DEFECTOS CARDIACOS CONGÉNITOS PRONÓSTICO Y
Anuncio

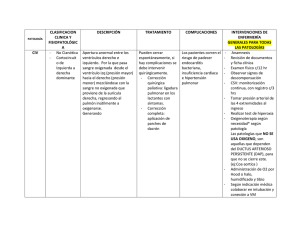
DEFECTOS CARDIACOS CONGÉNITOS PRONÓSTICO Y TRATAMIENTO DE LAS CARDIOPATÍAS CONGÉNITAS MM RODRÍGUEZ VÁZQUEZ DEL REY F. PERIN C. BRIALES CASERO INTRODUCCIÓN Tras el diagnóstico de una cardiopatía congénita en un feto viene la parte no menos importante de informar a los padres. Para transmitir la información, es nece‐ sario un conocimiento profundo y actualizado de la patología diagnosticada, pues éste es uno de los campos de la medicina que más ha cambiado en los últimos años y está continuamente progresando1. Es fundamental: Un diagnóstico lo más exacto posible de la malformación y sus implicaciones. Hay que contemplar todas las evoluciones, la más habitual, pero también la mejor y la peor de las posibles. Como es lógico cada información tendrá detalles específicos que dependerán del diagnóstico y de la seguridad en dicho diagnóstico, pues no siempre pode‐ mos valorar todos los detalles que queremos. Es importante valorar como puede va‐ riar el pronóstico la información que falta y si es importante, repetir el estudio en poco tiempo o consultar con un colega; de la edad gestacional. Hay cardiopatías evolutivas a lo largo de la gestación y estas pueden modificar el pronóstico a peor. de la asociación con otras malformaciones extracardíacas. ‐ El pronóstico debe ser real, incluyendo además en él las posibles secuelas neurológicas que pueden sufrir estos pacientes a lo largo de las cirugías, o por su propia condición2. ‐ Posibles opciones terapéuticas y manejos alternativos si los hay. [1] Actualización Obstetricia y Ginecología 2011 ‐ Actitud obstétrica: salvo en casos excepcionales, el parto deberá ser a térmi‐ no y por vía vaginal. La misión del informador es ayudar a que los padres, una vez informado co‐ rrectamente, hagan la “mejor” decisión para sus circunstancias y su familia. Es imprescindible comunicar a los padres: lo que va a pasar cuando su hijo nazca (síntomas al nacimiento), la necesidad o no de programar el parto en un centro terciario con cuidados intensivos (dependencia ductal y/o de cirugía urgente); tratamiento quirúrgico, morbi‐mortalidad a corto y a largo plazo. Antes de explicar el tratamiento y pronóstico de algunas lesiones cardiacas, vamos a aclarar tres conceptos claves: SINTOMATOLOGÍA DE LAS CARDIOPATÍAS La fecha del comienzo y el cuadro sintomático acompañante dependen de la naturaleza y la gravedad del defecto anatómico y de los efectos del cierre del ductus arterioso y de la caída de las resistencias pulmonares. Las manifestaciones clínicas de las innumerables formas heterogéneas de cardiopatía se resumen fundamentalmente en dos cuadros clínicos: la CIANOSIS (hipoxemia y coloración azulada de la piel y mu‐ cosas) y la INSUFICIENCIA CARDIACA (dificultad para respirar, alimentarse, fallo de medro). DEPENDENCIA DUCTAL En algunas cardiopatías graves, la supervivencia del recién nacido depende de la persistencia en la vida postnatal del ductus arterioso para mantener el flujo a nivel PULMONAR o SISTÉMICO o para lograr suficiente MEZCLA circulatoria. Esto se consi‐ gue mediante la administración intravenosa de prostaglandinas E1 (PGE). Tras estabi‐ lizar al paciente y con la finalidad de poder suspender la infusión de PGE, todos estos niños necesitarán un procedimiento a corto plazo, bien una fístula sistémico‐ pulmonar para sustituir el ductus, implantación de stent ductal mediante cateterismo para mantenerlo abierto a largo plazo o cirugía correctora definitiva. [2] Medicina Materno‐Fetal CORRECCIÓN COMPLETA VERSUS PALIACIÓN (VÍA UNIVENTRICU‐ LAR) Existen lesiones que pueden ser corregidas totalmente y otras que sólo pue‐ den ser paliadas. En este último grupo se encuentran formas heterogéneas de car‐ diopatías para las cuales, bien por la existencia de un solo ventrículo desarrollado o por dificultades técnicas que imposibilitan le reparación, la única vía quirúrgica posi‐ ble es la UNIVENTRICULAR. La vía univentricular se basa en el principio de Fontan según el cual el retorno venoso sistémico, gracias a las presiones más bajas en el cir‐ cuito pulmonar, se puede hacer circular a través de los pulmones sin la ayuda de una bomba ventricular. Mediante cirugía se separan las circulaciones sistémicas y pulmo‐ nares, y se realiza una conexión directa del retorno venoso sistémico a la circulación pulmonar. Se suele conseguir en dos estadíos: conexión de la vena cava superior a la arteria pulmonar alrededor de los 4‐6 meses de vida (Glenn), para completar poste‐ riormente con conexión de la vena cava inferior al circuito pulmonar alrededor de los 2‐4 años (Fontan). La calidad de vida de pacientes con paliación univentricular es aceptablemente buena, aunque a largo plazo (3 o 4 décadas) pueden surgir complica‐ ciones, es frecuente que precisen reintervenciones, y finalmente acaban precisando trasplante cardiaco. TRATAMIENTO ESPECÍFICO Y PRONÓSTICO DE ALGUNAS CARDIO‐ PATÍAS Siguiendo el esquema del capítulo anterior, clasificaremos las cardiopatías según el riesgo y la morbi/mortalidad que conllevan. Riesgo bajo: lesiones simples, sin necesidad de programar el parto en un cen‐ tro terciario, con excelente resultado quirúrgico, totalmente curables. C OMUNICACIÓN INTERVENTRICULAR : Las CIV pequeñas‐moderadas suelen cerrarse espontáneamente y cursar de forma asintomática. La clínica de las CIV grandes es de insuficiencia cardiaca progre‐ siva conforme van bajando las resistencias pulmonares y aumenta el cortocircuito izquierda‐derecha por lo que no darán síntomas en las primeras semanas de vida y por lo tanto no precisan nacer en un hospital terciario. El tratamiento quirúrgico por lo común se planifica y rara vez hay que realizarlo de manera urgente. La mortalidad es extremadamente baja (<1%). [3] Actualización Obstetricia y Ginecología 2011 E STENOSIS VALVULAR PULMONAR LEVE - MODERADA La estenosis valvular pulmonar con flujo pulmonar no dependiente del ductus es una lesión que no suele dar síntomas hasta que progresa al grado de severa (y a menudo ni siendo severa da síntomas). Presenta muy buenos resultados a largo plazo y el tratamiento de elección es la VALVULOPLASTIA pulmonar, mediante cateterismo cardiaco. Riesgo moderado: lesiones con buenos resultados quirúrgicos pero con posi‐ ble morbilidad a largo plazo y necesidad de más intervenciones futuras: C ANAL AURÍCULO - VENTRICULAR COMPLETO Un recién nacido con canal AV completo estará asintomático, empezando con un cuadro de insuficiencia cardiaca progresiva alrededor del mes/dos meses de vida dependiendo del momento en que bajen las resistencias pulmonares. No es una car‐ diopatía ductus‐dependiente. La intervención quirúrgica (septación y creación de dos válvulas separadas) se suele llevar a cabo alrededor entre los 3 y los 9 meses de vida. La mortalidad atribuible a la reparación quirúrgica en la mayoría de las series es infe‐ rior a un 5%. Los resultados a medio‐largo plazo son buenos, dependiendo en gran medida del grado de competencia que se haya podido conseguir en el funcionamien‐ to de las válvulas AV. Aproximadamente entre un 5 y un 10% de los pacientes requie‐ ren a largo plazo revisión quirúrgica o reemplazo valvular por disfunción de la válvula AV izquierda (sobre todo los niños sin cromosomopatía). T ETRALOGÍA DE F ALLOT (TOF) La sintomatología al nacimiento dependerá del grado de estenosis pulmonar y subpulmonar (CIANOSIS). Es excepcional que sea necesaria la administración de PGE1 para mantener permeable el ductus arterioso y en general los pacientes con este dia‐ gnóstico pueden ser dados de alta pronto sin necesidad de algún procedimiento en la época neonatal. Los niños afectos de TOF pueden presentar cianosis (progresivamen‐ te más severa conforme va aumentando el grado de obstrucción al flujo pulmonar) pero por lo demás suelen estar asintomáticos y tienden a crecer de manera normal hasta el momento de la cirugía. La fecha recomendada para la reparación (corrección completa: cierre de la CIV y ampliación del tracto salida ventrículo derecho) es alre‐ dedor de los 6 meses de vida. La mortalidad de la cirugía en manos expertas es me‐ nor del 5%. Es frecuente la necesidad de reintervenciones a lo largo de 20 o 30 años de seguimiento (sobre todo por la existencia de una estenosis y/o regurgitación resi‐ dual de la válvula pulmonar). [4] Medicina Materno‐Fetal T RANSPOSICIÓN COMPLETA DE GRANDES VASOS ( D -TGA) Al nacimiento la forma más usual de presentación es la cianosis que no res‐ ponde a administración de oxígeno. Va a precisar en la mayor parte de los casos tra‐ tamiento con PGE1 para mantener el ductus arterioso permeable y en algunas oca‐ siones realizar una atrioseptostomía con balón (Rashkind) en la que se agranda la comunicación interauricular para mejorar la mezcla de sangre intracardiaca con un catéter balón (se puede hacer en UCIN guiado por ECO). La cirugía (switch arterial o técnica de Jatene) se realiza en el primer mes de vida. Los dos grandes vasos son conectados con sus respectivos ventrículos y las arte‐ rias coronarias son anastomosadas a la nueva raíz aórtica. En un mismo tiempo se realiza cierre de la CIA y de la CIV (si está presente, 40‐50%) Con tratamiento médico‐quirúrgico se obtienen unos resultados de supervi‐ vencia del 88‐98% de los pacientes3,4 . D RENAJE VENOSO PULMONAR ANOMALO TOTAL (DVPAT): La presentación clínica depende de la existencia o no de obstrucción del drena‐ je venoso. En casos de que exista, el paciente puede deteriorarse rápidamente con aparición de cianosis severa e insuficiencia cardiaca precisando una intervención quirúrgica MUY urgente. El DVPAT obstructivo es casi la única cardiopatía que repre‐ senta una verdadera emergencia quirúrgica. En los casos no obstructivos la clínica consiste en un grado variable de cianosis e insuficiencia cardiaca. La cirugía en estos casos debe realizarse en los primeros meses de vida, conectando el colector a la aurí‐ cula izquierda. Actualmente la mortalidad quirúrgica varía entre el 2 y el 20%. E STENOSIS VALVULAR PULMONAR CRÍTICA : El cuadro clínico está dominado por la cianosis. Existe una estenosis tan severa de la válvula pulmonar que es necesario el ductus para mantener el flujo sanguíneo pulmonar y por lo tanto perfusión de prostaglandinas al nacimiento. En la ecocardio‐ grafía fetal se habrá detectado flujo reverso en arco ductal (de aorta a pulmonar). Dichos pacientes necesitan nacer en un centro con UCIN para administración de PGE1 y realización de valvuloplastia pulmonar por cateterismo. El resultado suele ser bue‐ no salvo la necesidad, en algunos casos, de reemplazar la valvula pulmonar por re‐ gurgitación severa. A TRESIA PULMONAR CON SEPTO INTEGRO (APSI) Según el desarrollo del ventrículo derecho, la válvula tricúspide y la presencia de anomalías coronarias, las opciones terapeúticas serán totalmente distintas: perfo‐ [5] Actualización Obstetricia y Ginecología 2011 ración de la válvula mediante cateterismo o cirugía para obtener una circulación bi‐ ventricular o bien elección de la vía univentricular. La supervivencia global a 5 años se sitúa en torno al 60‐80%. A TRESIA TRICÚSPIDE Cuadro clínico dominado por la cianosis. Según la presencia de una CIV necesi‐ tará o no perfusión de PGE1. La única solución terapeútica es la vía univentricular. La mortalidad suele ser baja y los resultados mejores que en otras formas de cardiopat‐ ías monoventriculares. E STENOSIS AÓRTICA CRÍTICA En este caso el recién nacido va a presentar una insuficiencia cardiaca severa en los primeros días de vida. Es necesaria la infusión de PGE1 para mantener el duc‐ tus permeable. El tratamiento de elección, en el caso de tener un ventrículo izquierdo bien desarrollado, es la valvuloplastia por cateterismo, incluso en la vida fetal. La supervivencia global es de más del 75% en 8 años. Sobre un tercio de estos pacientes requerirán una reintervención por reestenosis5 o intervención quirúrgica por regurgitación aórtica (que de ser necesaria en época del lactante conlleva morbi‐ mortalidad elevadas). En la vida adulta todo son candidatos, en el mejor de los casos, a reemplazo valvular aórtico. C OARTACIÓN DE AORTA (C O A O ) La clínica, en los casos de coartación de aorta severa, consiste en insuficiencia cardiaca que aparece en las primeras semanas de vida, al cerrarse el ductus. En estos casos los pacientes precisan tratamiento con PGE1. Los resultados actuales de la ci‐ rugía son buenos, especialmente en casos de coartación aórtica aislada, con una mor‐ talidad quirúrgica <2% y una supervivencia >95% al año y >90% a los 5 y 10 años. El pronóstico es peor si asocia hipoplasia del arco aórtico, que es el tipo de coartación que suele diagnosticarse en la vida fetal. El cateterismo cardiaco con angioplastia (con o sin implatanción de stent) es el tratamiento de elección en el caso de recoar‐ tación. V ENTRÍCULO DERECHO DE DOBLE SALIDA (VDDS) La clínica varía enormemente según la situación de la CIV y la presencia o no de estenosis pulmonar. Las distintas cirugías correctoras pretender alcanzar la fisio‐ logía normal del corazón. Dentro del grupo de pacientes con VDDS y CIV no relacio‐ nada puede ser necesario realizar una corrección univentricular (Fontan). [6] Medicina Materno‐Fetal V ENTRÍCULO ÚNICO (VU) La clínica es también variable según el tipo, pudiendo predominar la cianosis si hay estenosis pulmonar asociada o la insuficiencia cardiaca si no está presente. Cuando la estenosis pulmonar es muy severa puede ser necesario tratar con PGE1 intravenosas. En todos los casos la paliación va a ser univentricular. Riesgo elevado: lesiones con alta mortalidad quirúrgica y elevada morbilidad a largo plazo S ÍNDROME DEL VENTRÍCULO IZQUIERDO HIPOPLÁSICO (SVIH) Es una cardiopatía de mal pronóstico. El paciente va a desarrollar en los prime‐ ros días de vida clínica de insuficiencia cardiaca y shock cardiogénico. Se debe instau‐ rar infusión de PGE. Para esta cardiopatía la única opción viable es la univentricular, precisando 3 intervenciones. En la etapa neonatal se realiza la cirugía de Norwood (reconstrucción de la aorta utilizando el tronco pulmonar y restablecimiento del flujo pulmonar con una fístula sistémico pulmonar o una conducto desde el ventrículo de‐ recho). Posteriormente cirugía de Glenn (2º estadio) y luego tercer estadío, cirugía de Fontan. Hace 15 años la mortalidad de este tipo de cardiopatía era del 100%; hoy día sigue siendo una de las cardiopatías de más alto riesgo. La mortalidad varía según factores de riesgo propios de la cardiopatía, como foramen oval permeable restricti‐ vo, y según los centros (oscilando la mortalidad publicada entre el 58%6 y el 12% tras el primer estadío). La mortalidad de las siguientes cirugías suele ser más baja. T RUNCUS ARTERIOSO : Las manifestaciones clínicas van a aparecer en las primeras semanas de vida y consisten en un cuadro de insuficiencia cardiaca con mayor o menor grado de ciano‐ sis. No es una cardiopatía dependiente del ductus pero, por su gravedad, es conve‐ niente el nacimiento en un centro terciario. La cirugía debe realizarse en los primeros 3 meses de vida. La intervención consiste en cierre de la comunicación interventricu‐ lar de manera que el vaso truncal se relacione solamente al ventrículo izquierdo y conexión del ventrículo derecho con las ramas pulmonares a través de un conducto protésico. Posteriormente precisará al menos uno o dos recambios quirúrgicos del conducto. La mortalidad hospitalaria para el truncus oscila entre el 5 y el 10% en cen‐ tros especializados en cirugía neonatal, y la mortalidad para las formas complejas que asocian más malformaciones cardiacas, se sitúa en el 20%, con una tasa de supervi‐ vencia estimada a los 10 años de aproximadamente el 65‐70%.7 [7] Actualización Obstetricia y Ginecología 2011 CONCLUSIÓN Los padres quieren tener un hijo sano y con una larga supervivencia. No les sir‐ ve que sobreviva 5 años ni siquiera 30 años. Sin embargo no estamos totalmente en situación de ofrecerles información acerca de los pacientes a largo plazo. Las técnicas quirúrgicas para el tratamiento de las cardiopatías congénitas se han desarrollado con éxito en los últimos años y los datos a largo plazo que tenemos son de pacientes sometidos a cirugía hace 40 años, con técnicas y circunstancias distintas, por lo que no podemos extrapolar esos datos a los que tratamos hoy día. Es probable que la ca‐ da vez más frecuente detección prenatal de las cardiopatías congénitas8, las mejorías en el tratamiento y el refinamiento de técnicas quirúrgicas culminen en mejores re‐ sultados a corto y largo plazo. Anatomía Síntomas Dep. ductal Intervención inicial Mortalidad quirúrgica (%) CIV Riesgo ICC No Reparación <1% Bajo EPV no crítica Asintomático No Valvuloplastia <1% Bajo Canal AV ICC No Corrección Qx 5% Moderado TOF Cianosis Rara Corrección 5% Moderado EPV critica Cianosis Si Valvuloplastia 1‐5 Moderado APSI con buen VD Cianosis Si Perforación valvu‐ la 1‐3 Moderado APSI Cianosis Si Fístula Ao‐P + UniV 20‐40% E.V.Ao crítica ICC Si Valvuloplastia 3‐5% Moderado CoAo ICC Si Cirugía <1% Bajo‐ Mod SVIH ICC Si Norwood+UniV 30‐58 Elevado D‐TGA Cianosis Si Cirugía 2‐10% Moderado TAPVR Cianosis/ICC No Cirugía 2‐20% Moderado Truncus Cianosis o ICC No Cirugía 5‐10% Elevado o Cianosis [8] Elevado Medicina Materno‐Fetal BIBLIOGRAFIA Allan L.D, Cook A.C, Huggon I.C. Fetal Echocardiography. A Practical Guide. Cambridge, Uni‐ ted Kingdom: Cambridge University Press;2009 Hagan E, Feldman H, Neurodevelopmental Outcomes in Patients with Hypoplastic Left Heart Syndrome. The Internet Journal of Allied Health Sciences and Practice. April 2010. Volume 8 Number 2. García‐Hernández JA, Montero‐Valladares C, Martínez‐Lopez AI, Gil‐Fournier M. Praena‐ Fernández JM, Cano‐Franco J, Loscertales‐Abril M. Valoración pronostica del switch arterial en la transposición de grandes arterias, Anales de ped, in press, available online 28 dic 2010. Freed DH, Robertson CM, Sauve RS, Joffe AR, Rebeyka IM, Ross DB, Dyck JD; Western Cana‐ dian Complex Pediatric Therapies Project Follow‐up Group. Intermediate‐term outcomes of the arte‐ rial switch operation for transposition of great arteries in neonates: alive but well? J Thorac Cardio‐ vasc Surg 2006; 132: 845‐52. Keane JF, Fyler DC. Aortic Outflow Anormalities. En: Keane JF, Lock JE, Fyler DC (eds.). Nadas’ Pediatric Cardiology. 2nd ed. Philadelphia: Elsevier; 2006. p. 581‐601. Caffarena Calvar JM. Truncus. Capítulo 12 de los protocolos de la Sociedad española de Car‐ diología Pediátrica y Cardiopatías Congénitas. www.secardioped.es García‐Hernández JA, González‐Rodríguez JD, Martínez‐López A, Canalejo‐González D, Rome‐ ro‐Parreño A, Santos de Soto J, Loscertales‐Abril M, Cayuela‐Domínguez A,Gil Fournier‐Carazo M. Resultados de la intervención de Norwood para el síndrome del corazón izquierdo hipoplásico, Rev Esp Cardiol. 2007; 60:732‐8. Tworetzky W, McElhinney MB, Reddy VM, Brook MM, Hanley FL, Silverman NH, Improved surgical outcome after fetal diagnosis of hypoplastic left heart syndrome Circulation. 2001 Mar 6;103 (9):1269‐73 [9]