][S ] − k 2 [ES ] (32) Dado que la concentración del complejo enzima-sustrato [ES] no puede medirse, es necesario expresarla en función de variables que puedan medirse. Con la excepción de la etapa inicial de la reacción, que por lo general dura milisegundos en los que se mezcla E y S, [ES] permanece prácticamente constante hasta que el sustrato está casi agotado. En consecuencia, la velocidad de producción de ES es igual a la velocidad de su consumo durante la mayor parte del curso de la reacción. O sea rES = k1 [E ][S ] − k 2 [ES ] − k3 [ES ] = 0 (33) Despejando [ES] [ES ] = k1 [E ][S ] k 2 + k3 (34) y sustituyendo la ecuación (34) en la (32) y simplificando − rS = k1k 3 [E ][S ] k2 + k3 (35) Esta expresión aun no puede utilizarse dado que [E] no puede medirse; sin embargo si puede medirse la concentración total de enzima. En ausencia de desnaturalización de la enzima, la concentración total de enzima (Et) es constante e igual a la suma de las concentraciones de enzima libre y la enzima unida al sustrato: [E ]t = [E ] + [ES ] (36) [E ]t = [E ] + k1 [E ][S ] (37) k2 + k3 Despejando [E] 18 Reactores Químicos y Biológicos [E ] = [E ]t (k2 + k3 ) k3 + k1 [S ] + k 2 Catálisis Heterogénea (38) Sustituyendo esta expresión en la ecuación (35): − rS = k1k 3 [E ]t [S ] k1 [S ] + k 2 + k 3 (39) Definiendo kcat=k3 y multiplicando y dividiendo la ec.(39) por k1, podemos obtener la ecuación de Michaelis-Menten: − rS = k cat [E ]t [S ] [S ] + K M (40) k cat + k 2 k1 (41) Donde KM = Si definimos Vmax como la máxima velocidad de reacción para una dada concentración total de enzima: Vmax=kcat [E]t, la ecuación de Michaelis-Menten toma la forma mas comúnmente conocida: − rS = Vmax [S ] [S ] + K M (42) 19