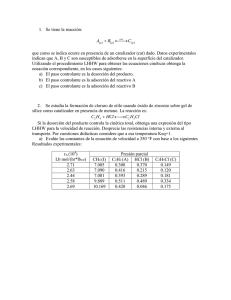
QUÍMICA FÍSICA III Tema 6 CATÁLISIS HETEROGÉNEA Y
Anuncio

QUÍMICAFÍSICAIII Tema6 CATÁLISISHETEROGÉNEAYCINÉTICA ELECTRÓDICA DepartamentodeQuímicaFísica UniversidaddeValencia QFIII Tema 6 1 Índice 6.1.‐Introducción 6.2.‐Característicasdelosfenómenoscatalíticos 6.2.1.‐MecanismoGeneraldelaCatálisis 6.2.2.‐Característicasdelacatálisisheterogénea 6.2.3.‐Etapasdelacatálisisheterogénea 6.2.4.‐Ejemplosdecatálisis 6.3.‐IntroducciónalaCinéticaElectródica 6.3.1.Mecanismogeneral 6.3.2.Leydevelocidaddelatransferenciaelectrónica 6.3.3.Relaciónentrecorrienteyvelocidaddereacción.EcuacióndeButler‐Volmer. 6.3.4.Formasaproximadasdelarelaciónentrecorrienteyvelocidaddereacción. 6.4.‐Ejerciciosadicionales QFIII Tema 6 2 6.1.Introducción En las lecciones de Cinética Macroscópica estudiadas en cursos anteriores se ahondó enlainfluenciaquesobrelasvelocidadesdereacciónejercíanlosparámetrosdecantidady los parámetros energéticos (energía térmica, energía luminosa). El conocimiento de las dependencias de las velocidades de reacción con todos los parámetros permite diseñar experienciasparaobtenerproductosaunavelocidaddeterminada.Sinembargo,haymuchos procesos en los que aun extremando las condiciones experimentales: concentraciones y temperaturas altas, por ejemplo, transcurren a velocidades lentas. Si son productos de gran consumo, la pequeña velocidad de sus síntesis los hará prohibitivos. Afortunadamente, la Industria Química emplea en la mayor parte de procesos unas sustancias denominadas catalizadores, en cuya presencia aumentan significativamente las velocidades de reacción, pudiendo ahora sí sintetizar grandes cantidades de productos en tiempos razonables y económicos. Entre los procesos catalíticos más importantes por el volumen de las materias sintetizadas se encuentran: las síntesis de amoniaco, de ácido sulfúrico (producto que se tomabacomoíndicedelgradodedesarrollodeunpaís),lahidrogenacióndeaceitesygrasas vegetales para consumo alimenticio, los procesos de la industria petroquímica como hidrotratamientos,craqueo,hidrocraqueo,alquilación,reformadocatalítico,oxidación,…Yno menosimportantes,aunquefuerayadelaesferadelasíntesisquímicaindustrial,setienenlos convertidorescatalíticos,presentesenlostubosdeescapedelosvehículosautomóvilespara reducir la emisión de gases contaminantes, las enzimas, los catalizadores biológicos, que regulanlavelocidaddegrannúmerodelosprocesosmetabólicosdelosseresvivos. Tanto la catálisis heterogénea como los procesos electródicos tienen importantes aplicacionestecnológicasencamposmuydiversos:almacenamientoyproduccióndeenergía, síntesisdepolímeros,síntesisdegasesindustriales,síntesisdefármacos,recubrimientosde superficies,inhibicióndelacorrosiónmetálica,descontaminación,etc.Enestetemaseaborda en primer lugar la catálisis heterogénea y, en una segunda parte, la cinética de procesos electródicos. 6.2.‐Característicasdelosfenómenoscatalíticos Lavelocidaddelasreaccionesquímicaspuedemodificarsepordiferentescausascomo: concentracióndelosreactivos temperaturaypresión eldisolventeylafuerzaiónicadelmedio A éstas es necesario añadir el fenómeno de la catálisis, término propuesto por Berzeliusen1836paradeterminarelaumentodelavelocidaddereacciónporadicióndeuna sustancia que se recupera sin alterar al final de la reacción. Esta sustancia se denomina catalizador. Berzelius escribía para describir los experimentos que había realizado sobre la descomposición del recién descubierto compuesto que era el agua oxigenada, QFIII Tema 6 3 1 H 2 O 2 H 2 O O 2 , muy estable a temperatura ambiente, pero cuya descomposición se 2 acelerabaenpresenciadediversassustancias: “LassustanciasquecausanladescomposicióndelH2O2noloconsiguenporincorporarsea los nuevos compuestos (H2O y O2); en todos los casos permanecen sin cambio, actuando por medio de una fuerza inherente de naturaleza todavía desconocida… . Mientras permanezca oculta la naturaleza de la nueva fuerza, será útil para nuestras investigaciones y discusiones sobreelasunto,sileponemosunnombre.Yolavoyallamarfuerzacatalíticadelassustanciasy a la descomposición que ejerce catálisis. La fuerza catalítica se refleja en la capacidad que tienenalgunassustanciasparadespertaractividadesenmoléculasqueaunatemperaturadada parecendormidas”. Existenvariasdefinicionesdecatalizador.Entreellasencontramoslasdebidasa: • Ostwald (1901) define catalizador como cualquier sustancia que modifica la velocidad de unareacciónquímicasinexperimentarcambioalgunoensímisma. •Bell(1941)catalizadordeunareacciónescualquiersustanciacuyaconcentraciónfiguraen la ecuación de velocidad elevada a una potencia superior a la que le correspondería en la ecuaciónestequiométricadedichareacción. Existenvariostiposdecatálisis - catálisis homogénea: el catalizador y el sistema reactivo forman un sistema homogéneo con una sola fase. Son reacciones en fase gas o en disolución, por ejemplocatálisisácido‐base. - catálisis heterogénea: la reacción se produce en una región interfacial. Así, para una reacción donde los reactivos están en fase gas o disolución el catalizadorsesuelepresentarenformadesólido. - catálisisenzimática:reaccionesbioquímicascuyavelocidadseincrementapor la acción de las enzimas, que son proteínas formando una dispersión coloidal. Aunque formalmente es homogénea, presenta características propias de la catálisisheterogénea,comolaexistenciadecentrosactivos. Losfenómenosdecatálisispresentanunaseriedecaracterísticascomunes: a) El catalizador se recupera al completarse la reacción sin haberse consumido. Esta característicanosiempresecumpleensutotalidadyaqueelcatalizadorpuederecuperarse como una especie distinta, por ejemplo formando un complejo con los productos de la reacción. Ejemplo:i)alquilacióndeFriedel‐Crafts. C6 H6 RCl AlCl 3 C6 H5 R HCl partedelcatalizador,AlCl3,setransformaenAlCl4‐ ii)halogenacióndecetonasenmedioácido: QFIII Tema 6 4 H R C CH3 I2 R C CH2I HI II II O O R C CH2I II OH Lo importante es que la formación de este complejo sea reversible, de forma que el catalizador pueda quedar de nuevo disponible. Esta característica se verifica mejor en los catalizadoresheterogéneos. b)Pequeñascantidadesdecatalizadorproducenunaumentoimportantedelavelocidadde reacción,lacuales,engeneral,directamenteproporcionalalaconcentracióndecatalizador (ordenuno). Alahoradeexpresarlavelocidaddeunprocesohemosdetenerencuentaquelavelocidad no es cero en ausencia de catalizador en muchos casos, por lo que hemos de introducir un términoindependientedelaconcentracióndecatalizador.Así,lavelocidadsepuedeescribir comosumadelavelocidaddelareacciónsincatalizarmásladelareaccióncatalizada.Para unareacciónconreactivosA…L: v=F0+F[C]=k0[A]…[L]+kcat[A]…[L][C],dondek0eslaconstantedevelocidaddelprocesono catalizado y kcat la del proceso catalizado. [C] es la concentración de catalizador y [L] la de sustratoelevadaasuordendereacción. Para que exista catálisis kcat >>k0, con lo que la velocidad de reacción normalmente puede expresarsesimplementecomo: vkcat[A]…[L][C] Las dos constantes de velocidad tienen una dependencia con la temperatura según la ecuación de Arrhenius, k=Aexp(‐Ea/RT), por lo que es necesario, para que haya un cambio importante en la constante de velocidad, que la reacción catalizada tenga una energía de activación menor (Figura 6.1). Por ejemplo, a 298 K, una disminución de 4 kcal/mol en la energíadeactivaciónaumentalaconstantedevelocidad1000veces. partedelácidoH+quedacomo QFIII Tema 6 5 Figura6.1‐Diagramaenergéticodeunareaccióncatalizadaysincatalizar Además existen sustancias que disminuyen la velocidad de reacción. Son los catalizadores negativosoinhibidores.Estosnofuncionanaumentandolaenergíadeactivación,yaqueen esecasolareaccióntranscurriríaporsuscaucesordinarios(sikinh<<k0,vk0[A]…[L])sino bloqueandoelcaminodelareacción.Porejemplolahidroquinonainhibelasreaccionesque transcurrenmedianteradicaleslibresyaquereaccionaconellosrompiendolacadena. c)Loscatalizadoresnovaríanlaconstantedeequilibriodelareacción,K. Dadoqueelestadoinicialyfinaldelareacciónsonidénticosconosincatalizador,lavariación de energía libre de la reacción, ∆G°reac, no varia: Greac iG , como i K exp( Greac / RT ) ,laconstantedeequilibrio,K,seráindependientedelmecanismodela reacción. Aunque un catalizador no modifica la constante de equilibrio, lo que si puede realizar un catalizadorhomogéneoescambiarlacomposicióndeequilibrio.Laconstantedeequilibrioen función de las fracciones molares, xi, se puede expresar como: K (ai ,eq ) i , siendo la i actividad:ai=ixi. Un catalizador que se encuentre en la misma fase puede modificar los coeficientes de actividad idereactivosyproductos.Amenosqueloscambiosen isecompensenentresi,el catalizadorvariarálasfraccionesmolaresdelosreactivosyproductosenelequilibrio.Dado que el catalizador esta presente en pequeñas cantidades, su efecto en la composición del equilibrio es, en general, pequeño. Esta característica de los catalizadores puede venir matizadaporsucapacidaddeformarcomplejosconlosreactivosoproductosdelareacción. QFIII Tema 6 6 Estacaracterísticadelacatálisistieneunaconsecuenciaobviaqueesnecesarioresaltar.Dado queparaunequilibrioelementallaconstantedeequilibriopuedeexpresarsecomoelcociente entrelasconstantesdevelocidaddirectaeinversa(K=k1/k‐1),todocatalizadordelareacción directaloserátambiéndelainversa.Estoeslógico,yaquelasbarrerasdeenergíaasuperar sereducenparaelcaminodirectoeinverso. d)Laaccióndeloscatalizadorespuedesergeneraloespecífica. Laaccióngeneralcorrespondeacatalizadoresqueactúansobremuchasreaccionesdelmismo o distinto tipo y es un comportamiento típico de la catálisis homogénea, ejemplo: catálisis ácido‐base. La acción específica es más propia de la catálisis heterogénea o enzimática. Por ejemplo: los alcoholes pueden reaccionar por deshidratación o deshidrogenación. Son reacciones termodinámicamente espontáneas pero que transcurren a velocidades muy pequeñas. CH3CH2OHCH2=CH2+H2O CH3CH2OHCH3‐CHO+H2 Losóxidosmetálicoscomolaalúmina(Al2O3)catalizanlaprimerareacción,mientrasquelos metales(Cu)favorecenlasegunda. 6.2.1.‐Mecanismogeneraldelacatálisis Paraserefectivo,uncatalizadordebecombinarseconunoomásreactantesoconuna especieintermediaimplicadaenlareacción.Despuésdequelareaccióntengalugar,selibera el catalizador y se puede combinar con otro reactante o intermedio en una reacción subsecuente. El catalizador no se consume durante la reacción, de forma que una pequeña cantidaddecatalizadorpuedeparticiparennumerosasreacciones. Supongamos una reacción con un solo reactivo (S). Un esquema simple para este mecanismogeneraldereaccióncatalíticaes: k1 S C SC ,etapa(1) k1 k 2 SC P C ,etapa(2) (6.1) (6.2) donde S representa al reactante o sustrato, C es el catalizador y P el producto. El complejo sustrato‐catalizador se representa por SC y es una especie intermedia del mecanismo. La expresióndevelocidaddereaccióndiferencialparalaformacióndeproductoes: v dP k 2 SC dt (6.3) AplicandolaaproximacióndeestadoestacionarioalintermedioSC: QFIII Tema 6 7 dSC k1 S C k 1 SC k 2 SC 0 dt (6.4) dedondeseobtieneque: SC k1 S C k 1 k 2 (6.5) Comoelcatalizadornoseconsumelohabitualesexpresarlasecuacionescinéticasenfunción de la concentración inicial de catalizador [C]0. La concentración que existe de complejo catalizador‐sustratolapodemosdeterminarempleandolacondicióndeestadoestacionarioy teniendoencuentaquelaconcentracióndecatalizadorlibre[C]esigualalainicialmenosla queestáformandocomplejo([C]=[C]0‐[SC]): d [ SC ] 0 k 1 [ S ][ C ] k 1 [ SC ] k 2 [ SC ] k 1 [ C ]0 [ SC ] [ S ] k 1 [ SC ] k 2 [ SC ] dt (6.6) dedonde: k 1 [ C ]0 [ S ] [ SC ] (6.7) k 1 [ S ] k 1 k 2 ysustituyendoenlaexpresióndelavelocidad(6.3)queda: k k [ C ]0 [ S ] v 1 2 k 1 [ S ] k 1 k 2 Estaecuaciónpuederescribirsedividiendonumeradorydenominadorpork1 k 2 [ C ]0 [ S ] k [ C ]0 [ S ] v 2 k 1 k 2 [ S ] Km [S] k1 (6.8) (6.9) dondeKm=(k‐1+k2/k1).Seobservaquevvaríalinealmentecon[C]0,siendoladependenciacon [S] más complicada. La velocidad de la reacción (ecuación 6.9) es de orden uno respecto al catalizador,mientrasquesepuedenobservardossituacioneslímiterespectoalsustrato(ver figura6.2.): QFIII Tema 6 8 v [S] Figura6.2.‐Variacióndelavelocidaddereacciónconlaconcentraciónde sustrato. k [ C ]0 [ S ] si se desprecia [S] del denominador y por si [S]<<Km se obtiene que v 2 Km tanto determinaríamos en estas condiciones una cinética orden 1 respecto del sustrato. si [S]>>Km se desprecia Km llegando a v k 2 [ C ]0 determinando por tanto una cinéticadeorden0respectoalsustrato. Elvalorlímitedelavelocidadvienedadoporlacantidaddecatalizadorpresente.Cuandola [S] es tan grande que todo el catalizador está como SC (equilibrio (1) desplazado completamentealaderecha)unaumentoadicionaldesustratonocambialavelocidadinicial. Laecuación(6.9)sepuedelinealizarobteniendo: Km 1 1 1 (6.10) v k 2 [ C ]0 [ S ] k 2 [ C ]0 si representamos 1/v frente a 1/[S] se obtiene de la ordenada en el origen 1/k2[C]0 y de la pendienteKm/(k2[C]0),loquepermitedeterminarlasconstantesdevelocidadydeequilibrio. Unavezdeterminadaslasconstantescinéticas,podemosestudiarcómocambiarálavelocidad delareacciónconlatemperatura,esdecir,cuálserálaenergíadeactivaciónparaelproceso catalizado.Aligualqueelordendereacciónobservadovaríaenfuncióndelascondicionesde la reacción, la energía de activación observada también puede cambiar. Esta energía de QFIII Tema 6 9 activacióndelprocesocatalizado(Ea,c)seráfuncióndelasenergíasdeactivacióndecadauna delasetapasindividuales: k1 (1) S C SC k 1 k 2 (2)SC P+C Si cada una de las constantes de velocidad depende de la temperatura según la ecuaciónde Arrhenius,seobtiene: k 1 A1 exp( E a ,1 / RT ) k 1 A1 exp( E a ,1 / RT ) (6.11) k 2 A2 exp( E a ,2 / RT ) En cualquier caso la energía de activación será siempre menor que la del proceso no catalizado (Ea,0). Analicemos esta Ea,c a partir de la ecuación de velocidad que hemos k k [ C ]0 [ S ] determinado(ecuación6.8): v 1 2 k 1 [ S ] k 1 k 2 Vamosadistinguirdostiposdesituacioneslímite: a) complejo tipo Arrhenius, la transformación del complejo SC en productos es la etapa dterminantedelavelocidaddereacción(k‐1>>k2).Puestoquelaetapa(2)esmáslentaque las etapas que componen el equilibrio (1), entonces Ea,1 y Ea,‐1 serán menores que Ea,2, tal y comoapareceenlafigura6.3.a.Simplificandolaecuacióndevelocidadinicialanteriorqueda: k k [ C ]0 [ S ] v 1 2 (6.12) k1 [ S ] k 1 Dependiendo de los valores relativos de los dos sumandos del denominador podemos distinguirdossituaciones: •Sik1[S]<<k‐1,seobtienesimplificando: k k v 1 2 [ S ][ C ]0 (6.13) k 1 Laconstantedevelocidadqueseobservaríaparaelprocesocatalizadoseríaiguala: k k kc 1 2 (6.14) k 1 yaplicandolaecuacióndeArrheniusacadaunadelasconstantesdevelocidaddelaexpresión anteriorseobtiene: Ac exp( E a ,c / RT ) A1 exp( E a ,1 / RT ) A2 exp( E a ,2 / RT ) / A1 exp( E a ,1 / RT ) (6.15) Yteniendoencuentaqueenelproductodeexponencialesesigualalaexponencialdelasuma delosexponentes,podemosllegara: QFIII Tema 6 10 (6.16) E a ,c E a ,1 E a ,1 E a ,2 Ypuestoqueladiferenciaentrelasenergíasdeactivaciónensentidodirectoeinversoesigual alaentalpíadelareacción: E a ,c H r ,1 E a ,2 (6.17) Estasituacióneslaqueapareceenlafigura6.3.a.indicandolaenergíadeactivacióncomouna flechaverde. •Sik1[S]>>k‐1,simplificandoqueda: v k 2 [ C ]0 (6.18) porloquekc=k2yportanto Ea,c=Ea,2. (6.19) Estasituacióneslaqueapareceenlafigura6.3.a.indicandolaenergíadeactivacióncomouna flechaazul. De una manera cualitativa podemos decir que Ea,c corresponde con la diferencia de energía entreelpuntodepartidayelestadodetransicióndelabarreramayor(etapamáslenta)que quede por sobrepasar. Si la [S]0 es alta o k1 >>k‐1, el equilibrio (1) está desplazado hacia la derechayelpuntodepartidadelareaccióneselcomplejoSCyEa,c=Ea,2.Si,porelcontrario, [S]0 o k‐1>>k1, el equilibrio (1) está desplazado a la izquierda y el punto de partida es el reactivoycatalizadorseparados.Entonceslabarreraasuperaresladiferenciadeenergíacon elestadodetransicióndeaetapa2,esdecirHr,1+Ea,2. b) complejo de Van’t Hoff. La transformación de complejo en productos no es la etapa limitante. (k2≈k‐1 o k2>>k‐1)En este caso Ea,1 y Ea,‐1 serán parecidas o mayores que Ea,2. La velocidadenestecasonopuedesimplificarseapriori: k k [ C ]0 [ S ] v 1 2 k 1 [ S ] k 1 k 2 Y un posible perfil energético correspondiente a esta situación será el representado en la figura6.3.b. Enfuncióndelvalordelprimersumandopodemossimplificarlaexpresióndelavelocidad. •Sik1[S]>>k‐1+k2sellegaa: v k 2 [ C ]0 porloque (6.20) kc=k2 (6.21) (6.22) yEa,c=Ea,2. Estasituacióneslaqueapareceenlafigura6.3.b.indicandolaenergíadeactivacióncomouna flechaazul. •Sik1[S]<<k‐1+k2songrandessellegaa: k k v 1 2 [ S ][ C ]0 (6.23) k 1 k 2 QFIII Tema 6 11 Enestecaso,debidoaquekcnoesunproductoococientedeconstantescinéticaselementales nopodemosobtenerunaexpresiónsencilladeEa,cenfuncióndelasenergíasdeactivaciónde lasdistintasetapas,exceptosik2>>k‐1.Enestecaso: v k 1 [ C ]0 [ S ] (6.24) (6.25) yEa,c=Ea,1 Estasituacióneslaqueapareceenlafigura6.3.b.indicandolaenergíadeactivacióncomouna flecha verde. De nuevo podemos ver que cualitativamente la energía de activación correspondeconladiferenciadeenergíaentreelpuntodepartida(SCóS+C)yelestadode transicióndelabarreramayor(etapamáslenta)quequedeporsobrepasar. a) b) Ea,c Ea,c Ea,c Ea,c Figura6.3.‐Diagramaenergéticodeunareaccióntipoquetranscurremediante: a)uncomplejodeArrhenius,b)uncomplejoVan’tHoff 6.2.2.‐Característicasdelacatálisisheterogénea La importancia económica de la catálisis heterogénea es tal que casi el 80% de los procesosindustrialesutilizanenalgunadesusetapasunareaccióncatalizadaporunsólido. Los productos sintetizados mediante procesos catalíticos heterogéneos son muy variados, tantoensunaturalezaquímicacomoenlacantidadproducidaysucosteporunidaddepeso. Desde los combustibles líquidos o el ácido sulfúrico, que son substancias de estructura química simple y se producen en cantidades enormes a un preciorelativamente bajo, hasta diferentestiposdefármacos,fragancias,productosagroquímicosypolímeros,algunosdelos cuales presentan estructuras químicas extremadamente complejas y que, en general, se producenencantidadesrelativamentepequeñas,perocuyoprecioporunidaddepesoes,en algunoscasos,muyelevado. Enlacatálisisheterogéneaelfenómenocatalíticoestárelacionadoconlaspropiedades químicasdelasuperficiedelsólidoquesehaelegidocomocatalizador,siendoporsupuesto estas propiedades superficiales un reflejo de la química del sólido. Para que un catalizador sólido sea efectivo, uno o más de los reactivos deben quimisorberse sobre el mismo, dando QFIII Tema 6 12 lugarauncomplejosuperficialsustrato‐catalizadorqueseráelcausantedeladisminuciónde la energía de activación. La adsorción física es solamente importante en la catálisis heterogéneaenalgunoscasosmuyparticulares,comoenlarecombinaciónderadicales. Quizás la reacción catalizada heterogénea mejor entendida es la oxidación del CO sobre catalizadoresdePtoPd(reacciónimportanteenloscatalizadoresempleadosenlostubosde escapedelosautomóvilesparaeliminarlasespeciestóxicasformadasdurantelacombustión incompletadelagasolina): CO ( g) M CO M (6.26) O 2 ( g ) 2 M 2O M (6.27) O M CO M CO 2 ( g ) 2 M (6.28) Mrepresentaunaposicióndeadsorciónsobrelasuperficiedelcatalizador. Lassustanciasmásutilizadascomocatalizadoresheterogéneosson: Metalesdetransición:Fe,Co,Ni,Pd,Pt,Cr,W,Ag,Cu….yaquedisponendeorbitalesd parcialmenteocupadosquepuedenparticiparenlaformacióndeenlacesdelaespecie quimiadsorbida. Óxidosmetálicossemiconductores:NiO,ZnO,V2O5,Fe2O3,MgO Óxidosaislantes:Al2O3,SiO2 Agentesbiológicos(enzimas,anticuerpos,microorganismos,DNA,…) Laefectividaddeuncatalizadorpuedemedirseporlacantidaddeproductoformadopor unidad de tiempo y unidad de área superficial de catalizador. En principio, el efecto de un catalizador es proporcional a su área superficial, por ello es deseable que sea lo mayor posible.Estosepuedeconseguirdisminuyendoeltamañodelaspartículasdelcatalizadorya quelacantidaddemateria(númerodeátomos)superficialaumentaaldisminuiraltamañode las partículas. Estas partículas se suelen extender sobre la superficie de un soporte poroso (propagador),siendolosmásutilizadoselgeldesílice(SiO2),laalúmina(Al2O3)yelcarbón activo. Unodelosproblemasquepuedesufriruncatalizadorparticularesquesesinterice(se aglutine)formandopartículasdemayortamaño,enlasquesereduceeláreasuperficialy,con ello,laactividadcatalítica,aumentandoelcostodeproducción.Paraevitarlasinterización,se añade una pequeña cantidad de una sustancia conocida como promotor que actúa como barrera, impidiendo que las partículas de catalizador se aglutinen. Por ejemplo, pequeñas QFIII Tema 6 13 cantidades de Al2O3 evitan el agrupamiento de los cristales de hierro utilizados como catalizadoresenlasíntesisindustrialdeNH3apartirdeN2yH2. Otropeligroquecorrenloscatalizadoressólidosessuenvenenamiento,esdecir,cuando una sustancia se enlaza fuertemente a las posiciones de adsorción, dejando de estar disponibles para la reacción catalítica. Estas sustancias pueden ser bien impurezas que acompañenalosreactivosolosmismosproductosdelareacciónysuelentenercompuestos deS,NyP,conparesdeelectronessolitarios,yalgunosmetales(Hg,Ag,Pb…).Debidoaqueel plomoesunvenenocatalítico,debeusarsegasolinasinplomoencochesconcatalizador(la gasolinaconplomocontienealquilosdeplomoparaaumentarsuoctanaje). Si es posible regenerar el catalizador se habla de venenos reversibles. El SH2 o PH3 formanenlacesmuyfuertesconPtyloinhabilitanpermanentemente,mientrasqueenelcaso del CO o CS2 es posible recuperar el catalizador. En algunos casos, la acción de los venenos puedeserútilparafavorecerunareaccióneinhibirotra.Porejemplo,elThO2(óxidodetorio) que cataliza la deshidratación de alcoholes a olefinas y su deshidrogenación a aldehídos o cetonas es un buen ejemplo de su utilidad. Con vapor de agua pueden bloquearse los iones oxígeno de la superficie (por formación de puentes de hidrógeno) inhibiendo así la deshidratación; mientras que la deshidrogenación puede seguir produciéndose sobre los átomosdetorio. La cantidad de veneno necesaria para inhabilitar un catalizador es menor que la requeridaparacubrirtodasusuperficie,loqueindicaquelaactividadcatalíticaselocalizaen algunasposicionesespecíficasdelasuperficiequerecibenelnombredecentrosactivos. 6.2.3.Etapasdelacatálisisheterogénea Paraquelareacciónentreelsistemagaseosoreactivoyelcatalizadorsólidotengalugar esnecesarioquesedenlassiguientesetapas: 1.Transportedelasmoléculasdereactivoshastalasuperficiedelsólido. 2. Quimisorcióndealmenosunadelasespeciesreactivassobrelasuperficie. 3. Reacciónquímicaentrelosreactivosadsorbidosoentreunreactivoadsorbidoylas moléculasnoadsorbidasquechocancontralasuperficie. 4. Desorcióndelosproductosdelasuperficie. 5. Transportedelosproductosdesdelasuperficiedelsolido. Untratamientogeneraldeestemecanismoimplicaelestudiodelasvelocidadesdelas cinco etapas, lo cual es complicado. Sin embargo, en muchos casos, una de estas etapas es mucho más lenta que todas las demás, y, a efectos cinéticos, solamente es necesario considerarlavelocidaddeestaetapadeterminante. QFIII Tema 6 14 Lasetapasdetransporte,enausenciadegradientesmecánicos,suelenserdedifusión, aunquetambiénpuedenestarafectadasporgradientesdepotencialeléctrico.Porejemplo:en algunas reacciones que se dan entre dos moléculas adsorbidas se puede incluso dar la migracióndelasmismassobrelasuperficieentrelasetapas2y3. Lasetapas1y5songeneralmenteetapasdedifusiónydependendelatemperatura,de la presión, de la viscosidad del medio. Como se ve en la Figura 6.4, nos encontramos habitualmente con condiciones experimentales de catálisis en las que estas etapas son rápidas,aunquetambiénpodríadarseloscasosdondealgunadelasetapasdedifusiónfuera la etapa determinante de la reacción. Estas situaciones, bastante inusuales, se denominan procesos controlados por difusión; un ejemplo es la reacción entre los iones hidronio e hidroxiloenagua.Lasetapas2a4quedanrecogidasenlafiguraquerepresentalavariación delaenergíaenfuncióndelacoordenadadereacción. La etapa de adsorción (etapa 2) suele ser rápida. El proceso de adsorción va acompañado de una liberación de calor debido a la formación del complejo sustrato‐ catalizador. Ea,adsR Reactivos(g) Transcurso de la reacción Figura6.4.‐.Esquemaenergéticodelmecanismogeneral delacatálisisheterogénea QFIII Tema 6 15 Enprincipio,cuantomásnegativasealaentalpíadeadsorciónmásrápidoseproducirá el proceso de catálisis (Figura 6.4). Si ∆Hads es pequeña se producirá poca adsorción y por tantolareaccióndelaetapa3serálenta,habrápocosreactivos.Si,porelcontrario, ∆Hadses muy grande entramos en el límite de quimisorción fuerte: los reactivos se mantienen firmementeunidosaloscentrosdeadsorciónytienenpocatendenciaareaccionar(límitede quimisorción fuerte, la velocidad del proceso catalítico disminuye al aumentar ∆Hads). Este dobleefectovienerecogidoenlascurvasvolcán(llamadasasíporsuforma)comoseobserva en la Figura 6.5. La reacción representada en las curvas volcán ocurre por quimisorción de reactivos formando un enlace M‐S con la superficie. Representando la eficiencia del catalizador frente a la entalpía de formación de enlace S‐catalizador se observa una mayor actividadcatalíticaparalosvaloresintermedios.Sielenlaceesdébilhaypocaquimiadsorción y la reacción es lenta. Por el contrario, si el enlace es demasiado fuerte, el reactivo quimiadsorbido forma un complejo muy estable con inercia a reaccionar, con lo que la velocidadestambiénpequeña. Figura6.5.‐Gráficadelaevolucióndelavelocidaddecatálisisvs.∆Hads Comosehadichoanteriormente,lasespeciesadsorbidassonestablesenlascondiciones experimentales de trabajo por lo que su desorción suele implicar una cierta energía de activación.Aunquenosueleserlaetapamáslenta,puedeproducirciertoretrasooinhibición sielcatalizadorquedabloqueadotemporalmenteporlosproductosnodesorbidos. Puesto que estamos suponiendo que las velocidades de adsorción y desorción son mucho mayores que la de la reacción química superficial, se considera que el equilibrio de adsorción‐desorción se mantiene durante el tiempo que transcurre la reacción; y podemos, por tanto, suponer que la isoterma de Langmuir es válida para describir el equilibrio de adsorciónprevioalaetapadeterminante. La etapa 3 es propiamente la etapa de reacción, porque en ella se produce la transformacióndereactivosenproductosysueleconllevarunaenergíadeactivaciónmayor altenerunamayorenergíadeactivación,sueleserlaetapadeterminantedelavelocidad.Si QFIII Tema 6 16 estaetapatienelugarentreespeciesadsorbidassobrelasuperficiedelcatalizador,sediceque lareaccióntienelugaratravésdeunmecanismotipoLangmuir‐Hishelwood.Encambio,sila etapa 3 involucra una especie adsorbida que reacciona con otra especie no adsorbida, el mecanismo se denomina de Eley‐Rideal. Los mecanismos de Lagmuir‐Hishelwood son más comunesquelosdeRideal‐Eley. Vamosanalizarelcasodeprocesoscontroladosporlaetapadereacciónconsiderando, además,estaetapacomoelementaleirreversible.Distinguiremosdostiposdereacciones: a) reaccionesunimoleculares A(ads)Productos b) Reaccionesbimoleculares A(ads)+B(ads)Productos;seexplicaporelmecanismodeLangmuir‐Hinshelwood A(ads)+B(g)Productos;seexplicaporelmecanismodeEley‐Rideal Puestoquesuponemosquelaetapalentaesladereacción,lasvelocidadesdeadsorción ydesorciónseránmuchomayoresyelequilibrioadsorción‐desorciónsemantendráalolargo de la reacción. Para describir esta adsorción (quimisorción) usaremos normalmente la isoterma de Langmuir. Esta suposición de que una de las etapas es significativamente más lenta que las demás, permite obtener con facilidad la ley de velocidad de cualquier mecanismo.Además,permiteobtenerexpresionessencillassinqueseanecesariorecurriral método de velocidades iniciales. Debemos señalar que existen bastantes ejemplos donde la velocidad de reacción está controlada por la adsorción, por ejemplo la síntesis de NH3 catalizadaporhierroestácontroladaporlaadsorcióndeN2oporladesorcióndeproductos, veremoseltratamientodeestoscasosenlosproblemas. Para el caso de la catálisis heterogénea se define la velocidad de reacción como la velocidaddeconversiónporunidaddesuperficiedelcatalizador: 1 1 dnA (6.29) A A dt si no se conoce A, puede definirse por unidad de masa de catalizador o referirnos siempreaunamismacantidaddelcatalizador.Deestaformapodremoscompararresultados donde cambia la cantidad de catalizador. Para establecer comparaciones también será necesario que el catalizador se haya preparado de forma similar, para que la superficie que presenteporunidaddemasasealamisma. Si la etapa de reacción es la etapa lenta, vs será proporcional al número de moléculas adsorbidasporunidaddeáreay/oalafraccióndecentrosocupadospormoléculareactivaA: vs=kA. vs QFIII Tema 6 17 a)Reaccionesunimoleculares Supongamos una reacción elemental e irreversible de una especie A adsorbida y que ningunodelosproductosquedaadsorbido: KA k A( g) A( ads ) productos( g) (6.30) La velocidad de reacción por unidad de superficie será proporcional al número de moléculas adsorbidas o, lo que es equivalente, a la fracción de centros ocupados por moléculasdeA: v s k A (6.31) dondekvieneexpresadaenmolm‐2s‐1(S.I.). K APA y sustituyendo en la ecuación 6.31 se Según la isoterma de Langmuir: A 1 K APA obtiene: vs kK A PA 1 K APA (6.32) Para un caso de este tipo, al estudiar la dependencia de vs con la presión del reactivo (PA) obtendríamos una gráfica como la Figura 6.6. Para presiones bajas se cumple que v s kK A PA , ecuación de primer orden respecto A; mientras que para presiones altas se cumpleque v s k ,ecuacióndeordencero. Porejemplo,ladescomposicióndelafosfina(PH3)a700°Csobrewolframioapresiones menoresde10‐2torresdeorden1respectoa PPH3 yporencimade1torresdeordencero. Por supuesto el tipo de cinética depende también del valor de KA, la constante del equilibriodeadsorción: Si tenemos adsorción débil, KA será pequeña: se mantendrá un comportamiento de cinéticadeorden1hastapresionesmásaltas.Lapresióndereactivonecesariaparapoder despreciarelunoeneldenominadordelaleydevelocidadserámasgrande.Unejemplode estetipodecinéticasesladescomposicióndeHIsobrePt. Si tenemos adsorción intensa, KA será grande: el comportamiento de orden cero aparecerádesdepresionesmásbajas.AlserKAgrande,podremostomarellímite1<<KAPA conpresionesmáspequeñas.Unejemplodeestetipodecinéticasesladescomposiciónde HIsobreAu. QFIII Tema 6 18 vs vs=k vs=kKAP PA (Atm) Figura 6.6.‐ Evolución de la velocidad de conversión por unidad de superficie del catalizador en función de la presión parcial de A para un mecanismo tipo Langmuir‐ Hinshelwood. Ejercicio 6.1.- La descomposición de la fosfina sobre wolframio es de primer orden a presiones bajas y de orden cero a presiones elevadas. Justifique este comportamiento. Solución.K PH 3 (g) Pr od(g) PH 3 (ads) k La velocidad de reacción vendrá dada por: dPPH 3 dt sigue la isoterma de Langmuir, se transforma en k PH 3 , que, considerando que se dPPH 3 dt k PH 3 k KPPH 3 1 KPPH 3 . A presiones muy bajas, 1+ KPPH 3 ≈1 con lo que: dPPH 3 dt k KPPH 3 1 KPPH 3 kKPPH 3 k ' PPH 3 (orden 1 en PH3) A presiones altas, 1+ KPPH 3 ≈ KPPH 3 con lo que: dPPH 3 dt k (orden 0 en PH3). Lacinéticasecomplicasialgunodelosproductosquedaadsorbidosobrelasuperficie, reduciendoeláreadelcatalizadordisponibleparaA. QFIII Tema 6 19 Teniendo en cuenta que A K APA (adsorción competitiva) y 1 K APA K C PC K D PD sustituyendoen vs k A ,seobtiene: vs kK APA 1 K A PA K C PC K D PD (6.33) Silosproductosestándébilmenteadsorbidos,esdecir,KCPc+KDPD..<<1+KAPA,seobtiene que: vs kK A PA 1 K A PA (6.34) Siunoovariosdelosproductosestáfuertementeadsorbidoseobtieneque: kK A PA kK P o v s A A (6.35) vs K C PC 1 K C PC Por ejemplo en la descomposición del NH3 sobre Pt el H2 queda muy fuertemente k' PNH3 adsorbidoyseobtieneque v s . PH 2 Ejercicio6.2.‐Enalgunasreaccionescatalíticas,losproductossepuedenadsorbermás fuertemente que el gas reaccionante. Este es el caso de la descomposición catalítica del amoniaco sobre platino a 1000°C. Como primer paso del análisis cinético de este proceso, muestrequelavelocidaddedescomposicióndelamoniacodebeseguirlaecuación: dPNH3 kc PNH3 dt PH2 Solución.Lareacciónglobalqueestamosestudiandoes:NH3(g)→1/2N2(g)+3/2H2(g) Sihaycatálisisheterogénea,tendráquehaberadsorcióndealmenosunreactivo.Enestecaso, habiendo sólo uno, es claro que el amoniaco tiene que quimiadsorberse. El mecanismo más sencilloposibleseríaentonces: NH3(g)↔NH3‐M(sup) NH3‐M(sup)→1/2 N2(g)+3/2 H2‐M(sup) 3/2 H2‐M(sup)↔3/2H2(g) QFIII Tema 6 20 Enestecaso,puestoqueenlaleyexperimentalapareceelhidrógenotendremosquesuponer que éste queda adsorbido y por lo tanto hemos de añadir la etapa de desorción del mismo Igualmente, comenzamos suponiendo que la reacción química es una etapa elemental e irreversibleyqueeslaetapalenta.Laadsorción/desorciónessiemprereversibleyalsermás rápidaqueladereacciónpodremosasumirqueestaránsiempreenequilibrio. Al haber una etapa lenta podemos escribir la velocidad de la reacción teniendo en cuenta únicamenteestaetapa.Apartirde(2): v k2 · NH 3 Y como la adsorción de amoniaco e hidrógeno estánenequilibrio, utilizando la isotermade Langmuirdeacuerdocon(1)y(3)lafraccióndecentrosocupadosporamoniacoseráiguala: K NH 3PNH 3 NH 3 1 K NH 3PNH 3 K H 2PH 2 Sustituyendo nos queda la velocidad de reacción enfunción de la presión deamoniaco, una magnitudquepodemosseguirduranteeltrascursodelareacción: k2K NH 3PNH 3 v 1 K NH 3PNH 3 K H 2PH 2 Si suponemos que la adsorción del hidrógeno es más fuerte que la de amoniaco, en el denominadorpodremossimplificar 1 K NH 3PNH 3 K H 2PH 2 1 K H 2PH 2 Siademáspodemossuponerquelaadsorcióndelhidrógenoessuficientementeintensa comoparadespreciarlaunidadfrentealtérminoK·P 1 K NH 3PNH 3 K H 2PH 2 K H 2PH 2 Conloquelaleydevelocidadquedaría: k2K NH 3PNH 3 kK P P v 2 NH 3 NH 3 kc NH 3 1 K NH 3PNH 3 K H 2PH 2 K H 2PH 2 PH 2 Si los productos quedan adsorbidos, entonces a medida que transcurre la reacción, la velocidad es menor ya que la porción de área del catalizador bloqueada por el producto es mayor. Otro ejemplo de este tipo de comportamiento es la descomposición de N2O sobre Mn3O4 para dar O2 y N2. En este caso el O2 puede quedar quimiadsorbido disociativamente, mientras que el N2 no lo hace de forma significativa. La ley de velocidad que corresponde, asumiendolaetapadereaccióncomoetapalenta,es:(verejercicio6.) k ' PN2O (6.36) vs 1/2 1 bPN2O cPO2 QFIII Tema 6 21 b)Reaccionesbimoleculares Sonreaccionesdeltipo:A+Bproductosysepuedenexplicarporlosdosmecanismos anteriormenteindicados. i) Mecanismo de Langmuir‐Hinshelwood: A y B reaccionan cuando están adsorbidos sobrelasuperficie: A(g)+B(g)A(ads)+B(ads)productos(g) Esquemáticamentelareacciónsedaríaatravésdeunestadodetransicióndeestetipo: Las moléculas adsorbidas sobre la superficie pueden difundirse de un centro activo a otro hasta que se encuentren en posiciones vecinas y reaccionen. Asumiendo como etapa lentalareacciónyquelaadsorciónseencuentraenequilibrio,lavelocidaddereacciónserá proporcional al producto de las concentraciones superficiales o de forma equivalente, al productodelasfraccionesdecentrosocupados,delaforma: kK AK B PAPB vs k A B (6.37) 1 K APA K B PB 2 si ambas especies están débilmente adsorbidas, KA y KB pequeñas, y/o nos encontramos a presionessuficientementebajas,PAyPBpequeñas,laecuacióndevelocidadsereduceauna cinéticadesegundoorden: vs=k’PAPBconsiderandoquek’=kKAKB SiunodelosdosreactivosseadsorbemuyfuertementeKBPB>>1+KAPA,quedando: kK P (6.38) vs A A K B PB es decir, el reactivo B inhibe la reacción. Esto ocurre porque a medida que aumenta PB, la fracción de superficie ocupada por A (A=KAPA/(1+KAPA+KBPB)) tiende a cero. Si representamos la ecuación de velocidad completa frente a PB, siendo PA constante, la velocidadprimeroaumentaydespuéstiendeacero,pasandoporunmáximo(Figura6.7). QFIII Tema 6 22 Figura 6.7.‐ Evolución de la velocidad de conversión por unidad de superficie del catalizador en función de la presión parcial de B para un mecanismo tipo Langmuir‐ Hinshelwood. Un ejemplo de reacciones de este tipo es la reacción 2CO+O22CO2 catalizada por platino,dondeelCOseadsorbemuchomasfuertementequeelO2.UnexcesodeCO,enlugar deaumentarlavelocidaddelareacciónprovocaríaunacaídadelamisma.Estacompetencia entreelCOyelO2porloscentrosdeadsorciónsedatambiénenlahemoglobina,enlazándose elCOmásfuertementealátomodehierroeimpidiendoeltransportedeO2. ii) MecanismodeEley‐Rideal Supongamoslasreacciones: A(g)A(ads) A(ads)+B(g)productos(g) Esquemáticamente, la reacción tendría lugar a través de un estado de transición de la forma: De acuerdo con el mecanismo, y asumiendo la etapa de reacción como etapa lenta, podemosescribirvs=kAPBdedonde: kK P P (6.39) vs A A B 1 K A PA Tal y como se aprecia en la Figura 6.8, en el límite de adsorción débil, KAPA<<1, se obtiene:vs≈kKAPAPB,ecuacióndeordendosglobalyenellímitedeadsorciónfuerte,KAPA>>1, vs≈kPB,ecuacióndeordenuno. QFIII Tema 6 23 kKAP B Figura6.8.‐Evolucióndelavelocidaddeconversiónporunidaddesuperficie delcatalizadorenfuncióndelapresiónparcialdeAparaun mecanismotipoEley‐Rideal. Por ejemplo la combinación de dos átomos de H para dar hidrógeno molecular, 2HH2. Experimentalmenteseobservaqueesdeorden1respectoalapresióndehidrógenoatómico atemperaturasbajasydeorden2atemperaturasaltas. Estaobservaciónsepuedeexplicarconunmecanismo: H(g)H(ads) H(ads)+H(g)H2(g) Laleydevelocidadquecorrespondeaestemecanismoes: 2 vs kK H PH 1 K H PH (6.40) Como ∆Hads<0, temperaturas bajas, KH es grande y vs≈kPH, (orden 1); mientras que a temperaturasaltasvs≈k’PH2(orden2). Ejercicio6.3.‐LareacciónentreNO(g)yCO(g)paradarN2(g)yCO2(g)estácatalizadapor unasuperficiesólidadeRodio.SabiendoquelaadsorcióndelNOescondiciónnecesariapara quetengalugarlareacciónyquelosproductosnoseadsorbensignificativamente:a)deducir si la reacción responde a un mecanismo del tipo Langmuir‐Hinshelwood (dos moléculas adsorbidas)odeEley‐Rideal(unaadsorbidaotraenfasegaseosa)sabiendoqueenellímite depresionesparcialesdeCOmuyaltaslaleydevelocidadexperimentalesv=k’PNO/PCO;b)si secambiaelcatalizadorutilizandounoenelquenohayadsorciónsignificativadeCO¿Cual seráelordencinéticototalenellímitedemuybajastemperaturas? Solución: NO(g)+CO(g)→N2(g)+CO2(g) a) MecanismodeLangmuir‐Hinshelwood: QFIII Tema 6 24 NO(g)↔NO(ads) CO(g)↔CO(ads) k 1/2N2(g)+CO2(g) NO(ads)+CO(ads) Etapalenta: v k NOCO Ads.NOenequilibrio NO K NO PNO 1 K NO PNO K CO PCO v Ads.COenequilibrioCO PCOmuyalta v kK CO K NO PNO PCO 1 K NO PNO K CO PCO 2 K CO PCO 1 K NO PNO K CO PCO P kK CO K NO PNO PCO kK NO PNO k ' NO 2 PCO K CO PCO K CO PCO MecanismodeEley‐Rideal NO( g) NO(ads ) 1 k CO( g) NO(ads ) N2( g) CO2( g) 2 v kPCO NO Etapalenta kK NO PNO PCO v 1 K NO PNO Ads.NOenequilibrio K NO PNO NO 1 K NO PNO b) SinohayadsorcióndeCOtendremosunmecanismodeEley‐Rideal: kK NO PNO PCO v (1 K NO PNO ) SilaTesmuybajaKNO>>1 kK P P v NO NO CO kPCO K NO PNO QFIII Tema 6 25 6.2.4.‐Ejemplosdecatálisis Se estima que el 90% de todos los productos químicos producidos comercialmente involucran catalizadores en alguna etapa del proceso de su fabricación. Los procesos catalíticos generan más de 900.000 millones de dólares por año en productos de todo el mundo.Ahíradicaunodelosprincipalesmotivosquehacenindispensableelestudioteórico detodosestoscatalizadores. Algunosdelossectoresenlosquelacatálisistieneespecialinterésson: i)Elpetróleo. LospetróleoscrudosqueseextraendelosdiferentescampospetrolíferosdelaTierra son de naturaleza muy variada incluso en su apariencia externa. El petróleo bruto es una mezcla de diferentes hidrocarburos (la mayor parte saturados). El gas natural, por ejemplo, consisteenunamezclademoléculasligerascomometano,etano,propanoybutano.Además en su composición podemos encontrar ácido sulfhídrico que es corrosivo, por lo que es necesariaunapurificaciónparaeliminarlo.Tradicionalmenteelgasnaturalesutilizadocomo combustible para uso doméstico e industrial. Sin embargo, en la última década su consumo paralaproduccióndehidrógenosehaelevado.Elhidrógenopuedeserutilizadoenprocesos como la síntesis del amoniaco o como combustible no contaminante en motores de combustióninterna. Elpetróleo,unavezextraído,esenviadoporoleoductoshacialasrefinerías.Loscrudos sonseparadosinicialmentepordestilación.Enesteprocesoaproximadamenteel75%delos compuestos son volátiles, quedando un residuo llamado asfáltico en el fondo. La fracción volátil se separa como sigue en orden decreciente de punto de ebullición: 1) hidrocarburos gaseosos(metanoobutano),2)gasolinaligera,3)gasolinapesadaonafta;4)queroseno,5) gasóleoligero,y6)gasóleopesado.Generalmente,losproductosobtenidosenesteprocesono son suficientes en calidad ni cantidad para los requerimientos actuales de consumo. Por lo tanto, se requiere transformar estos productos en otros de uso más conveniente mediante procesosquímicosquehabitualmenteconllevanelempleodecatalizadores. Elobjetivodelosprocesoscatalíticosdelpetróleoeseldemodificarlasfraccionesdel mismo para la obtención de productos en cantidad y calidad acordes con los requisitos del mercado.Podemosdestacarlossiguientes: a)Fragmentación.Muchasdelasmoléculasorgánicasdepequeñotamañoutilizadasen la industria química se obtienen cortando hidrocarburos de cadena larga procedentes del petróleo. La fragmentación de estos hidrocarburos inducida catalíticamente se denomina crackingyserealizasobrealúmino‐silicatos(SiO2/Al2O3,zeolitas)paraformarmoléculasmás QFIII Tema 6 26 ramificadas. Estos isómeros, más cortos, se queman de forma más suave y eficaz en los motoresdecombustióninterna. b) Reforming.Eslatransformacióndelaestructuramoleculardelasnaftas.Lasnaftas extraídasdirectamentedeladestilaciónprimariasuelentenermoléculaslinealesporloque tiendenadetonarporpresión.Así,elreformingseencargadetransformardichasmoléculas lineales en ramificadas y cíclicas, que, al ser más compactas, no detonan por efecto de la presión.Elreformingutilizauncatalizadordedoblefunción,mezcladeplatinoyalúmina.El platino proporciona la función metal, catalizando la hidrogenación y deshidrogenación y la alúmina aporta la función ácida. Primero el hidrocarburo se quimisorbe sobre el platino. El hidrocarburopierdedosátomosdehidrógenoformandounalqueno.Elalquenomigraauna posiciónácidadondeaceptaunprotónyseunealasuperficiecomoióncarbonio.Esteiónse puede romper, isomerizarse a formas más ramificadas o formar anillos. El efecto generales que el producto contiene hidrocarburos con formas moleculares más compactas que dan valores más altos de octanaje que los hidrocarburos presentes en la nafta. Estos productos sonpartedelagasolinadealtooctanajeogasolinacomercial. ii)Biocombustibles Los biocombustibles son combustibles de origen biológico alternativos al petróleo, obtenidos a partir de restos orgánicos. Estos restos orgánicos proceden habitualmente del azúcar, trigo, maíz o semillas oleaginosas. El uso de biocombustibles tiene impactos ambientales negativos y positivos. Los impactos negativos hacen que, a pesar de ser una energía renovable, no sea considerada por muchos expertos como una energía no contaminantey,enconsecuencia,tampocounaenergíaverde.Unadelascausasnegativases que,apesardequeinicialmenteensuproducciónsóloseusanrestosdeactividadesagrícolas, con su generalización en los países desarrollados, muchos países subdesarrollados, especialmenteelsuresteasiático,estándestruyendosusespaciosnaturales,incluyendoselvas ybosques,paracrearplantacionesparabiocombustibles.Laconsecuenciadeestoesjustola contrariadeloquesedeseaconseguirconlosbiocombustibles:losbosquesyselvaslimpian máselairedeloquelohacenloscultivosqueseponenensulugar. Los biocombustibles líquidos proporcionan actualmente la energía equivalente a 20 millones de toneladas de petróleo. Los biocombustibles que más se utilizan son el biodiesel (ésteresmetílicosdeácidosgrasos)yelbioetanol(etanoldeorigenbiológico).Elbiodieseles unbiocombustiblesintéticolíquidoqueseobtieneapartirdelípidosnaturalescomoaceites vegetalesograsasanimales,nuevasousadas,medianteprocesosindustrialesdeesterificación y transesterificación y que se aplica en la preparación de sustitutos totales o parciales del petrodiéselogasóleoobtenidodelpetróleo.Losvegetalesmásutilizadosparalaobtencióndel bioetanol suelen poseer elevadas cantidades de sacarosa (caña de azúcar, remolacha, …), almidón(maíz,patata,…)ocelulosa(madera,residuosagrícolas,...).Elprocesodefabricación delbioetanolapartirdelalmidónesmáscomplejoqueapartirdelasacarosa,pueselalmidón QFIII Tema 6 27 debeserhidrolizadopreviamenteparaconvertirloenazúcares.Paraellosemezclaelvegetal trituradoconaguayconunaenzima(ocatalizador),ysecalientalapapillaobtenidaa120‐ 150°C.Posteriormente,secuelalamasaenunprocesollamadoescarificaciónyseenvíaalos reactores de fermentación. La fermentación de los azúcares es llevada a cabo por microorganismos (levaduras o bacterias) y produce etanol, así como grandes cantidades de CO2.Además,produceotroscompuestosoxigenadosindeseablescomo elmetanol,alcoholes superiores,ácidosyaldehídos. Elprocesodefabricaciónapartirdecelulosaesaúnmáscomplejo,yaqueprimerohay quepre‐tratarlamateriavegetalparaquelacelulosapuedaserluegoatacadaporlasenzimas hidrolizantesquelaconviertenenlosazúcaresprecursoresdelbioetanol.Elpre‐tratamiento puede consistir en una combinación de trituración, pirólisis y ataque con ácidos y otras sustancias.Estoesunodelosfactoresqueexplicanelporquélosrendimientosenetanolson altosparalacañadeazúcar,mediocresparaelmaízybajosparalamadera. iii)Productosquímicosdesíntesis Algunos de los productos químicos obtenidos a granescala se producen a través de la oxidacióncatalítica,amenudousandooxígeno.Algunosejemplossonelácidonítrico(apartir de amoníaco), el ácido sulfúrico (a partir de dióxido de azufre a trióxido de azufre por el proceso de las cámaras de plomo) y el acrilonitrilo a partir de propano y amoníaco. No obstante, otros muchos productos químicos son generados por reducción a gran escala, a menudo a través de la hidrogenación. Un ejemplo de estos procesos de reducción es la fabricación industrial del metanol a partir de monóxido de carbono. La reacción global, catalizadamedianteunamezcladeNiO/Cu,es: CO( g ) 2H 2 ( g ) CH 3 OH ( g ) (6.41) EsteprocesosigueformalmenteunmecanismodetipoLangmuir‐Hinshelwood,donde tanto el CO como el H2 sufren quimisorción (ésta última disociativa). Los reactivos quimisorbidosreaccionanenunprocesoquecontienevariasetapas: CO( g ) CO( ads ) (6.42) H 2 ( g ) 2 H ( ads ) (6.43) CO( ads ) 2H ( ads ) CHOH( ads ) (6.44) CHOH ( ads ) 2 H ( ads ) CH 3OH ( ads ) (6.45) Este mismo proceso puede conducir a la formación de hidrocarburos, dependiendo de las condiciones experimentales. Si el CO adsorbido reacciona con otras moléculas de CO QFIII Tema 6 28 adsorbido genera carbono adsorbido. Éste puede reaccionar fácilmente con el hidrógeno monoatómico para dar una cadena de reacciones que conducen a la formación de hidrocarburos: CO( ads ) CO( ads ) C ( ads ) CO 2 ( ads ) (6.46) C ( ads ) H ( ads ) CH( ads ) (6.47) CH ( ads ) H ( ads ) CH 2 ( ads ) (6.48) iv)Químicafina Muchos productos de química fina también se preparan a través de la catálisis. Son procesos especializados que serían prohibitivamente caros a gran escala, tales como la síntesisdealgunosfármacosyreactivosbiomédicos. Debidoaquelamayoríadeloscompuestosbioactivossonquirales,muchosproductos farmacéuticos son producidos mediante procesos catalíticos asimétricos. El primer proceso catalíticoasimétricocomercializadofuelasíntesisdelaL‐DOPA(L‐3,4‐dihidroxifenilalanina) porhidrogenacióncatalíticaasimétrica.LaL‐DOPAeselaminoácidoprecursorinmediatode la dopamina y el fármaco disponible más eficaz en el tratamiento de la enfermedad de Parkinson. En el tratamiento del Parkinson se administra L‐DOPA en lugar de dopamina porque la dopamina no puede atravesar la barrera hematoencefálica (la que forman las meningesentrelosvasossanguíneosyellíquidocefalorraquídeo),encambiolaL‐DOPAsíque puedeatravesarla.WilliamS.KnowlesyRyojiNoyorirecibieronelPremioNobeldeQuímica en 2001 por sus estudios sobre las reacciones de hidrogenación mediante catalizadores quirales,laconocidacomohidrogenacióncatalíticaasimétrica.Elmecanismodelareacciónde hidrogenacióncatalíticaasimétricaempleadaenlasíntesisdelaL‐DOPAvienedescritoenla Figura6.8. Figura6.9.‐Mecanismodecatálisisasimétricaempleadoen lasíntesisdelaL‐DOPA QFIII Tema 6 29 v)Procesamientodealimentos Un ejemplo de las aplicaciones de la catálisis en el sector alimentario es la hidrogenación de las grasas. Frecuentemente se usa níquel metálico como catalizador para producir la margarina. En este caso, la hidrogenación de los dobles enlaces se utiliza para obtenergrasascomestiblesapartirdeaceitesvegetales. Figura6.10.‐Mecanismogeneraldehidrogenación El mecanismo de este proceso está detallado en la Figura 6.9, donde el grupo químico alqueno (1) se adsorbe formando dos enlaces con la superficie (2), sobre la cual se ha producidotambiénquimisorcióndisociativadeH2.Cuandoseformaunenlacealqueno‐Hse rompeunodelosenlacesalqueno‐superficie(3,4).Laformacióndeunnuevoenlacealqueno‐ Hliberalagrasaconlosdoblesenlacessaturados(5). 6.3.IntroducciónalaCinéticaElectródica Si bien las etapas, por ejemplo, las de transporte, que se observan en la catálisis heterogénea, se pueden dar también en los procesos electródicos, la casuística de éstos es muchomásgrande,tantocomoparanopoderproponerunesquemareactivogeneralcomose ha planteado anteriormente en la catálisis. La razón fundamental es que muchos procesos electródicos afectan también al propio material del electrodo. Por ejemplo, en la corrosión electroquímica de los aceros es el propio electrodo el que se transforma químicamente a diferencia de los catalizadores sólidos que prácticamente permanecen inalterados. Este mismotipodefenómenoseplanteaporejemploenelanodizadodelaluminio,yaqueeneste caso el electrodo de aluminio se cubre con una capa pasiva de óxido de aluminio con fines decorativosylimitadoresdelavancedelapropiacorrosióndelmaterial. Sonmúltipleslosejemplosenlosquesobreelpropiomaterialdelelectrodosepueden depositar capas modificando sus propiedades. Por ejemplo, en la formación superficial de capas de polímeros conductores, porque se han depositado en el proceso de su síntesis electroquímica.Despuésestospolímerosmuestranotrosprocesoselectródicos,manifestando electrocromismooinclusoelectrocatálisisparareaccionesdeinterésenbiosensoresyotras QFIII Tema 6 30 interesantesaplicaciones,enlasquelasetapasdeinsercióndeionesymoléculasneutrasson muy importantes para describir su cinética de actuación. Otro ejemplo de formación de multicapas, como consecuencia de procesos electródicos de interés tecnológico, es el recubrimiento electrolítico por metales, tal y como sucede en los circuitos impresos, en bisuteríaoenmuchosbienesdeconsumo.Otrosprocesosquesedanenlosseresvivosson aún más difíciles de modelizar que los precedentes, por ejemplo, el del transporte de informaciónenelsistemanerviosoolosimpulsoseléctricosenórganoscomoelcorazón,ya que funciona a través de procesos electródicos en el que intervienen sistemas celulares de gran complejidad químico‐física. Pero por muy complicado que sea el proceso electródico para su interpretación, siempre se pretende descomponerlo en procesos más elementales paralosquesepuedanplantearmecanismoscinéticosapartirdelasherramientasteóricasy experimentalesqueofrecelaQuímicaFísica. Losprocesoselectródicospuedenimplicartransferenciadeelectronesentreelelectrodo ylasespeciespresentesenelmediovecinoquesoncapacesdereducirseodeoxidarse.Son los llamados procesos faradaicos. En estos casos, los electrones actúan como reactantes o productosdelasreaccioneselectroquímicasqueseproducen.Sonmuyconocidasalgunasde estasreacciones:síntesisdehidrógeno,delejía,deamoniaco,decisteína,etc,o,simplemente, larecuperacióndemetalesoeliminacióndecontaminantesdeaguasfluviales,sinmencionar las que se dan con objetivos electroanalíticos. Un ejemplo de proceso faradaico es el correspondiente a la oxidación o reducción de materiales electroactivos depositados sobre electrodos.SialelectrodoasímodificadoseleaplicaunpotencialE(t),quevaríelinealmente coneltiempo,larespuestaeléctricai(t)queseregistreporelexteriordelaceldadependerá directamente de la extensión y velocidad en la que se produce el cambio de estado de oxidacióndelfilmdepositado. También hay otros procesos electródicos que no implican reacciones electroquímicas, sonlosllamados procesosnofaradaicos,quesedanporejemploenlaelectrodiálisis,enla electroósmosis,enlasmedidasconductimétricas,enlamedidastensimétricas,etc.,enlasque la presencia de electrodos causan efectos físicos por el hecho de introducir un gradiente de potencial eléctrico en los respectivos sistemas. En general, por el hecho de perturbar un sistemaelectrodo/disoluciónmedianteunacorrienteeléctrica,nonecesariamenteseproduce un proceso faradaico que implique transferencia de electrones entre el electrodo y las sustanciaselectroactivas.Siempre,hayaonoreacciónelectroquímica,seproduciráelpasode la propia perturbación a través del sistema, con la consiguiente reestructuración de las especiescargadasenlaregióninterfacial,asícomosutransporteforzadoporelinteriordela celda electroquímica. En ese caso, se puede interpretar la respuesta eléctrica mediante un circuito eléctrico sencillo: un condensador eléctrico conectado en serie con una resistencia eléctrica Enlossiguientesapartadosconsideraremosquemedianteunafuenteeléctricaseaplica unadiferenciadepotencialE(t)entreloselectrodosdeunaceldaelectroquímica,lacualcausa una reacción electroquímica cuya velocidad se relaciona con la intensidad de corriente i(t) que fluye por un circuito eléctrico externo. El proceso electródico se describe mediante un conjunto de etapas químicas o físicas que ocurren en la celda debido a esta perturbación QFIII Tema 6 31 eléctrica.Resultandoquelaintensidaddecorrienteesunamedidadelavelocidadglobaldel proceso,porloquelaexpresióndeleycinéticaasociadaaundeterminadoprocesoelectródico consistirá en una relación matemática entre la intensidad de corriente i(t), las concentracionesdelasespecies,loscoeficientesdevelocidaddelasetapasfísicaoquímicas postuladas,lasconstantesdeequilibriodelasreaccionesreversiblesacopladassilashubiera y,también,deladiferenciadepotencialaplicada,E(t). 6.3.1.‐MecanismoGeneral El mecanismo general de un proceso faradaico es similar al mecanismo general de la catálisisheterogéneaeincluyelassiguientesetapas: 1. Movimientodelosreactivosdesdeelsenodeladisoluciónalelectrododetrabajo.Este movimientotienelugarmedianteunmecanismodetransportequeconllevaladifusión y migración de las especies. El proceso se describe normalmente mediante un coeficientedetransferenciademasaodedifusión,kd 2. Reorganizacióndelreactante,susligandos(siloshay)ysuesferadesolvatación 3. Transferenciaelectrónica 4. Relajaciónenelsentidoinverso Los pasos del 2 al 3 son normalmente incluidos en las constantes de velocidad de transferenciadecarga,ka(constanteanódica,oxidación)okc(constantecatódica,reducción), eincluyenlaadsorcióndelosreactivossobrelasuperficiedelelectrodo,quesedesorberán posteriormente si el producto es soluble. El paso 2 puede ser vistos como un tipo de pre‐ equilibrio necesario antes de producirse la transferencia electrónica, etapa que consideraremos como la limitante de la velocidad de reacción. Durante esta transferencia, etapa 3, se cumple el principio de Franck‐Condon, el cual establece que las transiciones electrónicassonprocesosinstantáneossisecomparanconlaescaladetiemposenlaquese producenlosmovimientosnucleares. Ensuformamássimple,asumiendoquenohayningunaetapaquímica,quelamigración está anulada por la presencia de una gran cantidad de electrolito y que no hay agitación, el mecanismodeunprocesoelectródicoserá: difusión O Oo Oo ne Ro difusión Ro R dondeelsubíndiceoeindicanrespectivamenteelplanodeHemholtzyelsenodela disolución. QFIII Tema 6 32 6.3.2.‐Leydevelocidaddelatransferenciaelectrónica Consideremos el caso sencillo de un equilibrio en que el control cinético en ambos sentidossealatransferenciadeelectronesentreelelectrodoyunaespecieoxidadapresente enladisolución,ensentidodereduccióny,ensentidoinverso,enlaoxidacióndelaespecie reducidaRyenlaqueportantoladifusiónpuedaconsiderarsemuyrápidadeformaquelas concentraciones[O]0=[O]y[R]0=[R].Elprocesoquedaentoncessimplificadodelasiguiente forma: kc O ne R ka (6.49) De esta manera, la velocidad del proceso catódico o reducción, es directamente proporcional a la concentración de especie oxidada [O]. La expresión de la velocidad superficialdelareaccióncatódicasepuedeexpresarcomo: vc kc [ O ] (6.50) Delmismomodo,laexpresiónparalareaccióndeoxidaciónes: va k a [ R ] (6.51) porlotanto,lavelocidaddereaccióntotales: vtotal vc va kc [ O ] ka [ R ] (6.52) La teoría del complejo activado (Tema 3) nos indica que las constantes cinéticas, k, se puedenescribirenfuncióndelosparámetrosdeactivación(energíalibre,entropíayentalpía) delaforma G ‡ S ‡ H ‡ k A' e RT A' e R e RT (6.53) dondeA’tieneencuentaelfactordefrecuenciayeltérminodebidoalestadoestándar. QFIII Tema 6 33 Energía Libre Gc0 ‡ Ga0 ‡ Gc‡ Ga‡ Energía Libre nF( E E 0' ) O + ne‐ R ( 1 )nF ( E E 0' ) Coordenada de Reacción Figura6.11.‐Efectodeuncambioenelpotencialaplicadosobrelaenergíalibredeactivación paralosprocesosdeoxidaciónydereducción.LascurvasapotencialE0’aparecenenazul(O + e) y rojo (R). Al cambiar a potencial E la curva de los reactivos se desplaza y aparece representada en verde. El cuadro inferior es un aumento de la zona señalada en el cuadro superior. La peculiaridad de las reacciones redox es que podemos modificar la energía libre de activacióncambiandoelpotencialaplicadoyaqueésteafectaalaenergíalibredeunodelos reactivos,loselectrones.Efectivamenteeltrabajoeléctricoescargaporpotencial,conloque la energía libre de los n electrones vendrá afectada por un término igual a –nF·E. Los electronesprovienenosecedenalelectrodocuyopotencialesE.Paraanalizarlacinéticade las transferencias electrónicas vamos a suponer una forma simple para el perfil de energía libre a lo largo de la coordenada de reacción. Esta coordenada puede ser un movimiento complejo de la esfera de solvatación de las especies O y R, de forma que se estabilice o desestabilicelacarasobreellas.Elperfildeenergíalibre,correspondienteaunciertovalor del potencial del electrodo (E=E0’), aparece representado en la figura 6.11 donde se han utilizado dos parábolas centradas en los valores de la coordenada correspondientes a las formasestablesdeOyR.Estasparábolassecruzanenelestadodetransiciónyladiferencia entrelosmínimosyestecrucenosdanlasenergíasdeactivaciónparalareaccióncatódicay QFIII Tema 6 34 G0‡ G0 ‡ anódica ( c y a ). Si el potencial del electrodo se modifica hasta un valor E la curva correspondientealosreactivossedesplazarárespectodelainicialenunacantidadiguala– nF·(E‐E0’)mientrasqueladelosproductospermaneceráinalterada. Enlafigura6.11seveclaramentecomoalalterarelpotencialaplicadoycambiarelnivel energéticodeloselectronesseproduceunamodificacióndelasenergíalibresdeactivación paraelprocesodeoxidaciónydereducción.Enelcasorepresentado,dondeseproduceuna variación positiva del potencial, con la correspondiente estabilización de los reactivos, la energía libre de activación para el proceso de oxidación Ga‡ disminuye respecto al valor G0 ‡ anterior a .Elvalorconcretodeladisminucióndependedelaformaconcretaquetengala regióndeinterseccióndelascurvasquerepresentanlavariacióndeenergíalibredereactivos yproductos.Engeneralestadisminuciónseráunafracciónoporcentajedelavariacióndela energía total de los reactantes. A esta fracción se la llama 1 , donde , el coeficiente de transferenciadecarga,puedevariardesdeceroauno,dependiendodelaformadelaregión de intersección. Por lo que se puede escribir la siguiente ecuación para el proceso de oxidación: Ga‡ Ga0‡ ( 1 )nF ( E E 0 ' ) (6.54) Paraelprocesodereducciónhemosdeconsiderartambiénqueelestadodetransición disminuyeen(1‐)nF(E‐E0’)peroademásquelosreactivosdisminuyenennF(E‐E0’)porloque lanuevaenergíalibredeactivaciónserá: Gc‡ Gc0‡ ( 1 )nF ( E E 0' ) nF ( E E 0' ) Gc0‡ nF ( E E' 0 ) (6.55) EnambasexpresionessehaconsideradoquelaenergíalibredeGibbsvaríalinealmente conelpotencialaplicado.Partedelaenergíaaplicadaconunafuenteeléctrica,setraduceen incrementarlaenergíadeactivaciónensentidodereducciónenunafraccióndelaenergía aplicada,mientrasqueenelsentidooxidación,laenergíadeactivaciónhadisminuidoen(1‐ ).Elvalordedependedecadareacción,aunquenormalmenteseencuentranvaloresentre 0,3y0,7;porloquepuedeseraproximadoa0,5sinosedisponedeunamedidaexperimental. Al combinar las ecuaciones (6.54) y (6.55) con la (6.53) encontramos la relación existente entre las constantes cinéticas de una transferencia electródica y el potencial aplicado: QFIII Tema 6 35 Ga0‡ ( 1 )nF ( E E 0' ) Ga0‡ ( 1 )nF ( E E 0' ) Ga‡ RT A' e RT e RT ka A' e R A' e o' ( 1 )nF ( E E ) 0 RT ka e (6.56) Gc0‡ nF ( E E 0' ) Gc0‡ nF ( E E 0' ) Gc‡ RT A' e RT e RT kc A' e R A' e (6.57) nF ( E E 0' ) RT kc0 e 0' El potencial de referencia E E se elige de forma que las constantes de velocidad 0' k 0 kc0 k 0 anódica y catódica sean iguales, es decir a . De esta manera, E es el potencial dondelasvelocidadesdereacciónanódicaycatódicatienenelmismovalorenunadisolución con [O ] [ R ] yporlotantolareacciónseencuentraenequilibrio.Estevalordelaconstante 0 develocidadsedenominaconstantedevelocidadestándar, k ylasconstantesdevelocidada 0 otrospotencialespuedenserexpresadassimplementeentérminosde k : ka k e 0 RT nF E E 0 ' RT 0 kc k e 1 nF E E 0 ' (6.58) (6.59) Así,lavelocidaddereaccióntotalpuedepuesexpresarsecomo: vtotal nF ( E E 0' ) ( 1 )nF ( E E 0' ) RT [ O ] k 0e RT k 0e [R] (6.60) QFIII Tema 6 36 6.3.3.‐Relaciónentrecorrienteyvelocidaddereacción.EcuacióndeButler‐Volmer Teniendo en cuenta que la velocidad total se define por unidad de superficie del electrodoyquecadamolquereaccionaconducealintercambiodenelectrones(carga‐n·F),la intensidad de corriente (la carga que circula por unidad de tiempo) vendrá dada por la expresión: i nFAvtotal ia ic ( 1 )nF ( E E 0' ) RT nFAk 0 e [R] nFAk 0 e nF ( E E 0' ) RT [O ] (6.61) dondeAeseláreadelelectrodo,iaeicsonlasintensidadesdecorrientedelosprocesos anódico y catódico. Este resultado es conocido comúnmente como la ecuación de Butler‐ Volmer, en honor a los pioneros en esta área, John Alfred Valentine Butler y Max Volmer. Nótesequeelcriteriodesignospuedediferiryencontraselaecuaciónanteriorconelsigno cambiado. La inclusión del signo en la carga nos permite seguir las recomendaciones de la IUPAC, que define la corriente catódica (de reducción) como negativa y la anódica (de oxidación) como positiva (véase la Figura 6.12). También es posible encontrar la ecuación anteriorenformadedensidaddecorrienteocargaquecirculaporunidaddetiempoyunidad deárea(j=i/A). Para un determinado valor del potencial Eeq la velocidad de reacción electródica y por tantolaintensidaddecorrientetotalseanula,loquequieredecirquelasvelocidadesdelos procesoscatódicosyanódicosseigualan.Aestevalordeia(ode–ic)seledenominacorriente deintercambioi0: i0 nFAk0e ( 1 )nF ( E eq E 0' ) RT ne Electrodo nF ( E eq E 0' ) RT [O] (6.62) Proceso catódico O + ne → R [ R ] nFAk0e O electrones i<0 corriente R Proceso anódico R → O + ne ne R Electrodo electrones i>0 corriente O Figura6.12.‐Criteriodesignosparalaintensidaddecorriente QFIII Tema 6 37 Ejercicio6.4.Demostrar,apartirdelaigualdad(6.62)quelaecuacióndeButler‐Volmer escompatibleconlaecuacióndeNernst. Solución:Apartirdelaecuación(6.60)podemosdespejarelpotencialenelequilibrio: nFAk0e ( 1 )nF ( E eq E 0' ) RT [ R ]eq nFAk0e nF ( E eq E 0' ) RT [ O ]eq Despejandoelcocientedeconcentraciones: [ R ]eq [ O ]eq e nF ( E eq E 0' ) RT ( 1 )nF ( E eq E 0' ) RT e Operandoconlosexponentes: [ R ]eq [ O ]eq e nF ( E eq E 0' ) ( 1 )nF ( E eq E 0' ) RT RT e nF ( E eq E 0' ) RT Tomandologaritmosneperianosenamboslados: ln [ R ]eq [ O ]eq nF( Eeq E 0' ) RT Finalmente,despejandoelpotencialenelequilibriollegamosalaecuacióndeNernst: Eeq E 0' RT [ R ]eq ln nF [ O ]eq La ecuación de Butler‐Volmer suele expresarse en función de esta corriente de intercambioydelsobrepotencial ( E E eq ).Sisustituimosenlaecuación(6.61)elvalor deE=+Eeq,queda: i nFAk 0 e ( 1 )nF ( E eq E 0' ) RT [ R ] nFAk 0 e nF ( E eq E 0' ) RT [O ] 0' nF nF ( E eq E ) ( 1 )nF ( 1 )nF ( E eq E ) RT RT nFAk 0 e RT e [ R ] nFAk 0 e RT e [O] 0' ( 1 )nF nF RT i0 ·e i0 ·e RT (6.63) Odeformaequivalente: nF ( 1 )nF e RT ; i i0 · e RT nF ( 1 )nF e RT j j0 · e RT QFIII Tema 6 (6.64) 38 queeslaformamáshabitualdelaecuacióndeButler‐Volmerexpresadacomointensidadde corriente(i)odensidaddecorriente(j). El comportamiento predicho por la ecuación anterior se muestra en la Figura 6.13. La línea negra muestra la corriente total, que es la suma de las componentes ic e ia, dibujadas como líneas de color azul y rojo, respectivamente. Para sobrepotenciales muy negativos, la componente anódica es despreciable y, por lo tanto la corriente total depende casi exclusivamentedelacorrientecatódica.Porotraparte,asobrepotencialespositivos,sedael casocontrarioylacorrientedependecasiexclusivamentedelacorrienteanódica.Enambos casoslasrespectivascorrientesosusdensidadesdecorrientevaríanexponencialmenteconel sobrepotencial. A temperatura constante, las corrientes superpuestas de reducción y oxidación dependen del número de moles de electrones transferidos n y del coeficientes de transferencia. C V 50 C i > 0 V ia i0 ·e C V ‐0.1 < 0 C V 1 nF RT i0 i0 i ic ia nF > 0 ic i0 ·e RT C V i < 0 ‐50 C V Figura 6.13.‐ Dependencia de la corriente electródica con el sobrepotencial predicha por la ecuación de Butler‐Volmer. La intensidad total se representa en negro, la catódica en azulylaanódicaenrojo. Alex y Hickling propusieron un método para extraer los parámetros de la cinética electródica i0 y linealizando la ecuación de Butler‐Volmer. La ecuación (6.64) puede ser reescritacomo: QFIII Tema 6 39 nF nF i e RT · e RT 1 i0 (6.65) Despejandolacorrientedeintercambiopodemosescribir: i nF RT 1 e i0 ·e nF RT (6.66) (6.67) Ytomandologaritmosneperianosnosqueda: ln i nF RT e 1 ln i0 nF RT · Esdecirqueunarepresentacióndelogaritmoneperianoqueaparecealaizquierdadela ecuaciónanteriorfrentealsobrepotencialnosproporcionaunalínearectadecuyapendiente podemosconocerydelaordenadaenelorigeni0. 6.3.4.‐Formasaproximadasdelarelaciónentrecorrienteyvelocidaddereacción. Si el sobrevoltaje tiende a cero, es decir, si el sistema se desplaza muy poco de la situacióndeequilibrio,sepuedeencontrarunaformaaproximadadelarelacióni=i().Para ellobastacondesarrollarlaecuacióndeButler‐VolmerenseriesdeTaylor: x2 x3 e 1 x 2! 3 ! x nF 1 Y utilizando sólo los dos primeros términos (válido cuando RT , es decir para sobrevoltajespequeños)laecuación(6.64)queda: i ( 1 )nF nF nF 1 1 i0 RT RT RT QFIII Tema 6 (6.68) 40 Esta ecuación muestra como la corriente neta está relacionada linealmente con el sobrepotencialenunintervalocercanoalEeq.Larelación/itieneunidadesderesistenciay,a menudo,esdenominadacomolaresistenciadetransferenciadecarga,Rct. i Rct RT nFi0 (6.69) Rct se puede evaluar directamente a partir de la pendiente de la representación i‐, comoserepresentaenlafigura6.14yseutilizacomounparámetroparamedirlafacilidad conlaquetienelugarlareacciónelectródica.Esteparámetroesimportanteporejemplopara seleccionar un buen electrodo de referencia y depende de la corriente de intercambio. Si el valordei0 eselevadoentonceslaresistenciatiendeaceroyportantolascargasatravesarán la interfase del electrodo sin aumentar la separación de cargas y por tanto el potencial. La interfase no se polariza (no cambia el potencial) y por eso se califica como idealmente no polarizable.Estetipodeinterfacessonlasquecaracterizanunbuenelectrododereferencia, aquelenelqueelpotencialpermanececonstante(elsobrepotencialescercanoacero).Siel valor de i0 es pequeño, entonces la interfase presenta una elevada resistencia al paso de la corrienteyportantolacargaseacumulaalosladosdelainterfaseaumentandoelpotencial.A través de estas interfaces por tanto no circula corriente y se denominan idealmente polarizablesy.Evidentementelasinterfacesrealessesitúanentreambosextremos. C V 50 C i > 0 V 1 nFi0 Rct RT CV ‐0.08 < 0 CV > 0 C V i < 0 ‐50 C V Figura6.14.‐FormalinealdelaecuacióndeButler‐Volmerasobrepotenciales pequeños.Delapendientedelarepresentaciónseobtienelaresistenciadetransferenciade carga. Para grandes valores de (positivos o negativos), uno de los términos de la ecuación (6.63)devienedespreciable.Porejemplo,agrandessobrepotencialesnegativos: QFIII Tema 6 41 ( 1 )nF e RT nF e RT y,portanto,únicamenteseregistraríacorrientecatódica.Enesecasolaecuación(6.64) sepuedeescribircomo: nF i e RT i0 (6.70) Ytomandologaritmosdelmódulodelaintensidadenestaexpresión ln i nF i0 RT (6.71) Ydespejandoelsobrepotencial: RT RT ln i0 ln i nF nF (6.72) Agrandessobrepotencialespositivossoloseregistraríacorrienteanódicayseobtendría unasrelacionesequivalentesala(6.70)y(6.72): 1 nF i e RT i0 RT RT ln i lni 1 nF 1 nF 0 (6.73) Mediante las ecuaciones (6.72) y (6.73), el modelo planteado de cinética electródica justifica la dependencia de la corriente eléctrica con el sobrepotencial que fue deducida experimentalmente por Julius Tafel en 1905. Esta ecuación es conocida como ecuación de Tafel( a b ln i ).LadependenciadescritaporTafeltienelugarcuandolareaccióninversa contribuyeenmenosdel1%alacorrientetotal.Estepuntoponederelieveelhechodequeel comportamientodeTafelesunindicadordeunprocesoirreversible.Aaltossobrepotenciales, elprocesoelectródicoesunidireccionaly,portanto,químicamenteirreversible. Eldiagramadeln|i|vs.,conocidocomodiagramadeTafel,esutilizadoparaevaluarlos 1 nF RT parámetroscinéticos.Engeneral,hayunaramaanódicacuyapendientees yuna nF RT .ComomuestralaFigura6.15,laextrapolaciónde ramacatódicacuyapendientees ambos segmentos lineales proporciona el valor de lni0. Los segmentos se apartan de la linealidad a potenciales cercanos al Eeq, ya que las reacciones inversas dejan de ser QFIII Tema 6 42 despreciables. Cuandosepuedaaplicar,elcoeficientedetransferenciadecargaylacorriente deintercambiopuedensercalculadosfácilmenteapartirdeestetipoderepresentación. lni Pendiente nF Pendiente RT ( 1 )nF RT lnio < 0 ‐0.5 0 0 = 0 > 00.5 Figura6.15.‐DiagramadeTafelln|i|vs..Lalíneaazulrepresentalacorrientecatódica ylarojaaanódica.Lanegraeslacorrientetotal. QFIII Tema 6 43 6.4.‐Ejerciciosadicionales Ejercicio 6.5.- En algunas reacciones catalíticas los productos se adsorben más fuertemente que el gas reaccionante. Este es el caso de la descomposición catalítica del amoniaco sobre platino a 1000 ºC ya que el hidrógeno producido se adsorbe más fuertemente que los otros gases presentes en el reactor. a) Deduzca una posible ley de velocidad. b) A partir de los datos de la tabla en la que figura la presión del amoniaco P en distintos tiempos de la reacción, calcule un valor aproximado de la constante de velocidad de la etapa de descomposición del amoniaco adsorbido. t/s 0 30 60 100 160 200 250 P/Torr 100 88 84 80 77 74 72 Para ello puede emplear una representación lineal de F(t)=(P0/t)·ln(P/P0) en función de G(t)=(PP0)/t. Solución.-a) Inicialmente tan sólo hay gas amoniaco y ejerce una presión P0=1000 torr. Se puede suponer que la primera etapa corresponde a su adsorción reversible sobre el catalizador: NH 3 (g) NH 3 (ads) y que la etapa irreversible posterior: 1 3 k2 NH 3 (ads) N 2 (g ) H 2 ( g ) 2 2 es la determinante de la velocidad del proceso, por lo que se puede proponer que: v dPNH 3 dt k 2 NH 3 k 2 K NH 3 PNH 3 1 K H 2 PH 2 K NH 3 PNH 3 k2 K NH 3 PNH 3 K H 2 PH 2 kC PNH 3 PH 2 Esta expresión incluye la drástica aproximación de que en el denominador la contribución del hidrógeno es tal que K H 2 PH 2 1 K NH 3 PNH3 . Por la estequiometria de la reacción es fácil deducir que la presión del hidrógeno producido 3 a cada tiempo t es PH 2 (P0 P) , considerando P PNH 3 , por lo que la ley de velocidad 2 se puede expresar como una función de P: dPNH 3 dt kC P 3 ( P0 P ) 2 k' C P ( P0 P ) Integrando, entre los límites t = 0 y t, se obtiene que: P k' C t f ( P ) P0 (ln 1 ) P P0 QFIII Tema 6 44 Para ajustar los datos experimentales a esta ecuación conviene buscar una forma de linealizarla. Para ello podemos seguir la recomendación del enunciado e introducir las funciones F(t) y G(t). Reagrupando la ecuación anterior: P P0 ·ln ( P P0 ) k' c ·t P0 P0 P P P0 ·ln k' c t P0 t Por lo tanto, si el mecanismo propuesto explica las observaciones experimentales, la representación de F(t) frente a G(t) debería dar una línea recta de pendiente unidad y de cuya ordenada en el origen podemos obtener k’c. Con los datos experimentales podemos obtener la siguiente tabla t(s) P (torr) F(t) (torr/s) G(t) (torr/s) 0 100 0/0 0/0 30 88 -0.426 -0.400 60 84 -0.291 -0.267 100 80 -0.223 -0.200 160 77 -0.163 -0.148 200 74 -0.151 -0.130 250 72 -0.131 -0.112 QFIII Tema 6 45 Efectivamente el ajuste es bueno y la pendiente es la unidad, por lo que daremos por bueno el mecanismo. Respecto a la constante: kc 3 0.0172 torr·s 1 0.0258 torr·s 1 2 Ejercicio 6.6.- a) Deducir, indicando las hipótesis asumidas, la ley de velocidad de una reacción catalizada que sigue el siguiente mecanismo: 1) A↔Aad B↔Bad Adsorción rápida 2) Aad+Bad→Dad Reacción lenta 3) Dad↔D Desorción rápida donde A y B son gases reactantes, C es un catalizador sólido y D es el gas producido. b) En el caso de que ni B ni D se adsorban, ¿Cuál es la ley de velocidad esperada?¿Podría darse el caso de que la reacción fuera de pseudo-primer orden? c) Si un gas inerte H se adsorbe sobre el catalizador ¿Cómo quedarían afectadas las leyes de velocidad en los casos de los apartados a) y b)? d) Si A y B fueran especies disueltas en un líquido y la reacción sigue el mismo mecanismo (a la misma presión y temperatura) que en el caso a),¿Cuál será la ley de velocidad?¿Las constantes de adsorción y coeficientes de velocidad tendrán el mismo valor que en el caso anterior?¿Es cierto que la velocidad de reacción catalizada en este caso no depende del disolvente que se utilice? Ejercicio 6.7.- Una reacción entre gases A(g) y B(g), que sigue el modelo cinético de Langmuir-Hinshelwood, supone una etapa controlante en la que ambas especies están adsorbidas sin disociarse, por lo que la velocidad del proceso es v = k θA θB. , en las que θA y θB son las fracciones de recubrimiento superficiales respectivas. Ambas sustancias se adsorben competitivamente con sendas constantes de adsorción KA y KB (siguiendo la isoterma de Langmuir) sobre un catalizador de gran superficie específica que no se envenena con el producto de la reacción. ¿Cuál es las siguientes leyes cinéticas se ajusta a las premisas enunciadas? ¿A qué corresponden las otras dos expresiones? Justifique las respuestas a) v kK A K B p A pB (1 K A p A K B pB ) 2 b) v kK A p A pB 1 K A pA c) v k K A K B p A pB (1 K A p A K B pB ) 2 Solución.- La ecuación a) es la correcta, la b) corresponde al mecanismo de Eley-Rideal y la c) corresponde al caso con adsorción disociativa de las especies A y B. QFIII Tema 6 46 Ejercicio 6.8.- La descomposición del óxido de nitrógeno N2O, sobre Pt a 750°C, se produce con una adsorción fuerte del oxígeno que se produce y sin adsorción del nitrógeno producido. Calcular la constante cinética aparente a partir de los datos experimentales siguientes donde se da la presión de N2O frente al tiempo t (s) 0 315 750 1400 2250 3450 5150 P (Torr) 95 85 75 65 55 45 35 Integre la ecuación cinética usando las funciones F(t) y G(t) definidas anteriormente (problema 5) Solución.- Estrictamente no se puede calcular la constante cinética aparente pues el oxígeno se adsorbe fuertemente. Deduciendo la ley cinética integrada y linealizándola como en el ejercicio 5, se pueden calcular las pendientes y ordenadas del origen. Con esas dos magnitudes, se pueden calcular estos los valores: K O 2 6,52x10 2 Torr y K N 2 O k 2 4,41x10 4 Torr s 1 Ejercicio 6.9.- Se sabe que el CO y el O2 se adsorben sobre una superficie de Pt a 370 °C según la isoterma de Langmuir, si bien el O2 lo hace con disociación: La constante de adsorción del CO es 1.359x10-3 Pa-1. En la siguiente tabla se indican los resultados obtenidos al hacer pasar O2 sobre una malla de Pt a la temperatura de 370 °C: P (Pa) O 2 100 0.605 200 0.685 400 0.755 700 0.800 1000 0.830 1300 0.850 1800 0.865 Calcular la constante de velocidad de la etapa de reacción, k a 370 °C para la reacción de oxidación: 2CO (g) + O2 (g) 2 CO2 (g) llevada a cabo sobre una superficie de Pt sabiendo que cuando las presiones parciales de CO y de O2 son de 500 y 200 Pa, respectivamente, la velocidad de formación de CO2 es de 0.0025 Ms-1. Solución.- k2=0,0252 M s-1. Ejemplo 6.10.- Se estudió experimentalmente la reacción C2H4 + H2 C2H6 sobre un catalizador de níquel, proponiéndose como etapa controlante la reacción del etileno adsorbido con el hidrógeno en fase gas. Los resultados cinéticos obtenidos se relacionaron según la siguiente expresión (con T en K): 6050 D ·e T PH 2 PC 2 H 4 11100 1 B· e T PC 2 H 4 a) ¿Esta ecuación es consistente con el mecanismo propuesto? b) ¿ Cómo explicaría que los exponentes de las exponenciales sean positivos?. Calcular la energía de activación de la etapa de reacción y la entalpía de adsorción. c) Se observó una energía de activación de 41.9 kJ/mol a 100°C y de 26.8 kJ/mol a 150°C. Estos valores son consistentes con la ley de velocidad? QFIII Tema 6 47 d) Cuando la reacción se realiza en presencia de algunos catalizadores se observa un máximo, mientras que en presencia de otros catalizadores la velocidad siempre aumenta con la temperatura. ¿A qué se debe este comportamiento? Solución.- La primera etapa es un equilibrio de adsorción reversible con constante K: K C 2 H 4 (g ) C 2 H 4 (ads) La segunda etapa es la reacción controlante de la velocidad: k 2 C 2 H 4 (ads) H 2 (g) C2H6 Admitiendo que el etileno se adsorbe según la isoterma de Langmuir: K PH 2 PC 2 H 4 k C 2 H 4 PH 2 k 2 1 K PH 2 PC 2 H 4 Comparando esta ecuación con la del enunciado, se concluye que: 6050 k2K D e T ; 11100 K Be T Y dividiendo la primera ente la segunda: 5050 D k2 e T B Teniendo en cuenta que K es una constante de equilibrio: K e Gads S ads H ads RT e R e RT de donde se deduce que: S ads B e R y H ads 11100 T e e RT Por otra parte, como la segunda reacción se supone que es una reacción elemental se debe cumplir la ecuación de Arrhenius y: Ea ,2 5050 D k 2 A2 ·e RT e T B H 0 11000 e e se deduce que T De la igualdad RT y teniendo en cuenta que -1 -1 -1 R=8,314 kJ K mol , se obtiene H0 = -92 kJ mol . 11000 T Hads RT Análogamente, por comparación de los exponentes de la ecuación: QFIII Tema 6 48 Ea ,2 5050 RT e e T se deduce: -1 Ea,2 = -R·5050, de donde se calcula Ea,2 = 42 kJ mol . Ejercicio 6.11.- En la tabla siguiente se ha tabulado la velocidad inicial de una reacción A(g) B(g) (con K ∞) para la cual se sospecha que puede transcurrir a través de tres etapas de equilibrio sucesivas: k1 k2 k3 k 1 k 2 k 3 A (g ) A (ads) B(ads) B(g ) T/ºC P/atm 0,5 380 1,0 2,0 5,0 0,5 400 1,0 v0/mol/h g(cat) T/ºC -3 1,70x10 -3 2,60x10 800 P/atm v0/mol/h g(cat) 0,5 3,83 2,0 3,85 -3 5,0 3,75 -3 10,0 3,79 -3 0,5 4,20 2,0 4,18 3,55x10 4.50x10 3,31x10 -3 5,18x10 825 -3 2,0 7,25x10 5,0 4,15 5,0 9,50x10-3 10,0 4,25 a) Para esta reacción, esboce las curvas teóricas de v0 = f(P), siendo P la presión inicial de A, considerando cada uno de los pasos (adsorción de A, reacción superficial y desorción de R) que controlan la reacción. b) ¿Cuál es la etapa controlante a temperaturas bajas? Determine las constantes de la función v0 = f(P), así como la energía de activación de la etapa limitante y la entalpía de adsorción. c) Repita el apartado b) a temperaturas altas y calcule la energía de activación. d) Explique por qué hay alteraciones de la etapa controlante al variar las temperaturas. e) ¿Qué ocurriría a una temperatura de 540 °C? Solución.- Si la etapa A(ads) B(ads) fuera la más lenta porque los equilibrios de adsorción de A(g) y de desorción de B(ads) son relativamente rápidos , la velocidad del proceso sería: v k 2A k 2 K A PA 1 K A PA K B PB En condiciones iniciales PB 0 , pudiéndose escribir: QFIII Tema 6 49 v0 k 2 K A PA 1 K A PA o lo que es lo mismo: v 0 v 0 K A PA k 2 K A PA y reordenando términos: 1 1 1 v 0 k 2 K A PA k 2 que es la ecuación de una recta a la que se ajustan los datos de la tabla a 380ºC (653 K) y 400 ºC 1 (673 K). A partir de los datos de la tabla, como inicialmente PA = P, representando frente a v0 1 , se obtienen los valores a las dos temperaturas más bajas: P 3 380 1 k 380 2 5,52 x10 mol / h g (cat ) y K A 0,887 atm 2 400 1 k 400 2 1,199 x10 mol / h g (cat ) y K A 0,765 atm Suponiendo que k2 depende de la temperatura conforme a la ecuación de Arrhenius, se cumplirá que: T k 1 E 1 1 ln 2 a ,2 T R T2 T1 k 22 y sustituyendo los valores calculados: T E a ,2 k 2 R ln 2 T k 21 1 1 T1 T2 R ln 1,199 x10 2 5 ,52 x10 3 142 kJ mol 1 1 1 653 673 También, como la adsorción de A se produce según un equilibrio cuya K1=KA ,se supone que su variación con la temperatura sigue la ecuación de vant’Hoff: d ln K A H ads dT RT 2 QFIII Tema 6 50 Integrando entre ambas temperaturas y reordenando la expresión, se calcula la entalpía de adsorción en este intervalo de temperaturas: T K A2 0,765 0,887 H ads 27 kJ mol 1 1 1 1 1 T1 T2 653 673 R ln T k A1 8,314 ln A temperaturas altas, la reacción global es de pseudo-orden cero respecto a las especies en fase gaseosa, ya que no depende de la presión, y deben haberse alcanzado rápidamente los equilibrios precedentes al de desorción de la especie B, por lo que la velocidad podría expresarse como: v k 3 B k 3 PB (1 A B ) Como inicialmente PB 0 , la ley de velocidad se simplifica, de manera que v k 3 B y K 2 K APA ,0 como K 2 B , v o k 3 1 K APA ,0 K 2 K APA ,0 A 1 PA ,0 KR vo k 3 k3 k3 1 1 1 1 K A· PA ,0 K 2 K A · PA ,0 K 2K A· PA ,0 KR KR KR KR K 2 ·K A · 1 PA ,0 KR K 2 ·K A · Esta ecuación corresponde con el comportamiento observado a temperatura altas. Por lo tanto, promediando los valores experimentales obtenidos a cada una de las dos T se obtienen los siguientes valores T 1073.15 K v0 k 3 3.805 mol /( h· gcat ) T 1098.15 K v0 k 3 4.195 mol /( h· gcat ) 1 Y de acuerdo con la ec. de Arrhenius: E a ,3 38.25 kJ ·mol Se producen cambios en la etapa controlante porque las energía de activación son muy diferentes y por lo tanto el efecto de la T sobre cada etapa es distinto. A T altas la k2 se hace mayor que k3 y la etapa (3) controla la velocidad. A T bajas, k3 sería mayor y la etapa (2) controla. A T intermedias las constantes de velocidad serían similares y no habría una única etapa controlante de la velocidad QFIII Tema 6 51 Ejercicio 6.12.- Se desea calcular el sobrevalor del potencial respecto del valor de equilibrio en un proceso catódico que transcurre a una cierta velocidad, para lo cual se aplica una densidad de corriente global de 0,1 A cm-2. Para este proceso, j0 vale en un caso 0,1 A cm-2, y en otro caso vale 10-3A cm-2. La temperatura se mantuvo constante a 25 °C y el valor de α= 0,5. Solución: En un proceso catódico se tiene La densidad de corriente total será la suma algebraica de las densidades de corrientes anódica y catódica: j=ja+jc. En el potencial del equilibrio, Eeq, la corriente que circula es nula por lo que j=0. Esto significa que : j=ja+jc=0 por lo que ja=-jc. A este valor de ja (o de –jc) se les conoce como densidad de corriente de intercambio, j0. Así pues, la ecuación de Butler-Volmer se suele expresar en función de esta densidad de corriente de intercambio, j0, y del sobrepotencial, n=E-Eeq, o sea ecuación 6.64: nF ( 1 )nF j j0 e RT e RT (1 )nF nF pues = 0,5. RT RT exp( x ) exp( x ) nF Teniendo en cuenta que: senh( x ) senh 2 RT Quedando la ecuación de Butler-Volmer como: 0 ,5 F j 2 j0 senh ; en el que se ha tomado n=1. RT Despejando el valor de la sobretensión para los casos que nos dice el problema se tiene: Si realizamos la simplificación: x j 8 ,314( JK 1mol 1 ) 298 K RT 0 ,1 arcsenh arcsenh 2 0 ,025V 1 0 ,5 F 2 0 ,1 96485Cmol 2 j0 Si j0= 10-3 Acm-2, el valor obtenido para = -0,230 V. El potencial del sistema se desplaza 25 mV o 230 mV en sentido catódico (i<0) según sea i0. QFIII Tema 6 52 Ejercicio 6.13.- Si se aplica un sobrevoltaje de -0,020V a la reacción catódica: Ox+ e R que transcurre a 25 °C con un valor de j0 = 10-3 A cm-2 y α = 0,4, calcule la densidad de corriente haciendo uso de la ecuación de Butler-Volmer. Solución: El proceso que se nos indica es un proceso catódico. nF ( 1 )nF e RT con = 0,4, por lo que podemos La corriente que circula es: j j0 e RT sustituir directamente: 1 0 ,496485 Cmol 1 ( 0 ,02V ) ( 10 ,4 )96485 Cmol ( 0 ,02V ) 3 2 1 e j 10 ( Acm ) e 8 ,314 JK 298 K 8 ,314 JK 1 298 K -4 -2 J=-7,39x10 A cm . La aproximación de campo bajo proporciona un valor de: 7,78 A cm-2. Ejercicio 6.14.- Considere la reacción electroquímica en la que se intercambia un electrón que ocurre en un electrodo de 1 cm2, a 25º C y en las condiciones siguientes: a) las concentraciones de especie reducida, CR* y oxidada, CO*, correspondientes al seno de la disolución sean iguales a 103 M; b) la constante de velocidad estándar, k0 sea igual a 10-7 cm s-1 y c) el coeficiente de transferencia de carga, α = 0,3. Calcule: (1) la densidad de corriente de intercambio, j0. (2) Represente gráficamente la curva densidad de corriente frente al sobrepotencial (=E-E0) por debajo de los 600 A m-2 sin considerar los posibles efectos de transporte. (3) Represente las curvas de Tafel catódica y anódica para este rango de intensidad. Solución: (1) Cálculo de la densidad de corriente de intercambio, j0: En nuestro caso (proceso catódico) i<0, los valores C R* C0* C 103 M . ja nFkaC R* y jc nFkc C0* , por lo tanto: j ja jc 0 , en el potencial de equilibrio, Eeq: ja jc j0 Con lo que el valor de la densidad de corriente de intercambio se obtiene de: j0 nFk 0 C 96485 10 9 1 9 ,6485 10 5 A ·m2 j0 ( C ·mol 1 )( m· s 1 )( mol·m3 ) A·m2 (2) Si el límite de la densidad de corriente es de 600 mA·m-2 significa que las intensidades de corrientes catódica (j<0) y anódica (j>0) no deberán sobrepasar dicha cantidad, por lo que los sobrepotenciales, , tendrán unos determinados límites. Dichos límites de se calculan a partir de la ecuación Butler-Volmer haciendo: QFIII Tema 6 53 nF ( 1 )nF j j0 e RT e RT 600 A·cm2 por lo que = -0,15663 V nF ( 1 )nF j j0 e RT e RT 600 A·cm2 por lo que = 0,06957 V. Si sustituimos los datos que nos da el problema: A= 0,30; F= 96485 Cmol-1; R= 8,31451 JK-1mol-1; T= 298 K y j0= 9,6485x10-5 A·m-2 obtenido anteriormente en (1) Observar la asimetría de la curva, j frente a dado el valor de = 0,30 y también que se extiende entre los valores límite de para los cuales la densidad de corriente sea 600 mAm-2. En la figura se observa que un sobrepotencial (por ejemplo el positivo) no elimina totalmente la componente catódica (curva azul cuya j<0) hasta alcanzar un cierto valor. Asimismo sucede con la componente anódica (curva roja con j>0). Es interesante calcular cuando la componente anódica (o la catódica) no participa significativamente en la densidad de corriente global, j, o sea cuando ja >>jc por lo que j~ja. De la ecuación Butler-Volmer: nF Si observamos que jc j0 e RT 0 ( 1 )nF 0 Si observamos que ja j0 e RT Se acepta que un término puede despreciarse frente a otro cuando sea unas 100 veces menor. (i) ( 1 )nF nF RT ln(100) 100 j0 Para que ja>>jc → j0 e RT 0,118V RT → e nF ( 1 )nF nF RT ln(100) j0 Para que jc>>ja → 100 j0 e RT 0,118V RT → e nF Conclusión: cuando el sobrepotencial del proceso sea mayor que 0,118 V, el término catódico de la ecuación Butler-Volmer puede ser despreciado. Cuando < -0,118 V puede despreciarse la componente anódica de la densidad de corriente. (ii) (3) Proceso anódico: j j0 e (1 )nF RT QFIII Tema 6 54 nF Proceso catódico: j j0 RT Tomando logaritmos decimales en ambos procesos y reordenando adecuadamente tendremos: e Obsérvese que al ser =0,30 se tiene un valor para (1-) =0,70 en consonancia con las pendientes de la figura adjunta. 6.5.‐Bibliografía ‐J.BertrányJ,Núñez(coords),“QuímicaFísica(Vol.2)”,ArielCiencia.1ªedición,2002. ‐J.K.Vemulapali,“PhysicalChemistry”,Prentice‐HallInternacionalInc,1993. ‐E.Gileadi,“ElectrodeKinetics”,V.C.H.PublishersInc,2008. QFIII Tema 6 55