Introducción Generalidades
Anuncio

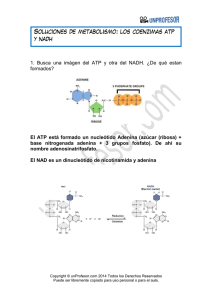
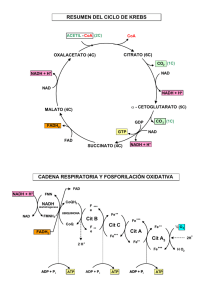
Introducción Generalidades Las células se clasifican en autótrofas o heterótrofas según el método de obtener la energía para su metabolismo. Las células autótrofas poseen cloroplastos, organelos citoplasmáticos que pueden efectuar la fotosíntesis, mediante la cual logran capturar energía solar, llamándose por esto células endergónicas. El proceso que realizan puede resumirse en la ecuación : E 6CO2 + 6H2O ----> C6H12O6 + 6O2 Las mitocondrias son organelos citoplasmáticos muy plásticos y móviles. Con microcinematografía se las ha visto que cambian constantemente de forma, se mueven por el citoplasma y a menudo se asocian con microtúbulos. El tamaño es variable con un ancho de alrededor 0.5 µm y una longitud de ± 7.0 µm. Las células heterótrofas son las que liberan energía (exergónicas) mediante el proceso de fosforilación oxidativa, que se efectúa en las mitocondrias. El proceso puede resumirse en la ecuación siguiente : 20 minutos C6H12O6 + 6O2 ----> 6CO2 + 6H2O E Fotosíntesis Fosforilación Con luz Periódica Usa CO2 y H2O Libera O2 Rompe H2O Endergónica Capta energía En cloroplasto Sin luz Contínua Usa O2 Libera CO2 Forma H2O Exergónica Libera energía En mitocondria La distribución de las mitocondrias es generalmente uniforme en el citoplasma, aunque hay células que muestran una distribución especial, como es el caso de las células musculares estriadas esqueléticas, que se ubican en su periferia; o en las células del tubo proximal del riñón, que se encuentran en el citoplasma basal; o en la pieza intermedia de la cola del espermatozoide. Mitocondria La orientación de las mitocondrias, especialmente las largas, es paralela al eje de la célula, cuando ésta es cilíndrica. Si la célula es esférica, tienden a ubicarse en posición radial a los centríolos. La cantidad de mitocondrias en una célula varía. Hay células, como las parietales de la mucosa del estómago, que cerca del 50 % del volumen del citoplasma está ocupado por las mitocondrias. En los hepatocitos, la cantidad de mitocondrias varía según la posición que ellos tengan en el lobulillo acinar: en los centrales hay cerca de 1.600 mitocondrias, en los mediales unas 1.300 y en los periféricos, 1.060; además el diámetro de las mitocondrias aumenta desde las centrales hacia las periféricas, pero disminuye la longitud. Estructura de la mitocondria La mitocondria está conformada por una membrana externa de unos 6 nm, una cámara externa de unos 6-8 nm, llamada también espacio intermembranal, una membrana interna que proyecta hacia el interior unos pliegues denominados crestas mitocondriales y una cámara interna llamada matriz mitocondrial. Las crestas mitocondriales son tabiques incompletos que no interrumpen la continuidad de la cámara interna, la cual se encuentra llena de un gel denso con altas concentraciones de proteínas solubles y pequeñas moléculas. Microfotografía de una mitocondria de una célula gástrica mostrando las crestas laminares. Coloca los nombres de cada parte de la mitocondria señalada por las flechas. Mitocondria Mediante la técnica de tinción negativa, se ha visto que la superficie interna es totalmente asimétrica por la presencia de unas partículas de unos 9 nm, unidas a la membrana por un pequeño tallo de 3 x 5 nm. Estas partículas se llaman partículas elementales ó partículas F0-F1, las que se encuentran espaciadas a intervalos de unos 10 nm. Cada una de ellas representa una ATPasa especial en donde ocurre la producción de moléculas de ATP. Por último, la estructura interna de la mitocondria varía según los diferentes tipos celulares. Así, el número de crestas mitocondriales por unidad de volumen es alto en las células musculares y bajo en las células germinales. También, las crestas pueden presentarse como láminas (célula parietal en estómago)(ver foto ME en página anterior) o como túbulos (célula de Leydig en testículo)(ver foto ME abajo) Organización molécular de las membranas mitocondriales Se ha logrado separar la membrana externa de la mitocondria mediante la digitonina, produciéndose unas estructuras llamadas mitoplastos, constituidos por la membrana interna que encierra la matriz mitocondrial. Estos mitoplastos son capaces de realizar la fosforilación oxidativa. La membrana externa posee un 40% de lípidos contra un 20 % de la membrana interna y contiene colesterol. Esto significa que La relación lípido–proteína es de 0.8 para la membrana externa y de un 0.3 para la membrana interna. La membrana externa posee una proteína llamada porina de 29 kD, sintetizada en ribosomas libres e integrada a la membrana en forma postranscripcional, concede una resistencia a la digestión por tripsina y parece formar canales para el paso de solutos. porina Microfotografía de una mitocondria de una célula de Leydig (testículo) mostrando las crestas tubulares. Mitocondria La membrana externa contiene, entre otras proteínas, un sistema enzimático específico : la monoamino-oxidasa, la cual viene a constituirse en una molécula marcadora de la membrana externa. La membrana interna, represen.tada por los mitoplastos, posee un 20 % de lípidos y un 80 % de proteínas, carece de colesterol y posee un alto contenido de ácidos grasos insaturados, todo lo cual explica su baja fluidez (o alta viscosidad). La importancia de esta situación es que la transferencia de electrones entre los diferentes componentes de la cadena transportadores de electrones, podría ser mediada por difusión lateral y por colisiones en el plano de la membrana. Entre las proteínas de la membrana interna se encuentran los componentes de la cadena respiratoria y de la fosforilación oxidativa. Además, contiene varios transportadores (carriers) para varias moléculas. Los transportadores más importantes son los que se relacionan con el pasaje de ATP, ADP y PO4. Corresponde a un transportador que mueve ATP hacia el citosol y ADP hacia la matriz, es decir, es del tipo antiporte. Entre los lípidos de la membrana interna hay uno muy especial : la cardiolipina. Este lípido posee 4 cadenas de ácidos grasos, que se considera que reduce la permeabilidad de la bicapa a los protones hidrógenos. Función de la mitocondria. Proteínas, lípidos y carbohidratos constituyen la mayor parte de los alimentos que consumimos y que deben ser degradados en moléculas pequeñas (monómeros) para que puedan proporcionar energía a la célula. La degradación es lograda por acción enzimática y se llama catabolismo, el cual se considera que ocurre en 3 etapas: (a) etapa 1, ocurre en el tracto digestivo cuando moléculas grandes son reducidas a sus monómeros; (b) etapa 2, ocurre dentro de la célula cuando los monómeros son degradados hasta piruvato que entra a la mitocondria y se convierten en acetil CoA; y (c) etapa 3, ocurre dentro de la matriz de la mitocondria, cuando el acetil CoA es degradado completamente a CO2 y H2O liberándose energía en forma de ATP. Mitocondria Glucólisis La glucólisis corresponde a la degradación de la glucosa para producir ATP en ausencia de O2 (anaerobiosis). En este proceso, la glucosa de 6 C se convierte en dos moléculas de piruvato de 3 C cada una. Esta conversión comprende una secuencia de 9 pasos enzimáticos que produce intermediarios fosfatados. La célula hidroliza 2 moléculas de ATP para conducir los pasos iniciales y produce 4 ATP en los pasos finales, con una ganancia neta de 2 ATP por cada molécula de glucosa. ALIMENTOS Proteínas Carbohidratos Etapa Aminoácidos 1 Monosacáridos Glucólisis anaeróbica Etapa 2 Lípidos Ac. grasos ATP ADP Piruvato Acetil CoA ciclo de Krebs Etapa 3 NADH Transporte 3 ATP de electrones 3 ADP O2 NH3 H2 O CO2 Productos de desecho La secuencia de reacciones puede ser dividida en 3 etapas: (a) etapa 1, pasos del 1 al 4: la glucosa de 6C se convierte en 2 moléculas de gliceraldehido 3 fosfato de 3C, estas reacciones requieren de 2 ATP; (b) etapa 2, pasos 5 y 6: el grupo aldehido se oxida a grupo carboxilo liberando ATP y reduciendo una molécula de NAD+ (transportadora de electrones); y (c) etapa 3, pasos del 7 al 9: los fosfatos de la molécula son transferidos al ADP para producir ATP. En resumen, la glucólisis es el paso previo, en células aeróbicas, para entrar a la mitocondria. En células anaeróbicas es la principal fuente de ATP. Convierte una molécula de glucosa de 6C en 2 moléculas de piruvato de 3C cada una, utiliza 2 ATP y produce 4 ATP, ganando 2 ATP. Además reduce una molécula de NAD+. Mitocondria GLUCOSA ATP 1 6C ADP + Pi G-6-P 6C 2 Fructuosa-6-P 3 ATP 6C ADP + Pi Fructuosa-1,6-diP 6 C Con algunas excepciones, este proceso de glucólisis anaeróbica, que lleva la glucosa a piruvato y a acetil CoA, ocurre en todas las células. En el caso de contracciones musculares bruscas, el piruvato puede ser transformado en lactato, mediante un tipo de reacción química llamada fermentación. Este proceso se produce en situaciones de hipoxia (poco oxígeno), cuando por contracciones bruscas el aporte de O2 es insuficiente para cubrir las necesidades de todas las fibras musculares. 4 Gliceraldehido-3-P 3 C 5 Pi NAD NADH 1,3 difosfoglicerato 3 C 6 ADP ATP 3 fosfoglicerato 3C 2 fosfoglicerato 3C 7 8 fosfoenolpiruvato 3 C 9 ADP ATP PIRUVATO 3C La acumulación de lactato produce en el músculo contracciones dolorosas y puede llegar a producir tetanización (paralización). Sólo con un aporte suficiente de O2, el lactato puede revertir a piruvato y seguir así la vía metabólica normal. En el caso de los eritrocitos (G.R.), la glucólisis anaeróbica termina siempre en lactato, porque las mitocondrias que contienen las enzimas para la oxidación aeróbica del piruvato no están presentes. En el caso de las levaduras, el piruvato es llevado hasta alcohol etílico (etanol) por el proceso llamado fermentación alcohólica. Este proceso no ocurre en el humano por la ausencia de la enzima alcohol deshidrogenasa. Mitocondria En resumen, en condiciones aeróbicas (con aporte de O2) la glucosa es degradada hasta piruvato, que al unirse con la CoA se transforma en acetil–CoA que entra al ciclo de Krebs (que ocurre en la matriz); en condiciones anaeróbicas (con deficiencia de O2), la glucosa es degradada hasta piruvato y luego en lactato. NADH NAD es una abreviación de nicotinamida adenina dinucleótido, lo que deduce que una parte de la molécula es un AMP y otro es un anillo de nicotinamida, con capacidad para captar un protón hidrógeno con dos electrones (ión hidruro, H–, H :). Se encuentra en 2 estados: oxidado NAD+ ó reducido NADH. En su forma reducida es muy inestable con gran tendencia a transferir el ión H– a otras moléculas. La acción de la forma oxidada es captar 2 átomos de H de un sustrato : un H con dos e– (H : –) es añadido al NAD y otro H como protón es liberado al medio circundante. Importante, casi toda la energía disponible de la combustión de carbohidratos, lípidos y otros compuestos en las etapas tempranas de la oxidación es guardada en forma de e– de alta energía, captados por NAD+ y FAD. Ciclo del ácido cítrico El ciclo de Krebs (por su creador Sir Hans Krebs) llamado también ciclo del ácido cítrico (por ser el primer intermediario) o ciclo de los ácidos carboxílicos (por sus 3 primeros intermediarios) ocurre en la matriz mitocondrial de las células eucarióticas o en el citosol de las procarióticas. El ciclo de Krebs se produce cuando hay O2 molecular disponible y el piruvato. Aunque este ciclo no usa O2 directamente, lo hace a través de las enzimas deshidrogenasas NAD (nicotinamida adenin dinucleótido) y FAD (flavin adenin dinucleótido). Las principales funciones del ciclo de Krebs son : (a) descarboxilación, es decir, pérdida de carbonos mediante la producción de CO2: (b) protonización : liberación de protones H+ que serán captados por las enzimas NAD y FAD. El ciclo comienza con la reacción entre el acetil CoA y el oxalacetato (4C) para formar el citrato (6C), luego una serie de reacciones enzimáticas logran desprender 2 C (en forma de CO2 ) del citrato para convertirlo nuevamente en oxalacetato, que repetirá el ciclo. El CO2 producido difunde de la mitocondria y abandona la célula. En este ciclo, átomos de H son transferidos a moléculas transportadoras ( NAD y Mitocondria FAD). En cada ciclo 3 moléculas de NAD+ son convertidas en NADH y una molécula de FAD en FADH2. La energía almacenada en estos electrones es aprovechada posteriormente en las reacciones de fosforilación oxidativa que ocurre a nivel de moléculas ubicadas en la membrana interna de la mitocondria. (ver esquema en la siguiente página) La cadena respiratoria está conformada por una serie de macromoléculas ubicadas en la membrana interna de la mitocondria y cuya función es movilizar los e– recibidos del NADH y entregarlos al O2; además, bombea H+ desde la matriz hacia el espacio intermembranal. Fosforilación oxidativa La fosforilación oxidativa es la última etapa en el catabolismo y es donde se libera la mayor cantidad de energía. En este proceso, la moléculas de NADH y FADH2 entregan sus electrones al O2 para formar H2O. Parte de la energía liberada es utilizada para producir ATP y el resto se libera como calor. Cada molécula de NADH lleva un ión hidruro (H :, H-, un átomo de H con un electrón extra). El proceso de transporte de e– comienza cuando el ión hidruro es removido del NADH regenerando el NAD+ y entregando un H+ al medio y dos e– a la cadena respiratoria. NADH : Õ NAD+ + 2e– + H+ Las macromoléculas se agrupan en 3 complejos : (a) complejo deshidrogenasa NADH, es el complejo más grande (800 Kd y más de 22 cadenas polipeptídicas), acepta los 2 e– del NADH, los pasa a la ubiquinona y bombea el H+ al espacio intermembranal (b) complejo citocromo b–c1, un poco más pequeño que el anterior (500 Kd y 8 cadenas polipeptídicas), recibe los e– de la ubiquinona, los pasa al citocromo c y bombea otro H+ hacia el espacio; y (c) el complejo citocromo oxidasa (a, a3) el más pequeño (300 Kd y 18 cadenas polipeptídicas), acepta los e– del citocromo c, los entrega al O2 y bombea otro H+ al espacio intermembranal. (ver esquema en la página 204) Mitocondria Glucosa 2NAD+ oxalacetato 2NADH- 2NAD+ Matriz citoplasm malato Piruvato M. Ext. M. Int. 2NAD+ Matriz mitocond 2NADH2NAD+ 2NADH- malato Ac CoA 2NADH2NAD+ 2NAD+ 2NADH- ciclo de ácido cítrico oxalacetato CO2 2FADH22FAD+ 2NAD+ 2NADHCO2 10 NADH ---> 30 ATP 2 FADH2 ----> 4 ATP Mitocondria 3 ATP (NADH) 2 ATP (FADH) NADHFADH2 FAD+ NAD+ O2 F1 c M. Int. Espacio intermembranal I II u III 4H+ IV V F0 4H+ 2H+ 10H+ (NADH) 4H+ 2H+ 6H+ (FADH) M. Ext. De lo anterior se deduce que la transferencia de e– está acoplada con una liberación orientada de H+. El movimiento de H+ trae 2 consecuencias : (a) genera una gradiente de pH a través de la membrana interna, con un pH mayor en la matriz que en el espacio intermembranal (equivalente al citosol, cerca de 7, ya que la membrana externa es permeable a los iones); y (b) genera una gradiente de voltaje (potencial de membrana), negativa hacia la matriz y positiva hacia el espacio. En conjunto, se dice que ambas gradientes conforman el llamado gradiente protónica electro-química, la cual ejerce una fuerza que trata de mover H+. Complejos : I - NAD deshidrogenasa II- succinato deshidrog. III- citocromo b-c1 IV- citocromo oxidasa V- ATP sintasa -------------U- ubiquinona C- citocromo c dirección mov. Electr. membrana interna de mitocondria H+ + + + potencial de membrana UV matriz mitocondrial - - - H+ membrana interna de mitocondria H+ acido gradiente de concentración UpH alcalino matriz mitocondrial H+ Mitocondria Producción de ATP Una proteína muy compleja, ubicada en la membrana interna de la mitocondria, denominada ATPasa F0-F1 o simplemente ATP sintasa, es responsable de la producción de ATP.Este complejo es visible al ME, como una masa oscura abultada (9 nm) dirigida hacia la matriz y unida a la membrana por una unión más estrecha. Este complejo posee dos componentes principales : F0 y F1. El componente F0 es un complejo proteico intrínseco en la membrana interna, integrado por varias unidades de proteínas a, b y c; en conjunto, forma un canal a través del cual los protones pueden fluir desde el espacio intermembranal hacia la matriz. El componente F1 es un complejo de 9 cadenas polipeptídicas (3 α, 3β, γ, δ y ε), las cadenas γ, δ y ε permiten la unión con el complejo F0. En las cadenas β ocurre alternativamente la síntesis de ATP desde ADP y PO4, cuando el H+ pase por el centro de las cadenas. Se sabe que el complejo F1 solo sintetizará ATP cuando se encuentre unido al F0, pues al estar separado realiza la acción contraria: hidroliza ATP. α β α β Matriz Membrana interna Espacio inter membranal Recordemos en este punto que 10 protones son bombeados a través de la membrana por cada par de electrones entregados por una molécula de NADH. Así, la oxidación de una molécula de NADH conduce a la síntesis de casi 3 moléculas de ATP (recordar : 3 H+ produce una molécula de ATP). Por otro lado, la oxidación de una molécula de que resulta en la FADH2, transferencia de 6 protones, dirige la síntesis de solo 2 moléculas de ATP. Mitocondria Translocación de proteínas mitocondriales La información genética para las proteínas mitocondriales se encuentra en dos sitios diferentes : ADN nuclear y ADN mitocondrial. La mayor proporción de proteínas mitocondriales poseen su información en el ADN nuclear; por consiguiente, de allí derivarán los ARNm, que junto con los ribosomas citosólicos las sintetizarán en el citosol. Estas proteínas deberán atravesar las membrana mitocondriales (externa e interna) para llegar a su destino final. En proceso de translocación es similar al descrito para las proteínas del RER. Tomaremos primero el caso de una proteína de la matriz, por ejemplo, una enzima del ciclo de Krebs. Es sintetizada en el citosol, sus primeros aminoácidos (20 a 80) conforman la secuencia señal, la cual dispone de un receptor en la membrana externa que la moviliza hacia el translocador. Éste es una serie de proteínas ubicadas en la membrana externa e interna y que contactan de tal manera que puede evidenciarse al ME en los llamados puntos de contacto de las membranas. La translocación del precursor de la proteína de la matriz ocurre en un solo paso a través de las dos membranas. Sin embargo, hay dos momentos en este proceso que deben aclararse : (1) la inserción del precursor en el translocador requiere de chaperoninas citosólicas (Hsp 70) que ayudan a mantener desplegada la proteína, mientras se inicia la translocación y de una gradiente electroquímica mantenida por el bombeo de H+ desde la matriz hacia el espacio intermembranal (cámara externa); Mitocondria (2) la translocación a través del translocador, requiere de hidrólisis de ATP para liberar la cadena de las chaperoninas citosólicas y de otras chaperoninas mitocondriales (Hsp 60) que mueven vectorialmente la cadena hacia la matriz. Una vez en la matriz, el precursor pierde la secuencia señal por acción de una peptidasa señal, la cual la deja libre en la matriz y se libera de las chaperoninas mitocondriales por hidrólisis de ATP. Otras chaperoninas colaboran luego con el plegamiento definitivo de la proteína mitocondrial haciéndola activa. Las proteínas que tienen su residencia en el espacio intermembranal (cámara externa) son translocadas por el mismo mecanismo que las proteínas de la matriz; sin embargo, hay medios por los cuales ellas no se quedan en la matriz. Uno de ellos es que al eliminarse el péptido señal por acción de la peptidasa señal, queda evidente una segunda secuencia señal hidrofóbica que dispone de un translocador en la cara que mira hacia la matriz de la membrana interna. Así, ocurre la translocación desde la matriz hacia la cámara externa en donde existe una nueva peptidasa señal que la libera en el espacio intermembranal. Mitocondria ADN mitocondrial Hace años atrás, cuando unos científicos estaban detectando ADN en células eucarióticas, además de encontrar ADN en los núcleos, detectaron unas manchas en el citoplasma. Pensaron que era una falla de la técnica (artefacto). Sin embargo, al aplicar con más precisión se dieron cuenta que la detección del ADN citoplasmático correspondía a las . . . ¡mitocondrias! La pregunta clave aquí es ¿de dónde provino? Otras preguntas podrían ser ¿cómo es? y ¿qué hace allí? Después de muchos estudios, la respuesta la tiene la Evolución. Parece ser que hace millones de años atrás, una célula que vivía tranquilamente en el primitivo caldo terrestre, vio con envidia que cerca de ella existía un organismo más pequeño que producía más energía. Ni tonta ni perezosa, la engulló (fagocitó). Así quedó formada la mitocondria, la cual tiene dos membranas : una de la célula primitiva y otra del organismo fagocitado. Por eso es que hay diferencias químicas entre las dos membranas de la mitocondria, la interna posee la cardiolipina, un lípido con 4 cadenas de ácidos grasos, en tanto que todos los lípidos de la célula poseen sólo tres. Volviendo al ADN, no debemos olvidar que el organismo fagocitado, aunque carecía de núcleo visible (procarionte), tenía su propio ADN, que es diferente al ADN de la célula primitiva. De hecho, el ADN de la mitocondria es circular con dos hebras una pesada y otra liviana, con más contenido de G y C. Ahora bien, este ADN mitocondrial posee la información hereditaria para 13 proteínas pertenecientes a los complejos de la membrana interna. Teoría simbiótica Membrana externa : del organismo que fagocitó (eucarionte) Membrana interna : del organismo fagocitado (procarionte) Mitocondria Complejo Nº proteínas Nombre del complejo I 7 (de 22) NADH deshidrogenasa (reductasa) III 1 (de 8) Citocromo b-c1 (reductasa) IV 3 (de 7) Citocromo c (oxidasa) V 2 (de 13) F0 Total 13 proteínas El genoma mitocondrial humano codifica para 13 proteínas comprometidas en el transporte de electrones y la fosforilación oxidativa. Además, codifica para los dos ARNr : el 12 S de la subunidad pequeña y el 16 S para la subunidad grande del ribosoma. Por otro lado, codifica para 22 ARNt que son necesarios para la traducción de las proteínas mitocondriales. El cuadro mostrado arriba detalla las proteínas mitocondriales codificadas en el genoma mitocondrial, por cada complejo. Casi todas las mitocondrias del cigoto son aportadas por el ovocito más que por el espermatozoide. Al igual que el genoma nuclear es susceptible de sufrir mutaciones, también lo es el genoma mitocondrial. Ahora bien, las mutaciones en el ADN mitocondrial son transmitidas a la siguiente generación por la madre. ATP sintasa Aplicación clínica La enfermedad hereditaria de Leber, que se manifiesta por una neuropatía del nervio óptico, es una rara enfermedad que deviene en ceguera, debido a una degeneración del nervio óptico. La visión se pierde alrededor de los 20 años, siendo más frecuente en los varones. En 1988, Douglas Wallace identificó una mutación en el ADN mitocondrial causante de esta enfermedad. Esta mutación afecta una de las subunidades del complejo I en la cadena transportadora de electrones, que resulta en la sustitución de una histidina por una arginina. La mutación reduce la capacidad de la mitocondria para realizar el transporte de electrones y la fosforilación oxidativa, con la consecuencia de disminuir la producción de ATP. Esto tiene grandes efectos en los tejidos muy dependientes de la fosforilación oxidativa, como es el tejido nervioso incluyendo el nervio óptico.