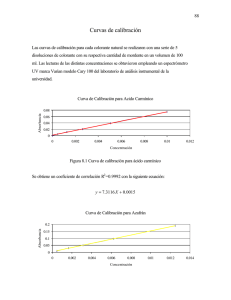
Guia Labo y Problemas QA
Anuncio

Análisis Instrumental Módulo Analítico Guía Laboratorio y Problemas Actualizada 2do Cuatrimestre 2016 1 IMPORTANTE – Nuevas correlatividades. A partir de Abril de 2013 las correlatividades vigentes para cursar los TP y para dar Final de Análisis Instrumental serán: Para cursar 1 - Con examen final aprobado: Química Analítica, Química Orgánica II y Química Biológica. 2 - Con los TP aprobados: Química Física I Para dar el final Las indicadas en los puntos 1 y 2 anteriores, más el examen final aprobado de Física II. Los alumnos que hubieran aprobado los trabajos prácticos con anterioridad al ciclo lectivo 2013 tendrán la posibilidad de rendir los correspondientes exámenes finales con las correlatividades exigidas en el momento de su cursada. Desde el año 2011 los temas de final de la parte de QB son muy diferentes, y los de QA han cambiado levemente. Las teóricas correspondientes están en esta página web: www.qi.fcen.uba.ar/materias/ai/ Introducción y objetivos Durante esta materia, y en particular en el Modulo Analítico - Biológico, se verán varias de las principales técnicas instrumentales de medición de parámetros químicos y biológicos. Se estudian cuestiones básicas como ruido, adecuación de la señal, selectividad y sensibilidad, robustez, parámetros de mérito, como también temas aplicados, incluyendo técnicas usuales de biología molecular. El Módulo Analítico - Biológico consta de dos clases semanales teóricas de 2 horas de duración cada una y asistencia optativa (aunque recomendable), y dos clases semanales de laboratorio o problemas de asistencia obligatoria. Turno Día y Hora Teo Lab Mañana Lab Tarde Lab Noche Ma y Ju de 18 a 20 Hs Ma y Ju de 8:30 a 12:30Hs Ma y Ju de 13:30 a 17:30Hs Mi y Vi de 18 a 22Hs Se formaran 6 grupos, de 3 o 4 alumnos, los cuales rotaran por distintas actividades. Practicas a desarrollar: Cromatografía Gaseosa – Análisis Cualitativo Cromatografía Gaseosa – Análisis Cuantitativo Cromatografía Liquida de Alta Resolución Espectroscopia de Absorción y Emisión Atómica Imaging de pH en dos dimensiones. Problemas de Análisis Instrumental pag. 7 pag. 12 pag. 14 pag. 20 pag. 25 pag. 34 Luego del primer parcial, todos juntos desarrollaran los siguientes dos TPs: Espectroscopia de Emisión Molecular - Problemas pag. 30 pag. 46 2 Seguridad en el laboratorio Las medidas de Seguridad en Laboratorios son un conjunto de normas preventivas destinadas a proteger la salud de los que allí se desempeñan frente a los riesgos propios derivados de la actividad, para evitar accidentes y contaminaciones tanto dentro de su ámbito de trabajo, como hacia el exterior. Las reglas básicas aquí indicadas son un conjunto de prácticas de sentido común realizadas en forma rutinaria. El elemento clave es la actitud proactiva hacia la seguridad y la información que permita reconocer y combatir los riesgos presentes en el laboratorio. Será fundamental la realización meticulosa de cada técnica, pues ninguna medida, ni siquiera un equipo excelente puede sustituir el orden y el cuidado con que se trabaja. 1- Se deberá conocer la ubicación de los elementos de seguridad en el lugar de trabajo, tales como: matafuegos, salidas de emergencia, mantas ignífugas, lavaojos, gabinete para contener derrames, accionamiento de alarmas, etc. 2- No se permitirá comer, beber, fumar o maquillarse. 3- No se deberán guardar alimentos en el laboratorio, ni en las heladeras que contengan reactivos. 4- Se deberá utilizar vestimenta apropiada para realizar trabajos de laboratorio y cabello recogido (guardapolvo preferentemente de algodón y de mangas largas, zapatos cerrados, evitando el uso de accesorios colgantes). 5- Es imprescindible mantener el orden y la limpieza. Cada persona es responsable directa de la zona que le ha sido asignada y de todos los lugares comunes. 6- Las manos deben lavarse cuidadosamente después de cualquier manipulación de laboratorio y antes de retirarse del mismo. 7- Se deberán utilizar guantes apropiados para evitar el contacto con sustancias química o material biológico. Toda persona cuyos guantes se encuentren contaminados no deberá tocar objetos, ni superficies, tales como: teléfono, lapiceras, manijas de cajones o puertas, cuadernos, etc. 8- No se permitirá pipetear con la boca. 9- No se permitirá correr en los laboratorios. 10- Siempre que sea necesario proteger los ojos y la cara de salpicaduras o impactos se utilizarán anteojos de seguridad, viseras o pantallas faciales u otros dispositivos de protección. Cuando se manipulen productos químicos que emitan vapores o puedan provocar proyecciones, se evitará el uso de lentes de contacto. 11- No se deben bloquear las rutas de escape o pasillos con equipos, máquinas u otros elementos que entorpezcan la correcta circulación. 12- Todo material corrosivo, tóxico, inflamable, oxidante, radiactivo, explosivo o nocivo deberá estar adecuadamente etiquetado. 13- No se permitirán instalaciones eléctricas precarias o provisorias. Se dará aviso inmediato a la Secretaría Técnica en caso de filtraciones o goteras que puedan afectarlas instalaciones o equipos y puedan provocar incendios por cortocircuitos (Interno 355). 14- Se requerirá el uso de mascarillas descartables cuando exista riesgo de producción de aerosoles (mezcla de partículas en medio líquido) o polvos, durante operaciones de pesada de sustancias tóxicas o biopatógenas, apertura de recipientes con cultivos después de agitación, etc. 15- Las prácticas que produzcan gases, vapores, humos o partículas, aquellas que pueden ser riesgosas por inhalación deben llevarse a cabo bajo campana. 16- Se deberá verificar la ausencia de vapores inflamables antes de encender una fuente de ignición. No se operará con materiales inflamables o solventes sobre llamas directa o cerca de las 3 mismas. Para calentamiento, sólo se utilizarán resistencias eléctricas o planchas calefactoras blindadas. Se prestará especial atención al punto de inflamación y de autoignición del producto. 17- El material de vidrio roto no se depositará con los residuos comunes. Será conveniente ubicarlo en cajas resistentes, envuelto en papel y dentro de bolsas plásticas. El que sea necesario reparar se entregará limpio al taller. 18- Será necesario que todo recipiente que hubiera contenido material inflamable, y deba ser descartado sea vaciado totalmente, escurrido, enjuagado con un solvente apropiado y luego con agua varias veces. 19- Está prohibido descartar líquidos inflamables o tóxicos o corrosivos o material biológico por los desagües de las piletas, sanitarios o recientes comunes para residuos. En cada caso se deberán seguir los procedimientos establecidos para la gestión de residuos. Consultar al Servicio de Higiene y Seguridad (Interno 275). 20- Cuando sea necesario manipular grandes cantidades de materiales inflamables (más de 5 litros.) deberá tenerse a mano un extintor apropiado para ese material en cuestión. 21- Cuando se trasvase material combustible o inflamable de un tambor a un recipiente más pequeño, realice una conexión con una cadena del tambor a tierra y con otra entre el tambor y el recipiente de manera de igualar potenciales y eliminar la posible carga estática. 22- Al almacenar sustancias químicas considere que hay cierto número de ellas que son incompatibles pues almacenadas juntas pueden dar lugar a reacciones peligrosas. Ante dudas consultar al Servicio de Higiene y Seguridad (Interno 275). 23- No almacene en estantes sobre mesadas sustancias corrosivas, hágalo en estantes bajo mesadas y en caso de ácidos o álcalis concentrados (mayor de 2N) deben ser mantenidas dentro de lo posible en bandejas de material adecuado. 24- Los cilindros de gases comprimidos y licuados deben asegurarse en posición vertical con pinzas, grampas y correas o cadenas a la pared en sitios de poca circulación, protegidos de la humedad y fuentes de calor, de ser posible en el exterior. 25- Los laboratorios contarán con un botiquín de primeros auxilios con los elementos indispensables para atender casos de emergencia. 26- Se informará al Dpto. de Seguridad y Control cuando se necesiten dejar equipos funcionando en ausencia del personal del laboratorio. ¡Recuerde!: USTED también es responsable de la seguridad en su lugar de trabajo. 4 Procedimientos ante emergencias: Emergencias médicas Si ocurre una emergencia tal como: cortes o abrasiones, quemaduras o ingestión accidental de algún producto químico, tóxico o peligroso, se deberá proceder: 1- A los accidentados se les proveerán los primeros auxilios. 2- Simultáneamente se tomará contacto con el Servicio Médico (Interno 482), o al Servicio Médico de Deportes (4784-4351 / 3948) 3- Avise al Jefe de Laboratorio o autoridad del Departamento, quienes solicitarán asistencia de la Secretaría Técnica (interno 380) para que envíen personal del Dpto. de Mantenimiento, Seguridad y Control o Servicios Generales según correspondan. 4- El Jefe de Departamento notificará el accidente al Servicio de Higiene y Seguridad para su evaluación e informe, donde se determinarán las causas y se elaborarán las propuestas para modificar dichas causas y evitar futuras repeticiones. 5- Centros para requerir ayuda médica: S.A.M.E. Teléfono 107 Hospital Pirovano Av. Monroe 3555 Tel.4542-5552 / 9279 INTOXICACIONES: Hospital de Niños. Dr. R. Gutiérrez Sánchez de Bustamante 1399. Capital Federal.Tel: 4962-6666. Hospital de Niños. Dr. P. de Elizalde Av. Montes de Oca 40 Tel. 4307-7491 Toxicología 4300-2115 QUEMADURAS: Hospital de Quemados P.Goyena 369 Tel. 4923-4082 / 3022 OFTALMOLOGÍA Hospital Santa Lucía San Juan 2021 Tel. 4941-7077 Hospital Dr. P. Lagleyze Av. Juan B. Justo 4151 Tel. 4581-0645 / 2792 Incendio: 1- Mantenga la calma. Lo más importante es ponerse a salvo y dar aviso a los demás. 2- Si hay alarma, acciónela. Si no grite para alertar al resto. 3- Se dará aviso inmediatamente al Dpto. de Seguridad y Control (Interno 311) informando el lugar y las características del siniestro. 4- Si el fuego es pequeño y sabe utilizar un extintor, úselo. Si el fuego es de consideración, no se arriesgue y manteniendo la calma ponga en marcha el plan de evacuación. 5- Si debe evacuar el sector apague los equipos eléctricos y cierre las llaves de gas y ventanas. 6- Evacúe la zona por la ruta asignada. 7- No corra, camine rápido, cerrando a su paso la mayor cantidad de puertas. No utilice ascensores. Descienda siempre que sea posible. 8- No lleve consigo objetos, pueden entorpecer su salida. 9- Si pudo salir por ninguna causa vuelva a entrar. Deje que los equipos especializados se encarguen. Teléfonos útiles BOMBEROS Teléfono 100 5 DIVISIÓN CENTRAL DE ALARMA: 4381-2222 / 4383-2222 / 4304-2222. CUARTEL V DE BELGRANO: Obligado 2254 Capital Tel. 4783-2222 BOMBEROS DE VICENTE LÓPEZ Av. Maipú 1669 Vicente López. Tel. 4795-2222 BOMBEROS DE SAN ISIDRO: Santa Fe 650 Martínez. Tel. 4747-2222 Derrame de productos químicos 1- Atender a cualquier persona que pueda haber sido afectada. 2- Notificar a las personas que se encuentren en las áreas cercanas acerca del derrame. Coloque la cinta de demarcación para advertir el peligro. 3- Evacuar a toda persona no esencial del área del derrame. 4- Si el derrame es de material inflamable, apagar las fuentes de ignición, y las fuentes de calor. 5- Evite respirar los vapores del material derramado, si es necesario utilizar una máscara respiratoria con filtros apropiados al tipo de derrame. 6- Ventilar la zona. 7- Utilizar los elementos de protección personal tales como equipo de ropa resistente a ácidos, bases y solventes orgánicos y guantes. 8- Confinar o contener el derrame, evitando que se extienda. Para ello extender los cordones en el contorno del derrame. 9- Luego absorber con los paños sobre el derrame. 10- Deje actuar y luego recoger con pala y colocar el residuo en la bolsa roja y ciérrela. 11- Comuníquese con el Servicio de Higiene y Seguridad para disponer la bolsa con los residuos. 12- Si el derrame es de algún elemento muy volátil deje dentro de la campana hasta que lo retire para su disposición. 13- Lave el área del derrame con agua y jabón. Seque bien. 14- Cuidadosamente retire y limpie todos los elementos que puedan haber sido salpicados por el derrame. 15- Lave los guantes, la máscara y ropa. Declaro haber leído las medidas de seguridad que aparecen en la guía de Trabajos Prácticos de Análisis Instrumental - Módulo Analítico bajo los títulos SEGURIDAD EN EL LABORATORIO y PROCEDIMIENTOS ANTE EMERGENCIAS. Fecha: .................................. L.U. Nº: ................................ Turno de Laboratorio:...................... Firma:....................................... Aclaración:....................................... 6 CROMATOGRAFÍA GASEOSA INTRODUCCION La cromatografía es un método físico de separación basado en la distribución de los componentes de una mezcla entre dos fases inmiscibles, una fija y otra móvil. En cromatografía gaseosa, la fase móvil es un gas que fluye a través de una columna que contiene a la fase fija. Esta fase fija puede ser un sólido poroso (cromatografía gas-sólido o CGS), o bien una película líquida delgada que recubre un sólido particulado o las paredes de la columna (cromatografía gas-líquido o CGL). En el primer caso, el proceso que produce la separación es la adsorción de los componentes de la mezcla sobre la superficie sólida y en el segundo, la partición de los mismos entre las fases líquida y gaseosa. Por ser la más utilizada, la discusión que sigue se refiere a CGL. INSTRUMENTAL El instrumental utilizado en la cromatografía gaseosa se puede resumir en el siguiente esquema 1: Esquema 1: esquema de un CG típico (a) y del equipo real utilizado en esta práctica (b). El gas portador (fase móvil) proviene de cilindros provistos con válvulas reductoras de presión. La muestra se introduce en el inyector con una microjeringa a través de un septum de goma. Allí se produce la vaporización instantánea de la misma y su introducción en la corriente de gas. La columna se halla dentro de un horno de temperatura variable, lugar donde se realiza la separación de los componentes de la muestra. El detector produce una señal eléctrica proporcional a la cantidad de materia a medida que cada componente separado fluye a través de él. Esa señal es enviada al registrador que realiza un gráfico de intensidad en función del tiempo (cromatograma) del tipo, ver esquema 2. 7 Esquema 2: Análisis de un cromatograma (a) y un cromatograma típico (b) Idealmente, se trata de picos gaussianos y cada pico corresponde a un componente de la muestra original. El integrador (o el software de control) calcula además el área correspondiente a cada pico, la cual es proporcional a la cantidad de analito. Parámetros relevantes Tiempo de retención (tr) de un dado componente de la muestra es el tiempo transcurrido desde la inyección de la misma hasta la aparición del máximo correspondiente al pico de ese componente. Un caso especial lo constituye un componente que no es retenido por la fase fija (no se reparte entre fases), el cual saldrá de la columna antes que cualquier sustancia a un tiempo llamado tiempo muerto (t0). De esta manera, es posible definir t’r como el tiempo que un cierto componente permanece retenido en la fase estacionaria: tr = t0 + t’r El factor de capacidad (k’) de un dado compuesto se define como: que, en función de la constante de partición (K), del volumen de la fase fija (VL) y del volumen de la fase móvil (VG) se puede escribir como: y en función de los tiempos de retención: Con respecto a la columna, se define como plato teórico a una capa estrecha de la misma en donde se produce el equilibrio de partición de un compuesto entre la fase fija y la móvil. Para una columna de longitud L, el número de platos teóricos (N) es: 8 siendo H la altura de plato teórico, uno de los parámetros que determina la capacidad del proceso cromatográfico para separar dos compuestos dados, y W el ancho de los picos extrapolada a la altura de la línea de base como se muestra en el esquema 2. A partir de las definiciones anteriores, es posible definir parámetros asociados a la separación de dos sustancias A y B que caracterizan la eficiencia de una separación. Siendo B la sustancia con mayor tr estos parámetros son: Factor de selectividad Resolución Teniendo en cuenta que k’, α, N y H dependen de la temperatura, tipo de fase fija, longitud de columna y flujo de fase móvil, es posible mejorar una separación optimizando estos parámetros experimentales. Etapas del análisis de una muestra 1. Optimización de las condiciones instrumentales. 2. Obtención de los cromatogramas en condiciones óptimas (muestras y patrones). 3. Calibración (diferentes técnicas). 4. Cuantificación del/los componentes presentes en la muestra. 5. Tratamiento de los resultados: A: Se busca obtener como respuesta instrumental: • Picos bien resueltos. • Buena relación señal/ruido. • Línea de base horizontal (sin deriva). • Picos no distorsionados. B: Normalmente suele realizarse una calibración por estándar externo o utilizando un estándar interno. i. Estándar externo: Se preparan muestras de patrones de concentración conocida que se analizan y permiten construir una curva de calibración para cada componente a cuantificar. Con este método es 9 necesario medir exactamente los volúmenes inyectados tanto de los patrones como de la muestra incógnita. ii. Estándar interno: En este caso se agrega a la muestra una cantidad conocida de un compuesto que no está presente originalmente y que posea tr diferente al de los componentes de la muestra. En este caso, la calibración se realiza analizando patrones de concentración conocida para cada componente a cuantificar a los cuales se le agrega la misma cantidad del estándar interno que a la muestra incógnita. La curva de calibración se construye graficando el cociente entre la señal del analito incógnita y el estándar interno en función de la concentración del analito incógnita. Este método elimina el error cometido al no poder reproducir el volumen inyectado. C: Tanto para la medición de la muestra como de los patrones, es conveniente estudiar la reproducibilidad de la señal analítica y la de los tiempos de retención del/los analitos. Para ello se deben realizar varias inyecciones (n) de cada muestra y cada uno de los patrones y utilizar los valores medios. D: Se realizará el tratamiento de resultados siguiendo las indicaciones dadas en la guía de trabajos prácticos. Consultar el apunte correspondiente. PARTE EXPERIMENTAL Primera parte - análisis cualitativo: Separación de alcoholes Objetivos • Analizar la influencia de los diversos parámetros instrumentales en la identificación y separación de una mezcla de alcoholes. • Seleccionar las condiciones instrumentales óptimas para lograr dicha separación (temperatura / rampa de temperatura, flujo del gas carrier) • Determinar la identidad de los componentes de la muestra utilizando patrones de cada analito y los tiempos de retención obtenidos para cada uno de ellos en idénticas condiciones operativas. • Analizar el orden de elusión de todos los componentes, en función de su estructura y punto de ebullición. Materiales a emplear (provisto por el plantel docente) • Cromatógrafo de gases Shimadzu GC/17 A con un integrador Shimadzu CR5A. Detector: FID. 10 • Columna capilar marca Agilent J&W HP-INNOWAX, longitud: 30 m, diámetro interno: 0,250 mm, espesor de film: 0,25 µm. Rango de temperatura de trabajo: 40°C a 250°C TR ABAJAR SI O SI EN ESTE INTERVALO • Gas natural, para calcular t0 • Patrones individuales de metanol, n-propanol, iso-propanol, n-butanol, iso-butanol, npentanol y acetonitrilo al 1% en agua. • Mezcla de todos los componentes al 1% en agua. • Microjeringa de 10 µL. Análisis cualitativo de la muestra. Para identificar los componentes de la muestra se realizarán distintas corridas cromatograficas de una mezcla sintética de alcoholes que contiene acetonitrilo (como patrón interno) todos en la misma concentración (1%), hasta optimizar la resolución variando las distintas condiciones instrumentales (temperatura del horno y flujo del gas portador). El objetivo de este día es que se interiorice del funcionamiento y modificaciones que ocurren en la corrida cromatográfica con cada parámetro ajustado. El flujo puede ser variado desde 0.1 a 4 mL/min (¡PRUÉBELO!), y la temperatura entre 40 y 230 ºC (¡PRUÉBELO!). Utilice todas las combinaciones que le sean necesarias para comprender como se ven afectados las posiciones y formas de los picos. Optimice ambos parámetros para lograr la separación de todos los componentes de la mezcla, no olvide que pueden realizarse rampas de temperatura. En las condiciones de flujo y temperatura optimizados, inyecte un compuesto que no interactúe con la columna para determinar t0. En este caso, se utilizara gas natural (metano). Se analizarán luego patrones de cada componente por separado para proceder a su identificación a través de los tiempos de retención obtenidos en idénticas condiciones operativas. Análisis de los resultados Realizar un análisis crítico de los parámetros modificados y el efecto sobre la posición de los picos de cada uno. Informar las condiciones experimentales optimizadas y los tiempos de retención de cada componente de la mezcla. Obtenga los parámetros que caracterizan la eficiencia de cada columna para estos analitos en las condiciones de trabajo (k’, α, N y H). Efectuar el análisis estadístico apropiado. 11 Segunda parte – Análisis cuantitativo de bromoalcanos Materiales a emplear (provisto por el plantel docente) • Cromatógrafo de gases Shimadzu GC/17 A con un integrador Shimadzu CR5A. Detector: ECD • Columna capilar marca Supelco, longitud: 30 m, diámetro interno: 0,250 mm, espesor de film: 0,20 µm. Rango de temperatura de trabajo: 40°C a 250°C TR ABAJAR SI O SI EN ESTE RANGO • Patrones individuales de bromopropano, bromooctano, bromodecano, bromododecano todos al 1% en metanol. • Patrones conteniendo mezclas de los cuatro bromoalcanos en metanol entre 1% y 5% en cada componente, además en todos los casos agregar bromoacetonitrilo al 1%, como patrón interno. • Muestra artificial de composición desconocida de los bromoalcanos, con el patron interno al 1%. • Microjeringa de 10 µL. • NUNCA INYECTAR AGUA, ya que se arruina el detector. Experimentos a realizar • Análisis cuantitativo. Se utilizarán las metodologías de cuantificación descriptas anteriormente: i) Estándar externo ii) Estándar interno Primeramente debe optimizar nuevamente los parámetros para lograr una separación de todos los componentes de la mezcla (utilice un flujo entre 1 – 2 ml/min y una temperatura máxima de 230 ºC). Luego comience a cuantificar, para ello inyecte mínimo 2 veces (3 si le da el tiempo) cada patrón y la muestra incógnita de a una “tanda” por vez (tanda= 5 patrones + MI). Cada “tanda” debe ser realizada por un operario distinto. Analice los resultados por ambos métodos, considerando primero cada “tanda” por separado y luego todos los datos juntos. Interprete críticamente los resultados obtenidos (vea la dispersión de datos, por ejemplo) → ¿Qué diferencias hay entre ambos detectores? ¿Porqué no se utiliza metano para obtener el t0 de esta nueva columna? ¿Qué compuesto se podría usar? 12 Observaciones y cuidados experimentales: Ya habrá notado que el vástago de la jeringa Hamilton es muy fino, tenga mucho cuidado cuando realiza las inyecciones y los lavados. Los lavados de la jeringa Hamilton deben realizarse con metanol HPLC, tenga cuidado de no dañar el vástago. No utilice agua. Tenga en cuenta que ese metanol puede terminar en el cromatograma si no lava bien con la solución de muestra. Cuando realice la carga de la jeringa, cuidar bien de que no queden burbujas de aire dentro, realice con cuidado las maniobras necesarias para eliminarlas. El operador debe sincronizar muy bien la inyección y presión en el botón de START, para dar el comienzo a la corrida. Análisis de los resultados Discutir los resultados obtenidos mediante las distintas metodologías utilizadas, analizando las ventajas y desventajas de cada una de ellas. Informar las condiciones experimentales, los tiempos de retención de cada componente de la mezcla y la composición porcentual de cada bromoalcano en la muestra incógnita. Efectuar el análisis estadístico apropiado. Obtenga los parámetros que caracterizan la eficiencia de cada columna para estos analitos en las condiciones de trabajo (k’, α, N y H). Para la columna del segundo día, utilice t0 = 1.9 minutos. Estimar el limite de detección de la técnica empleando todas las estrategias posibles (discutir previamente este punto con el docente). BIBLIOGRAFÍA 1. “Principios de Análisis Instrumental”. D. A. Skoog, F. J. Holler y T. A. Hieman, 5a edición, Mc Graw Hill, madrid 2001. 2. “Métodos Instrumentales de Análisis”. Willard, Merritt y otros. VI edición. 13 CROMATOGRAFIA LIQUIDA DE ALTA RESOLUCION INTRODUCCION La cromatografía es un método físico de separación basado en la distribución de los componentes de una mezcla entre dos fases inmiscibles, una fija o estacionaria y otra móvil. En cromatografía líquida, la fase móvil es un líquido que fluye a través de una columna que contiene a la fase fija. La cromatografía líquida “clásica” se lleva a cabo en una columna generalmente de vidrio, la cual está rellena con la fase fija. Luego de sembrar la muestra en la parte superior, se hace fluir la fase móvil a través de la columna por efecto de la gravedad. Con el objeto de aumentar la eficiencia en las separaciones, el tamaño de las partículas de fase fija se fue disminuyendo hasta el tamaño de los micrones, lo cual generó la necesidad de utilizar altas presiones para lograr que fluya la fase móvil. De esta manera, nació la técnica de cromatografía líquida de alta resolución (HPLC), que requiere de instrumental especial que permita trabajar con las altas presiones requeridas. Dependiendo del tipo de fase fija y del tipo de fenómeno físico que provoca la separación, la cromatografía líquida de alta resolución puede ser: 1. Cromatografía de adsorción. La fase fija es un sólido y se utiliza casi exclusivamente sílice (sílica) y en menor medida alúmina. 2. Cromatografía de reparto. En casi todos los casos, como fase estacionaria se utilizan compuestos unidos químicamente a un soporte sólido de sílica. Se la subdivide en cromatografía en fase normal y fase reversa. En la cromatografía en fase normal, la fase fija es polar (como por ejemplo agua o trietilenglicol) y los compuestos menos polares eluyen primero. En la cromatografía en fase reversa, el compuesto unido químicamente es un hidrocarburo alifático y se emplean fases móviles polares. En este caso, las sustancias más polares eluyen primero. 3. Cromatografía iónica. Se utilizan columnas rellenas con resinas de intercambio iónico para separar y determinar iones. 4. Cromatografía de exclusión por tamaño. La fase fija está formada por partículas poliméricas o de sílice que contienen una red uniforme de poros y llevan a cabo un fraccionamiento relacionado con el tamaño molecular. Las moléculas de tamaño mayor son excluídas y eluyen primero, mientras que las más pequeñas que penetran en los poros son retenidas más tiempo. 14 INSTRUMENTAL Un equipo para cromatografía líquida de alta resolución puede representarse por el siguiente esquema: La fase móvil puede ser un solvente puro o una mezcla de solventes. Cuando se trata de una mezcla, puede programarse la bomba para que tome solventes de diferentes botellas en una proporción determinada y realice la mezcla en una cámara de mezclado. Cuando durante toda la separación se utiliza siempre el mismo solvente, se denomina isocrática, sin embargo es normal realizar un gradiente de composición del solvente a lo largo de la cromatografía para mejorar la eficiencia y acortar la duración del proceso. Estos gradientes de solvente también son realizados en forma automática por las bombas. La bomba envía al solvente a través de caños de diámetro pequeño, generalmente de acero inoxidable, hacia la válvula inyectora. Esta consiste en una válvula de seis vías que permite introducir en el flujo de solvente, la muestra contenida en un aro o loop de volumen calibrado. Luego de que se produzca la separación en la columna, los componentes de la mezcla pasan por el detector. Este produce una señal eléctrica proporcional a la cantidad de materia y esa señal es enviada al registrador que realiza un gráfico de intensidad en función del tiempo (cromatograma): 15 Idealmente, se trata de picos gaussianos y cada pico corresponde a un componente de la muestra original. El integrador calcula además el área correspondiente a cada pico, la cual es proporcional a la cantidad de sustancia. Dado que los detectores de HPLC son no destructivos, es posible recuperar los productos que salen de él. De esta manera, dependiendo del tamaño del loop de inyección y de la columna, y del tipo de bomba, es posible realizar además de separaciones analíticas, cromatografías preparativas. Parámetros relevantes Los parámetros teóricos que caracterizan las separaciones cromatográficas por HPLC son idénticos a los descriptos en cromatografía gaseosa. En HPLC, en lugar de variar la temperatura de la columna para mejorar la resolución del cromatograma, se cambia la composición de la fase móvil a lo largo de la separación utilizando mezclas de entre dos y cuatro solventes. Etapas análisis de una muestra Son idénticas a las puntualizadas para cromatografía gaseosa. En lo que respecta a la optimización de las condiciones experimentales, en este caso es posible realizar ensayos preliminares orientativos utilizando cromatografía en capa delgada con la misma fase fija que contiene la columna. Por otra parte es necesario tener en cuenta que dado que se utilizan altas presiones, es imprescindible evitar la presencia de partículas que puedan obstruir los caños y la formación de burbujas que puedan deteriorar el relleno de las columnas y que produzcan inestabilidad en la señal del detector. Para evitar las obstrucciones, los solventes y las muestras a inyectar se filtran con membranas de 0,45 a 0,22 µm. Para evitar la formación de burbujas, los equipos de HPLC cuentan con desgasadores de solvente por vacío o por burbujeo con He y, en el caso de no contar con los mismos, se deben desgasar los solventes por medio de ultrasonido o agitación bajo vacío antes de utilizarlos como fase móvil. 16 PARTE EXPERIMENTAL Objetivos • Realizar curvas de calibración para distintos analitos. • Cuantificar el contenido de los mismos en muestras sintéticas y comerciales. Materiales a emplear • Equipo para HPLC marca Thermo Separations compuesto por una bomba binaria modelo Spectra System P2000, una válvula inyectora Reodyne provista de un loop de inyección de 10 µl, un detector de absorción UV-visible de longitud de onda variable modelo Spectra 100 y un integrador registrador modelo Datajet. • Columna analítica marca Alltech econosphere C18 (fase reversa unida químicamente formada por cadenas de hidrocarburo lineal de 18 átomos de carbono) de 150 mm de longitud por 4,6 mm de diámetro interno. • Buffer fosfato 0,025 M; pH = 3; calidad HPLC. • Metanol calidad HPLC. • Patrones de aspartamo, cafeína y ácido benzoico en agua, de diferentes concentraciones. • Muestra incógnita sintética que contiene los tres analitos. • Muestras comerciales que contengan uno o varios de los analitos mencionados (¡mejor los tres!). Ej.: gaseosas light (con aspartamo), gaseosas cola, edulcorantes a base de aspartamo (equalsweet), etc. Todas las gaseosas tienen benzoato como conservante. • Jeringas de 1mL. • Jeringa de 5 mL con soporte para membranas de 0,45 ó 0,22 micrones. Filtros de nylon de 0,45 ó 0,22 micrones. Procedimiento Cuando se desarrolla una técnica analítica por HPLC, normalmente se elige en primer lugar una fase fija adecuada y una fase móvil con una composición tal que sea compatible con la fase fija, que disuelva los componentes a analizar y que permita una buena separación. Luego se optimizan las condiciones de flujo de solvente, cantidad de muestra a inyectar y, en el caso de un detector de absorción como el que se empleará en este caso, se determinará la/s longitud/es de onda de detección. Para el análisis de la mezcla de aspartamo, ácido benzoico y cafeína se utilizará una columna de fase reversa C18 y, como fase móvil, una mezcla formada por 45% de metanol y 55% de buffer fosfato (acuoso) 0,025 M de pH = 3. Se inyectarán 10 µl de cada solución. 17 → Buscar en bibliografía los valores de pKa para los tres compuestos y, en base al estado de ionización de los mismos al pH de trabajo, predecir el orden de elución. A continuación se presentan los espectros de absorción en las condiciones de corrida de aspartamo; ácido benzoico y cafeína. Teniendo en cuenta los espectros, elegir la longitud de onda a utilizar en el análisis. Curva de Calibración y análisis de muestras 1. Se realizará la curva de calibración para cada analito inyectando 1 mL de cada solución patrón mezcla de modo de saturar y limpiar el loop del inyector. Graficar luego el área de pico en función de la concentración y verificar que se está trabajando dentro del ámbito lineal. Idealmente cada punto de la curva de calibración se realizará por duplicado 2. Realizar el análisis cromatográfico de la muestra sintética y de las muestras comerciales. La muestra sintética no requiere tratamiento previo mientras que las comerciales deben agitarse y sonicarse (especialmente las de bebidas gaseosas) y filtrarse antes de ser inyectadas. El filtrado se lleva a cabo con una jeringa de 5 mL unida a un soporte conteniendo un filtro de nylon de 0,45 ó 0,22 micrones; se descarta el primer mL de filtrado y luego se recoge el líquido en un recipiente limpio. 3. Si fuera necesario, diluir la muestra para obtener valores dentro del intervalo de calibración. Para el caso de muestras sólidas, preparar soluciones de concentración adecuada y verificar que el material se disuelva por completo. 4. Para uno de los patrones mezcla utilizados en la curva de calibración, realizar variaciones en la longitud de onda del detector. Discutir la longitud de onda de trabajo. 18 Análisis de los resultados • Una vez obtenidas las curvas de calibración, comparar la sensibilidad del método para la determinación de cada compuesto en las condiciones de trabajo. Relacionar la misma con los espectros de absorción y proponer mejoras en las condiciones experimentales para hacer más eficiente el análisis. • Informar las concentraciones de las muestras-problema con su incertidumbre. Comparar con los valores informados en literatura y con los declarados por los fabricantes de las muestras comerciales. • Proponer un método para evaluar el límite de detección. • Discutir la necesidad de utilizar otros métodos (estándar interno, agregado patrón) para realizar la cuantificación. Consultar “Calibración y Límite de Detección en Técnicas Instrumentales”. • Estimar el limite de detección de la técnica empleando todas las estrategias posibles (discutir previamente este punto con el docente). BIBLIOGRAFÍA General de cromatografía: 1. “Análisis Instrumental”. Skoog y Leary. 2. "Principios de Análisis Instrumental", Skoog, Holler, Nieman. 3. “Métodos Instrumentales de Análisis”. Willard, Merritt y otros. 4. “Cromatografía líquida de alta presión”. H. M. McNair y B. Esquivel. Monografía científica de la OEA. Determinación de cafeína, aspartamo y benzoato por HPLC: 1. K. Helrich (Editor). Official Methods of the Association of Official Analytical Chemistry (AOAC Official Methods of Analysis), 15 th. Ed., Vol. 2, pág. 752 (1990). 2. M. F. Delaney, K. M. Pasko, D. M. Mauro, D. S. Gsell, P. C. Korologos, J. Morawski, L. J. Krolikowski, F. V. Warren Jr. J. Chem Ed., 62, 618 (1985). 3. J. E. DiNunzio. J. Chem Ed., 62, 446 (1985). 4. N. Strohl. J. Chem Ed., 62, 447 (1985). 5. V. L. McDevitt, A. Rodríguez, K. R. Williams. J. Chem Ed., 75, 625 (1998). 19 ESPECTROSCOPIA DE EMISION Y ABSORCION ATOMICA INTRODUCCIÓN Espectroscopía es la medición e interpretación de la radiación electromagnética absorbida, dispersada o emitida por átomos, moléculas u otras especies químicas. Estos fenómenos están asociados con cambios en los estados de energía de las diferentes especies. Por consiguiente, dado que cada especie posee estados energéticos característicos, la espectroscopía puede utilizarse para identificarlas y cuantificarlas. La espectroscopía constituye la base del análisis espectroquímico, en el que la interacción de la radiación electromagnética con la materia se utiliza para obtener información cualitativa y cuantitativa acerca de la composición de una muestra. Dentro del análisis espectroquímico, la espectroscopía atómica estudia la absorción y emisión de la radiación por especies atómicas, iónicas y moleculares libres. Estas especies son generadas y examinadas en un medio gaseoso de alta energía, que constituye una fuente de vaporización-atomización-ionización-excitación. Técnicas de espectroscopía atómica con llama. Cuando una solución es introducida por aspiración en una llama, la mayor parte de los componentes inorgánicos son vaporizados y convertidos en su forma elemental, y los átomos son excitados térmicamente por la energía generada mediante reacciones químicas. En un átomo en su estado de menor energía (estado fundamental), uno de los electrones de valencia correspondiente a un orbital externo es transferido a un estado electrónico de mayor energía. Al retornar al estado fundamental o a uno de menor energía, los átomos pierden su energía de excitación en forma de calor o como radiación electromagnética de longitud de onda característica. → Explique el origen y las características de los espectros atómicos, moleculares y continuos. La absorción de energía térmica generada en una llama seguida por la emisión de toda o parte de esa energía en forma de una línea espectral discreta se denomina emisión atómica. La longitud de onda y la intensidad de las líneas de emisión constituyen la base del análisis cualitativo y cuantitativo por espectrometría de emisión atómica (EEA). Los átomos neutros gaseosos en su estado fundamental pueden también absorber radiación a longitudes de onda específicas, correspondientes a las energías de las transiciones electrónicas en sus orbitales externos. Este fenómeno se denomina absorción atómica. La medición de la magnitud de esa absorción atómica y su aplicación al análisis cuantitativo constituyen la espectrometría de absorción atómica (EEA). La fuente primaria de radiación luminosa es generalmente una lámpara de cátodo hueco del elemento de interés. Los factores principales que determinan la magnitud de la emisión y absorción son: • la distribución energética de niveles excitados. • probabilidades de transición de emisión-absorción • el coeficiente de absorción atómica. • las características de la celda de atomización → Indique las expresiones fundamentales que relacionan las señales medidas en emisión y absorción atómica con la concentración de átomos emisores o absorbentes. 20 INSTRUMENTACIÓN Se utilizará un espectrómetro de absorción / emisión atómica Perkin Elmer (Analyst 200), provisto de un quemador de flujo laminar para llama de aire-acetileno y una lámpara de cátodo hueco de níquel como fuente de radiación. Los componentes básicos de un sistema espectrométrico para la medición de emisión y absorción atómicos son los siguientes: Explique el funcionamiento de cada uno de los componentes, e indique cuáles se utilizan para cada técnica. Identifique las características específicas del instrumento que utilizará en la práctica. Selección de las condiciones de operación Los procesos que sufre el analito en la celda de atomización son esencialmente comunes a ambas metodologías, con lo cual en ambos casos es necesario optimizar: • la selección del tipo y condiciones de la llama (relación oxidante/combustible) • la región de observación en la llama como así también • el paso de banda espectral del monocromador empleado para la selección de la longitud de onda • la velocidad de aspiración de la solución • los parámetros del sistema de detección y lectura y en el caso de absorción atómica • la intensidad de corriente de la lámpara de cátodo hueco → Discuta para cada una de las técnicas, la influencia de las variables indicadas sobre la sensibilidad, la relación señal - ruido, la presencia de interferencias espectrales y el límite de detección. Interferencias 21 11Interferencia es el efecto de un concomitante presente en la muestra sobre la señal generada por el analito. La presencia de un interferente conduce en todos los casos a un error sistemático. Existe una interferencia cuando el resultado de la medición sobre una dada solución de muestra difiere del obtenido con la misma concentración de analito en la misma combinación química y solvente, pero en ausencia del interferente. En EEA y EAA con llama se producen esencialmente las mismas interferencias, aunque con magnitudes diferentes. Pueden clasificarse en cuatro grupos: 1. Espectrales, incluyendo efectos de emisión o absorción de fondo. 2. Físicas, asociadas con el transporte y dispersión de la muestra en la llama. 3. Químicas, relacionadas con la vaporización del soluto. 4. De ionización, relacionadas con la variación de la concentración de átomos neutros emisores o absorbentes en la llama provocada por el fenómeno de ionización térmica. Explique de qué manera son afectadas por las interferencias espectrales las mediciones en EEA y EAA con llama. Discuta procedimientos para corrección por fondo espectral. Indique cuáles son aplicables al instrumento que utilizará en la práctica. Calibración Como todas las técnicas instrumentales, la espectrometría de llama es una técnica relativa. Por consiguiente, es necesario establecer experimentalmente una curva (o función matemática) que relacione la señal analítica obtenida con la concentración del elemento analito en las soluciones a analizar. En el caso más directo, la concentración de una solución incógnita se obtiene por interpolación gráfica a partir de una curva de señal (A o IE) vs. c , obtenida con varios estándares (patrones) adecuadamente espaciados, que cubren el ámbito de concentraciones requerido. → Discuta las características de las curvas de calibración obtenidas en emisión y absorción. Defina sensibilidad y límite de detección. En la mayoría de los casos es necesario que la composición de las soluciones patrón sea similar a la de la muestra a analizar. En el caso de soluciones muestras muy complejas, en las que la presencia de elementos concomitantes puede afectar la respuesta obtenida para el analito, existen otros procedimientos de calibración, tales como el método de agregado patrón de analito (simple o múltiple). 11En términos estadísticos, el principal inconveniente es que se trata de un método de extrapolación; por lo tanto, menos preciso que los de interpolación. Cualquier variación en la pendiente de la recta por errores indeterminados se traduce en una variación apreciable en el resultado. En este sentido, influye el tamaño de la adición, por lo que hay que procurar que el tramo incierto no sea demasiado grande. Por otra parte, el método puede ser difícil de automatizar, y suele necesitar cantidades de muestra mayores que otros. → Describa otros procedimientos para calibración y discuta las posibles fuentes de error. PARTE EXPERIMENTAL 22 El primer día cada grupo debe traer un protocolo de trabajo de las 2 prácticas a realizar. En el mismo debe diseñar cada curva de calibración y/o preparación de muestra correspondiente. Cuenta con matraces de diversos tamaños (25 – 250 mL) y pipetas automaticas (20 µl – 10 mL). El volumen para las mediciones es de 100 mL. ESPECTROSCOPIA DE ABSORCION ATOMICA Determinación de Níquel en una muestra de acero Objetivos Evaluar la importancia de la selección de la longitud de onda y las posibles correcciones del fondo en espectroscopia de absorción atómica. Aplicar esta técnica a la determinación de níquel en una aleación metálica. Preparación de la muestra. Pesar exactamente alrededor de 0,1g de muestra (realizar el tratamiento por duplicado), añadir 5 mL de HCl (c) y 2,5 mL de HNO3 (c) y calentar suavemente sobre tela metálica hasta disolución completa. Dejar enfriar y trasvasar a un matraz aforado de 100,0 mL. completando hasta el enrase con agua destilada. Pipetear 10,00 mL de esta solución y diluir a 100,0 mL en un matraz aforado. Realizar en paralelo un blanco de reactivos (¿cuál es?). Calibración Preparar estándares para calibración conteniendo respectivamente 0, 1, 2, 4, 5, 6, 7, 8 y 10 ppm de Ni en una solución base que contenga 0,5 % (v/v) de HCl y 0,25 % (v/v) de HNO3. Dispone de un patrón de 1000 ppm de niquel y preparará 100mL de solución en todos los casos. Procedimiento 1) Con la ayuda del personal docente y el manual de instrucciones del espectrómetro seleccionar las condiciones generales de trabajo para la determinación de níquel (longitud de onda, ancho de ranura, corrección de fondo). 2) Utilizando las soluciones patrones obtener curvas de calibración analítica (absorbancia vs concentración) a dos longitudes de onda (232 nm y 341,5 nm), para la longitud de onda menor realizarlo con y sin “corrección de fondo”. ¿Qué es? ¿Por qué no se hace con la de 341 nm la corrección de fondo? 3) Medir la absorbancia para la solución de la muestra y calcular el valor de concentración por interpolación a partir de la curva analítica. Tratamiento de resultados A. Informar un único valor de níquel en la muestra. En el anexo, calcular la concentración de níquel en la aleación metálica utilizando todos los gráficos de calibración obtenidos (cuatro en total). Comparar críticamente. Expresar los resultados con sus correspondientes límites de confianza. B. Estimar el limite de detección de la técnica empleando todas las estrategias posibles (discutir previamente este punto con el docente). ESPECTROSCOPIA DE EMISION ATOMICA CON LLAMA 23 Primera Parte: Determinación de Calcio en una muestra de agua mineral Objetivos Comparar dos métodos de calibración para la determinación de calcio en muestras de agua mineral comercial. Evaluar la influencia del agregado de agentes liberadores y buffers de ionización en el mecanismo de atomización de calcio en una llama de aire/acetileno. Calibración Se compararán dos métodos de calibración: curva de calibración estándar con patrones acuosos y método de agregado patrón de analito. A. Preparar soluciones patrón de calcio de concentraciones 2, 4, 6, 8 10, 15 y mg/l a partir de una solución estándar de 1000 mg Ca/l. En todos los casos preparar 100 mL de solución. B. Preparar una solución diluyendo adecuadamente la muestra de agua mineral comercial de tal manera que caiga en el ámbito lineal de la curva de calibrado. Aproximademente 5 ppm C. Repita la preparación de esta solución (dilución de la muestra), con el agregado de nitrato de lantano (2000 mg/l) pero para un volumen final de 25mL. D. Teniendo en cuenta el ámbito de linealidad y la concentración de calcio en la muestra (ver en el envase que traerán) diseñar una curva de agregado patrón con un mínimo de cinco puntos teniendo en cuenta de no salirse del ámbito lineal visto anteriormente. Procedimiento 1) Con la ayuda del personal docente y el manual de instrucciones del espectrómetro, utilizar una de las soluciones patrón (10 mg Ca/L) para seleccionar las condiciones generales de trabajo para la determinación de calcio (longitud de onda, ancho de ranura). Asimismo optimizar la relación combustible-comburente de la llama y la altura de observación. 2) Una vez seleccionados los parámetros óptimos medir la intensidad de emisión para todas las soluciones. Tratamiento de resultados - Informar un único valor de calcio en la muestra. En el anexo, calcular la concentración de calcio en el agua mineral por ambas metodologías (curva de calibración externa y método de agregado patrón). Comparar críticamente. Expresar los resultados con sus correspondientes límites de confianza. - Comparar los resultados obtenidos en ausencia y presencia nitrato de lantano. Justificar la diferencia si la hubiera. Estimar el limite de detección de la técnica empleando todas las estrategias posibles (discutir previamente este punto con el docente). 24 IMAGING DE pH EN DOS DIMENSIONES: APLICACIÓN A MONITOREO DE SISTEMAS EN FLUJO OBJETIVOS Utilizar un indicador acido-base para mapear el pH en una superficie en forma ultrarrápida (nanosegundos), utilizando toma de datos con una cámara digital convencional. INTRODUCCION El Calcón es una substancia utilizada normalmente como indicador complejométrico. El hecho de poseer hidroxilos fenólicos permite también utilizarlo como indicador acido-base con pKa cercano a 7. Figura 1. Estructura del Calcón. Este indicador posee un absorción máxima alrededor de los 515 nm (verde) en su forma ácida que se corre a 610-640 nm (rojo) al deprotonarse en medio básico. Los pKa de los hidroxilos fenólicos son 7.40 y 13.50. Los espectros de absorción entre pH 3 y 11 se muestran en la figura 2. Figura 2: Espectros del Calcón entre pH=3 y pH=11. 25 Setup experimental Por un capilar circula una solución conteniendo un complejo de Rutenio que es fotoactivo, y el indicador Calcón. El flujo es continuo, de la misma forma en que lo hace en un sistema de análisis por inyección en flujo (FIA). En un punto del capilar, un pulso de laser de 405 nm provoca la ruptura fotoquímica del complejo de rutenio el cual libera butilamina al medio, basificándolo y aumentando el pH de 4 a 10 aproximadamente.1 Esta variación in situ de pH provoca los cambios colorimétricos en el indicador Calcón, lo cuales van a ser medidos. El detector no es de los usados habitualmente (UV-Vis, IR, FID, o MS), sino que se utilizó una cámara fotográfica digital para tomar imágenes. Dichas imágenes corresponden a microfotografías del tubo capilar, en la zona de la irradiación del laser. Para ello se coloca el objetivo del microscopio en la zona a examinar, el cual está conectado a la cámara fotográfica. El esquema del sistema completo se muestra en la figura 3. Para más detalles experimentales y de la técnica utilizada, leer el trabajo original.1 Figura 3. A) entrada de solución. B) dirección del flujo. C) descarte. D) objetivo del microscopio. E) Laser 405nm enfocado sobre el capilar. Consideraciones de la adquisición de datos de este “detector” Para poder medir el pH en todos los puntos a la vez, se utilizó una cámara fotográfica digital. Las cámaras digitales convencionales poseen un sensor de semiconductor (CCD o CMOS) capaz de leer la cantidad de luz en forma independiente en cada uno de los puntos que componen la imagen. El número de celdas de CCD o CMOS que lo componen determina la resolución en megapixels. Valores de hasta 15 Mp son habituales en las cámaras digitales actuales. 26 Si bien existen cámaras monocromáticas para usos especiales (microscopías, astronomía, etc.) las cámaras comerciales tienen la matriz de CCD dividida en los 3 colores primarios que detecta el ojo humano: rojo, verde y azul. Este sistema se conoce como RGB por sus iniciales en inglés y su distribución es casi siempre la misma, la cual se conoce como “Bayer”. En una matriz de filtros Bayer hay 2 pixeles verdes por cada uno rojo o azul, como se ve en la figura 4. Figura 4. Filtro tipo Bayer, y la respuesta de cada uno de los filtros. Una vez tomada la imagen, el firmware contenido en la máquina digital calcula 3 imágenes completas para eliminar el pattern Bayer. De esta forma se obtienen las 3 imágenes R, G y B, las cuales se combinan para dar la imagen color. Normalmente esta imágen color se comprime y el usuario obtiene una fotografía en formato Jpeg. Como se vé claramente, las respuestas de los filtros son muy anchas, de más de 100 nm. Esto implica que la Ley de Lambert-Beer, que sólo es válida al utilizar luz monocromática, no se cumplirá si se pretende medir absorbancia directamente con este dispositivo. Afortunadamente, existen formas de minimizar este error. Una de ellas es iluminar la muestra con luz laser (estrictamente monocromática) o de LED (20 nm de ancho). Las tomas de las imágenes que se utilizarán en esta práctica son realizadas con iluminación de LEDs verde (535 nm) y rojo (635 nm), cuyos espectros se observan en la figura 5. Figura 5 – Espectros de emisión de los LEDs utilizados para la toma de datos. Debido a esta iluminación, la imágen correspondiente al color azul practicamente no existe, mientras que las imágenes verde y roja corresponden a la luz transmitida por el indicador a 535 nm y 635 nm. Para la manipulacion matemática de las imágenes se utilizará el programa de acceso libre ImageJ.2 27 PARTE EXPERIMENTAL Toma de imágenes Las imágenes del capilar del sistema FIA fueron adquiridos utilizando un microscopio invertido Nikon TS-100 con iluminación de campo brillante. El sistema de iluminación del microscopio fue reemplazado con un sistema de 2-LED, con irradiación de luz verde (535 ± 16 nm FWHM) y rojo (635 ± 11 nm FWHM) y se coloca en el mismo punto focal a través de un espejo dicroico. Las imágenes fueron tomadas con una cámara digital compacta (Casio Exilim EX-FC100) que hizo foco a través de un ocular normal y un adaptador personalizado y ajustado a sensibilidad ISO 100. Los análisis de imágenes y videos se realizaron con el software público acceso ImageJ.2 Las imágenes se toman a 30 cuadros/segundo y se graban en un archivo de formato AVI. Posee cuadros de 1280x720 pixeles, de los cuales los primeros corresponden a la medición, y los últimos a un campo fijo a pH bajo para tener una línea de base útil para el cálculo. Carga de las imágenes Para abrir el archivo de video basta con correr el programa ImageJ y arrastrar el video hasta la parte superior del software. Una vez cargado el video es conveniente cortar (crop) la imágen de forma de no tener que utilizar las partes que no corresponden a la medición y hacer los cálculos más rápidos. Una vez obtenido el stack de imágenes con el que se va a trabajar es conveniente grabarlos como imágenes TIFF de 32 bits. De esta forma no se pierde información por redondeo al efectuar operaciones matemáticas. Nota: En la página de la materia cuenta con el video y el manual del Image J. Al terminar llevarse los datos en un pendrive y borrarlos de la computadora, ya que ocupan cientos de MB. Tratamiento de las imágenes Considere que cada pixel de cada imagen es un valor de intensidad de luz completamente independiente de los demás (esto es una aproximación válida). De esta forma, es en principio posible, conociendo la absorbancia [-log(I/I0)] en cada punto a ambas longitudes de onda, obtener la fracción molar de la especie deprotonada y protonada del indicador, y por lo tanto el pH. Sin embargo, es importante considerar algunas características de la medición: 1) no se cuenta con un “blanco” que permita determinar en forma directa las absorbancias 2) la absorbancia es una función no lineal de la intensidad de luz 3) puede existir alinealidad en la toma de datos por la cámara y la posterior digitalización en formato jpg. Este último problema (3) no puede solucionarse salvo conociendo la respuesta de la cámara a intensidades de luz patrón, por lo cual se considerará respuesta lineal (esta aproximación es correcta dentro del 5% en las condiciones de la medición). Las dificultades provenientes de (1) y (2) pueden ser subsanadas a través de manejo matemático de las imágenes (lo cual será explicado por el docente). Al conocer los espectros de absorción de las especies básica y ácida, es posible reconstituir las absorbancias correspondientes a las zonas ácidas (flujo sin irradiar, intensidad del pixel rojo) y básicas (irradiación máxima, intensidad del pixel verde) correspondientes a cerca del 0 y 100% de fracción molar de las especies del indicador. De esta forma, y utilizando las funciones matemáticas 28 del ImageJ (ver manual) es posible calcular el pH correspondiente a cada punto. Considere el pKa = 7.40. Informe En el informe deberá incluir necesariamente: - Una foto de cada uno de los cuadros “notables” del video, indicando los pH correspondientes. - Gráficos de línea obtenidos de esas fotos indicando pH vs. longitud, a tiempo fijo. - Gráficos de línea obtenidos para una zona importante fija de la medición, en los que se grafiquen pH vs. tiempo. - Análisis y discusión de la forma geométrica del perfil obtenido en la pluma de pH del sistema de flujo. - Análisis del error en pH en zonas de la imagen, e informe maneras de minimizar ese error. - Utilizando una imágen en escala de fraccion molar, estimar el limite de detección de la técnica empleando las funciones estadísticas del ImageJ. Ejemplos: BIBLIOGRAFIA 1. Oscar Filevich, Guillermo Carrone, Victoria Andino Pavlovsky y Roberto Etchenique, Anal. Chem, 2012, 84, 5618−5624 2. Abramoff, M. D.; Magalhaes, P. J.; Ram, S. J. Biophotonics Int. 2004, 7, 36−42. 29 CIFRAS DE MÉRITO EN ESPECTROMETRÍAS MOLECULARES FLUORESCENCIA MOLECULAR Y ABSORCIÓN MOLECULAR. DETERMINACION DE RIBOFLAVINA EN UNA PREPARACION FARMACEUTICA OBJETIVOS • Aplicación de la espectrometría de emisión molecular (fluorescencia). • Realizar la determinación de riboflavina (vitamina B2) en solución, utilizando dos métodos de cuantificación distintos (absorción molecular y fluorescencia molecular). Estimar la incertidumbre de las determinaciones. • Evaluar las cifras de mérito de cada una de las metodologías. Estimar el límite de cuantificación (concentración límite). • A partir del análisis de las curvas de calibración, analizar los factores instrumentales y de medición que afectan, entre otras cosas, al ruido de la medición. INTRODUCCIÓN Principios A temperatura ambiente, la riboflavina (vitamina B2) puede ser promovida a varios niveles de vibración excitados, en el primer estado excitado electrónico. La transición está en la energía correspondiente al UV y parte del espectro azul (350-450 nm). La existencia de esta transición se corresponde con la presencia de una banda de absorción en la región azul del espectro visible, la cual puede ser utilizada para cuantificar la concentración de riboflavina mediante la espectroscopia de absorción molecular. El proceso de absorción se produce en aproximadamente el 10-15 femtosegundos. La absorción es seguida por la relajación vibracional, luego de la cual se establece una distribución de la población térmica de moléculas excitadas de tal manera que se acumulan en los niveles más bajos de vibración del estado electrónico excitado. Esta relajación vibracional concluye dentro de los 10 picosegundos posteriores a la absorción. Las moléculas electrónicamente excitadas "relajan" a los distintos niveles de vibración del estado fundamental a través de diversos procesos. En las substancias no fluorescentes, el mecanismo dominante es la relajación vibracional, que disipa en forma de calor la energía del estado excitado. Algunas substancias (la riboflavina entre ellas), pueden relajar la energía a través de un proceso de emisión espontánea en el rango de las decenas a centenas de nanosegundos. Este tipo de emisión se denomina fluorescencia. El espectro de fluorescencia está desplazado hacia longitudes de onda más largas que el de absorción. (¿Por qué?). En el caso de la riboflavina, la emisión fluorescente se observa de color verde. La espectroscopia de fluorescencia es ampliamente utilizada en los análisis biomédicos, ya que tiene varias ventajas con respecto a la espectrometría de absorción. Algunas de estas ventajas son: 1 - Es más selectiva, ya que sólo un pequeño subconjunto de las moléculas que absorben son fluorescentes, (lo que también reduce la posibilidad de aplicación). 2 - Es más sensible, ya que el detector sólo detecta la radiación de fluorescencia, entre “cero” y un cierto 30 valor, mientras que en la medición de la absorción el detector debe detectar pequeñas diferencias entre la absorción del blanco y de la muestra. A bajas concentraciones, se puede disminuir el límite de detección de la determinación por fluorescencia colectando más luz o utilizando una fuente de luz de mayor potencia. En cambio, en la absorciometría, la única posibilidad es elevar la sensibilidad del detector, para que permita diferenciar entre pequeñas variaciones, lo cual es mucho más difícil de lograr. Por supuesto, la relación de mérito entre el método de fluorescencia y el de absorción dependerá de varios factores, entre ellos: a) la eficiencia cuántica de fluorescencia del analito en cuestión. b) su absortividad molar c) la precisión (en miliabsorbancias) del espectrofotómetro disponible d) la sensibilidad del fluorómetro e) el posible quenching de la fluorescencia por diversos factores En esta práctica se compararán ambos métodos, utilizando un mismo instrumento con configuración de espectrofotómetro y fluorímetro. En una segunda etapa, se estudiarán las condiciones de medición de espectrofluorometría con dicho instrumento (espectrofluorómetro de matriz de diodos) PARTE EXPERIMENTAL A - Muestra incógnita Se deberá informar el contenido de riboflavina en una muestra provista por el docente. Para ello cada grupo preparará una dilución 1:10 de la misma según: medirá exactamente 5.00 ml y llevará a 50 ml en matraz, por agregado de aproximadamente 40 ml de solución de ácido acético 20 mM (¿por qué no agua?). Enrase el matraz con piseta. A esta solución la llamaremos Solución 1. Cada grupo debe determinar la concentración de riboflavina en cada una de las “Soluciones 1” preparadas por cada uno de los grupos de su turno (entre cuatro y seis) y calcular la concentración de riboflavina en la solución original. En particular, para las determinaciones por fluorescencia se deberá efectuar una dilución 1:10 adicional de las “Soluciones 1”. A esta nueva dilución la llamaremos “Solución 2”. B - Determinación por espectrometría de absorción molecular Recibirá los patrones para realizar la curva de calibración de parte de los docentes (NO tiene que prepararlos, solo medirlos). Los mismos se realizaron con una muestra de riboflavina pura recién disuelta, en ácido acético 20 mM. Se prepararon 6 diluciones entre 0 y 62.5µM de riboflavina a partir de una solución madre de 125 µM. Aproximadamente, se utilizaran las siguientes concentraciones: 0, 6.25, 12.5, 25, 50, 62.5 µM, aunque pueden ser otras. Tome los espectros de absorción de estas soluciones, elija convenientemente el tiempo de integración, manteniendo el AVERAGE y el BOXCAR WIDTH en 10. Sálvelos para trabajar posteriormente con ellos, en el modo TAB DELIMITED, NO HEADER. Elija la longitud de onda adecuada (máximo de absorbancia), y calcule la recta de calibración correspondiente, utilizando regresión por cuadrados mínimos. Anote en el cuaderno TODOS los parámetros instrumentales conque realizó esta medición. 31 - Determinación espectrofotométrica: Determine la absorbancia de las “Soluciones 1” preparadas por cada uno de los grupos, en la longitud de onda elegida. Repita el mismo procedimiento para cada una de ellas. Determine la concentración molar de riboflavina (en µM) y estime la incertidumbre de dos maneras distintas: 1) Dispersión de los replicados de muestra (soluciones madre) Desviación estándar de la muestra s = (Σ (x i – x m)2 / (N-1)) 1/2 t para 95% de confianza, y N-2 grados de libertad. 2) a partir de la curva de calibración estimada por cuadrados mínimos t para 95% de confianza, y N-2 grados de libertad. Debe informar dos resultados de concentración en la muestra original, con sus respectivas incertidumbres. Compare las incertidumbres obtenidas en cada resultado (la que resulta de considerar las diluciones (Soluciones 1) preparadas por cada grupo, con la que resulta de considerar su propia curva de calibración). C - Determinación por espectrometría de emisión por fluorescencia molecular Recibirá los patrones para medir la curva de calibración de parte de los docentes (NO tiene que prepararlos, solo medirlos). Los mismos se realizaron con una muestra de riboflavina pura recién disuelta en ácido acético 20 mM. Se prepararon 6 diluciones entre 0 y 12.5 µM de riboflavina. (Note que se usan concentraciones menores. ¿Por qué?) Coloque el espectrofotómetro en configuración de medición de fluorescencia, teniendo en cuenta que debe mover la llave selectora hasta que encienda el LED violeta que se encuentra a 90 grados de la entrada del espectrofotómetro. Esta configuración minimiza la luz espúrea del LED que entra directamente al equipo. Con el programa de toma de datos en modo SCOPE y utilizando el patrón mas concentrado (12.5 µM) elija convenientemente el tiempo de integración, manteniendo el AVERAGE y el BOXCAR WIDTH en 10. El máximo del espectro de emisión no debe saturar. No es importante si satura o no la luz espurea de excitación. Con la excitación apagada o introduciendo una cubeta que bloquee completamente la excitación tome un espectro “DARK” que corresponde al cero de fluorescencia. Este espectro debe tomarse con los mismos 32 parámetros (integracion, boxcar, average) con que tomó el espectro de máxima concentración. Cliquee el ícono correspondiente para dejar grabado ese espectro. A partir de ahora no deberá cambiar los parámetros de medición. Tome los espectros de emisión de las soluciones patrón. Sálvelos para trabajar posteriormente con ellos, en el modo TAB DELIMITED, NO HEADER. Anote en su cuaderno los parámetros con que tomó los espectros. Elija una longitud de onda adecuada y calcule la recta de calibración (borrador preliminar) correspondiente, utilizando regresión por cuadrados mínimos Tome 10 ml con pipeta aforada de la muestra incógnita del matraz conteniendo “Solución 1” y lleve a 100 ml en un matraz aforado con ácido acético 20 mM, enrasando al final con la piseta. Cada grupo realizará esta dilución, a la que llamamos “Solución 2”, y todos los grupos realizarán la determinación de todas las “Soluciones 2” de su turno. Mezcle la solución invirtiendo el matraz un mínimo de 5 veces. Sobre estas soluciones “diluidas” tome las medidas de fluorescencia de la misma forma que hizo la curva de calibración, a la misma longitud de onda. Determine la concentración molar de riboflavina en la muestra original, y estime la incertidumbre, utilizando las mismas dos estimaciones consideradas en el caso de absorción molecular. Debe informar dos resultados de concentración en la muestra original, con sus respectivas incertidumbres. Compare las incertidumbres obtenidas en cada resultado (la que resulta de considerar las diluciones (Soluciones 2) preparadas por cada grupo, con la que resulta de considerar su propia curva de calibración). Estimación del límite de detección y del límite de cuantificación en la determinación por absorción molecular y por fluorescencia molecular Desde el gráfico de la recta de calibración pueden calcularse los valores de LD y LC. En particular, utilizaremos las hipérbolas de confianza según: El LD es la concentración correspondiente al punto de la curva de calibración que resulta de la intersección horizontal de la hipérbola superior con el eje de coordenadas (señal). El LC es la concentración correspondiente al punto de la hipérbola inferior que resulta de la intersección horizontal de la banda de predicción superior con el eje de coordenadas (señal). En la bibliografía se discute si deben considerarse las bandas de confianza o las bandas de predicción (que consideran los replicados de cada medición). Teniendo en cuanta que no hemos realizado replicados de los puntos de la curva de calibración, utilizaremos las bandas de confianza. Considerando entonces la intersección con la banda de predicción inferior, podemos calcular al LC (C Q) como se indica en el Manual de Incertidumbre y Cifras de Mérito del curso. Compare al menos 3 diferentes estimaciones del valor del Límite de cuantificación, y discuta sobre las discrepancias que encuentra entre los valores. EVALUACIÓN DEL RUIDO EN FLUORESCENCIA MOLECULAR Con el programa de toma de datos Spectra Suite en modo “emisión minus dark” (recuerde medir correctamente el “dark” antes de las muestras), elija convenientemente el tiempo de integración, manteniendo el SCANS AVERAGE y el BOXCAR WIDTH en 1. Tome los espectros de emisión en las mismas soluciones de la curva de calibración que midió en el ítem anterior. Sálvelos para trabajar posteriormente con ellos, en el modo TAB DELIMITED. De esta forma son fáciles de importar por cualquier programa de manejo de datos, como Excel, Origin, SigmaPlot o Matlab. 33 Calcule nuevamente la curva de calibración en dos situaciones distintas: 1) máximo de intensidad de fluorescencia 2) área del espectro en un intervalo que contenga los valores de espectro por sobre el 20% del máximo, y una despreciable cantidad de la luz de excitación. Compare el Límite de Cuantificación en las siguientes condiciones: a) SCANS AVERAGE= 1 BOXCAR WIDTH = 1 b) SCANS AVERAGE= 1 BOXCAR WIDTH = 9 c) SCANS AVERAGE=16 BOXCAR WIDTH = 1 Guía Informe El informe contendrá los siguientes resultados y análisis: 1) Molaridad de la solución de Riboflavina analizada, según las dos metodologías (AM y FM). Discusión de la incertidumbre calculada con los dos métodos de evaluación, en cada metodología. 2) Análisis de los límites de cuantificación de Riboflavina en AM y FM, y según los tres métodos de evaluación propuestos. 3) Análisis del ruido de los espectros de fluorescencia obtenidos en diferentes condiciones y relación con la robustez de los datos informados y con el límite de cuantificación. BIBLIOGRAFIA - Análisis Químico Cuantitativo, Daniel C. Harris, 2da edición (o posterior). Editorial Reverté. http://omlc.ogi.edu/spectra/PhotochemCAD/html/riboflavin.html 34 GUIA DE PROBLEMAS CROMATOGRAFIA GASEOSA 1. Enumerar los tipos de detectores habitualmente empleados y evaluar su ámbito de aplicación. 2. Discutir la elección del gas portador a utilizar en relación al tipo de detector con que se cuenta. ¿Qué características debe reunir el gas elegido? 3. Discutir la aplicación de la ecuación de Van Deemter para modificar la eficiencia de un análisis. 4. ¿De qué manera influyen las condiciones instrumentales en el cromatograma? 5. ¿Qué condiciones debe reunir un componente para ser utilizado como estándar interno? 6 . Discutir las ventajas y desventajas de cada uno de los métodos de cuantificación utilizados. 7. Discutir la conveniencia de considerar como “señal” la altura o el área del pico obtenido como respuesta de la muestra. 8. Una muestra contiene las sustancias X e Y. Se desea determinar cuantitativamente X utilizando el método del estándar interno. Se probaron los compuestos 1, 2 y 3 como posibles estándares internos, obteniéndose el siguiente cromatograma: a. ¿Cuál elegiría como estándar y qué condiciones variaría en el cromatograma para su uso? b. Una vez conseguida las condiciones óptimas, ¿cómo procede para la determinación? 9. Ejercicio de Parcial Utilizando las siguientes condiciones: flujo 1 ml/min; L= 30 mts, se realizó la separación de 6 compuestos (ver tablas y figura) Tabla 1 # Compuesto Puntos de ebullición (°C) tr (min) Isoterma 100º 1 2 3 4 5 6 Octano Nonano Fenol Decano Naftaleno Dodecano 125 151 182 174 219 216 3,2 4,2 5,7 6,5 9,1 9,7 35 Figura 1 – Cromatograma de los compuestos listados en la tabla según su # Tabla 2 Muestra Área Octano Área Nonano Área Fenol Área Decano Área Naftaleno Área Dodecano 1% 2% 3% 4% 5% 6% 72404 130952 264199 304840 355134 452120 81454,9 147321 297224 342944,5 399526 508634,5 77468 63938 86091 75001 70304 82231 90505 163690 330249 381049 443918 565149 85742 70153 95879 83645 78338 92479 68488 135924 226711 301952 389958 466062 Incógnita 180453 203010 60503 225567 67670 246001 a) Justifique la elución de los compuestos y explique por qué algunos no cumplen el orden de los puntos de ebullición. ¿Cómo obtendría el t0? b) Si se quisieran cuantificar TODOS los hidrocarburos, ¿qué compuesto de la tabla 1 elegiría como patrón interno? Calcule N, k`, α y R para el compuesto elegido respecto de su hidrocarburo más próximos. ¿Con que criterio hizo la elección? Asuma W = 0.05 min para todos y t0= 1,5 min. c) Utilizando la misma metodología que en el TP se obtuvieron los datos de la Tabla 2, cuantifique la cantidad de Nonano presente en una muestra incógnita. 10. Ejercicio de Parcial El mentol es ampliamente utilizado en la preparación de formulaciones para la tos e inhalaciones nasales. A continuación se presenta un protocolo para la determinación de mentol en el aceite esencial de menta por cromatografía gaseosa con columna HP-5, 30 m, 0,32 mm x 0,25 mm (5 %fenilmetilpolisiloxano) y corrida isotérmica a 110 °C. a. Pesar 10 mg de mentol patrón y llevar a volumen con etanol en un matraz aforado de 10,00 mL (Solución A) b. Pesar 10 mg de estandar interno (octanol) y llevar a volumen con etanol en un matraz aforado de 10,00 mL (Solución B) c. Tomar 1,00 mL de Solución A y 1,00 mL de Solución B y llevar a volumen con etanol en un matraz aforado de 5,00 ml (Solución C) d. Pesar 10 mg de aceite esencial y llevar a volumen con etanol en un matraz aforado de 10,00 ml (Solución D) e. Tomar 1,00 mL de la Solución D y 1,00 mL de la Solución B y llevar a volumen con etanol en un matraz aforado de 5,00 ml (Solución M) f. Inyectar 1 µL de la Solución C en el cromatógrafo. Repetir para 1 µL de la Solución M 36 En base a los datos de la siguiente tabla, determine la concentración de mentol en el aceite esencial expresando el resultado en % m/m Solución Area Mentol Area Octanol C 95.000 98.000 M 100.000 120.000 Si en las condiciones de la corrida la resolución entre el mentol y el octanol es de 1,5, qué condiciones variaría para mejorarla? 11) Problema Parcial En una planta industrial se cuenta con un CGL equipado con una columna capilar polar para realizar controles de calidad sobre los productos obtenidos en la misma. En particular, se utiliza para cuantificar la concentración de alcohol (etanol) en vodka. Los patrones y la muestra incógnita -diluida 1:10- se inyectan (1 µL) a 50ºC en el CGL, el gas carrier utilizado es N2. Los resultados obtenidos son: Concentración Solución Componente (m/m) Area Metanol 1.00 42009 Patrón 1 Etanol 1.00 60900 Metanol 1.00 42504 Patrón 2 Etanol 2.00 117587 Metanol 1.00 44600 Patrón 3 Etanol 3.00 210172 Metanol 1.00 44979 Patrón 4 Etanol 4.00 290570 Metanol 1.00 41283 Patrón 5 Etanol 5.00 320105 Metanol 1.00 41300 Vodka (1:10) Etanol -255570 Las regresiones lineales de dos curvas de calibración (y=mx+b) posibles son: m=1.63, b=-0.26, Sx=0.01, t=12.7 m= 69139.3, b= -7551.1, Sx=0.08, t=12.7 La medición de la muestra incógnita se ha realizado por triplicado y el valor informado en la tabla indica el promedio de los valores obtenidos. a)Determine a partir de los datos cuál es la concentración de etanol en la muestra de vodka sin diluir. Recuerde que debe informarlo junto con el error correspondiente. (Utilice la curva de calibración que considere adecuada indicando a qué tipo de calibración pertenece –puede usar los datos de la tabla para identificar a qué tipo de calibración pertenece cada regresión lineal-.) b) Indique qué tipo de detector puede ser utilizado para esta muestra. Justifique c) ¿Cuál de los compuestos tiene el menor tiempo de retención para este tipo de columna? ¿Porqué? d) Indique qué parámetro relevante de la cromatografía (k,α,R) utilizaría usted para determinar las condiciones óptimas de separación de ambos componentes. ¿ Qué condiciones puede variar -o ha variado en el laboratorio- para obtener un cromatograma con la separación de picos deseada ? 37 HPLC 1. 2. Cual(es) es(son) los criterios de elución para cada una de las siguientes cromatografías: a.- líquida con columna con fase estacionaria inversa b.- iónica con columna de intercambio aniónico. Los parabenos se utilizan en la industria cosmética como conservantes. Su fórmula general es dada en la figura 1, donde R es un grupo alquílico. Un fabricante de columnas de fase reversa C18 recomienda las siguientes condiciones de uso: Fase móvil: 40/60 Acetonitrilo/agua, buffer fosfato pH 7.0 con los resultados observados en la Figura 1. i.- Cuando se intentó hacer un cambio en la fase móvil usando 40/60 Acetonitrilo/agua, buffer pH 4.0 se obtuvo el cromatograma de la figura 2. Qué efecto tiene en la corrida una disminución del pH. ii.- Como reduciría el tiempo de la corrida si tiene la posibilidad de aplicar un gradiente de solventes. 3. Ejercicio de Parcial El siguiente cromatograma corresponde a una mezcla de cuatro compuestos separados sobre una columna de 25 cm longitud x 4.6 mm diámetro interno. La fase móvil utilizada fue methanol:etanol 80:20 y el flujo 0.9 mL/min. Si un compuesto no retenido en la columna tarda 1 minuto en salir, calcular: a) El tiempo de retención, tiempo de retención corregido y el factor de retención para todos los analitos. b) El factor de separación de la columna para todos los analitos. c) El número de platos teóricos y la altura equivalente de plato teórico de la columna, calculados a partir del analito mas retenido. d) La resolución de la columna para los analitos. 4. Ejercicio de Parcial a) Se ha llevado a cabo un estudio de determinación de 4 antidepresivos tricíclicos por HPLC utilizando tres columnas de polaridades diferentes: C8, con grupos fenilo terminales y con grupos ciano terminales. A continuación se muestran las estructuras de los cuatro compuestos estudiados, las constantes de acidez correspondientes al grupo mas básico, y los tres cromatogramas obtenidos en las mismas condiciones de flujo y composición de fase móvil. 38 Nortriptilina pKa: 10,1 Doxepina 9,76 Amitriptilina 9,4 Trimipramina 9,42 A partir de los cromatogramas anteriores, asigne cada uno de ellos a cada tipo de columna utilizada, justificando adecuada y brevemente su respuesta. b) Se estudia la separación de dos compuestos A y B por HPLC utilizando una columna C-18. La fase móvil es una fase binaria compuesta por agua y acetonitrilo. Se ha encontrado una relación lineal entre el logaritmo del factor de retención y el % de acetonitrilo en la mezcla utilizada. Se obtuvieron una serie de cromatogramas a composiciones variables, obteniéndose dos rectas cuyas ecuaciones son: Para el compuesto A: log KA = - 6.075x10-3 (%CH3CN) + 1.3283 Para el compuesto B: log KB = - 0.0107 (%CH3CN) + 1.5235 i) ii) Encuentre la composición de la fase binaria para la cual el factor de selectividad = 1. Teniendo en cuenta su respuesta anterior, ¿cómo debería ser la composición de la fase móvil para obtener una mejor separación? c) Un fabricante presenta los resultados de la separación de dos compuestos A y B utilizando una nueva columna, para la cual no muestra los cromatogramas a distintas composiciones de fase móvil, sino una tabla de valores de k, α y R. i) ¿Qué es y qué representa cada uno de los parámetros mencionados? ii) ¿Cuál de ellos es el mejor para determinar la eficiencia de separación de esta columna? Justifique adecuadamente su respuesta. ESPECTROSCOPIA DE EMISION ATOMICA 39 1) Discuta la influencia de la temperatura de la llama sobre las señales obtenidas en EEA y en EAA. 2) Analice ventajas y desventajas de la utilización de una llama de óxido nitroso-acetileno frente a una llama de aire-acetileno en el trabajo práctico. Podría utilizar una llama de aire-butano? 3) Analice la influencia de cantidades sub-, sobre- y estequiométricas de fosfato, con respecto a Ca, en la muestra a analizar. 4) ¿Utilizaría el procedimiento del trabajo práctico para determinar Ca en agua de mar? Discuta. 5) Discuta la conveniencia de utilizar el método del agregado patrón frente al método de la curva de calibración en determinaciones por espectrometría de emisión por llama. 6) Discuta brevemente cómo espera que varíe la sensibilidad en la determinación de Ca en agua mineral cuando se aumenta la ranura del monocromador. 7) Sugiera posibles explicaciones para las siguientes observaciones efectuadas cuando se determina Ba en solución acuosa por espectrometría de emisión en llama: i) La emisión a la longitud de onda analítica, para bajas concentraciones, es despreciable si la llama es de aire-acetileno, pero apreciable cuando la llama es de óxido nitrosoacetileno. ii) El Ba presenta líneas de emisión a 553.55 nm y 455.40 nm. La intensidad observada en una llama laminar de óxido nitroso-acetileno para una solución de 10 m/ml es apreciable a ambas longitudes de onda. Pero si se agregan a la solución cantidades crecientes de cesio, la emisión de la primera línea se incrementa hasta alcanzar un valor constante, en tanto que la emisión de la segunda disminuye. iii) Con la llama de óxido nitroso-acetileno, al variar la altura de observación sobre el quemador de 4 mm a 20 mm, la intensidad de emisión disminuye abruptamente, pero vuelve a aumentar al incrementar ligeramente el flujo de acetileno. 8) Para determinar Ca por emisión atómica, 5 g. de una muestra de origen geológico se disuelven en un volumen dado de una mezcla ácida, se filtra y se disuelve a 100 ml. Para estimar la concentración residual de Ca se prepara un blanco de reactivos con un volumen de mezcla ácida igual al doble del anterior, diluido a 100 ml. Muestra y blanco se analizan por el método del agregado patrón, con los siguientes resultados (las concentraciones indicadas corresponden a mg/ml de Ca agregado en cada caso, a alícuotas de 20 ml, despreciando la variación de volumen). Calcular el % de Ca en la muestra original. MUESTRA BLANCO Agregado (mg/ml) 0 0.1 0.2 0.3 Apromedio 0.100 0.160 0.220 0.290 Agregado (mg/ml) 0 0.02 0.04 0.06 Apromedio 0.100 0.170 0.290 0.380 9) En la llama de N2O - C2H2 el Ba(I) presenta una línea de emisión a 553.55 nm que se superpone a una banda molecular correspondiente a CaOH. El espectro observado para 0.5 mg Ba/mL en presencia de concentraciones elevadas de Ca es el siguiente: Cómo determinaría en este caso la relación de intensidades línea fondo? Indique qué parámetros instrumentales podrían variarse para mejorar ese valor. Justifique su respuesta. 10) Ejercicio de Parcial 40 El cloruro de rubidio (RbCl) es un excelente biomarcador no invasivo. Este compuesto se disuelve bien en agua y es fácilmente asimilado por el organismo. Una vez en el cuerpo, el ion Rb+ reemplaza el ion potasio (K+) en los tejidos debido a su similitud química (pertenece al mismo grupo de la tabla periódica). Un ejemplo de esto es el uso de 82RbCl, isótopo radiactivo, para evaluar la perfusión (llevar oxígeno y nutrientes a un tejido por medio de la sangre) del músculo del corazón, el miocardio. El rubidio tiene intensas líneas de emisión a 420.1 nm y 780.0 nm. Para hacer una determinación cuantitativa del analito se procede a hacer una calibración externa con un estándar certificado del mismo en cada una de esas líneas de emisión. Los resultados obtenidos se detallan en la siguiente tabla. El valor informado para cada patrón es el obtenido de integrar durante 10 segundos la señal para cada solución. Línea 780,0 nm Línea 420,1 nm ppm Rb Intensidad ppm Rb Intensidad 0 47 0 5 0 44 0 45 0 46 0 30 0 45 0 16 0 43 0 54 0,2 61 10 155 1,0 124 20,0 280 2,0 206 25,0 602 10,0 845 35,0 468 25,0 2045 40,0 531 40,0 2429 60,0 780 50,0 2534 80,0 1030 60,0 2606 100,0 1055 1. Se quiere cuantificar una solución de rubidio. Se toman dos alícuotas de la muestra y se mide su emisión a las dos longitudes de onda obteniéndose los siguientes resultados. Cuantifique. Explique. Alícuota 1 2 780,0 nm - Intensidad 1361 1369 420,1 nm - Intensidad 238 234 Ayuda: Haga un boceto de ambas rectas. 2. El cloruro de rubio presenta polimorfismo dependiendo de las condiciones de cristalización. Se quiere determinar el porcentaje de rubidio en un mineral para ello se toman dos porciones del residuo (Masa Mineral), se disuelven completamente en agua milliQ, se lleva a volumen en matraz de 50 mL y se mide su emisión con un equipo similar al utilizado en la práctica. Cuantifique. Porción 1 2 Masa Mineral / g 1,0483 1,14538 780,0 nm - Intensidad 2419 2483 420,1 nm - Intensidad 515 575 3. Explique las siguientes observaciones a. Cuando se realiza la misma calibración en el rango de bajas concentraciones agregando KCl o CsCl respectivamente se obtienen las siguientes curvas de 41 calibración. Asigne cuál curva corresponde a KCl y cuál a CsCl. Explique brevemente. Ayuda: Mr Cl = 35.35 gr/mol ; Mr K = 39.09 gr/mol ; Mr Rb = 85.46 gr/mol ; Mr Cs = 132.91 gr/mol b. Cuando se realiza la misma experiencia que el punto 1, pero cambiando las condiciones de la llama (FA: flujo acetileno ; FO: flujo oxidante) se observa el siguiente fenómeno. Explíquelo e indique en que llama tiene mayor temperatura. c. A continuación se observa un grafico de Intensidad (c.a) vs. longitud de onda (nm). 420.1 nm 415.1 nm 425.1 nm 42 I. II. III. Que tipo de interferencia espectra para este sistema. ¿Como la remedia? El analista percibio la interferencia y para contrarrestarla realizó una curva de agregado patron, para lo cual tomó 5 matraces de 50 ml y agregó 1 ml de una solucion de 1000 ppm de cloruro de rubidio en cada uno. Como resultado de la cuantificacion obtuvo 25 ppm de Rb. ¿Es correcto lo realizado? ¿Ud informaria ese valor? Si se quisiera medir la misma muestra por absorción atómica (con la lámpara de catodo hueco correspondiente), tendría sentido medir utilizando corrección de fondo con una lámpara de deuterio?? En las 2 lineas?? 11) Ejercicio de Parcial Se desea cuantificar el contenido de Ca en 30 muestras de orina y para ello se compararon tres metodologías distintas en una misma muestra. En todas ellas se utilizaron los siguientes parámetros: Línea 422,7 nm. Ranura 0,7 µm. Llama aire-acetileno estequiométrica. a) Absorción atómica con corrección de fondo espectral Curva de Calibración con 5 soluciones entre 0 y 3 mg Ca/l, diluciones con agua milliQ. A = (0.047±0.001) . (Ca) + 0.001 r2= 0.9997 con (Ca) en mg/l Dilución 1:100 de la muestra A = 0.031 b) Emisión atómica en llama b.1 Curva de Calibración con 5 soluciones entre 0 y 12 mg Ca/l, diluciones con solución 0.5% de LaCl3 y 0.1% de KCl Ie = (377±9) . (Ca) + 28 r2= 0.998 con (Ca) en mg/l Dilución 1:25 de la muestra Ie = 1350 b.2 Agregados patrón de 2, 4 y 6 mg Ca/ l en la dilución final, sobre 2 ml de muestra llevados a 50 ml con solución 0.5% de LaCl3 y 0.1% de KCl Ie = (380±10) . (Ca) + 1390 r= 0.9996 con (Ca) en mg/l a) Seleccione una de las tres metodologías para analizar las 30 muestras y justifique su elección. b) Cuantifique el contenido de Ca en la muestra analizada c) Explique, si fuera posible, cómo podría mejorar la exactitud de las dos metodologías que no eligió. 12) Ejercicio de Parcial Se desea cuantificar Cr en acetona para lo cual se prueban los siguientes cuatro protocolos experimentales: a) Se preparan patrones de cromo en solución acuosa cubriendo un rango de concentraciones entre 2 y 8 ppm, se selecciona como longitud de onda de trabajo 357,9 nm y se emplea una llama estequiométrica de aire-acetileno, una intensidad de lámpara de 5 mA y un ancho de ranura de 0,5 nm. Los parámetros de mérito obtenidos fueron: A = a*[Cr] + b con a = 0,078 +/- 0,001 ppm-1 b = 0,090 +/- 0,009 r2 = 0,9975 Una dilución 1/5 de la muestra en agua destilada arrojó una lectura de absorbancia de 0,610 43 b) Se realizó una curva de agregado patrón con las mismas condiciones experimentales sobre alícuotas diluídas 1/5 de la muestra en agua destilada, obteniéndose las siguientes figuras de mérito: A = a*[Cr] agregado + b con a = 0,086 +/- 0,001 ppm-1 b = 0,608 +/- 0,009 r2 = 0,9895 c) Se preparan patrones de cromo en solución acuosa cubriendo un rango de concentraciones entre 10 y 40 ppm, se selecciona como longitud de onda de trabajo 425,4 nm y se emplea una llama estequiométrica de aire-acetileno, una intensidad de lámpara de 5 mA y un ancho de ranura de 0,5 nm. Los parámetros de mérito obtenidos fueron: A = a*[Cr] + b con a = 0,025+/- 0,002 ppm-1 b = 0,000 +/- 0,004 r2 = 0,9901 Una dilución 3/5 de la muestra en agua destilada arrojó una lectura de absorbancia de 0,508 y la muestra tal cual un valor de 0,924 d) Se realizó una curva de agregado patrón con las mismas condiciones experimentales del protocolo 3 sobre alícuotas diluídas 3/5 de la muestra en agua destilada, obteniéndose las siguientes figuras de mérito: A = a*[Cr] agregado + b con a = 0,0282 +/- 0,002 ppm-1 b = 0,510 +/- 0,004 r2 = 0,9877 Se pide: a) Cuantifique el contenido de cromo en la muestra a través de cada uno de los protocolos experimentales tanto para las muestras diluídas como para la muestra sin diluir b) Justifique las diferencias entre cada uno de los valores encontrados c) Indique cual de los protocolos seleccionaría para analizar nuevas muestras d) ¿Qué variables experimentales cambiaría para realizar la determinación por emisión atómica? Justifique e) Calcule el límite de detección y cuantificación de la metodología utilizada. 13) Ejercicio de Parcial Verdadero o Falso. Justifique su respuesta a) El tiempo muerto depende principalmente de la temperatura de trabajo. b) Si se obtiene una buena recta por calibracion externa, no es necesario usar un patron interno. c) Para determinar los parametros cromatograficos se utiliza tr, ya que es el que mide el tiempo total de residencia del analito en la columna. 14) Ejercicio de Parcial El piruvato de sodio (Fórmula molecular: C3H3NaO3; Mr = 110,04 g/mol) se utiliza comúnmente para la preparación de soluciones reguladores en medios de cultivo. Para determinar la pureza de la sal el laboratorio de control de calidad en el que usted es analista utiliza como metodología analítica Espectrometría de Emisión Atómica. Para ello se desarrolla el siguiente protocolo: se disuelven por quintuplicado 0,5000 g de muestra en HNO3 al 1 % y se trasvasan a matraces aforados de 250,0 mL completándose el volumen con agua Milli-Q. De estas soluciones se toman alícuotas de 0,10 mL, se transfieren a matraces aforados de 100,0 mL y se completa el volumen. Estas soluciones se rotulan como M1, M2, M3, M4 y M5, respectivamente. Por otra parte se preparan patrones de Na de 0,1, 0,2, 0,4, 0,6, 0,8 y 1,0 ppm. 44 Los patrones de calibrado y las muestras se miden en un espectrómetro de emisión atómica con llama de aire/acetileno a 589,0 nm usando un paso de banda de 0,1 nm (el sodio tiene un doblete de emisión a 589,6 nm). Los parámetros de regresión y la curva de calibración se muestran al final. Paralelamente se realiza un blanco metodológico obteniéndose los siguientes valores de intensidad de emisión: 0, 1, 2, 3, 1, 2, 0,2, 1 y 2. a) Explique a qué se deben las diferencias entre los dos sets de parámetros de regresión b) ¿Cuál es la mínima concentración de sodio detectable por esta metodología? c) Las intensidades de emisión de las muestras fueron de 73, 74, 72, 71 y 75, respectivamente. De acuerdo a las especificaciones técnicas del producto el contenido mínimo de piruvato de sodio debe ser de 99,5 +/- 0,5. ¿Qué decisión adoptaría con respecto a la aprobación o rechazo de la partida? (el valor del t de Student para n-1 grados de libertad y 95 % de confianza es de 2,78) d) ¿ Cómo espera que se modifiquen las figuras de mérito del análisis si utiliza un ancho de banda de 1 nm?. e) En caso de que decidiera implementar una metodología basada en absorción atómica, discuta si los siguientes argumentos resultarían convincentes para que el laboratorio adquiera una lámpara de cátodo hueco de Na, i) las medidas por absorción atómica son más exactas y sensibles, ii) las medidas por absorción atómica son más precisas 200 Intensidad 150 y = 162.41x + 2.0877 R² = 0.9679 100 50 0 0 0.5 1 Na, ppm 1.5 Fórmula auxiliar: X +/- t.s / n1/2 Parámetros de regresión sin considerar los patrones de 0,1 y 1,0 ppm: y = 178,1x + 0,5005 R2 = 0,9984 ESPECTROSCOPIA ABSORCION ATOMICA 1) Discuta las ventajas y desventajas que reporta trabajar con intensidades de lámpara más altas que las recomendadas por el fabricante. 2) ¿Por qué si la lámpara de cátodo hueco es una fuente de líneas es necesario utilizar un monocromador? 3) ¿Es posible utilizar la instrumentación de espectrofotometría molecular para realizar medidas en absorción atómica? 4) En la determinación de níquel el ancho de banda espectral juega un rol fundamental cuando se trabaja a una longitud de onda de 232 nm, ya que si se pasa de un ancho de banda de 0,1 nm a 0,5 nm la sensibilidad y el ámbito dinámico lineal disminuyen. Sin embargo, cuando se determina Cu a una longitud de onda de 324,7 nm, el mismo cambio de ancho de banda no 45 produce prácticamente variación de sensibilidad ni de los ámbitos de linealidad. Discuta las causas de este comportamiento diferente. 5) Discuta la conveniencia de corregir el fondo espectral en las mediciones realizadas en el trabajo práctico. 6) Discuta críticamente las siguientes afirmaciones: a) La absorción atómica es más sensible que la emisión atómica. b) Modular electrónicamente la lámpara de cátodo hueco permite corregir la absorción inespecífica de los componentes de la llama. c) En absorción atómica el ámbito de linealidad depende del tipo de llama utilizada. 7) Una muestra de NaCl contiene Cu (II) en una concentración de aproximadamente 0,003 %. Diseñe un protocolo de análisis que permita la cuantificación del cobre indicando: masas, volúmenes, reactivos a utilizar, preparación de la muestra y patrones, procedimiento general y cálculos. 8) Ejercicio de Parcial El agua de consumo que recorre cañerías de plomo puede presentar niveles peligrosos de este metal. La ingesta continua de aguas contaminadas con plomo provoca intoxicaciones por elevada plumbemia (nivel de plomo en sangre). El valor límite de plumbemia que considera a los individuos fuera de peligro es de 50 µg/L. Se tomaron muestras de sangre (M1 a M4) a cuatro habitantes de un edificio muy antiguo del centro porteño, las cuales fueron sometidas a análisis por espectrometría de absorción atómica de llama. Para esto se ensayaron diferentes métodos utilizando una lámpara de cátodo hueco del metal, una ranura estrecha, y se realizó una digestión ácida que trajo aparejada la dilución al medio en los cuatro casos. El blanco de reactivos fue realizado y restado antes de confeccionar toda curva de calibración. Se utilizaron dos líneas de plomo diferentes, λ1 = 217 nm y λ2 = 283,3 nm. La curva de calibración para todos los casos fue externa, de cinco patrones entre 50 y 500 µg/L. Las variaciones entre métodos fueron: A) λ1 sin corrección de fondo. B) λ1 con corrección de fondo con lámpara de deuterio. C) λ2 con corrección de fondo con lámpara de deuterio. Los resultados obtenidos fueron: Método Regresión Señal M1 Señal M2 Señal M3 Señal M4 A y=0,003x+0,200 0,520 0,295 0,987 0,241 B y=0,003x+0,001 0,307 0,079 0,751 0,031 C y=0,0015x+0,001 0,144 0,037 0,383 0,013 Nota: considerar el error en los parámetros de regresión en la última cifra indicada. Los valores de x se deben tomar en µg/L. a) ¿qué método seleccionarías y por qué? b) ¿se pueden identificar interferencias de matriz? ¿de qué tipo? c) Cuantificá plumbemia para todos los individuos. ¿qué le recomendarías a cada uno? 9) Ejercicio de Parcial Se desea cuantificar el contenido de aluminio en una muestra de Titanio Grado 5 (contiene aluminio y vanadio como aleantes), utilizando espectrometría de absorción atómica. Se mide la señal de absorbancia a 309.3 nm., utilizando llama de N20- C2H2 y corrección de fondo con lámpara de deuterio. 46 Para ello se pesan 380 mg., se disuelven en una mezcla ácida y se llevan a 250 ml en matraz (Solución A). Se toma una alícuota de 10 ml y se lleva a 100 ml previo agregado de solución de Cs y La (concentración final de ambos 1000 ppm). La Absorbancia medida es 0.079. Se realizan las siguientes curvas: a) curva de calibración sin agregado de Cs y La. b) curva de calibración con agregado de Cs y La (concentración final de ambos 1000 ppm). c) método de agregado patrón con 10 ml de Solución A) y Volumen final 100 ml, con agregado de Cs y La. (ppm) Curva 1 (A) Curva 2 (A) Agregado patrón (ppm) Curva 3 (A) 0 5 0.002 0.021 0.003 0.052 0 0.079 10 0.074 0.099 5 0.132 20 0.162 0.214 10 0.171 30 0.239 0.295 20 0.261 50 0.402 0.511 30 50 0.348 0.435 a) Justifique cuál de las curvas utilizará para realizar la cuantificación, explicando detalladamente las diferencias observadas entre ellas. b) Estime el ámbito dinámico lineal en las condiciones de operación elegidas. c) Calcule el % de Al en la muestra. 10) Ejercicio de Parcial Se quiere determinar la concentración de Zn en una muestra de orina utilizando un espectrómetro de absorción atómica sin corrección de absorción de fondo. Se supone que la muestra de orina puede presentar interferencias espectrales y químicas, las mismas de la orina sintética. Para ello se realizan los siguientes Protocolos de análisis: 1) Curva de Calibración a 213.9 nm (ranura 0.7 nm), con patrones preparados en matriz de orina sintética, intervalo 0.1 a 1.2 mg Zn/ litro: A = 0.25 C (mg/l) + 0.060 r = 0.9996 Medición de la muestra sin diluir A = 0.209 2) Curva de agregado patrón a 213.9 nm (ranura 0.7 nm), con agregado de solución patrón de Zn a alícuotas de una dilución 1:5 de la muestra: A = 0.21 C (mg/l) + 0.042 r = 0.09997 a) Justifique el valor de la ordenada al origen en la curva de calibración b) Justifique si puede emplear únicamente la Curva de agregado patrón para realizar el análisis. c) Calcule la concentración de Zn en la muestra. ESPECTROSCOPIAS OPTICAS (segundo parcial) Los 4 ejercicios son de parciales 1) Indique V o F y justifique a) Un instrumento con una fuente de luz monocromatica capaz de emitir más intensidad de luz aumentará la sensibilidad de una absorciometria y de una fluorimetria. b) Se mide un espectro de fluorescencia en una solución acuosa de riboflavina y se obtiene un archivo con el valor de la emisión cada 0.1 nm. Si se hace un promedio de ventana con un ancho de 10 mediciones se obtiene un espectro con menos ruido sin que cambie apreciablemente la forma del espectro obtenido. c) en el espectro obtenido en el punto (b) mediante el promedio de ventana de 10 mediciones, el ruido se reduce aproximadamente a la decima parte. d) para medir valores de absorbancia por medio de una cámara fotográfica digital no es necesario utilizar fuentes de luz de alta monocromaticidad, ya que la misma cámara contiene filtros capaces de discriminar las diversas longitudes de onda. 47 (el siguiente punto NO ES un Verdadero o Falso) e) se toman durante un día fotografías aéreas de una laguna donde navegan continuamente lanchas y botes para determinar mediante el análisis de la reflexión a ciertas longitudes de onda la presencia de contaminantes y de esa forma hacer una mapa 2D del estado de la laguna. Se puede tomar un fotograma cada 1 minuto. Explique en menos de 10 renglones cual es el primer paso que efectuaría en ImageJ (o cualquier programa similar) para obtener una imagen inicial con el menor ruido posible, incluyendo eliminar las embarcaciones (que también son ruido). 2) Para determinar la concentración de un analito, se marca este con una sonda fluorescente cuyo fluoroforo es Rodamina 123. Los espectros de absorcion y de emision normalizados se ven en la figura superior. (Max abs = 45000 M-1cm-1 a 513 nm, Max emi. a 533 nm) Para efectuar la determinacion se utilizará un especrofluorómetro de matriz de diodos con excitacion a LED. Se dispone de 3 LEDS a 405, 473 y 510 nm cuyos espectros se muestran en la figura inferior. Las cubetas tienen 1cm de paso óptico, de 4 caras pulidas y medición en la parte central de la misma, a 90 grados respecto de la excitación. Elija el LED mas adecuado para medicion de fluorescencia. Justifique brevemente. a) Explique brevemente la razon por la cual el espectro 1.2 de emision está corrido a la derecha y tiene una 1 forma similar al de absorcion. b) Sabiendo que el detector tiene un rango de 0 a 4096 0.8 unidades, y un error σ=43 u.a. constante en todo el 0.6 rango, y que una concentración de analito de 1 µM 0.4 produce una salida de 3040 u.a.,.estime los límites 0.2 de detección y de cuantificación para dicho analito. 0 c) Sabiendo que se puede reducir la sensibilidad del 350 450 550 650 detector, y que éste es lineal en todas las condiciones de medida ¿ A que concentración estima Ud. que las medidas de fluorescencia 1 comenzarán a ser 0.5 alineales ? Justifique en menos de 5 renglones. 0 350 450 550 650 3) Es necesario determinar la concentración de Hg2+ en una muestra incógnita. Para ello se dispone de dos métodos alternativos: uno absorciométrico y uno fluorométrico. En el método de absorción, 1 mL de la muestra se mezcla con 2 mL de un reactivo colorimétrico que genera un complejo coloreado de estequiometría 1:1 que tiene el espectro que se indica en la figura 1. Al medir la muestra se encuentra que la absorbancia es 0.195 a 435 nm en una cubeta de 1cm de paso óptico. Cuando se lo utiliza del modo adecuado con este método analítico, la medición espectrofotométrica tiene una desviación estándar experimental que se ve en la figura 2. En el método fluorométrico, 1 mL de la muestra incógnita se mezcla con 2mL de otro reactivo que produce un compuesto fluorescente. Al medir repetidamente la emisión en un espectrofluorómetro a 566 nm se miden 7620 u.a. (promedio) con un desvío estándar σf = 27. a) Estime el límite de detección y de cuantificación para Hg2+ con el método de absorbancia. b) Calcule la concentración de Hg2+ en la muestra. c) Estime cuál de los dos métodos presenta menos incertidumbre para determinar Hg2+ en las condiciones indicadas. Justifique su respuesta en base a calculos simples. 48 (Suponga linealidad en el ámbito de aplicación, y ausencia total de interferencias y desviaciones a la ley de Lambert Beer) 8000 0.001 Figura 1 7000 1 0.0008 sigma (absorbancia) 6000 epsilon / M-1cm-1 Figura 2 0.0009 5000 4000 3000 2000 0.0007 0.0006 0.0005 0.0004 0.0003 0.0002 1000 0 250 0.0001 0 350 450 550 650 0 lambda / nm 0.1 0.2 0.3 0.4 0.5 0.6 0.7 absorbancia 4) Indicar Verdadero o Falso, justificando la respuesta. a) Una imagen de 600 x 200 pixeles contiene la información sobre la concentración de un fármaco en la superficie de un comprimido. Cuando se toma un cuadrado de 10x10 pixeles, la medición da 3.2 ± 0.6 mg/cm2 (95%). Si se quiere reducir la incertidumbre a ± 0.3 mg/cm2, basta con aumentar la superficie de la medición a 20x20 pixeles. b) En una medición de emisión fluorescente, es necesario colocar en la cubeta una solucion de absorbancia baja, debido a que a absorbancias altas el corrimiento de Stokes se reduce, impidiendo separar correctamente la luz de excitacion de la de emisión. La emision de un compuesto se utiliza para determinar su concentracion. La forma del espectro de emision de concentraciones crecientes 0, 1, 2, 4,10, 15 nM se muestra en la figura 1 y los valores numericos se muestran en la tabla. c / nM 0 1 2 4 10 15 Integración 480-580nm -11117 167632 318521 656125 1617965 2420588 medida a 537 nm -111.6 714.2 2336.2 4012.3 9757.5 14212.0 • Protocolo 1: integracion de la emision entre 480 y 480 580 680 780 580 nm y [u.a.] = 161668 c[nM] - 611.75 r2 = 0.9999 • Protocolo 2: medicion a la lambda maxima de emision, a 537 nm, y[u.a.] = 954.07 c[nM] + 65.048 r2 = 0.998 La medicion de 10 blancos conteniendo solo la matriz dio a 537 nm dio promedio = -52 unidades con σ = 302 unidades. La misma medicion integrando entre 480 y 580 nm dio un promedio = -4508 unidades con σ = 3900 unidades. a) Justifique en menos de 10 renglones cual de los métodos (medicion en el maximo a 537 nm vs integracion entre 480 y 580nm) le parece mas adecuado. b) Estime el limite de cuantificacion para ambos metodos. Explique en no mas de 5 renglones la diferencia que encuentra entre estas cifras de merito. c) una impureza fluorescente que se forma durante el tratamiento tiene un espectro de emision entre 540 y 600 nm. Indique en este caso como modificaria el protocolo de medicion. 49 d) la absortividad molar maxima de la substancia medida es ε456 = 16000 M-1cm-1. Explique en base a este parámetro por qué su medición por fluorescencia presenta un menor limite de deteccion y cuantificacion. ¿existen condiciones instrumentales en que la medicion de absorbancia seria mejor? 50