POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE. A
Anuncio

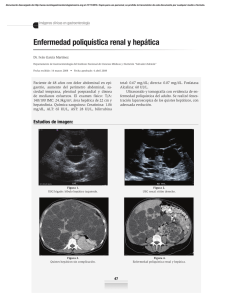
POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE. A PROPÓSITO DE UN CASO. AUTOR: ELENA BERNUÉS. TUTOR: FRANCISCO J. LAVILLA. UNIVERSIDAD DE NAVARRA - 2014 1.INTRODUCCIÓN. 2.METODOLOGÍA. La enfermedad de poliquistosis renal (PKD) del adulto está incluida en las enfermedades hereditarias que causan pérdida irreversible de la función renal. Puede ser autosómica dominante o recesiva. La autosómica dominante (ADPKD) es la causa genética más común de ERC. Tiene una prevalencia de 1:400 a 1:1000. Es causada por las mutaciones del PKD1 en el cromosoma 16 (85%) o del PKD2 en el cromosoma 4 (15%). Descripción de un caso clínico y revisión de la literatura. FIGURA 1. Riñones con numerosos quistes en paciente con ADPKD. Los pacientes presentan hipertensión, hematuria, insuficiencia renal o dolor en el flanco atribuible a una hemorragia renal, cálculos o infección del tracto urinario. También presentan manifestaciones extrarenales, entre las que destacan los aneurismas intracraneales, enfermedad diverticular del colón y quistes en otros órganos. 3. RESULTADOS. CASO CLÍNICO. Hombre de 65 años, con antecedentes de madre fallecida por PKD y 3 de sus 7 hermanos con PKD en la actualidad. Presenta enfermedad poliquística hepática y renal con enfermedad renal secundaria terminal, que precisó de hemodiálisis en 1991; Primer trasplante renal en octubre 1997 que se extirpa por nefropatía del injerto en 2002 entrando nuevamente en diálisis , hasta el 4 de mayo de 2004 en que se realiza retrasplante y posteriormente presenta nefropatía del injerto nuevamente. FIGURA 3 y 4. Evolución de creatinina y potasio. Obsérvense los dos picos que coinciden con entrada en hemodiálisis. FIGURA 5. Evolución hemoglobina, curva espejo de las anteriores. Se han identificado varios factores de riesgo que contribuyen a la severidad de la enfermedad. Estos son la genética (PKD1 vs PKD2), la hipertensión, la aparición temprana de los síntomas, el género masculino, el incremento del tamaño renal y el bajo peso al nacer, entre otros. El paciente desarrolla complicaciones asociadas a la enfermedad, como son la cardiopatía hipertensiva e isquémica, la enfermedad diverticular del colón, y la anormalidad ventilatoria restrictiva moderada de origen multifactorial (obesidad, poliquistosis hepatorrenal e insuficiencia cardíaca). El diagnostico se realiza mediante la visualización de los quistes en la ecografía. Desde el 2010, empeora precisando varios ingresos por insuficiencia respiratoria, infecciones, agudizaciones de la insuficiencia cardíaca y una corrección de eventración abdominal. En diciembre del 2012, muere en su domicilio por complicación cardiovascular. FIGURA 2. Ecografía de paciente con ADPKD, donde se observan los quistes. FIGURA 6. TAC del paciente donde se aprecian quistes hepáticos y renales. FIGURA 7. Urografía intravenosa. Ambos riñones están aumentados y existe desplazamiento de los cálices. FIGURA 8. Rx del paciente que muestra cardiomegalia. ICC. 4. CONCLUSIONES. Debe prestarse especial atención a la detección precoz de la enfermedad, mediante los antecedentes familiares y los factores de mal pronóstico. Como ocurre en este caso, los pacientes presentan diversas complicaciones multisistémicas, por lo que el tratamiento está dirigido a aliviar los síntomas y a la prevención de dichas complicaciones, mediante un estricto control de la HTA con IECAS o ARA II , estatinas, y dieta hiposódica, entre otros. No hay un tratamiento específico que haya sido probado para prevenir o disminuir la progresión de la ADPKD. Aunque hay terapias muy prometedoras que están siendo evaluadas, como son los antagonistas de los receptores de vasopresina , el incremento del consumo de agua, la somatostatina y los inhibidores del mTOR. Cuando los pacientes con ADPKD progresan al estadio final de la enfermedad requieren terapia sustitutiva renal, con hemodiálisis o trasplante renal.