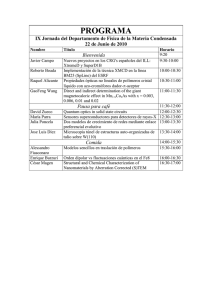
1 TEMA 1 - INTRODUCCION A LA QUIMICA DE POLIMEROS
Anuncio

TEMA 1 - INTRODUCCION A LA QUIMICA DE POLIMEROS INDICE. 1.- Introducción. Generalidades. 2.- Historia. 3.- Usos e importancia de los materiales plásticos. 4.- Mercado actual. Producción y consumo de plásticos. 5.- Definición y estructura de los polímeros. 5.1.- Introducción 5.2.- Estructura química. 5.3.- Fuerzas de enlace interatómicas. 5.4.- Forma molecular. Longitud y esbeltez molecular de los polímeros 6.- Estructura molecular de las cadenas. 7.- Configuraciones moleculares de las cadenas. 8.- Copolímeros. Elastómeros termoplásticos. 9.- Peso molecular y grado de polimerización medio. 10.- Métodos para la determinación del peso molecular medio. 11.- Cristalización. Polímeros cristalinos. 11.1.- Introducción. 1 1 . 2 . - Grado de cristalinidad. Métodos para su determinación. 11.2.1.- Introducción. 11.2.2.- Métodos de difracción de rayos X 11.2.3.- Métodos volumétricos. 11.2.4.- Métodos calorimétricos 11.3.- Cristales poliméricos. 11.3.1.- Introducción. 11.3.2.- Modelo de micela con flecos. 11.3.3.- Modelo de cadenas plegadas. 11.4.- Factores que favorecen el estado cristalino. 11.5.- Nucleación y crecimiento de los cristales. 11.6.- Cristalización por estirado. 11.7.- Celda unidad de los polímeros cristalinos. 11.8.- Comportamiento mecánico de los polímeros cristalinos. 12.- Clasificación de los polímeros industriales. 13.- Relaciones entre la estructura y las propiedades de los polímeros. Propiedades generales de los Polímeros 1 1.- Introducción. Generalidades. A lo largo de cientos de años se han utilizado polímeros naturales procedentes de plantas y animales. Estos materiales incluyen madera, caucho, lana, cuero y seda. Otros polímeros naturales tales como las proteínas, los enzimas, los almidones y la celulosa tienen importancia en los procesos bioquímicos y fisiológicos de plantas y animales. Desde principios del siglo XX, la moderna investigación científica ha determinado la estructura molecular de este grupo de materiales y ha desarrollado numerosos polímeros, sintetizados a partir de pequeñas moléculas orgánicas. Muchos plásticos, cauchos y materiales fibrosos son polímeros sintéticos. Desde el fin de la Segunda Guerra Mundial, el campo de los materiales se ha visto revolucionado por la llegada de polímeros sintéticos. Las síntesis suelen ser baratas y las propiedades conseguidas comparables, y a veces superiores, a las de los análogos naturales. En algunas aplicaciones, los metales y la madera se sustituyen por polímeros, que tienen propiedades idóneas y se pueden fabricar a bajo coste. Los materiales plásticos actualmente en nuestra sociedad son, sin lugar a dudas, uno de los materiales más utilizados en casi todos los sectores industriales como consecuencia de las buenas propiedades que poseen. Si se comparan polímeros como el polietileno y el nylon con materiales que son utilizados más comúnmente por los ingenieros, de inmediato se encontrarán algunas diferencias importantes. Unas pruebas simples muestran que los polímeros: 1.- Tienen resistencia mecánica y rigidez (Módulo elástico) bajas, 7-100 MPa y 1-4 GPa respectivamente. Sin embargo, tienen una buena relación resistencia peso. 2.- Su uso, frecuentemente, está limitado por condiciones de temperatura (< 150 °C). 3.- Los ensayos mecánicos, por ejemplo, los de tracción, muestran que se deforman cuando se someten por un tiempo a una carga, o sea, sus propiedades dependen del tiempo, y esta es su característica mecánica más significativa. Las propiedades mecánicas también dependen marcadamente de la temperatura. 4.- Expansión térmica elevada (30 - 200xl0-6 K-1) 5.- Problemas de disposición y reciclado. Las características anteriores representan desventajas en comparación con los metales, la madera, los materiales cerámicos, etc. Obviamente, los polímeros también deben de poseer ventajas, ya que son muy utilizados e, incluso, substituyen a los materiales comunes en muchas áreas importantes. ¿Cuáles son las ventajas que tienen estos materiales que provocaron una expansión tan rápida en su uso?. La mayoría de éstas es: 1.- Versatilidad. 2.- Los materiales poliméricos, tanto los plásticos como los cauchos, son fácilmente conformados a bajas temperaturas y bajas presiones, lo que hace que los procesos industriales de fabricación de objetos por moldeo con plásticos se abaraten sensiblemente respecto a aquéllos en los que se emplean otros materiales. Permite la obtención de formas complejas (Posibilidad de diseño de geometrías intrincadas) con un mínimo de operaciones de fabricación y acabado (facilidad de fabricación, baja energía para moldearlos y conformarlos, es decir menor energía de procesamiento). 3.- Fácil maquinabilidad, es decir, se trabaja fácilmente con herramientas que produzcan arranque de material como fresadoras, tornos y limas. 4.- Su baja densidad da como resultado productos ligeros (800 – 1500 kg/m3), lo que los hace idóneos para piezas y componentes para la industria del transporte como en aviones, barcos, automóviles o trenes. 5.- Alto grado de inalterabilidad ante agentes químicos y elevado grado de resistencia a la corrosión, lo que los hace muy adecuados para revestimientos en industrias químicas, conducciones de fluidos, objetos a la intemperie o en ambientes corrosivos. 6.- Su conductividad térmica y eléctrica es muy baja por lo que se emplean como aislantes en la mayoría de los componentes o materiales eléctricos y como aislantes térmicos en cámaras frigoríficas o termostáticas, en los muros de las casas, etc. En algunos casos, pueden ser conductores eléctricos 2 7.- Durables y tenaces 8.- La flexibilidad natural de los polímeros los hace útiles. Esto es especialmente cierto para los cauchos. 9.- Aunque los valores de la resistencia mecánica absoluta y del módulo de elasticidad de los polímeros son bajos, los valores específicos por unidad de peso o volumen, son con frecuencia favorables. De aquí el uso de materiales poliméricos que se utilizan especialmente en proyectos aeroespaciales. 10.- Los cauchos, los cuales se usan en resortes y montajes absorbentes de impacto por sus cualidades de elasticidad y amortiguamiento. 11.- Posibilidad de transparencia u opacidad. Acabado superficial óptimo y buena posibilidad de coloración (metalizado), lo que origina, para cualquier aplicación, un acabado muy estético. 12.- Bajo coste y bajo precio. Los polímeros conductores eléctricos surgieron a raíz de una afortunada equivocación en el año 1977 de un estudiante dirigido por el doctor Hideki Shirakawa (Instituto Tecnológico de Tokio). Debido a tal error, se obtuvo por primera vez un polímero, el poliacetileno, con una conductividad eléctrica mil millones de veces mayor que la esperada. El motivo para tan extraordinario resultado fue que dicho estudiante agregó mil veces más catalizador (yodo) que el requerido en las instrucciones, lo cual generó cambios sorprendentes en la estructura del polímero. Por este descubrimiento, Shirakawa, MacDiarmid y Heeger ganaron el Premio Nobel de química en el año 2000. Desde entonces se ha podido emplear el dopaje en diversos polímeros, como las polianilinas, polipirroles y politiofenos, logrando nuevamente un aumento considerable de la conductividad eléctrica (Tabla 1.1). Tabla 1.1.- Conductividades eléctricas de algunos polímeros conductores dopados con diferentes moléculas. Polímero Material dopante Poliacetileno I2, Br2, Li, Na, AsF5 Polipirrol BF–4 , ClO–4 , tosilato Politiofeno BF–4 , ClO–4 , tosilato, FeCl–4 Poli(3-alquiltiofeno) BF–4 , ClO–4 , FeCl–4 Polifenilen sulfuro AsF5 Polifenilenvinileno AsF5 Politienilenvinileno AsF5 Polifenilen AsF5, Li, K Poliisotianafteno BF–4 , ClO–4 Polifurano BF–4 , ClO–4 Polianilina HCl Polietileno Aluminio Cobre Acero inoxidable -1 (*)1 Siemens/cm = 1 (µΩ.cm) Conductividad aproximada (S/cm) (Siemens/cm) (*) 10000 500-7500 1000 1000-10000 500 10000 2700 1000 50 100 200 377000 596000 20000 Una de las aplicaciones más prometedoras de los PC son las baterías recargables totalmente poliméricas. En ellas los electrodos serán de PC, siendo más ligeras, flexibles y con una gran capacidad de aceptar y donar carga eléctrica reversiblemente. Además, tendrán una larga vida, utilizarán potenciales de alrededor de los 3V y producirán una densidad de energía varias veces mayor que las baterías actualmente disponibles, de níquel/cadmio y plomo/ácido. En la tabla 1.2 pueden verse comparadas algunas propiedades de los plásticos y del acero. 3 TABLA 1.2.- Comparación de algunas propiedades de los plásticos y del acero. PROPIEDAD PLASTICOS ACERO DENSIDAD 800/1000 kg/m 3 7900 kg/m 3 MODULO ELASTICO 1 - 4 GPa 210 GPa RESISTENCIA 7 - 100 MPa 400 - 1000 MPa VENTAJA DESVENTAJA TEMPERATURA DE DISTORSION 50-150°C TEMPERATURA FUSION <350 °C 1700 °C EXPANSIÓN TÉRMICA 30-200xl0-6K-1 11xl0-6K-1 CONDUCTIVIDAD TERMICA 0.1 - 0.4 W/m.K 63 W/m.K VENTAJAS DESVENTAJAS RESISTENCIA A LA CORROSION BAJA RIGIDEZ AISLAMIENTO ELECTRICO (REVESTIMIENTO DE HILOS CONDUCTORES) RUIDO DE VIBRACION REDUCIDO BAJA RESISTENCIA BAJA CONDUCTIVIDAD TERMICA BAJA TEMPERATURA DE DISTORSION DURABLES Y TENACES VARIACION DE LAS PROPIEDADES MECANICAS CON EL TIEMPO PROBLEMAS DE DISPOSICIÓN Y RECICLADO BAJA DENSIDAD EXPANSION TERMICA ELEVADA BAJA ENERGIA PARA SU CONFORMADO OBTENCIÓN DE FORMAS COMPLEJAS EN UNA SOLA PIEZA (MOLDEO POR INYECCION) FACILIDAD PARA DARLES COLORACION 4 En las figuras 1.1 a 1.5 puede verse una comparación de los valores de diferentes propiedades para los cuatro tipos de materiales, entres los cuales están los poliméricos. Figura 1.1.- Densidad de los distintos tipos de materiales. Figura 1.2.- Módulo elástico de los distintos tipos de materiales 5 Figura 1.3.- Tensión de fluencia de los distintos tipos de materiales Figura 1.4.- Tenacidad de los distintos tipos de materiales 6 Figura 1.5.- Temperatura de fusión de los distintos tipos de materiales En la tabla 1.3 puede verse la energía requerida para la fabricación de diversos materiales. 3 Tabla 1.3.- Energía requerida para la fabricación de diversos materiales (kWh/m ). y en la figura 1.6 la energía requerida para la fabricación de diversos productos con distintos materiales, en toneladas de petróleo. 7 Figura 1.6.- Energía requerida para la fabricación de diversos productos con distintos materiales. En la figura 1.7 puede verse el precio del material base y del producto acabado para diversos tipos de materiales. Figura 1.7.- Precio del material base y del producto acabado para diversos tipos de materiales. En la figura 1.8 se da la conductividad térmica de una serie de materiales y en la figura 1.9 el coeficiente de dilatación térmica de metales y plásticos 8 Figura 1.8.- Conductividad térmica de una serie de materiales. Figura 1.9.- Coeficiente de dilatación térmica de metales y plástico. Al principio las propiedades especiales de los plásticos y los cauchos eran mal interpretadas y muchos de los fallos existentes eran porque los ingenieros de diseño no conocían la importancia (y muchas veces, ni siquiera sabían de la existencia) de la variación de estas propiedades con el tiempo. En la actualidad, dichas propiedades están bien determinadas y se aplican en el diseño de componentes para tener un buen comportamiento y una larga vida de servicio. Además, en los años recientes se han obtenido muchos polímeros nuevos de alta eficiencia, los cuales superan a las deficiencias de los primeros. Estos son los polímeros de ingeniería, los cuales tienen propiedades físicas superiores y mejor tolerancia a la temperatura. Así, la fácil moldeabilidad y sus cualidades aislantes y de resistencia a la corrosión hacen a los polímeros sumamente utilizables. Sin embargo poco se sabe acerca de ellos: de su naturaleza, de sus métodos de fabricación, de sus propiedades, de sus infinitas aplicaciones, muchas de ellas todavía por encontrar, así como de su incidencia medioambiental. 9 De esta última tan sólo se conoce «la gran cantidad de plásticos que hay como residuos», y se presenta la problemática de QUÉ HACER CON ELLOS. Al ser los plásticos productos de síntesis de laboratorio, la Naturaleza en la mayoría de los casos es incapaz de hacerlos desaparecer, por lo que comparativamente con otros materiales su presencia en los vertederos es mayor, es decir permanecen durante más tiempo. Así, los plásticos son unos materiales que ocupan un lugar indiscutible en la sociedad actual. Su presencia pasa casi desapercibida entre nosotros al ser unos materiales de uso diario en los que apenas recapacitamos acerca de su naturaleza. Basta hacer una revisión de todos los artículos que utilizamos en nuestra vida cotidiana para darnos cuenta de las múltiples aplicaciones que tienen estos materiales. La implantación creciente de los plásticos en distintos sectores industriales, llegando incluso a sustituir a otros materiales tradicionales, se debe fundamentalmente a las interesantes propiedades que presentan. Destacan entre otras la baja densidad, la gran resistencia a la corrosión, la gran capacidad aislante, el bajo precio y sobre todo la característica que les hace ser más apreciados es la capacidad que presentan de ser «fabricados a medida» tanto con propiedades determinadas como en formas geométricas definidas por muy complicadas que éstas sean, con un bajo coste económico, mínimo esfuerzo de fabricación y la posibilidad de fabricar grandes series. La palabra plástico tiene dos acepciones diferentes según sea utilizada como adjetivo o como nombre. Cuando se utiliza como adjetivo se refiere a la propiedad que presentan algunos materiales de ser deformados cuando una fuerza externa actúa sobre ellos, manteniendo dicha deformación aun después de cesar la fuerza. El ejemplo más representativo de este fenómeno se encuentra en un bloque de plastilina o una plancha de metal. Cuando plástico se utiliza como nombre, el plástico o los plásticos, se refiere a un material con una determinada naturaleza química que le confiere unas propiedades y características específicas. El nombre científico correcto de estos compuestos es el de polímeros o macromoléculas y cuando se les adicionan ciertas sustancias (aditivos, cargas, etc.) que modifican sus propiedades y pueden ser comercializados, se denominan plásticos. La palabra polímero se deriva del griego poli y meros, que significan mucho y partes respectivamente. Algunos científicos prefieren usar el término macromolécula, o molécula grande, en lugar de polímero. Otros sostienen que los polímeros naturales, o biopolímeros, y los polímeros sintéticos deberían estudiarse en cursos distintos. Sin embargo, los mismos principios son de aplicación a todos los polímeros. Si se descartan los usos finales, las diferencias entre los polímeros, incluyendo los plásticos, las fibras, y los elastómeros o cauchos, vienen determinadas principalmente por las fuerzas intermoleculares (entre moléculas) e intramoleculares (dentro de cada molécula individual) y por los grupos funcionales presentes. El plástico es un producto no natural que se obtiene en la industria a través de reacciones químicas. Es por tanto un producto de síntesis de laboratorio o un producto sintético. Las propiedades finales del material son muy diferentes según sea la naturaleza del producto de partida y el procedimiento seguido en su obtención. Por ello debe hablarse de «plásticos» y no del plástico, precisamente por la diversidad existente de los mismos. En la Naturaleza existen compuestos de naturaleza «polímero» utilizadas desde hace siglos por el hombre. Provienen tanto del reino vegetal como del animal: madera, caucho, algodón, lana, cuero y seda son buenos ejemplos de ellos. Otros polímeros naturales como las proteínas, enzimas, almidón y celulosa desempeñan procesos biológicos y fisiológicos importantes en los procesos vitales. Los polímeros desarrollados por el hombre en el laboratorio se denominan sintéticos, mientras que los que se encuentran en la Naturaleza son polímeros naturales o biopolímeros. En la figura 1.10 puede verse la estructura de la industria de los plásticos y en la figura 1.11 como se obtienen los polímeros. 10 Figura 1.10.- Estructura de la industria de los plásticos Figura 1.11. Proceso de obtención de los polímeros. 11 2.- Historia. Durante los últimos cincuenta años la industria de las materias plásticas ha tenido un desarrollo de proporciones gigantes, superando al de la industria del acero. Después del 1945 poliestireno, polietileno, cloruro de polivinilo, poliamidas, polimetilmetacrilato y sucesivamente polipropileno han entrado en las casas de todos nosotros, independientemente de la condición social, en las ciudades más remotas como en las grandes ciudades, en los países industrializados como en las economías agrícolas. Ha sido un fenómeno - que no se había verificado nunca en la historia del ser humano en proporciones tan grandes y con una dinámica tan rápida - de sustitución progresiva de los materiales tradicionales con las nuevas substancias sintéticas y de reproyección formal de las estructuras y de las formas ergonómicas de las herramientas y de los objetos de los cuales el ser humano se circunda e utiliza. La misma bandeja, fabricada con el mismo material, en los mismos colores y con las mismas formas la podemos encontrar tanto en Manila como en Boston, en Moscú como en París o en Lagos. Los primeros plásticos comerciales —«de síntesis»—, se obtuvieron por modificación de productos naturales mediante reacciones químicas. Así desde la aparición del celuloide —obtenido por reacción del ácido nítrico sobre la celulosa—, o la galatita obtenida por tratamiento de la caseína de la leche con formaldehído, hasta los plásticos utilizados actualmente en ingeniería con propiedades muy especiales, se ha recorrido un gran camino en un tiempo relativamente corto, pues los plásticos como tales empezaron a desarrollarse a partir del año 1930. Al modificar químicamente los productos naturales se observó que mejoraban en muchos aspectos sus propiedades. Esto facilitó su introducción en diversos sectores industriales en los que hasta entonces no habían tenido aplicación. Este hecho originó una demanda creciente de este tipo de materiales que condujo a realizar diferentes procesos sintéticos para obtener materiales de características similares a estos productos naturales modificados. Además de ser la base de la vida en sí, las proteínas se usan como fuente de aminoácidos y de energía. Antiguamente se degradaban o despolimerizaban las proteínas de las carnes duras envejeciéndolas o cocinándoles, y también la albúmina de huevo se desnaturalizaba calentando o añadiendo vinagre. Los primeros seres humanos aprendieron a procesar, teñir y tejer fibras proteínicas naturales, como la lana y la seda, y fibras de hidratos de carbono, como el lino y el algodón. Las primeras civilizaciones sudamericanas como la Azteca utilizaban caucho (Hevea brasiliensis) para fabricar artículos elásticos o impermeabilizar tejidos. Siempre hubo abundancia de fibras naturales y elastómeros pero no de materiales plásticos. Los hombres primitivos emplearon técnicas rudimentarias de plásticos para curtir las proteínas de las pieles animales para hacer cuero y para modelar caparazones de tortuga mediante calor. También se utilizaron alquitranes de origen natural como materiales de calafateado así como lacas extraídas a partir de excrementos de un pequeño insecto denominado cochinilla (Coccus lacca). Hasta que Wóhler sintetizó urea partiendo de compuestos inorgánicos en 1828, progresó poco la química orgánica desde que los alquimistas se concentrasen en la transmutación de metales básicos en oro y creyeran en la teoría de la fuerza vital. A pesar de tan importante descubrimiento, se hicieron pocos avances en el campo de la química orgánica hasta la década de 1850, cuando Kekulé desarrolló las técnicas de representación de fórmulas estructurales aceptadas hoy en día. Sin embargo, los científicos dedicados a los polímeros manifestaron un talento especial para realizar descubrimientos empíricos antes de que esta ciencia se desarrollase. Así, mucho antes de que Kekulé desarrollara su técnica para escribir las fórmulas, Charles y Nelson Goodyear transformaron el caucho de la hevea, un material termoplástico pegajoso, en un elastómero de utilidad (caucho vulcanizado) o un plástico termoestable (ebonita) calentándolo con cantidades pequeñas o grandes de azufre, respectivamente. 12 De la misma manera, Schónbein combinaba celulosa con ácido nítrico y Menard, en 1846, fabricaba colodión disolviendo nitrato de celulosa producto de la reacción anterior en una mezcla de etanol y éter etílico. El colodión, que se utilizó como líquido para engomar el tafetán, y también fue utilizado en la década de l860 como reactivo por Parks y Hyatt para obtener celuloide, fue el primer termoplástico artificial y el reactivo usado por Chardonnet para fabricar la seda artificial. El desarrollo de estas sustancias se inició en 1860, cuando el fabricante estadounidense de bolas de billar Phelan and Collander ofreció una recompensa de 10000 dólares a quien consiguiera un sustituto aceptable del marfil natural (en esa época se utilizaban tanto marfil, que se sacrificaba 12000 elefantes anualmente para cubrir la demanda). Una de las personas que optaron al premio fue el inventor estadounidense Wesley Hyatt, quien desarrolló un método de procesamiento a presión de la piroxilina, un nitrato de celulosa de baja nitración tratado previamente con alcanfor y una cantidad mínima de disolvente de alcohol. Si bien Hyatt no ganó el premio, su producto, patentado con el nombre de celuloide, se utilizó para fabricar diferentes objetos, desde placas dentales a cuellos de camisa. El celuloide tuvo un notable éxito comercial a pesar de ser inflamable y de su deterioro al exponerlo a la luz. Aunque la mayoría de estos descubrimientos iniciales fueran empíricos, pueden ser utilizados para explicar parte de la teoría y de la terminología de la ciencia de los polímeros moderna. Es importante tener en cuenta que todos estos inventores, como los hombres primitivos, utilizaron un producto que se encontraba en la naturaleza para obtener un producto de mayor utilidad. Así, Charles Goodyear transformó el caucho de la hevea, un material termoplástico y reblandecible al calor en un producto menos sensible al calor utilizando azufre para formar un número relativamente pequeño de uniones de conexión o enlaces cruzados entre las moléculas de cadena larga del poliisopreno. Nelson Goodyear usó azufre para generar muchos enlaces cruzados entre las cadenas de poliisopreno de manera que el material no fuera ya termoplástico sino termoestable. Los termoplásticos son moléculas bidimensionales que pueden ablandarse con el calor y volver a su estado inicial al enfriarse, mientras que los plásticos termoestables son polímeros de red tridimensional que no pueden moldearse por calentamiento. Tanto la celulosa como el nitrato de celulosa son polímeros lineales, o bidimensionales, pero el primero no puede reblandecerse a causa de los numerosos puentes de hidrógeno que hay entre las cadenas moleculares. Cuando se usa como explosivo, el nitrato de celulosa está completamente nitrado y es básicamente trinitrato de celulosa. Por otro lado, Parks y Hyatt utilizaron dinitrato de celulosa, o nitrato de celulosa secundario, el cual contenía numerosos puentes de hidrógeno, además de ser altamente inflamable. Parks añadió aceite de ricino para plastificar -o atenuar el efecto de- los enlaces de hidrógeno. Hyatt utilizó alcanfor con la misma finalidad. El conde Hilaire de Chardonnet hizo pasar el colodión de Menard a través de orificios muy pequeños llamados hileras y obtuvo filamentos al evaporar la mezcla de disolventes. La inflamabilidad de estos filamentos se redujo mediante la desnitrificación con bisulfito sódico. Es interesante recalcar que los hermanos Goodyear convirtieron un elastómero termoplástico en un elastómero termoestable y en un plástico duro termoestable generando cantidades pequeñas o grandes de enlaces cruzados de azufre. Schónbein redujo el número de enlaces de hidrógeno intermoleculares presentes en la celulosa haciéndola reaccionar con ácido nítrico, y mientras que ni la celulosa ni el dinitrato de celulosa son solubles en etanol o en éter etílico, Menard pudo disolver el último de aquellos dos en una solución equimolar de esos dos disolventes. 13 Hyatt ablandó el inflamable dinitrato de celulosa añadiendo alcanfor, que reduce la efectividad de los enlaces de hidrógeno intermoleculares. Chardonnet transformó la celulosa en filamentos continuos. Después de una desnitrificación y un estirado, dichos filamentos poseían todas las propiedades físicas y químicas de la celulosa original. Sin embargo, puesto que nadie sabía en aquel entonces lo que era un polímero, nadie tenía una idea clara de los complicados cambios que se habían producido en las primeras obtenciones de caucho, plásticos y fibras utilizables. Aún hoy, algunos químicos orgánicos tienen dificultades para visualizar estas grandes macromoléculas. Hace más de un siglo, Graham acuñó el término "coloide" para los agregados de -9 -7 dimensiones entre 10 a 10 m. Desafortunadamente, el tamaño de muchos polímeros se sitúa en este intervalo, pero es importante señalar que al contrario que los coloides, los polímeros son moléculas individuales cuyo tamaño no puede reducirse sin romper los enlaces covalentes que mantienen los átomos unidos en estas moléculas de cadena. En 1860 se preparó un oligómero, polímero de peso molecular muy bajo del etilenglicol al que se le asignó la estructura correcta [HO(OCH2CH2).8OH] . Sin embargo, Fittig y Engelhorn asignaron una estructura cíclica incorrecta al ácido polimetacrilico [CH2C(CH3)COOH] que obtuvieron en 1880. Utilizando las leyes de Raoult y Van't Hoff, varios químicos obtuvieron pesos moleculares elevados para estos y otros polímeros lineales, pero como no podían imaginarse la existencia de las macromoléculas, concluyeron que la técnica de Raoult no era aplicable a la determinación del peso molecular de estas moléculas. En el siglo XIX estaba generalmente aceptado por los químicos orgánicos punteros que el fenol podía condensarse con formaldehído. Puesto que no conocían el concepto de funcionalidad, Baeyer, Michael y Kleeber produjeron inútiles mezcolanzas y amasijos de sustancias con enlaces cruzados volviendo a sus investigaciones sobre reacciones con reactivos monofuncionales. Sin embargo, usando gran exceso de fenol, Smith, Luft y Blumer fueron capaces de obtener productos de condensación termoplásticos. Aunque no existen pruebas de que Baekeland conociera la existencia de macromoléculas, él entendía el concepto de funcionalidad, y así en 1907 utilizando cantidades controladas de fenol y formaldehído, produjo resinas termoplásticas que podían ser convertidas en plásticos termoestables. El acuñó el término resol en fase A para describir la baquelita termoplástica producida por condensación de exceso de formaldehído y fenol en condiciones alcalinas. Este resol en fase A se transformaba en baquelita fase C con enlaces cruzados termoestable (infusible) por calentamiento o avance de la resina. Baekeland también preparó resinas termoplásticas llamadas novolacas por condensación de fenol con una pequeña cantidad de formaldehído en solución ácida. Las novolacas se transformaban en termoestables añadiendo formaldehído a la hexametilentetramina. Aunque antes de 1910 se habían sintetizado otros polímeros en el laboratorio, la baquelita fue el primer plástico verdaderamente sintético. El hecho de que los métodos que se usan hoy en día sean prácticamente los mismos que se describen en las patentes originales prueba la ingeniosidad y el conocimiento de Baekeland de la química de condensación del fenol trifuncional con el formaldehído bifuncional. Antes de la Primera Guerra Mundial, se encontraban ya a disposición del público plásticos como el celuloide, la laca, el Galalith (caseína), la baquelita, acetato de celulosa y el caucho de la hevea; fibras como el algodón, la lana, la seda y el rayón; y resinas como los recubrimientos de poliéster denominados gliptales, el asfalto o bitumen, las resinas de cumarona-indeno y las resinas de petróleo. No obstante, como demuestra la evidencia cronológica de la tabla 2.1, se produjeron pocos desarrollos adicionales en la tecnología de los polímeros antes de la Segunda Guerra Mundial debido a la falta de conocimientos de ciencia de los polímeros. 14 El premio Nobel Hermann Staudinger estableció las bases de la moderna ciencia de polímeros en los años veinte cuando demostró que los polímeros naturales y sintéticos no eran agregados como los coloides o compuestos cíclicos como el ciclohexano, sino moléculas de cadena larga con grupos terminales característicos. A el se le debe en 1930 la descripción de la estructura de estos materiales introduciendo el concepto de macromolécula. Sus colegas le aconsejaron: "Querido colega, deje tranquilo el concepto de macromoléculas.... esas cosas no pueden existir. " Desafortunadamente, algunos científicos actuales (que no se dedican a los polímeros), que ya no creen en el concepto de la fuerza vital, todavía dudan de la existencia o de la importancia de las macromoléculas, al igual que los colegas de Staudinger. A partir de este momento se desarrolló la investigación dirigida a la síntesis de macromoléculas o polímeros con aplicaciones industriales, al saber relacionar ya la estructura con las propiedades finales del material. De esta manera se obtuvieron «productos a medida» según la naturaleza del producto de partida, de los aditivos incorporados, y de la manera en que se había sintetizado el producto y del método seguido en la transformación. En 1928, Meyer y Mark utilizaron técnicas de rayos X para determinar las dimensiones de las cristalitas en la celulosa y el caucho natural. Al año siguiente, Carothers sintetizó y caracterizó los poliésteres alifáticos lineales. Puesto que no eran materiales apropiados para ser usados como fibras, sintetizó las poliamidas que se conocen con el nombre genérico de nilones. Es interesante recalcar que las fibras proteínicas de 'la seda y la lana naturales también son poliamidas. Otro hito revolucionario en la historia de los plásticos se debe a los trabajos realizados sobre el etileno por el químico alemán K. Ziegler en 1952, quien junto con el químico italiano G. Nana, que continuó sus trabajos, recibieron en 1964 el premio Nobel de Química. La aportación al mundo de los polímeros por estos dos investigadores fue la obtención de unas macromoléculas con una ordenación espacial muy regular, de gran cristalinidad, que presentaban unas prestaciones muy interesantes. Los expertos más avanzados en polímeros en los años treinta estaban de acuerdo en que los polímeros eran moléculas de cadena y que la viscosidad de las disoluciones de estas macromoléculas dependía del tamaño y de la forma de las moléculas en las disoluciones. Si bien es cierto que la producción de muchos polímeros en gran escala se aceleró con la Segunda Guerra Mundial, debe admitirse que la elaboración de estos productos esenciales se debió en gran parte a los conceptos desarrollados por Staudinger, Carothers, Mark y otros científicos punteros en el campo de los polímeros. El desarrollo de la tecnología de los polímeros a partir de la década de 1940 ha sido extremadamente rápido. En algunos casos, tales como el de la polimerización en sistemas de emulsión acuosa, la técnica se ha adelantado a la teoría; no obstante, se ha desarrollado abundante teoría, de forma que la ciencia de los polímeros actualmente es ya una ciencia aplicada y no una tecnología meramente empírica. El conocimiento de la existencia de compuestos polímeros se debe a Simon, quien en 1866 observó que el estireno, líquido incoloro y poco viscoso, se transformaba en una resma cuando se exponía a la acción de la luz o el calor durante algún tiempo. Berthelot denominó a este fenómeno polimerización, pues comprobó que los compuestos estireno (químicamente vinilbenceno) y la resma que se obtenía, tenían la misma fórmula empírica aunque distinto peso molecular. 15 Tabla 2.1.- Desarrollo cronológico de los polímeros comerciales hasta 1970. 16 Tabla 2.1.- Desarrollo cronológico de los polímeros comerciales hasta 1970 (Continuación). 17 Comportamientos similares se observaron en varias sustancias que poseían insaturaciones o dobles enlaces en la molécula, como el cloruro de vinilo, acetato de vinilo y otros derivados acrílicos. La investigación continuó al detectarse que este comportamiento también lo presentaban las moléculas que contenían grupos funcionales, como alcoholes, aminas, ácidos carboxílicos, etc. La tendencia en la síntesis de nuevos productos de naturaleza polímero continúa por modificación de los ya conocidos mediante aplicación de los diferentes tipos de reacciones habituales utilizados en los laboratorios de química: modificación de la estructura, formación de copolímeros, mezclas o aleaciones poliméricas o bien por adición de todo tipo de elementos de refuerzo o relleno. Desde las materias plásticas de masa a los tecnopolímeros Después del descubrimiento del PVC, del polietileno, de las poliamidas (Nylon), del poliestireno, el mejor conocimiento de los mecanismos de la polimerización ha contribuido, en los últimos veinticinco años, a la creación de otros materiales plásticos con características físicas y mecánicas y de resistencia al calor tan elevadas que sustituyen a los metales en aquellas utilizaciones que una vez se consideraban insustituibles. Estos materiales son denominados tecnopolímeros o polímeros para ingeniería. Para algunos de ellos se ha creado el término de superpolímeros. De los tecnopolímeros es posible recordar el policarbonato, el polimetilpentano, las resinas acetalicás, el óxido de polifenileno, los ionómeros, los polisofon, las poliinmidas, el sulfuro de polifenileno y el polibutilentereltalato. El policarbonato, aún teniendo una historia de laboratorio que nace en el siglo pasado (1898), se produce en cantidades comerciales solamente desde 1959 en Alemania y, aproximadamente en los mismos meses, en los Estados Unidos. Hoy en día el policarbonato es considerado un tecnopolímero con prestaciones superiores a la media y es utilizado, entre otras cosas para la producción de los cascos espaciales de los astronautas, las lentes cornéales, que substituyen los anteojos, los escudos antiproyectiles. El polimetilpenteno o TPX es un compuesto individualizado y polimerizado por Julio Natta pero desarrollado sucesivamente por ICL. Hoy en día la sociedad japonés Mitsui lo ha valorizado, sobretodo para la producción de artículos para laboratorios clínicos, por cuanto resiste en modo fantástico a la esterilización y tiene una perfecta transparencia. También las poliinmidas se mantienen estables si se someten por períodos muy largos, que pueden llegar hasta las cinco mil horas, a temperaturas del orden de 300° C. Estas resinas termofraguantes pueden dar una idea del nivel de prestaciones alcanzadas hoy por las materias plásticas en lo concerniente la resistencia mecánica, térmica y a la fatiga. Efectivamente las poliinmidas han substituido a los metales especiales en la producción de palas para turbinas de aviones y otras partes de los motores de los aviones a reacción y en la producción de pistones y juntas para automóviles. Estamos ya cerca al motor compuesto por materiales poliméricos En la tabla 2.2 se da desarrollo cronológico de los polímeros comerciales hasta 1995, señalando su descubridor y en la figura 2.1 varios de los hitos más destacables de la historia de los plásticos hasta 1985. 18 Tabla 2.2.- Historia de los polímeros. 19 Figura 2.1. - Hitos más destacables de la historia de los plásticos hasta 1985. En la figura 2.2 puede verse la evolución de la importancia de los materiales a lo largo del tiempo 20 Figura 2.2.- Evolución de la importancia de los materiales a lo largo del tiempo. 3.- Importancia y usos de los materiales plásticos. En la época actual resultaría difícil imaginar que alguno de los sectores de nuestra vida diaria, de la economía o de la técnica, pudiera prescindir de los plásticos. Sólo basta con observar a nuestro alrededor y analizar cuántos objetos son de plástico para visualizar la importancia económica que tienen estos materiales. Dicha importancia se refleja en los índices de crecimiento que, mantenidos a lo largo de algunos años desde principios de siglo, superan a casi todas las demás actividades industriales y grupos de materiales. En 1990 la producción mundial de plásticos alcanzó los 100 millones de tonelada y para el año 2000 llegará a 160 millones de toneladas. Los avances conseguidos por ésta en los últimos 30 años no habrían sido posibles sin estos materiales que son fundamentales en sectores como la agricultura, la industria, la alimentación, la medicina, las telecomunicaciones o el transporte. Las actividades cotidianas giran en torno a artículos de plástico como jarras de leche, gafas, teléfonos, medias de nylon, automóviles, cintas de vídeo, etc. Sin embargo, hace cien años escasos, el plástico que hoy en día nos parece algo tan normal no era un material habitual o cotidiano. Mucho antes del desarrollo de los plásticos comerciales algunos materiales existentes presentaban características singulares. Los había resistentes, translúcidos, ligeros y moldeables, pero muy pocos combinaban a un tiempo estas propiedades. En la actualidad, estos materiales se denominan plásticos naturales y constituyen el punto de partida en la historia de los materiales plásticos. Los polímeros sintéticos, comúnmente denominados plásticos, han sustituido en numerosas aplicaciones al papel, algodón, lana, cuero, acero, vidrio, etc. siendo los más usados los termoplásticos que representan el 80 % del volumen total en peso de la producción, con extensivas aplicaciones en los embalajes y en las fibras sintéticas, entre las más importantes. Esta última aplicación de los polímeros sintéticos es, por sí sola, de una importancia industrial tan considerable, que constituye un sector industrial claramente diferenciado en la estructura económica y de la producción de todos los países desarrollados, 21 El total de las primeras materias plásticas consumidas en España, en 2001, se distribuye entre los siguientes sectores: el de envases que representa el 36 %, el de la construcción un 14 %, el de mobiliario un 9 %, la industria del automóvil un 8 %, la agricultura un 5 %, el sector eléctrico un 4%, el textil y el calzado un 4 %, y las colas y adhesivos también un 4 %. Envases y embalajes. Los envases y embalajes plásticos constituyen un factor de desarrollo y progreso ya que son ligeros, suponiendo un ahorro de materia prima y combustible en el transporte de los productos envasados y, por tanto, una disminución de la contaminación atmosférica. Además, son reciclables y representan una fuente de energía alternativa que equivale a la de otros combustibles. En Europa, principalmente en los países de la Unión, el 50 % de los alimentos se envasa en plásticos, si bien está cantidad tan sólo supone entre el 10 y el 15 % en peso del total de los residuos de envases domésticos. Muchos de estos envases pueden reutilizarse, ya que tienen una vida útil prolongada, como es el caso de las cajas de botellas, bolsas, etc. Cada español consumió en 2001 un total de 37.4 kilos de envases plásticos. Igualmente estos envases son duraderos y prácticamente irrompibles, reduciendo así los posibles accidentes domésticos. Por otro lado, son muy versátiles en sus formas, pudiendo conformarse en envases rígidos o flexibles, de paredes gruesas o delgadas, adoptando las formas más variadas que aseguran la máxima protección con la mínima cantidad de material, siendo higiénicos y seguros, ayudando a evitar la contaminación por gérmenes en los alimentos envasados. En 2001 se produjeron un total de 1502519 toneladas de envases de los cuales se exportaron 311602 toneladas. Las primeras materias plásticas más consumidas el año pasado en España en el mercado del envase y embalaje han sido el Polietileno de Alta Densidad, el Polietileno de Baja Densidad, el Polietileno Lineal, el PET, el Polipropileno, el Poliestireno, y el PVC. El destino final de las primeras materias plásticas consumidas en España en este mercado ha sido para la elaboración de bolsas y sacos con un 27 %, botellas con un 24 %, Filme 22 %, Bidones 4 %, Tapones 3 % y Cajas un 4 %. Sector médico. La esperanza de vida y la mejor calidad de esta se debe en gran parte a la utilización de los plásticos. En España, 125000 personas disfrutan de un mejor nivel de vida gracias a un marcapasos fabricado sobre plástico, según datos facilitados por la Asociación Nacional de Cardiología. Además, otros productos del área sanitaria tienen al plástico como principal componente. Las jeringuillas, lentillas, prótesis, cápsulas, envases de productos farmacéuticos, bolsas de sangre y suero, guantes, filtros para hemodiálisis, válvulas, tiritas, gafas, e incluso, el acondicionamiento de cada una de las salas de un hospital se construye con materiales plásticos. Concretamente, el sector hospitalario en España consumió en 2001, 83.000 toneladas de plásticos. Sector agrícola. La producción en el campo se ha triplicado gracias a la Plasticultura, o cobertura de los cultivos agrícolas con plásticos para protegerlos de los agentes externos. Esta practica supone en nuestro país el 4.7 % del consumo de primeras materias plásticas. Un ejemplo de la puesta en marcha de este método, es la provincia de Almería donde, en los últimos 26 años, la aplicación de los plásticos en la agricultura ha favorecido su crecimiento económico pasando de tener la renta per cápita más baja del país a ocupar ahora el quinto lugar con renta per cápita más elevada de España según datos facilitados por el Comité Español de Plásticos en Agricultura, CEPLA. 22 En el caso de Almería esta provincia es la quinta en el mundo en cultivo bajo plásticos. Andalucía es la región española donde más extendido está el concepto de la Plasticultura, y dentro de ella, Sevilla, Almería y Huelva, destacan, cada una de ellas, en una especialidad de cultivo forzado: la primera en acolchado, la segunda en invernaderos, y la tercera en túneles. En 2001 se destinaron 196255 toneladas de Primeras Materias Plásticas para la fabricación de productos con destino al sector agrícola. Las aplicaciones más extendidas de la Plasticultura son: acolchamiento de suelos, túneles de cultivo, invernaderos, tuberías para conducción de agua y drenaje, filmes para ensilar, cortavientos, láminas para embalses y cordelería. El Polietileno es el plástico más consumido con 115.380 toneladas, seguido del PVC con 56065 t. La resistencia al impacto y al rasgado, la transparencia a la radiación solar, la dispersión de la luz y la reducción del riesgo de heladas, son entre otros, los beneficios que ofrecen los plásticos en la agricultura. Sector del transporte y comunicaciones. La fabricación de aviones, barcos, cohetes, trenes, motocicletas, globos, coches, bicicletas, teléfonos, antenas parabólicas, cámaras e incluso las nuevas redes de cable, se hace con plásticos. Al ahorro de combustible y a la disminución de la contaminación atmosférica contribuyen de forma decisiva los plásticos. De las 5000 piezas que lleva un automóvil fabricado en España, 1700 son de plásticos. Cada automóvil lleva incorporados, en media, unos 123 kilos de plásticos, que han sustituido a 300 de otros materiales, permitiendo, con la disminución de peso, un ahorro de combustible que alcanza los 750 litros durante la vida útil del vehículo. El tipo de plásticos más utilizado en el sector del automóvil es el Polipropileno, le siguen el Poliuretano, el ABS, Poliamidas y PVC. De esta forma los coches son cada vez más ligeros, lo cual se transforma en disminución del gasto y protección del medio ambiente. Además, permiten un ahorro de energía, que en Europa supone 3.5 millones de toneladas de combustible. Los plásticos contribuyen a que el transporte sea cada vez más seguro con innovaciones tecnológicas tales como el "airbag". Sector de la construcción. La mayoría de los edificios públicos, nuestras viviendas, nuestros lugares de trabajo, ya sean fábricas u oficinas, los edificios destinados al ocio y servicios, hospitales, etc., tienen a los plásticos como elemento común. La razón es que estos permiten un abaratamiento de los costes en la producción de grandes series de piezas para la construcción, a la vez que facilitan el ahorro de energía por su bajo peso, sus grandes prestaciones y su alto poder aislante. En España el sector de la construcción consumió en 2001 un total de 602042 toneladas de plásticos. Alrededor de 300 empresas fabrican en España materiales plásticos destinados al sector de la construcción. El tipo de plástico más utilizado en el sector de la construcción es el Policloruro de Vinilo o PVC con un consumo de 317735 t en 2001, lo que representa el 53 % de todos los plásticos empleados en este sector. A más distancia está el Polietileno con un 17 %, y el Poliestireno con un 10 %. En la actualidad, la industria europea de la construcción utiliza más de 5 millones de toneladas anuales de plásticos, ya que prácticamente todos los edificios construidos a partir de 1950 contienen plásticos en tuberías, ventanas, tejados, suelos, revestimiento de cables, conducciones y aislamientos. Sector de la electricidad y la electrónica. El empleo de los plásticos ha permitido mejorar sensiblemente las comunicaciones, ya que por un lado contribuye al ahorro de los combustibles y por otro, su capacidad como aislante, protege de los agentes externos. Los plásticos han contribuido notablemente a la evolución de la denominada "Era de la Información". Internet, comunicaciones por satélite, cables, ordenadores personales, telefonía fija y móvil, etc.Todos contienen plásticos en su diseño. 23 En 2001 el sector de la Electricidad y la Electrónica consumió en España, 80450 toneladas de plásticos. El área más importante de consumo en este sector es la de equipamientos electrónicos. El PVC, utilizado para el recubrimiento de cables eléctricos, ha sido el más utilizado, ocupando el 44 % del conjunto de plásticos más usados Otros sectores de aplicación de los plásticos. - Mobiliario, que representa el 8.8 % del total del sector, con la fabricación de tableros a la cabeza, y utilizando las Colas de Urea como plástico más empleado, con un 61% del total. - Textil y Calzado, con una cuota de mercado del 3.7 % del total del sector, siendo el Polipropileno el plástico más consumido en este mercado, con un 54 %. - Electrodomésticos, destino del 2.8 % de los plásticos. Frigoríficos y televisiones marcan la pauta, siendo Poliestireno y Poliuretano los plásticos más utilizados. - Menaje, con el Polipropileno como plástico más empleado, con un 86 % del total.- Juguetes, Ocio y Deporte. Poliestireno, Polipropileno y PVC son los plásticos más utilizados por este sector. Plásticos más utilizados Entre los más utilizados en España, destacan el Polietileno de Alta Densidad (PEAD) con 14.85 kilos por habitante, el Polipropileno (PP) con 14.12 kilos, el Policloruro de Vinilo (PVC) con 14.04 kilos, el Polietileno de Baja Densidad (PEBD) con 11.48 kilos, el PET con 6.94 kilos y el Poliestireno (PS) con 6.32 kilos. PEBD (Polietileno de Baja Densidad) Es uno de los más empleados y se utiliza sobre todo en filmes, bolsas, tanto comerciales como de saco, bolsas para basura, bidones, etc. Es también un importante componente de juguetes, menaje, agricultura, piezas para la industria y para la construcción. PEAD (Polietileno de Alta Densidad) Es el más consumido. Se utiliza en la fabricación de bolsas, cables, cajas, bidones y depósitos, cascos de seguridad para la construcción, depósitos de gasolina para automóviles y botellas. PE lineal Se emplea básicamente en la fabricación de filme estirable y filme para uso agrícola. También se utiliza como mezclas con los otros polietilenos para fabricar sacos y bolsas. PP (Polipropileno) Es el segundo plástico más utilizado en España. Es el plástico de los automóviles. Aunque también se utiliza en el menaje, hilos, cordelería, tarrinas de margarina, envoltorios para galletas, bolsas para patatas fritas, etc. PS (Poliestireno) Los electrodomésticos, especialmente los frigoríficos, tienen a este plástico como protagonista. También se emplea en teléfonos, juguetes, menaje, etc. 24 PVC (Policloruro de Vinilo) Es, junto con el Polipropileno, el segundo plástico más consumido. Se utiliza en tuberías, cables, envases, carpintería, calzado, usos hospitalarios, tarjetas de crédito, etc. EPS (Poliestireno Expandido) Se utiliza en la construcción como aislamiento y aligerante de encofrados y en el envase y embalaje. PET (Polietilentereftalato) Se emplea para botellas para productos alimenticios (agua, bebidas carbónicas, aceites) y productos de droguería y cosmética; también se utiliza para tarrinas para productos alimenticios. Además de todos estos, hay otras 23 clases de plásticos, que se emplean para todo tipo de productos industriales, sanitarios, alimentarios y sociales. Las resinas duroplásticas encuentran aplicaciones en la industria de bienes de consumo y en la de bienes de equipo, aunque su producción es del orden del 16% de la de los plásticos. En combinación con otros materiales, tales como la fibra de vidrio, proporcionan materiales compuestos o «composites» que presentan características extraordinarias, en competencia con los materiales metálicos. En las figuras 3.1, 3.2 y 3.3 se puede ver el consumo de plásticos por usos en Estados Unidos, en Europa del Este y en España, respectivamente. Figura 3.1.- Consumo de plásticos por usos en Estados Unidos. 25 2000 2003 2004 Figura 3.2.- consumo de plásticos por usos en la Europa del Este. 26 2003 2004 2005 Figura 3.3.- Consumo de plásticos por usos en España (2002-2005). 27 En la figura 3.4 puede verse la evolución del consumo de los mercados analizados durante el período que va desde 1990 al 2005. Figura 3.4.- evolución del consumo de los mercados analizados (Periodo 1990-2005) En la tabla 3.1 se da consumo anual estimado, porcentaje de participación y variación respecto al ejercicio anterior Tabla 3.1.- consumo anual estimado, porcentaje de participación y variación respecto al ejercicio anterior. CONSUMO ANUAL ESTIMADO, PORCENTAJE DE PARTICIPACION Y VARIACION RESPECTO AL EJERCICIO ANTERIOR. Toneladas MERCADOS ENVASE Y EMBALAJE(a) 2004 2005 % total 05/04% var. 1.778.175 1.881.200 46,8 5,8 CONSTRUCCION 549.990 604.907 15,1 1,7 AUTOMOCION 397.455 378.134 9,4 -4,9 MOBILIARIO 235.495 237.790 5,9 1,0 AGRICULTURA 235.483 236.802 5,9 0,6 ELECTRONICA 155.850 151.620 3,8 -2,7 PINTURAS (b) 108.445 108.830 2,7 0,4 ELECTRODOMESTICOS 96.520 94.160 2,3 -2,4 PIEZA INDUSTRIAL 82.820 82.470 2,1 -0,4 JUGUETES Y OCIO 76.454 68.304 1,7 -10,7 MENAJE 64.710 65.930 1,6 1,9 ARTICUL. PAPELERIA 25.360 23.660 0,6 -6,7 CALZADO 19.000 14.900 0,4 -21,6 APLICAC. MEDICAS(c) 16.800 16.900 0,4 0,6 Sin clasificar (d) 56.571 53.443 1,3 5,5 3.934.627 4.019.050 100,0 1,9 TOTAL (e) (a) Los datos recogen todos los materiales destinados a envase y embalaje aunque se utilicen en otros mercados analizados. (b) Se incluyen todos los materiales, prescindiendo de su utilización en mercados determinados. (c) Incluidos todos los tipos de monturas de gafas. (d) Aquellos materiales que no se puede determinar o se desconoce con exactitud el mercado de destino. (e) En las cifras que se señalan en el cuadro, no quedan recogidas las importaciones, exportaciones, materiales reciclados, cargas y refuerzos ni plastificantes. Teniendo en cuenta estas magnitudes, el mercado de plásticos o ventas interiores se estima en 4.889.360 toneladas. 28 4.- Mercado actual. Producción y consumo de plásticos. El interés y la aceptación de los materiales plásticos se reflejan en los datos de la figura 4.1, en la que se muestra el crecimiento en la producción de los mismos hasta el año 2000. La demanda mundial de plásticos fue de 235 millones de toneladas en 2005, pero se prevé un aumento cercano al 30 por ciento en los próximos cinco años para superar los 300 millones. Europa consume el 25 por ciento del total y España ocupa el cuarto lugar entre los países europeos en demanda de estos materiales. La demanda en nuestro país crecerá a un ritmo del 5.8 %, aunque todavía estamos algo por debajo del incremento europeo que en media superará el 5.3 % anual durante el próximo quinquenio. El plástico superó en producción al acero en 1989. Figura 4.1.- Evolución de la producción mundial de plásticos en millones de toneladas. En la tabla 4.1 se muestra la producción mundial por países (%) en el año 2000 y la previsión para el 2010, por zonas mundiales. De las aproximadamente 110 millones de toneladas que se fabricaron en el año 1995 en el mundo (Tabla 4.2), los principales países productores y consumidores fueron Estados Unidos, Japón y Alemania, situados a una gran distancia de los demás, haciéndose patente la gran diferencia existente con los países poco desarrollados, como Colombia, Hungría y Turquía. 29 Por su parte, en la figura 4.2 se da el índice de crecimiento de la producción de plásticos, con respecto al acero y al aluminio, tomando como base el año 1970. La producción de plásticos se ha multiplicado por 5, la de aluminio por 2.4 y la del acero por 1.4. En la figura 4.3 se da la producción de plásticos en los países de la Europa del Oeste (Año 2003). En las figuras 4.4 y 4.5 se muestran lo datos de consumo de termoplásticos y termoestables, respectivamente, por tipo en Europa del Oeste (Año 2003). Tabla 4.1.- Producción mundial de plásticos por países (%) en el año 2000. Previsión para el 2010. Tabla 4.2.- Producción y consumo mundial de plásticos en 1995 en miles de t (Datos ANAIP). 30 Figura 4.2.- Índice de crecimiento de la producción de plásticos, con respecto al acero y al aluminio. 31 Figura 4.3.- Producción de plásticos en los países de la Europa del Oeste (Año 2000). 32 Figura 4.4.- Datos de consumo de termoplásticos por tipo en Europa del Oeste 33 Figura 4.5.- Datos de consumo de termoestables por tipo en Europa del Oeste 34 En España la situación ha seguido el mismo hábito creciente. Desde las 6500 t que se produjeron en el año 1955, hasta alcanzar las 3934627 toneladas en el año 2004 y de 3958162 en el año 2005, el crecimiento fue muy rápido al principio, manteniéndose después un crecimiento más constante. El consumo aparente se ha incrementado un 4.9 % respecto al año anterior, hasta las 4386808 toneladas. El grupo con mayor porcentaje de participación sigue correspondiendo a los termoplásticos de gran consumo, cuyo consumo se ha situado en las 3063.19 toneladas y ha explicado por si solo el 78,6 % del consumo de materiales plásticos. El consumo per cápita de este grupo de materiales se ha situado en los 70,9 Kg. Los materiales termoestables han presentado un consumo de 633309 toneladas, creciendo un 5,6 % con relación al año anterior. El consumo medio por habitante ha sido de 14.7 Kg. Los plásticos técnicos han experimentado un ligero retroceso del 1.1 %, hasta las 500608 toneladas, lo que ha generado un consumo de 11.6 Kg por habitante en el año 2004. El grupo de las materias plásticas varias ha supuesto el consumo aparente de 189772 toneladas, un 22.7 % superior al ejercicio precedente y un consumo de 4.4 Kg por habitante. El consumo siempre ha ido por encima de la producción (Figura 4.6), cifrándose que por habitante y año cada español ha consumido 101.6 kg de materiales plásticos en 2004. España ocupa el séptimo lugar mundial en el consumo de plásticos por habitante y año. En la tabla 4.3 se refleja la evolución de la producción, comercio exterior y consumo aparente de las primeras materias plásticas. Figura 4.6.- Evolución general de las primeras materias plásticas (Miles t). 35 TABLA 4.3.- EVOLUCION DE LA PRODUCCION, COMERCIO EXTERIOR Y CONSUMO APARENTE DE LAS PRIMERAS MATERIAS PLASTICAS. Miles de toneladas. AÑOS PRODUCCION % IMPORTACION EXPORTACION CONSUMO APARENTE % (kg/habitante) 1955 6,5 - , - -,- 2,5 0,0 9,0 -,- 0,3 1960 28,0 34,0 11,0 0,0 39,0 34,0 1,3 1965 100,7 29,0 94,0 2,0 192,0 38,0 6,0 1970 404,3 32,0 149,6 20,0 533,9 23,0 15,9 1975 723,1 -13,1 153,6 39,5 837,3 -12,6 23,6 1980 1197,0 -7,7 176,3 219,3 1154,0 -7,0 30,9 1985 1502,0 7,1 315,6 608,1 1210,0 -0,9 31,3 1990 2053,9 4,6 825,7 730,9 2148,7 5,6 54,5 1991 2006,6 -2,3 904,9 816,6 2094,3 -2,5 53,2 1992 2039,0 1,6 899,2 839,1 2099,1 0,2 53,3 1993 2044,3 0,3 875,7 938,9 1981,0 -5,6 50,7 1994 2387,6 16,8 1029,7 1141,5 2275,8 14,9 58,1 1995 2461,8 3,1 1158,7 1199,4 2421,2 6,4 61,6 1996 2475,6 0,6 1306,2 1136,9 2644,9 9,2 67,3 1997 2736,1 10,5 1474,9 1314,3 2896,8 9,5 73,0 1998 3040,6 11,1 1623,2 1440,8 3223,1 11,3 81,0 1999 3235,8 6,4 1952,9 1676,9 3511,8 9,0 88,2 2000 3450,6 6,6 1795,9 1758,7 3487,8 -0,7 87,0 2001 3441,9 -0,3 1961,4 1699,3 3703,9 6,2 91,94 2002 3604,9 4,7 2341,8 1817,0 4129,8 11,5 98,7 2003 3715,1 3,1 2523,9 2057,7 4181,3 1,2 97,9(1) 2004 3894,7 4,8 2615,6 2123,6 4386,8 4,9 101,5 (1) 2005 3958,1 1,6 2667,7 2114,3 4511,5 2,8 102,3 (1) (1) Población española según revisión del padrón 2005 elaborado por el INE = 44.108.530 habitantes Producción. La producción de las empresas fabricantes de primeras materias plásticas instaladas en España en el año 2005 ha sido de 3958162 toneladas, dato que supone un tímido incremento del 1.6 % respecto a la del año anterior. De la evolución de los cuatro grandes grupos de materiales en el año analizado se ha observado el siguiente comportamiento: - La producción de termoplásticos de gran consumo ha sido de 2370170 toneladas. Este volumen es superior en un 0.1 % a los resultados del año anterior. El porcentaje de participación de este grupo de materiales respecto a la producción total de materias plásticas es del 59.9 %. - Se han producido 729721 toneladas de materias plásticas termoestables, lo que representa un descenso anual de la producción del 0.2 %. El porcentaje de participación de este tipo de materiales respecto a la producción total de primeras materias plásticas en el año 2005 asciende al 18.4 %. 36 - La producción total de plásticos técnicos en este ejercicio ha ascendido a las 673200 toneladas, con un incremento del 10.6 % respecto al año anterior. Su participación respecto al total de materiales plásticos producidos en España ha supuesto el 17.0 %. - Finalmente, la producción de los materiales recogidos en el grupo de las materias plásticas varias se cifra en 185071 toneladas, un 4.7 % sobre la producción total y es un 0.1 % superior a los resultados del año anterior. En la tabla 4.4 se muestra la producción por tipo principales de plásticos en España. Tabla 4.4.- Producción de plásticos en España. AÑO TIPO DE PLASTICO TERMOPLÁSTICOS TERMOESTABLES PLÁSTICOS TÉCNICOS PLÁSTICOS VARIOS TOTAL 2000 2001 3441915 2002 2003 2004 2005 3604998 2244926 691973 612755 165450 3715104 2369726 731540 608559 184954 3894779 2370170 729721 673200 185071 3958162 En la tabla 4.5 se muestra la producción de primeras materias plásticas por varios tipos de plásticos en España durante los años 2000 y 2001 Tabla 4.5.- Producción de primeras materias plásticas en España. 37 Comercio exterior. En el año 2005 el valor de las exportaciones ha presentado un incremento del 3.8 %. El valor de las importaciones también se ha incrementado y lo ha hecho en un 4.6 %. En efecto, expresado en euros, frente a unas ventas al exterior de 2328.53 millones de euros, las importaciones han alcanzado los 3034.83 millones de euros, resultando una tasa de cobertura del comercio exterior del 76.7 %. Este ratio se viene deteriorando año tras año y cabe recordar que este indicador en el año 2000 se situaba en el 90.3 %. En cuanto a los diferentes grupos de materiales y por lo que respecta a las exportaciones, todos presentan resultado modesto aunque de índole positiva. Las exportaciones de termoplásticos de gran consumo han crecido un 3.5 %, alcanzando los 993.81 millones de euros y las de termoestables han ascendido un 2.6 %, siendo las exportaciones de 425.47 millones de euros. Las exportaciones de materiales plásticos técnicos han alcanzado un valor de 665.80 millones de euros, un 3.2 % más que en el año 2004 y el grupo de materias plásticas varias ha alcanzado los 243.45 millones de euros, un 9.1 % superior al ejercicio precedente. Respecto a las importaciones, todos los grupos de materiales han presentado también valores superiores a los del ejercicio anterior. El grupo de los termoestables ha presentado unas compras por valor de 423.92 millones de euros, un 4.1 % por encima del año 2004. El grupo de los termoplásticos de gran consumo es el que sigue presentando un mayor volumen de importaciones, con un valor de 1719.58 millones de euros, con un incremento del 5.0 %. Respecto a los plásticos técnicos, las compras procedentes del exterior han alcanzado un valor de 561.80 millones de euros, un 2.2 % por encima del año anterior. En la tabla 4.6 se dan las exportaciones españolas de Primeras Materias Plásticas durante los años 2000 y 2001. Tabla 4.6.- Exportaciones españolas de Primeras Materias Plásticas. 38 Consumo aparente. En el año 2005 ha ascendido un 7.7 %, hasta los 4696.56 millones de euros. El grupo de los termoplásticos de gran consumo se ha incrementado un 5.9 %, hasta los 2855.51 millones de euros. Los termoestables se han incrementado un 5.1 %, hasta los 629.31 millones de euros, con un valor unitario de 0.98 €/Kg. El grupo de los plásticos técnicos ha ascendido un 18.4 %, alcanzando los 832.97 millones de euros y un valor unitario de 1.51 €/Kg, significando una mejora del 7.5 % respecto al ejercicio precedente. El valor unitario del consumo aparente se ha situado en los 1.04 €/Kg, suponiendo una recuperación en el valor del 4.8 % respecto al del ejercicio anterior. Resultados globales expresados en euros. Los precios unitarios de las materias primas en el año 2005 se han caracterizado por las fuertes oscilaciones que han experimentado como consecuencia de las variaciones de los precios de las materias primas y de las fluctuaciones de los tipos de cambio. En el primer trimestre los precios han experimentado una clara tendencia alcista, retrocediendo en el segundo trimestre, para experimentar en el tercer trimestre del año un intenso repunte que ha quedado ligeramente atenuado por el descenso del cuarto trimestre. En términos interanuales, el precio medio unitario del consumo aparente se ha incrementado un 7.7 %. Producción. La producción total de materias plásticas se cifra en el año 2005 en los 3990.26 millones de euros suponiendo un incremento del 7.8 % respecto al ejercicio precedente, manteniendo la tendencia al alza de la producción expresada en valor que se inició en el ejercicio anterior, en el cual se habían recuperado los retrocesos experimentados en los años anteriores. La producción de los plásticos de gran consumo se ha situado en 2129.74 millones de euros, creciendo un 5.4 %. El precio medio unitario de producción ha alcanzado los 0.90 €/Kg registrándose un incremento del 5.4 %. El grupo de los plásticos termoestables, cuya producción ha sido de 630.86 millones de euros, ha presentado un incremento del 4.1 % respecto a la del año anterior. El precio unitario de producción se ha situado en los 0,86 €/Kg, un 4.3 % superior al año anterior. La producción total de plásticos técnicos expresada en millones de euros ha alcanzado los 936.97, lo que ha significado un incremento del 17.3 % respecto al ejercicio anterior. El precio unitario de producción ha sido de 1.39 €/Kg, representado un incremento del 6.0 % respecto al ejercicio anterior. Finalmente, el valor de producción de los materiales que se recogen en las materias plásticas varias ha alcanzado los 292.69 millones de euros, creciendo un 6.1 %, con un incremento del valor unitario de producción del 6.1 %, hasta 1.58 €/Kg. En la figura 4.7 se da la evolución de los resultados globales en euros (Miles de millones de € constante) de la industria transformadora (Año base = 1994) y en la figura 4.8 la evolución de los resultados globales en euros (Miles de millones de € constantes) de los termoplásticos de gran consumo. (Año base = 1994). 39 Figura 4.7.- Evolución de los resultados globales en euros (Miles de millones de € constantes) de la industria transformadora. (Año base = 1994). Figura 4.8.- Evolución de los resultados globales en euros (Miles de millones de € constantes) de los termoplásticos de gran consumo. (Año base = 1994). En la tabla 4.7 se dan los datos sectoriales de la industria productora y transformadora de los plásticos. 40 Tabla 4.7.- Datos sectoriales de la industria productora y transformadora de los plásticos. PRODUCTORA TRANSFORMADORA Más de 95000 personas trabajan en nuestro país en el sector de los plásticos, que está integrado por 4215 empresas que producen y transforman este material. En la tabla 4.8 se da el numero de personas ocupadas en la industria transformadora de los plásticos en España (Año 2000) por comunidades autónomas. Cataluña, Valencia, Madrid, País Vasco, este es el orden de las Comunidades Autónomas en cuanto a implantación de la industria transformadora de plásticos en España. En estas cuatro comunidades se factura el 71.4 % de la cifra de negocio y se da trabajo a 60315 personas. 41 En 2001 esta industria produjo 4083559 toneladas, lo que ha supuesto una facturación de más de 13800 millones de euros. Sin embargo, la industria quiere aumentar esta cifra en los próximos años y superar el séptimo puesto mundial en consumo y el octavo en producción de plásticos. Actualmente está por encima, en producción, USA., Japón, Alemania, Francia, Países Bajos, Italia y Canadá, situándose por delante de países como Australia, México y Polonia. La facturación de las empresas supone el 2.1 % del Producto Interior Bruto, ya que cada ciudadano español utiliza una media de 101.6 kilos de plásticos al año, algo menos de lo consumido por los alemanes. Todas las empresas españolas del sector están plenamente preparadas para crecer y para elaborar cualquier producto o artículo, por complejo que parezca, con los medios actuales, ya que cuentan con una capacidad tecnológica ilimitada para la producción y posterior transformación de los plásticos. En el pasado ejercicio, para atender las necesidades del mercado interno y de las exportaciones, la industria de los plásticos utilizó un 90 % del total de su capacidad de producción. Tabla 4.8.- Número de personas ocupadas en la industria transformadora de los plásticos en España (Año 2000) por comunidades autónomas. COMUNIDAD AUTÓNOMA ANDALUCÍA ARAGÓN ASTURIAS BALEARES CANARIAS CANTABRIA CASTILLA-LEÓN CASTILLA-LA MANCHA CATALUÑA EXTREMADURA GALICIA LA RIOJA MADRID MURCIA NAVARRA PAÍS VASCO VALENCIA TOTAL PERSONAS 4.654 3.387 557 467 737 1.320 2.901 2.044 29.690 195 3.916 439 8.978 2.679 1.409 7.306 14.341 85.020 El industrial español de plásticos atiende, prioritariamente, a las necesidades del mercado interior. Pero no olvida que también puede actuar en el exterior. Los plásticos nacionales se venden en 101 países diferentes, especialmente en Francia, Italia, Alemania, Portugal, Reino Unido, Países Bajos, Estados Unidos, Marruecos, Turquía y México, siendo el país galo el primer destinatario de nuestros plásticos. 42 Productores y transformadores de plásticos. En la actualidad existen 30 tipos de plásticos con las correspondientes fórmulas, todas ellas diferentes. Las industrias de plásticos que en España producen más del 90 % del producto nacional son las siguientes: • • • • • • • • • AISCONDEL, S.A. BASF ESPAÑOLA, S.A. BAYER POLÍMEROS, S.L. CIBA DOW CHEMICAL IBÉRICA, S.A. ATOFINA ESPAÑA, S.A. HISPAVIC INDUSTRIAL, S.A. REPSOL YPF BASELL POLIOLEFINAS IBÉRICA, S.A. En los próximos años, la industria española de los plásticos continuará creciendo, aunque se prevé que sea a un ritmo inferior al 10 % conseguido en la anterior década para el consumo. Parte de este crecimiento futuro se apoyará en el desarrollo de las exportaciones, las cuales, de seguir al ritmo actual de crecimiento, dentro de dos años, pueden superar ampliamente los 3 millones de toneladas. La industria de los plásticos dedicará en los próximos años sus esfuerzos en investigación a las siguientes áreas: ● Atención al medioambiente, a través de la investigación y la búsqueda de nuevas técnicas y materias que protejan y ayuden a preservar aún más el entorno ambiental. ● Formación constante de los trabajadores. ● Mejora de la seguridad en las fábricas. ● Incremento de la calidad de los productos. ● Modernización y actualización de las plantas. ● Renovación de la infraestructura tecnológica. ● Búsqueda de nuevas aplicaciones para los plásticos reciclados. Como resumen en la tabla 4.9 se dan las cifras de la industria de los plásticos en España. Tabla 4.9.- Cifras de la industria de los plásticos en España. Facturación: Millones de euros Número de empleados Inversión total realizada:Miles de euros Número de empresas Exportación total (Toneladas) Exportación: Productos terminados y Semielaborados (t.) Consumo habitante (kg/año) Consumo Aparente (Toneladas) (*) Estimación ANAIP 2000 13809 95000 600253 4215 2598629 615496 100.6 3947536 2001 13837 95000 (*) 570240 4215 (*) 2675034 743346 101.6 4083559 En la figura 4.9 puede verse la distribución geográfica de la industria transformadora de los plásticos en España (Año 2000) por comunidades autónomas. 43 Figura 4.9.- Distribución geográfica de la industria transformadora de los plásticos en España (Año 2000) por comunidades autónomas. Finalmente, en la tabla 4.10 se da una lista de los fabricantes más importantes de productos químicos, los cuales están relacionados directa o indirectamente con el mundo de los polímeros 44 Tabla 4.10.- Principales fabricantes de productos químicos según el volumen de ventas (x106 $). 45 Tabla 4.10.- Principales fabricantes de productos químicos según el volumen de ventas (x106 $)(Cont.). 46 5.- Definición y estructura molecular de los polímeros. 5.1.- Introducción. El conjunto de materiales conocido como plásticos posee unas características estructurales comunes que le confieren propiedades definidas, y que ha originado la denominación genérica anterior. La característica común a todo ellos es que poseen naturaleza de polímero. Un polímero, como indica su denominación, es una sustancia formada por muchas (del griego poli) unidades iguales (del griego meros). Así, esencialmente, un polímero está formado por la unión repetitiva, mediante enlaces covalente, de unidades pequeñas de agrupaciones atómicas denominadas unidades monoméricas o unidades de monómero, formando largas y flexibles cadenas cuyo esqueleto es una hilera de átomos de C, por lo común de varios millares de átomos de longitud. La transformación monómero/polímero se lleva a cabo mediante las reacciones de POLIMERIZACIÓN. Esas moléculas de gran tamaño se denominan macromoléculas. El término "unidad monomérica" se refiere a la unidad que se repite en una cadena de un polímero, mientras que "monómero" se usa en el contexto de una molécula que consiste en una sola unidad monomérica. Dentro de cada molécula, los átomos están unidos mediante fuertes enlaces interatómicos covalentes. MONOMERO ⇒ MONOMERO-MONOMERO-MONOMERO-MONOMERO (POLIMERO) La característica necesaria para que un monómero polimerice es que exista en el monómero un doble enlace, generalmente un enlace doble C = C (Monómeros vinílicos, representados de forma general como CH2 = CHX, donde X representa a un sustituyente) o que el monómero sea multifuncional en el que existan al menos dos grupos funcionales (bifuncional) distintos capaces de reaccionar entre sí mediante clásicas reacciones de condensación como las que se dan entre grupos ácido y alcohol o grupos ácido y amina.. Esto significa que la molécula que constituye el monómero debe contener dos o más grupos reactivos que le permitan realizar la unión y formar la macromolécula. Los polímeros tienen ciertas características y difieren unos de otros, según sea la naturaleza química y física de sus unidades repetitivas en las cadenas. Cuando a un material polímero se le incorporan ciertas sustancias—denominadas aditivos— para modificar sus propiedades, facilitar su transformación y mejorar su resistencia, el producto obtenido se denomina plástico. Por tanto podemos decir que un plástico es un polímero aditivado. POLIMERO + ADITIVO = PLASTICO 47 El tamaño y la forma de los monómeros y el modo en que se empaquetan y se disponen en el material sólido final determinan, de forma significativa, las propiedades mecánicas y las características del procesado de los plásticos. Conviene aclarar que no todas las moléculas grandes, de alto peso molecular se consideran macromoléculas. Por ejemplo, las moléculas de clorofila y de hemoglobina tienen un peso molecular elevado y no son macromoléculas, ya que su estructura no está formada por unidades más pequeñas repetidas un gran número de veces. Para que una molécula de gran tamaño sea considerada polímero o macromolécula, además de cumplir la condición de estar formada por una serie de unidades pequeñas unidas secuencialmente, debe tener un peso molecular por encima de 10.000. Las moléculas formadas por la unión de monómeros pero que no alcanzan pesos moleculares elevados, es decir son de pequeño tamaño, se denominan oligómeros. Gráficamente podría representarse un polímero como un collar en el que cada eslabón o cuenta representa una unidad monómerica (Figura 5.1.1). (a).- POLITETRAFLUORETILENO (PTFE), (b).- CLORURO DE POLIVINILO (PVC), (c).- PROPILENO (PP) Figura 5.1.1.- Esquema de la estructura de la unidad monómerica y de la macromolécula. Estas macromoléculas, se comportan de modo diferente a las moléculas pequeñas y existen tres aspectos por los cuales los polímeros actúan de modo distinto a las moléculas pequeñas. Y las razones son un poco más complicadas que decir simplemente "porque son más grandes". Los tres aspectos son: 1.- Enredo de cadena 2.- Adición de fuerzas intermoleculares 3.- Escala de tiempo del movimiento 48 Enredo de Cadena. La mayoría de los polímeros son polímeros lineales, es decir, son moléculas cuyos átomos se encuentran unidos en una larga línea, formando una inmensa cadena. Generalmente, aunque no siempre, esta cadena no es ni recta ni rígida, sino flexible. Se tuerce y se dobla formando una enredada maraña. Las cadenas tienden a torcerse y envolverse entre sí, de modo que las moléculas del polímero formarán colectivamente una enorme maraña enredada (Figura 5.1.2). Figura 5.1.2.- Maraña de macromoléculas Cuando un polímero se funde, las cadenas se comportan como espaguetis enredados en un plato. Si se trata de retirar uno del plato, éste se deslizará sin mayores problemas. Pero cuando los polímeros se enfrían o permanecen en estado sólido, actúan como si fueran un ovillo de hilo. Pero no un ovillo nuevo, prolijamente enrollado, sino de un ovillo enmarañado y viejo, constituido por hilos que se han ido juntando durante años. Intentar sacar una hebra de este ovillo, es un poco más complicado y se terminaría haciendo un gran nudo. Los polímeros en el estado sólido son así. Las cadenas se encuentran tan enrolladas entre sí, que es difícil desenrollarlas. Esto es lo que hace tan fuertes a muchos polímeros en materiales como plásticos, pinturas, elastómeros y materiales compuestos. Adición de fuerzas intermoleculares. Todas las moléculas, tanto las pequeñas como las poliméricas, interactúan entre sí promoviendo la atracción electrostática. Algunas moléculas se atraen más que otras y las polares lo hacen más que las no polares. Por ejemplo, el agua y el metano poseen pesos moleculares similares, el del metano es 16 y el del agua 18. A temperatura ambiente, el metano es un gas y el agua un líquido. Esto es porque el agua es muy polar, lo suficiente como para que sus moléculas se mantengan unidas como líquido, mientras que el metano es no polar y por lo tanto, sus moléculas no permanecen unidas en absoluto. Las fuerzas intermoleculares afectan tanto a los polímeros como a las moléculas pequeñas. Pero en los polímeros, estas fuerzas son más intensas y cuanto más grande sea la molécula mayor será la fuerza intermolecular. Aún cuando sólo las débiles fuerzas de Van de Waals estén en juego, pueden resultar muy fuertes en la unión de las distintas cadenas poliméricas. Esta es otra razón por la cual los polímeros pueden ser muy resistentes como materiales. El polietileno, por ejemplo, es muy apolar. Sólo intervienen fuerzas de Van der Waals, pero es tan resistente que es utilizado para la confección de chalecos a prueba de balas. Escala de tiempo del movimiento. Los polímeros se mueven más lentamente que las moléculas pequeñas. Imaginemos un maestro cuya tarea es llevar a sus alumnos del aula a comer, sin perder ninguno de ellos y produciendo el menor daño posible en el trayecto. Mantenerlos en fila va a ser difícil. A los chicos pequeños les encanta correr por todos lados, brincando y gritando aquí y allá. Una manera de frenar este movimiento caótico es hacer que todos los chicos se tomen de sus manos cuando se les conduce a comer. 49 Una vez que se consigue que todos se tomen de sus manos, su habilidad para correr se verá severamente restringida. Por supuesto, su movimiento aún será caótico. La cadena de niños se curvará y serpenteará aquí y allá a lo largo de su trayecto. Pero el movimiento será mucho más lento. Si uno de los chicos pretendiera adelantarse en una dirección, no lo podrá hacer porque será arrastrado por el peso de todos los demás chicos a los cuales está unido. Seguramente, el chico puede desviarse de su camino y hacer que otros chicos hagan lo mismo, pero esa desviación será mucho menor que si los chicos no estuvieran unidos. Lo mismo ocurre con las moléculas. Un grupo de moléculas pequeñas puede moverse mucho más rápido y más caóticamente cuando éstas no se encuentran unidas entre sí. Si se las une a lo largo de una extensa cadena, se desplazarán más lentamente, al igual que los niños cuando forman una cadena. Entonces ¿cómo influye esto para que un material polimérico sea diferente de un material compuesto por moléculas pequeñas? Esta lenta velocidad de movimiento hace que los polímeros hagan cosas inusuales. Para empezar, si usted disuelve un polímero en un solvente, la solución resultará mucho más viscosa que el solvente puro. De hecho, la medición de este cambio de viscosidad se emplea para estimar el peso molecular del polímero. 5.2.- Estructura química. La mayoría de los polímeros son compuestos orgánicos macromoleculares, es decir están formados por largas cadenas lineales, que consisten en una cadena principal o “esqueleto” a la cual se unen grupos laterales. La cadena principal suele estar compuesta por átomos de carbono unidos entre sí un gran número de veces. Forman largas cadenas, como consecuencia de la capacidad que tiene el átomo de carbono para enlazarse consigo mismo y con otros elementos (Macromoléculas formadas por secuencias de átomos de carbono y otros elementos). También se encuentran presentes o se pueden intercalar, entre otros, los siguientes elementos: Valencia 1: H, F, Cl, Br, I Valencia 2: O, S Valencia 3: N Valencia 4: Si Valencia 5: P unidos por enlaces interatómicos covalentes, constituyendo lo que se denomina una cadena molecular. La existencia de tantos compuestos orgánicos de diferentes tamaños se debe principalmente a: 1.- La capacidad del átomo de carbono para formar enlaces con otros átomos de carbono. 2.- La facilidad con que el átomo de carbono puede formar cadenas lineales, ramificadas, cíclicas, con enlaces sencillos, dobles o triples. 3.- El átomo de carbono, puede formar enlaces en las tres dimensiones del espacio. En la tabla 5.2.1 se da una clasificación simple de los distintos tipos de cadenas principales que pueden presentarse y en la tabla 5.2.2 los grupos laterales más frecuentes. 50 Tabla 5.2.1.- Distintos tipos de cadenas principales que pueden presentarse. CADENA PRINCIPAL EJEMPLOS Saturada POLIETILENO No saturada PP, PS, PVC, PB, PMMA, PTFE, etc POLIBUTADIENO Más insaturada IR, CR, etc. POLIACETILENO Solamente átomos de carbono POLIOXIMETILENO (POM) Átomos de carbono y de oxígeno POLIETILENOXIDO (PEO) Átomos de carbono y de nitrógeno VARIOS TIPOS DE PA (Nylon) Átomos de Carbono, oxígeno y nitrógeno VARIOS TIPOS DE POLIURETANOS POLICARBONATO (PC) POLIETILENO TEREFTALATO (PETP) POLIBUTILENO TEREFTALATO (PBTP) Anillos de carbono (PEEK) POLIFENILENO ETER (PPE) 51 Tabla 5.2.1.- Distintos tipos de cadenas principales que pueden presentarse (Continuación). CADENA PRINCIPAL EJEMPLOS POLIFENILENO SULFURO (PPS) Combinación con átomos se azufre POLISULFONA (PSU) POLIETER SULFONA (PES) Átomos de silicio y de oxígeno solamente POLIDIMETILSILOXANO (GOMA DE SILICONA) Anillos múltiples POLIIMIDA (PI) 52 Tabla 5.2.2.- Distintos tipos de grupos laterales que pueden presentarse. GRUPO LATERAL EJEMPLOS POLIETILENO POLIBUTADIENO Átomo de hidrogeno solamente POLIACETILENO POLIOXIMETILENO (POM) Polímeros vinílicos Polímeros vinildienos R=F Polidienos 53 2 2 2 El átomo de carbono tiene una configuración electrónica 1s 2s 2p , es decir tiene 4 electrones en su órbita mas externa, 2 en 2s y 2 en 2p, lo que le confiere valencia 4 y le permite establecer hasta cuatro enlaces con otros tantos átomos adyacentes. Generalmente, el número de enlaces que forma un átomo depende de la cantidad de electrones desapareados. El carbono muestra en su estado fundamental dos electrones de esta clase y en consecuencia su capacidad de enlace es de dos. Así se comporta cuando forma compuestos como el monóxido de carbono, CO. Sin embargo, en los compuestos orgánicos, el carbono no forma dos sino cuatro enlaces, lo cual significa que debe poseer cuatro electrones desapareados. ¿Cómo hace el carbono para cumplir tal requisito? Para dar respuesta a la interrogante anterior, el químico Linus Pauling formuló la teoría de la hibridación. Dicha teoría afirma que: "En el momento de combinarse, los átomos alcanzan un estado de excitación, como consecuencia de la energía que ganan. En tal estado, algunos electrones saltan de un orbital inferior a uno inmediatamente superior". Para el carbono, debemos suponer que el electrón del orbital 2s salta al orbital 2pz que en el estado fundamental se encontraba se encontraba vacío, quedando la siguiente estructura: Esta distribución constituye el estado excitado del carbono. En ella se observan cuatro electrones desapareados que justifican su valencia de cuatro. Al promocionar un electrón desde el orbital 2s al 2p el átomo de carbono tiene disponibles cuatro electrones para formar cuatro enlaces covalentes y de esta forma puede conseguir la configuración electrónica de gas noble. 54 La formación de un enlace covalente produce un descenso de energía en el sistema. Por ejemplo, la formación de un enlace C-H produce un descenso de energía de 364 kJ/mol. Por tanto la formación de dos enlaces covalentes más en el átomo de carbono provoca un descenso de 728 kJ/mol de energía que compensa sobradamente los 401 kJ/mol que se requieren para promover al átomo de carbono desde el estado fundamental al estado excitado. Esto explica por qué el átomo de carbono tiende a ser tetravalente en lugar de divalente. Con dicha estructura se esperaría la formación de cuatro enlaces, tres de los cuales serían más energéticos que el restante, puesto que los orbitales p son más energéticos que el orbital s (Tres enlaces perpendiculares entre sí, mediante los orbitales 2px, 2py, y 2pz) y el otro no dirigido, más débil, con el orbital 2s y no se explicaría la forma tetraédrica de la molécula de metano. Si se admite que el átomo de carbono en la molécula de metano participa con el orbital 2s y los tres orbitales 2p, hay que concluir que se formarían tres enlaces covalentes por solapamiento C2p-H1s, y el cuarto enlace covalente se formaría por solapamiento C2s-H1s. Esto significaría que tres de los ángulos H-C-H serían de 90º, y los otros quedarían indeterminados. Sin embargo, los resultados experimentales demuestran que los cuatro enlaces que forma el carbono en compuestos como el metano (CH4), por ejemplo, son perfectamente equivalentes y sus direcciones forman ángulos de 109 grados, como si el átomo de carbono estuviese en el centro de un tetraedro regular y la dirección de los enlaces fuese la de cada uno de los vértices Esto implica que si se trata de romper dichos enlaces, la energía necesaria para hacerlo, es igual en cada uno de ellos. ¿Cómo explicar esta realidad experimental?. La solución es aceptar la formación de orbitales híbridos o mezclados: el electrón del orbital 2s y los tres electrones de los orbitales 2p, sumarían sus energías y la redistribuirían entre sí por partes iguales. Así, los orbitales del átomo de carbono pueden hibridarse (combinarse) de tres maneras diferentes dando lugar 3 2 a orbitales híbridos sp , sp y sp, según se hibride el orbital 2s con los tres orbitales 2p, con dos orbitales 2p o con un orbital 2p respectivamente. 3 Hibridación sp . Matemáticamente los orbitales 2s y 2p del carbono pueden combinarse y reproporcionarse para darnos 4 3 orbitales híbridos equivalentes no simétricos, que se denominan orbitales sp , cada uno de los cuales puede aceptar un electrón y que tienen una energía intermedia entre los s y los p de los cuales derivan. La aportación de energía es del 25 % y del 75 % de cada uno de los orbitales participantes. La geometría molecular que se obtiene es tetraédrica con un ángulo de 109.5° y la podemos observar en los elementos de los grupos IV y VI de la tabla periódica. La geometría de estos orbitales está representada en la figura 5.2.1. 55 Los orbitales atómicos de valencia (2s, 2p) del átomo de C central que se muestran en la figura 5.2.1, no apuntan a las direcciones que se encuentran ocupadas por los H de la molécula de metano, en las circunstancias que son las que realmente contienen los electrones. Así, es necesario que cambien su "dirección" mediante interacciones entre sí, formando nuevos orbitales atómicos conocidos como híbridos, esta vez formados como combinación lineal entre los orbitales atómicos originales, OA(Hibrido sp 3 ) = a 2s + b 2 px + c 2 py + d 2 pz donde los " coeficientes de mezcla " a,b,c,d indican el aporte de cada uno de los orbitales atómicos originales a la dirección tetraédrica requerida. Si bien no haremos uso de los valores de estas constantes de mezcla, es conveniente presentar los resultados para cada una de las mezclas de 2s con 2px, 2py, 2pz, en total 4 posibilidades: sp 3 = 1 sp 3 = 1 sp 3 = 1 sp 3 = 1 2s + 2 p x + 2 p y + 2 p z 2 2s − 2px − 2py + 2pz 2 2s + 2px − 2py − 2pz 2 2s − 2px + 2py − 2pz 2 Lo que se logra entonces, es una mezcla de un 2s con tres 2p, lo que genéricamente se denomina hibridación 3 sp como se presenta la figura 5.2.1. 56 3 Figura 1.5.1.1.- Geometría y formación de los orbitales sp . 57 2 Hibridación sp . Esta hibridación se logra cuando se combina un orbital s con dos orbitales p. La aportación de energía es del 33.3 % y del 66.6 % de cada uno de los orbitales participantes. Son no simétricos y tienen una energía 2 intermedia entre los s y los p de los cuales derivan. El resultado de esta hibridación son 3 orbitales sp y la geometría molecular que se obtiene es plana trigonal con un ángulo de 120°. 3 Los orbitales sp , que se construyen para formar ángulos de 109.5° entre enlaces, no sirven para el caso en que dichos ángulos sean de 120°. Un conjunto de tres orbitales formando 120° entre sí, centrados en cada C, puede lograrse combinando un orbital 2s con dos orbitales tipo 2p, específicamente 2px y 2py, como lo muestra la figura 5.2.2, en la que se muestra la geometría de dichos orbitales. Sin embargo una combinación lineal del tipo: OA(Hibrido sp 2 ) = a 2s + b 2 px + c 2 py esto es, un orbital 2s más los orbitales 2px, 2py produce la dirección deseada, combinados de la forma: sp 2 = 1 ( 2)2s + 2px , 6 sp 2 = 2 2(2s ) − 2px + ( 3)2 py 6 , sp 2 = 3 2(2s ) − 2px − ( 3)2 py 6 Se generan tres orbitales híbridos en el plano xy, dirigidos a 120° entre si como se muestra en la figura 5.2.2. 2 Esta hibridación sp , aplicable a todas las estructuras trigonales planas, permite que se enlacen otros tres átomos formando 120° entre sí. Obsérvese que aún queda libre un orbital atómico 2pz original que no participa en esta hibridación trigonal, por lo que efectivamente la forma final es la que se muestra, con el 2pz 2 perpendicular a los dos sp . 58 2 Figura 5.2.2.- Geometría y formación de los orbitales sp . 59 2 Los tres orbitales sp forman tres enlaces σ y el 2pz un enlace π. El enlace σ se origina por el 2 3 solapamiento superposición de un orbital s puro con un orbital hibrido del tipo: sp, sp y sp , o por solapamiento de los orbitales híbridos. Las combinaciones quedan de la siguiente manera: Orbital s con sp. 2 Orbital s con sp 3 Orbital s con sp Orbital sp con sp. 2 2 Orbital sp con sp . 3 3 Orbital sp con sp . Este tipo de enlace es muy fuerte y origina simples ligaduras y, por lo mismo, es un enlace muy estable. Por su parte, el enlace π se forma por superposición de orbitales p puros cada uno con un electrón. Este tipo de enlace es muy reactivo y genera dobles o triples ligaduras. Para que se pueda formar este enlace se necesita la formación de orbitales con este mismo nombre, los cuales se generan por la unión de electrones en orbitales p. Estas estructuras se conocen como insaturadas pues presentan el átomo de carbono unido a tres elementos, en lugar de a cuatro que es el máximo de elementos con que puede unirse. Hibridación sp. Esta hibridación se logra cuando se combina un orbital s con un orbital p. La aportación de energía es del 50 % de cada uno de los orbitales participantes. También son no simétricos y tienen una energía intermedia entre los s y los p de los cuales derivan. La geometría molecular que se obtiene es lineal con un ángulo de 180°. El resultado de esta hibridación son dos orbitales sp. 60 Cuando la geometría resultante es lineal, solo requiere que se formen dos orbitales híbridos sp, mediante una combinación lineal del tipo: OA(Hibrido sp ) = a 2s + b 2 p x esto es, un orbital 2s se combina con uno de los orbitales 2p, por ejemplo el 2px, dando: [sp ]1 = 2s + 2px , 2 [sp ]2 = 2s − 2 px 2 Donde √2 es el coeficiente de normalización. Así se generan formas lineales de enlaces, a 180° entre sí, como puede verse en la figura 5.2.3, en la que se muestra la geometría de estos orbitales. Además podemos decir que hay dos orbitales tipo 2p libres que no participan en la hibridación 2py y 2pz. Figura 1.5.1.3.- Geometría y formación de los orbitales sp . Los dos orbitales sp forman dos enlaces σ y los dos p dos enlaces π. 61 El grado de participación de carácter s en un orbital hibrido puede usarse para explicar muchas propiedades de los enlaces covalentes. Así, cuanto mayor es la aportación de s en el orbital hibrido, este estará más próximo al núcleo y la longitud del enlace será más corta y la resistencia más grande. La tendencia general es: 3 -La longitud del enlace C-C disminuye desde sp hasta sp. 3 - La resistencia del enlace C-C aumenta desde sp hasta sp. 3 - La longitud del enlace C-H disminuye (ligeramente) desde sp hasta sp. 3 - La resistencia del enlace C-H aumenta desde sp hasta sp. En la tabla 5.2.3 puede verse una comparación entre las longitudes y las energías de los enlace C - C y C H para el etano, eteno y etino. Tabla 5.2.3.- Comparación entre las longitudes y las energías de los enlace C - C y C - H para el etano, eteno y etino. 62 En la figura 5.2.4 se puede ver, finalmente las geometrías de los diferentes orbitales híbridos del átomo de carbono Figura 5.2.4.- Geometrías de los diferentes orbitales híbridos del átomo de carbono. Según el tipo de orbital que utilice el átomo de carbono para enlazarse consigo mismo, da lugar a compuestos orgánicos de diferente estructura y geometría, denominados compuestos saturados e insaturados. En los compuestos saturados los átomos están unidos con enlaces simples, mientras que en los insaturados existen enlaces múltiples: dobles y triples donde se comparten dos o tres pares de electrones respectivamente. 3 2 Cada tipo de hibridación (sp , sp y sp) explica la existencia de familias de hidrocarburos: alcanos, cuatro enlaces sencillos; alquenos, dos enlaces sencillos y uno doble carbono-carbono, y alquinos un enlace sencillo y uno triple carbono-carbono. 3 Cuando el carbono presenta hibridación sp , al unirse consigo mismo y con átomos de hidrógeno da lugar a hidrocarburos saturados, también conocidos como parafinas (Alcanos), que carecen de grupos funcionales y, por lo tanto, son sustancias relativamente apolares y muy poco reactivas. La polaridad es debida a la diferencia de electronegatividad entre dos átomos unidos entre si. Dada la geometría del enlace se forman largas cadenas en zig-zag como las representadas en la figura 5.2.5, con ángulos entre sucesivos enlaces de 109.5° y distancias entre átomos de carbono iguales a 154 pm. (154 • 10-12 m.) ------PLANO DEL PAPEL - - - - HACIA ATRÁS DEL PAPEL ← SALE DEL PLANO DEL PAPEL Figura 5.2.5.- Cadenas carbonadas de compuestos saturados. 63 El representante más sencillo de los hidrocarburos saturados es el metano CH4, cuya estructura puede verse en la figura 5.2.6. El átomo de hidrógeno sólo tiene un electrón "enlazante". Figura 5.2.6.- Representación esquemática del enlace covalente de una molécula de metano (CH4). No obstante, el etano C2H6 da una mejor idea de las características de este tipo de hidrocarburos. La molécula de etano está compuesta por dos átomos de carbono y seis átomos de hidrógeno que se unen entre sí mediante enlaces covalentes sencillos. Desde un punto de vista puramente geométrico se puede representar la molécula de etano mediante dos tetraedros contiguos y opuestos por uno de sus vértices, en donde los dos átomos de carbono ocupan los centros de los respectivos tetraedros, y los de hidrógeno los vértices libres. 3 El etano CH3-CH3 está compuesto por dos grupos metilo. Cada átomo de carbono presenta una hibridación sp y se une a los átomos de hidrógeno mediante un enlace σ formado por solapamiento del orbital 1s del 3 hidrógeno con un orbital sp del carbono. Además, existe un enlace σ(C-C) formado por el solapamiento de 3 3 un orbital sp de un carbono con el otro orbital sp del otro átomo de carbono. Los dos grupos metilo no están fijos en una posición determinada, sino que pueden girar con relativa libertad alrededor del enlace sigma que mantiene unidos a los dos átomos de carbono. El enlace σ mantiene el solapamiento lineal cuando giran los átomos de carbono Todos los enlaces C - H tienen la misma longitud igual a 1.06 Å, mientras que el enlace C- C, de características electrónicas diferentes, presenta un valor superior e igual a 1.54 Å. El resto de los compuestos de esta serie de hidrocarburos de cadena abierta puede obtenerse intercalando en el etano sucesivamente grupos –CH2. En la figura 5.2.7 pueden verse la estructura del etano. 64 Figura 5.2.7.- Estructura del etano. El enlace carbono-carbono se forma por superposición sigma de dos 3 orbitales híbridos sp del carbono. 2 Cuando el carbono presenta hibridación sp , origina al unirse consigo mismo y con átomos de hidrógeno, unas sustancias denominadas hidrocarburos insaturados u olefinas, y tienen la característica de poseer un doble enlace entre los átomos de carbono. Este tipo de compuestos —las olefinas— son la base de un tipo de materiales polímeros formados por reacciones de adición. La geometría de las moléculas es lineal (Figura 5.2.8), y son compuestos muy reactivos porque pueden admitir más átomos en la molécula transformándose en compuestos saturados. Figura 5.2.8.- Geometría de las cadenas con dobles enlaces. 65 Las moléculas que tienen dobles y triples enlaces covalentes se denominan insaturadas (Alquenos y alquinos). Esto significa que cada átomo no está unido al número máximo (cuatro) de otros átomos especificados por su valencia. Un doble enlace de una molécula insaturada se puede interpretar como dos enlaces sencillos. El cambio de posición de uno de estos enlaces sencillos alrededor del átomo de carbono permite la adición de otro átomo o grupo de átomos a la molécula original. En un hidrocarburo saturado, todos los enlaces son sencillos (y saturados) y no se pueden añadir otros nuevos átomos sin previa eliminación de los ya enlazados. En la figura 5.2.9 puede verse una representación esquemática del enlace covalente de una molécula de 2 etileno (Eteno) C2H4, en la cual pueden verse los cuatro enlaces de tipo σ(sp -s) entre el carbono y el 2 2 hidrógeno, el enlace σ(sp - sp ) carbono-carbono y el enlace tipo π(2p-2p) también carbono-carbono. LONGITUD DEL ENLACE C-C: 1.33 Å LONGITUD DEL ENLACE C-H: 1.076 Å ANGULO DEL ENLACE C-H: 121.7 º Figura 5.2.9.- Representación esquemática del enlace covalente de una molécula de etileno. Cuando el carbono presenta hibridación sp, origina al unirse consigo mismo y con átomos de hidrógeno, unas sustancias denominadas alquinos (hidrocarburos insaturados), que poseen un triple enlace. En la figura 5.2.10 puede verse una representación esquemática del enlace covalente de una molécula de acetileno (Etino), C2H2, en la cual pueden verse los dos enlaces de tipo σ(sp-s) entre el carbono y el hidrógeno, el enlace σ(sp-sp) carbono-carbono y los dos enlaces tipo π(2p-2p) también carbono-carbono. 66 Figura 5.2.10.- Representación esquemática del enlace covalente de una molécula de acetileno. Tipos de unidades monoméricas. Hay dos tipos: 1.- Las olefinas o alquenos que poseen un solo grupo reactivo con dos enlaces activos que les permiten unirse covalentemente a otras unidades monómericas. Estas se denominan bifuncionales y pueden enlazarse con otras dos unidades para formar estructuras moleculares bidimensionales en forma de cadenas. En la tabla 5.2.4 se dan algunos de los monómeros más comunes, pertenecientes a este tipo, utilizados en la fabricación de productos plásticos. 67 Tabla 5.2.4.- Monómeros tipo olefinas utilizados en la fabricación de productos plásticos. 2.- Los compuestos funcionalizados que son sustancias orgánicas que tienen en la molécula —en la parte que interviene en la formación de la macromolécula— elementos como oxígeno, nitrógeno, cloro, azufre, etc. formando determinadas funciones orgánicas reactivas. Poseen al menos dos grupos reactivos en la molécula, siendo capaces, por tanto, de formar estructuras moleculares tridimensionales. Si en la molécula de etano se reemplaza un átomo de hidrógeno por el grupo OH, llamado hidroxilo, se obtiene un nuevo compuesto: el etanol. Este compuesto, debido a la presencia del grupo OH, es mucho más reactivo que el etano y que los demás alcanos. Igual que el etanol, muchas moléculas orgánicas constan de una parte poco reactiva, formada por átomos de carbono e hidrógeno, unida a un átomo o grupo de átomos, que se denomina grupo funcional, cuya reactividad es mucho mayor. 68 Un grupo funcional es un átomo o grupo de átomos presente en una molécula orgánica, que determina las propiedades químicas de dicha molécula. Una molécula puede tener un grupo funcional repetido varias veces, o incluso tener más de un grupo funcional. Al identificar los grupos funcionales presentes en la molécula siguiente: CH 3 − C H − CH 2 − CH 2 − OH I NH2 se puede observa que posee el grupo funcional –NH2 (Amina) y el -OH (Hidroxilo). La molécula dada puede considerarse un derivado del butano: CH 3 − CH 2 − CH 2 − CH 3 en el que se ha sustituido un átomo de hidrógeno del carbono 1 por un grupo OH, y otro átomo de H del carbono 3 por el grupo NH2. La olefina más sencilla es la que posee dos átomos de carbono y se denomina etileno, que es un gas a temperatura y presión ambientales. El gas etileno, en presencia de un catalizador y en las condiciones apropiadas de temperatura y presión, se transforma en polietileno (PE), que es un material polimérico sólido. Este proceso se realiza por polimerización por adición que comprende tres etapas: (1).- Apertura del enlace no saturado y formación de un radical mediante la absorción de energía. Se distinguen las iniciadas por: (i).- Radicales libres (R) (ii).- Iónicas (catiónicas y aniónicas) (iii).- Catalizadores complejos (Z-N, Ziegler-Nata) (2).- Reacción de adición del radical con otro monómero, formándose un nuevo radical y repitiéndose el proceso (3).- Terminación del proceso INICIACION: Se inicia al generarse una unidad monomérica reactiva por reacción entre un radical libre (R.) que hace de iniciador y el monómero etileno. Puede entenderse por radical libre a un grupo de átomos que teniendo un electrón desapareado puede unirse covalentemente a un electrón desapareado de otra molécula. Un ejemplo sería el peróxido de hidrógeno. El proceso se puede representar del siguiente modo: 69 El radical libre actúa en este caso como catalizador iniciador para la polimerización de etileno. CRECIMIENTO O PROPAGACION: El nuevo radical libre activará a otra unidad monomérica de etileno, de la misma forma en que lo hizo el catalizador, y luego a otra y así sucesivamente. El centro activo, o electrón desapareado (designado por • ), se transfiere sucesivamente a la unidad monomérica del final de la cadena. Las cadenas mantienen un crecimiento continuo debido a que la suma de las energías de los polímeros producidos es menor que la suma de las energías de las unidades monoméricas que producen los polímeros. Esquemáticamente se representa así: El grado de polimerización de los polímeros producidos por la polimerización en cadena varía dependiendo del material polimérico usado TERMINACION: La terminación puede suceder por la adición de un radical libre finalizador o cuando dos cadenas se combinen entre si. Otra posibilidad es que pequeñas cantidades de impurezas presentes catalicen la reacción de finalización de la cadena polimérica. 70 Algunos de los métodos de polimerización radical a escala industrial más importante son los siguientes: Polimerización en bloque o en masa (Figura 5.2.11): El monómero y el activador se mezclan en un reactor, el cual es calentado y enfriado según se requiera. Ocurre autoaceleración y la eliminación de calor es importante. La principal ventaja es la alta pureza óptica de los polímeros, ya que están libres de contaminantes. Ejemplos: PS, PMMA, PUC. Este proceso es de amplia utilización en polimerización por condensación cuando un monómero se ha cargado en el reactor y el otro se va adicionando poco a poco. El proceso puede ser empleado globalmente para muchas condensaciones de polímeros por sus bajas temperaturas de reacción. Figura 5.2.11.- Polimerización en bloque o en masa. Polimerización por solución: El monómero se disuelve en un solvente no reactivo que contiene un catalizador. La presencia de solvente facilita la transferencia de calor y reduce la viscosidad del medio. El calor desprendido por la reacción es absorbido por el solvente y se disminuye la velocidad de reacción. Existen reacciones de transferencia de cadena, Ejemplos: PS, PMMA, PAN. Polimerización por suspensión: El monómero se mezcla con un catalizador, que es soluble en él, y se produce una suspensión e agua de un monómero insoluble en agua, a través de pequeñas gotas (0.01 cm) obtenidas por una agitación violenta. En este proceso el calor desprendido por la reacción es absorbido por el agua. Después del proceso, el producto polimerizado se separa y deshidrata. Corresponde a un tipo de polimerización en masa, pero que evita el efecto de viscosidad y transferencia de calor. Polimerización por precipitación. Se utiliza un solvente en el cual sea soluble el monómero, pero insoluble el polímero. Se producen reacciones laterales. Por ejemplo acrilonitrilo en agua (también , Vinilacetato, etileno). Polimerización por emulsión (Figura 5.2.12). La mezcla de reacción consiste en: monómero, agente emulsificante, agua, iniciador soluble en agua. Las partículas que se forman son de alrededor de 0.05 a 5 µm de diámetro. Se forman agregados micelares del detergente (0.1 a 0.3 µm). Las gotas de monómero permanecen en la fase acuosa, pero algunas moléculas ingresan a la micela. Los radicales iniciados en la fase acuosos difunden, hacia el interior de la micela donde ocurre la polimerización. Ejemplos: Polímeros acrílicos, PUC, PUA, copolímeros, etc. 71 Figura 5.2.12.- Polimerización por emulsión. 4 6 Al unirse el etileno un gran número de veces (Usualmente 10 , pero puede ser tan alto como 10 y tan bajo 3 como10 ), siempre por los carbonos que soportan la insaturación, origina la macromolécula que se denomina anteponiendo el prefijo poli al nombre de la olefina. Así el etileno forma el polietileno (PE). Se puede observar que al formarse un polímero a partir de una olefina se forma un hidrocarburo saturado de gran tamaño molecular. 72 En la figura 5.2.13.a se muestra parte de la macromolécula de polietileno. Esta representación no es del todo correcta, ya que los enlaces sencillos entre átomos de carbono no están en un plano. Un modelo tridimensional más real es el de átomos en posiciones zigzagueantes (Figura 5.2.13.b) con una longitud de enlace C - C de 0.154 nm. En la figura 5.2.13.c se da una representación en 3-D. (a) (b) (c) Figura 5.2.13.- Polietileno:(a).-Representación esquemática de las estructuras de la unidad monomérica y de la cadena (b).- Perspectiva de la cadena indicando la estructura de la espina dorsal en zigzag y (c).-Representación en 3-D 73 Los enlaces C – C son muy rígidos, pero las unidades pueden rotar, obteniéndose diferentes conformaciones ( CONFORMACIONES = Todas las disposiciones de átomos que se pueden obtener o alterar por simple rotación alrededor del ángulo de enlace θ de un enlace sencillo). En la figura 5.2.14.a se da una representación de los conos de revolución disponibles para el tercer y cuarto enlaces de una cadena simple de carbono con un ángulo de enlace fijo θ. Para establecer la posición del carbono 2 con respecto al primero basta con precisar únicamente la longitud de enlace l, que si bien es variable, fundamentalmente debido a la existencia de vibraciones, podemos considerarla prácticamente constante. Para establecer la posición del carbono 3 necesitamos ya establecer dos valores, la longitud de enlace 1 y el ángulo de enlace θ, prácticamente constante e igual al ángulo en el centro de un tetraedro. Sin embargo, para establecer la posición del carbono 4, y todos los posteriores, se necesitará fijar, además de la longitud y del ángulo de enlace, un denominado ángulo diedro (entre dos planos) de rotación interna Φ para cada nuevo enlace definido. Este ángulo presenta una gran variabilidad y sus posibles valores son difíciles de establecer con precisión en cada caso. Debido a esto último una macromolécula tendrá múltiples modos de disponerse espacialmente y esa disposición puede variar de una molécula a otra dependiendo de factores como los efectos estéricos, el disolvente, la temperatura, etc. Por tanto, el polietileno forma largas cadenas retorcidas de conformación aleatoria y de baja rigidez, que pueden moverse bajo el efecto de pequeñas fuerzas. Sin embargo, si las cadenas de polímero se alinean como en el caso de la producción de fibras, el módulo elástico en la dirección paralela al eje de la fibra puede ser tan alto como 200 Gpa, en comparación con valores menores de 1 Gpa para el polímero global. (a) (c) (b) Figura 5.2.14.- (a).- Conos de revolución disponibles para el tercer y cuarto enlaces de una cadena simple de carbono con un ángulo de enlace fijo θ (b).- Conformaciones por rotación alrededor de un enlace sencillo (c).- Ilustración del modelo de enrollamiento aleatorio 74 La capacidad de una macromolécula de ovillarse más o menos depende de la capacidad que tienen los átomos que constituyen la cadena de girar alrededor de los enlaces que los unen Del modelo de enrollamiento aleatorio se pueden sacar las siguientes consecuencias: 1.- El enmarañamiento de las cadenas impide la cristalización, la cual solo ocurre de forma parcial. 2.- El enmarañamiento hace que los polímeros fundidos tengan una elevada viscosidad. Las cadenas pueden hacerse más rígidas adicionando grupos laterales no tan simples como el hidrógeno (Figura 5.2.15). Así, en el caso del poliestireno (PS) se unen a la cadena principal de átomos de carbono anillos bencénicos (6 átomos de C), los cuales interfieren con la rotación de la cadena principal por efecto estérico, haciéndola menos flexible y, por tanto, más rígida. Figura 5.2.15.- Presencia de grupos laterales de gran tamaño previene el movimiento relativo de las cadenas adyacentes, de lo que resulta un incremento de la rigidez. Los dobles enlaces también impiden la rotación por el efector del enlace π. Por otra parte, si en el esqueleto existen más de un tipo de átomos (cadenas heteroatómicas) se facilita la rotación, por ejemplo: -- C – O – C -- 75 No todos los polímeros poseen cadenas ovilladas. El óxido de polifenileno (PPO) tiene un esqueleto formado por átomos de oxígeno alternándose con anillos bencénicos, lo que da como resultado una cadena mucho menos flexible. POLIESTIRENO POLIFENILENO (PPO) CADENA MUCHO MENOS FLEXIBLE El polietileno se puede representar por: Φ1 − (CH 2 − CH 2 )GP − Φ 2 donde GP es el grado de polimerización (número de unidades monoméricas que se repiten para formar la molécula de polímero) y Φ1 y Φ2 son los grupos pequeños finales, los cuales tienen poca influencia sobre las propiedades. Multiplicando el grado de polimerización por el peso molecular de la unidad monomérica se obtiene el peso molecular de la macromolécula. Si en el polietileno se reemplazan todos los átomos de hidrógeno por átomos de flúor, se obtiene el politetrafluoroetileno (PTFE). Su unidad monomérica y la estructura de su cadena se muestran en la figura 5.1.1.a. El politetrafluoroetileno (cuyo nombre comercial es Teflón) tiene un alto punto de fusión (327 ºC) y pertenece a la familia de los polímeros denominados fluocarburos. Se utiliza para fabricar sartenes donde no se pegue la comida, y todo aquello que requiera de tales características. El PTFE también se utiliza para tratar alfombras y telas para hacerlas resistentes a las manchas. Y lo que es más, es también muy útil en aplicaciones médicas. Dado que el cuerpo humano raramente lo rechaza, puede ser utilizado para hacer piezas artificiales del cuerpo. Resistencia química El PTFE es prácticamente inerte a todos los agentes o compuestos químicos. Es solamente atacado por un pequeño número de productos químicos inusuales como los metales alcalinos, trifloruro de cloro y fluoruros a altas temperaturas y presiones. El PTFE es insoluble en todos los solventes hasta temperaturas arriba de 300°C. Algunos aceites altamente fluorinados, pueden absorber y disolver el PTFE a temperaturas cercanas al punto de fusión cristalino. Resistencia al medio ambiente El PTFE tiene muy buena resistencia a la luz y medioambiente y es idóneo para uso en exteriores. No se requieren estabilizadores de UV para mejorar sus propiedades a la luz y medioambiente. Resistencia al fuego. Bajo condiciones normales de operación, es incombustible y no-inflamable, el oxigeno acelera la degradación del PTFE a temperaturas arriba de los 400°C. Si la temperatura y presión se incrementan al mismo tiempo, puede ocurrir una degradación del polímero rápidamente, particularmente en oxigeno puro, con la descomposición de contaminantes peligrosos, el PTFE tiene un coeficiente de transmisión térmica muy bajo. El flúor es un elemento muy extraño. Cuando forma parte de una molécula, no le agrada estar alrededor de otras moléculas, incluso cuando éstas contengan átomos de flúor. Menos aún cuando se trata de otras clases de moléculas. De modo que una molécula de PTFE, estando tan repleta de átomos de flúor como está, quisiera estar lo más alejada posible de otras moléculas. Por esta razón, las moléculas en la superficie de un trozo de PTFE rechazarán cualquier cosa que intente acercárseles. Esta es la razón por la cual nada se pega al PTFE. 76 Otro polímero común, el cloruro de polivinilo (PVC) tiene una estructura que resulta ser una ligera variante de la del polietileno, pues uno de cada cuatro hidrógenos es reemplazado por un átomo de cloro. Existen dos tipos de cloruro de polivinilo, el flexible y el rígido. Ambos tienen alta resistencia a la abrasión y a los productos químicos. El PVC flexible o también llamado plastificado, constituye el 50% de la producción. En este tipo de PVC, se emplea un polímero de suspensión o masa y aditivos que hacen procesable el material como son plastificantes que imparten al producto terminado flexibilidad, dependiendo de la proporción del plastificante usado. Este tipo de PVC es destinado para hacer manteles, cortinas para baño, muebles, alambres y cables eléctricos, tapicería de automóviles, etc. El PVC rígido utiliza un polímero o resina de PVC de suspensión o masa y que se encuentra integrado con un gran número de aditivos como modificadores de flujo, de impacto, estabilizadores, colorantes, entre otros, pero que no contiene plastificantes que modifiquen la flexibilidad del material. Se usa en la fabricación de tuberías para riego, juntas, techado, botellas, y también en partes de automóviles. Por otro lado, la sustitución de los átomos de cloro del PVC por el grupo CH3, genera el polipropileno (PP). En la tabla 5.2.5 se dan las estructuras de la unidad monomérica de algunos de los polímeros más comunes y de los correspondientes polímeros a que dan lugar. Algunos de estos polímeros, como el nylon (o nailon), el poliéster y el policarbonato son relativamente complejos. 77 Tabla 5.2.5.- Estructuras de la unidad monomérica de algunos de los polímeros más utilizados. POLIMERO UNIDAD MONOMERICA PM Polietileno (PE) 28 Polipropileno (PP) 42 Cloruro de polivinilo (PVC) 62.5 Politetrafluoroetileno (PTFE) 100 Poliestireno (PS) 104 Polimetacrilato de metilo (PMMA) 100 Poli-isopreno 68 Poli-isobutileno (PIB) 56 Policarbonato (PC) 254 OH Fenol formaldehído (Baquelita) CH2 CH2 133 CH2 Polihexametilenadipamida (Nylon 66) 226 Tereftalato de polietileno (PET) (Un Poliéster) 192 Polibutadieno 54 Poli(óxido de fenileno), 78 Tabla 5.2.5.- Estructuras de la unidad monomérica de algunos de los polímeros más utilizados. POLIMERO UNIDAD MONOMERICA PM Nylón, 6 CH2 Policloropreno CH2 C C Cl H n Poli(ácido acrilico) O Policetonas Resinas epoxy CH2 CH2 C O CH2 CH CH2 O n CH3 O O CH2 CH C CH2 CH3 Poli(sulfuro de fenileno) (PPS) S n Polisulfona (PSU) Polieter sulfona (PES) Poliimida (PI) Poli(óxido de metileno) (POM) Poli(óxido de etileno) (PEO) Tereftalato de polibutadieno (PBTP) 79 Las unidades monoméricas funcionalizadas son aquellas que poseen al menos dos grupos reactivos en la molécula para que de esta manera la reacción pueda transcurrir repetidas veces y puedan unirse un número importante de unidades entre sí. Estos grupos funcionales suelen ser ácidos dicarboxílicos (dioicos), diaminas, dialcoholes, etc., aunque también en la misma molécula puede tener dos grupos reactivos diferentes como hidroxi-ácido, amino-ácido, lactonas (Esteres cíclicos), etc. En la tabla 5.2.6 se presenta una selección de los grupos funcionales más corrientes (hidroxilo, carbonilo, carboxilo, etc), la clase de compuestos a los que dan lugar y un ejemplo. Tabla 5.2.6.-.- Grupos funcionales más comunes. COMPUESTO GRUPO FUNCIONAL R-X HALOGENUROS DE ALQUILO X = F, Cl, Br ó I ALCOHOLES ETERES EJEMPLO R-OH GRUPO HIDROXILO R-O-R’ GRUPO ALCOXI O ARILOXI R-CHO ALDEHIDOS GRUPO CARBONILO (C=O) R-CO-R’ CETONAS GRUPO CARBONILO (C=O) R-COOH ACIDOS GRUPO CARBOXILO ANHIDRIDOS DE ACIDO R-COOR’ ESTERES GRUPO ALCOXICARBONILO O ARILOXICARBONILO R-CONR’R’’ AMIDAS R-NR’R’’R’’’ AMINAS GRUPO AMINO: –NH2 R-CN NITRILOS 80 El símbolo R (que denota “radical” o “residuo”) se usa comúnmente para describir un fragmento molecular derivado de un hidrocarburo. Tales fragmentos se denominan grupos ALQUILO. Los diferentes grupos alquilo pueden distinguirse añadiendo primas a la letra R, es decir R’, R’’, R’’’, etc. En la tabla 5.2.7 se dan grupos de alquilo más corrientes. Por su parte, los grupos fenilo Tabla 5.2.7.- Grupos alquilo corrientes (R-) y fragmentos relacionados GRUPO NOMBRE ABREVIACION Fenilo 81 Los halogenuros de alquilo son derivados hidrocarbonados en los que uno, o más, enlaces C-H han sido sustituidos por enlaces C-X (X= F, Cl, Br, I). El enlace C-halógeno es el resultado del solapamiento de un 3 orbital sp del carbono con un orbital híbrido del halógeno. Los alcoholes son compuestos que tienen grupos hidroxilo (-OH) unidos a átomos de carbono saturados, con hibridación sp3 Pueden considerarse como los derivados orgánicos del agua, donde uno de los hidrógenos es sustituido por un grupo orgánico: H–O–H R–O–H La estructura de un alcohol se asemeja a la del agua puesto que un alcohol procede de la sustitución formal de uno de los hidrógenos del agua por un grupo alquilo. En el agua el ángulo del enlace H-O-H es de 104.5º y el ángulo que forman los dos pares de electrones no 3 compartidos es de 114º. Estos ángulos de enlace se pueden explicar admitiendo una hibridación sp en el átomo de oxígeno. Ahora bien, no hay ninguna razón para que un átomo (oxígeno, nitrógeno, carbono, etc) forme un conjunto de orbitales híbridos equivalentes cuando no todos los orbitales se van a utilizar del mismo modo. 82 3 En el agua los orbitales híbridos sp que se van a emplear en los enlaces con los átomos de hidrógeno tienen un menor carácter s, lo que explica la disminución del ángulo de enlace tetraédrico desde 109.5º a 104.5º. Por 3 otra parte, los dos orbitales híbridos sp , que contienen a los dos pares de electrones no enlazantes, tienen un mayor carácter s, lo que explica el aumento del ángulo de enlace desde 109.5º a 114º. El aumento del ángulo de enlace entre los pares de electrones no compartidos tiene un efecto estabilizante al disminuir la repulsión electrónica entre los mismos. En el metanol el ángulo del enlace C-O-H es de 108.9º. Este ángulo es mayor que en el agua debido a la presencia del grupo metilo, mucho más voluminoso que el átomo de hidrógeno, que contrarresta el efecto de compresión del ángulo de enlace que provocan los dos pares de electrones no enlazantes. Las longitudes de enlace O-H son aproximadamente las mismas en el agua que en los alcoholes, pero la distancia de enlace C-O es bastante mayor (1.4 Å) debido al mayor radio covalente del carbono en comparación con el del hidrógeno. Un alcohol puede clasificarse como primario, secundario o terciario, dependiendo del número de sustituyentes de carbono unidos al átomo de carbono que tiene el grupo hidroxilo. Los éteres son compuestos de fórmula R-O-R´ en la que R y R´ pueden ser grupos alquilo o arilo (fenilo). Los éteres podrían considerarse derivados del agua, por simple sustitución de los átomos de hidrógeno por grupos alquilo. En la siguiente figura se indican, a modo de comparación, las estructuras del agua, el metanol y el dimetil éter. 83 Los éteres se caracterizan por su falta de reactividad química lo que les hace ser muy empleados como disolventes en un gran número de reacciones orgánicas. El éter de mayor importancia comercial es el dietil éter, llamado también éter etílico o simplemente éter. El éter etílico se empleó como anestésico quirúrgico pero es muy inflamable y con frecuencia los pacientes vomitaban al despertar de la anestesia. Actualmente se emplean varios compuestos que son menos inflamables y se toleran con más facilidad, como el óxido nitroso (N2O) y el halotano (BrClCHCF3). Los aldehídos y cetonas, junto con los ácidos carboxílicos, los ésteres, las amidas y los cloruros de ácido, se caracterizan por contener en su estructura el grupo funcional carbonilo (C=O). El grupo carbonilo, (C = O), es uno de los grupos funcionales más importantes. El átomo de carbono y el átomo de oxígeno que forman el grupo carbonilo se encuentran unidos mediante dos enlaces: uno σ y otro π. El átomo de carbono del grupo carbonilo presenta hibridación sp2 y está enlazado al átomo de oxígeno y a otros dos átomos mediante tres enlaces σ coplanares, separados entre sí 120°. El segundo enlace entre el carbono y el oxígeno, el enlace π, se forma por solapamiento del orbital 2p no hibridizado del carbono con un orbital 2p del átomo de oxígeno. El oxígeno es más electronegativo que el carbono y su orbital 2p es de menor energía que el orbital 2p del carbono, por tanto el orbital molecular π enlazante se formará con más de un 50 % del orbital atómico 2p del oxígeno y con menos del 50 % del orbital atómico 2p del carbono. 84 El doble enlace entre el carbono y el oxígeno es semejante en su estructura orbitálica al doble enlace de los alquenos, aunque el doble enlace del grupo carbonilo es un poco más corto y fuerte. Se puede considerar a los aldehídos y cetonas como derivados de los alcoholes, a los cuales se les ha eliminado dos átomos de hidrógeno, uno de la función hidroxilo y otro del carbono contiguo, apareciendo, por tanto, el grupo carbonilo. Los aldehídos son compuestos de fórmula general R-CHO y las cetonas son compuestos de fórmula general R-CO-R’, donde los grupos R y R´ pueden ser alifáticos o aromáticos. Ambos tipos de compuestos se caracterizan por tener el grupo carbonilo por lo cual se les suele denominar como compuestos carbonílicos. En los aldehídos el grupo carbonilo está unido a un átomo de carbono y a un átomo de hidrógeno y en las cetonas el grupo carbonilo está unido a dos átomos de carbono. El grupo carbonilo es muy polar de manera que la mayor parte de los aldehídos y cetonas son más solubles en agua que los correspondientes hidrocarburos análogos. De hecho, tanto la acetona como el acetaldehído son miscibles con agua. 85 Los ácidos carboxílicos se caracterizan por poseer en su estructura al grupo funcional carboxilo (carbonilo+hidroxilo). Algunos ácidos alifáticos se conocen desde hace cientos de años y sus nombres comunes reflejan sus orígenes históricos. El ácido carboxílico más simple, el ácido fórmico, es el causante de la irritación causada por la picadura de las hormigas (del latín formica, hormiga). El ácido acético se aisló del vinagre, cuyo nombre en latín es acetum (agrio). El ácido propiónico se consideró como el primer ácido graso, y su nombre deriva del griego protos pion (primera grasa). El ácido butírico se obtiene por oxidación del butiraldehído, que se encuentra en la mantequilla (en latín butyrum). Los ácidos caproico, caprílico y cáprico se encuentran en las secreciones cutáneas de las cabras (capri en latín). En la tabla 5.2.8 que se da a continuación aparecen los nombres comunes y los nombres IUPAC de los ácidos carboxílicos simples. Tabla 5.2.8.- Nombres comunes y los nombres IUPAC de los ácidos carboxílicos simples. NOMBRE IUPAC ácido 2-hidroxi-propanoico NOMBRE COMUN FORMULA ácido láctico ácido 2-hidroxi-propano tricarboxilico ácido citrico ácido benzeno- carboxilico ácido benzoico ácido 2-hidroxi-benzoico ácido salicilico 86 El grupo carboxilo, que es característico de los ácidos carboxílicos, es un grupo funcional que resulta de la unión de un grupo hidroxilo (-OH) al átomo de carbono perteneciente a un grupo carbonilo (-C=O). Se representa como: O II −C = O El grupo hidroxilo, en el cual un átomo de oxigeno está asociado a uno de hidrógeno, es el que confiere casi todas las propiedades químicas al grupo funcional (pérdida o reemplazo del hidrógeno el cual cede al oxígeno su electrón y confiere a la molécula que lo posee un marcado carácter ácido). Estas propiedades del grupo hidroxilo sólo son posibles por la presencia del grupo carbonilo en este grupo funcional. Los ácidos dicarboxilicos son los que tiene dos grupos carboxilo, como por ejemplo: O O II II OH − C − C − OH (Acido oxálico o etanodioico) O O II II OH − C − CH 2 − C − OH (Acido malónico o propanodioico) A partir de los ácidos carboxílicos, se pueden formar varios grupos funcionales relacionados que presentan el grupo carbonilo pero el hidroxilo, que junto al carbonilo conforma el grupo carboxilo, es reemplazado por un elemento o grupo de elementos aceptores de hidrógeno. Entre ellos que se encuentran los haluros de ácido (RCOCl), los anhidridos de ácido (RCOOCOR), los ésteres (RCOOR´), y las amidas (RCONH2). 87 Los haluros de ácido más utilizados y fáciles de preparar son los cloruros. Los ácidos carboxílicos reaccionan con el cloruro de tionilo para dar haluros de ácido. El grupo funcional está formado por un carbonilo + cloro Se pueden preparar haluros de ácido con los demás halógenos. Los cloruros de ácido y anhídridos son el punto de partida para la obtención de fluoruros de ácido, extremadamente reactivos y muy difíciles y peligrosos de manejar. Los bromuros de ácido se obtienen por reacción de adición-eleiminación del tribromuro de fósforo sobre un ácido carboxílico. Los ioduros de ácido se obtienen mediante la reacción del iodo y el fósforo rojo, que forman in situ trioduro de fósforo, con un ácido carboxílico. 88 Los cloruros de ácido, que son los derivados de ácido más reactivos, se pueden convertir fácilmente en los otros derivados de ácido menos reactivos. A continuación se indica gráficamente las transformaciones de los cloruros de ácido en los otros derivados de ácido. Los anhídridos de ácido se preparan por reacción de los ácidos carboxílicos con cloruros de ácido en presencia de una base no nucleofílica, como la piridina. Los ésteres se forman al sustituir el hidrógeno del grupo hidroxilo por una cadena hidrocarbonada. Se nombran cambiando la terminación -oico por la terminación -ato, y añadiendo el nombre de la cadena hidrocarbonada acabada en -ilo. Una representación del acetato de metilo (CH3-CO-O-CH3) es la siguiente: 89 Los ésteres se forman en una reacción de gran importancia denominada esterificación (reacción de esterificación de Fischer), que consiste en hacer reaccionar un ácido carboxílico con un alcohol, obteniéndose un éster y agua por ejemplo: En la Naturaleza aparecen dos grupos de ésteres de gran importancia, tanto para la química como para la vida: los aceites y las grasas Las amidas y diamidas son aquellas que contienen los grupos funcionales: I − N Amino : I y Carbonilo : C = O La estructura general es: O II R − C − N − R '(H ) I R ''(H ) O II CH 3 − CH2 − CH 2 − C − NH 2 BUTANAMIDA A las amidas se les puede considerar como derivados del amoníaco o de amidas primarias o secundarias por sustitución del hidrógeno por un radical acilo (R-CO-) o por sustitución del oxidrilo del carboxilo de un ácido por un grupo (-NH2). Cuando el nitrógeno de la función está unido con dos hidrógenos la amida es primaria, si uno de ellos es radical alquilo (R), la amida es secundaria y si ambos son radicales alquilo la amida es terciaria. 90 Entre las amidas merece citarse la carbodiamida o urea, que puede considerarse como la diamida del ácido carbónico: H2N − CO − NH2 Es el producto de excreción nitrogenado más importante, tanto en el hombre como en los animales. Por otra parte, tiene también un gran interés técnico, por su utilización como abono, así como en la industria farmacéutica, y sobre todo para la fabricación de resinas y materiales plásticos. Las amidas son los derivados de ácido menos reactivos. Las aminas se pueden considerar como compuestos nitrogenados derivados del amoniaco (:NH3) en el que uno o más grupos alquilo o arilo están unidos al nitrógeno. El átomo de nitrógeno de la molécula de amoniaco contiene un par electrónico libre, de manera que la forma de esta molécula, considerando en ella al par de electrones no enlazantes, es tetraédrica ligeramente distorsionada. El par aislado de electrones no enlazantes ocupa una de las posiciones tetraédricas. El ángulo del enlace H-N-H del amoniaco es de 107°, y tanto la forma de la molécula como el valor anterior se pueden explicar admitiendo una hibridación sp3 en el átomo de nitrógeno. El par electrónico libre provoca una compresión del ángulo que forman entre sí los orbitales híbridos sp3, reduciéndolo de 109° a 107° grados. En las aminas, como la trimetilamina ((CH3)3N:), el ángulo del enlace C-N-C no está tan comprimido como en el amoniaco porque los grupos alquilo, más voluminosos que los átomos de hidrógeno, abren ligeramente el ángulo, como se muestra a continuación: Las aminas se pueden clasificar según el número de grupos alquilo o arilo que están unidos al nitrógeno. Si sólo hay uno, la amina es primaria. Si hay dos grupos, la amina es secundaria y si hay tres es terciaria. 91 La propiedad más característica de las aminas es su olor a pescado descompuesto. Algunas diaminas son especialmente pestilentes y sus nombres comunes describen correctamente sus olores. Como clase, las aminas comprenden algunos de los compuestos biológicos más importantes que se conocen. Las aminas funcionan en los organismos vivos como biorreguladores, neurotransmisores, en mecanismos de defensa y en muchas otras funciones más. Debido a su alto grado de actividad biológica muchas aminas se emplean como medicamentos. A continuación, se muestran las estructuras y los usos de algunas aminas biológicamente activas. 92 La adrenalina y la noradrenalina son dos hormonas secretadas en la médula de la glándula adrenal y liberadas en el torrente sanguíneo cuando un animal se siente en peligro. La adrenalina causa un aumento de la presión arterial y de las palpitaciones, lo que prepara al animal para la lucha. La noradrenalina también causa un incremento de la presión arterial y está implicada en la transmisión de los impulsos nerviosos. La dopamina y la serotonina son neurotransmisores que se encuentran en el cerebro. Los niveles anormales de dopamina se asocian con muchos desórdenes psiquiátricos, incluyendo la enfermedad de Parkinson. La esquizofrenia se debe a la presencia de niveles anormales de serotonina en el cerebro. La acetilcolina es una molécula pequeña e iónica y por tanto altamente soluble en agua. Las moléculas de 4 acetilcolina son liberadas por la membrana presináptica en grupos de 10 moléculas, difundiéndose en la región de contacto entre las prolongaciones nerviosas de dos neuronas adyacentes y dando lugar a la transmisión del impulso nervioso de una célula nerviosa a otra. Los alcaloides son un grupo importante de aminas biológicamente activas, que son biosintetizadas por algunas plantas para protegerse de insectos y otros animales depredadores. A continuación, se indican las estructuras de algunos alcaloides representativos. Aunque en medicina se utilizan algunos alcaloides, principalmente como analgésicos, todos son tóxicos y causan la muerte si se ingieren en grandes cantidades. El filosofo griego Sócrates fue envenenado con coniina. Los casos benignos de intoxicación por alcaloides pueden producir alucinaciones o efectos psicológicos que se asemejan a la tranquilidad o a la euforia. El alcaloide histrionicotoxina se encuentra en la piel de unas ranas que habitan en la selva amazónica colombiana. La histrionicotoxina provoca la muerte por parálisis de los músculos respiratorios. 93 Las diaminas son sustancias que contienen dos grupos amina, por ejemplo: H2NCH2CH2CH2CH 2NH 2 ,1 − 4 Butanodiamina Los grupos hidroxi – acido son aquellos que contienen los grupos funcionales: - Hidroxilo : −OH como por ejemplo : O II y - Carboxilo : − C − OH HOCH 2CH2CH2CH 2COOH y desarrollándola: H H H H O I I I I II OH − C − C − C − C − C − OH I I I I H H H H Los grupos amino– acido son aquellos que contienen los grupos funcionales: I Amino: − N I y O II carboxilo : C − OH Los aminoácidos son las unidades estructurales básicas de las proteínas. Un aminoácido (libre, sin polimerizar) siempre tiene: (A).- Un grupo amino: - NH2 (B).- Un grupo carboxilo: - COOH (C).-Un hidrógeno: -H (D).- Una cadena lateral: - R 94 Esos cuatro elementos están unidos entre sí a través de un carbono central, conocido como carbono α (Figura 5.2.16). Figura 5.2.16.- Estructura de un aminoácido. Existen 20 aminoácidos diferentes y todos ellos tienen una parte común en su molécula que consisten en un grupo amino (NH3) y un grupo ácido, (COOH) como puede verse en la tabla 5.2.9, en la que se observa la fórmula de ellos, en color negro la parte común, mientras que en color azul puede verse la parte variable, que da a los aminoácidos distinto comportamiento. Tabla 5.2.9.- Formula de los aminoácidos. color marrón= aminoácidos hidrófobos; color verde= aminoácidos polares; color fuchsia = aminoácidos ácidos; color turquesa = aminoácidos básicos Las unidades monoméricas funcionalizadas que dan lugar a los plásticos de mayor consumo se representan en la tabla 5.2.10. 95 Tabla 5.2.10.- Unidades monoméricas funcionalizadas componentes de los plásticos más comunes. A lo largo de las cadenas moleculares se reconocen agrupaciones atómicas características, entre las que se pueden destacar las siguientes: Los enlaces laterales de los átomos de carbono (que no participan en la secuencia de la cadena principal) están saturados, las más de las veces, por átomos de hidrógeno, pero también por átomos de cloro, flúor o agrupaciones atómicas (grupos sustituyentes), como las siguientes: 96 Los grupos funcionales intermedios pueden encontrarse ya en los monómeros como tales u originarse en las reacciones de polimerización a partir de grupos funcionales reactivos, así, por ejemplo: Si la funcionalidad de las unidades monoméricas es 2 (Cada una de ellas reacciona con otras dos) el polímero que se obtiene a partir de ellas será lineal, formando estructuras moleculares bidimensionales en forma de cadenas, mientras que si es mayor presentan la oportunidad de formar redes (Estructuras moleculares tridimensionales) o cadenas enlazadas transversalmente (Figura 5.2.17). Figura 5.2.17.- Red polimérica lineal y reticulada. Supongamos que un monómero A, con funcionalidad 2, se mezcla con una pequeña proporción de un monómero B, con funcionalidad 4. Entonces se producirá una red enlazada transversalmente, donde 4 cadenas del monómero A se encontrarán en cada monómero B. Representamos por A y B grupos funcionales de un monómero y por R y R’ la parte hidrocarbonada del mismo. La reacción siempre ocurre de tal manera que un extremo A de un monómero se une a un extremo B de otro. 97 5.3.- Fuerzas de enlace interatómicas. 5.3.1.- Introducción. Hay que distinguir dos tipos de enlace: intramolecular e intermolecular (Figura 5.3.1.1). Se tienen fuerzas entre átomos que dan lugar a los enlaces químicos y fuerzas entre moléculas o fuerzas intermoleculares (de van der Waals y puentes de H). Figura 5.3.1.1.- Enlace intramolecular e intermolecular (H2O y HCl). 5.3.2.- Enlace intramolecular. El enlace intramolecular es covalente y es característico de los materiales poliméricos, en los cuales la estructura molecular fundamental es una larga cadena de átomos de carbono enlazados covalentemente entre sí mediante dos de los cuatro enlaces disponibles por átomo. Los dos enlaces restantes normalmente participan en la unión covalente con otros átomos o grupos de átomos. Así, los enlaces intramoleculares, tipo C-C y C-H, son fuertes enlaces primarios de tipo covalente. En 1916, Lewis propuso que la formación de un enlace covalente se produce por la compartición de dos electrones entre átomos vecinos y se cumpla que ninguno de los átomos: - Tiene suficientemente poca energía en su capa de valencia como para perder sus electrones - Es suficientemente atractivo para quitarle al otro sus electrones por tanto, tendrán que compartir sus electrones. Es el caso más típico es el de dos átomos del mismo elemento. 98 Dependiendo de su capa de valencia dos átomos pueden formar un enlace simple, compartiendo un solo par de electrones (Enlace covalente sencillo), o enlaces múltiples (doble, triple, etc.), compartiendo dos o más pares. El número de pares electrónicos compartidos se llama orden o multiplicidad de enlace. En el enlace covalente los electrones compartidos pertenecen por igual a los dos átomos que forman el enlace. Esta circunstancia permite contabilizar a los electrones de enlace tanto en un átomo como en el otro, por lo que contribuyen a completar los electrones de la capa de valencia hasta alcanzar el octeto, lo que da gran estabilidad a cada átomo en la molécula. Cada par de electrones comunes a dos átomos se llama doblete electrónico. Este tipo de enlace se encuentra en todas las moléculas constituidas por elementos no metálicos o combinaciones binarias que estos elementos forman entre sí, tales como: hidruros gaseosos y en la mayoría de compuestos de carbono. El enlace covalente ocurre entre no metales porque la diferencia de electronegatividades entre los átomos es cero o muy pequeña- Los orbitales de las capas de valencia de ambos átomos se combinan para formar uno solo que contiene a los dos electrones. Muchas moléculas de elementos no metálicos (H2, Cl2, F2, etc), así como muchas moléculas que contienen átomos diferentes (CH4, H2O, HNO3, HF, etc) tienen enlaces covalentes. Además, este tipo de enlace aparece en sólidos elementales, tales, como diamante (carbono), silicio, germanio, y en compuestos sólidos formados por elementos localizados a la derecha de la tabla periódica, tales como arseniuro de galio (GaAs), antimoniuro de iridio (IrSb) y carburo de silicio (SiC). La energía del enlace viene dada por: UTOTAL = − A rm + B rn con m<n y m>2 El numero de enlaces covalentes posibles para un átomo particular depende del numero de electrones de valencia. Para N’ electrones de valencia, un átomo puede enlazarse covalentemente, como máximo, con 8-N’ átomos. Por ejemplo, para el cloro N’=7 y, por tanto, 8 - N’=1, esto significa que un átomo de cloro puede enlazarse con un solo átomo, como en la molécula de Cl2. Análogamente, para el carbono N’=4, por lo que 8-N’=4 y así cada átomo de carbono tiene cuatro electrones para compartir. El diamante es la estructura del carbono interconectada en tres dimensiones, donde cada átomo de carbono se une covalentemente con otros cuatro átomos de carbono. Los enlaces covalentes pueden ser muy fuertes, como en el caso del diamante, que es un material muy duro y tiene una temperatura de fusión muy elevada, Tf > 3550 ºC (6400 °F), pero también pueden ser muy débiles, como es el caso de los enlaces del bismuto, que funde a 270 °C (518 °F). Si los átomos que forman el enlace son del mismo elemento los electrones están compartidos por los dos átomos en igual proporción y se denomina enlace covalente no polar. Por el contrario, si los átomos son de diferentes elementos, y éstos tienen diferentes electronegatividades, los electrones serán atraídos más fuertemente por el átomo del elemento más electronegativo, por tanto no serán compartidos en igual proporción por los dos átomos que forman el enlace, y tendremos un enlace covalente polar. El enlace covalente no polar es un enlace covalente en el que la densidad electrónica está distribuida simétricamente. 1.- Se da entre dos átomos con iguales electronegatividades. 2.- Los electrones compartidos están distribuidos simétricamente. 3.- Los electrones compartidos son atraídos por igual por ambos núcleos. 4.- Se da entre dos no metales. 99 Ejemplos: : H2: Estructura de Lewis: H H . El par de electrones compartidos es atraído por igual por ambos núcleos porque los dos átomos tienen electronegatividades iguales. :: O2: Estructura de Lewis: O O. Los dos pares de electrones compartidos están distribuidos simétricamente porque son atraídos por igual por ambos átomos que tienen iguales electronegatividades. El enlace covalente polar es un enlace covalente en el que hay una distribución asimétrica de la densidad electrónica. 1.- Se da entre dos átomos con electronegatividades diferentes. 2.- Los electrones compartidos están distribuidos asimétricamente. 3.- Los electrones compartidos están más cercanos al átomo más electronegativo porque los atrae con mayor fuerza que el átomo menos electronegativo 4.- Se da entre dos no metales. Ejemplos: : Cloruro de hidrógeno HCl, Estructura de Lewis: H:Cl. Fluoruro de hidrógeno HF, Estructura de Lewis: H F, El par de electrones compartidos está más cerca del átomo más electronegativo (F, Cl). Esta situación origina un dipolo. La polaridad se mide a través del momento bipolar µ del enlace, que es una magnitud vectorial. Su módulo viene dado por el producto de la carga Q por la distancia r entre el baricentro de las cargas: µ = Qr Enlace covalente puro. En este tipo de enlace se unen dos no metales que tienen la misma electronegatividad, aportando cada uno electrón por cada par compartido, el número de pares compartidos es igual a la cantidad de electrones que faltan para tener ocho electrones (configuración de gas noble).Ejemplos: I2, O2, N2. Elemento: Hidrógeno, H2 (Figura 5.3.2.1). 100 Figura 5.3.2.1.- Enlace covalente puro del hidrógeno. Elemento: Yodo. En la figura se observa que el par de electrones se encuentra a la mitad ya que ambos átomos tienen la misma electronegatividad. Como ambos elementos poseen siete electrones sólo comparten un par aportando cada uno un electrón. Elemento: Oxígeno. La molécula de O2, se forma por dos átomos de oxígeno a cada uno le faltan dos electrones, para completar su octeto. Estos elementos comparten dos pares de electrones. Los dos pares de electrones se encuentran a la mitad. Debido a que ambos átomos poseen la misma electronegatividad, por esa razón cada par de electrones se encuentra ubicado a la mitad. 101 Elemento: Nitrógeno. En la formación del N2, ambos átomos tienen 5 electrones en su última capa y presentan la misma electronegatividad, estos átomos necesitan compartir tres pares de electrones para completar cada uno su octeto y estar así estables. Enlace covalente coordinado o dativo. Es el enlace que se produce cuando dos átomos comparten una pareja de electrones, pero dicha pareja procede solamente de uno de los átomos combinados. El átomo que aporta la pareja de electrones recibe el nombre de donante, y el que los recibe, aceptor. Cuando queremos simplificar la formula electrónica se pone una flecha que va del donante al aceptor, es decir: N →H En este enlace también se combinan los orbitales de las capas de valencia de ambos átomos para formar uno solo que contiene a los 2 electrones; la diferencia con el anterior es que sólo uno de los átomos aporta los 2 electrones y queda con carga positiva. Este puede ser normal cuando cada uno de los átomos aporta un electrón o coordinado (o dativo), cuando solo los aporta un átomo. La configuración electrónica estable del enlace covalente se consigue compartiendo electrones entre átomos vecinos. Dos átomos unidos covalentemente contribuyen cada uno al enlace con al menos un electrón. Los electrones compartidos se consideran de ambos átomos. Son enlaces direccionales. Parámetros de los enlaces covalentes. Se llaman parámetros de enlace a aquellas propiedades características de los enlaces que dependen de los átomos específicos que se enlazan pero que varían poco de compuesto a compuesto. Longitud de enlace. La longitud de un enlace covalente puede ser estimada aproximadamente mediante la o o o suma de los radios covalentes: rc (C ) = 0.77 A , rc (H ) = 0.37 A ⇒ d (C − H ) = 1.14 A . Este valor de distancia C-H es prácticamente independiente del compuesto considerado. Sin embargo, la longitud de un enlace disminuye al aumentar el orden de enlace, lo que se utiliza como criterio para estimar la multiplicidad del enlace (Tabla 5.3.2.1). o ( ) Tabla 5.3.2.1.- Longitudes medias de enlaces intramoleculares (simples y múltiples) en A 10−10 m . Entalpía de enlace. La entalpía de enlace ( ∆H E ) es la puesta en juego en el proceso de ruptura de enlaces en el estado gaseoso. Así, la entalpía de enlace para el hidrógeno es la correspondiente al proceso H 2 (g ) ⇒ 2H (g ) 102 La entalpía de un enlace aumenta al aumentar la multiplicidad del enlace (Tabla 5.3.2.2) Tabla 5.3.2.2.- Entalpías medias de enlace (kJ/mol). Los demás tipos de enlaces primarios: iónicos y metálicos normalmente están ausentes en los polímeros. o En la tabla 5.3.2.3 se presentan las energías y las longitudes de los enlaces intermoleculares (1 A = 100 pm.). o ( ) Tabla 5.3.2.3.- Energías y longitudes de enlaces intermoleculares en A 10−10 m . Con estos datos es fácil demostrar que todas las reacciones de polimerización son exotérmicas. Por ejemplo, la polimerización del polietileno se escribe: CH 2 = CH 2 + CH 2 = CH 2 ⇒ −CH 2 − CH 2 − CH 2 − CH 2 − es decir: 2 (C = C ) ⇒ 4 (C − C ) Luego dos dobles enlaces dan lugar a cuatro enlaces sencillos entre átomos de carbono. Se aportan, por tanto, 2x612 =1224 kJ /mol para romper los dobles enlaces y se desprenden 4x348 kJ /mol = 1392 kJ/mol en la formación de los enlaces simples. La polimerización es entonces espontánea y genera 168 kJ/mol. 103 5.3.3.- Fuerzas intermoleculares. 5.3.3.1.- Introducción. El fuerte enlace covalente es la unión que explica el mantenimiento de la unidad estructural de un compuesto orgánico, en nuestro caso es el que mantiene unidos a los átomos que constituyen la cadena del polímero. Además de este enlace intramolecular se pueden dar interacciones entre las moléculas, que son mucho más débiles que los enlaces covalentes, pero que a menudo son las responsables de las propiedades físicas de los compuestos orgánicos. Este tipo de interacciones intermoleculares son de especial importancia en el estado sólido y en el estado líquido, situaciones en las que las moléculas están en íntimo contacto. Los puntos de fusión, de ebullición y las solubilidades de los compuestos orgánicos muestran los efectos de estas fuerzas. Los tipos principales de interacciones intermoleculares que hacen que las moléculas se asocien para formar sólidos y líquidos se dan en la tabla 5.3.3.1.1. Tabla 5.3.3.1.1.- Tipos de fuerzas intermoleculares. ION-DIPOLO DIPOLO-DIPOLO FUERZAS FUERZAS DE FUERZAS DE ION-DIPOLO INDUCIDO INTERMOLECULARES VAN DER DISPERSION DIPOLO-DIPOLO INDUCIDO WAALS DE LONDON DIPOLO INDUCIDO-DIPOLO INDUCIDO PUENTE DE HIDROGENO Todas estas fuerzas intermoleculares resultan de la atracción mutua de cargas opuestas o la repulsión mutua de cargas iguales. El valor de la fuerza de estas interacciones es inversamente proporcional a alguna potencia de r y, por tanto, dependen en gran medida de la distancia entre moléculas que interaccionan. Así, muchas propiedades físicas de los polímeros dependen fundamentalmente, tanto de la conformación (disposiciones relacionadas con la rotación en torno a enlaces sencillos) como de la configuración (disposiciones relacionadas con el enlace químico de un átomo dado), ya que ambas afectan a la proximidad que una cadena puede tener respecto a otra. Hay tres contribuciones de tipo atractivo: - El efecto de la orientación, o interacción entre los dipolos permanentes; - El efecto de la inducción, o interacción entre un dipolo permanente y un dipolo temporal; - El efecto de la dispersión o la fuerza de London, interacción entre los dipolos temporales y los dipolos inducidos. Además hay fuerzas repulsivas que aparecen cuando las moléculas se acercan unas a otras. Efecto de la orientación (dipolo-dipolo). La energía de interacción entre dos dipolos permanentes depende de su orientación relativa, y se puede esperar que su valor global sea nulo para un compuesto si todas las orientaciones son posibles. Esto sería verdad si las moléculas pueden rotar de forma totalmente libre, pero esto no se cumple ya que algunas orientaciones tienen preferencias sobre otras. La energía de la interacción 6 7 varía con (1/r ), la fuerza entre los dipolos como (1/r ). En un sólido la energía de la interacción varía 4 con (1/r ), donde r es la separación entre las cadenas. La energía del enlace disminuye drásticamente al aumentar la separación entre las cadenas, por ejemplo, por calentamiento (Expansión térmica) o por la incorporación de plastificantes. 104 Efecto de inducción (dipolo permanente - dipolo inducido). En este caso la energía de la interacción 6 también varía con (1/r ). Su magnitud depende de la mayor o menor polarización de la molécula. Efecto de la dispersión o la fuerza de London (dipolo temporal - dipolo inducido). Esta es la única fuerza de interacción entre moléculas no polares, y surge de un dipolo temporal que induce un dipolo complementario en una molécula adyacente. Esos dipolos siempre están cambiando de lugar, pero se inducen en fase y dan una atracción neta. Dependen también de la mayor o menor polarización de la 6 molécula, y varían con (1/r ). Las fuerzas repulsivas son de un rango mucho más corto que las atractivas. Un modelo (Potencial de 12 Lennard – Jones, Figura 5.3.3.1.1) utiliza una variación con (1/r ), que no siempre esta muy de acuerdo con la experiencia práctica, existiendo la alternativa de expresiones exponenciales que proporcionan una aproximación mejor. Figura 5.3.3.1.1.- Modelo de potencial de Lennard – Jones. Las fuerzas de Van der Waals son muy importantes en estructuras en capas, como son las arcillas, grafito y nitruro de boro hexagonal. Estas fuerzas mantienen las capas unidas. Todas estas estructuras tienen un fácil deslizamiento entre capas, por lo que pueden utilizarse como materiales de baja fricción y superficies autolubricables en sellos, así como lubricantes sólidos y recubrimientos en capa delgada. Los enlaces secundarios o físicos son débiles en comparación con los primarios o químicos (Tabla 5.3.3.1.2), pero tienen gran importancia dado que en la mayoría de los casos, la fuerza necesaria para romper el polímero es la requerida para separar las moléculas, es decir, para vencer los enlaces intermoleculares. La deformación plástica de un polímero, principalmente un termoplástico, ocurre por el rompimiento de estos enlaces, lo que permite el deslizamiento de una cadena sobre otra (Figura 5.3.3.1.2). Tabla 5.3.3.1.2.- Intensidad de las fuerzas intermoleculares e intramoleculares. 105 Figura 5.3.3.1.2.- Rompimiento de los enlaces de Van der Waals de un polímero, lo que permite el deslizamiento de una cadena sobre otra Las energías de enlace características son del orden de 10 kJ/mol (0.1 eV/átomo). En realidad, existen enlaces secundarios entre todos los átomos de las moléculas, pero su presencia puede pasar desapercibida si concurre alguno de los tres tipos de enlaces primarios. Este tipo de enlace, el secundario o físico, es evidente en los gases inertes, que tienen configuración electrónica estable, y, además, entre moléculas cuyos átomos estén unidos covalentemente. Las fuerzas de enlace secundario surgen de los dipolos atómicos o moleculares. En esencia, un dipolo aparece si hay alguna separación entre las regiones positiva y negativa de un átomo o molécula. Ciertas partículas y moléculas no presentan momento dipolar intrínseco pero son susceptibles de polarizarse (desplazamiento de cargas) por efecto de un campo eléctrico, en cuyo caso decimos que se trata de un dipolo inducido. Cuando el campo eléctrico que polariza a la molécula tiene su origen en la presencia de una carga eléctrica próxima aparecerá una interacción electrostática entre la carga y el dipolo inducido. La figura 5.3.3.1.3 trata de representar el fenómeno de la inducción de un momento bipolar. 106 Figura 5.3.3.1.3.- Representación del fenómeno de la inducción de un momento bipolar. También pueden producirse dipolos instantaneos en moléculas polarizables debido a las fluctuaciones de la carga nuclear y de la nube electrónica (Figura 5.3.3.1.4). Figura 5.3.3.1.4.- Formación de dipolos instantaneos en moléculas polarizables En la situación A de la figura anterior se consideran dos moléculas polarizables sin momento dipolar intrínseco muy próximas entre sí. La carga electrónica y nuclear fluctuan y en un instante dado un desplazamiento de carga asimétrico produce un dipolo instantáneo en una de las moléculas (situación B). Este dipolo instantáneo induce un dipolo en la otra molécula vecina apareciendo una fuerza atractiva entre ambos dipolos instantáneos (situción C). El enlace es el resultado de la atracción entre el extremo positivo de un dipolo y la región negativa del vecino, como indica la figura 5.3.3.1.5. Interacciones dipolares ocurren entre dipolos inducidos y dipolos permanentes (que poseen las moléculas polares). El enlace por puentes de hidrógeno, es un tipo especial de enlace secundario, aparece entre moléculas que tienen átomos de hidrógeno en su constitución. Seguidamente se discuten los mecanismos de estos enlaces. 107 Figura 5.3.3.1.5.- Ilustración esquemática del enlace de tipo Van der Waals entre dos dipolos. 5.3.3.2.- Fuerzas ión-dipolo (Figura 5.3.3.2). En un dipolo un lado de la molécula tiene un exceso neto de electrones y una carga parcial negativa (δ-) mientras que el otro lado es deficiente de electrones y tiene una carga parcial positiva (δ+). Se forma por la diferencia de electronegatividad entre dos átomos de una molécula polar. Figura 5.3.3.2.1.- Fuerza ión-dipolo. La fuerza ión-dipolo resulta de interacciones eléctricas entre un ión y las cargas parciales en una molécula polar. La orientación de una molécula polar en presencia de iones es el terminal positivo del dipolo cerca de un anión y el terminal negativo del dipolo cerca del catión. La interacción depende de la carga y del tamaño del ion como así también del momento dipolar y del tamaño de la molécula. La hidratación es un ejemplo de interacción ion – dipolo. En la figura 5.3.3.2.2 se presenta un esquema de este tipo de interacción. 108 Figura 5.3.3.2.2.- Esquema de hidratación. 5.3.3.3.- Fuerzas dipolo-dipolo. Las fuerzas dipolo–dipolo se presentan en moléculas polares como resultado de las interacciones eléctricas entre dipolos de moléculas vecinas. Las fuerzas pueden ser de atracción o repulsión dependiendo de la orientación de las moléculas (Figura 5.3.3.3.1). Figura 5.3.3.3.1.- Fuerzas dipolo-dipolo. Así, la asociación se establece entre el extremo positivo (densidad δ+) de una molécula y el extremo negativo (densidad δ-) de otra. El origen es netamente electrostático. La figura 5.3.3.3.2 esquematiza la interacción entre moléculas de ClCH3 (clorometano, de naturaleza orgánica). Puede observarse la disposición que adquieren las moléculas con el fin de aumentar la atracción entre los dipolos. 109 Figura 5.3.3.3.2.- I nteracción entre moléculas de ClCH3 (clorometano). La fortaleza de la interacción dipolo-dipolo depende del tamaño de los momentos dipolares envueltos. Mientras más polar sea una sustancia, mayor será la fortaleza de las interacciones dipolo-dipolo. Existe una relación entre el momento dipolar y el punto de ebullición. Mientras más alto es el momento dipolar mas fuerte es la fuerza intermolecular y más grande es la cantidad de calor que se debe añadir para sobrepasar las fuerzas y la sustancia poder pasar a fase gaseosa (vapor). Por lo que sustancias con alto momento dipolar tienen alto punto de ebullición (Tabla 5.3.3.3.1) Tabla 5.3.3.3.1.- Puntos de ebullición en función del momento dipolar. 5.3.3.4.- Fuerzas de dispersión de London. Todos los átomos y moléculas polares y no-polares experimentan estas fuerzas de dispersión London que resultan del movimiento de los electrones. Se forman de dipolos instantáneos (Figura 5.3.3.4.1) en un átomo que puede afectar la distribución electrónica en átomos vecinos e inducir dipolos temporales en esos átomos vecinos. Como resultado se desarrollan fuerzas de atracción débiles. 110 Figura 5.3.3.4.1.- Formación de dipolos instantáneos (Dipolo instantáneo-dipolo inducido). Su magnitud depende de la facilidad con la cual la nube electrónica de una molécula puede ser distorsionada por un campo eléctrico cercano, propiedad conocida como polarizabilidad. Moléculas pequeñas o átomos livianos tienden a ser menos polarizables y tienen fuerzas de dispersión pequeñas porque tienen pocos electrones a diferencia de una molécula grande o átomo pesado que posea muchos electrones que serán más polarizables y sus fuerzas de dispersión serán mayores. Otro factor importante en la determinación de la magnitud de fuerzas de dispersión es la forma, moléculas con formas más dispersas maximizan el área de superficie molecular lo que permite mayor contacto entre moléculas y fuerzas de dispersión altas que las que poseen formas mas compactas. Enlace ión-dipolo inducido. La fuerza de enlace ión-dipolo inducido se presenta entre un ión y un dipolo inducido que resulta del movimiento de los electrones (Figura 5.3.3.4.2) Figura 5.3.3.4.2.- Fuerza de enlace ión-dipolo inducido Enlace dipolo inducido-dipolo (molécula polar). Las fuerzas dipolo–dipolo inducido se dan entre una molécula polar y otra no polar. La molécula polar induce un dipolo en la no polar. En algunas moléculas existen dipolos permanentes como consecuencia de la distribución asimétrica de las regiones cargadas positiva y negativamente, son las denominadas moléculas polares. La figura 5.3.3.4.3 es la representación esquemática de una molécula de fluoruro de hidrógeno, en la cual el momento dipolar permanente se origina a partir de las cargas positiva y negativa asociada a los extremos del hidrógeno y del flúor en la molécula de HF (La parte de la molécula donde se encuentra el flúor será negativa mientras la del hidrógeno será positiva). Las moléculas polares pueden inducir dipolos en las moléculas apolares próximas, y el enlace es la resultante de las fuerzas de atracción entre dos moléculas. Además, la magnitud de este enlace aumenta con los dipolos inducidos fluctuantes. 111 Figura 5.3.3.4.3.- Representación esquemática de una molécula polar de fluoruro de hidrógeno Enlace con dipolos permanentes Enlace dipolo inducido-dipolo inducido. En una molécula que normalmente es simétrica eléctricamente (Figura 5.3.3.4.4.a) se puede crear un dipolo inducido por la distribución espacial de los electrones respecto a los núcleos cargados positivamente, como se muestra en la figura 5.3.3.4.4.b. Todos los átomos están vibrando constantemente y pueden causar distorsiones instantáneas en la simetría eléctrica de los átomos y moléculas, creando pequeños dipolos eléctricos, como se muestra en la figura 5.3.3.4.4.b. A su vez, los dipolos suelen desplazar la distribución electrónica de átomos o moléculas próximas y generar otro dipolo que luego se enlaza débilmente al primero. Estas fuerzas atractivas que se originan entre gran número de átomos o moléculas son temporales y su magnitud fluctúa con el tiempo. Este tipo de enlace es el responsable de la condensación y, a veces, de la solidificación de los gases inertes y de otras moléculas eléctricamente neutras y simétricas, tales como H2 y Cl2. En aquellos materiales en los cuales predominan enlaces debidos a dipolos inducidos, las temperaturas de fusión y ebullición son extremadamente bajas. Son los enlaces intermoleculares más débiles. Figura 5.2.5.- Representación esquemática de: (a).- Atomo electricamente simétrico. (b).- Dipolo atómico inducido. Las energías de enlace asociadas a las fuerzas dipolo inducido-dipolo inducido son significativamente menores que las de los enlaces entre dipolos inducidos. 112 5.3.3.5.- Enlace de puente de hidrogeno. El tipo de enlace secundario más fuerte y que merece especial atención es el enlace por puente de hidrógeno, que es un caso particular del enlace de molécula polar covalente como el agua o el amoníaco, que poseen átomos de hidrógeno unidos a elementos muy electronegativos como el flúor, oxígeno o nitrógeno (Es una interacción primordialmente de tipo dipolo–dipolo especialmente fuerte). Tiene lugar entre moléculas con el hidrógeno unido covalentemente al flúor (como en el HF), al oxígeno (como en el H2O) y al nitrógeno (como en el NH3). Para cada enlace H - F, H - O o N - H, el electrón solitario del hidrógeno es compartido con otro átomo. Se da entre un átomo de hidrógeno con carga parcial positiva y un átomo electronegativo pequeño (normalmente N, O ó F). De ese modo, el extremo hidrógeno del enlace es esencialmente un simple protón cargado positivamente, sin electrones que lo apantallen. Este extremo de la molécula cargado más positivamente es capaz de generar una elevada fuerza de atracción con el extremo negativo de una molécula adyacente, pudiendo dar lugar a enlaces de tipo iónico con otros grupos semejantes como se muestra en la figura 5.3.3.5.1 para el HF y para el H2O. En esencia, este simple protón forma un puente entre dos átomos cargados negativamente. La magnitud del enlace por puente de hidrógeno es generalmente mayor que la asociada a otros tipos de enlaces secundarios y puede llegar hasta 51 kJ/mol (0.52 eV/molécula). Como consecuencia del enlace por puente de hidrógeno, las temperaturas de fusión y ebullición del fluoruro de hidrógeno (HF) y del agua H2O son normalmente altas, comparadas con sus pesos moleculares. Figura 5.3.3.5.1.- Representación esquemática del enlace de puente de hidrógeno en el HF y en el agua. 113 En el caso particular del agua, H2O (Figura 5.3.3.5.2), el par electrónico del enlace O-H pertenece casi totalmente al oxigeno, altamente electronegativo, constituyéndose en el polo negativo (δ-) de la molécula. Por su parte, el hidrógeno queda reducido prácticamente a su núcleo, es decir, forma el polo positivo (δ+) de la misma molécula. Dicho átomo de hidrógeno, deficiente en electrones, tratará de conseguirlos a través de la interacción con el par de electrones del átomo de oxígeno de otra molécula de agua. Así, el átomo de hidrógeno establece un puente entre dos moléculas. De ahí el nombre de este tipo de fuerza intermolecular. Este ordenamiento de las moléculas determina que en el espacio, cada molécula de agua se encuentre unida a otras. El número de moléculas de agua que permanecen unidas por enlace puente de hidrógeno puede diferir y es precisamente lo que determina el estado de agregación del agua en distintas condiciones de presión y temperatura. Figura 5.3.3.5.2.- Enlace de puente de hidrógeno en el agua. Longitudes de enlace y disposición 114 La presencia de enlace de hidrogeno en el H2O, NH3 y HF, justifica las anormalidades encontradas en sus puntos de fusión (Figura 5.3.3.5.3). Es también el responsable de la alta capacidad calorífica molar del agua líquida, así como de sus elevados calores de vaporización y de fusión. Figura 5.3.3.5.3.- Temperaturas de ebullición de algunos compuestos simples de hidrógeno. Las líneas conectan moléculas que contienen átomos del mismo periodo. Obsérvese el efecto del enlace de hidrógeno en los puntos de ebullición del agua, fluoruro de hidrógeno y amoníaco. La tabla 5.3.3.5.1 ordena los tipos de interacciones por su fortaleza. La energía de las interacciones dipolo– dipolo y de London disminuye muy rápidamente con la distancia. Tabla 5.3.3.5.1.- Energía de las interacciones interiónicas e intermoleculares En la tabla 5.3.3.5.2 se desglosa la contribución aproximada de cada tipo de fuerza a la energía total de interacción intermolecular para algunas sustancias. La contribución de las fuerzas de London puede ser mayor que la de las fuerzas dipolo–dipolo incluso en moléculas polares. 115 Tabla 5.3.3.5.2.- Contribuciones aproximadas a la energía total de interacción entre moléculas en kJ/mol. En la figura 5.3.3.5.4 se pueden ver las fuerzas intermoleculares entre las cadenas adyacentes de un polímero debidas a la atracción entre grupos cargados con distinto signo. Figura 5.3.3.5.4.-Fuerzas atractivas entre grupos cargados con distinto signo en cadenas adyacentes. Las moléculas no polares como el etano CH3-CH3 y el polietileno se atraen entre sí mediante fuerzas de dispersión de London débiles, que resultan de la interacción dipolo inducido - dipolo inducido. Los dipolos temporales o transitorios en el etano o a lo largo de la cadena de polietileno se deben a las fluctuaciones instantáneas de densidad en las nubes de electrones. Las energías de estas fuerzas se sitúan en torno a las 8.36 kJ por unidad, tanto en polímeros polares como no polares, siendo esta fuerza independiente de la temperatura. Las fuerzas de London son las principales fuerzas presentes entre las cadenas de polímeros fundamentalmente no polares, que se hallan en los elastómeros y en los plásticos blandos. 116 Es interesante recalcar que el metano, el etano y el etileno son todos gases, el hexano, el octano y el nonano son todos líquidos (a temperatura ambiente), mientras que el polietileno es un sólido céreo. Esta tendencia se debe fundamentalmente tanto al aumento de la masa molecular como al aumento de las fuerzas de London por molécula a medida que se incrementa la longitud de cadena. Suponiendo que la atracción entre unidades de metileno o metilo es de 4.36 kJ/mol, se calcula que la interacción entre moléculas de metano es igual a 8.36 kJ/mol, entre las moléculas de hexano es igual a 50.16 kJ/mol, y para un mol de cadenas de polietileno de mil unidades es igual a 16720 kJ/mol. ___________________________________________________________________________________ Problema. Determinar la energía de interacción aproximada presente en una cadena de polietileno de 1500 unidades de repetición en una solución de hexano liquido, suponiendo que la energía de interacción de una unidad 23 de metileno es de 8.36 kJ/mol. Hay 6x10 unidades por mol, así la energía de interacción por unidad de metileno es: 8360 J mol = 1.393x10−20 J 23 unidades mol 6 x10 mol Hay dos unidades de metileno por unidad de repetición de manera que habrá: 2 x1500 = 3000 unidades de metileno La energía de interacción que tendrá el polietileno será entonces de: 3000 unidades x1.393x10−20 J unidad = 4.18 x10−17 J ____________________________________________________________________________ Las moléculas polares tales como el cloruro de etilo CH3-CH2Cl y el cloruro de polivinilo [-(CH2-CHCl)n], PVC, (Figura 5.3.3.5.5), se atraen entre sí mediante interacciones dipolo - dipolo, que resultan de la atracción electrostática entre los átomos de cloro de una molécula y los átomos de hidrógeno de otra. Puesto que esta interacción dipolo-dipolo, cuya energía varia entre 4.36 y 25.08 kJ por mol de unidad de repetición en la molécula, depende de la temperatura, estas fuerzas disminuyen a medida que se aumenta la temperatura en los procesos de fabricación de los polímeros. Mientras que las fuerzas de London son típicamente más débiles que las fuerzas dipolo-dipolo, éstas también se hallan en los compuestos polares, tales como el cloruro de etilo y el PVC. Las fuerzas dipolo dipolo son características de muchos plásticos. Las moléculas altamente polares, como el etanol o el alcohol polivinílico y la celulosa se atraen entre sí mediante un tipo especial de interacción dipolo - dipolo denominada enlace de hidrógeno, en el cual los átomos de oxígeno o nitrógeno de una molécula son atraídos hacia los átomos de hidrógeno de otras moléculas. Estas son las fuerzas intermoleculares más potentes y alcanzan energías de hasta 41.8 kJ/mol por unidad de repetición. Los enlaces de hidrógeno intermoleculares pueden encontrarse habitualmente en fibras como el algodón, la lana, la seda, el nilón, los poliacrílicos los poliésteres y los poliuretanos. Los enlaces de hidrógeno intramoleculares son responsables de las configuraciones en hélice, que se observan en el almidón y en las proteínas. 117 Figura 5.3.3.5.5.- Interacciones dipolo-dipolo típicas entre moléculas de cloruro de metilo y segmentos de cadenas de policloruro de vinilo y poliacrilonitrilo. Es importante remarcar que el elevado punto de ebullición del nilón-66 (265 °C, Figura 5.3.3.5.6) es el resultado de una combinación de fuerzas de enlaces de hidrógeno, fuerzas de London y de dipolo - dipolo entre las cadenas de poliamidas. Los enlaces de hidrógeno disminuyen en número cuando se sustituyen los átomos de hidrógeno de los grupos amido del nilón por grupos metilo, y cuando se esterifican los grupos hidroxilo de la celulosa. Figura 5.3.3.5.6.- Enlaces de hidrógeno entre átomos de hidrógeno y oxígeno o nitrógeno en el nilón-66. Además de la influencia de las fuerzas intermoleculares, el enmarañamiento de las cadenas es también un factor determinante de las propiedades físicas de los polímeros. Aunque la cera de parafina y el HDPE son homólogos de peso molecular , relativamente alto, la longitud de cadena de la parafina es demasiado corta para permitir enmarañamiento y, por tanto, carece de la resistencia y de otras propiedades físicas del HDPE. La longitud crítica de cadena necesaria para que se produzca enmarañamiento depende de la polaridad y de la forma del polímero. Así, el número de átomos de la longitud crítica en la cadena del poli(metacrilato de metilo) (PMMA), el poliestireno (PS) y el poliisobutileno son de 208, 730 y 610 respectivamente. 118 5.4.- Forma molecular. Longitud y esbeltez molecular de los polímeros. No existe razón para suponer que las cadenas de las moléculas de los polímeros son estrictamente rectas, independientemente de la disposición en zig-zag del esqueleto atómico (Figura 5.4.1.b). Las cadenas con enlaces sencillos son capaces de rotar y curvarse en tres dimensiones. Al considerar la cadena de átomos de la figura 5.4.1.a se aprecia que el tercer átomo puede encontrarse en cualquier punto de la circunferencia de la base de un cono de semiángulo 70.5 º y su enlace forma un ángulo de 109.5 ° con el enlace de los otros dos átomos. (a) (c) Figura 5.4.1.- Representaciones esquemáticas de cómo la posición de los átomos de carbono (círculos llenos) en el esqueleto atómico influye en la forma de las cadenas de polímeros. En (a) el átomo que está más a la derecha puede hallarse en cualquier punto de la circunferencia discontinua y formar siempre un ángulo de, aproximadamente, 109 º con el enlace de los otros dos átomos. Se generan segmentos de cadena rectos y torcidos cuando los átomos se sitúan como en (b) y (c). La colocación de sucesivos átomos en la cadena origina segmentos de cadenas rectas, como indica la figura 5.4.1.b. Por otro lado, las cadenas también pueden curvarse y retorcerse cuando los átomos situados en otras posiciones de la cadena rotan, como se indica en la figura 5.4.1.c. De este modo, una simple cadena molecular compuesta de muchos átomos puede adquirir una forma parecida a la representada esquemáticamente en la figura 5.4.2, con multitud de dobleces, torceduras y pliegues. En esta figura también se indica la distancia entre los extremos de la cadena del polímero, r. Esta distancia es mucho menor que la longitud total de la cadena. 119 Figura 5.4.2.- Representación esquemática de una cadena molecular de un polímero simple, que tiene numerosos pliegues producidos por las rotaciones de los enlaces. Algunos polímeros consisten en un gran número de largas cadenas de moléculas que pueden doblarse, enrollarse y plegarse de modo parecido al esquematizado en la figura 5.4.2. Este comportamiento hace que las cadenas vecinas se entremezclen y se enreden extensamente. Muchas características importantes de los polímeros se deben a esta maraña molecular, como, por ejemplo, la gran elasticidad del caucho. Alguna de las características mecánicas y térmicas de los polímeros son función de la capacidad de los segmentos de cadenas para rotar en respuesta al esfuerzo aplicado o a las vibraciones térmicas. La flexibilidad rotacional depende de la estructura y de la química de la unidad monomérica. Por ejemplo., la rotación está dificultada en una región de un segmento de cadena con doble enlace (C = C). La substitución de átomos por grupos atómicos también restringe el movimiento rotacional de las cadenas. Por ejemplo, las moléculas de poliestireno, que tienen anillos bencénicos son más resistentes al movimiento rotacional que las cadenas de polietileno. Calculemos las dimensiones de una macromolécula cuyo peso molecular vale 700000 g/mol formada a partir del monómero etileno. Como el peso molecular del etileno vale 28 g/mol, resulta: GP = 700000 = 25000 28 (5.4.1) El número de enlaces de la cadena total será: N = 2(GP) = 50000 Y la longitud de la cadena extendida se calculará (Figura 5.4.3): L = Ndsen (θ ) (5.4.2) y sustituyendo 109.5 6 L = Ndsen = 50.000x154 x sen(54.75) = 6.28x10 pm. = 6.28 micras 2 120 Por otro lado, el diámetro o espesor de la cadena, D, será: D = 2rC + 4rH + y = 2rC + 4rH + d cos(54.75) (5.4.3) donde : rC = radio del átomo de carbono rH = radio del átomo de hidrógeno Sustituyendo en esta expresión sus respectivos valores de 77 y 37 pm, resulta: D = 2.77 + 4.37 + 90.6 = 391pm. luego: L 6.28x106 = = 16037 D 391.6 Lo que nos pone de manifiesto la esbeltez de las cadenas que se parecerían a hilos muy largos. Figura 5.4.3.- Cálculo de la longitud y esbeltez molecular de un polímero. Sin embargo, en los cálculos anteriores hemos considerado que la larga cadena del polímero estaba extendida adoptando la disposición de una línea quebrada, lo que solamente ocurre muy raras veces, ya que como cualquier enlace entre átomos de carbono permite la rotación completa de la molécula alrededor de su eje, resulta que la disposición habitual es la de una cadena muy enrollada. Comprobémoslo considerando el caso más sencillo de la molécula de butano (4 átomos de carbono). La situación del cuarto carbono respecto a los otros tres, supuestos fijos, podrá ser cualquier punto de la base de un cono formado al rotar el enlace C3-C4 sobre el eje C2-C3. La longitud de la generatriz del cono será 154 pm y su semiángulo θ igual a (180° - 109.5 º) 121 Cualquiera de todas estas posibilidades tiene igual probabilidad de existencia y se denominan conformaciones. La conformación es la geometría específica de una molécula. Las conformaciones se diferencian entre si por la extensión de la rotación sobre un enlace sencillo. La disposición del carbono C5 en la molécula con 5 carbonos seguirá un esquema constructivo similar. Considerando ahora las cadenas muy largas de los polímeros (50000 átomos de carbono en el ejemplo que presentábamos al principio), el número de conformaciones diferentes que podría adoptar la cadena sería infinito y, en principio, no hay razón alguna para suponer que existirá una cualquiera de ellas con preferencia a las demás. La cadena lineal extendida que habíamos supuesto en el cálculo de la longitud no sería entonces mas que una conformación particular y por ello su existencia sería muy improbable. Para estimar la distancia entre el principio y el final de la cadena, r, se asumira, en primera aproximación, que los segmentos de la cadena pueden moverse libremente, uno con respecto a otro, en todas las direcciones. La cadena estaría compuesta por N segmentos de longitud l. Se situara el principio de la cadena en el origen del sistema de coordenadas (xyz) y se construira la cadena paso a paso con ángulos (α, β, γ) aleatorios, siendo α, β, γ los ángulos que forman los segmentos de cadena con las direcciones positivas de los ejes Ox, Oy y Oz, respectivamente. La posición del final de la cadena vendra dada por: N x = l cos α1 + l cos α 2 + ................. + l cos α N = l ∑ cos α i i =1 N y = l cos β1 + l cos β 2 + ................. + l cos β N = l ∑ cos βi i =1 N z = l cos γ 1 + l cos γ 2 + ................. + l cos γ N = l ∑ cos γ i i =1 y la distancia principio-fin será. r = x2 + y 2 + z 2 2 2 2 2 2 N N r = x + y + z = l ∑ cos α i + l 2 ∑ cos βi + l 2 ∑ cos γ i i =1 i =1 i =1 2 2 N 2 ( l) = ∑ cos 2 α i + ∑∑ cos α i cos α j + ∑ cos 2 βi + ∑∑ cos βi cos β j + ∑ cos 2 γ i + ∑∑ cos γ i cos γ j ( l) = ∑ cos 2 α i + cos 2 βi + cos 2 γ i + ∑∑ cos α i cos α j + cos βi cos β j + cos γ i cos γ j ( l) =N+ r2 r2 r2 2 2 2 N N N N N N N N N i =1 i =1 j =1 j ≠i i =1 i =1 j =1 j ≠i i =1 i =1 j =1 j ≠i N ( i =1 N N ) i =1 j =1 j ≠i ( ) 1 N N ∑∑ cos αi − α j + cos αi + α j + cos βi − β j + cos βi + β j + cos γ i − γ j + cos γ i + γ j 2 i =1 j =1 ( ) ( ) ( ) ( ) ( ) ( ) j ≠i El segundo sumando del segundo miembro es cero debido a que (αi ± α j), (βi ± β j) y (γi ± γj) se distribuyen aleatoriamente. Por tanto: r 2 = Nl 2 o bien : r = l N (5.4.4) En una segunda aproximación se introduce un valor fijo para el ángulo entre dos enlaces de la cadena de átomos de carbono (Figura 5.4.4) y se supone que la rotación es libre alrededor de dicho ángulo. 122 Figura 5.4.4.- Rotación libre con ángulo de valencia fijo. En este caso se obtiene: r 2 = Nl 2 1 − cos θ 1 − cos θ , r =l N 1 + cos θ 1 + cos θ (5.4.5) La formula anterior no es válida cuando el valor de es proximo a 0 y a 180 º. El factor de corrección de la formula (5.4.5) con respecto a la (5.4.4) toma los valores: En una tercera aproximación se limita la rotación libre. Esto puede ilustrase con el etano y tiene en cuenta que cuando esta molécula gira alrededor del enlace C-C, tienen que superarse barreras de potencial como resultado de las interacciones entre los átomos de H (Figura 5.4.5). Para el polietileno la curva de potencial es similar. Figura 5.4.5.- Curva de potencial para la rotación. 123 El impedimento en la rotación hace necesario la introducción de un factor de corrección adicional en la formula (5.4.5) que nos da el valor de r. La formula teniendo en cuenta la limitación en la rotación: r 2 = Nl 2 1 − cos θ 1 + cos ϕ 1 − cos θ 1 + cos ϕ . , r =l N . 1 + cos θ 1 − cos ϕ 1 + cos θ 1 − cos ϕ (5.4.6) El factor nuevo contiene la cantidad <cosφ>, que es el valor medio de los cosenos de los ángulos de rotación. En el caso de rotación libre se tiene que <cosφ>= 0 (Todos los valores de son igualmente probables), pero difiere de cero cuando estan presentes las barreras de potencial. La figura 5.4.6, nos muestra el aspecto real que presentaría una molécula de butano que contiene 4 enlaces C - C que pueden rotar libremente Figura 5.4.6.- Modelo de molécula de butano que contiene 4 enlaces C - C que pueden rotar libremente. Las posiciones de los sucesivos enlaces son completamente aleatorias. En el caso de la cadena de polietileno compuesta por N (en el caso expuesto 50000) vectores de longitud L (154 pm ) la distancia princio-fin aplicando la formula (5.4.4) sería: r = 154 50000 = 34435 pm. y aplicando la formula (5.4.5) el doble, es decir 68870 pm. Resulta, en realidad, una longitud de cadena dos órdenes de magnitud inferior a la correspondiente de la conformación extendida, como resultado del arrollamiento habitual. Esta disposición habitual, muy arrollada, permite explicar las grandes elongaciones en el estirado de muchos polímeros: sus macromoléculas se estiran bajo una carga por rotación de los enlaces entre carbonos. 124 6.- Estructura molecular de las cadenas. 6.1.- Introducción. Es de destacar que aún con composiciones idénticas, con las mismas agrupaciones atómicas presentes y con la misma distribución estadística de pesos moleculares, los polímeros de diferentes estructuras o configuraciones de las cadenas moleculares ofrecen diferencias en sus propiedades y comportamientos característicos (por ejemplo las ramificaciones dificultan un buen empaquetamiento molecular, disminuyendo la densidad y las reticulaciones limitan el deslizamiento relativo bajo esfuerzos de tracción, impidiendo la termoplasticidad). Las técnicas modernas de síntesis de polímeros permiten un gran control sobre varias posibilidades de estructuraa moleculares, entre ellas las lineales, lineales ramificadas, entrecruzadas y reticuladas, y también varias configuraciones isoméricas. Las unidades monoméricas al enlazarse secuencialmente formando las macromoléculas dan lugar a estructuras diferentes por la diversidad de formas de unión que pueden realizarse entre las mismas durante la reacción de polimerización y por la diferente estructura que presentan. Así, por ejemplo, se forman estructuras de cadenas lineales y de cadenas ramificadas, representadas esquemáticamente en la figura 6.1.1. Un polímero lineal es una molécula polimérica en la cual los átomos se arreglan en una larga cadena (En las reacciones de polimerización las unidades estructurales se colocan en forma de una cadena totalmente lineal). Tal es el caso de polímeros como el polietileno, el polipropileno, el policloruro de vinilo o el poliestireno. Dicha cadena se denomina cadena principal. Por lo general, algunos de estos átomos de la cadena están enlazados a su vez, a pequeñas cadenas de átomos. Estas cadenas pequeñas se denominan grupos pendientes. Las cadenas de grupos pendientes son mucho más pequeñas que la cadena principal. Normalmente tienen unos pocos átomos de longitud, sin embargo la cadena principal posee generalmente cientos de miles de átomos. Cuando hablamos de polímeros nos estamos refiriendo a moléculas con pesos moleculares de cientos de miles, o aún millones. 125 Figura 6.1.1.- Tipos de cadenas macromoleculares lineales: (a).- Sencilla (b).- Ramificada La aparición de una macromolécula ramificada puede ocurrir, por ejemplo al polimerizar butadieno, CH2=CH-CH=CH2. Un posible proceso, con el catalizador adecuado, implica la ruptura y el crecimiento de la cadena principal a través del doble enlace 1-2. Pero al quedar intacto el enlace 3-4, éste tiene la posibilidad de generar cadenas laterales a partir de cualquier punto que lo contenga. Otra posibilidad la constituyen las reacciones de condensación en las que uno de los monómeros sea trifuncional. Al reaccionar con otro bifuncional de estructura adecuada puede igualmente generar estructuras ramificadas. 6.2.- Polímeros lineales. Los polímeros con estructuras lineales, tanto sencillas como ramificadas, se conocen con el nombre de TERMOPLÁSTICOS. Al calentar estos polímeros les comunicamos la energía suficiente para romper los débiles enlaces intermoleculares existentes entre las cadenas, posibilitándose los deslizamientos entre si de las cadenas componentes del polímero en virtud de la libre rotación alrededor de los enlaces entre carbonos. 126 Los termoplásticos, duros y compactos a temperatura ambiente, se reblandecen progresivamente por acción del calor hasta convertirse en líquidos, a temperaturas relativamente bajas (100 – 300 °C), lo que permite darles forma mediante moldeo por inyección, extrusión, etc, forma que posteriormente se estabiliza al enfriarlos. No les afecta la repetición de los procesos de calentamiento, fusión y enfriamiento (Son reciclables). Esta característica permite su manejo y facilita el conformado de las piezas, pero limita su utilización como elemento estructural a temperaturas muy moderadas. En un polímero lineal las unidades monoméricas se unen unas a otras formando cadenas sencillas. Estas largas cadenas son flexibles y se comportan como una masa de fideos. Las cadenas de los polímeros lineales no están unidas mediante enlaces químicos, sino que se unen entre sí mediante enlaces secundarios débiles de tipo físico (fuerzas de van der Waals), debido a lo cual se pueden mover unas con respecto a las otras bajo la influencia de tensiones externas relativamente pequeñas. En consecuencia, la rigidez de este tipo de polímeros es baja. Por el contrario, las propiedades de flujo estan fuertemente influenciadas por el enmarañamiento de las cadenas a su vez enrolladas (Las cadenas normalmente no están presentes en la forma extendida, sino formando un ovillo al azar), como puede verse en la figura 6.2.1, lo que provoca que los polímeros fundidos muestran viscosidades altas. Figura 6.2.1.- Ilustración del enmarañamiento de las cadenas a su vez enrolladas. La eficacia del empaquetamiento de la cadena se reduce con las ramificaciones con lo que se incrementa el volumen y, por tanto, también disminuye la densidad del polímero. Por su parte, la linealidad proporciona resistencia, mientras que la ramificación suministra tenacidad. Los polímeros ramificados tendrán más dificultad en interpenetrarse y por tanto fluirán más fácilmente que los que no lo son. Por otra parte, cuanto más largas sean las cadenas más fácilmente se enroscarán unas alrededor de otras dificultando el movimiento de las mismas y provocando mayor rigidez. Una propiedad muy interesante de este tipo de polímeros es que existe una temperatura más o menos definida a la cual las cadenas adquieren suficiente energía para desplazarse o deslizarse unas con respecto a las otras. A esta temperatura se la denomina temperatura de transición del estado vítreo y se representa por Tg. (Figura 6.2.2) 127 Figura 6.2.2.- Temperatura de transición del estado vítreo de los polímeros. Los polímeros termoplásticos son rígidos, duros y quebradizos (como el vidrio) por debajo de Tg y deformables (blandos y dúctiles) por encima de esa temperatura. Es importante tener presente que si la aplicación del polímero exige que posea rigidez a temperatura ambiente (por ejemplo, si va a utilizarse para construir tuberías o envases), debe cumplirse que Tg > TAMBIENTE. No obstante, no va a interesar tampoco que Tg sea demasiado grande pues esto dificultaría el procesado del polímero. Las teclas plásticas del teclado de un ordenador son rígidas, pero el plástico que recubre los cables de la misma computadora es blando. La Tg es distinta para cada plástico. A temperatura ambiente, algunos plásticos se encuentran por debajo de sus Tg, por lo tanto son rígidos. Otros plásticos se encuentran por encima de su Tg a temperatura ambiente y son blandos. La mayoría de los plásticos usados en ingeniería son termoplásticos: Polietileno, polipropileno, cloruro de polivinilo (PVC), poliestireno, poli(metacrilato de metilo), nilón y fluorocarbonos son algunos polímeros de estructura lineal. Las ramificaciones pueden ser de varios tipos: en árbol, en estrella, etc, como puede verse en la figura 6.2.3. Los polímeros con un alto grado de ramificación reciben el nombre de dendrímeros. Figura 6.2.3.- Diferentes tipos de ramificaciones. 6.3.- Polímeros entrecruzados. En los polímeros entrecruzados, las cadenas principales lineales adyacentes se unen transversalmente en varias posiciones mediante otras cadenas secundarias formando enlaces covalentes, como se representa en la figura 6.3.1.a. El entrecruzamiento se realiza durante la síntesis o por reacciones químicas irreversibles que normalmente ocurren a elevada temperatura. A menudo el entrecruzamiento va acompañado por la adición mediante enlace covalente de átomos o moléculas a las cadenas. Muchos de los materiales elásticos de caucho están entrecruzados. 128 Como se muestra en la figura 6.3.1.b y c la densidad de enlaces cruzados puede variar desde la baja densidad, por ejemplo, del caucho vulcanizado, hasta la alta densidad que se observa, por ejemplo, en la ebonita. Cuando se forman unos pocos enlaces fuertes intermoleculares, el resultado es la familia de polímeros denominada ELASTOMEROS. Si el polímero se procesa de tal forma que se produce una estructura ligeramente entrecruzada (densidad de enlaces cruzados baja) con largas longitudes de cadena en la unión transversal, entonces permanece lo bastante blando a alta temperatura como para permitir el movimiento de las cadenas, estando en estado gomoso. El caso más típico lo constituye la goma natural o poli-isopreno. El latex de caucho natural extraído del árbol no sirve de mucho. Gotea y se pone pegajoso cuando se lo calienta, y se endurece volviéndose quebradizo cuando se enfría. Las cubiertas para autos hechas de este latex, no serían muy buenas, a menos que uno viva en algún lugar donde la temperatura se mantuviera en unos 25 grados durante todo el año. La estructura entrecruzada tiende a ser mas estable, tanto térmica como mecánicamente, que la estructura de cadenas enmarañadas de los termoplásticos. La unión puede ser directa si se forma directamente entre las mismas cadenas o puede ser a través de una tercera molécula, cuando se hace con un injerto (Figura 6.3.2) (a) (b) (c) (d) Figura 6.3.1.-Representación de polímeros amorfos.(a).- Diferencia entre un grupo de cadenas poliméricas no entrecruzadas y una red entrecruzada (b).- Polímero entrecruzado (c).-Polímero reticular con densidad de enlaces cruzados baja. (d).- Polímero reticular con densidad de enlaces cruzados alta. 129 DIRECTO INJERTO Figura 6.3.2.- Entrecruzamiento directo o por medio de injertos. El monómero isopreno tiene dos dobles enlaces de los que únicamente gasta uno en su polimerización hasta la estructura lineal. El poli-isopreno sería entonces un termoplástico lineal no saturado (aún posee dobles enlaces que posibilitan una unión fuerte entre cadenas). La unión entre sus macromoléculas se realiza por adición de azufre en el proceso denominado de vulcanización. Los puentes formados por cadenas cortas de átomos de azufre unen una cadena de poli-isopreno con otra, hasta que todas las cadenas quedan unidas en una supermolécula gigante. Estos entrecruzamientos mantienen unidas a las moléculas poliméricas. Debido a ello, cuando el caucho se calienta, no pueden deslizarse una encima de la otra, ni siquiera una alrededor de la otra. Por esa razón el caucho no funde. Y también debido a que todas las moléculas están unidas, no pueden separarse unas de otras. Esto explica por qué el caucho vulcanizado no se vuelve quebradizo cuando se enfría Cuando la cantidad de azufre añadida es baja, sólo se forman unos pocos enlaces fuertes intermoleculares y el resultado es un ELASTOMERO. El proceso de formación de la goma natural se esquematiza en la figura 6.3.3. Los elastómeros son materiales muy elásticos. Al aplicarles una carga se origina un gran alargamiento (pueden ser estirados hasta muchas veces su propia longitud,) en virtud del estirado de las largas moléculas, muy enrolladas, que lo constituyen. Esta elasticidad es debida a la gran flexibilidad de las largas cadenas poliisoprénicas (hasta 5000 unidades), las cuales, unidas entre sí a través de fuerzas de van der Waals, apenas ofrecen resistencia. Al ser descargados los enlaces intermoleculares dispersos de azufre actúan como resortes y se recupera instantáneamente la forma original enrollada. Este efecto de carga y descarga se representa también en la figura 6.3.3. 130 (ISOPRENO) → Figura 6.3.3.- Esquema del proceso de formación de la goma natural. 131 Figura 6.3.3.- Esquema del proceso de formación de la goma natural. Otra característica notable de los elastómeros es el hinchamiento por absorción de líquidos. Las moléculas de los líquidos se introducen en la estructura de la goma cuyas macromoléculas se desenrollan para permitir su alojamiento, conforme se representa en la figura 6.3.4. Hay que tener en cuenta por otro lado que el comportamiento elástico típico de los elastómeros se pierde si en el proceso de vulcanización introducimos demasiado azufre, ya que aumentarían los fuertes enlaces intermoleculares y resultaría una estructura final tridimensional. Así, si la proporción de azufre se aumenta en 30-50 % se obtiene un polímero rígido, duro y frágil que se denomina ebonita y que se utiliza en la construcción de instrumentos musicales. (a) (b) (c) Figura 6.3.4.- Expansión de una red con enlaces covalentes cruzados en el hinchamiento. (a).- Sin hinchamiento (b).- Moderado grado de hinchamiento (c).- Alto grado de hinchamiento No todos los polímeros amorfos son elastómeros, ya que algunos son termoplásticos. Que un polímero amorfo sea termoplástico o elastómero, depende de su temperatura de transición vítrea, Tg. Si un polímero amorfo tiene una Tg por debajo de la temperatura ambiente, será un elastómero, porque es blando y elástico a temperatura ambiente, pero si tiene una Tg por encima de la temperatura ambiente, será un termoplástico, ya que a dicha temperatura es duro y quebradizo. De modo que, por regla general para los polímeros amorfos, tenemos que los elastómeros poseen bajas Tg y los termoplásticos poseen altas Tg. Hay que tener presente, que ésto sólo es aplicable para polímeros amorfos y no para polímeros cristalinos. 132 Entre los polímeros que son elastómeros se encuentran el poliisopreno o caucho natural, el polibutadieno, el poliisobutileno, y los poliuretanos. En el curado de resinas de poliéster no saturadas también se forman puentes entre las cadenas, en este caso con la ayuda del poliestireno. Un inconveniente de los polímeros entrecruzados es que no pueden ser fácilmente reciclados. De modo que, con el interés de preservar el medio ambiente, se ha sugerido una nueva propuesta, el elastómero termoplástico. La idea detrás de los elastómeros termoplásticos es la noción de un entrecruzamiento reversible. 6.4.-Polímeros reticulares o reticulados. Las unidades monoméricas trifuncionales, que tienen tres enlaces covalentes activos, forman redes tridimensionales (Figura 6.4.1), en lugar de las cadenas lineales generadas por las unidades monoméricas bifuncionales. Se forman así los polímeros reticulados que se caracterizan por la presencia de fuertes enlaces covalentes intermoleculares que constituyen una gigantesca estructura reticulada. En ellos también tiene lugar el enmarañamiento de las cadenas y los movimientos locales de los segmentos de las cadenas, pero el flujo no es posible. Polímeros tales como las resinas epoxy y poliesteres insaturados (Usadas como matrices en plasticos reforzados) y melamina formaldehído y resinas fenolicas (usadas en componentes eléctricos) pueden formar redes tridimensionales de cadenas cortas durante la reacción de curado, que en muchos casos (pero no en todos) ocurre a temperaturas elevadas. Esas uniones transversales son enlaces primarios covalentes, que pueden realizarse por la acción del calor como es el caso de los polímeros fenólicos, que se fabrican por moldeo por compresión en caliente o químicamente mezclando una resina y un catalizador, como es el caso de polímeros epóxicos, que se fabrican por procesado en estado liquido, con o sin aplicación de calor. Los polímeros compuestos por unidades trifuncionales (o más) se denominan polímeros reticulados. Un polímero con un grado de entrecruzamiento elevado, prácticamente, se puede clasificar también como polímero reticulado. Estos materiales tienen propiedades mecánicas y térmicas específicas. Los polímeros epoxy y los fenol-formaldehído pertenecen a este grupo. Figura 6.4.1.- Representación esquemática de una estructuras molecular reticulada (Los círculos representan unidades monoméricas). 133 Conviene recordar que algunos polímeros no pertenecen a un solo grupo. Por ejemplo, un polímero predominantemente lineal puede tener algún número limitado de ramas y de entrecruzamiento o de reticulación tridimensional. Los polímeros tridimensionales se denominan TERMOESTABLES. Por lo general, a los polímeros reticulados se los moldea y se les da la forma antes de reticularlos. Una vez que se produce el reticulamiento, usualmente a altas temperaturas, al material ya no se le puede dar forma. Dado que generalmente es el calor el que causa el reticulamiento que da una forma permanente, es por lo que a estos materiales se les denomina termoestables, termofijos o termorrígidos. Dicha denominación se diferencia de los termoplásticos, que no son reticulados y puede volver a dárseles forma una vez que fueron moldeados. Curiosamente, el primer termorrígido fue otra vez, el poliisopreno. Cuando más azufre se agregue al poliisopreno, más rígido se volverá éste. Si está poco entrecruzado, es un caucho flexible. Pero si se encuentra densamente entrecruzado, es decir formando prácticamente una red, es un termorrígido duro. Una vez que el material ha polimerizado la red tridimensional (3 - D) no puede romperse, a no ser que se llegue a la destrucción del polímero. Al calentar un polímero termoestable sus moléculas no deslizan entre si el polímero no fluye - debido a los fuertes enlaces intermoleculares. Solamente a temperaturas mayores suministramos la energía necesaria para romper cualquier enlace primario y el polímero se degrada sin fundir (se descompone a la temperatura de descomposición). En contraste, con los termoplásticos un polímero termoestable no puede ablandarse por la aplicación del calor. Esto hace que los polímeros termoestables sean más fuertes, más rígidos y capaces de aguantar temperaturas más altas que los termoplásticos (Cadenas ovilladas), debido a que las cadenas están ancladas unas a otras formando moléculas inmensas que impiden el deslizamiento entre ellas. Sin embargo, tienden a ser más frágiles. No son solubles, porque todas las cadenas poliméricas se encuentran unidas covalentemente. Pero pueden absorber solventes. Un material reticulado que ha absorbido gran cantidad de solvente, se denomina un gel. Un tipo de gel es la poliacrilamida reticulada. La poliacrilamida no reticulada es soluble en agua y las poliacrilamidas reticuladas pueden absorber el agua y se emplean para fabricar lentes de contacto blandas. El primer componente del epoxi es un polímero de bajo peso molecular con sendos grupos epoxi en cada extremo. Su estructura se parece a esto: Prepolimero con n variable entre 0 y 30. Para pegamentos n = 0, usualmente. La segunda parte es una diamina, similar a ésta: 134 Cuando se mezclan ambas partes, el diepoxi y la diamina reaccionan y se unen entre sí, de manera tal que se enlazan todas las moléculas del diepoxi y de la diamina, de esta forma: Lo que se forma, no es un polímero lineal, sino una red entrecruzada que se ve más o menos así: 135 Es decir que todas las moléculas de diamina y de epoxi se han convertido en una molécula gigantesca. Cuando ésto sucede, el resultado es una sustancia rígida que puede ser muy resistente, pero no procesable. No puede ser moldeada, ni siquiera fundida. Las resinas epoxi son excelentes pegamentos, siendo unos de los pocos que se pueden utilizar en los metales. Pero también se los utiliza como recubrimientos protectores, como materiales en objetos tales como tableros electrónicos y para emparchar agujeros en pavimentos de cemento. La baquelita es el polímero sintético más antiguo y fue descubierto por Baeckeland en 1907. Se prepara a partir de fenol y formaldehído. Presenta un gran número de enlaces entrecruzados y con grupos metileno en posiciones orto y para al grupo hidroxilo fenólico. La baquelita es un polímero termoestable, es decir, cuando se calienta se forman más enlaces cruzados, lo que produce mayor endurecimiento de este material resistente a la fusión. Este proceso es irreversible. Se obtiene por policondensación del fenol y formaldehído. Los monómeros que reaccionan tienen un grupo funcional reactivo en cada extremo de la molécula y la unión entre los monómeros requiere la pérdida de una molécula pequeña, normalmente agua. Son reacciones de equilibrio que requieren la separación de los productos secundarios (H2O,HCl, alcohol). Los pesos moleculares promedio no llegan a 100000 y cada cadena crece a una velocidad relativamente baja. Ejemplos de polímeros sintetizados por este mecanismo son poliésteres, poliamidas, poliuretanos, resinas urea-formaldehído. En la formación de la baquelita el primer paso es la sustitución electrofílica del fenol por el formaldehído en posiciones orto y para: En una segunda etapa, una parte del fenol hidroximetilado pierde H2O intramolecularmente: 136 En un tercer paso ocurre una reacción de adición con una molécula de fenol: La repetición sucesiva de los pasos 2 y 3 conduce a un compuesto de alto peso molecular, con muchas uniones covalentes transversales y, por consiguiente, con una estructura rígida: 7.- Configuraciones moleculares de los polímeros. 7.1.- Introducción. En los polímeros que tienen más de un átomo o grupo de átomos enlazados a la cadena principal, la regularidad y simetría de la disposición de este grupo repercute significativamente en las propiedades. Las moléculas con un carbono asimétrico (Monómeros asimétricos con átomo de carbono unido a un hidrógeno y a otro grupo diferente) pueden presentar diferentes morfologías denominadas configuraciones (El término configuración se refiere a la disposición de las unidades a lo largo del eje de la cadena, o a las posiciones de los átomos que sólo se alteran por escisión y posterior formación de enlaces primarios). Es el caso de los polímeros vinílicos obtenidos en la polimerización de monómeros vinílicos (CH2 = CHR), donde R, es un átomo o grupo atómico cualquiera (- Cl, - OH, - CH3 , etc.), diferente del hidrógeno. Son pequeñas moléculas conteniendo dobles enlaces carbono-carbono, que constituyen una gran familia de polímeros. El polímero vinílico más simple, es el polietileno, el cual se obtiene a partir del monómero etileno, llamado también eteno. Cuando polimeriza, las moléculas de etileno se unen por medio de sus dobles enlaces, formando una larga cadena de varios miles de átomos de carbono conteniendo sólo enlaces simples entre sí. 137 Los polímeros vinílicos más complejos se obtienen a partir de monómeros en los cuales uno o más de los átomos de hidrógeno del etileno han sido reemplazados por otro átomo o grupo atómico, pudiendo obtener un gran número de plásticos comunes, como son: Polipropileno Poliestireno Poli(cloruro de vinilo) Reemplazando dos átomos de hidrógeno, sobre el mismo átomo de carbono, podemos obtener poliisobutileno, que es un tipo de caucho. Poli (metacrilato de metilo) 138 No muchos monómeros en los cuales se hayan reemplazado los átomos de hidrógeno en ambos átomos de carbono son capaces de polimerizar. Pero un polímero que se obtiene a partir de un monómero sustituido en ambos átomos de carbono es el politetrafluoroetileno, que fabrica Du Pont y lo llama Teflon. Los polímeros vinílicos se obtienen a partir de monómeros vinílicos por medio de una gran variedad de rutas sintéticas, tales como: 1.- Polimerización vinílica por radicales libres 2.- Polimerización vinílica aniónica 3.- Polimerización vinílica catiónica 4.- Catálisis de Ziegler-Natta 5.- Polimerización catalizada por metalocenos Los monómeros asimétricos —tipo vinílico CH2 = CHR, como pueden ser el propileno o el cloruro de vinilo— pueden enlazarse de diferentes maneras entre ellos según se realice la unión por las posiciones denominadas cabeza o cola (Figura 7.1.1). Figura 7.1.1.- Uniones cabeza-cabeza, cabeza-cola en monómeros vinílicos. En la mayoría de los polímeros predomina la configuración cabeza-cola, pues en la configuración cabeza-cabeza aparece repulsión polar entre grupos R. Los grupos pendientes se sitúan cada dos átomos de carbono de la cadena. Las moléculas de polímeros que tienen la misma composición y varias configuraciones atómicas constituyen el fenómeno denominado isomería. Dos subclases isoméricas, la estereoisomería y la isomería geométrica, se van a describir a continuación. 139 7.2.- Estereoisomería. Tacticidad. La estereoisomería o tacticidad indica la situación de los átomos enlazados en el mismo orden (cabeza-cola) pero con diferente disposición espacial, es decir es la forma en la que se encuentran dispuestos los grupos pendientes a lo largo de una cadena polimérica. En el caso de enlazarse las unidades monoméricas vinílicos cabeza-cola, se obtienen otros tipos de estructuras macromoleculares, cuya diferencia radica en la disposición espacial que ocupan los sustituyentes. La incorporación de las unidades monoméricas a la cadena en crecimiento puede hacerse siempre conservando la misma disposición espacial, es decir cada unidad monomérica se incorpora de idéntica manera a la cadena, con lo que existirá una gran ordenación espacial. De esta manera todos los sustituyentes -CH3 o -Cl del propileno o del cloruro de vinilo respectivamente, puestos como ejemplo anteriormente, se encuentran en el mismo lado del plano que contiene la cadena principal, representado en la figura 7.2.1 en el caso (a). Esta estructura del polímero se denomina isotáctica. (a) (b) (c) Figura 7.2.1.- Estructuras que pueden adoptar las macromoléculas formadas por olefinas asimétricas. Estereoregularidad de un polímero vinilico con tres configuraciones (a) isotactica (b) sindiotáctica y (c) atáctica. La forma molecular puede cambiarse, en su zig-zag planar, por rotación alrededor de los enlaces C-C. Esto cambiará la conformaqcion de la molécula, pero no cambia la configuración, que se establece en el instante de la polimerización. Si la incorporación de las unidades monoméricas a la cadena en crecimiento se hace de manera alternante en cuanto a su posición espacial, como en el caso (b), los sustituyentes de unidades monoméricas contiguas en la cadena se encuentran en lados opuestos del plano de la misma. La estructura resultante se denomina sindiotáctica. Por último, si las unidades monoméricas se incorporan de manera aleatoria a la cadena en crecimiento como en el caso (c) sin mantener ninguna ordenación, se obtiene una estructura totalmente irregular denominada atáctica. La conversión de un estereoisómero en otro (por ejemplo, isostático a sindiotáctico) no es posible por la simple rotación del enlace de cadena simple. Estos enlaces primero se deshacen y, después de una rotación apropiada, se vuelven a constituir. En realidad, los polímeros específicos no presentan una sola configuración, pero la forma predominante depende del método de síntesis. Para ilustrar la tacticidad, vamos a usar un polímero vinílico como es el poliestireno. 140 Muchas veces el poliestireno es representado como una figura plana similar a ésta: Pero los polímeros no son en realidad así de planos. Los átomos de carbono no se encuentran en línea recta como esa, ni tampoco los hidrógenos y los grupos fenilo están situados de modo que forman ángulos rectos. La cadena carbonada constituye más bien un zigzag como éste: Los grupos pendientes tienden a dirigirse fuera de la cadena, de este modo: Se puede apreciar que los grupos fenilo se encuentran dispuestos del mismo lado de la cadena polimérica. Pero no tienen por qué hacerlo así, ya que los grupos fenilo pendientes pueden estar tanto a la izquierda como a la derecha de la cadena. Si todos los grupos fenilo se encuentran del mismo lado de la cadena, decimos que el polímero es isotáctico. Si los grupos fenilo aparecen alternadamente a ambos lados de la cadena, se dice que el polímero es sindiotáctico. Si los grupos fenilo están distribuidos al azar a izquierda y derecha, sin ningún ordenamiento particular, decimos que el polímero es atáctico. 141 Cuando los polímeros tienen un ordenamiento regular en sus átomos, tal como vemos en el poliestireno isotáctico y sindiotáctico, les resulta sumamente fácil empaquetarse en forma de cristales y fibras. Pero si no existe ordenamiento, como en el caso del poliestireno atáctico, el empaquetamiento no se produce. Esto es porque las moléculas se agrupan mejor con otras moléculas de la misma forma. El poliestireno atáctico es un plástico rígido y completamente amorfo. No cristaliza en absoluto. Fue entonces como apareció la polimerización vinílica catalizada por metalocenos y fue posible obtener el poliestireno sindiotáctico. No sólo es cristalino, sino que además funde a los 270 oC. En la figura 7.2.2 pueden verse las formulas de esqueleto del polipropileno (PP) isotactico, sindiotactico y atactico. Figura 7.2.2.- Formulas de esqueleto del polipropileno (PP) isotáctico, sindiotáctico y táctico. El PP isotactico es un polimero altamente cristalino con un punto de fusion de 160 °C, mientras que el isomero atactico es un polimero amorfo (no cristalino), blando y pegajoso, no muy resistente y con un punto de fusion de 75 °C. Es un polímero no demasiado útil. El termino eutactico se usa para describir polimeros tanto isotacticos, como sindiotacticos, asi como a las mezclas de ambos. 142 Mientras que la mayoria de los polimeros contienen un solo centro quiral o asimetrico en la unidad de repeticion, es posible que haya diisotacticidad cuando se tienen dos radicales (R y R') en los centros quirales. Estos isomeros se denominan isomeros eritro- y treodiisotacticos y eritro- y treosindiotacticos y se muestran en la figura 7.2.3 Figura 7.2.3.- Formulas de esqueleto de isomeros ditacticos. 7.3.- Isomería geométrica. En las unidades monoméricas que tienen dobles enlaces entre átomos de carbono son posibles otras importantes configuraciones de la cadena: los isómeros geométricos. Los átomos o radicales unidos a los carbonos que intervienen en el doble enlace pueden situarse al mismo lado o a lados opuestos de la cadena. Al considerar la unidad monomérica del isopreno con la estructura se aprecia que el grupo CH3 y el átomo H se sitúan al mismo lado de la cadena. Esta estructura se denomina cis y el isómero resultante es el cis-isopreno o caucho natural. La alternativa, con el CH3 y el H en lados opuestos de la cadena, es la estructura trans. El trans- isopreno, a veces denominado gutapercha, tiene propiedades muy diferenciadas del caucho natural como consecuencia de la alteración de la configuración. La conversión de trans a cis, o viceversa, no es posible, ya que el doble enlace es excesivamente rígido y no permite la rotación de la cadena. 143 Las configuraciones " cis " y " trans " se ilustran para el poliisopreno de la figura 7.3.1. La diferencia entre éstas estriba en la posición de los grupos hidrógeno ( - H ) y metilo ( - CH3) respecto del doble enlace no giratorio: en la configuración cis (goma natural) ambos grupos se sitúan a un mismo lado respecto del doble enlace, mientras que en el poliisopreno trans(gutapercha) se disponen en lados opuestos. Como se puede observar el isomero trans tiene una estructura más regular lo que permite su cristalización, como resultado la gutapercha es dura y rígida. Por su parte, el isómero cis posee una estructura menos simétrica, lo que no permite su cristalización, con lo que la goma natural es amorfa y elástica. Finalmente conviene diferenciar claramente los conceptos conformación y configuración. Mientras el paso de una conformación a otra es muy fácil gracias a la libertad de giro de los enlaces sencillos de carbono, el cambio de configuración exige la rotura de los enlaces intramoleculares. Resumiendo lo dicho en las secciones precedentes, las moléculas poliméricas se caracterizan por el tamaño, la forma y la estructura. El tamaño molecular se especifica por el peso molecular (o grado de polimerización). La forma molecular se relaciona con el grado de torsión, doblado y plegado de la cadena. La estructura molecular depende del modo de unión de las unidades estructurales entre sí. Son posibles, por un lado, las estructuras lineal, ramificada, entrecruzada y reticulada y, por otro, varias configuraciones isoméricas: isotáctica, sindiotáctica, atáctica, cis y trans. Estas características moleculares se presentan en el cuadro sinóptico de la figura 7.3.2. Se observa que algunos elementos estructurales no son excluyentes y por este motivo se necesita especificar la estructura molecular en función de más de un elemento. Por ejemplo, un polímero lineal también puede ser isotáctico. trans Figura 7.3.1.- Estructuras moleculares de : (a) goma natural (cis - poliisopreno) , (b) gutapercha (trans poliisopreno). o Grupos metilo • Carbonos. Los átomos de hidrógeno no se muestran. 144 Figura 7.3.2.- Esquema de la clasificación de las características de las moléculas poliméricas. 8.- Copolímeros. Elastómeros termoplásticos. 8.1.- Copolimeros. Si en la formación de la macromolécula durante la polimerización ha intervenido solamente un tipo de monómero, el polímero que se forma se denomina homopolímero. Por el contrario, si han sido dos, tres o más monómeros los que han formado la macromolécula, el polímero se denomina copolímero y terpolimero respectivamente y las unidades estructurales, comonómeros. La obtención de copolímeros resulta una práctica muy habitual en el sector de los plásticos para conseguir estructuras con propiedades específicas que cada homopolímero por separado no es capaz de ofrecer. Variando las proporciones relativas de cada monómero pueden obtenerse características mecánicas y de procesado adecuadas para aplicaciones específicas. Las propiedades que pueden controlarse por cambios en la composición del copolímero incluyen: el módulo elástico, la tenacidad, la viscosidad del fundido y la estabilidad térmica. Además, la distribución de los respectivos monómeros dentro de las cadenas del polímero también influyen en las propiedades del copolímero. 145 La combinación de monómeros para formar copolímeros puede compararse con la mezcla de metales para formar soluciones sólidas, que es la base de la formación de las aleaciones. Considérese un copolímero compuesto por dos unidades monoméricas, representadas por un circulo negro y por uno rojo en la figura 8.1.1. Figura 8.1.1.- Representación esquemática de copolímeros. (a).- Al azar (b).- Alternados (c).- En bloque (d).- De Injerto Dependiendo del proceso de polimerización y de las fracciones de las unidades monoméricas, es posible obtener diferentes tipos de secuencias en las cadenas de polímeros. Si las dos unidades monoméricas están distribuidas aleatoriamente a lo largo de la cadena, como representa la figura 8.1.1.a, se denominan copolímeros al azar. En los copolímeros alternados, como indica su nombre, las dos unidades monoméricas se van alternando en las posiciones de la cadena, como muestra la figura 8.1.1.b. En los copolímeros en bloque las unidades monoméricas (comonómeros) se encuentran separadas en largas secciones de la cadena polimérica principal. Cada una de estas secciones, llamadas bloques, se ve como si fuera una especie de homopolímero. (Figura 8.1.1.c). Finalmente, en los copolímeros de injerto, la cadena principal está formada por un solo tipo de unidad monomérica y todas las cadenas laterales están constituidas por el otro tipo de unidad monomérica (figura 8.1.1.d). Copolimeros de etileno y butileno, sintetizados a, relativamente, bajas presiones dan lugar a los polietilenos 3 de baja densidad lineal (LLDPE), en los que ρ = 0.92-0.95 g/cm . 146 En la figura 8.1.2 se dan ejemplos de copolímeros y se comparan con un homopolímero. Cloruro de polivinilo Estireno-Isopreno Butadieno-Estireno (SBR, GRS) Figura 8.1.2.- Homopolímero. Ejemplos de copolímeros Un copolímero en bloque conocido, siempre y cuando se usen zapatos, es el caucho SBS (estireno-butadienoestireno). Se emplea para las suelas de los zapatos y también para las cubiertas de los neumáticos. La cadena principal de dicho copolímero está constituida por tres segmentos. El primero es una larga cadena de poliestireno, el del medio es una cadena de polibutadieno, y el último es otra larga sección de poliestireno. 147 Si se pudiera extender una cadena de SBS, se vería como: El poliestireno es un plástico duro y resistente y le da al SBS su durabilidad. El polibutadieno es un material parecido al caucho y le confiere al SBS sus características similares al caucho. Además, las cadenas de poliestireno tienden a agruparse formando grandes masas. Cuando un grupo estireno de una molécula de SBS se une a una de estas masas y la otra cadena de poliestireno de la misma molécula de SBS se une a otra masa, las diversas masas se ensamblan entre sí con las cadenas similares al caucho del polibutadieno. Esto le da al material, la capacidad de conservar su forma después de ser estirado. 148 Un tipo de copolímero de injerto es el poliestireno de alto impacto, abreviado en inglés como HIPS. Consta de una cadena principal de poliestireno y cadenas de polibutadieno injertadas en dicha cadena principal. El poliestireno le confiere resistencia al material, en tanto que las cadenas del elastómero polibutadieno le otorgan la elasticidad suficiente como para lograr que sea menos quebradizo. Así las cadenas elastómericas colgando de la cadena principal son altamente beneficiosas para el poliestireno. Los homopolímeros de polibutadieno y poliestireno no se combinan entre sí. De modo que las ramas de polibutadieno tratan de provocar una separación de fases y forman pequeñas bolitas 149 Pero esas pequeñas bolitas siempre estarán unidas a la fase de poliestireno y, por lo tanto, ejercen un efecto sobre ese poliestireno. Actúan para absorber energía cuando el polímero es golpeado con algo, confiriéndole una resistencia que el poliestireno normal no posee. Este copolímero de injerto permite que la tensión sea transferida de la fase poliestireno a la fase polibutadieno. Dado que el polibutadieno es elástico, disipa la energía que de otra forma causaría la ruptura de la frágil fase de poliestireno. Esto lo hace más fuerte, no quebradizo y capaz de soportar impactos más violentos, sin romperse como el poliestireno normal. El HIPS puede ser mezclado con un polímero llamado poli(óxido de fenileno), o PPO. Esta mezcla de HIPS y PPO es manufacturada por General Electric (GE) y comercializada como NorylTM En las síntesis de cauchos se suelen utilizar copolímeros. Las unidades monoméricas empleadas en alguno de estos cauchos se indican en la tabla 8.1.1. Tabla 8.1.1.- Unidades monoméricas utilizadas como copolímeros del caucho 150 El caucho al nitrilo (NBR) es un copolímero al azar, compuesto de acrilonitrilo y butadieno y es altamente elástico y resistente al hinchamiento frente a los disolventes orgánicos. Se usa para fabricar mangueras para conducir gasolina. CH2 CN CN CH CH CH2 CH2 C C CH2 H H n n ´ ACRILONITRILO BUTADIENO Las propiedades generales del NBR son: - Conocidos como caucho nitrilos o nitrílicos. - Polimerización en emulsión a bajas temperaturas (5 - 8 oC). - Mayor resistencia a los disolventes hidrocarbonados, en especial a aceites y solventes. - Se disuelve en: acetona, ésteres y aminas. - Baja permeabilidad al aire y otros gases. - Debido al acrilonitrilo se logra ganar resistencia a la abrasión, dureza y resistencia al calor. - Baja resistencia al ozono y a la intemperie. - Buenas propiedades mecánicas, como tracción, compresión, flexión. - Moderado envejecimiento. - Buena adhesividad al acero. Sus usos son: Mangueras (gasolina, jardin, etc), guantes, calzados industriales, rodillos de imprenta, mangos, juntas, diafragmas, saturantes de papel, acabados de textiles y pieles, adhesivos, ligantes de fibras no tejidas. 151 Como ejemplo de terpolimero se tiene al ABS (Acrilonitrilo – butadieno – estireno), que es un plástico muy fuerte y muy poco pesado, que se usa para parachoques de coche. CH2 CN CH2 CH CH2 m CH CH CN m´ CH2 CH CH CH2 CH2 CH m m´ n Los copolímeros y terpolímeros de mayor aplicación en la industria son los siguientes: SAN. Copolímero de estireno-acrilonitrilo en los que el contenído de estireno varía entre 65 y 80 %. Estos materiales tienen buena resistencia a los aceites lubricantes, a las grasas y a las gasolinas. Asimismo, tiene mejores propiedades de impacto, tensión y flexión, que los homopolímeros del estireno. Los copolímeros son transparentes, pero con un ligero color amarillo que se vuelve más oscuro a medida que aumenta el contenido en acrilonitrilo. Al mismo tiempo mejora la resistencia química, la resistencia al agrietamiento ambiental y la resistencia térmica al aimentar el porcentaje en acrilonitrilo. El SAN se usa cuando se requieren partes rígidas, con buena estabilidad dimensional y buena resistencia térmica, por ejemplo, en partes de las máquinas lavaplatos y en piezas para radios u televisores. Se lo emplea en grandes cantidades en la industria alimenticia. los copolímeros con 30 % estireno y 70 % acrilonitrilo, son excelentes barreras contra el oxígeno, el CO2 y la humedad. 152 ABS. Terpolímero acrilonitrilo-butadieno-estireno. Son materiales heterogéneos formados por una fase homogénea rígida y una elastomérica. El ABS es un plástico más fuerte que el poliestireno debido a la presencia de los grupos nitrilo en sus unidades de acrilonitrilo. Los grupos nitrilo son muy polares, así que se atraen mutuamente. Esto permite que las cargas opuestas de los grupos nitrilo puedan estabilizarse Esta fuerte atracción sostiene firmemente las cadenas de ABS, haciendo el material más fuerte. Originalmente se mezclaban emulsiones de los dos polímeros de SAN y polibutadieno. La mezcla era coagulada para obtener ABS. Ahora se prefiere polimerizar estireno y acrilonitrilo en presencia de polibutadieno. De esa manera, una parte del estireno y del acrilonitrilo se copolimerizan formando SAN y otra porción se injerta sobre las moléculas de polibutadieno. El ABS se originó por la necesidad de mejorar algunas propiedades del poliestireno de alto impacto. Este material tiene tres desventajas importantes: - Baja temperatura de ablandamiento. - Baja resistencia ambiental. - Baja resistencia a los agentes químicos. La incorporación del acrilonitrilo en la fase continua, imparte mayor temperatura de ablandamiento y mejora considerablemente la resistencia química. Sin embargo, la resistencia ambiental se vuelve todavía menor, pero este problema se resuelve empleando aditivos. Las propiedades del ABS son suficientemente buenas para varias aplicaciones: - Artículos moldeados, - Artículos extruidos. 153 Resina Noryl GTX® PPE/PA Noryl GTX® es una mezcla de PPE/PA. Un producto de poliamida (PA) reforzado con el polímero éter de polifenileno (PPE). Esta tecnología combina la estabilidad dimensional, baja absorción de agua y resistencia al calor (ventajas inherentes al polímero PPE) con la resistencia química y fluidez del polímero de nylon. El resultado es un material de extraordinaria resistencia química con la rigidez, resistencia al impacto y el rendimiento térmico que requiere la pintura en línea. La baja densidad de la resina Noryl GTX sin carga puede generar ahorros en el peso de las piezas de hasta un 25 % con respecto a las resinas con carga de vidrio o mineral. Las resinas Noryl GTX ofrecen amplia resistencia ambiental a los combustibles, grasas y aceites de uso corriente en automóviles. Además, estas resinas son resistentes a los detergentes, alcoholes, hidrocarburos alifáticos y aromáticos y agentes químicos alcalinos. Cuando una aplicación requiere que el producto esté expuesto a, o sumergido en estos u otros entornos agresivos, deben probarse prototipos o muestras del material sometidas a una tensión adecuada, en condiciones reales de funcionamiento. Las resinas Noryl GTX ofrecen: 1.- Resistencia a impactos a baja temperatura. 2.- Alta resistencia al calor 3.- Resistencia química 4.- Baja contracción en el molde. 5.- SE pueden pintar en línea. 6.- Capacidad térmica a corto plazo superior a 204 ºC. 7.- Buena estabilidad dimensional 8.- Aspecto superficial (Clase A). 9.- Absorción lenta de agua Entre las aplicaciones habituales de las resinas Noryl GTX® cabe citar: 1.- Tapacubos para automóviles. 2.- Paneles de carrocería verticales exteriores para automóviles. 3.- Carcasas de espejos y componentes de ornamentación exterior para automóviles 4.- Componentes de sistemas de tratamiento de materiales. 5.- Componentes de tratamiento de fluidos Copolímeros estireno-butadieno. Éstos son los hules sintéticos que han sustituído practicamente en su totalidad al natural, en algunas aplicaciones como las llantas para automóviles. Los hules sintéticos contienen 25 % de estireno y 75 % butadieno; sus aplicaciones incluyen en oreden de importancia: - Llantas, - Espumas, - Empaques, - Suelas para zapatos, - Aislamiento de alambres y cables eléctricos, - Mangueras. Los copolímeros de estireno-butadieno con mayor contenido de batadieno, hasta de 60 %, se usan para hacer pinturas y recubrimientos ahulados. Para mejorar la adhesividad, en ocasiones se incorpora el ácido acrílico o los ésteres acrílicos, que elevan la polaridad de los copolímeros. 154 Otros copolímeros del estireno. MBS. Es un terpolímero de metacrilato de metilo-butadieno-estireno y se obtiene injertando metacrilato de metilo o mezclas de metacrilato y estireno, en las cadenas de un hule de estireno-butadieno. Acrílicos. Copolímeros de metacrilato-butilacrilato-estireno o de metacrilato-hexilacrilato-estireno. Otros copolímeros importantes del estireno, se realizan polimerizando en suspensión, estireno en presencia de divinil-benceno, para obtener materiales entrecruzados, que por sulfonación y otras reacciones químicas se convierten en las conocidas resinas de intercambio iónico. Poliestireno de alto impacto. Para hacer este material, se dispersa un elastómero en una matríz que puede ser de poliestireno o de algunos de sus copolímeros. Las variables importantes de la fase continua son: - Distribución de pesos moleculares. - Composición, cuando se trata de un copolímero. Las variables importantes de la fase elastomérica son: - Número, tamaño, distribución de tamaños y formas de las partículas dispersadas. - Composición, si es un copolímero. - Grado de entrecrusamiento en el elastómero. Existen dos procedimientos para obtener poliestireno de alto impacto: - Mezclar poliestireno directamente con el elastómero. - Mezclar estireno, el elastómero, el catalizante y el acelerante y se produce la polimerización. CPE. Los polietilenos clorados se obtienen clorando polietileno de alta densidad con 30 % a 40 % de cloro. Tiene baja cristalinidad y baja temperatura de transición vítrea. Un nivel de cloro del 36 % resultó experimentalmente para un buen balance al impacto-dispersabilidad-procesabilidad. EVA. Copolímero del etileno y acetato de vinilo, con 30 % a 50 % del acetato, posee propiedades elastoméricas. Puede ser utilizado para mejorar la flexibilidad, resistencia y rigidez de otras resinas. Entres sus aplicaciones se pueden citar las siguientes: adhesivos, selladores y laminados, cables, envases flexibles, calzado y asfalto La cadena siguiente representa seis unidades monoméricas: 155 8.2.- Elastómeros termoplásticos. 8.2.1.- Introducción. Los polímeros entrecruzados no pueden ser reciclados fácilmente. De modo que, con el interés de preservar el medio ambiente han surgido los elastómeros termoplásticos. La idea que subyace detrás de los elastómeros termoplásticos es la noción de un entrecruzamiento reversible. Los polímeros entrecruzados normalmente no pueden ser reciclados porque no funden, ya que el entrecruzamiento mantiene unidas las cadenas, impidiendo que el material sea capaz de fluir. Aquí es donde interviene el entrecruzamiento reversible. Los retículos normales son covalentes, uniendo químicamente a las cadenas poliméricas en una sola molécula. El entrecruzamiento reversible emplea interacciones secundarias no covalentes para unir entre sí a las cadenas. Estas interacciones incluyen los enlaces por puente de hidrógeno y los enlaces iónicos. Al emplear interacciones no covalentes para formar retículos es que cuando al calentar el material se rompen los retículos. Esto permite que dicho material pueda ser procesado, y lo más importante, reciclado. Cuando se enfría, los retículos vuelven a formarse. Se han intentado dos métodos, ionómeros y copolímeros en bloque. 8.2.2.- Ionómeros. Tal como podría suponerse por su nombre, un ionómero es un polímero que contiene un ión. Pero un ionómero es algo más que un polímero con grupos iónicos. Cualquier polímero con grupos iónicos se denomina un polielectrolito. Los electrolitos son componentes que tienden a separarse. Si se los coloca en agua, se separarán en iones positivos y negativos. Por ejemplo, la sal de mesa es un electrolito. Cuando se la disuelve en agua, se separa en iones positivos sodio y en iones negativos cloruro. 156 Los polielectrolitos son polímeros que hacen lo mismo, se separan en el agua. Así, el (poli)ácido acrílico Si se lo coloca en contacto con el agua, los hidrógenos del ácido se apartarán con las moléculas de agua, formando iones H3O+: y el polímero quedará en cambio con un cúmulo de grupos con carga negativa. Esto ocasiona un extraño comportamiento de la cadena polimérica. Cuando están en solución, las moléculas neutras tienden a enrollarse formando ovillos al azar. Pero cuando las cadenas poliméricas se encuentran pobladas de cargas negativas (lo cual produce su repulsión mutua) el polímero no puede enrollarse. Por lo tanto la cadena se estira, de esta forma: Esto hace que la solución se vuelva más viscosa (estamos hablando de polielectrolitos en solución). Cuando la cadena de polielectrolito se expande, ocupa más espacio y se hace más resistente a fluir a través de las moléculas de solvente que la rodean. Por lo tanto la solución se vuelve espesa. Pero existe una forma para evitar ésto. Si se parte de una solución de polielectrolito en agua y se agrega bastante NaCl se separará en iones Na+ y Cl-. En el caso de un polielectrolito con cargas negativas, como el poli(ácido acrílico), los iones Na+ cargados positivamente se interpondrán entre las cargas negativas del polímero y las neutralizarán. Cuando esto sucede, la cadena polimérica colapsa y vuelve a formar ovillos al azar. 157 Pero un ionómero es un tipo especial de polielectrolito. En primer lugar, es un copolímero, que está formado por unidades repetitivas no iónicas y pequeñas cantidades de unidades repetitivas conteniendo iones. Así, los ionómeros son un tipo de copolímeros en los cuales una pequeña porción de unidades repetitivas posee grupos iónicos pendientes (menos del 15 % del polimero). Por lo general, la cadena polimérica principal es no polar. Un ejemplo de un ionómero es el poli(etileno-co-ácido metacrílico). Este polímero es una sal sódica o una sal de zinc (que aporta los iones) de copolímeros derivados del etileno y del ácido metacrílico. Las atracciones iónicas que se manifiestan, ejercen una gran influencia en las propiedades del polímero Las cadenas principales no polares se agrupan entre sí y los grupos iónicos polares pendientes lo hacen por su lado (Figura 8.2.2.1). Pero cuando estas agrupaciones o "clusters" de grupos iónicos quieran separarse completamente de las cadenas apolares, no podrán. De modo que el resultado final es que estos clusters de grupos iónicos servirán para mantener juntas a esas cadenas principales, tal como lo haría un entrecruzamiento normal. Las atracciones iónicas que se manifiestan, ejercen una gran influencia en las propiedades del polímero. 158 Figura 8.2.2.1.- Ionómero. Agrupación de los grupos iónicos polares No obstante, los ionómeros no son polímeros entrecruzados, sino un tipo de termoplástico llamado entrecruzante reversible. Existe una pequeña diferencia. Si se calientan estos ionómeros, ocurre algo muy conveniente y es que los clusters iónicos se rompen. Cuando se calientan, los grupos iónicos dejan de atraerse y las cadenas comienzan a moverse libremente (Figura 8.2.2.2). A medida que la temperatura aumenta, las cadenas se mueven más y más rápido y los grupos ya no pueden quedarse en sus lugares de partida y, por lo tanto, se rompen. Ahora el ionómero ha perdido su reticulación y puede ser procesado y reciclado como un polímero común. Si lo enfriamos nuevamente, los clusters se formarán una vez más y actuará otra vez como un polímero entrecruzado. El polímero adquiere las propiedades de un elastómero y la facilidad de procesado de un termoplástico. Estos ionómeros son conocidos a veces con el nombre de elastómeros termoplásticos. Figura 8.2.2.2.- Calentamiento de un ionómero 159 Hasta ahora sólo se ha hablado de los llamados ionómeros al azar. Un ionómero al azar es aquél cuyos grupos iónicos están unidos a la cadena principal a intervalos erráticos. Sin embargo, existen otros tipos de ionómeros. Están aquéllos que se emplean como membranas semi-permeables. Una membrana semi-permeable es una muy delgada pieza de material que permite el paso de ciertas sustancias, mientras que otras permanecen dentro. Las membranas constituidas por ionómeros se denominan específicamente membranas selectivas de iones. Funcionan dejando pasar el agua, pero no los iones metálicos. Estas membranas selectivas de iones, reducen en los residuos acuosos, los niveles de iones metálicos como por ejemplo el plomo, que ninguno de nosotros quiere beber. Estas membranas también pueden ser usadas para recuperar metales valiosos de soluciones diluidas, eliminar el zinc de desperdicios textiles y "ablandar" las aguas duras. Una membrana selectiva de iones sumamente específica es un ionómero perfluorosulfonato, que la DuPont llama Nafion. Todos los ionómeros perfluorosulfonato poseen excelente estabilidad química y térmica, así como también la gran habilidad de absorber increíbles cantidades de agua. Las membranas de Nafion pueden fabricarse en forma de films o tubos, y ser empleadas en muchos procesos cáusticos y peligrosos, como por ejemplo la producción de cloro, la regeneración de ácidos, separaciones en el procesado químico, y también en electrodiálisis y celdas de combustible. 8.2.3.- Copolímeros en bloque. Podemos hacer un elastómero termoplástico de otro modo. Ese otro modo es mediante un copolímero en bloque. Un elastómero termoplástico muy común que es un copolímero en bloque es el caucho SBS. Pero resulta sumamente difícil mezclar dos polímeros diferentes, aún cuando sean muy similares. Esto es aplicable tanto a los bloques de nuestro SBS como a otros polímeros. Por lo tanto los bloques de poliestireno tienden a agruparse entre sí, al igual que los bloques de polibutadieno. Pero cada bloque de polibutadieno posee un bloque de poliestireno en cada extremo y los distintos bloques de poliestireno de la misma molécula de SBS no forman parte necesariamente del mismo cluster. Esto quiere decir que los distintos clusters de poliestireno se mantendrán unidos entre sí por los bloques de polibutadieno (Figura 8.2.3.1). Los clusters de poliestireno actúan como reticulantes para los bloques de polibutadieno. Y al igual que los clusters iónicos de los ionómeros, las agrupaciones de poliestireno se romperán cuando el SBS es calentado, de modo que puede ser procesado y reciclado como un polímero no entrecruzado. Figura 8.2.3.1.- Bloques de polibutadieno unidos por clusters de poliestireno. 160 También se puede fabricar un elastómero termoplástico empleando un copolímero en bloque a partir de una sola clase de monómero. Se puede fabricar polipropileno en el cual existan bloques de distinta tacticidad. Puede hacerse polipropileno a partir de bloques atácticos empleando una polimerización catalizada por metalocenos, de este modo: Los bloques se separan tal como lo hacen en el caucho SBS. Y ésto es posible porque los bloques isotácticos forman cristales, mientras que los atácticos son amorfos. El resultado es algo similar a lo que se muestra en la figura 8.2.3.2. Se comporta como un elastómero por las mismas razones que el caucho SBS. Figura 8.2.3.2.- Polipropileno con bloques de distinta tacticidad. 9.- Peso molecular y grado de polimerización medio. En materiales simples, como la sal, el fenol o el alcohol, todas las moléculas son iguales y tienen una masa molar característica. Una peculiaridad de los materiales plásticos (y en particular de los termoplásticos) es que el peso molecular de las macromoléculas que lo constituyen no es el mismo (contiene macromoléculas de diferentes tamaños), debido a que el número de moléculas que se agrupan en la reacción de polimerización (grado de polimerización) queda determinado por circunstancias casuales. Así cualquiera que haya sido el mecanismo seguido en la polimerización, el crecimiento de las cadenas poliméricas no se detiene siempre al alcanzar una determinada longitud, produciendo, por tanto, cadenas con números distintos de unidades de repetición (hay que tener en cuenta que no todas las cadenas se inician a la vez ni tampoco terminan su crecimiento al mismo tiempo), las cuales están intimamente entremezcladas. Un ejemplo es el polietileno el cual puede haber más de 10 tipos de masa molecular, difiriendo los extremos en un factor de 20. Dentro de cada tipo las cadenas pueden diferir en su longitud en un factor de 100 o incluso de 1000. 161 La resistencia mecánica de las cadenas varía significativamente dependiendo de cual sea su peso molecular, como pone de manifiesto la figura 9.1. Figura 9.1.- Efecto de la masa molecular relativa sobre la resistencia al impacto del polietileno lineal. La polimerización finaliza de forma estadística regida por múltiples factores determinantes del medio. Si la finalización del crecimiento de la cadena en una polimerización tipo radicálica fuese por desproporción, la molécula tendría el tamaño de la especie activa. Pero si la terminación es por choque entre dos especies activas, el tamaño de la macromolécula es mucho mayor. Existe, por tanto, una distribución estadística de pesos moleculares de las macromoléculas (longitudes de la cadena) que constituyen una muestra de polímero, debiendo definirse un peso molecular medio, un grado de polimerización medio y un índice de heterogeneidad, que sirvan de referencia para caracterizarle. En la figura 9.2 se muestran algunas formas comunes de distribución de pesos moleculares. Figura 9.2.- Ejemplos de distribuciones de masas molares: (a).- distribución bimodal ( b).-distribución simétrica amplia ( c).-distribución simétrica estrecha. (d).- distribución asimetrica 162 De ellas una forma común es la distribución asimétrica que se encuentra representada ella sola en la figura 9.3. Figura 9.3.- Distribución de tamaños moleculares. Se pueden describir varios momentos matemáticos o valores promedio mediante la curva de distribución diferencial o de frecuencias, con las correspondientes ecuaciones. El promedio puede ser calculado de diferentes maneras, y cada una tiene su propio valor. El primer momento de la distribución se denomina peso molecular medio en número, M n y es el peso de la muestra de polímero (peso total de todas las moléculas poliméricas contenidas en una muestra) dividido por el número de moléculas o cadenas en esa muestra (relaciona la longitud total de la cadena de polímero con el número de cadenas individuales presentes). Así, M n , para tres moléculas de pesos moleculares 1.00 x 105, 2.00 x 105, y 3.00 x 105 sería: 6.00 x 105 = 2.00 x 105 3 La expresión matemática es: Peso total de la muestra W Mn = = = Numero de moleculas N ∑ i i ∑W ∑ M i N ∑ M n = = N N ∑ ∑ ∑n i i i i i i i i i i i (9.1) i i donde: W = Peso total de la muestra. Ni = Número de cadenas de polímero en la fracción con un peso molecular medio Mi. ni = Número de moles de polímero de peso molecular medio Mi. Wi = NiMi = Masa de las cadenas de la fracción i 163 Otra manera de obtener el peso molecular medio numérico es distribuyendo las cadenas en una serie de intervalos de tamaño y luego determinando la fracción del número total de cadenas correspondiente a cada intervalo de tamaño (Figura 9.4.a). Este peso molecular medio numérico se expresa como: M n = ∑ xi M i (9.2) I donde Mi representa el peso molecular medio (intermedio) del intervalo de tamaño i, y xi es la fracción del número total de cadenas dentro del correspondiente intervalo de tamaño. La mayoría de las propiedades termodinámicas, por ejemplo las coligativas, están relacionadas con el número de partículas y dependen por tanto de M n . Los valores de M n , son independientes del tamaño molecular y son muy sensibles a la presencia de moléculas pequeñas en la mezcla. Los valores de M n se determinan por los métodos que siguen la ley de Raoult, que dependen de las propiedades coligativas, como pueden ser la ebulloscopía (elevación del punto de ebullición), crioscopía (descenso del punto de congelación), osmometría. Además se emplea también el análisis de grupos terminales. Figura 9.4 .- Hipotéticas distribuciones de tamaños de las moléculas poliméricas basadas en las fracciones : (a).- numéricas y (b).- másicas de las moléculas El peso molecular medio en peso, M w , se define como la fracción en peso Wi de las cadenas del polímero de peso molecular Mi dividido por el peso total de la muestra (relaciona la longitud total de la cadena con la fracción másica de las cadenas individuales). Está basado en el hecho de que una molécula más grande contiene más de la masa total de la muestra polimérica que las moléculas pequeñas. 164 Se determina a partir de experimentos en los que cada molécula o cadena contribuye a la medida final de acuerdo con su tamaño. Esta media es más sensible al número de moléculas pesadas que el número de peso molecular medio, el cual sólo depende del número total de partículas. El peso molecular medio en peso, M w , es el momento de segundo orden o media cuadrática y se expresa matemáticamente como sigue: ∑W M i Mw = ∑N M M = ∑W i i i i i i W ∑N M = ∑N M i 2 i i i i i i (9.3) i Así, el peso molecular medio en peso, para el ejemplo que se ha usado para calcular M n , sería 2.33 x 105, ya que: (1.00 x 105 ) + ( 2.00 x 105 ) + ( 3.00 x 105 ) 2 2 2 6.00 x 105 14.00 x 1010 = = 2.33 x 105 5 6.00 x 10 Otra manera de obtener el peso molecular medio másico se basa en el peso de la fracción de moléculas incluidas dentro de varios intervalos de tamaños (Figura 9.4.b). Se calcula según: Mw = ∑ w i M i (9.4) i donde ahora Mi denota el peso molecular dentro del intervalo de tamaño i, mientras wi significa el peso de la fracción de moléculas situadas dentro del correspondiente intervalo de tamaños. Las propiedades de la masa que se asocian con grandes deformaciones, tales como la viscosidad y la tenacidad, se hallan particularmente afectadas por el valor de M w . Los valores de M w , se determinan mediante métodos de dispersión de luz y de ultracentrifugación. Sin embargo, la elasticidad en estado fundido depende en mayor grado de M z - la media z del peso molecular, que también puede obtenerse por medio de técnicas de ultracentrifugación. M z es el tercer momento o media cúbica (también se denomina peso molecular medio centrifugal o «Z») y se expresa matemáticamente como: ∑i Ni M 13 Mz = ∑ Ni M i2 i Por tanto, el valor medio del peso molecular M z en el ejemplo utilizado para calcular M n y M w sería 2.57 x 105, ya que : (1.00 x 105 ) + ( 2.00 x 105 ) + ( 3.00 x 105 ) = 36.00 x 1015 = 2.57 x 105 2 2 2 10 (1.00 x 105 ) + ( 2.00 x 105 ) + ( 3.00 x 105 ) 14.00 x 10 3 3 3 165 Peso Molecular Promedio en Viscosidad, M v . El peso molecular también puede calcularse a partir de la viscosidad de una solución polimérica. El principio es muy simple: las moléculas poliméricas más grandes forman una solución más viscosa que las moléculas más pequeñas. Obviamente, el peso molecular obtenido por medición de la viscosidad, es distinto al peso molecular promedio en número o en peso. Pero se acerca más al promedio en peso que al promedio en número. Aunque se pueden calcular medias z + 1 y superiores del peso molecular, las más interesantes son los anteriores, que como se muestra en la figura 9.3 se hallan en orden de magnitud creciente. Veamos unos ejemplo de cálculo de los distintos pesos moleculares promedio. EJEMPLO 1: 166 EJEMPLO 2: 167 Mw La relación entre el promedio en peso y el promedio en número, , que siempre es mayor que la unidad, Mn nos da una idea de la amplitud de la desviación, o anchura de la distribución, por lo que se utiliza para caracterizar la dispersión de pesos moleculares de una muestra (medida de la distribución del peso molecular) y se denomina índice de heterogeneidad, o polidispersidad o dispersividad . Es igual a la unidad únicamente cuando todas las cadenas del polímero tienen la misma longitud, o sea, cuando el polímero es monodisperso. Valores próximos a 1 designan una gran homogeneidad de pesos moleculares (campana cerrada), y valores altos determinan una gran dispersión (campana muy abierta). Una polidispersidad baja del orden de 1.0 a 1.3 es aceptable para realizar determinaciones sobre la muestra. Deben ser rebajados los valores mayores de la polidispersidad. Para ello se somete a la muestra a un fraccionamiento por tamaños utilizando un sistema de dos disolventes inmiscibles entre si con efecto disolvente/precipitante del polímero. De esta manera se puede disponer de muestras de tamaño más homogéneo. Los polímeros comunes que se usan en el comercio nunca son monodispersos, sino que tienen una distribución del peso molecular que varía de acuerdo al método de manufactura y al producto terminado, así como a las necesidades de la transformación. La dispersividad de los llamados “polímeros vivientes”, obtenidos por polimerización aniónica, es, con frecuencia, casi igual a 1, y estos polímeros son importantes corno productos especiales (Tabla 9.1) Tabla 9.1.- Polidispersividad para diferentes esquemas de reacción. 168 EJEMPLOS: Polydispersity = M w 10000 = =5 2000 Mn 169 Una forma alternativa para expresar el tamaño medio de cadena de un polímero es el grado de polimerización medio GP , que representa el número medio de unidades monoméricas en una cadena. Existen tanto el grado de polimerización medio numérico GP n , como el grado de polimerización medio másico GP w : GP n = Mn m , GP w = Mw m donde M n y M w son el peso molecular medio numérico y medio másico antes definidos y m es el peso molecular de la unidad monomérica. En un copolímero con dos o más unidades monoméricas diferentes, m se calcula del modo siguiente: m = ∑fjmj j donde fi y mi son la fracción de cadenas y el peso molecular de la unidad monomérica j respectivamente. La magnitud del peso molecular afecta a varias características del polímero. El peso molecular medio y la distribución de los pesos moleculares tienen una gran importancia en la determinación de todas las propiedades físicas de los plásticos, para una misma estructura molecular, debido a que las fuerzas de atracción entre las moléculas aumentan con su tamaño, es decir, con su longitud. Concretamente la resistencia a la tracción y el índice de refracción, varían según una ley, en función del peso molecular medio M , del tipo: ( ) f M =a− b M existiendo siempre un peso molecular crítico, por debajo del cual las propiedades del polímero varían bruscamente y su comportamiento resulta muy distinto. La temperatura de fusión o la temperatura de ablandamiento es otra de las características que depende del peso molecular del polímero. Para polímeros M con superior a 100000 g/mol, la temperatura de fusión aumenta al incrementarse el peso molecular. Los polímeros de cadena muy corta, con pesos moleculares del orden de 100 g/mol, son líquidos o gases a temperatura ambiente. Los de pesos moleculares de aproximadamente 1000 g/mol son sólidos cerosos (cera de parafina) o resinas blandas. Los polímeros más interesantes como materiales son los de pesos moleculares comprendidos entre 10000 y varios millones g/mol. A veces se denominan altos polímeros. La temperatura de fusión se incrementa al aumentar las fuerzas intermoleculares. En general, al prolongarse la longitud de la cadena se incrementa el grado de enlace entre las moléculas. Los enlaces intermoleculares generalmente son del tipo van der Waals y/o de hidrógeno. Por otra parte, para la mayoría de los usos industriales, existe también un límite máximo de peso molecular, pues, por encima de éste, el material presenta unas características desfavorables. Así, en el caso de los polímeros naturales (como la celulosa y el caucho) se requiere llevar a cabo una despolimerización (molienda en refinadores, en el caso de la celulosa, y una masticación, en el caso del caucho), que reduce el tamaño de las macromoléculas. En la figura 9.5 se representa la variación de diversas propiedades de los polímeros en función del peso molecular. Asimismo, en la figura 9.6 puede verse la relación entre el peso molecular y las propiedades físicas y facilidad de procesado, observándose que existe una zona óptima. 170 Figura 9.5.- Variación de diversas propiedades de los polímeros en función del peso molecular. Figura 9.6- Relación entre el peso molecular y las propiedades físicas y facilidad de procesado 171 10.- Métodos de determinación del peso molecular medio. 10.1.- Introducción. Existen dos clases de métodos para determinar los pesos moleculares medios; unos proporcionan valores absolutos y otros relativos. Evidentemente estos últimos deben ser contrastados con los resultados que se obtienen usando determinados patrones, para cuantificar el valor del peso molecular medio. En la tabla 10.1.1 se presentan algunas técnicas típicas de determinación del peso molecular. Los métodos más utilizados se explicarán brevemente. Tabla 10.1.1.- Métodos para la determinación del peso molecular de los polímeros. Todos los métodos clásicos de determinación de pesos moleculares requieren que el polímero se halle en disolución. Para minimizar las interacciones polímero - polímero se utilizan disoluciones de menos de l g de polímero por 100 ml de disolución. Para minimizar aún más las interacciones del soluto se efectúa la extrapolación de las medidas para dilución infinita. 172 10.2.- Cromatografía de permeación en gel (GPC). Los métodos coligativos y de dispersión de luz permiten el cálculo de los pesos moleculares absolutos. Estos métodos a menudo necesitan de tiempos largos y pueden ser costosos. Para análisis rutinarios, frecuentemente se acostumbra a determinar el peso molecular empleando valores calibrados relacionados con la viscosimetría y columnas de cromatografía de permeación en gel. La cromatografía de permeación en gel (GPC), que se ha denominado de tamices moleculares o filtración en gel, es un tipo de cromatografía sólido-líquido que separa los polímeros polidispersos en fracciones por tamizado mediante un gel de poliestireno con enlaces cruzados u otro de características semejantes. El gel de poliestireno, que sirve de fase estacionaria, se puede obtener comercialmente en una gran variedad de tamaños 6 de poro (1 a 10 nm). Puesto que las moléculas pequeñas penetran más fácilmente en las partículas de gel, las fracciones de más alto peso molecular se separarán antes. De esta manera, la GPC separa las fracciones de acuerdo con su tamaño. Como se ve en la figura 10.2.1, en dos columnas separadas se bombea por un lado una disolución con un disolvente como el tetrahidrofurano (THF) y por otro el disolvente (THF) con un caudal de 1 ml/min. Las diferencias en el indice de refracción entre el disolvente y la disolución se determinan mediante un refractómetro diferencial y se registran automáticamente. Cada unidad de columna debe calibrarse utilizando polímeros de peso molecular conocido. Figura 10.2.1.- Esquema de flujo de la disolución y del disolvente en un cromatógrafo de permeación en gel (GPC). 173 En la figura 10.2.2 se muestra una columna en detalle. Figura 10.2.2.- Esquema de una columna de GPC rellena con un polímero reticulado e hinchado por el disolvente El rendimiento de estas columnas comprimidas puede determinarse calculando su altura (en pies) equivalente a un plato teórico (HETP), que es el inverso del número de platos por pie (P). Como muestra la expresión (10.2.1), P es directamente proporcional al cuadrado del volumen de elución (Ve) e inversamente proporcional a la altura de la columna en pies y el cuadrado de la línea de base (d). Como se presenta en la figura 10.2.3 esta última magnitud es la anchura de la base de un pico ideal que se obtiene dibujando las líneas tangentes a las pendientes de un pico real de GPC en forma de campana de una sustancia pura, como THF. 16 V p= e f d 2 (10.2.1) El peso molecular de un polímero es función del volumen efectivo ocupado por la cadena del polímero en disolución, o volumen hidrodinámico (V). Por ejemplo, para el poliestireno en THF a 25 °C, V es igual a: 1.70 1.5 x10 −2 M w Además de fraccionar polímeros y proporcionar información sobre el tamaño de los mismos, la GPC puede combinarse con otros instrumentos, como los espectrofotómetros de luz ultravioleta, para el análisis1de las fracciones separadas. El movimiento de entrada y salida en la fase estacionaria depende de una serie de factores, entre ellos el movimiento Browniano, el tamaño de las cadenas y la conformación. Los dos últimos son función del volumen hidrodinámico de la cadena del polímero, es decir, el volumen excluido real ocupado por la cadena del polímero. La velocidad de flujo de una cadena de un polímero está relacionada con su tamaño y con su volumen hidrodinámico. Se ha sugerido que es posible generar, para una columna específica, una curva sencilla de [n]M en función del volumen de elución para muestras de polímeros de peso molecular conocido, pudiéndose entonces emplear esta curva de calibración para obtener [n]M, y posteriormente M cuando se determine [n]; es decir, [n]M = Volumen hidrodinámico = Volumen de elución. 174 Figura 10.2.3.- Esquema del ancho (d) de un pico de GPC 10.3.- Osmometría. Las medidas de una cualquiera de las propiedades coligativas de una disolución de un polímero implica el recuento de las moléculas de soluto (polímero) en una cantidad dada de disolvente y proporciona una media en número de moléculas. La única propiedad coligativa que se mide sin problema para los polímeros de alto peso molecular es la presión osmótica. Dicha determinación se basa en la utilización de una membrana semipermeable a través de la cual pasan libremente las moléculas de disolvente peso no lo hacen las moléculas del polímero. Las membranas reales sólo ofrecen una versión aproximada de la semipermeabilidad ideal, siendo la limitación principal el paso a través de la membrana de cadenas del polímero de bajo peso molecular. Existe una tendencia termodinámica a la dilución de la disolución del polímero mediante un flujo neto de disolvente hacia la célula que contiene el polímero. Esto ocasiona un aumento de la cantidad de líquido en dicha célula que causa un aumento en el nivel de líquido del tubo de medida correspondiente. El aumento del nivel de líquido se opone e iguala a una presión hidrostática que resulta de la diferencia en los niveles de líquido de los dos tubos de medida - la diferencia está directamente relacionada con la presión osmótica de la disolución del polímero. Así, las moléculas de disolvente tienden a pasar a través de la membrana semipermeable para alcanzar un equilibrio "estático", como se ve en la figura 10.3.1. Las membranas semipermeables pueden fabricarse con celofán (Zim-Myerson), caucho de hevea, polialcohol vinílico o nitrato de celulosa. Puesto que la presión osmótica depende de las propiedades coligativas, es decir, del número de partículas, la medida de esta presión (osmometría) podrá aplicarse a la determinación de la presión osmótica de disolventes en relación con disoluciones de polímeros. La diferencia de alturas , ∆h , de las columnas de líquido se transforma en presión osmótica , π , multiplicando por la gravedad , g, y la densidad de la disolución , ρ , es decir, π = ρ g ∆h (10.3.1) En un osmómetro de membrana automático, como el que se muestra en la figura 10.3.2, el ascenso capilar no limitado de una disolución de polímero se mide de acuerdo con la ecuación modificada de Van't Hoff : π= RT C + BC 2 Mn (10.3.2) 175 Figura 10.3.1.- Diagrama esquemático que muestra la variación con el tiempo del efecto de la presión que ejerce un disolvente separado de una disolución que contiene un material no transportable (polímero) por una membrana semipermeable, donde t1 representa la medida inicial de los tubos, t2 el nivel al cabo de un tiempo, y t3 los niveles cuando hay equilibrio"estático". Figura 10.3.2.- Osmómetro de membrana automático. 176 Como muestra la figura 10.3.3, el inverso de la media aritmética del peso molecular curva π RTC 1 es el corte de la Mn en función de C cuando se extrapola a concentración cero. Figura 10.3.3.- Curva de π RTC en función de C que se usa para determinar 1 en osmometría. Mn Las medidas de la presión osmótica estática generalmente requieren varios días o semanas antes de que se alcance un equilibrio apropiado que permita una medida significativa de la presión osmótica. El tiempo necesario para alcanzar el equilibriose acorta a unos minutos o una hora en la mayoría de los instrumentos comerciales que utilizan técnicas dinámicas. La osmometría clásica es útil y ampliamente utilizada para la determinación de los valores de M n en un intervalo de 5x104 a 2x106. Los osmómetros dinámicos modernos amplían este intervalo de 2x104 a 2x106. El peso molecular de los polímeros de pesos moleculares más bajos, que pueden pasar a través de una membrana semipermeable, se determinan por osmometría de presión de vapor (VPO) o destilación isotérmica. En el método de VPO se colocan unas gotas de disolvente y de disolución en una cámara aislada cerca de sondas provistas de un termistor. Puesto que las moléculas de disolvente se evaporan más rápidamente en el disolvente que en la disolución, se registra una diferencia de temperaturas ( ∆T ). Así, la molalidad (M) puede determinarse utilizando: RT 2 ∆T = (1.10.3.3) M 100λ si se conoce el calor de vaporización por gramo dedisolvente ( λ ). En la figura 10.3.4 se muestra un esquema de un osmómetro de presión de vapor. 177 Figura 10.3.4.- Esquema de osmómetro de presión de vapor. 10.4.-Ebulloscopía y crioscopía. Estos métodos, basados en la ley de Raoult, se parecen a los que se usan para los compuestos clásicos de bajo peso peso molecular y dependen de la sensibilidad de la termometría disponible. El peso molecular medio en número M n en los dos casos (ebulloscopía y crisocopía) se basa en la ecuación de Clasius-Clapeyron utilizando la elevación del punto de ebullición y la disminución del punto de congelación (∆T), de un disolvente en presencia de las moléculas del polímero (soluciones diluidas), como se ve a continuación: Mn = RT 2V C ∆H ∆T C →0 (10.4.1) Los resultados que se obtienen utilizando la ecuación de Clasius-Clapeyron, en la que T es la temperatura Kelvin y ∆H es el calor de transición, deben extrapolasse para una concentración cero. Los métodos ebulloscópicos y crioscópicos sólo son aplicables a polímeros de peso molecular relativamente 4 bajo (< 2-5x10 ), pues, en caso contrario, el error de medida en ∆T origina errores importantes en M n . -4 Usando termistores sensibles hasta 1 x 10 °C es posible medir valores del peso molecular de hasta 40000 a 50000, aunque el limite típico es de aproximadamente 5000. 10.5.- Método viscosimétrico. El método viscosimétrico esté basado en que la viscosidad intrínseca de una solución de un polímero es función del peso molecular medio Mw , según la ecuación empírica: η − η0 = K Mw c → 0 cη 0 η = lim ( ) a (10.5.1) siendo K y a constantes que dependen de la naturaleza del polímero, del disolvente y de la temperatura. 178 La viscosidad intrínseca se determina por extrapolación de los valores de una serie de soluciones diluidas del polímero, de diferente concentración c, para c→0, siendo: η0 = Viscosidad del disolvente puro η = Viscosidad de la solución ambas expresadas como el tiempo empleado por cierto volumen del líquido en fluir a través de un tubo capilar de dimensiones fijas (viscosímetros de Ostwald- Fenske, o de Ubbelohde). Se usa para pesos moleculares mayores de 50000. 10.6.- Método de dispersión de la luz. El método de difusión de la luz está basado en que cualquier partícula extraña en un medio homogéneo transparente, en el cual se propaga un rayo de luz, produce una dispersión de la luz en todas direcciones y, en consecuencia, una disminución de la intensidad del haz luminoso inicial. La iluminación de partículas de polvo es un ejemplo de dispersión de luz, y no de reflexión. La reflexión es la desviación de la luz incidente de tal manera que el ángulo de incidencia es igual al ángulo de reflexión. La dispersión consiste en la desviación de la luz en todas direcciones. Así, cuando se observa un "rayo de luna" las partículas de polvo dirigen los rayos hacia el observador sin importar el ángulo que forme con el rayo incidente. La energía que se dispersa por segundo (flujo de dispersión) está relacionada con el tamaño y la forma de la partícula dispersante y con el ángulo de dispersión. La medida de la dispersión de luz de moléculas de polímeros en disolución es una técnica ampliamente utilizada para la determinación de valores absolutos de óptica de las disoluciones de polímeros. Mw . Este método, aue se basa en la heterogeneidud Para las medidas de dispersión de luz, la cantidad total de luz dispersada se deduce a partir de la disminución de la intensidad del rayo de luz incidente, Io, al pasar a través de una muestra de polímero. Esto se puede representar en función de la ley de Beer de la absorción de luz de la siguiente manera: I = e −τ e I0 (10.6.1) siendo: I0 y I = Intensidades luminosas para el disolvente puro y para la solución del polímero, respectivamente. e = Espesor de la lámina de líquido atravesada por la luz τ = Turbidez de la solución (es la medida de la disminución de la intensidad del rayo incidente por unidad de longitud de un disolución dada), 1 = Transparencia de la solución τ La intensidad de luz dispersada, o turbidez (τ), es proporcional al cuadrado de la diferencia entre los indices de refracción (n) de la disolución que contiene el polímero y del disolvente, al peso molécular medio del polímero ( M ), y al inverso de la cuarta potencia de la longitud de onda de la luz utilizada (λ). De esta manera se tiene: Hc τ = 1 1 + 2Bc + Cc 2 + ........) ( M w Pθ (10.6.2) 179 donde la expresión para la constante H será: ( 2 dn 32π n0 dc H= 4 3 λN 3 ) 2 (10.6.3) siendo: n0 = Indice de refracción del disolvente n = Indice de refracción de la disolución c = Concentración, B, C, etc, = Constantes viriales que son función de la interacción en el disolvente, Pθ = Factor de dispersión de la partícula N= Número de Avogadro. dn = Incremento de refracción específica y se determina a partir de la pendiente de la curva dc construida con medidas del indice de refracción en función de la concentración del polímero. ( ) En la determinación de la media ponderada del peso molecular de las moléculas de un polímero en una disolución sin polvo, se mide la intensidad de luz dispersada de una lámpara de arco de mercurio o un laser para distintas concentraciones y distintos ángulos (θ), normalmente 0, 90, 45 y 135° (Figura 10.6.1). La luz incidente emite una envolvente de dispersión que tiene cuatro cuadrantes equivalentes. La relación de dispersión para 45° y para 135° se llama factor de disimetría o relación de disimetría Z. El factor de disimetría reducido Z0 es el valor de la curva de Z en función de la concentración extrapolada a una concentración nula. Figura 10.6.1.- Envolventes de dispersión de luz. La distancia desde la partícula dispersante a los límites de la envolvente representa la magnitud de la luz dispersada en función del ángulo. Para disoluciones de polímeros de peso molecular moderado o bajo, Pθ es 1 y la expresión (10.6.2) se reduce a: Hc τ = 1 1 + 2Bc + Cc 2 + ........) ( Mw (10.6.4) Para bajas concentraciones de polímeros en disolución, (10.6.4) se reduce a la ecuación de una recta: Hc τ = 1 2Bc + Mw Mw (y = mx + n) (10.6.5) Cuando se representa en función de la concentración el cociente de la concentración c y la turbidez τ (relacionada con la intensidad de dispersión a 0 y 90°) multiplicado por H, el valor extrapolado de la curva para c = 0 es el inverso de M w , siendo la pendiente igual a la constante virial B como se muestra en la figura 10.6.2 Z0 está directamente relacionado con Pθ , siendo ambos función tanto del tamaño, así como de la forma de las partículas dispersantes. 180 Cuando el tamaño de las cadenas del polímero se acerca aproximadamente a la veinteava parte de la longitud de onda de la luz incidente, se produce interferencia de dispersión con una envolvente que ya no es simétrica. Aquí la dependencia de la dispersión con el peso molecular se explica con la relación dada en (10.6.2), y así, la extrapolación para una concentración cero del polímero de la curva Hc τ en función de c proporciona el 1 1 Pe, y no de . El peso molecular en estos casos se determina normalmente utilizando uno o M w Pθ Mw dos métodos. valor Figura 10.6.2.- Curva típica utilizada para determinar 1 a partir de medidas de dispersión de luz. Mw 2 2 2 dn Para 546 nm: H = 6.18x10−5 n 2 = 6.18x10−5 (1.475 ) (1.0 ) = 1.34 x104 dc g g o como unidades de concentración en la fotometría de dispersión. Para la ml cc 0.14..g g disolución que nos atañe, la concentración es = 0.0014 100..ml ml Normalmente se utilizan 1.34 x10−4 x1.4 x10−3 = ≅ 8.6 x10−5 −3 τ 2.18 x10 Hc -5 4 El peso molecular aparente será entonces el inverso de 8.6x10 o 1.2x10 . Se llama aparente porque es para un solo punto y no se ha extrapolado para c = 0. La primera técnica se denomina método o enfoque disimétrico porque utiliza la determinación de función de Pθ como función de la forma del polímero. M w se determina a partir del valor de corte de sustituyendo un valor determinado de Z0 en 1 M w Pθ Pθ . El punto débil de este enfoque es la necesidad de suponer una forma del polímero en una disolución particular. Para valores pequeños de Z0 , un error al escoger la forma correcta para el polímero conduce a una 181 error pequeño, pero para valores de Z0 mayores, el error puede ser importante, o sea, mayor que un 30 %. La característica positiva del método es que el enfoque disimétrico es sencillo comparado con el segundo enfoque, o método de Zimm, en el que no se necesita suponer ninguna forma para el polímero. El método de Zimm utiliza la curva de Zimm, que es una extrapolación doble para concentración cero y Hc ángulo de dispersión cero. La curva utiliza una redistribución de los factores designando esta τ combinación como Kc . En la figura 10.6.3 se puede ver un diagrama de Zimm típico. R Figura 10.6.3.- Diagrama de dispersión de luz de Zimm típico. Se encuentran disponibles fotómetros de dispersión de luz de ángulo pequeño y multiángulo que permiten no sólo la determinación de la media ponderada del peso molecular, sino también una serie de valores adicionales en ciertas condiciones. Por ejemplo, los nuevos instrumentos multiángulo suministran datos utilizando una serie de detectores como los que se muestran en la figura 10.6.4. Con los datos obtenidos por estos instrumentos se construye un diagrama típico de Zimm como se muestra en la figura 10.6.5, que proporciona tanto la media ponderada del peso molecular como el radio medio independientemente de la conformación o ramificación molecular. La forma real de los diagramas de Zimm que se muestran en las figuras 10.6.3 y 10.6.5 depende de varias constantes de ajuste como el "100" utilizado en la figura 10.6.3 como multiplicador de "c" y el "1000" utilizado en la figura 10.6.5 como multiplicador de "c". La obtención de "buenos " diagramas puede ser una labor de rutina mediante el uso de ciertos programas de ordenador. Una de las mayores causas de error en la determinación del peso molecular medio en peso utilizando dn fotometría de dispersión de luz es la determinación de , puesto que un error en su determinación se dc amplifica al ser utilizado al cuadrado en la ecuación que relaciona la dispersión con el peso molecular, tal como la (10.6.3). 182 Figura 10.6.4.- Disposición de los detectores que representa una muestra rodeada por una serie de detectores que recogen luz laser dispersada por la muestra. Figura 10.6.5.- Diagrama de Zimm para un polímero completado con un coeficiente de conc entración negativo para mejorar la apariencia y accesibilidad del gráfico. 10.7.- Método de ultracentrifugación. El método de ultracentrifugación está basado en el equilibrio de sedimentación del polímero en un disolvente, que se establece cuando se igualan la fuerza centrífuga y la fuerza impulsora de la difusión debida al gradiente de concentración resultante. 183 Puesto que la energía cinética de las moléculas de disolventes es mucho mayor que la fuerza de sedimentación gravitacional, las moléculas de los polímeros se hallan en suspensión en las disoluciones. Sin embargo, este campo gravitacional tradicional, el cual permite el movimiento browniano, puede ser vencido aumentando la fuerza por medio de fuerzas centrífugas elevadas como las que se originan en la ultracentrifugación. Se hace girar una cubeta con la disolución del polímero a alta velocidad, ya que, para contrarrestar el movimiento browniano, se requieren grandes velocidades angulares. Tanto M w , como M z pueden determinarse mediante la acción de fuerzas de ultracentrifugación de alta velocidad en disoluciones diluidas de polímeros en disolventes adecuados. Se usan para ello disolventes de densidades e índices de refracción distintos a los de los polímeros para asegurar el movimiento de los polímeros. Para detectar la situación de la interfase disolución-disolvente se utiliza un sistema óptico que mide el índice de refracción a lo largo de la cubeta. En función de las variaciones del índice de refracción con la distancia al centro de rotación, se determina el valor de M z . Como para alcanzar el equilibrio de sedimentación se requiere mucho tiempo (varías semanas), se puede determinar M z midiendo la velocidad inicial de sedimentación pero, en este caso, debe conocerse el valor de la difusividad. El método es complejo, válido para polímeros de peso molecular entre 104 y 107, y es más adecuado para materiales biológicos formados por moléculas protéicas compactas. En los experimentos de velocidad de sedimentación se utiliza la ultracentrifugadora a velocidades extremadamente altas de hasta 70000 rpm para transportar las moléculas de los polímeros a través de un disolvente menos denso hacia la parte inferior del tubo de ensayo o a la zona superior, si la densidad del disolvente es mayor que la del polímero. El movimiento de contorno durante la ultracentrifugación puede seguirse utilizando medidas ópticas para controlar el cambio brusco del índice de refracción (n) entre el disolvente y la disolución. La velocidad de sedimentación se define mediante la constante de sedimentación s que es directamente proporcional a la masa m, la densidad de la disolución ρ , y el volumen específico del polímero V , e inversamente proporcional a la velocidad angular de rotación ω , la distancia desde centro de rotación al punto de observación en el tubo de ensayo r, y el coeficiente de fricción f el cual se halla inversamente relacionado con el coeficiente de difusión D extrapolado para dilución infinita. Estas relaciones se muestran en las ( ) siguientes ecuaciones en las que 1 − V ρ se llama factor de flotabilidad puesto que determina la dirección del transporte macromolecular en el tubo de ensayo. s= 1 dr m 1 − V ρ = ω 2 r dt f ( ) D= RT Nf y mN = M D RT = s M ω 1−V ρ ( ) (10.7.1) (10.7.2) (10.7.3) La determinación de la velocidad de sedimentación es dinámica y se puede realizar en un corto período de tiempo. Es particularmente útil para los sistemas monodispersos y proporciona datos cualitativos además de información sobre la distribución de pesos moleculares en sistemas polidispersos. 184 El método del equilibrio de sedimentación proporciona resultados cuantitativos, aunque son necesarios largos períodos de tiempo para la centrifugación y velocidades relativamente bajas para establecer un equilibrio entre sedimentación y difusión. Como se muestra en la siguiente ecuación, C 2RTLn 2 C1 Mw = 1 − V ρ ω ( r22 − r12 ) ( ) (10.7.4) el peso molecular medio en peso M w es directamente proporcional a la temperatura T y el logaritmo C neperiano del cociente de concentraciones 2 a distancias r1 y r2 del centro de rotación al punto de C1 observación en el tubo de ensayo, e inversamente proporcional al factor de flotabilidad, el cuadrado de la velocidad angular de rotación y la resta de los cuadrados de las distancias r1 y r2 . Es de destacar que todos los métodos de determinación del peso molecular, excepto el viscosimétrico, son absolutos, mientras que este es relativo. 11.- Cristalización. Polímeros cristalinos. 11.1.- Introducción. Los polímeros cristalinos tienen unas características peculiares que los diferencian de las sustancias cristalinas de bajo peso molecular. Los polímeros son sólo parcialmente cristalinos, es decir, no alcanzan un orden cristalino completo y, en la mayoría de ellos, no se pueden conseguir cristales macroscópicos, sino que forman microcristales de una gran imperfección. Los cristales poliméricos tienen una morfología compleja, pero puede decirse que prácticamente en todos ellos existe una unidad cristalina básica que es la laminilla, en la cual se aloja la cadena de una forma más o menos ordenada. Los sólidos pueden clasificarse, desde un punto de vista estructural, en cristalinos y amorfos. La diferencia entre ambos puede ponerse de manifiesto mediante un experimento típico de difracción de rayos X. Cuando un haz de rayos X (RX) incide sobre un sólido ordenado regularmente, se produce una difracción descrita por la ley de Bragg, según la ecuación: nλ = 2dsenθ (11.1) donde 2θ es el ángulo formado por el rayo incidente y el difractado, λ es la longitud de onda de la radiación utilizada, n es el llamado orden de difracción y d es la distancia entre los planos cristalinos implicados en la difracción. Tanto la distancia d como los ángulos θ están determinados por la clase, número y localización de los átomos que constituyen la celdilla unidad, esto es, la porción de estructura que repetida periódicamente en las tres direcciones del espacio genera la estructura total del cristal. Los cristales grandes y casi perfectos como los de la mayoría de las sustancias de bajo peso molecular, orgánicas o inorgánicas, suelen dar lugar a diagramas de difracción en los que aparecen picos muy estrechos con máximos de intensidad de radiación difractada a ángulos θ bien definidos y característicos de cada material (Figura 11.1), a partir de los cuales y de sus intensidades se puede reconstruir la organización del material. Sin embargo, el desorden presente en los materiales amorfos, donde no existe una estructura repetitiva a largas distancias, se traduce en difractogramas que están constituidos por un amplio pico o banda ancha que abarca un gran intervalo de ángulos θ. 185 Figura 11.1.- Diagrama de difracción de rayos X del Al2O3. Los difractogramas de los materiales poliméricos presentan picos cristalinos, más anchos que los correspondientes a las sustancias sencillas, superpuestos sobre otro pico amplio con forma similar al presentado por los materiales amorfos. En la figura 11.2.a se muestra el diagrama de difracción de una muestra de poli(cloruro de vinilo) sindiotáctico. En el difractograma aparecen dos picos relativamente anchos, los cuales descansan sobre otro más amplio, trazado con línea interrumpida, que recuerda al difractograma presentado por las sustancias amorfas. Este tipo de difractograma mixto presentado por los polímeros cristalinos es indicativo de la coexistencia de regiones cristalinas, formadas por cristales de pequeño tamaño con cierto grado de imperfección, y regiones amorfas en un mismo material polimérico. Esta es una de las razones por la que es preferible denominar a estos materiales como semicristalinos. Figura 11.2.- Diagrama de difracción de rayos X del poli(cloruro de vinilo) sindiotáctico resuelto en picos cristalinos y banda amorfa 186 Antes de 1920, los investigadores químicos más punteros no sólo afirmaban que las macromoléculas no existían, sino que también los productos llamados macromoleculares como las proteínas, los elastómeros de la hevea y la celulosa no podían existir en forma cristalina. Sin embargo, a principios de los años 20, Haworth utilizando técnicas de difracción de rayos X demostró que la celulosa estirada era un polímero cristalino constituido por unidades repetitivas de celobiosa. En 1925, Katz, casi de broma, colocó una goma de caucho natural estirada en un espectrómetro de rayos X y para su sorpresa, comprobó que presentaba un patrón de interferencia típico de una sustancia cristalina. Los polímeros generalmente poseen estructura amorfa (desordenada) como consecuencia del mecanismo seguido en la polimerización que generalmente es radicálico. Sin embargo, bien por la composición química del monómero o por el procedimiento seguido en la polimerización -coordinación y en ocasiones aniónico- el estado cristalino también puede existir en los polímeros. Mientras la cristalinidad en los metales y en las cerámicas implica disposición de átomos e iones, en los polímeros implica la ordenación de moléculas y, por tanto, la complejidad es mayor. La cristalinidad polimérica puede considerarse como el empaquetamiento de cadenas moleculares para producir una disposición atómica ordenada. La estructura cristalina se especifica en términos de celdillas unidad, que ordinariamente son complejas Las substancias moleculares constituidas por pequeñas moléculas (por ejemplo, agua y metano) generalmente son totalmente cristalinas (en estado sólido) o totalmente amorfas (en estado líquido). Las moléculas poliméricas, como consecuencia de su tamaño y de su complejidad, suelen ser parcialmente cristalinas (o semicristalinas) con regiones cristalinas dispersas dentro de un material amorfo. En la región amorfa aparecen cadenas desordenadas o desalineadas, condición muy común debido a las torsiones, pliegues y dobleces de las cadenas que impiden la ordenación de cada segmento de cada cadena. Otros efectos estructurales repercuten en la extensión de la cristalinidad. Las zonas cristalinas son las responsables de la resistencia mecánica y las amorfas están asociadas a la flexibilidad y elasticidad del material Solamente unas pocas familias de polímeros poseen la regularidad estructural suficiente como para cristalizar (orden a larga distancia) y, aún en estos casos, nunca es posible lograr un 100 % de estructura cristalina y habrá que definir el grado de cristalización como la fracción del polímero que presenta estructura cristalina con relación al polímero total. El resto será amorfa y, por ello, poseerá una temperatura Tg. Aunque algunos polímeros, muy regulares, pueden mostrar grados de cristalización tan altos como el 90 %, normalmente, rara vez se supera el 50 %. El grado de cristalinidad de los materiales poliméricos puede variar desde completamente amorfo a casi enteramente cristalino (hasta, aproximadamente, un 95 %). Las muestras metálicas casi siempre son totalmente cristalinas, mientras que las cerámicas son o totalmente cristalinas o totalmente amorfas. Los polímeros semicristalinos tienen analogía con las aleaciones metálicas bifásicas. Al igual que ocurre en las sustancias de bajo peso molecular, la ordenación que tienen los átomos en las regiones cristalinas de un polímero puede ser determinada mediante la técnica de RX. Sin embargo, debido a que en materiales poliméricos no se pueden conseguir cristales únicos de tamaño macroscópico, esta determinación resulta bastante más difícil que en el caso de las sustancias de bajo peso molecular. A pesar de ello, las características de la celdilla unidad, tales como el sistema cristalino, sus dimensiones y las posiciones de los átomos en la misma, han sido determinadas para una gran variedad de polímeros. En todos ellos la celdilla unidad no contiene a la molécula completa. Este es un hecho que diferencia a los polímeros de las sustancias de bajo peso molecular. 187 1 1 . 2 . - Grado de cristalinidad. Métodos para su determinación. 11.2.1.- Introducción. Puesto que los polímeros cristalinos no cristalizan al 100 % sino que son sólo parcialmente cristalinos, parece oportuno definir una magnitud que cuantifique la extensión del proceso de cristalización. Se define así el grado de cristalinidad como la fracción en masa (o en volumen) de polímero que se encuentra en estado cristalino. Las propiedades físicas y mecánicas de los polímeros son altamente dependientes de su grado de cristalinidad. Este hecho ha llevado a considerar el grado de cristalinidad como un dato fundamental en la caracterización de un material polimérico susceptible de cristalizar. Así, la determinación de la proporción del sólido que es cristalino (su cristalinidad) tiene, a menudo, una considerable importancia práctica, ya que una cristalinidad creciente tiene el efecto útil de mejorar propiedades tales como la resistencia y la rigidez, la resistencia a la disolución y la estabilidad dimensional (ablandamiento térmico). Los métodos para calcular el grado de cristalinidad suponen a los polímeros como sistemas formados por regiones cristalinas y amorfas bien delimitadas. Sin embargo, se sabe que esto no es así, pues existe una región interfacial entre las zonas cristalinas y amorfas en la que participan la zona de plegamientos de la cadena, cadenas interlaminares, etc. Además, existen defectos intracristalinos que pueden dar lugar a diferentes grados de ordenación. En cuanto a los métodos para determinar el grado de cristalinidad, la mayoría están basados en técnicas que miden propiedades relacionadas con él, por lo que la cristalinidad determinada por distintas técnicas no tiene por qué dar necesariamente el mismo valor. La única técnica que, en principio, puede proporcionar una medida directa del orden tridimensional que conlleva la cristalinidad es la difracción de rayos X. 11.2.2.- Métodos de difracción de rayos X. La determinación del grado de cristalinidad mediante la difracción de rayos X se basa en que cuando un haz de rayos X incide sobre un conjunto de átomos, la dispersión total producida por éstos es independiente de su estado de orden o desorden. Por consiguiente, si en el diagrama de difracción de rayos X puede identificarse y separarse la dispersión producida por las regiones cristalinas y la producida por las regiones amorfas, podría calcularse la fracción de material cristalino. La figura 11.2.2.1 muestra un diagrama de rayos X para un polímero semicristalino, donde puede verse la intensidad de los rayos X dispersados frente al ángulo de difracción 2θ. Los picos relativamente más estrechos son debidos a la dispersión producida por las regiones cristalinas, mientras que el pico más ancho (llamado halo amorfo), sobre el que los demás picos descansan, es debido, principalmente, a la dispersión producida por las zonas amorfas. En principio, a partir de las áreas bajo los picos cristalinos, Ac, y bajo el ancho pico amorfo, Aa, debería ser posible calcular el grado de cristalinidad, wc (en tanto por uno), mediante la expresión: wc = Ac Ac + Aa (11.2.2.1) 188 Figura 11.2.2.1.- Diagrama de difracción de rayos X de polipropileno, resuelto en picos cristalinos y halo amorfo. En la práctica, sin embargo, es difícil resolver el difractograma obtenido para un polímero semicristalino en las áreas correspondientes a cada fase ya que, en cierto grado, la dispersión correspondiente a las zonas cristalinas y amorfas está superpuesta, pues las regiones cristalinas exhiben también una dispersión debida a las vibraciones térmicas y a las imperfecciones en la red cristalina que aparece en el pico ancho difuso, con la correspondiente pérdida de intensidad en las reflexiones cristalinas. 11.2.3.- Métodos volumétricos. Cuando un polímero cristaliza se produce un aumento de su densidad. La densidad de un polímero cristalino, ρc, es mayor que la de un polímero amorfo, ρa, del mismo material y peso molecular, ya que las cadenas de la estructura cristalina están más empaquetadas. Es de esperar que la densidad de un polímero semicristalino esté comprendida entre la densidad del polímero completamente amorfo y la del polímero totalmente cristalino. El método supone que la muestra está formada por dos fases, cristalina y amorfa, y que las propiedades de las dos fases son aditivas. Se puede calcular el grado de cristalinidad como fracción en volumen de cristales, Φc, o como fracción en peso, wc. La medida, por cualquier método, es más convenientemente realizarla a la temperatura ambiente, por ejemplo, 20 °C. Si Vc es el volumen de los cristales y Va es el volumen del material amorfo, entonces el volumen total de la muestra, V, está dado por: V = Va + Vc (11.2.3.1) De la misma forma el peso de la muestra, W, está dado por: W = Wa + Wc (11.2.3.2) donde Wc y Wa son los pesos de material cristalino y amorfo, respectivamente. 189 Teniendo en cuenta la relación entre el peso y la densidad, la ecuación (11.2.3.2) se transforma en: ρV = ρ aVa + ρcVc (11.2.3.3) Sustituyendo Va de la ecuación (11.2.3.1) en la (11.2.3.3) y ordenando se obtiene: Φc = Vc ρ − ρa = V ρc − ρ a (11.2.3.4) De forma análoga, el grado de cristalinidad expresado como fracción en peso será: Wc = Wc ρcVc Vc ρ − ρ a ρc ρ − ρ a = = = = W ρV V ρc − ρ a ρ ρc − ρa (11.2.3.5) donde ρ es la densidad de la muestra para la cual se pretende determinar el porcentaje de cristalinidad, ρa la densidad del polímero totalmente amorfo, ρc la densidad del polímero totalmente cristalino. Hay muy pocos polímeros para que los que falla el método volumétrico. Uno es el PTFE. La ecuación (11.2.3.5) relaciona el grado de cristalinidad, expresado como fracción en peso, de una muestra de densidad con las densidades de los polímeros amorfo y cristalino, respectivamente. La densidad de las regiones cristalinas, ρc, puede ser calculada a partir de las dimensiones de la celda unidad determinadas por rayos X. La densidad de las regiones amorfas, ρa, puede ser medida directamente si el polímero puede obtenerse en estado completamente amorfo, por ejemplo, por enfriamiento brusco del polímero a partir del polímero fundido. Si esto no es posible, ρa puede determinarse por extrapolación de la densidad del fundido hasta la temperatura de interés o a partir de muestras semicristalinas, de cristalinidad y densidad conocidas, extrapolando la densidad para cristalinidad cero. En la práctica, debido a que las cadenas en estado amorfo están empaquetadas al azar, la densidad del polímero en estado amorfo puede ser diferente dependiendo del tratamiento térmico que haya sufrido la muestra. La densidad de una muestra problema, ρ, puede ser determinada por varios métodos; entre ellos: columna de gradiente de densidades, picnometría, dilatometría, etc. El método de la columna de gradiente de densidades es un método muy utilizado para determinar densidades en polímeros. En este método un gradiente lineal de densidades es generado mediante la mezcla de dos líquidos miscibles de diferente densidad en un vaso largo y cilíndrico. Los líquidos son mezclados en continuo, variando las proporciones de los mismos a lo largo de la longitud de la columna. El gradiente es calibrado 3 con flotadores de densidad conocida con una precisión alrededor de ± 0,2 mg/cm . Una vez de que el gradiente está establecido, la muestra se deja caer en la columna y por su nivel de flotación puede conocerse su densidad. 190 11.2.4.- Métodos calorimétricos. Uno de los métodos más utilizados para medir el grado de cristalinidad es la medida del calor de fusión mediante calorimetría diferencial de barrido, DSC. El grado de cristalinidad en fracción en peso puede determinarse mediante la expresión: ∆H m wc = (11.2.4.1) ∆H m0 0 donde ∆Hm es el calor de fusión por unidad de masa de la muestra de cristalinidad desconocida. ∆Hm es el calor de fusión por unidad de masa del polímero 100 % cristalino. Teniendo en cuenta que el calor de fusión es directamente proporcional al área de la endoterma (Figura 11.2.4.1). ∆Hm se determina de la siguiente forma: a partir del área de la endoterma de fusión Ar de una muestra patrón cuyo peso mr y calor de fusión por gramo AHr se conocen previamente, se determina la constante del calorímetro K. Habitualmente suele tomarse como muestra patrón un peso conocido del metal indio. K= mr ∆H r Ar (11.2.4.2) Si ahora se determina el área de la curva de fusión de la muestra problema, Ap, de peso conocido, mp,, puede usarse la constante K para determinar el calor de fusión de la muestra problema, AHm: ∆H m = KAp mp (11.2.4.3) Puesto que el calor de fusión se obtiene a partir del área de la endoterma, el trazado de la línea base tiene importancia en el resultado. Si el termograma, después de la endoterma, regresa a la misma línea base que tenía antes de la endoterma, el cálculo del área es fácil y puede realizarse trazando una línea recta que una la parte anterior y posterior a la endoterma de fusión. Sin embargo, no siempre después de la endoterma se regresa a la misma línea base, pues las propiedades térmicas del material antes y después de la transición no tienen por qué ser las mismas. En cualquier caso, el trazado de la línea base es subjetivo y puede dar lugar a errores importantes, sobre todo, si los picos de fusión son muy anchos. Figura 11.2.4.1.- Determinación del calor de fusión ∆Hm de una muestra de poli(óxido de etileno) para calcular su grado de cristalinidad. 191 11.3.-Cristales poliméricos. 11.3.1.- Introducción. Hasta el año 1957 no se obtuvieron los primeros cristales de polietileno con un tamaño suficiente para ser estudiados. Estos cristales, obtenidos por precipitación de una solución muy diluida, y observados al microscopio electrónico, tenían la forma de discos de muy pequeño espesor (10 - 20 nm.). Sorprendentemente la dirección de alineamiento de las cadenas de polímero coincidía con la del espesor del cristal, luego, como las cadenas son muy largas (por ejemplo, 5000 nm.), deberían doblarse múltiples veces en la formación del cristal. Hay que tener en cuenta además que las cadenas de los polímeros cristalinos son muy flexibles: el polietileno permite un doblado de 180° con sólo tres enlaces sucesivos (5 átomos de carbono) implicados. En los polímeros cristalinos reales tenemos siempre mezclas de zonas cristalinas y amorfas. La existencia de zonas amorfas es inevitable, en mayor o menor grado, por el propio proceso de cristalización. Las macromoléculas de los polímeros cristalinos a alta temperatura (estado líquido) tienen alta movilidad y se encuentran en un estado muy desordenado y entrelazado, existiendo una gran proporción de volumen libre entre las cadenas. Al enfriar el polímero hasta su temperatura de fusión, Tf, y comenzar la cristalización, sus macromoléculas comenzarán a reordenarse en una estructura regular y las cadenas llegan a alinearse localmente y empaquetarse en formaciones cristalinas regulares, alrededor de núcleos de cristalización Estas regiones cristalinas se denominan cristalitas y estan separadas unas de otras por regiones amorfas, ya que entre los núcleos cristalinos en crecimiento siempre quedarán atrapados segmentos de cadenas desordenados. Las estructuras cristalinas son asimilables a celdas unitarias a menudo muy complejas. El proceso anterior es bastante lento, mucho mas que la cristalización de un metal, pues el movimiento de las largas cadenas del polímero es mucho más difícil que el correspondiente a los átomos aislados de aquel. Digamos finalmente que las regiones cristalinas de los polímeros poseen defectos muy similares a los existentes en otros sólidos cristalinos (metales y cerámicos). Existen defectos puntuales (lugares vacantes, extremos de cadenas), dislocaciones y también defectos superficiales. 11.3.2.- Modelo de micela con flecos. El modelo más simple que describe el proceso anterior es el MODELO de MICELAS A FRANJAS O CON FLECOS, que se puede ver en la figura 11.3.2.1 y que fue propuesto por Hermans en 1930. El sólido consiste en una mezcla íntima de regiones cristalinas pequeñas (cristalitas o micelas) con cadenas de polímeros alineadas, embebidas en una matriz amorfa compuesta de moléculas orientadas al azar, (cristales ordenados y regiones amorfas distribuidas aleatoriamente). La longitud de los cristales (por ejemplo, 100 átomos de C) es menor que la de las moléculas (por ejemplo, 10000 átomos de C), por tanto, la macromolécula puede serpentear de un cristal a otro a través de regiones amorfas muchas veces (matriz amorfa en la que se encuentran inmersos los cristalitos). Todos los cristales y las regiones amorfas adyacentes están así firmemente entretejidas entre si por medio del hilado de las largas macromoléculas. 192 Figura 11.3.2.1.- Modelo de micelas con flecos de un polímero semicristalino, mostrando las regiones cristalina y amorfa. Este tipo de modelo fue particularmente útil para explicar un amplio abanico de propiedades de los materiales cristalinos, por ejemplo, la dureza o flexibilidad de un determinado sólido polimérico puede así explicarse sobre la base de los porcentajes de partes cristalinas o amorfas del material. Cuanto mayor sea el porcentaje de cristalinidad o grado de cristalinidad, mayor será la dureza intrínseca del material. Y cuanto más zonas amorfas tenga, más flexible será el sólido y más capacidad de absorción de golpes tendrá, debido al carácter más elástico de las zonas amorfas. Aunque el modelo de la micela con flecos servía para explicar ciertas propiedades, estaba en desacuerdo con otras como, por ejemplo, las propiedades ópticas de polímeros cristalinos, por lo que fue necesario continuar estudiando los cristales poliméricos a fin de proponer otros modelos. Para aislar a los cristales poliméricos de las zonas amorfas y facilitar así su estudio, existen procedimientos experimentales que consisten en atacar con un ácido diluido las partes amorfas, ya que las partes cristalinas son más resistentes, debido a la intensidad de las fuerzas intermoleculares que las mantienen unidas. De ese modo, se consigue carbonizar las zonas amorfas y liberar un polvo cristalino constituido por los cristales. Este procedimiento fue aplicado, ya en 1945, por Ranby y otros investigadores, pero los cristales aislados de esta forma resultan parcialmente dañados por la acción del ácido, impidiendo establecer con seguridad la naturaleza de los mismos. Esto llevó a intentar preparar cristales poliméricos por otros procedimientos. Estudios de microscopía óptica y de difracción de Rayos X han probado que el modelo de la micela con flecos no es del todo correcto, excepto para polímeros de excepcional baja cristalinidad, como por ejemplo, el PVC. Así, este modelo es válido para grados de cristalización inferiores al 50 %. 11.3.3.- Modelo de cadenas plegadas. Los conceptos sobre la naturaleza de los cristales en polímeros sintéticos sufrieron un brusco cambio cuando en 1957 Keller consiguió obtener cristales aislados (monocristales) de polietileno, esto es, cristales libres de conexiones con otros cristales a través de la zona amorfa. Vistos al microscopio electrónico, los monocristales aparecen como láminas planas que, frecuentemente, tienen estrías centrales (Figura 11.3.3.1). 193 Figura 11.3.3.1.- Fotomicrografia de un monocristal de polietileno obtenido a partir de una disolución en xileno. Estría central formada por derrumbamiento de la pirámide hueca Dependiendo de las condiciones de cristalización, temperatura, disolvente, concentración, etc. se obtienen cristales de diversas formas. La forma simple en monocapa, anteriormente expuesta, suele encontrarse a temperaturas de cristalización altas. Sin embargo, a bajas temperaturas de cristalización, aparecen predominantemente cristales en multicapas que se originan como consecuencia de dislocaciones tipo tornillo en el crecimiento del cristal (Figura 11.3.3.2). Figura 11.3.3.2.- Fotomicrografia de un cristal multicapa de poli(óxido de metileno) El estudio por difracción de electrones de los monocristales mostraba que las cadenas poliméricas tienen su eje colocado perpendicularmente a las bases del monocristal. Y para explicar el hecho de que las laminillas cristalinas encontradas eran mucho mas delgadas, unos 100 Ǻ, que la longitud de la cadena de polímero extendida, unos 10000 Ǻ, Keller postuló que las cadenas no podían hacer otra cosa que plegarse en dirección perpendicular a las caras más extensas del cristal. Estudios posteriores, realizados tanto sobre polietileno como sobre otros polímeros cristalizados a partir de la disolución, proporcionan evidencias en favor del concepto de cadena plegada. El MODELO de CADENAS PLEGADAS, que se puede ver en la figura 11.3.3.3, muestra que las regiones cristalinas toman la forma de placas delgadas (o laminillas), denominadas cristalitas, en las cuales las cadenas están alineadas perpendicularmente a las caras planas de las láminas y plegadas repetidamente. El espesor de las cristalitas es de, aproximadamente, 10 o 20 nm (En el caso del polietileno, las cadenas se extienden alrededor de 100 Ǻ antes de plegarse). , pero las dimensiones laterales alcanzan valores de 10 µm. Frecuentemente estas laminillas forman una estructura de multicapa, como muestra la micrografía electrónica 194 de un monocristal de polietileno, (Figura 11.3.3.4). Se teoriza que cada laminilla está formada por cadenas que se pliegan una y otra vez sobre sí mismas, que los dobleces de las cadenas se encuentran en las caras de la laminilla. Cada laminilla contendrá varias moléculas, pero la longitud media de las cadenas será muy superior al espesor de la laminilla Figura 11.3.3.3.- Estructura de cadenas plegadas para una cristalita polimérica laminar. Figura 11.3.3.4.- Micrografía electrónica de un monocristal de polietileno (x20000). Los monocristales no se consideran exentos de parte amorfa, constituida fundamentalmente por los bucles en los que la cadena se dobla para volver entrar en la fase cristalina. El grado de cristalinidad de los monocristales es, sin embargo, muy elevado y suele rondar el 90 %. La reentrada de la cadena en el cristal, tras el bucle correspondiente, ha sido cuestión de discusión. Keller y otros, basándose en el hecho de que los monocristales obtenidos tenían aristas y caras bien definidas, propusieron que las cadenas se doblan de forma absolutamente regular, entrando en el cristal en una posición adyacente a la de salida, minimizando así las irregularidades en el cristal (Figura 11.3.3.5.a). Por otro lado, teniendo en cuenta que la densidad de los cristales es bastante más baja que la calculada a partir de las dimensiones obtenidas por RX para la celdilla unidad, lo que supone un mayor contenido en parte amorfa que el postulado por el modelo anterior, Flory y otros han propuesto que los bucles dan lugar a extensiones más largas de las cadenas, en una entrada no adyacente, similar a la que se puede contemplar en una vieja centralita de teléfonos (Figura 11.3.3.5.b). Probablemente ambos planteamientos son casos extremos o ideales de una situación intermedia en la que pueden darse, tanto entradas adyacentes regulares como entradas al azar más distantes, que implican longitudes mayores de cadena. 195 Figura 11.3.3.5.- (a).- Modelo de entrada adyacente en las laminillas (b).- Modelo de entrada no adyacente en las laminillas Cuando los polímeros fundidos, con capacidad de cristalizar, se colocan a temperaturas inferiores a su temperatura de fusión, la mayoría de ellos cristalizan formando esferulitas. Como su nombre indica, cada esferulita crece en forma de esfera. En la figura 11.3.3.6 se muestran esferulitas de caucho natural observadas mediante microscopía electrónica de transmisión. Figura 11.3.3.6.- Micrografía electrónica de transmisión mostrando la estructura esferulítica de una probeta de caucho natural. Las cristalitas laminares de cadenas plegadas se extienden radialmente desde el centro, apareciendo como líneas blancas. Las esferulitas consisten en un agregado de cristalitas o láminas delgadas de cadena plegada(similar a los monocristales obtenidos desde la disolución) de, aproximadamente, 10 nm de espesor que parten desde un núcleo central (A menudo diminutas partículas de impureza), orientándose de forma radial. Pueden tener unas dimensiones (diámetro) que suele variar entre la micra y el milímetros. 196 Algunas laminillas se extienden desde el centro de la esferulita hasta el final de la misma. Sin embargo, la mayoría de ellas, mediante algún mecanismo de dislocación tipo tornillo, generan ramas que divergen propagándose también radialmente, originando así la forma esférica. En resumen, la parte cristalina de una esferulita consiste en multitud de láminas delgadas que parten desde un punto central ramificándose. La estructura detallada de la esferulita se esquematiza en la figura 11.3.3.7, donde se aprecia que los cristales laminares están separados por material amorfo. Por consiguiente, es probable que las macromoléculas individuales unan las regiones amorfas y cristalinas como sugiere el modelo de micelas a franjas (Las moléculas de unión conectan las laminillas contiguas a través de regiones amorfas). Así, algunas cadenas comienzan en una lámina, atraviesan la región amorfa y finalmente se unen a otra lámina. Dichas cadenas reciben el nombre de moléculas vínculo. Figura 11.3.3.7.- Representación esquemática del detalle de la estructura de una esferulita. En realidad, las esferulitas tienen simetría esférica sólo en las primeras fases de la cristalización. Dependiendo de la temperatura de cristalización, que condiciona el número de esferulitas que aparecen por unidad de tiempo, llega un momento en el que las esferulitas colisionan unas con otras y los extremos de las esferas adyacentes se tocan formando límites más o menos planos destruyéndose así su apariencia redondeada, como indica la figura 11.3.3.8, que corresponde a una micrografía del polipropileno utilizando luz polarizada. 197 Por otro lado, cuando lo que se cristaliza es una lámina delgada de polímero, las entidades cristalinas que se forman son discos que pueden ser considerados como una sección transversal de la esferulita. Figura 11.3.3.8.- Esferulitas de polipropileno isotácticos cristalizando isotérmicamente a 140 °C. Fotografías realizada en un microscopio óptico con polarizadores cruzados. Arriba: esferulitas en crecimiento en cuyo interior puede observarse la característica cruz de Malta. Entre unas esferulitas y otras se observa un fondo negro que corresponde al material que aún no ha cristalizado. Abajo: esferulitas después de completar el crecimiento. En el interior de las mismas puede observarse su textura fina. Como consecuencia de la interacción de la luz polarizada con la estructura real del cristal se produce, cuando la observación se hace entre polarizadores cruzados (Luz polarizada y prismas cruzados de NICOL), la conocida cruz de Malta, indicativa de que las esferulitas son entidades birrefringentes con simetría esférica; sin embargo, el medio que las rodea aparece oscuro, como consecuencia de que el polímero que permanece sin cristalizar es isótropo. Las esferulitas de los polímeros equivalen a los granos de los metales policristalinos y de las cerámicas. Sin embargo, cada esferulita está formada por diferentes cristales laminares y por algún material amorfo. En una porción de polímero cristalino, existen varios millones de esferulitas. Polietileno, polipropileno, cloruro de polivinilo, politetrafluoroetileno y nilón tienen estructura esferulítica al cristalizar a partir de un líquido. En la figura 11.3.3.9 puede verse una representación esquemática del proceso de nucleación y crecimiento de esferulitas durante el enfriamiento de un polímero fundido. Figura 11.3.3.9.- Nucleación y crecimiento de esferulitas durante el enfriamiento de un polímero fundido. 198 El mecanismo de formación de las esferulitas se esquematiza en la figura 11.3.3.10. La estructura que se forma inicialmente es un cristal sencillo con láminas de cadena plegada (Primer cristalito precursor, figura 11.3.3.10.a), del que emana progresivamente (b) una fina textura radial formada por laminillas que crecen por las puntas. Esto conduce rápidamente a la formación de estructuras en forma de haz o gavilla (Figura 11.3.3.10.d) que se llaman axialitas o hedritas. A medida que se desarrolla el crecimiento, las láminas se desarrollan a los dos lados de un plano central de referencia. Las láminas continúan su expansión ocupando secciones cada vez mayores mediante la formación de más láminas en puntos de ramificación apropiados. El resultado es la formación de esferulitas. Figura 11.3.3.10.- Representación esquemática de las sucesivas etapas en la formación de una esferulita. El tamaño relativo de la esferulita puede predecirse de lo que es conocido de la cristalización de sólidos monoatómicos. La solidificación a baja temperatura da lugar a esferulitas de pequeño tamaño porque la velocidad de nucleación es alta y la de crecimiento baja. Recíprocamente, la solidificación a alta temperatura da lugar a esferulitas de gran tamaño porque la velocidad de nucleación es baja y con relación a la de crecimiento. 199 La velocidad de crecimiento radial, G, es constante con el tiempo. R = Gt (11.3.3.1) La figura 11.3.3.11 muestra la evolución del radio de las esferulitas con el tiempo para el polipropileno a varias temperaturas de cristalización. Figura 11.3.3.11.- Radio de las esferulitas en función del tiempo para una muestra de polipropileno cristalizado isotermicamente a partir del fundido a varias temperaturas de cristalización. Igualmente puede observarse que la velocidad de crecimiento, G, tiene una gran dependencia de la temperatura de cristalización y presenta un máximo, cuando se representa frente a ella, tal como se observa en la figura 11.3.3.12 para el caso de un poliestireno isotáctico. Figura 11.3.3.12.- Velocidad de crecimiento frente a la temperatura de cristalización para esferulitas de poliestireno isotáctico. La curva más baja corresponde a una muestra de Mw=190000 y baja tacticidad y la más alta a una muestra de Mw=1380000 y alta tacticidad 200 Cristales obtenidos mediante orientación. Cuando un polímero, fundido o en disolución, se enfría bajo la acción de una fuerza unidireccional cristaliza desarrollando una morfología diferente a la obtenida en ausencia de ella. Muchos procesos tecnológicos, como la formación de fibras o el moldeo, llevan consigo la aplicación de fuerzas orientacionales durante el proceso de cristalización. Durante estos procesos las cadenas son estiradas y cristalizan, dando lugar a una morfología de tipo fibroso. Pero en este proceso de estiramiento no es posible orientar completamente a todas las cadenas en su conjunto. Así, mientras unas son orientadas en la dirección del estiramiento, otras quedan más flojas, orientadas más o menos al azar. Las cadenas más orientadas se encuentran prácticamente extendidas, alineadas en la dirección de la fuerza formando un cristal fibrilar central. Las cadenas no orientadas se depositan sobre las orientadas formando laminillas de cadenas plegadas también paralelas a la dirección de la fuerza. Este conjunto fibralaminilla tiene forma de brocheta; un esquema puede verse en la figura 11.3.3.13. Figura 11.3.3.13.- Modelo de la morfología “brocheta”. En la figura 11.3.3.14 se muestra un ejemplo típico de la morfología desarrollada por el polietileno cuando es cristalizado a partir de una disolución en xileno al 5 % mientras es agitada rápidamente. Figura 11.3.3.14.- Microfotografía tomada mediante microscopía electrónica de polietileno cristalizado a partir de una disolución con agitación. 201 Este tipo de cristalización tiene importantes consecuencias tecnológicas, pues la morfología fibrosa resultante es tenaz a lo largo de la dirección del eje de fibra, ya que los enlaces de valencia de la cadena principal están orientados en esta dirección. 11.4.- Factores que favorecen el estado cristalino. Los factores determinantes de la cristalinidad de un polímero son aquellos que permiten el empaquetamiento de las cadenas poliméricas favoreciendo la cristalinidad. Así, el grado de cristalización de un polímero depende principalmente de dos factores: la mayor o menor flexibilidad de sus cadenas y la regularidad, tanto química como estructural, de las mismas. Los homopolímeros presentan mayor grado de cristalinidad que los copolímeros. La flexibilidad incide sobre la movilidad de las moléculas para reordenarse y constituir el cristal. La regularidad es absolutamente necesaria para constituir el bloque repetitivo (celda unidad) constructor del cristal. La condición de regularidad, que una condición necesaria pero no suficiente, puede lograrse de varias formas, entre las cuales se pueden citar: 1.- Cuando los grupos laterales a ambos lados del átomo de la cadena principal son iguales, de modo que la cadena es simétrica. Ejemplos de tales cadenas son: 202 2.- Cuando los grupos laterales son lo bastantes pequeños para poder ajustarse en una estructura cristalina. Ejemplos de tales cadenas son: 3.- Un solo grupo lateral en un átomo de la cadena lateral. Ejemplos son : 203 4.- La regularidad de la disposición de los grupos laterales de la cadena (isotacticidad) y la linealidad originan altos grados de cristalinidad al permitir una gran aproximación y empaquetamiento de las mismas. En el polietileno de alta densidad, PEAD(Figura 11.4.1) se alcanza hasta un 90-95 % de cristalinidad. En cambio en el polietileno ramificado, PEBD (Figura 11.4.1) (copolimerizado con otras α-olefinas) no se ha conseguido cristalizar el material, y por ello es amorfo. La existencia de ramificaciones en las cadenas dificulta el ajuste perfecto de la estructura cristalina. Figura 11.4.1.- Polietileno de alta densidad (PEAD) y de baja densidad PEBD (Ramificado). La cristalización es muy fácil en los polímeros lineales, puesto que no existen restricciones al alineamiento de las cadenas. Las ramas interfieren la cristalización, ya que no pueden ser incorporadas en el retículo cristalino y quedan como dominios amorfos reduciendo fuertemente la cristalinidad, con lo que los polímeros ramificados nunca son totalmente cristalinos. De hecho, una ramificación excesiva puede prevenir toda cristalización. El polietileno polimerizado a presión, LDPE, tiene, aproximadamente, 3 ramas colgantes (–CH3, – C2H5, etc.) por cada 100 unidades de cadena y alcanza un grado de cristalinidad entre el 40-50 %. Por el contrario, el polietileno obtenido a baja presión, HDPE, tiene 3 grupos colgando por cada 1000 unidades y alcanza un grado de cristalinidad del 90-95%. 204 Los polímeros reticulados son casi totalmente amorfos, mientras que los entrecruzados tienen varios grados de cristalinidad. Los anclajes introducidos en la vulcanización de la goma -aunque pequeños en número- tienen este mismo efecto además de dificultar el movimiento de las moléculas: el grado de cristalización disminuye. La flexibilidad de las cadenas dificulta el empaquetamiento de las mismas, en consecuencia disminuye la cristalinidad. Tal es el caso de las siliconas, que son polímeros inorgánicos, es decir, no contienen átomos de carbono en su cadena principal (Figura 11.4.2). Figura 11.4.2.- Estructura de una cadena de silicona. Esta es una cadena alternada de átomos de silicio y de oxígeno. Cada átomo de silicio tiene dos grupos unidos a él, que pueden ser orgánicos. Por lo tanto las siliconas pueden soportar altas temperaturas sin descomponerse, pero tienen temperaturas de transición vítrea muy bajas. La figura 11.4.2 muestra grupos metilo unidos a los átomos de silicio. Este polímero se llama polidimetilsiloxano, que es la silicona más común. Las siliconas constituyen buenos elastómeros porque las cadenas macromoleculares principales son sumamente flexibles. Los enlaces entre un átomo de silicio y los dos átomos de oxígeno unidos a él, son fuertes pero a su vez altamente flexibles. El ángulo formado por estos enlaces, puede abrirse y cerrarse como si fuera una tijera, sin demasiados problemas. Esto hace que toda la cadena principal sea flexible. El polidimetilsiloxano hace algo realmente extraño cuando se lo mezcla con ácido bórico, B(OH)3. La mezcla es suave y dúctil, y puede ser moldeada fácilmente con los dedos. Pero también es muy elástica. Más aún, si se toca suavemente se desliza, pero si se golpea fuertemente con un martillo se quiebra . Si se esparce sobre un papel de diario y se aprieta, el texto del diario queda impreso en ella. No se ha encontrado una aplicación industrial para este maravilloso material, pero se han vendido toneladas del mismo a través del juguete llamado Silly Putty. La capacidad de un polímero para cristalizar está influida por la química molecular y por la configuración de la cadena. La cristalización no está favorecida en los polímeros constituidos por unidades monoméricas químicamente complejas (por ejemplo: poliisopreno). La cristalización tampoco se evita fácilmente en polímeros químicamente sencillos, como polietileno y politetrafluoroetileno, incluso a velocidades de enfriamiento muy rápidas. En cuanto a los estereoisómeros, los polímeros atácticos son difíciles de cristalizar, mientras que los polímeros isotácticos y sindiotácticos cristalizan más fácilmente debido a que la regularidad de las posiciones de los grupos laterales contribuye al proceso de ordenación de las cadenas contiguas. Cuanto mayor es el tamaño de los grupos substituyentes, menor es la tendencia a la cristalización. Dentro de los polímeros vinílicos sólo las configuraciones isotáctica y sindiotáctica tienen la regularidad suficiente como para cristalizar. La configuración atáctica con el grupo lateral (- R) dispuesto de manera aleatoria nunca podrá cristalizar. Así, el polipropileno isotáctico tiene un grado de cristalización del 50-60% mientras que el atáctico es amorfo. 205 Otros polímeros vinílicos con grupos sustitutivos mayores (PVC, PMMA, etc) no cristalizan nunca -ni siquiera su configuración isotáctica- en virtud de la distorsión que introducen los citados grupos laterales además de por la pérdida de libertad de movimientos de las cadenas; a este respecto también influye la mayor o menor polaridad de estos grupos. También la simetría del monómero favorece la cristalinidad. La introducción de un sustituyente en el etileno como por ejemplo un grupo fenilo (estireno), disminuye la cristalinidad. En este ejemplo también hay que considerar el gran tamaño del sustituyente que impide un total acercamiento de las cadenas. La copolimerización reduce el grado de cristalización porque el segundo monómero no podrá ajustarse adecuadamente en la red cristalina del primero y, caso de ser muy diferentes, la cristalización podría verse completamente impedida por falta de regularidad estructural. Mientras las estructuras cis y trans del poliisopreno cristalizan respectivamente un 20-30 % y un 55-70 %, la mezcla de ambas es un polímero amorfo. Por regla general, los copolímeros con unidades monoméricas más irregulares y libremente dispuestas tienen mayor tendencia al estado amorfo. Los copolímeros alternos y en bloque siempre presentan cristalización. En los copolímeros de bloque, la disparidad en la longitud de los bloques dificulta la cristalización. Los copolímeros al azar y con injertos normalmente son amorfos. Si los copolímeros son al azar, la dificultad para cristalizar aumenta al aumentar la cantidad del comonómero que se encuentre en menor proporción. Si uno de los comonómeros no puede cristalizar, queda confinado en dominios amorfos. La existencia de fuerzas intermoleculares entre las cadenas favorece el empaquetamiento y alineación de las mismas. La presencia de grupos polares entre los que se puedan establecer puentes de hidrógeno, como en las poliamidas, incrementa de manera notable la cristalinidad . Por el contrario, la disminución de estas fuerzas moleculares originadas por la incorporación de plastificantes al polímero, disminuye la cristalinidad pero tiene la ventaja de que facilita el procesado del material. Finalmente, es importante tener en cuenta que como el proceso de cristalización es bastante lento pues la libertad de movimientos de las largas cadenas del polímero no es total, el grado de cristalización aumentará al disminuir la velocidad de enfriamiento del polímero, o manteniendo a éste un cierto tiempo en el rango de temperaturas donde la velocidad de cristalización es mayor. Durante la cristalización, al enfriar hasta la temperatura de fusión, las cadenas enmarañadas y situadas al azar en el líquido viscoso asumen una configuración ordenada. Para que esto ocurra, las cadenas necesitan suficiente tiempo para moverse y alinearse. El grado de cristalinidad de un polímero depende de la velocidad de enfriamiento durante la solidificación y de la configuración de la cadena. En general, cuanto menor es la velocidad de enfriamiento mayor es la cristalinidad y más grande es el tamaño de las esferulitas. Reciprocamente, si el enfriamiento es más rápido, la cristalinidad será menor, así como el tamaño de las esferulitas. La cristalización ocurre más rápidamente a una temperatura alrededor del 80 % del punto de fusión. En la tabla 11.4.1 se resumen los factores determinantes de la cristalinidad. 206 Tabla 4.1.- Factores determinantes de la cristalinidad. FAVORECEN DISMINUYEN Regularidad (PE, PP isotáctico) Ramificaciones Simetría Sustituyentes voluminosos Rigidez Flexibilidad Fuerzas intermoleculares Plastificantes 11.5.- Nucleación y crecimiento de los cristales. Como en la mayoria de los procesos de cristalizacion, si no hay nucleacion, existe un periodo de induccion durante el que se desenredan las cadenas. Este proceso viene seguido de un crecimiento cristalino lento. Sin embargo, la velocidad de cristalizacion aumenta y luego disminuye cuando se acerca al final del proceso de cristalizacion. La velocidad de crecimiento cristalino puede seguirse mediante dilatometria utilizando la ecuacion de Avrami, la cual fue desarrollada para seguir la velocidad de cristalizacion de los metales: Vt − V f V0 − V f = e− K n (11.5.1) Como se indica en mediante la ecuacion (11.4.1), el cociente de la diferencia entre el volumen especifico Vt en el instante t y el volumen final Vf, dividido por la diferencia entre el volumen especifico inicial V0 y el volumen final Vf es igual a una expresion experimental en la que K es una constante cinetica relacionada con la velocidad de nucleacion y crecimiento y n es un entero relacionado con la nucleacion y crecimiento de cristales. El valor de n puede situarse entre 1 y 4, siendo igual a 4 para el crecimiento de cristales tridimensionales. La cristalización de un polímero a temperaturas inferiores a la de fusión (Tf) puede seguirse fácilmente a partir de medidas de su densidad. Sabemos que cualquier proceso de cristalización induce un brusco cambio en la densidad del material como consecuencia de la mayor compacidad de la estructura cristalina en relación al estado líquido. En la figura 11.5.1, se da la variación del volumen específico en función del tiempo durante la cristalización de la goma natural a temperatura constante. La goma presenta un grado de cristalización máximo del 30%. Estas curvas muestran la forma típica de los procesos de nucleación y crecimiento (lento al principio, aceleración intermedia y, de nuevo, lento al final). Figura 11.5.1.- Variación del volumen específico en función del tiempo durante la cristalización (T = cte). 207 La influencia de la temperatura se refleja mas claramente en la figura 11.5.2. En ella vemos que la cristalización no comienza hasta los 28° C (temperatura de fusión, Tf), se acelera para temperaturas inferiores, pasa por un máximo (a la temperatura de 25 °C) y luego el proceso se ralentiza y, finalmente, ya no tiene lugar para temperaturas inferiores a -73 °C. Esta última temperatura coincide con la T, de la goma natural, donde la imposibilidad del movimiento intermolecular impide la cristalización. Figura 11.5.2.- Influencia de la temperatura en la velocidad de cristalización. 11.6.- Cristalización por estirado. La figura 11.6.1 refleja los cambios de densidad que tienen lugar en la goma natural, mantenida a 0°C, espontaneamente y, en otros dos casos, actuando un cierto grado de estirado. La variación expresada de densidad nos indica que la goma está cristalizando (estamos por debajo de su Tf ). El efecto de incrementar el grado de estirado es similar al de disminuir la temperatura, en ambos casos aumenta la velocidad de cristalización. Nótese que para grados de estirado muy grandes (500 %) la cristalización es tan rápida que ocurre simultaneamente con la extensión. El aumento del grado de estirado también incrementa el grado de cristalización logrado (mayor variación final de densidad ). Figura 11.6.1.- Cambios de densidad que tienen lugar en la goma natural en función del tiempo. El efecto del estirado se explica fácilmente al tener en cuenta la morfología que adoptan las cadenas del polimero en su estado cristalino. La figura 11.6.2.(a), muestra la estructura de la goma cristalizada sin estirar. Las regiones cristalinas presentan las macromoleculas alineadas en perfecto orden mientras que en las zonas amorfas existe un completo desorden molecular. Si entonces estiramos el polimero, sus macromoléculas se alinearán en la dirección del estirado lo que, obviamente, facilita la formación de núcleos cristalinos por precisarse menores movimientos para la ordenación de las cadenas. Otro hecho importante, consecuencia de lo anterior, es que las cadenas que constituyen todos los cristales han quedado alineadas según una única dirección, la dirección de estirado, como queda de manifiesto en la figura 11.6.2.(b) . 208 Figura 11.6.2.- Estructura de la goma cristalizada (a).- Sin estirar (b).- En estado estirado. Los dominios ordenados representan cristales individuales. 11.7.- Celda unidad de los polímeros cristalinos. Como el resto de los sólidos cristalinos su estructura se describe en términos de su celda unidad o bloque que se repite a lo largo de toda la estructura cristalina. Por esta razón, la cristalización de un polímero exige que sus cadenas posean la regularidad suficiente para constituir la celda unidad. La figura 11.7.1 presenta la celda unidad constitutiva del polietileno cristalizado. Consta de 5 cadenas, una central y cuatro laterales alineadas. Las dimensiones de la celda también se incluyen en la citada figura. Nótese que el parámetro " c " corresponde al doble de la distancia vertical entre dos átomos de carbono en la conformación estirada. Los polímeros con estructuras más complicadas muestran celdas unidad bastante mas complejas. Figura 11.7.1.- Ordenación de las cadenas en la celda elemental del polietileno 209 11.8.- Comportamiento mecánico de los polímeros cristalinos. Los polímeros cristalinos, como todos los sólidos cristalinos, son duros, resistentes y tenaces. Recuerdese que la existencia de defectos lineales (dislocaciones) permite la deformación plástica del material. También hay que tener en cuenta que estos polímeros presentan además zonas amorfas cuyo comportamiento mecánico es bien diferente y que además en ellas este comportamiento depende de si estamos por encima o por debajo de su temperatura Tg. La figura 11.8.1, muestra la variación del módulo elástico, E, con la temperatura, del poliestireno (PS) cristalino; para comprobar se presenta también la correspondiente variedad amorfa. El poliestireno cristalino (configuración isotáctica) revela un lento y progresivo descenso del módulo E con el aumento de la temperatura hasta la brusca caida que tiene lugar en la región correspondiente al punto de fusión. Con respoecto al comportamiento del polímero amorfo han desaparecido las regiones viscoelástica y elastómera, dado que, incluso a alta temperatura, la estructura cristalina impide totalmente el movimiento molecular. Figura 11.8.1.- Variación del módulo elástico E con la temperatura, del poliestireno (PS) cristalino y amorfo. Sobre la figura 11.8.1, se ha representado también la curva correspondiente al polímero (poliestireno, también) de estructura tridimensional. Nótese el descenso del módulo E en la zona de transición vítrea. Por encima de Tg los segmentos de cadena entre dos puntos de anclaje (enlaces intermoleculares) podrán moverse. Sin embargo estos movimientos nunca serán grandes y el módulo E mantiene su valor intermedio. Por otro lado estos polímeros mantienen su rigidez a temperaturas bastante elevadas debido a sus fuertes enlaces intermoleculares. 210 12.- Clasificación de los polímeros industriales. 12.1.- Introducción. Además de los polímeros clásicos que se producen y comercializan desde hace años, cada día aparecen otros nuevos, provenientes de las investigaciones científicas y tecnológicas que se desarrollan en todo el mundo. Por lo que, dada la gran variedad de materiales poliméricos existentes, se hace necesario agruparlos según sus características, facilitando así el entendimiento y el estudio de las propiedades. Se han hecho numerosos intentos de proporcionar a los Polímeros Industriales una clasificación definitiva, teniendo en cuenta sus estructuras químicas, sus distintas propiedades mecánicas, eléctricas y ópticas, el comportamiento frente el calor, tipos de aplicaciones, escala de producción, o aún otras características. Existen, pues, diferentes posibilidades de clasificación de los polímeros. Entre las más habituales se pueden encontrar las que siguen. 12.2.- Clasificación según el tipo de Estructura Química. Se pueden agrupar en tres divisiones: 12.2.1. -Según la cantidad de meros diferentes en el polímero. Un polímero puede ser constituido por una única unidad monomérica (Cadena homogénea) o por dos o más unidades monomérica (cadena heterogénea). Cuando la cadena es homogénea, se llama homopolímero, y cuando es heterogénea copolímero. Los copolímeros constituidos de tres unidades monoméricas diferentes se denominan terpolímeros. Un ejemplo típico es el ABS, es decir, el terpolímero de acrilonitrilo-butadieno-estireno. La reacción química que forma los copolímeros se conoce como copolimerización, y los monómeros de comonómeros. Cuando se cambian los comonómeros o la cantidad relativa de cualquiera de ellos, el material que se obtiene presenta propiedades diferentes, tanto químicas como físicas. 12.2.2.- Con relación a la estructura química de los meros que constituyen el polímero. Otra posibilidad es contemplar la similitud química de los productos, obteniéndose un gran número de familias. Esta clasificación se basa en el grupo funcional al cual pertenecen los meros. Así tenemos como ejemplos: - POLIOLEFINAS – POLIPROPILENO, POLIBUTADIENO, POLIESTIRENO. - POLIÉSTERES – POLI(TEREFTALATO DE ETILENO), POLICARBONATO. - POLIÉTERES – POLI(ÓXIDO DE ETILENO), POLI(ÓXIDO DE FENILENO). - POLIAMIDAS – NYLON, POLIIMIDA. - POLÍMEROS CELULOSICOS – NITRATO DE CELULOSA, ACETATO DE CELULOSA. - POLÍMEROS ACRÍLICOS – POLI(METACRILATO DE METILO), POLIACRILONITRILO. - RESINAS TERMOPLASTICAS : POLIACETALES Y POLIHALOETILENOS -POLIURETANOS, DENOMINACIÓN GENÉRICA PARA LOS QUE SON DERIVADOS DE ISOCIANATOS - RESINAS FORMALDEHIDO – RESINA FENOL-FORMOL, RESINA UREA-FORMOL. - FENOPLASTOS, AMINOPLASTOS, TIOPLASTOS - RESINAS DUROPLASTICAS : POLIESTER INSATURADO, POLIURETANOS, - SILICONAS - POLIVINILIDÉNICOS - DERIVADOS DE LA CELULOSA 211 12.2.3.- Con relación a la forma de la cadena polimérica. Las cadenas macromoleculares pueden ser: Lineales – en que no tienen ramificaciones. Ramificadas – todas las moléculas contienen ramificaciones, es decir pequeñas cadenas laterales. Entrecruzadas – los polímeros poseen estructura tridimensional, donde las cadenas están unidas unas a otras por enlaces químicos. 12.3.- Constitución química. Además de la similitud, se puede hacer una clasificación según la constitución química. Como la forma de obtención de un polímero condiciona las características químicas de un producto, la clasificación según la constitución incluye la clasificación según el mecanismo de polimerización que se mencionó anteriormente. Por esta razón, no se detallan dentro de esta clasificación. Según este criterio se pueden encontrar cuatro tipos de materias plásticas: (i).- Materias plásticas naturales transformadas. Como por ejemplo los derivados de la celulosa (acetato de celulosa, celuloide, etc.), derivados del caucho (ebonita, pliofilm, etc.), entre otros. (ii).- Materias plásticas polimerizadas. (iii).- Materias plásticas policondensadas. (iv).- Siliconas.Constituyen un tipo de plásticos en los que el papel del carbono lo desempeñan átomos de silicio. La unión con otras moléculas se realiza a través de átomos de oxígeno. 212 12.4.- Mecanismo de polimerización. Atendiendo al mecanismo de polimerización mediante el que se han obtenido, se tienen: (i).- Polímeros de adición. Pueden ser derivados de monómeros vinilicos: polietileno, polipropileno, poliestireno, cloruro de polivinilo; también derivados del butadieno: polibutadieno, caucho natural, teflón; etc. (ii).- Polímeros de condensación. Éstos incluyen varias familias, como poliamidas, poliésteres, poliuretanos, resinas fenólicas, resinas epoxi, poliéteres, etc. Algunos ejemplos son: nylon, tergal, melamina, baquelita, araldite, etc. 12.5.- Clasificación conforme el comportamiento frente la temperatura. La clasificación más extendida, y que hasta el momento no se ha logrado encontrar ninguna más conveniente, está relacionada con la estructura macromolecular del polímero, la cual condiciona de forma importante sus propiedades físicas y químicas, entre ellas el comportamiento frente la temperatura. Así pues, pueden diferenciarse los siguientes grupos: - TERMOPLÁSTICOS. - DUROPLÁSTICOS O TERMOENDURECIBLES O TERMOESTABLES O TERMOFIJOS - ELASTÓMEROS. TERMOPLASTICOS. Un polímero termoplástico tiene una estructura molecular mayoritariamente lineal con o sin ramificaciones (Figura 12.5.1). Las moléculas quedan enmarañadas unas con otras, pero sin que se entrelacen o reticulen entre si con enlaces covalentes (enlaces químicos). La elevación de la temperatura hace que la fuerza de los enlaces secundarios de debilite (porque la movilidad molecular aumenta) y esto facilita el movimiento relativo de las cadenas adyacentes al aplicar un esfuerzo, fluyendo como un líquido altamente viscoso. Un termoplástico reblandece (se ablanda) llegando a fluir cuando se somete a un calentamiento y vuelve a ser sólido y rígido (se endurecen) cuando baja la temperatura. Estos procesos son totalmente reversibles y pueden repetirse de forma reiterada. Este comportamiento permite que el plástico sea moldeado un número indefinido de veces, por efecto combinado de la presión y la temperatura, pues basta calentarlo para que reblandezca, se haga viscoso y sea introducido en un molde para que al enfriarse adquiera la forma del mismo. La degradación irreversible se produce cuando la temperatura de un termoplástico fundido se eleva hasta el punto que las vibraciones moleculares son tan violentas que pueden romper los enlaces covalentes. Los termoplásticos son relativamente blandos y dúctiles. La mayoría de los polímeros lineales y los que tienen estructuras ramificadas con cadenas flexibles son termoplásticos. TERMOESTABLES. Por el contrario, un polímero termoestable es aquel que no reblandece ni fluye por mucho que se eleve la temperatura, es más llega antes a descomponer que a fluir. No pueden ser los plásticos termoestables moldeados repetidas veces por esta razón. Los polímeros termoestables se endurecen al calentarse y no se ablandan al continuar calentando. El término termoestable puede inducir a error en cuanto a la estabilidad térmica de un polímero. Hay plásticos termoplásticos formados a base de poliamidas que son estables a temperaturas superiores a los 400 °C. Por tanto es más correcto llamar a los polímeros que no fluyen por acción del calor termofijos, ya que mantienen su forma fija aunque se eleve la temperatura. 213 Los polímeros termoestables (duroplásticos) tienen una estructura altamente reticulada (curados) (Figura 12.5.1) con una longitud de cadena pequeña entre las uniones transversales. Son duros y rígidos aún a temperaturas relativamente altas y no funden por efecto del calor. La denominación termoestable, en la actualidad, puede resultar confusa, ya que se emplea también para materiales poliméricos (termoplásticos y duroplásticos, indistintamente) que resisten temperaturas en el intervalo de los 200-300 ºC y aún superiores. Tal comportamiento es debido a la existencia de reticulaciones entre cadenas moleculares que se producen en las reacciones de curado, que se favorecen aumentando la temperatura (de ahí el nombre de termoendurecibles), formando una red espacial, en la que los movimientos de los segmentos moleculares quedan muy restringidos. Al iniciar el tratamiento térmico se origina entrecruzamiento covalente entre cadenas moleculares contiguas. Estos enlaces dificultan los movimientos de vibración y de rotación de las cadenas a elevadas temperaturas. Generalmente el entrecruzamiento es extenso: del 10 al 50% de las unidades monoméricas de las cadenas están entrecruzadas. Sólo el calentamiento a temperaturas excesivamente altas causa rotura de estos enlaces entrecruzados y degradación del polímero. Los polímeros termoestables generalmente son más duros, más resistentes y más frágiles que los termoplásticos y tienen mejor estabilidad dimensional. La mayoría de los polímeros entrecruzados y reticulados, como el caucho vulcanizado, los epoxi y las resinas fenólicas y de poliéster, son termoestables. ELASTOMEROS. Los elastómeros tienen una estructura poco reticulada (vulcanizados), distancia entre nudos muy grande. y admiten altas deformaciones de tipo elástico, es decir, que cesando los esfuerzos que originan la deformación, recuperan, al menos en parte, su forma original. Su estructura es también reticulada (Figura 12.5.1), pero en mucha menor extensión que los materiales duroplásticos, de modo que admiten relativamente grandes deformaciones con recuperaciones elásticas. La longitud de las cadenas es grande entre las uniones transversales. Los elastómeros son los componentes fundamentales de los cauchos. El diferente comportamiento frente a la temperatura así como frente a otros agentes físicos de los diferentes tipos de polímeros descritos anteriormente, es consecuencia de la estructura que presentan las cadenas poliméricas entre sí. En los plásticos termoplásticos, las cadenas macromoleculares se encuentran desordenadas, enrolladas sobre sí mismas pero independientes unas de otras. Solamente las mantienen unidas fuerzas electrostáticas débiles tipo Van der Waals. Estas fuerzas son lo suficientemente débiles como para que desaparezcan por acción de la temperatura al aumentar los movimientos moleculares. Estos movimientos adquieren mayor importancia a medida que aumenta la temperatura, ocasionando el deslizamiento de unas moléculas sobre otras. Estos materiales se caracterizan también porque pueden disolverse en determinados disolventes orgánicos. En este caso el disolvente penetra primeramente en la maraña de las cadenas poliméricas debilitando las fuerzas de unión entre ellas y de esta manera las separa, para disolverlas posteriormente. El proceso de disolución de una macromolécula requiere más tiempo que el que es necesario para disolver compuestos de bajo peso molecular, como consecuencia de la complejidad del proceso. Por eso, para probar la solubilidad de un determinado polímero en un disolvente deben transcurrir al menos varias horas. Las fuerzas de enlace de Van der Waals entre las moléculas son las responsables de no sólo el comportamiento térmico del material, sino también del resto de las propiedades mecánicas: resistencia y dureza, es decir de la propia cohesión del material. 214 En los polímeros termoestables o termofijos, las cadenas macromoleculares se encuentran unidas unas a otras tridimensionalmente por fuertes enlaces covalentes, denominándose nudos los puntos comunes a varias cadenas. Aunque aumente la temperatura, las cadenas pueden moverse exclusivamente en la extensión que les permita la distancia entre los puntos de unión de las cadenas. Nunca hay desplazamientos efectivos de unas cadenas sobre otras, llegando el material a descomponer si la temperatura a la que se le somete es muy elevada. Por tanto no fluyen ni se disuelven en disolventes orgánicos. Pueden llegar a retener determinados disolventes en su estructura, aumentando de tamaño, es decir, se hinchan. Figura 12.5.1.- Ilustración esquemática de los diferentes tipos de polímeros. 12.6.- Clasificación según la Escala de Fabricación. Uno de los últimos intentos realizados en este sentido ha pretendido dividir a los Polímeros Industriales en dos grandes grupos, uno en el que se incluyen un corto número de plásticos, pero que, por sus buenas propiedades y bajo precio relativo, se consumen masivamente y que se denomina plásticos de comodidad («COMMODITY PLASTICS»), por ejemplo, polietileno, polipropileno, poliestireno, etc. y otro constituido por un numeroso grupo de productos de alto precio, pequeño consumo, pero de importantes aplicaciones específicas, por ejemplo, Poli(óxido de metileno) y poli(cloruro de vinilideno). Como la mayoría de estas aplicaciones estaban en el campo de la construcción, componentes de máquinas y equipos de plantas químicas, aeronaves, etc., se les ha denominado Plásticos de Ingeniería («ENGINEERING PLASTICS»). Sin embargo, no se ha llegado a un acuerdo de incluir en uno u otro grupo a materiales de uso 215 tan frecuente como pueden ser las resinas ABS, las resinas de poliester o incluso las poliamidas para moldeo. Por ello, parece preferible mantener la tradicional clasificación que tiene en cuenta la estructura molecular del material. 12.7.- Clasificación según el Comportamiento Mecánico (Figura 127.1). Plásticos (del griego: adecuado al modelado, moldeo) – son materiales poliméricos estables en las condiciones normales de uso, pero que durante alguna etapa de su fabricación estuvieron fluidos. Esta propiedad les permite ser moldeados por calentamiento, por presión o por ambos. Por ejemplo: polietileno, polipropileno, poliestireno. Elastómeros (o cauchos) – son materiales poliméricos que pueden ser tanto de origen natural como sintética. Después de sufrir una deformación bajo la acción de una fuerza, recuperan la forma original rápidamente, por más que la deformación haya sido grande o aplicada por bastante tiempo. Por ejemplo: polibutadieno, caucho nitrílico, poli(estireno-co-butadieno). Fibras – las fibras tienen una relación muy elevada entre la longitud y el diámetro (gran esbeltez). Generalmente son constituidas de macromoléculas lineales y se mantienen orientadas longitudinalmente. Por ejemplo: poliésteres, poliamidas y poliacrilonitrilo Figura 12.7.1.- Curvas tensión deformación caracterizando el comportamiento mecánico. 12.8.- Clasificación según el tipo de aplicación. Un plástico puede tener una aplicación general o ser un plástico de ingeniería. Plásticos de uso general – son polímeros muy versátiles, utilizables en las más variadas aplicaciones. Como el polietileno, el polipropileno, el estireno, el (metacrilato de metilo), el poli(cloruro de vinilo), la baquelita, etc. Tecnopolímeros - plásticos para ingeniería – son polímeros empleados en sustitución de materiales tradicionalmente utilizados en la ingeniería, como la madera y varios metales. Por ejemplo: poliacetal, policarbonato y poli(tetraflúor-etileno). 216 Además de las clasificaciones descritas, el término resina se emplea muy a menudo en la industria de los plásticos. Las resinas naturales son compuestos orgánicos AMORFOS secretados por algunas plantas u insectos; generalmente son insolubles en agua, pero solubles en diversos disolventes orgánicos. Las resinas sintéticas se describen como un grupo de sustancias sintéticas cuyas propiedades se asemejan a las de las resinas naturales. Generalmente, a la temperatura ambiente las resinas presentan un aspecto de líquido bastante viscoso o aspecto pastoso que se reblandece gradualmente al calentarlo. También se nombran resinas termofijas a los plásticos termofijos. 13.- Relaciones entre la estructura y las propiedades de los polímeros. Las propiedades de los polímeros, como en el caso de los metales y de las cerámicas, están relacionadas con la estructura elemental del material. Además, no sólo están relacionadas con su naturaleza química, sino también con factores tales como la distribución y magnitud de la cristalinidad, la distribución de longitudes de cadena, la naturaleza y cantidades de aditivos, como puedan ser las cargas, refuerzos y plastificantes, por nombrar sólo unos cuantos. Los factores anteriores modifican en alguna medida prácticamente todas las propiedades de los polímeros tales como la dureza, la resistencia a desgarro, la inflamabilidad, la resistencia a la intemperie, la resistencia química, las respuestas biológicas, la confortabilidad, apariencia, facilidad de teñido, punto de reblandecimiento, cuando el material es estirado, las cadenas aleatorias se ven obligadas a ocupar posiciones más ordenadas. En las tablas 13.1 y 13.2 puede verse un resumen de algunas relaciones entre propiedades y estructura. Tabla 13.1.- Relaciones ESTRUCTURA – PROPIEDADES 217 Tabla 13.2.- Relaciones generales PROPIEDADES – ESTRUCTURA. Las propiedades generales de los polímeros pueden resumirse como sigue: 1.- La naturaleza hidrocarbonada de las cadenas del polímero es la responsable de su baja densidad (típicamente 800 - 1500 kg/m3). 2.- Baja conductividad térmica (0.1 W/m.K). 3.- Alto coeficiente de expansión térmica (50 - 200x106 K-1). 4.- En ausencia de uniones transversales de tipo químico (termoplásticos) las macromoléculas se mantienen juntas por medio de débiles enlaces secundarios de tipo físico como las fuerzas de Van der Waals, los cuales pueden romperse mediante calentamiento. Por consiguiente, los puntos de fusión de los termoplásticos son bajos, normalmente menores de 250 °C. 5.- La estructura molecular de largas cadenas da como resultado una alta viscosidad del material fundido, lo que representa problemas durante el procesado. 6.- Las débiles fuerzas intermoleculares también explican el bajo valor del modulo de elasticidad (0.1 - 3 Gpa) y su baja resistencia (menor de 100 Mpa). 7.- Muchos polímeros cuando se usan a temperaturas relativamente próximas a su temperatura de fusión tienden tendencia a mostrar fluencia. 218 Las propiedades mecánicas, en particular la rigidez, pueden mejorarse controlando los siguientes aspectos de la estructura química y física 1.- Aumentando la longitud de las cadenas o el peso molecular (las parafinas simples y las ceras tienen una estructura química similar al polietileno (PE), excepto que este tiene unas cadenas con una longitud uno o dos órdenes mayores. 2.- Aumentando el alineamiento de las cadenas (fibras y film estirados). 3.- Aumentando la cristalinidad (El PEAD es más rígido que el PEBD). 4.- Aumentando las fuerzas atractivas entre las cadenas (enlaces de hidrogeno). 5.- Aumentando la complejidad de las cadenas e incrementando el tamaño de los grupos laterales En la tabla 13.3 de dan las propiedades fundamentales de los termoplásticos mas comunes . Se dan valores para el aluminio y el acero a efectos comparativos. Tabla 13.3.- Propiedades fundamentales de los polímeros termoplásticos Asimismo, en la tabla 13.4 de dan las propiedades fundamentales de los termoendurecibles más comunes . 219 Tabla .4.- Propiedades fundamentales de los polímeros termoendurecibles. Carswell y Nason (Figura 13.1) clasificaron los polímeros en cinco categorías según como sea su comportamiento mecánico. La clase (a) incluye polímeros blandos y débiles, entre ellos el poliisobutileno, que se caracterizan por un bajo valor del módulo de elasticidad, un bajo punto de fluencia y un moderado alargamiento en función del tiempo. El módulo de Poisson, es decir, la relación entre contracción y alargamiento, para polímeros de clase (a) es de 0.5, que es parecido al de los líquidos. Por otro lado, el módulo de Poisson de los polímeros duros y frágiles de la clase (b), como puede ser el poliestireno, se acerca, a 0.3. Los polímeros de clase (b) se caracterizan por un módulo de elasticidad alto, un punto de fluencia poco definido y una deformación pequeña antes de la rotura. Los polímeros de clase (c), como el PVC plastificado, tienen un bajo módulo de elasticidad, gran alargamiento, un módulo de Poisson de alrededor de 0.5-0.6 y un punto de fluencia bien definido. Puesto que los polímeros de clase (c) se alargan después del punto de fluencia, el área bajo la curva de tensión deformación que representa la tenacidad será mayor que para la clase (b). El PVC rígido es un exponente de los polímeros duros y resistentes de la clase (d). Estos polímeros tienen un alto módulo de elasticidad y una alta resistencia a la fluencia. La curva para los polímeros duros y tenaces de clase (e), como por ejemplo los copolimeros ABS, experimentan un alargamiento moderado antes del punto de fluencia seguido de una deformación irreversible. 220 En general, el comportamiento de todas las clases es hookeano antes del punto de fluencia. La deformación recuperable reversible antes del punto de fluencia, en el intervalo Ilamado elástico, es fundamentalmente el resultado de la flexión y alargamiento de los enlaces covalentes de la cadena principal del polímero. Esta parte útil de la curva tensión - deformación puede también comprender el desenrollamiento recuperable de algunas cadenas del polímero. Después del punto de fluencia, el mecanismo predominante es el deslizamiento irreversible de las cadenas de polímero. Valores elevados del módulo de Young indican que el material es rígido, resistente al alargamiento y estirado. Muchos polímeros sintéticos tienen su módulo de Young comprendido en el intervalo general alrededor de 105 psi (690 MPa), el cuarzo fundido tiene un módulo de Young de 106 psi ( 6897 MPA), la fundición de hierro, el wolframio y el cobre tienen valores del orden de 107psi ( 68965 Mpa) y el diamante de alrededor de 108 psi (689655 MPa). Dado que estas propiedades dependen del tiempo, los polímeros de clase (a) pueden comportarse como los de clase (d) si se aplican los esfuerzos rápidamente, y viceversa. Estas propiedades también dependen de la temperatura. Así, las propiedades de los polímeros de clase (c) se parecerán a las de los polímeros de clase (b) cuando disminuye la temperatura. Los efectos de la temperatura y los mecanismos de alargamiento se hallan resumidos en la figura 13.2 . Figura 13.1.- Curvas tensión – deformación típicas de los polímeros. 221 Figura 13.2.- Efectos característicos de la temperatura en las propiedades de un polímero. En la figura 13.3 puede verse el efecto de la temperatura sobre la curva tensión deformación del polimetacrilato de metilo (PMMA). El aumento de la velocidad de deformación tiene el mismo efecto que la disminución de la temperatura. Figura 13.3.- Efectos de la temperatura sobre la curva tensión deformación del PMMA. 222