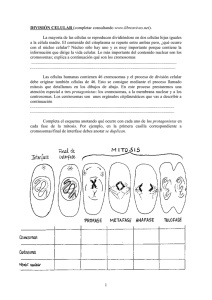
Licenciatura en Genética CATEDRA DE CITOGENETICA GENERAL
Anuncio

Licenciatura en Genética CATEDRA DE CITOGENETICA GENERAL GUIA DE TRABAJOS PRACTICOS Coordinación General María C. Pastori DOCENTES CITOGENETICA GENERAL Alberto S. Fenocchio María C. Pastori Héctor Alberto Roncati Jacqueline D. Caffetti 2012 I- Cariotipo e identificación de cromosomas Objetivos ESTABLECER las diferencias entre cariograma e idiograma. ADQUIRIR las habilidades necesarias para el armado de un cariotipo. RECONOCER la utilidad del cariotipo como herramienta en citotaxonomia, evolución y diagnóstico. Cariograma, Cariotipo e Idiograma Una vez fotografiada una metafase, los cromosomas deben ser recortados del papel y ordenados en pares de homólogos, formando un cariograma; esta ordenación se hace de acuerdo con el tamaño del cromosoma, la posición del centrómero y otras características como la existencia de constricciones secundarias, satélites, etc. (Ford, 1961). La denominación de cariotipo, en forma estricta, se da al complemento cromosómico de una especie. El idiograma, corresponde a la representación gráfica del cariotipo en pares de homólogos o del set haploide. Sin embargo, el uso de la palabra cariotipo se ha generalizado para indicar un complemento cromosómico ordenado en pares, sea a partir de una fotografía o de un dibujo con cámara clara de una metafase, teniendo en cuenta también los datos obtenidos de la medición de los cromosomas y por lo tanto la relación de brazos o el índice centromérico de cierto número de metafases (por lo regular, de 20 a 30). 1- Cariotipo. Montaje del Cariotipo El método más útil consiste en emplear cinta adhesiva por sus dos caras. Se colocan varias tiras de dicha cinta, de unos 2 cm de ancho, sobre un papel plastificado, para evitar que las fibras del papel sean arrancadas por el adhesivo. Ordenar los cromosomas pegando sólo una punta de los mismos; una vez apareados los mismos y ordenado el cariotipo, pegar completamente los cromosomas sobre la cinta. Arrancar la cinta adhesiva del papel y recortar los cromosomas de modo que su cara posterior quede recubierta por la capa adhesiva. Para el armado definitivo del cariotipo y/o cariograma pegar los cromosomas en forma definitiva sobre una cartulina, también plastificada, de modo que sea fácil despegar los cromosomas si fuera necesario, para variar el orden de los cromosomas. No es obligatorio recortar los cromosomas siguiendo su contorno, ésto lleva un tiempo considerable. Basta recortarlos en forma de rectángulos o cuadrados, según su tamaño. Las consideraciones realizadas en el párrafo anterior hacen referencia al armado de un cariotipo de manera manual, hasta podría señalarse “artesanal”. Dado el avance tecnológico y la disponibilidad de medios informatizados que posibilitan el uso de programas tales como Corel Draw o Photo Shop, el 2 armado de un cariotipo se realiza directamente utilizando esta herramienta. Para ello debemos partir de metafases digitalizadas. Con dichos programas los cromosomas se recortan y ordenan por orden decreciente de tamaño o bien con las mediciones correspondientes, se agrupan por pares de homólogos siempre que el material lo permita. De esta manera los cariogramas se organizan muy rapidamente y además se mejoran las imperfecciones difíciles de corregir con el armado manual. 2- Identificación de cromosomas La identificación individual de cromosomas es, en muchos casos, problemática; ello se debe a la similitud morfológica entre varios pares cromosómicos. Esto es especialmente cierto si se refiere a cromosomas profásicos o anafásicos. Así, en el cariotipo humano, por combinación de datos puramente morfológicos, de coloración convencional y autorradiográficos, es posible identificar en forma individual solamente 15 de los 23 pares que constituyen el complemento cromosómico. Si se emplean criterios morfológicos exclusivamente, sólo 6 de los 23 pares son identificables. A los datos morfológicos y autorradiográficos pueden sumarse los resultados obtenidos de la medición de los cromosomas, aunque la evaluación de dichos datos es también problemática. Los patrones más frecuentemente empleados son la relación de brazos, obtenida de dividir la longitud del brazo largo por la del brazo corto; y el índice centromérico, que viene dado por el resultado de dividir la longitud del brazo corto por la longitud total del cromosoma, multiplicado por cien. 2.1- RELACION DE BRAZOS= LONGITUD DEL BRAZO LARGO LONGITUD DEL BRAZO CORTO 2.2- I.C = LONGITUD BRAZO CORTO x 100 LONGITUD TOTAL CROMOSOMA Los problemas más importantes, en relación a la validez de estos datos son: 1- El grado de contracción de los cromosomas no siempre es el mismo, lo que resulta en mediciones distintas, no sólo entre homólogos, sino también entre los distintos brazos de un mismo cromosoma. 2- En una metafase, los cromosomas situados en la periferia tienden a estar más desespiralizados y por lo tanto ser más largos que sus homólogos en el centro de la metafase. 3- Si las mediciones se efectúan sobre fotografías, los cromosomas son fotografiados a distintos aumentos según su distancia del eje focal. 4- La determinación de la posición del centrómero, o de la zona terminal de los segmentos eucromáticos de los brazos del cromosoma es imposible en muchos casos. 3 5- Al microscopio, los cromosomas no muestran un borde definido, sino que aparecen rodeados de un halo de difracción por lo que la localización de los puntos terminales de los brazos cromosómicos es difícil. 6- La medición de zonas cromosómicas es pues, también dificil. 7- La medición de zonas tan pequeñas como los brazos cortos de un cromosoma de los D o G humanos es prácticamente imposible. Sin embargo, dichas mediciones llevadas a cabo en un número suficiente de células por un mismo investigador, perrmiten la confección de un idiograma que puede considerarse representativo de una especie determinada. Lo que no puede pretenderse, como lo indica Patau (1965) es diferenciar basándose únicamente en mediciones (Lejeune, 1964), pares cromosómicos muy parecidos morfológicamente, y que no presenten patrones de marcado radiactivo diferenciales o de otro tipo de bandeos cromosómicos. Tal es el caso del grupo C en el hombre. Para la identificación de cromosomas es necesario basarse en datos morfológicos, autorradiográficos, de medición de cromosomas en metafase de la mayor calidad posible y principalmente en bandeos cromosómicos (C, G, R). Metafases con superposiciones cromosómicas o con los cromosomas excesivamente contraídos o superpuestos no deben ser empleadas a efectos de identificación. 3-Automatización del análisis citogenético (Tomado de Lacadena, 1996) Implica el análisis mediante un sistema informatizado: computador de imágenes bidimensionales de microscopía óptica. Implica los siguientes pasos: 3.1-Captura de la imagen: luz transmitida a través de la preparación microscópica es enfocada sobre la diana de una cámara electrónica y muestreada sobre una rejilla bidimensional. Valores de intensidad de brillo (pixel) resultantes son almacenados con números en la memoria de la computadora. Los valores pixel y su posición geométrica constituyen los datos básicos para el análisis subsiguiente. 3.2-Segmentación: posibilidad de aislar el conjunto de valores correspondientes a un cromosoma respecto a la de otros objetos del campo y de otros cromosomas. 3.3-Medición: las mediciones de las características de cada región de la imagen tales como tamaño del cromosoma, densidad relativa, eje medio, orientación, polaridad, perfil, centrómero, forma, bandeo, etc. se hacen aplicando fórmulas matemáticas al conjunto de pixeles. 3.4-Clasificación: la clasificación de las regiones de imagen se hace aplicando un tratamiento estadístico al conjunto de mediciones de las características realizadas. 3.5-Modelo: la interpretación del significado de la imagen digitalizada se realiza mediante comparación con un modelo o conjunto de principios generales que 4 predicen cómo cualquier entidad biológica –el cromosoma metafásico en este caso- estará representado en valores pixel. 4-Análisis citogenético molecular: (Tomado de Lacadena, 1996) 4.1Citogenética de flujo: aplicar técnicas de citometría de flujo permite separar y purificar cromosomas específicos en pequeñas pero suficientes cantidades (Gray, 1989). El análisis bivariado de cromosomas (por ej., humanos) en flujo utilizando dos fluorocromos con especificidad de bases A-T y G-C, permite que cromosomas con doble tinción puedan ser separados en citometría de flujo no sólo por su contenido de ADN sino también por la proporción de sus bases (Gray, 1979; Carter, 1974). Una vez aislados, los cromosomas pueden ser analizados citogenéticamente o ser utilizados para construir bibliotecas de ADN específicas de un cromosoma determinado (Krumlauf et al., 1982; Van Dilla and Deaven, 1990; Fantes et al., 1994), o ser preparados para el empleo de pintura cromosómica junto con la técnica de Hibridación in situ con Fluorescencia (FISH). El uso de estas técnicas es sumamemte valioso en la identificación y caracterización de cromosomas individuales tanto en metafase como en interfase como también en la identificación de translocaciones cromosómicas (Lichter et al., 1988; Cremer et al., 1988). 4.2 Análisis electroforético del cariotipo: Cariotipo electroforético es un término introducido por Carle y Olson (1985) y representa la utilización de la técnica de electroforesis en gel en campo pulsante (PFGE) para separar cromosomas completos de organismos unicelulares. Mediante electroforesis convencional en gel de agarosa se puede separar hasta un límite de 20 kb, la PFGE puede llegar hasta un límite de entre 6-12 Mb., incluyendo, por lo tanto, el tamaño de los cromosomas de organismos unicelulares tales como Hongos y Protozoos. Si bien el cariotipo electroforético no es un cariotipo convencional, es de utilidad pues permite aislar e individualizar el ADN de los diferentes cromosomas del organismo. 4.3 Hibridación in situ en la identificación cromosómica: (Tomado de Lacadena, 1996) En sus inicios, las técnicas de Hibridación in situ (ISH) de ácidos nucleicos que fueron introducidas por Ritosa y Spiegelman (1965), Gall and Pardue (1969), Pardue and Gall (1969) servían para localizar sobre los cromosomas información genética correspondiente a ADN o ARN, que se utilizaba como sonda marcada radiactivamente. También se puede utilizar una sonda de localización previamente conocida para identificar el cromosoma o región cromosómica correspondiente en la placa metafásica analizada. ISH, mediante el uso de isótopos radiactivos se aplicó durante las décadas de los setenta y ochenta. El marcaje radiactivo de la sonda fue sustituído por marcado no radiactivo como ser mediante el uso de biotina y digoxigenina, dando lugar a la hibridación in situ con fluorescencia (FISH), (Lichter and Ward, 1990; Lichter 5 and Cremer, 1991; Lichter et al., 1991; Lichter and Ried, 1994). La utilización simultánea de dos sondas marcadas con biotina y digoxigenina permite que la fluorescencia de distintos colores en los cromosomas sirva para identificar y analizar alteraciones cromosómicas en metafase e interfase. Cuando la sonda se obtiene a partir de ADN total de un cromosoma aislado por citometría de flujo (Lichter et al., 1988), se produce lo que se denimina pintura cromosómica (Pinkel et al., 1988). Una innovación a posteiori fue la microdisección de cromosomas utilizando micropipetas que permiten cincelar cromosomas individuales o partes de cromosomas a partir de la preparación citológica y utilizar el material extraído en técnicas de PCR para construir bibliotecas genómicas o pinturas cromosómicas (Ludecke et al., 1989; Meltzer et al., 1992). En plantas, Vega et al., (1995) utilizaron la micromanipulación para aislar cromosomas individuales en plantas y obtener a partir de ellos la pintura cromosómica. Aislaron el isocromosoma 5BL de trigo en células madre de polen en metafase I. La pintura cromosómica (Chromosome painting) puede extenderse a todos los cromosomas procedentes de uno de los parentales en combinaciones híbridas interespecíficas vegetales y sus derivados, pasando a constituir la hibridación in situ genómica (GISH) (Heslop-Harrison et al., 1988; Schwarzacher et al., 1989; Le et al., 1989; Anamthawat-Jónsson et al., 1990) debido a que las sondas utilizadas proceden del ADN genómico de una de las especies parentales. 5- Consideraciones generales para la identificación de cromosomas humanos Los cromosomas humanos son clasificados de acuerdo con su morfología y tamaño en 7 grupos: A, B, C, D, E, F y G. los cromosomas se agrupan en pares y se ubican en orden decreciente de tamaño, numerándose del 1 al 22. Esta forma de agrupación fue consensuada en varias reuniones de estandarización (conferencias de Denver, 1960; Londres, 1963; Chicago, 1966; París, 1971) y además hay un Sistema Internacional de Nomenclatura Citogenética Humana (ISCN, 1981). Este sistema internacional permite describir los patrones de Bandeo de Alta Resolución. GRUPO A: Pares 1, 2 y 3. Son los de mayor tamaño del complemento humano. El par 1 es metacéntrico (M), el 2 sub-metacéntrico (SM) y el par 3 M. GRUPO B: Pares 4 y 5. Cromosomas de tamaño grande, SM. El tamaño de los brazos cortos representa aproximadamente 1/3 del de los brazos largos. Ambos pares son de difícil distinción morfológica. GRUPO C: Pares 6 a 12 y cromosomas X. Son 16 cromosomas en la mujer y 15 en el hombre. Se clasifican todos como SM de tamaño medio. La identificación es dificil por su parecido morfológico. Generalmente los cromosomas del par 6 y el X son los mayores del grupo. 6 GRUPO D: Pares 13 a 15. Son cromosomas A de tamaño medio con satélites visibles en los brazos cortos. Los pares 13 y 14 son de dificil distinción. El par 15 es menor. GRUPO E: Pares 16 a 18. Cromosomas de tamaño medio. El par 16 es SM casi M y el mayor del grupo el par 17 es un SM y el 18 SM de menor tamaño pudiendo presentarse como Acrocéntrico (A). GRUPO F: individual Pares 19 y 22 y cromosomas pequeños y M. Identificación dificil con coloración convencional. GRUPO G: Pares 21, 22 y cromosomas Y. Cuatro cromosomas en la mujer y cinco en el hombre. Son los menores cromosomas del cariotipo y son todos (A). Los pares 21 y 22 presentan satélites en sus brazos cortos. 7 II- Metodologías de obtención de preparaciones cromosómicas En este apartado se abordará el aprendizaje de metodologías que incluyen el manejo de técnicas de obtención de preparaciones cromosómicas directas e indirectas, a partir de las cuales se pueden aplicar metodologías de coloración de distinta índole. 1- Preparaciones cromosómicas directas (Técnica de Aplastado) 1.1- Cromosomas metafásicos en hembras de Ortópteros Objetivos LOGRAR el manejo de la técnica de aplastado (Squash). OBTENER preparaciones cromosómicas directas a partir de gónadas femeninas y ciegos gástricos de Ortópteros. RECONOCER a través del M.O. los tipos cromosómicos que caracterizan al grupo de estudio (cromosomas metafásicos mitóticos tratados con colchicina). Técnica 1- Inyectar colchicina al 0,05% (inyección intraabdominal) hasta que el abdomen se hinche. Dejar actuar alrededor de 6 a 12 hs. 2- Sacrificar al animal con éter sulfúrico y hacer un corte a lo largo del abdomen, a la altura de la unión de los segmentos. 3- Extraer los ovarios (generalmente de color amarillo intenso) y ciegos gástricos. Fijarlos en alcohol metílico-ácido acético glacial en proporción 3:1 (fijador Farmer) (algunas veces es preferible realizar antes de la fijación una leve hipotonía con ClK 0,075M, de 5 a 10 minutos). 4- Tomar una pequeña porción de ovario o ciego gástrico y macerar con una o dos gotas de orceína lacto-propiónica. Colocar el cubreobjetos, dejar actuar el colorante unos minutos y luego hacer squash. 5- Examinar el preparado y si está en buenas condiciones sellar con cera o pegamento para cámara de bicicletas. 6- El preparado puede ser guardado en la heladera, durante unos pocos meses. . 8 1.2- Cromosomas metafásicos en hembras de Hemípteros Objetivos OBTENER preparaciones cromosómicas directas a partir de gónadas femeninas de Pyrrhochoris sp. RECONOCER a través del M.O. los tipos cromosómicos que caracterizan al grupo de estudio (cromosomas mitóticos metafásicos tratados con colchicina). Técnica 1- Incubar las gónadas (ovariolas), luego de sacrificar y disecar al organismo, en una solución de colchicina al 0,05% durante 8 a 10 hs. 2- Fijar las gónadas en fijador Farmer (algunas veces es preferible realizar antes de la fijación una leve hipotonía con ClK 0,075M, de 5 a 10 minutos). 3- Tomar una pequeña porción de ovario y macerar con una o dos gotas de orceína lacto-propiónica. Colocar el cubreobjetos, dejar actuar el colorante unos minutos y luego hacer squash. 4- Examinar el preparado y si está en buenas condiciones, sellar con cera o con pegamento para cámara de bicicletas. 5- El preparado se debe guardar en la heladera. 9 1.3- Cromosomas metafásicos mitóticos en vegetales Objetivos OBTENER preparaciones cromosómicas en células vegetales mediante pretratamiento. EVALUAR el uso del pretratamiento sobre la calidad de los preparados. OBSERVAR metafases y determinar el número diploide. ESTABLECER las características cromosómicas del material estudiado. Técnica 1- Se corta la raíz, se coloca en pretratamiento durante 3 hs. con 8oxiquinoleína 0,002N o en frío a 0 ºC 24 a 72 hs. dependiendo el tiempo, del material. 2- Fijar las raíces de 2 a 24 hs. en alcohol metílico-ácido láctico en proporción 5:1. Pasar luego a conservador: alcohol 70%. 3- Colorear las raíces mediante coloración de Feulgen: a) Hidrolizar con ác. clorhídrico 1N durante 8’ a 60 ºC en baño termostático. b) Colocar las raíces en reactivo de Schiff durante 20’ en cámara oscura. 4- Cortar una porción de tejido meristemático que se coloreó mediante la reacción de Feulgen (color rojo purpúreo) y colocar sobre un portaobjetos con unas gotas de orceína (lacto-propiónica) o con ácido acético al 45% y disgregar bien el material. 5- Colocar el cubreobjetos y sostenerlo con la punta del dedo y, con un lápiz con una goma en el extremo, golpear sobre el cubre varias veces en círculo, luego, hacer squash. 6- Sellar, y si se desea realizar preparaciones permanentes proceder de la siguiente manera: Colocar el preparado en una Caja de Petri con fijador; este debe quedar suspendido sobre unas varillas con el cubre hacia abajo. Debe marcarse antes el contorno del cubreobjetos. Cuando se desprende, dejar secar el preparado y agregar una gota de medio de montaje y colocar el cubre. Cubrir con un papel de filtro, colocar unas pesas y dejar secar.7- Observar al microscopio y contar los cromosomas. Soluciones 8-oxiquinoleína 0,002 N Pesar 0,29 g de 8-oxiquinoleína y diluir en 1 litro de agua destilada. Colocar 2 hs. en agitador magnético y luego filtrar. HCI 1N Preparar la solución como se detalla a continuación: - Agua destilada 200 cc 10 - HCI - Agua destilada 20,8 cc 30 cc 11 Reactivo de Schiff - Fucsina básica 2,2 g - Metabisulfito de K o de Na 3 g - Agua destilada 200 cc - HCI 1 N 30 cc - Carbón activado 0,5 cc o 1/2g Carbón vegetal. Calentar agua destilada hasta punto de ebullición y agregar en ese momento Fucsina básica, agitando bien con una varilla. Enfriar a 50ºC y filtrar. Agregar HCl. Después de una hora o al llegar a 25 ºC poner el metabisulfito. El líquido se pone amarillo a las 24 hs. Luego agregar 1/2 gr de Carbón vegetal. Filtrar y guardar en botella oscura en heladera. Orceína Lacto-propiónica - Acido Láctico - Acido Propiónico - Orceína 50 cc 50 cc 2g Disolver la orceína en una mezcla en proporciones iguales de ácido láctico y ácido propiónico Filtrar y guardar en frasco oscuro. Dejar madurar el colorante durante al menos una semana, antes de usar. Volver a filtrar. 12 1.4-Cromosomas metafásicos en vertebrados (Técnica de Suspensión Celular) 1.4.1- Cromosomas metafásicos mitóticos en Peces (Según Bertollo et al., 1978) Objetivos ADQUIRIR habilidad en el manejo de la técnica directa de obtención de cromosomas mitóticos en vertebrados. RECONOCER la morfología cromosómica que caracteriza a cada grupo estudiar. a ESTABLECER las ventajas y desventajas de las técnicas de obtención de preparaciones cromosómicas directas por aplastado y por suspensión celular. Técnica 1- Inyectar intraabdominalmente entre las aletas pectorales y ventrales, solución acuosa de colchicina 0,05 % en proporción de 1 ml/100 gr de peso del animal. 2- Dejar el pez en un acuario bien aireado, durante 1/2 a 1 h. Posteriormente sacrificar el animal y retirar el órgano deseado: porciones cefálica y dorsal del riñón. 3- Lavar rápidamente el riñón en una solución hipotónica de KCl 0,075 M. 4- Transferir el material a una pequeña cuba de vidrio, que contenga 8-10 ml de solución hipotónica de KCl 0,075 M. 5- Fragmentar bien el material con auxilio de pinzas de disección, complementando este proceso con una jeringa hipodérmica sin aguja. A través de movimientos leves de aspiración y espiración del material, se facilita la separación de las células obteniéndose una suspensión celular homogénea. 6- Colocar la suspensión obtenida a 36-37 ºC o Tº ambiente, durante 20-40’. 7- Resuspender cuidadosamente el material con auxilio de una jeringa o pipeta Pasteur y transferir la suspensión a un tubo de centrífuga. Prefijar con unas gotas de Farmer. Los trozos de tejidos no disgregados, se descartan. 8- Centrifugar durante 10’ a 900-1.000 r.p.m., descartando el sobrenadante con pipeta Pasteur. 9- Adicionar 5-6 ml de fijador Farmer recién preparado, dejando escurrir a través de las paredes del tubo. 10- Resuspender el material cuidadosamente con una pipeta Pasteur. 11- Repetir los ítems 8-10 dos veces. Después de la última centrifugación y eliminación del sobrenadante, adicionar 1 ml de fijador y resuspender el material. 12- Gotear, 2 a 3 gotas de la suspensión final sobre un portaobjetos limpio y desengrasado. Dejar secar. 13- Colorear con Giemsa al 5-10% durante 10-20 minutos. Enjuagar, dejar secar y observar al M.O. 13 NOTA: Los puntos 1 y 2 de la técnica se pueden realizar, sacrificando al animal con posterior extracción del riñón, disgregación e incubación de la células renales en colchicina, utilizando tiempos variables que pueden transcurrir entre 20-50’ (a 30ºC). 14 1.4.2- Médula ósea de Vertebrados (anfibios, reptiles, aves y pequeños mamíferos) Animales colchicinados Egozcue, J. (1971) 1- 1 1/2 hs. (en algunos casos debe realizarse un tratamiento más prolongado) antes de sacrificar al animal se le inyecta, intraperitonealmente con una jeringa tipo tuberculina, solución de colchicina al 0,05% (porcentaje variable) en proporción 1:100 v/p). 2- Pasado ese tiempo, se sacrifica por dislocación cervical o con éter (dependiendo ésto de qué organismo estemos estudiando). 3- Se extraen los fémures, se descarnan y se cortan las epífisis. La médula se extrae con una jeringa que contiene solución de Hank’s sin Ca ++ ni Mg++ y se coloca en un tubo de centrífuga. Se llevan a 7 u 8 ml con Hank’s, se resuspende bien con una pipeta Pasteur y se centrifuga a 1.000 r.p.m. durante 5’. 4- Se descarta el sobrenadante, se agregan 7-8 ml de solución hipotónica de KCl 0,075M y se incuba a 37 ºC en baño termostático durante 30-40’, previa resuspensión. 5- Agregar 4 a 5 gotas de fijador Farmer (prefijación) centrifugar 8’a 800 r.p.m. 6- Descartar el sobrenadante, agregar nuevamente 7-8 ml de fijador Farmer recién preparado, resuspender y centrifugar 5’a 900-1.000 r.p.m. 7- Descartar el sobrenadante y agregar nuevamente 7-8 ml fijador y resuspender. Centrifugar 5’a 1.000 r.p.m. 8- Descartar el sobrenadante y agregar fijador. A partir de este paso es posible guardar el material en heladera 12 o más horas o continuar el procesado. 9- Centrifugar 5’ a 1.000 r.p.m. Descartar el sobrenadante y agregar fijador Farmer fresco hasta 1 o 2 ml para establecer la concentración final, que debe tener color lechoso. 10- Sobre portaobjetos enfriados en la heladera, previamente desengrasados, gotear 2-3 gotas del material. Dejar dispersar y secar al aire o flamear con mechero. 11- Colorear en una solución de Giemsa diluído en tampón fosfato al 3-4% durante 5 a 10’ dependiendo el tiempo y la concentración del colorante de la calidad del material. 12- Dejar secar al aire y observar al microscopio óptico. 15 1.4.3- Preparaciones por aplastado 1- Debe seguirse la técnica anterior hasta el paso número 5 sin realizar prefijación. 2- Centrifugar a 1.000 r.p.m. durante 5’. Descartar el sobrenadante. 3- Añadir 5 ml de ácido acético o ácido propiónico al 50% sin romper el botón celular. Dejar fijar durante 20’. 4- Descartar el fijador. Añadir de 1 a 3 gotas de orceína acética al 2%. Resuspender las células con pipeta Pasteur. 5- Colocar una gota de suspensión sobre el porta. Cubrir con un cubreobjetos. Aplastar vigorosamente. 6- Flamear en mechero de modo que la orceína no llegue a hervir. Esto favorece la tinción y extensión de los cromosomas. Inmediatamente aplastar de nuevo con fuerza. 7- Sellar la preparación con cera, esmalte de uñas u otro pegamento adecuado. 8- Pueden también realizarse preparaciones permanentes como en el caso explicado para vegetales. 16 1.4.4- Preparaciones de médula no colchicinada (Según Tjio y Whang, 1965) Para estudiar material humano o procedente de animales a los que no es posible inyectar colchicina, se utiliza un procedimiento de incubación de las células en una solución que contiene un inhibidor del huso mitótico. De esta manera se acumulan así metafases suficientes para el análisis cromosómico. El único fallo posible podrá deberse a que la punción medular obtenida contenga pocas células en división. 1- Por punción esternal, femoral, tibial, ilíaca o isquiática (Switzar, 1967), aspirar 0,5 ml de médula ósea. 2- Pasar la médula a 2-3 ml de medio de cultivo con un veneno mitótico (colcemid o colchicina) para médula ósea. 3- Transferir los pedazos de médula a un segundo cambio de 2-3 ml de medio de cultivo adicionado con colcemid o colchicina, limpiándolos de coágulos sanguíneos. 4- Incubar durante 1-2 hs. a 37 ºC. 5- Centrifugar a 500-700 r.p.m. durante 5’. Descartar el sobrenadante. 6- Resuspender las células, añadir 5 ml de solución hipotónica. 7- Incubar 20’ a 37 ºC. 8- Seguir la técnica descrita para animales cochicinados a partir del paso 5. 17 1.4.5- Preparaciones directas de tejidos sólidos Esta técnica puede aplicarse a varios órganos, como ser bazo, hígado, intestino, riñón, etc. 1- En un animal colchicinado, tomar el órgano a utilizar y colocarlo en una placa de Petri que contenga suficiente solución hipotónica a 37 ºC para cubrir la pieza. 2- Con una tijera de disección, cortar la pieza en pequeños trozos, de 0,5 mm aproximadamente. 3- Transferir los trozos de tejido a una caja de Petri que contenga solución hipotónica. 4- Cubrir la caja de Petri e incubar a 37 ºC. 5- Transferir las piezas de tejido a una caja de Petri que contenga orceína acética al 2%. Es importante drenar las piezas de la mayor cantidad posible de solución hipotónica, pero sin dejar que se sequen. Esto se consigue arrastrando las piezas por la superficie de una caja de Petri seca. 6- Cubrir la caja de Petri para evitar la evaporación de la orceína, dejar en frío a 4ºC durante 1-2 hs. 7- Tomar un pedazo de órgano y colocarlo sobre un porta. No dejar secar. Cubrirlo con una gota de orceína, colocar un cubre y aplastar en la forma descrita. 8- Observar al microscopio. 18 1.4.6- Consideraciones prácticas sobre el aplastado o squash final. 1- Colocar un porta limpio sobre un papel de filtro doble o triple. Evitar las arrugas en el papel que puedan producir roturas de porta o cubre. Las preparaciones deben llevarse a cabo sobre una superficie lisa. Si la mesada del laboratorio es embaldosada, el porta debe situarse en el centro de una baldosa, no en la unión de dos de ellas. 2- Situar una pequeña cantidad de material en el centro del porta. 3- Cubrir el material con una gota de orceína. Si se trabaja con una suspensión celular en orceína este paso es innecesario. 4- Colocar un cubreobjetos verticalmente, de modo que uno de sus bordes se apoye en el portaobjetos, tocando el colorante. Este se extenderá por capilaridad, a lo largo del borde del cubre. Dejar caer el cubreobjetos suavemente sobre el material, evitando la formación de burbujas de aire. Si es posible, utilizar cubres pequeños que pueden ser totalmente cubiertos con el pulgar. 5- Colocar sobre la preparación un papel de filtro de doble o triple grosor para absorver el exceso de colorante. 6- Colocar la yema del pulgar en la zona correspondiente al material. Ejerciendo una presión vertical para evitar el deslizamiento del cubre, iniciar el aplastamiento con un movimiento balanceante del pulgar de delante atrás y de atrás adelante, aumentando la presión progresivamente. El aplastamiento debe ser realmente vigoroso, cargando el peso del cuerpo sobre el dedo. El movimiento balanceante del pulgar de uno de sus bordes al otro hace que el exceso de colorante vaya siendo absorbido, y evita las corrientes de líquido hacia el centro de la preparación. 7- Sellar la preparación y conservarla en frío hasta el momento de observar. 19 2- Preparaciones Cromosómicas suspensión celular Meióticas mediante aplastado y 2.1- Meiosis en Ortópteros Objetivos OBTENER cromosomas meióticos de Ortópteros por aplastado. RECONOCER las distintas fases de la meiosis. COMPARAR la morfología de los cromosomas mitóticos y meióticos. Técnica 1- Sacrificar con éter un ejemplar macho de Ortóptero. 2- Extraer los testículos, cortando la parte dorsal del abdomen a la altura de los dos primeros segmentos. 3- Fijarlos en fijador Farmer, durante por lo menos 15 minutos. 4- Tomar 1 o 2 folículos y colocarlos sobre un portaobjetos. 5- Agregar una o dos gotas de orceína lacto-propiónica y macerar lo suficiente como para disgregar bien los folículos. 6- Colocar el cubreobjetos, dejar colorear unos minutos y hacer squash. 7- Observar al microscopio y si el material está bien disgregado y coloreado, sellar con cera o goma para cámaras de bicicletas. 20 2.2- Meiosis en Hemípteros Objetivos OBTENER cromosomas meióticos de Hemípteros por aplastado RECONOCER fases meióticas y el tipo cromosómico que caracteriza a Pyrrhochoris sp. DISTINGUIR a través de las fases meióticas el significado de meiosis invertida COMPARAR la morfología de cromosomas holocéntricos con la de los monocéntricos Técnica 1- Sacrificar con éter, un ejemplar macho de Pyrrhocoris sp. 2- Extraer los testículos, cortando la parte dorsal del abdomen. Los testículos son órganos pares, generalmente, de color blanquecino. 3- Fijarlos en fijador Farmer durante por lo menos 15 minutos. 4- Tomar uno o dos folículos y colocar sobre un portaobjetos, agregar una o dos gotas de orceína lacto-propiónica y macerar lo suficiente como para disgregar bien los folículos. 5- Colocar el cubreobjetos, dejar colorear unos minutos y hacer squash. 6- Observar al M.O. y si el material está bien disgregado y coloreado, sellar con cera o goma para cámara de bicicletas. 7- Analizar las preparaciones a través de M.O. 21 2.3- Meiosis en Vegetales Objetivos OBTENER cromosomas meióticos vegetales por aplastado. RECONOCER las distintas fases de la meiosis. ESTABLECER el número diploide a través de la observación de células en metafase I. COMPARAR la morfología de los cromosomas mitóticos y meióticos. Técnica 1- Tomar el botón floral y colocarlo en fijador durante 2-24 hs. El fijador puede ser: Farmer o alcohol metílico-ácido láctico 5:1; la ventaja del último es que se puede tener preparado y su tiempo de duración es de tres meses. 2- Luego de fijado el material durante 24 hs. se pasa a alcohol al 70 % (conservador). 3- Si se desea realizar el preparado en el momento, se toma la antera y se la coloca sobre el portaobjetos. 4- Macerar bien el material con unas gotas de orceína lactoacética o lactopropiónica, o con hematoxilina propiónica. Colocar el cubreobjetos y realizar squash. 5- Una vez examinado el preparado en el M.O., si se desea se pueden realizar preparaciones permanentes, de la forma descrita en la técnica correspondiente a cromosomas metafásicos mitóticos en vegetales. 22 2.4- Meiosis en Peces (Según Kligermann and Bloom, 1977) Objetivos OBTENER preparaciones meióticas en Peces por suspensión celular. RECONOCER las fases meióticas correspondientes. Técnica 1- Sacrificar al animal y extraer los testículos. 2- Seccionar las gónadas en fragmentos bien pequeños de 2 mm aproximadamente y colocarlos en solución hipotónica de KCl 0,075M durante 15 a 20’ a temperarura ambiente. 3- Transferir el material a fijador recién preparado, fíjándolos durante 30’. 4- Repetir el proceso de fijación durante 2 veces más. El material puede guardarse en refrigerador a 4ºC o trabajarse de la siguiente manera: 5- Retirar el material del fijador, secándolo con papel de filtro para remover el exceso de fijador. 6- Transferir algunos fragmentos de órganos a una placa cerrada, adicionando 2-3 gotas de ácido acético al 5%. 7- Fragmentar el material con cuidado, para obtener una suspensión celular y con una pipeta Pasteur de punta fina, colocar una gota de suspensión sobre una lámina limpia, calentada en una placa a más o menos 36-40ºC reaspirándola inmediatamente. 8- Repetir la operación en 2-3 campos del mismo portaobjetos. 9- Colorear con Giemsa al 5% diluído en tampón fosfato pH 6,8 por 5-10’ 10- Lavar con agua corriente, secar al aire y observar. 23 2.5- Meiosis en Anfibios, Reptiles y Mamíferos Objetivos OBTENER habilidad en el manejo de la técnica. RECONOCER en el material de estudio las fases de la meiosis. Técnica 1- Sacrificar el animal y extraer las gónadas. Quitar la túnica albugínea y disgregar el material en Hank’s con pinzas y tijeras de disección. 2- Colocar la suspensión obtenida en un tubo de centrífuga y centrifugar 5’ a 1.000 r.p.m. 3- Descartar el sobrenadante y agregar 7-8 ml de solución de KCl 0,075M. Resuspender y colocar en estufa a 37º C durante 20’. Agregar 3 a 4 gotas de fijador Farmer. 4- Centrifugar 10’ a 1.000 r.p.m. 5- Descartar el sobrenadante y agregar 8-10 ml de fijador Farmer, resuspender y centrifugar 10’ a 1.000 r.p.m. 6- Repetir este paso 2-3 veces o más. 7- Descartar el sobrenadante y agregar 2-3 ml de fijador 3:1 para lograr la concentración final del material biológico. 8- Gotear sobre un vidrio limpio 2-3 gotas de la fijación final, dejar secar y colorear con Giemsa al 5-10% en tampón fosfato pH 6,8 durante 10-20’. Enjuagar bien y dejar secar. 9- Observar las preparaciones al M.O. 24 3- Preparaciones cromosómicas indirectas a partir de células cultivadas 3.1- Cultivo de células de riñón de Peces (Según Fenocchio et al., 1991) Objetivos LOGRAR el manejo de la técnica de obtención de preparaciones cromosómicas mediante el cultivo de células de riñón de Peces. ESTABLECER las diferencias entre las técnicas de obtención de preparaciones cromosómicas directas e indirectas. RECONOCER la morfología cromosómica del modelo biológico utilizado. CONTABILIZAR y OBTENER mediante el conteo de placas metafásicas el número cromosómico modal. Técnica El material a utilizar debe encontrarse en condiciones de asepsia adecuada . 1- Sacrificar el ejemplar y extraer el riñón (porciones dorsal y cefálica). 2- Disgregarlo y cultivar en una caja de Petri pequeña que contenga de 8-10 ml de medio de cultivo completo (medio TC199-Suero humano inactivado al 20%-Antibiótico). 3- Cultivar de 6 a 8 hs. en estufa a 28-30ºC. 4- Antes de realizar la cosecha de las células, agregar 3 a 4 gotas (0,1 ml) de colchicina al 0,05 % durante 15 a 20’. 5- Cosechar las células y colocar el material en un tubo de centrífuga. Centrifugar a 1.000 r.p.m. durante 10’ 6- Descartar el sobrenadante y agregar 8-10 ml de Hank's, disgregar el botón celular y centrifugar a 1.000 r.p.m. durante 10’ 7- Descartar el sobrenadante y agregar 8-10 ml de KCl 0,075 M. Resuspender y colocar los tubos en estufa a 30 º C durante 20-30’. Centrifugar 10’ a 1.000 r.p.m. Antes de centrifugar agregar 3 a 4 gotas de fijador Farmer (prefijación). 8- Descartar el sobrenadante y agregar de 8-10 ml de fijador Farmer. Resuspender el botón de células y centrifugar a 1.000 r.p.m. durante 10’. Repetir este paso dos veces más. 9- Descartar el sobrenadante y agregar fijador de acuerdo con el tamaño del pellet para preparar la suspensión final a utilizar. 10- Gotear de la solución final con una pipeta Pasteur sobre un portaobjetos limpio y desengrasado. Dejar secar al aire. 11- Colorear con Giemsa en tampón fosfato al 3-5% durante 5 a 20’. Enjuagar con agua de canilla y destilada. Dejar secar y observar. NOTA: esta misma técnica puede emplearse urilizando médula ósea de otros vertebrados. 25 3.2- Cultivo de linfocitos en sangre periférica Objetivos LOGRAR el manejo de la técnica de obtención cromosómicas mediante cultivo de sangre periférica. de preparaciones ESTABLECER las diferencias entre las técnicas de obtención de preparaciones cromosómicas directas e indirectas. EVALUAR las ventajas e inconvenientes de cada una de ellas. RECONOCER la morfología cromosómica del modelo biológico utilizado. CONTABILIZAR Y OBTENER mediante el conteo de placas metafásicas el número cromosómico modal. 3.2.1- Cultivo de linfocitos humanos El material a utilizar debe ser estéril y el cultivo realizarse en condiciones asépticas. 1- Extraer en forma estéril 10ml de sangre venosa con una jeringa previamente heparinizada (10 UI/ml de sangre). 2- En un frasco de cultivo que contiene 10 ml de Medio, agregar aproximadamente 0,5 ml de sangre, mezclar con movimientos circulares y colocar en estufa a 37ºC durante 72 hs. 3- Una hora antes de la cosecha de las células, agregar 2 gotas de colchicina al 0,05 %. 4- Pasar el contenido del frasco a un tubo de centrífuga y centrifugar a 1.000 r.p.m. durante 5’. 5- Descartar el sobrenadante y agregar 7-8 ml de solución hipotónica de KCl 0,075M, resuspender con una pipeta Pasteur y colocar a 37 ºC en estufa durante 30’. 6- Agregar 2-3 gotas de fijador y centrifugar durante 10’ a 1.000 r.p.m. 7- Descartar el sobrenadante y agregar 7-8 ml de fijador, resuspender nuevamente y centrifugar 5’a 1.000 r.p.m. 8- Descartar el sobrenadante y agregar fijador y dejar en la heladera de 12 a 24 hs . 9- Resuspender y centrifugar a 1.000 r.p.m. 5’, descartar el sobrenadante. 10- Agregar fijador 7-8 ml y centrifugar nuevamente 5’a 1.000 r.p.m. 11- Descartar el sobrenadante y agregar fijador de acuerdo con el tamaño del pellet para preparar la suspensión final. 12- Gotear material con una pipeta Pasteur sobre un portaobjetos limpio y desengrasado. Dejar secar al aire. 13- Colorear con Giemsa en tampón fosfato al 5% (95 ml de buffer y 5 de colorante) durante 5’. Enjuagar bien con agua de canilla y destilada. Dejar secar y observar. 26 Para preparar 100 cc de Medio de Cultivo para Sangre periférica se puede seguir con este procedimiento: 1) Agua bidestilada estéril 2) Medio de Cultivo concentrado (10 X) 3) Glutamina 200 X 4) Suero Fetal Bovino 5) Bicarbonato de Na 1M 6) Fitohemaglutinina 75 cc 10 cc 2 cc 15 cc 1 cc 2 cc La misma técnica y los mismos medios de cultivo pueden ser empleados, con pequeñas modificaciones y adaptaciones, a otros vertebrados como por ej. Peces. 3.2.2- Material de vidrio a utilizar La limpieza del material de vidrio debe ser rigurosa. Existen una gran variedad de detergentes para su utilización. Las normas que deben seguirse son las siguientes: Sumergir el material de vidrio en la solución de limpieza. Debe tenerse la precaución de que queden bien sumergidos. Cuando quedan restos de grasa y cera, éstos deben quitarse con tetracloruro de carbono. Los detergentes actúan mejor en soluciones calientes. Una vez lavado, el material debe ser bien enjuagado, como mínimo 10 veces en agua de canilla y 5 veces en agua destilada. 3.2.3- Esterilización del material de vidrio e insumos Se utilizan los métodos de esterilización en horno, autoclave y por filtración. La mayoría de las soluciones y en parte el material de vidrio se esterilizan en autoclave. En general, se utiliza una temperatura de 120-121ºC (15 libras) durante 20’. El tiempo se cuenta sólo a partir que la temperatura llega a 120ºC. No debe olvidarse al autoclavar soluciones que las tapas de los recipientes deben quedar flojas. La esterilización por calor seco, realizada en horno, es utilizada para el material de vidrio, excepto cuando se incluyen partes de goma o plástico. La esterilización se realiza a 180ºC durante 2 hs. Tanto en el caso de esterilización con horno o con autoclave se recomienda ubicar el material de manera de permitir el pasaje de calor adecuadamente. Los filtros de esterilización remueven en general bacterias y hongos de las soluciones, pero no remueven la mayoría de los virus. Comunmente se utilizan 4 tipos de filtro: de asbesto; de membranas; de vidrio sintetizado y de porcelana. El diámetro de poro del filtro debe tener 0,5 micras o menos (0,2 para medios de cultivo). Un filtro saturado de organismos e impurezas no debe ser utilizado. Soluciones viscosas o que contienen proteínas, como suero, necesitan presión al ser filtradas. La velocidad de la filtración debe ser apropiada para no perjudicar el filtro. Frecuentemente se utiliza un filtro dosificante que se inserta antes del esterilizante, que limpia parcialmente la solución de impurezas. Actualmente, se trabaja con sueros y medios de cultivo que vienen totalmente esterilizados, como también la fitohemaglutinina. 27 Existen filtros de asbesto y de membrana apropiados para ser adaptados en jeringas hipodérmicas comunes. Son muy ventajosos para filtrar pequeños volúmenes de solución. Los filtros de asbesto deben ser descartados después de su utilización. Los filtros de membrana son más ventajosos que los de asbesto para filtrar pequeños volúmenes, pues se pierde menos material absorbido en el filtro. Una vez preparado el medio de cultivo completo, se fracciona en alícuotas de 10 ml. y se almacenan en freezer a aproximadamente –10ºC. Las alícuotas de medio completo se fraccionan en frascos de vidrio estériles o bien en tubos cónicos de centrífuga, también estériles (esta última opción es la más adecuada dado que se trabaja con tubos estériles adquiridos en el mercado). 3.2.4- Antibióticos Antibióticos selectivamente tóxicos para diferentes agentes microbianos pueden ser de gran auxilio en cultivos de tejidos. Se utilizan siempre que se trabaje con material que puede ser contaminado o en cultivos en gran escala. En trabajos experimentales con cultivo de tejidos, deben usarse sólo cuando son necesarios. El criterio usado para determinar la toxicidad de los antibióticos son alteraciones en la morfología o en el patrón de crecimiento de las células cultivadas. El uso continuo de antibióticos puede enmascarar infecciones por bacterias u hongos y llevar a infecciones de tipo crónico o latente. Si un cultivo está contaminado, es dificil suprimir definitivamente la contaminación con antibióticos. Frecuentemente, la contaminación se manifiesta nuevamente, cuando el antibiótico es suprimido. La combinación de antibióticos más usada para controlar la contaminación bacteriana es: Penicilina (50 ug/ml de medio de cultivo) y Estreptomicina (50 ug/ml), con o sin adición de nistatina (antimicótico, 30 ug/ml). Otros antibióticos son Tetraciclina (5 ug/ml), Neomicina (25 ug/ml) y Kanamicina (50 ug/ml). Estos dosajes sirven como ejemplo, debiendo acertarse para cada caso en particular. Así por ej. para medios que contienen suero es recomendable doblar las cantidades citadas. Por otro lado, actualmente se utilizan combinaciones comerciales especialmente formuladas. 3.2.5- Asepsia Los cuidados de asepsia varían según el material y problema en estudio. Aunque muchas veces no es necesaria una cámara aséptica (banco o mesa de flujo laminar), ésta es recomendable. Cuando no se dispone de cámara aséptica, el lugar en que se realiza el cultivo debe ser relativamente aislado, sin corrientes de aire y exento de polvo. En este último caso se trabaja con el auxilio de un mechero a gas o alcohol. 28 III- Técnicas de tinción diferencial de cromosomas A partir de preparaciones cromosómicas se emplean técnicas de coloración diferencial de los cromosomas. Esto permite identificar regiones de bandeo simple o múltiple, dependiendo del tipo de tratamiento y de coloración se realice. En este sentido es importante esclarecer qué objetivos se persiguen con estas tinciones diferenciales. Por ejemplo, si se desea conocer o estudiar el patrón de distribución de heterocromatina, las regiones donde se localizan genes organizadores nucleolares, las alteraciones involucradas en alguna región del cromosoma o bien cromosomas enteros que participan de alteraciones. Objetivos ADQUIRIR habilidad en el manejo de algunas de las distintas técnicas de bandeo cromosómico en animales y vegetales. ESTABLECER la utilidad de las técnicas de bandeo en cada modelo biológico utilizado. RECONOCER a través del M.O. los patrones característicos de cada tipo de bandeo cromosómico básicos (G, NOR y C ). COMPARAR a través del M.O. las difierencias en el número de bandas que se obtiene al aplicar bandeo de Alta Resolución con respecto al bandeo G convencional. ADOPTAR criterios que permitan aplicar técnicas de bandeo de cromosomas adecuadas al modelo biológico utilizado y al problema a resolver. 1- Introducción al bandeo cromosómico Pueden utilizarse técnicas especiales para obtener patrones de coloración diferencial, característicos de los cromosomas, los que permiten su identificación con mayor facilidad. Los cromosomas se obtienen por los métodos usuales y luego se colorean por fluorocromos o colorantes como el Giemsa, que, con diversos tratamientos previos, producen en las preparaciones cromosómicas patrones de bandeos (Q, C, G, R, NOR, etc.). Las técnicas de fluorescencia y bandeos estructurales o por incorporación de análogos de base, tienen gran aplicabilidad en la detección citológica de anomalías estructurales de los cromosomas y en la localización de regiones cromosómicas específicas, así como, en algunos casos, genes en experiencias de hibridazión "in situ" con sondas de ADN. 29 1.1- Bandeo Q Se observó mediante varios experimentos que los fluorocromos se ligan diferencialmente a los cromosomas metafásicos de plantas y animales. Los cromosomas tratados con esas sustancias muestran, al microscopio de fluorescencia, un patrón característico de estriación transversal (longitudinal). Los patrones más evidentes son los obtenidos con los derivados de acridina, quinacrina y la mostaza de quinacrina. Como los patrones son bastantes estables y reproducibles, pueden utilizarse para facilitar la identificación de los cromosomas Y, los que aparecen en interfase como puntos con fluorescencia intensa. Es también visible en espermatocitos que contienen al cromosoma Y. La fluorescencia intensa del Y, permite pues la identificación de las células masculinas en interfase, en raspados de mucosa bucal o en espermatozoides, siendo esto designado como cromatina sexual masculina o cromatina sexual Y. También en otros animales hay patrones característicos de fluorescencia. Así por ej. en las serpientes cuyo mecanismo de determinación del sexo en el macho es ZZ y en la hembra, ZW, el cromosoma sexual W presenta una fluorescencia característica en la interfase somática designada como cromatina sexual W. Técnica 1- Usar preparaciones citológicas de cromosomas fijados, y secados al aire o según otra técnica. No acercar las láminas a la llama para secar las preparaciones . 2- Rehidratar las preparaciones en serie de alcoholes (95, 70 , 50 y 30 %) y agua destilada. 3- Enjuagar en solución tampón fosfato pH 7,0. 4- Colorear en solución de quinacrina por 20’. 5- Lavar cuidadosamente 3 veces en tampón fosfato pH 7,0. 6- Montar en solución fosfato y si se desea conservar por más tiempo sellar con parafina. Nota: Si se trata de distintos tipos de preparados, la coloración puede variar de 5’(espermatozoides) a 20’(cromosomas metafásicos). Soluciones 1- Quinacrina o Mostaza de quinacrina: preparar una solución que contenga 0,05 mg de quinacrina por ml de ácido cítrico-tampón fosfato pH 7,0. Puede guardarse en heladera durante varias semanas. 2- Acido cítrico-tampón fosfato pH 7,0: A- 0,2M Na2HPO4-2H 2 0 (36,6 g/l de agua destilada). B- 0,1M ácido cítrico (21,014 g/l de agua destilada). Para obtener 400 ml de tampón pH 7,0 mezclar 330 ml de solución A con 700 ml de solución B. 30 Fotografía de fluorescencia Los cromosomas se observan inmediatamente después de la coloración. Se fotografían las mejores metafases en el microscopio de fluorescencia. Se recomienda mantener la luz U.V. (lámpara de mercurio) con una intensidad mínima, antes de fotografiar, procurando hacerlo en contraste de fase. Este cuidado es necesario para evitar el exceso de irradiación de los cromosomas que pierden con esto fluorescencia y nitidez de las bandas. Análisis cariotípico Se realiza una fotografía por ampliación del negativo de la metafase. Como la fluorescencia total del cromosoma depende en gran parte de lo que se encuentra alrededor (núcleos, cromosomas, residuos), debe realizarse más de una ampliación con tiempos de revelación distintos. Posteriormente la preparación puede recuperarse y nuevamente utilizarse para colorear con Giemsa, bandas C o autorradiografía. Puede recolorearse también para fluorescencia, pero en este caso sólo las metafases sin irradiar por U.V. presentarán bandeo nítido. Preparaciones de cultivo de linfocitos Es esencial que las preparaciones cromosómicas sean de alta calidad, esto es, que tengan un mínimo de citoplasma alrededor. Los cromosomas deben estar bien dispersos y no muy contraídos. Fijar las preparaciones con fijador Farmer recién preparado. Cambiar tres veces el fijador y usar láminas y cubreobjetos bien limpios. Coloración 1- Colorear las láminas durante 6-8’, en di-hidroclorato de quinacrina (Sigma) o Atebrin (Winthrop) a 0,5% (p/v) en agua deionizada pH 5,5-6,0 a temperatura ambiente. 2- Lavar durante 2-3’en agua de canilla. 3- Enjuagar con agua deionizada. 4- Secar las láminas (opcional). 5- Cubrir con un cubreobjetos, en agua deionizada. 6- Remover el exceso de agua. Sólo debe quedar una película de agua entre porta y cubreobjetos, para evitar la difusión. 7- Sellar las láminas. 8- De preferencia, examinar el mismo día. Si hubiese mucho brillo, remover el cubreobjetos y montar nuevamente. Microscopia Examinar en microscopio de fluorescencia con lámparas de mercurio, filtro de exitación BG 12, filtro de barrera de 510nm y objetivo de inmersión de 40 X a 100 X. 31 Fotografía Pueden ser utilizadas diversas películas de grano fino y alto contraste, entre ellos films Kodak Panatomic, Tri-X; Plus X T-Max 100 ASA, también AGFAPAN A.H.U. 25 ASA u otros. Mucosa bucal 1- Fijar en metanol 30’. 2- Secar. 3- Colorear durante 5’con el colorante preparado como se describió antes. Continuar como para linfocitos (2-8 anterior). Raspados de mucosa bucal pueden guardarse luego de fijados y secados, pero deben ser examinados el mismo día en que fueron coloreados. Espermatozoides A) 1- Esparcir una gota de semen en el portaobjetos como si fuera un frotis de sangre. 2- Fijar en metanol: ác. acético (3:1) durante 30’. 3- Colorear durante 3’con una solución de quinacrina. 4- Lavar en agua de canilla. 5- Enjuagar con agua deionizada. Montar como antes o: B) 1- Centrifugar el semen y descartar el sobrenadante (plasma seminal). 2- Resuspender en KCl durante 8’. 3- Centrifugar y descartar el sobrenadante. 4- Adicionar fijador Farmer lentamente, recién preparado, agitando al mismo tiempo. 5- Centrifugar, descartar el sobrenadante, resuspender en volumen reducido de fijador fresco. 6- Gotear en un porta limpio (una gota) y secar al aire. 7- Colorear como se describió anteriormente en A (3-5). Es preferible realizar la técnica B puesto que con A el plasma seminal también colorea, lo que resulta en un fondo fluorescente. 32 Cromomicina A3 y DAPI (Di-amino-fenilindol) (tomado del Lab. de Genética de la Univ. Estadual de Londrina) Sobre preparaciones cromosómicas, sin teñir previamente: 1-Colocar 30 ul de solución de CMA3 o DAPI y cubrir con un cubreobjetos. 2-Dejar colorear en lugar oscuro durante aproximadamente 90 minutos. 3-Lavar con agua destilada y dejar secar a oscuras 15’. 4-Montar la preparación con 30 ul de medio de montaje compuesto por: 5 ml. de tampón McIlvaine 5 ml. de glicerol pro análisis apto para fluorescencia. 5 ul. de solución de Cloruro de Magnesio 2,5 M. 5-Dejar estabilizar la coloración durante algunos días antes de analizar. 1.2-Bandeo C En general, es posible reconocer la heterocromatina en núcleos interfásicos o profásicos debido a su heteropicnosis. Es sabido que un segmento cromosómico puede ser heteropicnótico y no ser heterocromático. En los análisis de metafases con coloración convencional, los cromosomas se colorean intensamente, salvo las regiones en los que se encuentran las constricciones, no pudiéndose de esta manera visualizar los bloques de heterocromatina. Debido a esto varios citogenetistas procuraron desarrollar técnicas que permitieran detectar la ubicación de los bloques heterocromáticos en los cromosomas metafásicos. Existen varias técnicas que permiten la localización de las regiones heterocromáticas, entre ellas: * tratamiento con frío en algunos vegetales. * autorradiografía. * hibridación "in situ". * bandeo C. *fluorocromos específicos para AT o GC. Bandeo C. Procedimiento (Según Sumner, 1972, con modificaciones) 1- Hidrolizar en una solución de 0,2N de HCI, a Tº ambiente durante 15’. 2- Lavar con agua destilada deionizada. 3- Sumergir los preparados en una solución de Hidróxido de Bario al 5%, durante aproximadamente 1 a 2’ a 60º C o temperatura ambiente. El tiempo como la concentración de la Sn. de Bario pueden variar de acuerdo con el material biológico utilizado. 4- Lavar en una Sn. de HCL 0,2 N y con abundante agua de canilla. 5- Incubar las láminas cubiertas en 2xSSC a 60º C durante 1 hora. 6- Dejar secar las preparaciones al aire. 7- Colorear con Giemsa al 2% en tampón fosfato 0,01 M, pH 7,00 durante 1015’. 8- Lavar los portaobjetos en agua de canilla, secar y montar. 33 Solución 2xSSC Cloruro de Na (0,3 M) 17,53 g Citrato trisódico (0,03 M) 8,82 g Completar con agua deionizada o destilada hasta 1 litro. Solución de HCL 0.2N 1,6 ml. de HCL 0.2N 100 ml. de agua destilada Solución de Hidróxido de Bario 5 grs. de Hidróxido de Bario 100 ml. de agua destilada Agitar enérgicamente, se prepara una solución sobresaturada. 1.3-Bandeos G, R y de Alta Resolución (AR) Luego del desarrollo de las técnicas de bandeo C, fue posible lograr otras técnicas que permitieron producir varios tipos de bandas a lo largo del cromosoma. En un principio, se pensó que estas bandas claras y oscuras, se producían por acción del colorante Giemsa. De allí la denominación de bandas G. Actualmente sabemos que las bandas G o segmentos que reaccionan positivamente a la técnica de bandeo G (o sea las regiones oscuras), representan los segmentos cromosómicos que se condensan más temprano en la profase; en cambio las regiones claras o segmentos negativos para bandas G se condensan más tardíamente en profase. El patrón de bandas G por ej. en humanos, es similar al que se observa al realizar bandeo Q. Esto se debe al hecho de que el colorante mostaza de Quinacrina tiene mayor afinidad por el complejo DNA-proteína de las regiones cromoméricas que por el de las intercromoméricas. Pero existen fluorocromos con afinidad inversa, donde las regiones intercromoméricas se colorean más brillantemente. Cuando esta coloración se realiza, se obtiene un patrón de bandas reverso al que observamos con el bandeo G o Q. De allí la denominación de bandeo R. 1.3.1.BANDEO DE ALTA RESOLUCION (AR) 1-Sembrar sangre fresca (recién extraída), como si se tratara de un cultivo convencional. 2-Cosechar las células en cultivo a las 48 o 72 Hs. Se advierte sobre la importancia de cuidar la esterilidad hasta el momento de la cosecha. 3-A las 48 Hs., de cultivo, agregar 0,1ml de FUDR y envolver los frascos o tubos con papel de aluminio. 4-Prolongar el cultivo 15 o 17 Hs. (hasta completar 66 Hs. de cultivo) y adicionar 0,1ml de BUDR. 5-Agregar 2 gotas de colchicina (0,005%), una hora antes de cumplirse las 72 horas de cultivo y cosechar las células como un cultivo corriente. 1.4-BANDAS G 34 Las preparaciones deben estar secas y no debe utilizarse la llama en el proceso de secado. Se recomienda dejar secar los preparados durante una noche o al menos de 2-4 hs. en estufa a 65ºC. En muchos laboratorios se utilizan preparaciones envejecidas durante alrededor de 20 días. Técnica número 1 1- Cubrir las preparaciones en una solución de tripsina, de 30 a 90’ a temperatura ambiente. 2- Lavar las preparaciones con solución salina. 3- Dejar las láminas en etanol 70 % durante 5’. 4- Colocar las preparaciones en etanol 95% durante 5’. 5- Dejar secar al aire. 6- Colorear las láminas en Giemsa-tampón fosfato- 0,01 M pH 7,0. 7- Lavar las láminas en agua de canilla, secar y montar. Solución salina: Consiste en solución de Hank’s sin Ca++ ni Mg++. Solución de tripsina: Solución stock de tripsina 0,025% 10 ml Solución salina de Hank’s, sin Ca++ ni Mg++ 50 ml Como la tripsina contiene 189 ug/g de actividad, la concentración final de tripsina usada es de cerca de 8 ug/ml. Técnica número 2 1- Incubar las preparaciones citológicas en solución (3:1) de urea 8 M en tampón Sörensen (M/15 KHPO/NaHPO) pH 6,8 a 37ºC, por 10’. 2- Lavar en agua de canilla. 3- Colorear las preparaciones en solución de Giemsa-fosfato (1:50) pH 6,8 por 10’. Técnica número 3 1- Incubar las láminas durante 60´ a 60ºC, en solución de 2xSSC. 2- Lavar en agua destilada o deionizada. 3- Colorear en solución de Giemsa-tampón fosfato (1:50) 4- Lavar las láminas,secar y montar 5- Lavar las láminas en agua deionizada o destilada, secar y montar. Técnica número 4 (tomada del Lab. de Citogenética y Genética Humana Convenio UNaM-IPS) 1-Envejecer las preparaciones durante, aproximadamente, 20 días. 2-Sumergirlos en sn. de Tripsina al 0,1 % durante tiempos variables entre 15’’ a 1,5’. 3-Lavar con sn. fisiológica. 4-Colorear con Giemsa al 5% durante 4 a 10’ 5-Enjuagar con abundante agua de corriente 6-Dejar secar y observar al MO. 35 Solución de Tripsina 1- Preparar Tripsina 1:250 al 1% en agua bidestilada (diluir 1 gr. de Tripsina en 100 ml de agua bidestilada, sn. stock). Agitar dicha solución durante 2 Hs. y luego filtrar con papel de filtro. Conservar en freezer en alícuotas de 3 a 5 cc. 2-Diluir: 1 ml de Tripsina (sn. stock). 4,5 ml. de Buffer Fosfato disódico 0,2N. 4,5 ml. de CLNA al 0,9%. Esta solución de Tripsina al 0.1% es la que se utiliza para realizar los bandeos. Si esta solución digiere en exceso, los cromosomas, se la puede disolver tantas veces sea necesario hasta lograr el bandeo deseado. Solución de Buffer Fosfato Disódico (PO4HNa2) 0,2N 1-Diluir 14,2 grs. de Fosfato Disódico en 1L. de agua destilada Solución de Buffer Sörensen pH 6.8 Esta Solución se utiliza para diluir el colorante Giemsa. 1-PO4H2K 0,067M 9,08 grs./L. 2-PO4HNa2 (2 H2O) 0,067M 11,88 grs./L. (Sn. A) (Sn. B) 3-solución de uso: 50 ml. de sn. A y 50 ml. de sn. B, llevar a pH 6,8. A esta mezcla agregar de 3 a 4 ml. de colorante Giemsa. NOTA: La sn. B puede prepararse con PO4HNa2 (12 H2O) diluyendo 23,99 grs. en 1L. de agua destilada. 1.5-BANDAS R 1- Las preparaciones citológicas puden ser obtenidas por los métodos usuales de cultivo de linfocitos. La solución hipotónica usada es la siguiente: Suero fetal bovino diluído 1/6 15 vol. MgCl2 en agua (3,39 %) 1 vol. Hialuronidasa 2,5 UI/ml 2- Esparcir algunas gotas de suspensión celular en láminas recién sacadas del agua helada. Dejar secar al aire. 3- Utilizar los vidrios 3 o más días después de realizar la preparación citológica. Colocar las láminas en tampón fosfato pH 6,5 a 86-87 ºC,durante 10’. 4- Enjuagar en agua de canilla, por algunos segundos. 36 5- Colorear con solución de Giemsa: Giemsa 4 ml; tampón fosfato 4 ml; agua bidestilada 92 ml. Si no se observan modificaciones en los cromosomas, será necesario un envejecimiento más prolongado. La visualizaciòn de retracción pericentromérica, que ocurre antes del bandeo, es una buena indicación para prolongar el tiempo de envejecimiento. Nota: No pueden usarse láminas previamente coloreadas. Las láminas a ser usadas para los preparados deben estar bien limpias. El secado de los preparados al aire debe realizarse en ambiente no muy seco o caliente. 37 1.6-Bandeo NOR Los cromosomas que presentan constricción secundaria, son frecuentemente observados, como asociados al nucléolo (durante la profase). De allí la denominación de esta región como región organizadora del nucléolo (NOR o RON). Esta asociación se debe al hecho de que en las constricciones secundarias se situan los genes que producen RNA ribosómico, que constituirá gran parte del nucléolo. Las constricciones secundarias pueden colorearse fuertemente con nitrato de Plata el que presenta afinidad por las proteínas nucleolares. Probablemente algunas proteínas nucleolares persisten en la región de la constricción secudaria durante todo el ciclo celular, permitiendo colorear los nucléolos y las constricciones secundarias en metafase. Técnica (Según Howell & Black, 1980) 1- Sobre un vidrio previamente goteado con material de interés, colocar una gota de gelatina a pH 4-4,5 y dos de nitrato de Plata al 2%. Mezclar bien con el extremo de un cubreobjetos. 2- Colocar sobre el colorante un cubreobjetos de 40 x 22 mm y llevar la preparación, en cámara húmeda a una estufa o baño termostático a 60ºC. 3- Colorear durante el tiempo necesario hasta que la coloración tome color amarillo ámbar. 4- Enjuagar con agua de canilla, y dejar caer el cubreobjetos. Secar y observar. 1.7-FISH (Fluorescence in situ hibridization) (Tomada del Lab. de la Univ. Estadual de Londrina) Para realizar esta técnica es necesario disponer de una sonda previamente marcada. Esta sonda marcada puede adquirirse lista para ausar o bien podemos marcarla en el laboratorio. Hay varias técnicas de marcaje. A continuación se describe la de Nick translation. 1.1-Marcaje de la sonda mediante Nick translation 1-Colocar tubos de microcentrífuga en hielo. 2-Pipetear 5 ul. 10X dntp ul. (1 ug.) DNAxx 5 ul. 10X mix de enzima ul. H2O destilada para completar 45 ul. xx Cálculo de la cantidad de DNA a ser colocado en la reacción: a) cuantificar DNA (valor X). La cantidad a tomar será: cuántas veces el valor cuantificado se debe tomar para darnos 1000 ng: para 1000 debo sacar aprox. 11 veces o sea 11 ul. 3-Centrifugar 5’’ (spin) 38 4-Incubar a 16ºC durante 1-2hs. en máquina de PCR (en programa Nick: 90’ a 16ºC) 5-Este paso es opcional: agregar 5ul. de Stop Buffer. 5-Precipitar con acetato de Na 3M 1:10 Para 45 ul. de Sn. de reacción que contiene DNA, colocar 4,5 ul. de Acetato (se puede considerar a la suma de 45+4,5=49,5 ul. igual a 50 ul.). 6-Agregar 2 volúmenes de Etanol 100% helado (100 ul.) 7-Homogeneizar suavemente y colocar en hielo. 8-Precipitar en freezer –80ºC durante 15’ o –20ºC durante 2 Hs. o se puede dejar hasta el día siguiente. 9-Centrifugar 15 rpm durante 10’. 10-Retirar el sobrenadante y dejar el pellet. 11-Adicionar 30-50 ul. TE (puede ser H2O esterilizada) y dejar en la heladera overnight. 12-Repetir los pasos 5 a 9 y luego de algunas horas cuantificar. 1.2-Técnica de FISH (Tomada del Lab. de la Univ. Estadual de Londrina) Se realiza en cuatro etapas a saber: 1er. Etapa: 1-Colocar 50 ul. de RNAsa en cada preperado y cubrir con un cubreobjetos de plástico (blando) y llevar a estufa a 37ºC durante una hora. RNAsa diluída 1:100 (solución stock de RNAsa: 2XSSC) –0.5ul. de RNAsa : 49.5 ul. de 2XSSC= 50ul. 2-Lavar las láminas en 2XSSC por 10 minutos a temperatura ambiente en agitador (luego de 1-2 min.) retirar los cubreobjetos que estarán boyando. 3-Descartar el 2XSSC (en la pileta) y en la misma cubeta descartar paraformaldehído 4% a Tº ambiente, por 10 min. en agitador. Paraformaldehído 4% (bajo campana de vidrio): 4 grs. de paraformaldehído despolimerizado en 98 ml. de agua destilada a 60ºC en agitador. Agregar 2ml de NaOH 1M. 4-Descartar el paraformaldehído en frascos de descarte apropiado y colocar nuevamente 2XSSC, por 10’ en agitador. 5-Descartar el 2XSSC y colocar alcohol etílico 70% por 5’ en agitador. 6-Descartar el alcohol 70% y colocar 100% durante 5’. No se necesita agitar. 7-Descartar el alcohol 100% y dejar secar las láminas por 1-3 horas. 8-En este intervalo preparar la mezcla de hibridación por lámina: colocar en un eppendorff con punteras nuevas: ** Todas las soluciones están congeladas y deberán retirarse y colocar al inicio en hielo batido. Formamida 100% Dextran 50% 20XSSC 15 ul 0,6 ul. 0,3 ul. 39 DNA de bloqueo 0,1 ul. (timo de becerro o placenta). Nota: en lugar de Dextran 50%, también se puede utilizar Polietilenglicol 50% SDS 10% Sonda Agua 0,1 ul. hasta 0.5 ul. hasta completar 30-31 ul. Mezclar en agitador. 9-Desnaturalizar la mezcla de hibridación (ISH) a 70ºC durante 10 min. en baño termostático. 10-Incubar en hielo al menos 5’ y como máximo 2 horas. 11-Colocar 30 ul. de mezcla de ISH por preparado y cubrir con cubreobjetos plásticos y llevar al termociclador: 90ºC por 10’; 48ºC por 10’; 38ºC por 5’ y 37ºC hasta infinito. 12-Colocar los preparados en cámara húmeda a 37ºC durante la noche. Nota: colocar la cámara húmeda en estufa mientras los vidrios se encuentran en el termiciclador. 13-colocar las soluciones de 2XSSC; 0,1XSSC y 4XSSC y 0,2% Tween en baño termostático durante toda la noche a 42ºC. 2da. Etapa: Baños poshibridización (Sonda DNAr 18S) 1-Sacar los vidrios de la estufa, dejando la cámara húmeda en la misma. Hacer baños poshibridización. Preparar formamida deionizada al 20%: 20 ml. de formamida deionizada diluir en 80 ml. de 0’1XSSC. 2-Lavar en 2XSSC a 42ºC por 5’, en agitador. los cubreobjetos de plástico boyan y se retiran de las cubetas. 3-Descartar 2XSSC, en la pileta, y en la misma cubeta agregar formamida 20% a 42ºC durante 10’ en agitador. 4-Descartar la formamida, en vidrios apropiados, y colocar en la misma cubeta 0,1XSSC a 42ºC por 5’ en agitador. 5-Descartar el 0,1XSSC y agregar, en la misma cubeta, 2XSSC a 42ºC por 5’ en agitador. 6-Descartar el 2XSSC y agregar 4XSSC, 0,2% Tween a 42ºC por 5’ en agitador. 7-Descartar 4XSSC, 0,2% Tween a 42ºC y colocar 4XSSC, 0,2% Tween a temperatura ambiente, en agitador durante 5’. 2da. Etapa: Baños poshibridización (Sonda DNAr 5S) 1- Sacar los vidrios de la estufa, dejando la cámara húmeda en la misma. Hacer baños poshibridización. 40 2- 2-Lavar en 6XSSC a Tº ambiente por 10 (15) minutos en agitador. Retirar los cubreobjetos que boyan en la cubeta. 3- Descartar 6XSSC en la pileta y en la misma cubeta colocar 6XSSC a Tº ambiente por 10 a 15’ en agitador. 4- Descartar la solución de 6XSSC y colocar en la misma cubeta 6XSSC a 42ºC por 3’ sin agitar. 5- 5-Descartar 6XSSC y en la misma cubeta colocar 4XSSC/Tween a Tº ambiente por 5’ en agitador (Optativo) 3er. Etapa: Detección de la Hibridación 1-Retirar los vidrios y sacudir cuidadosamente en papel de filtro y con los vidrios aún mojados gotear 50 ul. de BSA 5% por 5’ y cubrir con cubreobjetos de plástico. Nota: Retirar el BSA del freezer mientras se realizan los lavados poshibridización. 2-Retirar los cubreobjetos colocando el preparado hacia abajo. De ser necesario ayudarse con una pinza. 3-Colocar la solución de detección y cubrir con cubreobjetos de plástico e incubar a 37ºC por 1 hora en cámada húmeda. Para un vidrio: Avidina-FITC 1 ul. (cerca de la heladera) y BSA 5%, 49 ul. Hacerlo en papel un eppendorff envuelto en papel de aluminio. Agitar. 4-Lavar las láminas en 4XSSC-0,2% Tween por 10’ a Tº ambiente en agitador, a oscuras. Retirar los cubreobjetos luego de unos minutos. 5-Descatar la solución y repetir el mismo lavado una vez más. (Opcional). 4ta. Etapa: Coloración: 1-Retirar las láminas del 4XSSC-0,2% Tween, sacudir cuidadosamente en papel de filtro y humedacidas hacia abajo colocar 25 ul. de solución de Ioduro de Propídeo y cubrir con un cubreobjetos de vidrio. Retirar el exceso entre papel de filtro. Solución de Ioduro de Propídeo: 25 ul. de Antifading y 0,1 ul. de Ioduro de Propídeo (50 ug/ml). SOLUCIONES DE USO Paraformaldehído 4%: 4 grs. de paraformaldehído 98 ml. de agua destilada a 60ºC 2 ml. de Hidróxido de Sodio 1M Hidróxido de Sodio 1M: 41 1M= peso molecular en 1000 ml. 40 grs. en 1000 ml. ó 4 grs. en 100 ml. 20XSSC pH 7,0: Cloruro de Sodio 175,6 grs. Citrato de Sodio 2H2O 88,2 grs. Diluir en 500 cc de agua, en agitador, y luego llevar a 1L. Nota: Para la mezcla de hibridización preparar separadamente con agua destilada y deionizada, siendo el volumen de 10 ml. separar en alícuotas y congelar. 2XSSC: 20XSSC 1000 ml. 2XSSC X X= 100 ml de 20XSSC + 900 ml. de agua 4XSSC: 20XSSC 1000 ml. 4XSSC X X= 200 ml de 20XSSC + 800 ml. de agua 4XSSC/0,2 % Tween: 20XSSC 1000 ml. 4XSSC X X= 200 ml de 20XSSC + 800 ml. de agua + 2 ml. de Tween 0,2% 0,1XSSC: 20XSSC 1000 ml. 0,1XSSC X X= 5 ml de 20XSSC + 995 ml. de agua FORMAMIDA DIONIZADA: 450 ml. de Formamida 2 cucharadas de té de resina Resina AG 501/ X8 de Bio-Rad. Colocar en erlenmeyer cubierto con papel de aluminio y llevar al agitador toda la noche ó hasta que tome color dorado. SULFATO DE DEXTRAN 50%: 5 grs. de dextran 10 ml. de agua mili Q colocar en alícuotas (en eppendorffs) y congelar. 42 SDS 10%: 1 gr. de SDS 10 ml. de agua mili Q BSA 5% (Suero de Albúmina de Bovino): Lavar muy bien, un frasco con agua mili Q y preparar una solución de: 2 ml. de 20XSSC + 8 ml. de H2O mili Q + 20 ul. de Tween Gibco= 10 ml Observación: solución A= 4XSSC/Tween 0,2% 0,5 grs. de BSA 10 ml de Solución A Disolver y separar en alícuotas en eppendorffs y congelar. RANsa (10 mg/ml) Sn. Stock 0,01 gr. (10 mg) de RNAsa 1 ml de agua mili Q RANsa de Uso 0,5 ul de RANsa de Sn. Stock 49,5 ul de 2XSSC AVIDINA-FITC/SOLUCIÓN STOCK (500 ug/ml): 5 mg (0,005 gr) 10 ml de BSA 5% en 4XSSC/Tween 20 AVIDINA-FITC/SOLUCIÓN DE USO (5 ug/ml): 10 ul de solución stock 990 ul de BSA 5% Sn. stock = 500 ug/ml Sn. de uso = 5 ug/ml para cada vidrio 43 IV- Cromosomas sexuales Objetivos - IDENTIFICAR y RECONOCER cromosomas sexuales observación de preparaciones citológicas y fotomicrografías. mediante la - IDENTIFICAR y PROPONER diversos sistemas de determinación cromosómica del sexo (mecanismos simples y múltiples) a través del montaje de cariotipos hipotéticos. Recortar las siguientes cuatro páginas por la línea punteada y armar los cariotipos de las especies hipotéticas tal como lo indica el 2º objetivo de este trabajo práctico. (Tomado de Guerra, com. Pers.) 44 V-Mutagénesis: Ensayos Citogenéticos de Genotoxicidad en la evaluación de Mutagénesis Ambiental. En este apartado se incluye información básica para introducir conceptos de mutagénesis y su importancia en ensayos de genotoxicidad y monitoreo ambiental. Mutaciones génicas y cromosómicas causan un serio impacto en la economía y salud humanas. La transformación de la biósfera por acción del hombre es la causa principal del incremento de alteraciones génicas, ya que las actividades industriales, el exceso de desperdicios producidos por grandes concentraciones urbanas, el empleo masivo de agentes químicos en agricultura y las modificaciones de los cursos de agua naturales (construcción de embalses y represas) contribuyen a que se agreguen continuamente al medio ambiente sustancias potencialmente mutagénicas. Cuando ocurre daño genético, aumenta la carga de afecciones hereditarias en las generaciones futuras. Esto da lugar al incremento de un amplio rango de anormalidades (malformaciones congénitas, abortos espontáneos, enfermedades físicas y mentales) que como se producen en generaciones subsiguientes a aquella que recibió el daño, impiden establecer una relación causa-efecto precisa y en consecuencia, su aumento se hace practicamente irreversible. Se conoce que la mayor parte de los agentes carcinogénicos poseen acción mutagénica. En consecuencia cualquier contaminante ambiental, capaz de inducir mutaciones, es un factor potencial de carcinogénesis. Las investigaciones genéticas sobre contaminantes ambientales deben abarcar aquellas sustancias que por su empleo masivo pueden constituir un riesgo serio para la salud humana. Por ende, resultan de interés los contaminantes de origen industrial, los aditivos de alimentos, los plaguicidas, los medicamentos, etc. Otro aspecto importante en esta guía representa el conocimiento de las distintas pruebas de genotoxicidad que pueden emplearse para evaluar mutagénesis y/o carcinogénesis. Estas pruebas pueden agruparse en cuatro niveles a saber: a) Nivel Primario: comprenden el estudio de mutaciones inducidas en bacterias u otros microorganismos tanto de manera directa como con el agregado de microsomas de hígado de rata o ratón para inducir la metabolización “in vitro” de la sustancia ensayada. b) Nivel Secundario: ensayos de mutagénesis en sistemas celulares “in vitro”, también de manera directa o indirecta con el agregado de microsomas. Estos estudios incluyen cultivo de linfocitos humanos. c) Nivel Terciario: Ensayos de mutagénesis “in vivo” utilizando plantas, animales de laboratorio, cuyos resultados pueden extrapolarse en algunos casos al ser humano. d) Nivel Cuaternario: estudios epidemiológicos de mutagénesis en el ser humano. 45 1-Técnicas Citogenéticas de Mutagénesis Ambiental: 1.1-Test de Allium: (Según Rank & Nielssen, 1994 en Andrioli & Mudry, 2002) Las técnicas de citogenética vegetal que se transcriben a continuación han sido tomadas del trabajo de Andrioli & Mudry, (2002). 1-Poner a germinar los bulbos de cebolla en agua de control por 48 horas cambiando el agua cada 24 horas y con aireación. 2-Pasar los bulbos a sendas soluciones de prueba, que son de muestras tomadas en sitios diferentes de un determinado cuerpo de agua o bien dosis de algún compuesto a probar. Rotular. 3-Luego de 24 horas de exposición, cortar tres raicillas de cada bulbo (el resto dejarlas crecer 5 días más). 4-Enjuagar con agua destilada y colocar en fijador Farmer (Etanol-Ac. Acético glacial en proporción 3:1) de 12 a 24 horas. 5-Enjuagar nuevamente y almacenar en etanol 70% en heladera. 6-En el momento de realizar el preparado, colocar las raicillas en vidrio de reloj con ácido clorhídrico 10% para disolver el cemento péctico, durante 5’. 7-Enjuagar con agua destilada. 8-Cortar ápices blancos y colocarlos en un portaobjetos con una gota de colorante orceína lactopropiónica. 9-Colocar el cubreobjetos y hacerlo girar sobre su eje para disgregar el tejido y separar las células. Otra forma consiste en macerar el tejido con el colorante y luego agregar el cubreobjetos. 10-Envolver el preparado en papel de filtro y aplastar con los dedos para realizar el aplastado (“sqash”). 11-rotular los preparados según corresponda y sellar con parafina o adhesivo para cámara de caucho. 12-Observar al microscopio óptico. Otra posibilidad de indagación consiste en realizar monitoreo ambiental y en este sentido el mismo ensayo (arriba descrito) nos permite abordarlo. Para monotoreo ambiental se hace necesario: El ensayo consiste en medir raicillas que se dejaron crecer durante 5 días más. 1-Por cada bulbo, medir 10 raicillas. 2-Establecer el promedio para cada muestra y el control. 3-Hacer un gráfico comparativo (gráfico de barras) con las medias de longitud de las raíces en el eje de ordenadas y la estación control en el eje de abscisas. 4-Establecer la comparación entre las distintas muestras, analizando si el crecimiento de las raíces en las muestras 1 y 2 poseen diferencias mayores al 50% en relación al control. 5-realizar una curva colocando las muestras en orden creciente de acuerdo a los valores de longitud de las raíces procediendo luego a la interpretación de la curva, relacionando las características de la muestra con los valores de crecimiento de las raíces. 46 1.2.1-Ensayo de Micronúcleos (en ratón): (Según Schmid, 1975) 1-administrar por vía intraperitoneal o “per os”, el compuesto a ser examinado. El tiempo de acción dependerá de si se trata de un ensayo agudo o crónico. 2-Sacrificar al animal mediantes vapores de éter o bien por dislocación cervical. 3-Extraer fémures, descarnarlos y cortar las epífisis a efectos de localizar el canal medular. 4-Extraer (por presión) la médula ósea mediante la ayuda de una aguja que contiene 0,2 ml de suero bovino fetal. 5-Lograr una fina suspensión entre el suero y la médula, resuspendiendo. 6-Colocar la suspensión en un tubo de centrífuga y centrifugar a 1000 rpm. Durante 5’. Extraer el sobrenadante con una pipeta Pasteur. 7-Mezclar las células, cuidadosamente, y realizar los frotis de la siguiente manera: 8-Gotear sobre un portaobjetos la suspensión obtenida y con otro portaobjetos extenderla de manera homogénea, como si se tratara de un frotis sanguíneo. 9-Dejar secar 24 horas y luego colorear con coloración diferencial de MayGrunwald/Giemsa. 10-Colorear con May-Grunwald durante 3’; luego 2’ con May-Grunwald diluído con agua destilada en proporción 1:1 y posteriormente con Giemsa diluído en agua destilada en proporción 1:6 durante 10’. Una vez secos los preparados se montan para luego ser analizados al microscopio óptico. 11-Analizar 1000 células por animal. Un 8% de las células que se observen serán eritrocitos, de los cuales la mitad deben ser eritrocitos policromáticos. 1.2.2-Ensayo de Micronúcleos en Peces: (Tomado de Ale, 2003) Cabe señalar que en el monitoreo ambiental del medio acuático, varios modelos biológicos pueden ser utilizados. Entre ellos se destacan como muy efectivos Peces, Reptiles y Moluscos. Uno de los más usados son los Peces, sea en ensayos de laboratorio como en la evaluación directa del ambiente acuático (monitoreo ambiental). En el caso de evaluar el potencial mutagénico de algún contaminante en particular, los experimentos se realizan de manera controlada. En primer lugar debe mantenerse en cuarentena a los animales motivo del experimento. Con esto se logra la detoxificación de los animales a ser tratados y a los que se tomará como grupo control. Durante este lapso se les debe administrar alimento, cambiar el agua de los acuarios y controlar la aireación. Para los experimentos se aconseja el uso de 10 animales por lote experimental (a los efectos de garantizar una muestra estadística mínima). El tiempo de acción del mutágeno dependerá de si se trata de una exposición aguda o crónica y las dosis deberán ser mínimas a los efectos de afectar al material genético sin producir mayor efecto tóxico. La técnica consiste en: 1-Obtener sangre por punción caudal con aguja y jeringa heparinizada. 2-Realizar un frotis y dejar secar al aire (hacer 2 o 3 preparados por animal). 3-Fijar con Alcohol metílico durante 10 a 15’. 4-Colorear con Giemsa al 10% y enjuagar. 47 5-Observar y analizar 2000 eritrocitos por individuo. Nota: los eritrocitos de los peces son nucleados, con el citoplasma de forma ovalada. El núcleo se ubica en el centro y los micronúcleos pueden posicionarse en cualquier lugar de la célula pero presentan coloración y consistencia propios del material nuclear. 1.2.2-Ensayo de Micronúcleos en Humanos: (Tomado de Kornoski, 2006) Incubación Se trabaja con cultivo de linfocitos humanos con bloque de la citocinesis. Para obtener linfocitos binucleados, se adiciona a cada tubo de cultivo 0,15 ml de Citocalasina B, a las 48 Hs. de iniciada la siembra de sangre. Cosecha 1-Al completar 72 Hs., el cultivo es interrumpido sometiéndolo a centrifugación a 800 rpm durante 10 minutos, luego se descartó el sobrenadante y se agregó a cada tubo 8 ml de solución hipotónica a la que se dejó actuar en estufa a 37ºC durante 5 minutos, a continuación se interrumpió el cultivo, incorporando a cada tubo 0,5 ml de fijador Farmer, seguidamente se procedió a la centrifugación a 800 rpm durante 10 minutos y se descartó el sobrenadante. 2-Posteriormente, a cada tubo se agregó 7 ml de fijador, en esta etapa se guardaron las muestras en freezer durante tiempo indeterminado o bien se dejó a temperatura ambiente durante 20 minutos para realizar los lavados posteriores. 3-Los lavados consistieron en centrifugar el material a 800 rpm durante 10 minutos, se descartó el sobrenadante, nuevamente se agregó aproximadamente 4 ml de fijador resuspendiendo el pellet, luego se centrifugó a 800 rpm durante 10 minutos, descartándose el sobrenadante y así sucesivamente se realizaron tres o más lavados hasta obtener un precipitado limpio con una adecuada suspensión celular. 4-Preparación de láminas: los portabojetos deben estar bien limpios para realizar el goteo; se agregan tres o cuatro gotas, se secan sobre el calor del mechera. Coloración Las láminas se introdujeron en un copling cuyo interior contenía solución de colorante Giemsa (diluído en buffer Sorensen), durante 8 minutos. Mediante esta coloración se pueden diferenciar claramente, el contraste en el material nuclear respecto al citoplasma, y así distinguir la presencia o no de micronúcleos. Finalmente, una vez coloreados, se procedió al análisis de 1000 linfocitos binucleados (LB), por cada individuo expuesto y no expuesto. 1.3-Alteraciones Cromosómicas en Metafase: 48 En este caso y tomando los recaudos planteados en el punto 1.2.2 de este apartado, la técnica de suspensión celular mediante cultivo de células renales o bien procesadas de forma indirecta, es la que se utiliza para analizar alteraciones cromosómicas. Esta técnica es la misma que fuera descripta en el apartado II. Sea se trate de Peces o de Roedores. 1.4-Intercambio de Cromátidas Hermanas: (Según Korenberg and Freedlander, 1974) Generalmente, esta técnica se realiza en condiciones de cultivo de células de sangre entera, sea de animales domésticos, humanos, etc. 1-Adicionar 5-BrdU (2 mg/ml) al cultivo de linfocitos, agitando el tubo o frasco, suavemente. 2-Dejar actuar durante 18-22 hs. 3-Cosechar las células como se describió en el apartado III, ítem 3.2.1. 4-Dejar envejecer las láminas durante 5-7 días en un lugar oscuro y seco. 5-Trancurrido este tiempo cubrir las láminas con una solución de Hoechst 33258 (1 mg. De Hoechst/1 ml de metanol/100 ml. de 0,5 XSSC) y exponer a la luz ultravioleta cerca de una hora. 6-Lavar en agua destilada y tratar con tampón fosfato pH 8 a 87-89ºC, durante 20’’ a 3’, dependiendo de la preparación. 7-Colorear con solución de Giemsa a 4% durante 3 a 5’. 8-Obesrvar al microscopio óptico y realizar el conteo de los intercambios producidos. 1.5-Ensayo Cometa: (Según Singh et al., 1988) Esta técnica descripta por SINGH et al., (1988), con algunas modificaciones, consiste en varios pasos: 1.5.1-Preparación de las láminas con agarosa normal : 1-Disolver agarosa normal 1,5% (300mg. en 20ml de PBS) en un erlenmeyer dejándose en agitación por dos horas, para hervirla después. 2-Dejar solidificar a temperatura ambiente., para ser totalmente cortada y nuevamente llevada a hervir en microondas. Repetir esta etapa tres veces. Al final de esta operación mantener la agarosa en baño maría (60C). 3-Sumergir las láminas en agarosa caliente y retirar luego de esta solución. Uno de los lados de la lámina debe limpiarse con papel. 4-Dejar las láminas en una superficie plana y a temperatura ambiente overnight para solidificar la capa de agarosa y luego guardarlas en la heladera. Nota: estos pasos pemiten preparar los portaobjetos unos dias antes. Para realizar el ensayo proceder de la siguiente manera: 49 1.5.2-Toma de muestra: 1-Extrae sangre por vía caudal con aguja y jeringa heparinizada colocándola en un microtubo ependorff. 2-Recolectar 10l de sangre de cada tubo y mezclar con 1ml de medio de cultivo; de esta mezcla extraer una alícuota de 10l. 1.5.3-Siembra de la alícuota con LMP en láminas con agarosa normal : 1-Preparar agarosa de bajo punto de fusión (LMP) al 0,5% (100mg. en 20 ml PBS), hervir y luego mantener en baño maría a 37C. 2-Mezclar y homogeneizar en un ependorff 120l agarosa de bajo punto de fusión con 10l de la alícuota, sembrar esta mezcla sobre el portaobjeto con agarosa normal y cubrir con un cubreobjetos. Refrigerar durante 10 min. 3-Retirar los cubreobjetos. 1.5.4-Lisis : 1-Colocar los preparados en coplines y adicionar la solución de lisis (2.5 M NaCl, 100mM EDTA, 10mM de Tris, NaOH pH 10, 10% de DMSO y 1% de Triton X-100). 2-Refrigerar después en heladera (4C), durante una hora. 3-Remover los preparados y transferirlos a una cuba de electroforesis horizontal. 1.5.5-Desnaturalización y Electroforesis: 1.Depositar la cuba en una caja plástica para permitir la refrigeración con hielo, el cual se coloca a su alrededor, para mantener todo el proceso de desespiralización del ADN y la propia corrida a una temperatura de 4C. 2-Dentro de la cuba de electroforesis, adicionar la solución de electroforesis (300mM NaOH y 1 mM EDTA pH13) de manera de cubrir todas las láminas con una fina capa de solución. 3-Antes del inicio de la corrida, dejar las láminas en la solución de electroforesis por 25 minutos, para permitir la desespiralización del ADN. 4-Corrida electroforética: a 25 v y 300mA. durante 25 min. 5-Neutralizar los preparados con 5 ml de solución de neutralización (0.4 M Tris pH: 7.5). en posición horizontal por 5 min., este procedimiento se repite tres veces. 6-Secar las láminas a temperatura ambiente. 7-Fijar con etanol por 5 min. y secar a temperatura ambiente. 1.5.6-Coloración y Análisis: 1-Colocar 100l de bromuro de etídio al 10% (20g/ml) en cada lámina, y se cubrir con un cubreobjeto. 2-Observar en aumento 400x usando microscopio de fluorescencia equipado con filtro de excitación de 515-560 nm y un filtro de barrera de 590 nm. De acuerdo con la longitud de la cola se clasifican en cuatro clases: clase 0- sin 50 detección de daño en el ADN ni cola; clase 1- una cola con una longitud menor que el diámetro del núcleo; clase 2- una cola con una longitud entre uno y dos veces el diámetro del núcleo; clase 3- una cola con una longitud dos veces mayor que el diámetro del núcleo. Se procede entonces a cuantificar los tipos de daños en cada lámina y la atribución de valores de frecuencia en cada clase. Las frecuencias se obtienen multiplicando el número de cometas encontrados en cada clase por el valor de la clase. Se realiza un preparado por tilapia, analizándose en cada uno 100 células. 1.5.7-Análisis Estadístico: a los resultados obtenidos en ambos tests se aplican los estadísticos usuales en este tipo de estudios, media, desvío estándar y análisis de la varianza. 51 BIBLIOGRAFIA Ale, E. (2003): Análisis de un modelo de ensayo de genotoxicidad en Peces (Oreochromis niloticus, Cichlidae) con (NO3)2Pb como contaminante. Tesis de Licencitura, págs. 1-87. FCEQyN. Univ. Nac. de Misiones. Posadas, Misiones. Argentina. Ale, E.; Fenocchio, A.S.; Pastori, M.C.; Oliveira Ribeiro, C.; Cestari, M.M.; Zacharzewski, C. (2004): Evaluation of effects of ((NO3)2Pb) on Oreochromis niloticus (Pisces, Cichlidae) by means of Cytogenetic Techniques. Cytologia 69 (4): 453-458. Andrioli, N.; Mudry, M.D. (2002): El uso de bioindicadores en el diagnóstico de la calidad ambiental: su aplicación en la práctica escolar. Vol. 5, Nº 27-33. Edit. Científica Universitaria, Universitas, Córdoba. Barch,M.J. ed. (1991): The act cytogenetics laboratory manual. Second edition. Raven Press. E.E.U.U. Beçak, W. e Paulete, J. (1976): Técnicas de Citologia e Histología. Vol. 1ª ed. Livros técnicos e científicos. Editora SA. Río de Janeiro. Brasil. Bertollo, L. A. C., Takahashi, C. S. and Moreira Filho, O. 1978. Cytotaxonomy considerations on Hoplias lacerdae (Pisces: Erythrynidae) Brazil. J. Genet., 2: 17-37. Dulout, F.N.; Pastori, M.C.; Olivero, O.A.; Von Guradze, H.N. (1982): Los ensayos citogenéticos in vivo en estudios de mutagénesis química. Actas V Congreso Latinoam. de Genética, págs. 209-225. Egozcue, J. (1971): Técnicas en citogenética. Edit. Espaxs. Barcelona. España. Green, D.M. and Sessions, S.K. eds. (1991): Amphibian cytogenetics and evolution. Academic Press , Inc. E.E.U.U. Guerra, M. (1988): Introduçao à citogenética Geral. 1ª ed. Editora Guanabara SA. Río de Janeiro, RJ Brasil. Guerra, M; De Souza, M.J. (2002): Como observar Cromossomos. Um guia de técnicas em Citogenética Vegetal, Animal y Humana. Editora Funpec. Ribeirão Preto. Brasil. Korenberg, J.R. and Freedlander, E.F. (1974): Giemsa Technique for the detection of sister chromatid exchanges. Chromosoma, 48: 355-360 Obe, G. and Basler A. eds. (1987): Cytogenetics. Springer-verlag. Berlin Heidelberg. Germany. 52 Ribeiro, L:R; Salvadori, D.M.F.; Marques, E.K. (2003): Mutagênese Ambiental. Editora da ULBRA, Canoas, RS. Brasil. Rooney, D.E. and Czepulkowski, B.H. eds. (1992): Human cytogenetics. A practical approach, vol. 1,second edition. Oxford University Press. N.Y. Schmid, W. (1975): The micronucleus test. Mutation Research, 31: 9-15. Singh, N. P., Mc Coy, M.T., Tice, R.R. and Scheider, E.L..(1988): A simple technique for quantitation of low levels of DNA damage in individuals cells. Exp. Cell. Res. 237 (3-4) pp 123-130 Verma, S., Babu, M. (1995): Human Chromosomes. Principles and techniques. 2d ed. Mc Graww Hill 53