trombocitosis hereditaria
Anuncio

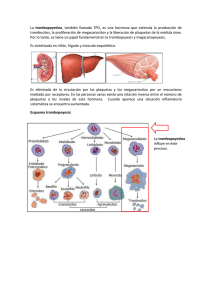
TROMBOCITOSIS DEFINICIONES Trombocitosis: recuento de plaquetas > 450 x 109 / L Trombocitosis Primaria: trombocitosis causada por una alteración en las células hematopoyéticas; el nivel sérico de trombopoyetina (Tpo), la principal citocina responsable de la producción de plaquetas, es bajo o normal. La trombocitosis primaria puede ser hereditaria (infrecuente) o adquirida (más frecuente, ver neoplasias mieloproliferativas). Trombocitosis Secundaria: trombocitosis con causa externa; el nivel sérico de TPO es ,normalmente, elevado. Normalmente, la trombocitosis secundaria es adquirida: algunas de las enfermedades que cursan con un elevado número de plaquetas son: la inflamación, la deficiencia de hierro, el asplenismo y los síndromes mielodisplásicos. La trombocitosis secundaria hereditaria (familiar) es infrecuente. Trombocitemia: neoplasia mieloproliferativa “Trombocitemia Esencial (TE)”. caracterizada por trombocitosis-ver Trombocitosis Hereditaria: trombocitosis primaria o secundaria debida a alteraciones genéticas que se pueden transmitir a la descendencia -con una causa genética familiar o hereditaria. Figure 1. Classification (courtesy of Eric Lippert). TROMBOCITOSIS HEREDITARIA CONOCIMIENTO ACTUAL En los últimos años ha habido un gran avance en el conocimiento de la biología de la trombocitosis hereditaria. Se ha descrito que mutaciones en los genes THPO y MPL pueden causar trombocitosis hereditaria: -THPO, gen que codifica para la trombopoyetina (trombocitosis secundaria con niveles altos de Tpo) -MPL, gen que codifica para el receptor de la trombopoyetina, expresado por los progenitores hematopoyéticos( trombocitosis primaria con niveles bajos o normales de Tpo). El análisis de estas mutaciones ha ayudado a conocer la regulación fisiológica de la homeostasis de las plaquetas (Ghilardi et al. 1998). Sin embargo, es probable que la trombocitosis hereditaria también esté causada por alteraciones en otros genes, ya que no todos los pacientes tienen mutaciones en los genes THPO o MPL. CONSIDERAR EL PACIENTES CON: ESTUDIO DE TROMBOCITOSIS HEREDITARIA EN * Recuento de Plaquetas > 450 x10^9/L Y * No trombocitemia Esencial (ausencia de la mutación JAK2V617F) Y * No causa identificada de trombocitosis secundaria: inflamación, deficiencia de hierro, asplenia, síndrome mielodisplásico (biopsia de médula ósea o aspirado; cariotipo) * Pacientes jóvenes * Historia familiar de trombocitosis REFERENCIAS - Review: Skoda RC. Hereditary myeloproliferative disorders. Haematologica 2010; 95: 6-8. - Ghilardi N, Wiestner A, Skoda RC. Thrombopoietin production is inhibited by a translational mechanism. Blood 1998; 92: 4023-30. MUTACIONES EN THPO Los pacientes con mutaciones en THPO presentan niveles elevados de trombopoyetina en sangre. Estudios de desequilibrio alélico han permitido el descubrimiento mutaciones en el gen de la trombopoyetina (THPO) en varias familias. La trombopoyetina es la principal citocina implicada en la producción de plaquetas y a su vez tiene un papel crítico en el mantenimiento y el destino de los progenitores mieloides tempranos. Las mutaciones de THPO detectadas en familias con trombocitosis se localizan en la región 5’ UTR del mRNA o en las fronteras exón/intrón, produciendo alteraciones en el splicing alternativo (Wiestner et al.1998; Ghilardi et al. 1999a; Ghilardi et al. 1999b). Estudios in vitro han mostrado que estas alteraciones aumentan la traducción de la trombopoyetina. Figure 2. THPO mutations in Hereditary Thrombocytosis (courtesy of Radek Skoda). En la región 5’ UTR previa al codón de inicio de la THPO, existe otro codón ATG que codifica para una región uORF, la traducción de la cual impide la traducción de la trombopoyetina. Todas las mutaciones detectadas en el gen de la THPO hasta el momento, alteran esta uORF de manera que se altera la represión de la traducción de la trombopoyetina, y se produce un aumento de expresión de la trombopoyetina, niveles elevados de trombopoyetina en sangre periférica y un aumento en la producción de plaquetas. REFERENCIAS - Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nature Genet 1998; 181: 49–52. - Ghilardi N, Wiestner A, Kikuchi M, Ohsaka A, Skoda RC. Hereditarythrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br J Haematol 1999a; 107: 310-316. - Ghilardi N, Skoda RC. A single-base deletion in the thrombopoietin (TPO) gene causes familial essential thrombocythemia through a mechanism of more efficienttranslation of TPO mRNA. Blood 1999b; 94:14801482. MUTACIONES EN MPL Los pacientes con mutaciones en MPL presentan niveles bajos de trombopoyetina en sangre. También se han descrito casos de trombocitosis familiar con alteraciones en el receptor de la trombopoyetina (Mpl). En esos casos, las alteraciones pueden ser polimorfismos que afectan a un único nucleótido, restringido a grupos étnicos específicos (Moliterno et al. 2004; El Harith et al. 2009) u otras mutaciones como la S505N (Ding et al. 2004). Figure 3. MPL mutations in Hereditary Thrombocytosis (courtesy of Radek Skoda) Cabe destacar, que las mutaciones en el aminoácido W515 de MPL se detectan, mayoritariamente, en neoplasias mieloproliferativas adquiridas. Mutaciones en éste aminoácido están presentes en un 5% de las mielofibrosis primarias y un 1% de las trombocitemias esenciales (Pikman et al. 2006; Pardanani et al. 2006). Recientemente, se ha reportado un caso de un paciente con una neoplasia mieloproliferativa que presentaba en la misma célula, coexistencia de las mutaciones S505N y W515L. Todas las mutaciones de MPL reportadas hasta el momento, producen la activación constitutiva del receptor de la trombopoyetina (MPL). REFERENCIAS - Moliterno AR, Williams DM, Gutierrez-Alamillo LI et al. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci USA 2004; 101: 11444-11447. - El-Harith HA, Roesl C, Ballmaier M et al. Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br J Haematol 2009; 144: 185-194. - Ding J, Komatsu H, Wakita A et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood 2004; 103: 4198-4200. - Boyd EM, Bench AJ, Goday-Fernandez AA et al. Clinical utilityof routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br J Haematol. 2010; 149:250-257. - Pikman Y, Lee BH, Mercher T et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270 - Pardanani AD, Levine RL, Lasho T et al. MPL515 mutations in myeloproliferative myeloid disorders: a study of 1182 patients. Blood 2006; 108: 3472-3476. and other