El tumor del estroma gastrointestinal
Anuncio

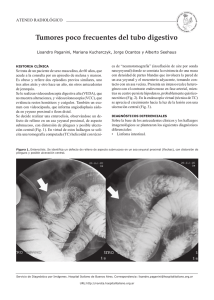
Tumor del estroma gastrointestinal. Tumor del estroma gastrointestinal Imagen histopatológica de tumor del estroma gastrointestinal de estómago. Tinción con hematoxilina eosina. Clasificación y recursos externos OMIM 606764 DiseasesDB 33849 MeSH D046152 Imagen Endoscópica de un GIST en el fondo gástrico, mostrado en retroflexion. El mismo GIST observado desde la vista anterior del endoscopio, mostrándolo recubierto por un coagulo. El tumor del estroma gastrointestinal (GIST del inglés gastrointestinal stromal tumors), es uno de los tumores mesenquimales más frecuentes del tracto gastrointestinal (1-3% de todas los cánceres malignos gastrointestinales). Estos son típicamente definidos como tumores cuyo comportamiento esta inducido por mutaciones en el gen Kit del PDGFRA, y puede o no teñirse positivamente para el Kit.1 Contenido 1 Signos y síntomas 2 Diagnóstico 3 Radiología 4 Patofisiología 5 Tratamiento 6 Epidemiología 7 Referencias Signos y síntomas los pacientes presentan disfagia, hemorragia gastrointestinal o metástasis (principalmente en le hígado). La obstrucción intestinal es rara, debido a su patrón extraluminal de crecimiento. Con frecuencia, existe una historia de dolor abdominal vago o malestar y el tumor se hace mucho más grande por el tiempo que toma hacer el diagnóstico. Generalmente el diagnóstico definitivo es hecho con una biopsia, la cual puede ser obtenida por vía endoscópica, por vía percutanea dirigida por TAC o Ultrasonido o al momento del procedimiento quirúrgico. Diagnóstico Como parte de los análisis también se realizan, muestras de sangre y TAC. Un espécimen para biopsia podría ser analizado bajo el microscopio. La histopatología identifica las características de los GISTs (células fusiformes en el 70-80%, de aspecto epiteloide en el 20-30%). Tumores más pequeños pueden ser usualmente encontrados en la capa muscular propia de la pared intestinal. Aquellos con mayor crecimiento, lejos de la pared del intestino, llegan al punto de exceder el aporte de sangre y necrosarse en el centro, formando una cavidad que puede eventualmente comunicarse con el lumen del intestino. Cuando existe sospecha de GIST los patólogos pueden usar inmunohistoquímica (anticuerpos específicos que marcan la molécula CD117 (también conocida como c-kit)). El 95% de todos los GISTs son CD117 positivos (otros posibles marcadores incluyen CD34, desmina, vimentina y otros). Otras células que muestran positividad para CD117 son los mastocitos. Si el marcador CD117 es negativo y aun existen sospechas que el tumor es a GIST, el nuevo anticuerpo DOG-1 (Descubierto en GIST-1) puede ser usado. También la secuenciación de Kit y PDGFRA puede ser usado para probar el diagnóstico. Radiología Los exámenes fluoroscópicos con bario, de las vías digestivas altas y las vías digestivas bajas (tránsito intestinal) y el TAC son los más frecuentemente usados para el diagnóstico de pacientes con dolor abdominal superior. Ambos son adecuados para hacer el diagnóstico de GIST, sin embargo los tumores pequeños pueden permanecer ocultos, especialmente en casos de exámenes poco exhaustivos. Los GISTs pequeños aparecen como masas intramurales. Cuando crecen (> 5 cm), la mayoría de ellos, lo hacen con mayor frecuencia hacia afuera del intestino. Pueden existir calcificaciones. Cuando disminuye el aporte sanguíneo para el tumor, este puede hacer necrosis interna, creando una cavidad central llena de fluido que eventualmente puede ulcerarse en el lúmen del intestino o el estómago. El tumor puede invadir directamente estructuras en el abdomen. El sitio más común de extensión es al hígado. Extensión al peritoneo puede ser vista. A diferencia del adenocarcinoma gástrico o el linfoma gástrico o de intestino delgado, las adenopatías malignas (inflamación de nódulos linfáticos) son poco comunes (<10%). Patofisiología los GISTs son tumores del tejido conectivo, ej. sarcomas; a diferencia de la mayoría de los cánceres intestinales, no son de tipo epitelial. El 70% ocurre en el estómago, 20% en el intestino delgado y menos del 10% en el esófago. Los tumores pequeños son en general, benignos, especialmente cuando la tasa de mitosis es baja, pero los tumores grandes se diseminan al hígado, omento y cavidad peritoneal. Raramente invaden otros órganos abdominales. Algunos tumores del estómago e intestino delgado llamados leiomiosarcomas (tumores malignos del músculo liso) podrían se reclasificados más frecuentemente como GISTs, basados en los marcadores inmunohistoquímicos. Los GISTs son originados a partir de las células intersticiales de cajal,2 las cuales normalmente hacen parte del sistema nervioso autónomo del intestino, funcionando como mensajeras para el control de la motilidad intestinal. La mayor parte de los GISTs, entre el (50-80%), se originan en una mutación en el gen llamado c-kit. Este gen codifica un receptor trasmembrana de un factor de crecimiento llamado scf (factor de células madre). El receptorc-kit/CD117 es expresado por las células intesticiales de Cajal y una gran cantidad de otras células, principalmente de la médula ósea, mastocitos, melanocitos y algunas otras. En el intestino, sin embargo, una masa que marque positiva para CD117 probablemente es un GIST, originado de las células intersticiales de Cajal. La molécula c-kit comprende un gran dominio extracelular, un segmento trasmembrana, y una porción intracelular. Las mutaciones generalmente ocurren en el ADN codificado en la parte intracelular (exon 11), el cual actúa como una tirosina quinasa para activar otras enzimas. Las mutaciones hacen la función de la c-kit independiente de la activación por scf, propiciado una alta tasa de división celular y posiblemente inestabilidad del genoma. Es probable que mutaciones adicionales sean «requeridas» por una célula con una mutación c-kit para convertirse en un GIST, pero tal mutación de c-kit es probablemente el primer paso dentro del proceso. La función de la tirosina quinasa de c-kit es vital para la terapia del GISTs. Tratamiento El tamaño del tumor, la tasa de mitosis, y la localización pueden ser usados para predecir el riesgo de recurrencia en los pacientes con GISTs. Los tumores menores de 2 cm con una tasa de mitosis de 5/50 HPF han mostrado tener menor riesgo de recurrencia que aquellos más grandes o más agresivos. no obstante, a todos los GISTs podría considerárseles con potencial de malignidad y los tumores no GIST pueden ser correctamente clasificados como «benignos». 3 La cirugía es la base del tratamiento el GISTs no metastásico. Las metástasis en nódulos linfáticos es rara y la remoción de rutina de los nódulos linfáticos usualmente no es necesaria. La resección de márgenes ámplias tampoco es necesaria. La cirugía laparoscópica ha mostrado se efectiva para la resección de estos tumores sin requerir grandes incisiones.4 Hasta hace poco tiempo, los GISTs eran notorios por ser resistentes a la quimioterapia, con un porcentaje de éxito menor al 5%. Recientemente los c-kit inhibidores de tirosina quinasa como el imatinib (Glivec/Gleevec), un medicamento inicialmente comercializado para el tratamiento de la leucemia mieloide crónica, demostró ser útil para para el tratamiento de los GISTs, elevando a un 40-70% el porcentaje de respuesta de metástasis o casos inoperables. Datos presentados en 2007 mostraron que el tratamiento con imatinib subsiguiente a la resección quirúrgica del GISTs, puede reducir significativamente el riesgo de recurencia de la enfermedad (6% de recurrencia con Imatinib vs. 17% sin tratamiento en 12 meses). La duración óptima del tratamiento abyudante es actualmente desconocida; los estudios actuales están evaluando la duración de los tratamiento por 1, 2, y 3 años. Los pacientes que desarrollan resistencia al imatinib pueden responder a inhibidores múltiples de la tirosina quinasa como el sunitinib (comercializado como sutent). La efectividad del imatinib y el sunitinib depende del genotipo.5 Epidemiología Los GIST ocurren en aproximadamente 10-20 casos por cada millón de habitantes. La verdadera incidencia podría ser mal alta, en la medida que los nuevos métodos de laboratorio sean mucho más sensibles para el diagnóstico del GISTs. Por todos hay 3500-5000 casos de GIST por año en los Estados Unidos. Esto hace al GIST el tipo más común de sarcoma, los cuales conforman un grupo de más de 70 tipos de cáncer, pero en todas sus formas constituyen menos de 1% de todos los tumores. Referencias. 1. ↑ Miettinen M, Lasota J (October 2006). «Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis». Arch. Pathol. Lab. Med. 130 (10): pp. 1466–78. PMID 17090188. http://journals.allenpress.com/jrnlserv/?request=get-abstract&issn=00039985&volumen=130&page=1466. 2. ↑ Miettinen M, Lasota J (2006). «Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis». Arch Pathol Lab Med 130 (10): pp. 1466–78. PMID 17090188. 3. ↑ Raut, Chandrajit and Dematteo, Ronald (March 2008). «Evidence-Guided Surgical Management of GIST: Beyond a Simple Case of Benign and Malignant». Ann. Surg. Onc. 15 (5): pp. 1542. doi:10.1245/s10434-0089817-1. 4. ↑ Nguyen SQ, Divino CM, Wang JL, Dikman SH (May 2006). «Laparoscopic management of gastrointestinal stromal tumors». Surg Endosc 20 (5): pp. 713–6. doi:10.1007/s00464-005-0435-8. PMID 16502196. 5. ↑ «News: Genetic Variations in GI Tumors Determine Which Medications Are Efficacious.». Genetic Engineering & Biotechnology News (13 Nov 2008). 6. de Silva CM, Reid R (2003). «Gastrointestinal stromal tumors (GIST): C-kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib». Pathol Oncol Res. 9 (1): pp. 13–9. doi:PAOR.2003.9.1.0013 (inactivo desde 2008-12-07). PMID 12704441.