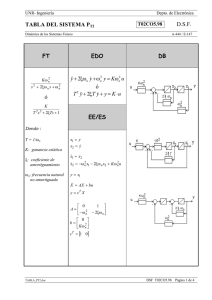
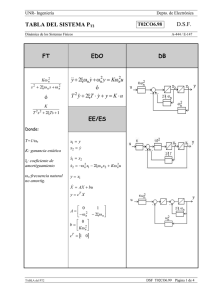
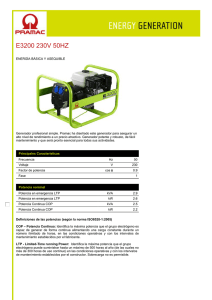
PRIONES. ¿LA ÚLTIMA FRONTERA? La llamada encefalopatía
Anuncio

PRIONES. ¿LA ÚLTIMA FRONTERA? La llamada encefalopatía espongiforme bovina (EEB, BSE del inglés bovine spongiform encephalopathy) forma parte de un grupo de enfermedades denominadas genéricamente como encefalopatías espongiformes transmisibles (EET, TSE del inglés Transmissible Spongiform Encephalopathies), cuyos agentes causales parecen presentar características comunes. Este conjunto de enfermedades provoca alteraciones irreversibles en el cerebro, dando lugar a un aspecto literalmente similar al de una esponja, y todas son inevitablemente mortales. Las TSE incluyen a un amplio grupo de enfermedades presentes tanto en diversas especies de animales como en el propio hombre. Entre las que afectan a especies animales cabe citar al scrapie, que afecta a ovejas y a cabras, la encefalopatía transmisible de los visones y la enfermedad debilitante crónica de los mulos, de los ciervos y de los alces de las Montañas Rocosas. Por lo que se refiere a las EET humanas, las más conocidos (aun asumiendo que son muy raras) son la enfermedad de Creutzfeldt-Jakob, el síndrome de Gerstmann-StraussierScheinker, el insomnio familiar letal y el kuru. Entre las características comunes que presentan las EET, una de las más importantes es su período de incubación, que suele ser muy prolongado (hasta treinta años, en algunos casos humanos extremos). Un antecedente, el scrapie El scrapie es quizás la mejor conocida de las EET animales . Es citada ya en la literatura francesa del siglo XIX, donde aparece descrita como una enfermedad mortal de la oveja denominada "tremblante au mouton" y que se caracterizaba por un temblor de todo el cuerpo y un rascado incesante, con pérdida de lana en diversas partes del cuerpo. En 1829 Berger publica "Observations de paraplégie sur deux moutons " proporcionando una extensa descripción clínica y que popularizó el término de "tremblante" u oveja temblorosa. Fue en 1898 cuando se constataron las alteraciones neuronales (detectándose la vacuolización de las astas anteriores de la médula espinal). A principios del siglo XX, Stockman firma un trabajo denominado "Scrapie: an obscure disease of sheep"; en el que el autor no sólo caracteriza patológicamente la enfermedad sino que describe la cronología de aparición en Gran Bretaña, señalando la existencia de brotes durante 1767 en Northumberland. Es este autor quien señala la principal característica de la enfermedad: el picor y el rascado (en inglés to scrape, rascar). Gaiger estudia en el año 1924 las vías de transmisión, aunque sin resultados positivos. Curiosamente, indica que consumió carne infectada, “sin notar diferencia de sabor” con la sana. La muerte de Gaiger por una patología neurovegetativa (con demencia), no se pudo relacionar con sus investigaciones. En Alemania se había descrito ya en 1755 una enfermedad ovina denominada "traberkrankheit", entre cuyos signos clínicos aparecen los clásicos de ataxia, prurito y temblor. Mucho más recientes son los casos descritos en Estados Unidos y Canadá, que datan de mediados del siglo XX. La constatación de su carácter infeccioso se realizó en 1948 en Escocia, cuando en un vano intento de vacunar corderos contra la enfermedad denominada del "looping-ill virus" mediante una suspensión de un extracto formolizado de cerebro y bazo de ovino contaminados, se provocó la muerte de 1.500 de los animales vacunados, dos años más tarde. Esa denominación encubre a una enfermedad de patología similar, incluso probablemente la misma. Actualmente el scrapie es una enfermedad endémica para las ovejas de prácticamente todos los países del mundo, con excepción de Australia y Nueva Zelanda. Se estima que un tercio de todos los rebaños de ovejas británicos presentan casos de scrapie y en Estados Unidos un 1% de toda la cabaña ovina está infectada, hasta el punto de que no es una enfermedad de declaración obligatoria en ese país. Generalmente, número de cabezas infectadas en cada rebaño no supera el 5%. La enfermedad es transmisible a través de la placenta o por contacto con la misma, si bien la transmisión materno-fetal ha sido y es discutida. Actualmente, se considera que el scrapie es una enfermedad es una enfermedad infecciosa adquirida mediante el consumo de pasto o de piensos que han estado o contienen tejido placentario contaminado. Tiene un período de incubación de entre dos y cinco años. El agente casual del scrapie puede ser recuperado a partir de diversas localizaciones orgánicas, tales como el tejido linfático, las amígdalas, el bazo o el timo, así como en todo el sistema nervioso central. No parece presentar ningún riesgo de infección para el hombre, incluso para las personas que cuidan directamente el ganado enfermo. Encefalopatía espongiforme bovina (EEB). La encefalopatía espongiforme bovina es una enfermedad relativamente nueva, que se observó por primera vez en Gran Bretaña en abril de 1985, siendo confirmado su diagnóstico en noviembre de 1986, como un proceso neurodegenerativo bovino similar al de la enfermedad ovina del scrapie. Su origen se establece por el aumento en la exposición del ganado al agente infeccioso por el cambio de alimentación, al incluir harinas obtenidas a partir de carne y restos óseos de oveja (en un extraño y antinatural "canibalismo animal"), con objeto de abaratar los costes de forraje y optimizar así la producción ganadera. Apenas cinco años más tarde, el número oficialmente reconocido por las autoridades británicas de reses afectadas superaba las 30.000 y en el año 2001 la cifra se aproxima en toda la Unión Europea a las 200.000. El resto de la historia es sobradamente conocido (¿o quizás no?). La enfermedad presenta los mismos signos clínicos que el scrapie en la oveja, con ataxia, temblor, picores que inducen a un rascado frenético y un comportamiento anormal del animal, caracterizado por la tendencia a la embestida y mugidos compulsivos, lo que justifica sobradamente el término popular de "vacas locas" (mad cows, en inglés). Encefalopatías espongiformes humanas En los seres humanos no es hasta hace relativamente poco cuando se pudo establecer una similitud entre ciertas patologías y las encefalopatías espongiformes animales. Concretamente en 1959, Hadlow establece una similitud anatomopatológica entre el scrapie y una enfermedad neurovegetativa humana denominada kuru. Se trata de una patología descubierta por Vincent Zigas en 1951 en la población Foré de Nueva Guinea Papúa (zona montañosa oriental), posteriormente descrita y estudiada por Gibbs y Gajdusek, lo que le valió el Premio Nobel de medicina en 1976. En lengua nativa kuru significa sacudida y se caracteriza por un temblor discapacitante, demencia y ataxia progresivas, que se manifiesta principalmente en niños y mujeres, con características anatomopatológicas de vacuolización e hipergliosis cerebral y talámica. Dicha enfermedad se manifestaba en la población citada como consecuencia de un ritual religioso de canibalismo. Este consistía en que los vencedores de los combates entre tribus ingerían el encéfalo y algunas vísceras, dándoselas principalmente a mujeres embarazadas y lactantes, para que heredasen las características del muerto. Posiblemente, también colaborase como medio de transmisión la contaminación derivada de la técnica ritual de limpieza del muerto, mediante la cual las mujeres limpian los huesos de vestigios de tendones y separan el encéfalo del cráneo mediante el uso de sus propias uñas. Esto último hace que las mujeres presenten múltiples escoriaciones y lesiones cutáneas pequeñas, por las que podría entrar el agente infeccioso. De todas maneras por sí sola esta costumbre no justifica la gran incidencia de la enfermedad en la foblación Foré, por lo que la predisposición genética posiblemente tenga un papel muy relevante. Actualmente esta enfermedad ha desaparecido casi por completo, como consecuencia de la erradicación del canibalismo ritual en estas zonas, aunque todavía persisten ciertas prácticas necrofílicas difíciles de controlar. El síndrome de Creutzfeld-Jakob (ECJ, CJD de sus siglas inglesas) fue definido por Creutzfeld en 1920 y por Jakob (tres casos) entre 1921 y 1923. Se trata de una deme ncia progresiva de curso rápido, de tipo presenil y con deterioro intelectual. Se acompaña de temblores neurológicos complejos y va asociada a un síndrome cerebeloso extrapiramidal y mioclonías con evolución mortal en menos de un año desde la aparición sintomá- tica, aunque su generación y evolución es de larga duración hasta la aparición explosiva de los síntomas. Aparece bajo tras formas, la hereditaria, la adquirida y la forma esporádica. La forma heredada es de carácter autosómico dominante; es decir, sólo requiere la mutación de una de las dos copias del gen priónico humano, tal como se analizará más adelante. La forma adquirida consiste mayoritariamente en un ECJ iatrogénico, proveniente de la utilización de somatropina (hormona del crecimiento) humana de origen extractivo procedentes de hipófisis humanas extraídas de pacientes. Esta última forma ha desaparecido prácticamente por completo desde el abandono de la somatropina extractiva en favor de la forma recombinante (obtenida mediante técnicas de bioingeniería). No obstante, también puede ser transmitida en transplantes de córnea y de duramadre, así como por prácticas forenses inadecuadas. La incidencia media mundial del ECJ es muy baja, en torno a 1 caso por cada millón de habitantes. En España es aun menor, con 0,19 casos, frente al 4,9 por millón de Túnez ó 1,07 por millón en Israel, ó 0,38 por millón de Francia. Sin embargo sí existe una clara influencia geográfica y sobre todo étnica, con casos tan llamativos como los judios de origen libio, con una tasa de 44,41 por millón, o poblaciones de Orava y Lucenec en Eslovaquia o en grupos familiares chilenos. El 85% de los casos de ECJ se producen de forma aleatoria y tiene un origen desconocido, de ahí que se agrupe a estos casos como la forma esporádica de la enfermedad. Esta forma aparece en pacientes mayores de 55 años y presenta una demencia rápidamente progresiva, mioclonía y cambios característicos en el electroencefalograma. La muerte suele sobrevenir como consecuencia de una neumonía hipostática. A mediados de los años 60 se demostró que esta enfermedad era susceptible de ser transmitida experimentalmente. De hecho, se ha demostrado la existencia de una forma simiesca de la enfermedad, tras la inoculación del agente causal del ECJ a chimpancés, que presenta una clara similitud con la enfermedad humana, aunque de diferente evolución. También se ha podido infectar a otros animales (hámster y gatos incluidos). La enfermedad de Gertsmann-Straussier-Scheinker consiste en una ataxia cerebelosa crónica, predominantemente familiar (aunque también existe una forma esporádica), cuyos signos de demencia aparecen muy posteriormente a los síndromes cerebelosos. Fue descrita en 1936 y su carácter hereditario quedó demostrado en la década de los 60. Aunque todavía no se ha conseguido conocer cuál es el gen mutante responsable del proceso, sí se sabe que tiene un carácter autosómico y dominante. Su incidencia es cincuenta veces inferior (apenas un 2%) de la de ECJ, de la que se diferencia en que aparece de forma más temprana entre los pacientes (entre los 40 y los 50 años de edad). El curso de la enfermedad es más prolongado que el de ECJ, alcanzando varios años. Aún más infrecuente es la enfermedad denominada insomnio letal familiar, una patología caracterizada por una grave y selectiva atrofia del tálamo, que fue descrita por vez primera hace apenas 15 años (en 1986). Finalmente, se utiliza el término de síndrome de Alpers a patologías de este tipo (priónico) manifestadas en niños. Globalmente, estas enfermedades neurodegenerativas se caracterizan por: • Vacuolización o espongiosis en el citoplasma de neuronas y células gliales. Esto mismo sucede en las enfermedades de Alzheimer y de Huntington, aunque en estos casos la vacuolización se produce en el espacio extracelular, como resultado de la pérdida de neuronas. • Gliosis (crecimiento exagerado de las células gliales), en ausencia de inflamación. • Pérdida de neuronas. Estos síntomas pueden observarse en cualquier área y lámina de la corteza cerebral, en el putamen, en el núcleo caudado, en el tálamo y en la capa molecular de la corteza cerebelosa. La sustancia blanca no presenta cambios patológicos. Las vacuolas tienen un tamaño entre 20 y 200 micras, aunque pueden fusionarse formando grandes vacuolas que distorsionan la célula. La espongiosis cortical puede ir acompañada de procesos análogos en ganglio basal, tálamo y corteza cerebelosa. También pueden observarse depósitos amiloides de PrP S c (insolubles) en forma de placas amiloides, que se tiñen debido a su afinidad por la eosina. Están presentes sobretodo a nivel del cerebelo y, minoritariamente, a nivel del tálamo, ganglios basales y corteza cerebral. Aunque estas placas son un buen indicador de la infección priónica, aparentemente no son causa importante de la enfermedad. Estudios ultraestructurales han demostrado una relación de la microglía con la formación de estas placas. Se ha comprobado que forma fisiológica (PrP) se localiza en lugares distintos a los que se encuentran la forma patológica (PrP S c). Esta última está presente (en los enfermos) en la materia blanca y en los ovillos de axones mielinizados, lo que demuestra que los priones se mueven a través de los axones de las neuronas, y la infectividad de (PrP S c) se basa en un patrón de transporte retrógrado a lo largo del axón. La variante de la enfermedad de Creutzfeldt-Jakob: el miedo a las vacas locas. En marzo de 1996 en el Reino Unido se registraron diez casos de personas menores de 45 años con una enfermedad con sintomatología similar a la relativa a las encefalopatías espongiformes transmisibles. Al principio se pensó en la ECJ, pero los análisis científicos posteriores revelaron diferencias sintomáticas y patológicas de los tejidos cerebrales en comparación con los de los enfermos de ECJ. Se dedujo, sin embargo, que estos diez casos correspondían a una variedad de una encefalopatía espongiforme transmisible y se denominó esta alteración como variante de ECJ (vECJ). La vECJ presenta varias diferencias con respecto a la ECJ. La vECJ tiene un curso más prolongado que ECJ. Asimismo, los pacientes afectados con vECJ experimentan síntomas psiquiátricos más tempranos, como depresión, pérdida prematura de la coordinación y posteriormente demencia, además de ocurrir hasta la fecha en personas menores de 55 mientras que ECJ típicamente ocurre en personas mayores de esta edad. En febrero de 2001, el número total casos oficialmente aceptados de vECJ no llega al centenar en todo el Europa. Eso es tanto como decir en todo el mundo, ya que por el momento esta forma variante de la ECJ está exclusivamente circunscrita a Europa. Las investigaciones en el Reino Unido han demostrado que vECJ se produce por el mismo agente que causa la encefalopatía espongiforme bobina (EEB). La probabilidad de contagio al comer material de riesgo infectado por EEB viene determinada por la predisposición genética y la cantidad de material de riesgo ingerida. Todas las proteínas de la dieta son degradadas en aminoácidos por los ácidos y protesas gástricos. No obstante, existe la posibilidad de que este proceso degradativo no sea completo y una pequeña cantidad de proteínas sobreviva al contenido gástrico y sea absorbida en el intestino, pasando a circulación sanguínea. La probabilidad de que esto ocurra será mayor cuanta más proteína haya sido consumida. El riesgo que representa la ingestión de tejido muscular es ínfimo, ya que es muy escaso el tejido nervioso que contiene. Por tanto, si el riesgo es de por sí bajo para el tejido nervioso puro, se considera como prácticamente nulo para el tejido muscular. El tiempo de incubación hasta que se manifiestan los síntomas es variable y depende de los factores anteriormente nombrados. El máximo tiempo registrado está entre dos y tres años. Existe la incertidumbre de si es posible la transmisión de vECJ a través de la sangre de humanos en periodo de incubación asintomático. Esto ha sido causa de algunos titulares, no demasiado afortunados, en algunos medios de comunicación. Los bioensayos con sangre humana solamente pueden ser llevados a cabo en especies animales, por obvios motivos éticos, lo que supone importantes limitaciones de la validez de los análisis. Una forma de evitar la diferencia de especie es la transmisión de la sangre en un modelo animal apropiado de encefalopatía espongiforme transmisible. Las ovejas infectadas con EEB albergan la infección en los tejidos periféricos lo que presentan similitudes con los humanos que padecen vECJ. El razonamiento supone la base para la realización de un estudio en el que se transfundió sangre de ovejas alimentadas con cerebro de ganado afectado por EEB a ovejas sanas. Los resultados preliminares encontraron signos clínicos y patológicos de EEB en una oveja receptora de otra infectada con EEB. Los ensayos inmunocitoquímicos confirmaron la infección. Esta observación es consistente con la experiencia anterior en roedores experimentalmente infectados y además amplía el conocimiento relativo a la infectividad sanguínea en modelos experimentales en animales con encefalopatía espongiforme transmisible más parecidos a la situación de vECJ en humanos. También supone la primera transfusión con éxito de sangre de animales con EEB en el periodo de incubación más importante. Este resultado es parte de un estudio más amplio que incluye animales de control positivo y negativo además de ovejas aún sanas y en varios estadíos prematuros del periodo de incubación. La importancia de los resultados preliminares de este estudio, aparte de la sugerencia de la posibilidad de desarrollo de un test diagnóstico basado en muestras sanguíneas, radica en la posibilidad de la identificación de las células que están infectadas y del análisis de la eficacia de la leucodeplección, medida tomada por el Servicio Nacional de Transfusiones Sanguíneas del Reino Unido, junto con la importación de plasma y derivados de países libres de EEB, para prevenir nuevos casos de vECJ. ¿Qué son los misteriosos priones? Las encefalopatías espongiformes transmisibles (EET) son causadas por un agente con capacidad para infectar o contagiar, que no es ni hongo, ni protozoo, ni helminto, ni bacteria, ni virus, ni nada que contenga en su interior un ácido nucleico (ADN o ARN). Antes de conocer con detalle la estructura de esta agente infectante, existía la convicción de que se trataba de una forma especial de virus, denominados virus lentos, debido a que los primeros estudios se centraron sobre la posibilidad del origen vírico de estas infecciones (al ser eleme ntos filtrables) y con un marcado neurotropismo. Tras excluirse la presencia de ácidos nucleicos, se acuñó el término de proteína autorreplicante, al constatarse la presencia de formaciones túbulo-fibrilares en los tejidos afectados (confirmando su potencial infeccioso) y demostrando que se trataba de depósitos amiloides cuyo principal componente es proteico, con una estrecha similitud con las placas que aparecen en la enfermedad de Alzheimer. Finalmente se aisló y purificó el agente en 1982 por Stanley y Prusiner, a partir de tejido cerebral de hámster. El término prión, acuñado por Prusiner, es un acrónimo de "small proteinaceous infectious particles" (pequeñas partículas proteináceas infecciosas). Por consiguiente, el concepto de PRIÓN consiste en una proteína normal (PrP) aunque con una conformación aberrante (PrP SC ), que se hace extremadamente resistente al calor y a las técnicas habituales de esterilización. El gen que codifica a esta proteína está presente en la mayor parte de las especies animales de mamíferos, aunque por ahora de desconoce cuál es su función biológica. Algunos autores han sugerido que la forma fisiológica de la proteína prión (PrP) podría estar implicada en la transducción de señales celulares. De hecho, se ha observado que el plasminógeno (que a su vez está implicado en mecanismos de excitoxicidad neuronal) se une de forma selectiva a la proteína prión fisiológica (PrP), pero no a la patológica (PrP SC ). Esto último ha sido propuesto como base para elaborar métodos bioquímicos de análisis. Asimismo, se le ha atribuido papeles biológicos en la regulación y distribución de receptores colinérgicos, así como en procesos de adhesividad celular. Algunos autores han apuntado una actividad de tipo superóxido dismutasa, implicada en la captación y neutralización de radic ales libres de tipo superóxido (O2 •). Fisiológicamente parece colaborar en los mecanismos de supervivencia de las neuronas de Purkinje, en el cerebelo. Como es sabido, este órgano tiene importantes funciones coordinadoras Se trata de en una proteína glucosilada de masa molecular de 27.000-30.000 daltons (27-30 Kda), resistente a la proteasa y que deriva de una molécula mayor (de 33-35 Kda), sensible a proteinasa K. La PrP SC 33-35 se acumula dentro de la célula con posibilidades de agregación, mientras que la forma proteasa-resistente PrP SC 27-30 se acumula extracelularmente y, de hecho, constituye la base para la prueba bioquímica de detección priónica denominada Prionics®. Se sabe que ambas isoformas pueden encontrarse ancladas en la superficie de la célula. De hecho, su transformación a en la forma patológica (PrP SC ) sólo tiene lugar cuando la cuando la forma fisiológica (PrP) alcanza dicho lugar. Estos lugares de anclaje son denominados GPI (Glucoinositol fosfolípido). La secuencia de aminoácidos en ambas formas de la proteína priónica son idénticas (se trata de una cadena de 256 aminoácidos). Esto significa que la única diferencia entre la forma normal o fisiológica y la aberrante o patológica en su conformación (la forma tridimensional) y que esta modificación conformacional se produce después de la síntesis de PrP. En concreto la forma normal de la PrP presenta una disposición en el espacio de tipo helicoidal, mientras que la isoforma patológica tiene un aspecto aplanado. Este simple cambio conformacional hace que la PrP SC adquiera una capacidad catalítica capaz de facilitar su autopropagación mediante la formación de una reacción en cadena similar a la que se produce en un reactor nuclear. El resultado es la formación de agregados proteicos de PrP SC que acaban por precipitar formando placas. La isoforma PrP SC es, además, muy resistente a otros enzimas biodegradativos, especialmente a proteasas, así como a la mayor parte de los métodos físicos y químicos de esterilización. En especial, es muy resistente al calor; en este sentido, se estima que un simple hervido (100º C) no afecta prácticamente a la capacidad de infectar, requiriéndose no menos de 30 minutos a 130º C para inactivarla. Los priones (PrP SC ) mantienen prácticamente todo su poder de “contagio” a las proteínas PrP normales en condiciones normalmente esterilizante para cualquier agente infeccioso (virus, bacterias, etc), tales como radiación UV, formol, pH extremos, disolventes orgánicos no polares, etc. Su pequeño tamaño impide emplear técnicas de ultrafiltración, ya que pasan fácilmente por filtros de 0,1 µm, cuando un simple filtro con poros de 2 µm sirve retener la mayor parte de las bacterias. ¿Qué les hace volverse malas? La transformación de PrP a PrP SC no es un proceso que ocurra de forma espontánea. Es decir, desde un punto de vista bioquímico, se trata de un proceso termodinámicamente desfavorable. Por ello, se sospecha que esta transformación espontánea (sin PrP SC previa) podría deberse a una mutación en el gen que codifica la proteína fisiológica (PrP). De hecho, se han descrito hasta el momento más de 20 mutaciones diferentes de este gen priónico que conducen a la síntesis de una proteína fisiológica (PrP) fácilmente transformable en condiciones normales a la isoforma patológica (PrP SC ), con capacidad infectante. ¿Origen infeccioso, genético o mixto? Aunque la capacidad transformadora de la isoforma patológica (PrP SC ) de su confórmero fisiológico (PrP) ha quedado ampliamente demostrada, no está tan claro el mecanismo patogénico por el que se desarrolla la encefalopatía espongiforme en los animales y en el hombre. Se ha sugerido que, al margen de la hipótesis infecciosa (literalmente, una proteína infectante), posiblemente se requiriese la existencia o la coparticipación de una mutación o modificación genética, que facilitase la infectividad de la isoforma patológica (PrP SC ) en los indiv iduos. La predisposición genética de un ser humano a padecer vECJ parece radicar en el codon 129 del gen que codifica la proteína prión PrP. Todos los casos de vECJ estudiados son homocigóticos (las dos copias del gen son iguales) para el aminoácido metionina codificado por ese codon (M/M 129). Sin embargo, las personas homocigóticas para valina en ese codon (V/V 129) o heterocigóticas (M/V 129) parecen ser relativamente resistentes a la infección, o bien tener tiempos de incubación más largos o presentar síntomas diferentes. Sea como fuere, la isoforma patológica (PrP SC ), ingerida puede ser absorbida en el intestino en las placas de Peyer, que forman parte del llamado tejido linfoide asociado a las mucosas. Justamente, es a través de este tejido linfoide por donde se presentan los priones infectantes al sistema inmune, facilitando la respuesta de éste. Según esto, las células linfoides fagocitarían las partículas priónicas, que viajarían a través del sistema linfático a otras localizaciones no digestivas, como las amígdalas, el bazo o los nódulos linfáticos, replicándose los priones en estos sitios. En este sentido, existen datos que demuestran que las células dendríticas foliculares son los lugares principales para la replic ación priónica en el bazo. Estas localizaciones están inervadas y, eventualmente, serían capaces de alcanzar el cerebro a través de los axones de las fibras nerviosas y de la propia médula espinal, donde se acumularían, depositándose en forma de placas amiloides. ¿Y, además, hay “cepas” diferentes de un mismo tipo de prión? Por si la situación no fuese lo suficientemente complicada, resulta que se ha descrito la existencia de “cepas” diferentes (por utilizar la terminología típica de Microbiología) de proteínas prión patológicas en algunas otras formas de encefalopatía espongiforme animal, como el scrapie. Esto sugiere que el mecanismo patogénico no es, aparentemente, tan simple como el indicado anteriormente, e implicaría la participación de agentes patógenos que contengan ácidos nucleicos, ya que, en principio, sólo estos últimos son susceptibles de formas “cepas” diferentes. Concretamente, se ha postulado la participación de virus o incluso de “virinos” (pequeñas partículas víricas con un ácido nucleico muy corto). Sin embargo, la ausencia de ácidos nucleicos “extraños” en el material biológico procedente de animales y humanos ha ido postergando la hipótesis co-viral de la enfermedad, a favor de otras posibilidades. Una de ellas, es que las mencionadas “cepas” se correspondan con diferencias en la estructura química o en la conformación de la proteína prión patológica. Esto último implicaría que las conformaciones posibles de la PrP serían múltiples. Sea como fuere, las diferentes cepas detectadas de scrapie son capaces de producir diferencias ostensibles en el tipo de alteraciones patológicas que producen, en especial de la degeneración vacuolar del cerebro. Algunos datos recientes apuntan la posibilidad de que la variante de la enfermedad de Creutzfeldt-Jakob sea una cepa priónica estrechamente relacionada con la proteína prión de la encefalopatía espongiforme bovina. ¿Cómo de infecciosos son los priones? Admitiendo completamente la vía digestiva como posible vía de entrada al organismo de la isoforma patológica del prión, ésta vía no parece que tenga una elevada capacidad de transmisión, ya que en estudios en animales de experimentación se ha observado que la dosis de cerebro bovino con encefalopatía espongiforme requerida para transmitir la enfermedad es entre 125.000 y 200.000 veces superior a la precisa para transmitirla mediante una inyección intracerebral. En este sentido, se estima que 0,5-1 g de cerebro infectado es suficiente para infectar (por inoculación intracererbral). ¿Todo esto qué quiere decir? Básicamente, que la forma patológica de los priones (PrP SC ) tiene una elevada capacidad contagiosa, pero sólo cuando se le pone en el seno del tejido adecuado (el nervioso) y en personas con una determinada predisposición genética. La ingestión, por sí sola, parece tener una capacidad muy limitada de transmisión de la enfermedad. ¿Transmisión entre especies animales y... el hombre? Las proteínas prión fisiológicas (PrP) son específicas de cada especie animal (hombre incluido), aunque guardan estrechas relaciones estructurales entre todas ellas. Por eje mplo, el gen que codifica al prión en el ratón difiere sólo en 16 de los 254 codones del correspondiente al hámster. Diferentes ensayos han demostrado que cuanto más parecida es la estructura de las proteínas prión fisiológicas entre especies animales diferentes, tanto mayor es el riesgo de que un prión patológico de una especie sea capaz de serlo para otra, transformando la proteína prión fisiológica (PrP) en la isoforma patológica (PrP SC ). El caso del prión patológico asociado a la encefalopatía espongiforme bovina, sólo difiere en siete aminoácidos del correspondiente al de las ovejas, lo que justifica la infectividad del pienso obtenido a partir de vísceras de ovejas utilizado en la alimentación de las vacas de la cabaña británica. Por el contrario, las correspondientes secuencias de las PrP bovinas y humanas difieren notablemente, en más de 30 aminoácidos (más del 10% del total de la proteína). Lo cual está en consonancia con la enorme desproporción observada entre el número de casos de vECJ (menos de 100) y el de EEB (cerca de 200.000), y todo ello pese a haber estado consumiéndose material de alto riesgo por gran número de ciudadanos británicos durante varios años. No obstante, es posible que la capacidad transformadora de la isoforma patológica (PrP SC ) no requiera una absoluta identidad y que sólo sea precisa la similitud en determinadas zonas de las moléculas. Esto facilitaría, sin duda alguna, la rotura de la llamada “barrera de las especies animales”, permitiendo en mayor o menor medida la transmisión de las enfermedades priónicas de una especie animal a otra. Para entender mejor este mecanismo, se ha propuesto la hipótesis de la participación de un característico tipo de proteínas presentes en todas las especies de mamíferos. Se trata de las chaperonas. Las chaperonas son proteínas de gran trascendencia biológica, ya que participan muy activamente en el proceso de la síntesis proteica, ya que su actuación facilita que la proteína en fase de formación vaya adquiriendo su conformación natural de forma adecuada. Según una de las hipótesis manejada, las chaperonas serían “engañadas” por la parte común de la cadena de la isoforma patológica del prión (PrP SC ) proveniente de otra especie animal, facilitando la conversión de la proteína prión fisiológica (PrP) de la especie huésped en su isoforma patológica (PrP SC ) en el propio proceso de su síntesis. ¿Existe algún tratamiento para las enfermedades priónicas? Lamentablemente, hoy sólo existe una respuesta escueta: No. El panorama terapéutico que se plantea frente a las enfermedades asociadas a los priones es en estos momentos desolador: Son partículas infectantes extraordinariamente resistentes a los métodos habituales de esterilización, incluyendo el calor. Los priones son proteínas “normales” y, como tales, reconocidas por el propio organismo, de tal manera que no provocan reacciones inflamatorias o alérgicas que alerten o permitan detectar su presencia mediante signos o síntomas clínicos (salvo cuando la enfermedad está intensamente arraigada). Ninguna de las herramientas farmacológicas actualmente disponibles (antibacterianos, antivirales, antifúngicos, etc) producen el más mínimo efecto sobre las formas patológicas de los priones. Su patogénesis recuerda en no pocos aspectos a la de otras enfermedades neurodegenerativas humanas, como la enfermedad de Alzheimer o la escleosis lateral amiotrófica. Ninguna de estas últimas tiene actualmente tratamiento curativo y los paliativos son bastante cuestionables y no mejoran significativamente la supervivencia. La transformación de una proteína buena (PrP), aunque sin función biológica conocida, en otra patológica (PrP SC ) con capacidad para destruir las células nerviosas, es un proceso que se produce en el interior celular, de muy difícil acceso para sustancias administradas exógenamente (como un medicame nto). Las neuronas, dianas patológicas de los priones, no tienen capacidad de división mitótica, por lo que la destrucción de cada una de ellas no es compensada por la producción de nuevas neuronas. Frente a esta auténtica cascada de malas noticias hay que oponer algunos razonamientos que, aunque no compensan completamente lo anterior, permiten vislumbrar cierta luz al final del túnel: Las encefalopatías espongiformes de carácter epidémico en animales están relacionadas mayoritariamente con desastrosas intervenciones humanas en la cadena de alimentación animal (empleo de piensos de origen animal, placentas y otros materiales biológicos de riesgo, etc). Tales intervenciones, por reprochables que sean (y lo son, porque han demostrado niveles inimaginables de codicia y me z- quindad), pueden ser evitadas o, al menos, reducidas mediante intervención de las autoridades públicas (que sean competentes, claro). Los riesgos reales de contagio humano a partir de animales se ha demostrado excepcionalmente bajo, incluso en condiciones claramente favorecedoras, tal como se ha demostrado en Gran Bretaña. Las formas esporádicas de las enfermedades priónicas tienen una incidencia verdaderamente ínfima. No por ello, como es obvio, puede ni debe prescindirse de la búsqueda de tratamientos eficaces para estas patologías que entran claramente dentro del calificativo de “raras”. Se han propuesto algunos mecanismos biológicos que permitirían impedir o frenar la conversión de la forma fisiológica de la proteína prión (PrP) en su isoforma patológica (PrP SC ) o, incluso revertir este proceso. Entre estos hipotéticos mecanismos biológicos o farmacológicos se han propuesto los siguientes: Reducción de la síntesis de proteína prión fisiológica (PrP), es decir, frenar la expresión del gen humano o animal correspondiente, ya que se ha comprobado que la sobreexpresión de este gen facilita el desarrollo de este tipo de enfermedades. Estabilización de la conformación de la proteína prión fisiológica (PrP) mediante su unión a determinadas sustancias químicas, impidiendo su transformación por la isoforma patológica (PrP SC ). Desnaturalización de la isoforma patológica (PrP SC ) o su reversión a la forma fisiológica (PrP). Son numerosos los estudios en fase de desarrollo actualmente en la búsqueda de soluciones para las enfermedades priónicas. En línea con los mecanismos hipotéticos antes indicados, merece la pena citar los trabajos de un grupo de investigadores de la Universidad de Nueva York, que han sintetizado un péptido capaz de interactuar con la proteína prión patológica (PrP SC ), induciendo la reversión a la forma fisiológica (PrP). Es decir, las láminas beta de PrP SC son desplegadas por acción de este péptido sintético. El estudio fue llevado tanto en modelos animales (ratones enfermos), como en muestras humanas procedentes de pacientes con ECJ. Los resultados demostraron que la administración in vivo de este péptido a los ratones vivos experimentalmente enfermos presentaban un retraso en la aparición de los síntomas clínicos de la enfermedad y una disminución de la capacidad infectiva de cerca del 95%. Atendiendo a la capacidad del péptido beta amiloide presente en las placas neurofibrilares de los enfermos de Alzheimer y la proteína prión patológica (PrP SC ) tienen capacidad para formar agregados fibrilares, se ha sugerido que esta condición podría constituir una posible diana farmacológica, mediante la utilización de pequeñas moléculas capaces de inhibir el proceso de agregación fibrilar. BIBLIOGRAFÍA - - - Aguzzi A. The genetics of prions – a contradiction in terms? Lancet 1999; 354: 22-5. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379(6563): 339-43. Brown P. BSE and transmission through blood. Lancet. 2000; 356:955-6. Cutlip RC. Intracerebral transmision of scrapie to cattle. J Infect Dis 1994; 169(4): 814-20. Domich L. Patologías tipo prión. Rev Neurol 1996;24:125. Esteban JM. De la encefalopatía espongiforme ("vacas locas") a la enfermedad de Creutzfeld-Jacob. Panorama Actual Med 1996; 22(193): 256-65. Fischer MB, Roeckl C, Parizek P, et al. Binding of disease-associated prion protein to plasminogen. Nature 2000; 408: 479-83. Foster JD. Transmission of bovine spongiform encepahlopathy to sheep and goats. Vet Rec 1993;133(14): 339-41. Gabizon R, Telling G, Meiner Z, Halimi M, Kahana I, Prusiner SB. Insoluble wild-type and prote ase-resistant mutant prion protein in brains of patients with inherited prion disease. Nat Med 1996; 2(1): 59-64. Harris DA, Gorodinsky A, Lehmann S, Moulder K, Shyng SL. Cell biology of the prion protein. Curr Top Microbiol Immunol 1996; 207: 77-93. Houston F, Foster J, Chong A, et al. Transmission of BSE by blood transfusion in sheep. Lancet. 2000; 356:999-1000. Kuner P, Bohrmann B, Tjernberg LO, et al. Controlling polymerization of beta-amyloid and prionderived peptides with synthetic small molecule ligands. J Biol Chem 2000; 275(3): 1673-8. - - Montrasio F, Frigg R, Glatzel M, et al. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 2000; 288: 1257-9. Mouillet-Richard S, Eermonval M, Chebassier C, et al. Signal transduction through prion protein. Science 2000; 289: 1925-8. Pattison J. The Emergence of Bovine Spongiform Encephalopathy & Related Diseases. Emerging Infectious Disease 1998; 4(3): 390-4. Prusiner SB. Human prion diseases and neurodegeneration. Curr Top Microbiol Immunol 1996; 207: 1-17. Prusiner SB. Human prion disease. Ann Neurol 1994;35(4):385-95. Prusiner SB. Prion encephalopathies of animals and humans. Dev Biol Stand 1993;80:31-44. Soto C, Kascsak RJ, Saborio GP, et al. Rerversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 2000; 355(9199): 192-7. Taylor, DM. Bovine spongiform encephalopathy and its association with the feeding of rumiantderived protein. Dev Biol Stand 1993;80:215-24.