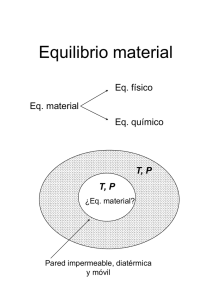
Capitulo 4. - la web de julián moreno mestre
Anuncio

Ejercicios y problemas de Termodinámica I CAPÍTULO 4º Potenciales termodinámicos. Relaciones de Maxwell. Resumen de teoría: Funciones termodinámicas: Energía interna: U dU = TdS − pdV Entalpía: H = U + pV dH = TdS + Vdp Función de Helmholtz: F = U − TS dF = − SdT − pdV G = U − TS + pV dG = − SdT + Vdp Función de Gibbs: Relaciones de orden cero: ⎛ ∂F ⎞ ⎛ ∂U ⎞ ⎟ = −S ⎟ =T ⎜ ⎜ ⎝ ∂T ⎠V ⎝ ∂S ⎠V ⎛ ∂F ⎞ ⎜ ⎟ = −p ⎝ ∂V ⎠ T ⎛ ∂G ⎞ ⎜ ⎟ =V ⎝ ∂p ⎠T ⎛ ∂H ⎞ ⎜ ⎟ =V ⎝ ∂p ⎠ S ⎛ ∂U ⎞ ⎜ ⎟ = −p ⎝ ∂V ⎠ S ⎛ ∂H ⎞ ⎜ ⎟ =T ⎝ ∂S ⎠ p ⎛ ∂G ⎞ ⎜ ⎟ = −S ⎝ ∂T ⎠ p Condición de Schwartz (Solo sirve para ecuaciones diferenciales exactas): ⎛ ∂M ⎞ ⎛ ∂N ⎞ ⎟⎟ = ⎜ dz = M ( x, y )dx + N ( x, y )dy ⇒ ⎜⎜ ⎟ ⎝ ∂y ⎠ x ⎝ ∂x ⎠ y Relaciones de primer orden (relaciones de Maxwell): ⎛ ∂T ⎞ ⎛ ∂V ⎞ ⎛ ∂p ⎞ ⎛ ∂T ⎞ ⎜ ⎟ =⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ ⎝ ∂S ⎠V ⎝ ∂V ⎠ S ⎝ ∂p ⎠ S ⎝ ∂S ⎠ p ⎛ ∂p ⎞ ⎛ ∂S ⎞ ⎟ ⎟ =⎜ ⎜ ⎝ ∂V ⎠ T ⎝ ∂T ⎠V ⎛ ∂S ⎞ ⎛ ∂V ⎞ ⎜⎜ ⎟⎟ = −⎜ ⎟ ⎝ ∂T ⎠ p ⎝ ∂p ⎠ T 78 Julián Moreno Mestre Problemas: 1º Demuestre para un gas ideal las siguientes igualdades: CV ⎛ ∂U ⎞ ⎛ ∂F ⎞ ⎛ ∂G ⎞ a) ⎜ b) ⎜ c) ⎜ ⎟ = −∞ ⎟ = ⎟ =1 ⎝ ∂H ⎠U ⎝ ∂F ⎠ S CV − S ⎝ ∂F ⎠ H ⎛ ∂H ⎞ d) ⎜ ⎟ =γ ⎝ ∂U ⎠ S Solución: a) Partiendo de: dU = TdS − pdV dF = − SdT − pdV y dividiendo entre si las dos ecuaciones: dU TdS − pdV = dF − SdT − pdV y considerando que es a entropía constante, esto implica que el dS es cero: ⎛ ∂V ⎞ 1 −p⎜ ⎟ − pdV − pdV ⎛ ∂U ⎞ ⎝ ∂T ⎠ S dT · = = ⎜ ⎟ = ⎛ ∂V ⎞ ⎝ ∂F ⎠ S − SdT − pdV − SdT − pdV 1 −S − p ⎜ ⎟ dT ⎝ ∂T ⎠ S Partiendo de la siguiente relación de Maxwell: ⎛ ∂S ⎞ ⎛∂p⎞ ⎛ ∂T ⎞ ⎛ ∂V ⎞ ⎟ ⎜ ⎟ = −⎜ ⎟ →⎜ ⎟ = −⎜ ⎝ ∂S ⎠V ⎝ ∂V ⎠ S ⎝ ∂T ⎠ S ⎝ ∂p ⎠V y de la siguiente ecuación a V constante: ⎛ ∂S ⎞ µ TdS = λ dV + µ dp → TdS = µ dp → ⎜ ⎟ = ⎝ ∂p ⎠V T y sabiendo que el coeficiente calorimétrico µ es: ⎛ ∂T ⎞ µ = CV ⎜ ⎟ ⎝ ∂p ⎠V Derivando parcialmente ahora la ecuación de estado del gas ideal llegamos a la conclusión que: ⎛ ∂T ⎞ ⎛ ∂S ⎞ C CV 1 V V V ⎛ ∂V ⎞ → µ = CV → ⎜ ⎟ = CV = V →⎜ ⎜ ⎟ = ⎟ =− nR ⎝ ∂p ⎠V T nR p p ⎝ ∂T ⎠ S ⎝ ∂p ⎠V nR y por tanto con esto demostramos que: ⎛ C ⎞ −p⎜− V ⎟ ⎛ ∂U ⎞ ⎝ p ⎠ = CV ⎜ ⎟ = ⎛ C ⎞ CV − S ⎝ ∂F ⎠ S −S − p ⎜ − V ⎟ ⎝ p ⎠ b) Partiendo de: dU = TdS − pdV dF = − SdT − pdV dH = TdS + Vdp Nos basaremos en un procedimiento parecido al apartado anterior. Pero antes: dF − dU = − SdT − pdV − TdS + pdV dH − dU = TdS + Vdp − TdS + pdV dF = dU − TdS − SdT dH = dU + Vdp + pdV Dividiendo entre si: dF dU − TdS − SdT = dH dU + Vdp + pdV Si tomamos como constante la energía interna, implica además que es constante la 79 Ejercicios y problemas de Termodinámica I temperatura ya que los gases ideales su energía interna depende exclusivamente del cambio de su temperatura. −TdS ⎛ ∂F ⎞ ⎜ ⎟ = ⎝ ∂H ⎠U Vdp + pdV Dividiendo ahora por dS numerador y denominador: 1 −TdS −T −T ⎛ ∂F ⎞ · dS = = ⎜ ⎟ = 1 dp dV ⎛ ∂p ⎞ ⎛ ∂V ⎞ ⎝ ∂H ⎠U Vdp + pdV V +p V ⎜ ⎟ + p⎜ ⎟ dS dS dS ⎝ ∂S ⎠T ⎝ ∂S ⎠T Utilizando las relaciones de Maxwell y derivando después la ecuación de estado del gas ideal: p V ⎛ ∂p ⎞ ⎛ ∂T ⎞ ⎛ ∂V ⎞ ⎛ ∂T ⎞ ⎟ = ⎜ ⎟ = −⎜ ⎟ =− ⎜ ⎟ =⎜ nR ⎝ ∂S ⎠T ⎝ ∂V ⎠ p ⎝ ∂S ⎠T ⎝ ∂p ⎠V nR Sustituyendo: −T −T −1 ⎛ ∂F ⎞ = = = −∞ ⎜ ⎟ = 0 ⎝ ∂H ⎠U V ⎛ − p ⎞ + p ⎛ V ⎞ −T + T ⎜ ⎟ ⎜ ⎟ ⎝ nR ⎠ ⎝ nR ⎠ c) Partiendo de: dF = − SdT − pdV dG = − SdT + Vdp dH = TdS + Vdp y restando entre si: dG − dH = − SdT + Vdp − TdS − Vdp → dG = dH − SdT + Vdp − TdS − Vdp dividiendo ahora: dG dH − SdT + Vdp − TdS − Vdp dH − SdT − TdS = = dF − SdT − pdV − SdT − pdV Considerando que la entalpía es constante esto implica que también será constante la temperatura ya que para un gas ideal sabemos que esta depende únicamente de la temperatura, esto implicará que dS = 0 y dT = 0, por tanto: −TdS T ⎛ ∂S ⎞ ⎛ ∂G ⎞ = ⎜ ⎜ ⎟ = ⎟ ⎝ ∂F ⎠ H − pdV p ⎝ ∂V ⎠T Partiendo de la relación de Maxwell y derivando: nR ⎛ ∂S ⎞ ⎛ ∂p ⎞ ⎜ ⎟ =⎜ ⎟ = ⎝ ∂V ⎠T ⎝ ∂T ⎠V V Sustituyendo: T ⎛ ∂S ⎞ T nR ⎛ ∂G ⎞ =1 ⎜ ⎟ = ⎜ ⎟ = ⎝ ∂F ⎠ H p ⎝ ∂V ⎠T p V d) Partiendo de: dU = TdS − pdV Dividiendo entre si: dH = TdS + Vdp dH TdS + Vdp = dU TdS − pdV Considerando la entropía constante: 80 Julián Moreno Mestre Vdp −V ⎛ ∂p ⎞ V ⎛ ∂p ⎞ ⎛ ∂T ⎞ ⎛ ∂H ⎞ =− ⎜ ⎟ = ⎜ ⎟ =− ⎜ ⎟ ⎜ ⎟ p ⎝ ∂V ⎠ S p ⎝ ∂T ⎠ S ⎝ ∂V ⎠ S ⎝ ∂U ⎠ S − pdV Partiendo de la relación de la Maxwell: ⎛ ∂S ⎞ ⎛ ∂p ⎞ ⎛ ∂S ⎞ ⎛ ∂T ⎞ ⎜ ⎟ =⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ ⎝ ∂V ⎠ S ⎝ ∂T ⎠ S ⎝ ∂V ⎠ p ⎝ ∂p ⎠V Partiendo de: Cp Cκ TdS = dV + v T dp βV β Dado que es diferencial exacta, de la misma podemos deducir las relaciones: Cp 1 ⎛ ∂S ⎞ CV κ T βT ⎛ ∂S ⎞ ⎛ ∂p ⎞ →⎜ ⎟ = ⎜ ⎟ = ⎜ ⎟ = βT ⎝ ∂V ⎠ p β TV ⎝ ∂S ⎠V CV κ T ⎝ ∂p ⎠V Sustituyendo: 1 1 Cp V ⎛ ∂p ⎞ ⎛ ∂T ⎞ V ⎛ ∂p ⎞ ⎛ ∂S ⎞ V βT Cp 1 ⎛ ∂H ⎞ =− ⎜ ⎟ =− ⎜ ⎟ ⎜ ⎟ =− ⎜ ⎟ ⎜ ⎟ =− p ⎝ ∂T ⎠ S ⎝ ∂V ⎠ S p ⎝ ∂S ⎠V ⎝ ∂V ⎠ p p CV κ T β TV p κ T CV ⎝ ∂U ⎠ S Y partiendo de la definición del coeficiente de compresibilidad isoterma y derivando a partir de la ecuación de estado del gas ideal: 1 ⎛ ∂V ⎞ 1 nRT 1 κT = − ⎜ = ⎟ =− V ⎝ ∂p ⎠T V p2 p Y sustituyendo conseguimos demostrar la igualdad: 1 Cp Cp ⎛ ∂H ⎞ = =γ ⎜ ⎟ =− p p CV CV ⎝ ∂U ⎠ S 2º Demuéstrese que: Cp ⎛ ∂S ⎞ ⎜ ⎟ =− TS ⎝ ∂G ⎠ p CV ⎛ ∂S ⎞ ⎜ ⎟ =− TS ⎝ ∂F ⎠V Solución: Partiendo de: ⎛ ∂G ⎞ dG = − SdT + Vdp → p ≡ cte → dG = − SdT → ⎜ ⎟ = −S ⎝ ∂T ⎠ p ⎛ ∂F ⎞ dF = − SdT − pdV → V ≡ cte → dF = − SdT → ⎜ ⎟ = −S ⎝ ∂T ⎠V Operando ahora en los diferenciales: ⎛ ∂G ⎞ ⎛ ∂G ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ 1 ⎛ ∂S ⎞ ⎛ ∂H ⎞ 1 = −⎜ ⎜ ⎟ = −S → ⎜ ⎟ ⎜ ⎟ = −S → ⎜ ⎟ = −⎜ ⎟ ⎟ ⎜ ⎟ ⎝ ∂T ⎠ p ⎝ ∂S ⎠ p ⎝ ∂T ⎠ p ⎝ ∂G ⎠ p ⎝ ∂T ⎠ p S ⎝ ∂H ⎠ p ⎝ ∂T ⎠ p S ⎛ ∂F ⎞ ⎛ ∂F ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ 1 ⎛ ∂S ⎞ ⎛ ∂U ⎞ 1 = −⎜ ⎜ ⎟ = −S → ⎜ ⎟ ⎜ ⎟ = −S → ⎜ ⎟ = −⎜ ⎟ ⎟ ⎜ ⎟ ⎝ ∂T ⎠V ⎝ ∂S ⎠V ⎝ ∂T ⎠V ⎝ ∂F ⎠V ⎝ ∂T ⎠V S ⎝ ∂U ⎠V ⎝ ∂T ⎠V S Recordando la definición de los calores a presión y volumen constante: ⎛ ∂U ⎞ ⎛ ∂H ⎞ ⎜ ⎟ = CV ⎜ ⎟ = Cp ⎝ ∂T ⎠V ⎝ ∂T ⎠ p y las relaciones de Maxwell de orden cero: 81 Ejercicios y problemas de Termodinámica I 1 1 ⎛ ∂U ⎞ ⎛ ∂S ⎞ ⎛ ∂H ⎞ ⎛ ∂S ⎞ ⎜ ⎟ =T →⎜ ⎟ = ⎜ ⎟ =T →⎜ ⎟ = ⎝ ∂S ⎠V ⎝ ∂U ⎠V T ⎝ ∂S ⎠ p ⎝ ∂H ⎠ p T Conseguimos demostrar las igualdades del ejercicio: Cp ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂H ⎞ 1 ⎛ ∂S ⎞ →⎜ ⎜ ⎟ = −⎜ ⎟ ⎜ ⎟ ⎟ =− TS ⎝ ∂G ⎠ p ⎝ ∂H ⎠ p ⎝ ∂T ⎠ p S ⎝ ∂G ⎠ p CV ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂U ⎞ 1 →⎜ ⎟ =− ⎜ ⎟ = −⎜ ⎟ ⎜ ⎟ TS ⎝ ∂F ⎠V ⎝ ∂F ⎠V ⎝ ∂U ⎠V ⎝ ∂T ⎠V S 3º La ecuación fundamental, en la representación entrópica, de cierto sistema hidrostático viene dada por: 1/ 3 S = 3 ( anVU ) donde a es una constante. Halle la ecuación térmica de estado así como CV, Cp, β y κT del sistema. Solución: Sabiendo que es un sistema hidrostático, el cual depende de sus variables: ⎛ ∂U ⎞ ⎛ ∂U ⎞ dU = ⎜ ⎟ dS + ⎜ ⎟ dV = TdS − pdV ⎝ ∂S ⎠V ⎝ ∂V ⎠ S De la cual sabemos enseguida que: ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎜ ⎟ = −p ⎜ ⎟ =T ⎝ ∂V ⎠ S ⎝ ∂S ⎠V Y por tanto: S3 1/ 3 S = 3 ( anVU ) → S 3 = 27anVU → U = 27 anV Y derivando siguiendo las relaciones de Maxwell de orden cero: 3S 2 −S 3 ⎛ ∂U ⎞ ⎛ ∂U ⎞ = = T → S = 9 TanV = = − p → S = 3 27 panV 2 ⎜ ⎟ ⎜ ⎟ 2 ⎝ ∂S ⎠V 27anV ⎝ ∂V ⎠ S 27anV S = 3 27 panV 2 = 9TanV → ( 27 panV 2 ) = ( 9TanV ) → 27 2 p 2 a 2 n 2V 4 = 93 T 3 a 3 n3V 3 2 3 p 2V = anT 3 Partiremos de de la definición de los coeficientes o de expresiones útiles para deducirlos: Calor a volumen constante: ⎛ ∂U ⎞ CV = ⎜ ⎟ ⎝ ∂T ⎠V ( ) 3 S3 ⎫ 9TanV ⎪ = T 3 anV 27 anV ⎬ ⇒ U = 27 anV S = 9TanV ⎪⎭ S 3 ⎛ ∂U ⎞ CV = ⎜ TanV → CV = ⎟ = 2 ⎝ ∂T ⎠V 2 Calor a presión constante: ⎛ ∂p ⎞ ⎛ ∂V ⎞ Cp = T ⎜ ⎟ ⎜ ⎟ + CV ⎝ ∂T ⎠V ⎝ ∂T ⎠ p U= 82 Julián Moreno Mestre 3 anT ⎛ ∂p ⎞ ⎜ ⎟ = ⎝ ∂T ⎠V 2 V Cp = T 3anT ⎛ ∂V ⎞ ⎜ ⎟ = p2 ⎝ ∂T ⎠ p 2 3 anT 3anT 2 S 3 anTV 3anT 2 S S 3anT 3 S 3S S + = T + = + = + 2 V p2 2 2 V p2 2 2 p 2V 2 2 2 C p = 2S Coeficiente de dilatación isobárico: 1 ⎛ ∂V ⎞ 3anT 2 3anT 3 1 3 = = = β= ⎜ ⎟ V ⎝ ∂T ⎠ p Vp 2 Vp 2 T T S2 S = 9TanV → T = 9anV 27anV β= S2 Coeficiente de compresibilidad isotermo: 1 ⎛ ∂V ⎞ κT = − ⎜ ⎟ V ⎝ ∂p ⎠T ⎛ ∂V ⎞ −2anT 3 S3 2 3 S = 27 panV → p = ⎜ ⎟ = 27anV 2 p3 ⎝ ∂p ⎠T 1 −2anT 3 2anT 3 anT 3 2 = = κT = − V p3 Vp 3 Vp 2 p κT = 4º 54anV 2 S3 Un mol de gas ideal realiza un proceso isotermo desde un estado inicial caracterizado por la temperatura 27 ºC y la presión 10 atmósferas, hasta un estado final en el que ocupa un volumen doble del inicial. Hallar los incrementos de energía libre de Helmholtz y de entalpía libre de Gibbs que experimenta el gas en este proceso. Solución: Partiendo de: dF = − SdT − pdV Al ser un proceso isotermo esto implica que: dF = − pdV dG = − SdT + Vdp dG = Vdp Dado que es un mol de gas ideal que obedece a la ecuación de estado: pV = RT los estados final e inicial son: pi = 10 atm Vi = 2.46 L p f = 5 atm V f = 4.92 L Por tanto la energía libre de Helmholtz y la entalpía libre de Gibbs es: Vf Vf ∆F = − ∫ pdV = − ∫ V0 ∆G = 83 V0 pf pf p0 p0 ∫ Vdp = ∫ RT dV = − RT ln 2 = −1727.3 J V RT dp = RT ln 2 = 1727.3 J p Ejercicios y problemas de Termodinámica I 5º Un gas perfecto se comprime isotérmicamente a 300 K desde 2 a 500 atmósferas. Calcular la variación de la entalpía libre G y de la energía libre F para un mol de gas. Solución: Partiendo de: dG = − SdT + Vdp dF = − SdT − pdV Al ser un proceso a temperatura constante dT = 0: RT RT pi = 2 atm → Vi = = 12.3 L p f = 2 atm → V f = = 4.92·10−2 L pi pf dG = Vdp → ∆G = pf pf pi pi ∫ Vdp = RT ∫ ⎛p ⎞ dp = RT ln ⎜ f ⎟ = 135.83 atm·L = 13.76 KJ p ⎝ pi ⎠ Vf Vf Vi Vi dF = pdV → ∆F = − ∫ pdV = − RT ∫ 6º ⎛V ⎞ dV = − RT ln ⎜ f ⎟ = 135.83 atm·L = 13.76 KJ V ⎝ Vi ⎠ Determinar el incremento de energía libre de Helmholtz que experimenta un mol de gas ideal con cv cuando se lleva desde una situación de equilibrio inicial caracterizado por las variables T0 y V0 hasta otro estado de equilibrio final a Tf y Vf. Solución: Partiendo que: dF = − SdT − pdV Para un mol gas ideal, la ecuación de la entropía es: dT dV dS = cv +R T V Dado que es un proceso a volumen constante: dT dS = cv T Integrando desde la temperatura inicial hasta una temperatura final T: T ⎛T ⎞ ⎛T ⎞ dT ∆S = ∫ ncv = ncv ln ⎜ ⎟ → S − S0 = ncv ln ⎜ ⎟ T ⎝ T0 ⎠ ⎝ T0 ⎠ T0 ⎛T ⎞ S = S0 + ncv ln ⎜ ⎟ ⎝ T0 ⎠ Con esta ecuación de la entropía, ya estamos en condiciones de calcular el cambio de energía libre de Helmholtz: Tf Vf ⎛ ⎛ T ⎞⎞ RT ∆F = − ∫ ⎜⎜ S0 + ncv ln ⎜ ⎟ ⎟⎟ dT − ∫ dV V ⎝ T0 ⎠ ⎠ T0 ⎝ V0 ⎛ Vf ⎞ ∆F = − (T f − T0 ) ( S0 − ncv ln T0 + ncv ) + ncvT0 ln T0 − ncvT f ln T f − RT ln ⎜ ⎟ ⎝ V0 ⎠ 84 Julián Moreno Mestre 7º El punto de ebullición del mercurio bajo la presión normal es 357 ºC y el calor latente de vaporización 72.78 cal/g. Se hace hervir 1 Kg de mercurio bajo la presión atmosférica. Determinar: a) El calor absorbido. b) Trabajo realizado por el mercurio contra la presión atmosférica suponiendo que es un gas perfecto. c) Incremento de energía interna. d) Incremento de entalpía. e) Incremento de entropía. f) Incremento de energía libre de Helmholtz. g) Incremento de entalpía libre de Gibbs. Dato: ρ Hg = 13.55 kg/L Solución: Vayamos por apartados. a) El calor absorbido es: Q = Lvap ·m = 72780 cal b) Determinamos el volumen inicial y final del mercurio: nRT 1000 0.082·630 m Vf = = = 257.5 L Vi = = 7.38·10−2 L 200.6 1 p ρ Hg El trabajo realizado por el mercurio contra la presión atmosférica es: Vf W= ∫ pdV = p(V f − Vi ) = 6239 cal Vi c) El cambio de energía interna es: ∆U = Q − W = 66541 cal d) Para calcular el incremento de entalpía, partiremos de: dH = TdS + Vdp Dado que es un proceso a presión constante: dH = TdS Sabiendo que además: δQ dS = T Por tanto: δQ = δ Q → ∆H = Q = 72780 cal dH = T T e) El incremento de entropía es: ∆S = 85 Q 72780 = = 115.5 cal/K T 630 Ejercicios y problemas de Termodinámica I f) Calcularemos el incremento de energía libre de Helmholtz: dF = − SdT − pdV Dado que es un proceso a isotermo hervir el mercurio: Vf dF = − pdV → ∆F = − ∫ pdV = − p (V f − Vi ) = −6239 cal Vi g) Calcularemos el incremento de entalpía libre de Gibbs mediante: dG = − SdT + Vdp Dado que es un proceso isotermo hervir el mercurio y transcurre a presión constante, el cambio de entalpía libre de Gibbs es nulo ya que dT = 0 y dp = 0. 8º El potencial de Gibbs molar de cierto sistema termodinámico cerrado está dado por la expresión ⎛ cT ⎞ g (T , p) = g 0 + aT − bT ln T − cT ln ⎜ ⎟ ⎝ p ⎠ donde g0, a, b y c son constantes. Derivando la expresión anterior con respecto a las variables adecuadas, las veces que sean necesarias, hállese: a) la ecuación de estado. b) la expresión de la entropía en función de T y v. c) el coeficiente de compresibilidad isoterma. d) el calor específico a presión constante. Solución: a) Partiendo de la relación de Maxwell de orden cero: ⎛ ∂g ⎞ cT = v → cT = vp ⎜ ⎟ =v→ p ⎝ ∂p ⎠T b) Partiendo de la relación de Maxwell: ⎛ cT ⎞ ⎛ ∂g ⎞ ⎜ ⎟ = − s → − s = a − b ln T − b − c ln ⎜ ⎟ − c → s = c + b − a + b ln T + c ln v ⎝ ∂T ⎠ p ⎝ p ⎠ c) Partiendo de la expresión que define el coeficiente de dilatación isoterma: 1 1 ⎛ ∂v ⎞ 1 cT 1 κ = = → κT = − ⎜ ⎟ = T p v ⎝ ∂p ⎠T v p 2 p d) Partiendo de: Cp ⎛ ∂S ⎞ dT dp ⎛ ∂S ⎞ ⎛ ∂S ⎞ −R =⎜ ⎟ dT + ⎜ ⎟ dp → ⎜ ⎟ = T p ⎝ ∂T ⎠ p ⎝ ∂T ⎠ p T ⎝ ∂p ⎠T ⎛ ∂S ⎞ ⎛b c⎞ Cp = T ⎜ ⎟ = T ⎜ + ⎟ = b + c → Cp = b + c ⎝ ∂T ⎠ p ⎝T T ⎠ dS = C p 86 Julián Moreno Mestre 9º Determinar las relaciones de orden cero y de primer orden (relaciones de Maxwell) de un hilo metálico que cumple: dU = TdS + ZdL Donde Z es la fuerza de contracción o estiramiento, L su longitud y T su temperatura. Solución: Determinamos las expresiones correspondientes a la entalpía, entalpía libre de Gibbs y energía libre de Helmholtz. dH = TdS − LdZ dG = − SdT − LdZ dF = − SdT + LdZ De aquí se deducen las relaciones de orden cero: ⎛ ∂H ⎞ ⎛ ∂H ⎞ ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎜ ⎟ =T ⎜ ⎟ = −L ⎜ ⎟ =T ⎜ ⎟ =Z ⎝ ∂S ⎠ L ⎝ ∂Z ⎠ S ⎝ ∂S ⎠ Z ⎝ ∂L ⎠ S ⎛ ∂G ⎞ ⎛ ∂G ⎞ ⎛ ∂F ⎞ ⎛ ∂F ⎞ ⎜ ⎟ = −L ⎜ ⎟ = −S ⎜ ⎟ = −S ⎜ ⎟ =Z ⎝ ∂Z ⎠T ⎝ ∂T ⎠ L ⎝ ∂T ⎠ Z ⎝ ∂L ⎠T Las correspondientes relaciones de Maxwell son: ⎛ ∂T ⎞ ⎛ ∂Z ⎞ ⎛ ∂S ⎞ ⎛ ∂Z ⎞ ⎛ ∂T ⎞ ⎛ ∂L ⎞ ⎛ ∂S ⎞ ⎛ ∂L ⎞ ⎜ ⎟ =⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ ⎜ ⎟ =⎜ ⎟ ⎝ ∂L ⎠ S ⎝ ∂S ⎠ L ⎝ ∂L ⎠T ⎝ ∂T ⎠ L ⎝ ∂Z ⎠ S ⎝ ∂S ⎠T ⎝ ∂Z ⎠T ⎝ ∂T ⎠ Z 10º La ecuación térmica de estado de un hilo metálico ideal es Z = T·φ(L) y la energía interna del sistema es solamente función de la temperatura. Demuéstrese que: Z 2 ⎛ ∂L ⎞ a) CZ − CL = ⎜ ⎟ T ⎝ ∂Z ⎠T C b) es un proceso reversible y adiabático L dT = φ ( L)dL T Solución: Para resolver este problema nos basaremos en las relaciones de Maxwell determinadas en el problema anterior. Y partiremos además que: ⎛ ∂U ⎞ ⎛ ∂H ⎞ CL = ⎜ CZ = ⎜ ⎟ ⎟ ⎝ ∂T ⎠ L ⎝ ∂T ⎠ Z a) Partiendo de la expresión de la entalpía: H = U − ZL Determinando sus relaciones diferenciales a Z y L constantes: ⎛ ∂H ⎞ ⎛ ∂U ⎞ ⎛ ∂L ⎞ ⎜ ⎟ =⎜ ⎟ −Z⎜ ⎟ = CZ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎛ ∂H ⎞ ⎛ ∂U ⎞ ⎛ ∂Z ⎞ ⎛ ∂Z ⎞ ⎛ ∂H ⎞ ⎛ ∂Z ⎞ ⎜ ⎟ =⎜ ⎟ − L⎜ ⎟ = CL − L ⎜ ⎟ → CL = ⎜ ⎟ + L⎜ ⎟ ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L Restando las relaciones diferenciales entre si: ⎛ ∂U ⎞ ⎛ ∂L ⎞ ⎛ ∂H ⎞ ⎛ ∂Z ⎞ CZ − C L = ⎜ ⎟ −Z⎜ ⎟ −⎜ ⎟ − L⎜ ⎟ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L Y desarrollando las derivadas parciales con la entalpías y la energía interna: ⎡⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎛ ∂S ⎞ ⎤ ⎛ ∂L ⎞ ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎛ ∂L ⎞ ⎜ ⎟ =⎜ ⎟ ⎜ ⎟ = ⎢⎜ ⎟ +⎜ ⎟ ⎜ ⎟ ⎥⎜ ⎟ ⎝ ∂T ⎠ Z ⎝ ∂L ⎠ Z ⎝ ∂T ⎠ Z ⎣⎝ ∂L ⎠ S ⎝ ∂S ⎠ L ⎝ ∂L ⎠ Z ⎦ ⎝ ∂T ⎠ Z ⎡⎛ ∂H ⎞ ⎛ ∂H ⎞ ⎛ ∂S ⎞ ⎤ ⎛ ∂Z ⎞ ⎛ ∂H ⎞ ⎛ ∂H ⎞ ⎛ ∂Z ⎞ ⎜ ⎟ =⎜ ⎟ ⎜ ⎟ = ⎢⎜ ⎟ +⎜ ⎟ ⎜ ⎟ ⎥⎜ ⎟ ⎝ ∂T ⎠ L ⎝ ∂Z ⎠ L ⎝ ∂T ⎠ L ⎣⎝ ∂Z ⎠ S ⎝ ∂S ⎠ Z ⎝ ∂Z ⎠ L ⎦ ⎝ ∂T ⎠ L 87 Ejercicios y problemas de Termodinámica I Con las relaciones de Maxwell del ejercicio anterior: ⎡ ⎛ ∂U ⎞ ⎛ ∂S ⎞ ⎤ ⎛ ∂L ⎞ ⎛ ∂L ⎞ ⎛ ∂S ⎞ ⎛ ∂L ⎞ ⎛ ∂L ⎞ ⎛ ∂S ⎞ ⎜ ⎟ = ⎢Z + T ⎜ ⎟ ⎥ ⎜ ⎟ =Z⎜ ⎟ +T ⎜ ⎟ ⎜ ⎟ =Z⎜ ⎟ +T ⎜ ⎟ ⎝ ∂T ⎠ Z ⎣ ⎝ ∂L ⎠ Z ⎦ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂L ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎡ ⎛ ∂H ⎞ ⎛ ∂S ⎞ ⎤ ⎛ ∂Z ⎞ ⎛ ∂S ⎞ ⎛ ∂Z ⎞ ⎛ ∂Z ⎞ ⎛ ∂S ⎞ ⎛ ∂Z ⎞ ⎜ ⎟ = ⎢− L + T ⎜ ⎟ ⎥⎜ ⎟ =T ⎜ ⎟ ⎜ ⎟ − L⎜ ⎟ =T⎜ ⎟ − L⎜ ⎟ ⎝ ∂T ⎠ L ⎣ ⎝ ∂Z ⎠ L ⎦ ⎝ ∂T ⎠ L ⎝ ∂Z ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L Sustituyendo: ⎛ ∂L ⎞ ⎛ ∂S ⎞ ⎛ ∂L ⎞ ⎛ ∂S ⎞ ⎛ ∂Z ⎞ ⎛ ∂Z ⎞ CZ − C L = Z ⎜ ⎟ +T ⎜ ⎟ −Z⎜ ⎟ −T ⎜ ⎟ + L⎜ ⎟ − L⎜ ⎟ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎝ ∂T ⎠ L ⎡⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎤ ⎛ ∂S ⎞ ⎛ ∂S ⎞ CZ − C L = T ⎜ ⎟ −T ⎜ ⎟ = T ⎢⎜ ⎟ −⎜ ⎟ ⎥ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ L ⎣⎝ ∂T ⎠ Z ⎝ ∂T ⎠ L ⎦ Y partiendo que: ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂S ⎞ ⎛ ∂L ⎞ ⎜ ⎟ −⎜ ⎟ =⎜ ⎟ ⎜ ⎟ ⎝ ∂T ⎠ Z ⎝ ∂T ⎠ L ⎝ ∂L ⎠T ⎝ ∂T ⎠ Z ⎛ ∂S ⎞ ⎛ ∂L ⎞ CZ − C L = T ⎜ ⎟ ⎜ ⎟ ⎝ ∂L ⎠T ⎝ ∂T ⎠ Z Nuevamente recurriendo a las relaciones de Maxwell del ejercicio anterior: ⎛ ∂Z ⎞ ⎛ ∂L ⎞ CZ − CL = −T ⎜ ⎟ ⎜ ⎟ ⎝ ∂T ⎠ L ⎝ ∂T ⎠ Z Donde: Z ⎛ ∂Z ⎞ ⎜ ⎟ = φ ( L) = T ⎝ ∂T ⎠ L Por tanto: ⎛ ∂L ⎞ CZ − C L = − Z ⎜ ⎟ ⎝ ∂T ⎠ Z ⎛ ∂L ⎞ ⎛ ∂L ⎞ ⎛ ∂Z ⎞ ⎛ ∂L ⎞ Z ⎜ ⎟ = −⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ ⎝ ∂T ⎠ Z ⎝ ∂Z ⎠T ⎝ ∂T ⎠ L ⎝ ∂Z ⎠T T CZ − C L = Z 2 ⎛ ∂L ⎞ ⎜ ⎟ T ⎝ ∂Z ⎠T Demostrado. b) Partiendo de: dU = TdS + ZdL Y que tratamos un proceso reversible, en cuyo caso dS = 0. dU = ZdL La energía interna obedecerá a una expresión del tipo: dU = CL dT Por tanto: CL dT = T φ ( L)dL CL dT = φ ( L)dL T Demostrado. 88 Julián Moreno Mestre 11º Un cilindro contiene 100 g de agua a 15 ºC. Mediante un pistón se aumenta la presión sobre el agua isotérmica y reversiblemente desde 1 a 100 bar. Calcúlense ∆U , ∆H , ∆F y ∆G en dicho proceso. Datos: β = 1.5·10−4 K −1 , κ T = 0.47·10−9 pas −1 Solución: Partiendo de las relaciones diferenciales de los coeficientes: 1 ⎛ ∂V ⎞ 1 ⎛ ∂V ⎞ β= ⎜ κT = − ⎜ ⎟ ⎟ V ⎝ ∂T ⎠ p V ⎝ ∂p ⎠T Procedemos integrando a buscar la ecuación de estado del mercurio: 1 ⎛ ∂V ⎞ ∂V 1 ⎛ ∂V ⎞ ∂V β= ⎜ → −κ T ∂p = κT = − ⎜ ⎟ → β∂T = ⎟ V ⎝ ∂T ⎠ p V V ⎝ ∂p ⎠T V ln V = β T + f ( p) ln V = −κ T p + g (T ) Podemos deducir ahora la ecuación de estado más un término constante: ln V = β T − κ T p + k O bien expresado como: V = ke β T −κT p Suponiendo que 100 g de agua a 1 atm = 101.3·103 pas y a la temperatura de 273.15 K ocupase un volumen de 1 L, establecemos esto como condición inicial, valiendo por ello la constante: −4 6 −9 0.1 = ke273.15·1.5·10 −101.3·10 ·0.47·10 → k = 0.10067 L = 0.10067·10−3 m3 Procedemos ahora a calcular la energía interna mediante: dU = TdS − pdV Valiéndonos de: ⎛ ∂p ⎞ TdS = CV dT + T ⎜ ⎟ dV ⎝ ∂T ⎠V Y sustituyendo: ⎛ ∂p ⎞ dU = CV dT + T ⎜ ⎟ dV − pdV ⎝ ∂T ⎠V Al ser expansión isoterma, y derivando la ecuación de estado: ⎛Tβ ⎞ β dU = T dV − pdV = ⎜ − p ⎟ dV κT ⎝ κT ⎠ Derivando a temperatura constante la ecuación de estado: dV = − kκ T e β T −κT p dp Sustituyendo e integrando: ⎛Tβ ⎞ dU = − ⎜ − p ⎟ kκ T e β T −κ p dp = k (κ T p − T β ) e β T −κ p dp ⎝ κT ⎠ T T 107 ∆U = k ∫ (κ T p − T β ) e β T −κ p dp = −41.35 J T 5 10 Calculamos el incremento de la entalpía con: dH = TdS + Vdp 89 Ejercicios y problemas de Termodinámica I Usamos la ecuación: ⎛ ∂V ⎞ TdS = C p dT − T ⎜ ⎟ dp ⎝ ∂T ⎠ p Por tanto el diferencial de entalpía es: ⎛ ∂V ⎞ dH = C p dT − T ⎜ ⎟ dp + Vdp ⎝ ∂T ⎠ p Dado que es a temperatura constante, y derivando la ecuación de estado: dH = −Tk β e β T −κ p dp + ke β T −κ p dp = k (1 − β T )e β T −κ p dp Integrando: T T T 107 ∆H = k (1 − β T ) ∫ e β T −κ p dp = 993.31 J T 105 Calculamos el cambio de energía libre de Helmholtz: dF = − SdT − pdV Al ser un proceso isotermo, eliminamos el término dT, sustituimos el dV por un dp siguiendo la ecuación de estado tal y como hicimos anteriormente e integramos: dF = − pdV = kκ T pe β T −κ T p dp → ∆F = kκ T 107 ∫ pe β T −κ p dp = 2.46 J T 5 10 Calculamos el cambio de la entalpía libre de Gibbs: dG = − SdT + Vdp Al ser un proceso isotermo, eliminamos el término dT, sustituimos el V siguiendo la ecuación de estado e integramos: dG = Vdp = ke β T −κ T p 107 dp → ∆G = k ∫ e β T −κ p dp = 1038.18 J T 105 12º El coeficiente de dilatación térmica y el de compresibilidad isoterma de cierto gas real vienen dados por las expresiones V −b V −b β= κT = TV pV –2 siendo b = 3.64·10 L/mol. Calcúlense los valores de ∆U , ∆H , ∆F y ∆G , cuando manteniendo la temperatura constante en 300 K, se comprime 1 mol de dicho gas desde 1 a 2 bar. Solución: Partiendo de las relaciones diferenciales de los coeficientes: 1 ⎛ ∂V ⎞ 1 ⎛ ∂V ⎞ β= ⎜ κT = − ⎜ ⎟ ⎟ V ⎝ ∂T ⎠ p V ⎝ ∂p ⎠T Procedemos integrando a buscar la ecuación de estado del mercurio: 1 ⎛ ∂V ⎞ V −b ∂V ∂T 1 ⎛ ∂V ⎞ V − b ∂V ∂p → = β= ⎜ = → =− κT = − ⎜ ⎟ = ⎟ V ⎝ ∂T ⎠ p TV V −b T V ⎝ ∂p ⎠T pV V −b p ln (V − b ) = ln T + f ( p ) ln (V − b ) = − ln p + g (T ) Podemos deducir ahora la ecuación de estado más un término constante: ln (V − b ) = ln T − ln p + k 90 Julián Moreno Mestre O bien expresado como: p (V − b ) = kT Podemos darnos cuenta que la constante en realidad es R, constante del gas ideal. p (V − b ) = RT Determinamos ahora el cambio de energía interna partiendo de: ⎛ ∂p ⎞ dU = TdS − pdV TdS = CV dT + T ⎜ ⎟ dV ⎝ ∂T ⎠V ⎛ ∂p ⎞ dU = CV dT + T ⎜ ⎟ dV − pdV ⎝ ∂T ⎠V Y como es un proceso a temperatura constante: ⎛ ∂p ⎞ dU = T ⎜ ⎟ dV − pdV ⎝ ∂T ⎠V Y derivando la ecuación de estado, integramos para calcular el cambio de energía interna: R R ⎛ ∂p ⎞ dV − pdV = pdV − pdV = 0 → ∆U = 0 → dU = T ⎜ ⎟ = V −b ⎝ ∂T ⎠V V − b Calculamos el valor de la entalpía con: ⎛ ∂V ⎞ TdS = C p dT − T ⎜ ⎟ dp ⎝ ∂T ⎠ p ⎛ ∂V ⎞ dH = C p dT − T ⎜ ⎟ dp + Vdp ⎝ ∂T ⎠ p dH = TdS + Vdp Y como es a T constante: ⎛ ∂V ⎞ dH = −T ⎜ ⎟ dp + Vdp ⎝ ∂T ⎠ p Derivando ahora la ecuación de estado, e integrando: R dH = −T dp + Vdp = −(V − b)dp + Vdp = bdp → ∆H = p 2·105 ∫ bdp = 3640 pas·L/mol = 3.64 J 105 Calculamos el valor de la energía libre de Helmholtz y de la entalpía libre de Gibbs con: dF = − SdT − pdV dG = − SdT + Vdp A temperatura constante: dF = − pdV dG = Vdp Despejando y derivando la ecuación de estado: RT TR V= +b dV = − 2 dp p p Sustituyendo e integrando: 2·105 TR TR dp dF = − pdV = − p 2 dp = − dp → ∆F = −TR ∫ = −1728.8 J p p p 1·105 5 2·10 ⎛ RT ⎞ ⎛ RT ⎞ dG = ⎜ + b ⎟ dp → ∆G = ∫ ⎜ + b ⎟ dp = 1732.4 J p ⎝ p ⎠ ⎠ 105 ⎝ 91 Ejercicios y problemas de Termodinámica I 13º Sea un gas real de coeficientes de dilatación cúbica y el de compresibilidad isoterma: 3aT 3 b β= κT = V V –8 3 –1 donde b = 1.97·10 m ·pas y a es una constante. Sea realiza con el gas una compresión isoterma a la temperatura de 300 K, con un volumen inicial de 10 L y variándose la presión desde 1 bar hasta 2 bar. a) Calcular ∆G y ∆F. b) ¿Puede tener lugar el proceso anterior a energía interna constante? Solución: Partiendo de las ecuaciones: dU = TdS − pdV dF = − SdT − pdV ⎛ ∂p ⎞ TdS = CV dT + T ⎜ dG = − SdT + Vdp ⎟ dV ⎝ ∂T ⎠V Y sabiendo que es un proceso a temperatura constante: dU = TdS − pdV dF = − pdV ⎛ ∂p ⎞ TdS = T ⎜ dG = Vdp ⎟ dV ⎝ ∂T ⎠V Sustituyendo en la expresión diferencial de la energía interna el término TdS: ⎛ ⎛ ∂p ⎞ ⎞ ⎛ ∂p ⎞ dU = T ⎜ ⎟ dV − pdV = ⎜ T ⎜ ⎟ − p ⎟ dV ⎝ ∂T ⎠V ⎝ ⎝ ∂T ⎠V ⎠ Deducimos ahora la expresión de la ecuación de estado con los coeficientes elásticos: 1 ⎛ ∂V ⎞ 3aT 3 3aT 4 ⎛ ∂V ⎞ 3 = → = 3 aT → V = + f ( p) β= ⎜ ⎟ ⎜ ⎟ V ⎝ ∂T ⎠ p V 4 ⎝ ∂T ⎠ p ⎛ ∂V ⎞ 1 ⎛ ∂V ⎞ b ⎜ ⎟ = →⎜ ⎟ = −b → V = −bp + g (T ) V ⎝ ∂p ⎠T V ⎝ ∂p ⎠T 3aT 4 V= − bp + k 4 Deducimos el valor de k usando las condición inicial: Vi = 10 L = 0.01 m3 pi = 1 bar = 105 pas Ti = 300 K κT = − 3·a·3004 3·a·3004 − 1.67·10−8 ·105 + k → k = 0.01167 − 4 4 Las ecuaciones quedan finalmente así para responder uno por uno los apartados: ⎛ ⎛ ∂p ⎞ ⎞ dF = − pdV dG = Vdp dU = ⎜ T ⎜ ⎟ − p ⎟ dV ⎝ ⎝ ∂T ⎠V ⎠ 0.01 = a) Calcularemos la energía libre de Helmoltz integrando la expresión dF y sustituyendo el dV por un dp mediante: ⎛ ∂V ⎞ 1 ⎛ ∂V ⎞ b κT = − ⎜ ⎟ = →⎜ ⎟ = −b → dV = −bdp V ⎝ ∂p ⎠T V ⎝ ∂p ⎠T 2·105 dF = − pdV = bpdp → ∆F = b ∫ 105 2·105 ⎡ p2 ⎤ pdp = b ⎢ ⎥ = 295.5 J ⎣ 2 ⎦105 92 Julián Moreno Mestre Calculamos la entalpía libre de Gibbs utilizando la ecuación de estado: ⎛ 3aT 4 3·a·3004 ⎞ dG = Vdp = ⎜ − bp + 0.01167 − ⎟ dp 4 ⎝ 4 ⎠ Como es a temperatura constante y de valor 300 K: ⎛ 3a·3004 3·a·3004 ⎞ dG = ⎜ − bp + 0.01167 − ⎟ dp = ( 0.01167 − bp ) dp 4 4 ⎝ ⎠ 2·105 ∆G = ∫ 105 2·105 ⎡ p2 ⎤ ( 0.01167 − bp ) dp = ⎢0.01167 p − b ⎥ = 871.5 J 2 ⎦105 ⎣ b) Buscaremos en este apartado si el dU = 0. Partiendo de: ⎛ ⎛ ∂p ⎞ ⎞ dU = ⎜ T ⎜ ⎟ − p ⎟ dV ⎝ ⎝ ∂T ⎠V ⎠ Calculando la derivada: 3aT 3 ⎛ ∂p ⎞ ⎜ ⎟ = b ⎝ ∂T ⎠V Reemplazando: ⎛ 3aT 3 ⎞ 1 dU = ⎜ T − p ⎟ dV = ( 3aT 4 − bp ) dV b b ⎝ ⎠ 1⎛ 3aT 4 3a·3004 ⎞ dU = ⎜ 3aT 4 + V − − 0.01167 + ⎟ dV 4 4 ⎠ b⎝ Como la temperatura es constante e igual a 300 K: 1⎛ 3a·3004 3a·3004 ⎞ dU = ⎜ 3a·3004 + V − − 0.01167 + ⎟ dV 4 4 ⎠ b⎝ 1 ( 3a·3004 + V − 0.01167 ) dV b El dU no se anula, luego este gas real su energía interna cambia en procesos isotermos. No es posible tener el proceso isotermo del ejercicio a energía interna constante. dU = 14º Determine ∆U , ∆H , ∆F y ∆G en la comprensión isoterma reversible, a 25 ºC, de 0.9 Kg de agua desde 1 a 100 atm, sabiendo que en este intervalo de presiones el volumen molar, a 25 ºC, viene dado por: v = 18 − 7.15·10−4 p + 4.6·10−7 p 2 (cm3/mol) y que ⎛ ∂v ⎞ 3 −3 −6 ⎜ ⎟ = 4.5·10 + 1.4·10 p (cm /mol·K) ⎝ ∂T ⎠ p donde p se mide en atmósferas. Solución: Con las relaciones diferenciales: du = Tds − pdv dh = Tds + vdp dg = − sdT + vdp 93 df = − sdT − pdv ⎛ ∂v ⎞ Tds = c p dT − T ⎜ ⎟ dp ⎝ ∂T ⎠ p Ejercicios y problemas de Termodinámica I Y sabiendo que es en todo momento un proceso isotermo: du = Tds − pdv dh = Tds + vdp df = − pdv dg = vdp ⎛ ∂v ⎞ Tds = −T ⎜ ⎟ dp ⎝ ∂T ⎠ p Remplazando los términos Tds: ⎛ ∂v ⎞ ⎛ ∂v ⎞ du = −T ⎜ dh = −T ⎜ df = − pdv ⎟ dp − pdv ⎟ dp + vdp ⎝ ∂T ⎠ p ⎝ ∂T ⎠ p Diferenciando: dv = ( −7.15·10−4 + 9.2·10−7 p ) dp dg = vdp Integrando ahora una por una: ⎛ ∂v ⎞ −3 −6 −4 −7 du = −T ⎜ ⎟ dp − pdv = −298.15 ( 4.5·10 + 1.4·10 p ) dp − p ( −7.15·10 + 9.2·10 p ) dp ⎝ ∂T ⎠ p 100 ( ) ∆u = − ∫ 298.15 ( 4.5·10−3+ 1.4·10−6 p ) + p ( −7.15·10−4 + 9.2·10−7 p ) dp = −138.79 atm·mL/mol 1 ⎛ ∂v ⎞ dh = −T ⎜ ⎟ dp + vdp ⎝ ∂T ⎠ p dh = −298.15 ( 4.5·10−3+ 1.4·10−6 p ) dp + (18 − 7.15·10−4 p + 4.6·10−7 p 2 ) dp 100 ( ) ∆h = − ∫ 298.15 ( 4.5·10−3+ 1.4·10−6 p ) − (18 − 7.15·10−4 p + 4.6·10−7 p 2 ) dp = 1 ∆h = 1643.7 atm·mL/mol df = − pdv = − p ( −7.15·10−4 + 9.2·10−7 p ) dp 100 ∆f = − ∫ p ( −7.15·10−4 + 9.2·10−7 p )dp = −3.88 atm·mL/mol 1 dg = vdp = (18 − 7.15·10−4 p + 4.6·10−7 p 2 ) dp 100 ∆g = ∫ (18 − 7.15·10 −4 p + 4.6·10−7 p 2 ) dp = 1778.6 atm·mL/mol 1 Teniendo en cuenta que un mol de agua pesa 18 g, y que tenemos 900 g de agua, tendremos 900/18 = 50 moles de agua. Multiplicando las energías molares por el número de moles, y pasando a julios obtendremos: ∆h = 8325.3 J ∆f = −19.65 J ∆g = 9008.6 J ∆u = −703.53 J 94 Julián Moreno Mestre 15º Cierto gas cumple la ecuación de estado: pV = n( RT − ap) –1 con a = 0.523 L·mol . Inicialmente, el gas se encuentra en equilibrio a presión una atmósfera ocupando un volumen de 22.11 L·mol–1 y se lleva a la presión de 3 atm donde el volumen de equilibrio vale 7.02 L·mol–1. Determinar el incremento específico de energía libre de Helmholtz que acompaña a ese proceso. Solución: Reescribimos la ecuación de estado en una forma más sencilla y que mejor permita despejar variables, y además convirtiendo el volumen en volumen molar: pV = n( RT − ap ) → p ( v + a ) = RT Determinamos las temperaturas de equilibrio final e inicial: p f (v f + a) p (v + a) Ti = i i = 276 K Tf = = 276 K R R Es un proceso isotermo. Partiendo de la relación diferencial y considerando que es isotermo: dF = − SdT − pdV → dF = − pdV Sustituyendo la presión e integrando: 7.02 RT ⎛ 7.02 + 0.523 ⎞ ∆F = − ∫ dV = −0.082·276 ln ⎜ ⎟ = 2520 J/mol v + a) ⎝ 22.11 + 0.523 ⎠ 22.11 ( 16º Entre 250 K y 300 K un material tiene un valor de la entropía específica dado por: s = a + bT –1 –1 donde a = 4.76 cal·mol ·K y b = 3.02·10 – 3 cal·mol–1·K–2, y una ecuación constitutiva: κT ·v = C siendo v el volumen molar, κT el coeficiente de compresibilidad isoterma y siendo C = 0.28 L·atm–1·mol–1. Sabiendo que a 1 atm el volumen molar del material vale 1.72 L·mol–1, hallar la diferencia de entalpía libre de Gibbs entre el estado T1 = 300 K y p1 = 10 atm y el estado T2 = 250 K y p2 = 1 atm. Solución: Partiendo de: dg = − sdT + vdp Y sabiendo que: ⎛ ∂v ⎞ C 1 ⎛ ∂v ⎞ = − ⎜ ⎟ → C = − ⎜ ⎟ → C∂p = −∂v → Cp = −v + k v v ⎝ ∂p ⎠T ⎝ ∂p ⎠T Aplicando la condición inicial que a 1 atm el volumen molar es de 1.72 L/mol: 0.28·1 = −1.72 + k → k = 2 L/mol Sustituyendo e integrando ahora: dg = − ( a + bT ) dT + ( k − Cp ) dp κT ·v = C → κ T = 250 1 ⎡ ⎡ T2 ⎤ p2 ⎤ ∆g = − ∫ ( a + bT ) dT + ∫ ( k − Cp ) dp = − ⎢ aT + b ⎥ + ⎢ kp − C ⎥ = 179 cal/mol 2 ⎦ 300 ⎣ 2 ⎦10 ⎣ 300 10 250 95 1 Ejercicios y problemas de Termodinámica I 17º Determine el cambio experimentado en los potenciales termodinámicos de 1 Kg de agua en el proceso de ebullición de la misma a la presión atmosférica normal. Considérese el vapor como gas ideal y que Vl << Vg (∆Hebullición = 536 cal/g). Solución: Partiendo de: dU = TdS − pdV dH = TdS + Vdp dF = − SdT − pdV dG = − SdT + Vdp Al ser una transición de fase, el proceso es isotermo, y además se realiza a la presión exterior constante, lo que implica un proceso isóbaro, por tanto: ∆H = T ∆S ∆G = 0 ∆U = T ∆S − p∆V ∆F = − p∆V Ya sabemos tras este primer paso la variación de la entalpía libre de Gibbs. El enunciado nos facilita el cambio experimentado por la entalpía. Por tanto: ∆H = T ∆S = 536000 cal ∆F = − p∆V ∆U = T ∆S − p∆V El agua ebulle a 100 ºC (= 373.15 K) a 1 atmósfera de presión. Un kilogramo de agua tiene cerca de 1000/18 = 55.5 moles de agua. El volumen en fase vapor será de: nRT Vg = = 1699 L p Dado que el volumen final es mucho mayor que el inicial, el incremento de volumen es prácticamente de 1699 L. Por tanto: ∆U = 536000 − 41174 = 494826 cal ∆F = −41174 cal 18º Cierta sustancia gaseosa posee un coeficiente de compresibilidad isoterma que vale 2.81·10–2 atm–1 a 300 K. Determina los incrementos de energía libre de Helmholtz y de entalpía libre de Gibbs que experimenta un mol de ese gas cuando partiendo de un estado de equilibrio a una 1 atm y 29.8 L duplica su volumen a temperatura constante de 300 K. Solución: Partiendo de: dF = − SdT − pdV dG = − SdT + Vdp Y que es un proceso isotermo: dF = − pdV dG = Vdp A partir del coeficiente de compresibilidad isoterma: 1 ⎛ ∂V ⎞ dV dV = −Vdp → = −κ T dp → ln V = −κ T p + k κT = − ⎜ ⎟ → κT V ⎝ ∂p ⎠T V Utilizando las condiciones iniciales de 1 atm y 29.8 L: ln 29.8 = −2.81·10−2 ·1 + k → k = 3.4226 Por tanto: 3.4226 − ln V ln V = −κ T p + 3.4226 → p = κT Enseguida podemos calcular el incremento de la entalpía y energía libre: 59.6 dV dV dG = Vdp = − → ∆G = − ∫ = 1060 atm·L = 107.4 kJ κT dF = − pdV = ln V − 3.4226 κT dV → ∆F = 29.8 59.6 ∫ 29.8 κT ln V − 3.4226 κT dV = 384.6 atm/L = 38.96 kJ 96 Julián Moreno Mestre 19º Teniendo en cuenta las relaciones de Maxwell, demostrar que: ⎛ ∂U ⎞ ⎛ ∂V ⎞ ⎛ ∂V ⎞ ⎜ ⎟ = −T ⎜ ⎟ ⎟ − p⎜ ⎝ ∂T ⎠ p ⎝ ∂p ⎠T ⎝ ∂p ⎠T Solución: Partiendo de: dU = TdS − pdV Dividiendo entre dp: dU dS dV =T −p dp dp dp Tomando como constante la temperatura llegamos a: ⎛ ∂U ⎞ ⎛ ∂S ⎞ ⎛ ∂V ⎞ ⎜ ⎟ = T ⎜ ⎟ − p⎜ ⎟ ⎝ ∂p ⎠T ⎝ ∂p ⎠T ⎝ ∂p ⎠T Y por la relación de Maxwell: ⎛ ∂S ⎞ ⎛ ∂V ⎞ ⎜⎜ ⎟⎟ = −⎜ ⎟ ⎝ ∂T ⎠ p ⎝ ∂p ⎠ T Queda pues demostrado: ⎛ ∂U ⎞ ⎛ ∂V ⎞ ⎛ ∂V ⎞ ⎜ ⎟ = −T ⎜ ⎟ ⎟ − p⎜ ⎝ ∂T ⎠ p ⎝ ∂p ⎠T ⎝ ∂p ⎠T 20º La tensión superficial de una lámina de agua líquida es una función exclusiva de a temperatura: σ (T ) = a − bT 3 –4 3 con a = 0.145 J/m y b = 2.5·10 J/m ·K. a) Determine la variación de energía interna de la lámina cuando a temperatura constante de 300 K su superficie A aumenta en 100 cm2. b) Suponiendo constante la capacidad calorífica, CA = 4.25 J/K, calcula la variación de temperatura de la lámina, si el aumento de superficie es isoentrópico. c) ¿Cuál hubiera sido la variación de energía interna en el proceso isotermo a) si la tensión superficial variase en la forma: T σ (T , A) = c A siendo c constante? Solución: a) Partiendo de las relaciones diferenciales: dU = TdS + σ dA Sustituyendo: ⎛ ∂σ ⎞ TdS = C A dT − T ⎜ ⎟ dA ⎝ ∂T ⎠ A ⎛ ∂σ ⎞ dU = C A dT − T ⎜ ⎟ dA + σ dA ⎝ ∂T ⎠ A Retirando CA dT ya que es un proceso a temperatura constante: ⎛ ∂σ ⎞ dU = −T ⎜ ⎟ dA + σ dA ⎝ ∂T ⎠ A 97 Ejercicios y problemas de Termodinámica I Y sustituyendo en la expresión, e integrando, calculamos el cambio de energía interna: dU = −T ( −b ) dA + ( a − bT ) dA = adA → ∆U = 10−2 ∫ adA = aA = 1.45·10−3 J 0 b) Partiendo únicamente de: ⎛ ∂σ ⎞ TdS = C A dT − T ⎜ ⎟ dA ⎝ ∂T ⎠ A Al ser isoentrópico significa que TdS = 0: CA ⎛ ∂σ ⎞ dT = −bdA 0 = C A dT − T ⎜ ⎟ dA → T ⎝ ∂T ⎠ A Integrando ahora: Tf 10−2 ⎛ Tf ⎞ CA −4 −2 ∫300 T dT = − ∫0 bdA → ln ⎜⎝ 300 ⎟⎠ = −2.5·10 ·10 → T f = 299.9998235 ∆T = −1.76·10−4 K c) Procediendo como en el apartado a): ⎛ ∂σ ⎞ ⎛ ∂σ ⎞ Isotermo dU = C A dT − T ⎜ → dU = −T ⎜ ⎟ dA + σ dA ⎯⎯⎯⎯ ⎟ dA + σ dA ⎝ ∂T ⎠ A ⎝ ∂T ⎠ A Sustituyendo: c T dU = −T dA + c dA = 0 → ∆U = 0 A A 21º La fuerza recuperadora de una varilla elástica estirada está relacionada con su longitud y temperatura por la expresión: F = aT 2 ( L − L0 ) donde a y L0 son constantes positivas (L0 es la longitud de la varilla sin estirar). Para L = L0, la capacidad calorífica de la varilla, medida a longitud constante, es CL(L0,T) = bT, siendo b una constante. Determine: a) La expresión general de la capacidad calorífica CL(L,T). b) El cambio de entropía de la varilla cuando se aumenta su longitud, reversible e isotérmicamente, desde L0 a 3L0/2. c) La entropía S(L,T), a cualquier longitud y temperatura, supuesta conocida S(L0,T0). d) La temperatura final Tf, cuando, estando la varilla adiabáticamente aislada y partiendo del estado (Li,Ti), se ejerce una tracción reversible hasta que la varilla alcanza una longitud Lf. Solución: Recordemos que en el problema, F no significará energía de Helmholtz a) Partiendo de: ⎛ ∂2 F ⎞ ⎛ ∂CL ⎞ ⎜ ⎟ = −T ⎜ 2 ⎟ = −T 2a ( L − L0 ) → dCL = −T 2a ( L − L0 ) dL ⎝ ∂L ⎠T ⎝ ∂T ⎠ L Integrando, a T constante: ⎛ L2 ⎞ CL = ∫ T 2a ( L − L0 ) dL = T 2a ⎜ − L0 L ⎟ + f (T ) ⎝ 2 ⎠ 98 Julián Moreno Mestre Aprovechando que CL(L0,T) = bT llegamos a la conclusión que: ⎛ L2 ⎞ CL = bT − T 2a ⎜ − L0 L ⎟ + g (T ) ⎝ 2 ⎠ Siendo el parámetro k: ⎛ L2 ⎞ ⎛ L2 ⎞ CL ( L0 , T ) = bT − T 2a ⎜ 0 − L20 ⎟ + g (T ) = bT → −T 2a ⎜ 0 − L20 ⎟ + g (T ) = 0 ⎝ 2 ⎠ ⎝ 2 ⎠ g (T ) = Ta ( L20 − 2 L20 ) = −TaL20 Por tanto: ⎛ L2 ⎞ CL ( L, T ) = bT − T 2a ⎜ − L0 L ⎟ − TaL20 = bT − Ta ( L2 − 2 L0 L + L20 ) ⎝ 2 ⎠ CL ( L, T ) = bT − Ta ( L − L0 ) 2 b) Partiendo de: CL ⎛ ∂L ⎞ ⎛ ∂L ⎞ TdS = CL dT + T ⎜ dT + ⎜ ⎟ dF → dS = ⎟ dF T ⎝ ∂T ⎠ F ⎝ ∂T ⎠ F Como es isotermo el proceso, y conociendo la derivada: 2F 2F ⎛ ∂L ⎞ ⎛ ∂L ⎞ dS = ⎜ ⎜ ⎟ =− 3 ⎟ dF = − 3 dF aT aT ⎝ ∂T ⎠ F ⎝ ∂T ⎠ F Sabiendo que: dF = aT 2 dL Sustituyendo e integrando entre L0 hasta 3L0/2: 3 L0 / 2 2aT 2 ( L − L0 ) 2 dS = − aT dL = − 2 aT L − L dL → ∆ S = − 2 ( 0) ∫L aT ( L − L0 ) dL aT 3 0 ∆S = − a 2T 2 L20 4 c) Utilizando expresiones usadas o determinadas en el apartado anterior como: C 2F ⎛ ∂L ⎞ ⎛ ∂L ⎞ dS = L dT − ⎜ dF = aT 2 dL ⎟ dF ⎜ ⎟ =− 3 T aT ⎝ ∂T ⎠ F ⎝ ∂T ⎠ F Sustituyendo: bT − Ta ( L − L0 ) 2aT 2 ( L − L0 ) 2F 2 2 dS = dT − 3 aT dL = b − a ( L − L0 ) dT − dL T aT T 2 ( ) ( ) dS = b − a ( L − L0 ) dT − 2aT ( L − L0 ) dL 2 Integrando entre T0 y T, y entre L0 y L: T ( ) L ∆S = ∫ b − a ( L − L0 ) dT − ∫ 2aT ( L − L0 ) dL = b (T − T0 ) − a ( L − L0 ) ( 2T − T0 ) 2 T0 2 L0 Tomando: ∆S = S ( L, T ) − S ( L0 , T0 ) Llegamos a la conclusión que: S ( L, T ) = S ( L0 , T0 ) + b (T − T0 ) − a ( L − L0 ) ( 2T − T0 ) 2 99 Ejercicios y problemas de Termodinámica I d) Partiendo de: CL ⎛ ∂L ⎞ dT − ⎜ ⎟ dF T ⎝ ∂T ⎠ F Dado que es adiabático reversible, el dS = 0: dS = bT − Ta ( L − L0 ) 2aT 2 ( L − L0 ) 2 CL ⎛ ∂L ⎞ dT = ⎜ dT = aT dL ⎟ dF → T T aT 3 ⎝ ∂T ⎠ F 2a ( L − L0 ) dT 2 b − a ( L − L0 ) dT = 2aT ( L − L0 ) dL → = dL 2 T b − a ( L − L0 ) E integrando desde (Li,Ti) hasta (Lf,Tf): Tf Lf ⎛ b − a ( L − L )2 ⎞ ⎛ b−a L −L 2 ⎞ 2a ( L − L0 ) ⎛ Tf ⎞ ( i 0) dT 0 f ⎜ ⎟ ∫T T = L∫ b − a ( L − L )2 dL → ln ⎜⎝ Ti ⎟⎠ = − ln ⎜ b − a ( L − L )2 ⎟ = ln ⎜⎜ b − a L − L 2 ⎟⎟ ( f 0) ⎠ 0 0 i i i ⎝ ⎠ ⎝ 2 ( ) Tf Ti = b − a ( Li − L0 ) 2 b − a ( L f − L0 ) 2 → T f = Ti b − a ( Li − L0 ) 2 b − a ( L f − L0 ) 2 22º Una película delgada de área A0 se encuentra a temperatura T0. La película se estira de forma isoterma hasta duplicar su área inicial. Durante el proceso el cambio de tensión superficial viene dado por: dσ aT 2 =− 2 dA A a es una constante. Si el coeficiente de dilatación superficial de la lámina es βA = 2T, determinar ∆S y ∆G . Solución: La derivada del enunciado es en un proceso isotermo: aT 2 aT 2 ⎛ ∂σ ⎞ = − → d = − dA σ ⎜ ⎟ A2 A2 ⎝ ∂A ⎠T Sabemos que el coeficiente de dilatación lineal es: 1 ⎛ ∂A ⎞ ⎛ ∂A ⎞ β A = ⎜ ⎟ = 2T → ⎜ ⎟ = 2TA A ⎝ ∂T ⎠σ ⎝ ∂T ⎠σ Calcularemos el cambio de entropía mediante: aT 2 2aT 3 ⎛ ∂A ⎞ ⎛ ∂A ⎞ TdS = Cσ dT − T ⎜ dA ⎟ dσ → dS = − ⎜ ⎟ dσ = 2TA 2 dA = A A ⎝ ∂T ⎠σ ⎝ ∂T ⎠σ ∆S = 2 A0 ∫ A0 2aT 3 dA = 2aT 3 ln 2 → ∆S = 2aT 3 ln 2 A El cambio de energía libre de Gibbs es: dG = − SdT − Adσ → dG = − Adσ → dG = A ∆G = 2 A0 ∫ A0 aT 2 aT 2 dA dA = A2 A aT 2 dA → ∆G = aT 2 ln 2 A 100 Julián Moreno Mestre 101