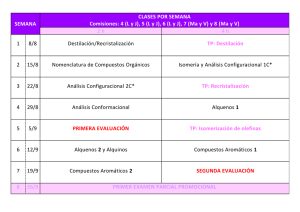
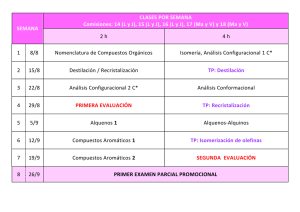
Descargar archivo adjunto - Almagro
Anuncio

QUÍMICA ORGÁNICA GUÍA DE TRABAJOS PRÁCTICOS ESCUELA TÉCNICA ORT 5° QUÍMICA 2015 PROF: ROMINA TORRES PURIFICACIÓN DE COMPUESTOS ORGÁNICOS SÓLIDOS RECRISTALIZACIÓN INTRODUCCIÓN Hoy día esta técnica se mantiene como el procedimiento más adecuado para la purificación de sustancias sólidas. En general, la purificación por recristalización se basa en el hecho de que la mayoría de los sólidos son más solubles en un disolvente en caliente que en frío. El sólido que se va a purificar se disuelve en el disolvente caliente, generalmente a ebullición, la mezcla caliente se filtra en caliente para eliminar todas las impurezas insolubles en caliente, y entonces la solución se deja enfriar para que se produzca la cristalización. En el caso ideal, toda la sustancia deseada debe separarse en forma cristalina y todas las impurezas solubles deben quedar disueltas en las aguas madres. Finalmente, los cristales se separan por filtración al vacío y se dejan secar. Si con una recristalización sencilla no se llega a una sustancia pura, el proceso puede repetirse empleando el mismo u otro disolvente. • Elección del disolvente La mejor forma de encontrar un disolvente adecuado para la recristalización de una sustancia determinada es ensayar experimentalmente distintos disolventes. No obstante, algunas generalizaciones, razonablemente válidas, pueden ayudar a simplificar la búsqueda. 1. Los compuestos iónicos (sales) se disuelven en disolventes polares y los compuestos no iónicos (orgánicos) en disolventes no polares. 2. Los compuestos no iónicos se pueden disolver en agua si sus moléculas se ionizan en solución acuosa o puedan asociarse con moléculas de agua a través de puentes de hidrógeno. Por este motivo, los hidrocarburos y sus derivados halogenados son prácticamente insolubles en agua, pero los compuestos en cuyas moléculas existen grupos funcionales tales como alcohol (-OH), aldehído (-CHO), cetona (R-CO-R), ácido carboxílico (-COOH) y amida (-CONH2)], que pueden formar puentes de hidrógeno con agua, son solubles en este disolvente, a menos que la relación del número total de átomos de carbono al de tales grupos funcionales en la molécula sea superior a 4 ó 5. 3. Los disolventes hidroxílicos asociados como metanol, etanol, ácido acético, presentan un orden intermedio entre agua y el éter etílico o benceno. Son buenos disolventes para los compuestos orgánicos que pueden asociarse. Un disolvente ideal para una recristalización debe poseer las siguientes características: a) Un coeficiente de temperatura elevado para la sustancia que se va a purificar, esto es, debe disolver una gran cantidad de la misma a su temperatura de ebullición y sólo una pequeña cantidad a la temperatura ambiente o ligeramente por debajo de ella. Disolver en caliente la muestra. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 2 b) No disolver las impurezas en caliente. c) Al enfriarse debe suministrar rápidamente cristales bien formados del compuesto que se purifica, de los cuales debe ser fácilmente separable. d) No debe reaccionar con el soluto. e) Su utilización no debe ser peligrosa (inflamable). f) Debe ser barato • Preparación de la solución Como regla general, el objetivo es disolver el soluto en la mínima cantidad de disolvente a su temperatura de ebullición. Se recomienda el siguiente procedimiento (Figura 4): el compuesto a recristalizar, finamente pulverizado, se coloca en un balón de fondo redondo del tamaño adecuado al que se acopla un refrigerante de reflujo. Se echa un trocito de plato poroso para evitar las proyecciones del solvente y se cubre el sólido con un volumen del disolvente elegido que se juzgue todavía insuficiente para disolverlo totalmente. Sobre un baño de agua (o directamente sobre la placa calefactora si el disolvente tiene un punto de ebullición mayor que el del agua) se calienta la mezcla hasta ebullición, agitando constantemente al comunicar al líquido un movimiento de giro. A la solución hirviente se añade más disolvente en pequeñas porciones y continuando la agitación. Se continúa la adición de disolvente por la parte superior hasta que todo el soluto se ha disuelto a la temperatura de ebullición. Figura 4. Disolución del sólido, aparato de calentamiento a reflujo • Decoloración Frecuentemente la solución se colorea con impurezas orgánicas de peso molecular elevado que acompañan al producto natural deseado o que se han formado como productos de descomposición o subproductos en el proceso de síntesis. En estos casos el color se puede eliminar hirviendo la solución durante cinco o diez minutos con una pequeña cantidad de carbón adsorbente activado. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 3 Filtración de la solución caliente La solución caliente se debe filtrar de tal forma que no cristalice nada de soluto ni en el papel de filtro ni en el embudo. Generalmente, para ello se requiere una filtración rápida con un mínimo de evaporación en el embudo de vástago corto, previamente calentado en una estufa, y provisto de un filtro de pliegues para aumentar la velocidad de filtración (Figuras 5 y 6). Figura 5. Filtración en caliente a través de un filtro de pliegues QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 4 • Enfriamiento Durante el enfriamiento de la solución caliente se pretende que cristalice la máxima cantidad de la sustancia deseada con un mínimo de impurezas. El proceso se realiza en un matraz erlenmeyer. Generalmente, es preferible que los cristales tengan un tamaño medio, porque los cristales grandes pueden incluir gran cantidad de disolvente, el cual lleva impurezas disueltas, y los cristales pequeños presentan una gran superficie sobre la que éstas quedan adsorbidas. El tamaño de los cristales se puede controlar por la velocidad de cristalización; una cristalización rápida favorece la formación de cristales pequeños y una cristalización lenta origina cristales grandes. Generalmente lo mejor es dejar que el enfriamiento de la disolución sea lento o al menos moderado. Si la cristalización es demasiado lenta, se puede favorecer rascando con una varilla de vidrio la superficie interior del erlenmeyer (para que se formen pequeñísimos fragmentos de vidrio que actúen como núcleos de cristalización), o bien, añadiendo, durante el enfriamiento y de vez en cuando, un pequeño cristal del producto para sembrar la solución y provocar su cristalización. • Separación de los cristales En este paso se pretende separar los cristales formados, quitándoles la mayor cantidad posible de aguas madres, con una evaporación mínima. Generalmente esto se consigue empleando un embudo Büchner unido a un kitasato, que a su vez se conecta a la trompa de vacío (Figura 7). Los kitasatos deberán sujetarse mediante unas pinzas a un soporte. El Büchner debe ser de tamaño adecuado, eligiéndose el más pequeño que permita la recogida con holgura de toda la masa cristalina sin que ésta llegue a rebosar el borde superior del embudo. Figura 7. Filtración en frio a través de un embudo Buchner El papel de filtro debe cubrir por completo todos los orificios de la placa del Büchner, pero su diámetro debe ser ligeramente inferior al de esta placa. Al colocarlo debe quedar QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 5 completamente liso y sin arrugas para que no pueda pasar nada de sólido por sus bordes. Esto se consigue fácilmente humedeciendo el papel con disolvente y haciendo succión. Después, sin succión, o mejor, sólo con una ligera succión, para evitar evaporaciones innecesarias, se echa la mezcla (o parte de ella) dentro del embudo. Entonces se aplica todo el vacío (o el máximo deseado). Se debe utilizar una varilla de vidrio o una espátula para que, ayudándose con ella, se pueda pasar lo más rápidamente posible toda la masa cristalina al embudo. Si algunos cristales quedan adheridos a las paredes del erlenmeyer, se pueden lavar y echar en el embudo con pequeñas cantidades del disolvente frío. Tan pronto como la masa sólida se hace suficientemente rígida, se presiona, con cuidado pero con firmeza, con un corcho o tapón de frasco invertido. Cuando cesa el paso de líquido a través del filtro se interrumpe la succión. En este momento, si el filtrado tiene valor, se deberá transferir a otro recipiente. Entonces se procederá al lavado de los cristales para eliminar todo el disolvente que llevan adherido (que, desde luego, contendrá impurezas solubles). Sin succión, se cubrirán los cristales con una pequeña cantidad de disolvente puro y frío. En este momento conviene agitar la mezcla cuidadosamente, para no romper el papel de filtro, con una espátula o varilla de vidrio roma para asegurar que todos los cristales se humedecen con el disolvente. Entonces se aplica de nuevo la succión y los cristales se presionan con un tapón como antes. Este proceso se puede repetir varias veces. Con frecuencia, por concentración de las aguas madres (filtrado) se puede obtener una nueva cantidad de cristales. Sin embargo, éstos son casi siempre algo menos puros que los cristales obtenidos en primer lugar. • Secado de los cristales Como paso final de la recristalización, los cristales obtenidos deben quedar libres del disolvente adherido mediante un secado. Entonces se pasan los cristales a un vidrio de reloj limpio o una cápsula plana. En estas condiciones se pueden dejar secar al aire a la temperatura ambiente o se pueden introducir en un desecador de vacío sobre un desecante que sea eficaz para eliminar el disolvente usado. El secado a temperaturas superiores a la ambiente se puede realizar en una estufa. Se debe tener en cuenta que las muestras al principio están humedecidas con el disolvente y que, por tanto, fundirán a una temperatura inferior a la del punto de fusión de la sustancia pura. Tambien se pueden dejar secar cerca de un mechero con el trípode y tela metálica, se coloca la muestra cerca de la parte inferior del mechero y se deja secar. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 6 • Recristalización de una sustancia empleando una mezcla de disolventes Con frecuencia se encuentra que una sustancia es demasiado soluble en un disolvente y demasiado poco soluble en otro para realizar una recristalización de la misma. Entonces se pueden utilizar, frecuentemente con buen resultado, pares de disolventes tales como alcohol metílico-agua, alcohol etílico-agua, éter-acetona y benceno-ligroína. En estos casos, el compuesto se disuelve en el disolvente, en el que es muy soluble (a su temperatura de ebullición o ligeramente por debajo de ésta), y entonces se añade, gota a gota y caliente, el otro disolvente en el que la sustancia es sólo ligeramente soluble, hasta que aparece una tenue turbidez persistente. Se añaden entonces unas gotas del otro disolvente para eliminar la turbidez y la solución se deja enfriar de la forma habitual. • Puntos de fusión El punto de fusión de un sólido es la temperatura a la que el sólido se transforma en líquido a la presión de una atmósfera. En una sustancia pura el cambio de estado es muy rápido y la temperatura es característica. Por eso, el punto de fusión es un dato utilizado en la identificación de un sólido. Además, debido a que el punto de fusión se altera sensiblemente por la presencia de impurezas (rango mayor y menor valor), esta constante constituye un valioso criterio de pureza. En general, un sólido puro (rango de 0,5-1°C) funde en un intervalo muy pequeño de temperatura y con un límite superior muy próximo al punto de fusión verdadero. El punto de fusión mezcla consiste en mezclar la sustancia (en proporción 9:1), 9 partes de una sustancia pura que yo sospecho que es mi muestra y 1 parte de la muestra recristalizada. Sólo se mantendrá la constancia del punto de fusión en el caso de que se trate de la misma sustancia, pues las que son diferentes actuarán como impurezas al determinar los puntos de fusión, éstos serán más bajos y de rango mayor. • Determinación experimental de puntos de fusión La determinación de puntos de fusión se lleva a cabo en un balón Kjeldhal (marcianito) o en un aparato de punto de fusión eléctrico. Para determinar el punto de fusión de un sólido se introduce una pequeña muestra (ver Figura 8) en un capilar cerrado de un extremo, se coloca el capilar cerca del bulbo del termómetro, se calienta el baño de glicerina lentamente y se observa la temperatura a la que la fusión comienza y se completa. El intervalo de fusión para una muestra pura suela estar comprendido entre 1-2 ºC. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 7 Balón de Kjeldhal (marcianito) La muestra sólida deberá estar seca y finamente dividida. El sólido sólo deberá ocupar en el capilar una altura aproximada de medio centímetro. Los capilares para punto de fusión deben ser de paredes finas, de 1 ó 2 mm de diámetro y cerrados por un extremo. En caso contrario, la determinación del punto de fusión va acompañada de un considerable error. En el caso de un producto conocido, la calefacción será rápida al principio hasta llegar a unos 10 ºC por debajo del punto de fusión y después lenta, de modo que la velocidad de calentamiento sea inferior a 2 ºC/min. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 8 TRABAJO PRÁCTICO N °1 RECRISTALIZACIÓN E IDENTIFICACIÓN DE UNA SUTANCIA ORGÁNICA SÓLIDA Este Trabajo Práctico consta de tres partes: en la primera se seleccionará el solvente adecuado para la recristalización de una sustancia X y se tomará su punto de fusión. Una vez seleccionado el solvente, segunda parte, se realizará la recristalización (purificación) de una cantidad significativa de la sustancia incógnita. Finalmente, tercera parte, se identificará la sustancia tomando el punto de fusión luego de recristalizada la muestra y comparar dicho punto con tablas. Parte A: Elección del solvente de recristalización Materiales: 1 mechero 1 tela metálica 1 trípode varilla de vidrio 4 tubos de ensayo 1 gradilla 1 vaso de precipitados Reactivos: 5 – 10 ml de cada uno de los solventes a ensayar. Características de los solventes Agua Etanol acetona tolueno p.e. 100ºC p.e. 78ºC p.e. 58ºC p.e. 111ºC Inflamable Inflamable inflamable Descripción de la técnica: - Identificar con letras los tubos. Colocar una punta de espátula (10-20 mg) de la sustancia a ensayar en un tubo de ensayo, agregar gota a gota 1-2 ml de solvente agitando con varilla de vidrio. Observar si se disuelve total o parcialmente. Si no se disuelve totalmente calentar en un baño María con agua caliente a una temperatura cercana a su punto de ebullición mantener lejos de cualquier llama cuando se emplean solventes inflamables. Una vez que hierve el solvente retirarlo rápidamente del baño María. En el caso en que la sustancia se disuelva en caliente, enfriar y observar si precipita, raspar con cuidado las paredes del tubo con la varilla de vidrio si es necesario. Expresión de los resultados: Para cada sustancia ensayada pueden anotarse los resultados de esta manera: (COMPLETAR s=soluble, ps=poco soluble, i=insoluble) Sustancia “X” Agua Etanol tolueno acetona En frio En caliente Precipita al enfriar QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 9 El mejor solvente será aquel que tenga las siguientes condiciones para la muestra Buena solubilidad en caliente Baja solubilidad en frío Facilidad de eliminación por lavado o evaporación Formación de buenos cristales al enfriar - Tomar el p.f. de la sustancia incógnita. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 10 Parte B: Recristalización Materiales: 1 tubo de crioscopia o vaso de 150 ml 1 vaso de precipitados 1 mechero 1 tela metálica 1 trípode varilla de vidrio embudo y papel de filtro soporte metálico capilares para punto de fusión plato poroso Reactivos: Sustancia a recristalizar. Solvente elegido: cantidad necesaria Técnica: Colocar la sustancia a recristalizar en el tubo de crioscopia o balón, agregar solvente hasta cubrir la muestra. Agitar el tubo sumergido en un baño de agua (la un temperatura del baño debe estar unos grados por debajo del p.e. del solvente) si el solvente es agua se puede calentar a fuego directo. Si se utiliza el aparato de calentamiento a reflujo conectar el balón al refrigerante. Si es necesario agregar un poco más de solvente mezclando luego de cada agregado hasta lograr disolver toda la muestra en la menor cantidad de solvente posible. Filtrar en caliente. Enfriar y filtrar al vacío, lavar el filtrado con una pequeña cantidad de solvente en frío. Secar unos mg de sustancia para tomar p.f., bajo el mechero, comparar con el valor inicial. Repetir la recristalización hasta obtener p.f. constante después de dos recristalizaciones sucesivas. Comparar el p.f. con el de tablas para establecer cuál es la sustancia incógnita. Si fuese necesario confirmar con un p.f. mezcla. Presentación de los resultados: p.f. inicial: p.f. 1ª recristalización: Sustancia incógnita: p.f. de tablas: Mezcla de solventes: Hay sustancias que cristalizan con dificultad cuando se encuentran impurificadas y al enfriar sus soluciones tienden a dar líquidos viscosos aceites. Salvo casos raros solo ocurre con sólidos de p.f. inferiores a 100 ºC. Para conseguir que cristalicen a partir de estos aceites, puede disolverse la muestra a temperatura ambiente o como máximo 30-35 ºC utilizando tanto solvente como sea necesario. Agregar entonces gota a gota y agitando otro solvente en el cual la muestra sea insoluble. Cuando la turbidez se mantenga raspar las paredes del tubo con una varilla y enfriar a baño de QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 11 hielo. Filtrar la solución La sustancia a purificar puede contener distintos tipos de impurezas: a. Impureza incolora soluble en el solvente frió y en caliente b. Impureza incolora insoluble en caliente c. Impureza soluble coloreada En el caso (a) la técnica se realiza sin necesidad del filtrado en caliente pues las impurezas permanecen en las aguas madres. En el caso (b) y (c) se hace necesaria la filtración en caliente para separar las impurezas del compuesto que se encuentra en solución. En particular el caso (c) se hace necesaria la filtración con carbón activado. Medios filtrantes Existen diversos tipos de medios filtrantes, el más común de los cuales es el papel de filtro que puede encontrarse en el comercio con distintos grados de porosidad según el tipo de precipitado a filtrar. Puede utilizarse también lana de vidrio que es útil en el caso de tener cristales grandes suspendidos en un líquido que podría atacar el papel de filtro (por ej. NaOH (c) o Ácidos (c)). Se pueden utilizar crisoles de Gooch (crisol filtrante) y filtrar por succión. Ambos métodos se utilizan cuando lo que se desea es el líquido filtrado pues resulta difícil separar los cristales. Otro de los medios de filtración consiste en placas porosas de vidrio fritado o porcelana que se utilizan ajustadas a un embudo especial. Vienen con distintos grados de porosidad y son inertes a reactivos químicos. Lamentablemente no son adecuados para precipitados gelatinosos pues se utilizan con succión y los poros se obturan. Una desventaja adicional consiste en su difícil limpieza. Métodos de filtración en caliente La filtración en caliente se utiliza cuando se desea separar las impurezas insolubles en el solvente utilizado de la solución de la sustancia a purificar. Filtración en caliente: a) Simple para impurezas sólidas filtrables por papel de filtro. b) Lecho de celite para impurezas que podrían pasar por el papel de filtro (soluciones turbias, impurezas en suspensión, etc.) c) Con carbón decolorante (activado) impurezas coloreadas solubles. El lecho se utiliza para retener el carbón. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 12 Recomendaciones Para el filtrado en caliente debe tenerse en cuenta la precaución de calentar el embudo antes de filtrar. Para ello se lo puede sumergir en un baño de agua hirviendo o bien colocarlo en una estufa eléctrica a temperatura adecuada de donde se lo retira en el momento de filtrar. (Si no se utiliza agua como solvente el embudo debe secarse). Mantener la solución a filtrar caliente durante toda la operación y una vez pasado todo el líquido enjuagar el recipiente con pequeñas porciones del solvente caliente. Filtración por succión. Recomendaciones. Puede utilizarse el embudo Buchner o el Hirsch, dependiendo si el volumen de líquido es grande o chico respectivamente. Al aplicar succión puede producirse una rápida evaporación del solvente provocando la recristalización del compuesto en el vástago del embudo. Por ello colocar una cantidad adicional de solvente en la solución antes de filtrar. El filtrado se calienta nuevamente y se redisuelven los cristales; de esta manera entran en solución aquellos que quedaron adheridos en las paredes del recipiente. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 13 CUESTIONARIO 1- ¿Qué es el punto de fusión de una sustancia? 2- ¿Puede utilizar el punto de fusión como criterio de pureza? ¿Y como criterio de identidad? 3- Describa el balón de Kjeldhal modificado para determinar punto de fusión. 4- ¿Qué se entiende por recristalización? 5- ¿Qué tipo de solventes disuelven sustancias polares? Justifique. ¿Y sustancias no polares? 6- ¿Qué condiciones debe reunir un solvente para ser utilizado en una recristalización? 7- ¿Cómo procede para seleccionar el solvente adecuado? 8- Describa detalladamente el procedimiento para recristalizar una sustancia una vez seleccionado el solvente a utilizar. 9- ¿En qué casos se utiliza una mezcla de solventes para recristalizar? ¿Qué condiciones deben cumplir los solventes a mezclar? 10- ¿En qué casos utiliza carbón activado? ¿En qué cantidad debe agregarlo? 11-¿Cómo procede para recristalizar una muestra si el solvente adecuado es inflamable? 12-¿Qué funciones cumple la filtración en caliente? ¿Por qué utiliza en este caso papel plegado? 13-¿Cómo debe efectuarse el enfriamiento de la solución filtrada para una óptima cristalización? 14-¿Cómo procede si en la solución filtrada no se produce cristalización? 15-¿Cómo procede para separar los cristales obtenidos de las aguas madres? 16-¿Cómo procede a lavar los cristales? ¿Con qué solvente lava? 17-¿Cómo procede para secar el sólido obtenido? 18-Indique si estas afirmaciones son V o F: a. La filtración al vacío se utiliza para recuperar el líquido filtrado. b. Para filtrar en caliente es conveniente calentar previamente el embudo. c. De ser posible la elección de dos solventes para una recristalización, conviene elegir el menos volátil. 19- Qué solvente elegiría para purificar la sustancia A de la siguiente lista. Justifique su elección. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 14 Solvente 1 2 3 A frío i. caliente p.s. frío i. caliente s. frío s. caliente s. 20- ¿Cómo procedería en una recristalización si al calentar la muestra con el solvente obtiene: a. una solución coloreada sin impurezas sólidas en caliente, si la sustancia a recristalizar es incolora? b. una solución coloreada con impurezas sólidas en caliente, si la sustancia a recristalizar es coloreada? c. una solución incolora con impurezas sólidas en caliente? d. una solución coloreada sin impurezas sólidas en caliente, si la sustancia a recristalizar es coloreada? e. una solución coloreada con impurezas sólidas en caliente, si la sustancia a recristalizar es incolora? QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 15 DESTILACIÓN INTRODUCCIÓN La destilación constituye el método más frecuente e importante para la purificación de líquidos. Se utiliza siempre en la separación de un líquido de sus impurezas no volátiles y, cuando ello es posible, en la separación de dos o más líquidos. Líquidos puros Cuando un líquido puro se introduce en un recipiente cerrado y vacío parte del mismo se evapora hasta que el vapor alcanza una determinada presión, que depende solamente de la temperatura. Esta presión, que es la ejercida por el vapor en equilibrio con el líquido, es la presión de vapor del líquido a esa temperatura. Cuando la temperatura aumenta, la presión de vapor también aumenta regularmente hasta que llega un momento en que la presión de vapor alcanza el valor de 760 mm, entonces, si el líquido está en contacto en el exterior, comienza a hervir. La temperatura a la que esto ocurre recibe el nombre de punto de ebullición normal del líquido en cuestión, y es una constante característica para cada líquido. Mezclas de líquidos Cuando se calienta una solución o una mezcla de dos o más líquidos, el punto de ebullición normal es entonces la temperatura a la cual la presión de vapor total de la mezcla es igual a la presión atmosférica (760 mm). La presión de vapor total de una mezcla es igual a la suma de las presiones de vapor parciales de cada componente. En las soluciones ideales, las únicas que vamos a considerar, se cumple la ley de Raoult, que se expresa en los siguientes términos: "La presión parcial de un componente en una disolución a una temperatura dada es igual a la presión de vapor de la sustancia pura multiplicado por su fracción molar en la solución". PT = Px + Py =Pxo Nx + Pyo Ny De la ley de Raoult se puede deducir las siguientes conclusiones: 1ª) El punto de ebullición de una mezcla depende de los puntos de ebullición de sus componentes y de sus proporciones relativas; 2ª) En una mezcla cualquiera de dos líquidos, el punto de ebullición está comprendido entre los puntos de ebullición de los componentes puros; 3ª) El vapor producido será siempre más rico en el componente de punto de ebullición más bajo (más volátil). Siempre que se tenga una mezcla de dos o más componentes que se diferencien suficientemente en sus puntos de ebullición, se podrá separar en sus componentes por destilación. Se pueden distinguir tres tipos principales de destilación: destilación sencilla, destilación fraccionada y destilación a vacío. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 16 • Destilación simple Para la destilación simple se utiliza el aparato representado en la Figura 1 montado sobre dos soportes. Consta de un balón de destilación, provisto de un termómetro. El balón descansa sobre una placa calefactora. El balón de destilación va unido a un refrigerante con camisa de refrigeración por la que circula agua en contracorriente. Finalmente el extremo inferior del refrigerante se une a una alargadera que conduce el destilado al matraz colector. Figura 1. Aparato de destilación simple. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 17 El líquido que se quiere destilar se pone en el matraz (que no debe llenarse mucho más de la mitad de su capacidad) y se calienta con la placa calefactora. Cuando se alcanza la temperatura de ebullición del líquido comienza la producción apreciable de vapor, condensándose parte del mismo en el termómetro y en las paredes del balón. La mayor parte del vapor pasa a través de la tubuladura lateral de la cabeza de destilación al refrigerante donde se condensa debido a la corriente de agua fría que asciende por la camisa de este. El destilado (vapor condensado) escurre al matraz colector a través de la alargadera. Durante la destilación el extremo superior del bulbo termométrico debe quedar justamente a la altura de la horizontal que pasa por la parte inferior de la tubuladura lateral de la cabeza de destilación (figura 1), de tal forma que todo el bulbo sea bañado por el vapor que asciende. La destilación debe hacerse con lentitud pero sin interrupciones, manteniendo para ello la calefacción adecuada. Casi todos los líquidos tienden a sobrecalentarse (alcanzar una temperatura algo superior al punto de ebullición). Se encuentran entonces en un estado metaestable que se interrumpe periódicamente al formarse súbitamente una gran burbuja de vapor en el seno del líquido. Se dice entonces que este hierve a saltos. Para evitar esto, antes de iniciar la destilación se añaden al líquido uno o dos trocitos de porcelana porosa, cuyos pequeños poros constituyen un lugar adecuado para la formación de núcleos de burbujas, hirviendo así el líquido normalmente al alcanzarse la temperatura de ebullición. Si el líquido se abandona cierto tiempo a una temperatura inferior a su punto de ebullición, entonces los poros de la porcelana se llenan de líquido y ésta pierde su efectividad. Para la adición de un nuevo trocito, el líquido debe enfriarse por debajo de su punto de ebullición; la adición de un trocito de material poroso a un líquido sobrecalentado provoca una ebullición repentina que puede ser violenta (peligro de incendio y quemaduras). La existencia de una capa de sólido en el fondo del matraz de destilación puede ser causa de violentos saltos durante la destilación, especialmente si se utiliza una calefacción local fuerte en el fondo del matraz. La calefacción de un matraz que lleva cierta cantidad de sólido depositado en el fondo se debe realizar siempre mediante un baño líquido. Mediante la destilación sencilla que se acaba de describir se pueden separar mezclas de dos componentes que hiervan con una diferencia de puntos de ebullición de al menos 60-80°C, para averiguar el pto. de ebullición de una sustancia o para mezclas de un sólido con un líquido. Mezclas de sustancias cuyos puntos de ebullición difieren de 30-60°C se pueden separar por destilaciones sencillas repetidas, recogiendo durante la primera destilación fracciones enriquecidas en uno de los componentes, las cuales se vuelven a destilar. Tales mezclas se separan mucho mejor por destilación fraccionada. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 18 • Destilación fraccionada Es una técnica que permite la realización de una serie de destilaciones sencillas en una sola operación continua. Una columna sencilla como la representada en la Figura 2, puede rellenarse con cualquier tipo de sustancia inerte que posea gran superficie, por ejemplo anillos o hélices de vidrio, alambre, trocitos de arcilla, fragmentos de porcelana o de carborundo, etc. a) b) Figura 2. Columnas de destilación: a) Columna de relleno sencilla.; b) Columna Vigreux . A medida que los vapores calientes suben a través del relleno, se van condensando en todas las zonas de la columna. El condensado gotea a través del relleno; al gotear y descender tiene lugar un intercambio de calor continuo con los vapores calientes, que continúan ascendiendo por toda la superficie del relleno. Si el condensado acepta en algún punto calor de los vapores se reevapora y el vapor formado será más rico en el componente más volátil que el condensado, a la vez, el vapor al haber perdido calor por habérselo cedido al condensado, se condensa parcialmente. Este condensado es más rico en el componente menos volátil. Cuando este proceso se repite muchas veces a través de toda la altura de una columna eficaz, acaba por producir vapor puro del componente de menor punto de ebullición, que pasa a través de la cabeza de destilación hacia el refrigerante. El residuo en el matraz de destilación se va enriqueciendo, mientras tanto, en el componente de mayor punto de ebullición de una manera continua. El componente de menor punto de ebullición continúa pasando a su temperatura de ebullición hasta que se separa completamente de la mezcla. Entonces, la temperatura de los vapores que destilan se eleva hasta el punto de ebullición del componente menos volátil de forma que este empieza a llegar al refrigerante. Se denomina destilación fraccionada a la totalidad del proceso. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 19 Aparato de destilación simple Aparato de destilación fraccionada • Destilación de vacío Es una forma de destilación (sencilla o fraccionada) que se efectúa a presión reducida. Muchas sustancias no pueden purificarse por destilación a presión atmosférica porque se descomponen antes de alcanzar sus puntos de ebullición normales. Otras sustancias tienen puntos de ebullición tan altos que su destilación es difícil o no resulta conveniente. En estos casos se emplea la destilación a presión reducida. Como ya se ha indicado un líquido comienza a hervir a la temperatura en que su tensión de vapor se hace igual a la presión exterior, por tanto, disminuyendo esta se logrará que el líquido destile a una temperatura inferior a su punto de ebullición normal. ROTAVAPOR QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 20 TRABAJO PRÁCTICO N° 2 DESTILACIÓN Objetivos generales Que el alumno: - Adquiera manualidad en el armado de los diferentes equipos utilizados para esta operación unitaria - Aplique los principios de la destilación para purificar sustancias líquidas. - Comprenda la diferencia entre purificación y aislamiento de una sustancia - Compare diferentes tipos de destilación para el aislamiento y purificación de diferentes sustancias líquidas. DESTILACIÓN Objetivos Destilación I Que el alumno: - Se entrene en el armado del aparato de destilación simple. - Realice una destilación simple de una mezcla de sustancias. Material a emplear Destilación simple Balón (100 mL) Embudo Doble nueces Cabezal de destilación Soporte universal Tubos de ensayo Termómetro Gomas Gradillas Refrigerante Plato poroso Probetas Erlenmeyer Agarraderas Grasa siliconada Placa calefactora Sulfato de cobre agua Procedimiento: - Medir 50 mL de mezcla agua/sulfato de cobre con una probeta. - Introducir en el balón de destilación. - Armar el equipo de destilación. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 21 - Encender el fuego y calentar la solución. No dejar que el balón se seque completamente. Responda las siguientes preguntas: 1- ¿Para qué realiza la destilación de la sustancia pura? 2- Explique las diferencias entre las destilaciones simples y fraccionadas. 3- ¿Resulta la destilación fraccionada más efectiva para separación de componentes? CUESTIONARIO 1- ¿En qué consiste el proceso de destilación? ¿Cuál es su utilidad? 2- Enuncie los diferentes tipos de destilación. Explique brevemente en qué consiste cada una y cuál es su utilidad. 3- ¿Por qué el bulbo del termómetro debe estar enteramente por debajo de la salida al refrigerante? 4- ¿Por qué el agua en el refrigerante debe circular desde el final (alargadera) hacia el cabezal? 5- ¿Puede aplicar una destilación simple para separar cualquier mezcla de líquidos? ¿Por qué? 6- ¿Cuál es la función de una columna de fraccionamiento?¿Qué función cumple el relleno de una columna? ¿Qué materiales se emplean con este fin? Fundamente brevemente su respuesta. 7- ¿Cómo procedería para purificar una sustancia líquida de punto de ebullición 220º C que se descompone a 190º C? 8- ¿Para qué realiza la destilación de la sustancia pura? 9- Explique las diferencias entre las destilaciones simples y fraccionadas. 10- ¿Resulta la destilación fraccionada más efectiva para separación de componentes? QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 22 EXTRACCIÓN La extracción es la técnica empleada para separar un producto orgánico de una mezcla de reacción o para aislarlo de sus fuentes naturales. Puede definirse como la separación de un componente de una mezcla por medio de un disolvente. En la práctica es muy utilizada para separar compuestos orgánicos de las soluciones o suspensiones acuosas en las que se encuentran. El procedimiento consiste en agitarlas con un disolvente orgánico inmiscible con el agua y dejar separar ambas capas. Los distintos solutos presentes se distribuyen entre las fases acuosas y orgánicas, de acuerdo con sus solubilidades relativas. De este modo, las sales inorgánicas, prácticamente insolubles en los disolventes orgánicos más comunes, permanecerán en la fase acuosa, mientras que los compuestos orgánicos que no forman puentes de hidrógeno, insolubles en agua, se encontrarán en la orgánica. • Coeficiente de reparto Ciertos compuestos orgánicos, como los alcoholes, aldehídos, cetonas, ácidos, ésteres, aminas, etc., capaces de asociarse con el agua a través de puentes de hidrógeno, son parcialmente solubles en este disolvente y en los orgánicos; en estos casos pueden ser necesarias varias extracciones sucesivas para eliminar la sustancia orgánica de la fase acuosa. Cuando se agita una solución acuosa de una sustancia con un disolvente orgánico en el que la sustancia es al menos algo soluble, el compuesto se disuelve parcialmente en cada disolvente. La relación de las concentraciones en ambos (CO y CA)- proporcionales a las solubilidades respectivas, SO y SA-, cuando se alcanza el estado de equilibrio a una temperatura determinada, se llama coeficiente de distribución o de reparto, KD. CO KD = SO = CA SA En general, para un volumen determinado de líquido extractivo, la eficacia de la extracción aumenta con el número de las mismas. En la práctica, no obstante, deben tenerse en cuenta también el tiempo y el trabajo requeridos por las repetidas extracciones y los inconvenientes que presenta la manipulación de muy pequeñas cantidades de disolvente. Como norma práctica puede indicarse que para solutos mucho más solubles en el disolvente extractivo que en el agua, debe utilizarse en cada extracción un volumen de aquél igual a la tercera parte de ésta. Disolventes orgánicos muy utilizados son el tolueno (C6H5-CH3), el éter de petróleo (mezcla de alcanos de baja magnitud molecular), el cloruro de metileno (CH2Cl2), el cloroformo QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 23 (CHCl3), el tetracloruro de carbono (CCl4), el acetato de etilo (CH3-COOC2H5) y el alcohol nbutílico (CH3CH2CH2CH2OH). La elección del disolvente se realiza en cada caso teniendo en cuenta la solubilidad en el mismo de la sustancia a extraer y la facilidad con que puede separarse ésta del disolvente. El éter dietílico es el más utilizado por la gran solubilidad en el mismo de la mayor parte de los compuestos orgánicos y por su bajo punto de ebullición (35°). Sin embargo, su gran volatilidad y su fácil inflamabilidad exigen manejarlo con las precauciones debidas. • Equipo y procedimiento El aparato utilizado en las extracciones es la ampolla de decantación que se muestra en la figura. El tapón y la llave, que deben estar bien ajustados, se lubrican con una grasa adecuada antes de cada uso. Figura 9. Extracción. Como indica la Figura 9, la ampolla de decantación debe manejarse con ambas manos; con una se sujeta el tapón -asegurándolo con el dedo índice- y con la otra se manipula la llave. Se invierte la ampolla y se abre la llave para eliminar la presión de su interior; se agita con suavidad durante uno o dos segundos y se abre de nuevo la llave. Cuando deja de aumentar QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 24 perceptiblemente la presión en el interior, se aseguran tapón y llave y se agita enérgicamente durante uno o dos minutos. Se pone de nuevo en contacto con la atmósfera a través de la llave, se vuelve a cerrar ésta y se apoya, ya en posición normal, en un aro metálico con unos trozos de tubo de goma que lo protegen de roturas. Se destapa y se deja en reposo hasta que sea nítida la separación entre las dos capas de líquido. En la parte inferior debe tenerse siempre un vaso de precipitados de gran tamaño con objeto de poder recoger todo el líquido en caso de que el embudo se rompiese por accidente. Después de separadas ambas fases, se saca el inferior por la llave y la superior por la boca; así se previenen posibles contaminaciones. El número de extracciones necesarias en cada caso particular depende del coeficiente de reparto y de los volúmenes relativos de agua y de disolvente. La posición relativa de las capas acuosa y orgánica depende de sus densidades. En caso de duda puede determinarse la identidad de cada una de ellas ensayando la solubilidad en agua de unas gotas de la misma. Es una medida prudente, en especial cuando se trata de reacciones nuevas, conservar todos los extractos y líquidos residuales hasta comprobar que se obtiene el producto final con el rendimiento esperado; sólo entonces debe procederse a la limpieza. • Emulsiones Con frecuencia, sobre todo cuando se trabaja con soluciones alcalinas, se forman emulsiones durante el proceso de extracción. Estas pueden romperse, de ordinario, mediante: 1) un movimiento de giro suave al líquido de la ampolla de decantación, mantenido en su posición normal; 2) agitación vigorosa de la capa emulsionada con una varilla de vidrio; 3) saturación de la capa acuosa con sal común; 4) centrifugación. El método 3, de saturación con sal, tiene una doble ventaja: hace disminuir la solubilidad en agua de la mayor parte de los solutos y de los disolventes orgánicos. Su nombre es efecto salino. • Extracción con ácidos y álcalis Con frecuencia se consiguen separaciones muy netas de compuestos orgánicos, utilizando soluciones ácidas o alcalinas capaces de convertir dichas sustancias en sales, solubles en agua e insolubles en éter. Una solución de hidróxido sódico al 5-10 % convierte, por ejemplo, los ácidos carboxílicos, R-COOH, en sus sales sódicas, R-COO-, Na+. Los compuestos fenólicos experimentan una transformación semejante con el mismo reactivo. Por esta causa puede utilizarse una solución de hidróxido sódico para extraer un ácido carboxílico o un compuesto fenólico de su solución en un disolvente orgánico o, recíprocamente, liberar estos tipos de compuestos de sus impurezas orgánicas por extracción de sus soluciones alcalinas con un disolvente adecuado. Las soluciones acuosas de bicarbonato sódico convierten también los ácidos carboxílicos en sus respectivas sales sódicas, pero no son lo suficientemente básicas para formar sales con los compuestos fenólicos. Esta es la base de un elegante método de separación QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 25 de ácidos carboxílicos y fenoles: el ácido se extrae en primer lugar de su solución en un disolvente orgánico con una solución de bicarbonato sódico y, posteriormente, el fenol con solución de sosa. Los ácidos inorgánicos se eliminan con facilidad de los disolventes orgánicos por extracción con una solución de hidróxido, carbonato o bicarbonato sódicos. El ácido clorhídrico diluido se emplea con frecuencia para la extracción de sustancias básicas de sus mezclas con otras neutras o ácidas, o bien para eliminar impurezas básicas. El ácido diluido convierte la base, p.ej., amoniaco o una amina orgánica (R3N), en el correspondiente hidrocloruro (R3NH+, Cl-), soluble en agua. Por otra parte, las impurezas orgánicas que acompañan a una amina pueden eliminarse por extracción de las mismas con un disolvente orgánico de una solución ácida de aquella. Las sales sódicas de los ácidos carboxílicos y de los fenoles son fácilmente convertibles en los compuestos de partida por tratamiento de ácido sulfúrico o fosfórico. Los hidrocloruros de las aminas se transforman de nuevo en éstas por adición de una solución de hidróxido sódico. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 26 AGENTES DESECANTES Importancia del secado Pequeñas cantidades de humedad inhiben la cristalización de muchos sólidos. Además, muchos líquidos, cuando destilan en presencia de agua, reaccionan con ésta (se hidrolizan) o destilan con el agua (se arrastran) a temperaturas bastantes distantes de sus puntos de ebullición. Por estas razones, el paso final, antes de la recristalización de un sólido o de la destilación de un líquido, es la eliminación del agua que lleva consigo mediante algún proceso de secado. En general, como mejor se seca una sustancia orgánica es en solución (generalmente en el disolvente de extracción). El proceso de secado se puede realizar por medios mecánicos o por medios químicos. Los desecantes químicos se utilizan más que los procedimientos de secado mecánicos. Un buen desecante químico debe reunir las siguientes condiciones: 1) No reaccionar con la sustancia a secar; 2) tener una gran eficacia o poder desecante, esto es, eliminar el agua completamente o casi completamente; 3) tener una gran capacidad de desecación, es decir, eliminar una gran cantidad de agua por unidad de peso de desecante; 4) secar rápidamente y 5) ser fácilmente separable de la sustancia una vez seca. Clasificación de los desecantes Desecantes no reversibles Está formado por aquellos que reaccionan químicamente con el agua en un proceso no reversible dando lugar a un nuevo compuesto. Con este tipo de desecantes el producto secado se separa generalmente por destilación. • El anhídrido fosfórico (P2O5) elimina el agua con mucha eficacia y muy rápidamente. Es caro. Solamente se emplea cuando se necesita un alto grado de desecación y sólo después de un secado preliminar con un agente menos caro y eficaz. Se emplea par secar hidrocarburos y sus derivados halogenados sencillos, éteres y nitrilos, pero nunca para alcoholes, cetonas, aminas y ácidos. • El sodio metálico (Na) es un agente muy eficaz, especialmente cuando se utiliza en forma de un hilo muy fino, pero se puede utilizar solamente para secar éteres, alcanos e hidrocarburos aromáticos. Su utilización debe siempre ir precedida por un secado previo con cloruro cálcico, sulfato magnésico o anhídrido fosfórico. • El hidruro de calcio (CaH2) es un desecante poderoso y de gran capacidad de desecación. Su eficacia aumenta enormemente al elevar la temperatura. El hidruro cálcico se recomienda para eliminar trazas de humedad de gases y de éteres y aminas terciarias. • El óxido cálcico se utiliza corrientemente para el secado de alcoholes de peso molecular bajo. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 27 Desecantes que forman hidratos Estos desecantes actúan combinándose con el agua para formar hidratos. Se separan siempre de la sustancia seca por filtración o decantación, pues por destilación una gran parte del agua de hidratación pasaría con el destilado. Asimismo, el secado se realiza a muy bajas temperaturas para conseguir la máxima eficacia del agente de desecación. Frecuentemente, la agitación, que ayuda a alcanzar el equilibrio, acelera la velocidad de secado. El cloruro cálcico anhidro (CaCl2) se utiliza mucho por ser de gran capacidad y relativamente barato. Sin embargo, es bastante lento y moderadamente eficaz. Es particularmente adecuado para secados preliminares, pero se recomienda solamente para hidrocarburos y sus derivados halogenados y para éteres. Es generalmente inadecuado para compuestos ácidos, tales como ácidos carboxílicos y fenoles, porque con frecuencia contiene algo de cal, y para alcoholes, cetonas, aminas, aminoácidos, amidas y algunos aldehídos y ésteres por la formación de complejos. Las sales anhidras neutras: son inertes e insolubles en los líquidos orgánicos por lo que se pueden utilizar para secar cualquier tipo de compuestos. o El sulfato sódico (NaSO4) es barato y presenta una gran capacidad, sin embargo, es lento y, debido a su baja eficacia, es casi siempre inservible para disolventes tales como el benceno, tolueno y cloroformo, en los que el agua se disuelve muy poco. Es recomendable como agente de secado preliminar para la eliminación de cantidades grandes de agua, especialmente en las soluciones etéreas. o El sulfato magnésico anhidro (MgSO4) es un desecante excelente para todos los fines. Presenta gran capacidad y eficacia y es barato y bastante rápido. o El sulfato cálcio anhidro (CaSO4) (Drierita) es muy rápido y eficaz, pero tiene una capacidad de secado pequeña. Con frecuencia se utiliza después de un desecante primario, como el sulfato sódico. El hidróxido sódico anhidro, y especialmente el hidróxido potásico anhidro, son los desecantes más adecuados para el secado de aminas. Debido a su fuerte basicidad, no se utilizan para el secado de otros compuestos, excepto en los desecadores en los que el desecante no se pone en contacto con el producto a secar. Carbonato potásico anhidro (K2CO3) es de capacidad y eficacia moderadas. Actúa lentamente. Se utiliza con cierta frecuencia para el secado de cetonas, ésteres, alcoholes y aminas, especialmente como agente de secado previo. Es un reactivo básico y por tanto es incompatible con compuestos de carácter ácido como ácidos carboxílicos y fenoles. Agentes adsorbentes Actúan por adsorción de agua en su superficie. Son una forma de sílice especialmente tratada llamada gel de sílice y una serie de silicatos de calcio y sodio, cristalinos y muy porosos, que han sido previamente tratados para eliminar su agua de hidratación, llamados tamices moleculares. Estos agentes son extraordinariamente eficaces para eliminar el vapor de agua de los gases. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 28 CROMATOGRAFÍA DE ADSORCIÓN: APLICACIÓN A LA SEPARACIÓN DE COMPUESTOS ORGANICÓS INTRODUCCIÓN La cromatografía es una técnica que permite separar los componentes de una mezcla. Esta separación se logra utilizando un sistema bifásico: fase estacionaria donde se retienen los compuestos a separar y fase móvil que desplaza de forma diferencial los compuestos a través de la fase estacionara. Dependiendo de la naturaleza de las fases, se pueden distinguir distintos tipos de cromatografía: Cromatografía sólido-líquido: la fase estacionaria es un sólido y la móvil un líquido. Cromatografía líquido-líquido: ambas fases son líquidos y en la estacionaria el líquido se ancla a un soporte sólido. Cromatografía líquido-gas: la fase estacionaria es un líquido no volátil sobre soporte sólido y la fase móvil un gas. Cromatografía sólido-gas: la fase estacionaria es un sólido y la móvil un gas. La mezcla a separar se deposita sobre la fase estacionaria y la fase móvil atraviesa el sistema desplazando los componentes de la mezcla a distintas velocidades que dependen de la afinidad de los mismos por cada una de las fases (Figura 11). Se denomina elución a la migración de los componentes de la mezcla a lo largo de la fase estacionaria impulsados por la móvil. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 29 Existen otras clasificaciones para los distintos tipos de cromatografía: A) En función de la interacción que se establece entre los componentes de la mezcla y las fases móvil y estacionaria: * Cromatografía de adsorción: se producen interacciones de tipo polar siendo la fase estacionaria un sólido. * Cromatografía de partición: la separación se basa en las diferencias de solubilidad de los componentes de la mezcla entre las dos fases siendo ambas líquidas. Cuando la estacionaria es menos polar que la móvil se denomina cromatografía en fase inversa. * Cromatografía de intercambio iónico: se producen intercambios entre iones presentes en la fase estacionaria y los del compuesto orgánico solubilizado e ionizado en la fase móvil B) En función del tipo de soporte empleado para la fase estacionaria: * Cromatografía en columna: utiliza como soporte una columna de vidrio * Cromatografía en capa fina: el soporte es una placa de vidrio, aluminio o plástico La cromatografía se puede emplear para: -Conocer el número de componentes de una mezcla e identificarlos por comparación con patrones, Cromatografía Analítica -Separar mezclas de compuestos y como método de purificación, Cromatografía Preparativa CROMATOGRAFÍA DE ADSORCIÓN La cromatografía de adsorción emplea una fase estacionaria sólida de carácter polar, ADSORBENTE y una fase móvil líquida, ELUYENTE. Se utiliza tanto con fines analíticos como preparativos y la separación de los componentes de la mezcla viene determinada por las interacciones polares de los componentes de la misma con las fases estacionaria y móvil. Por lo tanto, los compuestos más difíciles de separar mediante este tipo de cromatografía, serán aquellos que tengan una polaridad muy similar. FASE ESTACIONARIA: ADSORBENTE Está constituida por un sólido poroso, finamente granulado, con centros activos polares en su superficie donde se adsorben las moléculas de los compuestos que se van a cromatografiar. Cuanto menor sea el tamaño de partícula de este material mayor será la capacidad de adsorción. La adsorción se debe a interacciones intermoleculares del tipo dipolo-dipolo, dipolodipolo inducido o enlaces de hidrógeno entre el adsorbente y el soluto. El adsorbente más utilizado es la gel de sílice (Figura 12) donde las interacciones tienen lugar entre los grupos Si—OH y Si—O—Si; también se emplea con relativa frecuencia alúmina (Al2O3). QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 30 I I Figura 12.- Estructura de la gel de sílice e interacciones con algunos compuestos polares El adsorbente debe ser inerte con las sustancias a cromatografiar. El gel de sílice presenta carácter ácido y la alúmina puede adquirirse con carácter neutro, ácido o básico. FASE MOVIL: ELUYENTE Es un disolvente en el que los componentes de las mezcla son, al menos, parcialmente solubles. Al aumentar la polaridad del disolvente aumenta la velocidad de elución de los compuestos de la mezcla. Se puede utilizar un único disolvente o una mezcla de disolventes e incluso llevar a cabo la elución con un gradiente de polaridad aumentando progresivamente la proporción del disolvente más polar. RETENCIÓN Las moléculas de soluto S se adsorben en los centros polares de la fase estacionaria X y, a medida que se produce la elución, van siendo desplazadas por las moléculas de disolvente/s que constituyen la fase móvil M. La retención de un soluto se puede justificar por la competencia que se establece entre S y M por adsorberse a los centros polares X, es decir, depende de los valores de las constantes de los equilibrios: —X + S ----------- ► —X...S —X + M------------ ► —X...M que están en función de: Polaridad del compuesto a eluir que depende de sus grupos funcionales Naturaleza del adsorbente Naturaleza del disolvente QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 31 ORDEN DE POLARIDAD DE LOS COMPUESTOS ORGÁNICOS: Ácidos carboxílicos > Fenoles > Alcoholes y Tioles > Aminas > Ésteres > Aldehídos y Cetonas > HC. Aromáticos > HC. Halogenados > Éteres > Hidrocarburos Insaturados > Alcanos ■=> Cuanto más polar sea un compuesto mas se retendrá en la fase estacionaria. Por ejemplo, se retiene más un alcohol que un hidrocarburo ORDEN DE POLARIDAD DE LOS ELUYENTES MÁS HABITUALES: H20 > CH3-OH > (CH3)2CH-OH > CH3-CN > Dioxano > CH3COOEt > THF > CH2CI2 ( Cloruro de metileno) > CHCI3 (Cloroformo) > CCI4 (Tetracloruro de carbono) > CH3(CH2)4CH3 ■=> Para un mismo compuesto, un aumento en la polaridad de la fase móvil hace que se se desplace con más facilidad de la fase estacionaria. Por ejemplo, se eluirá más rápidamente una amina en acetonitrilo que en hexano > Para compuestos poco polares, que se retienen poco en el adsorbente, se utilizan eluyentes apolares o poco polares como, por ejemplo, hexano > Para compuestos muy polares, que quedan muy retenidos en el adsorbente, se emplean eluyentes muy polares como, por ejemplo, metanol o mezclas metanoldiclorometano > Para compuestos de polaridad media se emplean eluyentes de polaridad intermedia y son muy aconsejables en estos caso las mezclas en distintas proporciones de hexanoacetato de etilo CROMATOGRAFÍA ANALÍTICA EN CAPA FINA (TLC) La fase estacionaria (adsorbente) se encuentra depositada, formando una capa fina de un espesor uniforme sobre una placa de vidrio, plástico o una lámina metálica. La mezcla a analizar se deposita con un capilar a una pequeña distancia del borde inferior de la placa (Figura 13) y se introduce en una cubeta donde está la fase móvil (eluyente), que ascenderá a lo largo de la capa por capilaridad eluyendo a los componentes de la mezcla a distintas velocidades, lo que provoca su separación (Figura 14). Cuando el frente del disolvente se encuentra a ª 0.5 cm del borde superior, se saca la placa de la cubeta, se marca hasta donde ha llegado el eluyente y se deja secar para, a continuación, proceder al la visualización de las manchas correspondientes a los productos cromatografiados. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 32 Figura 13. Aplicación de la muestra Figura 14. Desarrollo de la placa Determinación del Rf Se conoce como Rf (rate factor) la relación entre las distancias recorridas por un compuesto y por el disolvente desde el origen del cromatograma. Rf = Distancia recorrida por el compuesto Distancia recorrida por el disolvente Cada compuesto en unas condiciones cromatográficas determinadas: adsorbente, disolvente, temperatura, etc..., tiene un valor constante y característico de Rf. Sin embargo, solo se pueden establecer comparaciones entre los Rf de dos compuestos cuando los dos se eluyan juntos en la misma placa. La distancia recorrida por el compuesto se mide desde el centro de la mancha (Figura 15) QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 33 Visualización del Cromatograma ■=> Con luz UV (ultravioleta).- Las placas suelen llevan incorporado un indicador fluorescente que absorbe luz UV y emite luz visible, generalmente verde. Cuando se cromatografían sustancias que absorben en el UV donde se encuentra el compuesto no absorbe el indicador y como resultado vemos una mancha que indica su presencia. ■=> Con un agente revelador.- Se emplea cuando las sustancias no absorben radiación UV. El revelador reacciona con los productos absorbidos dando lugar a compuestos coloreados y por tanto se utilizará uno u otro en función del compuesto que se quiera visualizar. Algunos ejemplos: H2SO4 concentrado en etanol para azúcares, ninhidrina para aminoácidos y yodo para compuestos insaturados y aromáticos. Aplicaciones de la TLC (CCF) La CCF es una técnica cualitativa muy utilizada en los laboratorios de Química Orgánica para: * Determinar el número de componentes de una muestra: Precaución si solo hay una mancha muy retenida o aparece junto al frente del disolvente hay que probar otro eluyente (Figura 16). a) b) QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS c) 34 Comprobar la pureza de un compuesto: aparecerá una sola mancha. Se debe comprobar que solo hay una mancha utilizando distintos eluyentes. * Determinar la identidad de dos compuestos: Hay que tener precaución, pues valores de Rf iguales (o prácticamente iguales) para dos compuestos en una misma placa no garantizan inequívocamente su identidad. En la misma placa hay que cromatografiar una mezcla de los compuestos cuya identidad se quiere comprobar (Figura 17). * Seguir la evolución de una reacción: cada cierto tiempo se cromatografían en una misma placa los reactivos y la mezcla de reacción (Figura 18). * QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 35 * determinar el disolvente más apropiado para llevar a cabo una separación mediante una cromatografía preparativa (en columna o en placa) * Seguir el progreso de una cromatografía en columna (CC) (Figura 19) Tanto para determinar la pureza como la identidad de los compuestos se debe realizar la corrida en 3 sistemas diferentes, variando fase móvil o fase estacionaria. CROMATOGRAFÍA EN COLUMNA (CC) Es el método más utilizado para la separación y purificación de compuestos orgánicos, sólidos o líquidos, a escala preparativa. La fase estacionaria (adsorbente) se coloca en una columna de vidrio que termina en un estrechamiento con una llave y se impregna con el eluyente (fase móvil). La mezcla a separar se deposita en la parte superior de la columna y la fase móvil atraviesa el sistema (Figura 20). Los compuestos se van eluyendo disueltos en el eluyente, se van recogiendo ordenadamente en fracciones de pequeño volumen (1,2,….) y se analizan por CCF (Figura 18). Los productos van saliendo de la columna según su polaridad, primero salen los menos polares que son los menos retenidos por el adsorbente y los últimos los más polares por su mayor retención en la fase estacionaria. El adsorbente más utilizado para este tipo de cromatografía es el gel de sílice y en segundo lugar la alúmina. El tamaño de partícula del adsorbente es importante para la separación y su elección depende en gran medida de que la elución de la cromatografía se realice por gravedad o a media presión (flash chromatography). En una elución a media presión se pueden emplear tamaños de partícula más pequeños, que permiten una separación más eficaz. Sin embargo, en una elución por gravedad el uso de tamaños de partícula pequeños impediría el flujo del disolvente. Antes de realizar una separación en cromatografía de columna hay que elegir adecuadamente el disolvente haciendo ensayos en CCF. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 36 QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 37 TRABAJO PRÁCTICO N° 3 EXTRACCIÓN DE CAFEÍNA Materiales: Ampolla de decantación de 100ml Aro de hierro Cloroformo 2 Vasos de ppdos baño maría bebida de cola Procedimiento: Coloca 50ml de coca-cola en una ampolla de decantación y agrega 50 ml de cloroformo. Agitar 10 veces, abrir la llave para liberar los gases después de cada extracción. Dejar reposar hasta que se separen las fases, recoger el cloroformo que contiene la cafeína en un vaso de ppdo. . Evaporar el cloroformo calentando en un baño maría o directamente en campana hasta sequedad.( los vapores de cloroformo son muy tóxicos) Identificar el residuo con cromatografía en capa delgada con solvente acetato de etilo y otro con alcohol. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 38 TRABAJO PRÁCTINO N° 4 EXTRACCIÓN POR ARRASTRE DE VAPOR DE AGUA EUGENOL OH OCH3 CH2 – CH = CH2 Eugenol La esencia de clavos de especia (EUGENIA CARIOPHYLLATA), que se obtiene por arrastre con vapor, esta formada casi exclusivamente por el eugenol, compuestos de este tipo son muy abundantes en la naturaleza; por ejemplo, la esencia de pimienta de Jamaica tiene hasta 80% de eugenol. Aparte de su uso en odontología por sus propiedades antisépticas se lo utiliza como punto de partida en la obtención de vainillina. El rendimiento del eugenol a partir del producto seco varia entre 10 y 17% según el estado de conservación de los clavos, pues el fenol se pierde lentamente por oxidación y/o volatilización. Aislamiento de eugenol a partir de clavos de olor Se parte de 3 gr de clavos sin moler que se lo colocan en un balón de 200 ml a 300 ml junto con unos trozos de material poroso y 150 ml de agua. Al balón con salida lateral se le adapta, un refrigerante y una alargadera que desemboca en una probeta. Se lleva a ebullición sobre tela metálica y se destilan unos 80 ml aproximadamente ( alrededor de 1 h ) El destilado se alcaliniza con 10 ml de una solución de NaOH 20%. Se extrae con 30 ml de cloruro de metileno en ampolla de decantación. La fase orgánica se desecha y la fase acuosa se acidifica con HCl 20% (papel pH) y luego se extrae con 30 ml de cloruro de metileno, desechándose la fase acuosa. La solución no polar se seca sobre sulfato de sodio anhidro y se concentra por evaporación. El residuo (aprox. 230 mg), está constituido casi totalmente por el eugenol (4-alil- 2- metoxifenol). Se siembra una microplaca de silicagel con 1 o 2 gotas de solución de eugenol. Se utiliza como solvente de desarrollo alcohol. Las placas de silicagel se observan con luz U.V. y luego se pueden revelar con I2. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 39 TRABAJO PRÁCTICO N°5 EXTRACCIÓN ÁCIDO-BASE Objetivos: - Que el alumno comprenda los alcances y limitaciones de la técnica de extracción aplicada al aislamiento de compuestos orgánicos a partir de una mezcla, teniendo en cuenta la solubilidad y las propiedades ácido-base de las mismas. - Que el alumno se familiarice con los contenidos procedimentales y actitudinales inherentes a esta metodología. Material a emplear Ampolla de Probetas Erlenmeyer Pipetas Vaso de precipitados Varilla Embudo Papel de filtro Kitasato Vidrio de reloj Hirsch Aparato de punto de decantación fusión Drogas Solución de NaHCO3 5 % Na2SO4 anh. Solución de NaOH 10 % HCl cc 0.4 g de Salicilamida 0.4 g de Ácido Benzoico Cl3CH o Cl2CH2 0.4 g de Acetanilida Técnica Mida con una probeta 20 ml de la “Mezcla de Extracción” y viértalos en una ampolla de decantación. Agregue 3 ml de solución NaHCO3 5%, agite la ampolla y luego abra el robinete sujetando el tapón; cierre el robinete y coloque la ampolla en el aro nuevamente. Espere hasta observar la separación de las dos fases y luego decante. Recuerde SACAR el tapón. Coloque la FASE ACUOSA I en un vaso de precipitados e introduzca la fase QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 40 orgánica en la ampolla. Repita este procedimiento dos veces y luego lave la fase orgánica con 5 ml de agua. Realice luego la extracción con 4 ml de solución NaOH 10 % y obtenga la FASE ACUOSA II. Repita una vez este procedimiento (si desea, podrá comprobar con papel indicador de pH que dos alícuotas son suficientes). Lave la fase orgánica con 5 ml de agua. Coloque la fase orgánica remanente en un erlenmeyer SECO y agregue Na2SO4 anh.; filtre con papel plegado en el balón. Proceda a remover el CH2Cl2, ya sea por destilación simple ó empleando evaporador rotatorio. Pese el sólido obtenido y determine su punto de fusión. Acidifique la FASE ACUOSA I con 3 ml de HCl cc., mientras mezcla con una varilla. Filtre el sólido obtenido a presión reducida (recuerde lavarlo con agua) y proceda a secarlo. Acidifique la FASE ACUOSA II con 3 ml de HCl cc., mientras mezcla con una varilla. Filtre el sólido obtenido a presión reducida (recuerde lavarlo con agua) y proceda a secarlo. Nota Al realizar extracciones NO descarte ninguna fase hasta el final del trabajo. Cuando use un agente desecante la espátula debe estar SECA. Mantenga tapados todos los frascos de reactivos. Recuerde recuperar los productos sólidos en los frascos correspondientes como así también el solvente. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 41 CUESTIONARIO EXTRACCIÓN 1- ¿Qué entiende por extracción? ¿En qué fenómenos se basa? 2- Enuncie la ley de partición. 3- ¿Qué se entiende por lavado? 4- ¿Cómo se elige el solvente de extracción? 5- ¿Cómo se rompe una emulsión? 6- ¿Qué es el efecto “salting out” y para qué se usa en extracción? 7- Tipos de agentes desecantes. Ejemplos. 8- Teniendo una mezcla de ácido benzoico, fenol y tolueno en solución clorofórmica. Qué compuesto extraerá con: a) NaHCO3 al 5%. ¿Qué indica el cese de desprendimiento de gas al extraer con NaHCO3?¿Qué gas es? b) NaOH al 10% c) ¿Qué compuesto queda en la solución clorofórmica? d) ¿Por qué se trata esta fase orgánica con Na2SO4 anh.?¿Qué otra sustancia podría usar? 9- Se desea extraer de una fase acuosa un compuesto con un bajo coeficiente de reparto entre el agua y el disolvente de extracción. a) ¿Qué tipo de extracción utilizaría?¿Porqué? Ventajas y desventajas. b) ¿Qué aparato utilizaría si usa cloroformo como solvente de extracción? ¿Y si usa benceno? QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 42 CUESTIONARIO CROMATOGRAFÍA 1- ¿Qué métodos de separación conoce? 2- Defina cromatografía. ¿Cuáles son los distintos métodos cromatográficos y qué fundamentos tiene cada uno? 3- ¿Cuál es la diferencia entre absorción y adsorción? Nombre adsorbentes para TLC. 4- ¿Cuáles son los componentes de un sistema cromatográfico? 5- ¿Qué es el soporte y qué es el adsorbente en una TLC? Mencione ejemplos. 6- a- ¿Qué diferencias existen entre cromatografía en placa delgada, en placa preparativa y en columna? 7- ¿Qué importancia tiene la saturación de la cuba con el solvente a usar en el desarrollo antes de realizar la cromatografía en placa? 8- ¿Qué es la relación de frente (Rf) y de qué factores depende? ¿Puede usarse como criterio de identificación y/o pureza? 9- ¿Qué es el desarrollo y elución de un cromatograma? 10- ¿Qué es el revelado de un cromatograma? ¿Qué es un revelador universal? ¿Qué es un revelador específico? ¿En qué casos se utilizan reveladores? 11- ¿La cromatografía en capa delgada puede tener utilidad para cuantificar una sustancia? 12- ¿Cuál es la importancia de la homogeneidad en el armado de la fase fija en la cromatografía en columna? 13- Si las fracciones eluídas mediante cromatografía en columna son incoloras, ¿cómo procedería para saber en qué tubos se encuentra/n la/s sustancia/s y si la separación fue efectiva? QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 43 TRABAJO PRÁCTICO N° 6 CROMATOGRAFÍA Cromatografía preparativa en columna Objetivos - Que el alumno comprenda el fundamento y las aplicaciones de las diferentes técnicas cromatográficas. - Que el alumno se familiarice con los contenidos procedimentales y actitudinales inherentes a esta metodología. Material a emplear Columna Cuba Pinza cromatográfica de Mohr Vaso de Erlenmeyers Probeta Agarradera Papel precipitados Soporte universal de filtro Doble nuez Algodón Balanza Drogas Rojo de Sílicagel 60 (0.063-0.200mm) para metilo columna Azul de Metanol metileno Cloruro de metileno COOH N X- S N CH3 N N N CH3 ROJO DE MET ILO CH3 N CH3 CH3 CH3 AZUL DE MET ILENO QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 44 Técnica Armado de la columna: Coloque en el fondo de la columna una pequeña torunda de algodón con la ayuda de una varilla de vidrio. Sostenga la columna con pie universal, doble nuez y agarradera de modo que la columna quede en posición vertical. En un erlenmeyer coloque 5g de sílica gel y luego 25ml de Cl2CH2. Homogenice la suspensión sílica-Cl2CH2 moviendo el erlenmeyer en forma circular y vierta dentro de la columna de forma tal que comience a drenar el Cl2CH2 de la suspensión (con la pinza de Mohr abierta y un erlenmeyer debajo), mientras da golpecitos suaves a la columna con los dedos para que la sílica se distribuya de manera uniforme, evitando la formación de canales o burbujas de aire. Si quedara sílica en el erlenmeyer, resuspenda con el Cl2CH2 que ha eluído, e introdúzcala en la columna. Una vez colocada toda la sílica deje que continúe el goteo de solvente hasta que quede justo por encima del material de relleno, procurando que no se seque. (Si se seca, puede agregar unas gotas de solvente y volver a golpear suavemente, para que no queden burbujas en el seno del material de relleno). Siembra: Tome con una pipeta 0,5ml de la mezcla de colorantes a separar disuelta en el solvente apropiado, introduzca en la columna haciendo deslizar la muestra cuidadosamente por las paredes internas de la misma, dejando que se deposite en la superficie del material de relleno. Este paso de debe hacer con delicadeza para evitar que se estropee la superficie donde se siembra. Luego abra la llave de paso inferior para que eluya ligeramente la fase móvil de manera que no se vea líquido en la superficie y evitando a su vez que se seque totalmente. Agregue otra torunda de algodón sobre la sílica (LUEGO DE HABER SEMBRADO) para evitar que por agregado de la fase móvil se resuspenda la sílica. Desarrollo: Tome 30 ml de la primera fase móvil (Cl2CH2:MeOH, 30:1) en una probeta graduada y agregue en porciones a la columna. Deje eluir recogiendo el eluato 1 en un erlenmeyer y agregando siempre fase móvil para que no se seque la parte superior de la columna. Luego tome 40 ml de la segunda fase móvil (Cl2CH2:MeOH, 30:10) en la probeta graduada y agregue en porciones a la columna del mismo modo que en el paso anterior, QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 45 recogiendo el eluato 2 en otro erlenmeyer o vaso de precipitados. Como los compuestos son coloreados, las bandas observadas en la columna, que avanzan con diferente movilidad, se ven a simple vista. Por lo tanto el eluato se recoge fraccionándolo en erlenmeyers de acuerdo a la forma en que los compuestos van saliendo de la columna. Tome tres fracciones: la que corresponde al primer colorante (Eluato 1), la fracción intermedia entre ambos y la que corresponde al segundo colorante (Eluato 2). Analice mediante cromatografía en capa delgada (TLC) Cromatografía analítica en capa delgada Identificación de los colorantes por TLC. Fase estacionaria: Cromatofolio cubierto de sílica gel sin indicador de fluorescencia. Fase móvil: Cl2CH2:MeOH (9:1). Siembra: a) Testigo de Rojo de metilo, b) Testigo de Azul de metileno, c) Eluato 1 y d) Eluato 2. Revelado: Luz visible. Dibuje el resultado obtenido y analice los resultados Figura 1: Placa Cromatográfica Figura 2: Cuba Cromatográfica QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 46 TRABAJO PRÁCTICO N°7 Preparación de Ácido Benzoico. Propiedades físicas. Un resto alquilo unido a un grupo aromático se puede oxidar dando un ácido carboxílico con buen rendimiento. OH- ArCH3 + KMnO4 ArCOO- + H2O + MnO2 [O] Procedimiento: En un balón de 250 ml se calientan a reflujo 2,6 ml de tolueno, 120 ml de agua, 2 ml de NaOH al 10% y 7 gr de permanganato de potasio. Conviene agregar unos trozos de piedra porosa para evitar los sobresaltos durante la ebullición. Después de lo cual se mantiene la ebullición durante 1 hora, agitando cada 5 minutos a efectos de que entren en contacto todos los reactivos. Luego se deja enfriar la mezcla a temperatura ambiente y se agregan, por la parte superior del refrigerante de 5 a 10 ml de etanol, a los efectos de destruir el exceso de permanganato de potasio presente. Luego se filtra el precipitado a través de un Buchner y se lava con 2 ml de agua bien caliente. El contenido del kitasato se pasa a un vaso de precipitados grande o un recritalizador y se concentra la solución a un volumen de 10-20 ml. La solución se enfría a temperatura ambiente y se acidifica con H2SO4 50%, agregando el ácido gota a gota y agitando continuamente la solución, se verifica con papel tornasol. Se filtra el ácido benzoico así obtenido y se recristaliza en la menor cantidad de agua a ebullición. Se filtra, seca y se determina el punto de fusión. Cuestionario 1) ¿Porque agregar etanol es necesario y no interfiere en los resultados? 2) ¿En qué casos debe hacerse la recristalización del benzoico? 3) Mencione dos propiedades físicas del ácido benzoico. 4) Teniendo en cuenta que se forma 1 mol de ácido por mol de tolueno. ¿qué cantidad de ácido se hubiera formado con un rendimiento del 100 %? QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 47 REACCIONES DE CARACTERIZACIÓN COMPORTAMIENTO DE GRUPOS FUNCIONALES Como es sabido, la presencia de un grupo funcional en una molécula orgánica proporciona un comportamiento que se debe a la naturaleza y características de los enlaces presentes en el grupo funcional. Por tanto, el análisis funcional no permite establecer la composición de la molécula orgánica, pero garantiza la presencia o ausencia de los diversos grupos funcionales en moléculas orgánicas. Se pretende en esta práctica dar una orientación del comportamiento de algunos de los grupos funcionales más comunes. Todos los ensayos que se describen a continuación, se realizarán en tubos de ensayo que deben estar perfectamente limpios y además secos. REACCIONES DE CARACTERIZACIÓN DE GRUPOS FUNCIONALES ¿Qué es una reacción de caracterización? Es una reacción química para poner en evidencia la presencia de un grupo funcional. Para que una reacción pueda ser usada con este fin debe dar como resultado un cambio visible en el medio de la reacción, por ejemplo un cambio de color, el desprendimiento de un gas, la formación de un precipitado o la separación de fases si partimos de una solución. El resultado no es definitivo ya que se pueden obtener resultados falsos positivos o falsos negativos. Para comparar la reacción de la muestra se realizan: blanco o testigo negativo que consiste en realizar la reacción con todos los componentes excepto la sustancia a analizar testigo positivo que se realiza reemplazando la muestra con una sustancia que contiene el grupo funcional a evaluar. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 48 TRABAJO PRÁCTICO N° 8 REACCIONES DE CARACTERIZACIÓN Reacciones de alcoholes, aldehídos y cetonas Materiales: 6 tubos de ensayo 1 gradilla 1 pipeta graduada 1 vaso de precipitados 1 cristalizador 1 erlenmeyer chico 1 mechero 1 tela metálica 1 trípode Reactivos: alcoholes 1º, 2 º ,3 º ; aldehídos, cetonas y ácidos carboxílicos como muestras incógnitas Solución sulfocrómica: se emplearán unas gotas de solución (aproximadamente 1 ml de solución para todos los alcoholes a ensayar). Solución de KMnO4 Na: un trocito en forma de cubo de unos 2 mm de lado para cada alcohol a ensayar, que será provisto en el momento por el docente. NaOH 5%: 5 ml H2SO4 conc.: gotas Ac. Acético: 10 ml Solución de carbonato de sodio 10%: 100 ml Solución de 2,4-dinitrofenilhidracina Reactivo de Tollens Reactivo de Fehling Procedimiento: 1- Solubilidad en agua: Colocar aproximadamente 0,5 ml de cada alcohol a ensayar en un tubo de ensayo rotulado y 1 ml de agua. Agitar y observar o no disolución, total o parcial. Comparar los resultados. 2- Oxidación con CrO3 / H2SO4:. Colocar 1 ml de cada uno de los alcoholes en cada tubo de ensayo rotulado, agregar 2 a 3 gotas de solución sulfocrómica. Observar: un cambio de color del anaranjado del CrO3 a verde del Cr+3 indica resultado positivo. Sacar conclusiones acerca de las distintas reacciones según sean alcoholes primarios, secundarios y terciarios. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 49 3- Oxidación con KMnO4: Colocar 1 ml de cada uno de los alcoholes en un tubo de ensayo rotulado, acidificar con una gota de H2SO4 10% y agregar 2 a 3 gotas de solución de KMnO4, agitar enérgicamente y observar. Calentar a baño maría y observar nuevamente. La decoloración total del KMnO4 cambios de color indican resultado positivo. Anotar los resultados y escribir las reacciones correspondientes. 4- Propiedades ácido / base: Reacción con Naº: Colocar 2 ml de cada uno de los alcoholes a ensayar en tubos de ensayo perfectamente secos. Agregar un trocito de sodio (de superficie brillante) de 2-3 mm de lado. En un vaso de precipitados de 250 ml de agua, agregar una cantidad similar de Naº. Observar el desprendimiento de H2. comparar la velocidad de reacción con el Naº de los diferentes alcoholes entre sí, y con el agua. Escribir las ecuaciones correspondientes. 5- Formación de ésteres: Colocar en un erlenmeyer chico 1 ml de ác. acético, 2 ml de etanol y agregar con cuidado 10 gotas de H2SO4 concentrado. Calentar suavemente durante 2 minutos. Enfriar y verter cuidadosamente sobre una solución de carbonato de sodio contenida en un cristalizador y oler inmediatamente, tratar de reconocer el oler del compuesto formado. Repetir el ensayo con el alcohol amílico. Formular las reacciones correspondientes. 6- Ensayo con 2,4-dinitrofenilhidracina: Agregar 3-5 gotas de reactivo a 2-3 ml de sustancia a probar, preferentemente recién preparado, agitar enérgicamente. Si no se forma precipitado inmediatamente, dejar reposar 5 – 10 min. Un precipitado amarillo, anaranjado o rojo correspondiente al derivado 2,4-dinitrofenilhidrazona indica la presencia de un aldehído o una cetona. X1, X2 = H, R ó Ar X1 NO2 C=O+ NO2 NHNH2 NO2 NO2 X2 X1 NHN=C X2 7- Ensayo del Yodoformo: Disolver 0,1 gr ó 4 – 5 gotas del compuesto en 2 ml de agua. Agregar 2 ml de NaOH al 5% y luego gota a gota y con agitación, una solución de Lugol hasta que se observe exceso de yodo (el color pardo persiste), dejar reposar 2 – 3 minutos. Si no aparece el precipitado amarillo del yodoformo, calentar unos minutos a baño maría a unos 60 ºC; si durante el calentamiento se decolora la solución, agregar unas gotas más de Lugol. Esta reacción da positiva con metil cetonas, acetaldehído y alcoholes que por oxidación den estos compuestos. RCOCH3 + 3I2 +3NaOH RCOONa + I3CH (ppdo amarillo) + NaCl + H2O QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 50 Propiedades reductoras de los aldehídos: Los aldehídos se oxidan muy fácilmente en medio alcalino. Esta propiedad permite diferenciarlos de las cetonas. Los ensayos que se emplean son los de Fehling y Tollens. 8- Ensayo de Tollens: En un tuvo limpio colocar 3-5 gotas de reactivo de Tollens a 2-3 ml de sustancia a probar. Si no hay reacción en frío calentar a baño maría. El ensayo es positivo cuando se forma un espejo de plata e indica la presencia de un reductor como el aldehído. Una vez realizado el ensayo lavar el tubo con ácido HNO3 diluido: el espejo de Ag puede quedar contaminado con fulminato de plata, que cuando se seca es explosivo. H RC = O + Ag(NH3)2 OHRCOO- + Ag º + 2 NH3 9- Ensayo de Fehling: En un tubo de ensayo mezclar 3-5 gotas de reactivo de Tollens a 2-3 ml de sustancia a probar. Si no hay reacción en frío calentar a baño maría. El ensayo es positivo cuando se forma un precipitado rojizo de Cu2O e indica la presencia del aldehído reductor. H RC = O + Cu+2 (sol. azul) OHRCOO- + Cu2O Nota: Toda las cantidades requeridas son aproximadas. Preparación de los reactivos: Solución de 2,4-dinitrofenilhidracina: suspender 2 gr de 2,4-dinitrofenilhidracina en 100 ml de metanol; agregar con cuidado 2 ml de H2SO4 concentrado. La mezcla se calienta y el sólido generalmente se disuelve totalmente, en caso contrario, filtrar. Reactivo de Tollens: mezclar volúmenes iguales de AgNO3 al 10% y NaOH al 10%, se observara la formación de un precipitado pardo de óxido de plata. Agregar NH3 (1:1) gota a gota hasta que se redisuelva el óxido de plata. Reactivo de Fehling: sol A: disolver 7 gr de sulfato de cobre pentahidratado en agua acidulada con H2SO4 y llevar con agua hasta 100 ml. Sol. B: disolver 12 gr de NaOH y 36 gr de tartrato de Na y K en agua, llevar hasta 100 ml; filtrar si es necesario. Solución sulfocrómica: Mezclar 250 ml de H2SO4 con 750 ml de agua. Disolver en ésta 100gr de Na2Cr2O7. Solución de KMnO4 : Preparar una solución al 5% en agua. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 51 BIODIESEL Tanto en el transporte como en la industria está muy difundido el uso de motores Diesel. Se trata de un motor térmico de combustión interna en el cual el encendido se logra por la temperatura elevada, producto de la compresión del aire en el interior del cilindro. El combustible más utilizado por estos motores es una fracción de destilación del petróleo crudo que se recoge en el rango de 175 – 370 °C den ominada gasóleo. Esta fracción contiene hidrocarburos como parafina, naftalenos, olefinas y compuestos aromáticos conteniendo moléculas de 20 carbonos. Sin embargo, el motor inventado y patentado por Rudolf Diesel en 1892, utilizaba originalmente un biocombustible: aceite de Palma, o de coco. El biodiesel es el monoalquil éster de un ácido graso de cadena larga derivado de aceites vegetales o de grasas animales que se utilice en motores de ignición por compresión (llamados Diesel). Se obtiene por transesterificación de aceites vegetales. La transesterificación consiste en la conversión de los triglicéridos, esteres de ácidos grasos y glicerol, que conforman los aceites vegetales en metil o etil esteres de los mismos ácidos grasos. Esto se puede lograr tratando los aceites vegetales con metanol o etanol en medio ácido o alcalino y la mezcla obtenida corresponde al biodiesel. Los aceites vegetales más utilizados provienen de soja, girasol y de colza. H2C – O – CO– ácido graso HC – O – CO– ácido graso + CH3OH H2C – O – CO– ácido graso Ácido graso–COOCH3 + glicerol Aceite vegetal El biodiesel produce una cantidad de energía similar al diesel de petróleo pero es un combustible más limpio que el diesel regular y puede ser utilizado por cualquier tipo de vehículo diesel (vehículos de transporte, embarcaciones, naves turísticas y lanchas), solo o en solución con aditivos para mejorar la lubricidad del motor. Actualmente en varios países el biodiesel es utilizado en mezclas con porcentajes diversos. El producto de la transesterificación es una mezcla de esteres, glicerol, alcohol, catalizador (acido o álcali) y mono-, di- triglicéridos. Otro de los subproductos que se obtiene junto con los mencionados son jabones. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 52 TRABAJO PRÁCTICO N° 9 Síntesis de Biodiesel y determinación de la densidad del producto Materiales: · metanol o isopropanol · Aceite de girasol sin usar y usado · catalizadores: NaOH · Balón y tubo refrigerante · Agitador magnético · Ampolla de decantación · Probetas, pipetas, balanza, vasos de precipitados Procedimiento: 1. Colocar 130 g de aceite de girasol en un balón de destilación de 250 cm3 2. Pesar 37,5 g de metanol o equivalente en moles y 1,3 g de hidróxido de sodio sólido en un vaso de precipitados, mezclar con un agitador magnético hasta obtener una solución. 3. Agregar la solución al aceite de girasol en el balón. 4. Calentar a reflujo en baño de agua a 65 °C y agitando continuamente durante 1 hora. 5. Luego de este intervalo de tiempo se retira el glicerol formado utilizando una pipeta y se calienta durante una hora más. 6. Transferir la mezcla de reacción a una ampolla de decantación donde se deja por un día. 7. Separar la capa de glicerol de la de Biodiesel, midiendo el volumen de ambas con una probeta. 8. Colocar la capa de Biodiesel en la ampolla de decantación y lavar con agua destilada hasta que el agua de lavado esté neutra (se controla esta etapa utilizando papel pH universal). 9. Romper la emulsión formada agregando a la ampolla de decantación 75 cm3 de solución saturada de cloruro de sodio y dejando la mezcla por 5 días. 10. Separar ambas capas y lavar la capa de Biodiesel con pequeñas porciones de agua, secar son sulfato de sodio anhidro. (se puede utilizar nitrato de plata para comprobar que no quedó nada de cloruro de sodio). 11. Guardar la muestra de Biodiesel en una botella oscura tapada. 12. Determinar la densidad de las muestras, midiendo 5 cm3 de Biodiesel en una probeta del mismo volumen previamente pesada. Pesar nuevamente la probeta con Biodiesel y calcular la densidad. 13. Medir la densidad de todos los reactivos utilizados. 14. Repetir el experimento en iguales condiciones pero utilizando aceite de girasol usado. Datos: La calidad del biodiesel obtenido se determina según las siguientes propiedades: _ El calor calorífico: es una medida de la energía disponible en el combustible. _ La viscosidad: es importante ya que afecta el flujo del combustible en las tuberías y el inyector. _ La densidad. _ Punto de nube: es la temperatura a la cual el biodiesel forma una nube cuando es enfriado, es una medida del punto de congelación. _ El “flash point”: es una medida de la volatilidad del combustible _ El número de cetano: es una medida de la calidad de ignición del combustible. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 53 PREPARACIÓN DE LA ASPIRINA (ÁCIDO ACETILSALICÍLICO) La aspirina es el fármaco que más se ha empleado en la sociedad moderna. El nombre de aspirina deriva de su estructura, ácido acetilsalicílico. Antiguamente al ácido salicílico se le conocía como ácido spiraerico (de la Spiraea ulmaria) y por lo tanto la aspirina era el ácido acetilespiraerico, de donde derivó su nombre. La aspirina es, aún actualmente, uno de los medicamentos de mayor uso y consumo mundial por su conocida acción analgésica, antipirética y antiinflamatoria sobre el organismo. La aspirina, actúa inhibiendo la biosíntesis de prostaglandinas, compuestos que inducen el dolor, la inflamación y la fiebre. Asimismo, la aspirina pose un moderado efecto anticoagulante derivado de la inhibición que ejerce en la biosíntesis del tromboxano, un agregador plaquetario, y que ha llevado a su utilización en la prevención del infarto de miocardio y de ataques al corazón por formación de trombos. El propio ácido salicílico es un analgésico. Este es el producto que se extrae de varias plantas y es capaz de aliviar el dolor. Inicialmente, este fármaco se administró en forma de sal sódica. Sin embargo, el uso del salicilato sódico producia molestos efectos secundarios y se buscó una modificación del fármaco que retuviese las propiedades terapéuticas sin presentar los efectos secundarios indeseables. Por tratamiento de ácido salicílico con anhídrido acético se obtiene el ácido acetil salicílico, un compuesto tan eficaz como el salicilato sódico, pero de reducidos efectos secundarios. Este mismo tipo de estrategia se empleó mas tarde para la modificación de un potente analgésico, la morfina. En este caso el problema consistía en su capacidad de crear adicción y, con la idea de solventarlo, se acetiló la morfina, obteniéndose la heroína, no cabe duda que en este caso la estrategia no tuvo el éxito que en el ácido salicílico. El producto de partida para la fabricación de la aspirina es el ácido salicílico que, a su vez, se prepara como sal sódica por tratamiento del fenóxido sódico con dióxido de carbono a unas 5 atm de presión y a una temperatura de 125ºC (síntesis de Kolbe). El ácido acetilsalicílico se prepara fácilmente en el laboratorio por esterificación del grupo hidroxilo del ácido salicílico (ácido 2-hidroxibenzoico). La formación de un éster a partir de un ácido carboxílico y un alcohol (esterificación de Fischer) es un proceso que sólo se produce si se utiliza como catalizador un ácido fuerte: ácido orto-fosfórico, ácido sulfúrico, etc. Es una reacción de equilibrio que puede evolucionar en ambas direcciones dependiendo de las condiciones empleadas. Los estudios realizados para conocer el mecanismo de esta reacción, han puesto de manifiesto que el –OH del ácido (del grupo –COOH) y el –H del alcohol (del grupo – OH) son los que forman la molécula de H2O. QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 54 La reacción se puede desplazar hacia la formación del éster eliminando el agua a medida que se va formando y/o utilizando un exceso de uno de los dos reactivos (generalmente de alcohol). Aunque se pueden obtener esteres de ácido por reacción directa del ácido con el alcohol, se suele utilizar un derivado de ácido como puede ser un anhídrido o un cloruro como agente acilante, ya que estos permiten obtener los ésteres a una velocidad mucho mayor. En esta práctica, se prepara el ácido 2-acetoxibenzoico por reacción entre el ácido ortohidroxibenzoico y el anhídrido acético utilizando ácido sulfúrico como catalizador: T.R. SNacilo QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 55 TRABAJO PRÁCTICO N° 10 SÍNTESIS DE ASPIRINA Obtención de ácido acetilsalicílico (Aspirina) Introducción La aspirina es uno de los medicamentos de uso más general. Desde que en 1859 fue introducida por primera vez por Dreser como analgésico y antipirético suave, su empleo fue creciendo hasta llegar a convertirse en la principal defensa contra pequeñas indisposiciones. En la actualidad se le han encontrado nuevas aplicaciones, y se la utiliza en múltiples situaciones, ya no necesariamente vinculadas a dolencias menores. El producto de partida para la fabricación de la aspirina es el ácido salicílico. Este ácido también es ampliamente usado en medicina, dado que produce una muy buena respuesta fisiológica en el organismo cuando se absorbe a través de la membrana intestinal (la aspirina también es absorbida a nivel del intestino). El problema es que el ácido salicílico resulta desagradablemente irritante cuando se toma por vía oral, debido a su fuerte carácter ácido. Por lo tanto se lo usa preferentemente en aplicaciones externas, ya sea en forma ácida o como salicilato de metilo, por ejemplo en pomadas para dolores musculares o golpes, muchas de estas son a base de estas drogas, en algunos casos asociadas con ácido benzoico. En el caso del ácido acetilsalicílico, cuando atraviesa los fluidos ácidos del estómago no tiene transformaciones químicas, pero cuando llega al intestino sufre una reacción química, llamada hidrólisis, que la vuelve a transformar en ácido salicílico, que es la droga realmente efectiva. La aspirina es un ácido pero es más débil que el ácido salicílico. Fundamentos de la técnica de obtención El ácido salicílico reacciona muy lentamente con el anhídrido acético a ebullición, pero si se agregan unas pocas gotas de ácido sulfúrico concentrado, la reacción comienza a temperatura ambiente y se desarrolla rápidamente con considerable desprendimiento de calor. Materiales necesarios: Tubos de ensayo Pipeta de 2 ml Vaso de precipitados Erlenmeyer de 125 ml de capacidad Varilla de vidrio Embudo Papel de filtro QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 56 Reactivos necesarios Ácido salicílico Anhídrido acético Ácido sulfúrico concentrado Solución alcohólica al 30% v/v Procedimiento 1. Colocar en un tubo de ensayos 3 g de ácido salicílico y añadir 10 ml de anhídrido acético. 2. Agregar muy cuidadosamente 5 gotas de ácido sulfúrico concentrado. 3. Calentar el tubo de ensayos a baño maría, a 50 º C aproximadamente (controlar con termómetro que la temperatura no sobrepase los 60 º C). Esperar al menos 5 minutos hasta que se disuelva el sólido y se complete la reacción. 4. Verter la solución sobre 30 ml de agua helada contenida en un erlenmeyer. 5. Filtrar el sólido precipitado. 6. Recristalizar el sólido en 30 ml de etanol caliente hasta redisolver el precipitado. 7. Una vez disuelto volcar sobre agua tibia y esperar la precipitación siempre agitando con varilla. 8. Filtrar el sólido obtenido y secarlo. 9. Pesar y tomar el punto de fusión. 10. Recristalizar. Datos: Punto de fusión del ácido salicílico: 160 º C Punto de fusión de la aspirina: 132 ºC QUÍMICA ORGÁNICA-GUÍA TRABAJOS PRÁCTICOS 57