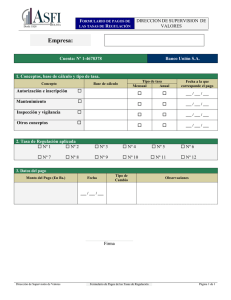
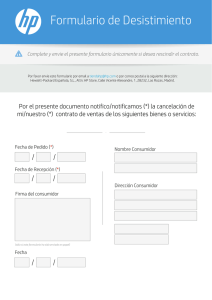
a tracto alim en tario ym etabo lism o
Anuncio

A02 DROGAS PARA DESÓRDENES RELACIONADOS CON ÁCIDO Hidróxido de Aluminio / Magnesio Omeprazol Ranitidina Sucralfato A04 ANTIEM…TICOS Y ANTINAUSEOSOS Ondansetrón A06 LAXANTES Glicerol (Glicerina) Glicósidos del Senna (Senósidos A y B) Ispaghula (Psyllium) Lactulosa Macrogol 3350 (Polietilén Glicol 3350) A07 ANTIDIARRÉICOS, AGENTES ANTIINFLAMATORIOS / ANTIINFECCIOSOS INTESTINALES Nistatina (Antiinfeccioso Intestinal) Sales de Rehidratación Oral (SRO) Sulfasalazina A10 DROGAS USADAS EN DIABETES Glibenclamida Insulina de Acción Rápida (Insulina Cristalina Humana Recombinante) Insulina de Acción Intermedia (Insulina N.P.H. Humana Recombinante) Insulina de Acción Intermedia (Insulina Premezcla Cristalina / N.P.H. Humana Recombinante (30/70)) Metformina A TRACTO ALIMENTARIO Y METABOLISMO A03 DROGAS PARA DES”RDENES FUNCIONALES GASTROINTESTINALES Atropina (Desórdenes Funcionales Gastrointestinales) Metoclopramida A A11 VITAMINAS Ácido Ascórbico (Vitamina C) A12 SUPLEMENTOS MINERALES Citrato de Calcio* (Citrato Tricálcico) Gluconato de Calcio 2ed. 2004 * Formulario TerapÈutico Nacional 1 HIDRÓXIDO DE ALUMINIO / MAGNESIO FARMACOLOGÍA Actúa neutralizando la acidez de la secreción gástrica produciendo un incremento del pH del medio y, en consecuencia, una disminución de la actividad proteolítica de la pepsina. Fortale-ce la barrera de la mucosa gástrica y aumenta el tono del esfínter esofágico. FARMACOCINÉTICA Luego de la administración oral se produce un efecto antiácido inmediato que persiste por 20-40 minutos si se administra con el estómago vacío, y por 2-3 horas si se administra con las comidas. El aluminio y el magnesio de la formulación se absorben en porcentajes inferiores al 20% y son posteriormente excretados por la orina. La fracción no absorbida es excretada en las heces. reducir la velocidad y la magnitud de la absorción gastrointestinal de numero- A sos fármacos, por modificación del pH del medio o por formación de complejos con ellas. Para evitarlo, se recomienda evitar la administración conjunta y, de ser posible, separar la toma de antiácidos y otros medicamentos por lo menos por 1-2 horas. Fármacos con cubierta entérica: El fármaco puede ser liberado prematuramente en el estómago. La administración de los fármacos debe ser separada por lo menos por 1 hora. CONTRAINDICACIONES/ PRECAUCIONES RÉGIMEN DE DOSIFICACIÓN Cinco a 10 ml después de o entre las comidas y antes de acostarse. Usar con precaución en pacientes con enfermedad renal severa, en ancianos, en el embarazo y durante la lactancia. En pacientes con insuficiencia renal, el hidróxido de aluminio puede ocasionar hiperaluminemia y el hidróxido de magnesio puede conducir a hipermagnesemia con manifestacio-nes de hipotensión, náusea, vómitos, trastornos cardiovasculares, depre-sión respiratoria y del sistema nervioso central. No es recomendable el uso por períodos mayores de 2 semanas. REACCIONES ADVERSAS Los antiácidos de aluminio y magnesio, por lo general, son seguros y bien tolerados por la mayoría de los pacientes. Sin embargo, ocasional- SOBREDOSIS Dado que la absorción gastrointestinal es muy limitada, la sobredosis aguda no representa un riesgo de importancia. INDICACIONES Alivio sintomático de la acidez gástrica, reflujo gastroesofágico y pirosis. 2ed. 2004 * Formulario TerapÈutico Nacional 3 gía inusual durante el tratamiento, en especial diarrea o constipación. No u- el consecuente aumento de la vida media. OMEPRAZOL INDICACIONES Tratamiento de la úlcera gastroduodenal, esofagitis por reflujo y el síndrome de Zollinger-Ellison. FARMACOLOGÍA Inhibe la actividad de la enzima H+ / K+ ATPasa localizada en la superficie secretora de la célula parietal; por tanto, inhibe la bomba de protones, bloqueando la síntesis del ácido clorhídrico. También inhibe la anhidrasa carbónica gástrica. FARMACOCINÉTICA Se absorbe en el tracto gastrointestinal sólo en un 30-40% como fármaco intacto debido a un intenso efecto metabólico de primer pasaje. Luego de la administración, se observa una res-puesta terapéutica inicial a los 60 minutos, la cual se hace máxima a las 2 horas y se mantiene por aproximadamente 3 días. Con la administración continua se alcanza el estado estable al cuarto día. Se une a las proteínas plasmáticas en un 95%. Se elimina como, por lo menos, 6 metabolitos inactivos en un 77% por la orina y el resto por vía biliar. Aunque la vida media plasmática es de 30-60 minutos, al suspender la dosificación la secreción gástrica retorna a sus valores basales en 3-5 días, debido al efecto irreversible sobre la enzima H+ / K+ ATPasa. En sujetos con insuficiencia hepática, la biodisponibilidad 4 RÉGIMEN DE DOSIFICACIÓN La dosis usual por vía oral en adultos para el tratamiento de la úlcera duodenal y la esofagitis por reflujo, sin lesiones erosivas, es de 20 mg/día hasta por 4 semanas, y en pacientes con esofagitis erosiva, 20 mg/día por 4 a 8 semanas; en la úlcera gástrica: 40 mg/día por 4-8 semanas y en el síndrome de Zollinger-Ellison: 60 mg/día por el tiempo que sea necesario. REACCIONES ADVERSAS Cardiovasculares: Dolor anginoso, palpitaciones, elevación de la presión arterial, bradicardia, edema periférico. Gastrointestinal: Dolor abdominal, náusea, vómito, diarrea, flatulencia, decoloración de las heces, atrofia de la mucosa de la lengua, perversión del gusto, constipación, anorexia, pancreatitis. Hematológicas: Discrasias sanguíneas. Hepáticas: Elevación de los niveles de las enzimas hepáticas, falla hepática. Neurológicas: dolor de cabeza, fatiga, astenia, mareo, tinitus, depresión, confusión, nerviosismo, vértigo. Reacciones de hipersensibilidad: Erupción, eritema multifor- Formulario Terapéutico Nacional * 2ed. 2004 dolor en las piernas, ginecomastia, hipoglicemia. INTERACCIONES Prolonga la eliminación del diazepam, de la warfarina y de la fenitoína. Por su efecto modificador del pH gástrico, puede alterar la absorción gastrointestinal de fármacos para los cuales el pH gástrico es importante, tales como el ketoconazol, la ampicilina, las sales de hierro y el fenobarbital. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al fármaco o a cualquier componente de la formulación. Usar con precaución en pacientes con falla hepática. Se ha descrito casos de gastritis atrófica en pacientes sometidos a tratamiento por períodos prolongados. La respuesta sintomática al tratamiento con omeprazol no descarta la presencia de una malignidad gástrica. Dado que no se conoce con precisión la seguridad del uso en el embarazo, se recomienda limitar su uso en esta condición a situaciones de verdadera necesidad en las que el beneficio supere al riesgo potencial para el feto. Se desconoce la seguridad de su uso durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado cápsula de liberación prolongada no debe ser abierta, masticada o triturada A y debe tomarse completa. Este medicamento no se puede tomar con antiácidos. Informar al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. También debe informar al médico si RANITIDINA FARMACOLOGÍA Inhibe competitivamente la acción de la histamina sobre los receptores H2 de histamina, incluyendo aquellos presentes en la célula parietal, reduciendo así la secreción del ácido gástrico. In-hibe tanto la secreción del ácido gás-trico basal nocturna como aquella esti-mulada por la comida, por betazol y por la pentagastrina, aunque parece ser menos sensible que aquella inducida por la comida y por la pentagastrina. FARMACOCINÉTICA Aunque se absorbe rápidamente después de la administración oral, sólo un 50% de la dosis alcanza la circulación sistémica como fármaco intacto debi-do al efecto de primer pasaje. Alcanza concentraciones plasmáticas pico a las 2-3 horas. Se une a las proteínas plasmáticas en un 10-19% y se distribuye ampliamente en el organismo. Se metaboliza en el 2ed. 2004 * Formulario TerapÈutico Nacional 5 materna. La vida media de eliminación es de 1,7-3,2 horas y se incrementa en pacientes con alteración de la función renal. INDICACIONES Tratamiento de la úlcera gástrica, úlcera duodenal, esofagitis de reflujo, condiciones hipersecretorias asociadas a la hospitalización y a la cirugía, y del síndrome de Zollinger-Ellison. R…GIMEN DE DOSIFICACI”N Se administra por vía oral y por vía IV. La dosis oral recomendada en adultos para el manejo de la úlcera gástrica y duodenal es: 150 mg dos veces diarias o, alternativamente, dosis única diaria de 300 mg después de la cena o antes de dormir. Para la terapia de mantenimiento, se recomienda administrar la dosis al acostarse. En niños de 1 mes16 años: 2-4 mg/kg dos veces al día, hasta un máximo de 300 mg/día. Para el tratamiento del reflujo gastroesofágico en adultos: 150 mg dos veces al día; en niños de 1 mes-16 años: 2,55,0 mg/kg. Para el síndrome de Zollinger-Ellison en adultos: 300-600 mg/día en dosis divididas. Cuando se administra por vía IV, la dosis recomendada es de 150 mg/día en dosis divididas o en infusión continua por 24 horas; en caso necesario, se puede incrementar la dosis pero sin exceder 400 mg/día. 6 pasmo, fiebre, erupción y eosinofilia. INTERACCIONES Puede incrementar los niveles séricos del triazolam, procainamida, glipizida y nifedipina. En tratamientos prolongados puede afectar la absorción gastrointestinal de la vitamina B12. Los antiácidos pueden interferir en la acción de la ranitidina. Si se decide administrarlos conjuntamente, debe hacerse con un intervalo de 2 horas entre los dos medicamentos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al fármaco. La respuesta sintomática a la ranitidina no descarta la presencia de una malignidad gástrica. Usar con precaución en pacientes con alteración de la función renal y/o hepática, historia de porfiria, en ancianos y durante la lactancia. En la terapia IV por más de 5 días, se recomienda vigilar la función hepática. SOBREDOSIS Vaciamiento gástrico, uso de carbón activado y un catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. La ranitidina es dializable. SUCRALFATO FARMACOLOGÍA El sucralfato reacciona con el ácido Formulario Terapéutico Nacional * 2ed. 2004 do viscoso que se adhiere a la superficie lesionada constituyendo una película insoluble y estable, la cual actúa como barrera protectora que adsorbe y forma complejos con la pepsina y los ácidos biliares, a la vez que bloquea el acceso de los iones H+ a la zona erosionada. En el tiempo, permite la regeneración tisular y, en consecuencia, la curación de la lesión. FARMACOCINÉTICA Debido a la escasa solubilidad y elevada polaridad, luego de la administración oral, el sucralfato permanece casi en su totalidad en el tubo digestivo sin absorberse y adherido a la superficie erosionada hasta por 6 horas. Se estima que sólo un 3-5% de la dosis se absorbe sistémicamente y es posteriormente excretado intacto por la orina en 48 horas. INDICACIONES Tratamiento de la úlcera duodenal acti-va. RÉGIMEN DE DOSIFICACIÓN Se administra solamente por vía oral. La dosis usual en adultos es de 1 g 4 veces al día por 4-8 semanas. La dosis de mantenimiento recomendada para adultos es 1 g dos veces al día. INTERACCIONES Puede reducir la absorción gastroin- A testinal de cimetidina, ranitidina, digoxina, ketoconazol, quinolonas, tiroxina, fenitoína, quinidina, tetraciclinas y teofilina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Ante la posibilidad de acumulación e intoxicación por el aluminio contenido en la molécu-la, se debe usar con precaución en pa-cientes con insuficiencia renal crónica. Dado que no se conoce si la pequeña cantidad absorbida sistémicamente a-traviesa la placenta o se distribuye a la leche materna, se recomienda usar el sucralfato con precaución en tales circunstancias. SOBREDOSIS Debido a que no se absorbe bien en el tracto gastrointestinal, la sobredosis de sucralfato no reviste un riesgo importante de toxicidad. Las manifestaciones descritas en tales circunstancias incluyen dispepsia, dolor abdominal, náusea y vómito. INFORMACIÓN AL PACIENTE Tomar 1 hora antes o 2 horas después de las comidas. Si el paciente toma REACCIONES ADVERSAS Gastrointestinales: Constipación, dia- algún otro medicamento, se debe adrrea, náusea, vómito, indigestión, ministrar éste 2 horas antes del sucralfato. Informar al médico si se 7 2ed. 2004 * Formulario TerapÈutico Nacional ATROPINA fotofobia, aumento de la presión in(DesÛrdenes Funcionales Gastrointestinales) traocular. Reacciones de hipersensibilidad: Erupción, urticaria y anafilaxis. FARMACOLOGÍA Antagonista competitivo de la acetil- Otras: Retención urinaria, anhidrosis, c o l i n a e n l o s r e c e p t o r e s impotencia. muscarínicos. Inhibe las acciones muscarínicas de la acetilcolina sobre INTERACCIONES Presenta efectos anticolinérgicos adilas estructuras iner-vadas por los tivos con procainamida, quinidina, dinervios postgangliona-res sopiramida, meperidina, fenotiazinas, colinérgicos. antidepresivos tricíclicos, agentes antiparkinsonianos y algunos antihistaFARMACOCINÉTICA Se absorbe bien por vía IM y SC. Se mínicos. El retardo del vaciamiento distribuye ampliamente en el organis- gástrico producido por el fármaco puemo y atraviesa la barrera hematoence- de favorecer la degradación gástrica fálica. Se une a las proteínas de la levodopa y disminuir sus niveles p l a s m á t i - c a s e n u n 1 8 % . S e plasmáticos. Puede antagonizar los m e t a b o l i z a e n e l h í - g a d o efectos de la metoclopramida y de la transformándose en productos cisaprida. inactivos que se excretan, junto a un 30-50% de fármaco intacto, principalmente por la orina. La eliminación es bifásica, con una vida media de 2 horas en su fase inicial y de 12,5 horas en su fase terminal. INDICACIONES Trastornos funcionales de la motilidad gastrointestinal. R…GIMEN DE DOSIFICACI”N En adultos: 0,4-0,6 mg IM, IV ó SC cada 4-6 horas; en niños: 0,01 mg/kg IM, IV ó SC cada 4-6 horas (hasta un máximo de 0,4 mg/dosis). REACCIONES ADVERSAS 8 CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con neuropatía autonómica, glaucoma de ángulo cerrado, hipertrofia prostática, enfermedad gastrointestinal obstructiva, colitis ulcerativa, íleo paralítico y miastenia gravis. Usar con precaución en pacientes con enfermedad hepática y/o renal, fiebre, reflujo gastroesofágico, hipertensión, hipertiroidismo, taquiarritmias, insuficiencia cardiaca congestiva, enfermedad coronaria, enfermedad pulmonar crónica, daño cerebral, síndrome de Down, en ancianos, en niños y en el Formulario Terapéutico Nacional * 2ed. 2004 jo sintomático y terapia de soporte. En casos severos se puede usar neostigmina para revertir los efectos anticolinérgicos. INFORMACIÓN AL PACIENTE METOCLOPRAMIDA FARMACOLOGÍA Agente procinético. Estimula la motilidad del tracto gastrointestinal superior sin afectar la secreción gástrica, biliar o pancreática. Incrementa el tono de reposo del esfínter esofágico inferior, el tono y la amplitud de las contracciones del estómago, particularmente de la porción antral, relaja el esfínter pilórico e incrementa la peristalsis, dando lugar a una aceleración del vaciamiento gástrico y del tránsito intestinal. Parece que a su actividad contribuye una sensibilización de los tejidos a la acción de la acetilcolina. El efecto sobre la motilidad no es dependiente de una inervación vagal intacta, pero puede ser abolida por agentes anticolinérgicos. Ejerce, además, efectos antagonistas sobre los receptores dopaminérgicos centrales y periféricos involucrados en el mecanismo del vómito. Como otros antagonistas de los receptores dopaminérgicos, el cual puede estar asociado con una A retención transitoria de fluido. FARMACOCINÉTICA Se absorbe rápida y casi completamente en el tracto gastrointestinal, 80% por el efecto metabólico del primer pasaje. Genera un inicio de acción a los 30-60 minutos de la administración oral, a los 15-30 minutos de la inyección IM y a los 1-3 minutos por vía IV, que se mantiene (independiente-mente de la vía de administración) por 1-2 horas. Se une a las proteínas plas-máticas en un 30% y se distribuye am-pliamente en el organismo. Se excreta en un 85% por la orina (la mitad como fármaco intacto) y el resto en las he-ces. La vida media es de 5-6 horas. INDICACIONES Gastroparesis del diabético, reflujo gastroesofágico y prevención de las náuseas y vómitos post-operatorios o inducidos por la quimioterapia antineoplásica; como ayuda en procedimientos diagnósticos. RÉGIMEN DE DOSIFICACIÓN - Gastroparesis del diabético: 10 mg por vía oral 30 minutos antes de cada comida y al acostarse, durante 2-8 semanas, para síntomas leves. Para síntomas severos, una infusión lenta de 1 a 2 minutos en un esquema igual que para la vía oral 2ed. 2004 * Formulario TerapÈutico Nacional 9 lización de la cirugía. Se puede repe-tir cada 4 a 6 horas, según sea nece-sario. - Prevención de las náuseas y los vómitos inducidos por la quimioterapia antineoplásica: 1-2 mg/kg por infusión IV lenta 30 minutos antes del inicio de la quimioterapia; repetir 2 veces más a intervalos de 2 horas y luego cada 3 horas hasta 3 dosis. REACCIONES ADVERSAS Cardiovasculares: Hipotensión o hipertensión, bradicardia o taquicardia. Gastrointestinales: Acalasia, diarrea. Genitourinarias: Frecuencia urinaria, incontinencia, amenorrea. Hematológicas: Agranulocitosis, neutropenia, leucopenia. Neurológicas: Cefalea, mareos, fatiga, debilidad, somnolencia o insomnio, depresión, ansiedad, confusión, trastornos visuales, reacciones extrapiramidales como distonía, síntomas tipo Parkinson, diskinesia tardía, protrusión de la lengua, opistótonos, convulsiones. Raras veces puede aparecer el síndrome neuroléptico maligno. Reacciones de hipersensibilidad: Erupción, prurito, angioedema. Respiratorias: Estridores y disnea. Otras: Galactorrea, ginecomastia, porfiria. INTERACCIONES Puede tener un efecto aditivo cuando se administra conjuntamente con otros 10 ticos dependientes de insulina. Puede reducir la absorción gastrointestinal de la digoxina, y aumentar la del acetaminofén, litio, ácido acetilsalicílico, diaze-pam, levodopa, tetraciclina y etanol. Los fármacos anticolinérgicos antago-nizan su acción. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, hemorragia, obstrucción o perforación gastrointestinal, feocromocitoma, historia de convulsiones y en combinación con medicamentos que pueden causar reacciones extrapiramidales. Usar con precaución en pacientes con hipertensión, con feocromocitoma, en niños, ancianos, en el embarazo y durante la lactancia. No debe ser usada cuando la estimulación de la motilidad intestinal puede ser peligrosa, esto es, en presencia de hemorragia gastrointestinal, obstrucción mecánica o perforación. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. La difenhidramina, por su actividad anticolinérgica central, puede re-sultar útil para el control de las reaccio-nes extrapiramidales. INFORMACIÓN AL PACIENTE Formulario Terapéutico Nacional * 2ed. 2004 puede alterar la capacidad física y mental requerida para tareas con cierto riesgo, como operar maquinaria o conducir automóviles, se debe advertir ONDANSETRÓN FARMACOLOGÍA Agente antiemético cuya actividad parece ser debida a la inhibición selectiva de los receptores tipo 3 de serotonina (5-HT 3 ) centrales y periféricos, pre-sentes, respectivamente, en la zona quimiorreceptora gatillo del área postrema y en los terminales nerviosos vagales. Aunque no se sabe si el efecto antiemético es central o periférico, se sabe que la terapia antineoplásica está asociada con la liberación de serotonina de las células enterocromafines. La liberación de serotonina puede estimular los nervios aferentes vagales a través de los receptores 5-HT3 e iniciar el reflejo del vómito. Es posible que el bloqueo tanto de los receptores centrales como de los periféricos contribuyan al efecto antiemético. FARMACOCINÉTICA Aunque se absorbe bien en el tracto gastrointestinal sólo un 56% de la dosis alcanza la circulación sistémica co-mo fármaco intacto, por el efecto de primer pasaje. Se une a las proteínas plasmáticas en un 70-76%. Se meta- incrementa en pacientes con insufiA ciencia hepática. INDICACIONES Prevención y tratamiento de náuseas y vómitos inducidos por la quimioterapia antineoplásica, la radioterapia y post-operatorios. R…GIMEN DE DOSIFICACI”N Se administra por vía oral o en infusión IV durante 15 minutos. - PrevenciÛn de náuseas y vómitos inducidos por la quimioterapia antineoplásica en adultos y niños mayores de 4 años, por infusión IV, en 15 minutos: Iniciar con 0,15 mg/kg 30 minutos antes de la quimioterapia y repetir a las 4 y 8 horas después de la primera dosis. Alternativamente, en adultos: Dosis única de 32 mg 30 minutos antes de la quimioterapia, durante 15 minutos. - PrevenciÛn de las náuseas y vómitos inducidos por la quimioterapia en adultos y niños mayores de 12 años, por vía oral: Iniciar con 8 mg 30 minutos antes de la quimioterapia. Luego, 8 mg 8 horas después de la primera dosis, y repetir cada 12 horas hasta por 1-2 días después de completar la terapia. En niños de 4-11 años: Iniciar con 0,15 mg/kg 30 minu-tos antes de la quimioterapia y repetir a las 4 y 8 horas; a partir de allí, 4 mg cada 8 horas hasta por 1-2 2ed. 2004 * Formulario TerapÈutico Nacional 11 post-operatorios en adultos, por vía IV (sin diluir) en 2-5 minutos: 4 mg inmediatamente antes de la inducción de anestesia o al finalizar la cirugía si el paciente manifiesta náusea o vómito después de terminada la operación. En niños de 2-12 años con más de 40 kg se usa la misma dosis que en el adulto. En niños de 212 años con menos de 40 kg: 0,1 mg/kg. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, hipotensión, dolor anginoso (raro), alteraciones del ECG, trastornos de la conducción. Gastrointestinales: Constipación, diarrea, dolor abdominal. Hepáticas: Alteración de las enzimas hepáticas. Neurológicas: Cefalea, malestar, mareos, sedación, fatiga, ansiedad, parestesia, reacciones extrapiramidales. Reacciones de hipersensibilidad. Otras: Mialgia, retención urinaria, trastornos ginecológicos, visión borrosa, dolor musculoesquelético, crisis oculo-gíricas, sensación de frío, disnea, hi-poxia. INTERACCIONES Reduce la exposición sistémica de la ciclofosfamida en pacientes con cáncer. Los inductores o los inhibidores de las enzimas del citocromo P450 pueden alterar la vida media del ondansetrón. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso GLICEROL (GLICERINA) FARMACOLOGÍA Laxante hiperosmolar que, administrado por vía rectal, ejerce un efecto higroscópico y lubricante. La retención de agua estimula el peristaltismo, hi-drata las heces y se produce la evacuación. FARMACOCINÉTICA La absorción por vía rectal es escasa y carente de importancia clínica. El efecto laxante se evidencia a los 15-30 minutos de su administración. INDICACIONES Condiciones en las que se requiere una evacuación rápida del recto. RÉGIMEN DE DOSIFICACIÓN Se administra por vía rectal como supositorio. Adultos y niños mayores de 6 años: Dosis única de 2-3 g. En niños menores de 6 años: Dosis única de 1-1,5 g. REACCIONES ADVERSAS Molestia rectal, irritación, sensación CONTRAINDICACIONES/ 12 Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Ninguna significativa. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con obstrucción intestinal, impactación fecal, abdomen quirúrgico agudo, presencia de dolor abdominal agudo, náuseas, vómitos u otros síntomas que sugieran la posibilidad de una apendicitis, hipersensibilidad al medicamento. No usar en forma crónica. SOBREDOSIS Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE No usar en presencia de dolor abdominal, náuseas o vómitos. En caso de sangramiento rectal o GLICÓSIDOS DEL SENNA (SEN”SIDOS A Y B) FARMACOLOGÍA Agente laxante, perteneciente al grupo de las antraquinonas. Los derivados antraquinónicos y sus glicósidos cons-tituyen 3 a 5% del sen. Los senósidos A y B son los principales componentes. Altera la permeabilidad de las células intestinales produciendo una secre-ción activa de iones hacia la luz intes-tinal que conduce a la acumulación de fluido, inducción de la peristalsis y la consiguiente evacuación. Actúa fundamentalmente a nivel de la pared FARMACOCINÉTICA Luego de la administración por vía A oral se absorbe sólo muy poco en el intestino delgado. Al llegar al colon, sufre hidrólisis por la acción de las bacterias presentes, transformándose en la forma farmacológicamente activa. El efecto laxante, por lo general, ocurre 6-12 horas después, aunque en algunos casos podría presentarse a las 24 horas. La fracción absorbida sis-témicamente es metabolizada en el hí-gado y excretada como metabolitos y fármaco intacto en las heces y en la orina. INDICACIONES Tratamiento de corta duración de la constipación aguda. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos y niños mayores de 12 años: 12-50 mg 1-2 veces al día por no más de 7 días. En niños de 6-11 años: La mitad de la dosis de los adultos. En niños de 2-5 años: Un tercio de la dosis de los adultos. REACCIONES ADVERSAS Gastrointestinales: Se ha reportado casos de diarrea, irritación gastrointes-tinal, náusea, malestar abdominal y depleción de fluidos y electrolitos. El uso prolongado o en dosis excesivas puede producir diarrea persistente, pérdida de 2ed. 2004 * Formulario TerapÈutico Nacional 13 medicamentos de administración oral, disminuyendo así la absorción y, en consecuencia, la eficacia terapéutica. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, cuadros de obstrucción intestinal y en presencia de dolor abdominal agudo, n·useas, vÛmitos u otros síntomas que sugieran la posibilidad de una apendicitis. El uso crónico puede producir pérdida de la función normal del intestino, malabsor-ción, constipación y dependencia. Usar con precaución en niños peque-ños, ancianos y durante la lactancia. SOBREDOSIS Vaciamiento gástrico si la ingestión es reciente. No administrar carbón activa-do ni catárticos salinos. Manejo sinto-mático y terapia de soporte. INFORMACI”N AL PACIENTE Se debe acompañar la terapia con una ingesta adecuada de fibra y líquidos y ISPAGHULA (PSYLLIUM) FARMACOLOGÍA Laxante formador de volumen. Constituye una fibra no digerible que absorbe agua y se expande para aumentar el volumen y el contenido de humedad de las heces, estimulando la 14 tránsito intestinal. FARMACOCINÉTICA No se absorbe desde el tracto gastrointestinal. Aunque se puede observar una acción laxante inicial a las 1224 horas, el efecto completo requiere de 2-3 días de tratamiento continuo. INDICACIONES Tratamiento de corta duración de la constipación aguda. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Adultos y niños mayores de 12 años: Hasta 30 g/día, divididos en 4-8 dosis. En niños de 6-11 años: Hasta 15 g/día, divididos en 4-8 dosis. Debe administrarse, co-mo mínimo, un vaso de agua con cada dosis. REACCIONES ADVERSAS Poco frecuentes si se emplea en las dosis apropiadas y por períodos cortos. Se han reportado casos de obstrucción esofágica e intestinal cuando se administra con una cantidad insuficiente de agua. Se puede producir cólicos abdominales si el estreñimiento es severo. Con el uso excesivo puede presentarse náusea, dia-rrea y vómitos. INTERACCIONES Al incrementar la motilidad intestinal se reduce el tiempo de tránsito de otros Formulario Terapéutico Nacional * 2ed. 2004 nistración por lo menos por 3 horas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento, cuadros de obstrucción intestinal y en presencia de dolor abdominal agudo, náuseas, vómitos u otros síntomas que sugieran la posibilidad de una apendicitis, adhe-rencias incapacitantes, y en caso de dificultad para tragar. Los laxantes for-madores de bolo no son adecuados cuando se requiere una evacuación in-testinal rápida. SOBREDOSIS Manejo sintomático y terapia de soporte. Administrar abundante agua. INFORMACIÓN AL PACIENTE Se debe acompañar la terapia con una ingesta adecuada de fibra, líquidos y ejercicios. No usar en presencia de dolor abdominal, náuseas o vómitos. Si después de una semana de uso continuo la condición persiste, suspender la administración y consultar al médico. Debe tomarse el medicaLACTULOSA mento con suficiente cantidad de líqui-do. Los pacientes diabéticos FARMACOLOGÍA deben asegurarse que el psyllium Disacárido sintético análogo de la que lacingiere no contenga azúcar. El uso tosa, constituido por galactosa y frucprolongado o excesivo puede conducir tosa. Reduce la concentración a s adepen-dencia. nguí-nea de amoníaco y disminuye el grado de encefalopatía rias intestinales y transformado en productos que inducen un aumento de la A presión osmótica y una ligera acidifica-ción del contenido del colon, ocasio-nando un aumento de la cantidad de agua en el lumen intestinal y heces blandas. Al ser el contenido del colon más ácido que la sangre, el amoníaco migra desde el torrente sanguíneo al colon donde es convertido en amonio no absorbible, que resulta rápidamen-te eliminado con las heces como con-secuencia del efecto laxante. FARMACOCINÉTICA Administrada por vía oral se absorbe menos de un 3%. La fracción absorbida no sufre metabolismo y se excreta intacta por la orina. La lactulosa no absorbida (97%) es metabolizada por las bacterias del colon y transformada en ácido fórmico y acético que se excretan en las heces. INDICACIONES Constipación. Prevención y tratamiento de la encefalopatía portalsistémica. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - ConstipaciÛn: La dosis usual en adultos: 10-20 g/día en una sola toma o dividida en 3-4 dosis. En niños: 0,20,4 g/kg/dosis 3-4 veces al día. Puede requerirse 24-48 horas para 2ed. 2004 * Formulario TerapÈutico Nacional 15 distensión abdominal, eructos, flatulencia, cólicos abdominales y diarrea. Dosis excesivas pueden producir diarrea, con la consecuente pérdida de fluidos y electrolitos. INTERACCIONES La administración conjunta de neomicina u otros agentes antiinfecciosos puede eliminar la flora bacteriana del colon, necesaria para metabolizar la lactulosa. Debe evitarse el uso conjunto de estos medicamentos. Los antiácidos pueden interferir con la acción de la lactulosa. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes que requieren una dieta baja en galactosa. Usar con precaución en niños, ancianos, diabéticos, en el embarazo, durante la lactancia y en pacientes que requieren electrocoagulación durante la colonoscopia y la proctoscopia. Cuando se usa en la encefalopatía portal sistémica puede provocar desequilibrios hidro-electrolíticos. Debe ser usada con precaución en pacientes diabéticos. La eficacia y la seguridad en pacientes pediátricos no ha sido establecida. SOBREDOSIS Puede ocasionar diarrea y cólicos abdominales. Interrumpir la dosificación y proceder al reemplazo de fluido 16 flatulencia, cólicos abdominales o diaMACROGOL 3350 (POLIETIL…N GLICOL 3350) FARMACOLOGÍA Laxante osmótico. Produce una retención de agua en el lumen intestinal que conduce a la acumulación de fluidos, inducción de la peristalsis y consecuente evacuación. La administración oral, en combinación con una adecuada proporción de electrolitos en la formulación, produce la evacuación neta del contenido intestinal sin alterar el equilibrio hidro-electrolítico. FARMACOCINÉTICA No se absorbe. Después de iniciada la administración, el efecto se observa en aproximadamente 1 hora y provoca la evacuación completa del tubo intesti-nal a las 3,5-4 horas. INDICACIONES Preparación o limpieza del tubo intestinal previo a la colonoscopia o al enema de bario para la exploración por rayos X. R…GIMEN DE DOSIFICACI”N Se administra por vía oral o por sonda nasogástrica. Disolver el contenido de un sobre del producto (60 g de polietilénglicol 3350) en 1 litro de agua. En adultos: Administrar 240 ml de la solución cada 10 minutos hasta lograr evacuaciones claras y libres de materia Formulario Terapéutico Nacional * 2ed. 2004 tal, cólicos, flatulencia, rinorrea, arritmias, dermatitis y urticaria. Con la administración nasogástrica se ha descrito casos de pneumonitis por aspiración y edema pulmonar. INTERACCIONES Puede impedir la absorción gastrointestinal de medicamentos administrados dentro de los 60 minutos siguientes al inicio del procedimiento. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a los constituyentes del producto y en pacientes con íleo paralítico, colitis, obstrucción o perforación in-testinal y megacolon. Evitar la inges-tión de alimentos sólidos dentro de las 3-4 horas previas al uso del producto. Se debe esperar al menos una hora después de la última evacuación clara antes de iniciar la técnica invasiva. Usar con precaución en ancianos. No se conoce la seguridad del uso durante el embarazo. No es recomendable el uso en niños. SOBREDOSIS No aplicable. INFORMACIÓN AL PACIENTE Notificar al médico si durante el procedimiento se presenta alguna sintomatología o reacción distinta a la NISTATINA (Antiinfeccioso Intestinal) A FARMACOLOGÕA Ver NISTATINA en ANTIINFECCIOSOS GINECOLÓGICOS. FARMACOCINÉTICA La absorción por vía oral es mínima y se excreta intacta por las heces. INDICACIONES Candidiasis orofaríngea e intestinal. RÉGIMEN DE DOSIFICACIÓN Candidiasis orofaríngea en neonatos e infantes prematuros: 100.000 unidades (1ml) 4 veces al día. En infantes: 200.000 unidades (2 ml) 4 veces al día. En adultos y niños: 400.000600.000 unidades (4-6 ml) 4 veces al día. La suspensión debe mantenerse en la boca, sin tragar, por el mayor tiempo que sea posible. Candidiasis intestinal: 500.000-1.000.000 unidades en tabletas 3 veces al día. La duración de la terapia es variable y dependiente de la severidad de la infección. Por lo general dura 14 días. Para garantizar erradicación completa se debe mantener el tratamiento hasta 48 horas después de desaparecidos los síntomas de la infección. REACCIONES ADVERSAS Con dosis elevadas se puede presentar malestar gastrointestinal, náuseas, vómitos y diarrea. Se ha descrito 2ed. 2004 * Formulario TerapÈutico Nacional 17 CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del producto. Aunque no se ha descrito efectos adversos sobre el feto, se recomienda usarlo con precaución en el embarazo y sólo cuando exista una verdadera necesidad. También debe tomarse precaución durante la lactancia. Este medicamento no de-be ser usado para infecciones sistémi-cas por hongos. con pérdida significativa de dichos elementos, como la diarrea aguda. La presencia de glucosa favorece la absorción intestinal de sodio, alterada como consecuencia de la diarrea. El cloruro de potasio restituye las pérdidas de potasio previniendo la hipopotasemia. El bicarbonato de sodio y el citrato de sodio promueven la absorción de sodio y cloruro, a la vez que corrigen la acidosis metabólica. El cloruro contribuye, con el sodio, a la absorción y retención de agua. SOBREDOSIS Dado que su absorción gastrointestinal es despreciable, no es factible la intoxicación sistémica ni necesarias medidas de tratamiento. FARMACOCINÉTICA No aplicable. INFORMACIÓN AL PACIENTE Para que el tratamiento resulte efectivo, se debe mantener la suspensión en la boca, sin tragar, el mayor tiempo que sea posible. No suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Mantener el SALES DE REHIDRATACIÓN ORAL (SRO) FARMACOLOGÍA Solución constituida por una mezcla de glucosa, cloruro de sodio, cloruro de potasio y bicarbonato o citrato de sodio en proporciones definidas que, diluida en agua, constituye una fuente de fluido y electrolitos para el manejo, 18 INDICACIONES Reemplazo de fluido y electrolitos en la prevención y tratamiento de la deshidratación por diarrea aguda. RÉGIMEN DE DOSIFICACIÓN Se usa como patrón la formulación recomendada por la Organización Mundial de la Salud (OMS), que se presenta como polvo (en sobres) constituido por: 3,5 g de cloruro de sodio, 2,5 g de bicarbonato de sodio o 2,9 g de citrato de sodio, 1,5 g de cloruro de potasio y 20 g de glucosa anhidra. Se disuelve el contenido de un sobre en agua potable cantidad suficiente para 1 litro, agitando por 2-3 minutos. La dosificación se establece con base en el peso y la edad del paciente y el grado de deshidratación. Se considera que existe Formulario Terapéutico Nacional * 2ed. 2004 gua) y pérdida de la elasticidad cutánea (retracción disminuida). En tales casos, se administra SRO por un período de 4 horas en cantidades que pueden estimarse utilizando la siEdad (*) Peso Volumen en ml < 4 meses < 5 kg 200-400 4-11 meses 5-7,9 kg 400-600 12-23 meses 8-10,9 kg 600-800 2-4 años 11-15,9 kg 800-1200 5-14 años 16-29,9 kg 1200-2200 > 15 años > 30 kg 2200-4000 (*) Usar la edad del paciente cuando no sea posible conocer su peso. Luego de la administración por 4 horas, se examina nuevamente al paciente a objeto de estimar el grado de deshidratación: 1. Si hubo mejorÌa en relación con los signos y síntomas previos, se debe administrar alimentos sólidos (cereales, frutas no cítricas, pescado fresco, carne, vegetales), fluidos (agua, sopa, jugos de fruta, gaseosas, agua de arroz, más SRO) y mantener al paciente en observación. 2. Si la condición se mantiene igual se repite el curso de 4 horas con SRO, pero incorporando alimentación sóli-da y otros fluidos (como en el caso anterior), y se evalúa al paciente después de 4 horas. 3. Si l a c o ndiciÛn empe ora, se man- por vía IV. A REACCIONES ADVERSAS Vómito ocasional, especialmente al inicio de la terapia. INTERACCIONES No se han descrito en la literatura. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en caso de alteraciones de la función renal (anuria, oliguria), deshidratación asociada al shock, vómito abundante y persistente, malabsorción de glucosa y perforación intestinal. Durante la terapia se debe examinar periódicamente al paciente a objeto de estimar el grado de deshidratación; de ser posible, se debe determinar la presión arterial, la variación del peso corporal, los electrolitos y el pH sanguíneo. Después de la reconstitución, la solución puede conservarse bajo refrigeración hasta por 24 horas; transcurrido ese tiempo, debe descartarse la porción no usada. SOBREDOSIS Puede ocurrir hipernatremia. Manejo sintomático y terapia de soporte. INFORMACI”N AL PACIENTE Usar solamente el volumen recomendado; evitar la ingesta excesiva. No añadir a la preparación más agua de la necesaria. Si durante el uso ocurre 2ed. 2004 * Formulario TerapÈutico Nacional 19 SULFASALAZINA FARMACOLOGÍA Conjugado de ácido 5-aminosalicílico y sulfapiridina. Aunque no se conoce bien su mecanismo de acción en la colitis ulcerativa, se ha propuesto que la sulfasalazina podría actuar como vehículo que transporta y libera ácido 5-aminosalicílico y sulfapiridina al colon en concentraciones superiores a las que se logran cuando se administran por separado. Una vez en el colon, el efecto terapéutico sería el resultado de la acción antibacteriana de la sulfapiridina o de la actividad antiinflamatoria del ácido 5aminosalicíli-co. Otros mecanismos que podrían ex-plicar su efecto incluyen modificacio-nes de la flora intestinal, inhibición de la síntesis de prostaglandinas, altera-ciones en la secreción y/o absorción de fluidos y electrolitos e inmunomodu-lación. FARMACOCINÉTICA Después de la administración oral, se absorbe un 10-15% en el tracto gastrointestinal. Una porción del fármaco absorbido es excretado por vía biliar. La fracción no absorbida pasa intacta al colon donde se degrada a sulfapiridina y a ácido 5-aminosalicílico por acción de la flora bacteriana. La sulfapiridina se absorbe casi por completo desde el colon, mientras que el ácido 20 un 66% de sulfapiridina y un 20-33% de ácido 5-aminosalicílico. El resto de la sulfasalazina y del ácido 5-aminosalicílico no absorbido se excreta en las heces. Los efectos terapéuticos se observan a las 3-4 semanas. La vida media de eliminación, después de dosis múltiples, de la sulfasalazina es de 7,6 horas, de la sulfapiridina 10,4 horas y la del ácido 5-aminosalicílico 0,6-1,4 horas. INDICACIONES Colitis ulcerativa. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. En adultos: Iniciar con 3-4 g/día divididos en 3-4 dosis iguales. Se ha sugerido dosis iniciales de 1-2 g/día para reducir la intolerancia gastrointestinal. Una vez que el paciente ha mostrado tolerancia a la terapia inicial, mantener con 2 g/día divididos en 4 dosis. La duración del tratamiento es por tiempo indefi-nido. En niños: Iniciar con 40-60 mg/kg/día divididos en 3-6 dosis, y una dosis de mantenimiento de 30 mg/kg/día divididos en 4 dosis. REACCIONES ADVERSAS Cardiovasculares: Taquicardia. Gastrointestinales: Náuseas, vómitos, anorexia, diarrea, flatulencia, dolor abdominal, estomatitis y trastornos del gusto. Hematológicas: Leucopenia, neutropenia, anemia macrocítica, Formulario Terapéutico Nacional * 2ed. 2004 nio, depresión, mareo, ataxia, alucinaciones y convulsiones, neuropatía periférica. Reacciones de hipersensibilidad: Síndrome de Stevens-Johnson, miopatías, lupus eritematoso sistémi-co, enfermedad del suero, poliarteritis nodosa, edema periorbital, anafilaxis. Renales: Nefrosis tóxica, anuria, he-maturia, proteinuria, cristaluria, oligu-ria, síndrome urémico hemolítico y coloración anaranjada de la orina. Respiratorias: Neumonía, infiltrados pulmonares. Otras: Deficiencia de ácido fólico, miopatías, infertilidad masculina, coloración anaranjada de la piel. guíneas, falla renal y/o hepática y con deficiencias de la enzima glucosa-6- A fosfato deshidrogenasa. Es recomendable una hidratación adecuada para prevenir la cristaluria y la formación de cálculos. Durante el tratamiento se debe vigilar la función hematológica, hepática y renal. Se recomienda suspender la lactancia durante el tratamiento. INTERACCIONES La administración de antibióticos puede alterar el efecto de la sulfasalazina al ejercer cambios en la flora intestinal. Forma un quelato con el hierro alterando su distribución en el lumen intestinal y su absorción. Inhibe la absorción del ácido fólico e interfiere en su metabolismo. Disminuye la absorción gastrointestinal de la digoxina. Las sulfonamidas interfieren el metabolismo hepático de los anticoagulantes orales incrementando la respuesta hipoprotrombinémica. Altera la respuesta inmunológica a la vacuna contra la fiebre tifoidea. Disminuye la eficacia de los anticonceptivos orales. INFORMACI”N AL PACIENTE Durante el tratamiento se deben ingerir abundantes cantidades de líquido. No alterar la dosis ni suspender el trata-miento antes del tiempo previsto, aun-que hayan desaparecido los CONTRAINDICACIONES/ SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y terapia de soporte. GLIBENCLAMIDA FARMACOLOGÍA Agente del grupo de las sulfonilúreas de segunda generación con actividad hipoglicemiante. Actúa sobre las células beta de los islotes de Langerhans en el páncreas estimulando la secreción de insulina y generando, en consecuencia, una reducción de los niveles de glucosa en sangre. También se han postulado otros mecanismos tales como: Disminución de la gluconeogé- 2ed. 2004 * Formulario TerapÈutico Nacional 21 la acción de la insulina. rética. FARMACOCINÉTICA Se absorbe rápida y completamente en el tracto gastrointestinal, alcanzando niveles plasmáticos pico en aproximadamente 4 horas y una acción hipoglicemiante que persiste por 24 horas. Se une a las proteínas plasmáticas en un 90% y se metaboliza en el hígado a productos acti-vos e inactivos que se excretan, en iguales proporciones, por vía biliar y por la orina. La vida media de elimina-ción es de 5-10 horas. INTERACCIONES Los bloqueantes de los receptores beta-adrenérgicos, particularmente los no selectivos como el propranolol, esteroides anabolizantes, cimetidina, ranitidina, clofibrato, cloranfenicol, warfarina, metildopa, etanol (ingesta aguda), fenfluramina, guanetidina, insulina, miconazol (oral), AINEs, sulfonamidas y acidificantes urinarios, pueden aumentar la acción hipoglicemiante del medicamento. Por el contrario, los bloqueantes de los canales de calcio, fenobarbital, etanol (uso crónico), anticonceptivos orales, tiazidas, aminas simpatomiméticas, glucocorticoides, fenotiazinas y los alcalinizantes urinarios, pueden disminuir el efecto terapéutico del medicamento. INDICACIONES Coadyuvante en el tratamiento de la diabetes mellitus tipo II. R…GIMEN DE DOSIFICACI”N En adultos: Iniciar con 2,5-5 mg/día. En caso necesario, incrementar la dosis en 2,5 mg/día a intervalos semanales hasta un máximo de 20 mg/día. En pacientes geriátricos, debilitados, malnutridos o en insuficiencia renal y/o hepática: Iniciar el tratamiento con 1,25 mg/día. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómito, plenitud, pirosis. Hematológicas: Leucopenia, anemia hemolítica, tromboci-topenia, agranulocitosis, pancitopenia. Hepáticas: Alteración de la función hepática. Neurológicas: 22 CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al fármaco o a otros derivados de la sulfonilúrea, diabetes juvenil, diabetes inestable, cetosis, acidosis, trauma severo, coma o infección severa. Usar con precaución en pacientes con insuficiencia adrenal, náuseas, vómito, debilidad generalizada o malnutriciÛn, fiebre, alteraciÛn de la función renal o hepática y trastornos endocrinos. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los Formulario Terapéutico Nacional * 2ed. 2004 ministración de carbón activado y un catártico salino, si la ingestión es reciente. La sobredosis se manifiesta principalmente por hipoglicemia; en casos severos puede presentar convulsiones, coma y muerte. Casos leves pueden ser manejados con glucosa oral, ajustes dietéticos y reducción o suspensión temporal del medicamento. Cuadros severos requieren hospitalización inmediata y administración IV de glucosa al 50%, seguida por infusión continua de glucosa al 10% a una velocidad tal que permita mantener la glicemia sobre 100 mg/dl. El paciente debe ser sometido a observación por 24-48 horas. INFORMACI”N AL PACIENTE Se debe explicar al paciente la importancia de respetar la dieta, realizar una rutina de ejercicios y realizar periódica-mente los exámenes pertinentes al control de su condición. Evitar el con-sumo de alcohol. Informar al médico si se presenta alguna reacción o sinto-matología aminoácidos. La insulina tiene acciones anabólicas y catabólicas en A muchos tejidos. En el músculo y otros tejidos (excepto el cerebro) causa un rápido transporte de glucosa y de aminoácidos intracelularmente, promueve el anabolismo e inhibe el catabolismo de las proteínas. En el hígado promueve la captación y almacenamiento de la glucosa en forma de glucógeno, inhibe la gluconeogénesis y promueve la conversión del exceso de glucosa en grasa corporal. Administrada en dosis apropiadas y a intervalos regulares mantiene la glucosa sanguínea en niveles adecuados, evita la glucosuria y previene la formación de cuerpos cetónicos, la acidosis y el coma diabético. FARMACOCINÉTICA Después de la administración SC se produce el inicio de acción a los 30-60 minutos, alcanzando la intensidad pico a las 2-5 horas y se mantiene por 6-10 horas. Se distribuye rápidamente a través de los líquidos extracelulares; se metaboliza en el hígado, principalmente, en el riñón y en los músculos; se filtra en el glomérulo y se reabsorbe INSULINA DE ACCIÓN RÁPIDA (INSULINA casi completamente (98%) en el túbulo CRISTALINA HUMANA RECOMBINANTE) proximal. Un 40% de la cantidad reabsorbida regresa a la sangre venosa, FARMACOLOGÍA Insulina humana de acción rápida ob- mientras que el 60% restante se metatenida por tecnología de ADN recom- boliza en las células cercanas al binante. La farmacología es similar a túbulo proximal. Un porcentaje inferior la de la hormona endógena. Estimula al 2% se excreta en la orina como el metabolismo de la glucosa y de los fármaco intacto. 23 2ed. 2004 * Formulario TerapÈutico Nacional R…GIMEN DE DOSIFICACI”N de 0,15-1 U/kg cada 1-2 horas. Las dosis se expresan en unidades (U) USP; se establecen con base en las REACCIONES ADVERSAS determinaciones de glucosa en la Reacciones de hipersensibilidad: Erisangre y en la orina, y deben ser tema, edema y prurito; reacciones cuidadosamente ajustadas a las ne- alérgicas con manifestaciones de cesidades particulares de cada pa- erupción, sibilancias, dificultad respiciente. Se administra por vía SC o IV. ratoria, hipotensión y taquicardia (raLa insulina cristalina es la única que ras). Otras: Hipoglicemia, que se manipuede ser usada por vía IV, reser- fiesta con sudoración, mareo, palpitavándose esta ruta para el manejo de ciones, tremor, cansancio, hormigueo emergencia de pacientes con colapso en las manos, los pies, los labios o la circulatorio, cetoacidosis diabética o lengua, náusea, dificultad para la coma. En tales casos, se emplea concentración, dolor de cabeza, trasdiluida en solución de cloruro de sodio tornos del sueño, ansiedad, visión borrosa, dificultad de expresión, depreal 0,9%. - Para iniciar la terapia en pacientes sión, irritabilidad, inestabilidad emocon diagnóstico reciente de diabetes cional y, en casos severos, desorientasevera, diabetes inestable o diabe- ción, inconsciencia y convulsiones. Lites con complicaciones, la dosis podistrofia en el sitio de la inyección. usual en adultos, por vía SC, es de 5-10 U y en niños 2-4 U, 15-30 INTERACCIONES minutos antes de las comidas y al Los fármacos que pueden potenciar el a c o s t a r s e . L a s d o s i s d e efecto hipoglicemiante de la insulina, mantenimiento y la frecuencia de incluyen: naproxeno, litio, teofilina, caladministración se defi-nen según la cio, bromocriptina, mebendazol, piridoxina, pentamidina, esteroides anarespuesta del paciente. - Para el tratamiento de la cetoaci- bolizantes, disopiramida, fibratos, fluodosis diabética severa o el coma en xetina, bloqueantes de los receptores adultos, iniciar con 100-200 U dividi- beta-adrenérgicos, inhibidores de la das en 2 dosis iguales, una por vía monoaminooxidasa, hipoglicemiantes IV y la otra por vía SC. En pacientes orales, salicilados, sulfonamidas, inhicon colapso circulatorio, administrar bidores de la enzima convertidora de la dosis inicial total por vía IV. En angiotensina, octreótida y el alcohol. niños, iniciar con 1-2 U/kg divididas Agentes que antagonizan la actividad en 2 do-sis iguales, una IV y la otra de la insulina y que pueden disminuir SC. Las dosis de mantenimiento se su efecto hipoglicemiante incluyen: 24 Formulario Terapéutico Nacional * 2ed. 2004 isoniazida, fenotiazinas, epinefrina, salbutamol, terbutalina, tiazidas, furosemida, ácido etacrínico y las hormonas tiroideas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con hipoglicemia. La insulina puede usarse sin problemas en el embarazo y durante la lactancia. daños locales. No administrar otros medicamentos sin conultar previa- A mente al médico. Evitar el consumo de alcohol. Notificar de inmediato al médico si se presenta alguna INSULINA DE ACCIÓN INTERMEDIA (INSULINA N.P.H. HUMANA RECOMBINANTE) FARMACOLOGÍA Insulina humana de acción intermedia obtenida por tecnología de ADN recombinante. Ver: INSULINA SOBREDOSIS La sobredosificación puede ocasionar CRISTALINA HUMANA RECOMBIhipoglicemia severa y coma. En tales NANTE. casos, se debe administrar solución FARMACOCINÉTICA de glucosa al 50% por vía IV. En Luego de la administración SC, el pacientes con depósitos adecuados inicio de la acción tiene lugar en 1-3 de glicó-geno puede resultar efectiva horas, alcanza su intensidad pico a las la admi-nistración de 1-2 U de 6-14 horas y se mantiene por 16-24 glucagon por vía SC, IM ó IV. Para horas. Ver: INSULINA CRISTALINA prevenir reacciones hipoglicémicas HUMANA RECOMBINANTE. secundarias debe mantenerse la infusión IV continua de glucosa al 10- INDICACIONES 15% o administrar car-bohidratos por Tratamiento de la hiperglicemia en el vía oral, si la condición lo permite. paciente con diabetes mellitus. INFORMACI”N AL PACIENTE El paciente debe ser adecuadamente instruido en relación con su enfermedad, técnica adecuada de administración, los síntomas de la hipoglicemia o la cetoacidosis, adhesión a la dieta, programa de ejercicios, higiene personal y prevención de infecciones. Se debe usar siempre el mismo tipo o marca de inyectadora, a objeto de R…GIMEN DE DOSIFICACI”N Las dosis se expresan en unidades (U) USP; se establecen con base en las determinaciones de glucosa en sangre y orina y deben ser cuidadosamente individualizadas. Se administra sólo por vía SC. La dosis usual en adultos es de 7-26 U 30-60 minutos antes del desayuno. En algunos pacientes po- 2ed. 2004 * Formulario TerapÈutico Nacional 25 semanales, según la respuesta. REACCIONES ADVERSAS Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. INTERACCIONES Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. CONTRAINDICACIONES/ PRECAUCIONES No debe usarse por vía IV. La insulina N.P.H. no es adecuada para el manejo del coma diabético o en situaciones de emergencia en las que se requiere de una acción rápida. Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. SOBREDOSIS Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. FARMACOCINÉTICA Después de la administración SC, la acción se inicia a los 30 minutos, alcanza su intensidad pico a las 2-12 horas y se mantiene por aproximadamente 24 horas. Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. INDICACIONES Tratamiento de la hiperglicemia en el paciente con diabetes mellitus. R…GIMEN DE DOSIFICACI”N Las dosis se expresan en unidades (U) USP; se establecen con base en las determinaciones de glucosa en sangre y orina y deben ser cuidadosamente individualizadas. Se administra sólo por vía SC. REACCIONES ADVERSAS Ver: INSULINA CRISTALINA HUMANA RECOMBINANTE. INSULINA DE ACCIÓN INTERMEDIA (INSULINA PREMEZCLA CRISTALINA / N.P.H. INTERACCIONES Ver: INSULINA CRISTALINA HUMAHUMANA RECOMBINANTE (30/70)) NA RECOMBINANTE. FARMACOLOGÍA Suspensión estéril constituida por CONTRAINDICACIONES/ 30% de insulina cristalina humana y PRECAUCIONES 70% de insulina N.P.H. humana, No debe usarse por vÌa IV. La preambas de ori-gen en el ADN mezcla no es adecuada para el manejo recombinante. Combina el rápido del coma diabético o en situaciones de inicio de acción de la insulina cristalina emergencia en las que se requiere de con el efecto prolongado de la insulina una acción rápida. Ver: INSULINA N.P.H. Ver: INSULINA CRISTALINA CRISTALINA HUMANA RECOMBINANTE. 26 Formulario Terapéutico Nacional * 2ed. 2004 INFORMACI”N AL PACIENTE Ver: INSULINA CRISTALINA HUMAMETFORMINA FARMACOLOGÍA Agente del grupo de las biguanidas con efecto antihiperglicémico. Disminuye la absorción intestinal de glucosa, reduce la producción hepática de glucosa e incrementa la sensibilidad tisular a la insulina, por aumento de la captación y utilización de la glucosa por el músculo y los adipocitos. FARMACOCINÉTICA Se absorbe en un 50-60% en el tracto gastrointestinal. Los alimentos reducen su absorción. Luego de la adminis-tración oral, alcanza niveles plasmáti-cos pico en 1-3 horas y produce una respuesta terapéutica que se inicia a los 2-4 días y se hace máxima aproxi-madamente en 2 semanas. Se distri-buye ampliamente en los tejidos cor-porales. No se une a las proteínas plasmáticas, no sufre metabolismo, no tiene excreción biliar y se elimina intacta mediante secreción tubular en la orina. Un 90% de la dosis se excreta dentro de las primeras 24 horas. Exhibe una vida media de eliminación inicial de 3-6 horas y una terminal de 8-20 horas que parece corresponder a una pequeña fracción (menor del 5%) de fármaco INDICACIONES En combinación con un programa de A dieta y ejercicios para el control de la hiperglicemia en pacientes con diabetes mellitus tipo II. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. En adultos: Iniciar con 500 mg dos veces al día o con una dosis única de 850 mg/día e incrementar 500 mg a intervalos de 1-2 semanas, hasta lograr la respuesta deseada, sin exceder la dosis máxima de 2550 mg/día. Una respuesta adecuada, por lo general, no se alcanza con dosis menores de 1500 mg/día. En niños mayores de 10 años: Iniciar con 500 mg dos veces al día e incrementar 500 mg a intervalos de 1-2 semanas, hasta lograr la respuesta deseada, sin exceder la dosis máxima de 2000 mg/día. En pacientes mayores de 65 años y en pacientes debilitados, la do-sis debe ser conservadora en previ-sión de una disminución de la función renal. REACCIONES ADVERSAS Gastrointestinales: Diarrea, náusea, vómito, flatulencia, dolor abdominal, anorexia, indigestión, sabor desagradable y metálico, constipación. Hematológicas: Anemia megaloblástica. Hepáticas: Elevación de los niveles de las enzimas hepáticas. Neurológicas: Astenia, mareos, dolor de cabeza. Reacciones de hipersensibilidad: Der- 2ed. 2004 * Formulario TerapÈutico Nacional 27 que reciben hipoglicemiantes orales en forma concomitante, se puede presentar casos de hipoglicemia. INTERACCIONES La cimetidina y el nifedipino aumentan los niveles séricos de la metformina. La administración concomitante con furosemida lleva a cambios en los parámetros farmacocinéticos de ambos fármacos, aumento de la concentración plasmática de furosemida y disminución de la concentración máxima y el AUC de la furosemida. El uso de medicamentos nefrotóxicos aumenta el riesgo de acidosis láctica. El consumo de alcohol en cantidades excesivas favorece la acidosis láctica. Los fármacos que producen hiperglicemia (corticosteroides, bloqueantes de los canales de calcio, niacina, estrógenos, anticonceptivos orales, isoniazida, fenotiazinas, simpatomiméticos, hor-monas tiroideas y las tiazidas) podrían, teóricamente, disminuir la respuesta farmacológica de la metformina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento y en pacientes con falla renal o insuficiencia cardiaca tratada con medicamentos. Dado que la insuficiencia hepática ha sido asociada con acidosis láctica, la metformina está contraindicada en 28 niveles de vitamina B12 (cada 2-3 aÒos). Pacientes con estados hipÛxicos (colapso cardiovascular, insuficiencia cardiaca congestiva aguda, infarto mioc·rdico) son propensos al desarrollo de acidosis l·ctica. Como los medios de contraste yodados pueden ocasionar falla renal y han sido asociados con el desarrollo de acidosis l·ctica en pacientes que reciben metformina, el uso del medicamento debe ser suspendido 48 horas antes de un procedimiento que implique el uso de tales productos, y restablecido 48 horas despuÈs, luego de comprobar el funcionamiento renal. Igualmente, la metformina debe suspenderse temporalmente en pacientes que se someten a cirugÌa mayor, y reiniciarse una vez restablecida la ingesta normal de alimentos y verificada la funciÛn renal. Usar con precauciÛn en pacientes con estados hipermetabÛlicos (infecciÛn, trauma, fiebre), en ancianos y en el embarazo. No es recomendable el uso durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. La ingestión de dosis elevadas no produce hipoglicemia pero puede generar acidosis láctica, en cuyo caso se recomienda hemodiálisis para remover el exceso del fármaco y, junto con medidas adecuadas de so- Formulario Terapéutico Nacional * 2ed. 2004 tratamiento se debe evitar el consumo de alcohol. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial: Malestar ge-neral, mialgia, hiperventilación, som-nolencia, molestias abdominales, hipotermia, ÁCIDO ASCÓRBICO (VITAMINA C) FARMACOLOGÍA Vitamina hidrosoluble que interviene en reacciones de óxido-reducción y en la respiración celular. Se requiere una fuente exógena de ácido ascórbico para la formación de colágeno y la reparación tisular. Tiene efecto antioxidante. Participa en el metabolismo de carbohidratos, hierro y tirosina, en la conversión de ácido fólico en ácido folínico, en la síntesis de lípidos y pro-teínas y en la resistencia a las infe-cciones. La deficiencia de ácido ascór-bico produce escorbuto. La estructura del colágeno es afectada primaria-mente, y se desarrollan lesiones en los huesos y los vasos sanguíneos. La administración de ácido ascórbico revierte todos los síntomas de defi-ciencia del mismo. FARMACOCINÉTICA Se absorbe con facilidad por vía oral, alcanzando niveles séricos pico a las 2-3 horas después de la administración. Se une a las proteínas forma activa de ácido dihidroascórbico y una pequeña cantidad se metaboliza A a productos inactivos que son excretados en la orina. Cuando los tejidos corporales se saturan y las concentraciones sanguíneas de la vitamina superan el umbral renal, se excreta por la orina. No obstante, cuando los niveles tisulares y plasmáticos son bajos, la excreción es escasa o nula. INDICACIONES Prevención y tratamiento del escorbuto. Como suplemento nutricional cuando la ingesta de alimentos es pobre o inadecuada, y en condiciones caracterizadas por reducción de la absorción o por aumento de la demanda (embarazo, lactancia, hemodiálisis, nutrición parenteral, fiebre o trauma severo). Como acidificante urinario. La administración parenteral es deseable en pacientes con deficiencia aguda y en aquellos cuya absorción oral es incierta. R…GIMEN DE DOSIFICACI”N Se administra usualmente por vía oral. Cuando la administración oral no es posible o se sospecha malabsorción, se puede administrar IV, IM o SC, prefiriéndose la administración IM. La dosis usual en estados de deficiencia es de 100-500 mg 1-2 veces al día por varios días. Como acidificante urinario se usa en 2ed. 2004 * Formulario TerapÈutico Nacional 29 de cabeza, fatiga, hiperoxaluria y precipitación de cálculos de oxalato, cistina o urato en el tracto urinario. En pacientes con deficiencia de glucosa-6fosfato deshidrogenasa, la inyección IV de dosis elevadas puede producir hemólisis. La administración IV produce dolor en el sitio de la inyección. INTERACCIONES En combinación con ácido acetilsalicílico disminuye la excreción renal del salicilado e incrementa la de la vitamina. El ácido ascórbico puede disminuir la acción anticoagulante de la warfarina. La acidificación de la orina inducida por el ácido ascórbico puede alterar la eliminación renal de otros fármacos. SOBREDOSIS No se conocen casos de intoxicación por sobredosis de ácido ascórbico. INFORMACIÓN AL PACIENTE No exceder las dosis recomendadas. Evitar el uso de dosis elevadas durante CITRATO DE CALCIO (CITRATO TRICÁLCICO) FARMACOLOGÍA El citrato de calcio se utiliza como fuente de calcio. El calcio es indispensable para el mantenimiento de la integridad funcional del sistema nervioso, muscular y esquelético, así como de la permeabilidad de las membranas celulares y de la vasculatura capilar. Participa como regulador en los procesos de almacenamiento y liberación de neurotransmisores y hormonas. Es un activador importante de numerosas reacciones enzimáticas y es esencial para la transmisión del impulso nervioso, la contracción del músculo liso, estriado y cardiaco, la función renal, la respiración, la coagulación sanguínea y constituye un elemento fundamental en los procesos de regeneración y remodelación ósea. CONTRAINDICACIONES/ PRECAUCIONES Usar con precaución en pacientes con hiperoxaluria. Los pacientes diabéticos, aquellos con tendencia a la formación de cálculos renales, con sangre oculta en heces y aquellos con restricción dietética de sal o con terapia anticoagulante deben evitar las dosis excesivas de Vitamina C por tiempo prolongado, ya que esto puede conducir a la inducción de su propio metabo- FARMACOCINÉTICA lismo y ocasionar escorbuto cuando la Luego de la administración oral, el calingesta del ácido ascórbico es redu- cio se ioniza en el tracto gastrointesticida a niveles normales. Los diabéti- nal y es absorbido mediante mecaniscos que toman más de 500 mg al día mos de transporte activo y difusión pueden tener falsos valores de gluco30 Formulario Terapéutico Nacional * 2ed. 2004 ral, mientras el 1% restante se distribuye libremente entre los líquidos extracelular e intracelular. Las concentraciones séricas fisiológicas son de 910,4 mg/dl (4,5-5,2 mEq/L). Se excreta principalmente por las heces (vía biliar y a travÈs del jugo pancre·tico) y en escasa proporción por la orina. INDICACIONES Suplemento nutricional. Deficiencias de calcio. Coadyuvante en el tratamiento de la osteoporosis en mujeres postmenopáusicas. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. Las dosis se expresan en mg de calcio elemental (1 g de citrato de calcio contiene 211 mg de calcio elemental) y dependen de los requerimientos individuales de cada paciente según su condición. La dosis usual como suplemento nutricional en adultos es de 1 g/día y en niños 45-65 mg/kg/día. Para el tratamiento de esta-dos deficitarios: 12 g/día. Como coad-yuvante en el tratamiento de la osteo-porosis: 1-1,5 g/día. REACCIONES ADVERSAS Los efectos adversos más comunes son irritación gastrointestinal y constipación. La administración de dosis ele-vadas en pacientes con insuficiencia renal crónica puede causar hiper-calcemia, que se INTERACCIONES El uso en pacientes en terapia con A digital incrementa el riesgo de arritmias cardíacas. Si se administra conjuntamente con tetraciclinas forma un complejo que impide la absorciÛn gastrointestinal del antibiÛtico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con cálculos renales, fibrilación ventricular o hipercalcemia. En pacientes con terapia crónica, se recomienda el control periódico de los niveles séricos de calcio (no deben exceder de 10,4 mg/dl o 5,2 mEq/L). Usar con precaución en casos de sarcoidosis, enfermedad cardiaca o renal y en pacientes en terapia con digitálicos. SOBREDOSIS No existen reportes de intoxicación por sobredosis de calcio. La ingestión agu-da de una dosis elevada no representa un problema de consideración. INFORMACIÓN AL PACIENTE Administrar preferiblemente con las GLUCONATO DE CALCIO FARMACOLOGÍA El calcio es esencial para el mantenimiento de la integridad funcional de los sistemas nervioso, muscular y esquelético, la membrana celular y la per31 2ed. 2004 * Formulario TerapÈutico Nacional meabilidad capilar. Es un activador de numerosas reacciones enzimáticas y elemento esencial para un número de procesos fisiológicos que incluye la contracción del músculo cardíaco liso y esquelético, la transmisión del impulso nervioso, la respiración, la coagulación sanguínea y la función renal. Ejerce un papel regulador en la liberación y almacenamiento de los neurotransmisores, en la captación y fijación de los aminoácidos, en la absorción de la vitamina B12 y la secreción de gastrina. FARMACOCINÉTICA Luego de la administración IV el calcio alcanza el líquido extracelular y de allí se distribuye al tejido óseo, donde se deposita. Las concentraciones basa-les de calcio se ubican entre 9 y 10,4 mg/dl (4,5-5,2 mEq/L). Se une a proteí-nas plasmáticas en un 40-45%. Se excreta principalmente por las heces y en escasa proporción por la orina y a través del sudor. INDICACIONES Hipocalcemia. REACCIONES ADVERSAS La administración IV rápida puede causar bradicardia, vasodilatación, hipotensión, arritmias cardíacas, síncope y paro. La extravasación puede causar necrosis severa. Puede causar hormigueo, sensación de opresión u ondas de calor o sabor a tiza. INTERACCIONES El calcio puede aumentar el efecto de los digitálicos y precipitar arritmias; forma complejos con las tetraciclinas y las inactiva. No deben mezclarse las soluciones. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en la hipercalcemia, en la fibrilación ventricular y en pacientes digitalizados. Usar con precaución en el embarazo. Debe usarse solamente por vía IV. Solamente debe administrarse a mujeres embarazadas si es claramente necesario. Precaución en las mujeres lactantes. SOBREDOSIS Suspender la infusión, evaluar al pa- RÉGIMEN DE DOSIFICACIÓN La dosis se establece en función de las necesidades individuales de cada paciente, considerando la edad, los niveles séricos de calcio y la condición clí32 Formulario Terapéutico Nacional * 2ed. 2004 B01 AGENTES ANTITROMB”TICOS Ácido Acetilsalicílico (Inhibidor de la Agregación Plaquetaria) Enoxaparina Sódica Estreptoquinasa Heparina Sódica Warfarina (Warfarina Sódica) B03 PREPARADOS ANTIAN…MICOS Ácido Fólico Cianocobalamina (Vitamina B12) Eritropoyetina (Epoetin Alfa) Sulfato Ferroso (Hierro Elemental) B05 SUSTITUTOS DEL PLASMA Y SOLUCIONES PARA PERFUSI”N Agua Estéril para Irrigación* Agua para Inyección* Albúmina Humana Amino Ácidos Bicarbonato de Sodio Cloruro de Potasio Cloruro de Sodio Cloruro de Sodio / Glucosa Dextrán (Dextrano 70) Emulsiones Grasas (Lípidos) Fosfato Monobásico de Potasio Glucosa (Dextrosa) Sulfato de Magnesio (Hipomagnesemia) Solución para Nutrición Parenteral Soluciones Electrolíticas (Ringer Lactato) Soluciones Electrolíticas (Ringer) Soluciones para Diálisis Peritoneal Soluciones para Hemodiálisis 2ed. 2004 * Formulario TerapÈutico Nacional B SANGRE Y ÓRGANOS FORMADORES DE SANGRE B02 ANTIHEMORR¡GICOS Ácido Tranexámico Factor VIII de la Coagulación Factor IX de la Coagulación Fitomenadiona (Vitamina K) B 33 ÁCIDO ACETILSALICÍLICO (Inhibidor de la AgregaciÛn Plaquetaria) FARMACOLOGÍA Ejerce una acción antiagregante plaquetaria por inhibición selectiva e irreversible de la isoenzima ciclooxigenasa 1 (COX-1) que interviene en la síntesis de prostaglandinas y del trom-boxano A2, factor inductor de la agre-gación plaquetaria. FARMACOCINÉTICA Aunque el ácido acetilsalicílico se absorbe bien después de su administración oral, sólo un 50-75% de la dosis alcanza la circulación sistémica como fármaco intacto debido al efecto del metabolismo del primer pasaje y a la hidrólisis que sufre en la mucosa del tracto gastrointestinal, que da lugar a su conversión en ácido salicílico. El ácido acetilsalicílico y el ácido salicílico se unen a las proteínas plasmáticas en un 33% y 90%, respectivamente, y se distribuyen rápida y ampliamente a los tejidos y fluidos corporales. El ácido acetilsalicílico absorbido es rápidamente hidrolizado en el plasma, los eritrocitos, el hígado y el líquido sinovial, transformándose en más ácido salicílico que, junto al formado durante el proceso de absorción, es conjugado en el hígado generando metabolitos inactivos que se excretan por la orina en un 75%, conjuntamente con un 10% de ácido salicílico intacto INDICACIONES Como antiagregante plaquetario en la prevención del infarto agudo del miocardio y su recurrencia, de los ataques B isquémicos transitorios y del accidente cerebro-vascular isquémico por trom-boembolismo. R…GIMEN DE DOSIFICACI”N Se administra por vía oral en dosis de 100 mg/día. REACCIONES ADVERSAS Reacciones de hipersensibilidad: Angioedema, broncoespasmo, urticaria, edema de la laringe, anafilaxis. Cardiovasculares: Hipotensión, taquicardia. Gastrointestinales: Náuseas, vómito, dispepsia, gastritis, ulceración, hemorragia gastrointestinal. Hematológicas: Trombocitopenia, prolongación del tiempo de protrombina. Hepáticas: Elevación de los niveles de las enzimas hepáticas, hepatitis. Neu-rológicas: Agitación, confusión, mareo, letargia. Respiratorias: Edema pulmo-nar, taquipnea. Renales: Falla renal. Otras: Edema cerebral, acidosis me-tabólica, alcalosis respiratoria, alte-raciones de la glicemia e hiperpota-semia. INTERACCIONES El ácido acetilsalicílico puede desplazar de su unión a proteínas a las sulfonilúreas, warfarina, ácido valpróico, di-fenilhidantoína y sulfonamidas e 2ed. 2004 * Formulario TerapÈutico Nacional 35 anticoagulante de la warfarina y de la heparina, disminuir la actividad de los inhibidores de la enzima convertidora de angiotensina, antagonizar la acción uricosúrica del probenecid y reducir el efecto natriurético de la espironolactona. La combinación con AINEs aumenta el riesgo de sangramiento y de falla renal. Los corticosteroides y el elevado consumo de alcohol predisponen a la ulceración gastrointestinal. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a los salicilatos o a otros antiinflamatorios no esteroideos (AINEs), úlcera péptica activa, trastornos hemorrágicos, dengue, varicela (lechina), tratamiento simultáneo con anticoagulantes y alteración severa de la función hepática y/o renal. El uso en niños o adolescentes con infección viral o enfermedades eruptivas ha sido asociado con el síndrome de Reye. Usar con precaución en pacientes con antecedentes de hipoprotrombinemia, deficiencias de vitamina K, enfermedad renal y/o hepática leve a moderada, asma, insuficiencia cardiaca congestiva, hipertensión, edema y, en general, aquellas condiciones que pueden agravarse por la retención de fluidos. Se debe evitar el uso en el embarazo a término. No se recomienda el uso durante la lactancia. 36 alcalina favorece la eliminación del medicamento. En situaciones graves se puede recurrir a la hemodiálisis, la diálisis peritoneal o, en casos extremos, a la exanguino-transfusión. INFORMACIÓN AL PACIENTE Administrar con las comidas o con leche para minimizar las molestias gastrointestinales. Evitar el consumo de alcohol. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial ENOXAPARINA SÓDICA FARMACOLOGÍA Heparina de bajo peso molecular con propiedades antitrombóticas. La acción principal es debida a la inactivación del factor X activado (Fxa) y la consecuente alteraciÛn de la transformaciÛn de la protrombina en trombina. Adicionalmente, aunque en menor grado, acelera la actividad inhibitoria de la antitrombina III sobre la trombina, impidiendo asÌ la influencia sobre la conversiÛn de fibrinÛgeno en fibrina. FARMACOCIN…TICA Luego de la administraciÛn SC, se absorbe en un 92% y produce la m·xima actividad inhibitoria sobre el factor Xa y la antitrombina III a las 3-5 horas. La inhibiciÛn del factor Xa en el plasma persiste por 12-24 horas. Se Formulario Terapéutico Nacional * 2ed. 2004 antifactor Xa) es de 4,5 horas y se prolonga en pacientes con insuficiencia renal. INDICACIONES Prevención de la trombosis venosa profunda asociada a la cirugía ortopédica, o cirugía abdominal en pacientes con riesgo de complicaciones tromboembólicas. orales, inhibidores de la agregación plaquetaria (AINEs y salicilados), dipiridamol, ticlopidina y corticosteroides sistémicos, aumenta el riesgo B de hemorragia. Con plicamicina y ácido valpróico puede causar hipoprotrombinemia e inhibición de la agregación plaquetaria. CONTRAINDICACIONES/ PRECAUCIONES RÉGIMEN DE DOSIFICACIÓN Contraindicado en casos de hipersenSe administra por vía SC. sibilidad al medicamento, a la heparina - Prevención de tromboembolismo en o a otras heparinas de bajo peso la cirugía ortopédica: Iniciar el molecular, así como en pacientes con tratamiento con 30 mg cada 12 procesos hemorrágicos activos o con horas, comenzando 12-24 horas trombocitopenia asociada a una después de la intervención y prueba positiva de anticuerpos mantenerlo por 7-10 días. Alterna- antiplaquetarios en presencia de tivamente, el tratamiento se puede enoxaparina. El uso de enoxaparina iniciar con 40 mg 12 horas antes de la en pacientes con anestesia epidural o operación y mantenerlo con 40 espinal ha ocasionado hematoma mg/día por 3 semanas. neuraxial resultante en parálisis - Prevención del tromboembolismo en prolongada o permanente. La la cirugía abdominal: Iniciar el enoxaparina no puede intercambiarse, tratamiento con 40 mg 2 horas antes unidad por unidad, con la heparina o de la intervención y mantenerlo con con otras heparinas de bajo peso 40 mg/día por 7-10 días. molecular. Durante el tratamiento se debe practicar, periódicamente, una REACCIONES ADVERSAS hematología completa, incluyendo el Los efectos adversos más comunes contaje plaquetario, así como incluyen: Hemorragia, trombo- uroanálisis y determinación de sangre citopenia, fiebre, náusea, edema oculta en las heces. Si se presenta periférico, anemia, elevación de las disminución del contaje plaquetario, transaminasas y reacciones en el sitio en un 30-50% de su valor previo al de la inyección como: Irritación, dolor, i n i c i o d e l t r a t a m i e n t o , d e b e hematoma, equimosis, eritema, suspenderse la administración de la 37 2ed. 2004 * Formulario TerapÈutico Nacional ancianos y en el embarazo. No se conoce la eficacia ni la seguridad de su uso en niños. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS La sobredosificación puede provocar sangramiento de magnitud, severidad y duración variables, según la cantidad administrada. Manejo sintomático y tratamiento con sulfato de protamina (solución al 1%) administrada lentamente. INFORMACIÓN AL PACIENTE No usar por vía IM. Notificar de inmediato al médico si se presenta ESTREPTOQUINASA FARMACOLOGÍA Agente trombolítico que activa el sis- nolítica (la reperfusión miocárdica se evidencia entre 20 minutos y 2 horas, promedio 45 minutos, después de la administración) que se mantiene mien-tras dura la infusión y persiste hasta por 4 horas después de finalizada la misma, aunque el efecto sobre el pro-ceso de coagulación (medido por el tiempo de protrombina) puede durar hasta 12-24 horas. Esto último está asociado a una disminución de los ni-veles plasmáticos de fibrinógeno y a un incremento de los productos de degradación de la fibrina. La estreptoquinasa se elimina en forma bifásica, mostrando una vida media de eliminación en la fase inicial de 18 minutos (debido a la acción de los anticuerpos) y una vida media de eliminación en la fase lenta de 83 minutos (en ausencia de anticuerpos). No se conoce la ruta de excreción. tema fibrinolítico endógeno a través de un mecanismo de interacción directa con la molécula de plasminógeno, dando lugar a la formación de un complejo activador que transforma a otras moléculas de plasminógeno en plasmina, enzima hidrolítica que de-grada al coágulo de fibrina, a la vez que hidroliza al fibrinógeno y a otras proteínas plasmáticas, incluyendo a los factores procoagulantes V y VIII. 38 INDICACIONES Infarto agudo del miocardio, embolismo pulmonar, trombosis venosa profunda, embolismo arterial y para la desobstrucción de cánulas arterio-venosas cuando la heparina es inefectiva. R…GIMEN DE DOSIFICACI”N Se administra por infusión IV. - Infarto agudo del miocardio: 1.500.000 UI en 60 minutos, tan pronto como sea posible después Formulario Terapéutico Nacional * 2ed. 2004 - Trombosis venosa profunda: Dosis inicial de 250.000 UI en 30 minutos, seguido por una dosis de mantenimiento de 100.000 UI/hora por 72 horas. - Trombosis o embolismo arterial: Dosis inicial de 250.000 UI en 30 minutos, seguido por una dosis de mantenimiento de 100.000 UI/hora por 24-72 horas. - OclusiÛn de cánulas arterio-venosas: 250.000 UI en 2 ml de solución. Se instila lentamente en la cánula; la solución es mantenida allí por 2 horas, luego la solución es aspirada y finalmente es lavada con solución salina fisiológica y luego la cánula es reconectada. REACCIONES ADVERSAS Cardiovasculares: Arritmias ventriculares, hipotensión. Hematológicas: Hemorragia, hemorragia cerebral, leucocitosis. Hepáticas: Alteración de la función hepática. Neurológicas: Depresión, alucinaciones, agitación, con-fusión, neuralgia amiotrófica. Reaccio-nes de hipersensibilidad: Broncoes-pasmo, erupción y anafilaxis. Renales: Falla renal. Otras: Depresión res-piratoria (rara), dolor de espalda durante la infusión. persensibilidad al medicamento, aneurisma, malformación arterio-venosa, tumor cerebral, sangramiento activo, hipertensión severa no controlada, B accidente cerebro-vascular reciente (2 meses) y cirugía intracraneana o intraespinal reciente (2 meses). Usar con precaución en condiciones con predisposición al sangramiento, trastornos de la coagulación, endocarditis bacteriana, trastornos hepáticos o renales, tratamiento con medicamentos anticoagulantes o inhibidores de la agregación plaquetaria, sangramiento gastrointestinal reciente (6 meses), colitis ulcerativa o diverticulitis, infección estreptocócica reciente (10 días) y biopsia hepática o renal reciente (10 días). El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales a la madre superen al posible riesgo para el feto. No se conoce la seguridad del uso durante la lactancia. HEPARINA SÓDICA FARMACOLOGÍA Agente anticoagulante. La heparina se combina con antitrombina III (cofactor de la heparina) e inhibe la trombosis por inactivación del factor X activado e INTERACCIONES El uso concomitante de medicamentos inhibición de la conversión de anticoagulantes o de agentes que alte- protrombina en trombina. Una vez ran o inhiben la agregación plaqueta- activada la trombosis, mayores 39 2ed. 2004 * Formulario TerapÈutico Nacional pueden inhibir, adicionalmente, la coagulación inactivando la trombina e inhibiendo la conversión de fibrinógeno en fibrina. En presencia de heparina, la antitrombina III también neutraliza a los factores de coagulación IX, X, XII activados y a la plasmina. FARMACOCINÉTICA Luego de la administración por bolo IV, se produce un efecto anticoagulante inmediato, mientras que por la inyección SC, el inicio de acción se produce a los 20-60 minutos. La duración del efecto es variable y dependiente de la dosis. Una vez absorbida, la heparina se distribuye en el plasma y se une en un elevado porcentaje a las globulinas, a las lipoproteínas de baja densidad y al fibrinógeno. Es removida de la circulación por el sistema retículo endotelial y se cree que es parcialmente metabolizada en el hígado. Se excreta por la orina. La vida media de eliminación es de 1-2 horas y ésta se prolonga al incrementar la dosis y en pacientes con falla hepática o renal. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección SC profunda, IV intermitente o por infusión IV continua. - Inyección SC: Iniciar con 10.00020.000 U (precedido por una dosis IV de carga de 5000 U) y continuar con 8.000-10.000 U cada 8 horas, o 15.000-20.000 U cada 12 horas. - Inyección IV intermitente: Iniciar con 10.000 U y continuar con 5.00010.000 U cada 4-6 horas. - Infusión IV continua: Iniciar con 5.000 U y mantener con 20.00040.000 U/día. - Dosis pediátrica: Iniciar con infusión IV continua de 50 U/kg y mantener con 100 U/kg/dosis cada 4 horas. - Profilaxis del tromboembolismo venoso post-operatorio: 5.000 U vía SC 2 horas antes de la cirugía y repetir cada 8-12 horas hasta que el paciente esté en condición ambulatoria. - Cirugía cardiovascular: Infusión IV de 300 U/kg para procedimientos de corta duración (menor de 60 minutos) y 400 U/kg para procedimientos largos (mayores de 60 minutos). - Heparinización de muestras de sangre: 70-150 U para 10-20 ml de sangre completa. INDICACIONES Profilaxis y tratamiento de la trombosis venosa profunda, trombosis cerebral, embolismo (pulmonar y arterial periférico) y de la fibrilación auricular REACCIONES ADVERSAS con embolización y para el diag- Gastrointestinales: Náuseas, vómitos. nóstico y tratamiento de la 40 Formulario Terapéutico Nacional * 2ed. 2004 urticaria. Otras: Irritación, eritema, dolor moderado, hematoma, ulceración asociada a la inyección SC profunda y necrosis cutánea o subcutánea, osteoporosis, insuficiencia renal, alopecia, priapismo e hiperlipidemia de rebote al descontinuar la terapia. INTERACCIONES Los digitálicos, las tetraciclinas, la nicotina y los antihistamínicos pueden interferir con la acción anticoagulante de la heparina. El uso de ácido acetilsalicílico, fenilbutazona, ibuprofeno, indometacina, hidroxicloroquina, dextrano, dipiridamol y demás agentes que interfieren con la agregación plaquetaria (principal defensa hemostática del paciente heparinizado) puede generar sangramiento. También interactúa con los anticoagulantes orales, dicumarol y warfarina, impide la formación de un coágulo estable de fibrina mediante la inhibición de la activación del factor estabilizador de la fibrina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, trombocitopenia severa o sangramiento incontrolable y en individuos que no pueden realizarse las pruebas de coagulación a los intervalos requeridos. Usar con precaución en tinales, diverticulitis, menstruación, enfermedades hepáticas con alteraciones de la hemostasis, enfermedad biliar o renal severa, tuberculosis B activa, carcinoma visceral, abuso de alcohol, y durante o después de la anestesia espinal o cirugía del cerebro, de la médula espinal o de los ojos. Durante el tratamiento es recomendable realizar periódicamente pruebas de coagulación, contaje de plaquetas, determinación del hema-tocrito y de sangre oculta en heces. No se conoce la seguridad del uso en el embarazo; solamente debe admi-nistrarse a la mujer embarazada si es claramente necesario. Como no se excreta en la leche materna, es segura durante la lactancia. Si un paciente desarrolla una nueva trombosis en asociación con trombocitopenia (“Síndrome del coágulo blanco”), debe suspenderse la administración de heparina. También puede presentarse resistencia a la heparina en casos de fiebre, trombosis, tromboflebitis, infecciones con tendencia a la trombosis, infarto del miocardio, cáncer y en pacientes posquirúrgicos. Es importante la elevación significativa en las aminotransferasas observada con la heparina ya que ayuda al diagnóstico diferencial del infarto del miocardio, la enfermedad hepática y el embolismo pulmonar. 2ed. 2004 * Formulario TerapÈutico Nacional 41 INFORMACIÓN AL PACIENTE Informar sobre los signos de sobredosificación e importancia del control periódico de la coagulación. Advertir la necesidad de notificar de inmediato al médico la ocurrencia de cualquier reacción o sintomatología inusual WARFARINA (WARFARINA SÓDICA) FARMACOLOGÍA Interfiere con la síntesis hepática de los factores de coagulación II, VII, IX y X dependientes de la vitamina K y de las proteínas inhibidoras de la coagulación: Proteína C y proteína S. Aunque no ejerce una acción directa sobre el trombo una vez establecido, ni revierte el daño al tejido isquémico, puede prevenir la extensión adicional del coágulo formado y las complicaciones tromboembólicas secundarias que pueden resultar en secuelas serias y posiblemente fatales. (vía citocromo P450 y a través de enzimas reductasas) en metabolitos con mínima o nula actividad farmacológica que son posteriormente excretados, en su mayoría, por la orina y en escasa proporción por vía biliar. La vida media de eliminación es de 20-60 horas (promedio 40 horas). INDICACIONES Prevención y tratamiento de la trombosis venosa y su extensión, embolismo pulmonar, complicaciones tromboembólicas asociadas a la fibrilación auricular y/o al reemplazo valvular y reducción del riesgo de eventos tromboembólicos posteriores al infarto del miocardio. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral una vez al día. La dosis inicial recomendada es de 2-5 mg con ajustes subsiguientes basados en la determinación del tiempo de protrombina (PT). Se considera que una respuesta anticoagulante es adecuada cuando se corresponde con un PT de 1,2-2,0 veces su valor basal, lo cual podría requerir de 5 a 10 días. Las dosis usuales de mantenimiento se ubican en el rango de 2-10 mg diarios. En ancianos y/o pacientes debilitados se deben emplear dosis menores al inicio y durante el mantenimiento. Previo al inicio de la terapia se debe practicar FARMACOCINÉTICA La warfarina es una mezcla racémica de los enantiómeros R y S, mostrando el enantiómero S una actividad anticoagulante 2-5 veces mayor que la del R en humanos, pero tiene una depuración más rápida. Se absorbe completamente en el tracto gastrointestinal y genera niveles plasmáticos pico en aproximadamente 4 horas. Aunque la actividad comienza a evidenciarse a las 36-72 horas de la 42 Formulario Terapéutico Nacional * 2ed. 2004 determinaciones se hacen cada 1-2 meses, según la uniformidad de sus respuestas. Los métodos más precisos para el ajuste de la dosis se basan en la determinación del INR (International Normalized Ratio o Relación Internacional Normalizada), que equivale a la relación entre el PT observado y el PT control o basal, elevada al valor del Índice Internacional de Sensibilidad (ISI) de la tromboplastina usada para la determinación. ISI PT observado INR = PT control Se considera que una respuesta anticoagulante es adecuada cuando se corresponde con un INR de 2-3 para la mayoría las indicaciones. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, anorexia, diarrea, cólicos abdominales. Hematológicas: Hemorragia, agranulocitosis, leucopenia, eosinofilia. Hepáticas: Hepatitis, colestasis, elevación de los niveles de las aminotransferasas e ictericia. Neurológicas: Astenia, cefalea, parestesias. otros medicamentos puede alterar la respuesta anticoagulante del paciente. Entre los agentes que han demostrado incrementar la respuesta de la B warfarina se incluyen: acetaminofén, alcohol (en altas dosis), alopurinol, amiodarona, esteroides anabolizantes, hidrato de cloral, cloranfenicol, cimetidina, clofibrato, trimetoprima/sulfametoxazol, danazol, eritromicina, AINEs, quinolonas, vacuna contra la influenza (virus vivos), metronidazol, miconazol, pentoxifilina, propoxifeno, propiltiouracilo, quinidina, salicilados, estreptoquinasa, tiazidas, uroquinasa, sulfonamidas, tamoxifeno, agentes tiroideos, tetraciclinas, antidepresivos tricíclicos y vitamina E. Entre los medicamentos que podrían disminuir la respuesta anticoagulante de la warfarina se incluyen: Alcohol (crónico), barbitúricos, carbamazepina, corticosteroides, corticotropina, glutetimida, griseofulvina, mercaptopurina, nafcilina, anticonceptivos orales estrogénicos, rifampicina, espironolactona, trazodona, sucralfato y vitamina K. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en individuos con hipersensibilidad al medicamento, tendencias hemorrágicas, hemofilia, púrpura trombocitopénica o leucemia con tendencia al sangramiento, 43 2ed. 2004 * Formulario TerapÈutico Nacional pericárdica, endocarditis bacteriana sub-aguda, carcinoma visceral, alcoholismo y durante el embarazo. Se debe considerar la presencia de factores endógenos que pueden aumentar o reducir la acción anticoagulante de la warfarina. Entre los que aumentan la acción se incluyen: Carcinoma, trastornos hepáticos, deficiencia de vitamina K, hipertiroidismo, esteatorrea, insuficiencia cardiaca congestiva, diarrea, enfermedad del colágeno, desnutrición y discrasias sanguíneas. Entre los que pueden disminuir el efecto se cuentan: Síndrome nefrótico, edema, hiperlipidemia, hipotiroidismo y la resistencia hereditaria a los anticoagulantes. Debido a sus múltiples posibilidades de interacción se recomienda consultar fuentes especializadas antes de asociar warfarina a cualquier otro medicamento. Durante el tratamiento se debe suspender la lactancia. La dosis debe ser controlada mediante determinaciones periódicas del tiempo de protrombina (PT)/International Normalized Ratio (INR) u otras pruebas de coagulación adecuadas. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. La hipoprotrombinemia excesiva puede controlarse con fitome44 de otros tratamientos durante el uso de warfarina debe ser consultado previamente con el médico. Evitar el uso de alcohol y los cambios drásticos en la dieta habitual. Notificar de inmediato al médico si se presenta alguna reacción o síntoma inusual durante el tratamiento, en especial manifestaciones hemorrágicas o ÁCIDO TRANEXÁMICO FARMACOLOGÍA Agente antifibrinolítico. Inhibe competitivamente la activación del plasminógeno, disminuyendo su conversión en plasmina e inhibiendo así la fibrinólisis. Es alrededor de diez veces más po-tente que el ácido aminocaproico. FARMACOCINÉTICA Se absorbe en un 30-50% en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico en 3 horas, las cuales persisten por 7-8 horas en el plasma y hasta por 17 horas en los tejidos. La biodisponibilidad no es afectada por los alimentos. Se distribuye ampliamente en el organismo y se une al plasminógeno en un 3%, sin unión apreciable a la albúmina. Sólo una pequeña fracción del fármaco es metabolizada. Después de la administración oral se excreta un 39% Formulario Terapéutico Nacional * 2ed. 2004 INDICACIONES Prevención y tratamiento de hemorragias post-quirúrgicas, hemorragias en pacientes hemofílicos y las generadas por hiperfibrinólisis generalizada. RÉGIMEN DE DOSIFICACIÓN Se administra por infusión IV lenta. - Hemorragias post-quirúrgicas: Dosis inicial de 10-15 mg/kg o 0,5-1 g durante la cirugía y luego cada 8 horas por 3 días. - Hemorragia por cirugía dental en hemofílicos: 25 mg/kg IV antes de iniciar la cirugía y luego, 10 mg/kg cada 6-8 horas por 2-8 días. - Hemorragias por hiperfibrinólisis: 15 mg/kg cada 6-8 horas hasta el cese del sangramiento o hasta lograr una fibrinólisis adecuada, según los valores de laboratorio. REACCIONES ADVERSAS Cardiovasculares: Infarto del miocardio, hipotensión, tromboembolismo, embolismo pulmonar. Gastrointestinales: Dolor abdominal, náuseas, vómito, diarrea. Neurológicas: Vértigo, can-sancio, visión borrosa e hidrocefalia. INTERACCIONES La administración de ácido tranexámico conjuntamente con estrógenos aumenta el riesgo de formación de trombos. Los agentes trombolíticos (riesgo de trombosis). Usar con cautela en pacientes con historia de o con predisposición a la trombosis, defectos adquiridos de la visión de co- B lores, falla renal y hematuria originada en el tracto urinario superior. La infusión IV rápida puede producir hipotensión, por lo cual la tasa máxima de inyección no debe superar 1 ml/min. No se conoce la seguridad de su uso en el embarazo y durante la lactancia. SOBREDOSIS Tratamiento sintomático y de soporte. Las formas inyectables pueden causar hipotensión y bradicardia. INFORMACIÓN AL PACIENTE FACTOR VIII DE LA COAGULACIÓN FARMACOLOGÍA Liofilizado concentrado de factor VIII de coagulación obtenido por tecnología de ADN recombinante. El factor VIII es necesario para la formación del coágulo y el mantenimiento de la homeostasis. Actúa como cofactor con el factor IX activado en la activación del factor X que, en presencia del factor V activado, convierte a la protrombina en trombina, la cual, a su vez, convierte al fibrinógeno en fibrina generándose el coágulo definitivo de la hemostasia, el cual es estabilizado por el factor XIII. FARMACOCINÉTICA La farmacocinética del factor VIII 45 2ed. 2004 * Formulario TerapÈutico Nacional que es similar a la del factor endógeno. Aunque la vida media de eliminación promedio varía según el producto empleado, por lo general se ubica entre 12 y 14 horas. Por ejemplo: En un niño de 25 kg, la dosis de factor VIII requerida para incrementar los niveles de factor VIII plasmático en un 30% es: Dosis Requerida = 25 x 0,5 x 30 = 375 UI INDICACIONES Prevención y control de los episodios hemorrágicos en pacientes con hemofilia A por deficiencia comprobada de factor VIII. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV. Las dosis varían con cada paciente y dependen del grado de deficiencia del factor, del nivel de factor deseado, del peso del paciente, del tipo y severidad de la hemorragia y de la presencia de inhibidores del factor antihemofílico. Por ello, las dosis deben individualizarse con base en los estudios de coagulación y en los niveles plasmáticos de factor VIII medidos antes de iniciar la terapia y a intervalos regulares durante su desarrollo. Para calcular el porcentaje de incremento aproximado que se espera de una dosis, así como la dosis requerida para alcanzar un determinado porcentaje de incremento, se emplean las siguientes fórmulas (que aplican sólo para pacientes sin inhibidores del factor antihemofílico): Incremento Deseado = 46 Dosis Administrada (en UI) x 2 La experiencia clínica permite estimar como dosis iniciales de factor VIII en hemofilia A para tratamiento precoz de hemorragias leves (hemartrosis, hematomas, episitaxis, gingivorragia o hematuria): 15 U/kg (nivel del Factor: 30%); hemorragias mayores (hemartrosis, hematomas con dolor o inflamación, hematomas del psoas, cuello, faringe o lengua, trauma craneal asintomático, traumas severos, exodoncia, dolor abdominal, hemorragia digestiva, compresión neurológica): 25 U/kg (nivel del Factor: 50%) y de hemorragias graves (intracraneanas, traumatismo severo, cirugías): 40-50 U/kg (nivel del Factor: 80-100%) REACCIONES ADVERSAS Cardiovasculares: Alteraciones de la presión arterial, opresión en el pecho, taquicardia. Gastrointestinales: Náusea, vómito, boca seca, sabor metálico. Hematológicas: Trombosis, anemia hemolítica, trombocitopenia. Neurológicas: Cefalea, mareo, despersonalización. Reacciones de Hipersensibilidad: Erupción, prurito y urticaria. Otras: Rubor, fiebre, Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES No se ha descrito casos de interacciones significativas con el factor VIII recombinante. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. Se debe vigilar periódicamente (cada 3-6 meses) la posibilidad de desarrollo de inhibidores del factor antihemofílico. No se conoce la seguridad de su uso en el embarazo ni durante la lactancia. SOBREDOSIS Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Es recomendable que el paciente porte una placa de idenFACTOR IX DE LA COAGULACIÓN FARMACOLOGÍA Liofilizado concentrado de los factores IX, II, VII y X de coagulación obtenidos de sangre fresca de donantes sanos. Una unidad (1 UI) del complejo contiene el promedio de actividad de los factores IX, II, VII y X presentes en 1 ml de plasma fresco de menos de 1 hora de extraído. El complejo promueve la coagulación en pacientes como en el caso de la hemofilia. FARMACOCINÉTICA La vida media plasmática es de B aproximadamente 24 horas. INDICACIONES Prevención y control de los episodios hemorrágicos en pacientes con hemofilia B causados por deficiencia de factor IX. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV. Las dosis se expresan en términos de factor IX. La dosis necesaria para lograr hemostasis en pacientes con deficiencias de factor IX, II, VII y X debe ser individualizada en función del grado de deficiencia de cada paciente, del nivel deseado de cada factor, del peso corporal y de la severidad de la hemorragia. Para calcular el porcentaje de incremento aproximado que se espera de una dosis, así como la dosis requerida para alcanzar un determinado porcentaje de incremento, se emplean las siguientes fórmulas: Incremento Deseado = (% de lo normal) Dosis Administrada (en UI) x 1,3 Peso Corporal (en kg) Dosis Requerida (en UI) = Peso Corporal (en kg) x 1,2 x Incremento Deseado (% de lo normal) 2ed. 2004 * Formulario TerapÈutico Nacional 47 Dosis Requerida = 70 x 1,2 x 50 = 4200 UI Tal método para calcular la dosis; sin embargo, constituye sólo una aproximación que no debe reemplazar a la observación clínica ni a los parámetros de laboratorio como indicadores para la determinación de la dosis necesaria para mantener la hemostasis. La experiencia clínica permite estimar como dosis iniciales de factor IX en hemofilia B para tratamiento precoz de hemorragias leves (hemartrosis, hematomas, episitaxis, gingivorragia o hematuria): 20 U/kg (nivel del factor: 20%); hemorragias mayores (hemartrosis, hematomas con dolor o inflamación, hematomas del psoas, cuello, faringe o lengua, trauma craneal asintomático, traumas severos, exodoncia, dolor abdominal, hemorragia digestiva, compresión neurológica): 40 U/kg (nivel del factor: 40%) y de hemorragias graves (intracraneanas, traumatismo severo, cirugías): 60-70 U/kg (nivel del factor: 60-70%) REACCIONES ADVERSAS Cardiovasculares: Alteraciones de la presión arterial, presión en el pecho, infarto del miocardio, coagulación intravascular diseminada, trombosis venosa y embolismo pulmonar. INTERACCIONES La administración conjunta de complejo de factor IX con ácido aminocapróico o con ácido tranexámico aumenta el riesgo de trombosis. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al producto, alteración de la función hepática y sospecha de coagulación intravascular o fibrinólisis. Como el producto se prepara a partir de un pool de plasma humano, se debe considerar la posibilidad de la presencia de agentes causantes de hepatitis viral u otras enfermedades virales. Se debe evaluar el balance riesgo-beneficio de su uso en pacientes post-quirúrgicos con predisposición a trombosis. No se conoce la seguridad de su uso en el embarazo ni durante la lactancia. Sólo puede administrarse a mujeres embarazadas si es realmente necesario. Se ha enfatizado que la vacunación contra la hepatitis B es esencial para los pacientes con hemofilia, inclusive al nacimiento o al diagnóstico. También debe aplicarse la vacuna contra la hepatitis A si el paciente es seronegativo para este virus. SOBREDOSIS Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE 48 Formulario Terapéutico Nacional * 2ed. 2004 FITOMENADIONA (VITAMINA K) FARMACOLOGÍA Promueve la síntesis hepática de los factores II, VII, IX y X, así como de las proteínas C y S que, en conjunto, conforman el grupo de proteínas plasmáticas vitamina K dependientes, esenciales para el proceso de coagulación. La vitamina K probablemente participa también como cofactor en la carboxilación de precursores inactivos de estos factores. En dosis adecuadas, revierte el efecto de los anticoagulantes orales. FARMACOCINÉTICA Es absorbida rápidamente en el tracto gastrointestinal por un proceso saturable dependiente de las sales biliares. Se absorbe muy bien después de la administración intramuscular. Luego de la administración oral produce un incremento en los factores de coagulación dentro de 6-10 horas, mientras que por vía parenteral produce un control del cuadro hemorrágico dentro de 3-6 horas y un nivel normal de protrombina a las 1214 horas. Se distribuye rápidamente a los fluidos y tejidos corporales y se metaboliza aparentemente en el hígado, aunque se sabe poco sobre su metabolismo. No se conoce bien la eliminación de la fitomenadiona. orales. Profilaxis y tratamiento de hemorragias del recién nacido. Tratamiento de hipoprotrombinemia inducida por fármacos. Es inefectiva B en el manejo de las hemorragias producidas por la heparina y en el tratamiento de la hipoprotrombinemia hereditaria y aquella causada por enfermedades hepáticas severas. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, SC ó IM. Si no es factible su administración por estas vías, puede usarse la vía IV mediante inyección lenta, sin exceder una velocidad de 1 mg/minuto. - En la hipoprotrombinemia inducida por fármacos y por síndrome de malabsorción en adultos: Iniciar con 2,5-25 mg VO, IM ó SC, y repetir, en caso necesario, a las 12-48 horas de la administración oral o a las 6-8 horas de la administración parenteral, previo control del tiempo de protrombina. En infantes: Iniciar con 1-2 mg y en niños con 5-10 mg VO, SC ó IM. - Para la prevención de la hipoprotrombinemia en la nutrición parenteral, la dosis usual en adultos es de 5- 10 mg 1 vez por semana y en niños 2-5 mg IM una vez por semana. - Para la profilaxis y tratamiento de hemorragias en el recién nacido, la dosis usual en los neonatos es de 2ed. 2004 * Formulario TerapÈutico Nacional 49 paración parenteral puede ser administrada por vía oral a pacientes neonatos, según la dosis indicada. REACCIONES ADVERSAS Aunque por lo general es segura y bien tolerada por la mayoría de los pacientes, su uso ha sido asociado con reacciones ocasionales de hipersensibilidad (anafilaxis), trastornos del sentido del gusto, eritema facial y dolor o edema en el sitio de la inyección. En recién nacidos, con dosis elevadas (10-20 mg) se ha reportado casos raros de hiperbilirrubinemia y anemia hemolítica severa. Con la administración IV se ha reportado casos de: Hiperhidrosis, dolor anginoso, trastornos cardiacos, broncoespasmo, disnea, cianosis, hipotensión transitoria, colapso circulatorio, shock, paro cardíaco y/o respiratorio y muerte. esperarse un efecto coagulante inmediato. Toma 1-2 horas una mejoría medible en el tiempo de protrombina. No se conoce la seguridad del uso en el embarazo ni durante la lactancia. Debe protegerse constantemente de la luz. SOBREDOSIS Manejo sintomático y terapia de soporte. ÁCIDO FÓLICO FARMACOLOGÍA El ácido fólico es requerido para la síntesis de nucleoproteínas y en el mantenimiento de la eritropoyesis normal. Es precursor del ácido tetrahidrofólico, el cual participa como cofactor en las reacciones de transferencia de átomos de carbono para la biosíntesis de purinas y timidilatos de los ácidos nucléicos. Se cree que los trastornos en la biosíntesis de los timidilatos, por deficiencia de ácido fólico, dan lugar a una síntesis defectuosa de ADN, a la formación de megaloblastos y, en consecuencia, a la ocurrencia de anemia megaloblástica y macrocítica. INTERACCIONES Los antiácidos disminuyen la absorción gastrointestinal de fitomenadiona. Los salicilados, dactinomicina, quinina, quinidina, sulfonamidas y los antibióticos de amplio espectro aumentan los requerimientos de fitomenadiona. La primaquina potencia sus efectos tóxicos. Cuando se usan altas dosis de vitamina K FARMACOCINÉTICA puede haber una resistencia temporal Luego de la administración oral se a los anticoagulantes, lo cual puede absorbe rápidamente en el tracto requerir un aumento de la dosis gastrointestinal, alcanzando niveles 50 Formulario Terapéutico Nacional * 2ed. 2004 lorraquídeo, los eritrocitos y en el hígado, donde se metaboliza por reducción y metilación, transformándose en ácido metiltetrahidrofólico, que constituye la principal forma de transporte y almacén de folatos en el organismo. Se excreta, en su mayoría, por la orina y en muy escasa proporción por las heces. INDICACIONES Tratamiento de la anemia megaloblástica por deficiencia de folato, en la cual se observan eritrocitos macrocíticos anormales. En el embarazo, para la prevención del desarrollo de anomalías del tubo neural (espina bífida, anencefalia y encefalocele) en el feto. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM profunda o IV en dosis similares. La dosis terapéutica usual en adultos y niños es de 0,25-1 mg/día. La dosis diaria de mantenimiento (una vez corregida la deficiencia de folato) es de 0,1 mg en infantes, de 0,3 mg en niños menores de 4 años, de 0,4 mg en adultos y niños mayores de 4 años y de 0,8 mg en mujeres embarazadas y madres que amamantan. Cuando se usa para la prevención de la anemia megalo-blástica del embarazo y del daño fetal (defectos del tubo neural) por deficiencia de folato, la dosis sión, irritabilidad, excitabilidad, trastornos del sueño, confusión y dificultad para la concentración. Reacciones de hipersensibilidad: Eritema, B erupción, urticaria, malestar general y dificultad respiratoria broncoespástica. INTERACCIONES El ácido fólico en altas dosis contrarresta el efecto de la fenitoína, el fenobarbital y la primidona y aumenta la frecuencia de las convulsiones en niños. El cloranfenicol puede antago-nizar la respuesta hematopoyética del ácido fólico. La trimetoprima, el triamtereno, los anticonceptivos orales y la pirimetamina pueden inducir deficiencia del ácido fólico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. El ácido fólico puede enmascarar el diagnóstico de la anemia perniciosa y permitir (mientras mejoran las manifestaciones hematológicas de la enfermedad) la progresión de las complicaciones neurológicas, lo que podría ocasionar un daño severo al SNC antes de llegar a establecerse un diagnóstico concreto. También es inapropiado su uso solo para el tratamiento de anemias megaloblásticas donde hay deficiencia de Vitamina B12. 2ed. 2004 * Formulario TerapÈutico Nacional 51 CIANOCOBALAMINA (VITAMINA B12) FARMACOLOGÍA Forma sintética de la vitamina B12 con actividad farmacológica similar. Es una coenzima que interviene en los proce-sos de síntesis de nucleoproteínas y mielina, metabolismo de lípidos y carbohidratos, crecimiento, reproducción celular y eritropoyesis. La deficiencia produce anemia megaloblástica. FARMACOCINÉTICA La absorción por vía oral es pobre y dependiente de la presencia del factor intrínseco y de los iones calcio. Por vía IM; sin embargo, se absorbe rápida y completamente alcanzando niveles séricos pico en 60 minutos, produ-ciendo el inicio de la respuesta (en ca-so de anemia megaloblástica) a las 8 horas, la cual se mantiene por 2-4 semanas. Una vez en la sangre, se une a la transcobalamina II (globulina beta), formando un complejo a través del cual es distribuida a los tejidos. Se almacena en el hígado, desde donde es liberada lentamente a la periferia cuando es requerida. Se excreta casi en su totalidad por la orina como fármaco intacto y en menor proporción por vía biliar. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM ó SC profunda. La dosis usual para el tratamiento de la anemia perniciosa o síndrome de malabsorción de vitamina B12 es de 100 mcg/día por vía IM ó SC profunda por 5-10 días, seguido por 100-200 mcg/mes hasta la remisión completa y, a partir de allí, dosis de mantenimiento de 100 mcg/mes de por vida. Alternativamente, pueden emplearse dosis de mantenimiento por vía oral de 1000 mcg/día. Cuando se usa como suplemento nutricional se considera suficiente una dosis de 1-25 mcg/día. REACCIONES ADVERSAS Es por lo general segura y libre de efectos adversos, aún en dosis elevadas. Sin embargo, se ha descrito casos de diarrea transitoria, trombosis vascular periférica, erupción, edema pulmonar, insuficiencia cardiaca congestiva, urticaria, dolor en el sitio de la inyección y shock anafiláctico. INTERACCIONES La colchicina, los anticonvulsivantes, los aminoglicósidos, el ácido aminosalicílico, el potasio por vía oral y la ingesta prolongada de alcohol pueden ocasionar malabsorción de vitamina B12. El cloranfenicol y varios antibióticos, el metotrexato y la pirimetamina invalidan los ensayos sanguíneos del ácido fólico y la vitamina B12. Puede INDICACIONES Tratamiento de la anemia perniciosa y 52 Formulario Terapéutico Nacional * 2ed. 2004 sibles al medicamento o al cobalto. La terapia con vitamina B12 puede enmascarar las deficiencias de ·cido fÛlico. Dada la posibilidad de hipopotasemia al inicio del tratamiento, es recomendable determinar los niveles sÈricos de potasio durante las primeras 48 horas. La respuesta a la cianocobalamina podrÌa ser inadecuada en presencia de infecciÛn, falla renal, tumores o deficiencias concurrentes de hierro o ·cido fÛlico. Debido al contenido de alcohol bencÌlico, no debe ser usado en niÒos reciÈn nacidos. SOBREDOSIS No se conocen casos de intoxicación aguda por sobredosis de cianocobalamina. INFORMACI”N AL PACIENTE No alterar la dosis ni suspender la terapia sin el conocimiento del médico. En casos de anemia perniciosa, se debe advertir al paciente que el trata-miento es de por vida y que su incum-plimiento puede ocasionar una reci-diva de la enfermedad, con la posibili-dad de daños irreversibles sobre los nervios y la médula espinal. Notificar de ERITROPOYETINA (EPOETIN ALFA) FARMACOLOGÍA Glicoproteína producida por tecno- tituida por 165 aminoácidos en secuencia idéntica a la de la eritropoyetina endógena humana y, como tal, con las mismas propiedades B biológicas de la hormona natural. Induce la eritropoyesis por estimulación de la división y diferenciación de las células eritroides precursoras en la médula ósea y promueve su posterior liberación al torrente sanguíneo donde finalmente maduran hasta convertirse en eritrocitos. Aumenta el contaje de reticulocitos y el hematocrito de una manera dosis-dependiente. La eritropoyetina humana es producida en el riñón. FARMACOCINÉTICA Posterior a la administración IV, alcanza concentraciones plasmáticas pico en aproximadamente 15 minutos (o 5-24 horas tras la administración subcutánea) que se mantienen hasta por 12-16 horas. Aún cuando su acción sobre los tejidos eritropoyéticos (aumento en el contaje de reti-culocitos) se inicia a los 7-10 días, son necesarias 2-6 semanas para obser-var un efecto clínicamente signifi-cativo (aumento en el contaje de eritrocitos, del hematocrito y de la hemoglobina) y hasta 2 meses para alcanzar el efecto máximo. La vida media de eliminación se ubica en el rango de 4-13 horas. 2ed. 2004 * Formulario TerapÈutico Nacional 53 RÉGIMEN DE DOSIFICACIÓN Las dosis recomendadas para adultos y niños a partir de los 2 años son: - Anemia por falla renal: Iniciar con 50100 U/kg IV ó SC 3 veces/semana hasta lograr un hematocrito de 3036%; si después de 8 semanas la respuesta no es satisfactoria, incrementar la dosis en 25 U/kg. - Anemia por terapia con zidovudina: Iniciar con 100 U/kg IV 3 veces/semana. Si después de 8 semanas la respuesta no es satisfactoria, incrementar la dosis en 50-100 U/kg hasta lograr un máximo de 300 U/kg 3 veces/semana. - Anemia por quimioterapia antineoplásica: Iniciar con 150 U/kg SC 3 veces/semana por 8 semanas o hasta que se alcance el nivel de hemoglobina adecuado; si después de 8 semanas la respuesta no es satisfactoria, incrementar la dosis sin exceder 300 U/kg 3 veces/semana. Si el hematocrito excede 40% debe suspenderse hasta obtener un hematocrito de 36%. - Para reducir la frecuencia de las transfusiones durante la cirugía no cardiovascular: 300 U/kg/día SC por 10 días antes de la cirugía, el día de la cirugía y por 4 días después de la misma. angioedema. Otras: Anemia por aplasia de la serie roja, artralgia, tos, sudoración, pirexia, infecciones respiratorias y del tracto urinario, dolor en el sitio de inyección. INTERACCIONES Debido al incremento en el contaje de los eritrocitos inducido por la eritropoyetina, es posible que se requiera mayores dosis de heparina en pacientes sometidos a hemodiálisis, a objeto de evitar la coagulación en el dializador o en el acceso vascular. Los pacientes con terapia antihipertensiva podrían requerir dosis mayores de ésta para contrarrestar los incrementos de presión arterial generados por la eritropoyetina. En pacientes sometidos a hemodiálisis y tratamiento con inhibidores de la enzima convertidora de angiotensina se ha observado disminución de la eficacia clínica de la eritropoyetina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento, a la albúmina humana u otros hemoderivados y en pacientes con hipertensión no controlada. Usar con precaución en pacientes con hipertensión previa, deficiencias de ácido fólico o de vitamina B12, hemólisis, infección, malignidad, trombosis, inflamación, hipercoagulabilidad, convulsiones y REACCIONES ADVERSAS Cardiovasculares: Hipertensión, dolor en el pecho, palpitaciones, edema. 54 Formulario Terapéutico Nacional * 2ed. 2004 Durante la terapia se debe evaluar periódicamente la presión arterial, el contaje plaquetario, el hematocrito, la función renal y el estado neurológico del paciente. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales a la madre superen el posible riesgo para el feto. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico si se presenta alguna reacción o síntoma inusual durante el tratamiento. Dentro de 2 horas de la inyección puede presentarse dolor en los miembros y la pelvis. Sensación de frío y sudoración son comunes. Los síntomas persisten por 12 horas y luego desaparecen. Debe tomarse una muestra de sangre cada semana para el contaje de células SULFATO FERROSO (HIERRO ELEMENTAL) FARMACOLOGÍA El hierro es un constituyente esencial de numerosas enzimas que participan en sistemas de transferencia de energía (citocromo-oxidasa, deshidrogenasa succínica), de enzimas metaloflavoproteínas como la oxidasa de xantinas y la oxidasa del alfa- zación de oxígeno (hemoglobina y mioglobina), de enzimas del heme como los citocromos, la peroxidasa y la catalasa. La administración del B sulfato ferroso corrige las anormalidades eritropoyéticas asociadas a la deficiencia del hierro. FARMACOCINÉTICA La absorción gastrointestinal del hierro es compleja y depende de factores entre los cuales se incluyen: La sal empleada, la dosis, los depósitos cor-porales, el grado de eritropoyesis y la dieta. En individuos sanos, la cantidad de hierro absorbida de la dieta es de aproximadamente un 5-10%, mientras que en sujetos con deficiencia de hie-rro puede alcanzar hasta 10-30%. Se cree que la absorción de hierro (como hierro ferroso) de la dieta ocurre a través de un mecanismo de transporte activo en el duodeno y en el yeyuno proximal, y por transporte pasivo cuan-do la dosis ingerida es superior a la que aporta una dieta normal. Después de la absorción, el hierro se combina con la transferrina (globulina-glicopro-teína beta1 plasmática) y es trans-portado a la médula ósea donde se in-corpora a la hemoglobina. Los exce-sos de hierro son oxidados al estado férrico en las células vellosas epi-teliales y éste se combina con la apo-ferritina para transformarse en ferritina en las 2ed. 2004 * Formulario TerapÈutico Nacional 55 renal, por la bilis y a través de la piel. La excreción promedio en sujetos sanos es de 0,5-2 mg diarios. Durante la menstruación se pierde 1-2 mg diarios. INDICACIONES Prevención y tratamiento de las anemias por deficiencia de hierro. R…GIMEN DE DOSIFICACI”N Las dosis de sulfato ferroso se expresan en términos de hierro elemental. El sulfato ferroso contiene un 20% de hierro elemental. La ingesta diaria recomendada (dieta) para adultos es de 10 mg para los hombres y 18 mg para las mujeres; durante el embarazo y la lactancia, 30-60 mg. Para la terapia de reemplazo en adultos y niños mayores de 12 años, se recomiendan dosis diarias de 100-200 mg o de 2-3 mg/kg de hierro elemental divididos en 3 dosis. En niños de 2-12 años: 3 mg/kg/día divididos en 3-4 dosis; niños de 6 meses-2 años: Hasta 6 mg/kg/día divididos en 3-4 dosis; y en infantes: 10-25 mg/día divididos en 3-4 dosis. La duración de la terapia es variable y depende de la causa y de la severidad de la deficiencia. Como suplemento se indica solamente en sujetos con factores de riesgo de deficiencia de hierro. Para la deficiencia nutricional moderada: 30 mg/día en adultos. 56 INTERACCIONES El sulfato ferroso puede interferir con la absorción gastrointestinal de penicilamina, quinolonas, hormonas tiroideas y tetraciclinas. Los antiácidos, la colestiramina y los alimentos ricos en metales divalentes, como los huevos, el yogurt y la leche, disminuyen la absorción del hierro. El cloranfenicol puede interferir con la acción farmacológica del hierro retrasando la respues-ta terapéutica. La vitamina C aumenta la absorción del hierro y la vitamina E disminuye la respuesta a la terapia con hierro. El café y el té tomados con o una hora después de la comida, pue-den interferir con la absorción del hierro. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en individuos con hipersensibilidad al medicamento, en la hemocromatosis, hemosiderosis, anemia hemolítica, úlcera péptica, enteritis regional y en la colitis ulcerativa. SOBREDOSIS La ingestión de dosis superiores a 60 mg/kg de hierro elemental puede resultar fatal. Si la ingestión es reciente se debe proceder al vaciamiento gástrico (preferiblemente emesis si el paciente coopera) y administrar un catártico salino. En pacientes con concentraciones de hierro sérico superiores a 300 mcg/dl Formulario Terapéutico Nacional * 2ed. 2004 INFORMACIÓN AL PACIENTE Tomar preferiblemente con el estómago vacío. No usar leche o antiá-cidos para minimizar las molestias gástricas. Ingerir las preparaciones lí-quidas diluidas y con pitillo para evitar manchas en los dientes. Se debe ad-vertir al paciente AGUA PARA INYECCIÓN INDICACIONES El agua estéril libre de pirógenos y de solutos empleada como solvente o B como vehículo de fármacos de administración IV ó IM. DOSIFICACIÓN Cantidad suficiente para la disolución AGUA ESTÉRIL PARA IRRIGACIÓN o suspensión del medicamento a administrar y según el tipo de INDICACIONES El agua estéril, libre de pirógenos y de inyección (IV ó IM). solutos, y con un pH de 5-7, es usada tópicamente para la irrigación, lavado PRECAUCIONES Use técnicas asépticas para el retiro y limpieza de tejidos y heridas y drenaje de abscesos. ALBÚMINA HUMANA RÉGIMEN DE DOSIFICACIÓN FARMACOLOGÍA Depende del procedimiento para el La albúmina es una proteína plasmácual se utiliza. tica globular altamente soluble, que tiene como función el mantenimiento REACCIONES ADVERSAS Como el agua para irrigación es hipo- de la presión coloidosmótica y el tónica (libre de solutos), la absorción t r a n s - p o r t e d e m e t a b o l i t o s sistémica en cantidades elevadas intermediarios producidos en las células. Representa el 50-60% de las pue-de causar hemólisis. proteínas plas-máticas y provee CONTRAINDICACIONES/ alrededor del 85% de la presión PRECAUCIONES coloidosmótica del plasma. Participa Dada la posibilidad de hemólisis se- en la regulación del volumen de cundaria a la absorción sistémica, no sangre circulante y su pérdida o es recomendable el uso del agua para disminución (como en los casos de irrigación en situaciones que hagan s h o c k h i p o v o l é m i c o ) r e s u l t a n factible la absorción en magnitudes críticas. En situaciones que cursan importantes (como en caso de procedi- con una disminución importante del mientos quirúrgicos). No usar por vía volumen plasmático, la inyección de parenteral. Una vez abierto el envase una can-tidad adecuada de albúmina 57 * Formulario TerapÈutico Nacional que la contiene se debe 2ed. usar 2004 tan pronto oncóticamente equivalente a un volumen igual de plasma, e incrementa el volumen de sangre en una cantidad aproximadamente igual al volumen de albúmina infundido. La infusión de albúmina al 25% es oncóticamente equivalente a un volumen 5 veces superior de plasma, e incrementa el volumen de sangre en una cantidad 3,5 veces superior al volumen de albúmina infundido. También es una proteína de transporte, y fija compuestos que ocurren naturalmente en el organismo, así como agentes terapéuticos y agentes físicos. FARMACOCINÉTICA No aplicable. INDICACIONES Manejo de situaciones que cursan con disminución importante del volumen circulante y/o con déficit oncótico. RÉGIMEN DE DOSIFICACIÓN La dosis se establece de acuerdo a la condición y necesidades particulares de cada paciente y a la respuesta hemodinámica que éste exhiba al tratamiento. La meta de la terapia es restituir la concentración total de albúmina plasmática a niveles de 2-3 g/dl y la presión oncótica a 20 mm Hg (equivalente a una concentración de 5,2 g/dl de proteínas séricas totales). Se administra por infusión IV sin diluir o diluida en solución salina normal o 58 exceder 5-10 ml/minuto, y en el caso de la albúmina al 25% no debe exceder 2-3 ml/minuto. REACCIONES ADVERSAS Casos poco frecuentes de hipotensión, taquicardia, fiebre, escalofríos, náuseas, erupción y urticaria. Si se administra rápidamente puede resultar en una sobrecarga vascular y en edema pulmonar. INTERACCIONES No se describen en la literatura. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al producto y en condiciones en las que una sobrecarga de fluido podría representar un riesgo para el paciente, como en la insuficiencia cardiaca, en el edema pulmonar o en la anemia severa. Usar con precaución en pacientes con hipertensión, con falla renal y en el embarazo. Las soluciones no deben ser usadas si están turbias o muestran un sedimento. No debe iniciarse la infusión 4 horas después de haber abierto el frasco. Los pacientes con deshidratación requieren la administración de fluidos adicionales. No debe administrarse a través de la misma vía que soluciones que contengan hidrolizados de pro-teínas o alcohol ya que estas combina-ciones pueden Formulario Terapéutico Nacional * 2ed. 2004 INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso AMINO ÁCIDOS FARMACOLOGÍA Soluciones con combinación de aminoácidos esenciales y no esenciales en concentración de 3-15%, empleadas por vía IV como fuente de elementos para la síntesis de proteínas en pacientes que requieren soporte nutricional. FARMACOCINÉTICA Luego de la administración IV, los aminoácidos, se distribuyen ampliamente a todos los tejidos corporales donde son posteriormente metabolizados. El metabolismo aumenta en presencia de trauma y sepsis y disminuye en estados de insuficiencia hepática. INDICACIONES Como parte del soporte nutricional en el tratamiento de condiciones que cursan con un balance nitrogenado negativo, o en las que los requerimientos proteicos están significativamente aumentados y no es factible la nutrición por vía oral. R…GIMEN DE DOSIFICACI”N La solución de aminoácidos se administra por infusión en la vena central. Los requerimientos varían en duras severas: 2-3 g/kg/día. En niños con estrés metabólico leve a moderado, la dosis es de 1-3 g/kg/día y en pacientes críticos o con quemaduras B graves: 2,5-3,5 g/kg/día. REACCIONES ADVERSAS Se ha descrito casos de tromboflebitis, encefalopatía, hiperamonemia y colestasis, estupor y coma. La hiperamonemia es particularmente importante en infantes. INTERACCIONES No se describen en la literatura. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a uno o más aminoácidos, enfermedad hepática severa, coma hepático o anemia. Utilizar con precaución en pacientes con insuficiencia renal aguda, insuficiencia hepática y con restricción de fluidos. La administración de este medicamento requiere el conocimiento del balance de fluidos y electrolitos así como también experiencia clínica en el reconocimien-to y tratamiento de las complicaciones que puedan ocurrir. En caso de hiperamonemia, el medicamento debe ser suspendido y el paciente reeva-luado. En caso de insuficiencia renal se requiere una evaluación clínica y de laboratorio 2ed. 2004 * Formulario TerapÈutico Nacional 59 ciente e instituir tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE No aplica; medicamento para uso hosBICARBONATO DE SODIO FARMACOLOGÍA Incrementa los niveles séricos de bicarbonato y actúa como buffer en condiciones de acidosis, elevando el pH sanguíneo. Revierte las manifestaciones clínicas de la acidosis. FARMACOCINÉTICA Se disocia en bicarbonato y sodio. El bicarbonato reacciona con los hidrogeniones (H+) convirtiéndose en ácido carbónico y finalmente en dióxido de carbono (CO2) que se excreta por vía respiratoria. En un adulto sano con función renal normal, prácticamente todo el bicarbonato filtrado es reabsorbido. Menos del 1% es excretado en la orina. INDICACIONES Acidosis metabólica. Alcalinizante urinario. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV. La dosificación debe ajustarse a las necesidades individuales de cada paciente, en función del pH sanguíneo y la presión de Co2. La dosis usual en adultos para el 60 REACCIONES ADVERSAS Alcalosis metabólica, que puede estar asociada a contracciones musculares, irritabilidad y tetania, hipernatremia, hiperosmolaridad, edema cerebral y hemorragia intracraneana. Se ha descrito casos de necrosis tisular por extravasación de soluciones hipertónicas. INTERACCIONES La alcalinización urinaria inducida por el bicarbonato puede incrementar la excreción renal del litio, metotrexato, tetraciclinas y de la clorpropamida y disminuir la de la pseudoefedrina, efedrina, anfetaminas, quinina y quinidina. La norepinefrina y la dobutamina son incompatibles con la solución de bicarbonato. Debe evitarse la adición de bicarbonato a las soluciones de calcio ya que puede formarse una turbidez o un precipitado. Puede producir la liberación prematura de fármacos con cubierta entérica en el tracto gastrointestinal. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con alcalosis metabólica o respiratoria, hipocalcemia e hipocloremia. Usar con precaución en pacientes con restricción de sodio, hipertensión, insuficiencia hepática, historia de insuficiencia cardiaca congestiva y/o insuficiencia renal y durante el embarazo. Formulario Terapéutico Nacional * 2ed. 2004 SOBREDOSIS Puede ocasionar alcalosis. Instituir un tratamiento orientado a corregir la alcalosis y el balance hidro-electrolítico. Casos moderados o severos pueden requerir gluconato de calcio IV. Casos más severos pueden requerir infusión IV de cloruro de amonio al 2,14% (excepto en pacientes con insuficiencia hepática). INFORMACIÓN AL PACIENTE CLORURO DE POTASIO FARMACOLOGÍA El potasio es el principal catión de la mayoría de los tejidos corporales, y es un elemento esencial para el mantenimiento del equilibrio ácido-base, la isotonicidad y las características electrodinámicas de la célula. Es un activador importante de numerosas reacciones enzimáticas, y fundamental para el desarrollo de diversos procesos fisiológicos como la transmisión del impulso nervioso, la contracción del músculo cardiaco y de la musculatura lisa y esquelética, el mantenimiento de la función renal normal, la síntesis de proteínas y el metabolismo de los carbohidratos. FARMACOCINÉTICA Los niveles fisiológicos de potasio se ubican en el rango de 3,5-5,5 mEq/L. Se excreta en un 85-90% por vía renal R…GIMEN DE DOSIFICACI”N Las dosis deben individualizarse de acuerdo a los requerimientos particulares de cada paciente según su B condición clínica y la respuesta a la terapia, con vigilancia frecuente del ECG y de los niveles séricos. El cloruro de potasio aporta 13,4 mEq/g de ión potasio. Se administra por vía IV, previamente diluido. La inyección di-recta del medicamento sin diluir puede provocar la muerte en forma instantá-nea. En pacientes adultos con nivel de potasio sérico mayor de 2,5 mEq/L, usar soluciones con concentración de potasio de 40 mEq/L y administrar a una velocidad de 10 mEq/hora, sin exceder la cantidad de 200 mEq tota-les en 24 horas. No debe administrarse más de 20 mEq por dosis. Si el nivel sérico es menor que 2 mEq/L, se debe usar concentraciones de 80 mEq/L e infundir la solución a una velocidad de 40 mEq/hora, sin exceder la cantidad de 400 mEq totales en 24 horas. La dosis en niños no debe exceder 3 mEq/kg/día. REACCIONES ADVERSAS Hiperpotasemia, que se manifiesta por parestesias en las extremidades, confusión mental, debilidad o pesadez de los miembros, parálisis flácida, arritmias, bloqueo cardíaco, posible paro cardíaco, cambios en el ECG, hipotensión, náuseas, vómitos, dolor 2ed. 2004 * Formulario TerapÈutico Nacional 61 que contienen potasio, aumenta el riesgo de hiperpotasemia. Usado en combinación con digitálicos, incrementa la cardiotoxicidad. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con insuficiencia renal severa, insuficiencia adrenocortical (enfermedad de Addison), deshidratación aguda, hiperpota-semia y daño tisular extenso (quema-duras severas). Los pacientes geriá-tricos son más susceptibles al desa-rrollo de la hiperpotasemia. FARMACOLOGÍA Aporta los iones sodio y cloruro necesarios para el restablecimiento y mantenimiento del balance hidro-electrolítico y de la presión osmótica de los fluidos corporales. El sodio, en asociación con cloruro y bicarbonato, participa en la regulación del equilibrio ácidobase. SOBREDOSIS La sobredosis produce hiperpotasemia. Las medidas de tratamiento incluyen vigilancia permanente del ECG y determinación de los niveles séricos de potasio. En pacientes con niveles séricos mayores a 6,5 mEq/L, administrar bicarbonato de sodio 40-60 mEq por infusión IV en 5 minutos y repetir cada 10-15 minutos mientras persistan las anormalidades en el ECG. En pacientes con ausencia de onda P o con ampliación del complejo QRS (si no reciben terapia digitálica), administrar gluconato de calcio 0,5-1 g (5-10 ml de solución al 10%) IV en 2 minutos para antagonizar la cardiotoxicidad del potasio. Administrar 300500 ml/hora de una solución de glucosa al 10% con 10-20 U/L de 62 CLORURO DE SODIO FARMACOCINÉTICA No aplicable. INDICACIONES Como fuente de sodio, cloruro y agua para hidratación en condiciones carac-terizadas por depleción aguda del lí-quido extracelular o deshidratación severa asociada a hiponatremia e hipocloremia (diarrea, vómito, sudora-ción profusa, hemorragia masiva, que-maduras severas o shock hipovolémi-co). R…GIMEN DE DOSIFICACI”N Se administra por vía IV. La dosis debe ajustarse a las necesidades individuales de cada paciente y en función de su edad, peso, condición clínica, balance hidro-electrolítico y equilibrio ácido-base. Para el cálculo del déficit de sodio corporal se emplea la fórmu- Formulario Terapéutico Nacional * 2ed. 2004 - Agua Corporal Total = Peso Corporal x 0,6 y sin exceder la velocidad de 100 ml/hora. Un litro de solución de cloruro de sodio al 0,9% aporta 154 mEq de sodio y 154 mEq de cloruro; 20 ml de la solución hipertónica (20%) aportan 68,4 mEq de sodio y 68,4 mEq de cloruro. SOBREDOSIS B Suspender la infusión, evaluar al paciente e instituir tratamiento sintomático y de soporte. INFORMACI”N AL PACIENTE REACCIONES ADVERSAS Hipervolemia, manifestaciones febriles, trombosis venosa o flebitis e infección en el sitio de inyección. Dosis excesivas pueden resultar en hipernatremia e hipercloremia. La hipercloremia puede generar pérdida de bicarbonato y, en consecuencia, acidosis metabólica. Si se administra muy rápidamente o en exceso puede agravar la insuficiencia cardiaca y causar edema pulmonar. INTERACCIONES No aplicable. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipernatremia y retención de fluidos. Usar con precaución en pacientes con insuficiencia cardiaca congestiva, insuficiencia circulatoria, alteración de la función renal, hipoproteinemia, enfermedad hepática, hipervolemia, obstrucción del tracto urinario y en pacientes sometidos a terapia con fármacos que producen retención de CLORURO DE SODIO / GLUCOSA FARMACOLOGÍA Ver monografías individuales de CLO-RURO DE SODIO y GLUCOSA. FARMACOCINÉTICA Ver monografías individuales de CLO-RURO DE SODIO y GLUCOSA. INDICACIONES Fuente de agua, calorías y electrolitos en el manejo de condiciones que cursan con depleción aguda o en caso de incremento en la demanda de tales elementos. R…GIMEN DE DOSIFICACI”N Las dosis dependen de los requerimientos particulares de cada paciente según su edad, peso, equilibrio ácidobase, balance hidro-electrolítico y con-dición clínica. REACCIONES ADVERSAS Sobrecarga de fluidos, edema, alteraciones hidro-electrolíticas y reacciones vinculadas a la técnica de 2ed. 2004 * Formulario TerapÈutico Nacional 63 INTERACCIONES Ver monografías individuales de CLO-RURO DE SODIO y GLUCOSA. dada a glucosa en los tejidos y posteriormente metabolizada a dióxido de carbonoy agua. CONTRAINDICACIONES/ PRECAUCIONES Ver monografías individuales de CLO-RURO DE SODIO y GLUCOSA. INDICACIONES Expansión de volumen en el tratamiento del shock hipovolémico por cirugía u otros traumas, hemorragia o quemaduras severas. SOBREDOSIS Ver monografías individuales de CLO-RURO DE SODIO y GLUCOSA. INFORMACI”N AL PACIENTE DEXTRÁN (DEXTRANO 70) FARMACOLOGÍA Polímero sintético de glucosa con peso molecular promedio de 70.000 daltons y propiedades coloidosmóticas similares a las de la albúmina. La administración IV del dextrán produce un efecto expansor del volumen plasmático como resultado del transporte de fluidos desde los espacios intersticiales al compartimiento vascular. FARMACOCINÉTICA Produce un efecto expansor máximo a los 60 minutos de la infusión IV, el cual da lugar a un incremento de la volemia y a una mejoría substancial del estado hemodinámico que persiste hasta por 24 horas. Se distribuye rápidamente a través del sistema vascular y se excreta en un 50% intacto por la 64 R…GIMEN DE DOSIFICACI”N Se administra solamente por infusión IV, en soluciones al 6%. Las dosis y la velocidad de infusión deben individualizarse de acuerdo a la magnitud de la pérdida de fluido y del grado de hemoconcentración resultante. En adultos y niños, la dosis total diaria no debe exceder 1,2 g/kg (20 ml/kg) durante las primeras 24 horas, ni 0,6 g/kg (10 ml/kg) en los días subsiguientes. Para una expansión rápida del volumen en adultos, se recomienda iniciar con 30 g (500 ml) a una velocidad de 1,2-2,4 g/minuto (20-40 ml/minuto). La dosis no debe exceder 20 ml/kg de peso diariamente. Si la terapia continúa por más de 24 horas, no debe exceder 10 ml/kg/día y por no más de 5 días. REACCIONES ADVERSAS Gastrointestinales: Náuseas y vómitos. Genitourinarias: Estasis y bloqueo tubular, aumento de la viscosidad urinaria, oliguria, anuria. Hematológicas: Disminución de los niveles de hemoglobina (con dosis de 15 ml/kg), Formulario Terapéutico Nacional * 2ed. 2004 congestión nasal, sibilancias, disnea e hipotensión severa. Otras: Tromboflebitis, fiebre, artralgia, congestión nasal, sobrecarga de fluido, defecación involuntaria. INTERACCIONES La combinación con heparina, aspirina, abciximab, trombolíticos y warfarina produce un efecto aditivo que puede conducir a hemorragias severas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al dextrán y en pacientes con defectos hemostáticos, edema pulmonar, insuficiencia cardiaca congestiva y falla renal severa con oliguria o anuria. Previo a la administración se debe verificar y corregir, si fuese necesario, el balance hidro-electrolítico y posibles deficiencias de los fac-tores de coagulación. Al inicio de la infusión, se recomienda vigilar la presión venosa central a objeto de prevenir la ocurrencia de una sobrecarga de fluido. Durante la administración se debe evitar la caída del hematocrito por debajo del 30%. Se debe vigilar constantemente al paciente y prestar especial atención a la posibilidad de reacciones de hipersensibilidad, falla renal o sangramiento. Usar con precaución en pacientes con insuficiencia embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales para la madre superen al posible riesgo para el feto. B No se conoce la seguridad del uso del dextrán durante la lactancia. SOBREDOSIS La sobredosificación puede provocar un estado de excesiva expansión de volumen y complicaciones congestivas. En tales casos se debe detener inmediatamente la infusión y practicar medidas correctivas, según la circunstancia clínica. EMULSIONES GRASAS (LÍPIDOS) FARMACOLOGÍA Constituyen emulsiones de partículas grasas, en concentraciones de 10 y 20%, compuestas por mezcla de triglicéridos naturales de ácidos grasos no saturados. Se emplean como fuente de calorías y de ácidos grasos esenciales en pacientes que requieren soporte nutricional. FARMACOCINÉTICA Luego de la administración IV, los trigli-céridos emulsificados son hidrolizados en la sangre a ácidos grasos libres y glicerol por la acción de las lipasas. Los ácidos grasos libres se distribu-yen a los tejidos corporales (prin-cipalmente al músculo 2ed. 2004 * Formulario TerapÈutico Nacional 65 utilizado para la síntesis de grasa corporal. La vida media de eliminación plasmática es de 30-60 minutos. INDICACIONES Como parte del soporte nutricional en el tratamiento de condiciones que cursan con deficiencia calórica y de ácidos grasos esenciales. R…GIMEN DE DOSIFICACI”N Se administran por vía IV como parte de una nutrición parenteral total en dosis que dependen de la edad del paciente, de su estado nutricional y metabólico y de su tolerancia venosa. La dosis usual en adultos no debe exceder de 2,5 g/kg/día, que equivale a 60% de la ingesta calórica diaria; el 40% restante lo aportan los carbohidratos y los aminoácidos. La dosis usual en niños no debe exceder los 3 g/kg/día. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y en aquellos con trastornos del metabolismo lipídico (hiperlipidemia grave, nefrosis lipoide o pancreatitis asociada a hiperlipidemia). Usar con suma precaución en pacientes con insuficiencia hepática, enfermedad pulmonar y trastornos de la coagulación. Durante el tratamiento se debe realizar pruebas periódicas de la coagulación sanguínea y de la función hepática. Además, se debe determinar el perfil lipídico. SOBREDOSIS Suspender la infusión, evaluar al paciente e instituir tratamiento sintomático y de soporte. FOSFATO MONOBÁSICO DE POTASIO FARMACOLOGÍA El potasio es el principal catión del líquido extracelular y elemento esencial para el mantenimiento del equilibrio ácido-base, isotonicidad y característi-cas electrodinámicas de la célula. Es un activador importante de numerosas reacciones enzimáticas y fundamen-tal para el desarrollo de diversos pro-cesos fisiológicos como: La transmi-sión del impulso nervioso, la contrac-ción cardiaca y de la musculatura lisa y esquelética, la Formulario Terapéutico Nacional * 2ed.renal, 2004la síntesis de proteínas función REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos. Hematológicas: Hipercoagulabilidad, trombocitopenia en recién nacidos (ra-ra), trombocitopenia, leucocitosis y leucopenia (tardías). Hepáticas: Aumentos transitorios en las pruebas de función hepática y hepatomegalia (tardías). Neurológicas: Cefalea, somnolencia, mareos, presión en los ojos, convulsiones focales (tardías). Reacciones de hipersensibilidad respiratorias: Disnea, cianosis. Otras: Enroje66 coagulante a la warfarina. FARMACOCINÉTICA Los niveles fisiológicos de potasio se ubican en el rango de 3,5-5,5 mEq/L. Se excreta en un 85-90% por vía renal y el resto por las heces. mentos o suplementos alimenticios que contienen potasio aumenta el riesgo de hiperpotasemia. En combinación con digitálicos, incrementa la B cardiotoxicidad. INDICACIONES Hipopotasemia. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con insuficiencia renal severa, insuficiencia adrenocortical (enfermedad de Addison), deshidratación aguda, hiperpotasemia y daño tisular extenso (quemaduras severas). Los pacientes geriátricos son más susceptibles al desarrollo de la hiperpotasemia. R…GIMEN DE DOSIFICACI”N Las dosis deben individualizarse de acuerdo a los requerimientos particulares de cada paciente según su condición clínica y respuesta a la terapia, con vigilancia frecuente del ECG y de los niveles séricos. El fosfato monobásico de potasio aporta 7,3 mEq/g de ión potasio. Se administra por vía IV, previamente diluido. La inyección directa del producto sin diluir puede provocar la muerte en forma instantánea. En pacientes adultos con un nivel de potasio sérico mayor que 2,5 mEq/L, usar soluciones con con-centración de potasio de 40 mEq/L y administrar a una velocidad de 10 mEq/hora, sin exceder la cantidad de 150 mEq totales en 24 horas. Si el nivel sérico es menor que 2 mEq/L, usar concentraciones de 80 mEq/L e infundir a una velocidad de 40 mEq/hora, sin exceder la cantidad de 400 mEq totales en 24 horas. La dosis en niños no debe exceder 3 mEq/kg/día. SOBREDOSIS La sobredosis produce hiperpotasemia. Las medidas de tratamiento incluyen vigilancia permanente del ECG y determinación de los niveles séricos de potasio. En pacientes con niveles séricos mayores de 6,5 mEq/L administrar bicarbonato de sodio 40-60 mEq por infusión IV en 5 minutos y repetir cada 10-15 minutos mientras persistan las anormalidades en el ECG. En pacientes con ausencia de onda P o ampliación del complejo QRS (si no reciben terapia digitálica) ad-ministrar gluconato de calcio 0,5-1 g (5-10 ml de solución al 10%) IV en 2 minutos para antagonizar la cardiotoxicidad del potasio. Administrar 300500 ml/hora de solución de glucosa al REACCIONES ADVERSAS 2ed. 2004 * Formulario TerapÈutico Nacional 67 INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso GLUCOSA (DEXTROSA) FARMACOLOGÍA Azúcar hidrosoluble en solución a diversas concentraciones, empleada por vía IV como fuente de calorías y fluidos en pacientes con condiciones de aumento de la demanda o como parte de una nutrición parenteral total. La administración de glucosa aumenta la glicemia, induce diuresis, disminuye las pérdidas de nitrógeno y proteína, promueve la deposición de glucógeno y previene la cetosis. FARMACOCINÉTICA Luego de la administración IV se distribuye a los tejidos corporales donde se almacena. Se metaboliza a dióxi-do de carbono y agua liberando ener-gía (3,4 kcal/g). Se elimina por filtra-ción glomerular pero es reabsorbida en un 90% en los túbulos renales. Al superarse el umbral de reabsorción tubular se elimina en la orina. INDICACIONES Manejo de condiciones que cursan con depleción de carbohidratos y fluidos en las que la reposición por vía oral resul-ta inadecuada o insuficiente y como fuente de carbohidratos en la nutrición parenteral total. 68 administración venosa central. La escogencia de la solución, según su concentración, varía con la condición a tratar: - Para reemplazo calórico y de fluidos: Soluciones al 2,5, 5 y 10%. Para reemplazo calórico con mínimo de fluidos: Solución al 20%. - Para hipoglicemia aguda en neonatos y niños mayores: Solución al 25%. - Para hipoglicemia inducida por insulina (hiperinsulinemia o shock insulínico): Solución al 50%. Las dosis dependen de las necesidades individuales de cada paciente y varían con el peso, la edad, la glicemia y la condición clínica. La dosis usual en adultos para el tratamiento de la hipo-glicemia es de 10- 25 g, en lactantes y niños mayores: 0,5-0,6 g/kg/dosis (10-12 ml de solución al 25%) y en neo-natos: 0,25- 0,5 g/kg/dosis (5-10 ml de solución al 25%). La velocidad máxima de infusión, sin producir glicosuria, es de 0,5 g/kg/hora. REACCIONES ADVERSAS Hiperglicemia, glicosuria, sobrecarga de fluidos y reacciones vinculadas a la técnica de administración, que incluyen fiebre, infección en el sitio de inyección, extravasación y necrosis tisular. Puede ocurrir un síndrome Formulario Terapéutico Nacional * 2ed. 2004 nicas se ha descrito casos de irritación venosa, flebitis y trombosis. La suspensión brusca o repentina de una solución altamente concentrada puede provocar una hipoglicemia de rebote. INTERACCIONES Si se administra simultáneamente con sangre a través de la misma línea de infusión puede ocurrir hemólisis. CONTRAINDICACIONES/ PRECAUCIONES No usar soluciones concentradas en pacientes con hiperglicemia o con coma diabético, anuria, hemorragia intracraneana o intraespinal, ni en presencia de delirium tremens. Usar con precaución en pacientes con diabetes o intolerancia a los carbohidratos. Posterior a la retirada abrupta de una solución altamente concentrada, se debe administrar una solución al 5 o al 10% para evitar la hipoglicemia de rebote. La hiperglicemia inducida por la solución de glucosa puede acentuar el daño neurológico en pacientes con isquemia cerebral aguda. Durante el uso prolongado pueden ocurrir déficits electrolíticos, particularmente de pota-sio y fosfato. Es necesario controlar los electrolitos. SOBREDOSIS Detener la administración, evaluar al paciente e instituir tratamiento sintomático y de soporte. Considerar la SULFATO DE MAGNESIO (Hipomagnesemia) FARMACOLOGÍA El magnesio es el cuarto catión más B abundante en el organismo y constituye un electrolito esencial como cofactor de numerosos sistemas enzimáticos que incluyen los relacionados con la transferencia de grupos fosfato, las reacciones que involucran ATP, los procesos de replicación y transcripción de DNA y translación de RNA-mensajero, mecanismos de estabilización de membrana, conducción nerviosa, transporte de hierro y actividad de los canales de calcio. FARMACOCINÉTICA Luego de la administración IV, se une a las proteínas plasmáticas en 2535% y se distribuye ampliamente a todos los tejidos y fluidos corporales. Se excreta, en su mayoría, por filtración glomerular y, en escasa proporción, por las heces. INDICACIONES Prevención y tratamiento de estados de hipomagnesemia. R…GIMEN DE DOSIFICACI”N Se administra por vía IV y por vía IM. Se usa como sal heptahidratada y la dosis se expresa en términos de magnesio, teniendo en cuenta que 1 g de 2ed. 2004 * Formulario TerapÈutico Nacional 69 mente 4-24 mEq/día) y de 0,25-0,60 mEq/kg/día (2-10 mEq/día) para niños. - En la hipomagnesemia leve a moderada: 1 g (8,1 mEq) IM cada 6 horas hasta por 4 dosis. En casos severos: 5 g (40,5 mEq) en solución de dextrosa al 5% o en solución fisiológica de NaCl al 0,9% por infusión IV en 3 horas o, alternativamente, 2 mEq/kg IM en 4 horas. REACCIONES ADVERSAS El efecto más importante es la hipermagnesemia que se puede manifestar (según los niveles séricos) con sudora-ción profusa, hipotensión, pérdida de los reflejos, parálisis flácida, hipoter-mia, colapso circulatorio, depresión cardiaca, central y respiratoria, coma y, en casos graves, muerte. La hiper-magnesemia se presenta con niveles séricos superiores a 2,5 mEq/L. INTERACCIONES Puede potenciar el efecto de los bloqueantes neuromusculares y la depresión central de los barbitúricos, opiáceos y otros depresores empleados en la anestesia general. Ejerce efecto aditivo con el nifedipino. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con bloqueo cardíaco, lesión miocárdica y 70 debe evitar su uso antes del parto (al menos 24 horas) para evitar la hipermagnesemia neonatal. SOBREDOSIS Suspender la administración del medicamento. Determinar los niveles séricos de magnesio, asistir la respiración, administrar gluconato de calcio 5-10 mEq por vía IV (solución al 10%) para revertir la depresión respiratoria y el bloqueo cardíaco y forzar la diuresis con manitol. En casos graves, practicar diálisis peritoneal o hemo-diálisis. SOLUCIÓN PARA NUTRICIÓN PARENTERAL FARMACOLOGÍA Están constituidas por mezclas de aminoácidos, carbohidratos, lípidos, electrolitos, vitaminas y minerales (oligoelementos) para administración IV que, en proporciones apropiadas, se emplean para la alimentación de pacientes en quienes la utilización de la vía oral no es posible o resulta inadecuada para sus requerimientos nutricionales. Los aminoácidos aportan los substratos necesarios para la síntesis de proteínas, reducen la tasa de catabolismo proteico, promueven la cicatrización de heridas y actúan como tampones en el líquido extracelular e intracelular. Los carbohidratos (glucosa) aportan calorías, reducen la Formulario Terapéutico Nacional * 2ed. 2004 vitaminas y minerales compensan las pérdidas sensibles e insensibles de tales elementos. FARMACOCINÉTICA No aplicable. INDICACIONES Nutrición de pacientes con condiciones que limitan, contraindican o imposibilitan la alimentación por vía oral y estados hipercatabólicos o de depleción severa que cursan con un incremento significativo de los requerimientos proteicos y energéticos. R…GIMEN DE DOSIFICACI”N Varía con cada paciente, según sus características y condición patológica particular. Para definir la dosis total diaria se debe determinar los requerimientos proteicos, calóricos, de agua, electrolitos, vitaminas y oligoelementos en forma individual y practicar ajustes periódicos de acuerdo a la respuesta clínica y metabólica. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad a alguno de los constituyentes de la solución y en pacientes con dis- B minución del volumen de sangre circulante, insuficiencia renal y/o hepática severa, desequilibrio hidro-electrolítico y desbalance ácido-base. Antes de iniciar la nutrición IV, y durante su administración, se debe realizar hematología completa y determinación periódica de electrolitos, gases arteriales, glicemia, creatinina sérica, proteínas totales, lípidos, ácido úrico, enzimas hepáticas, bilirrubina, nitrógeno ureico sanguíneo, amonemia, cloremia, hierro sérico, tiempo de protrombina, osmolaridad de la orina y cetonuria. Usar con precaución en pacientes con alteración de la función renal y/o hepática leve a moderada y durante el embarazo. Evitar la sobrecarga circulatoria. Tomar medidas para evitar infecciones. SOBREDOSIS REACCIONES ADVERSAS Cardiovasculares: Flebitis, trombosis venosa. Gastrointestinales: Náuseas. Hepáticas: Elevación de los niveles de las enzimas hepáticas. Otras: Fiebre, rubor, alcalosis y acidosis metabólica, hipofosfatemia, hipocalcemia, hiperglicemia, glicosuria, hipo e hipermagnesemia, diuresis osmótica, deshidratación, hipervolemia, hiperamo- SOLUCIÓNES ELECTROLÍTICAS (RINGER LACTATO) FARMACOLOGÍA Solución estéril no pirogénica de cloruro de sodio, cloruro de potasio, cloruro de calcio y lactato de sodio en agua para inyectables. Contiene 130 mEq/L de sodio, 4 mEq/L de potasio, 2ed. 2004 * Formulario TerapÈutico Nacional 71 cloruro y 26-29 mEq/L de lactato; tiene un pH de 6-7,5. Aporta fluido y electrolitos para restituir pérdidas, a la vez que constituye una fuente de bicarbonato a partir de la transformación hepática del lactato, para actuar como alcalinizante en condiciones de acidosis; de esta manera eleva el pH sanguíneo. FARMACOCINÉTICA Luego de la administración IV, los constituyentes de la solución se distribuyen a los tejidos y fluidos corporales. En el hígado, el lactato se transforma por oxidación en bicarbonato. En condiciones de adecuada actividad oxidativa, el proceso ocurre en 1-2 horas. INDICACIONES Estados de deshidratación asociados a acidosis metabólica leve a moderada. R…GIMEN DE DOSIFICACI”N Se administra por infusión IV. La dosis es individualizada y dependiente del grado de deshidratación, severidad de la acidosis, parámetros de laboratorio y condición clínica del paciente. Sin embargo, usualmente se administra 1,5 a 3 litros (2-6% del peso corporal), infundido durante 18 a 24 horas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con acidosis láctica, acidosis severa (en la que se requiere una rápida reposición del bicarbonato sérico), hipernatremia y en condiciones en las que una sobrecarga de sodio podría resultar riesgosa. Usar con precaución en pacientes con alcalosis metabólica o respiratoria, insuficiencia hepática severa, tratamiento prolongado con esteroides y edema de origen cardíaco, renal o hepático. Durante la terapia se debe vigilar periódicamente el balance hidro-electrolítico y el equilibrio ácidobase. SOBREDOSIS Suspender la administración del medicamento y practicar medidas de corrección del balance hidro-electroSOLUCIÓNES ELECTROLÍTICAS (RINGER) FARMACOLOGÍA Solución estéril no pirogénica de cloru-ro de sodio, cloruro de potasio y cloruro de calcio en agua para inyectables. Contiene 147,5 mEq/L de sodio, 4 mEq/L de potasio, 4,5 mEq/L de calcio y 156 mEq/L de cloruro; tiene un pH de 5-7,5 y una osmolaridad de 309 mOsm/L. Aporta fluido y electrolitos en situaciones deficitarias. REACCIONES ADVERSAS Alcalosis metabólica, sobrecarga FARMACOCINÉTICA hidro-electrolítica y reacciones 72 Formulario Terapéutico Nacional * 2ed. 2004 INDICACIONES Estados de deshidratación con pérdida moderada de electrolitos. R…GIMEN DE DOSIFICACI”N Se administra por infusión IV. La dosis varía según el grado de deshidratación y la condición clínica del paciente. La dosis usual en adultos es de 500-3000 ml (60-80 gotas por minuto) y en niños 50 ml/kg/día (6080 gotas por minuto). REACCIONES ADVERSAS Reacciones asociadas a la técnica de administración (infección en el sitio de inyección, fiebre, flebitis, trombosis y extravasación). En dosis elevadas puede provocar una sobrecarga hidroelectrolítica y acidosis hiperclorémica. INTERACCIONES Puede potenciar la toxicidad de los digitálicos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en estados edematosos en pacientes con insuficiencia cardíaca, hepática o renal. Usar con precaución en pacientes con hipertensión. Vigilar periódicamente el balance hidro-electrolítico. SOBREDOSIS SOLUCIONES PARA DIÁLISIS PERITONEAL FARMACOLOGÍA Soluciones electrolíticas estériles formuladas en concentraciones similares B a las del plasma, empleadas para el reemplazo o intercambio de electrolitos y remoción de líquidos, metabolitos y/o sustancias tóxicas del organismo. El intercambio iónico se realiza por difusión a favor del gradiente de concentración que se establece entre el compartimiento sanguíneo y la solución instilada y retenida en el espacio peritoneal, a través de la membrana filtrante que constituye la red vascular peritoneal. El exceso de líquido es removido por el gradiente osmótico generado por la presencia de glucosa en la solución. FARMACOCINÉTICA No aplicable. INDICACIONES Corrección del desequilibrio hidroelectrolítico y metabólico secundario a la insuficiencia renal aguda o crónica, tratamiento de intoxicaciones o sobre-dosificación con medicamentos diali-zables, edema intratable, hipercalce-mia, hiperpotasemia, azotemia, ure-mia y coma hepático. Las soluciones para hemodiálisis están constituidas por sodio, cloruro, bicarbonato (o lactato como precursor), calcio, magnesio y 2ed. 2004 * Formulario TerapÈutico Nacional 73 concentración de glucosa puede variar dependiendo del objetivo final del procedimiento: - Soluciones con glucosa al 1,5% (15 g/L): Se usan para la remoción de desechos metabólicos, pero no de fluidos. - Soluciones con glucosa al 2,5% (25 g/L): Se usan para filtrar desechos metabólicos y fluidos. - Soluciones con glucosa al 4,25% (42,5 g/L): Se usan sólo para remover fluidos. La escogencia de la solución, según su composición, dependerá de la condi-ción clínica del paciente y del objetivo terapéutico deseado. R…GIMEN DE DOSIFICACI”N La mayoría de los adultos toleran la infusión peritoneal de 2 litros de solución. La mayoría de los niños toleran hasta 45 ml/kg; sin embargo, se recomienda iniciar la infusión con volúmenes de 15-20 ml/kg. REACCIONES ADVERSAS Dolor abdominal, hemorragia gastrointestinal, peritonitis, íleo paralítico y her-nia abdominal o inguinal. En pacientes crónicos, las soluciones con concen-traciones elevadas de glucosa pueden generar hiperglicemia, hiperlipidemia e incrementos importantes en el peso corporal. renal son susceptibles a la remoción por la diálisis y, en consecuencia, podría ser necesario practicar ajustes en la dosificación de las mismas. La hipopotasemia generada por el procedimiento puede predisponer al paciente a la toxicidad de la digoxina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con cirugía abdominal reciente. Usar con precaución en pacientes con compromiso pulmonar, diabetes, enfermedad inflamatoria intestinal, perforación intestinal o colostomía y trastornos de la coagulación. SOBREDOSIS No aplicable. SOLUCIONES PARA HEMODIÁLISIS FARMACOLOGÍA Son soluciones electrolíticas estériles formuladas en concentraciones simi-lares a las del plasma, empleadas para el reemplazo o intercambio de electro-litos y remoción de líquidos, metaboli-tos y/o sustancias tóxicas del organis-mo mediante el empleo de un equipo automatizado conocido como dializador. El intercambio iónico ocurre por di-fusión a favor del gradiente de concentración que se establece entre el compartimiento sanguíneo y la solu- INTERACCIONES 74 Formulario Terapéutico Nacional * 2ed. 2004 electrolitos). El exceso de líquido es removido por ultrafiltración generada por los gradientes de presión. FARMACOCINÉTICA No aplicable. INDICACIONES Corrección del desequilibrio hidroelectrolítico y metabólico secundario a la insuficiencia renal aguda o crónica, tratamiento de intoxicaciones o sobre-dosificación con fármacos dializables, edema intratable, hipercalcemia, hi-perpotasemia, azotemia, uremia y co-ma hepático. Las soluciones para he-modiálisis están constituidas por so-dio, cloruro, bicarbonato (o un precur-sor), calcio, magnesio, glucosa y, ocasionalmente, potasio. La composición puede variar con la incorporación o exclusión de elementos que, según la circunstancia clínica, resulte conveniente conservar o no en el organismo. Por esta razón existe lo siguiente: - Soluciones con glucosa y con potasio - Soluciones con glucosa y sin potasio - Soluciones sin glucosa y con potasio - Soluciones sin glucosa y sin potasio La escogencia de la solución (según su composición) dependerá de la situación clínica y del objetivo terapéutico deseado. Las soluciones con glucosa y con potasio, por ejemplo, se indican en condiciones en las que no es Arterio-venosa, trombosis. Gastrointestinales: Vómito, náuseas. Hematológicas: Hemorragias. Reacciones de hipersensibilidad: ReacB ciones alérgicas atribuibles a los detergentes o agentes usados para la limpieza del equipo. Otras: Calambres musculares, embolismo aéreo. INTERACCIONES Aunque no existen interacciones atribuibles de manera directa a la solución, es importante tener en cuenta que los medicamentos de bajo peso molecular, alta solubilidad en agua, escaso volumen de distribución, baja unión a proteínas y elevada excreción renal son susceptibles a la remoción por la hemodiálisis y, en consecuencia, podría ser necesario practicar ajustes en la dosificación de las mismas. La hipopotasemia generada por el pro-cedimiento podría predisponer al pa-ciente a la toxicidad de la digoxina. CONTRAINDICACIONES/ PRECAUCIONES Usar con precaución en pacientes con enfermedad cardiovascular inestable, diabetes y en individuos con trastornos hemorrágicos. SOBREDOSIS No aplicable. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso 75 2ed. 2004 * Formulario TerapÈutico Nacional C01 TERAPIA CARDÕACA Amiodarona Atropina* (Terapia Cardíaca) Digoxina Dinitrato de Isosorbida Dobutamina Dopamina Lidocaína (Antiarrítmico) Mexiletina Trinitrato de Glicerilo (Nitroglicerina) C03 DIUR…TICOS Espironolactona Furosemida Hidroclorotiazida Manitol* C04 VASODILATADORES PERIF…RICOS Fentolamina Pentoxifilina C05 VASOPROTECTORES Prednisolona / Anestésico Local (Prednisolona Capronato / Anestésico Local) C07 AGENTES BETA-BLOQUEANTES Atenolol (Antihipertensivo y Tratamiento de Angina de Pecho) Metoprolol C SISTEMA CARDIOVASCULAR C02 ANTIHIPERTENSIVOS Clonidina Metildopa (Alfametildopa) Nitroprusiato (Nitroprusiato Sódico) C C08 BLOQUEANTES DE CANALES DE CALCIO Amlodipino (Amlodipina Besilato) Nifedipino Nimodipino Verapamilo C09 AGENTES QUE ACT⁄AN SOBRE EL SISTEMA RENINA-ANGIOTENSINA Enalapril Losartán 2ed. 2004 * Formulario TerapÈutico Nacional 77 C10 AGENTES QUE REDUCEN LOS LÕPIDOS S…RICOS Gemfibrozilo Simvastatina 78 Formulario Terapéutico Nacional * 2ed. 2004 AMIODARONA FARMACOLOGÍA Antiarrítmico clase III, con características electrofisiológicas de las otras clases de la clasificación Vaugham Williams. Disminuye la velocidad de conducción intraventricular por bloqueo de los canales de sodio, disminuye la frecuencia cardiaca e impide la conducción en el nodo sinoauricular por bloqueo de los receptores betaadrenérgicos y de los canales de calcio. Prolonga la repolarización auricular y ventricular por bloqueo de los canales de potasio. El efecto electrofisiológico más marcado de la amiodarona es la prolongación de la duración del potencial de acción, del tiempo de repolarización y del período refractario de todas las estructuras cardíacas, in-cluyendo el nodo sinusal, el nodo aurí-culo-ventricular, las aurículas, el haz de His, los ventrículos y las fibras de Purkinje. sión en casi todos los tejidos. Se metaboliza en el hígado, transformándose en N-desetilamiodarona, metabolito activo que se excreta, junto con el fármaco intacto, por vía biliar incluyendo la circulación enterohepática. La excreción renal es menor C que 1%. La vida media de eliminación se ubica en el rango de 26 a 107 días. Para el metabolito, la vida media de eliminación es, aproximadamente, 6 días. INDICACIONES Taquicardia y fibrilación ventricular severas que no responden al tratamiento con otros agentes. RÉGIMEN DE DOSIFICACIÓN Para el manejo de la arritmia ventricular por vía oral en adultos, iniciar con 800-1600 mg/día por 1-3 semanas, seguido por una disminución gradual de la dosis hasta 400 mg/día y mantener esta dosis por el tiempo que sea necesario. En niños, iniciar con 5-10 FARMACOCINÉTICA mg/kg/día por 10-14 días, seguido La absorción en el tracto gastrointes- por una disminución gradual hasta tinal es lenta e incompleta con una bio- 2,5-6 mg/kg/día y mantener por el disponibilidad del 50% (35-65%). Des- tiempo que sea necesario. Por vía IV, pués de iniciada la terapia oral, produ- iniciar con una infusión rápida de 150 ce un inicio de acción a las 1-3 sema- mg en 10 minutos (15 mg/minuto), nas, pero puede requerirse varios me- seguido por una infusión lenta de 360 ses para alcanzar una respuesta tera- mg en 6 horas (1 mg/minuto) y péutica óptima. Una vez en la sangre después mante-ner al paciente con se une a las proteínas plasmáticas en 540 mg durante 18 horas (0,5 un 95,6%. Se distribuye ampliamente mg/minuto) para completar 24 horas 79 2ed. 2004 * Formulario TerapÈutico Nacional se puede observar en el siguiente cuadro: Duración de la terapia IV Dosis inicial por vía oral < 1 semana 1-3 semanas > 3 semanas 800-1600mg 400-800 mg 400 mg REACCIONES ADVERSAS Cardiovasculares: Insuficiencia cardiaca congestiva, arritmias cardíacas, disfunción del nodo sinoauricular, taquicardia ventricular paroxística, bloqueo cardíaco. Dermatológicas: Dermatitis solar, fotosensibilidad, pigmentación azul de la piel, alopecia, erupciones. Gastrointestinales: Náuseas y vómitos, constipación, anorexia, dolor abdominal. Hematológicas: Equimosis espontánea, alteraciones de la coagulación. Hepáticas: Alteración de las pruebas de función hepática, trastornos hepáticos no específicos. Neurológicas: Malestar, fatiga, temblor, movimientos involuntarios anormales, falta de coordinación, marcha anormal, ataxia, mareos, parestesias, insomnio, cefalea, trastornos del sueño, neuropatía o neuritis óptica. Respiratorias: Inflamación o fibrosis pulmonar, síndrome de distrés respiratorio del adulto. Tiroideas: Hipotiroidismo, hipertiroidismo. Otras: 80 digoxina, quinidina, teofilina, procainamida, warfarina, dextrometorfano, ciclosporina, fenitoína, propranolol, diltiazem, verapamil, disopiramida, fentanil y metotrexato, originando incrementos en los niveles plasmáticos, con el consecuente riesgo de toxicidad. En combinación con bloqueantes de los receptores betaadrenérgicos y de los canales de calcio, puede provocar hipotensión y bradicardia por efecto aditivo. La amiodarona puede disminuir la depuración hepática o renal de otros antiarrítmicos, especialmente la flecaimida, procainamida, quinidina. Con antiarrítmicos como la mexiletina, propafenona, quinidina, disopiramida y procainamida puede inducir torsades de pointes (torcida de punta). La cime-tidina aumenta los niveles séricos de la amiodarona. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, shock cardiogénico, disfunción del no-do sinusal severa, bloqueo AV de se-gundo y tercer grado y durante el em-barazo. Antes de iniciar el tratamiento, y periódicamente durante su desa-rrollo, se debe realizar exámenes de la función pulmonar, tiroidea, hepática y visual. Previo al inicio de la terapia, se debe verificar los Formulario Terapéutico Nacional * 2ed. 2004 no respondan a un solo agente o no respondan a la amiodarona. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Vigilar el ritmo cardíaco y la presión arterial. La bradicardia puede ser tratada con agonistas de los recep-tores beta-adrenérgicos y, en casos graves, con un marcapaso. La hipoten-sión y el shock cardiogénico se pueden tratar con medicamentos vasopreso-res, agentes inotrópicos y expansores de volumen, según la severidad del caso. Ni la amiodarona ni su metabolito son dializables. INFORMACI”N AL PACIENTE Evitar la exposición prolongada al sol. Informar de inmediato al médico si se ATROPINA (Terapia Cardiaca) el organismo y atraviesa la barrera hematoencefálica. Se une a proteínas plasmáticas en un 18%. Se metaboliza en el hígado, transformándose en productos inactivos que se excretan, junto a un 30-50% del fármaco intacto, principalmente por la orina. La eliminación C es bifásica, con una vida media de 2 horas en su fase inicial y de 12,5 horas en su fase terminal. INDICACIONES En la resucitación cardio-pulmonar para el tratamiento de la bradicardia sinusal con compromiso hemodinámico y de la asístole ventricular. En cirugía, para la prevención de efectos coli-nérgicos cardíacos (arritmias, hipo-tensión, bradicardia) inducidos por reflejo vagal, medicamentos anestési-cos o para la manipulación quirúrgica. RÉGIMEN DE DOSIFICACIÓN - Tratamiento de la bradicardia duFARMACOLOGÍA rante la resucitación cardio-pulmoVer: ATROPINA en la sección SISnar, en adultos: 0,5-1 mg IV, con TEMA DIGESTIVO Y METABOLISrepeticiones cada 3-5 minutos hasta MO. alcanzar la frecuencia cardiaca deseada. En la asístole ventricular: FARMACOCINÉTICA 1 mg IV, con repeticiones cada 3-5 Se absorbe bien por vía IM y SC. Desminutos si la asístole persiste. En pués de la administración IM produce niños: 0,02 mg/kg IV, con una respuesta anticolinérgica inicial repeticiones cada 5 minutos en caso sobre el corazón después de 5-40 necesario. minutos, la cual se hace máxima a los MedicaciÛn preoperatoria, en adul20-60 minutos. Por vía IV, el efecto tos y niños con más de 20 kg: 0,481 2ed. 2004 * Formulario TerapÈutico Nacional REACCIONES ADVERSAS Ver: ATROPINA en la sección SISTEMA DIGESTIVO Y METABOLISMO. INTERACCIONES Ver: ATROPINA en la sección SISTEMA DIGESTIVO Y METABOLISMO. CONTRAINDICACIONES/ PRECAUCIONES Ver: ATROPINA en la sección SISTEMA DIGESTIVO Y METABOLISMO. SOBREDOSIS Ver: ATROPINA en la sección SISTEMA DIGESTIVO Y METABOLISMO. INFORMACIÓN AL PACIENTE DIGOXINA FARMACOLOGÍA La digoxina es uno de los glicósidos cardíacos, digitálicos, que tienen efectos específicos comunes en el miocardio. Es extraída de las hojas de la Digitalis lanata. La digoxina inhibe la ATPasa de sodio y potasio, una enzima que regula la concentración intracelular de estos iones. La inhibición de la enzima lleva a un aumento de la concentración intracelular de sodio y así (por estimulación del intercambio sodiocalcio) a un aumento de la concentración intracelular de calcio. Los efectos beneficiosos resultan de acciones directas e indirectas sobre el 82 AV; 2) sensibilización de los barorreceptores, que resulta en un aumento de la actividad aferente del sistema simpático y una reducción en la actividad del sistema nervioso simpático y del sistema reninaangiotensina para cualquier incremento en la presión arterial media. Las consecuencias farmacológicas de estos efectos directos e indirectos son: 1) Un aumento en la fuerza y la velocidad de contracción (acción inotrópica positiva); 2) una disminución en la activación del sistema nervioso simpático y del sistema renina-angiotensina (efecto desactivante neurohumoral) y 3) disminución de la frecuencia cardiaca y la velocidad de conducción a través del nodo AV (efecto vagomimético). Los efectos en la insuficiencia cardiaca son mediados por el efecto inotrópico positivo y de desactivación neurohumoral, y en las arritmias auriculares se asocia al efecto vagomimético. El aumento en la actividad simpática puede ser un factor importante en la toxicidad de la digital. FARMACOCINÉTICA La biodisponibilidad oral es variable y dependiente de la formulación (6080% para las tabletas, 70-85% para los elíxires y 90-100% para las cápsulas). La presencia de comida en el estómago reduce la tasa de absorción Formulario Terapéutico Nacional * 2ed. 2004 en 1,5-4 horas. Los niveles séricos terapéuticos se ubican en el rango de 0,8-2 ng/ml y se consideran tóxicos por encima de ese valor. Se une a las proteínas plasmáticas en un 20-25% y se distribuye ampliamente a los tejidos. Se metaboliza parcialmente (16%) en el hígado por sistemas no dependientes del citocromo P450 , transformándose en metabolitos activos que se excretan, junto con el fármaco intacto, en un 57-80% por la orina y en un 6-8% por la bilis. La vida media de eliminación es de 1,3-2,2 días y se incrementa en pacientes con falla renal. emplea el 25-35% de la respectiva dosis oral o IV usada como carga. REACCIONES ADVERSAS Cardiovasculares: Bloqueo cardíaco en pacientes con trastornos de la conducción SA y AV. Altas dosis C pueden causar una variedad de trastornos del ritmo cardíaco, bloqueo de primero a tercer grado, incluyendo asístole; taquicardia auricular con bloqueo; disociación AV; ritmo nodal acelerado; contracciones ventriculares prematuras, taquicardia y fibrilación ventricular. Prolongación del segmento PR y depresión del segmento ST. Gastrointestinales: Ano-rexia, náuseas, vómitos y diarrea. INDICACIONES Tratamiento de la insuficiencia Hematológicas: Trombocitopenia. cardiaca congestiva y la fibrilación Neurológicas: Trastornos visuales (visión borrosa o visión amarilla y auricular. 'flashes' de luz), escotomas, cefalea, RÉGIMEN DE DOSIFICACIÓN debilidad, mareos, apatía, confusión La dosis oral y la dosis IV para y trastornos mentales (ansiedad, digitalización en adultos es de 10-15 depresión, delirio y alucinaciones), mcg/kg de peso magro/día divididos vértigo, parestesia, fotofobia, en 2-4 dosis, seguidos por dosis de d i p l o p í a . R e a c c i o n e s d e mantenimiento de 0,125-0,5 mg/día h i p e r s e n s i b i l i d a d : E r u p c i ó n como dosis única, con ajustes acordes maculopapular y, raras veces, otras con las necesidades individuales de reacciones en la piel. Otras: cada paciente, las concentraciones Ginecomastia. Las reacciones séricas del digitálico, la función renal y adversas en los niños se inician en la respuesta clínica. La dosis oral para forma diferente a los adultos. En digitalización en niños es como sigue: niños, la manifestación más temprana y frecuente es la aparición de arritmias Neonatos prematuros 20-30 mcg/kg cardíacas, con o sin bloqueo, y Neonatos a término 25-35 mcg/kg taquicardia nodal. Niños de 1-2 años 35-60 mcg/kg 83 2ed. 2004 * Formulario TerapÈutico Nacional alcaloides de la vinca) pueden disminuir la absorción de la digoxina. La anfotericina B, los corticosteroides, los laxantes, el glucagon y dosis elevadas de dextrosa pueden predisponer a la toxicidad digitálica por la hipokalemia que producen, al igual que las alteraciones electrolíticas causadas por diuréticos como la furosemida, el ácido etacrínico y las tiazidas. Los anticolinérgicos, la eritromicina, la claritromicina, el itraconazol, las tetraciclinas, la hidroxicloroquina, la indometacina, el alprazolam, la quinidina, la amiodarona, el verapamilo y el nifedipino pueden aumentar los niveles séricos de la digoxina; la penicilamina puede disminuirlos. Los glicósidos cardíacos son sinergísticos con las sales de calcio, los alcaloides de la rauwolfia, la efedrina, la epinefrina y, posiblemente, con otros agentes adrenérgicos. Algunas plantas pueden incrementar la toxicidad de la digoxina [ regaliz (Glycirrhiza glabra), ginseng siberiano (Eleutherococcus senticosus), ruda (Ruta graveolens), nuez de areca (Areca catechu), semilla hindú (Thevetia peruviana) pueden aumentar la toxicidad de la digoxina]. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y en casos de fibrilación ventricular. La 84 digoxina puede empeorar la condición. Los pacientes con hipertiroidismo, pericarditis constrictiva y miocardio-patía amiloide son, por lo general, resistentes a la digoxina. El hipotiroidismo puede reducir los requerimientos de digoxina. Los pacientes con infarto del miocardio o con enfermedad pulmonar severa pueden resultar inusualmente sensibles a los efectos arritmogénicos de la digoxina. Durante la terapia se debe realizar controles periódicos de la función renal, de los electrolitos séricos y del ECG. Puede ser deseable reducir la dosis de digoxina 1 a 2 días antes de la cardioversión eléctrica de la fibrilación auricular. Usar con precaución en pacientes ancianos, con falla renal, enfermedad pulmonar, hipertensión, embarazo y durante la lactancia. SOBREDOSIS Suspender la administración de digoxina. Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Después de 2 horas de la administración, no debe inducirse vómitos ni pasar una sonda debido a la posible inducción de un episodio vagal. Manejo sintomático y terapia de soporte. Vigilar el ECG, el balance hidro-electrolítico, el equilibrio ácido-base y los niveles Formulario Terapéutico Nacional * 2ed. 2004 dure la terapia, evitar el uso de otros medicamentos sin consultar previamente al médico. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial náuseas, vómitos, latidos irregulares, dificultad para respirar, pérdida del apetito, diarrea, DINITRATO DE ISOSORBIDA FARMACOLOGÍA Agente vasodilatador. Aunque el mecanismo de acción antianginosa no está totalmente aclarado, se cree que es debido a su transformación en óxido nítrico en el interior de la célula vascular, donde produce la activación del GMPc y un efecto relajante sobre la fibra muscular. La relajación de la musculatura lisa vascular produce, en consecuencia, una vasodilatación sistémica que disminuye el retorno venoso, la presión venosa de llenado (precarga), el volumen y la presión diastólica final del ventrículo izquierdo. La relajación arteriolar reduce la resistencia vascular periférica, la presión arterial sistólica y la presión arterial media (postcarga); también ocurre dilatación de las arterias coronarias. FARMACOCINÉTICA Se absorbe en un 19-96% (promedio 58%) por vía oral o sublingual. Con la administración sublingual, el efecto se minutos, se hace máximo en 30-60 minutos y se mantiene por 6-8 horas. Se une poco (4%) a las proteínas plasmáticas. El rápido desarrollo de tolerancia hace que la determinación de los niveles séricos carezca de importancia clínica. Se metaboliza C extensamente (80-90%) en el hígado, con un alto metabolismo de primer pasaje en el hígado, transformándose en metabolitos activos (5-mononitrato y 2-mononitrato) que se eliminan por la orina y en muy escasa proporción por las heces. La vida media de eliminación del fármaco cuando se administra por vía oral es de 4 horas y de 60 minutos cuando se administra por vía sublingual; la vida media de eliminación del metabolito 5mononitrato es de 4-5 horas y la del 2mononitrato de 1-3 horas. La eliminación de una dosis oral es casi completa (80-100%) a las 24 horas. INDICACIONES Prevención y tratamiento de la angina de pecho. RÉGIMEN DE DOSIFICACIÓN La dosis usual para iniciar el tratamiento en adultos con tabletas de liberación convencional es de 5-20 mg dos o tres veces al día, y como dosis de mantenimiento 10-40 mg cada 6-8 horas. Se recomienda un intervalo diario de 14 horas sin medicamento para minimizar la tolerancia. Si se usan 2ed. 2004 * Formulario TerapÈutico Nacional 85 ataque agudo, iniciar con 2,5-5 mg sublingual y repetir, en caso necesario, a intervalos de 10-15 minutos, sin exceder 3 dosis en 15-30 minutos. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, taquicardia, palpitaciones, vasodilatación cutánea, edema del tobillo, desmayos. Gastrointestinales: Náuseas, vómitos. Dermatológicas: Enrojecimiento, erupciones. Neurológicas: Cefalea (algunas veces pulsátil), mareos, debilidad. Reacciones de hipersensibilidad. Otras: Sensación de quemadura sublingual. INTERACCIONES El efecto vasodilatador puede resultar aditivo con el de otros agentes con propiedades vasodilatadoras o hipotensoras, incluido el alcohol. Marcada hipotensión ortostática cuando se usa con bloqueantes de los canales de calcio. Los inhibidores de la fosfodiesterasa tipo 5 potencian la acción hipotensora de los nitratos. puede desencadenar angina de rebote, por lo que se recomienda un retiro gradual del medicamento. El uso repetido puede conducir al desarrollo de tolerancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Vigilar los gases arteriales y la metahemoglobina. Si ocurre hipotensión, colocar al paciente en posición Trendelenburg y administrar fluidos intravenosos. Los agentes vasopresores están contraindicados. Si se produce metahemoglobina, el tratamiento de soporte es azul de metileno 1 a 2 mg/kg IV. INFORMACIÓN AL PACIENTE Administrar 1 hora antes o 2 horas después de las comidas. Evitar el consumo de alcohol mientras durante el tratamiento. Informar al paciente que la terapia puede producir dolor de cabeza, mareos y enrojecimiento de la piel. Advertir al paciente que en caso de que se presenten tales efectos, no debe alterar la dosificación, ni suspender el tratamiento; el dolor de cabeza por lo general responde a la aspirina o al acetaminofén. En caso de prescripción por vía sublingual enseñar al paciente que la tableta debe colocarse bajo la lengua, sin tragarse, al presentarse el primer signo de un ataque de angina y antes de desa- CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a los nitratos. Usar con precaución en pacientes con hipotensión severa, infarto agudo del miocardio con baja presión de llenado ventricular izquierdo, miocardiopatía hipertrófica, aumento de la presión 86 Formulario Terapéutico Nacional * 2ed. 2004 DOBUTAMINA FARMACOLOGÍA Catecolamina sintética que ejerce un efecto inotrópico positivo por estimulación directa y selectiva de los receptores beta1-adrenérgicos del corazón. Ejerce, además, un leve efecto agonista sobre receptores beta2 -(vasodilatadores) y alfa1-(vasoconstrictores) adrenérgicos, el cual, por ser balanceado, no incide de manera importante en la vasculatura sistémica. A diferencia de la dopamina no produce liberación de norepinefrina endógena. FARMACOCINÉTICA Después de la administración IV, produce una respuesta en 1-2 minutos, la cual se mantiene por aproximadamente 10 minutos. Se metaboliza en el hígado y otros tejidos por acción de la catecol-o-metil transferasa dando origen a un metabolito inactivo (3-o-metildobutamina), y por conjugación con el ácido glucurónico. Un 66% de la dosis es excretada en 48 horas por la orina como glucurónido y 3-o-metildobutamina y el resto por las heces. La vida media de eliminación es de 1-2 minutos. INDICACIONES Como soporte inotrópico en el tratamiento de corta duración de pacientes con descompensación cardiaca de- IV. La velocidad de infusión y la duración de la terapia se ajustan a la respuesta que exhiba cada paciente según su circunstancia clínica particular en función de la presión arterial, la frecuencia cardiaca, el flujo urinario, la presencia de actividad ectópica, el C gasto cardíaco y la presión venosa central o la presión capilar pulmonar en cuña. La infusión se inicia con 0,5-1 mcg/kg/minuto y se va ajustando con incrementos graduales a intervalos de pocos minutos hasta alcanzar la respuesta deseada. La dosis con la cual se logra la estabilización del paciente (adultos y niños) se ubica usualmente en el rango de 2-20 mcg/kg/minuto. No obstante, se han usado dosis de hasta 40 mcg/kg/minuto. REACCIONES ADVERSAS Cardiovasculares: Aumento de la frecuencia cardiaca, hipertensión, contracciones ventriculares prematuras, angina, dolor torácico no específico, palpitaciones, hipotensión. Gastrointestinales: Náuseas, vómitos. Hemato-lógicas: Trombocitopenia. Respirato-rias: Disnea, episodios asmáticos. Neurológicas: Cefalea, parestesia. Reacciones de hipersensibilidad: Ana-filaxis. Otras: Flebitis, necrosis por ex-travasación. INTERACCIONES Los bloqueantes de los receptores be- 2ed. 2004 * Formulario TerapÈutico Nacional 87 da lugar a un mayor gasto cardiaco y a una menor presión en cuña que cuando se administran por separado. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en la estenosis subaórtica hipertrófica idiopática y en pacientes con hipersensibilidad al medicamento. Usar con precaución en pacientes que han sufrido un infarto reciente. Durante la administración se debe vigilar constantemente el ECG, la presión arterial y, de ser posible, la presión capilar pulmonar en cuña y el gasto cardíaco. En caso de hipovolemia, se debe corregir la misma antes de iniciar el tratamiento. Debe tenerse especial precaución a objeto de evitar la extravasación del fármaco. SOBREDOSIS Reducir o descontinuar la administración del medicamento hasta que el paciente se estabilice. Debe asegurarse una vía aérea libre y asegurar la oxigenación y la ventilación. Las taquiarritmias ventriculares severas responden al propranolol o a la lido-caína. Por lo DOPAMINA FARMACOLOGÍA La dopamina es una amina simpaticomimética vasopresora que se produce naturalmente y es precursora 88 neurotransmisor en ciertas ·reas del SNC. Estimula los receptores adrenÈrgicos del sistema nervioso simp·tico. Ejerce un efecto directo por estimulaciÛn de los receptores beta1adrenÈrgicos e indirecto por liberaciÛn de norepinefrina de sus sitios de almacenamiento. Interact˙a tambiÈn con receptores dopaminÈrgicos especÌficos a nivel renal, mesentÈrico, coronario e intracerebral, causando vasodilataciÛn. Tiene poca afinidad por los receptores beta2-adrenÈrgicos. La estimulaciÛn de los distintos tipos de receptores adrenÈrgicos depende de la velocidad de administraciÛn; la infusiÛn de 0,5-2 mcg/kg/minuto genera vasodilataciÛn en el lecho renal por estimulaciÛn dopaminÈrgica; la infusiÛn de 2-10 mcg/kg/minuto estimula los receptores beta1-adrenÈrgicos mejorando la contractilidad mioc·rdica, el gasto cardiaco y la conducciÛn de impulsos; la infusiÛn de 10-20 mcg/kg/minuto o m·s produce vasoconstricciÛn perifÈrica y aumento de la presiÛn arterial por estimulaciÛn de los receptores alfa-adrenÈrgicos, con reversiÛn de la vasodilataciÛn renal. FARMACOCINÉTICA Después de la administración IV, se observa una respuesta dopaminérgica en 5 minutos la cual se mantiene por aproximadamente 10 minutos. Se distribuye ampliamente en Formulario Terapéutico Nacional * 2ed. 2004 mientras que el 25% restante se transforma en norepinefrina en los terminales nerviosos. Se excreta en un 80% como metabolitos y en una pequeña proporción como fármaco intacto por la orina en 24 horas. La vida media plasmática es de 2 minutos. INDICACIONES Shock e hipotensión severa que no responde a la administración de fluidos o a la expansión de volumen. RÉGIMEN DE DOSIFICACIÓN Se administra por infusión IV continua a una tasa que oscila entre 1 y 50 mcg/kg/minuto. En adultos, iniciar con 1-5 mcg/kg/minuto con incrementos de 1-4 mcg/kg/minuto a intervalos de 1030 minutos, hasta obtener la respuesta deseada. La mayoría de los pacientes responden con dosis de 20 mcg/kg/minuto. En niños, la dosis recomendada es de 2-20 mcg/kg/minuto (titulada de acuerdo a la repuesta del paciente). Una vez logrados los efectos hemodinámicos requeridos, usar la mínima dosis necesaria para mantenerlos. La suspensión o el retiro del medicamento deben hacerse gradualmente. tisular con extravasación, piloerección. INTERACCIONES Los antidepresivos tricíclicos pueden potenciar la respuesta presora a la dopamina. Los inhibidores de la MAO y C otros fármacos con efectos similares (como la furazolidona y la selegilina) pueden prolongar e intensificar los efectos de la dopamina. Los bloqueantes de los receptores betaadrenérgicos antagonizan los efectos cardíacos de la dopamina y los bloqueantes alfa-adrenérgicos, la vasoconstricción periférica. El uso con anestésicos inhalatorios puede pro-vocar hipertensión y arritmias ventri-culares. Con la fenitoína, puede generar hipotensión y bradicardia. Con la ergotamina existe riesgo de isquemia vascular periférica y gangrena. Con la digoxina puede ocurrir un efecto inotrópico aditivo. Con la levodopa se incrementa el riesgo de arritmias. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con feocromocitoma, taquiarritmias o REACCIONES ADVERSAS fibrilación ventricular. Usar con Cardiovasculares: Latidos ectópicos, precaución en pacientes con enfertaquicardia, dolor anginoso, palpi- medad vascular oclusiva (ateroestaciones, hipotensión. Menos frecuen- clerosis, enfermedad de Raynaud, temente: Bradicardia. Ensanchamien- diabetes y otras) debido al riesgo de to del complejo QRS, trastornos de la necrosis periférica o gangrena. En conducción, vasoconstricción, hiper- casos de hipovolemia, hipoxia, 89 2ed. 2004 * Formulario TerapÈutico Nacional pulmonar en cuña y el gasto cardíaco. Debe tenerse especial precaución al administrar dopamina a objeto de evitar la extravasación del fármaco y con el uso en pacientes con enfermedad cardiaca isquémica. Cuando se detiene la administración del medicamento, debe tenerse cuidado de una caída súbita de la presión arterial. Debe disminuirse la dosis gradualmente para evaluar la estabilidad de la presión arterial. La dopamina es sensible a la luz; está disponible en frascos que la protegen de la luz. SOBREDOSIS Reducir o descontinuar la administración hasta que el paciente se estabilice. Por lo general, no requiere medidas especiales dado que la duración de la acción es corta. Si la estabilización no ocurre en pocos minutos, se debe considerar la administración de un bloqueante de los receptores alfa-adrenérgicos de LIDOCAÍNA (AntiarrÌtmico) FARMACOLOGÍA Agente antiarrítmico clase IB. Estabiliza la membrana neuronal mediante el bloqueo de los canales de sodio, ion cuyo influjo es requerido para el inicio y la conducción de los impulsos. Deprime la despolarización y la auto90 te la fase diastólica por acción directa sobre los tejidos, especialmente la red de Purkinje. Tiene un efecto mínimo sobre el tejido auricular y, en dosis terapéuticas, no afecta significativamente la contractilidad miocárdica ni la conducción AV. FARMACOCINÉTICA Después de la administración IV, produce una respuesta antiarrítmica a los 45-90 segundos, la cual se mantiene por 10-20 minutos. La concentración plasmática terapéutica necesaria para mantener el efecto se ubica en el rango de 1,5-6 mcg/ml. Concentraciones superiores se consideran tóxicas. Se une a las proteínas plasmáticas en un 3380% y se distribuye ampliamente a los tejidos corporales, sobretodo a los órganos de mayor perfusión (riñón, pulmones, hígado, corazón). La lidocaína atraviesa la barrera hematoencefálica y la placentaria. Se metaboliza rápida y extensamente (90%) en el hígado, transformándose en metabolitos activos que se excretan, junto con menos de 10% del fármaco intacto, por la orina. La vida media de eliminación después de la administración IV en bolo es de 1,5-2 horas y se prolonga en pacientes con insuficiencia hepática o insuficiencia cardiaca y en los ancianos. INDICACIONES Formulario Terapéutico Nacional * 2ed. 2004 ticiones cada 5-10 minutos hasta lograr el control de la arritmia, o hasta que se desarrollen efectos adversos, seguidos por una infusión IV de 20-50 mcg/kg/minuto (1-4 mg/minuto) como dosis de mantenimiento. En niños, iniciar con 0,5-1 mg/kg/dosis en bolo IV, con repeticiones cada 5-10 minutos hasta lograr la respuesta deseada o hasta un máximo de 5 mg/kg, seguidos por una infusión IV de 10-50 mcg/kg/minuto como dosis de mantenimiento. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, bradicardia, nuevas arritmias o empeoramiento de arritmias preexistentes, paro cardíaco. Neurológicas: Confusión, temblor, letargia, somnolencia, estupor, intranquilidad, ansiedad, alucinaciones, nerviosismo, aturdimiento, parestesia, contracciones musculares, convulsiones. Reacciones de hipersensibilidad: Anafilaxis. Respiratorias: Depresión y paro respiratorio. Otras: Tinitus, visión borrosa o doble, dolor en el sitio de inyección, sensación de frío, vómitos. INTERACCIONES La cimetidina y el propranolol pueden incrementar los niveles plasmáticos de la lidocaína, aumentando el riesgo de toxicidad. Cuando se administra con otros antiarrítmicos, aumenta la depresión miocárdica. La adminis- el tropisetrón aumentan el riesgo de arritmias ventriculares. Los antipsicóticos que prolongan el intervalo QT aumentan el riesgo de arritmias ventriculares. Fenobarbital, fenitoína, rifampicina aumentan la depuración y reducen los niveles plasmáticos de la C lidocaína. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, síndrome de Stokes-Adams o con bloqueo cardíaco. Usar con precaución en pacientes con falla hepática y/o renal, insuficiencia cardiaca, enfermedad neurológica preexistente, bradicardia sinusal, síndrome de WolffPar-kinson-White, hipovolemia, shock y en el embarazo. Durante el tratamiento se debe vigilar constantemente el ECG y el estado de conciencia del paciente. SOBREDOSIS Manejo sintomático y tratamiento de soporte. En caso de convulsiones, manejarlas con diazepam o con barbitúricos de acción ultracorta. Si se preMEXILETINA FARMACOLOGÍA Antiarrítmico clase IB con propiedades electrofisiológicas similares a las de la lidocaína. Inhibe 2ed. 2004 * Formulario TerapÈutico Nacional 91 sodio al interior de la célula cardiaca, reduciendo la tasa inicial del incremento del potencial de acción, fase 0, y el período refractario efectivo en las fibras de Purkinje. En sujetos con sistema de conducción normal, afecta muy poco la generación y propagación del impulso cardíaco; sin embargo, en pacientes con trastornos de conduc-ción preexistentes, se ha observado depresión de la frecuencia sinusal, prolongación del tiempo de recupe-ración nodal, disminución de la velo-cidad de conducción e incremento del período refractario efectivo del sistema de conducción intraventricular. FARMACOCINÉTICA Se absorbe bien en el tracto gastrointestinal (90%) alcanzando niveles séricos máximos en 1-4 horas. Una vez en la sangre, se une a las proteínas plasmáticas en un 50-70%. Se metaboliza extensamente en el hígado, trasformándose en productos con mínima o ninguna actividad farma-cológica que se excretan, junto con un 10% del fármaco intacto, en la orina. La vida media de eliminación es de 6-17 horas y en pacientes con insufi-ciencia hepática puede prolongarse hasta 25 horas. INDICACIONES Arritmias ventriculares. 92 antiácidos. Cuando se requiere un control rápido de las arritmias, se inicia con una dosis de 400 mg seguida, 8 horas más tarde, por 200 mg. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, dolor anginoso, nuevas arritmias o empeoramiento de las arritmias ventriculares, hipotensión, hipertensión, bradicardia, bloqueo AV y shock cardiogénico. Gastrointestinales: Náuseas, vómito, disfagia, dolor epigástrico, acedía, diarrea, constipación, cambios en el apetito, dolor abdominal, úlcera pép-tica y ulceración esofágica. Hematológicas: Discrasias sanguíneas. Hepáticas: Elevación de los niveles de las enzimas hepáticas y necrosis hepática. Neurológicas: Mareos, nerviosismo, cefalea, parestesias, debilidad, depresión, desmayos, alucinaciones, psicosis, temblores, convulsiones, aturdimiento, falta de coordinación, cambios en los hábitos de sueño, dificultad al hablar, confusión, diplopía. Respiratorias: Disnea. Otras: Tinitus, edema, artralgias, fiebre, diaforesis, caída del cabello, disminución de la libido, impotencia, hipo, síndrome lúpico. INTERACCIONES Los niveles plasmáticos pueden ser reducidos por la fenitoína, rifampicina y fenobarbital. Los analgésicos nar- Formulario Terapéutico Nacional * 2ed. 2004 ciones séricas de metilxantinas (cafeína, teofilina). Los alcalinizantes urinarios aumentan los niveles sanguíneos de la mexiletina, los acidificantes las disminuyen. Aumento del riesgo de arritmia con ritonavir. Los diuréticos de asa y las tiazidas pueden antagonizar el efecto de la mexiletina debido a la hipokalemia. ciones farmacológicas, se sugiere el uso de marcapaso cardiaco transvenoso, dependiendo de la condición clínica del paciente. INFORMACIÓN AL PACIENTE Administrar con las comidas. No C alterar la dosificación ni suspender el tratamiento sin la autorización del médico. Informar al médico si se presenta CONTRAINDICACIONES/ PRECAUCIONES TRINITRATO DE GLICERILO (NITROGLICERINA) Contraindicada en pacientes con shock cardiogénico, bloqueo AV FARMACOLOGÍA Produce vasodilatación venosa y arpreexistente de segundo o tercer terial sistémica por una acción directa grado (en ausencia de marcapaso) y sobre la musculatura lisa vascular, a en casos de hipersensibilidad al través de un mecanismo no depenmedicamento. Dado que la alcalidiente de la inervación autonómica. La nización de la orina puede prolongar la nitroglicerina forma radicales libres de vida media de eliminación, se óxido nítrico (ON), el cual activa la recomienda evitar la ingesta de guanilato ciclasa, que resulta en un medicamentos o alimentos que pueaumento del GMPc en el músculo liso dan modificar el pH urinario. Usar con y otros tejidos. Esto lleva a la precaución en pacientes con fosforilación de las cadenas ligeras de disfunción hepática, enfermedad de miosina, lo cual conduce a vasoParkinson, hipotensión, trastornos de dilatación. La dilatación venosa conducción cardiaca, insuficiencia resulta en disminución del retorno cardiaca congestiva, trastornos venoso, de la presión diastólica final convulsivos, insuficiencia hepática, del ventrículo izquierdo (precarga) y historia de discrasias sanguíneas y en de la presión capilar pulmonar en el embarazo. Debido a que en la leche cuña. La dila-tación arteriolar materna alcanza concentraciones disminuye la resis-tencia vascular similares a las plasmáticas, debe sistémica y la presión arterial usarse un método alternativo para la (poscarga). alimentación del recién nacido si el uso de la mexiletina es esencial. No se FARMACOCINÉTICA conoce con precisión la seguridad del 2ed. 2004 * Formulario TerapÈutico Nacional 93 en el hígado, en los glóbulos rojos y en las paredes vasculares a compuestos con menor actividad farmacológica. El mononitrato de glicerilo, el principal metabolito, es inactivo. Poste-riormente, los metabolitos son trans-formados a mononitratos inactivos y finalmente convertidos en glicerol y dióxido de carbono. Se excreta en un 22% intacta por la orina. La vida media de eliminación es de 3-4 minutos. INDICACIONES Tratamiento pre-operatorio de la hipertensión arterial, control de la insuficiencia cardiaca congestiva asociada al infarto miocárdico agudo, tratamiento de la angina de pecho en pacientes que no responden a otras medidas y manejo de la crisis hipertensiva durante y después de la cirugía. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV diluida en solución de cloruro de sodio al 0,9% o en glucosa al 5% para una concentración final de 100-400 mcg/ml. Evitar el uso de contenedores y catéteres de PVC (cloruro de polivinilo) a objeto de evitar la adsorción de la nitroglicerina a las paredes de los mismos. Iniciar con 5 mcg/minuto e incrementar en 5 mcg/minuto cada 3-5 minutos hasta obtener la respuesta deseada. Si se 94 REACCIONES ADVERSAS El uso de la nitroglicerina por vía IV puede provocar hipotensión grave, shock, bradicardia severa y empeoramiento de la angina de pecho. Aunque raros, se ha reportado casos de alergia al medicamento. INTERACCIONES El efecto vasodilatador puede resultar aditivo con el de otros agentes con propiedades vasodilatadoras o hipotensoras. Los inhibidores de la fosfodiesterasa tipo 5 potencian la acción hipotensora de la nitroglicerina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a los nitratos, pacientes con hipotensión, hipovolemia, miocardiopatía restrictiva, tapo-namiento pericárdico y pericarditis constrictiva. Usar con precaución en pacientes con miocardiopatía hiper-trófica, aumento de la presión intra-craneana, hipertiroidismo, trauma-tismo craneoencefálico y en el em-barazo. SOBREDOSIS CLONIDINA FARMACOLOGÍA Agonista de los receptores alfa2adrenérgicos ubicados en el tallo cerebral. Causa una reducción del Formulario Terapéutico Nacional * 2ed. 2004 simpático desde el SNC, lo que lleva a una disminución de la resistencia vascular periférica, de la resistencia renovascular, de la frecuencia cardiaca y de la presión arterial. Durante la terapia a largo plazo, el gasto cardíaco, que disminuye en 15-20%, tiende a volver a los valores control pero la resistencia periférica permanece baja. Se observa disminución de la frecuencia de pulso pero no se altera la respuesta hemodinámica normal al ejercicio. FARMACOCINÉTICA Se absorbe bien en el tracto gastrointestinal. Luego de su administración por vía oral produce el inicio de la respuesta hipotensora en 30-60 minutos. Esta se hace máxima en 2-4 horas y perdura por aproximadamente 8 horas. Posterior a la administración IV en pacientes con crisis hipertensivas, produce una acción hipotensora a los pocos minutos, la cual se hace máxima en 30-60 minutos y se mantiene por aproximadamente 4 horas. Una vez en la sangre, se une a las proteínas plasmáticas en un 2040% y se distribuye ampliamente a los tejidos corporales. Se metaboliza parcialmente (50%) en el hígado, transformándose en productos inactivos. Se excreta en un 40-60% como fármaco intacto por la orina y el resto por las heces. El principal me- INDICACIONES Hipertensión arterial y tratamiento de emergencia de la crisis hipertensiva. RÉGIMEN DE DOSIFICACIÓN La dosis debe individualizarse en función de la respuesta clínica y C tolerancia de cada paciente a la terapia. Para el tratamiento de la hipertensión arterial en adultos por vía oral iniciar con dosis de 0,0750,150 mg/día e incrementos graduales cada 2-3 días hasta obtener la respuesta adecuada, la cual se logra, generalmente, con 0,3001,200 mg/día en dosis divididas. Para el manejo de la crisis hipertensiva se recomienda 0,150-0,300 mg por vía IM o por infusión IV en 5 minutos. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, palpitaciones, taquicardia, bradicardia. Insuficiencia cardiaca congestiva, anormalidades en el ECG, síncope y fenómeno de Raynaud (reportados raras veces). Neurológicas: Cefalea, nerviosismo, agitación, ansiedad, debilidad, depresión mental, insomnio y pesadillas (raras veces). Gastrointestinales: Náusea, vómito, anorexia y constipación. Hepáticas: Alteración de las pruebas de función hepática. Dermatológicas: Erupción, prurito, ronchas, edema angioneurótico, urticaria y alopecia. Otras: Disminución de la libido, im- 2ed. 2004 * Formulario TerapÈutico Nacional 95 alcohol, barbitúricos, opiáceos y otros sedantes. Los antidepresivos tricíclicos pueden reducir el efecto antihipertensivo de la clonidina. La combinación con fármacos que afectan la función sinusal o la conducción AV como digitálicos, bloqueantes beta adrenérgicos y bloqueantes del calcio incrementa el riesgo de bradicardia y bloqueo AV. En combinación con amitriptilina se ha observado el desarrollo de lesiones corneales en animales de experimentación. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento. La suspensión abrupta del medicamento puede desencadenar un síndrome caracterizado por agitación, nerviosismo, cefalea y una rápida elevación de la presión arterial. Para evitarlo se debe retirar el medicamento lenta y gradualmente en un período de 2-4 días. Durante el tratamiento es recomendable practicar periódicamente exámenes oftalmológicos. Usar con precaución durante el embarazo y la lactancia. La eficacia y seguridad de su uso en niños no ha sido establecida. SOBREDOSIS Administración de fluidos intravenosos con dopamina para el tratamiento de la hipotensión. La bradicardia se puede tratar con sulfato 96 INFORMACIÓN AL PACIENTE No alterar la dosificación sin autorización del médico, ni suspender bruscamente la terapia por el riesgo de hipertensión de rebote. Debe tener precaución al realizar actividades que requieren estar alerta tales como manejar automóviles u operar maquinaria. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el trataMETILDOPA (ALFAMETILDOPA) FARMACOLOGÍA Agente antihipertensivo. La metildopa es un inhibidor de la descarboxilasa de aminoácidos aromáticos. Aunque su mecanismo de acción no ha sido completamente elucidado, se cree que el efecto antihipertensivo es debido a la descarboxilación del medicamento a metilnorepinefrina en el SNC, desde donde disminuye la presión arterial por estimulación de los receptores inhibitorios alfa2-adrenérgicos en el núcleo del tracto solitario en el SNC. También se ha postulado mecanismos que involucran falsa neurotransmisión, disminución de la actividad de la renina plasmática y de las concentraciones tisulares de serotonina, dopamina, norepinefrina y epinefrina. FARMACOCINÉTICA Se absorbe parcialmente (50%) en el tracto gastrointestinal alcanzando Formulario Terapéutico Nacional * 2ed. 2004 Se une débilmente a las proteínas plasmáticas. Se metaboliza extensamente en el hígado, dando lugar a productos conjugados, uno de los cuales, el mono-o-sulfato de metildopa, resulta el más abundante y con actividad farmacológica. La vida media de eliminación plasmática es de 105 minutos. Alrededor del 70% del fármaco absorbido se excreta por la orina como metildopa y su metabolito conjugado mono-o-sulfato, mientras la fracción no absorbida se elimina, en una pequeña proporción, por las heces. La metildopa atraviesa la barrera placentaria y aparece en el cordón umbilical y en la leche materna. INDICACIONES Hipertensión arterial e hipertensión durante el embarazo. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos, iniciar con 250 mg 2-3 veces al día durante las primeras 48 horas. Ajustar a intervalos de 2 días hasta lograr la respuesta adecuada. La dosis usual de mantenimiento es de 0,5-2 g/día divididos en 2-4 dosis. En niños, iniciar con 10 mg/kg/día divididos en 2-4 dosis y ajustar hasta alcanzar la respuesta adecuada, sin exceder 65 mg/kg/día o 3 g/día, lo que sea menor. sialadenitis, lengua negra o dolorosa, sequedad de la boca, diarrea, constipación, flatulencia, colitis y pancreatitis. Genitourinarias: Amenorrea, ginecomastia, disminución de la libido e impotencia. Hematológicas: Leucopenia, granulocitopenia, trombocito- C penia, anemia hemolítica, mielosupresión. Hepáticas: Elevación de las enzimas hepáticas, ictericia y hepatitis. Neurológicas: Somnolencia, cefalea, mareos, astenia, desvanecimientos, parestesias, disminución de la agudeza mental, movimientos invo-luntarios, parkinsonismo, pesadillas, psicosis, depresión, trastornos psí-quicos incluyendo pesadillas y psicosis leve reversible o depresión y aturdimiento. Reacciones de hiper-sensibilidad: Miocarditis, pericarditis, vasculitis, síndrome tipo lupus, fiebre, erupción, urticaria, eczema, prueba positiva para los anticuerpos antinu-cleares, células LE y de factor reumatoideo, prueba de Coombs positiva, necrólisis epidérmica tóxica. Renales: Aumento del BUN y de la creatinina sérica, retención de sodio y agua. Otras: Congestión nasal, ar-tralgia, mialgia, hiperprolactinemia. INTERACCIONES Los diuréticos y otros medicamentos hipotensores potencian el efecto hipotensor de la metildopa; las REACCIONES ADVERSAS 2ed. 2004 * Formulario TerapÈutico Nacional 97 dosis de los anestésicos (si se produce hipotensión durante la anestesia, usar agentes vasopresores). Los anti-psicóticos y la levodopa pueden incrementar los efectos adversos de la metildopa sobre el SNC. La metildopa puede incrementar los niveles séricos de litio y potenciar el efecto presor de las aminas simpaticomiméticas. Evitar el uso concomitante con ají ya que disminuye la eficacia antihipertensiva. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y en aquellos con enfermedad hepática activa. Antes de iniciar el tratamiento y posteriormente durante su desarrollo, se debe realizar exámenes hematológicos y pruebas de la función hepática. Usar con precaución en pacientes con historia de disfunción hepática y en tratamiento con inhibidores de la monoaminooxidasa (MAO). Debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Colocar al paciente en posición Trendelenburg y administrar fluidos intravenosos, con vigilancia permanente de la presión arterial, el gasto y la frecuencia cardíacos, el 98 INFORMACIÓN AL PACIENTE Notificar al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial cansancio general prolongado, fiebre y coloración amarilla de la piel y/o de las mucosas. El tratamiento puede alterar la capacidad para realizar actividades NITROPRUSIATO (NITROPRUSIATO S”DICO) FARMACOLOGÍA El nitroprusiato produce hipotensión debido a una vasodilatación venosa y arterial sistémica causada por una acción directa sobre la musculatura lisa vascular, a través de un mecanismo no dependiente de la inervación autonómica y que se cree es debido al grupo nitroso de la molécula. FARMACOCINÉTICA Después de la administración IV de una dosis única, se produce una respuesta en 30-60 segundos, la cual se hace máxima 1-2 minutos después de la administración y persiste por 110 minutos. Se metaboliza rápida y completamente en la sangre y los tejidos, liberando cianógeno (radical cianuro) que se transforma en tiocianato (inactivo) en el hígado por acción de la enzima tiosulfato sulfotransferasa, una enzima mitocondrial. Se excreta en un 100% por la orina como metabolitos. La vida media de eliminación plasmática del fármaco Formulario Terapéutico Nacional * 2ed. 2004 crisis hipertensivas. RÉGIMEN DE DOSIFICACIÓN Se administra solamente por infusión IV, diluido en glucosa al 5%; nunca por inyección IV directa. En adultos y niños, iniciar con una dosis de 0,250,3 mcg/kg/minuto e incrementar gradualmente a intervalos de pocos minutos hasta lograr el control de la crisis. No debe excederse el límite de 10 mcg/kg por 10 minutos. En neonatos, la dosis no debe exceder 3 mcg/kg/minuto. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, taquicardia, cambios electrocardiográficos. Dermatológicas: Enrojecimiento de la piel, erupción. Gastrointestinales: Náusea, dolor abdominal. Hematológicas: Disminución de la agregación plaquetaria. Neurológicas: Cefalea, mareos, pérdida de la conciencia, aumento de la presión intracraneana, intranquilidad, contracciones musculares. Renales: Aumento de la creatinina sérica. Otras: Acidosis, toxicidad por tiocianato, metahemoglobinemia, toxicidad por cianuro, irritación en el sitio de infusión, hipotiroidismo, marcaje de las venas. potensión controlada durante la cirugía en pacientes con circulación cerebral inadecuada, ni en la cirugía de emergencia en pacientes moribundos. El uso del nitroprusiato también está contraindicado en pacientes con encefalopatía, defi- C ciencia severa de vitamina B12, atrofia óptica congénita (de Leber) y ambliopía por tabaco (estos pacientes tienen una alta proporción de cianuro/tiocianato). En pacientes con daño hepático severo podría alterarse la transformación de cianógeno en tiocianato. En pacientes con falla renal puede acumularse el tiocianato. Usar con precaución en pacientes con aumento de la presión intracraneal, anemia e hipovolemia. No se conoce la seguridad de su uso durante el embarazo y la lactancia. SOBREDOSIS Administrar nitrito de amilo vía inhalatoria por 15-30 segundos, seguido por la administración IV de 4-6 mg/kg de solución de nitrito de sodio al 3% en 2-4 minutos, con vigilancia permanente de la presión arterial y medidas de rutina para su control. Administrar luego 150-200 mg/kg de solución de tiosulfato de sodio al 10 o al 25%. En caso necesario, repetir la inyección de nitrito de sodio y de INTERACCIONES tiosulfato de sodio a la mitad de la Los bloqueantes ganglionares, los dosis inicial, 2 horas más tarde. El anestésicos generales, los agentes tiocianato puede ser removido por 99 2ed. 2004 * Formulario TerapÈutico Nacional ESPIRONOLACTONA FARMACOLOGÍA Diurético ahorrador de potasio. Es un antagonista específico de la aldosterona. Se une a los receptores renales de aldosterona inhibiendo competitivamente los efectos de la misma y generando, en consecuencia, un incremento en la eliminación de sodio, cloruro y agua y una disminución de la excreción de potasio, magnesio, amonio, hidrogeniones y fosfato. Ejerce una acción hipotensora presumiblemente debida a un antagonismo del efecto de la aldosterona sobre la fibra de la musculatura lisa arteriolar. Posee, además, propiedades antiandrogénicas. FARMACOCINÉTICA Se absorbe en un 73% por vía oral y el inicio de la respuesta antialdosterona se observa en 2-4 horas, la cual se hace máxima a las 6-8 horas y persiste por 16-24 horas. La comida aumenta la biodisponibilidad de la espironolacto-na. El efecto diurético se inicia en forma gradual, alcanza su máximo al tercer día de la terapia y se mantiene hasta por 2-3 días después de sus-pendido el tratamiento. Se une a las proteínas plasmáticas en un 90%. Se metaboliza aparentemente en el híga-do, dando lugar a por lo menos 3 metabolitos activos, siendo la canrenona el más 100 INDICACIONES Edema idiopático y edema asociado a la insuficiencia cardiaca congestiva, el síndrome nefrótico y a la cirrosis hepática; diagnóstico y tratamiento preoperatorio y en el mantenimiento del hiperaldosteronismo primario; hipertensión arterial; hirsutismo. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. - Edema: Se recomienda una dosis inicial de 100 mg administrado como dosis única o dividida, pero puede estar en un rango de 25-200 mg/día por 5 días continuos. Si se logra una respuesta adecuada, mantener la dosis; en caso necesario, ajustar la dosis hasta alcanzar el nivel óptimo. Si no se logra la respuesta adecuada después de los 5 días iniciales, considerar la incorporación de otro diurético (de asa o tiazídico) al régimen, sin alterar la dosis de espironolactona. - HipertensiÛn: Iniciar con 25-100 mg/día (como dosis única o dividida) por 2 semanas y, posteriormente, mantener o ajustar, en caso nece-sario, según la respuesta del pacien-te. - Diagnóstico de hiperaldosteronismo primario: 400 mg/día por 3-4 semanas (prueba larga). Si se corrige la hipopotasemia y la hipertensión, el diagnóstico se considera positivo. Si se administra la dosis por sólo 4 Formulario Terapéutico Nacional * 2ed. 2004 varios días antes de la cirugía. En pacientes que no son candidatos a cirugía, iniciar la terapia con 100400 mg/día y ajustar, posteriormente, hasta lograr la mínima dosis efectiva de mantenimiento, según la res-puesta del paciente. - Hirsutismo: 50-200 mg/día por un período no menor de 6 meses. REACCIONES ADVERSAS Dermatológicas: Urticaria, erupciones maculopapulares. Endocrinas: Ginecomastia, disminución de la libido, menstruación irregular o amenorrea, sangrado posmenopáusico, dolor en las mamas. Gastrointestinales: Sangrado gástrico, diarrea, ulceración, có-licos, gastritis, vómitos. Hematológi-cas: Agranulocitosis. Reacciones de hipersensibilidad: Anafilaxis. Otras: Hi-perpotasemia, deshidratación, hipona-tremia, aumento transitorio del BUN, acidosis leve. INTERACCIONES Los inhibidores de la enzima convertidora de la angiotensina, la ciclosporina, la indometacina, la arginina, el triamtereno y el valsartán incrementan los niveles séricos de potasio, aumentando el riesgo de hiperpotasemia asociado a la espironolactona. La combinación con otros diuréticos o agentes hipotensores produce efectos efecto cicatrizante de úlceras de la regaliz (Glicirrhiza glabra) y los efectos tipo regaliz de la aldosterona. Los sustitutos de la sal que contienen potasio y los alimentos ricos en potasio (como las frutas cítricas y los tomates) aumentan el riesgo de hiperpotase- C mia. Aumento de la respuesta a los relajantes musculares. Potenciación de la hipotensión ortostática con alcohol, barbitúricos y narcóticos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con insuficiencia renal aguda, anuria, hiperpotasemia y en caso de hipersensibilidad al medicamento. Durante el tratamiento se debe realizar determinaciones periódicas del potasio séri-co. Usar con precaución en pacientes con falla renal leve a moderada y/o con enfermedad hepática. El uso en el embarazo debe limitarse a circuns-tancias de verdadera necesidad, cuan-do no exista otra alternativa terapéu-tica y previa consideración del balance riesgo-beneficio. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de 2ed. 2004 * Formulario TerapÈutico Nacional 101 tratamiento, en especial efectos enFUROSEMIDA FARMACOLOGÍA Diurético tipo sulfonamida y derivado del ácido antranílico. Interfiere con el mecanismo de transporte de iones a través del epitelio tubular renal. Inhibe la reabsorción de sodio y de cloruro en el túbulo proximal, en el túbulo distal y en el segmento ascendente del asa de Henle. Promueve la excreción de pota-sio, hidrogeniones, calcio, magnesio, amonio, bicarbonato y, posiblemente, fosfato. Produce un efecto hipotensor que resulta de la disminución del volu-men plasmático como consecuencia del efecto diurético. FARMACOCINÉTICA Se absorbe en un 60-80% en el tracto gastrointestinal. El efecto diurÈtico se inicia en 30-60 minutos, se hace m·ximo en 1-2 horas y persiste por 6-8 R…GIMEN DE DOSIFICACI”N - Tratamiento del edema: La dosis inicial por vía oral en adultos es 2080 mg como dosis única. Si el efecto resulta insuficiente, incrementar 2040 mg cada 6-8 horas hasta lograr la respuesta diurética deseada. En niños, iniciar con 2 mg/kg e incrementar, en caso necesario, 1-2 mg/kg cada 6-8 horas hasta obtener la respuesta adecuada. No se recomienda dosis mayores de 6 mg/kg de peso corporal. Para la terapia de mantenimiento en pacientes pediátricos, la dosis debe se ajustada al máximo nivel efectivo. La dosis por vía IV ó IM es de 20-40 mg con incrementos, en caso necesario, de 20 mg hasta lograr la respuesta adecuada, pero no antes de 2 horas de la dosis previa. A partir de allí, mantener la dosis 1-2 veces al día. - Tratamiento del edema pulmonar agudo: Administrar 40 mg IV en 1-2 minutos e incrementar, en caso necesario, a 80 mg en 1-2 minutos cada hora hasta obtener la respuesta deseada. En niños, iniciar con 1 mg/kg e incrementar, en caso necesario, 1 mg/kg cada 2 horas hasta obtener la respuesta de-seada. - Tratamiento oral de la hipertensión arterial en adultos: La dosis usual es de 80 mg, dividida en dos dosis de 40 mg. La dosis se ajusta de acuerdo a la respuesta. Si es necesario, horas. Luego de la administraciÛn IV, el efecto se inicia en 5 minutos, se hace m·ximo en 20-60 minutos y se mantiene por 2 horas. Se une a las proteÌnas plasm·ticas en un 95%. Se metaboliza poco en el hÌgado a un glucurÛnido de furosemida; se elimina por la orina en un 50-80% y el resto por las heces. La vida media de eliminaciÛn es de aproximadamente 2 102 Formulario Terapéutico Nacional * 2ed. 2004 trointestinales: Pancreatitis, anorexia, irritación oral y gástrica, cólicos, diarrea, constipación, náusea, vómitos. Hematológicas: Anemia aplásica (rara), trombocitopenia, agranulocitosis (rara), anemia hemolítica, leucopenia. Hepáticas: Ictericia (ictericia colestásica intrahepática). Neurológicas: Tinitus y pérdida de la audición, parestesia, vértigo, mareos, cefalea, visión borrosa, xantopsia. Reacciones de hipersensibilidad: Vasculitis, nefritis intersticial, angeítis necrotizante, dermatitis exfoliativa, eritema multiforme, urticaria, erupción. Otras: Hiperglicemia, glucosuria, hiperuricemia, espasmos musculares, debilidad, intranquilidad, espasmo de la vejiga urinaria, fiebre. INTERACCIONES La furosemida puede incrementar la acción hipotensora de los medicamentos antihipertensivos. Antagoniza el efecto de los relajantes musculares no despolarizantes y potencia la acción de la succinilcolina, incrementa las concentraciones séricas de litio y el riesgo de toxicidad de los salicilados. En pacientes que reciben digoxina, la hipopotasemia inducida por la furosemida puede favorecer la toxicidad digitálica. Los corticosteroides y la anfotericina B incrementan el efecto hipopotasémico de la furosemida. Los AINEs pueden reducir el efecto diurético de la furosemida. El nación con aloe (zábila) sistémico, se aumenta la pérdida de potasio inducida por la furosemida. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes anúricos C y en casos de hipersensibilidad al medicamento. Si durante el uso de la furosemida se desarrolla azotemia y/u oliguria se debe suspender la terapia. En la terapia prolongada se debe vigilar los electrolitos séricos, la glicemia (en pacientes diabéticos), la función renal, hepática, auditiva y hematológica. Usar con precaución en pacientes con falla renal y/o hepática, en diabéticos, en la terapia con digitálicos y en el em-barazo. Se recomienda suspender la lactancia durante la terapia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Los electrolitos séricos, el nivel de CO2 y la presión arterial deben ser determinados con frecuencia. HIDROCLOROTIAZIDA FARMACOLOGÍA Diurético tiazídico. Interfiere con el transporte de los iones a través del epitelio tubular renal por alteración del metabolismo celular, promoviendo la 2ed. 2004 * Formulario TerapÈutico Nacional 103 cantidades aproximadamente equivalentes. Indirectamente, la acción diurética de la hidroclorotiazida reduce el volumen plasmático, con aumento en la actividad de la renina plasmática, aumento de la secreción de aldos-terona y en la pérdida de potasio, y disminución del potasio sérico. Ejerce un efecto hipotensor posiblemente debido a una acción vasodilatadora arteriolar directa y a una reducción del volumen plasmático como resultado de la diuresis. FARMACOCINÉTICA Se absorbe en un 60-80% en el tracto gastrointestinal y el efecto diurético se inicia a las 2 horas, se hace máximo 3-6 horas después y se mantiene por 6-12 horas. Se une a las proteínas plasmáticas en un 40%. No sufre metabolismo y se excreta como fármaco intacto por vía renal. Atraviesa la barrera placentaria pero no la barrera hematoencefálica. Se secreta en la leche materna. La vida media de eliminación se ubica entre 5,6 y 14,8 horas. INDICACIONES Tratamiento del edema y de la hipertensión arterial. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis inicial para el tratamiento del edema en adultos es de 25-100 mg/día divididos 104 pediátricas usuales para diuresis y la hipertensión arterial son de 1-2 mg/kg/día como dosis única o divididos en 2 dosis, dentro de un rango de 12,5 a 37,5 mg/día en niños de 6 meses a 2 años y de 37,5 a 100 mg/día en niños de 2-12 años. REACCIONES ADVERSAS Cardiovasculares: Depleción de volumen y deshidratación, hipotensión ortostática, miocarditis alérgica, vasculitis. Dermatológicas: Dermatitis, fotosensibilidad, púrpura, alopecia. Gastrointestinales: Anorexia, náusea, pancreatitis, distrés epigástrico, vómitos, dolor abdominal, diarrea, constipación, sed. Hematológicas: Anemia aplásica, agranulocitosis, leucopenia, trombocitopenia, anemia hemolítica. Hepáticas: Ictericia. Reacciones de hipersensibilidad: Erupciones, anafilaxis. Renales: Poliuria, insuficiencia renal, nefritis intersticial. Neurológicas: Mareos, vértigo, cefalea, parestesia, debilidad, intranquilidad. Respiratorias: Distrés respiratorio, neumonitis. Otras: Hipokalemia, hiperuricemia asintomática; hiperglicemia y alteración de la tolerancia a la glucosa, desbalance de fluidos y electrolitos, incluyendo hiponatremia e hipocloremia por dilución, alcalosis metabólica, hipercalcemia, gota, calambres musculares. Formulario Terapéutico Nacional * 2ed. 2004 riesgo de nefrotoxicidad de los AINEs. En pacientes que reciben digoxina, la hipopotasemia inducida por la hidroclorotiazida puede favorecer la toxicidad digitálica. Los corticosteroides y la anfotericina B incrementan el efecto hipopotasémico de la hidroclorotiazida. Los depresores del SNC incrementan la factibilidad de hipotensión postural. El efecto hiperglicémico de la hidroclorotiazida puede interferir con la respuesta farma-cológica a los medicamentos anti-diabéticos. La colestiramina y el colestipol disminuyen su absorción. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con enfermedad renal severa, en casos de hipersensibilidad a las tiazidas y a las sulfonamidas y durante el embarazo. En caso de tratamiento prolongado se debe vigilar periódicamente los electrolitos séricos, la glicemia (en pacientes diabéticos), la función hepática, la función renal y la hematológica. Usar con precaución en pacientes con falla renal y/o hepática, en diabéticos, en la terapia con digitálicos, en pacientes con gota y durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia presenta alguna reacción o sintomatología inusual durante el trataMANITOL FARMACOLOGÍA Diurético osmótico. Incrementa la preC sión osmótica del filtrado glomerular impidiendo la reabsorción de agua y solutos en los túbulos renales. Eleva la osmolaridad plasmática resultando en un aumento del flujo acuoso al fluido extracelular. FARMACOCINÉTICA Después de la administración IV, indu-ce una respuesta diurética en 1-3 ho-ras, la cual perdura por 2-8 horas; una disminución de la presión intraocular en 30-60 minutos que se mantiene por 4-6 horas, y una reducción de la pre-sión intracraneana en 15 minutos que persiste por 3-8 horas. La distribución en el organismo se limita al espacio extracelular. Se metaboliza en el híga-do en muy escasa proporción. Es libre-mente filtrado por el glomérulo y reab-sorbido a nivel tubular en un por-centaje inferior al 10%. Se excreta en un 80% como fármaco intacto y tiene una vida media de eliminación de 100 minutos. INDICACIONES Prevención y tratamiento de la fase oligúrica de la insuficiencia renal agu- 2ed. 2004 * Formulario TerapÈutico Nacional 105 iniciar el tratamiento se recomienda usar una dosis de prueba de 0,2 g/kg en adultos como solución al 15 o al 20% en 3-5 minutos, y de 0,75 g/kg en niños. La excreción de un volu-men de orina de 30-50 ml/hora por 2-3 horas se considera una respuesta adecuada. Oliguria: Para la prevención, la dosis usual en adultos es de 50-100 g como solución al 5, 10 ó 15%. Para el tratamiento de la condición se usa 100 g como solución al 15 ó 20% por 24 horas. - Disminución de la presión intracraneana: En adultos, la dosis usual es de 0,25 g/kg cada 6-8 horas. En niños, la dosis es de 0,25 g/kg/dosis en bolo IV cada 5 minutos, según sea necesario. - IntoxicaciÛn por sobredosis de drogas: En adultos, usar una infusión IV continua de solución de manitol al 5-20%, en dosis suficiente para producir y mantener gasto urinario de 150-500 ml/hora. En niños, la dosis es de 2 g/kg como solución al 5 ó 10%, según sea necesario. - ReducciÛn de la presión intraocular: Se recomienda 1,5-2 g/kg en 3060 minutos como solución al 15 ó 20%. En niños, la dosis es de 1,5-2 g/kg/dosis infundidos en 30 minutos. - Tratamiento del edema: En adultos: 100 g por infusión IV como solución al 20% en un período de 26 horas. En niños, la dosis es de 1-2 106 cular. Gastrointestinales: Sed, boca seca, náusea, vómitos, diarrea. Genitourinarias: Retención urinaria. Otras: Desbalance de fluidos y electrolitos, deshidratación, dolor local, fiebre, es-calofríos, urticaria, visión borrosa, rinitis. INTERACCIONES Incrementa la excreción urinaria del litio. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes anúricos, en casos de edema o congestión pulmonar severa, insuficiencia cardiaca congestiva, edema asociado a fragilidad capilar o excesiva permeabilidad de las membranas, hemorragia intracraneana activa, deshidratación severa, daño renal progresivo o disfunción renal después de instituir la terapia con manitol y en pacientes con hipersen-sibilidad al medicamento. Previo a la administración de manitol, se debe corregir cualquier trastorno hidro-elec-trolítico pre-existente y establecer un flujo urinario adecuado. Durante el tra-tamiento se debe vigilar el gasto uri-nario, la volemia y las concentraciones séricas de electrolitos. Contraindica-do en pacientes con insuficiencia car-diaca o congestión pulmonar progre-siva después de la institución de la terapia con manitol. No se conoce la Formulario Terapéutico Nacional * 2ed. 2004 do a la temperatura corporal antes de administrarlo. No infundir las soluciones de manitol si contienen cristales. Cuando se trata de soluciones de manitol al 20-25%, el equipo de infusión debe incluir un filtro. Las soluciones al 15 ó 20% siempre deben ser filtradas. SOBREDOSIS Manejo sintomático y terapia de soporte. FENTOLAMINA FARMACOLOGÍA Antagonista competitivo de los receptores alfa1 y alfa2- adrenérgicos; causa vasodilatación, con la consecuente disminución de la resistencia vascular periférica y de la presión arterial. FARMACOCINÉTICA No se conoce bien su farmacocinética. Produce un efecto máximo de reduc-ción de la presión arterial a los 2 minu-tos de su administración IV y la presión retorna a los valores previos a los 15-30 minutos. Sólo un 10% de la dosis administrada se recupera en la orina como fármaco intacto. INDICACIONES Diagnóstico del feocromocitoma y prevención de la hipertensión paroxística previa y durante la es 2,5 mg (si la respuesta es negativa, puede probarse con 5 mg antes de concluir que la prueba es negativa) y en niños 1 mg. Se considera como respuesta positiva a la prueba, una reducción de 35 mm Hg en la presión sistólica y de 25 mm Hg en la presión C diastólica. Si la presión no cambia, se eleva o su disminución no alcanza los valores antes señalados, el diagnóstico se considera negativo. La dosis para el manejo de la hipertensión previa a la remoción quirúrgica del feocromocitoma en adultos es de 5 mg 1-2 horas antes de la operación y en niños 1 mg o 0,1 mg/kg (3 mg/m2). Repetir en caso necesario. Durante la cirugía se puede usar las mismas dosis, según sea necesario. REACCIONES ADVERSAS Cardiovasculares: Hipotensión aguda y prolongada, taquicardia, infarto del miocardio, arritmias, shock, hipotensión ortostática, enrojecimiento. Gastrointestinales: Náusea, vómitos, diarrea. Neurológicas: Mareos, debilidad, oclusión cerebrovascular, espasmo cerebrovascular. Otras: Congestión nasal. INTERACCIONES Antagoniza el efecto de la epinefrina, la norepinefrina y de la efedrina, produciéndose marcada hipotensión. 2ed. 2004 * Formulario TerapÈutico Nacional 107 pacientes con gastritis o úlcera péptica. El uso en el embarazo debe limitarse a circunstancias de verdadera necesidad en las que el potencial beneficio a la madre supere al posible riesgo para el feto. Suspender la lactancia durante el tratamiento. Los pacientes ancianos tienen mayor riesgo de desarrollar hipotermia. SOBREDOSIS Manejo sintomático y tratamiento de soporte. Usar norepinefrina para manejar la hipotensión. No es recomendable el empleo de epinefrina debido a que puede causar hipotensión paradójica. PENTOXIFILINA FARMACOLOGÍA Derivado de la metilxantina que posee propiedades hemorreológicas. Favorece el flujo sanguíneo tisular a través de los vasos de pequeño calibre por flexibilización de la pared del eritrocito y disminución de la viscosidad de la sangre. La capacidad para generar la deformación del glóbulo rojo, sin alterar su integridad, parece ser debida a un efecto inhibidor de la enzima fosfodiesterasa de la pared celular que da lugar a un incremento del AMPc. La reducción de la viscosidad sanguínea se cree que está asociada a una disminución de la concentración de 108 oxígeno en pacientes enfermedad arterial periférica. con FARMACOCINÉTICA Aunque se absorbe rápida y casi completamente por vía oral, sólo un 1050% de la dosis administrada llega a la circulación sistémica como fármaco intacto debido al efecto metabólico de primer pasaje. Después de iniciada la terapia por vía oral para el tratamiento de la claudicación intermitente, se observa un inicio del efecto a los 15-30 días, el cual se hace máximo en 8 semanas. Se une en un 45% a la membrana de los eritrocitos, donde es rápidamente metabolizada y convertida en el metabolito activo 5-hidroxihexildimetilxantina. Adicionalmente, el metabolismo a nivel hepático da origen a otros productos, de los cuales uno, el derivado 3-carboxipropil-dimetilxantina (también activo) contribuye junto con el anterior al efecto hemorreológico. En adultos con función renal normal, un 95% de la dosis es recuperado en la orina en 24 horas, fundamentalmente como metabolitos, mientras un 4% es excretado por las heces. La vida media de eliminación de la pentoxifilina es de 0,5-1 horas, la del metabolito 5-hidroxihexil es 0,83 horas y la del metabolito carboxipropil 1-1,5 horas. INDICACIONES Claudicación intermitente asociada a Formulario Terapéutico Nacional * 2ed. 2004 gastrointestinal y en el SNC son dependientes de la dosis, si el paciente desarrolla esos efectos, la dosis debe ser reducida a 800 mg/día. La dosis usual por vía IV es de 100 mg, diluidos en 250-500 ml de solución salina o glucosa al 5% por infusión lenta en 90180 minutos, con incrementos diarios de 50 mg hasta un máximo de 400 mg/día. REACCIONES ADVERSAS Cardiovasculares: Angina, dolor en el pecho, arritmias, palpitaciones, rubor. Gastrointestinales: Incomodidad abdominal, flatulencia, distensión abdominal, diarrea, dispepsia, náusea, vómitos, anorexia. Hematológicas: Discrasias sanguíneas, disminución de los niveles de fibrinógeno. Hepáticas: Aumento de las enzimas hepáticas. Neurológicas: Agitación, nerviosismo, mareos, somnolencia, cefalea, insomnio, temblor, visión borrosa. Reacciones de hipersensibilidad: Reacciones anafilactoides. INTERACCIONES Puede incrementar la toxicidad de la teofilina. Por su efecto antiagregante plaquetario podría incrementar la respuesta anticoagulante a los medicamentos cumarínicos. Aumenta el efec-to hipotensor de los antihipertensivos. En pacientes fumadores puede produ-cir vasoconstricción. con precaución en pacientes con falla hepática y/o renal, hipotensión, enfermedad coronaria, cirugía reciente, hemorragia digestiva y terapia anticoagulante. No debe usarse en el embarazo a menos que el beneficio potencial para la madre supere al C riesgo potencial para el feto. Como la pentoxifilina se secreta en la leche materna, debe tomarse la decisión de suspender la lactancia o el medicamento, dependiendo de la importancia del medicamento para la madre. Se recomienda la evaluación periódica de la función hematológica y la función cardiovascular. No se conoce bien la eficacia y seguridad de su uso en niños. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Vigilancia de la función car-diaca y de la función respiratoria. Aten-ción a la posibilidad de PREDNISOLONA / ANESTÉSICO LOCAL (PREDNISOLONA CAPRONATO / ANEST…SICO LOCAL) FARMACOLOGÍA La aplicación tópica de prednisolona en la mucosa ano-rectal para el manejo sintomático de las hemorroides produce un efecto antiinflamatorio, antiprurítico y vasoconstrictor a 2ed. 2004 * Formulario TerapÈutico Nacional 109 aclarados (ver farmacología de PREDNISOLONA en la sección ENDOCRINOS). La presencia de un anestésico local en la formulación contribuye al alivio de la afección. FARMACOCINÉTICA Después de la administración en forma de crema o de supositorio puede ocu-rrir absorción sistémica parcial del es-teroide (ver farmacocinética de la PREDNISOLONA en la sección ENDOCRINOS). INDICACIONES Hemorroides internas y externas, proctitis, prurito y fisura anal. RÉGIMEN DE DOSIFICACIÓN Se recomiendan 3 supositorios al día o 3-4 aplicaciones diarias del ungüento al 2%. INFORMACIÓN AL PACIENTE Aplicar preferiblemente después de la defecación, previo lavado de la zona. Si la condición se agrava o no mejora en 15 días, suspender el uso del meATENOLOL (Antihipertensivo y Tratamiento de Angina de Pecho) FARMACOLOGÍA Antagonista competitivo cardioselectivo de los receptores beta1adrenérgicos, inhibe la secreción de la renina plasmática y el sistema renina-angiotensina-aldosterona, y dismi-nuye la conducción a nivel del nodo aurículo-ventricular (AV). No posee actividad simpatomimética ni ejerce efectos significativos sobre los receptores beta2-adrenérgicos en dosis terapéuticas. FARMACOCINÉTICA Después de la administración oral, se absorbe rápida pero incompletamente (50-60%) en el tracto gastrointestinal. Se produce una respuesta inicial en 60 INTERACCIONES No se describen interacciones por esta minutos, la cual se hace máxima a las 2-4 horas y persiste por 24 horas. Se vía de administración. une a las proteínas plasmáticas en un 5-15% y se distribuye ampliamente a CONTRAINDICACIONES/ PRECAUCIONES los tejidos, excepto al SNC donde Contraindicado en casos de hipersen- penetra en muy escasa proporción. sibilidad a alguno de los constituyentes Puede llegar a alcanzar en el feto de la formulación. No usar en caso de concentraciones superiores al 50% de inflamación, dolor o prurito de origen la concentración sanguínea materna. infeccioso (bacteriano, viral, micótico No sufre metabolismo hepático, por lo que* 2ed. se excreta 110 Formulario Terapéutico Nacional 2004 en un 40-50% como REACCIONES ADVERSAS Con el tratamiento prolongado puede ocurrir atrofia cutánea. INDICACIONES Tratamiento de la hipertensión arterial y de la angina de pecho. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos, iniciar con 50 mg/día. Si no se logra una respuesta adecuada en una semana, incrementar la dosis a 100 mg/día. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, hipotensión, insuficiencia cardiaca, claudicación intermitente, extremidades frías. Dermatológicas: Erupciones. Gastrointestinales: Náuseas, diarrea. Hematológicas: Aumento del contaje de plaquetas. Hepáticas: Aumento de los niveles de las aminotransferasas, fosfatasa alcalina, deshidrogenasa láctica. Metabólicas: Aumento de los niveles séricos de potasio y de ácido úrico, aumento o disminución de los niveles de glucosa en pacientes diabéticos. Renales: Aumento del BUN y de la creatinina. Respiratorias: Disnea, broncoespasmo, sibilancias. Neurológicas: Fatiga, letargia, vértigo, somnolencia, mareos, depresión, cansancio, aturdimiento. Otras: Fiebre, dolor en las piernas. INTERACCIONES En combinación con bloqueantes de los canales de calcio u otros agentes hipotensores, puede producirse alterarse los requerimientos de dosis en pacientes diabéticos previamente estabilizados. Con agentes antiarrítmicos y con anestésicos generales se incrementa el riesgo de depresión cardiaca, bradicardia y bloqueo AV. C CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, bloqueo AV, shock cardiogénico, bradicardia sinusal y durante el embarazo. Usar con precaución en pacientes con falla renal, hipertiroidismo, enfermedad vascular periférica, insuficiencia cardiaca congestiva y enfermedad broncoespástica. Usar con precaución en pacientes diabéticos ya que puede enmascarar la taquicardia que ocurre con la hipoglicemia; también en pacientes con hipertiroidismo debe evitarse la suspensión abrupta del atenolol ya que puede precipitarse una tormenta tiroidea. La suspensión o interrupción abrupta de la terapia puede precipitar un síndrome anginoso e, inclusive, la posibilidad de un infarto del miocardio; por ello, se recomienda la suspensión gradual del medicamento en 1-2 semanas. No se conoce bien la eficacia y seguridad de su uso en niños. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS 2ed. 2004 * Formulario TerapÈutico Nacional 111 paciente con isoproterenol IV o con un marcapaso, según la severidad de la condición clínica. Si hay hipotensión, administrar fluidos IV y/o medicamentos vasopresores. Vigilar la presión arterial. Si ocurre insuficiencia cardiaca, tratar al paciente con digitálicos, diuréticos y oxígeno. El uso de glucagon ha demostrado ser útil. En casos extremos, usar la hemodiálisis. INFORMACIÓN AL PACIENTE No suspender el tratamiento abruptamente sin consultar al médico. No ingiera medicamentos antigripales o descongestionantes nasales. Informar al médico si presenta los siguientes síntomas: Dificultad para respirar, pulso lento, edema en las piernas y los tobillos. Este medicamento puede alterar los niveles de azúcar en la sangre, de particular interés en los METOPROLOL FARMACOLOGÍA Antagonista selectivo y competitivo de los receptores beta1-adrenérgicos en el corazón, suprime la actividad de la renina plasmática y disminuye la conducción nodal sino-auricular (SA) y aurículo-ventricular (AV). No posee actividad simpaticomimética intrínseca ni interactúa significativamente con los receptores beta 2 -adrenérgicos, a 112 FARMACOCINÉTICA Aunque se absorbe rápida y completamente en el tracto gastrointestinal, sólo un 50% de la dosis administrada alcanza la circulación sistémica como fármaco intacto debido a un intenso efecto de metabolismo de primer pasaje. Luego de iniciada la terapia, se observa una respuesta terapéutica máxima en aproximadamente 7 días. Se une a las proteínas plasmáticas en un 11-12% y se distribuye ampliamente a los tejidos, alcanzando concentraciones importantes en el corazón, el hígado, los pulmones, el SNC y los eritrocitos. El metabolismo hepático por la CYP2D6 origina productos inactivos que se excretan en un 95% por la orina, junto a un 5% del fármaco intacto. El metoprolol atraviesa la barrera hematoencefálica y alcanza concentraciones iguales al 78% de la plasmática. La vida media de eliminación es de 3-7 horas. INDICACIONES Tratamiento de la hipertensión arterial y de la angina de pecho. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos, iniciar con 50-100 mg/día en dosis única o dividida, e incrementar gradualmente, en caso necesario, a intervalos semanales hasta lograr la respuesta deseada. No exceder 400 mg/día. Formulario Terapéutico Nacional * 2ed. 2004 edema periférico. Gastrointestinales: Náusea, diarrea, boca seca, dolor de estómago, constipación, flatulencia y acedía. Hepáticas: Aumento de los niveles de las aminotransferasas y de la fosfatasa alcalina. Neurológicas: Fatiga, mareos, depresión, confusión mental, pérdida de la memoria a corto plazo, cefalea, tinitus, pesadillas e insomnio. Reacciones de hipersensibilidad: Erupciones, prurito, empeoramiento de la psoriasis. Respiratorias: Disnea, broncoespasmo. Otras: Aumento de los niveles de la deshidrogenasa láctica y del ácido úrico, dolor musculoesquelético, visión borrosa. INTERACCIONES El metoprolol, en combinación con otros medicamentos hipotensores puede causar efectos aditivos. Con los digitálicos, los bloqueantes del calcio no dihidropiridínicos y los anestésicos generales, se incrementa el riesgo de depresión cardiaca, bradicardia y blo-queo AV. El verapamilo aumenta significativamente la biodisponibilidad oral del metoprolol. La quinidina, la fluoxetina, la paroxetina y la propafenona pueden inhibir el metabolismo hepático del metoprolol e incrementar sus niveles plasmáticos. insuficiencia cardiaca congestiva y enfermedad broncoespástica. La suspensión o interrupción abrupta de la terapia puede precipitar un síndrome anginoso e, inclusive, la posibilidad de un infarto del miocardio. No se conoce bien la eficacia y seguridad de su C empleo en niños. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que el beneficio potencial supere al posible riesgo para el feto. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y de soporte. En casos de bradicardia severa tratar con atropina IV y si la condición persiste, usar isoproterenol IV cuidadosamente. Si se presenta hipotensión severa usar fármacos vasopresores y en casos refractarios, tratar con glucagon IV. Si ocurre falla cardiaca, tratar con digitálicos, diuréticos y oxígeno. En caso de broncoespasmo usar aminofilina o agonistas de los receptores beta2adrenérgicos. INFORMACIÓN AL PACIENTE No alterar la dosificación ni interrumpir CONTRAINDICACIONES/ la terapia sin el conocimiento del méPRECAUCIONES dico. Tomar el medicamento inmediaContraindicado en pacientes con hitamente después de una comida. 113 2ed. 2004 * Formulario TerapÈutico Nacional diato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, tal como dificultad AMLODIPINO (AMLODIPINA BESILATO) FARMACOLOGÍA Bloqueante de los canales de calcio derivado de la dihidropiridina. Inhibe el influjo de calcio extracelular a través de la membrana de las células de la musculatura lisa vascular y, en menor grado, del miocardio. Produce dilatación de las arterias periféricas, dando lugar a una disminución de la resistencia periférica y de la presión arterial. Además, se ha demostrado que la amlodipina bloquea la vasoconstricción y restaura el flujo sanguíneo en las arterias coronarias y las arteriolas en respuesta a iones y agonistas tales como: calcio, potasio, epinefrina, serotonina y análogos del tromboxano A2. FARMACOCINÉTICA Después de la administración oral se absorbe en un 64-90% alcanzando niveles séricos pico en 6-12 horas y concentraciones al estado estable a los 7-8 días de iniciada la terapia. Se une a las proteínas plasmáticas en un 93% y se metaboliza extensamente (90%) en el hígado, transformándose en productos inactivos que se excretan en un 60%, junto a un 10% de fármaco intacto por la orina. La vida media de 114 y la angina de pecho. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos iniciar con 2,5-5 mg/día e incrementar la dosis, según la respuesta y la tolerancia del paciente, a intervalos de 7-14 días hasta lograr la respuesta deseada, la cual se ubica, en la mayoría de los casos, en el rango de 5-10 mg/día. REACCIONES ADVERSAS Cardiovasculares: Arritmias (incluyendo taquicardia y fibrilación ventricular), bradicardia, taquicardia, hipotensión ortostática, palpitaciones, edema e isquemia periférica. Gastrointestinales: Náuseas, vómito, dolor abdominal, anorexia, disfagia, sequedad de la boca, sed, constipación, dispepsia, diarrea, flatulencia, hiperplasia gingival, ictericia y pancreatitis. Hematológicas: Leucopenia y trombocitopenia. Neurológicas: Dolor de cabeza, fatiga, somnolencia, insomnio, astenia, depresión, ansiedad, neuropatía periférica, hipoestesia, parestesia, temblor, vértigo, nerviosismo, pesadillas y despersonalización. Reacciones de hipersensibilidad: Erupción, prurito, dermatitis, eritema multiforme y angioedema. Otras: Artralgia, mialgia, disfunción sexual (en hombres y mujeres), disnea, diaforesis, epistaxis, trastornos visuales, frecuencia uri- Formulario Terapéutico Nacional * 2ed. 2004 vasodilatadores puede provocar un efecto hipotensor aditivo. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otros derivados de la dihidropiridina. Usar con precaución en pacientes con insuficiencia cardiaca congestiva, insuficiencia hepática, estenosis aórtica y en ancianos. En algunos pacientes con obstrucción coronaria severa sometidos a terapia con bloqueantes de los canales de calcio, se ha descrito agravamiento de la angina e infarto del miocardio. El uso durante el embarazo debe limitarse a circunstancias de verdadera necesidad en las que el beneficio a la madre supere al riesgo potencial para el feto. No se conoce la seguridad de su uso en niños. Durante el tratamiento se recomienda suspender la lactancia. Usar con precaución en pacientes con insuficiencia hepática. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Si ocurre hipotensión, manejar colocando al paciente en posición Trendelenburg, administrar fluidos IV, agentes vasopresores y/o sales de calcio IV, según la severidad del cuadro. NIFEDIPINO FARMACOLOGÍA Bloqueante de los canales de calcio, derivado de dihidropiridina. Inhibe el influjo de calcio extracelular a través de la membrana de las células del C miocardio y de la musculatura lisa vascular, causando una depresión del proceso de contractilidad cardiaca y dilatación de las arterias coronarias y arterias periféricas. Estos efectos dan lugar a una disminución de la resistencia periférica y de la presión arterial. Reduce el consumo de energía y los requerimientos miocárdicos de oxígeno. A diferencia del verapamilo, el nifedipino, en dosis terapéuticas, no deprime la conducción nodal SA y AV. FARMACOCINÉTICA Aunque se absorbe bien en el tracto gastrointestinal, sólo un 45-75% de la dosis alcanza la circulación sistémica como fármaco intacto debido a un efecto de metabolismo de primer pasaje. Se une a las proteínas plasmáticas en un 92-98% y se metaboliza extensamente en el hígado, transformándose en productos inactivos que se excretan en un 80% por la orina y el resto por las heces. La vida media de eliminación plasmática es de 2-5 horas y se incrementa en pacientes con insuficiencia hepática. INDICACIONES 2ed. 2004 * Formulario TerapÈutico Nacional 115 de la angina, la dosis inicial en adultos es 10-20 mg 3 veces al día si se usan formas de dosificación de liberación convencional o 30-60 mg/día si se usan formas de dosificación de liberación sostenida. En caso necesario, se pueden hacer incrementos graduales de la dosis a intervalos diarios o semanales hasta lograr la respuesta deseada. No se debe exceder 180 mg/día con las formas de liberación convencional o 90-120 mg/día con las formas de liberación sostenida. REACCIONES ADVERSAS Cardiovasculares: Edema periférico, hipotensión, palpitaciones, síncope, insuficiencia cardiaca, edema pulmonar. Dermatológicas: Erupción, prurito. Gastrointestinales: Náusea, diarrea, constipación, molestias abdominales. Hematológicas: Discrasias sanguíneas. Hepáticas: Aumento de las enzimas hepáticas. Neurológicas: Mareos, aturdimiento, cefalea, debilidad, nerviosismo. Respiratorias: Congestión nasal, disnea, tos, sibilancias. Otras: calambres musculares, hipokalemia, visión borrosa, rubor, fiebre, trastornos sexuales, cambios de humor. INTERACCIONES El nifedipino, en combinación con bloqueantes de los receptores betaadrenérgicos y en pacientes sometidos a anestesia con fentanilo, puede 116 con nifedipino y anticoagulantes cumarínicos. La cimetidina y el alcohol pueden incrementar las concentra-ciones séricas de nifedipino. La administración de nifedipino conjun-tamente con jugo de toronja (grape-fruit), aumenta marcadamente la biodisponibilidad del nifedipino y sus análogos. CONTRAINDICACIONES/ PRECAUCIONES El nifedipino está contraindicado en pacientes con hipersensibilidad al medicamento o a otros bloqueantes de los canales de calcio, estenosis aórtica avanzada, infarto miocárdico reciente (menos de 4 semanas) e insuficiencia hepática severa. Aún cuando el medicamento ha sido empleado para el manejo de la crisis hipertensiva por vía oral o sublingual, numerosos reportes de casos de hipotensión severa, infarto del miocardio y muerte, asociados al uso del nifedipino en dicha circunstancia, sugieren una contraindicación en tal sentido. Dado que al inicio del tratamiento y durante la fase de ajuste de la dosis han ocurrido casos de hipotensión excesiva, sobre todo en pacientes con terapia con bloqueantes de los receptores beta-adrenérgicos, se recomienda la vigilancia frecuente de la presión arterial durante ese período. Algunos pacientes con enfermedad coronaria obstructiva han Formulario Terapéutico Nacional * 2ed. 2004 puede provocar edema periférico en algunos individuos, es importante que en pacientes con angina complicada con insuficiencia cardiaca congestiva se establezca un diagnóstico diferencial que permita discriminar cuando esta manifestación no es debida a una disfunción ventricular izquierda. Usar con precaución en pacientes con insuficiencia cardiaca congestiva, enfermedad hepática leve a moderada, en el embarazo y durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y de soporte. Vigilar la función cardiaca y la respiratoria. Si ocurre hipotensión, colocar al paciente en posición Trendelenburg, administrar fluidos IV, agentes vasopresores y sales de calcio, según la severidad del cuadro. INFORMACIÓN AL PACIENTE Evitar el consumo de alcohol. Notificar al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial mareos, desmayos, hipotensión severa e inflamación de las manos y los pies. Advertir al paciente que al inicio del NIMODIPINO FARMACOLOGÍA Bloqueante de los canales de calcio. Inhibe el influjo de calcio extracelular a través de la membrana de la musculatura lisa vascular produciendo un efecto vasodilatador. A diferencia de C otros bloqueantes del calcio, el nimodipino actúa predominantemente sobre el lecho vascular cerebral en virtud de una elevada liposolubilidad que le permite atravesar con facilidad la barrera hematoencefálica y alcanzar altas concentraciones en el SNC. FARMACOCINÉTICA Se absorbe rápida y casi completamente por vía oral alcanzando concentraciones plasmáticas pico en 1 hora. Sin embargo, debido a un intenso efecto metabólico de primer pasaje, su biodisponibilidad es de alrededor de un 13% de la dosis administrada. Los alimentos reducen su absorción. Se une a las proteínas plasmáticas en un 95%. Se metaboliza extensamente en el hígado transformándose en productos inactivos que se excretan en la orina y las heces. La vida media de eliminación es de 1,7-9 horas cuando se administra por vía oral y de 1-1,5 horas tras la administración por vía IV. INDICACIONES Prevención del déficit neurológico secundario a espasmo vascular ce117 2ed. 2004 * Formulario TerapÈutico Nacional consecutivos, iniciando la terapia dentro de las 96 horas siguientes a la hemorragia subaracnoidea. Por vía IV, iniciar el primer día con una infusión continua de 0,5 mg/hora, seguido por 1 mg/hora el segundo día y por 2 mg/hora durante los días 3 al 12. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, edema, taquicardia, rubor. Gastrointestinales: Constipación, diarrea, náuseas, vómito, cólicos abdominales. Hepáticas: Elevación de los niveles de las enzimas hepáticas. Neurológicas: Dolor de cabeza, depresión, mareo, desmayo, confusión, psicosis, cansancio, somnolencia. Respiratorias: Disnea. Otras: Mialgia, hiperglicemia. INTERACCIONES Puede potenciar el efecto de los medicamentos antihipertensivos, así como el de otros bloqueantes de los canales de calcio. La cimetidina puede aumentar los niveles séricos de el nimodipino, por inhibición del metabolismo hepático. SOBREDOSIS Lavado gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y de soporte. En caso de hipotensión severa colocar al paciente en posición de Trendelenburg, administrar fluidos por vía IV o medicamentos vasopresores, según la severidad del caso. INFORMACIÓN AL PACIENTE En caso de administración oral, tomar el nimodipino una hora antes o 2 horas después de las comidas. Notificar al VERAPAMILO FARMACOLOGÍA Bloqueante de los canales de calcio, con efecto inotrópico negativo y propiedades antiarrítmicas. Inhibe el influjo del calcio extracelular a través de la membrana de las células del miocardio y de la musculatura lisa vascular, interfiriendo con el proceso de contractilidad cardiaca y dilatación de las coronarias y de las arterias periféricas, dando lugar a una disminución de la resistencia periférica y de la presión arterial. Esto disminuye el consumo de energía a nivel del miocardio y los requerimientos de oxígeno, lo cual probablemente explica la eficacia del verapamilo en el tratamiento de la angina de esfuerzo estable crónica. Ejerce un efecto directo sobre los CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a otros bloqueantes de los canales de calcio. Durante la terapia se recomienda vigilar periódicamente la presión arterial. Usar con precaución 118 Formulario Terapéutico Nacional * 2ed. 2004 FARMACOCINÉTICA Aunque se absorbe bien (90%) en el tracto gastrointestinal, sólo un 20-35% de la dosis alcanza la circulación sistémica debido al efecto de metabolismo de primer pasaje. Después de la administración oral, la respuesta antianginosa se inicia en 3090 minutos, y se hace máxima en 8 horas. La respuesta antihipertensiva se inicia en 1-2 horas y alcanza su máxima intensidad en 6-8 horas. Se une a las proteínas plasmáticas en un 90% y se distribuye rápidamente a los tejidos. Se metaboliza en el hígado casi por completo, transformándose en varios metabolitos, siendo solamente el norverapamilo relativamente activo. Se elimina en un 70% por la orina como metabolitos y 3-4% como fármaco intacto y en un 16% por las heces dentro de 5 días. La vida media de eliminación es de 4-12 horas y puede incrementarse en pacientes con falla hepática y/o renal. INDICACIONES Hipertensión arterial y angina de pecho, por vía oral, y taquicardia supraventricular, por vía IV. RÉGIMEN DE DOSIFICACIÓN Para el tratamiento por vía oral de la hipertensión arterial y de la angina con formas de dosificación de liberación convencional en adultos iniciar con 80 mg 3 veces al día e incrementos minutos, bajo control electrocardiográfico. Si no se logra la respuesta deseada, administrar 0,15 mg/kg adicionales, 30 minutos más tarde. En niños de 1-15 años iniciar con 0,1-0,3 mg/kg (sin exceder de 5 mg totales) y repetir, en caso necesario, 30 minutos C más tarde. En niños menores de 1 año administrar 0,1-0,2 mg/kg. REACCIONES ADVERSAS Cardiovasculares: Hipotensión transitoria, insuficiencia cardiaca, edema pulmonar, bradicardia, bloqueo AV de primero, segundo y tercer grado, asístole ventricular, fibrilación ventricular, angina, edema periférico, infarto del miocardio, síncope, accidente cerebrovascular. Dermatológicas: Equimosis o hematomas, hiperqueratosis, máculas, urticaria por sudoración. Gastrointestinales: Náusea, constipación, boca seca. Hepáticas: Aumento de las enzimas hepáticas. Neurológicas: Mareos, cefalea, astenia, trastorno del equilibrio, insomnio, parestesia, síntomas psicóticos, somnolencia. Reacciones de hipersensibilidad: Erupción de la piel, exantema, síndrome de StevensJohnson, eritema multiforme. Respiratorias: Disnea. Otras: Hiperplasia gingival, visión borrosa, tinitus, ginecomastia, galactorrea/hiperprolactinemia, poliuria, impotencia, manchado menstrual, artralgia. 2ed. 2004 * Formulario TerapÈutico Nacional 119 hipotensor aditivo. El verapamilo puede aumentar los niveles plasmáticos de la carbamazepina, la digoxina, la ciclosporina, la teofilina y el alcohol, incrementando los efectos y el riesgo de toxicidad. El uso con anestésicos volátiles favorece la posibilidad de depresión cardiovascular. Puede potenciar la actividad de los bloqueantes neuromusculares e incrementar la sensibilidad al litio, predisponiendo al paciente a la toxicidad por litio. Con la disopiramida, pueden aumentar los efectos inotrópicos negativos. Los suplementos de calcio, en cantidades elevadas, pueden antagonizar el efecto del verapamilo. La rifampicina reduce la biodisponibilidad del verapamilo y el fenobarbital promueve su eliminación. El verapamilo aumenta la eficacia de la doxorubicina y la absorción del verapamilo puede ser disminuida por varios antineoplásicos y por la prednisona. Administrado con yerba mate puede disminuir la depuración de las metilxantinas contenidas en la yerba y causar toxicidad. El verapamilo incrementa los efectos del alcohol. y trastornos de la transmisión neuromuscular. El uso en el embarazo debe limitarse a circunstancias de verdadera necesidad y previa consideración del balance riesgo/beneficio. Durante el tratamiento se recomienda la vigilancia periódica de la función hepática y renal. Se debe evitar la suspensión brusca de la terapia, debido al riesgo de crisis hipertensiva de rebote. Debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y de soporte. Si ocurre hipotensión, manejarla colocando al paciente en posición Trendelenburg, administrar fluidos IV, agentes vasopresores y/o sales de calcio por vía IV, según la severidad del cuadro. En caso de bradicardia o bloqueo AV de cualquier grado, usar atropina por vía IV, isoproterenol, norepinefrina, sales de calcio IV o colocar un marcapasos temporal. INFORMACIÓN AL PACIENTE Durante el tratamiento se debe evitar CONTRAINDICACIONES/ el consumo de alcohol. No se debe PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a ENALAPRIL otros bloqueantes de los canales de calcio, infarto miocárdico reciente, hi- FARMACOLOGÍA p o t e n s i ó n s e v e r a , s h o c k El enalapril es un pro-fármaco que, 120 Formulario Terapéutico Nacional * 2ed. 2004 una vez administrado, se hidroliza a enalaprilato, el cual inhibe la enzima convertidora de angiotensina (ECA), evitando la transformación de angiotensina I en angiotensina II (potente vasoconstrictor endógeno), impidiendo con ello la acción estimulante de ésta sobre la corteza suprarrenal, lo que daría lugar a la secreción de aldosterona. El resultado de esta supresión del sistema reninaangiotensina-aldosterona es una reducción de los niveles plasmáticos de angiotensina II y de la secreción de aldosterona, lo cual se traduce en una disminución de la resistencia vascular sistémica y de la presión arterial. La disminución de la secreción de aldosterona ocasiona un pequeño aumento en el potasio sérico. Los pacientes de etnia negra (población hipertensa usualmente con baja renina), muestran una menor respuesta promedio al enalapril como monoterapia, que los pacientes de otras etnias. Aún no se ha estudiado adecuadamente cómo afecta este aspecto étnico a la respuesta antihipertensiva de los IECA en poblaciones mestizas. FARMACOCINÉTICA Se absorbe en un 60% en el tracto gastrointestinal, produciendo una respuesta hipotensora inicial en 1-4 horas que se hace máxima en 4-8 transformándose en más enalaprilato. Se excreta principalmente como metabolito y, en menor proporción, como fármaco intacto en la orina (61%) y en las heces (33%). La vida media de eliminación del enalapril es de 1,3 horas y del enalaprilato 5,9-35 horas C (promedio 11 horas) y se prolonga en pacientes con insuficiencia renal severa. INDICACIONES Tratamiento de la hipertensión arterial e insuficiencia cardíaca izquierda. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis inicial en adultos es de 5 mg/día, con incrementos graduales a intervalos de 2-4 semanas hasta lograr la respuesta antihipertensiva adecuada, lo que se logra generalmente con dosis única de 10-40 mg/día o dividido en 2 dosis iguales. En niños, iniciar con 0,08 mg/kg/día (máximo 5 mg) con incrementos graduales hasta lograr la respuesta antihipertensiva adecuada, sin exceder 0,58 mg/kg/día. Para tratamiento de la insuficiencia cardíaca izquierda en adultos se inicia con 2,5 mg 1-2 veces al día, seguidos por incrementos graduales hasta lograr la respuesta adecuada, sin exceder de 20 mg 2 veces al día. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, hipotensión, síncope, dolor 121 2ed. 2004 * Formulario TerapÈutico Nacional depresiÛn de la mÈdula Ûsea. Hep·ticas: AlteraciÛn de las pruebas de funciÛn hep·tica y de la bilirrubina. NeurolÛgicas: Dolor de cabeza, mareos, fatiga, insomnio, nerviosismo, parestesia, astenia y depresiÛn. Reacciones de hipersensibilidad: Eritema, urticaria, prurito, alopecia y dermatitis. Renales: DisminuciÛn de la funciÛn renal (en pacientes con estenosis bilateral de la arteria renal o insuficiencia cardiaca), aumento de los niveles del BUN y de la creatinina. Respiratorias: Tos seca persistente, broncoespasmo, disnea. Otras: Hiperpotasemia, rinorrea, visiÛn borrosa, diaforesis, calambres, alteraciones del gusto, tinitus, disminuciÛn de la libido, impotencia, angioedema y anafilaxis. INTERACCIONES Los medicamentos con propiedades vasodilatadoras, los anestésicos y los diuréticos incrementan el efecto hipotensor del enalapril. Los AINEs pueden reducir el efecto antihipertensivo del enalapril. El enalapril puede disminuir la eliminación renal del litio y generar toxicidad. Los diuréticos ahorradores de potasio, los suplementos de potasio y los sustitutos de la sal aumentan el riesgo de hiperpotasemia. En caso de administración de insulina e hipoglicemiantes orales, aumenta el riesgo de hipoglicemia, especialmente al inicio 122 muerte fetal y neonatal cuando se administran a mujeres embarazadas. Cuando se detecta el embarazo, el medicamento debe ser suspendido lo más pronto posible. Durante el tratamiento se debe vigilar periódicamente la función hepática, la renal, la hematológica y los niveles séricos de potasio. Usar con precaución en pacientes con falla hepática y/o renal, insuficiencia cardiaca congestiva, estenosis aórtica, depleción de volumen, en ancianos y durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. El metabolito activo, enalaprilato, puede ser removido por hemodiálisis y ha sido removido de la circulación neonatal por diálisis peritoneal. INFORMACIÓN AL PACIENTE No alterar la dosificación ni suspender el tratamiento sin la autorización del médico. Notificar al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial fiebre recurrente o infecciones (sugestivo de neutropenia Formulario Terapéutico Nacional * 2ed. 2004 LOSARTÁN FARMACOLOGÍA Agente antihipertensivo. Antagoniza selectiva y competitivamente los receptores AT1 de la angiotensina II, encontrados en tejidos como el músculo liso vascular y la glándula adrenal. FARMACOCINÉTICA Aunque se absorbe bien por vía oral, sólo un 33% de la dosis alcanza la circulación como fármaco intacto debido a un intenso efecto metabólico de primer paso hepático. Dicho metabolismo, a través del citocromo P450, da lugar a un metabolito activo (E3174) con actividad farmacológica 1040 veces superior a la del losartán. El fármaco y su metabolito activo logran concentraciones séricas pico en 1-1,5 horas y 3-4 horas, respectivamente. La respuesta antihipertensiva inicial se observa en aproximadamente 6 horas, aunque se requieren hasta 3-6 semanas para observar el efecto máximo. Ambos compuestos se unen a las proteínas plasmáticas en un 98100%. Además del E-3174, el metabolismo hepático produce otros metabolitos, pero sin actividad farmacológica. Se excreta por la orina en un 35% (4% como fármaco intacto y 6% como E-3174) y en un 60% por vía biliar. La vida media de eliminación del losartán es de aproximadamente 2 horas y la del metabolito activo de 6 a 9 RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos, iniciar con 50 mg/día en dosis simple o divididos en 2 dosis iguales cada 12 horas, e incrementar a intervalos semanales hasta lograr la respuesta deseada, sin exceder la dosis diaria C total de 100 mg. REACCIONES ADVERSAS Cardiovasculares: Hipotensión postural, arritmias, edema, dolor anginoso y bloqueo AV de segundo grado. Gastrointestinales: Náuseas, vómitos, dolor abdominal, dispepsia y diarrea. Hematológicas: Anemia, disminución de la hemoglobina. Hepáticas: Elevación de las enzimas hepáticas, hiperbilirrubinemia y hepatitis (ocasionalmente). Neurológicas: Mareo, insomnio, somnolencia, fatiga, dolor de cabeza, astenia y depresión. Reacciones de hipersensibilidad: Erupción, prurito, eritema, fotosensibilidad, angioedema y anafilaxis. Renales: Aumentos menores del BUN y de la creatinina sérica, estenosis arterial renal y frecuencia urinaria. Respiratorias: Tos, congestión nasal, infecciones del tracto respiratorio alto, faringitis y sinusitis. Otras: Alopecia, calambres, mialgia, dolor de espalda y de las piernas, hiperpotasemia e hiponatremia. INTERACCIONES La combinación con diuréticos aho- 2ed. 2004 * Formulario TerapÈutico Nacional 123 activo, comprometiendo su eficacia terapéutica. El fenobarbital y la rifampicina pueden disminuir los niveles séricos de losartán por inducción de su metabolismo. El losartán aumenta los niveles plasmáticos y, por tanto, la toxicidad del litio. Los inhibidores de la CYP3A4 y de la CYP2C9 muestran una inhibición significativa de la formación del metabolito activo. Los AINEs antagonizan el efecto hipotensor del losartán y aumentan el riesgo de alteración renal. El uso concomitante con otros medicamentos antihipertensivos aumenta el efecto hipotensor. Los esteroides antagonizan el efecto hipotensor. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE El medicamento no debe ser usado durante el embarazo. En caso de embarazo o si una paciente planea quedar embarazada, discutir los riesgos con el médico. Debe evaluarse la presión arterial con regularidad. Notificar de inmediato al GEMFIBROZILO FARMACOLOGÍA Es considerado un agente con propiedades reguladoras de los lípidos. Es un derivado del ácido fíbrico. No se co-noce con precisión su mecanismo de acción. En pacientes con hiperlipopro-teinemia reduce las concentraciones séricas de triglicéridos y VLDL-colesterol, a la vez que eleva las de HDL-colesterol. Reduce la producción hepática de triglicéridos por inhibición de la lipólisis periférica y de la extracción hepática de ácidos grasos libres. También, inhibe la síntesis y aumenta la depuración de la apopro-teína B transportadora de VLDL, llevando a una disminución de la producción de VLDL. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y en el embarazo. Los medicamentos que actúan directamente sobre el sistema renina-angiotensina pueden causar morbilidad y muerte fetal. En los pacientes con depleción de volumen puede ocurrir hipotensión sintomática con losartán. Corregir la condición antes de usar losartán. Usar con precaución en pacientes con historia de alergia a otros medicamentos, con insuficiencia cardiaca congestiva, insuficiencia renal o hepática o estenosis de las arterias renales. Durante el tratamiento se debe vigilar 124 Formulario Terapéutico Nacional * 2ed. 2004 péutica a las 4-12 semanas. Se une a las proteínas plasmáticas en un 95% y se distribuye a los tejidos alcanzando concentraciones elevadas en el hígado y los riñones. Se metaboliza en el hígado y los metabolitos se excretan en un 70% por la orina como glucurónidos (menos de 2% como fármaco intacto) y un 6% por las heces. La vida media de eliminación es de 1,5 horas. INDICACIONES Coadyuvante en la prevención de la cardiopatía isquémica en pacientes con hiperlipidemia tipo IIb de Fredrikson, sin historia de enfermedad coronaria, que no responden adecuadamente a la dieta, ejercicios, reducción de peso y a otros agentes farmacológicos y que tienen un perfil lipídico caracterizado por bajos niveles séricos de HDL-colesterol y niveles elevados de triglicéridos y LDLcolesterol. Trata-miento de la hipertrigliceridemia (hi-perlipidemias tipo IV y V de Fredrikson) con riesgo de desarrollo de pan-creatitis que no responde adecua-damente a la dieta. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos es de 1200 mg/día dividida en 2 dosis iguales, 30 minutos antes del desayuno y de la cena. Algunos pacientes pueden requerir hasta 1500 mg/día. nia e hipoplasia medular. Hepáticas: Elevación de las enzimas hepáticas, ictericia, obstrucción del ducto biliar, colelitiasis y cólicos biliares. Músculoesqueléticas: Miopatía, mialgia, artral-gia, miastenia y, con estatinas, rabdo-miolisis. Neurológicas: Cefalea, C ma-reos, somnolencia, parestesia, neuritis periférica y depresión. Reacciones de hipersensibilidad: Erupción, prurito, ur-ticaria, dermatitis exfoliativa y angioe-dema. Otras: Disminución de la libido, disfunción eréctil, visión borrosa y tras-tornos del gusto. INTERACCIONES Puede potenciar el efecto de los anticoagulantes orales. Su asociación con inhibidores de la HMG-CoA reductasa (estatinas) puede provocar miopatía severa con rabdomiolisis. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con colecistopatía, disfunción hepática y falla renal severa. No debe combinarse con inhibidores de la HMG-CoA reductasa (estatinas) por el riesgo de rabdomiolisis. No se conoce la seguridad de su uso en niños, en el embarazo y durante la lactancia. Antes de iniciar la terapia debe practicarse medidas orientadas a corregir la hipercolesterolemia y la hipertrigliceri- 2ed. 2004 * Formulario TerapÈutico Nacional 125 determinaciones de los lípidos séricos, así como evaluaciones de la función hepática, renal y hematológica. Si al cabo de 3 meses no se ha logrado una respuesta satisfactoria, se debe sus-pender la terapia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Administrar 30 minutos antes del desayuno y de la cena. Respetar la dieta. Evitar el consumo de alcohol. Asistir al control mÈdico periÛdico. Notificar de inmediato al mÈdico si se presenta alguna reacciÛn o sintomatologÌa inusual durante el tratamiento, en especial dolor o debilidad muscular, o una fiebre no explicable. Notificar al SIMVASTATINA FARMACOLOGÍA Agente hipolipemiante perteneciente al grupo de las estatinas. Reduce la síntesis hepática de colesterol por inhibición competitiva de la enzima hidroximetilglutaril-CoA (HMG-CoA) reductasa, la cual cataliza la conversión de la HMG-CoA a mevalonato, que constituye un paso temprano en la síntesis del colesterol. 126 FARMACOCINÉTICA Aunque se absorbe bien en el tracto gastrointestinal, sólo un 5% de la dosis alcanza la circulación sistémica como fármaco intacto debido a un intenso efecto de metabolismo de primer pasaje. Con la administración continua, se observa una respuesta terapéutica inicial después de 1-2 semanas, la cual se hace máxima a las 4-6 semanas. La simvastatina y su metabolito beta-hidroxiácido se unen a las proteínas plasmáticas en un 95-99% y se distribuyen principalmente al hígado, donde se metabolizan a través del sistema microsomal del citocromo P450, trans-formándose en productos activos que se excretan en un 2-20% por la orina y 60-90% por las heces. La vida media de eliminación es de 0,5-3 horas. INDICACIONES Tratamiento de la hipercolesterolemia primaria y de la hipertrigliceridemia. Prevención y tratamiento de la cardiopatía isquémica en pacientes de alto riesgo. Reducción del riesgo de accidentes cerebro-vasculares isquémicos e isquemia cerebral transitoria. RÉGIMEN DE DOSIFICACIÓN La dosis inicial usual en adultos es de 20-40 mg/día con ajustes, en caso necesario, a intervalos de 4 semanas o más, hasta lograr la respuesta deseada. El rango de dosis es de 5-80 Formulario Terapéutico Nacional * 2ed. 2004 mento de las enzimas hepáticas y de bilirrubina, ictericia colestásica. Reacciones de hipersensibilidad: Síndrome de hipersensibilidad (raro), erupción, anafilaxis. Respiratorias: Infección del tracto respiratorio superior. Neurológicas: Cefalea, astenia, temblores, pérdida de la memoria, neuropatía periférica, trastornos psíquicos, ansiedad, insomnio, depresión. Otras: Mialgia, cataratas, calambres mus-culares, artralgia, rabdomiolisis, gine-comastia, disminución de la libido, anormalidades de la función tiroidea, fiebre. INTERACCIONES La combinación con fibratos (clofibrato o gemfibrozilo) y con niacina, incrementa de manera considerable el riesgo de miopatía y/o rabdomiolisis. La ciclosporina, el itraconazol, el ketoconazol, la eritromicina, la claritromicina, la amiodarona, el verapamilo, los inhibidores de proteasa, los bloqueantes de los canales de calcio y el jugo de toronja pueden inhibir el metabolismo hepático de la simvastatina incrementando, en consecuencia, los niveles plasmáticos y el riesgo de rabdomiolisis; la colestiramina y el colestipol los disminuyen. La simvastatina potencia los efectos anticoagulantes de la warfarina y puede aumentar las concentraciones séricas de la digo- tratamiento y periódicamente durante su desarrollo se debe practicar determinaciones de los lípidos sanguíneos, pruebas de la función hepática y las concentraciones séricas de la crea-tina quinasa (CK) y de la creatina fosfo-quinasa (CPK). No se conoce la C efica-cia y la seguridad en niños. Usar con precaución en ancianos. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Informar al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial dolor muscular no explicable, debilidad, fiebre, síntomas de gripe y malestar general, dado que podría tratarse del inicio de una miopatía o de rabdomiolisis. 2ed. 2004 * Formulario TerapÈutico Nacional 127 D01 ANTIF⁄NGICOS PARA USO DERMATOL”GICO Clotrimazol (Dermatológico) D04 ANTIPRURIGINOSOS, INCLUYE ANTIHISTAMÕNICOS, ANEST…SICOS, ETC Calamina* D06 ANTIBI”TICOS Y QUIMIOTER¡PICOS PARA USO DERMATOL”GICO Aciclovir (Dermatológico) Bacitracina Sulfadiazina de Plata D08 ANTIS…PTICOS Y DESINFECTANTES Etanol (Alcohol Etílico) Iodo Povidona (Polividona Iodada) D10 PREPARADOS ANTI-ACNÉ Peróxido de Benzoilo 2ed. 2004 * Formulario TerapÈutico Nacional D DERMATOLÓGICOS D07 PREPARADOS DERMATOL”GICOS, CORTICOESTEROIDES Betametasona (Betametasona Valerato) Desonida D 129 CLOTRIMAZOL (DermatolÛgico) FARMACOLOGÍA Antimicótico sintético derivado del imidazol. Es de amplio espectro. Como otros azoles, inhibe la síntesis del ergosterol de la membrana del hongo y, en consecuencia, la acumulación de C14-esteroles, disminuye la fluidez y la función de la membrana. En el caso de la Candida albicans, inhibe la transformación de los blastoporos en la forma micelial invasiva. Es fungistático, pero a altas concentraciones se comporta como fungicida in vitro. Presenta actividad in vitro contra Epidermophyton floccosum, Tri-chophyton metangrophytes, Tricho-phyton rubrum, Microsporum canis y algunas especies de Candida, incluyendo Candida albicans. FARMACOCINÉTICA Después de la administración tópica se absorbe en muy escasa proporción. Se ha detectado bajas concentraciones en el estrato córneo, el estrato espinoso y en la dermis reticular y papilar. INDICACIONES Infecciones micóticas de la piel causadas por hongos susceptibles. RÉGIMEN DE DOSIFICACIÓN Se emplea en formulaciones tópicas al 1%. Antes de usar el medicamento, se después de 4 semanas, se debe suspender el tratamiento y re-evaluar el diagnóstico. REACCIONES ADVERSAS Dermatológicas: Eritema, prurito, urticaria, ampollas, sensación punzante, ardor e irritación en el sitio de aplicación. En caso de absorción sistémica, pueden ocurrir cólicos D abdominales, aumento de la frecuencia urinaria y erupciones de la piel. INTERACCIONES Puede causar daño a materiales a base de látex como condones y diafragmas vaginales. El efecto es temporal y ocurre solamente durante el tratamiento. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del producto. Usar con precaución en el embarazo y durante la lactancia. No debe usarse en los ojos. SOBREDOSIS No se ha descrito problemas por sobredosificación con el medicamento. INFORMACIÓN AL PACIENTE Antes de usar el medicamento, lavar y secar bien el área afectada. No usar en 2ed. 2004 * Formulario TerapÈutico Nacional 131 pués de la desaparición de todos los síntomas. Durante el tratamiento evitar la exposición excesiva a la luz del sol. Si la condición persiste o empeora con el uso continuo, del producto. CALAMINA INFORMACIÓN AL PACIENTE Producto para uso externo. Agítese antes de usar. Previo a su aplicación se debe lavar bien el área afectada. Evitar el contacto con los ojos y las mucosas. No aplicar en heridas, lesiones o en la piel denudada. Si la condición no mejora con el uso FARMACOLOGÍA Suspensión constituida por una mezcla de óxido de zinc y una pequeña proporción de óxido férrico (que le confiere al producto un ligero color rosado), que posee propiedades astringentes y antipruríticas. FARMACOCINÉTICA La absorción sistémica tras la administración tópica es nula. INDICACIONES Alivio sintomático y temporal del prurito. RÉGIMEN DE DOSIFICACIÓN Se presenta comercialmente como loción al 8%. Aplicar con masaje suave, una capa fina de la loción 3-4 veces al día sobre el área afectada. REACCIONES ADVERSAS No se describen en la literatura consultada. INTERACCIONES No se describen en la literatura consultada. 132 SOBREDOSIS No se ha descrito problemas por sobredosificación del producto. ACICLOVIR (DermatolÛgico) FARMACOLOGÍA Ver: ACICLOVIR en la sección ANTIINFECCIOSOS. FARMACOCINÉTICA Después de la aplicación tópica, la absorción sistémica es clínicamente despreciable. INDICACIONES Episodios iniciales de la infección por herpes genital e infecciones mucocutáneas por herpes simplex. RÉGIMEN DE DOSIFICACIÓN Iniciar el tratamiento inmediatamente después de la aparición de los primeros signos de la infección. Aplicar cantidad suficiente de la formulación al 5% para cubrir toda la lesión, cada 3 Formulario Terapéutico Nacional * 2ed. 2004 REACCIONES ADVERSAS Dolor leve, sensación punzante, ardor, dermatitis por contacto, erupción y prurito en el sitio de aplicación. INTERACCIONES No se ha descrito interacciones por esta vía de administración. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. La terapia debe iniciarse tan pronto como sea posible después del inicio de los síntomas y signos de la infección herpética. No usar con fines profilácticos. Usar con precaución en el embarazo y durante la lactancia. SOBREDOSIS No se ha descrito problemas de sobredosificación con la administración tópica del medicamento. INFORMACIÓN AL PACIENTE No exceder la frecuencia y duración del tratamiento. No usar en los ojos. Aplicar con un hisopo para evitar la contaminación de otras áreas del BACITRACINA microorganismo. Inhibe la síntesis de la pared bacteriana al interferir con la incorporación de aminoácidos y nucleótidos al proceso y, además, parece que altera el transporte de mucopéptidos durante la formación de la pared. Es activa contra especies de Staphylococcus, Streptococcus, Clos-tridium, Corynebacterium y D contra algunos cocos anaeróbicos. FARMACOCINÉTICA La absorción a través de la piel intacta o con abrasiones, de heridas, o de las membranas mucosas es insignificante. INDICACIONES Sola o en combinación con otros agentes para la prevención y tratamiento de infecciones superficiales de la piel causadas por microorganismos susceptibles. RÉGIMEN DE DOSIFICACIÓN Aplicar una capa fina del medicamento sobre el área afectada 1-3 veces al día, previa limpieza de la zona. Posterior a su aplicación se puede cubrir el área con una gasa estéril o apósito apro-piado. REACCIONES ADVERSAS Aunque resulta bien tolerado por la mayoría de los pacientes, se ha reportado casos aislados de erupción, sensación de quemadura, enrojecimiento y reacciones anafilactoides 133 2ed. 2004 * Formulario TerapÈutico Nacional FARMACOLOGÍA Antibiótico polipeptídico producido por el Bacillus subtilis. Ejerce una acción bactericida o bacteriostática según la concentración que se logre CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del producto. Los individuos sensibles a la neomicina también pueden serlo a la bacitracina. El uso prolongado puede dar lugar al sobrecrecimiento de organismos no susceptibles, incluyendo hongos patógenos (particularmente Candida). No debe usarse durante el embarazo. SOBREDOSIS No se ha descrito problemas por sobredosificación del medicamento. INFORMACIÓN AL PACIENTE Antes de aplicar el medicamento se debe limpiar bien el área afectada. No usar en los ojos. No usar por más de una semana. Notificar al médico si se presenta alguna reacción o sintomatología inusual durante el trataSULFADIAZINA DE PLATA FARMACOLOGÍA La sulfadiazina impide el crecimiento y la multiplicación bacteriana por inhibición competitiva de la enzima que incorpora al ácido paraaminobenzoico (PABA) en el proceso de biosíntesis del ácido tetrahidrofólico, esencial para la síntesis del ADN. Se cree que la plata contenida en el fármaco contribuye a su acción bactericida. Es activa frente a algunas 134 trobacter, Enterobacter, Proteus, Serratia, Streptococcus, Escherichia coli y Corynebacterium diphteriae. FARMACOCINÉTICA Después de la aplicación, el fármaco reacciona lentamente con el cloruro de sodio, los grupos sulfhidrilo y las proteínas presentes en los fluidos y tejidos, liberando sulfadiazina y plata al medio. Si la lesión es extensa y/o profunda puede ocurrir absorción sistémica (hasta un 10%) de la sulfadiazina, la cual es excretada por vía renal. La absorción de la plata es insignificante. INDICACIONES Prevención y tratamiento de infecciones por quemaduras de segundo y tercer grado. RÉGIMEN DE DOSIFICACIÓN Se emplea en formulaciones tópicas al 1%. Cubrir la lesión con una cantidad suficiente de la crema, 1-2 veces al día, previa limpieza y eliminación de los restos de tejido y secreciones. Aplicar con guantes estériles. En caso de ser removida involuntariamente por la actividad del paciente, aplicar nuevamente la crema. El tratamiento debe mantenerse por el tiempo que sea necesario, mientras persista la posibilidad de infección, hasta lograr una cicatrización satisfactoria. REACCIONES ADVERSAS Formulario Terapéutico Nacional * 2ed. 2004 sulfonamidas, que incluyen: Trastornos hematológicos, hepáticos, renales y neurológicos (Ver: SULFAMETOXIPIRIDAZINA) INTERACCIONES Inactivación de enzimas proteolíticas de aplicación tópica. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a alguno de los constituyentes del producto y a las sulfonamidas. Debido a la posibilidad de ictericia y kernicterus, está contraindicada en el embarazo a término y en los neonatos. Cuando se usa en áreas extensas, se debe vigilar periódicamente la función renal, la hepática y la hematológica. Usar con precaución en pacientes con deficiencia de glucosa-6-fosfato-deshidrogenasa. Debe suspenderse la lactancia durante el tratamiento. Se debe considerar la posibilidad de sobrecrecimiento de microorganismos no susceptibles, incluyendo hongos. teroides ejercen una actividad antiinflamatoria, antiprurítica, vasoconstrictora local y antiproliferativa. Pueden disminuir la inflamación a través de varios mecanismos como la estabilización de las membranas lisosomales de los leucocitos, inhibición de la acumulación de macrófagos en el área inflamada, disminución de la D adhesión de leucocitos al endotelio capilar y formación de edema, antagonismo de la actividad histamínica, disminución de la proliferación de fibroblastos y de la deposición de colágeno, disminución de los componentes del sistema del complemento. También se piensa que actúan mediante la inducción de las lipocortinas, proteínas inhibidoras de la fosfolipasa A2. Se postula que estas proteínas controlan la biosíntesis de mediadores de la inflamación como las prosta-glandinas y los leucotrienos, mediante la inhibición de la liberación del ácido araquidónico por la fosfolipasa A2. FARMACOCINÉTICA La absorción percutánea es variable y puede aumentar por el uso de vendas oclusivas. Depende de la concenINFORMACIÓN AL PACIENTE tración del producto, del vehículo utilizado y de la integridad de la barrera BETAMETASONA (BETAMETASONA VALERATO) epidérmica. El uso de vendajes oclusivos, la inflamación u otros procesos FARMACOLOGÍA de enfermedad en la piel pueden Aplicados tópicamente, los corticosSOBREDOSIS No aplicable. 2ed. 2004 * Formulario TerapÈutico Nacional 135 como la cara, región genital, axilas, párpados y cuero cabelludo. La fracción absorbida se metaboliza en el hígado transformándose en productos inactivos que se excretan por la orina y sus metabolitos también se excretan en la bilis, en forma similar a los corticosteroides administrados sisté-micamente. INDICACIONES Alivio sintomático de las manifestaciones inflamatorias y pruríticas de las dermatosis agudas, subagudas y crónicas, que responden a esteroides tópicos. RÉGIMEN DE DOSIFICACIÓN Luego de lavar y secar bien el área afectada aplicar, con masaje suave, una capa fina de la formulación 1-4 veces al día. La duración del tratamiento no debe exceder 14 días. La dosis máxima es 45 g/semana o 50 ml de la loción. REACCIONES ADVERSAS Dermatológicas: Ardor, prurito, irritación, eritema, sequedad, foliculitis, hipertricosis, erupciones acneiformes, hipopigmentación, dermatitis perioral, dermatitis por contacto, infección secundaria, atrofia de la piel y estrías. La absorción sistémica puede ocasionar supresión reversible del eje hipotálamo-hipófisis-adrenal, manifes-taciones de síndrome de 136 CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a otros corticosteroides. No es recomendable el uso de la betametasona en infecciones micóticas, tuberculosis de la piel, varicela, herpes simplex o en zonas lesionadas o excoriadas. No debe utilizarse en dermatitis del área genital, incluida la pañalitis. Usar con precaución cuando es aplicado en cara, axila, niños menores de 12 años, en el embarazo y durante la lactancia. Debe usarse por corto tiempo y en áreas limitadas. SOBREDOSIS En caso de aplicación excesiva y/o absorción sistémica significativa, manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Evitar el contacto con los ojos. No usar vendajes oclusivos. Informar al médico si la condición persiste o empeora, si se presenta ardor, irritación o se desarrolla una infección. DESONIDA FARMACOLOGÍA Ver BETAMETASONA. La desonida es un esteroide de baja potencia, no fluorado, que puede usarse de forma más segura que la betametasona en niños y en áreas como la cara, las Formulario Terapéutico Nacional * 2ed. 2004 FARMACOCINÉTICA Ver: BETAMETASONA. Se absorbe en menor magnitud que la betametasona y de allí que la incidencia e intensidad de las reacciones adversas y los riesgos de toxicidad sistémica asociados a su uso sean menores. INDICACIONES Ver: BETAMETASONA. RÉGIMEN DE DOSIFICACIÓN Luego de lavar y secar el área afectada, aplicar con un masaje suave una capa fina de la formulación 2-4 veces al día. La duración del tratamiento no debe exceder 14 días. REACCIONES ADVERSAS Ver: BETAMETASONA. INTERACCIONES Ver: BETAMETASONA. CONTRAINDICACIONES/ PRECAUCIONES Ver: BETAMETASONA. ETANOL (ALCOHOL ETÍLICO) FARMACOLOGÍA Agente antiséptico con potente actividad bactericida, micobactericida, fungicida y virucida, pero ineficaz frente a esporas. Su acción germicida es debida a su capacidad para desnaturalizar FARMACOCINÉTICA La absorción a través de la piel intacta es insignificante. INDICACIONES La solución de etanol al 70% se usa como antiséptico para la limpieza de la piel antes de una inyección, en la punción venosa y en procedimientos quirúrgicos. D REACCIONES ADVERSAS La aplicación frecuente sobre la piel puede provocar irritación y sequedad. CONTRAINDICACIONES/ PRECAUCIONES No debe usarse para desinfectar instrumentos quirúrgicos o dentales, debido a su baja eficacia esporicida. IODO POVIDONA (POLIVIDONA IODADA) FARMACOLOGÍA Es un complejo de vinilpirrolidona polimerizada (polivinilpirrolidona) que transporta en su estructura yodo en una proporción de 9-12% que, al ser aplicada tópicamente, lo libera gradualmente al medio para ejercer un efecto antiséptico potente y sostenido, como resultado de su acción germicida. El yodo penetra la pared celular de los microorganismos y produce una serie de alteraciones bioquímicas, por reacción directa con moléculas fundamentales que provocan la muerte del 2ed. 2004 * Formulario TerapÈutico Nacional 137 rios, virus y algunas esporas. FARMACOCINÉTICA El yodo liberado del complejo se absorbe muy poco a través de la piel intacta. Sin embargo, cuando se aplica en áreas excoriadas, quemaduras severas, heridas profundas o sobre las mucosas, puede ocurrir absorción significativa y alcanzar la circulación sistémica. El yodo absorbido se transforma en yoduro y se almacena en la glándula tiroides bajo la forma de tiroglobulina. INDICACIONES Antiséptico de uso tópico en heridas menores, quemaduras leves no extensas y en lesiones superficiales con riesgo de infección. Preparación preoperatoria del campo quirúrgico y para el lavado de las manos del personal de quirófano involucrado en el proceso. RÉGIMEN DE DOSIFICACIÓN Se presenta como una solución tópica al 10% o como jabón líquido al 7,5%. Lavado de manos o aplicación local de la solución sobre el área afectada, según la circunstancia. REACCIONES ADVERSAS En pacientes con hipersensibilidad al yodo puede ocurrir irritación, dermatitis y urticaria. Se ha descrito casos raros de anafilaxis. La aplicación sobre áreas extensas, laceradas, en 138 vidona es reducida por los álcalis. La eficacia germicida puede inhibirse en presencia de materia orgánica como sangre, pus o esputo. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al yodo, en neonatos, pacientes con trastornos tiroideos, con insuficiencia hepática o renal, en la terapia con litio, en el embarazo y durante la lactancia. No usar en áreas extensas de la piel denudada de más del 20% de la superficie corporal, quemaduras severas, heridas profundas, en los ojos o en las mucosas. SOBREDOSIS En caso de administración excesiva, enjuagar con abundante agua; se puede usar solución de tiosulfato de sodio para inactivar al yodo. En caso de ingestión accidental o intencional se debe proceder al vaciamiento gástrico empleando una solución de almidón, a objeto de acomplejar el yodo e impedir su absorción. Manejo sintomático y terapia de soporte. INFORMACI”N AL PACIENTE PERÓXIDO DE BENZOILO FARMACOLOGÍA Agente con actividad antibacteriana, empleado para el tratamiento del Formulario Terapéutico Nacional * 2ed. 2004 Propionibacterium acnes. Se cree que el efecto bactericida es debido a una acción oxidativa sobre las proteínas del microorganismo. Además, se observa una reducción en los lípidos y en los ácidos grasos libres y una leve acción queratolítica con una reducción simultánea en los comedones y las lesiones del acné. quear el cabello. Debe usarse en el embarazo solamente si es claramente necesario. Administrar con precaución durante la lactancia. SOBREDOSIS La sobredosificación puede causar irritación excesiva, descamación, eritema y edema local. Se trata con emolientes, compresas frías y, en D casos severos, corticosteroides FARMACOCINÉTICA Se absorbe a través de la piel, donde tópicos. es metabolizado a ácido benzóico inactivo y excretado, posteriormente, INFORMACIÓN AL PACIENTE Producto para uso externo solamente. como benzoato por la orina. Antes de usarlo, lavar con jabón no medicado y secar bien el área de INDICACIONES Tratamiento del acné leve a aplicación. Evitar el contacto con los ojos y las mucosas. No aplicar en moderado. heridas, lesiones, áreas inflamadas o RÉGIMEN DE DOSIFICACIÓN sobre la piel denudada. Advertir al Se presenta comercialmente como paciente la posibilidad de enrojeciuna loción al 5-10%. Aplicar una capa miento y descamación como reacciofina de la loción sobre el área a tratar 1- nes normales de la terapia. Durante el 2 veces al día por 8-12 semanas. tratamiento se debe evitar el uso de otros agentes con propiedades REACCIONES ADVERSAS irritantes, a menos que el médico lo Irritación, dermatitis por contacto, indique, así como la exposición descamación, resequedad, eritema, edema, prurito y reacciones locales de hipersensibilidad. INTERACCIONES En combinación con tretinoína, compuestos sulfurados, resorcinol, ácido salicílico u otros agentes para el tratamiento tópico del acné puede causar irritación excesiva. 2ed. 2004 * Formulario TerapÈutico Nacional 139 G02 OTROS PRODUCTOS GINECOL”GICOS Cabergolina Fenoterol Metilergometrina G03 HORMOMONAS SEXUALES Y MODULADORES DEL SISTEMA GENITAL Danazol Estrógenos Conjugados Levonorgestrel / Estrógeno (Levonorgestrel / Etinilestradiol) Medroxiprogesterona (Medroxiprogesterona Acetato) (Tratamientos GinecolÛgicos) 2ed. 2004 * Formulario TerapÈutico Nacional G SISTEMA GENITOURINARIO Y HORMONAS SEXUALES G01 ANTIINFECCIOSOS Y ANTIS…PTICOS GINECOL”GICOS Clotrimazol (Ginecológico) Metronidazol (Ginecológico) Nistatina (GinecolÛgica) G 141 CLOTRIMAZOL (GinecolÛgico) prurito e irritación vulvar, urticaria, urgencia urinaria y dolor abdominal. FARMACOLOGÍA Antimicótico sintético derivado del imidazol. Es de amplio espectro. Como otros azoles, inhibe la síntesis del ergosterol de la membrana del hongo y, en consecuencia, la acumulación de C14-esteroles, disminuye la fluidez y la función de la membrana. En el caso de la Candida albicans, inhibe la transformación de los blastosporos en la forma micelial invasiva. Es fungistático, pero a altas concentraciones se comporta como fungicida in vitro. Presenta acti-vidad in vitro contra Epidermophyton floccosum, Trichophyton metangrophytes, Trichophyton rubrum, Microsporum canis y algunas especies de Candida, incluyendo Candida albicans. FARMACOCINÉTICA Luego de la administración vaginal se absorbe en un 3-10% a la circulación sistémica y se excreta como metabolitos por la orina. INDICACIONES Candidiasis vulvovaginal. RÉGIMEN DE DOSIFICACIÓN Se administra por vía vaginal. La dosis usual es de 1 óvulo o tableta vaginal de 500 mg como dosis única. Alternati-vamente, se puede INTERACCIONES Puede causar daño a materiales a base de látex como condones y diafragmas vaginales. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del producto. Aunque no se G ha descrito efectos adversos sobre el feto, se recomienda usarlo con precaución en el embarazo y sólo cuando exista una verdadera necesidad; sin embargo, se debe evitar su uso durante el primer trimestre. Usar con precaución durante la lactancia. SOBREDOSIS No se han descrito problemas por sobredosificación del medicamento. INFORMACIÓN AL PACIENTE Administrar preferiblemente en las noches, antes de dormir. No suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Si la METRONIDAZOL (GinecolÛgico) FARMACOLOGÍA Ver: METRONIDAZOL en ANTIPARASITARIOS. 2ed. 2004 * Formulario TerapÈutico Nacional 143 FARMACOCINÉTICA Después de la administración por vía vaginal, se absorbe en un 20-25% a la circulación sistémica. Una vez en la sangre, el patrón cinético es similar al descrito para el fármaco en la sección ANTIPARASITARIOS (ver: M E T R O N I - D A Z O L e n ANTIPARASITARIOS). INDICACIONES Vaginosis bacteriana (principalmente por Trichomona vaginalis y Gardnerella vaginalis). R…GIMEN DE DOSIFICACI”N Un óvulo o una tableta de 500 mg para administración vaginal, 1 vez al día por 7-10 días consecutivos. REACCIONES ADVERSAS Se han reportado casos de irritación, ardor y prurito vaginal. INTERACCIONES No se han descrito interacciones cuando se administra por esta vía. CONTRAINDICACIONES/ PRECAUCIONES Ver: METRONIDAZOL en ANTIPARASITARIOS. SOBREDOSIS No se han descrito casos de sobredosificación por esta vía de administración. 144 Evitar el consumo de alcohol mientras dure el tratamiento. Informar al médico si se presenta alguna reacción o sintomatología inusual NISTATINA (Ginecológica) FARMACOLOGÍA Antibiótico antifúngico producido por el Streptomyces noursei. Se une a los esteroles de la membrana celular del hongo alterando la permeabilidad y ocasionando con ello la pérdida de componentes intracelulares vitales para el hongo. Presenta actividad fungicida y fungistática in vitro contra Candida albicans, Candida guilliermondi, Candida krusei y Geotrichum lactis. Es inactiva frente a microorganismos que no contienen esteroles en su membrana, como las bacterias, los protozoarios y los virus. FARMACOCINÉTICA Después de la administración vaginal, la absorción sistémica es insignificante. INDICACIONES Candidiasis vulvovaginal. RÉGIMEN DE DOSIFICACIÓN Se administra por vía vaginal. La dosis usual es de 1 óvulo 1-2 veces al día, por 14 días consecutivos. REACCIONES ADVERSAS Aunque resulta bien tolerada por la Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES No se describen en la literatura. y 2- adrenÈrgicos, 5-HT1 y 5-HT2 de serotonina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del producto. Aunque no se ha descrito efectos adversos sobre el feto, se recomienda usarlo con precaución en el embarazo y solamente cuando exista una verdadera necesidad. FARMACOCINÉTICA Después de la administración oral, alcanza concentraciones plasmáticas pico en 2-3 horas e induce una respuesta terapéutica (reducción de los niveles de prolactina) a las 24 horas, la cual persiste por 14-21 días con la administración de una dosis única, o por 3-12 meses con dosis múltiples. G Se distribuye ampliamente en el organis-mo y se une a las proteínas plasmá-ticas en un 40-42%. Se metaboliza en el hígado a productos inactivos que se excretan, junto a menos de 4% del fármaco intacto, en un 20% por la orina y 60% por las heces. La vida media de eliminación es de 63-69 horas. SOBREDOSIS No se han descrito problemas por sobredosificación del producto. INFORMACI”N AL PACIENTE No suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. Si la condición persiste o empeora con el uso continuo, suspender la administración del medicamento CABERGOLINA FARMACOLOGÍA Agonista dopaminérgico de acción prolongada, con similitud estructural a los derivados del ergot y que tiene una alta afinidad por los receptores D2 de dopamina. Estimula los receptores dopaminérgicos hipotalámicos que regulan la liberación de prolactina, generando la supresión de la producción hipofisiaria de prolactina. INDICACIONES Tratamiento de trastornos hiperprolactinémicos y para la inhibición de la secreción láctea postparto o supresión de la misma una vez establecida. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Para el tratamiento de la hiperprolactinemia en adultos, iniciar con 0,50 mg semanales divididos en 2 tomas (por ejemplo: 0,25 mg el lunes y 0,25 mg el jueves) e incrementar 0,50 mg por semana a intervalos mensuales, divididos tam- 2ed. 2004 * Formulario TerapÈutico Nacional 145 REACCIONES ADVERSAS Cardiovasculares: Hipotensión postural, palpitaciones. Dermatológicas: Ac-né, prurito. Gastrointestinales: Náu-seas, constipación, dolor abdominal, dispepsia, vómitos, boca seca, fla-tulencia, diarrea. Neurológicas: Cefa-lea, mareos, parestesia, vértigo, somnolencia, depresión, nerviosismo, visión anormal, fatiga, alteración de la concentración. Otras: Astenia, calorones, dolor en las mamas, dismenorrea, irritación de la garganta, edema, dolor muscular, artralgia, rinitis. INTERACCIONES Los antagonistas dopaminérgicos (fenotiazinas, butirofenonas, tioxantinas y metoclopramida) disminuyen el efecto hipoprolactinémico de la cabergolina. La hipotensión inducida por la cabergolina podría incrementarse por la acción de agentes hipotensores. Los macrólidos, como la eritromicina, aumentan los niveles plasmáticos de la cabergolina por inhibición del citocromo P450, aumentando el riesgo de toxicidad. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otros derivados del ergot. Por su similitud estructural con otros derivados del ergot se recomienda usar con cautela en 146 Por su elevado metabolismo hepático se debe tener precaución cuando se usa en pacientes con insuficiencia hepática severa. Cuando se usa en casos de hiperprolactinemia, se debe vigilar periódicamente los niveles séricos de prolactina así como la presión arterial. No se conoce la seguridad de su uso en menores de 16 años. SOBREDOSIS La evidencia disponible revela un escaso grado de toxicidad, aun con dosis elevadas. Los signos y síntomas de una sobredosis son los que cabe esperar de una sobreestimulación de los receptores dopaminérgicos (náusea, vómito, dolor abdominal, hipotensión y trastornos de naturaleza central). Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Si es necesario, debe tomarse medidas para controlar la presión arterial. INFORMACIÓN AL PACIENTE El producto puede alterar la capacidad para realizar actividades que requieran concentración, alerta mental y coordinación motora. Durante la te-rapia se debe practicar métodos de anticoncepción a objeto de evitar el embarazo. Debe hacerse una prueba de embarazo si se sospecha el mismo y la continuación Formulario Terapéutico Nacional * 2ed. 2004 FENOTEROL FARMACOLOGÍA Agente simpatomimético agonista de los receptores beta2-adrenérgicos. Po-see propiedades tocolíticas que se evi-dencian por la disminución de la inten-sidad, la frecuencia y la duración de las contracciones uterinas. FARMACOCINÉTICA Posterior a la administración IV, se alcanzan niveles plasmáticos pico en 2 minutos, que se reducen en un 50% a los 12 minutos y se mantienen por 2 horas. Se une a las proteínas plasmáticas en un 45-50%. Se metaboliza en el hígado a productos inactivos que se excretan, junto a un 25% del fármaco intacto, en la orina y en las heces. La vida media de eliminación es de 7 horas. INDICACIONES Como tocolítico en la prevención del parto prematuro, a partir de la semana 20 de la gestación. RÉGIMEN DE DOSIFICACIÓN Se ha propuesto varios esquemas de dosificación: 10-40 mcg en bolo IV en 10 segundos, 60 mcg en bolo IV en 512 minutos, 1-8 mcg/minuto en infusión IV intermitente por 30 minutos, o infusión continua de 0,5-3 mcg/minuto, hasta la supresión de las contraccio-nes. En caso necesario, continuar con la terapia de pulmonar. Neurológicas: Temblor, ner-viosismo. Otras: Hipopotasemia. En recién nacidos de madres que han recibido fenoterol, se debe controlar la glicemia. INTERACCIONES Los inhibidores de MAO y los antidepresivos tricíclicos potencian la acción del fenoterol. Los simpatomiméticos incrementan las reacciones cardio-vasculares G adversas del fenoterol. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, hipertiroidismo y miocardiopatía hipertrófica. Usar con precaución en pacientes con diabetes mellitus, insuficiencia coronaria, disfunción renal y/o hepática, estenosis subaórtica hipertrófica idiopática, arritmias cardíacas de elevada frecuencia, asma intrínseca, bronquitis asmática, bronquitis enfisematosa. SOBREDOSIS Manejo sintomático y tratamiento de METILERGOMETRINA FARMACOLOGÍA Alcaloide derivado del ergot. Es el metabolito activo de la metisergida. Estimula las contracciones del músculo 2ed. 2004 * Formulario TerapÈutico Nacional 147 involucra la estimulación de la musculatura uterina a través de los receptores alfa-adrenérgicos. FARMACOCINÉTICA Después de la administración oral e IM, se absorbe rápidamente. Produce contracciones uterinas a los 5-15 minutos después de la administración oral, a los 2-5 minutos después de la inyección IM e inmediatamente cuando se administra por vía IV. El efecto persiste por 3 horas o más después de la administración oral o IM y por 45 minutos después de la administración IV. Se distribuye rápidamente a los tejidos. Sufre un extenso metabolismo de primer pasaje en el hígado y es excretada por las heces. La vida media de eliminación es de 0,5-2 horas. INDICACIONES Prevención y tratamiento de la hemorragia post-parto o post-aborto causada por atonía o sub-involución uterina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM e IV. La dosificación por vía IV debe usarse sólo en situaciones graves, diluida en 5 ml de solución salina (NaCl al 0,9%) y por inyección en no menos de 1 minuto. Después de la administración IM ó IV se debe cambiar a la vía oral tan pronto como sea posible. 148 - Por vía oral: 0,2-0,4 mg cada 6-12 horas, por 2-7 días. REACCIONES ADVERSAS Cardiovasculares: Hipertensión transitoria, palpitaciones, dolor torácico, tromboflebitis, espasmo coronario, infarto del miocardio, angina. Gastrointestinales: Náuseas, vómitos, diarrea. Neurológicas: mareos, tinitus, alucinaciones, parestesia, vértigo. Reacciones de hipersensibilidad: Reacciones alérgicas, incluyendo anafilaxis. Renales: Hematuria. Respiratorias: Disnea. Otras: Diaforesis, intoxicación hídrica, congestión nasal, calambres. INTERACCIONES Fármacos antivirales como el efavirenz, saquinavir, indinavir, nelfinavir, ritonavir y delavirdina pueden interferir con el metabolismo hepático del medicamento, aumentando los niveles séricos y el riesgo de ergotismo. La combinación con agonistas serotoninérgicos como el sumatriptán o el rizatriptán puede provocar una reacción vasoespástica aditiva prolongada. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipertensión no controlada, toxemia e hipersensibilidad al medicamento. Usar con precaución en el segundo y tercer período del trabajo de parto, en pacientes con enfermedad vascular Formulario Terapéutico Nacional * 2ed. 2004 reciente. La sobredosis puede provocar convulsiones, hipercoagulabilidad, gangrena, alteraciones de la presión arterial, disnea y coma. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE DANAZOL FARMACOLOGÕA Esteroide sintético derivado de la etisterona, con potente acción antigonadotrópica. Suprime el eje hipofisiarioovárico. La supresión probablemente es debida a una combinación de una respuesta hipotálamo-hipofisiaria deprimida debido a la disminución en la producción de estrógenos, la alteración del metabolismo de los esteroides sexuales y a la inhibición competitiva de la interacción de dichos esteroides con sus receptores en los órganos blanco. Inhibe la liberación de la hormona folículo-estimulante (FSH) y de la hormona luteinizante (LH). Además, posee una débil actividad androgénica. Disminuye significa-tivamente los niveles de IgG, IgM e IgA, en pacientes con endometriosis. Se sugiere que pudiera haber otro mecanismo para la regresión de la enfermedad. acción se inicia a las 3-6 semanas con la dosificación continua y persiste hasta por 8-12 meses después de suspendida la terapia. En la enfermedad fibroquística de la mama, la respuesta inicial se observa a las 4 semanas, se hace máxima a los 4-6 meses y se mantiene hasta por 1 año después de finalizado el tratamiento. No se conoce bien la cinética de distribución. Se metaboliza en el híga-do a hidroximetiletisterona, G como metabolito principal y se excreta por vía renal. La vida media de eliminación es de 4,5 horas. INDICACIONES Endometriosis. Enfermedad fibroquís-tica de la mama. Menorragia primaria (hemorragia uterina disfuncional). R…GIMEN DE DOSIFICACI”N Se administra por vía oral. - Endometriosis: Iniciar con una dosis de 800 mg/día dividida en 2 dosis y mantener la terapia hasta lograr amenorrea y alivio del dolor. A partir de allí, disminuir gradualmente hasta la mínima dosis necesaria para mantener la amenorrea. Para casos leves, iniciar con 200-400 mg/día, divididos en 2 dosis y ajustar la dosis según la respuesta del paciente. Mantener la terapia sin interrupción por 3-6 meses y, en caso necesario, FARMACOCIN…TICA Se absorbe bien en el tracto gastrointestinal y alcanza 2ed.niveles 2004 *séFormulario TerapÈutico Nacional 149 - Enfermedad fibroquística de la mama: Iniciar con una dosis de 100400 mg/día divididos en 2 dosis, según la respuesta del paciente. Usualmente, la eliminación de los nódulos requiere 4-6 meses de terapia. - Menorragia primaria: 200 mg/día divididos en 2 dosis iguales, por 3 meses. REACCIONES ADVERSAS Cardiovasculares: Aumento de la presión arterial. Gastrointestinales: Irritación gástrica, náuseas, vómitos, diarrea, constipación, cambios del apetito, gastroenteritis y, raras veces, pancreatitis. Genitourinarias: Hematuria, efectos hipoestrogénicos (rubor, diaforesis, vaginitis, incluyendo picazón, sequedad, sensación de quemadura, sangrado vaginal, nerviosismo, irregularidades menstruales), efectos androgénicos en las mujeres (aumento de peso, hirsutismo, ronquera, aumento del tamaño del clítoris, cambios en la libido). Hematológicas: Aumento en el contaje de los glóbulos rojos y las plaquetas. Se ha observado eritrocitosis, leucocitosis o policitemia. Hepáticas: Ictericia reversible, aumento de los niveles de las enzimas hepáticas, alteración de la función hepática, adenoma hepático. Reacciones de hipersensibilidad: Reacciones alérgicas, erupciones. 150 abdominal persistente, síndrome del túnel carpiano, escalofríos, piel y cuero cabelludo grasosos. INTERACCIONES El danazol potencia el efecto de los anticoagulantes orales e incrementa los requerimientos de insulina en pacientes diabéticos. Puede generar resultados anormales en la prueba de tolerancia a la glucosa. Incrementa los niveles séricos y el riesgo de toxicidad de la carbamazepina. Prolonga el tiempo de protrombina en pacientes estabilizados con warfarina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, hemorragia uterina de etiología desconocida, insuficiencia hepática, renal o cardiaca, en casos de porfiria y en el embarazo. Durante el tratamiento se debe evaluar periódicamente la presión arterial, la función hepática y prestar especial atención a la aparición precoz de signos de virilización. En los varones, se debe evaluar el semen cada 3-4 meses (volumen, viscosidad, contaje y motilidad). Usar con precaución en pacientes con condiciones que pueden agravarse por la retención o sobrecarga de fluidos como migraña, epilepsia y alteración de la función cardiaca o de la función renal. Durante el tratamiento Formulario Terapéutico Nacional * 2ed. 2004 carbón activado y un catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Durante el tratamiento se deben usar métodos anticonceptivos no hormonales. En caso de sospecha o certeza de embarazo, suspender el medicamento y notificar al médico. No ingiera danazol si está embarazada o amamantando. Se debe usar un método contraceptivo, diferente a los anticonceptivos orales, mientras dure el tratamiento. Prestar atención a la aparición precoz de signos de ESTRÓGENOS CONJUGADOS FARMACOLOGÕA Los estrógenos conjugados producen las mismas acciones fisiológicas que los estrógenos endógenos. En los tejidos que responden a los estrógenos (órganos genitales femeninos, mamas, hipotálamo, hipófisis), los estrógenos, como otros esteroides, dada su alta liposolubilidad, entran a la célula y son transportados al núcleo donde se unen al receptor nuclear, donde ocurre la síntesis de ARN y proteínas específicas, con los con-secuentes efectos de los estrógenos. Los estrógenos endógenos cons-tituyen hormonas esenciales para el normal desarrollo y crecimiento de los de la forma corporal característica, de la estructura esquelética y de la talla corporal definitiva de la hembra. Causan un incremento de las células y secreciones de la mucosa cervical, engrosamiento y cornificación de la mucosa vaginal, proliferación del endometrio y aumento del tono uterino. Ejercen un efecto anabólico débil que genera retención de sodio y fluidos; favorecen la deposición de calcio en los huesos y ejercen un G efecto positivo sobre los lípidos sanguíneos, reduciendo los niveles de LDL-colesterol y aumentando los de HDL-colesterol. FARMACOCIN…TICA Después de la administración oral, se absorben rápidamente. Se distribuyen ampliamente en el organismo y tienden a depositarse en el tejido adiposo, de donde se liberan lentamente. El estradiol y otros estrógenos naturales se unen a las proteínas plasmáticas en un 30-50%, principalmente a la globulina de unión a esteroides sexuales (SSBG) y, en menor grado, a la albúmina plasmática. El etinilestradiol se une principalmente a la albúmina. Se metabolizan principalmente en el hígado y en menor proporción en las gónadas, los riñones y en el tejido muscular, transformándose en metabolitos con menor actividad estrogénica que se excretan como conjugados 2ed. 2004 * Formulario TerapÈutico Nacional 151 atrófica, hipogonadismo femenino, insuficiencia ovárica primaria o castración, en la prevención de la osteoporosis y como tratamiento paliativo del carcinoma de mama y del carcinoma prostático. R…GIMEN DE DOSIFICACI”N Se administran por vía oral en regímenes de dosis continua o en forma cíclica (dosificación diaria por 3 semanas seguido por 1 semana de descanso) con repetición del ciclo según la evolución. - Síntomas vasomotores asociados a la menopausia: 1,25 mg/día. Se administra en forma cíclica dependien-do de la respuesta tisular del pacien-te individual. - Vaginitis atrófica: 0,3-1,25 mg/día. Se administra en forma cíclica. - Hipogonadismo femenino: 2,5-7,5 mg/día en dosis divididas por 20 días continuos, seguido por 10 días de descanso. Si no aparece sangrado al final de este período, repetir el esquema de dosificación. - DisfunciÛn ov·rica primaria o castración: 1,25 mg/día, en forma cí-clica. Ajustar la dosis de acuerdo a la respuesta de la paciente. - Tratamiento de la osteoporosis: 0,625 mg/día, en forma cíclica (tres semanas con el medicamento y una semana sin el mismo). En casos de fractura podría requerirse dosis 152 ces/día y evaluación periódica de la eficacia del tratamiento por determinaciones seriadas de la fosfatasa ácida sérica y de la mejoría sintomática del paciente. REACCIONES ADVERSAS Cardiovasculares: Hipertensión, tromboflebitis, tromboembolismo, edema, aumento del riesgo de accidente cerebrovascular, embolismo pulmonar, infarto del miocardio. Dermatológicas: Melanoma, hirsutismo o pérdida del cabello, eritema nodoso, dermatitis. Gastrointestinales: Náuseas, vómitos, cólicos abdominales, ictericia colestásica, distensión abdominal, anorexia, aumento del apetito, pancreatitis. Genitourinarias: Sangrado intermenstrual, alteración del flujo menstrual, aumento del riesgo de cáncer del endometrio, posibilidad de aumento del riesgo de cáncer de mama, erosión cervical, alteración de las secreciones cervicales, aumento del tamaño de los fibromas uterinos, candidiasis vaginal, ginecomastia, atrofia testicular, impotencia. Hepáticas: Adenoma hepático, enfermedad de la vesícula biliar. Neurológicas: Cefalea, mareos, movimientos coreicos, depresión, convulsiones. Reacciones de hipersensibilidad: Urticaria, eritema nodoso, eritema multiforme. Otras: Empeoramiento de la miopía o el astigmatismo, intolerancia a los lentes de contacto, cambio de peso, Formulario Terapéutico Nacional * 2ed. 2004 fenilbutazona, los barbitúricos, la fenitoína, la rifampicina y la primidona incrementan el metabolismo de los estrógenos y reducen sus niveles séricos. Los estrógenos pueden disminuir el efecto de los anticoagulantes orales. Los evacuantes intestinales reducen la absorción de los estrógenos. Los estrógenos aumentan la toxicidad de los antidepresivos tricíclicos y de los corticosteroides. Aumentan las concentraciones séricas de la cafeína. En pacientes fumadores aumenta el riesgo de efectos cardiovasculares. CONTRAINDICACIONES/ PRECAUCIONES Contraindicados en pacientes con hipersensibilidad al medicamento, carcinomas dependientes de estrógenos, sangramiento genital anormal no diagnosticado, antecedentes o presencia de tromboembolismo o tromboflebitis severa, daño hepático crónico o severo y durante el embarazo. Usar con precaución en pacientes con enfermedad cerebro-vascular o arterial coronaria, condiciones que puedan agravarse por la retención de fluidos (asma, trastornos convulsivos, migraña e insuficiencia cardiaca o renal) y en mujeres con antecedentes o historia familiar de cáncer de mama, de enfermedad fibroquística de mama, o con tendencia a la formación de nódulos. Durante la terapia se debe SOBREDOSIS No es factible la intoxicación aun con la ingestión de dosis elevadas; sin embargo, algunos pacientes podrían presentar náuseas y/o hemorragia vaginal. Tratamiento sintomático y de soporte. INFORMACI”N AL PACIENTE Se debe advertir a los pacientes sobre los efectos adversos más comunes asociados a la terapia con estrógenos, G así como la importancia de notificar de inmediato al médico la ocurrencia de cualquier manifestación o síntoma inusual que se presente durante el tratamiento. La paciente debe estar alerta por sangrado anormal por la vagina, dolor en las pantorrillas o en el pecho, falta de respiración, tos con sangre, cefalea severa, mareos, desmayos, nódulos en las mamas, ictericia y depresión mental. Se debe persuadir a los pacientes de practicarse, de manera regular, exámenes de la función hepática y hematológica, gliLEVONORGESTREL / ESTRÓGENO (LEVONORGESTREL / ETINIL ESTRADIOL) FARMACOLOGÕA La combinación del levonorgestrel y el etinilestradiol ejerce un efecto anticonceptivo por supresión de la función hipotálamo-hipofisiaria que regula la secreción de las hormonas folículoestimulante (FSH) y luteinizante (LH).También previene el transporte 153 2ed. 2004 * Formulario TerapÈutico Nacional Falopio. El estrógeno suprime la secreción de FSH, bloqueando el desarrollo folicular y la ovulación. El progestágeno suprime la secreción de la hormona luteinizante de tal manera que la ovulación no puede ocurrir aun cuando el folículo se desarrolle. Produce espesamiento del moco cervical, interfiere con la migración de los espermatozoides y produce cambios en el endometrio que impiden la implantación. FARMACOCIN…TICA Después de la administración oral, el levonorgestrel se absorbe en un 100% y el etinilestradiol, por el efecto de primer pasaje, sólo en un 40%. Una vez en la sangre, se unen a las proteínas plasmáticas (etinilestradiol 98% y levonorgestrel 94%) y se distribuyen ampliamente en el organismo. Se metabolizan en el hígado; el etinilestradiol por hidroxilación y el levonorgestrel por reducción. Ambos sufren posterior conjugación tanto con el ácido glucurónico como con el ácido sulfúrico. Los productos se excretan por la orina y las heces como metabolitos inactivos y, en pequeña proporción, como fármacos intactos. El etinilestradiol sufre circulación enterohepática. La vida media de eliminación del etinilestradiol es de 6-20 horas y la del levonorgestrel de 11-45 horas. 154 diaria por 21 días continuos comenzando el día 5 del ciclo menstrual, seguido por un descanso durante 7 días (sin medicación), al final de los cuales se reinicia un nuevo régimen. - Régimen de 28 días: Una tableta diaria por 28 días continuos (las últimas 7 tabletas son placebo), al final de los cuales se reinicia un nuevo régimen. REACCIONES ADVERSAS Cardiovasculares: Tromboembolismo, hipertensión, edema, embolismo pulmonar, accidente cerebrovascular, trombosis retiniana. Dermatológicas: Dermatitis, acné, hirsutismo, alopecia, eritema multiforme.Gastrointestinales: Náusea, malestar estomacal, cambios en el apetito, anorexia, cólicos, aumento de peso, pancreatitis, intolerancia a los carbohidratos. Genitourinarias: Amenorrea, dismenorrea, hipermenorrea, sangrado prolongado, manchado, inicio irregular del sangrado, inicio frecuente de sangrado, sangrado escaso, cervicitis, vaginitis, leucorrea, aumento de tamaño de los fibromas uterinos, candidiasis vaginal. Hepáticas: Enfermedad de la vesícula biliar, cálculos biliares, tumores hepáticos, ictericia colestásica. Neurológicas: Cefalea, nerviosismo, mareos, depresión, letargia, migraña. Otras: Aumento de tamaño de la glán- Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Los anticonceptivos orales pueden disminuir la eficacia del lorazepam y del oxazepam. Pueden aumentar el efecto del alcohol, de los bloqueantes de los receptores beta-adrenérgicos, los corticosteroides, el diazepam, el clordiazepóxido, la fenitoína y la teofilina. El efecto anticonceptivo puede ser disminuido por la fenitoína, los barbitúricos, la fenilbutazona, la carbamazepina, la primidona, la isoniazida, la rifampicina, el cloranfenicol, la neomicina, la nitrofurantoína, la griseofulvina, la ampicilina, la penicilina V, las sulfonamidas y las tetraciclinas. Pue-den disminuir el efecto de los anticoa-gulantes orales. El cigarrillo aumenta el riesgo de efectos adversos cardio-vasculares. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, trastornos graves de la función hepática, historia o antecedentes de tromboembolismo, tromboflebitis, enfermedad cerebro-vascular, embolismo pulmonar, enfermedad arterial coronaria, carcinoma de mama o del endometrio u otras neoplasias dependientes de estrógenos, diabetes con compromiso vascular, cirugía mayor con inmovilización prolongada, hemorragia vaginal anormal no diagnosticada, ictericia colestásica, sospecha o certeza de renal. Durante el tratamiento se debe evaluar periódicamente la función hepática y la oftalmológica, la presión arterial y el perfil lipídico. Suspender el uso del producto en caso de cefalea migrañosa aguda, trastornos de la visión, signos iniciales de tromboflebitis o de tromboembolismo, alteraciones hepáticas y aumento de la presión arterial. SOBREDOSIS G No es factible la intoxicación, aún con la ingestión de dosis elevadas. INFORMACI”N AL PACIENTE Se debe procurar tomar la dosis diaria siempre a la misma hora. En caso de olvido de alguna dosis, notificar al mé dico y tomar precauciones ante la posibilidad de falla del tratamiento. No tomar medicamentos por cuenta propia sin el conocimiento del médico. Asistir periódicamente al control médico para la evaluación correspondiente. No fumar mientras dure el tratamiento. Informar de inmediato al médico si se presenta alguna MEDROXIPROGESTERONA (MEDROXIPROGESTERONA ACETATO) (Tratamientos GinecolÛgicos) FARMACOLOGÍA Derivado de la progesterona. Es un progestágeno sintético con acciones similares a los de la progesterona en- 2ed. 2004 * Formulario TerapÈutico Nacional 155 renal. Durante el tratamiento se debe evaluar periódicamente la función hepática y la oftalmológica, la presión arterial y el perfil lipídico. Suspender el uso del producto en caso de cefalea migrañosa aguda, trastornos de la ovulación. Tiene efectos anabólicos y androgénicos discretos. Aparentemente carece de una actividad estrogénica significativa. Inhibe las contracciones uterinas espontáneas. FARMACOCINÉTICA Después de la administración oral, alcanza concentraciones séricas pico en 2-7 horas. Se une a las proteínas plasmáticas en un 90%. Se metaboliza en el hígado dando lugar a, por lo menos, 16 productos diferentes que se excretan en su mayoría por la orina y en menor proporción por las heces. La vida media de eliminación, tras la administración oral, es de 12-17 horas. INDICACIONES Tratamiento de la amenorrea secundaria, hemorragia uterina debida a trastornos hormonales sin patología orgánica de base y síntomas vasomotores asociados a la menopausia. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - Amenorrea secundaria: 5 a 10 mg/día por 5-10 días. La terapia puede comenzar en cualquier día 156 general, a los 7-10 días de finalizado el tratamiento. - Síntomas vasomotores asociados a la menopausia: 20 mg/día. REACCIONES ADVERSAS Cardiovasculares: Tromboflebitis, embolismo, tromboembolismo pulmonar, edema, accidente cerebrovascular. Dermatológicas: Erupción, acné, prurito, melanoma, alopecia, hirsutismo. Gastrointestinales: Náusea, dolor abdominal. Genitourinarias: Sangrado intermenstrual, dismenorrea, amenorrea, erosión cervical, secreciones anormales, cambios en el sangrado menstrual. Hepáticas: Ictericia colestásica. Neurológicas: Depresión, insomnio, somnolencia, mareos, cefalea. Reacciones de hipersensibilidad: Reacciones anafilactoides y anafilaxis. Otras: Exoftalmos, diplopía, sensibilidad y aumento de tamaño o secreción de las mamas, cambios de peso. INTERACCIONES La aminoglutetimida puede reducir la biodisponibilidad de la medroxiprogesterona y, por ende, su eficacia. La rifampicina, carbamazepina, fenobarbital y la fenitoína pueden inducir el metabolismo de la medroxiprogesterona y comprometer su eficacia terapéutica. La medroxiprogesterona puede disminuir el efecto hipoglicémico de la glibenclamida y la metformina, e Formulario Terapéutico Nacional * 2ed. 2004 puede requerir una terapia alternativa. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, hemorragia uterina no diagnosticada, antecedentes tromboembólicos, tromboflebitis, apoplejía o pacientes con historia pasada de estas condiciones. Disfunción o enfermedad hepática, carcinoma genital o de mama y durante el embarazo. Durante el tratamiento se debe evaluar frecuentemente la función oftalmológica. Dado que en pacientes diabéticos puede desarrollarse intolerancia a la glucosa, se debe practicar determinaciones periódicas de la glicemia. Usar con precaución en pacientes con diabetes mellitus, historia de depresión mental y enfermedades que pueden agravarse por la retención de líquido como: epilepsia, migraña, hipertensión, asma y disfunción cardiaca o renal. Durante la terapia se debe suspender la lac-tancia. Los pacientes con historia de depresión deben ser cuidadosamente vigilados después de suspender el medicamento, por una posible recaída de la depresión. tología inusual durante el tratamiento, en especial trastornos oculares, cefalea súbita y dolor en los miembros inferiores acompañado de aumento de volumen, temperatura y G SOBREDOSIS No es factible la intoxicación, aun con la ingestión de dosis elevadas. 2ed. 2004 * Formulario TerapÈutico Nacional 157 H02 CORTICOSTEROIDES PARA USO SIST…MICO Dexametasona (Sistémica) Hidrocortisona Prednisolona Prednisona (Sistémica) Triamcinolona (Triamcinolona Acetonita) H03 TERAPIA TIROIDEA Levotiroxina Sódica Liotironina (Triiodotironina) Propiltiouracilo Tiamazol (Metimazol) H PREPARADOS HORMONALES SIST…MICOS, EXCLUYE HORMONAS SEXUALES E INSULINAS H01 HORMONAS HIPOFISIARIAS E HIPOTAL¡MICAS Y SUS AN¡LOGOS Desmopresina (Desmopresina Acetato) Oxitocina 2ed. 2004 * Formulario TerapÈutico Nacional 159 H DESMOPRESINA (DESMOPRESINA ACETATO) FARMACOLOGÍA Análogo sintético de la hormona antidiurética (vasopresina) con una acción más específica, potente y prolongada que la de la hormona natural. Pro-mueve la absorsión de agua en los túbulos renales, disminuye el volumen de orina e incrementa su osmolaridad. Produce un aumento dosis-depen-diente en la concentración plasmática y la actividad del factor VIII (factor antihemofílico) FARMACOCINÉTICA Después de la administración de 0,025-0,4 mg en pacientes con diabetes insípida, se produce un efecto anti-diurético clínicamente significativo en 15-30 minutos. Dependiendo de la do-sis, el efecto puede durar de 8-12 ho-ras. Después de la administración IV, se genera un aumento en la actividad del factor VIII en 15-30 minutos, el cual se hace máximo después de 1,5-2 ho-ras. La vida media de eliminación terminal es de aproximadamente 9,7 (8,4-16) horas. Willebrand tipo I: 0,3 mcg/kg por infusión IV en 15-30 minutos. REACCIONES ADVERSAS Cardiovasculares: Aumento leve de la presión arterial en altas dosis, o hipotensión con taquicardia compensadora, eventos trombóticos (cerebrovascular, infarto del miocardio). Gastrointestinales: Náuseas, cólicos abdo-minales. Genitourinarias: Dolor vulvar. Neurológicas: Cefalea. Reacciones de hipersensibilidad: Anafilaxis. Otras: Ri-nitis, epistaxis, dolor de garganta, tos, rubor, eritema H local, hinchazón o sen-sación de quemadura después de la inyección, retención de fluidos, hipo-natremia. INTERACCIONES El litio, la heparina, la epinefrina (dosis elevadas), la demeclociclina y el alcohol pueden aumentar el riesgo de efectos adversos; la cloropromazina y la fludrocortisona pueden potenciarla. El clofibrato puede prolongar la duración del efecto antidiurético. La carbamaze-pina y la clorpropamida pueden poten-ciar los efectos de la desmopresina; se debe evitar el uso concomitante. Aun-que la actividad presora es muy baja, dosis altas (0,3 INDICACIONES Tratamiento de la diabetes insípida no mg/Kg) administradas conjuntamente nefrogénica, la hemofilia A y la enfer- con otros agentes pre-sores requiere medad de von Willebrand tipo I (leve a un control cuidadoso del paciente. La indometacina poten-cia los efectos de moderada). la desmopresina. 161 2ed. 2004 * Formulario TerapÈutico Nacional de fluido y desbalance electrolítico (fibrosis quística) e historia de trombosis. En niños y ancianos se debe restringir la ingesta de líquidos para disminuir el riesgo de intoxicación por agua y la hiponatremia. No debe utilizarse en la enfermedad de von Willebrand tipo II debido a que puede inducir agregación plaquetaria. Durante el uso en el manejo de la diabetes insípida, se debe controlar con frecuencia el volumen y la osmolaridad de la orina. En pacientes con hemofilia A se debe determinar los niveles del factor VIII y no usar el medicamento si son iguales o menores que 5%. No se debe usar para el tratamiento de la hemofilia tipo B, o en pacientes que tienen anti-cuerpos contra el factor VIII. No se conoce la seguridad de su uso en el embarazo ni durante la lactancia. SOBREDOSIS Reducir la dosis o suspender el tratamiento, según la severidad del caso. No hay un tratamiento específico. Si OXITOCINA FARMACOLOGÍA Hormona hipotalámica producida en la neurohipófisis, con actividad estimulante de la musculatura lisa uterina. Induce las contracciones uterinas al incrementar la permeabilidad de las miofibrillas del útero al ion sodio. 162 musculatura lisa del útero, particularmente hacia el final del embarazo, durante el parto e inmediatamente después del alumbramiento. Estimula las contracciones rítmicas del útero, aumenta la frecuencia y el tono de la musculatura uterina. La hormona sintética no posee los efectos cardiovasculares tales como aumento de la presión arterial como lo muestra la hormona natural, vasopresina. Tiene, además, un efecto antidiurético discreto. FARMACOCINÉTICA Después de la administración IV, produce una respuesta uterina inmediata que se mantiene por 1 hora, mientras que por vía IM, la respuesta se observa a los 3-5 minutos y persiste por 2-3 horas. Se distribuye en el líquido extra-celular; puede alcanzar concentracio-nes bajas en la circulación fetal. Se destruye en el hígado, en los riñones y en el plasma por acción de la enzima oxitocinasa. Una pequeña cantidad es excretada en orina como fármaco intacto. La vida media plasmática es de 3-5 minutos. INDICACIONES Estimulación o inducción del trabajo de parto, prevención de la hemorragia post-parto y manejo del aborto incompleto o inevitable. RÉGIMEN DE DOSIFICACIÓN Formulario Terapéutico Nacional * 2ed. 2004 tracciones similares a las de un trabajo de parto normal), sin evidencia de perturbación fetal. Hemorragia post-parto: 10 U por vía IM o por infusión IV a una velocidad de 20-40 mU/minuto. - Inducción de contracciones uterinas en aborto incompleto o inevitable: 10-20 mU/minuto (20-40 gotas/minutos), sin exceder 30 U en 12 horas. REACCIONES ADVERSAS En la madre: Cardiovasculares: Arritmia cardiaca, contracciones ventriculares prematuras. Gastrointestinales: Náuseas, vómitos. Hematológicas: Hemorragia posparto. Neurológicas: Hemorragia subaracnoidea (por la hipertensión), convulsiones, coma (por intoxicación hídrica). Reacciones de hipersensibilidad: Anafilaxis. Otras: Hematoma pélvico, contracciones ute-rinas tetánicas, abruptio placentae, aumento de la motilidad uterina, ruptura uterina. En el feto: Debidas a la motilidad uterina: Cardiovasculares: Bradicardia, contracciones ventriculares prematuras y otras arritmias. Neurológicas: Daño cerebral permanente. Respiratorias: Anoxia, asfixia. Otras: Muerte fetal. Debidas al uso de la oxitocina en la madre: Hemorragia retiniana neonatal. Bajo puntaje de Apgar a los 5 minutos. Ictericia neo- puede provocar hipertensión severa y persistente. El ciclopropano puede incrementar la hipotensión inducida por la oxitocina; también se ha observado bradicardia sinusal y ritmo aurículoventricular anormal en la madre. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y en circunstancias en las que no es recomendable el parto vaginal. No debe ser dada simultáneamente por más de una vía de administración. Durante la H administración se debe hacer seguimiento de las contracciones uterinas, la presión arterial materna, la frecuencia cardiaca materna y fetal y, de ser posible, la presión intrauterina. Si se presenta hiperactividad uterina se debe suspender de inmediato la administración. Evitar el uso de dosis elevadas por períodos prolongados. SOBREDOSIS DEXAMETASONA (SistÈmica) FARMACOLOGÍA Los corticosteroides se clasifican según su actividad farmacológica primaria en mineralocorticoides y glucocorticoides, cada uno con efectos y sitios de acción específicos. La separación de estos grupos; sin embargo, no es absoluta, toda vez que existen este163 2ed. 2004 * Formulario TerapÈutico Nacional grupos, como la hidrocortisona; otros, como la dexametasona, la prednisolona, la prednisona y la triamcinolona poseen actividad predominantemente glucocorticoide y débil actividad mineralocorticoide. Los corticosteroides interactúan con proteínas receptoras específicas en los tejidos blanco para regular la expresión de genes con capacidad de respuesta a los corticosteroides, lo cual modifica las cifras y la disposición de las proteínas sintetizadas. El tiempo requerido para estos cambios resulta en el hecho que pocas acciones de los corticoides son inmediatas, observándose, en general, un retardo en la aparición de dichas acciones. También hay ejemplos en los cuales los glucocorticoides disminu-yen la transcripción de los genes. Los mineralocorticoides afectan el balance hidroelectrolítico promoviendo la reab-sorción de sodio y la excreción de potasio e hidrógeno en los túbulos renales, lo cual conduce a edema e hipertensión. Los glucocorticoides disminuyen o previenen la respuesta tisular a los procesos inflamatorios y, además, suprimen la respuesta inmune. Otros efectos incluyen: inhibición de la secreción hipofisiaria de ACTH; incremento de la gluconeogénesis (que causa aumentos de la glicemia); catabolismo de proteínas; disminución de los procesos de 164 les solubles (fosfato sódico y succinato sódico) se absorben rápidamente, mientras que las sales liposolubles (acetato y acetónido) lo hacen más lentamente. La absorción sistémica posterior a la administración intraar-ticular, intrabursa, intrasinovial, intra-dérmica o en tejidos blandos, es lenta. Una vez absorbidos, los corticoste-roides abandonan rápidamente el torrente sanguíneo y se distribuyen a numerosos tejidos, en especial al músculo, hígado, piel, intestino y riñones. La unión a las proteínas plasmáticas es variable y dependiente del agente; por lo general, se unen a la transcortina (globulina transportadora de corticosteroides) y a la albúmina. Atraviesan la placenta y se distribuyen a la leche materna. Se metabolizan en todos los tejidos, principalmente en el hígado, dando origen a productos inactivos que son excretados, junto con pequeñas cantidades del fármaco intacto, en la orina y en escasa proporción por vía biliar. La vida media de eliminación de la dexametasona es de 1-2 horas y la duración de la ac-tividad antiinflamatoria (que equivale al tiempo de supresión del eje hipotálamo-hipófisis-adrenal) es de 2,25 días. INDICACIONES Los corticosteroides con escasa acti- Formulario Terapéutico Nacional * 2ed. 2004 - - - - - - en episodios agudos o exacerbación de espondilitis anquilosante, bursitis, tenosinovitis, artritis (gotosa, psoriática y reumatoidea), epicondilitis y osteoartritis post-traumática. Enfermedades del colágeno: Casos seleccionados de lupus eritematoso sistémico, carditis reumática aguda y dermatomiositis sistémica. Desórdenes dermatológicos: Pénfi-go, dermatitis bulosa herpetiforme, eritema multiforme severo, derma-titis seborreica severa, psoriasis severa, micosis fungoide, angioe-dema y dermatitis por contacto o atópica Estados alérgicos severos no controlados con otras medidas. Afecciones oftálmicas en procesos alérgicos o inflamatorios agudos y severos. Enfermedades respiratorias: Sarcoidosis, asma bronquial, síndrome de Loeffler no manejable con otras medidas, tuberculosis pulmonar di-seminada o fulminante, beriliosis, pneumonitis por aspiración y rinitis alérgica. D e s Ûr d e nes hematolÛgicos: Púrpura trombocitopénica idiopática y trombocitopenia secundaria en adultos, anemia hemolítica adquirida y anemia hipoplásica congénita (eritroide). Enfermedades neoplásicas: Manejo - Prevención de los vómitos inducidos por la quimioterapia. - Prevención del síndrome de distrés repiratorio en neonatos prematu-ros. - Misceláneas: Estados edematosos, shock refractario a medidas convencionales, exacerbación aguda de la esclerosis múltiple, síndrome nefrótico, meningitis tuberculosa con bloqueo sub-aracnoideo, colitis ulcerativa y enteritis regional. RÉGIMEN DE DOSIFICACIÓN Se presenta como dexametasona H base de uso oral y como fosfato de dexametasona de uso IV, IM, intraarticular, intrasinovial, intralesiones y en tejido blando. La dosis y la ruta de administración dependerán de la condición a tratar. - La dosis oral de dexametasona base para adultos es: 0,75-9 mg/día, dividida en 2-4 dosis; en niños: 0,024-0,34 mg/kg/día, dividida en 4 dosis. - La dosis de fosfato de dexametasona en el shock es: 1-6 mg/kg IV como dosis única o 40 mg IV cada 26 horas, según lo demande la situación. Alternativamente, puede iniciarse con 20 mg IV seguido por infusión continua de 3 mg/kg en 24 horas. Continuar solamente hasta que el paciente se estabilice (no más de 48-72 horas). 2ed. 2004 * Formulario TerapÈutico Nacional 165 - - - - curando cambiar a la administración oral tan pronto como la condición del paciente lo permita. En condiciones alérgicas agudas o en la exacerbación de trastornos crónicos: Iniciar el primer día con 48 mg IM; 3 mg por vía oral en el segundo y el tercer día en dosis divi-didas; 1,5 mg en el cuarto día en dosis divididas y finalizar en el quin-to y sexto día con una dosis única de 0,75 mg. En condiciones inflamatorias: Se administra por inyección intraarticular, intrasinovial, intralesión o en tejido blando, en dosis que dependen de la magnitud y localización de la inflamación. En las articulaciones grandes como la de la rodilla: 2-4 mg; en articulaciones pequeñas como las de los dedos: 0,8-1 mg; en la bursa: 2-3 mg; en el tejido blando: 0,4-1 mg o 2-6 mg en caso de infiltración. En la prevención de los vómitos inducidos por la terapia antineoplásica: 10-20 mg IV antes de la administración de la quimioterapia. En la prevención del síndrome de distrés respiratorio en infantes prematuros: 4 mg IM a la madre por 2 días consecutivos antes del parto. agravamiento de la hipertensión, ruptura miocárdica después de un infarto, insuficiencia cardiaca en pacientes susceptibles. Dermatológicas: Retardo en la cicatrización, adelgazamiento de la piel, petequias, equimosis, eritema, síndrome tipo lupus, atrofia grasa subcutánea, púrpura, hiperpigmentación, hirsutismo, erupciones y angioedema. Gastrointestinales: Náuseas, vómitos, aumento del apetito, esofagitis ulcerativa, distensión abdominal, úlcera péptica, perforación y hemorragia. Músculoesqueléticas: Debilidad muscular, ruptura de tendones, osteoporosis, necrosis aséptica, fracturas espontáneas, miopatía por esteroides, pérdida de masa muscular, compresión de vértebras, necrosis aséptica de las cabezas femoral y humeral. Neuro-lógicas: Cefalea, vértigo, insomnio, neuritis, parestesia, aumento de la presión intracraneana con papiledema (pseudotumor cerebral) usualmente después del tratamiento, agravamiento de condiciones psiquiátricas, psicosis por esteroides y convulsiones. Endocrinas: Irregularidades menstruales, síndrome de Cushing, retardo del crecimiento en niños, disminución de la tolerancia a los carbohidratos, hiperglicemia y supresión de la respuesta adrenocortical e hipofisiaria. Oftalmológicas: Catara- REACCIONES ADVERSAS El uso de corticosteroides en tratamientos de corta duración, por lo 166 Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Los fármacos inductores del metabolismo hepático como la fenitoína, los barbitúricos y la rifampicina, pueden acelerar la degradación de los corticosteroides. Los estrógenos pueden potenciar el efecto de los corticosteroides al incrementar los niveles de transcortina y disminuir así la cantidad de corticosteroides disponibles para el metabolismo. Con los AINEs, aumenta el riesgo de úlcera péptica. La asociación con amfotericina B o con diuréticos depletores de potasio incrementa la posibilidad de hipopotasemia. Los corticosteroides aumentan la toxicidad digitálica asociada a la hipopotasemia; disminuyen el efecto de los anticoagulantes orales; aumentan los requerimientos de insulina y de los hipoglicemiantes orales; reducen la eficacia de la isoniazida; inhiben el metabolismo hepático de la ciclofosfamida e interfieren con el efecto promotor de la hormona del crecimiento. Los anticonceptivos orales inhiben el metabolismo hepático de los corticosteroides. En pacientes que reciben corticosteroides, las vacunas de virus vivos pueden provocar reacciones severas por replicación del virus. Disminución de la respuesta a los antígenos en las pruebas dérmicas. El alcohol aumenta el riesgo de irritación gástrica y úlceras. camento o a otros esteroides. Se debe evitar, en lo posible, el uso prolongado en niños debido al riesgo de trastornos en el crecimiento. Usar con precaución en pacientes con úlcera péptica, colitis ulcerativa, diverticulitis, diabetes me-llitus, hipertensión, insuficiencia car-diaca congestiva, osteoporosis, sín-drome de Cushing, trastornos convul-sivos, trastornos hematológicos, car-cinoma metastásico, miastenia gravis, trastornos tromboembólicos, insuficiencia renal y/o hepática, ancianos y durante el embarazo. Se debe tener en H cuenta que los corticosteroides pueden enmascarar los signos de una infección y que es posible la aparición de nuevas infecciones durante el tratamiento. Los pacientes con hipotiroidismo pueden manifestar respuestas exageradas a la acción de los corticosteroides. Durante el tratamiento prolongado, el paciente debe ser evaluado periódicamente a objeto de detectar la posibilidad de edema, hipertensión, ganancia de peso, trastornos hidro-electrolíticos (en especial hipopotasemia), infecciones, alteración de la función cardiaca, trastornos hematológicos, trastornos endocrinos, hiperglicemia y problemas de visión. En personas sometidas a tratamiento prolongado está contraindicado el uso de vacunas de virus vivos. Durante el tratamiento se debe 2ed. 2004 * Formulario TerapÈutico Nacional 167 INFORMACIÓN AL PACIENTE No alterar la dosificación ni suspender el tratamiento sin el conocimiento del médico. Evitar el contacto con personas que padecen enfermedades infecciosas. Los pacientes susceptibles que están recibiendo dosis inmunosupresoras de esteroides deben evitar la exposición a la varicela o al sarampión. Notificar al médico si se presenta algún trastorno o reacción inusual durante el tratamiento, en especial signos y síntomas de infección HIDROCORTISONA FARMACOLOGÍA Corticosteroide de acción corta con ac-tividad glucocorticoide y mineralocor-ticoide. Ver: DEXAMETASONA. FARMACOCINÉTICA Ver: DEXAMETASONA. La vida media de eliminación es de 1-2 horas y la duración de la actividad antiinflamatoria es de 1,25-1,5 días (equivalente al tiempo de supresión del eje hipotálamo-hipófisis-adrenal). INDICACIONES Por su actividad mineralocorticoide y glucocorticoide, la hidrocortisona constituye el agente de elección como terapia de reemplazo en pacientes con insuficiencia adrenocortical. En algu168 supresor, se prefiere corticosteroides con propiedades de glucocorticoide y mínima actividad mineralocorticoide. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM e IV como succinato sódico, sal soluble en agua que resulta adecuada en situaciones de emergencia que requieren de un efecto rápido. La dosis para adultos es de 100-500 mg cada 2-10 horas, según sea necesario. En niños: 0,16-1 mg/kg 1-2 veces diarias. REACCIONES ADVERSAS Ver: DEXAMETASONA. INTERACCIONES Ver: DEXAMETASONA. CONTRAINDICACIONES/ PRECAUCIONES Ver: DEXAMETASONA. SOBREDOSIS Manejo sintomático y tratamiento de PREDNISOLONA FARMACOLOGÍA Corticosteroide de acción intermedia con acción predominantemente glucocorticoide y mínima actividad mineralocorticoide. Ver: DEXAMETASONA. FARMACOCINÉTICA Ver: DEXAMETASONA. La vida media Formulario Terapéutico Nacional * 2ed. 2004 ración de la actividad antiinflamatoria (que equivale al tiempo de supresión del eje hipotálamo-hipófisis-adrenal) es de 1,25-1,5 días. INDICACIONES Usada principalmente como agente antiinflamatorio y como agente inmunosupresor. Debido a su escasa actividad mineralocorticoide, resulta inadecuada para el manejo de la insuficiencia adrenocortical. Ver: DEXAMETASONA. RÉGIMEN DE DOSIFICACIÓN Se usa por vía oral, IM, IV, intraarticular. Independientemente de la sal que se emplee, la dosis se expresa en términos de prednisolona base. La dosis oral inicial en adultos es de 5-60 mg/día, dividida en 2-4 dosis; en niños, 0,14-2 mg/kg/día, dividida en 3-4 do-sis. La dosis IM ó IV en adultos es de 4-60 mg/día, dividida en 2-4 dosis; en ni-ños, 0,14-2 mg/kg/día, dividida en 3-4 dosis. La dosis intraarticular es de 4-60 mg/día. REACCIONES ADVERSAS Ver: DEXAMETASONA. INTERACCIONES Ver: DEXAMETASONA. CONTRAINDICACIONES/ PRECAUCIONES Ver: DEXAMETASONA. PREDNISONA (SistÈmica) FARMACOLOGÍA Corticosteroide de acción intermedia con acción predominantemente glucocorticoide y mínima actividad mineralocorticoide. Ver DEXAMETASONA. FARMACOCINÉTICA Ver DEXAMETASONA. La vida media de eliminación es de 2-3 horas y la duración de la actividad antiinflamatoria (que equivale al tiempo de supresión del eje hipotálamoH hipófisis-adrenal) es de 1,25-1,5 días. La pred-nisona es inactiva y en el organismo se transforma en prednisolona por las enzimas hepáticas. INDICACIONES Usada principalmente como agente antiinflamatorio y como agente inmunosupresor. Debido a su escasa actividad mineralocorticoide, resulta inadecuada para el manejo de la insuficiencia adrenocortical. Ver: DEXAMETASONA. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis en adultos es de 5-60 mg como dosis única o dividida en 2-4 dosis; en niños, 0,14-2 mg/kg/día (4-60 mg/m2), dividida en 4 dosis. La dosis de mantenimiento se da una vez al día o inter- 2ed. 2004 * Formulario TerapÈutico Nacional 169 CONTRAINDICACIONES/ PRECAUCIONES Ver: DEXAMETASONA. SOBREDOSIS Ver: DEXAMETASONA. INFORMACIÓN AL PACIENTE TRIAMCINOLONA (TRIAMCINOLONA ACETONITA) FARMACOLOGÍA Corticoesteroide de acción intermedia con acción predominantemente glucocorticoide y mínima actividad mineralocorticoide. Ver: DEXAMETASONA. FARMACOCINÉTICA Ver: DEXAMETASONA. La vida media de eliminación es de 2-3 horas y la duración de la actividad antiinflamatoria (que equivale al tiempo de supresión del eje hipotálamohipófisis-adrenal) es de 1,25-1,5 días. INDICACIONES Usada principalmente como agente antiinflamatorio y como agente inmunosupresor. Debido a su escasa actividad mineralocorticoide, resulta inadecuada para el manejo de la insuficiencia adrenocortical. Ver: DEXAMETASONA. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM, 170 0,2 mg/kg a intervalos de 1-7 días. La dosis por vía intraarticular, intrasinovial, intralesión y en tejidos blandos: 2,5-40 mg a intervalos de varias semanas, según la localización y el grado de inflamación del área afectada. Para las articulaciones grandes como la rodilla: 15-40 mg; para las articulaciones pequeñas como las de los dedos: 2,5-10 mg. Para los tejidos blandos: 2,5-10 mg. REACCIONES ADVERSAS Ver: DEXAMETASONA. INTERACCIONES Ver: DEXAMETASONA. CONTRAINDICACIONES/ PRECAUCIONES Ver: DEXAMETASONA. SOBREDOSIS Manejo sintomático y tratamiento de LEVOTIROXINA SÓDICA FARMACOLOGÕA Hormona tiroidea. La síntesis y secreción de las principales hormonas tiroideas, L-tiroxina y L-triyodotironina, es regulada por complejos mecanismos de retroalimentación del eje hipotálamo-hipófisis-tiroides. Las hormonas son secretadas en respuesta a la estimulación por la hormona estimulante de la tiroides (TSH). Las hormonas tiroideas, a su vez, actúan Formulario Terapéutico Nacional * 2ed. 2004 liberadora de tirotropina (TRH). El mecanismo de acción de las hormonas tiroideas no está claro. T4 y T3 son transportadas al interior de las células por mecanismos activos y pasivos. Ambas hormonas difunden al núcleo y se unen a las proteínas receptoras de hormona tiroidea, unidas al ADN. Esto lleva a la activación o a la represión de la transcripción del ADN, alterando las cantidades de ARN y de proteínas. Es-tos cambios son responsables por los cambios metabólicos en órganos y tejidos. Las hormonas tiroideas aumentan el consumo de oxígeno, la tasa metabólica basal y el metabolismo de carbohidratos, lípidos y proteínas. Es-tos efectos fisiológicos son producidos principalmente por la T3, la cual procede de la desyodación de la T4 en los tejidos periféricos. Alrededor de 7090% de la T3 periférica procede de la desyodación de la T4 en la posición 5'. de la T4 por dichas proteínas es mayor que la de la T3, lo que explica su mayor tiempo de permanencia en el organismo. Se inactiva en el hígado por conjugación con ácido glucurónico y con ácido sulfúrico y se excreta por la bilis. La vida media de eliminación de la T4 es de 6-7 días y la de la T3 de 1-2 días, aumentando ambas en pacientes con hipotiroidismo y disminuyendo en el hipertiroidismo. INDICACIONES Como terapia de reemplazo en el H manejo de condiciones que cursan con disminución de la función tiroidea. R…GIMEN DE DOSIFICACION Se administra por vía oral. En adultos, iniciar con 50-100 mcg/día e incrementar 25-50 mcg a intervalos de 2-3 semanas hasta obtener la respuesta adecuada. La mayoría de los pacientes se mantienen con 100-200 mcg/día. Los pacientes geriátricos requieren dosis menores. En niños, la dosis depende de la edad; niños de 06 meses: 25-50 mcg/día (5-6 mcg/kg); niños de 6-12 meses: 50-75 mcg/día (5-6 mcg/kg); niños de 1-5 años: 75100 mcg/día (3-5 mcg/kg); niños de 612 años: 100-150 mcg/día (4-5 mcg/kg) y niños mayores de 12 años: Más de 150 mcg/día (2-3 mcg/kg). FARMACOCIN…TICA La absorción en el tracto gastrointestinal, en el yeyuno proximal y medio, es variable (50-80%) y dependiente de la formulación; aumenta con el ayuno y puede disminuir por la presencia de ciertos alimentos. La acción terapéutica se observa 1-3 semanas después de iniciado el tratamiento. Se distribuye ampliamente a los tejidos. Se une REACCIONES ADVERSAS en un 95% a las proteínas 2ed. 2004 * Formulario TerapÈutico Nacional plasmáticas, incluyendo a la globulina 171 nes alérgicas de la piel. Otras: Pérdida de peso, diaforesis, intolerancia al ca-lor, fiebre, irregularidades menstrua-les, alopecia. cralfato interfieren con la absorción gastrointestinal de la levotiroxina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, tirotoxicosis, insuficiencia adrenal no compensada (Enfermedad de Addison) y en el tratamiento de la obesidad. Usar con extrema precaución en pacientes con trastornos cardiovasculares (incluyendo hipertensión, angina y enfermedad coronaria), diabetes, mixedema severo y en ancianos. Durante el tratamiento se debe practicar, periódicamente, determinaciones de T4 y T3. INTERACCIONES La levotiroxina aumenta la acción de las aminas simpatomiméticas, de los antidepresivos tricíclicos y de los anticoagulantes orales. En pacientes con diabetes puede incrementar los requerimientos de insulina y de los hipoglicemiantes orales. La fenitoína parece incrementar el metabolismo de la levotiroxina y puede desplazarla de su unión a las proteínas. Los siguientes agentes también pueden alterar la unión a las proteínas plasmáSOBREDOSIS ticas: Andrógenos y hormonas anaboSuspensión del medicamento. lizantes, asparaginasa, clofibrato, esVaciamiento gástrico, carbón activado trógenos, 5-fluorouracilo, furosemida, y catártico salino, si la ingestión es glucocorticoides, ácido meclofenáreciente. Manejo sintomático y terapia mico, metadona, perfenazina, fenilde soporte. butazona, salicilatos, tamoxifeno. El efecto de los anticoagulantes orales INFORMACIÓN AL PACIENTE puede ser potenciado. La amiodarona Administrar siempre a la misma hora y puede causar hipertiroidismo o hipo- con el estómago vacío, 1 hora antes o tiroidismo. Las acciones de algunos 2 horas después de las comidas. No bloqueantes de los receptores beta- se debe alterar la dosis ni suspender el adrenérgicos pueden alterarse tratamiento sin conocimiento del mécuando el paciente se hace eutiroideo. dico. Notificar al médico si se presenta Dis-minución de los efectos de los alguna reacción o síntoma inusual digitálicos. Aumenta el riesgo de durante el tratamiento, especialmente arritmias con la maprotilina. Dismi- dolor de cabeza persistente, dolor en nución de la captación de yodo el pecho, erupciones, diarrea, cambios radiactivo. Los agentes simpatico172 Formulario Terapéutico Nacional * 2ed. 2004 LIOTIRONINA (TRIIODOTIRONINA) FARMACOLOGÍA Ver: LEVOTIROXINA. semanas. Para el manejo del bocio simple (no tóxico) iniciar con 5 mcg/día e incrementar 5-10 mcg cada 1-2 se-manas, hasta alcanzar 25 mcg/día y, a partir de allí, incrementar 12,5-25 mcg cada 1-2 semanas hasta obtener la respuesta deseada. En niños con hipotiroidismo congénito, iniciar con 5 mcg/día e incrementar 5 mcg cada 3-4 días hasta lograr la respuesta desea-da. FARMACOCINÉTICA Ver: LEVOTIROXINA. Luego de la administración por vía oral se absorbe en un 95% en 4 horas. Debido a la débil unión a las proteínas plasmáticas, en comparación con levotiroxina, la ac-ción se inicia en 2472 horas y se man-tiene sólo por 72 horas. La vida media de eliminación es REACCIONES ADVERSAS de 1-2,5 días, se incrementa en Ver: LEVOTIROXINA. pacientes hipo-tiroideos y disminuye INTERACCIONES en los pacientes hipertiroideos. Ver: LEVOTIROXINA. INDICACIONES Como terapia de reemplazo en el manejo de condiciones que cursan con disminución de la función tiroidea. Es el medicamento de elección para pacientes con alteraciones en la conversión de T4 a T3. Por su efecto rápido y de corta duración, resulta adecuada cuando se requiere de un efecto inmediato o rápidamente reversible. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En el hipotiroidismo leve en adultos, iniciar con 25 mcg/día e incrementar 12,5-25 mcg a intervalos de 1-2 semanas, hasta obtener la respuesta deseada. La dosis usual de mantenimiento se ubica en el rango de 25-75 mcg/día. En el hipotiroidismo severo, iniciar con 5 H CONTRAINDICACIONES/ PRECAUCIONES Ver: LEVOTIROXINA. SOBREDOSIS PROPILTIOURACILO FARMACOLOGÍA Agente antitiroideo. Inhibe la síntesis de hormonas tiroideas impidiendo la incorporación del yodo en los residuos de tirosilo de la molécula de tiroglobulina; también interfiere con el acoplamiento de dichos residuos para formar la yodotironina. Puede interferir con la oxidación del ion yoduro y los grupos yodotirosilo. También parece que bloquea la conversión periférica de T4 en T3, por lo que resulta apro- 2ed. 2004 * Formulario TerapÈutico Nacional 173 hormonas tiroideas ya formadas ni la de las administradas exógenamente. FARMACOCIN…TICA Se absorbe rápidamente en el tracto gastrointestinal con una biodisponibilidad de 80-95%. Produce concentraciones plasmáticas máximas a las 1-1,5 horas y efectos terapéuticos en 24-36 horas. Se une a las proteínas plasmáticas en un 75-80%. Se metaboliza en el hígado por conjugación con ácido glucurónico, transformándose en productos inactivos que se excretan, junto con el fármaco intacto, por la orina. La vida media de eliminación es de 1-2 horas. INDICACIONES Tratamiento del hipertiroidismo. Es el medicamento de elección en las crisis tirotóxicas. dismo, iniciar con 50-150 mg/día y con 150-300 mg/día en niños mayores de 10 años. La dosis de mantenimiento se establece según la respuesta clínica. REACCIONES ADVERSAS Cardiovasculares: Vasculitis. Dermatológicas: Pigmentación de la piel. Gastrointestinales: Diarrea, náusea, vómitos, malestar epigástrico, aumento del tamaño de las glándulas salivales. Hematológicas: Agranulocitosis, leucopenia, trombocitopenia, anemia aplásica. Hepáticas: Ictericia, hepatotoxicidad. Reacciones de hipersensibilidad: Erupción, urticaria, síndrome tipo lupus, dermatitis exfoliativa, eritema nodoso. Neurológicas: Cefa-lea, somnolencia, vértigo, parestesia, neuritis, neuropatías, depresión. Renales: Nefritis. Otras: Trastornos visuales, artralgia, mialgia, fiebre, linfadenopatía, hipotiroidismo relacionado con la dosis (depresión mental, hipoprotrombinemia y sangramiento; intolerancia al frío, edema duro sin fovea), pérdida del sentido del gusto. RÉGIMEN DE DOSIFICACIÓN Se administra solamente por vía oral, en dosis diarias divididas en 3 tomas iguales a intervalos de 8 horas. En adultos con hipertiroidismo leve a moderado, iniciar con 300-450 mg/día o con 600-900 mg/día en pacientes con hipertiroidismo severo, hasta el control INTERACCIONES de la sintomatología, lo cual puede Puede potenciar la acción de los tomar hasta 2 meses. Una vez contro- anticoagulantes orales, de la heparina, lado el cuadro, ajustar la dosis de los agentes mielodepresores y de gradualmente hasta lograr la mínima medicamentos con potencial hepadosis efectiva posible que, por lo totóxico. Los yoduros y el litio general, se ubica en el rango de 100- potencian el efecto bociógeno del me174 Formulario Terapéutico Nacional * 2ed. 2004 CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y en el embarazo. Usar con suma precaución en pacientes mayores de 40 años debido a que aumenta el riesgo de agranulocitosis. Durante el tratamiento se debe realizar periódicamente evaluación hematológica y determinaciones del tiempo de protrombina. Se recomienda suspender la lactancia durante la terapia ya que el medicamento se excreta en la leche materna. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Protección de las vías aéreas, ventilación y perfusión de soporte. Debe controlarse la función de la médula ósea. INFORMACIÓN AL PACIENTE Informar de inmediato al médico si se presenta alguna reacción o síntoma TIAMAZOL (METIMAZOL) FARMACOLOGÍA Agente antitiroideo. Inhibe la síntesis de hormonas tiroideas al impedir la incorporación del yodo en los residuos de tirosilo en la molécula de tiroglobulina e interferir con el acoplamiento de dichos residuos para formar la de las administradas exógenamente. FARMACOCINÉTICA Después de la administración oral, se absorbe rápidamente en un 80-95%. Los alimentos alteran su absorción. La administración de una dosis única produce niveles séricos pico en 1 hora y efectos terapéuticos en 12-18 horas. No se une a las proteínas plasmáticas. Se metaboliza parcialmente en el híga-do, transformándose en metabolitos inactivos que se excretan, junto a un 12% del fármaco intacto, en la orina. H La vida media de eliminación es de 513 horas. INDICACIONES Tratamiento del hipertiroidismo. RÉGIMEN DE DOSIFICACIÓN Se administra solamente por vía oral en dosis diarias divididas en 3 tomas iguales a intervalos de 8 horas. En adultos con hipertiroidismo leve, iniciar con 15 mg/día; en el hipertiroidismo moderado con 30-40 mg/día y en el hipertiroidismo severo con 60 mg/día. Una vez logrado el control de los síntomas, mantener la dosis por 2 meses continuos y, a partir de allí, ajustar gradualmente hasta lograr la mínima dosis efectiva posible. Por lo general, la dosis de mantenimiento se ubica en el rango de 5-15 mg/día. En niños, la dosis inicial es de 0,4 2ed. 2004 * Formulario TerapÈutico Nacional 175 Hematológicas: Inhibición de la mielopoyesis (agranulocitosis, granulocitopenia y trombocitopenia), anemia aplásica, hipoprotrombinemia. Hepáticas: Hepatitis. Neurológicas: Parestesia, cefalea, somnolencia, neuritis, vértigo. Reacciones de hipersensibilidad: Síndrome tipo lupus, síndrome autoinmune a la insulina, erupciones en la piel, urticaria. Otras: Pigmentación de la piel, pérdida del sentido del gusto, pérdida del cabello, mialgia, linfadenopatía, fiebre medicamentosa. INTERACCIONES Ver: PROPILTIOURACILO. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y durante el embarazo. Usar con precaución en pacientes mayores de 40 años o en dosis mayores de 40 mg/día debido al riesgo de agranulocitosis. Durante la terapia, se debe verificar periódicamente el contaje leucocitario y practicar determinaciones del tiempo de protrombina y pruebas de la función hepática. Se debe suspender la 176 Formulario Terapéutico Nacional * 2ed. 2004 J02 ANTIMIC”TICOS PARA USO SIST…MICO Amfotericina B J ANTIINFECCIOSOS PARA USO SISTÉMICO J01 ANTIBACTERIANOS PARA USO SIST…MICO Amikacina Amoxicilina / Ácido Clavulánico Ampicilina Ampicilina / Sulbactam Azitromicina Aztreonam Bencilpenicilina Benzatínica (Penicilina G Benzatínica) Bencilpenicilina Procaínica (Penicilina G Procaínica) Bencilpenicilina (Benzatínica / Procaínica / Sódica) (Penicilina G (Benzatínica / Procaínica / Cristalina)) (6/3/3) Bencilpeniclina Sódica (Penicilina G Cristalina) (Penicilina G Sódica) Cefalexina Cefalotina Cefepima Cefoperazona / Sulbactam* Cefotaxima Ceftazidima Ceftriaxona Ciprofloxacino (Sistémico) Clindamicina Cloranfenicol Doxiciclina (Antibacteriano Sistémico) Eritromicina (Eritromicina Estolato) Gentamicina (Sistémica) Imipenem / Cilastatina Sódica Levofloxacino Metronidazol (Antibacteriano Sistémico) Nitrofurantoína (Nitrofurantoína Macrocristales) Oxacilina Oxitetraciclina Sulfametoxazol / Trimetoprima (Trimetoprim / Sulfametoxazol) Sulfametoxipiridazina Vancomicina 2ed. 2004 * Formulario TerapÈutico Nacional 177 J Etambutol Isoniazida Pirazinamida Rifampicina (Antileproso) Rifampicina (Antituberculoso) JJ05 ANTIVIRALES PARA USO SIST…MICO Abacavir Abacavir / Lamivudina / Zidovudina Aciclovir (Sistémico) Amprenavir Delavirdina Didanosina Efavirenz Estavudina (Stavudina) Ganciclovir Indinavir Lamivudina Nelfinavir Nevirapina Ritonavir Ritonavir / Lopinavir* Saquinavir Zalcitabina Zidovudina Zidovudina / Lamivudina J06 SUEROS INMUNES E INMUNOGLOBULINAS Antitoxina Tetánica (Antitoxina Tetánica Humana) Inmunoglobulina Anti-D (Rh) (Inmunoglobulina Rho (D) Humana) Inmunoglobulina Humana Normal Suero Antiescorpiónico* Suero Antiofídico (Suero Antiofídico Polivalente) Suero Antirrábico 178 Formulario Terapéutico Nacional * 2ed. 2004 J07 VACUNAS Toxoide Tetánico Vacuna Antituberculosa (BCG) Vacuna contra el Haemophilus Influenzae Tipo B (Antihaemophilus Influenzae Tipo B) Vacuna contra el Sarampión Vacuna contra el Sarampión, Rubeola y Parotiditis Vacuna contra la Difteria, Pertussis y Tétanos* (Difteria, Tosferina y Tétanos) Vacuna contra la Fiebre Amarilla (Antiamarílica) Vacuna contra la Hepatitis B (Antihepatitis B) Vacuna contra la Poliomielitis (Antipolio) Vacuna contra la Rabia (Antirrábica) J 2ed. 2004 * Formulario TerapÈutico Nacional 179 AMIKACINA FARMACOLOGÍA Antibiótico aminoglicósido con actividad bactericida. Inhibe la síntesis de proteínas en gérmenes susceptibles por unión a la subunidad ribosomal 30S. Es activa contra Acinetobacter spp., Citrobacter spp., Enterobacter spp., Proteus spp., Escherichia coli, Klebsiella spp., Pseudomonas spp., Providencia spp., Salmonella spp, Serratia spp., Shigella spp., Staphylococcus aureus y S. epidermidis. debidas a cepas susceptibles de bacterias Gram negativas incluyendo Pseudomonas spp., E. Coli, Proteus indol positivo e indol negativo, Providencia spp., Klebsiella, Enterobacter y Serratia spp. y Acinetobacter spp., infecciones del tracto respiratorio, huesos, articulaciones, piel y tejido blando, bacteriemia, septicemia, quemaduras; infecciones intraabdominales, intraoperatorias y urinarias complicadas y recurrentes. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV ó IM. La concentración sérica terapéutica se ubica en el rango de 10-30 mcg/ml. La dosis usual en adultos, niños e J infantes mayores de 1 mes es de 15 mg/kg/día divididos en 2-3 dosis iguales (máximo 1,5 g/día). En neonatos, iniciar con 10 mg/kg, seguidos por 7,5 mg/kg cada 12 horas. La duración de la terapia por lo general es de 7-10 días como mí-nimo, según la severidad de la infec-ción. La dosis IV se debe infundir en períodos de 0,5-1 hora en adultos y niños y de 12 horas en recién nacidos. La dosificación en pacientes con falla renal debe hacerse, en la medida de lo posible, con base en los niveles séricos del antibiótico. El ajuste de la dosis puede hacerse modificando los intervalos de dosificación o disminuyendo la dosis de acuerdo a la depuración de creatinina (Clcr) o de la creatinina 181 2ed. 2004 * Formulario TerapÈutico Nacional FARMACOCINÉTICA Los aminoglicósidos se absorben pobre y erráticamente en el tracto gastrointestinal, por lo que es necesaria su administración por vía parenteral (IV ó IM). La absorción por vía IM es rápida, alcanzando concentraciones plasmáti-cas máximas en 0,5-2 horas y que pueden persistir por 8-12 horas. Se une a las proteínas plasmáticas en muy escasa proporción. Se distribuye ampliamente en los fluidos y tejidos corporales, aunque difunden muy poco al sistema nervioso central, razón por la cual es necesaria su adminis-tración por vía intratecal o intraven-tricular para lograr concentraciones efectivas a ese nivel. Una pequeña cantidad se acumula en los tejidos, de donde es eliminada lenta y gradual-mente. No sufre metabolismo aprecia-ble y se excreta principalmente por filtración continuando con una dosis de mantenimiento cada 12 horas, calculada en función del valor de depuración de creatinina (Clcr) mediante la fórmula: Dosis de mantenimiento cada 12 horas (mg) = Clcr observado (ml/min) Clcr normal (ml/min) X dosis de carga (mg) Para ajustes más precisos, consultar la literatura especializada. INTERACCIONES El uso concurrente de otros medicamentos con potencial nefrotóxico y ototóxico (amfotericina B, aciclovir, ba-citracina, vancomicina, cisplatino, cefalosporinas, polimixina B y diuréticos potentes como el ácido etacrínico, furosemida, úrea y manitol) incrementa el riesgo de tales efectos. El uso de aminoglicósidos con anesté-sicos generales o bloqueantes neuro-musculares como la succinilcolina y la d-tubocurarina puede potenciar el e-fecto relajante muscular y causar pa-rálisis respiratoria. La clindamicina, el cloranfenicol y la tetraciclina antagonizan el efecto bactericida de los aminoglicósidos. La indometacina puede incrementar las concentraciones séricas de los aminoglicósidos cuando se administran conjuntamente en neona182 presión arterial y palpitaciones. Gastrointestinales: Náuseas, vómito, anorexia, pérdida de peso, sialorrea y alte-ración de las enzimas hepáticas. He-matológicas: Agranulocitosis, leucope-nia, leucocitosis, trombocitopenia, anemia, eosinofilia y pancitopenia. Músculo-esqueléticas: Temblor, es-pasmos musculares, debilidad muscu-lar y bloqueo neuromuscular. Neuro-lógicas: Confusión, depresión, cefa-lea, desorientación y parestesias. Ototoxicidad que se manifiesta por daño o lesión al VIII par nervioso craneal que puede dar lugar a tinitus, pérdida de la audición y síntomas vestibulares como vértigo, mareo y ataxia. Renales: Nefrotoxicidad que puede eviden-ciarse por necrosis tubular, proteinuria, hematuria, oliguria, azotemia e incre-mentos del BUN y de la creatinina séri-ca. Reacciones de hipersensibilidad: Erupción, urticaria, angioedema, der-matitis exfoliativa, alopecia y anafila-xis. Respiratorias: Depresión respira-toria, fibrosis pulmonar. Otras: Artral-gia, trastornos visuales, nistagmo, ambliopía, alteración de los electrolitos séricos (calcio, magnesio, sodio y potasio) y esplenomegalia. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a este medicamento o Formulario Terapéutico Nacional * 2ed. 2004 trastornos auditivos, ancianos, neonatos y durante la lactancia. La nefrotoxicidad y la ototoxicidad suelen ser más frecuentes y severas en ancianos, pacientes deshidratados, en la insuficiencia renal, con dosis elevadas y/o con tratamientos prolongados. Previo al inicio de la terapia y periódicamente durante su desarrollo se debe practicar evaluaciones de la función renal y auditiva. Se debe evitar el uso concurrente de medicamentos con potencial ototóxico, nefrotóxico y neurotóxico. El uso por períodos prolongados, además, puede ocasionar el sobrecrecimiento de microorganismos no susceptibles, incluyendo hongos. SOBREDOSIS Manejo sintomático y terapia de soporte. Es removible por hemodiálisis y por diálisis peritoneal. En el infante recién nacido puede considerarse la AMOXICILINA / ÁCIDO CLAVULÁNICO FARMACOLOGÍA La amoxicilina es un antibiótico semisintético del grupo de las penicilinas. Tiene acción bactericida. Inhibe la sín-tesis del mucopéptido en la pared bacteriana al unirse a las enzimas car-boxipeptidasas que participan en la fase final de la síntesis de la pared. El ácido clavulánico inhibe a las enzimas beta-lactamasas que inactivan a la amoxicilina por hidrólisis amoxicilina al incluir, dentro de su espectro de acciÛn, a microorganismos productores de beta-lactamasa usualmente resistentes a la amoxicilina sola. La combinaciÛn es activa contra bacterias aerobias Gram positivas como: Streptococcus spp., S. pneumoniae, Staphylococcus aureus (productor y no productor de beta-lactamasa) y bacterias aerobias Gram negativas como: Enterobacter spp., Klebsiella spp., Moraxella catarrhalis, Escherichia coli, P. mirabilis y Haemophilus influenzae (productores y no productores de beta-lactamasa). FARMACOCIN…TICA Ambos componentes se absorben J bien y casi completamente en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico 1-2 horas después de la administración oral. La administración con alimentos no interfiere con la absorción. La amoxicilina se une a las proteínas plasmáticas en un 17-20% y el ácido clavulánico en un 22-30%. Se distribuyen ampliamente en los fluidos y tejidos corporales, a excepción del SNC donde ninguno de los dos logra concentraciones clínicamente efectivas. La amoxicilina se metaboliza parcialmente por hidrólisis a ácido penicilóico (producto inactivo) que se excreta, junto con un 50-70% del fármaco intacto, por secreción tubular en la orina. 2ed. 2004 * Formulario TerapÈutico Nacional 183 ambas en pacientes con alteración de la función renal. INDICACIONES Infecciones del tracto respiratorio bajo, de la piel y sus estructuras, del tracto urinario, otitis media y sinusitis causadas por organismos sensibles. creción tubular renal de la amoxicilina, incrementando las concentraciones séricas y la vida media. La amoxicilina puede reducir la eficacia de los anticonceptivos orales, interferir con la respuesta inmunológica a la vacuna de la fiebre tifoidea y alterar la excreción renal de metotrexato, incrementando sus niveles plasmáticos y su toxicidad. El alopurinol puede incrementar la incidencia de erupciones en pacientes que reciben amoxicilina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Las dosis se expresan en términos de amoxicilina. En adultos: 500 mg de amoxicilina cada 12 horas en infecciones leves a moderadas o cada REACCIONES ADVERSAS 8 horas en in-fecciones severas. En Gastrointestinales: Náusea, vómito, niños menores de 3 meses: 30 diarrea, gastritis, indigestión, dolor abmg/kg/día divididos en dosis iguales dominal, anorexia, glositis, estomatitis, cada 12 horas. Niños mayores de 3 candidiasis mucocutánea y colitis meses con infecciones leves a pseudomembranosa. Hematológicas: moderadas: 25 mg/kg/día divi-didos Anemia, incluyendo anemia hemolítien dosis iguales cada 12 horas o 20 ca, trombocitopenia, eosinofilia, agramg/kg/día divididos en dosis igua-les nulocitosis y leucopenia. Hepáticas: cada 8 horas; en infecciones seve-ras: Alteración de los niveles de las 45 mg/kg/día divididos en dosis enzimas hepáticas. Neurológicas: Aniguales cada 12 horas o 40 mg/kg/día siedad, cambios de conducta, cefalea, divididos en dosis iguales cada 8 ho- mareo, insomnio, confusión, agitación ras. En niños con peso superior a 40 y convulsiones. Reacciones de hiperkg se administra la dosis de adultos. sensibilidad: Urticaria, prurito, erupPor lo general, la duración de la terapia ción, angioedema, eritema, síndrome depende del tipo y severidad de la de Stevens-Johnson, eritema multiinfección; el tratamiento debe durar el forme, síndrome similar a la enfertiempo que sea necesario para lograr medad del suero, necrólisis epidérla erradicación completa de la mica tóxica, resultados positivos de la infección y evitar la recidiva. En la prueba de Coombs, dermatitis exfomayoría de los casos debe mante- liativa y anafilaxis. Renales: Nefritis nerse hasta 48-72 horas después de intersticial y hematuria. 184 Formulario Terapéutico Nacional * 2ed. 2004 en pacientes con mononucleosis tratados con aminopenicilinas, no es prudente emplear el producto en esta condición. Usar con precaución en pacien-tes con historia de alergia a medica-mentos, disfunción renal, historia de colitis pseudomembranosa, en ancia-nos, en el embarazo y durante la lac-tancia. En la terapia prolongada se de-be vigilar la función renal, la hemato-lógica y la hepática. Su uso por perío-dos prolongados puede provocar el sobrecrecimiento de microorganismos no susceptibles, inclusive hongos pa-tógenos. Si ocurren superinfecciones debe suspenderse el tratamiento. A todos los pacientes tratados por gono-rrea se les debe hacer una prueba serológica para la sífilis. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Ambos fármacos son removibles por hemodiálisis. INFORMACIÓN AL PACIENTE Se recomienda administrar con las comidas para minimizar las molestias gástricas. No alterar la dosis ni suspender el tratamiento antes del tiempo AMPICILINA FARMACOLOGÍA Antibiótico semisintético derivado de penicilina con acción bactericida. Inhibe la síntesis de mucopéptido en la pa-red bacteriana al unirse a las enzimas carboxipeptidasas que participan en la fase final de la síntesis de la pared bacteriana. Es activa contra bacterias aerobias Gram positivas como: Sta-phylococcus spp., Streptococcus spp. (Incluyendo S. pneumoniae) y entero-cocos; bacterias aerobias Gram nega-tivas como: Neisseria gonorrhoeae (no productora de beta-lactamasa), Neisseria meningitidis, Haemophilus influ- J enzae (no productora de beta-lactamasa), Escherichia coli, Proteus mirabilis, Shigella y Salmonella spp. FARMACOCIN…TICA Se absorbe en un 30-55% en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico 1-2 horas después de la administración oral o 1 hora después de la administración IM. Se distribuye ampliamente a los tejidos y fluidos corporales, aunque en forma limitada al SNC (su penetración al SNC ocurre sólo en caso de meninges inflamadas). Se une a las proteínas plasmáticas en un 15-25%. Se metaboliza parcialmente por hidrólisis a ácido penicilóico (producto inactivo) que se excreta, junto con el fármaco 2ed. 2004 * Formulario TerapÈutico Nacional 185 media de eliminación es de 1 hora y se prolonga en pacientes con falla renal. INDICACIONES Tratamiento de infecciones del tracto respiratorio superior e inferior, la piel y sus estructuras, tracto gastrointestinal, tracto genitourinario y meningitis causadas por organismos susceptibles. 50-100 mg/kg/día por vía oral, IM ó IV divididos en dosis iguales cada 68 horas para el tratamiento de infecciones severas. Las dosis usadas en niños no deben superar las dosis de los adultos. En neonatos menores de 1 semana: 25 mg/kg IV ó IM cada 12 horas si pesan menos de 2 kg, o cada 8 horas si pesan más de 2 kg; en neonatos de 1-4 semanas: 25 mg/kg cada 8 horas si pesan menos de 2 kg o cada 6 horas si pesan más de 2 kg. Las dosis pediátricas no deben exceder las dosis de los adultos. La terapia debe ser continuada por 48 a 72 horas después que el paciente se hace asintomático o se haya obtenido evidencia de erradicación de la bacteria. RÉGIMEN DE DOSIFICACIÓN Se usa por vía oral, IM e IV. Aunque las dosis son similares por cualquier vía, se prefieren las rutas parenterales pa-ra infecciones severas. En adultos y niños con peso superior a 40 kg, la dosis usual para la mayoría de las infecciones causadas por gérmenes sensibles es 250-500 mg cada 6 horas. Para el tratamiento de infecciones del tracto INTERACCIONES gastrointestinal o urinario: 500 mg El probenecid puede bloquear la secada 6 horas; en casos graves creción tubular renal de la ampicilina, puede ser superior. En caso de incrementando las concentraciones septicemia o de meningitis bac- séricas y la vida media. El alopurinol teriana, 8-14 g/día o 150-200 puede incrementar la incidencia de mg/kg/día IV ó IM, divididos en 3-4 erupciones en pacientes que reciben dosis. Para la gonorrea: una dosis ampicilina. La ampicilina puede reducir única de 3,5 g en combinación con la eficacia de los anticonceptivos ora1 g de probenecid. les. - En niños con peso inferior a 40 kg, para infecciones respiratorias y de REACCIONES ADVERSAS la piel: 25-100 mg/kg/día, divididos Cardiovasculares: Edema y dolor en el en 4 dosis iguales, administradas pecho, tromboflebitis. Gastrointesticada 6 horas. Para infecciones del nales: Náusea, vómito, diarrea, gastracto gastrointestinal o urinario: 186 Formulario Terapéutico Nacional * 2ed. 2004 Neurológicas: Cefalea, mareo, confusión, fatiga y trastornos en la percepción olfativa y gustativa. Reacciones de hipersensibilidad: Urticaria, prurito erupción, angioedema, eritema, piel seca, dermatitis exfoliativa, eritema multiforme, síndrome de StevensJohnson y anafilaxis. Renales: Aumentos del BUN y de la creatinina sérica, nefritis intersticial, disuria y retención urinaria. Otras: Candidiasis, esca-lofríos, epistaxis, dolor subesternal y dolor en el sitio de la i n y e c c i ó n . INFORMACIÓN AL PACIENTE Se debe administrar 1 hora antes o 2 horas después de las comidas, para una absorción máxima. No alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Informar al médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial AMPICILINA / SULBACTAM FARMACOLOGÍA La ampicilina es un antibiótico semisintético derivado de penicilina. Tiene J acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipepti-dasas que participan en la fase final de la síntesis de la pared bacteriana. El sulbactam inhibe a las enzimas beta-lactamasas que inactivan la molécula de ampicilina por hidrólisis del anillo beta-lactámico. Así, el sulbactam potencia el efecto bactericida de la ampicilina al incluir dentro de su espectro de acción a microorganismos productores de betalactamasas usualmente resistentes a la ampicilina sola. La combinación es activa contra bacterias aerobias Gram positivas como: Staphylococcus aureus, S. epidermidis, S. saprophyticus, Streptococcus pneumoniae, S. pyoge-nes, S. faecalis 187 2ed. 2004 * Formulario TerapÈutico Nacional CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a las penicilinas o a otros antibióticos beta-lactámicos. Debido a la elevada incidencia de erupción eritematosa severa descrita en pacientes con mononucleosis tratados con aminopenicilinas, no es prudente emplear el medicamento en esta condición. Usar con precaución en pacientes con historia de alergia a medicamentos, alteración de la función renal, historia de colitis pseudomembranosa, en ancianos, en el embarazo y durante la lactancia. En caso de terapia prolongada se debe vigilar la función renal, la hematológica y la hepática. El uso por períodos prolongados puede provocar el sobrecrecimiento de microorganismos no susceptibles, mirabilis, P. vulgaris, Providentia rettgeri, P. stuartii, Morganella morganii y anaerobios como: Clostridium spp., Peptococcus spp., Peptostreptococcus spp. y Bacteroides spp. FARMACOCIN…TICA Después de la administración IM, la ampicilina y el sulbactam se absorben rápida y completamente, alcanzando concentraciones plasmáticas pico en 30-60 minutos. Ambos se distribuyen ampliamente en los tejidos y fluidos corporales, aunque en forma limitada al SNC. La ampicilina se une a las proteínas plasmáticas en un 15-28% y el sulbactam en un 38%. Tanto la ampicilina como el sulbactam se excretan en un 75-92% intactos por la orina en las primeras 8 horas de la administración, mediante secreción tubular (90%) y filtración glomerular (10%). La vida media de eliminación de ambos es de aproximadamente 1 hora y se prolonga en pacientes con alteración de la función renal. INDICACIONES Infecciones de la piel y sus estructuras, infecciones intra-abdominales y ginecológicas causadas por organismos susceptibles. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda, inyección IV lenta en 10-15 minutos y por infusión IV en 15-30 188 200 mg/kg/día por infusión IV divididos en dosis iguales cada 6 horas. Niños con peso superior a 40 kg se usa la dosis de adultos. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el trata-miento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. En la mayoría de los casos debe mantenerse hasta por 48-72 horas después que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. INTERACCIONES El probenecid puede bloquear la secreción tubular renal de la ampicilina, incrementando las concentraciones séricas y la vida media. El alopurinol puede incrementar la incidencia de erupciones en pacientes que reciben ampicilina. La ampicilina puede reducir la eficacia de los anticonceptivos orales. REACCIONES ADVERSAS Cardiovasculares: Edema, tromboflebitis y dolor en el pecho. Gastrointestinales: Náusea, vómito, diarrea, gastritis, lengua mega vellosa, dolor abdominal, flatulencia, glositis, estomatitis y colitis pseudomembranosa. Hematológicas: Disminución de la hemoglobina y del hematocrito, anemia, trombocitopenia, eosinofilia, Formulario Terapéutico Nacional * 2ed. 2004 caria, prurito, erupción, angioedema, eritema, piel seca, dermatitis exfoliativa, eritema multiforme, síndrome de Stevens-Johnson y anafilaxis. Renales: Aumentos del BUN y de la creatinina sérica, nefritis intersticial, disuria y retención urinaria. Otras: Candidiasis, escalofríos, epistaxis, dolor subesternal y dolor en el sitio de la inyección. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a las penicilinas o a otros antibióticos beta-lactámicos. Debido a la elevada incidencia de erupción eritematosa severa, descrita en pacientes con mononucleosis tratados con aminopenicilinas, no es prudente emplear el producto en esta condición. Usar con precaución en pacientes con historia de alergia a medicamentos, alteración de la función renal, historia de colitis pseudomembranosa, en ancianos, en el embarazo y durante la lactancia. En caso de terapia prolongada, se debe vigilar la función renal, la hematológica y la hepática. El uso por períodos prolongados puede provocar el sobrecrecimiento de microorganismos no susceptibles, inclusive hongos patógenos. Este medicamento no debe ser mezclado con amino-glicósidos debido a la inactivación in vitro de los INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso AZITROMICINA FARMACOLOGÍA Antibiótico macrólido, subclase azálido. Inhibe la síntesis de proteínas bac-terianas. Tiene actividad similar a la descrita para la eritromicina (ver: ERI-TROMICINA ESTOLATO). FARMACOCINÉTICA Se absorbe en un 37% en el tracto gastrointestinal. Se une a las proteínas plasmáticas en un 50-75%. Se distri-buye ampliamente a los tejidos y flui-dos corporales, J alcanzando concen-traciones elevadas y sostenidas (hasta por 4-7 días después de la última do-sis) en los tejidos infectados. Tal particularidad se cree que es debida a su fácil penetración a los fagocitos y posterior transporte, por quimiotaxis, a los focos infecciosos donde se libera y acumula. Se metaboliza en el hígado en un 35% transformándose en productos inactivos que se excretan, junto con un 50% del fármaco intacto, en su mayoría por vía biliar y en escasa proporción por la orina (12%). La vida media de eliminación terminal se ubica en el rango de 40-68 horas. INDICACIONES Infecciones del tracto respiratorio 2ed. 2004 * Formulario TerapÈutico Nacional 189 Rior e inferior, de piel y tejidos blandos en adultos: 500 mg/día por 3 días continuos; alternativamente se puede administrar una dosis inicial de 500 mg el primer día, seguido por dosis sucesivas de 250 mg/día por 4 días. En niños: 10 mg/kg/día por 3 días con-tinuos. En infecciones genitales provo-cadas por Chlamydia trachomatis o por Neisseria gonorrhoeae, en adultos: Dosis única de 1 g. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, dolor en el pecho. Gastrointestinales: Dispepsia, náusea, vómito, dolor abdominal, flatulencia. Hematológicas: Leucopenia, neutropenia, trombocitopenia. Hepáticas: Elevación de los niveles de las aminotransferasas, ictericia colestásica. Neurológicas: Mareo, cefalea, vértigo, somnolencia, fatiga. Reacciones de hipersensibilidad: Erupción, prurito, fotosensibilidad, angioedema y anafilaxis. Renales: Nefritis, elevación del BUN. Otras: Vaginitis. INTERACCIONES Los antiácidos reducen la velocidad (aunque no la magnitud) de la absorción gastrointestinal de azitromicina. La azitromicina incrementa los niveles plasmáticos de zidovudina. Su combinación con inhibidores de la HMG-CoA reductasa podría 190 sibilidad al fármaco o a otros medicamentos macrólidos. Usar con precaución en presencia de alteraciones de la función hepática y en pacientes con arritmias cardíacas. No se ha evaluado la seguridad de su uso en el embarazo y durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE No alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. El medicamento debe ser administrado una hora antes o dos horas después de las comidas. No hay restricción AZTREONAM FARMACOLOGÍA Antibiótico beta-lactámico monocíclico (monobactámico) con acción bacteri-cida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las proteínas fijadoras de penicilinas que intervienen en la fase final de la sínte-sis de la pared. Induce la formación de estructuras bacterianas filamentosas largas. Es resistente a muchas de las beta-lactamasas de las bacterias Formulario Terapéutico Nacional * 2ed. 2004 spp., Citrobacter spp., Pseudomonas aeruginosa, Escherichia coli, Haemophilus influenzae, Proteus mirabilis y Serratia spp. FARMACOCINÉTICA Después de la administración IM, se absorbe rápida y completamente, alcanzando concentraciones plasmáticas pico en 60 minutos. Se une a las proteínas plasmáticas en un 46-60% y se distribuye ampliamente a los tejidos y fluidos corporales. Se metaboliza parcialmente por hidrólisis inespecífica del anillo beta-lactámico dando lugar a productos sin actividad microbiológica que se excretan, junto a un 60-75% del fármaco intacto, por filtración glomerular y secreción tubular en la orina. Un 12% se excreta por las heces. La vida media de eliminación es de 1,3-2,2 horas y se prolonga en pacientes con falla renal. niños de 9 meses en adelante: 30 mg/kg IV cada 6-8 horas. La dosis total diaria no debe exceder 120 mg/kg. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. En la mayoría de los casos debe mantenerse hasta por 48-72 horas después que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, flebiJ tis, cambios en el ECG. Gastrointestinales: Náuseas, vómito, dolor abdominal, cólicos abdominales, trastornos del gusto, diarrea y colitis pseudomembranosa. Hematológicas: Eosi-nofilia, leucopenia, neutropenia, panci-topenia, anemia, INDICACIONES Infecciones del tracto respiratorio bajo, t r o m b o c i t o p e n i a , l e u c o c i t o s i s , infecciones de la piel y sus estructuras, trombocitosis e incre-mento de los del tracto urinario, intra-abdominales, tiempos de coagulación. Hepáticas: ginecológicas y septicemia causadas Aumento de los niveles de las enzimas hepáticas, ictericia y he-patitis. por organismos susceptibles. Neurológicas: Mareo, in-somnio, confusión, parestesia, dolor de RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM cabeza, debilidad, fatiga, trastornos profunda, por inyección IV lenta en 3-5 mentales y convulsiones. Reacciones minutos y por infusión IV en 20-60 mi- de hipersensibilidad: Erupción, urticanutos. La dosis usada en caso de in- ria, prurito, angioedema, broncoesfecciones del tracto urinario en adultos pasmo, fiebre, púrpura, eritema multiforme, dermatitis exfoliativa, ne191 2ed. 2004 * Formulario TerapÈutico Nacional INTERACCIONES No se describen interacciones de importancia clínica. Sin embargo, aztreo-nam es incompatible con las solu-ciones de nafcilina sódica, cefradina y metronidazol. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. Usar con precaución en pacientes con historia de alergia a otros medicamentos, de colitis pseudomembranosa o de discrasias sanguíneas, alteración de la función renal, en mujeres em-barazadas y durante la lactancia. Durante el tratamiento es recomenda-ble vigilar periódicamente la función renal, hepática y hematológica. El uso por períodos prolongados puede pro-vocar el sobrecrecimiento de microorganismos no susceptibles, inclusive hongos. SOBREDOSIS Manejo sintomático y tratamiento de BENCILPENICILINA BENZATÍNICA (PENICILINA G BENZATÕNICA) FARMACOLOGÍA Antibiótico beta-lactámico perteneciente al grupo de las penicilinas naturales. Las penicilinas tienen acción bactericida; inhiben la síntesis de mucopéptido en la pared 192 tidasas que participan en la fase final de la síntesis de la pared bacteriana. Es activa in vitro contra los estafilococos (excepto las cepas productoras de beta-lactamasa), estreptococos (grupos A, B, C, G, H, K, L y M), enterococos, Neisseria gonorrhoeae, Neisseria meningitidis, Corynebacterium diphtheriae, Listeria monocytogenes, Bacillus anthracis, Pasteurella multocida, Streptobacillus moniliformis, Actinomyces bovis, Actinomyces israelii, Treponema pallidum, Bacteroides spp., Clostridium spp., y Leptospira spp. FARMACOCINÉTICA Dado que la bencilpenicilina benzatínica es relativamente insoluble, la administración IM genera un depósito en el tejido muscular del cual es lentamente absorbida a la circulación sistémica e hidrolizada en el plasma a bencilpenicilina. La administración IM produce concentraciones plasmáticas de bencilpenicilina más sostenidas en el tiempo, aunque menores a las que se obtienen con dosis equivalentes de bencilpenicilina procaínica o de bencilpenicilina sódica. Luego de la administración IM se alcanzan niveles séricos pico en 13-24 horas y puede ser detectada en el plasma hasta 1-4 semanas después. Se distribuye ampliamente a los tejidos y fluidos corporales, alcanzando concentraciones elevadas en el riñón, pulmón, Formulario Terapéutico Nacional * 2ed. 2004 a las proteínas plasmáticas en un 60%. Se metaboliza parcialmente en el hígado a productos activos e inactivos que se excretan rápidamente por secreción tubular (90%) y en menor grado por filtración glomerular (10%) en la orina. Un 80-85% es eliminado como fármaco intacto. La vida media de eliminación es de 20-50 minutos y se incrementa en caso de falla renal. INDICACIONES Tratamiento de infecciones de intensidad leve a moderadamente severa causadas por organismos susceptibles a concentraciones bajas y persistentes de bencilpenicilina. RÉGIMEN DE DOSIFICACIÓN Se administra sólo por inyección IM profunda. En infecciones del tracto respiratorio superior, la dosis usual en adultos es 1.200.000 U como dosis única; en niños con peso superior a 27 kilos: 900.000 U; en niños con peso inferior a 27 kilos: 300.000-600.000 U; en neonatos: 50.000 U/kg. En la sífilis temprana (primaria, secundaria o latente de menos de 1 año de duración): 2.400.000 U como dosis única; en sífilis con más de 1 año de duración o en caso de sífilis primaria o se-cundaria en pacientes VIH positivos: 2.400.000 U una vez a la semana por 3 semanas; en caso de sífilis congénita en niños menores de 2 años: 50.000 U/kg como dosis INTERACCIONES Los antibióticos bacteriostáticos (cloranfenicol, eritromicina o tetraciclinas) pueden disminuir el efecto bactericida de las penicilinas. Las penicilinas pueden reducir la eficacia de los anticonceptivos orales y, en combinación con diuréticos ahorradores de potasio, pueden generar hiperpotasemia. El probenecid puede bloquear la secreción tubular renal de las penicilinas incrementando sus niveles séricos y vida media. Los AINEs podrían desplazar a las penicilinas de su unión a las proteínas. Incompatibilidad física y química con los aminoglicósidos. De-ben administrarse por separado. REACCIONES ADVERSAS Cardiovasculares: Taquicardia, hipotensión, vasodilatación, tromboflebitis, hipertensión pulmonar y paro cardíaco. Gastrointestinales: Náusea, vó-mito, diarrea, malestar epigástrico y colitis pseudomembranosa. Hematológicas: Anemia hemolítica, neutropenia, trombocitopenia, eosinofilia, agranulocitosis y leucopenia. Hepáticas: Elevación de los niveles de las enzimas hepáticas y la bilirrubina. Neurológicas: Cefalea, nerviosismo, astenia, fatiga, mareos, somnolencia, confusión y convulsiones. Reacciones de hipersensibilidad: Erupción, prurito, urticaria, angioedema, laringoespasmo, dermatitis exfoliativa, eritema mul- 2ed. 2004 * Formulario TerapÈutico Nacional 193 J foresis y dolor en el sitio de la inyección. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a las penicilinas. Usar con precaución en pacientes con historia de alergia a medicamentos, alteración de la función renal, historia de colitis pseudomembranosa, en ancianos, embarazo y durante la lactancia. En caso de terapia prolongada se debe vigilar la función renal, hematológica, cardiaca y los electrolitos. El uso de este medicamento por períodos prolongados puede ocasionar el sobrecrecimiento de organismos no susceptibles, inclusive hongos pató-genos. SOBREDOSIS Tratamiento sintomático y terapia de soporte. Es removible por hemodiálisis. INFORMACIÓN AL PACIENTE No alterar la dosificación ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los BENCILPENICILINA PROCAÍNICA (PENICILINA G PROCAÕNICA) FARMACOLOGÍA Ver: BENCILPENICILINA BENZATÍNI-CA. 194 FARMACOCINÉTICA Dado que la bencilpenicilina procaínica es relativamente insoluble, la administración IM genera un depósito en el tejido muscular desde el cual es lentamente absorbida a la circulación sistémica e hidrolizada en el plasma a bencilpenicilina. Luego de la administración se alcanzan niveles séricos pico en 1-3 horas y aún después de 1-2 días es posible detectar el fármaco en san-gre (o hasta 5 con dosis elevadas). La cinética de distribución, metabolismo y excreción es similar a la descrita para la bencilpenicilina b e n z a t í n i c a ( v e r : B E N C I L P E N I C I L I N A BENZATÍNICA). INDICACIONES Tratamiento de infecciones moderadamente severas causadas por gérmenes sensibles a las concentraciones de bencilpenicilina normalmente alcanzadas con esta dosificación. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda. La dosis usual en adultos y niños mayores de 12 años es 600.000-1.000.000 U/día por 7-10 días y en niños menores de 12 años: 25.000-50.000 U/kg/día, como dosis única o divididos en 2 dosis cada 12 horas. INTERACCIONES Formulario Terapéutico Nacional * 2ed. 2004 CA. las anteriores, se absorbe con mucha mayor lentitud, alcanzando niveles séSOBREDOSIS ricos pico después de 13-24 horas. Ver: BENCILPENICILINA Una vez en la sangre, las sales se BENZATÍNI-CA. hidrolizan liberando bencilpenicilina. Como la cinética de eliminación de INFORMACIÓN AL PACIENTE V e r : B E N C I L P E N I C I L I N A cada sal es proporcional a la velocidad con la que se absorben desde el músculo, la combinación de las 3 permite BENCILPENICILINA (BENZATÕNICA / PROCAÕNICA / S”DICA) lograr, rápidamente, una concen(PENICILINA G (BENZATÕNICA / PROCAÕNICA / tración efectiva, y a la vez sostenida, CRISTALINA)) (6/3/3) de bencilpenicilina en el plasma. La FARMACOLOGÍA cinética de distribución, metabolismo Formulación que contiene 600.000 U y excreción de la combinación es de bencilpenicilina benzatínica, similar a la descrita para la 300.000 U de bencilpenicilina procaí- bencilpenicilina benzatínica (ver: nica y 300.000 U de bencilpenicilina BENCILPENICILINA BENZATÍNICA) J sódica. Combina la rapidez de la bencilpenicilina sódica en lograr elevados INDICACIONES niveles sanguíneos con la acción Infecciones causadas por gérmenes inter-media de la bencilpenicilina sensibles en las que se requiere de procaínica y prolongada de la una acción rápida y prolongada de la bencilpenicilina benzatínica. El bencilpenicilina. espectro antibacteria-no de la combinación es similar al des-crito RÉGIMEN DE DOSIFICACIÓN para la bencilpenicilina benzatí-nica Se administra sólo por inyección IM (ver: BENCILPENICILINA BEN- profunda, como dosis única. En adultos: 1.200.000 U; en niños con peso ZATÍNICA). superior a 30 kilos: 600.000-1.200.000 FARMACOCINÉTICA U; en niños con peso inferior a 30 kilos: Luego de la administración IM, la ben- 600.000 U. cilpenicilina sódica se absorbe rápidamente, alcanzando niveles plasmá- INTERACCIONES ticos pico a los 15-30 minutos. La ben- V e r : B E N C I L P E N I C I L I N A cilpenicilina procaínica, en cambio, BENZATÍNI-CA. por ser relativamente insoluble tiende REACCIONES ADVERSAS 2ed. 2004 * Formulario TerapÈutico Nacional 195 SOBREDOSIS Ver: BENCILPENICILINA BENZATÍNI-CA. INFORMACIÓN AL PACIENTE Ver: BENCILPENICILINA BENCILPENICILINA SÓDICA (PENICILINA G CRISTALINA) (PENICILINA G S”DICA) FARMACOLOGÍA Ver: BENCILPENICILINA BENZATÍNI-CA. FARMACOCINÉTICA Se absorbe rápidamente después de la administración IM, alcanzando concentraciones séricas pico en 15-30 minutos. La cinética de distribución, metabolismo y eliminación es similar a la descrita para la bencilpenicilina benzatínica (Ver: BENCILPENICILINA BENZATÍNICA). INDICACIONES Infecciones severas causadas por microorganismos sensibles en las que se requieren niveles séricos elevados y en corto tiempo de bencilpenicilina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM ó IV. Las dosis en neonatos menores de 1 semana: 50.000-100.000 U/kg/día en dosis divididas cada 8-12 horas; en infecciones severas: 100.000-150.000 U/kg/día en dosis divididas cada 8-12 196 a 12 años: 25.000-50.000 U/kg/día en dosis divididas cada 6 horas; en infecciones severas: 100.000-400.000 U/kg/día en dosis divididas cada 4-6 horas. La duración del tratamiento es variable y dependiente del patógeno involucrado, del sitio y severidad de la infección y de las condiciones clínicas y tolerancia de cada paciente. En adultos la dosis usual en endocarditis por estreptococos es: 10-20 millones de U/día por infusión IV continua o en dosis divididas cada 4 horas por 2-4 semanas. En ántrax: 5 millones U/día IV ó IM en dosis divididas por el tiempo que sea necesario hasta lograr resolución de la infección. En infecciones causadas por Corynebacterium diphtheriae: 300.000-400.000 U/día IM en dosis divididas por 10-12 días. En meningitis por Listeria: 15-20 millones de U/día IV por 2 semanas. En endocarditis por Listeria: igual dosis, pero por 4 semanas. En infecciones por Erysipelothrix rhusiopathie: 2-20 millones U/día IV ó IM por 4-6 se-manas. Infecciones por Neisseria meningitidis: 20-30 millones U/día por infusión IV continua por 1014 días. Infecciones por Pasteurella multocida: 4-6 millones U/día IV ó IM por 2 semanas. Infecciones por Clostridium: 20 millones U/día IV ó IM. En actinomicosis cérvicofacial: 1-6 millones U/día IV por 3-6 semanas. En actinomicosis abdominal o pulmonar: Formulario Terapéutico Nacional * 2ed. 2004 niliformis causada por mordida de rata: 12-15 millones U/día IV ó IM por 3-4 semanas. En profilaxis de endocarditis bacteriana por procedimientos invasivos en adultos y niños con más de 27 kg: 2 millones de U IV ó IM una hora antes del procedimiento y 1 millón de U 6 horas más tarde; en niños con menos de 27 kg: 50.000 U/kg IV ó IM una hora antes y 25.000-50.000 U/kg 6 horas después. INTERACCIONES Ver: BENCILPENICILINA BENZATÍNI-CA. REACCIONES ADVERSAS Ver: BENCILPENICILINA BENZATÍNI-CA. CONTRAINDICACIONES/ PRECAUCIONES Ver: BENCILPENICILINA BENZATÍNI-CA. SOBREDOSIS Ver: BENCILPENICILINA BENZATÍNI-CA. CEFALEXINA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de primera generación. Posee acción bactericida; inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptida-sas que participan en positivas como: Staphylococcus aureus (productores y no productores de penicilinasas), Staphylococcus epidermidis (cepas sensibles a penicilina), Streptococcus pneumoniae y Streptococcus pyogenes y bacterias aerobias Gram negativas como: Escherichia coli, Haemophilus influenzae, Klebsiella pneumoniae, Moraxella catarrhalis y Proteus mirabilis. FARMACOCINÉTICA Se absorbe rápida y casi completamente en el tracto gastrointestinal, alcanzando concentraciones séricas pico a los 60 minutos después de la administración oral. Se distribuye ampliamente a los tejidos y fluidos J corporales. Se une a las proteínas plasmáticas en un 5-24%. No sufre metabolismo apreciable y se excreta casi en su totalidad como fármaco intacto en la orina por filtración glomerular y secreción tubular dentro de 8 horas. La vida media de eliminación es de 0,5-1,2 horas y se incrementa en pacientes con insuficiencia renal. INDICACIONES Tratamiento de infecciones del tracto respiratorio, otitis media, infecciones de la piel y estructuras de la piel, infecciones óseas e infecciones genitourinarias, incluyendo prostatitis aguda, causada por gérmenes sensibles. RÉGIMEN DE DOSIFICACIÓN 2ed. 2004 * Formulario TerapÈutico Nacional 197 por patógenos menos sensibles puede requerirse dosis mayores). La dosis máxima en adultos es 4 g. En niños de 1-15 años, la dosis usual es de 25-50 mg/kg/día dividida en dos dosis (en infecciones severas se puede duplicar la dosis) y para el tratamiento de la otitis media 75-100 mg/kg/día divi-didos en 4 dosis. En la mayoría de los casos, el tratamiento debe mantener-se hasta por 48-72 horas después de que el paciente se haga asintomático o haya evidencia de erradicación micro-biológica. En caso de infecciones por estreptococo betahemolítico, se debe continuar el tratamiento al menos por 10 días. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, dolor en el pecho. Gastrointestinales: Náusea, vómito, diarrea, dolor abdominal, tenesmo, dispepsia, gastritis, anorexia, flatulencia, trastornos del gusto, pirosis, candidiasis oral y colitis pseudomembranosa. Hematológicas: Anemia hemolítica, eosinofilia, neutropenia, trombocitopenia, leucocitosis, agranulocitosis, monocitosis, linfocitopenia, leucopenia, anemia aplásica y alteración del tiempo de coagulación. Hepáticas: Se han reportado casos raros de hepatitis transitoria e ictericia colestásica. Neurológicas: Mareos, dolor de cabeza, fatiga, pesadillas, vértigo, ansiedad, nerviosismo, debi198 tóxica, dermatitis exfoliativa, prueba de Coombs positiva, anafilaxis. Renales: Incrementos del BUN y de la creatinina sérica, alteración de la función renal, nefropatía tóxica y nefritis intersticial. Respiratorias: Efusión pleural, disnea, tos, rinitis. Otras: Fiebre, prurito anal y genital y alteraciones de la glicemia. INTERACCIONES El uso concurrente de fármacos nefrotóxicos incrementa el riesgo de falla renal aguda. El cloranfenicol puede antagonizar la acción de las cefalosporinas. El probenecid inhibe competitivamente la secreción tubular de las cefalosporinas e incrementa las concentraciones séricas con cefalexina. Efecto antabuse cuando se ingiere alcohol 48-72 horas después de la administración del antibiótico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a las cefalosporinas o a otros antibióticos beta-lactámicos. Usar con precaución en pacientes con historia de alergia a otros medicamentos, alteración de la función renal, historia de colitis pseudomembranosa, en ancianos, en el embarazo y durante la lactancia. Durante el tratamiento se debe vigilar periódicamente la función renal y el tiempo de protrombina. El uso del medicamento por períodos Formulario Terapéutico Nacional * 2ed. 2004 y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE No alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Informar al médico de inmediato si se presenta alguna reacción inusual durante el tratamiento, en especial manifestacioCEFALOTINA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de primera generación. Posee acción bactericida; inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptida-sas que participan en la fase final de la síntesis de la pared bacteriana. Es activa contra bacterias aerobias Gram positivas como: Staphylococcus aur-eus (productores y no productores de penicilinasas), Staphylococcus epider-midis, Streptococcus pyogenes (grupo A beta-hemolíticos), Streptococcus agalactiae y Streptococcus pneumoniae y bacterias aerobias Gram negativos como: Escherichia coli, Klebsiella pneumoniae, Moraxella catarrhalis, Proteus mirabilis, Shigella y Haemophilus influenzae. sitio de la inyección, generalmente se administra por vía IV. Luego de la administración IM, se alcanzan concentraciones séricas pico en 30 minutos. Se distribuye ampliamente a los tejidos y fluidos corporales. Se une a las proteínas plasmáticas en un 6580%. Se metaboliza parcialmente en el hígado y los riñones a desacetilcefalo-tina, la cual posee un 20-25% de la actividad antibacteriana de su precur-sora. Se excreta en 6 horas por secre-ción tubular en la orina, un 60-95% como fármaco intacto y un 27-54% como el metabolito desacetilado. La vida media de eliminación es de 30-60 J minutos y la de la desacetilcefalotina de 12 minutos; estos valores se incrementan en caso de insuficiencia renal. INDICACIONES Tratamiento de infecciones severas del tracto respiratorio, de la piel y sus estructuras y del tracto urinario; infecciones óseas, articulares, septicemia y endocarditis, causadas por gérmenes sensibles. Profilaxis perioperatoria en intervenciones quirúrgicas contaminadas y potencialmente contaminadas. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM y por vía IV. La dosis usual en adultos es de 5001000 mg IM ó IV cada 4-6 horas. En caso de infecciones severas, 2ed. 2004 * Formulario TerapÈutico Nacional 199 período post-operatorio; en niños: 2030 mg/kg, en los mismos intervalos de tiempo. La profilaxis debe descontinuarse a las 24 horas. Por lo general, la duración de la terapia depende del tipo y severidad de la infección. El tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. En la mayoría de los casos, el tratamiento debe mantenerse hasta por 48-72 horas después de que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. INTERACCIONES Ver: CEFALEXINA. REACCIONES ADVERSAS Ver: CEFALEXINA. CONTRAINDICACIONES/ PRECAUCIONES Ver: CEFALEXINA. SOBREDOSIS Tratamiento sintomático y terapia de CEFEPIMA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de cuarta generación. Posee acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptida-sas que participan en 200 tivo contra bacterias aerobias Gram negativas como: Enterobacter, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis y Pseudomonas aeruginosa y bacterias aerobias Gram positivas como: Staphylococcus aureus (sólo cepas susceptibles a meticilina), Streptococcus pneumoniae y S. pyogenes. Es estable a muchas betalactamasas. FARMACOCINÉTICA Luego de la administración IM se absorbe rápida y completamente, alcanzando concentraciones plasmáticas pico en 1-2 horas después de la administración. Se une a las proteínas plasmáticas en un 20% y se distribuye ampliamente a los tejidos y fluidos corporales. Se metaboliza poco en el hígado dando lugar a metabolitos inactivos que se excretan, junto a un 85% del fármaco intacto, por la orina. La vida media de eliminación es de 1,5-2,3 horas y se prolonga en pacientes con insuficiencia renal. INDICACIONES Infecciones del tracto urinario, de la piel y sus estructuras, infecciones intra-abdominales, neumonía y neutropenia febril (como terapia empírica) causadas por gérmenes sensibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM o por infusión IV en 30 minutos. Formulario Terapéutico Nacional * 2ed. 2004 - Infecciones no complicadas de piel y estructuras de la piel: 2 g por vía IV cada 12 horas por 10 días. - Neumonía: 1-2 g por vía IV cada 12 horas por 10 días. - Infecciones intra-abdominales (combinada con metronidazol): 2 g por vía IV cada 12 horas por 7-10 días. - Terapia empÌrica en pacientes neutropénicos: 2 g por vía IV cada 8 horas por 7 días o hasta la resolución de la neutropenia. En niños menores de 12 años: - Infecciones complicadas y no complicadas del tracto urinario, infecciones no complicadas de piel y sus estructuras, neumonía y como terapia empírica de la neutropenia: 50 mg/kg/dosis por vía IV cada 12 horas (cada 8 horas en caso de neutropenia) por el mismo tiempo indicado en el caso de pacientes adultos. Dosis máxima: 2 g. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, diarrea, moniliasis oral, colitis (incluyendo colitis pseudomembranosa). Hematológicas: Neutropenia, leucopenia, agranulocitosis, trombocitopenia, alteraciones del tiempo de coagulación. Hepáticas: Aumento de los niveles de las aminotransferasas y de la bilirrubina. Neurológicas: Cefalea, confusión, alucinaciones, estupor, coma, convulsiones. Reacciones de cefalosporinas (ver: CEFALEXINA). INTERACCIONES Ver: CEFALEXINA. Debe chequearse la función renal si se va a administrar altas dosis de aminoglicósidos con cefepima debido al aumento potencial de la nefrotoxicidad y la ototoxicidad de los aminoglicósidos. CONTRAINDICACIONES/ PRECAUCIONES Ver: CEFALEXINA. SOBREDOSIS Manejo sintomático y terapia de soporte. J CEFOPERAZONA / SULBACTAM FARMACOLOGÍA La cefoperazona es un antibiótico del grupo de las cefalosporinas de tercera generación. Posee acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptidasas que participan en la fase final de la síntesis de la pared bacteriana. El sulbactam inhibe a las enzimas beta-lactamasas que inactivan la molécula de cefoperazona por hidrólisis del anillo betalactámico. Así, el sulbactam potencia el efecto bactericida de la cefoperazona al incluir dentro de su espectro de acción a microorganismos produc- 2ed. 2004 * Formulario TerapÈutico Nacional 201 Staphylococcus aureus (productores y no productores de beta-lactamasa), Staphylococcus epidermidis, Streptococcus pyogenes (beta-hemolítico grupo A) y Streptococcus faecalis; bacterias aerobias Gram negativas como: Enterobacter spp., Klebsiella spp., Citrobacter spp., Pseudomonas spp., Escherichia coli, Haemophilus influenzae, Proteus mirabilis, Proteus vulgaris, Morganella morganii, Providentia stuartii, Providentia rettgeri, Serratia marcescens y Neisseria gonorrhoeae y algunas bacterias anaerobias como: Bacteroides spp., Clostridium spp. y cocos Gram positivos. FARMACOCIN…TICA Después de la administración IM, la cefoperazona se absorbe en un 5075%, alcanzando niveles plasmáticos pico en 1-2 horas. El sulbactam se absorbe casi completamente y produce niveles pico en 30-60 minutos. Ambos se distribuyen ampliamente a los fluidos y tejidos corporales, aunque en forma limitada en el SNC. En la bilis, las concentraciones alcanzadas por la cefoperazona son hasta 100 veces superiores a las plasmáticas. La cefoperazona se une a las proteínas plasmáticas en un 82-93% y el sulbactam en un 38%. Atraviesa la barrera placentaria. Debido a su escaso metabolismo, ambos se excretan en elevada proporción como 202 las heces. La vida media de eliminación de la cefoperazona es de 1,6-2,6 horas y la del sulbactam de 11,2 horas, elevándose ambos valores en pacientes con insuficiencia hepática. INDICACIONES Infecciones del tracto respiratorio, de la piel y sus estructuras, del tracto urinario, intra-abdominales, ginecológicas y septicemia causadas por organismos sensibles. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda y por infusión IV en 15-60 minutos. Las dosis se expresan en términos de cefoperazona. En adultos: 1-2 g ca-da 12 horas. En infecciones severas puede administrarse dosis mayores, 2-4 g cada 8 horas, o 3 g cada 12 horas, siempre que la dosis total diaria de sulbactam (según su proporción dentro de la combinación) no exceda 4 g. En pacientes con alteración de la función hepática, la dosis no debe exceder 4 g; en pacientes con alteración de la función renal y hepática, la dosis no debe exceder 2 g. En niños: 15-30 mg/kg cada 12 horas. En infecciones severas puede administrarse dosis mayores, siempre que la dosis total diaria de sulbactam (según la proporción dentro de la combina- Formulario Terapéutico Nacional * 2ed. 2004 necesario para lograr la erradicación completa de la infección y evitar la recidiva. En la mayoría de los casos debe mantenerse hasta 48-72 horas después de que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. En infecciones por Streptococcus pyogenes (beta-hemolítico grupo A) la terapia debe durar no menos de 10 días. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómito, diarrea, colitis pseudomembranosa. Hematológicas: Neutropenia, eosinofi-lia, hipoprotrombinemia y deficiencia de vitamina K. La cefoperazona puede producir hemorragia, como conse-cuencia de la presencia del grupo MTT (metiltiotetrazol). Reacciones de hipersensibilidad: Urticaria, prurito, erupción, prueba de Coombs positiva. Renales: Aumentos del BUN y de la creatinina sérica. Otras: Flebitis y dolor en el sitio de la inyección. Aunque no están descritas con la c o m b i n a c i ó n cefoperazona/sulbactam, se debe considerar la posibilidad de otras reacciones factibles en terapias con cefalosporinas (ver: CEFALEXINA). INTERACCIONES El uso concurrente de medicamentos nefrotóxicos, como los aminoglicósidos, incrementa el riesgo de falla renal hipersensibilidad a las cefalosporinas o a otros antibióticos beta-lactámicos. Usar con precaución en pacientes con historia de alergia a otros medicamentos, alteración de la función hepática, historia de colitis pseudomembranosa, en ancianos, en el embarazo y durante la lactancia. Durante el tratamiento se debe vigilar periódicamente la función renal, hepática y el tiempo de coagulación. Su empleo por períodos prolon-gados puede provocar el sobrecre-cimiento de microorganismos no sus-ceptibles, inclusive hongos patógenos. SOBREDOSIS J Manejo sintomático y terapia de soporte. Ambos fármacos son removibles CEFOTAXIMA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de tercera generación. Posee acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipepti-dasas que participan en la fase final de la síntesis de la pared bacteriana. Es activa contra bacterias aerobias Gram positivas como: Enterococcus spp., Staphylococcus aureus (productores y no productores de beta-lactamasa), Staphylococcus epidermidis, Strepto-coccus pneumoniae y Streptococcus 2ed. 2004 * Formulario TerapÈutico Nacional 203 influenzae, Haemophilus parainfluenzae, Klebsiella spp., Proteus mirabilis, Proteus vulgaris, Morganella morganii, Providentia stuartii, Providentia rettgeri, Serratia marcescens, Neisseria gonorrhoeae (productora y no productora de penicilinasa), Neisseria meningitidis y Enterobacter spp. y algunas bacterias anaerobias como: Bacteroides fragilis, Clostridium spp. (la mayorÌa de las cepas de C. difficile son resistentes), Fusobacterium spp., Peptococcus spp. y Peptostreptococcus spp. FARMACOCINÉTICA No se absorbe bien en el tracto gastrointestinal por lo que debe ser administrada por vía parenteral, IM ó IV. Luego de la administración IM se alcanzan concentraciones séricas pico en 30 minutos. Se distribuye ampliamente a los tejidos y fluidos corporales. Se une a las proteínas plasmáticas en un 13-38%. Se metaboliza par-cialmente en el hígado y en los riñones a desacetilcefotaxima, la cual tiene actividad antibacteriana, y a otros productos inactivos. Se excreta en 24 horas por filtración glomerular y secreción tubular en la orina, un 40-60% como fármaco intacto y un 24% como el metabolito desacetilado. Otros dos metabolitos (M2 y M3) contribuyen con 20-25%, pero son inactivos. La eliminación es bifásica, con una vida media 204 INDICACIONES Tratamiento de infecciones severas intra-abdominales, ginecológicas, óseas, articulares, del tracto respiratorio inferior, de la piel y sus estructuras, del tracto genitourinario y del sistema nervioso central, bacteriemia/septicemia, gonorrea y oftalmía blenorrágica causadas por microorganismos sensibles. Profilaxis perioperatoria en cirugía contaminada o potencialmente contaminada. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda, por bolo IV en 3-5 minutos y por infusión IV en 20-30 minutos. En adultos con infecciones no complicadas: 1 g cada 12 horas; en infecciones moderadas a severas: 1-2 g cada 6-8 horas y en infecciones severas: 2 g cada 4 horas (máximo 12 g/día). En caso de gonorrea no complicada: dosis única de 500 mg IM. En caso de gonorrea diseminada: 1 g IV cada 8 horas hasta 2 días después de la mejoría, seguida de cefixima o una quinolona oral por 7 días. En la profilaxis perioperatoria: 1 g IV 30-60 minutos antes de la cirugía. En caso de sección cesárea: 1 g IV inmediatamente después de pinzar el cordón umbilical, seguido por 1 g IV ó IM 6 y 12 horas después de la primera dosis. En niños mayores de 1 mes y hasta 12 años (con peso menor a 50 kg): 50-180 mg/kg/día por vía IM ó IV Formulario Terapéutico Nacional * 2ed. 2004 necesario para lograr la erradicación completa de la infección y evitar la recidiva. En la mayoría de los casos, el tratamiento debe mantenerse hasta 48-72 horas después de que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. INTERACCIONES Ver: CEFALEXINA. REACCIONES ADVERSAS Ver: CEFALEXINA. En caso de administración rápida (menos de 60 segundos), en bolo vía catéter central, se ha observado arritmias con posible riesgo de la vida. CONTRAINDICACIONES/ PRECAUCIONES Ver: CEFALEXINA. SOBREDOSIS Manejo sintomático y terapia de soporte. CEFTAZIDIMA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de tercera generación. Posee acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptida-sas que participan en la fase final de la síntesis de la pared bacteriana. La ceftazidima tiene un beta-lactamasas, lo que incrementa su actividad contra las pseudomonas y la disminuye para los organismos Gram positivos. Es activa contra bacterias aerobias Gram positivas como: Staphylococcus aureus (productor y no productor de beta-lactamasa), Streptococcus pneumoniae, Streptococcus pyogenes (grupo A beta-hemolítico) y Streptococcus agalactiae (grupo B); bacterias aerobias Gram negativas como: Enterobacter spp., Citrobacter spp., Escherichia coli, Haemophilus influenzae, Klebsiella spp., Proteus mirabilis, Proteus vulgaris, Pseudomonas spp., Serratia spp. y Neisseria meningitidis y algunas bacterias anaerobias como J Bacteroides spp. Muchas cepas de Bacteroides fragilis son resistentes. FARMACOCINÉTICA No se absorbe bien en el tracto gastrointestinal por lo que debe ser administrada por vía parenteral, IM ó IV. Luego de la administración IM se alcanzan concentraciones séricas pico en 60 minutos. Se distribuye amplia-mente a los tejidos y fluidos corpora-les. Se une a las proteínas plasmáticas en un 5-24%. No se metaboliza en for-ma apreciable. Se excreta en 24 horas por filtración glomerular en la orina en un 80-90% como fármaco intacto. La eliminación es bifásica, con una vida media en fase 2ed. 2004 * Formulario TerapÈutico Nacional 205 ciones, ginecológicas, intra-abdominales, del sistema nervioso central y septicemia, causadas por gérmenes sensibles. debe mantenerse hasta 48-72 horas después de que el paciente se haga asintomático o de evidencia de erradicación microbiológica. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda, bolo IV en 3-5 minutos y por infusión IV en 15-30 minutos. La dosis usual en adultos y niños mayores de 12 años es de 1 g IV ó IM cada 8-12 horas (máximo 6 g/día). En infecciones no complicadas del tracto urinario: 250 mg IV ó IM cada 12 horas y en infeccio-nes complicadas: 500 mg IV ó IM cada 8-12 horas. En caso de neumonía no complicada e infecciones de piel y sus estructuras: 500-1000 mg IV ó IM cada 8 horas. En infecciones óseas y de las articulaciones: 2 g IV cada 12 horas. En infecciones ginecológicas serias e intra-abdominales, meningitis e infecciones severas que ponen en peligro la vida, especialmente en pacientes inmunocomprometidos: 2 g IV cada 8 horas. En infecciones pulmonares causadas por Pseudomonas spp. en pacientes con fibrosis quística con función renal normal: 30-50 mg/kg IV cada 8 horas a un máximo de 6 g por día. Como terapia empírica en pacien-tes neutropénicos febriles: 100 mg/kg cada 8 horas. La dosis en niños me-nores de 1 mes es: 30 mg/kg IV cada 12 horas; y en infantes y niños INTERACCIONES Ver: CEFALEXINA. Es físicamente incompatible con vancomicina. Se recomienda que se administre por infusión IV intermitente, dándolas por separado y lavando las vías entre la administración de los dos agentes. 206 REACCIONES ADVERSAS Ver: CEFALEXINA. Se ha observado hiperbilirrubinemia. CONTRAINDICACIONES/ PRECAUCIONES Ver: CEFALEXINA. SOBREDOSIS Ha ocurrido en pacientes con insuficiencia renal. Las reacciones incluyen: convulsiones, encefalopatía, asterixis, excitabilidad neuromuscular y coma. Tratamiento sintomático y de soporte. Es removible por diálisis CEFTRIAXONA FARMACOLOGÍA Antibiótico del grupo de las cefalosporinas de tercera generación. Posee acción bactericida. Inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas carboxipeptidasas que participan en la Formulario Terapéutico Nacional * 2ed. 2004 bacteriana. Es activa contra bacterias aerobias Gram positivas como: Staphylococcus aureus (productor y no productor de beta-lactamasa), Staphylococcus epidermidis, Streptococcus pneumoniae, Streptococcus pyogenes (grupo A beta-hemolítico) y estreptococos del grupo Viridans; bacterias aerobias Gram negativas como: Acinetobacter calcoaceticus, Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella pneumoniae, Klebsiella oxytoca, Moraxella catarrhalis, Proteus mirabilis, Proteus vulgaris, Morganella morganii, Serratia marcescens, Neisseria gonorrhoeae (productores y no productores de penicilinasa) y Neisseria meningitidis y algunas bacterias anaerobias como: Bacteroides fragilis, Clostridium spp. y Peptostreptococcus spp. La mayoría de las cepas de C. difficile son resistentes a la ceftriaxona. FARMACOCINÉTICA No se absorbe bien en el tracto gastrointestinal por lo que debe ser administrada por vía parenteral, IM ó IV. Luego de la administración IM se alcanzan concentraciones séricas pico en 2-4 horas. Se distribuye amplia-mente a los tejidos y fluidos corpo-rales. La unión a las proteínas plas-máticas es dependiente de la dosis y disminuye en la medida en que ésta aumenta; con concentraciones filtración glomerular en la orina (67%), y el resto secretado en la bilis y, por tanto, encontrado en las heces. La vida media de eliminación es de 8,5-10 horas. INDICACIONES Tratamiento de infecciones del tracto respiratorio inferior, de la piel y sus estructuras, del hueso y articulaciones, infecciones intra-abdominales, del tracto urinario, otitis media, meningitis, septicemia, gonorrea no complicada y enfermedad inflamatoria pélvica aguda, causadas por gérmenes susceptibles. Profilaxis perioperatoria en cirugía contaminada o J potencialmente contaminada. RÉGIMEN DE DOSIFICACIÓN Se administra por inyección IM profunda o por infusión IV en 15-30 minutos. La dosis usual en adultos es 1-2 g/día por vía IM ó IV en dosis única o dividida en dos dosis cada 12 horas (máximo 4 g/día), dependiendo del tipo y severidad de la infección. La dosis diaria no debe exceder 4 g. En el tratamiento de la gonorrea no complicada: Dosis única de 125-250 mg por vía IM. En la profilaxis perioperatoria: 1 g por vía IM ó IV 30-90 minutos antes de la intervención. La dosis usual en niños es 50-75 mg/kg/día (máximo 2 g/día) por vía IM ó IV en dosis única o dividida en dos dosis cada 12 horas. Para el trata- 2ed. 2004 * Formulario TerapÈutico Nacional 207 horas después que el paciente se haga asintomático o haya evidencia de erradicación microbiológica. La duración usual de la terapia es de 4-14 días, aunque podría ser mayor en casos severos. En infecciones por Streptococcus pyogenes debe durar por lo menos 10 días. INTERACCIONES Ver: CEFALEXINA. REACCIONES ADVERSAS Ver: CEFALEXINA. Se ha observado prolongación del tiempo de protrombina. Ocasionalmente se ha observado diaforesis y enrojecimiento de la cara. CONTRAINDICACIONES/ PRECAUCIONES Ver: CEFALEXINA. SOBREDOSIS Manejo sintomático y terapia de soporte. CIPROFLOXACINO (SistÈmico) FARMACOLOGÍA Antibiótico del grupo de las fluoroquinolonas. Posee acción bactericida. Inhibe a las enzimas girasa del ADN y topoisomerasa IV, necesarias para la replicación, transcripción, reparación y recombinación del ADN bacteriano. Es activa in vivo contra: 208 dermidis, Staphylococcus saprophyticus, Streptococcus pneumoniae, Streptococcus pyogenes; bacterias aerobias Gram negativas como: Campylobacter yeyuni, Citrobacter spp., Enterobacter cloacae, Escherichia coli, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella pneumoniae, Legionella pneumophila, Moraxella catarrhalis, Morganella morganii, Neisseria gonorrhoeae, Proteus mirabilis, Proteus vulgaris, Providencia rettgeri, Providencia stuartii, Pseudomonas aeruginosa, Salmonella typhi, Serratia spp. y Shigella spp., Chlamydia pneumoniae, Mycoplasma pneumoniae y Bacillus anthracis. FARMACOCINÉTICA Después de la administración oral se absorbe en un 70-80% en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico en 1-2 horas. No sufre metabolismo de primer paso. Se une a las proteínas plasmáticas en un 20-40% y se distribuye ampliamente a los tejidos y fluidos corporales. Se metaboliza parcialmente en el hígado dando lugar a cuatro metabolitos con muy poca actividad farmacológica que se excretan, junto a un 40-50% (o 50-70% por administración IV) de fármaco intacto, por la orina y las heces. La vida media de eliminación es de 4-6 horas y se prolonga en pacientes con insuficiencia renal. Formulario Terapéutico Nacional * 2ed. 2004 RÉGIMEN DE DOSIFICACIÓN Se usa por vía oral y por infusión IV durante no menos de 60 minutos. - Por vía oral: - Sinusitis aguda: 500 mg cada 12 horas por 10 días. - Infecciones del tracto respiratorio inferior: 500 mg cada 12 horas por 7-14 días. - Infecciones óseas y en articulaciones: 500-750 mg cada 12 horas por 4-6 semanas. - Infecciones de piel y sus estructuras: 750 mg cada 12 horas por 7-14 días. - Infecciones no complicadas de tracto urinario: 100 mg por 3 días; infec-ciones severas: 250-500 mg cada 12 horas por 7-14 días. - Infecciones intra-abdominales (en combinación con metronidazol): 500 mg cada 12 horas por 7-14 días. - Diarrea infecciosa: 500 mg cada 12 horas por 5-7 días. - Infecciones gonocóccicas uretrales y cervicales: dosis única de 250-500 mg. - Por vía intravenosa: - Infecciones del tracto urinario: 200400 mg cada 12 horas. - Infecciones de la piel y sus estructuras, hueso y articulaciones: 400 mg cada 8 horas. - Infecciones del tracto respiratorio inferior: leve a moderada: 400 mg cuadro clínico y la respuesta del paciente. Se recomienda mantener la terapia por 48 horas después de desaparecidos los signos y síntomas de la infección. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, flebitis, prolongación del intervalo QTc, trombosis cerebral, arritmias, hipertensión, angina, infarto y paro cardiopulmonar. Gastrointestinales: Náuseas, vómito, constipación, diarrea, anorexia, dolor abdominal, dispepsia, flatulencia, alteración del gusto, pancreatitis y colitis pseudomembranosa. Hematológicas: Eosinofilia, incremento del tiempo de coagulación, J metahemoglobinemia, agranulocitosis y anemia hemolítica. Hepáticas: Elevación de los niveles de las enzimas hepáticas, ictericia colestásica y necrosis hepática. Neurológicas: Cefalea, mareos, nerviosismo, letargia, insomnio, ansiedad, pesadillas, depresión, parestesia, psicosis, manía, delirio, fobia, temblor y convulsiones. Reacciones de Hipersensibilidad: Prurito, erupción, urticaria, fiebre, fotosensibilidad, hiperpigmentación, eritema multiforme, síndrome de Stevens-Johnson, broncoespasmo, angioedema y anafilaxis. Renales: Incrementos del BUN y de la creatinina sérica, nefritis intersticial, hematuria, cálculos renales, insuficiencia renal aguda, retención urinaria, poliuria. 209 2ed. 2004 * Formulario TerapÈutico Nacional espalda, brazos y mandíbula, rigidez de las articulaciones, crisis de gota, mialgia, debilidad y dolor en el sitio de inyección. INTERACCIONES El ciprofloxacino produce efectos aditivos con los antibióticos betalactámicos, aminoglicósidos, clindamicina y metronidazol. Los antiácidos y los cationes divalentes o trivalentes, especialmente el hierro, interfieren con la absorción gastrointestinal del ciprofloxacino. El probenecid disminuye la depuración renal del ciprofloxacino e incrementa los niveles séricos. El ciprofloxacino puede aumentar las concentraciones plasmáticas de la teofilina y la fenitoína e incrementar el efecto de los anticoagulantes orales. La combinación con AINEs incrementa el riesgo de convulsiones. La administración concomitante con glibenclamida puede provocar hipoglicemia severa y cuando se administra con ciclosporina aumenta el riesgo de falla renal. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al fármaco o a otras quinolonas, en pacientes menores de 18 años y durante el embarazo. No debe usarse por vía IM. Usar con extrema precaución en pacientes con trastornos del SNC, arterioesclerosis 210 sanguíneas. Dado que se ha descrito la ocurrencia de cataratas con la administración de quinolonas, se recomienda la evaluación oftalmológica periódica. De igual manera, se debe vigilar frecuentemente la función renal, hepática, hematológica y cardio-vascular. Se han reportado casos de fotosensibilidad con la administración de quinolonas, por lo que es recomen-dable evitar, en lo posible, la expo-sición excesiva a la luz solar. Es recomendable mantener una adecua-da hidratación del paciente a objeto de prevenir la posibilidad de cristaluria. El uso del ciprofloxacino por períodos prolongados puede provocar el sobrecrecimiento de microorganismos no susceptibles, inclusive hongos patógenos. Solamente debe usarse en el embarazo si el beneficio potencial justifica el riesgo potencial para el feto. El ciprofloxacino se excreta en la leche materna, por lo que debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Mantener una adecuada hidratación. No es removible por diálisis peritoneal ni hemodiálisis. INFORMACIÓN AL PACIENTE Formulario Terapéutico Nacional * 2ed. 2004 o sintomatología inusual durante el tratamiento. Suspender la terapia de inmediato en caso de alergia o de dolor tendinoso, en especial del tendón de CLINDAMICINA FARMACOLOGÕA Antibiótico semisintético con acción bactericida o bacteriostática según la dosis y la sensibilidad del microorganismo. Inhibe la síntesis proteica en gérmenes susceptibles por unión a la sub-unidad ribosomal 50S. Es activa contra: especies de Staphylococcus, Streptococcus (excepto Streptococcus facealis), Actinomyces spp., Bacteroides, Eubacterium, Fusobacterium, Propionibacterium, Peptococcus spp. y Peptostreptococcus spp. FARMACOCIN…TICA Después de la administración oral, se absorbe en un 90% en el tracto gastrointestinal, alcanzando niveles plasmáticos pico en 45-60 minutos. Posterior a la administración IM, alcanza concentraciones máximas en 3 horas. Una vez en la sangre se une a las proteínas plasmáticas en un 93% y se distribuye a los tejidos y fluidos corporales, excepto el sistema nervioso central. Se metaboliza parcialmente a productos activos e inactivos que se excretan en la orina y en las heces. Solamente un 10% de la piratorio inferior, ginecológicas, de la piel y sus estructuras; óseas y articulares, intra-abdominales y septicemia causadas por organismos sensibles. R…GIMEN DE DOSIFICACI”N Por vía oral en adultos: 150-450 mg cada 6 horas; en niños: 8-25 mg/kg/día, divididos en 3-4 dosis iguales. Por vía IM ó IV en adultos: 600-2700 mg/día divididos en 2-4 dosis, sin exceder 600 mg IM por dosis, ni 1200 mg por infusión IV en 1 hora. La velocidad de infusión no debe exceder 30 mg/minuto. En infecciones muy severas puede incrementarse la dosis IV hasta un máximo de 4,8 J g/día. En niños mayores de un mes, por vía IM ó IV: 15-40 mg/kg/día divididos en 3-4 dosis iguales; en neonatos: 15-20 mg/kg/día IM ó IV divididos en 3-4 dosis; en infantes prematuros: 15 mg/kg. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Cardiovasculares: Hipotensión y paro cardiopulmonar por infusión IV rápida (a una velocidad de infusión mayor q u e 3 0 m g / m i n u t o ) . Gastrointestinales: Náuseas, vómitos, 2ed. 2004 * Formulario TerapÈutico Nacional 211 cia renal. Otras: Se ha reportado casos raros de poliartritis. INTERACCIONES Puede potenciar el efecto de los bloqueantes de la unión neuromuscular. El saquinavir puede incrementar los niveles séricos de la clindamicina, aumentando la posibilidad de efectos tóxicos. La eritromicina puede interferir con la acción de la clindamicina; debe evitarse el uso concomitante. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a la clindamicina. Usar con precaución en pacientes con trastornos de la función renal y/o hepática y en aquellos con historia de colitis o enteritis asociada al uso de antibióticos. El uso seguro del medicamento durante el embarazo no ha sido establecido. No se debe administrar por vía IV sin diluir. La velocidad de infusión no debe exceder 30 mg/minuto. El uso por períodos pro-longados puede favorecer el sobre-crecimiento de patógenos no suscepti-bles, especialmente levaduras. Debe suspenderse la lactancia durante el tratamiento ya que la clindamicina se excreta por la leche materna. Cuando ocurre una diarrea significativa debe suspenderse el 212 y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. No es removible por diálisis peritoneal ni por hemodiálisis. INFORMACI”N AL PACIENTE No alterar la dosificación ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Informar de inmediato al médico si ocurre alguna reacción o sintomatología inusual durante la terapia. Si se presenta diarrea CLORANFENICOL FARMACOLOGÕA Antibiótico con acción bactericida o bacteriostática según la dosis y la sensibilidad del microorganismo. Inhibe la síntesis de proteínas en gérmenes susceptibles por unión a la subunidad ribosomal 50S. El espectro de actividad incluye patógenos aerobios Gram positivos (Streptococus pneumoniae y otros estreptococos), bacterias aerobias Gram negativas (Haemophilus influenzae, Salmonella, Shigella, Neisseria meningitidis, Pseudomonas mallei, Pseudomonas cepacia, Proteus mirabilis, Vibrio cholerae, Francisella tularensis, Yersinia pestis y Brucella) y algunas bacterias anaerobias: Bacteroides melaninogenicus, B. fragilis, Fusobacterium, Clostridium, Veillonella, Rickettsia, Chlamidia y Formulario Terapéutico Nacional * 2ed. 2004 une a las proteínas plasmáticas en un 60% y se distribuye ampliamente a los fluidos y tejidos corporales, logrando su máxima concentración en el hígado y el riñón. En individuos con meninges no inflamadas, la concentración en el fluido cerebroespinal es equivalente al 2150% de la concentración plasmá-tica y de 45-89% en sujetos con las meninges inflamadas. Se metaboliza en el hígado, principalmente con ácido glucurónico, a productos inactivos que se excretan, junto a un 30% de droga intacta, por la orina. La vida media de eliminación es de 1,54,1 horas en adultos, de 24 horas o más en neo-natos y de 10 horas en niños de 10-16 días. Los tiempos se incrementan en pacientes con insuficiencia hepática. El cloranfenicol atraviesa la barrera placentaria y se distribuye hacia la leche materna. INDICACIONES Infecciones severas causadas por organismos susceptibles en las que otros agentes menos tóxicos resultan inefectivos o están contraindicados. R…GIMEN DE DOSIFICACI”N Se administra por vía IV. La dosis usual en niños y adultos por vía IV con función hepática y renal normales es de 50 mg/kg/día, divididos en dosis iguales cada 6 horas. La dosis máxima lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, diarrea, glositis, estomatitis, sabor desagradable, enterocolitis, prurito anal e ictericia. Hematológicas: Depresión de la médula ósea, anemia aplásica, ane-mia hipoplásica, trombocitopenia y granulocitopenia. Hepáticas: Ictericia. Neurológicas: Cefalea, depresión, confusión y neuritis óptica J que puede progresar hasta ceguera. Reacciones de hipersensibilidad: Erupción macu-lopapular y vesicular, fiebre, urticaria, angioedema, hemorragia cutánea o de las mucosas y anafilaxis. Otras: Síndrome del niño gris en prematuros y recién nacidos que reciben cloranfe-nicol o cuyas madres lo recibían los últimos días del embarazo, aunque se ha reportado casos en niños hasta de 2 años. Los síntomas incluyen: Dificul-tad para la alimentación, distensión abdominal con o sin émesis, cianosis pálida progresiva y colapso vasomotor frecuentemente acompañado de respiración irregular; muerte dentro de pocas horas del comienzo de los síntomas. 2ed. 2004 * Formulario TerapÈutico Nacional 213 puede prolongar el tiempo de protrombina en pacientes que reciben anticoagulantes al interferir con la producción de vitamina K por bacterias intestinales. Antagoniza la actividad bactericida de la penicilina y las cefa-losporinas. Puede reducir la efectivi-dad de la vitamina B12, sales de hierro y ácido fólico. La rifampicina y el feno-barbital disminuyen los niveles séricos del cloranfenicol al incrementar su metabolismo hepático. Con fármacos mielosupresores potencia el riesgo de depresión de la médula ósea. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. No se ha establecido la seguridad de su uso durante el embarazo. Usar con precaución en pacientes con enfermedad renal y/o hepática. Debido al estrecho margen terapéutico del cloranfenicol, se recomienda realizar determinaciones de los niveles séricos durante el tratamiento; la concentra-ción debe mantenerse dentro del rango de 5-20 mcg/ml para garantizar su eficacia y minimizar la posibilidad de efectos tóxicos. El uso prolongado puede provocar sobrecrecimiento de organismos no susceptibles, incluyen-do hongos patógenos. Si se presenta neuritis 214 SOBREDOSIS Manejo sintomático y terapia de soporte. La hemoperfusión con carbón activado es efectiva para remover el fármaco. No es removible por diálisis peritoneal ni por hemodiálisis. DOXICICLINA (Antibacteriano SistÈmico) FARMACOLOGÍA Antibiótico del grupo de las tetraciclinas, derivado de la oxitetraciclina. Posee acción bacteriostática. Inhibe la síntesis de proteínas en gérmenes sensibles por unión a la sub-unidad ribosomal 30S. Es activa in vivo contra: Neisseria gonorrhoeae, Haemophilus influenzae, H. ducreyii, Yersinia pestis, Francisella tularensis, Vibrio cholera, Bortonella bacilliformis, Brucella spp., Ricketsiae, Chlamydia trachomatis, C. psittaci, Mycoplasma pneumoniae, Ureaplasma urealyticum, Borrelia recurrentis, Treponema pallidum, Clostridium spp., Fusobacterium fusiforme, Actinomyces spp., Propionbacterium acnes, Plasmodium falciparum (forma asexual pero no contra los gametocitos), Bacillus anthracis y algunas cepas sensibles de Streptococcus pyogenes, S. pneumoniae, S. faecalis, estreptococos alfa-hemolíticos grupo viridans, Escherichia coli, Klebsiella spp., Formulario Terapéutico Nacional * 2ed. 2004 absorbe casi completamente (95%) en el tracto gastrointestinal, alcanzando concentraciones séricas pico en 1,5-4 horas. Los alimentos reducen su absorción en un 20%. Se une a las proteínas plasmáticas en un 25-93% y se distribuye ampliamente a los tejidos y fluidos corporales. Es parcialmente inactivada en el intestino por quelación con algunos iones divalentes y trivalentes. Se elimina intacta en un 20-26% por la orina y 20-40% por las heces. La vida media de eliminación con dosis única es de 16-18 horas y de 22-24 horas con la dosificación múltiple, incrementándose ambas en pacientes con insuficiencia renal severa. La doxiciclina atraviesa la barrera placentaria y llega a la circulación fetal y al cordón umbilical. En la leche materna alcanza concentraciones importantes. INDICACIONES Infecciones causadas por gérmenes sensibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos y niños mayores de 8 años con peso mayor a 45 kg es 100 mg cada 12 horas el primer día, seguido por 100 mg/día como dosis única o dividida en 2 dosis iguales cada 12 horas. En infecciones severas se recomienda 100 mg cada 12 horas. En niños mayores de 8 años con peso con peso superior a 50 kg se recomienda la dosis usada en adultos. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, diarrea, dolor abdominal, pancreatitis, glositis, disfagia, enterocolitis y lesiones inflamatorias en la región anogenital. Neurológicas: Mareos, vértigo, dolor de cabeza, ataxia, fatiga. Renales: Incrementos del BUN y de la creaJ tinina sérica. Hepáticas: Aumento de los niveles de las aminotransferasas. Hematológicas: Leucocitosis, neutropenia, anemia hemolítica, trombocitopenia, linfocitos atípicos. Reacciones de hipersensibilidad: Erupción maculopapular, urticaria, eritema multiforme, síndrome de Stevens-Johnson, fotosensibilidad, angioedema y anafilaxis. Pulmonares: Infiltrados pulmonares. Otras: Candidiasis, onicólisis, coloración de los dientes durante el desarrollo de los mismos (segunda mitad del embarazo, la infancia y la niñez), abultamiento de las fontanelas en infantes e hipertensión endocraneana en adultos. INTERACCIONES La administración con alimentos o me215 2ed. 2004 * Formulario TerapÈutico Nacional y pueden disminuir la eficacia de los anticonceptivos orales que contienen estrógenos. La combinación con metoxiflurano puede provocar nefrotoxicidad fatal. La fenitoína, los barbitúricos y la carbamazepina reducen los niveles plasmáticos de la doxiciclina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a las tetraciclinas, en niños menores de 8 años y durante el embarazo. Usar con precaución en pa-cientes con falla hepática y/o renal. El uso prolongado puede causar el sobrecrecimiento de organismos no susceptibles, incluyendo hongos patógenos. En caso de terapia prolongada se debe practicar exámenes periódicos de la función hematológica, hepática y renal. Dado el potencial fotosensibilizante del producto, se recomienda evitar la exposición prolongada a la luz solar. El uso de tetraciclinas después de la fecha de vencimiento puede provocar reaccio-nes renales severas como el síndrome de Fanconi. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. No es removible por 216 Cationes di o trivalentes (zinc, calcio, magnesio, aluminio o hierro). Durante la terapia evitar la exposición excesiva a la luz del sol. No alterar la dosificación ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. No usar el medicamento pasada la fecha de vencimiento. La doxiciclina debe ser ingerida con suficiente líquido y en ERITROMICINA (ERITROMICINA ESTOLATO) FARMACOLOGÍA Antibiótico macrólido de acción bacteriostática, pero puede ser bactericida a altas dosis. Inhibe la síntesis de proteínas en gérmenes sensibles por unión a la sub-unidad ribosomal 50S de los microorganismos sensibles. Es activa in vivo contra: Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma pneumoniae, Ureaplasma urealyticum, Treponema pallidum, Streptococcus pyogenes (beta-hemolítico del grupo A), Streptococcus pneumoniae, Staphylococcus aureus, Bordetella pertussis, Legionella pneumophila, Listeria monocytogenes, estreptococo alfa-hemolítico (grupo viridans) y Corynebacterium diphteriae. FARMACOCINÉTICA Formulario Terapéutico Nacional * 2ed. 2004 gre liberando la eritromicina. La administración de una dosis única alcanza concentraciones plasmáticas pico en 1-4 horas. Se une a las proteínas plasmáticas en un 96% y se distribuye ampliamente a los tejidos y fluidos corporales. Atraviesa la barrera placentaria pero los niveles plasmáticos fetales son bajos. Se metaboliza parcialmente en el hígado transformándose en productos inactivos que se excretan, junto con el fármaco en su mayoría intacto, por las heces y en escasa proporción por la orina. La vida media de eliminación es de 1,5-2 horas. INDICACIONES Infecciones causadas por gérmenes sensibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Las dosis del estolato de eritromicina se expresan en términos de eritromicina base. La dosis usual en adultos es 500 mg cada 6 horas; en infecciones severas puede incrementarse a 4 g/día o más. En niños, 30-50 mg/kg/día divididos en 4 dosis iguales cada 6 horas; en infec-ciones severas se puede duplicar la dosis. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección trointestinales: Náuseas, vómito, diarrea, colitis pseudomembranosa, dolor abdominal, anorexia, pancreatitis, melena, prurito anal. Hepáticas: Alteración de la función hepática con o sin ictericia, ictericia colestásica. Neurológicas: Ototoxicidad (con dosis de 4 g/día o más), tinitus, vértigo. Reacciones de hipersensibilidad: Erupción, urticaria y anafilaxis. INTERACCIONES La eritromicina puede inhibir el metabolismo e incrementar los niveles séricos de la carbamazepina, buspirona, digoxina, ciclosporina, cisaprida, fenitoína, clozapina, ebastina, alfentanil, atorvastatina, lovastatina, pra- J vastatina, benzodiazepinas, bromocriptina, ácido valpróico, tretinoína, ergotamina, sildenafil, prednisolona, felodipina, quinidina, disopiramida, fexofenadina, terfenadina, astemizol y warfarina. La eritromicina puede elevar las concentraciones plasmáticas de teofilina por disminución de su depuración. La administración con jugos de frutas, de tomate o bebidas gaseosas disminuye la biodisponibilidad de la eritromicina. Administrada con terfenedina o con cisaprida puede producir arritmias cardíacas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a los macrólidos, al- 2ed. 2004 * Formulario TerapÈutico Nacional 217 na, entre otras) por el riesgo de rabdomiolisis. El uso prolongado puede causar el sobrecrecimiento de organismos no susceptibles, incluyendo hongos patógenos. En caso de terapia prolongada, se debe practicar exámenes periódicos de la función hepática. El uso en el embarazo debe limitarse a condiciones de verdadera necesidad en las que los beneficios potenciales a la madre superen a los posibles riesgos para el feto. Usar con precaución durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. No es removible por diálisis peritoneal o hemodiálisis. INFORMACI”N AL PACIENTE No alterar la dosificación ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, tales como dolor abdominal severo, GENTAMICINA (SistÈmica) FARMACOLOGÍA Antibiótico aminoglicósido. Posee actividad bactericida. Inhibe la síntesis 218 bles por unión irreversible a la subunidad ribosomal 30S. Es activa contra: Enterobacter spp., Citrobacter spp., Proteus spp., Escherichia coli, Klebsiella spp., Pseudomonas spp., Serratia spp., Staphylococcus aureus y S. epidermidis. FARMACOCINÉTICA Ver: AMIKACINA. Después de la administración IM alcanza concentraciones séricas pico en 30-90 minutos. La vida media de eliminación es de 2-3 horas y se incrementa de manera significativa en pacientes con insuficiencia renal. INDICACIONES Tratamiento de infecciones severas del tracto respiratorio, urinario, gastrointestinal, sistema nervioso central, de la piel y tejidos blandos, óseas y septicemia, causadas por organismos susceptibles. RÉGIMEN DE DOSIFICACIÓN Se administra por infusión IV ó por vía IM. El nivel sérico terapéutico debe ubicarse en el rango de 4-12 mcg/ml de concentración pico (medida 30-60 minutos después de la administración de una dosis) y 1-2 mcg/ml de concentración valle (medida justo antes de la administración de una dosis). La dosis usual en adultos es de 3-5 mg/kg/día, divididos en 3 dosis iguales cada 8 horas. En neonatos, 5 Formulario Terapéutico Nacional * 2ed. 2004 en períodos de 0,5-2 horas. La dosificación en pacientes con insuficiencia renal debe hacerse, en la medida de lo posible, con base en los niveles séricos del antibiótico. El ajuste de la dosis puede hacerse modificando los intervalos de dosificación o disminuyendo la dosis de acuerdo a la depuración de creatinina (Clcr) o de la creatinina sérica (Cr). Para el ajuste del intervalo se inicia con la dosis de carga usual, continuando con la misma dosis a intervalos, en horas, calculados multiplicando el valor de creatinina sérica (Cr) por ocho (8). Para el ajuste de la dosis se inicia con la dosis de carga usual, continuando con una dosis de mantenimiento cada 8 horas, calculada dividiendo la dosis de carga usada entre el valor de creatinina sérica. Para ajustes más precisos, consultar literatura especializada. tienamicina producida por el Streptomyces cattleya. Tiene acción bactericida. Al igual que otros antibióticos beta-lactámicos, inhibe la síntesis de mucopéptido en la pared bacteriana al unirse a las enzimas que participan en la fase final de la síntesis de la pared bacteriana. Es muy resistente a la hidrólisis por las beta-lactamasas. La cilastatina inhibe competitivamente a la enzima dehidropeptidasa I presente en los túbulos renales proximales que metaboliza e inactiva al imipenem. Es activo contra bacterias aerobias Gram positivas como: el Staphylococcus aureus (productor y no productor de betaJ lactamasa), Staphylococcus epidermidis, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus pneumoniae y Enterococcus faecalis; bacterias aerobias Gram negativas como: Acinetobacter spp., EnterobacINTERACCIONES ter spp., Klebsiella spp., Citrobacter Ver: AMIKACINA. spp., Pseudomonas aeruginosa, Escherichia coli, Haemophilus influenREACCIONES ADVERSAS zae, Haemophilus parainfluenzae, Ver: AMIKACINA. Gardnerella vaginalis, Proteus vulgaris, Morganella morganii, Providentia CONTRAINDICACIONES/ rettgeri y Serratia spp.; bacterias PRECAUCIONES anaerobias Gram positivos como: Ver: AMIKACINA. Clostridium spp, Eubacterium spp., Peptococcus spp., PeptostreptocoIMIPENEM / CILASTATINA SÓDICA ccus spp. y Propionibacterium app. y FARMACOLOGÍA bacterias anaerobias Gram negativas El imipenem es un antibiótico del como: Bacteroides spp. y Fusobactegrupo de los carbapenems, derivado rium spp. 219 2ed. 2004 * Formulario TerapÈutico Nacional plasmáticas en un 13-21%. Aunque el metabolismo renal por la dehidropeptidasa I es inhibido por la cilastatina, un 20-30% del imipenem sufre un metabolismo no renal por hidrólisis inespecífica del anillo beta-lactámico, dando lugar a un producto sin actividad microbiológica que se excreta, junto a un 5075% de fármaco intacto, por filtración glomerular y secreción tubular en la orina. Por su parte la cilastatina es parcialmente metabolizada en el riñón y excretada en un 70-80% intacta, también por la orina. La vida media de eliminación del imipenem es de 0,851,3 horas y se incrementa en pacientes con insuficiencia renal. INDICACIONES Infecciones del tracto respiratorio inferior, de la piel y sus estructuras, del tracto urinario, intra-abdominales, ginecológicas, infecciones óseas y septicemia causadas por organismos susceptibles. por gérmenes susceptibles, la dosis usual es de 15-25 mg/kg/dosis cada 6 horas (dosis máxima diaria: 2 g). En infecciones por cepas de Pseudomonas aeruginosa moderadamente susceptibles, con riesgo de la vida, la dosis diaria máxima puede llegar hasta 4 g. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, hipotensión, taquicardia, tromboflebitis. Gastrointestinales: Náuseas, vómito, dolor abdominal, gastroenteritis, glositis, hipertrofia papilar de la lengua, pigmentación de los dientes y de la lengua, acedía, dolor faríngeo, aumento de la salivación, trastornos del gusto, diarrea, colitis hemorrágica y colitis pseudomembranosa. Hematológicas: Eosinofilia, leucopenia, neutropenia, agranulocitosis, pancitopenia, mielosupresión, anemia hemolítica, trombocitopenia, leucocitosis, monocitosis, linfocitosis y basofilia. Hepáticas: Aumento de las enzimas hepáticas, hiperbilirrubinemia, ictericia y hepatitis. Neurológicas: Mareos, somnolencia, confusión, vértigo, parestesia, dolor de cabeza, debilidad, astenia, encefalopatía, trastornos RÉGIMEN DE DOSIFICACIÓN La dosis se expresa en términos de imipenem. Se administra por infusión IV en 15-30 minutos (dosis iguales o menores que 500 mg) o en 40-60 minutos (dosis mayores de 500 mg). En adultos con infecciones causadas por gérmenes susceptibles, la dosis usual es de 250-500 mg cada 6-8 horas, según la severidad de la infección. Para infecciones causadas por 220 Formulario Terapéutico Nacional * 2ed. 2004 Renales: Incrementos del BUN y de la creatinina sérica, anuria, poliuria, coloración de la orina, proteinuria e insuficiencia renal aguda. Otras: Disnea, artralgia, hiperhidrosis, candidiasis, trastornos auditivos, hiponatremia, hiperpotasemia, hipercloremia, y dolor en el sitio de la inyección. INTERACCIONES El imipenem puede antagonizar la actividad antibacteriana de otros antibióticos beta-lactámicos como el aztreonam, las cefalosporinas y las penicilinas. La administración a pacientes que reciben ganciclovir puede generar convulsiones generalizadas. El probenecid puede incrementar los niveles plasmáticos y la vida media de eliminación del imipenem y la cilastatina. Debe evitarse el uso concomitante. No debe mezclarse con otros antibióticos, pero puede administrarse en forma concomitante con los aminoglicósidos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a otros antibióticos beta-lactámicos. No es recomendable su uso en caso de meningitis ni en pacientes con trastornos del sistema nervioso central (lesiones cerebrales o historia de convulsiones). Usar con precaución en pacien- madre supere al posible riesgo para el feto. Durante el tratamiento se debe vigilar periódicamente la función renal, hepática y hematológica. El uso por períodos prolongados puede provocar el sobrecrecimiento de microorga-nismos no susceptibles, inclusive hongos patógenos. SOBREDOSIS Manejo sintomático y tratamiento de soporte. Es removible por hemodiálisis, aunque la utilidad de este procedimiento en casos de sobredosis no ha sido plenamente demostrada. LEVOFLOXACINO J FARMACOLOGÍA Antibiótico del grupo de las fluoroquinolonas. Tiene acción bactericida. Inhibe a las enzimas girasa y topoisomerasa IV del ADN, necesarias para la replicación, transcripción, reparación y recombinación del ADN bacteriano. Es activa contra bacterias aerobias Gram positivas como: Enterococcus faeca-lis, Staphylococcus aureus, Staphylo-coccus saprophyticus, Streptococcus pneumoniae y Streptococcus pyoge-nes; bacterias aerobias Gram negativas como: Acinobacter spp., Enterobacter spp., Escherichia coli, Haemophilus influenzae, Haemophi-lus 2ed. 2004 * Formulario TerapÈutico Nacional 221 pneumoniae, Mycoplasma pnumoniae, Serratia marcescens, Morganella morganii, Providencia spp,; y bacterias anaerobias como el Clostridium perfringens. FARMACOCINÉTICA Después de la administración oral, se absorbe rápida y completamente en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico en 1-2 horas con una biodisponibilidad de 99%. Se une a las proteínas plasmáticas en un 24-38% y se distribuye ampliamente en los tejidos y fluidos corporales. Se metaboliza en el hígado en muy escasa magnitud dando lugar a metabolitos inactivos que se excre-tan, junto a un 87% del fármaco intacto, por secreción urinaria y menos de 4% en las heces. La vida media de eliminación plasmática es de 6-8 horas y se prolonga en pacientes con insufi-ciencia renal. INDICACIONES Infecciones del tracto urinario, de la piel y sus estructuras, del tracto respiratorio bajo y en la sinusitis maxilar aguda causada por organismos susceptibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral e IV. La dosis usual en adultos es de 250-500 mg por vía oral o por infusión IV en 60 minutos, cada 24 horas por 7-14 días. 222 500 mg/día, cada 24 horas por 7-14 días. Exacerbación aguda de la bronquitis crónica: 500 mg cada 24 horas por 7 días. Infecciones del tracto urinario y pielonefritis aguda: 250 mg cada 24 horas por 10 días. La duración de la terapia depende del tipo y severidad de la infección y debe ser determinada por la respuesta clínica y bacteriológica de cada paciente en particular. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, taquicardia, dolor anginoso, torcida de punta, flebitis, vasodilatación y falla circulatoria. Hematológicas: Eosinofilia, leucopenia, neutropenia, trombocitopenia, agranulocitosis, linfocitopenia, incremento del tiempo de coagulación, anemia hemolítica y pancito-penia. Gastrointestinales: Náuseas, vómito, constipación, diarrea, ano-rexia, dolor abdominal, dispepsia, flatulencia, alteración del gusto y colitis pseudomembranosa. Hepáticas: Ele-vación de las enzimas hepáticas, ictericia y hepatitis. Neurológicas: Cefalea, mareos, nerviosismo, fatiga, astenia, insomnio, somnolencia, ansiedad, pesadillas, encefalopatía, depresión, parestesia, psicosis, agitación, temblor y convulsiones. Reacciones de hipersensibilidad: Pru-rito, erupción, urticaria, fiebre, vas-culitis, eritema Formulario Terapéutico Nacional * 2ed. 2004 trastornos visuales, artralgia, mialgia, debilidad, fotosensibilidad y dolor en el sitio de inyección. INTERACCIONES Los metales divalentes pueden quelar la molécula de levofloxacino e interferir con su absorción gastrointestinal, por lo que se debe evitar su uso simul-táneo con antiácidos, sales de hierro, sales de calcio y productos que con-tengan zinc, sucralfato. El levofloxa-cino puede aumentar los niveles plasmáticos de hipoglicemiantes orales, teofilina y la respuesta hipoprotrombinémica de la warfarina por inhibición del citocromo P450. La combinación con AINEs incrementa el riesgo de convulsiones. Los alimentos prolongan el tiempo para alcanzar la concentración pico, el cual reducen hasta en un 14%. CONTRAINDICACIONES/ PRECAUCIONES Ver: CIPROFLOXACINO. METRONIDAZOL (Antibacteriano SistÈmico) FARMACOLOGÍA Ver: METRONIDAZOL ANTIPARA-SITARIOS. en FARMACOCINÉTICA Ver: METRONIDAZOL ANTIPARA-SITARIOS. en INDICACIONES Infecciones causadas por bacterias anaeróbicas susceptibles y profilaxis en la cirugía colorrectal. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral y por infusión IV lenta. - I nfecciones bacterianas por bacterias anaerobias: Iniciar con 15 mg/kg, seguido por 7,5 mg/kg cada 6 horas (máximo 4 g/día) por 7-10 días y en infecciones serias hasta por 2-3 semanas. Dosis por vía oral: 7,5 mg/kg cada 6 horas. - Profilaxis preoperatoria en cirugía colorrectal: 15 mg/kg por infusión lenta (30-60 minutos) 1 hora antes J de la intervención y en caso necesario, con 7,5 mg/kg a las 6 y 12 horas de la dosis inicial. REACCIONES ADVERSAS Ver: METRONIDAZOL ANTIPARA-SITARIOS. en INTERACCIONES Ver: METRONIDAZOL ANTIPARA-SITARIOS. en CONTRAINDICACIONES/ PRECAUCIONES Ver: METRONIDAZOL en ANTIPARASITARIOS. SOBREDOSIS Ver: METRONIDAZOL en ANTIPARA- 2ed. 2004 * Formulario TerapÈutico Nacional 223 NITROFURANTOÍNA (NITROFURANTOÕNA MACROCRISTALES) FARMACOLOGÍA Antibiótico derivado del nitrofurano. Posee actividad bactericida en la orina a dosis terapéuticas. Es reducido por las flavoproteínas bacterianas y convertido en un metabolito intermediario activo que inactiva o altera las proteínas ribosomales bacterianas y otras macromoléculas. Como resultado de esto, se inhiben dos procesos bioquímicos de síntesis de proteínas, metabolismo aeróbico, la síntesis del ADN, del ARN y de la pared celular. Este modo de acción explica la falta de resistencia a la nitrofurantoína. Es activa contra: Escherichia coli, Enterococcus faecalis, Staphylococcus aureus, estafilococos coagulasa-negativos, estreptococos grupo D y grupo viridans, Citrobacter spp. y Klebsiella spp. FARMACOCINÉTICA Se absorbe en un 90% en el tracto gastrointestinal. Los alimentos mejoran su absorción. Se une a las proteínas plasmáticas en un 20-60%. Alcanza niveles sub-terapéuticos en el plasma y los tejidos, pero elevadas concentraciones en el tracto urinario. Se metaboliza parcialmente en el hígado y se excreta en un 30-50% intacto por la orina. El pH ácido favorece su reabsorción tubular y 224 das por gérmenes susceptibles. No es efectiva en el tratamiento de la pielonefritis. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, preferiblemente con la comida para mejorar la absorción y la tolerancia. La dosis usual en adultos: 50-100 mg 4 veces al día. Para la supresión prolongada en infecciones recurrentes: Dosis única de 50-100 mg/día. En niños: 5-7 mg/kg/día dividido en 4 dosis iguales cada 6 horas, y para la supresión prolongada en infecciones recurrentes: Dosis única de 1 mg/kg/día o dividida en dos dosis. La terapia debe mantenerse hasta 3 días después de lograr que la orina se vuelva estéril. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, diarrea, dolor abdominal, anorexia, constipación y pancreatitis. Hematológicas: Agranulocitosis, trombocitopenia, leucopenia, metahemoglobinemia y anemia hemolítica en pacientes con deficiencia de glucosa-6-fosfato deshidrogenasa. Hepáticas: Aumento de las enzimas hepáticas, ictericia colestásica, hepatitis, hepatomegalia y necrosis hepática. Neurológicas: Cefalea, mareo, vértigo, astenia, somnolencia, nistagmo, neuropatía periférica, incluyendo neuritis óptica (en ocasiones con desenlace fatal), hipertensión intracraneana, neuritis retro- Formulario Terapéutico Nacional * 2ed. 2004 Agudas: Fiebre, escalofríos, tos, dolor torácico, disnea, infiltración pulmonar con consolidación con efusión pleural y eosinofilia (ocurren durante la primera semana de tratamiento y son reversibles al suspenderlo). Crónicas (ocurren en pacientes con tratamiento por 6 meses o más): Malestar, disnea al ejercicio, tos y alteración de la función pulmonar, pneumonitis y fibrosis. Otras: Cristaluria, coloración de la orina, alopecia transitoria, mialgia. ciales a la madre superen a los posibles riesgos para el feto. Durante la terapia debe suspenderse la lactancia. INTERACCIONES El probenecid y la sulfinpirazona inhiben la secreción renal, aumentando los niveles séricos y el riesgo de toxicidad. Los antiácidos reducen la absorción gastrointestinal del medicamento, el cual se adsorbe a la superficie del trisilicato de magnesio. La nitrofurantoína antagoniza in vivo el efecto antibacteriano de las quinolonas. INFORMACIÓN AL PACIENTE Administrar con las comidas para minimizar las molestias gástricas. No alterar la dosificación ni suspender el J tra-tamiento antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. El medicamento no debe ser tomado con antiácidos. El producto puede producir un color pardo en la orina. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, disfunción renal severa, deficiencia de glucosa-6-fosfato deshidrogenasa y en el embarazo a término. Usar con precaución en pacientes con falla renal leve a moderada, anemia, diabetes mellitus, trastornos hidroelectrolíticos y deficiencia de vitamina B (incre-menta el riesgo de SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Se recomienda la inducción del vómito. Manejo sintomático y terapia de soporte. Debe mantenerse una alta ingesta de fluido para promover la excreción urinaria. OXACILINA FARMACOLOGÕA Antibiótico semisintético derivado del ácido 6-aminopenicilánico. Posee actividad bactericida contra gérmenes sensibles productores de penicilinasa. El mecanismo de acción es similar al descrito para otros derivados de penicilina (ver: AMPICILINA). Los antibióticos de este grupo son altamente 2ed. 2004 * Formulario TerapÈutico Nacional 225 contra S. aureus productor y no productor de penicilinasa. FARMACOCIN…TICA Después de la administración IM alcanza niveles séricos pico en 30 minutos y en 5 minutos por vía IV. Se une a las proteínas plasmáticas en un 94% y se distribuye rápidamente a los fluidos y tejidos corporales; sin embargo, no logra niveles efectivos en el líquido cefalorraquídeo. Se metaboliza parcialmente en el hígado dando lugar a metabolitos activos e inactivos que se excretan, junto con un 40- 70% del fármaco intacto, en la orina. La vida media de eliminación es de 20-30 minutos. INDICACIONES Infecciones causadas por cepas de estafilococos productores de penicilinasa. R…GIMEN DE DOSIFICACI”N Se administra por vía IM ó IV, en dosis similares. - Infecciones leves a moderadas en adultos y niños con peso superior a 40 kg: 250-500 mg cada 4-6 horas; infecciones severas: 1-2 g cada 4 horas. - Infecciones leves a moderadas en niños con peso inferior a 40 kg: 50 mg/kg/día en dosis divididas cada 4-6 horas; infecciones severas: 100200 mg/kg/día en dosis divididas 226 REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos, diarrea, flatulencia y colitis pseudomembranosa. Hematológicas: Eosino-filia, neutropenia, leucopenia, trombo-citopenia, anemia hemolítica y agra-nulocitosis. Hepáticas: Alteración de las enzimas hepáticas y hepatome-galia. Neurológicas: Convulsiones (asociadas a dosis elevadas en pa-cientes con insuficiencia renal), neuro-patía, irritabilidad neuromuscular, letargo, alucinaciones, ansiedad, con-fusión, agitación, mareos y fatiga. Reacciones de hipersensibilidad: Erupción, prurito, fiebre, escalofríos, mialgia y anafilaxis. Renales: Aumento del BUN y de la creatinina sérica, nefritis intersticial y falla renal aguda. Otras: Dolor en el sitio de la inyección, flebitis y tromboflebitis. INTERACCIONES La oxacilina puede inactivar a los aminoglicósidos, disminuir la eficacia de los anticonceptivos orales y alterar la respuesta inmunológica a la vacuna tifoidea. La rifampicina y las tetraciclinas pueden antagonizar el efecto de la oxacilina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a las penicilinas. Usar con precaución en pacientes con en- Formulario Terapéutico Nacional * 2ed. 2004 tención a la posibilidad de colitis pseudomembranosa. Su uso por períodos prolongados puede ocasionar el sobrecrecimiento de organismos no susceptibles, incluyendo hongos patógenos. SOBREDOSIS Manejo sintomático y terapia de soporte. en adultos: 1-2 g/día divididos en 4 dosis iguales cada 6 horas. En niños mayores de 8 años: 25-50 mg/kg/día divididos en 4 dosis iguales cada 6 horas. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Ver: DOXICICLINA en ANIBIÓTICOS SISTÉMICOS. OXITETRACICLINA FARMACOLOGÍA Ver: DOXICICLINA en ANTIBIÓTICOS SISTÉMICOS. INTERACCIONES Ver: DOXICICLINA en ANIBIÓTICOS J SISTÉMICOS. FARMACOCINÉTICA Se absorbe en un 60-80% en el tracto CONTRAINDICACIONES/ gastrointestinal alcanzando concen- PRECAUCIONES traciones plasmáticas pico en 2-4 ho- Ver: DOXICICLINA en ANIBIÓTICOS ras. Los alimentos reducen su absor- SISTÉMICOS. ción en un 50% o más. La absorción SOBREDOSIS IM es errática e impredecible. Se une a Ver: DOXICICLINA en ANIBIÓTICOS las proteínas plasmáticas en un 10SISTÉMICOS. 40% y se distribuye ampliamente en los tejidos y fluidos corporales. Parece SULFAMETOXAZOL / TRIMETOPRIMA que no sufre metabolismo. Se excreta (TRIMETOPRIM / SULFAMETOXAZOL) en un 60-70% como fármaco intacto en FARMACOLOGÕA la orina. La vida media de eliminación Combina la actividad bactericida de la es de 6-10 horas y se incrementa de trimetoprima (TMP) y la acción bacmanera significativa en pacientes con teriostática del sulfametoxazol (SMX). insuficiencia renal. La TMP bloquea la producción de ácido tetrahidrofólico en el microorINDICACIONES ganismo por inhibición de la enzima Infecciones causadas por gérmenes dihidrofolato reductasa, mientras que 2ed. 2004 * Formulario TerapÈutico Nacional 227 ácido dihidrofólico por competencia con el ácido para-aminobenzóico que resulta en la inhibición de la pteroato sintetasa. La combinación bloquea 2 pasos consecutivos en la síntesis del ácido fólico, que resulta en la inhibición de la síntesis de proteínas y de los áci-dos nucléicos. La actividad antimicro-biana de la combinación comprende: Escherichia coli, Klebsiella pneumo-niae, Enterobacter spp., Proteus mira-bilis, Acinetobacter spp., Salmonella spp., Shigella spp., Haemophilus influenzae, H. ducreyi, Nocardia spp., Neisseria gonorrhoeae, Streptococcus pneumoniae y algunas cepas de Staphylococcus aureus. También es activa contra Pneumocystis carinii. FARMACOCIN…TICA La combinación se absorbe en un 90100% en el tracto gastrointestinal, alcanzando niveles séricos pico a las 14 horas. La TMP se une a proteínas plasmáticas en un 40-60% y el SMX en un 60-70%. Ambos se distribuyen a los tejidos y fluidos corporales, alcanzando concentraciones efectivas en el esputo, el humor acuoso, la efusión del oído medio, el fluido prostático, el fluido vaginal y el líquido cefalorraquídeo. Se metabolizan en el híga-do transformándose en productos inactivos que se excretan, junto con los fármacos intactos (TMP 228 causadas por cepas susceptibles de Enterobacter, Escherichia coli, Klebsiella spp. y Proteus, neumonías causadas por Pneumocystis carinii y enteritis causada por Shigella flexneri, Shigella sonnei y otros patógenos enterotoxigénicos. R…GIMEN DE DOSIFICACI”N Se administra por infusión IV en 60-90 minutos. La dosis se expresa en términos de la trimetoprima contenida en la formulación comercialmente disponible que combina 1 mg de TMP por cada 5 mg de SMX. La dosis por vía IV en adultos y niños para el tratamiento de neumonía por P. carinii es de 1520 mg/kg/día (basados en el componente trimetoprima) divididos en 3-4 dosis por 14 días; para infecciones severas del tracto urinario 8-10 mg/kg diarios divididos en 2-4 dosis por 14 días y para la shigellosis, la misma dosis, pe-ro por 5 días. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos, anorexia, diarrea, pancreatitis, glositis y colitis pseudomembranosa. Hematológicas: Agranulocitosis, trombocitopenia, leucopenia, neutropenia, eosinofilia, anemia hemolítica, anemia aplásica, metahemoglobinemia e hipoprotrombinemia. Hepáticas: Hepatitis, ictericia colestásica y necrosis hepática, aumento de los niveles de las aminotransferasas y de la bilirrubina. Formulario Terapéutico Nacional * 2ed. 2004 medicamentosa, necrólisis epidérmica, síndrome de Stevens-Johnson, en-fermedad del suero, miocarditis alér-gica, fotosensibilidad, eritema multi-forme, angioedema y anafilaxis. Rena-les: Aumento del BUN y de la crea-tinina sérica, insuficiencia renal aguda, cristaluria, nefrosis tóxica con oliguria, anuria y nefritis intersticial. Otras: Fie-bre, escalofríos, mialgia, artralgia, tos, hiperpotasemia e hiponatremia. INTERACCIONES La combinación de TMP/SMX interfiere con la secreción tubular de la digoxina aumentando el riesgo de toxicidad digitálica; interfiere con la respuesta inmunológica a la vacuna contra la fiebre tifoidea; incrementa el riesgo de discrasias sanguíneas de la pirimetamina; aumenta los niveles séricos de la procainamida incrementando el riesgo de reacciones cardíacas; inhibe el metabolismo de la warfarina y de la fenitoína predisponiendo al paciente a sus efectos tóxicos; puede aumentar el efecto mielotóxico del metotrexato y con inhibidores de la enzima convertidora de angiotensina puede generar hiperpotasemia severa. Los derivados del ácido para-aminobenzóico pueden antagonizar el efecto bacteriostático de las sulfonamidas. El SMX desplaza a las sulfonilúreas de su unión a las proteínas y aumenta el efecto Usar con precaución en pacientes con insuficiencia renal y/o hepática, asma, en ancianos, niños, en el embarazo y en deficiencia de folato o de glucosa-6-fosfato deshidrogenasa. Durante la terapia se debe vigilar periódicamente la función renal, hepática y hematoló-gica. Durante la terapia es recomenda-ble suspender la lactancia. Como en otros casos, si hay colitis pseudomem-branosa debe suspenderse el medi-camento. Cuando se trata de infusión intravenosa debe tenerse una ingesta adecuada de líquido. En pacientes ge-riátricos debe tenerse precaución ya que la supresión de la médula ósea J o una disminución específica de las plaquetas son muy frecuentes. El uso conjunto con leucovorina resulta en falla del tratamiento y en morbilidad. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Tomar el medicamento con un vaso completo de agua e ingerir abundante líquido durante el tratamiento. No alteSULFAMETOXIPIRIDAZINA FARMACOLOGÍA Agente antibacteriano del grupo de las 2ed. 2004 * Formulario TerapÈutico Nacional 229 sulfonamidas. Posee acciÛn bacteriost·tica. Impide el crecimiento y multiplicaciÛn bacteriana por inhibiciÛn competitiva de la enzima pteroato sintetasa que incorpora al PABA (·cido para-aminobenzóico) al proceso de biosÌntesis del ·cido tetrahidrofÛlico, esencial para la generaciÛn de los ·cidos nucléicos que intervienen en la sÌntesis del ADN. Es activa frente a algunas especies de Staphylococcus y Streptococcus, organismos Gram positivos como: Clostridium tetani y Clostridium perfringens, algunas cepas de Nocardia asteroides, Chlamydia trachomatis, Neisseria gonorrhoeae y Neisseria meningitidis y enterobacterias como: Enterobacter, Escherichia coli, Proteus mirabilis, Proteus vulgaris, Salmonella y Shigella. FARMACOCINÉTICA Después de la administración por vía oral se absorbe rápida y extensamente en el tracto gastrointestinal. Una vez en la sangre, se une a las proteínas plasmáticas en un porcentaje superior al 85% y se distribuye a todo el organismo. Se metaboliza en el hígado por acetilación y en menor grado por oxidación, dando lugar a productos inactivos que se excretan, junto con el fármaco intacto, por filtración glomerular. En los túbulos 230 INDICACIONES Infecciones causadas por gérmenes sensibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos, se recomienda iniciar con una dosis única de 1 g (2 tabletas de 500 mg) el primer día y continuar, a partir del segundo día, con dosis de mantenimiento de 500 mg/día. El tratamiento debe mantenerse hasta 48 horas después que el paciente se haya hecho asintomático o haya evidencia de erradicación de la infección. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómito, anorexia, pancreatitis, diarrea, colitis pseudomembranosa. Hematológicas: Metahemoglobinemia, agranulocitosis, leucopenia, neutropenia, eosinofilia, trombocitopenia, hipoprotrombinemia, anemia hemolítica y aplasia medular. Hepáticas: Hepatitis. Neurológicas: Cefalea, mareo, neuropatía periférica, depresión, sordera, hiper-tensión intracraneal, meningitis, convulsiones y síndrome de Guillain-Barré. Reacciones de hipersen-sibilidad: Erupción, prurito, urticaria, eritema, síndrome de Stevens-Johnson, necrólisis tóxica epidérmica, dermatitis exfoliativa, fotosensibilidad, fiebre, reacción similar a la enfer-medad del suero y Formulario Terapéutico Nacional * 2ed. 2004 a las proteínas plasmáticas. Los derivados del PABA, incluyendo algunos anestésicos locales, antagonizan la acción antibacteriana. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otras sulfonamidas, en pacientes con insuficiencia renal y/o hepática severa, porfiria, deficiencia de glucosa-6fosfato deshidrogenasa y en el embarazo. Usar con extrema precaución en pacientes con insuficiencia renal y/o hepática moderada y en caso de historia de discrasias sanguíneas o de colitis pseudomembranosa. Se debe garantizar una adecuada hidratación a objeto de evitar la cristaluria. Durante el tratamiento se debe vigilar la función renal, hepática, hematológica y practicar uroanálisis. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Evitar la exposición excesiva a la luz solar. Consumir abundantes líquidos durante el tratamiento. Informar al VANCOMICINA FARMACOLOGÕA Antibiótico con acción bactericida. Se une con gran afinidad a la terminación d-alanil-d-alanina en las sub-unidades precursoras de la pared celular bacteriana, inhibiendo la síntesis del peptidoglicano y de la pared bacteriana. Es activa contra: Streptococcus spp., Staphylococcus spp., Clostridium spp. y Corynebacterium spp. No tiene actividad contra organismos Gram negativos. FARMACOCIN…TICA Después de la administración IV alcanza niveles séricos pico en 5 J minutos. Se une a las proteínas plasmáticas en un 50-60%. Se distribuye ampliamente en los fluidos corporales y en las aurículas, pero en forma limitada en el líquido cefalorraquídeo; cuando las meninges están inflamadas alcanza concentraciones equivalentes al 15% de las plasmá-ticas. Tiende a acumularse en los tejidos luego de 2-3 días de adminis-tración continua. Se elimina en un 80-90% intacta por la orina en 24 horas. La vida media de eliminación es de 4-6 horas y se incrementa en pacientes con insuficiencia renal. INDICACIONES Infecciones severas causadas por 2ed. 2004 * Formulario TerapÈutico Nacional 231 en adultos: 500 mg cada 6 horas o 1 g cada 12 horas. En neonatos: iniciar con 15 mg/kg, seguidos por 10 mg/kg cada 12 horas en menores de 8 días y cada 8 horas en mayores de 8 días; en niños mayores de 1 mes: 40 mg/kg/día en dosis divididas. La dosis en niños no debe exceder 2 g/día. Por lo general, la duración de la terapia depende del tipo y severidad de la infección; el tratamiento debe durar el tiempo que sea necesario para lograr la erradicación completa de la infección y evitar la recidiva. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, colitis pseudomembranosa. Hematológicas: Eosinofilia, trombocitopenia, leucopenia, neutropenia. Neurológicas: Ototoxicidad. Reacciones de hipersensibilidad: Erupción, urticaria, disnea, reacciones anafilactoides, escalofrío y anafilaxis. Renales: Aumentos del BUN y la creatinina sérica, nefrotoxicidad. Otras: Fiebre, dolor en el sitio de la inyección y tromboflebitis. La infusión rápida puede causar el síndrome del cuello rojo (erupción maculopapular en la cara, el cuello, el tronco y las extremidades, prurito e hipotensión brusca y severa asociada a liberación de histamina). INTERACCIONES 232 muscular de la succinilcolina. Además, interfiere con la depuración de metformina. La administración concomitante con agentes anestésicos ha sido asociada con eritema y enrojecimiento similar al producido por la histamina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. No se debe usar por vía IM. Para minimizar el dolor y la tromboflebitis asociada a la administración IV, se debe diluir a con-centraciones de 2,5-5 g/L e infundir lentamente (1 hora mínimo). Usar con precaución en pacientes con insufi-ciencia renal, ajustando la dosis y/o el intervalo de dosificación. Durante la terapia se debe practicar evaluaciones periódicas de la función hematológica, auditiva y renal. El uso en el embarazo debe limitarse a circunstancias de verdadera necesidad y previa con-sideración del balance riesgo-beneficio. El uso prolongado puede ocasionar sobrecrecimiento de orga-nismos no susceptibles, incluyendo hongos patógenos. Durante el trata-miento se recomienda suspender la lactancia ya que la vancomicina se excreta en la leche materna. SOBREDOSIS Manejo sintomático y terapia de soporte. Se ha usado hemofiltración y Formulario Terapéutico Nacional * 2ed. 2004 AMFOTERICINA B la orina. FARMACOLOGÕA Agente antifúngico del grupo de los antibióticos poliénicos, producido por el Streptomyces nodosum con actividad fungistática o fungicida, según la dosis y la susceptibilidad del microorganismo. Se une a los esteroles de la membrana citoplasmática del patógeno alterando la permeabilidad y permitiendo la salida o pérdida de elementos intracelulares vitales. Es activo contra numerosas especies de hongos, que incluyen: Histoplasma capsulatum, Coccidioides immitis, Candida spp., Blastomyces dermatitis, Cryptococcus neoformans, Aspergillus fumigatus, Rhodotorula spp., Paracoccidioides brasilensis, Sporothrix schenckii y Mucor mucedo. FARMACOCIN…TICA Se absorbe pobremente por vía oral e IM, por lo que debe ser administrada por vía IV. Se une a las proteínas plas-máticas en un 90-95% y se distribuye ampliamente a los fluidos y tejidos corporales; sin embargo, la penetra-ción al sistema nervioso central es limitada, por lo que se hace necesaria la administración intratecal para lograr concentraciones efectivas a ese nivel. No se conoce bien su metabolismo. Se excreta en un 2-5% intacta por la orina. La vida media INDICACIONES Tratamiento de infecciones severas provocadas por patógenos susceptibles. R…GIMEN DE DOSIFICACI”N La administración IV de la amfotericina B debe hacerse por infusión lenta, en un período de 2-6 horas (dependiendo de la dosis). La concentración ideal de la solución debe ser 0,1 mg/ml (1 mg/10 ml). Bajo ninguna circunstancia la dosis diaria debe exceder 1,5 mg/kg. Previo al tratamiento se recomienda administrar una dosis de prueba de 1 mg diluida en J 20 ml de solución de dextrosa al 5% por infusión lenta (20-30 minutos), y con vigilancia de los signos vitales cada 30 minutos por 2-4 horas. Si el paciente tiene buenas condiciones cardio-renales y tolera adecuadamente la dosis de prueba, se puede iniciar la terapia con 0,25 mg/kg/día por infusión lenta con incrementos diarios de 0,5 mg/kg, hasta una dosis máxima diaria de 1,5 mg/kg, si el caso lo amerita. En pacientes con infección severa y de progreso rápido se puede iniciar con una dosis de 0,3 mg/kg/día. En pacientes con malas condiciones cardio-renales o que presentaron una reacción severa a la dosis de prueba, 2ed. 2004 * Formulario TerapÈutico Nacional 233 REACCIONES ADVERSAS Cardiovasculares: Arritmias, miocardiopatía, insuficiencia cardiaca congestiva, hipotensión, hipertensión, fenómeno de Raynaud y tromboflebitis. Gastrointestinales: Náusea, vómito, anorexia, dispepsia, dolor epigástrico, diarrea y melena. Hematológicas: Agranulocitosis, trombocitopenia, leucopenia, eosinofilia, anemia normocítica normocrómica y trastornos de la coagulación. Hepáticas: Hepatitis, ictericia, elevación de las enzimas hepáticas y de la bilirrubina, insuficiencia hepática aguda. Neurológicas: Neuralgia, parestesia, paraplejia, cefaleas, confusión, depresión, mareo, delirio, neuropatía periférica y convulsiones. Reacciones de hipersensibilidad: Erupción, prurito y anafilaxis. Renales: Falla renal aguda, aumentos del BUN y de la creatinina sérica, nefrocalcinosis y acidosis tubular renal, hipokalemia. Cuando se usa dosis muy altas puede haber alteración permanente de la función renal. Respiratorias: Edema pulmonar, sibilancia, disnea, taquipnea, broncoespasmo, hipertensión pulmonar e insuficiencia respiratoria, pneumonitis por hipersensibilidad. Otras: trastornos electrolíticos (hipopotasemia), trastornos visuales, tinitus, pérdida de la audición, dolor en el sitio de la inyección con o sin flebitis o tromboflebitis, dolor generalizado, inclu234 INTERACCIONES Cuando se usa en combinación con medicamentos nefrotóxicos se incrementa el riesgo de reacciones renales adversas. Dado que la amfotericina B tiene actividad depletora de potasio, puede predisponer al paciente al efecto tóxico de los digitálicos. La hipopota-semia inducida por la amfotericina B puede intensificar el efecto de los bloqueantes neuromusculares no despolarizantes. Los esteroides pue-den agravar el efecto hipopotasémico de la amfotericina B. Los antimicóticos imidazólicos y triazólicos pueden antagonizar los efectos de la amfotericina B. Puede aumentar el potencial de toxicidad renal, de producir broncoespasmo e hipotensión de los agentes antineoplásicos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes hipersensibles al medicamento. Debe ser usada bajo estricta observación de personal entrenado clínicamente. Usar con precaución en pacientes con falla renal y/o hepática. Durante el tratamiento se debe vigilar la función hematológica, hepática, renal y los electrolitos. No se ha establecido la seguridad de su uso en el embarazo. Se recomienda suspender la lactancia durante la terapia. Puede mejorarse la Formulario Terapéutico Nacional * 2ed. 2004 cardio-respiratorio. Debe suspenderse la terapia y evaluar la condición clínica del paciente. Manejo sintomático y terapia de soporte. INFORMACI”N AL PACIENTE No aplicable; medicamento para uso FLUCONAZOL FARMACOLOGÍA Antimicótico del grupo de los triazoles. Posee actividad fungistática. El principal efecto de los imidazoles y los triazoles es la inhibición de la esterol C14- -desmetilasa, enzima microsomal perteneciente al sistema del citocromo P450. La inhibición de esta enzima conduce a la inhibición de la síntesis del ergosterol de la membrana y a la acumulación de los C14- -me-tilesteroles. Esto conduce a la inhibi-ción de enzimas como la ATPasa y se inhibe la proliferación del hongo. Es activo contra: Candida spp., Crytoco-ccus neoformans, Histoplasma cap-sulatum, Blastomyces dermatitidis, Coccidioides immitis, Paracoccidioides brasiliensis, Epidermophyton, Microsporum y Trichophyton. FARMACOCINÉTICA Luego de la administración oral se absorbe rápidamente y en un porcentaje superior al 90% en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico en 1-2 horas. Se ces. La vida media de eliminación es de aproximadamente 30 horas (rango 20-50 horas) después de la administración oral y se incrementa en pacientes con falla renal. INDICACIONES Candidiasis orofaríngea, esofágica y vaginal e infecciones micóticas profundas y superficiales causadas por hongos susceptibles. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral o por infusión IV en dosis idénticas. La administración IV no debe exceder la velocidad de 200 mg/hora. Candidiasis orofaríngea o J esofágica en adultos: 200 mg el primer día, seguido por 100 mg/día (hasta 400 mg/día en casos severos). En la candidiasis orofaríngea la terapia debe mantenerse por 2 semanas y en la candidiasis esofágica por 3 semanas como mínimo y hasta 2 semanas después de la resolución de los síntomas. En niños, se recomienda 6 mg/kg el primer día, seguido por 3 mg/kg/día (hasta 12 mg/kg/día en casos severos) con la misma duración recomendada para los adultos. - Micosis profundas en adultos: 400 mg el primer día, seguido por 200400 mg/día (hasta 800 mg/día en casos severos) por 4 semanas 2ed. 2004 * Formulario TerapÈutico Nacional 235 mg/kg/día en casos severos) con la misma duración recomendada para adultos. - Candidiasis vaginal en adultos: dosis oral única de 150 mg. - Micosis dérmicas como tinea corporis, tinea cruris o tinea pedis en adultos: 150 mg semanal por 4 semanas; onicomicosis: 150 mg semanal por 3-12 meses, según la respuesta. REACCIONES ADVERSAS Cardiovasculares: Hipotensión. Gastrointestinales: Náuseas, vómito, diarrea, perversión del gusto, dolor abdominal. Hematológicas: Anemia, leucopenia, neutropenia, trombocitopenia. Hepáticas: Ictericia, hepatitis, aumento de ALT y AST. Neurológicas: Mareo, cefalea, somnolencia, parestesia, trastornos psiquiátricos, convulsiones. Reacciones de hipersensibilidad: Erupción, prurito, dermatitis exfoliativa, necrólisis epidérmica tóxica, síndrome de StevensJohnson y anafilaxis. Renales: Aumentos del BUN y de la creatinina sérica. Otras: Edema, hipercolesterolemia, hipo-potasemia, artralgia, mialgia, alopecia, fiebre. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con trastornos hepáticos. Usar con precaución en pacientes con alergia a otros azoles, falla renal, historia de enfermedad hepática y en el embarazo. Vigilar periódicamente la función hepá-tica. El uso por períodos prolongados puede provocar el sobrecrecimiento de microorganismos no susceptibles. Se debe suspender la lactancia duran-te el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE No alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los sínto-mas de la infección. Informar al médico de inmediato si se presenta alguna reacción inusual durante el tratamien-to, en especial anorexia, KETOCONAZOL FARMACOLOGÕA INTERACCIONES Antifúngico derivado del imidazol. PoEl fluconazol puede incrementar las see actividad fungistática o fungicida, concentraciones plasmáticas y la incisegún la dosis y la susceptibilidad del dencia de efectos adversos de la microorganismo. El principal efecto de rifabutina, carbamazepina, fenitoína, 236 Formulario Terapéutico Nacional * 2ed. 2004 inhibición de la esterol C14- -desmetilasa, enzima microsomal perteneciente al sistema del citocromo P450. La inhibición de esta enzima conduce a la inhibición de la síntesis del ergosterol de la membrana y a la acumulación de los C14- -metilesteroles. Esto conduce a la inhibición de enzimas como la ATPasa, y se inhibe la proliferación del hongo. Es activo contra: Paracoccidioides spp., Blastomyces dermatidis, Cryptococcus neoformans, Histoplasma capsulatum, Epidermphyton spp., Microsporum spp., Malassezia frufr˙ Candida spp., Aspergillus spp., Coccidioides spp, Trichophyton spp., Pityrosporum ovale y P. orbiculare. FARMACOCIN…TICA Se absorbe rápidamente en el tracto gastrointestinal en medio ácido y alcanza concentraciones plasmáticas pico en 1-2 horas. La hipoacidez gástrica reduce su absorción y la presencia de alimentos la incrementa. Una vez en la sangre, se une a las proteínas séricas en un 91-99% y se distribuye ampliamente a los fluidos y tejidos corporales, aunque no logra una buena penetración en el líquido cefalorraquídeo. Llega eficazmente a los queratinocitos. Se metaboliza parcialmente en el hígado generando productos inactivos que se excretan, junto con un 20-65% del fármaco intacto, en la bilis y por ende, por las heces. La eliminación plasmática es INDICACIONES Tratamiento de infecciones micóticas sistémicas causadas por organismos susceptibles. R…GIMEN DE DOSIFICACI”N La dosis usual por vía oral en adultos es de 200 mg/día y en niños mayores de 2 años: 3,3-6,6 mg/kg/día. En algunas condiciones, según el patógeno infectante y la severidad del cuadro, podría requerirse dosis de 400 mg/día. La duración de la terapia varía con el microorganismo, el sitio de la infección y su gravedad; en caso de candidiasis puede durar 1-4 semanas, en la paracoccidioidomicosis 6 meses y en la J cromomicosis o la blastomicosis, hasta 12 meses. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos, diarrea, dolor abdominal, flatulencia y constipación. Endocrinas: Ginecomastia bilateral, alteración del perfil lipídico, supresión de la síntesis de cortisol y de testosterona, hipotiroidismo, y oligospermia y azoospermia, irregularidades menstruales y disminución de la libido. Hematológicas: Anemia hemolítica, leucopenia y trombocitopenia. Hepáticas: Incremento de las enzimas hepáticas, ictericia y hepatitis, que a veces puede ser fatal. Neurológicas: Cefalea, somnolencia, mareo y nerviosismo. 237 2ed. 2004 * Formulario TerapÈutico Nacional bloqueantes de la bomba de protones y antimuscarínicos) y el sucralfato, reducen la absorción gastrointestinal de ketoconazol. El uso en combinación con fármacos hepatotóxicos incrementa el riesgo en tal sentido. La rifampicina reduce las concentraciones séricas de ketoconazol. La amfotericina B antagoniza el efecto antimicótico del ketoconazol. El ketoconazol puede incrementar el riesgo de miopatías inducidas por inhibidores de la HMG-CoA reductasa. Si se usa con alcohol puede producir reacciones tipo disulfiram. El ketoconazol inhibe el sistema microsomal del citocromo P450, pudiendo incrementar el nivel plasmático y la vida media de numerosos fármacos que se metabolizan a través de dicho grupo enzimático, como la terfenadina, astemizol, cisaprida, sulfonilúreas, ciclosporina, fenitoína, tretinoína, benzodiazepinas, sildenafil, algunos antivirales, corticosteroides, bloqueantes del calcio (dihidropiridinas) y warfarina. pacientes pediátricos a menos que el beneficio potencial sobrepase los riesgos. SOBREDOSIS Lavado gástrico con bicarbonato de sodio, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Tomar con las comidas. Evitar el alcohol y los antiácidos. No alterar la dosificación ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la infección. Notificar al médico si durante el tratamiento se presenta alguna reacción o sintomatología CLOFAZIMINA FARMACOLOGÍA Agente con actividad bactericida contra el Mycobacterium leprae (bacilo de Hansen). Aunque no se conoce con precisión su mecanismo de acción farmacológico, se cree que inhibe el crecimiento y la replicación del microorganismo al unirse irreversiblemente al ADN. También es activa contra Mycobacterium tuberculosis y otras especies de micobacterias. Tiene propiedades antiinflamatorias, las cuales ejerce para controlar las reacciones del eritema nodoso leproso. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a otros imidazoles. Debe evitarse el uso concomitante con terfenadina. Usar con precaución en pacientes con historia clínica o antecedentes de enfermedad hepática, alteración de la fun238 Formulario Terapéutico Nacional * 2ed. 2004 absorbe en un 45-70%. Los alimentos favorecen su absorción. Alcanza niveles séricos pico en 4-12 horas y concentraciones estables después de 30 días de terapia continua. Debido a su propiedad altamente lipofílica se deposita en el tejido adiposo y en las células del sistema retículoendotelial. No se conoce bien su metabolismo. Aunque la vida media de eliminación, desde los tejidos donde se acumula, se ha estimado en 70 días, es posible detectar fármaco intacto en la piel y los ganglios linfáticos hasta 2-4 años des-pués de finalizada la terapia. Se excre-ta principalmente por vía biliar y, por tanto, por las heces y muy poco en la orina. También se elimina una pequeña cantidad en el esputo y el sudor. INDICACIONES Tratamiento de la lepra (enfermedad de Hansen), en combinación con rifampicina y dapsona. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - Lepra multibacilar: - En adultos: Clofazimina (50 mg/día y 300 mg, una vez al mes) en combinación con rifampicina (600 mg, una vez al mes) y dapsona (100 mg/día) por 12 meses. - En niños de 10-14 años: Clofazimina (50 mg cada segundo día y 150 mg una vez al mes) en combinación con mg/día) por 12 meses. - Lepra paucibacilar resistente a la dapsona: Clofazimina (50-100 mg/día) en combinación con rifampicina (600 mg 1 vez al mes) por 6 meses, seguido por clofazimina sola (misma dosis) hasta lograr lesiones cutáneas negativas por 3 años en pacientes con lepra tuberculoide o indeterminada, o por 5 años en pacientes con lepra tuberculoide límite. REACCIONES ADVERSAS Dermatológicas: Pigmentación rosada a negra de la piel, resequedad, ardor, erupción, ictiosis, prurito, J irritación de la conjuntiva y la córnea, fotosen-sibilidad de la piel, erupciones acnei-formes. Gastrointestinales: Náuseas, vómito, dolor abdominal, diarrea, obs-trucción intestinal, hemorragia gástri-ca, constipación, anorexia, pérdida de peso, infarto esplénico. Hepáticas: Hepatitis, hepatomegalia. Neuroló-gicas: Mareo, dolor de cabeza, fatiga, depresión. Otras: Hiperglicemia, fiebre, linfadenopatía; coloración ocre de la orina, heces, sudor, lágrimas y esputo, que puede persistir por varias semanas, aún después de suspendida la terapia. INTERACCIONES La clofazimina puede reducir las concentraciones séricas de fenitoína. La 239 2ed. 2004 * Formulario TerapÈutico Nacional CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento. Usar con precaución en pacientes con trastornos gastrointestinales tales como dolor abdominal y diarrea. El médico debe estar pendiente de que los cambios de color de la piel pueden resultar en depresión del paciente. No se conoce la seguridad de su uso durante el embarazo. Se sabe que atraviesa la barrera placentaria y los infantes nacidos de madres que reciben clofazimina presentan marcada pigmentación. Sólo debe usarse en el embarazo si el beneficio potencial justifica el riesgo para el feto. La clofazimina se excreta en la leche materna. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Tomar con las comidas. No alterar la dosificación ni suspender el tratamiento, aunque hayan desaparecido los signos de la enfermedad. El medicamento puede colorear la piel de color rojo o marrón oscuro (ocre), la conjuntiva, las lágrimas, el sudor, las heces, el esputo y la orina y dicha coloración puede persistir por varios meses o años, inclusive, después de 240 DAPSONA FARMACOLOGÍA Sulfona con actividad bacteriostática contra Mycobacterium leprae. Se postula que actúa de manera similar a las sulfonamidas, interfiriendo con la síntesis de ácido fólico y, por ende, con la síntesis de proteínas y ácidos nucléicos en el microorganismo. La acción es antagonizada por el ácido paraami-nobenzóico. También es activa contra Mycobacterium tuberculosis y otras especies de micobacterias. Es un tratamiento primario para la dermatitis herpetiforme. FARMACOCINÉTICA Después de la administración oral, se absorbe casi por completo, alcanzando concentraciones séricas pico en 28 horas. Se une a las proteínas plasmáticas en un 70-90% y se distribuye ampliamente a los tejidos y fluidos corporales, acumulándose en la piel, el músculo, el hígado y el riñón. Se metaboliza en el hígado por acetilación e hidroxilación, transformándose en pro-ductos inactivos que se excretan, junto a 20% del fármaco intacto, en un 70-85% por la orina y el resto por la bilis. La vida media de eliminación es de 10-50 horas (promedio 30 horas). INDICACIONES Tratamiento de la lepra (enfermedad de Hansen), en combinación con ri- Formulario Terapéutico Nacional * 2ed. 2004 ANTILEPROSOS). Se recomienda que la dapsona sea mantenida a una dosis completa de 100 mg diarios sin interrupción. REACCIONES ADVERSAS Cardiovasculares: Taquicardia. Gastrointestinales: Náusea, vómito, dolor abdominal, pancreatitis, anorexia. Hematológicas: Agranulocitosis, metahemoglobinemia, anemia hemolítica (especialmente en pacientes con deficiencias de glucosa-6-fosfato deshidrogenasa), anemia aplásica, síndrome similar a la mononucleosis. Hepáticas: Hepatotoxicidad. Neurológicas: Psicosis, vértigo, dolor de cabeza, insomnio, tinitus, neuropatía periférica, debilidad muscular. Reacciones de hipersensibilidad: Erupción, urticaria, dermatitis exfoliativa, eritema multiforme, síndrome de Stevens-Johnson, necrólisis epidérmi-ca tóxica, eritema nodoso. Renales: Falla renal aguda, síndrome nefrótico, necrosis papilar renal. Otras: Fotosen-sibilidad, hipoalbuminemia, visión borrosa, fiebre, infertilidad masculina, lupus eritematoso inducido por medicamentos. INTERACCIONES El trimetoprim puede incrementar las concentraciones plasmáticas de la dapsona por inhibición del metabolismo hepático o por competencia por puede causar falla terapéutica de la dapsona. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al fármaco y en pacientes anémicos. Usar con precaución en pacientes con deficiencia de glucosa6-fosfato deshidrogenasa y en mujeres embarazadas. Sólo debe darse a mujeres embarazadas si es claramente necesario. Vigilar periódicamente la función hepática y la hematológica. La dapsona se excreta en cantidades sustanciales en la leche materna y pueden ocurrir reacciones hemolíticas en los neonatos. Si debe J suspenderse la lactancia o el medicamento dependerá de la importancia del medicamento para la madre. SOBREDOSIS Puede presentarse náuseas, vómitos e hiperexcitabilidad dentro de pocos minutos a 24 horas después de la administración. Vaciamiento gástrico, carbón activado y un catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Si se presenta metahemoglobinemia se debe tratar con azul de metileno (excepto en pacientes con deficiencia de glucosa-6-fosfato deshidrogenasa). Puede ocurrir hemólisis hasta 7-14 d í a s d e s p u é s d e l a sobredosificación. El fármaco es 2ed. 2004 * Formulario TerapÈutico Nacional 241 presenta alguna reacción o sintomaESTREPTOMICINA FARMACOLOGÕA Antibiótico aminoglicósido obtenido de cultivos de Streptomyces griseus. Tie-ne acción bactericida. Inhibe la sínte-sis de proteínas en gérmenes suscep-tibles por unión a la subunidad ribo-somal 30S. Es activa contra microor-ganismos aeróbicos Gram negativos como: Brucella, Calymmatobacterium granulomatis, Francisella tularensis, Pasteurella multocida, Yersinia pestis, Haemophilus influenzae y H. ducreyii; algunas bacterias Gram positivas como: Nocardia, Streptococcus faecalis y Erysipelothrix y contra micobacterias como: Mycobacterium tuberculosis, M. bovis, M. marinum y algunas cepas de M. kansaii, M. avium y M. leprae. FARMACOCIN…TICA La estreptomicina sigue un patrón farmacocinético similar al descrito para otros antibióticos aminoglicósidos (ver: AMIKACINA). Posterior a su administración IM alcanza concentraciones séricas máximas en 1-2 horas. Se distribuye a la mayoría de los tejidos corporales, excepto el cerebro y el ojo dada su naturaleza polar. Se detectan altas concentraciones en la corteza renal y en la endolinfa y 242 horas en pacientes con insuficiencia renal severa. INDICACIONES Tratamiento de la tuberculosis, en asociación con otros agentes antimicobacterianos. R…GIMEN DE DOSIFICACI”N Se usa sólo por vía IM. La dosis usual en adultos es de 1 g/día o 15 mg/kg/día y en niños: 20-40 mg/kg/día, hasta un máximo de 1 g/día. La administración de una sola dosis diaria parece ser menos tóxica e igualmente eficaz. El tratamiento debe mantenerse hasta obtener cultivos negativos. La dosis total por curso de terapia no debe exceder los 120 g. REACCIONES ADVERSAS Ver: AMIKACINA. INTERACCIONES Ver: AMIKACINA. CONTRAINDICACIONES/ PRECAUCIONES Ver: AMIKACINA. SOBREDOSIS Ver: AMIKACINA. INFORMACI”N AL PACIENTE No alterar la dosis ni suspender la te- Formulario Terapéutico Nacional * 2ed. 2004 ETAMBUTOL terianos como la isoniazida. FARMACOLOGÕA Agente antimicobacteriano sintético. Actúa bloqueando las transferasas de arabinosil involucradas en la biosíntesis de la pared celular. Interfiere con el metabolismo de las proteínas por inhibición de la síntesis de RNA en microorganismos susceptibles, impidiendo su multiplicación y provocando la muerte celular. Forma quelatos con los iones que intervienen en la síntesis de ADN y ARN. Es activo contra todas las cepas de Mycobacterium tuberculosis, M. kansasii, y otras del complejo M. avium. FARMACOCIN…TICA Se absorbe en un 75-80% en el tracto gastrointestinal, alcanzando niveles séricos pico a las 2-4 horas de su administración oral. Se une en un 630% a las proteínas plasmáticas. Se distribuye en la mayoría de los fluidos y tejidos corporales logrando concentraciones altas en eritrocitos, pulmón, riñón y saliva, y en menor magnitud, en fluido ascítico, pleural, en cerebro y líquido cefalorraquídeo. Se metaboliza en el hígado transformándose en pro-ductos inactivos (8-15%) que se excre-tan, junto con el fármaco intacto (50%), en la orina tanto por filtración glome-rular como por secreción tubular. El fármaco no absorbido (20%) es excre- R…GIMEN DE DOSIFICACI”N Se administra solamente por vía oral. La dosis usual en pacientes adultos que no han recibido tratamiento previo es de 15 mg/kg una vez al día; en pacientes tratados con anterioridad, iniciar con 25 mg/kg una vez al día por 2 meses y reducir después a 15 mg/kg/día. En pacientes con alteración de la función renal debe hacerse un ajuste de la dosis para evitar la acu-mulación del medicamento. La terapia debe mantenerse hasta lograr la má-xima mejoría clínica y la conversión J bacteriológica permanente. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, anorexia, dolor abdominal. Hematológicas: Leucopenia, neutropenia, trombocitopenia y eosinofilia. Hepáticas: Alteración de los niveles de las enzimas hepáticas. Neurológicas: El efecto adverso más importante es la neuritis óptica, que se manifiesta con disminución de la agudeza visual, reducción del campo visual, escotomatas y pérdida de la discriminación de los colores verde y rojo; cefalea, mareos, neuritis periférica, confusión, desorientación, alucinaciones. Reacciones de hipersensibilidad: Necrólisis epidérmica tóxica, dermatitis, 2ed. 2004 * Formulario TerapÈutico Nacional 243 intensificado por la isoniazida y la piridoxina. Las sales de aluminio pueden retardar y disminuir la absorción. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al etambutol y/o con neuritis óptica. Usar con precaución en pacientes con insuficiencia renal, con trastornos visuales y en el embarazo. Antes de iniciar la terapia, y frecuente-mente durante su desarrollo, se deben realizar pruebas de agudeza visual, de discriminación del color y de la función renal. Durante el tratamiento se debe suspender la lactancia. No se reco-mienda su uso en niños menores de 13 años. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. El medicamento es removible por diálisis peritoneal. INFORMACIÓN AL PACIENTE Administrar con las comidas. No alterar la dosis ni suspender el trataISONIAZIDA FARMACOLOGÍA Antimicobacteriano sintético con actividad bacteriostática contra los bacilos 244 teria se encuentra en fase de división. Se postula que actúa durante el crecimiento y multiplicación bacteriana interfiriendo con el metabolismo de carbohidratos, proteínas, lípidos y áci-dos nucléicos, e inhibiendo la síntesis de los ácidos micólicos, generando así la destrucción de la pared celular, con la consecuente muerte del patógeno. Es activa contra el Mycobacterium tuberculosis, M. bovis y algunas cepas de M. kansasii, aunque puede reque-rirse dosis muy altas para esta bacteria. FARMACOCIN…TICA Después de la administración por vía oral se absorbe rápida y completamente en el tracto gastrointestinal, alcanzando concentraciones plasmáticas pico en 1-2 horas. Los alimentos interfieren con la absorción de la isoniazida. Se une a las proteínas plasmáticas en un 4-30% y se distribuye ampliamente a los tejidos y fluidos corporales, alcanzando concentraciones en líquido cefalorraquídeo equi-valentes al 90-100% de las concentra-ciones plasmáticas. La isoniazida atraviesa la barrera placentaria y se excreta en la leche materna en con-centraciones similares a las del plas-ma. Se metaboliza parcialmente en el hígado por acetilación, dando origen a metabolitos inactivos que se excretan Formulario Terapéutico Nacional * 2ed. 2004 cada individuo. Los acetiladores lentos tienden a tener mayores concentraciones de isoniazida que los acetiladores rápidos, incrementándose la posibilidad de efectos tóxicos. La vida media de eliminación en los acetila-dores rápidos es de 0,7-2 horas y de 2,3-3,5 horas en los acetiladores lentos. Dichos valores aumentan en los pacientes con insuficiencia renal. INDICACIONES Prevención y tratamiento de la tuberculosis, en combinación con otros agentes antimicobacterianos. R…GIMEN DE DOSIFICACI”N Se usa en combinación con otros agentes antituberculosos. Se administra por vía oral como dosis única diaria. Para el tratamiento de la tuberculosis, la dosis usual en adultos es de 5 mg/kg, sin exceder 300 mg/día; en ni-ños: 10-20 mg/kg (según la severidad), sin exceder de 300 mg/día. Para la prevención de la tuberculosis en adul-tos, la dosis usual es de 300 mg/día por vía oral en una sola dosis, continuada por 6 meses a un año; en niños: 10 mg/kg sin exceder 300 mg/día conti-nuados por un año. De todas maneras, la terapia debe durar el tiempo que sea necesario, a juicio del médico, para erradicar la enfermedad y evitar la recidiva. notransferasas y de la bilirrubina. Neurológicas: Neuropatía periférica, neuritis y atrofia óptica, trastornos de la memoria, psicosis tóxica y convulsiones. Reacciones de Hipersensibilidad: Fiebre, erupción, vasculitis y linfadenopatía. Otras: Deplesión de piridoxina, ginecomastia, hiperglicemia, pelagra, acidosis metabólica, hipocalcemia, hipofosfatemia, síndrome reumá-tico y síndrome similar al lupus. INTERACCIONES La isoniazida puede inhibir el metabolismo de la carbamazepina y la fenitoína, incrementando los niveles J plasmáticos y el riesgo de toxicidad. Inhibición del metabolismo de las benzodiazepinas. La carbamazepina y el halotano aumentan el riesgo de hepatotoxicidad de la isoniazida. En pacientes que son acetiladores rápidos de la isoniazida y son tratados con enflurano puede ocurrir falla renal debido a concentraciones nefrotóxicas de flúor inorgánico. Puede aumentar el efecto de los anticoagulantes orales. El uso en combinación con otros fár-macos antituberculosos incrementa el riesgo de efectos adversos neuroló-gicos. Su eficacia es reducida por los corticosteroides. La ingesta de alcohol y la administración conjunta de rifampicina u otros fármacos potencialmen- 2ed. 2004 * Formulario TerapÈutico Nacional 245 CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y/o con daño hepático severo. Se debe usar con cautela en pacientes con falla renal, diabéticos, desnutridos y alcohólicos. Durante la terapia se debe vigilar periódicamente la función hepá-tica y la renal. Se recomienda la ad-ministración de suplementos de piridoxina, ante la posibilidad de deplesión inducida por la isoniazida. El uso en el embarazo debe limitarse a circunstancias de verdadera necesidad y previa consideración del balance riesgo-beneficio. Durante la terapia es recomendable suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Vía aérea permeable para una ventilación adecuada. Controlar el equilibrio hidroelectrolítico y el ácido-base. Manejar las convulsiones con diazepam IV. Si se desconoce la cantidad de isoniazida ingerida, puede administrarse piridoxina. Diuresis osmótica forzada. Hemodiálisis en casos severos; si no es posible, diálisis peritoneal en asociación con diuresis osmótica. 246 hol. No se debe alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la enfermedad. Notificar inmediatamente al médico si se presenta alguna reacción o síntoma inusual durante la terapia, en especial: Náuseas, vómito, ano-rexia, fatiga, debilidad, orina oscura y/o coloración amarillenta de la PIRAZINAMIDA FARMACOLOGÍA Antimicobacteriano. Es el análogo sintético pirazínico de la nicotinamida. Posee actividad bacteriostática o bactericida, según la dosis y la susceptibilidad del microorganismo. Aunque no se conoce con precisión su mecanismo de acción, se postula que el fármaco, al penetrar a la célula, es convertido en ácido pirazinóico por la acción de enzimas pirazinamidasas. Al parecer, el blanco es el gen que codifica por la sintetasa de ácidos grasos I micobacteriana comprendido en la biosíntesis del ácido micólico. Es activo a pH ligeramente ácido. Es activa contra el Mycobacterium tuberculosis. FARMACOCIN…TICA Después de la administración oral, se absorbe casi completamente en el tracto gastrointestinal, alcanzando niveles séricos pico en 1-4 horas. Se Formulario Terapéutico Nacional * 2ed. 2004 en el hígado por hidrólisis, transformándose en ácido pirazinóico, metabolito activo cuyos niveles séricos llegan a ser superiores a los de la pirazinamida a las 4-8 horas. Algo de ácido pirazinóico se forma también en el estómago y en la vejiga. El ácido pirazinóico es posteriormente hidroxilado y convertido en ácido 5hidroxipirazinóico, principal producto de excreción. El fármaco y sus metabolitos son eliminados en la orina (114% como pirazinamida intacta) por filtración glomerular. La vida media de eliminación es de 9-10 horas y la del ácido pirazinóico de 12,3 horas. Estos valores aumentan en pacientes con insuficiencia renal. rito. Gastrointestinales: Náuseas, vómitos. Hematológicas: Trombocitopenia y anemia sideroblástica. Hepáticas: Hepatotoxicidad (hasta en 15% de los pacientes, con 40-50 mg/kg o más). Otras: Hiperuricemia, gota, artralgia, porfiria, disuria, anorexia, fotosensibilidad. INTERACCIONES Reduce la eficacia del alopurinol y de la colchicina. Puede inducir el metabolismo de la ciclosporina, disminuyendo los niveles séricos y la eficacia terapéutica. Disminuye el metabolismo de la fenitoína, aumentando los niveles plasmáticos y el riesgo de toxicidad. Con etionamida incrementa J el riesgo de hepatotoxicidad. La zidovudina disminuye las concentraINDICACIONES Tratamiento de la tuberculosis, en ciones plasmáticas de la pirazinamida. combinación con otros antimicobac- Antagoniza los efectos de la sulfinpirazona. Posible reducción del terianos. efecto de los anticonceptivos orales. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. Se ha pro- CONTRAINDICACIONES/ puesto 2 esquemas de tratamiento, PRECAUCIONES aplicables por igual a pacientes Contraindicada en pacientes con hipersensibilidad al medicamento, adultos y niños: - Adultos y niños: Régimen de daño hepático severo y gota activa. administración diaria: 15-30 mg/kg, Usar con precaución en pacientes con una sola dosis diaria, hasta un insuficiencia renal, historia de gota, diabetes (dificulta el control) y en nimáximo de 2 g/dosis. - Adultos y niños: Régimen de 2 dosis ños. Antes y durante la terapia se debe por semana: 50-70 mg/kg, una sola vigilar la función hepática y los niveles dosis diaria, hasta un máximo de 3 de ácido úrico. Su uso en el embarazo debe limitarse a situaciones de 2ed. 2004 * Formulario TerapÈutico Nacional 247 y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. La pirazinamida es dializable. INFORMACI”N AL PACIENTE No modificar la dosis ni suspender la terapia antes del tiempo previsto, aunque hayan desaparecido los síntomas de la enfermedad. Notificar inmediatamente al médico si se presenta alguna reacción inusual, en especial: Fiebre, anorexia, náusea, vómitos, orina oscura y/o coloración amarillenta de piel y ojos (signos de hepatotoxicidad) o síntomas de artritis gotosa. RIFAMPICINA (Antileproso) FARMACOLOGÍA Ver: RIFAMPICINA en ANTITUBERCULOSOS. FARMACOCINÉTICA Ver: RIFAMPICINA en ANTITUBERCULOSOS. INDICACIONES Tratamiento de la lepra (enfermedad de Hansen), en combinación con dapsona y clofazimina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - Lepra multibacilar: - En adultos: rifampicina (600 mg 1 vez al mes) en combinación con 248 - En niños de 10-14 años: rifampicina (450 mg 1 vez al mes) en combinación con clofazimina (50 mg interdiariamente, y 150 mg 1 vez al mes) y dapsona (50 mg/día) por 12 meses. - En niños menores de 10 años: rifampicina (300 mg 1 vez al mes) en combinación con clofazimina (50 mg 2 veces por semana y 100 mg 1 vez al mes) y dapsona (25 mg/día) por 12 meses. - Lepra paucibacilar: - En adultos: rifampicina (600 mg 1 vez al mes) en combinación con dapsona (100 mg/día) por 6 meses. - En niños de 10-14 años: rifampicina (450 mg 1 vez al mes) en combinación con dapsona (50 mg/día) por 6 meses. - En niños menores de 10 años: rifampicina (300 mg 1 vez al mes) en combinación con dapsona (25 mg/día) por 6 meses. REACCIONES ADVERSAS Ver: RIFAMPICINA en ANTITUBERCULOSOS. INTERACCIONES Ver: RIFAMPICINA en ANTITUBERCULOSOS. CONTRAINDICACIONES/ PRECAUCIONES Formulario Terapéutico Nacional * 2ed. 2004 INFORMACIÓN AL PACIENTE Ver: RIFAMPICINA en ANTITUBERRIFAMPICINA (Antituberculoso) FARMACOLOGÍA Antibacteriano derivado de la rifamicina B, con actividad bactericida para microorganismos intracelulares y extracelulares. Inhibe la actividad de la ARN polimerasa dependiente de ADN en organismos susceptibles formando un complejo enzima-fármaco estable que suprime el inicio de la cadena en la síntesis de ARN. Es activa in vivo e in vitro contra: Mycobacterium tuberculosis, M. bovis, M. leprae, M. marinum, M. kansasii y algunas cepas de M. fortuitum, M. intracellulare y M. Avium. También presenta actividad in vitro contra bacterias Gram positivas y Gram negativas como: Neisseria meningitidis, Staphylococcus aureus, Haemophilus influenzae y Legionella pneumophila. FARMACOCINÉTICA Se absorbe rápida y casi completamente (90-95%) en el tracto gastrointestinal, alcanzando concentraciones séricas pico a las 2-4 horas de su administración oral. Los alimentos interfieren con su absorción. Se une a las proteínas plasmáticas en un 6090% y se distribuye ampliamente a los Se excreta en un 30% por la orina y en un 60% por las heces. Menos de la mitad del fármaco excretado está en forma intacta. La vida media de eliminación es de aproximadamente 3-4 horas, pero con la administración repetida puede disminuir a 2-3 horas, debido a la inducción del citocromo P450. INDICACIONES Tratamiento de la tuberculosis, en combinación con otros antimicobacterianos. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos: 10 mg/kg/día y en niños: 10-20 J mg/kg/día, sin exceder (en ambos casos) 600 mg/día, una hora antes o dos horas después de las comidas. El tratamiento debe durar el tiempo que sea necesario para erradicar la enfermedad y evitar la recidiva. REACCIONES ADVERSAS Cardiovasculares: Hipotensión. Gastrointestinales: Náuseas, vómito, dolor abdominal, anorexia, diarrea, flatulencia, dolor en la boca y en la lengua, pancreatitis y colitis pseudomembranosa. Hematológicas: Eosinofilia, trombocitopenia, leucopenia transitoria y anemia hemolítica. Hepáticas: Aumento de los niveles de las enzimas hepáticas, ictericia y hepatitis. Neu-rológicas: Dolor de 2ed. 2004 * Formulario TerapÈutico Nacional 249 proteinuria y hematuria. Otras: Aumento del ácido úrico, artralgia, debilidad muscular, mialgia, trastornos vi-suales, conjuntivitis exudativa, insufi-ciencia adrenocortical y coloración ro-ja de la orina, sudor, saliva, lágrimas, heces y esputo. INTERACCIONES La rifampicina puede disminuir la eficacia de los anticonceptivos orales. Inhibe el metabolismo de la carbamazepina, aumentando los niveles sanguíneos y el riesgo de toxicidad. El uso concomitante con isoniazida y halotano incrementa el riesgo de hepatotoxicidad. El alcohol también aumenta el potencial hepatotóxico. Induce el citocromo P450 y, por tanto, puede inducir el metabolismo y reducir la eficacia de la amiodarona, digitálicos, buspirona, bloqueantes de los canales de calcio, clofibrato, clozapina, bloqueantes de los receptores beta-adrenérgicos, antivirales, corticosteroides, cloranfenicol, sulfonilúreas, teofilina, metadona, morfina, benzodiazepinas, ciclosporina, fenitoí-na, ácido valpróico, tretinoína, rofe-coxib, warfarina, losartán, sildenafil, mexiletina, inhibidores de la enzima angiotensinaconvertasa, levotiroxina, dapsona, doxiciclina, antimicóticos azoles, quinidina, haloperidol y barbitúricos. El ketoconazol y el ácido 250 caución en pacientes con falla hepática y/o renal. Antes de iniciar el tratamiento, y periódicamente durante su desarrollo, se debe realizar un examen hematológico y pruebas de la función hepática y renal. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales a la madre superen los riesgos para el feto. Cuando se administra durante las últimas semanas del embarazo puede causar hemorragia post-natal en la madre y en el neonato. Se debe suspender la lactancia durante el tratamiento. Como el medicamento tiñe de rojo las lágrimas, puede manchar permanentemente los lentes de contacto. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. En caso de alteración severa de la función hepática, con más de 24-48 horas de evolución, podría requerirse drenaje biliar. INFORMACIÓN AL PACIENTE Tomar 1 hora antes o 2 horas después de las comidas. No alterar la dosis ni suspender el tratamiento antes del tiempo previsto, aunque hayan desaparecido los síntomas de la enfermedad. El medicamento puede colorear de rojo los fluidos corporales (orina, sudor, esputo y lágrimas). Los Formulario Terapéutico Nacional * 2ed. 2004 (sugestivo de hepatotoxicidad), diarrea persistente (posible colitis pseudomembranosa) o inflamación de las ABACAVIR FARMACOLOGÍA Agente anti-retroviral análogo sintético carboxicíclico de los nucleósidos. Inhi-be la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA Se absorbe rápida y extensamente en el tracto gastrointestinal. La biodisponibilidad es de 82-86%. Los alimentos no alteran su biodisponibilidad. Se une a las proteínas plasmáticas en un 50%. Se distribuye a los eritrocitos. Se une extensamente a los tejidos que con-tienen melanina (úvea, cabello, oído interno). Se metaboliza extensamente en el hígado, mediante la deshidroge-nasa del alcohol y la glucuronil transfe-rasa, transformándose en metabolitos inactivos que se excretan, junto a menos de un 2% del fármaco intacto, en la orina. Un 16% adicional se elimina por las heces. La vida media de eliminación es de aproximadamente 1,5 horas. INDICACIONES en adultos: 300 mg 2 veces al día. Niños de 3 meses-16 años: 8 mg/kg 2 veces al día, hasta un máximo de 300 mg por dosis. REACCIONES ADVERSAS Neurológicas: Dolor de cabeza, parestesia, fatiga, mareos, letargia y trastornos del sueño. Cardiovasculares: Hipotensión y edema. Respiratorias: Faringitis, disnea y tos. Gastrointestinales: Náuseas, vómito, diarrea y dolor abdominal. Hepáticas: Disfunción hepática y hepatomegalia con esteatosis. Otras: Fiebre, falla renal, linfadenopatías, mialgia, artralgia, hiperglicemia, acidosis J láctica, redistribución anormal de grasa corporal, lesiones en membranas mucosas, erupción máculopapular y urticarial, síndrome de Stevens-Johnson y anafilaxis. INTERACCIONES El abacavir aumenta la depuración de metadona. Con la ribavirina se incrementa el riesgo de acidosis láctica. El etanol puede aumentar el AUC y la vida media del abacavir. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática y en el embarazo. Duran-te el tratamiento, se debe vigilar 2ed. 2004 * Formulario TerapÈutico Nacional 251 dicamento y buscar atención médica inmediata. vudina y 300 mg de zidovudina (1 tableta) 2 veces diarias. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. REACCIONES ADVERSAS Ver las monografías individuales. INFORMACIÓN AL PACIENTE No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología ABACAVIR / LAMIVUDINA / ZIDOVUDINA FARMACOLOGÍA Combinación a dosis fijas de agentes anti-retrovirales nucleósidos inhibidores de la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA El comportamiento cinético de la combinación de estos fármacos es aquel que cabe esperar para cada uno de los constituyentes por separado (ver monografías individuales). INDICACIONES En combinación con otros anti-retrovirales para el tratamiento de la infección por virus de inmunodeficiencia humano. RÉGIMEN DE DOSIFICACIÓN 252 INTERACCIONES Ver las monografías individuales de los fármacos. CONTRAINDICACIONES/ PRECAUCIONES Ver monografías individuales de los fármacos. Contraindicado en casos de hipersensibilidad a alguno de los constituyentes del producto y en quienes requieren ajustes de dosificación. No usar en pacientes con depuración de creatinina menor o igual que 50 ml/minuto. SOBREDOSIS Ver las monografías individuales de los fármacos. Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Ver las monografías individuales de los fármacos. Este medicamento puede ser ingerido junto con las ACICLOVIR (SistÈmico) FARMACOLOGÍA Agente antiviral. Es un análogo del nucleósido acíclico guanina (purina) sin- Formulario Terapéutico Nacional * 2ed. 2004 tético. Interfiere con la síntesis del ADN viral por inhibición de la ADN polime-rasa e impide la replicación viral. Pre-senta actividad in vitro contra herpes humano, incluyendo herpes simplex virus tipo 1(HSV-1) y tipo 2(HSV-2), varicella-zoster (VZV), Epstein Barr (EBV) y citomegalovirus (CMV) (profi-laxis en pacientes inmunocomprome-tidos). FARMACOCINÉTICA La absorción desde el tracto gastrointestinal es variable e incompleta, estimándose en un 15-30% de la dosis administrada. La absorción disminuye según se aumenta la dosis. Posterior a la administración oral, se alcanzan concentraciones plasmáticas pico en 1,5-2,5 horas. Se une a las proteínas plasmáticas en un 9-33% y se distribuye ampliamente a los tejidos y fluidos corporales, se concentra en la leche materna y en el líquido amniótico. Se metaboliza parcialmente en el hígado y se elimina por vía renal como fármaco intacto (30-90%) y como metabolitos (1-14%) y escasamente (menos de 2%) por las heces. La vida media plasmática es de 2,5 a 3 horas y se incrementa hasta 20 horas en pacientes con falla renal. INDICACIONES Tratamiento de infecciones por herpes simplex, herpes genital y varicella- 4 horas durante las horas de vigilia (5 veces al día) por 7-10 días o hasta resolución clínica. En niños 1 g diario dividido en 35 dosis por 7-14 días, sin exceder de 80 mg/kg/día. - Tratamiento de los episodios iniciales de la infección por herpes genital, en adultos: 200 mg cada 4 horas durante las horas de vigilia (5 veces al día), por 7-10 días o hasta la resolución clínica de la condición. Tratamiento supresivo crónico de las recurrencias: 400 mg 2 veces al día hasta por 12 meses o más, sujeto a resul- J tados de la evaluación clínica. Tratamiento intermitente: 200 mg cada 4 horas (5 veces al día), iniciando el tratamiento al primer signo o síntoma (prodromo) de la recurrencia. - Tratamiento de la infección por varicella-zoster en adultos y niños de más de 40 kg: 800 mg 4 veces al día por 5 días; en niños de 2 años o más: 20 mg/kg 4 veces al día por 5 días; en ambos casos iniciando terapia al primer síntoma de la a p a r i c i ó n d e l a s manifestaciones cutá-neas. - Por vía IV: Se administra por infusión lenta, a velocidad constante y en un período nunca menor de 60 2ed. 2004 * Formulario TerapÈutico Nacional 253 - Tratamiento de los episodios iniciales de la infección por herpes genital: Las mismas dosis anteriores, pero por 5 días. - Encefalitis por herpes simplex en adultos y niños mayores de 12 años: 10 mg/kg cada 8 horas por 10 días; en niños menores de 12 años: 500 mg/m2 cada 8 horas por 10 días. - Tratamiento de infecciones por varicella-Zoster en pacientes inmunocomprometidos: las mismas dosis anteriores, pero por 7 días. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones. Dermatológicas: Erupción, prurito, vasculitis y reacciones locales en el sitio de inyección (dolor, irritación, eritema y flebitis). Gastrointestinales: Náusea, vómito, diarrea, anorexia y dolor abdominal. Hematológicas: Anemia, neutropenia, trombocitopenia, trombocitosis, leucopenia, linfadenopatía e hipoplasia medular. Hepáticas: Elevación de los niveles de las aminotransferasas. Neurológicas: Astenia, parestesia, mareo, fatiga, confusión, somno-lencia, psicosis, alucinaciones y ence-falopatía (caracterizada por letargia, desorientación, agitación, convulsio-nes y coma). Reacciones de hiper-sensibilidad: Anafilaxis. Renales: Au-mento del BUN, de la 254 INTERACCIONES La administración conjunta con zidovudina o con metotrexato puede incrementar el riesgo de neurotoxicidad. La combinación con medicamentos potencialmente nefrotóxicos aumenta el riesgo de falla renal aguda. La administración con probenecid aumenta la vida media y el AUC de concentracióntiempo. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con trastornos neurológicos, falla renal y/o hepática, deshidratados y en aquellos que reciben tratamiento con medicamentos potencialmente nefrotóxicos. Previo a la administración IV, se debe verificar el grado de hidratación del paciente y corregirlo en caso necesario, y garantizar el mantenimiento del flujo urinario, sobre todo durante las primeras 2 horas de la infusión. El período de infusión no debe ser menor de 60 minutos. Antes de iniciar el tratamiento, y periódicamente durante su desarrollo, se debe evaluar la fun-ción renal y el balance hidroelectro-lítico. No se conoce la seguridad de su uso en menores de 2 años. Usar con precaución en ancianos y en el em-barazo. Durante el tratamiento se recomienda suspender la lactancia. No debe excederse la Formulario Terapéutico Nacional * 2ed. 2004 de insuficiencia renal por daño tubular debido a precipitación del aciclovir cuando la solubilidad intratubular (2,5 mg/ml) es excedida. Es removible por hemodiálisis. INFORMACI”N AL PACIENTE Las dosis orales deben administrarse con abundante líquido y, de igual manera, mantener una ingesta adecuada de fluidos durante todo el tratamiento. Informar de inmediato al mé-dico si se presenta alguna reacción o sintomatología inusual durante el tra-tamiento. Las pacientes AMPRENAVIR FARMACOLOGÍA Agente anti-retroviral. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima proteasa viral. FARMACOCINÉTICA Se absorbe rápidamente a través del tracto gastrointestinal, alcanzando niveles plasmáticos pico en 1-2 horas y una respuesta terapéutica que se evidencia en 2-4 semanas después de iniciado el tratamiento. Los alimentos con alto contenido graso reducen la absorción. Se une a las proteínas plasmáticas en un 90%. Se metaboliza en el hígado a través del sistema microsomal del citocromo P 4 5 0 , 3% como fármaco intacto. La vida media de eliminación es de 7-10 horas. INDICACIONES Para el tratamiento de la infección por virus de inmunodeficiencia humano en combinación con otros agentes antiretrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - En adultos: 1200 mg 2 veces al día; o 1200 mg 1 vez al día en combinación con ritonavir 200 mg 1 vez al día; o 600 mg 2 veces al día en combinación con ritonavir 100 mg 2 J veces al día. - En niños de 4-12 años con peso inferior a 50 kg: 20 mg/kg 2 veces al día o 15 mg 3 veces diarias, si se usa en cápsulas; y 22,5 mg/kg 2 veces al día o 17 mg/kg 3 veces al día, si se usa solución. En niños de 13 años en adelante y peso superior a 50 kg: 1400 mg 2 veces al día, en solución. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, diarrea, dolor abdominal y trastornos del gusto. Hematológicas: Anemia hemolítica aguda. Neurológicas: Parestesias, depresión, trastornos de la conducta. Reacciones de hipersensibilidad: Erupción maculopapular, síndrome de Stevens-Johnson severo 2ed. 2004 * Formulario TerapÈutico Nacional 255 traciones plasmáticas y la incidencia de efectos adversos del ketoconazol, rifabutina, dapsona, warfarina, terfenadina, astemizol, inhibidores de la HMG-CoA reductasa, carbamazepina, bloqueantes de canales de calcio, dihidropiridinas, clozapina, pimozida, midazolam, triazolam, alprazolam, clorazepato, diazepam, flunazepam, alcaloides del ergot y derivados, ciclosporina, amiodarona, cisaprida y sildenafil. La combinación con claritromicina puede producir un aumento en los niveles séricos de ambos agentes. El amprenavir reduce la eficacia de los anticonceptivos orales. El fenobarbital, la fenitoína, la carbamazepina, el saquinavir, la rifampicina, la dexametasona y la hierba de San Juan (Hypericum perforatum) pueden reducir los niveles plasmáticos del amprenavir y comprometer su eficacia terapéutica. El ritonavir los aumenta. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y a otras sulfonamidas. Dado que la solución oral contiene una elevada proporción de propilénglicol en su formulación, su uso está contraindicado en niños menores de 4 años, en mujeres embarazadas y en pacientes con disfunción hepática y/o renal. Si se inicia tratamiento con esta formulación 256 lactancia durante el tratamiento. Pueden ocurrir interacciones serias y/o con riesgo de la vida entre amprenavir y amiodarona, lidocaína (sistémica), antidepresivos tricíclicos y quinidina. Se recomienda el control de los niveles plasmáticos de estos medicamentos. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Evitar la administración con comidas de alto contenido graso. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previamente al médico. El paciente debe informar al médico si es alérgico a las sulfas. Notificar al médico de inDELAVIRDINA FARMACOLOGÍA Agente anti-retroviral no nucleósido. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) al interferir con la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal alcanzando concen- Formulario Terapéutico Nacional * 2ed. 2004 nutos y una respuesta terapéutica que se evidencia a las 2 semanas de iniciado el tratamiento. Los alimentos no alteran la biodisponibilidad del fármaco. Se une a las proteínas plasmáticas en un 98-99%. Se metaboliza extensa-mente en el hígado a través del siste-ma microsomal del citocromo P450, por las isoenzimas CYP3A y CYP2D6, transformándose en productos inactivos que se excretan en un 44% por las heces y en un 51% por la orina, 5% como fármaco intacto. La vida media de eliminación es de 2-11 horas (promedio 5,8 horas). INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros antiretrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos y niños mayores de 16 años: 400 mg 3 veces al día. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, palpitaciones, hipotensión ortostática, síncope, taquicardia, vasodilatación. Dermatológicas: Alopecia, vasculitis leucocitoblástica, dermatitis, descamación, diaforesis, piel seca, alteraciones en las uñas, seborrea, nódulos en la piel. Gastrointestinales: Cólicos ab- nal, gingivitis, salivación, náusea, hepatitis no específica, pancreatitis, ede-ma o ulceración de la lengua, vómitos. Hematológicas: Anemia, equimosis, eosinofilia, granulocitosis, neutrope-nia, pancitopenia, petequias, PTT pro-longado, púrpura, trombocitopenia, trastornos del bazo. Hepáticas: Au-mento de los niveles de las transami-nasas. Neurológicas: Incoordinación, agitación, amnesia, ansiedad, al-teración cognitiva, confusión, depre-sión, desorientación, mareos, labilidad emocional, alucinaciones, cefalea, hiperestesia, hiperreflexia, insomnio, síntomas maníacos, migraña, ner- J viosismo, neuropatía, pesadillas, parálisis, síntomas paranoides, parestesia, intranquilidad, somnolencia, temblor, vértigo, debilidad. Reacciones de hipersensibilidad: Reacciones alérgicas, angioedema, eritema multiforme, erupción maculopapular, erupción petequial, erupción vesí-culobulosa, síndrome de Stevens-Johnson, quistes epidérmicos. Res-piratorias: Bronquitis, tos, laringitis, infección del tracto respiratorio su-perior. Otras: Blefaritis, conjuntivitis, diplopía, sequedad de los ojos, dolor de oído, epistaxis, nistagmo, faringitis, fotofobia, rinitis, sinusitis, perversión del gusto, tinitus, alopecia, artralgia, mialgia, astenia, dolor de espalda, 2ed. 2004 * Formulario TerapÈutico Nacional 257 INTERACCIONES Los antiácidos reducen la absorción gastrointestinal de la delavirdina. Puede incrementar las concentraciones plasmáticas y la incidencia de efectos adversos de saquinavir, ritonavir, nelfinavir, warfarina, inhibidores de la HMG-CoA reductasa, astemizol, terfenadina, claritromicina, dapsona, indinavir, rifabutin, alprazolam, midazolam, triazolam, bloqueantes de los canales de calcio, dihidropiridinas, alcaloides del ergot y derivados, cisaprida y sildenafil. La rifabutina, rifampicina, carbamazepina, fenobarbital, fenitoína y la hierba de San Juan (Hypericum perforatum) pueden reducir los niveles séricos de la delavirdina y comprometer su eficacia terapéutica. La fluoxetina y el fluconazol los incrementan. La administración conjunta de didanosina y delavirdina resulta en una reducción del 20% en el área bajo la curva de ambos medicamentos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática y en el embarazo. Ajustar la dosis en pacientes con falla hepá-tica. En lo posible, se debe evitar la lac-tancia. Resistencia cruzada con otros inhibidores de la transcriptasa reversa. 258 frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previamente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial de tipo dermatológico. Este medicamento no DIDANOSINA FARMACOLOGÍA Agente anti-retroviral análogo de los nucleósidos. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) al interferir con la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA La absorción gastrointestinal es variable y dependiente de la formulación que se emplee, del pH gástrico y de la presencia de alimentos. Por lo general, se ubica entre 21 y 43%. Se une a las proteínas plasmáticas en un 5%. Dada su naturaleza química (análogo estructural de la inosina), el metabolismo se presume similar al de las purinas endógenas; se transforma intracelularmente en productos activos e inactivos que se excretan en un 20% por la orina. La vida media de eliminación es de 1,3-1,5 horas. INDICACIONES En combinación con otros anti-retro- Formulario Terapéutico Nacional * 2ed. 2004 RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos con peso superior a 60 kg: 200 mg 2 veces al día o una dosis única de 400 mg/día; adultos con peso inferior a 60 kg: 125 mg 2 veces al día o una dosis única de 250 mg/día. En niños mayores de 8 meses: 120 mg/m2 2 veces al día; en niños de 2 semanas-8 meses: 100 mg/m2 2 veces al día. REACCIONES ADVERSAS Cardiovasculares: Hipertensión, edema, insuficiencia cardiaca. Gastrointestinales: Diarrea, náusea, vómitos, dolor abdominal, pancreatitis, boca seca, anorexia. Hematológicas: Leucopenia, anemia, granulocitosis, trombocitopenia. Hepáticas: Insuficiencia hepática, aumento de los niveles de las enzimas hepáticas. Reacciones de hipersensibilidad: Erupción, prurito, reacciones alérgicas. Respiratorias: Disnea, neumonía. Neurológicas: Cefalea, convulsiones, confusión, ansiedad, nerviosismo, pensamientos anormales, depresión, neuropatía periféri-ca, neuritis óptica, mareos, insomnio. Otras: Cambios retinianos, alopecia, astenia, dolor, infección, sarcoma, hi-peruricemia, escalofrío, fiebre, redistri-bución anormal de grasa corporal. INTERACCIONES El alopurinol y el ganciclovir aumentan absorción gastrointestinal del itraconazol, dapsona, ketoconazol, amprenavir, quinolonas y tetraciclinas. Cuando se administra con antiácidos, aumentan los efectos adversos de los componentes del antiácido (diarrea, constipación). CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática y/o renal, hiperuricemia, trastornos retinianos o neuritis óptica, diabetes mellitus, hemofilia, antecedentes de neuropatía periférica, histo-ria de pancreatitis y en el J embarazo. Durante el tratamiento, vigilar periódi-camente la función renal, la hepática, la hematológica y la glicemia. En pa-cientes con falla renal, ajustar la dosis. En lo posible, se debe evitar la lactan-cia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Es removible parcialmente por hemodiálisis, pero no por diálisis peritoneal. INFORMACIÓN AL PACIENTE Administrar 1 hora antes o 2 horas después de las comidas. No se debe modificar la dosis ni la frecuencia de 2ed. 2004 * Formulario TerapÈutico Nacional 259 EFAVIRENZ FARMACOLOGÍA Agente anti-retroviral no nucleósido. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH1) al interferir con la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA Se absorbe bien por vía oral, alcanzando niveles plasmáticos pico en 5 horas, concentraciones al estado estable a los 6-7 días y una respuesta terapéutica que se evidencia a las 2 semanas de iniciado el tratamiento. Los alimentos aumentan la biodisponibilidad del producto. Se une a las proteínas plasmáticas en un 99%. Se metaboliza a través del sistema microsomal hepático del citocromo P450, transformándose en productos inactivos que se excretan en un 16-61% por las heces y 14-34% por la orina. El efavirenz es capaz de inducir su propio metabolismo. Dado que el fármaco puede inducir su propio metabolismo, la vida media de eliminación, tras la administración de una dosis única, es de 52-76 horas y de 40-55 horas con la administración de dosis múltiples. INDICACIONES Para el tratamiento de la infección por virus de inmunodeficiencia humano en combinación con otros agentes antiretrovirales. 260 mg/día; de 15-<20 kg: 250 mg/día; de 20-<25 mg: 300 mg/día; de 25<32,5 kg: 350 mg/día; de 32,5-<40 kg: 400 mg/día y en niños de peso > o = a 40 kg: administrar la dosis de adultos: 600 mg diarios. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, rubor. Gastrointestinales: Constipación, malabsorción. Hepáticas: Aumento de los niveles de las enzimas hepáticas, insuficiencia hepática, hepatitis. Neurológicas: Incoordinación, ataxia, convulsiones, hipoestesia, parestesia, neuropatía, temblor, reacciones agresivas, delirio, labilidad emocional, manía, neurosis, paranoia, psicosis, ideas suicidas. Reacciones de hipersensibi-lidad: Eritema multiforme, síndrome de Stevens-Johnson, reacciones alérgi-cas, erupciones de la piel asociadas con flictenas, descamación húmeda o ulceración. Renales: Cálculos renales, hematuria. Respiratorias: Disnea. Otras: Ginecomastia, artralgia, mial-gia, miopatía, alteración de las uñas, despigmentación de la piel, visión anormal, tinitus, astenia, redistribución/acumulación anormal de grasa corporal, sudoración. INTERACCIONES El efavirenz puede incrementar las concentraciones plasmáticas y la incidencia de efectos adversos del Formulario Terapéutico Nacional * 2ed. 2004 pueden reducir las concentraciones séricas del efavirenz y comprometer su eficacia terapéutica. Con lopinavir disminuyen los niveles de ambos agentes y con ritonavir aumentan. Puede alterar las concentraciones plasmáticas y los efectos de la warfarina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática e historia de trastornos psiquiátricos. Las pacientes deben evitar quedar embarazadas mientras usan este medicamento. El alcohol y los fármacos psicoactivos potencian los efectos sobre el SNC. Durante el tratamiento, se debe vigilar periódicamente los niveles de lípidos en sangre y la función hepática. En lo posible, se debe evitar la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Administrar con las comidas. Se debe evitar comidas con alto contenido de grasa. Se debe informar a los pacientes que este medicamento no es una cura para el SIDA y que pueden seguir mo de alcohol durante el tratamiento. No se debe usar otros medicamentos sin consultar previamente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Este medicamento no ESTAVUDINA (STAVUDINA) FARMACOLOGÍA Agente anti-retroviral nucleósido que es análogo de la timidina. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima transcriptasa reversa viral. J FARMACOCINÉTICA Se absorbe en un 80-86% en el tracto gastrointestinal alcanzando concentraciones plasmáticas pico dentro de 1 hora y la respuesta terapéutica se evidencia a las 4-6 semanas de iniciado el tratamiento. Los alimentos no alteran la biodisponibilidad del medicamento. La unión a las proteínas plasmáticas es insignificante. El metabolismo no ha sido bien caracterizado. Se cree que la estavudina es fosforilada intracelularmente y convertida en la forma activa de trifosfato. Se excreta en un 40% por vía renal como fármaco intacto. La vida media de eliminación intracelular de la forma activa es de 3 horas, mientras que la vida media plasmática 2ed. 2004 * Formulario TerapÈutico Nacional 261 combinación con otros agentes antiretrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - En adultos con peso superior a 60 kg: 40 mg 2 veces al día; con peso inferior a 60 kg: 30 mg 2 veces al día. - En neonatos, desde el nacimiento hasta el día 13: 0,5 mg/kg 2 veces al día; a partir del día 14 y en niños con peso inferior a 30 kg: 1 mg/kg 2 veces al día; en niños con peso superior a 30 kg: administrar la dosis de adultos. la fosforilación intracelular de la estavudina, disminuyendo su eficacia terapéutica. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con falla hepática y/o renal, historia de neuropatía y en el embarazo. Debido a la aparición de acidosis láctica y hepatomegalia severa con estavudina o en combinación con otros agentes antiretrovirales, se debe usar en el embarazo solamente si el beneficio potencial supera los riesgos para el feto. Ajustar la dosis en pacientes con falla renal. En lo posible, se debe evitar la lactancia. REACCIONES ADVERSAS Cardiovasculares: Dolor en el pecho. Gastrointestinales: Dolor abdominal, diarrea, náusea, vómitos, anorexia, dispepsia, constipación, pancreatitis. SOBREDOSIS Hematológicas: Neutropenia, trombo- Vaciamiento gástrico, carbón activado citopenia, anemia. Hepáticas: Hepato- y catártico salino, si la ingestión es toxicidad. Reacciones de hipersensibi- reciente. Manejo sintomático y tratalidad: Erupción maculopapular. Neu- miento de soporte. Es removible por rológicas: Neuropatía periférica, hemodiálisis. cefa-lea, insomnio, ansiedad, depresión, nerviosismo, mareos. INFORMACIÓN AL PACIENTE Se debe informar al paciente la Otras: Malestar, pérdida de peso, importancia de reconocer los signos sudoración, prurito, mialgia, tempranos de acidosis láctica (maescalofrío, fiebre, astenia, do-lor de lestar abdominal, náusea, vómitos, faespalda, artralgia, disnea, contiga, disnea y debilidad motora), en juntivitis, debilidad motora severa (en presencia de los cuales se debe notipresencia de acidosis láctica), ficar al médico inmediatamente. Tamredistribución anormal de grasa corbién se les debe advertir que la neuroporal. 262 Formulario Terapéutico Nacional * 2ed. 2004 médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Este medicamento no previene la GANCICLOVIR FARMACOLOGÍA Agente antiviral sintético análogo estructural de la guanina. Interfiere con la síntesis de ADN viral mediante la inhibición de la ADN polimerasa, e impide la replicación viral al incorporarse como falso nucleótido en las cadenas de ADN viral en formación. FARMACOCINÉTICA Se absorbe pobremente en el tracto gastrointestinal, 5-9%. Alcanza concentraciones séricas pico a las 4,515,6 horas de su administración oral y 1 hora después de su inyección IV. Se une en 1-2% a las proteínas plasmáticas y se distribuye en cantidades significativas en el riñón y menores cantidades en el cerebro, hígado, pulmón, ojos y testículos. No sufre metabolismo apreciable, por lo que se excreta en su totalidad como fármaco intacto. Posterior a la administración oral, se excreta en un 86% por las heces y 5% por la orina; después de la administración IV, la excreción es casi exclusivamente (94-100%) renal. La vida media de eliminación es de 2,5-5 horas y se incrementa en pacientes con falla renal. órganos por transplante. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral e IV. La dosis oral se debe administrar con las co-midas y la dosis IV por infusión lenta a velocidad constante durante 1 hora. Las dosis usuales en adultos y niños son: - Para el tratamiento de la infección por CMV: Dosis IV de inducción de 5 mg/kg cada 12 horas por 14-21 días, seguido por una dosis IV de mantenimiento de 5 mg/kg/día o, alternativamente, una dosis oral de 1 g cada 8 horas. - Para la prevención de la infección por CMV en receptores de órganos J por transplante: 5 mg/kg IV cada 12 horas por 7-14 días, seguido por una dosis IV de mantenimiento de 5 mg/kg/día o, alternativamente, una dosis oral de 1 g cada 8 horas. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, arritmias cardíacas, alteraciones de la presión arterial. Gastrointestinales: Náuseas, vómito, diarrea, dolor abdominal. Hematológicas: Hemorragia, anemia, neutropenia, eosinofilia, trombocitopenia, pancitopenia. Hepáticas: Aumento de los niveles de las enzimas hepáticas. Neurológicas: Confusión, mareo, cefalea, agitación, nerviosismo, sueños anormales, pesadillas, 2ed. 2004 * Formulario TerapÈutico Nacional 263 HAD. Otras: Desprendimiento de la retina en pacientes con retinitis por citomegalovirus, fiebre, edema, mialgia, artralgia, escalofrío, sudoración, hipoglicemia, disnea, inhibición temporal o permanente de la espermatogénesis en varones, supresión de la fertilidad femenina, sordera, alopecia. INTERACCIONES El ganciclovir puede incrementar los niveles séricos de didanosina. Con zidovudina se incrementa el riesgo de toxicidad hematológica. Su combinación con imipenem aumenta la posibilidad de convulsiones. Con medicamentos potencialmente nefrotóxicos se incrementa el riesgo de reacciones adversas a nivel renal. Incrementa la factibilidad de mielosupresión al usarse en combinación con amfotericina B, dapsona, pentamidina, pirimetamina, flucitosina, vincristina, vinblastina, doxorubicina, trimetoprimsulfametoxazol y agentes inmunosupresores. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o al aciclovir y durante el embarazo. Usar con precaución en pacientes con citopenias preexistentes o historia de reacciones citopénicas a otros medicamentos u otros inductores, en pacientes con disfunción renal y en 264 lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Prestar especial atención a la posibilidad de neutropenia y disfunción renal. El fármaco es removible por hemodiálisis. INFORMACIÓN AL PACIENTE Administrar la dosis oral con las comidas. Durante el tratamiento se debe mantener una elevada ingesta de líquidos. A objeto de evitar el embarazo durante la terapia, se recomienda usar métodos INDINAVIR FARMACOLOGÍA Agente anti-retroviral. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima proteasa viral. La proteasa de VIH-1 es una enzima requerida para la descomposición proteolítica de los precursores poliproteicos virales en las proteínas individuales. La unión del indinavir al sitio activo de la enzima, inhibe su actividad y se produce la formación de partículas virales no in-fecciosas inmaduras. FARMACOCIN…TICA Se absorbe rápida, aunque par- Formulario Terapéutico Nacional * 2ed. 2004 do el tratamiento. Los alimentos reducen la biodisponibilidad. Se une a las proteínas plasmáticas en un 60%. Se metaboliza en el hígado a través del sistema microsomal del citocromo P450 por la isoenzima CYP3A4 y por glucuronidación, transformándose en siete productos inactivos que se excretan en un porcentaje no mayor de 20% por la orina (la mitad como fármaco intacto), y en un 83% por las heces. La vida media de eliminación es 1,8 horas. INDICACIONES Para el tratamiento de la infección por virus de inmunodeficiencia humano en combinación con otros agentes antiretrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos: 800 mg cada 8 horas. En niños de 4-17 años: 500 mg/m2 cada 8 horas, hasta un máximo de 800 mg por dosis. Debe ser administrado sin comida pero con agua 1 hora antes o 2 horas después de las comidas. REACCIONES ADVERSAS Cardiovasculares: Dolor torácico, palpitaciones. Gastrointestinales: Perversión del gusto, dolor abdominal, náusea, vómitos, diarrea, regurgitación, anorexia, boca seca. Dermatológicas: Erupciones, hiperpigmentación, alopecia. Hematológicas: Anemia, trombocitopenia, leucopenia, anemia he- maturia, nefrolitiasis, cristaluria, nefritis intersticial, insuficiencia renal. Respiratorias: Tos, disnea. Otras: Visión borrosa, dolor o hinchazón de los ojos, hiperbilirrubinemia, hiperglicemia, dolor en los flancos, malestar, dolor de espalda, aumento de los niveles séricos de la amilasa, redistribución anormal de grasa corporal. INTERACCIONES El indinavir puede incrementar las concentraciones plasmáticas y la incidencia de reacciones adversas de la rifabutina, pimozida, bloqueantes de los canales de calcio, terfenadina, astemizol, cisaprida, triazolam, midazoJ lam, alcaloides del ergot y derivados, inhibidores de la HMG-CoA reductasa, saquinavir y del sildenafil. Con nelfinavir aumentan los niveles de ambos agentes. El fenobarbital, fenitoína, car-bamazepina, dexametasona, didano-sina, efavirenz, rifampicina, rifabutina, nevirapina y la hierba de San Juan (Hy-pericum perforatum) disminuyen los niveles séricos y la eficacia terapéutica del indinavir; la zidovudina, ritonavir, delavirdina, itraconazol, ketoconazol claritromicina y la quinidina los au-mentan. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al fármaco. Usar con precaución en pacientes con disfunción 2ed. 2004 * Formulario TerapÈutico Nacional 265 SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Administrar 1 hora antes o 2 horas después de las comidas. Se recomienda ingerir abundante líquido (mínimo 1,5 litros diarios) durante el tratamiento. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previa-mente con el médico. Notificar al médico, de inmediato, si se LAMIVUDINA FARMACOLOGÍA Agente anti-retroviral análogo de nucleósidos sintéticos. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima transcriptasa reversa viral vía la terminación de la cadena del ADN viral. Intracelularmente, se fosforila a su metabolito activo 5'-trifosfato de lamivudina (L-TP), el cual también inhibe la actividad de la ADN polimerasa dependiente de ARN y ADN de la transcriptasa reversa. FARMACOCINÉTICA 266 manas de iniciado el tratamiento. Los alimentos no alteran la biodisponibilidad. Se une a las proteínas plasmáticas en un porcentaje inferior al 36%. La cantidad unida a los eritrocitos está alrededor de 53-57%. Se metaboliza poco en el hígado y se excreta principalmente por vía renal, en un 70% como fármaco intacto. La vida media de eliminación es de 3-7 horas y se incrementa en pacientes con falla renal. INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humano en combinación con otros agentes anti-retrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual para adultos con peso de 50 kg o más y niños de 12 a 16 años: 150 mg 2 veces al día. Adultos con peso menor de 50 kg: 2 mg/kg dos veces al día. En niños de 3 meses-12 años: 4 mg/kg 2 veces al día, hasta un máximo de 150 mg 2 veces al día. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos, anorexia, dolor abdominal, cólicos abdominales, dispepsia, pancreatitis (niños de 3 a 12 años). Hematológicas: Neutropenia, anemia, trombocitopenia. Hepáticas: Aumento de las enzimas hepáticas y de la bilirrubina, he- Formulario Terapéutico Nacional * 2ed. 2004 tomas nasales, malestar, fiebre, escalofríos, dolor musculoesquelético, mialgia, artralgia, rabdomiolisis, hiperglicemia, acidosis láctica, esplenomegalia, redistribución anormal de grasa corporal. INTERACCIONES La lamivudina puede antagonizar la actividad antiviral de la zalcitabina. Con la ribavirina se potencia la posibilidad de acidosis láctica. Trimetoprima-sulfametoxazol puede aumentar las concentraciones séricas de la lamivudina. Aumenta las concentraciones plasmáticas de zidovudina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática, diabetes y en el embara-zo. Ajustar la dosis en pacientes con insuficiencia renal. Precaución en ni-ños con historia de pancreatitis. Durante el tratamiento se debe vigilar periódicamente la función hepática y hematológica. En lo posible, se debe evitar la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE experimentando enfermedades asociadas con la misma, incluyendo las infecciones oportunísticas. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento. A los padres de pacientes pediátricos se les debe aconsejar controlarlos por signos y síntomas de pancreatitis. Este NELFINAVIR FARMACOLOGÍA Agente anti-retroviral. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la J enzima proteasa viral. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal alcanzando concentraciones plasmáticas pico en 2-4 horas. Los alimentos aumentan su biodisponibilidad. Se une a las proteínas plasmáticas en un 98-99%. Se metaboliza extensamente en el hígado a través del sistema microsomal he-pático del citocromo P450, particu-larmente la isoenzima CYP3A, transformándose en productos activos e inactivos. El principal metabolito oxidativo tiene actividad similar a la del compuesto padre in vitro. Se excreta en un 87% por las heces (78% como metabolitos y 2ed. 2004 * Formulario TerapÈutico Nacional 267 INDICACIONES Para el tratamiento de la infección por virus de inmunodeficiencia humano en combinación con otros agentes antiretrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos: 750 mg cada 8 horas o 1250 mg cada 12 horas. En niños de 213 años: 20-30 mg/kg cada 8 horas. REACCIONES ADVERSAS Dermatológicas: Dermatitis, foliculitis, dermatitis fúngica, sudoración, urticaria, prurito. Gastrointestinales: Náusea, diarrea, flatulencia, anorexia, dispepsia, dolor epigástrico, sangrado gastrointestinal, pancreatitis, ulceraciones de la boca, vómitos. Hematológicas: Anemia, leucopenia. Hepáticas: Hepatitis, alteración de las pruebas de función hepática. Neurológicas: Ansie-dad, depresión, mareos, labilidad emocional, hiperquinesia, insomnio, migraña, cefalea, parestesia, con-vulsiones, trastornos del sueño, som-nolencia, ideas suicidas. Reacciones de hipersensibilidad: Erupciones, reacciones alérgicas, dermatitis. Renales: Cálculos renales, anormalidad urinaria. Otras: Iritis, trastornos de los ojos, fiebre, malestar, disfunción sexual, dolor de espalda, artralgia, artritis, calambres, mialgia, miastenia, miopatía, deshidratación, hiper268 cadamente las concentraciones séricas y la incidencia de efectos adversos de la rifabutina, terfenadina, astemizol, cisaprida, saquinavir, amiodarona, quinidina, inhibidores de la HMG-CoA reductasa, midazolam, triazolam, ciclosporina, sildenafil y de los alcaloides del ergot y sus derivados, aumentando su toxicidad. Con indinavir, aumentan los niveles de ambos agentes. La rifabutina, rifampicina, carbamazepina, fenobarbital y la fenitoína pueden disminuir las concentraciones de nelfinavir y comprometer su eficacia terapéutica; el ketoconazol, ritonavir y el efavirenz los aumentan. El nelfinavir puede reducir la eficacia de los anticonceptivos orales y disminuir las concentraciones de amprenavir y metadona. La combinación con delavirdina aumenta el riesgo de neutropenia. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con falla hepática, historia de pancreatitis, hemofilia, diabetes mellitus y trastornos lipídicos. En el embarazo se puede usar sólo si es claramente necesario. Durante el tratamiento se debe vigilar periódicamente la función hepática, la hematológica y el perfil lipídico. Ajustar la dosis en pacientes con falla Formulario Terapéutico Nacional * 2ed. 2004 midazolam, triazolam). En lo posible se debe evitar la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Administrar con las comidas. Se debe informar a los pacientes que este medicamento no es una cura para el SIDA y que pueden seguir desarrollando complicaciones asociadas a la enfermedad, incluyendo las infecciones oportunísticas. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previamente al médico. Notificar al médico de NEVIRAPINA FARMACOLOGÍA Agente anti-retroviral no nucleósido. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH1) al interferir con la actividad de la enzima transcriptasa reversa viral. Bloquea la actividad de la polimerasa dependiente de ARN y ADN por alteraciones en el sitio catalítico de la enzima. FARMACOCINÉTICA puesta terapéutica que se evidencia a los 7 días de iniciado el tratamiento. Los alimentos no alteran su biodisponibilidad. Se une a las proteínas plasmáticas en un 50-60%. Se metaboliza a través del sistema microsomal hepático del citocromo P450, transfor-mándose en productos inactivos que se excretan principalmente por la orina. Dado que la nevirapina puede inducir su propio metabolismo, la vida media de eliminación, tras la ad-ministración de una dosis única, es de 45 horas y de 25-30 horas con la administración de dosis múltiples. Induce las isoenzimas CYP3A4 y la CYP2B6. J INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humano en combinación con otros agentes anti-retrovirales. Es usada también para prevenir la transmisión perinatal del VIH durante el parto. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos: 200 mg/día por 14 días, seguido por dosis de 200 mg 2 veces diarias. En niños menores de 8 años: 4 mg/kg diarios por 14 días, seguido por 7 mg/kg 2 veces diarias; en niños mayores de 8 años: 4 mg/kg diarios por 14 días, seguido por 4 mg/kg 2 veces diarias. Para reducir la transmisión perinatal: Dosis única de 2ed. 2004 * Formulario TerapÈutico Nacional 269 tiva. Hematológicas: Neutropenia, anemia. Hepáticas: Hepatitis, aumento de los niveles de AST, ALT, gammaglutamil transpeptidasa y de los niveles de bilirrubina total. Reacciones de hipersensibilidad: Erupciones, síndrome de Stevens-Johnson, forma-ción de ampollas, anafilaxis. Neuroló-gicas: Cefalea, parestesia. Otras: Fiebre, mialgia, redistribución y acumulación anormal de la grasa corporal. INTERACCIONES La nevirapina puede disminuir las concentraciones plasmáticas de lopinavir, ketoconazol y metadona. Además, puede disminuir las concentraciones plasmáticas de los fármacos extensamente metabolizados por la enzima CYP3A. Puede reducir la efectividad de los anticonceptivos orales y de otros inhibidores de la proteasa. La rifabutina, la rifampicina y la hierba de San Juan (Hypericum perforatum) pueden disminuir los niveles séricos de nevirapina y comprometer su eficacia terapéutica; la cimetidina y los macrólidos, por el contrario, los aumentan. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al fármaco. Usar con precaución en pacientes con disfunción hepática y/o renal, historia de síndrome 270 tratamiento por 14 días, seguido por la dosis usual 2 veces diarias. En caso de reaparición de la erupción o si se presenta hepatitis, descontinuar definitivamente el medicamento. Durante el tratamiento se debe vigilar periódicamente la función hepática. En lo posible se debe evitar la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Es removible por diálisis peritoneal. INFORMACIÓN AL PACIENTE Se debe informar a los pacientes que este medicamento no es una cura para el SIDA y que pueden seguir desarrollando complicaciones asociadas a la enfermedad, incluyendo las infecciones oportunísticas. No modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previaRITONAVIR FARMACOLOGÍA Agente anti-retroviral. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de las enzimas proteasas virales. La inhibición de la proteasa de VIH hace que Formulario Terapéutico Nacional * 2ed. 2004 precursor de poliproteína gag-pol, lo cual lleva a la producción de partículas virales inmaduras no infecciosas. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal. Alcanza concentraciones pico en 2-4 horas y genera una respuesta terapéutica que se evidencia a los 7-14 días de iniciado el tratamiento. Los alimentos aumentan su biodisponibilidad. Se une a las proteínas plasmáticas en un 98-99%. Se metaboliza en el hígado a través del sistema microsomal del citocromo P450 (CYP3A y CYP2D6), transformándose en productos activos e inactivos que se excretan en un 11,3% por vía renal (3,5% como fármaco intacto) y en 86,4% por las heces. La vida media de eliminación es de 3-5 horas. INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes anti-retrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis para adultos: Iniciar con 300 mg 2 veces al día e incrementar 100 mg 2 veces al día cada 2-3 días hasta alcanzar la dosis de mantenimiento de 600 mg 2 veces al día. Dosis para niños anorexia, constipación, diarrea, náusea, vómitos, dispepsia, flatulencia, cólicos. Genitourinarias: Disuria, hematuria, nocturia, poliuria, pielonefritis, uretritis hiperuricémica. Hematológicas: Disminución de los niveles de hemoglobina y del hematocrito, leucopenia, trombocitopenia, disminución del contaje de neutrófilos y eosinófilos, alteración del tiempo de protrombina (PT) y del INR. Hepáticas: Ictericia, aumento de la AST, ALT, GGT, fosfa-tasa alcalina, bilirrubina total. Neuro-lógicas: Astenia, cefalea, malestar, neuropatía periférica, mareos, in-somnio, somnolencia, pensamientos anormales, migraña. J Reacciones de hipersensibilidad: Erupciones, urtica-ria. Otras: Hiperlipidemia, hiperglice-mia, hiperkalemia, aumento de los triglicéridos, aumento de los niveles de CK, mialgia, sudoración, fiebre, irritación de la garganta, blefaritis, diplopía, fotofobia, perversión del gusto, redistribución anormal de grasa corporal. INTERACCIONES El ritonavir puede disminuir los niveles plasmáticos y la eficacia terapéutica de la warfarina, la metadona, la lamotrigina, la fenitoína y la teofilina. Puede incrementar las concentraciones y la incidencia de efectos adversos de la quinina, rifabutina, ampre- 2ed. 2004 * Formulario TerapÈutico Nacional 271 fetaminas, pimozida, antidepresivos tricíclicos, bupropión, nefazodona, tio-ridazina, perfenazina, risperidona, mi-dazolam, triazolam, alcaloides del er-got y derivados, cisaprida, corticosteroides, ciclosporina y del sildenafil. Con la claritromicina o con efavirenz pueden aumentar los niveles séricos de éstos y del ritonavir. El fluconazol, el ketoconazol y la fluoxetina producen aumentos en las concentraciones séri-cas del ritonavir; la rifampicina y la hierba de San Juan (Hypericum perforatum) las disminuyen. El ritonavir reduce la eficacia de los anticonceptivos orales. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con falla hepática, historia de pancreatitis, hemofilia, diabetes mellitus, trastornos lipídicos y en el embarazo. Durante el tratamiento se debe vigilar periódicamente la función hepática, la hematología y el perfil lipídico. En lo posible, se debe evitar la lactancia durante el tratamiento. El ritonavir está contraindicado con antiarrítmicos, antihistamínicos, derivados del ergot, cisaprida, pimozida, midazolam y triazolam. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es 272 y que pueden seguir desarrollando complicaciones asociadas a la enfermedad, inclusive las infecciones oportunísticas. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previamente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual duRITONAVIR / LOPINAVIR FARMACOLOGÍA Combinación, a dosis fijas, de agentes anti-retrovirales inhibidores de la actividad de proteasa del virus de inmunodeficiencia humano, previene la descomposición de la poliproteína gagpol, lo que resulta en la producción de partículas virales inmaduras, no infecciosas. El ritonavir incrementa los niveles plasmáticos del lopinavir al inhibir la isoenzima CYP3A, enzima metabolizante del lopinavir. FARMACOCINÉTICA El lopinavir exhibe una biodisponibilidad oral muy pobre como consecuencia de un intenso y rápido efecto metabólico a través del sistema microsomal del citocromo P450. Cuando se combina con una dosis baja de ritonavir, se produce una marcada inhibición del metabolismo del Formulario Terapéutico Nacional * 2ed. 2004 respuesta terapéutica a las 3 semanas de iniciado el tratamiento. Así, la pre-sencia del ritonavir en la combinación no responde propiamente a su ac-tividad antiviral sino a su efecto in-hibidor del metabolismo del lopinavir. Los alimentos aumentan la biodisponibilidad. Se une a las proteínas plasmáticas en un 98-99%. Se metaboliza en el hígado, transformándose en productos activos e inactivos que se excretan en la orina y en las heces, un 3% como fármaco intacto. La vida media de eliminación del lopinavir es de 5-6 horas. INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes anti-retrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - En adultos: 400 mg de lopinavir más 100 mg de ritonavir (3 cápsulas o 5 ml) 2 veces al día. - En niños con peso de 7-15 kg: 12 mg/kg de lopinavir más 3 mg/kg de ritonavir 2 veces al día. - En niños con peso de 15-40 kg: 10 mg/kg de lopinavir más 2,5 mg/kg de ritonavir 2 veces al día. nio, somnolencia, cefalea. Otras: Hiperglicemia, hipertrigliceridemia, hipercolesterolemia, redistribución anor-mal de grasa corporal, escalofrío. INTERACCIONES Las mismas descritas para el ritonavir (ver: RITONAVIR). CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a alguno de los constituyentes del medicamento. Usar con precaución en pacientes con falla hepática, historia de pancreatitis, hemofilia, diabetes mellitus, trastornos lipídicos y J en el embarazo. Durante el tratamiento se debe vigilar periódicamente la función hepática, hematológica y el perfil li-pídico. En lo posible, se debe evitar la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Administrar con las comidas. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. Notificar al médico de REACCIONES ADVERSAS inmediato si se presenta alguna reacGastrointestinales: Náuseas, vómitos, ción o sintomatología inusual durante 273 2ed. 2004 * Formulario TerapÈutico Nacional SAQUINAVIR FARMACOLOGÍA Agente anti-retroviral. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH-1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima proteasa viral. FARMACOCINÉTICA Después de la administración por vía oral, se absorbe muy poco como fármaco intacto (4% desde cápsulas rígidas y 3-4 veces superior desde cápsulas blandas) debido a un intenso efecto metabólico de primer pasaje. Los alimentos aumentan la biodisponibilidad del saquinavir. Produce una respuesta terapéutica que se evidencia a las 6 semanas de iniciado el tratamiento. Se une a las proteínas plasmáticas en un 98%. Se metaboliza en un 90% a nivel hepático del citocromo P450 (CYP3A), transformándose en productos inactivos que se excretan en un 81-88% en las heces y en 1-3% por la orina. La vida media de eliminación es de 13 horas. INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes anti-retrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual 274 eczema, fotosensibilidad. Gastrointestinales: Náusea, vómitos, dispepsia, malestar abdominal, hemorragia rectal, constipación, disfagia, gastritis, pancreatitis. Genitourinarias: Infecciones del tracto urinario, aumento del tamaño de la próstata, secreción vaginal. Hematológicas: Anemia, pancitopenia, trombocitopenia, microhemorragias. Neurológicas: Astenia, parestesia, neuropatía periférica, cefalea, mareos, confusión, convulsiones, depresión, irritabilidad, psicosis. Reac-ciones de hipersensibilidad: Anafilaxis. Respiratorias: Neumonía, bronquitis, tos, infecciones del tracto respiratorio superior. Otras: Hiperglicemia, pérdida o ganancia de peso, sudoración, deshidratación, pigmentación de los dientes, trastornos visuales, prurito, mialgia, artralgia, pérdida de la audición, redistribución anormal de la grasa corporal. INTERACCIONES El ketoconazol, indinavir, nelfinavir, ritonavir, delavirdina y el jugo de toronja incrementan los niveles plasmáticos del saquinavir. Los agentes que inducen la actividad de la CYP3A como la carbamazepina, la dexametasona, el fenobarbital, la fenitoína, la rifampicina, la rifabutina, el efavirenz, la nevirapina y la hierba de San Juan (Hypericum perforatum) disminuyen los Formulario Terapéutico Nacional * 2ed. 2004 de ambos medicamentos. Aumenta los niveles plasmáticos de las benzodia-zepinas, ergotamina, dihidroergotami-na, zolpidem (aumento de la probabili-dad de depresión respiratoria). También aumenta los niveles plasmáti-cos de la amiodarona, bupropión, cisa-prida, clozapina, encainida, flecainida, meperidina, piroxicam, quinidina, rifabutin. Disminuye la actividad de compuestos metabolizados por glucuronidación. Aumenta el riesgo de reacciones tipo antabuse. Disminuye los niveles séricos de los anticonceptivos orales. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. No se conoce la eficacia y seguridad de su uso en niños menores de 16 años. Usar con precaución en pacientes con falla hepática, hemofilia, diabetes y en el embarazo. Durante el tratamiento, vigilar periódicamente la función hepática y la hematología. En lo posible, se debe evitar la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previa-mente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual du-rante el tratamiento. Este ZALCITABINA FARMACOLOGÍA Agente anti-retroviral nucleósido. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima transcriptasa reversa viral. J FARMACOCINÉTICA Se absorbe en un 80-88% en el tracto gastrointestinal. Los alimentos reducen la biodisponibilidad. La unión a las proteínas plasmáticas es insignificante. Es convertida intracelularmente a su forma activa, trifosfato de zalcitabina. No sufre metabolismo hepático. Se excreta en un 62-75% por la orina en su mayoría como fármaco intacto y en un 10% por las heces. La vida media de eliminación es de 1-3 horas (promedio 2 horas). INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes an- 2ed. 2004 * Formulario TerapÈutico Nacional 275 niños: 0,015-0,040 mg/kg cada 6 horas. REACCIONES ADVERSAS Cardiovasculares: Miocardiopatía, insuficiencia cardiaca, dolor torácico, arritmias, hipertensión, palpitaciones. Gastrointestinales: Náusea, vómitos, diarrea, dolor abdominal, anorexia, constipación, estomatitis, úlcera esofágica, glositis, pancreatitis, úlceras orales. Hematológicas: Anemia, neutropenia, leucopenia, trombocitopenia. Hepáticas: Aumento de AST, ALT y de la fosfatasa alcalina, hepa-tomegalia severa con esteatosis, falla hepática, toxicidad hepática. Neuro-lógicas: Neuropatía periférica, cefalea, fatiga, mareos, confusión, convulsio-nes, alteración de la concentración, amnesia, insomnio, depresión, tem-blor, hipertonía, ansiedad, parestesia. Reacciones de hipersensibilidad: Erupción eritematosa, maculopapular o folicular. Respiratorias: Tos, secreción nasal. Otras: Faringitis, dolor ocular, visión anormal, ototoxicidad, mialgia, artralgia, fiebre, hipoglicemia, acidosis láctica, redistribución anormal de grasa corporal. INTERACCIONES Los antiácidos y la metoclopramida disminuyen la biodisponibilidad oral de la zalcitabina. La combinación con 276 ta el riesgo de acidosis láctica. La cimetidina y el probenecid interfieren con la depuración renal de la zalcitabina. No se recomienda el uso concomitante con lamivudina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con historia de pancreatitis, mielosupresión, miocardiopatía, falla hepática y/o renal, neuropatía periférica y en el embarazo. Si se desarrolla neuropatía o alguna otra reacción adversa severa durante el tratamiento, se debe interrumpir el tratamiento temporalmente hasta la resolución del cuadro y reiniciar con dosis de 0,375 mg cada 8 horas. En caso de reaparición de las manifestaciones adversas, descontinuar definitivamente el medicamento. Durante el tratamiento, se debe vigilar periódicamente la función renal, hepática y hematológica. Ajustar la dosis en pacientes con falla renal. Usar con precaución en el embarazo. En lo posible, se debe evitar la lactancia durante el tratamiento. La amfotericina B, el foscarnet y los aminoglicósidos aumentan el riesgo de neuropatía periférica. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es Formulario Terapéutico Nacional * 2ed. 2004 rrollando complicaciones asociadas a la enfermedad, incluyendo las infecciones oportunísticas. Se debe administrar 1 hora antes o 2 horas después de las comidas. No se debe modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previamente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Se le debe informar al paciente que la ZIDOVUDINA FARMACOLOGÍA Agente anti-retroviral del tipo de los nucleósidos, análogo de la timidina. Inhibe la replicación del virus de inmunodeficiencia humano tipo 1 (VIH1) y tipo 2 (VIH-2) al interferir con la actividad de la enzima transcriptasa reversa viral. En el interior de la célula es convertido a 5'-trifosfato de zidovudina (AztTP), metabolito activo, por la acción secuencial de las enzimas. AztTP también es un débil inhibidor de la alfa-polimerasa del ADN y de la gamma-polimerasa mitocondrial. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal, aunque sólo 52-75% de la dosis alcanza la circulación como plasmáticas en un porcentaje inferior al 38%. Se metaboliza extensamente en el hígado generando productos inactivos que se excretan, junto a un 15% de fármaco intacto, por la orina. La vida media de eliminación es de 0,5-3 horas y se incrementa en pacientes con falla renal. INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes anti-retrovirales. Es usada también pa-ra prevenir la transmisión del VIH materno-fetal durante la gestación y/o el parto. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral e IV. Dosis usual en adultos por vía oral: 200 mg 3 veces diarias o 300 mg 2 veces diarias. Para prevenir la transmisión maternofetal: Dosis oral de 100 mg 5 veces diarias desde la semana 14 de gestación hasta el inicio del parto; durante el trabajo de parto: 2 mg/kg por infusión IV en 1 hora, seguido por una infusión intravenosa continua de 1 mg/kg/hora hasta el pinzamiento del cordón umbilical. Dosis en neonatos por vía oral: 2 mg/kg cada 6 horas, comenzando a las 12 horas del nacimiento y continuando hasta la sexta semana. En casos de problemas o incapacidad para recibir la terapia oral: Los neonatos recibirán una 2ed. 2004 * Formulario TerapÈutico Nacional 277 J REACCIONES ADVERSAS Cardiovasculares: Miocardiopatía, insuficiencia cardiaca congestiva. Dermatológicas: Pigmentación de la piel, las uñas y la mucosa oral. Gastrointestinales: Náusea, vómitos, diarrea, dolor abdominal, dispepsia, constipación, pancreatitis, úlceras esofágicas. Hematológicas: Supresión severa de la médula ósea (resultante en anemia), agranulocitosis, trombocitopenia, leucopenia, pancitopenia. Hepáticas: Aumento de AST, ALT y de la fosfatasa alcalina, hepatomegalia severa con esteatosis, falla hepática. Reacciones de hipersensibilidad: Erupción, anafilaxis. Respiratorias: Tos, disnea. Neurológicas: Cefalea, fatiga, mareos, confusión, convulsiones, insomnio, somnolencia, vértigo, astenia, ansiedad, parestesia. Otras: Mialgia, rabdomiolisis, fiebre, perversión del gusto, acidosis láctica, ginecomastia, redistribución/acumulación anormal de grasa corporal. INTERACCIONES La zidovudina reduce los niveles séricos de pirazinamida. Inhibe la fosforilación intracelular de la estavudina, disminuyendo así su eficacia terapéu-tica. La administración con ácido val-próico o fenitoína puede provocar al-teración en los niveles plasmáticos tanto de la zidovudina como de los anticonvulsivantes. El 278 provocar incremento en los niveles séricos de tales agentes y/o de la zidovudina por inhibición competitiva de la glucuronidación. La combinación con medicamentos potencialmente mielosupresores o citotóxicos, como: amfotericina B, dapsona, doxorubicina, flucitosina, ganciclovir, interferón, pentamidina, vinblastina y vincristina incrementa el riesgo de toxicidad hematológica. El fluconazol, probenecid, co-trimoxazol y la metadona incrementan las concentraciones plasmáticas de zidovudina; el ritonavir, claritromicina, rifampicina, nevirapina y el nelfinavir las disminuyen. El dipiridamol y el molgramostim potencian su actividad antiviral. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con riesgo de desarrollo de acidosis láctica, con insuficiencia hepática y/o renal, mielosupresión y en el embarazo. Durante el tratamiento se debe vigilar periódica-mente la función renal, hepática y hematológica. Ajustar la dosis en pacientes con falla hepática y/o de-puración de creatinina menor de 15 ml/minuto. En lo posible, se debe evitar la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado Formulario Terapéutico Nacional * 2ed. 2004 el SIDA y que pueden seguir desarrollando complicaciones asociadas a la enfermedad, incluyendo las infecciones oportunísticas. No modificar la dosis ni la frecuencia de administración sin el conocimiento del médico. No usar otros medicamentos durante el tratamiento sin consultar previa-mente al médico. Notificar al médico de inmediato si se presenta alguna reacción o sintomatología ZIDOVUDINA / LAMIVUDINA FARMACOLOGÍA Combinación, a dosis fijas, de agentes anti-retrovirales nucleósidos inhibidores de la actividad de la enzima transcriptasa reversa viral. FARMACOCINÉTICA El comportamiento cinético de la combinación es el que cabe esperar de cada uno de los constituyentes por separado (ver las monografías individuales). INDICACIONES Para el tratamiento de la infección por el virus de inmunodeficiencia humana en combinación con otros agentes anti-retrovirales. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Dosis usual en adultos: 150 mg de lamivudina más 300 mg de zidovudina (1 tableta) 2 ve- uno de los constituyentes por separado (ver las monografías individuales). INTERACCIONES Las mismas que cabe esperar de cada uno de los constituyentes por separado (ver las monografías individuales). CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a alguno de los constituyentes del medicamento y en quienes requieren ajustes de dosificación (ver las monografías individuales). J SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. ANTITOXINA TETÁNICA (ANTITOXINA TET¡NICA HUMANA) FARMACOLOGÕA Solución estéril no pirogénica de anticuerpos (globulinas) purificados obtenidos de suero de caballos sanos hiperinmunizados con toxina o toxoide tetánico. Los anticuerpos neutralizan la exotoxina producida por el Clostridium tetani, organismo causante de la enfermedad. 2ed. 2004 * Formulario TerapÈutico Nacional 279 tección por aproximadamente 15 días. INDICACIONES Para proveer inmunidad pasiva contra el tétanos (profilaxis) en individuos con heridas factibles de generar la enfermedad y para tratamiento del tétano cuando no se dispone de la inmuno-globulina antitetánica. R…GIMEN DE DOSIFICACI”N La dosis profiláctica post-exposición en adultos y niños con peso mayor de 30 kg: 3.000-5.000 UI como dosis única por vía IM ó SC; en niños con peso menor de 30 kg: 1.500 UI IM ó SC. Para el tratamiento del tétanos: 50.000 a 100.000 UI, una parte por vía IV y la otra por vía IM. REACCIONES ADVERSAS Erupción y dolor en el sitio de la inyección, anafilaxis y enfermedad del suero, que puede presentarse hasta varias semanas después de la administración de la antitoxina con manifestaciones de urticaria, malestar general, linfadenopatía, nefritis, miocarditis, neuritis, broncoespasmo, poliartritis, uveitis, artralgia y fiebre. INTERACCIONES Aunque la antitoxina no interfiere con la respuesta inmune al toxoide tetánico, se debe tener cuidado (en caso de uso simultáneo) de no mezclar en 280 la terapia debe tenerse, a disposición inmediata, equipos para respiración asistida, epinefrina, antihistamínicos y esteroides. La preparación de antitoxina tetánica no debe ser usada si se dispone de inmunoglobulina humana antitetánica. Usar con precaución en pacientes con historia de asma o eczema infantil, o alérgico a otros medicamentos de este tipo. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación con el medicamento. INFORMACIÓN AL PACIENTE Advertir sobre la posibilidad de reacciones adversas hasta varios días después de la aplicación de la an-titoxina e indicar (si la naturaleza de INMUNOGLOBULINA ANTI-D (Rh) (INMUNOGLOBULINA Rho (D) HUMANA) FARMACOLOGÍA Contiene anticuerpos contra el antígeno Rho (D) de los glóbulos rojos. En personas Rho negativos expuestas a sangre Rho positiva suprime la respuesta inmune de formación de anticuerpos anti-Rho y posterior reacción antígeno-anticuerpo. Así, la administración post-parto a una mujer Rho (D) negativa cuyo infante es Rho (D) positivo, previene la formación de anticuerpos anti-Rho (D) que generarían he- Formulario Terapéutico Nacional * 2ed. 2004 previene la formación de anticuerpos anti-Rho (D) en personas Rho negativas que por error han recibido transfusión de sangre Rho positiva. El mecanismo a través del cual la inmunoglobulina Rho (D) humana ejerce dicho efecto no se conoce con precisión. FARMACOCINÉTICA La administración IM de la inmunoglobulina Rho (D) a personas Rho negativos genera concentraciones plasmáticas pico a los 5-10 días. La vida media de eliminación es de 24-30 días. INDICACIONES Prevención de la enfermedad hemolí-tica en un recién nacido Rho (D) po-sitivo de madre Rho (D) negativa expuesta con anterioridad a sangre Rho positiva. La inmunoglobulina Rho (D) humana se usa profilácticamente para suprimir la formación de anti-cuerpos anti-Rho (D) en mujeres Rho (D) negativas expuestas a sangre Rho positiva proveniente del feto durante el embarazo o el trabajo de parto, o como consecuencia de algún procedimiento obstétrico (amniocentesis, biopsia coriónica, toma de sangre umbilical) o trauma (aborto, hemorragia fetal) durante la gestación y en personas Rho (D) negativos que por error reciben siguientes al procedimiento. Si el procedimiento ocurre dentro de las primeras 12 semanas del embarazo, repetir la administración a las 26-28 semanas de gestación. - Profilaxis preparto después de trauma abdominal o abortos: Si ocurren dentro de las primeras 12 semanas del embarazo: 50 mcg. Para eventos que ocurren después de la semana 12 del embarazo: 300 mcg. Administrar siempre antes de las 72 horas siguientes al evento. - Profilaxis posparto (infante Rhopositivo): 300 mcg antes de las primeras 72 horas siguientes al parto. J - Transfusión de sangre Rho incompatible: La dosis es estimada para eliminar la posible cantidad de glóbulos rojos administrada: 25 mcg por cada mililitro de sangre transfundida. REACCIONES ADVERSAS Cardiovasculares: Hipotensión. Gastrointestinales: Náusea, vómitos. Reacciones de hipersensibilidad: Anafilaxis, reacciones alérgicas (eritema, pru-rito, erupción). Neurológicas: Cefalea, mareos, somnolencia. Otras: Escalo-fríos, fiebre, incomodidad y ligero ede-ma en el sitio de la inyección, mialgia, astenia, dolor de espalda, dolor abdo-minal, artralgia, palidez. 2ed. 2004 * Formulario TerapÈutico Nacional 281 tante, si la circunstancia impone la necesidad de la vacunación postparto, se debe administrar la vacuna y verificar, mediante una prueba serológica, 3 meses después, si hubo seroconversión. No debe mezclarse con otros medicamentos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. No se debe usar por vía IV. Debe administrarse sólo por vía IM profunda, teniendo la precaución de aspirar antes de inyectar para asegurar que la aguja no está en un vaso sanguíneo. El produc-to nunca debe ser administrado al neonato. No debe ser administrado a individuos Rho (D) positivo, incluyendo infantes; a mujeres Rho (D) negativo que están inmunizadas, cuando se evidencia por las pruebas correspon-dientes. Dado que el producto es derivado de plasma de donantes humanos, no se puede descartar la posibilidad, aunque remota, de la existencia de virus u otros agentes in-fecciosos. Este medicamento contiene trazas de IgA. Se debe pensar la posibilidad de reacciones de hiper-sensibilidad. Los individuos deficien-tes en IgA pueden desarrollar anti-cuerpos anti-IgA y reacciones ana-filácticas. Los individuos que han tenido reacciones 282 INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso INMUNOGLOBULINA HUMANA NORMAL FARMACOLOGÕA Solución estéril, no pirogénica, de glo-bulinas obtenidas de suero o plasma humano que contiene numerosos anticuerpos normalmente presentes en la sangre de humanos adultos. Las formulaciones de inmunoglobulinas IM (IGIM) e IV (IGIV) proveen inmunidad pasiva al incrementar los títulos de anticuerpos en el individuo, así como su potencial de reacción antígeno-anticuerpo. Se ha postulado que la IGIV satura los receptores Fc (frag-mento cristalizable) de las células del sistema retículo endotelial, dando como resultado una disminución de la fagocitosis de células cubiertas de anticuerpos mediada por los receptores Fc. Este bloqueo puede ocurrir en la médula ósea, el bazo y otras partes del sistema retículo endotelial a través de la competencia de inmunoglobuli-nas (IgG) o complejos inmunes por los receptores Fc. FARMACOCIN…TICA Después de la administración IM, se alcanzan concentraciones séricas pico de IgG en aproximadamente 2-5 días, mientras que con la Formulario Terapéutico Nacional * 2ed. 2004 nodeficiencias. En niños inmunodeficientes, se encontró una vida media de 21-42 días. La inmunidad proporcionada por el producto dura de 1-3 meses. INDICACIONES Para proveer inmunidad pasiva en individuos susceptibles expuestos a ciertas enfermedades infecciosas, cuando no hay disponibilidad de la vacuna para inmunización activa contra la enfermedad, o cuando no hay tiempo suficiente para que la inmunización activa estimule la producción de anticuerpos. Como terapia de reemplazo en individuos con síndromes de deficiencia de anticuerpos ocasionada por una síntesis defectuosa de los mismos. Púrpura trombocitopénica idiopática. RÉGIMEN DE DOSIFICACIÓN IGIV: - SÌndrome de inmunodeficiencia: 100-200 mg/kg por infusión IV 1 vez al mes; si no se alcanzan los niveles de IgG deseados, aumentar la do-sis (300-400 mg/kg) o la frecuencia de administración. - P˙rpura trombocitopénica idiopática: 400 mg/kg por 5 días consecutivos. IGIM: - Profilaxis de la hepatitis A: 0,02 ml/kg. - Profilaxis del sarampión: 0,2 ml/kg. REACCIONES ADVERSAS Reacciones locales en el sitio de inyección, urticaria, angioedema, vómitos, escalofríos, fiebre, hipotensión y reacciones anafilácticas (más frecuentes con la administración IV). Se ha descrito casos aislados de edema angioneurótico y síndrome ne-frótico. INTERACCIONES Los anticuerpos presentes en la formulación pueden interferir con la respuesta inmune a las vacunas de virus vivos. Para evitarlo, se debe administrar la vacuna 2 semanas antes o 3 meses después de la terapia con inmunoglobulinas (esto no aplica a J la vacuna de la fiebre amarilla ya que la inmunoglobulina normal no contiene anticuerpos contra este virus). CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al producto o a otras inmunoglobulinas. Usar con precaución en el embarazo. No se debe usar en pacientes con trastornos de la coagulación. La seguridad de la inmunoglobulina normal intramuscular en el embarazo no ha sido establecida. Los pacientes con riesgo de insuficiencia renal deben ser hidratados previo al tratamiento intravenoso y la función renal debe ser controlada. 2ed. 2004 * Formulario TerapÈutico Nacional 283 asociados al uso del producto e indicar las medidas terapéuticas requeridas SUERO ANTIESCORPIÓNICO FARMACOLOGÕA Solución estéril constituida por globulinas purificadas específicas obtenidas del suero de caballos hiperinmunizados con veneno de escorpiones del género Tityus. Las antiveninas presentes en 5 ml (1 ampolla) del suero neutralizan la acción de 1 mg de veneno de escorpión del género Tityus. INDICACIONES Tratamiento del emponzoñamiento por escorpiones del género Tityus. gotas/minuto. - En niños menores de 5 años: Iniciar diluyendo 2 ampollas en 50 ml de solución fisiológica (NaCl al 0,9%) y administrar por vía IV a razón de 20 gotas/minuto. De persistir los sínto-mas, administrar 2 ampollas adi-cionales. REACCIONES ADVERSAS Reacciones de hipersensibilidad aguda (dentro de las primeras 24 horas siguientes a la administración del suero): Eritema, prurito, angioedema, tos, disnea, vómito, cianosis, colapso cardiovascular y anafilaxis. Reacciones de hipersensibilidad retardada (se presentan a los 4-40 días de la administración del suero): Fiebre, urticaria, artralgia, proteinuria, linfadenopatías y neuropatías. R…GIMEN DE DOSIFICACI”N Dos factores determinan el tratamiento antiescorpiónico: a) Identificación del artrópodo agresor y b) severidad de INTERACCIONES los síntomas. Los signos y síntomas No se describen en la literatura. críti-cos ocasionados por el emponzoña-miento escorpiónico CONTRAINDICACIONES/ PRECAUCIONES incluyen: Dolor intenso, piel fría, Se debe usar solamente en ambientes palidez, sudoración, náuseas, hospitalarios y bajo estricta supervómitos, salivación, taqui-cardia, visión médica, a objeto de controlar la hipertensión, arritmias, dolor eventualidad de una reacción de epigástrico, dolor abdominal difuso. hipersensibilidad. No todas las picaAnte la presencia de tales síntomas das de escorpión en adultos requieren debe administrarse el suero. la administración del suero; es - En adultos y niños mayores de 5 indispensable una evaluación de la años: Iniciar diluyendo 2 ampollas severidad del accidente y determinar si de suero en 25 ml de solución fisioes necesaria la seroterapia. Como lógica. Administrar por vía IV a ra284 Formulario Terapéutico Nacional * 2ed. 2004 Laboratorio normal: Repetir los análisis de laboratorio a las 24 horas. - Pacientes con glicemia en 160-200 y amilasa normal: Repetir los análisis de laboratorio a las 4 horas. - Pacientes con amilasa elevada: Administrar el suero. - Pacientes sintomáticos sin posibilidad de realización de los análisis de laboratorio: Administrar el suero. Usar con precaución en pacientes con historia de alergia, o manifestaciones de hipersensibilidad por contacto frecuente con caballos, o pacientes que hayan recibido seroterapia. Antes de iniciar la terapia debe tenerse a disposición inmediata equipos para respiración asistida, epinefrina, antihistamínicos y esteroides. El uso de esteroides está contraindicado en presencia de pancreatitis, ya que puede agravar el cuadro; la epinefrina está contraindicada en la fase adrenérgica del cuadro clínico (taquicardia, hipertensión, arritmias). En ausencia de estos síntomas y frente a un shock anafiláctico, usar epinefrina con cautela y vigilar la función cardiorespiratoria constantemente. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación del suero antiescorpiónico. asistencia médica más cercano. SUERO ANTIOFÍDICO (SUERO ANTIOFÕDICO POLIVALENTE) FARMACOLOGÕA Solución estéril constituida por globulinas específicas purificadas obteni-das de suero de caballos hiperinmuni-zados con veneno de todas las espe-cies venezolanas de Bothrops (tigra mariposa, macagua y mapanare, entre otras) y de Crotalus (cascabel). Las antiveninas presentes en un frasco ampolla de suero polivalente neu-tralizan no menos de 20 g de veneno de Bothrops colombiensis y 15 mg de Crotalus J durissus cumanensis. INDICACIONES Tratamiento de emponzoñamiento por serpientes de los géneros Bothrops y Crotalus. R…GIMEN DE DOSIFICACI”N Iniciar el tratamiento con 5 ampollas de suero (cantidad suficiente para neutralizar 100 mg de veneno botrópico y 60 mg de veneno crotálico) por vía IM ó SC. En casos severos, administrar 10 frascos de suero, 5 por vía IM y 5 por vía IV, o todos por vía IV (2- 3 sin diluir y el resto diluidos en 500 ml de solución fisiológica). Si el tratamiento resulta in-suficiente, se puede administrar 5 am-pollas adicionales 2ed. 2004 * Formulario TerapÈutico Nacional 285 CONTRAINDICACIONES/ PRECAUCIONES Debe usarse solamente en ambientes hospitalarios y bajo estricta vigilancia médica, a objeto de controlar la eventualidad de una reacción de hipersensibilidad. Usar con extrema precaución en pacientes con historia de alergias, o manifestaciones de hipersensibilidad por contacto frecuente con caballos, o que hayan recibido seroterapia. Antes de iniciar la terapia debe tenerse, a disposición inmediata, equipos para respiración asistida, epinefrina, antihistamínicos y esteroides. SOBREDOSIS No se conocen casos de intoxicación SUERO ANTIRRÁBICO FARMACOLOGÕA Solución estéril, no pirogénica, de glo-bulinas purificadas obtenidas de suero de caballos sanos hiperinmunizados con vacuna antirrábica. Los anticuer-pos presentes en el suero antirrábico neutralizan el virus de la rabia impidiendo su diseminación en el organismo. INDICACIONES En combinación con la vacuna antirrábica para proveer inmunidad pasiva (por el suero) y desarrollar inmuni286 R…GIMEN DE DOSIFICACI”N Se administra por vía IM y por infiltración en la herida. El producto debe ser administrado tan pronto como sea posible después de la mordida (preferiblemente dentro de las primeras 24 horas). La dosis para adultos y niños es de 20 UI/kg por vía IM. La inyección debe aplicarse en el músculo deltoides o en la región anterolateral del muslo. Debido a la posibilidad de lesión del nervio ciático, la administración en los glúteos debe reservarse sólo para casos en los que se requieran grandes volúmenes del suero, o cuando una dosis elevada deba ser dividida en varias inyecciones. Hasta un 50% de la dosis del suero puede ser infiltrado en la herida y el resto administrado por inyección IM para un total de 40 UI/kg. No más de 5 ml deben ser inyectados en un solo sitio. Si la dosis es mayor, debe dividirse en tantas porciones como sea necesario para mantener el volumen inferior a 5 ml e inyectarlas en sitios diferentes. REACCIONES ADVERSAS Las reacciones más comunes incluyen: dolor en el sitio de la inyección, eritema y urticaria (hasta 10 días después de la inyección). Se puede presentar enfermedad del suero, que se manifiesta con erupción, prurito, artralgia y linfadenopatía. En casos severos, puede ocurrir dolor abdomi- Formulario Terapéutico Nacional * 2ed. 2004 terfieren con la respuesta de anticuerpos a la inmunización activa (vacuna) que se practica conjuntamente con la seroterapia. Aunque se ha demostrado que los anticuerpos contenidos en el suero antirrábico inhiben parcialmente la respuesta inmune a la vacuna an-tirrábica, es conveniente administrar el suero y la vacuna al mismo tiempo, pero nunca mezclados ni aplicados en el mismo sitio. CONTRAINDICACIONES/ PRECAUCIONES No administrar por vía IV. Usar con extrema precaución en personas con historia de alergia. Antes de iniciar la terapia, debe tenerse a disposición inmediata equipos para respiración asistida, epinefrina, antihistamínicos y esteroides. Además, debe realizarse una prueba de sensibilidad dérmica antes del uso. xido de aluminio, fosfato de aluminio o alumbre potásico. Promueve la inmunidad activa contra el tétanos por inducción de la producción de antitoxinas específicas. FARMACOCIN…TICA La administración de todas las dosis que comprende la inmunización primaria genera títulos efectivos de antitoxinas que persisten por 10 años o más. INDICACIONES Inmunización activa contra el tétanos. TOXOIDE TETÁNICO J R…GIMEN DE DOSIFICACI”N Se administra solamente por inyección IM profunda, preferiblemente en el deltoides o en la cara lateral de la por-ción media del muslo. La formulación disponible contiene 5 unidades Lf de toxoide tetánico por cada 0,5 ml. La inmunización primaria en adultos y niños de 7 años o más, comprende 2 inyecciones de 0,5 ml con un intervalo de 4-8 semanas y una tercera dosis de 0,5 ml a los 6-12 meses. Se puede dar una quinta dosis a los 4 años. Para mantener la protección se debe administrar un refuerzo de 0,5 ml cada 10 años. FARMACOLOGÕA Toxoide tetánico adsorbido en hidró- REACCIONES ADVERSAS Cardiovasculares: Taquicardia, hipo- SOBREDOSIS No se conocen casos de intoxicación por sobredosificación del suero antirrábico. INFORMACIÓN AL PACIENTE Advertir al paciente sobre la 2ed. 2004 * Formulario TerapÈutico Nacional 287 falopatía, alteraciones del EEG. Otras: Fiebre leve, escalofríos, malestar, en-rojecimiento de la cara, dolor muscu-loesquelético. INTERACCIONES Los agentes inmunosupresores pueden disminuir la respuesta inmune al toxoide. una reacción severa, informar de inVACUNA ANTITUBERCULOSA (BCG) FARMACOLOGÕA Liofilizado de bacilos vivos atenuados de la cepa Calmette-Guerin de Mycobacterium bovis. La cepa es inmunológicamente similar al Mycobacterium tuberculosis, por lo que la vacunación con BCG simula una infección natural con M. tuberculosis y promueve la inmunidad activa contra la tuberculosis. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o con procesos infecciosos activos. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios a la madre superen los riesgos potenciales para el feto. No usar durante el primer trimestre del embarazo. Evitar la administración en personas con hipogammaglobulinemia o que reciben terapia inmunosupresora. Usar con precaución en niños con daño cerebral, enfermedad neu-rológica o historia de convulsiones febriles. No es recomendable su uso en niños menores de 7 años. Debe usarse con extrema precaución en pacientes con trombocitopenia. Tam-bién es una contraindicación usarlo en individuos sensibles al timerosal. R…GIMEN DE DOSIFICACI”N Se administra por vía intradérmica, previa limpieza adecuada de la zona de aplicación. Después de reconstituida, la vacuna contiene 50-400 millones de unidades formadoras de colonias de BCG por mililitro. La dosis para adultos y niños mayores de 1 mes es de 0,2-0,3 ml. En niños menores de un mes, se usa la mitad de la dosis. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación con el toxoide. REACCIONES ADVERSAS Reacciones de hipersensibilidad: Anafilaxis, reacciones alérgicas. Otras: 288 FARMACOCIN…TICA Se estima que la protección proporcionada por la vacuna persiste por 7-10 años. INDICACIONES Prevención de la tuberculosis. Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Los medicamentos inmunosupresores pueden interferir con la respuesta inmune a la vacuna. Algunos agentes usados contra la tuberculosis, como la isoniazida, la rifampicina y la estreptomicina, inhiben la multiplicación de la BCG. El uso concomitante de esteroides puede causar infección sistémica. o con otro tuberculostático adecuado. INFORMACIÓN AL PACIENTE Mantener el sitio de la inyección limpio hasta la desaparición de la lesión local, la cual puede persistir hasta por 12 semanas. Advertir al paciente sobre los efectos adversos asociados a la vacunación e indicar (si la naturaleza de las reacciones lo permite) las medidas terapéuticas para su manejo ambulatorio. Si se presenta una reacción severa, acudir al centro de asistencia médica más cercano. El CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad a la vacuna, así como en personas con reacción positiva a la VACUNA CONTRA EL HAEMOPHILUS tuberculina, en tratamiento con medi- INFLUENZAE TIPO B J camentos inmunosupresores, inmuni- (ANTIHAEMOPHILUS INFLUENZAE TIPO B) zación antivariólica reciente, FARMACOLOGÍA pacientes quemados. También está Liofilizado de polisacárido capsular contraindi-cada en pacientes con (polirribosil-ribitol-fosfato, PRP) hipogamma-globulinemia, purificado de Haemophilus influenzae inmunodeficiencias congénitas, tipo b (Hib), cepa 1482, unido covasarcoidosis, linfoma, leu-cemia, lentemente al toxoide tetánico. cáncer generalizado, in-fecciones por Estimula la inmunidad activa contra VIH y otras condiciones que cursen infecciones por Hib al inducir la con compromiso inmuno-lógico. Las producción de anticuerpos especípersonas con enfermeda-des ficos. La conjugación con toxoide tetácrónicas de la piel deben recibir la nico tiene como objetivo estimular la inmunización por BCG en un área respuesta dependiente del timo a la sana de la piel. Evitar el uso durante el vacuna y generar una mayor duración embarazo, a menos que exista un del título de anticuerpos (por riesgo excesivo e inmediato de ex- d e s a r r o l l o d e l a r e s p u e s t a posición a la tuberculosis infecciosa. anamnésica). Pacientes con quemaduras o que reciben terapia con corticosteroides. FARMACOCINÉTICA 2ed. 2004 * Formulario TerapÈutico Nacional 289 sisten, como mínimo, por 1,5 - 3 años. INDICACIONES Inmunización activa contra infecciones causadas por Haemophilus influenzae tipo b. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM, preferiblemente en la porción anterolateral superior del muslo (en niños pequeños) o en el deltoides superior (en niños mayores). En pacientes con trombocitopenia u otros desórdenes sanguíneos se recomienda la administración por vía SC. Cada dosis de la vacuna consiste de 0,5 ml de la preparación que contiene 10 mcg de polisacárido capsular de Hib conjugado a 24 mcg de toxoide tetánico. Las dosis dependen de la edad del niño al momento de iniciar la inmunización: - NiÒos de 2-6 meses: 3 dosis (con un intervalo de 2 meses entre cada aplicación) y 1 refuerzo a los 15-18 meses. - Niños de 7-11 meses: 2 dosis (con un intervalo de 2 meses entre cada aplicación) y 1 refuerzo a los 15-18 meses. - Niños de 12-14 meses: 1 dosis y 1 refuerzo 2 meses después. - Niños de 15-69 meses: 1 dosis. reciben terapia con medicamentos inmunosupresores puede generar una respuesta inmunológica inadecuada a la vacuna. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a alguno de los constituyentes del medicamento. No es recomendable su uso en pacientes con fiebre o con procesos infecciosos agudos. Después de la administración, se recomienda mantener al paciente bajo observación por 30 minutos. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación de la vacuna. INFORMACIÓN AL PACIENTE Advertir a los familiares del paciente sobre los posibles efectos adversos asociados a la vacuna e indicar (si la VACUNA CONTRA EL SARAMPIÓN FARMACOLOGÕA Liofilizado de virus vivos atenuados de sarampión (cepa Schwartz), obtenidos en cultivos de células de embrión de pollo. Promueve la inmunidad activa contra el sarampión por inducción de la producción de anticuerpos espe-cíficos. REACCIONES ADVERSAS Por lo general son leves, transitorias y poco frecuentes. Las más comunes FARMACOCIN…TICA incluyen: Eritema e induración en el 290 Formulario Terapéutico Nacional * 2ed. 2004 semanas y persiste, como mínimo, por 8 años. En algunos individuos todavía se detectan anticuerpos después de 11-13 años de la primera vacunación. INDICACIONES Prevención del sarampión. R…GIMEN DE DOSIFICACI”N Cada dosis de 0,5 ml contiene no menos de 1000 DICT50 del virus atenuado. Se administra solamente por vía SC. La dosis usual en adultos y niños es de 0,5 ml, como dosis única. La edad recomendada para la primera vacunación es 12-15 meses. Para adultos: Las personas nacidas después de 1957 deben recibir 2 dosis con un intervalo de un mes. Para niños: Se recomienda un esquema de dos dosis, dando la primera a los 15 meses (12 meses en zonas de alto riesgo) y la segunda dosis a los 4-6 años o a los 11-12 años. REACCIONES ADVERSAS Cardiovasculares: Vasculitis. Dermatológicas: Erupción, eritema, edema y sensibilidad en el sitio de inyección. Hematológicas: Leucopenia, trombocitopenia, púrpura. Neurológicas: Con-vulsiones febriles en niños suscep-tibles, neuritis óptica. Se ha descrito casos de síndrome de Guillain-Barré, de encefalitis y sensibilidad cutánea a la tuberculina; por lo tanto, si se requiere pruebas de tuberculina las mismas deben ser hechas antes, simultáneamente o 6 semanas después de la vacunación con-tra el sarampión. Los medicamentos inmunosupresores pueden disminuir la respuesta a la vacuna y provocar la replicación viral. Las inmunoglobulinas y los hemoderivados pueden interferir con la respuesta inmune a la vacuna contra el sarampión; no usar vacunas por lo menos hasta 3 meses después de finalizado el tratamiento con estos productos. PRECAUCIONES/ CONTRAINDICACIONES Contraindicada en individuos con inmunodeficiencias primarias o con una respuesta inmune suprimida, ya sea por enfermedad o por terapia inmunosupresora; en pacientes con enfermedad respiratoria febril o condiciones infecciosas activas con manifestación febril. Contraindicada en caso de hipersensibilidad a cualquier componente de la vacuna, incluyendo la gelatina, en pacientes alérgicos a la neomicina, en el embarazo (las mujeres que reciben la vacuna deben evitar el embarazo durante por lo menos 3 meses después de la administración de la vacuna); en individuos hipersensibles a los huevos o al pollo. Si se 2ed. 2004 * Formulario TerapÈutico Nacional 291 J SOBREDOSIS No se conocen casos de intoxicación por sobredosificación de la vacuna. INFORMACIÓN AL PACIENTE Advertir a las mujeres que deben evitar el embarazo durante los tres meses siguientes a la vacunación, debido al riesgo de malformaciones congénitas. Informar al paciente o a su representante sobre los efectos adversos que pueden ocurrir después de la vacunación e indicar (si la naturaleza de dichos efectos lo permite) las medidas terapéuticas para su manejo VACUNA CONTRA EL SARAMPIÓN, RUBEOLA y PAROTIDITIS FARMACOLOGÕA Liofilizado de virus vivos atenuados de sarampión (cepa Schwartz), rubeola (cepa Wistar RA 27/3) y parotiditis (cepa Urabe AM-9). Genera inmunidad activa contra el sarampión, la rubéola y la parotiditis por inducción de la pro-ducción de anticuerpos específicos. INDICACIONES Prevención del sarampión, la rubéola y la parotiditis. R…GIMEN DE DOSIFICACI”N Cada 0,5 ml de la vacuna contiene no menos del equivalente a 1000 DICT50 de virus vivos atenuados de saram292 nuados de rubéola. Se administra por vía SC ó IM en la región externa y superior del brazo. La dosis usual para niños y adultos es de 0,5 ml. Para adultos: Se recomienda un esquema de dos dosis con un intervalo de un mes. Para niños: Se recomienda un esquema de dos dosis, dando la primera a los 15 meses (12 meses en zonas de alto riesgo) y la segunda dosis a los 4-6 años o a los 11-12 años. REACCIONES ADVERSAS Reacciones locales: eritema, ardor, induración, sensibilidad y dolor en el sitio de la inyección. Gastrointestinales: Diarrea. Reacción de hipersensibilidad: Erupciones, urticaria, vasculitis, anafilaxis. Otras: Artritis, artralgia, fiebre, linfadenopatía regional. Se han descrito casos raros de parálisis ocular, síndrome de GuillainBarré, encefalitis y encefalopatía, pero sin evidencia objetiva de relación causa-efecto. INTERACCIONES Los fármacos inmunosupresores pueden disminuir la respuesta inmune a la vacuna. Los hemoderivados (sangre, plasma e inmunoglobulinas) podrían contener anticuerpos que interfieren con la respuesta inmune a la vacuna. La vacuna puede disminuir temporalmente la sensibilidad dérmica a la tuberculina. Formulario Terapéutico Nacional * 2ed. 2004 ciosos activos con manifestación febril, en tratamiento con medicamentos inmunosupresores y durante el embarazo. Usar con precaución en pacientes con historia de convulsiones. No administrar por vía intravenosa. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación de la vacuna. INFORMACIÓN AL PACIENTE Advertir a los familiares del niño o al adulto sobre los efectos adversos que se asocian a la vacuna e indicar (si la naturaleza de dichos efectos lo permi- R…GIMEN DE DOSIFICACI”N Cada 0,5 ml de la formulación contiene no menos de 30 UI de toxoide diftérico, 60 UI de toxoide tetánico y 4 UI de Bor-detella pertussis. Se administra me-diante inyección IM profunda en la re-gión lateral media del muslo en los lactantes o en el deltoides en niños mayores. Para la inmunización prima-ria en niños de 6 semanas a 6 años de edad: 3 dosis de 0,5 ml, a intervalos de 4-8 semanas y una cuarta dosis de refuerzo a los 6-12 meses de la ter-cera dosis. Cada una de las dosis debe inyectarse en un lugar distinto. REACCIONES ADVERSAS Reacciones locales: Eritema, induraVACUNA CONTRA LA DIFTERIA, PERTUSSIS y TÉTANOS (DIFTERIA, TOSFERINA Y T…TANOS) ción, sensibilidad, inflamación y dolor en el sitio de la inyección. Reacciones FARMACOLOGÕA sistémicas: Elevación moderada de la Esta vacuna está constituida por la temperatura, malestar general, escavacuna contra la pertussis, el toxoide lofríos, irritabilidad, mareos, anorexia, diftérico y el toxoide tetánico adsorvómitos y llanto persistente. Se ha bidos en gel de fosfato de aluminio. La presentado casos poco frecuentes de administración induce la formación de reacciones severas (inclusive fatales) antitoxinas contra la difteria y el tétaa las 24-48 horas de la inyección, o nos y anticuerpos contra Bordetella hasta 7 días después, con manifestapertussis. ciones de fiebre, colapso con recuperación rápida o seguido de postraFARMACOCIN…TICA ción prolongada, episodios La administración de todas las dosis transitorios similares al shock, que comprende la inmunización prisomnolencia, con-vulsiones, maria produce una protección que encefalopatía con cambios en los persiste por 10 años. niveles de conciencia, púrpura 2ed. 2004 * Formulario TerapÈutico Nacional 293 J mayores de 7 años. No debe administrarse durante enfermedades agudas o cuando existan signos o síntomas neurológicos. Si se presentan reacciones adversas severas antes de finalizar la inmunización completa, no debe administrarse las dosis subsiguientes. No debe usarse en pacientes con hipersensibilidad a alguno de los constituyentes de la formulación, ni en pacientes inmunosuprimidos o que hayan recibido recientemente transfusiones de sangre, plasma o inmunoglobulinas. Está contraindicada en pacientes con tratamientos inmunosupresores, leucemia, linfoma o enfermedad maligna generalizada. No debe usarse para el tratamiento del tétanos, de la difteria o de la tosferina. No administrar por vía IV. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación del medicamento. INFORMACIÓN AL PACIENTE Informar a los familiares del niño sobre los efectos adversos asociados a la VACUNA CONTRA LA FIEBRE AMARILLA (ANTIAMARÕLICA) FARMACOLOGÕA Liofilizado de organismos vivos atenuados de la cepa 17D del virus de la fiebre amarilla, cultivados en em294 inmunidad activa contra la fiebre amarilla en individuos con riesgo de contraer la enfermedad. FARMACOCIN…TICA Después de la administración SC, la inmunidad se desarrolla a los 7-10 días y persiste por 30-35 años como míni-mo. INDICACIONES Inmunización de personas adultas y niños mayores de 9 meses que habiten o viajen a lugares donde la fiebre amarilla es endémica. Inmunización de personas con riesgo ocupacional o laboral de contraer la enfermedad. R…GIMEN DE DOSIFICACI”N Cada 0,5 ml de la vacuna contiene no menos de 2000 unidades DL50 en ratón. Se administra por vía subcutánea. La dosis usual es de 0,5 ml, como dosis única. Aunque el período de inmunidad puede durar hasta 30-35 años, las regulaciones internacionales sobre vacunación establecen la necesidad de refuerzos cada 10 años. REACCIONES ADVERSAS Por lo general son leves y poco frecuentes. Las más comunes incluyen fiebre y malestar, dentro de los 5-14 días posteriores a la vacunación; con menor frecuencia: mialgia, cefalea, reacciones de hipersensibilidad con Formulario Terapéutico Nacional * 2ed. 2004 y el desarrollo de la infección, en cuyo caso debe postergarse la vacunación hasta finalizar la inmunoterapia. El tiempo entre la finalización de la terapia y la vacunación no está claramente establecido; se estima entre 3 y 12 meses. La administración simultánea con la vacuna contra el cólera interfiere con la respuesta inmune a ambas vacunas; ellas deben ser administradas con un intervalo no menor de 3 semanas, a menos que el riesgo de contraer ambas enfermedades sea elevado. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en personas hipersensibles a los huevos o a las proteínas del embrión de pollo. No debe usarse en niños menores de 9 meses, ni durante el embarazo, excepto en áreas de alto riesgo y sólo cuando los beneficios superen los riesgos potenciales. Debido a que la replicación viral y el desarrollo de una infección con el virus de la vacuna pueden potenciarse por alteraciones del sistema inmune, la vacuna está contraindicada en individuos inmunocomprometidos, con hipoalbuminemia, o en aquellos que reciben terapia inmunosupresora. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación del producto. acudir al centro de asistencia médica VACUNA CONTRA LA HEPATITIS B (ANTIHEPATITIS B) FARMACOLOGÕA Suspensión estéril que contiene el antígeno de superficie de la hepatitis B (HBsAg) producido por la levadura Saccharomyces cerevisiae mediante tecnología de ADN-recombinante. La vacuna promueve la formación de anticuerpos (anti-HBs) que neutralizan al virus de la hepatitis B. FARMACOCIN…TICA Después de la administración IM, los anticuerpos anti-HBs aparecen en el J suero a las 2 semanas, alcanzan el nivel pico después de los 6 meses y persisten, como mínimo, por 3 años. _ 5,5 días. La vida media es 23,1 + INDICACIONES Inmunización activa contra la hepatitis B. R…GIMEN DE DOSIFICACI”N Cada dosis de 1 ml de la formulación para adultos contiene 20 mcg de HBsAg y cada dosis de 0,5 ml de la formulación para niños contiene 10 mcg de HBsAg. Se administra por vía IM solamente, preferiblemente en el músculo deltoides (en adultos) o en la región anterolateral del muslo (en 2ed. 2004 * Formulario TerapÈutico Nacional 295 ses después de la segunda dosis). - En niños, desde el nacimiento hasta los 19 años: 0,5 ml (10 mcg), siguiendo el mismo esquema descrito para los adultos. REACCIONES ADVERSAS Cardiovasculares: Hipotensión. Gastrointestinales: Náuseas, vómitos, anorexia, dolor abdominal, constipación, diarrea. Hematológicas: Trombo-citopenia. Neurológicas: Insomnio, somnolencia, irritabilidad, agitación, neuritis óptica, convulsiones. Reaccio-nes de hipersensibilidad: Síndrome ti-po enfermedad del suero, prurito, erupción, eritema multiforme, angioedema, anafilaxis. Otras: Fatiga, fiebre, malestar, debilidad, sudoración, sínto-mas similares a los de la gripe, escalo-fríos, artralgia, mialgia, trastornos vi-suales, dolor de oído. ción en individuos con inmunodeficiencia o en terapia con inmunosupresores, trastornos de coagulación o trombocitopenia, infecciones febriles severas, esclerosis múltiple, en el embarazo y durante la lactancia. No inyectar por vía IV o intradérmica. La inyección de la vacuna en los glúteos ha sido asociada con respuestas inmunológicas insuficientes. Los pacien-tes en hemodiálisis, por lo general, re-quieren dosis mayores para lograr un título adecuado de anticuerpos. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación de la vacuna. Las únicas manifestaciones que pudieran observarse, de acuerdo a la experiencia con otras globulinas, son dolor y sensibilidad en el sitio de inyección. INTERACCIONES Los agentes inmunosupresores pueden producir disminución de la respuesta a la vacuna. La vacunación con vacunas a virus vivos deben ser pospuestas hasta 3 meses después de esta vacuna. No debe ser mezclada con otros medicamentos. INFORMACIÓN AL PACIENTE Advertir al paciente o a sus familiares sobre los posibles efectos adversos asociados a la vacunación e informar (si la naturaleza de dichos efectos lo permite) las medidas para su manejo CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en individuos hipersensibles a las levaduras, a FARMACOLOGÕA Producto estéril del virus de la poliomielitis vivo atenuado (cepa Sabin) de los tipos 1, 2 y 3, propagados en cul- 296 VACUNA CONTRA LA POLIOMIELITIS (ANTIPOLIO) Formulario Terapéutico Nacional * 2ed. 2004 tivos primarios de tejidos de riñón de mono. Promueve inmunidad activa contra la poliomielitis por inducción de la producción de anticuerpos específicos. FARMACOCIN…TICA Después de la administración oral, el virus de la poliomielitis atenuado se multiplica en el tracto gastrointestinal, donde permanece por 4-6 semanas. La respuesta de anticuerpos se produce a los 7-10 días, se hace máxima a los 21 días y persiste, como mínimo, por 15 años. INDICACIONES Prevención de la poliomielitis. R…GIMEN DE DOSIFICACI”N Se administra solamente por vía oral. Cada dosis de la vacuna (2 gotas) contiene no menos de 1.000.000 DICT50 de virus del polio tipo 1, no menos de 100.000 DICT50 de virus del polio tipo 2 y no menos de 600.000 DICT50 de virus del polio tipo 3. La dosis usual es de 2 gotas en una serie de 4 tomas, iniciando la primera dosis al momento del nacimiento, la segunda dosis a los 2 meses de edad, la tercera dosis a los 4 meses y la cuarta dosis a los 6 meses. Se debe administrar una dosis de refuerzo a los 18 meses. nales: Disminución del apetito. Dermatológicas: Eritema, induración, dolor en el sitio de inyección. INTERACCIONES Los agentes inmunosupresores pueden disminuir la respuesta inmune a la vacuna y favorecer la replicación del virus. CONTRAINDICACIONES/ PRECAUCIONES Dado que la eficacia depende de la replicación de los virus en el tracto gastrointestinal, los pacientes con vómito o diarreas persistentes podrían manifestar una respuesta no satisfactoria a la vacuna. En tales circunstan- J cias se debe repetir la dosis una vez recuperado el paciente. La vacuna está contraindicada en pacientes con inmunodeficiencia primaria o que reciben terapia inmunosupresora. En caso de vacunación en adultos, particularmente en mujeres, no es recomendable la administración durante el embarazo. Bajo ninguna circunstancia, la vacuna debe administrarse por vía parenteral. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación con la vacuna. INFORMACIÓN AL PACIENTE Advertir a los familiares del paciente la REACCIONES ADVERSAS No se han reportado efectos adversos necesidad de informar al médico si severos por el uso de la vacuna; 297 2ed. 2004 *sin Formulario TerapÈutico Nacional VACUNA CONTRA LA RABIA (ANTIRR¡BICA) FARMACOLOGÕA Preparación estéril de virus inactivados de la rabia obtenidos en cultivos de células VERO. Induce el desarrollo de inmunidad activa contra la rabia en individuos expuestos a la enfermedad o al virus. FARMACOCIN…TICA Después de la administración IM, los anticuerpos contra la rabia comienzan a aparecer en la sangre a los 7-10 días; alcanzan el título máximo a los 30-60 días y persisten como mínimo por 1 año. INDICACIONES Para promover inmunidad activa contra la rabia en individuos expuestos a la enfermedad o al virus. R…GIMEN DE DOSIFICACI”N Se administra por inyección SC (preferiblemente en la fosa infraespinosa) o IM profunda (preferiblemente en el músculo deltoides). - Para la profilaxis pre-exposición, la dosificación se realiza en una serie de 3 inyecciones de 1 ml cada una. La primera dosis se administra en la fecha que se elija y la segunda y la tercera dosis a los 7 y 21 días, respectivamente, de la primera dosis. - Para la profilaxis post-exposición, la dosificación se realiza en una 298 tración de las 4 dosis restantes a los 3, 7, 14 y 28 días después de la pri-mera. La OMS recomienda una sexta dosis a los 90 días. REACCIONES ADVERSAS Dermatológicas: Dolor transitorio, eritema, edema o prurito y leve reacción inflamatoria en el sitio de inyección. Gastrointestinales: Dolor abdominal, náusea. Reacciones de hipersensibilidad: Reacción similar a la enfermedad del suero, anafilaxis. Otras: Mialgia, fiebre leve, fatiga. INTERACCIONES Los corticosteroides y los agentes inmunosupresores pueden interferir con el desarrollo de la inmunidad activa, y predisponer al paciente al desarrollo de la enfermedad. El uso concomitante de cloroquina ha sido asociado con una disminución de la respuesta a la vacuna. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad a la vacuna o a sus constituyentes. SOBREDOSIS No se conocen casos de intoxicación por sobredosificación de la vacuna. INFORMACIÓN AL PACIENTE Advertir sobre los posibles efectos asociados al uso de la vacuna e indicar Formulario Terapéutico Nacional * 2ed. 2004 L02 TERAPIA ENDOCRINA Flutamida Medroxiprogesterona (Medroxiprogesterona Acetato) (Antineoplásico) Tamoxifeno L AGENTES ANTINEOPL¡SICOS E INMUNOMODULADORES L01 AGENTES ANTINEOPL¡SICOS Asparaginasa Bleomicina Busulfano Carboplatino Ciclofosfamida Cisplatino Citarabina Clormetina (Mostaza Nitrogenada) Dacarbazina Dactinomicina (Actinomicina D) Daunorubicina Doxorubicina Etopósido Fluorouracilo (5-Fluorouracilo) Gemcitabina Hidroxicarbamida (Hidroxiurea) Ifosfamida Melfalán Mercaptopurina Metotrexato Paclitaxel Prednisona* (Antineoplásico) Procarbazina Vinblastina Vincristina L L03 INMUNOESTIMULANTES Filgrastim (G-CSF) Interferón Alfa-2B 2ed. 2004 * Formulario TerapÈutico Nacional 299 ASPARAGINASA FARMACOLOGÍA Agente antineoplásico; contiene a la enzima L-asparagina amidohidrolasa tipo EC-2, derivada de la Escherichia coli. En las células leucémicas, la enzima hidroliza al aminoácido asparagina a ácido aspártico y amonio, impidiendo su utilización para la síntesis de ADN y de proteínas esenciales. De esta manera, la depresión de la asparagina en las células leucémicas (incapaces de sintetizar por sí solas el aminoácido) impide la reproducción y provoca la muerte celular. La acción de la asparaginasa es específica sobre la fase G1 del ciclo celular. FARMACOCINÉTICA Como no se absorbe en el tracto gastrointestinal debe ser administrada por vía parenteral. Después de la inyección IV, alcanza niveles plasmáticos que se incrementan con la dosificación continua y produce una respuesta inicial que se evidencia a las 621 semanas. Debido a su elevado peso molecular, el fármaco difunde muy poco fuera del compartimiento vascular. En el estado de equilibrio, en el líquido cefalorraquídeo se ha detectado niveles de 0,5-1% de la concentración alcanzada en el plasma, y en el fluido linfático hasta de un 20%. Penetra fácilmente las células leucé- ni con el diagnóstico o extensión de la enfermedad. Se presume un metabolismo sistémico y una eliminación a través del sistema retículo-endotelial. En la orina se ha detectado sólo trazas. INDICACIONES En combinación con otros antineoplásicos para la inducción de la remisión en casos de leucemia linfocítica aguda. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV e IM. La dosis debe individualizarse con base en la respuesta clínica y la tolerancia de cada paciente. Se ha propuesto diver-sos regímenes de dosificación tales como: L - En niños (en combinación con prednisona y vincristina): - Infusión IV de 1.000 UI/kg/día por 10 días seguidos, comenzando el día 22 del ciclo de tratamiento. - Administración IM de 6000 UI/m2 en los días 4, 7, 10, 13, 16, 19, 22 y 28 del ciclo de tratamiento. - En adultos (como fármaco único): - Infusión IV de 200 UI/kg/día por 28 días, o 5.000-10.000 UI/m2/día por 7 días cada 3 semanas, o 10.000-40.000 UI cada 2-3 semanas. - Administración IM de 6.00012.000 UI/m2. 2ed. 2004 * Formulario TerapÈutico Nacional 301 somnolencia, depresión, alucinaciones, hemorragia intracraneal, fatiga, agitación, cefalea, letargia, coma. Reacciones de hipersensibilidad: Erupciones, urticaria, anafilaxis. Renales: Azotemia, insuficiencia renal, glucosuria, poliuria, nefropatía por ácido úrico, hiperamonemia. Otras: Escalofrío, hipertermia fatal, fiebre, hiperglicemia, hiperuricemia, muerte. INTERACCIONES La asparaginasa disminuye la actividad y la eficacia antineoplásica del metotrexato. La administración simultánea con prednisona o con vincristina incrementa, de manera importante, la toxicidad de la asparaginasa; para evitarlo se debe administrar siempre después del esteroide o de la vincristina, nunca antes o conjuntamente. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso BLEOMICINA FARMACOLOGÕA Antineoplásico de tipo antibiótico. Aunque no se conoce con exactitud su mecanismo de acción, se ha postulado que inhibe la síntesis de ADN y, en menor grado, la síntesis de ARN y de las proteínas. FARMACOCIN…TICA La administración IM produce concentraciones plasmáticas pico después de 30-60 minutos, las cuales se mantienen hasta por 4 horas; por vía IM, los niveles séricos son aproximadamente 1/3 de los obtenidos por vía IV. Se distribuye principalmente a la piel, pulmones, riñón, peritoneo y sistema linfático. En células tumorales de la piel y del pulmón logra concen-traciones superiores a las alcanzadas en el tejido hematopoyético. No se conoce con precisión su metabolismo. Se excreta en un 60-70% intacta por la orina. La vida media de eliminación es de aproximadamente 115 minutos y se incrementa en pacientes con falla renal. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con historia de pancreatitis. Previo al tratamiento, y periódicamente durante su desarrollo, se deben realizar evaluaciones de la función hepática, renal, hematopoyética, neurológica y pancreática, así como determinaciones de la amilasa y de la glicemia. Durante la terapia se debe INDICACIONES mantener una hidratación adecuada Sola o en combinación con otros agendel paciente. No se conoce la tes antineoplásicos, en el tratamiento 302 Formulario Terapéutico Nacional * 2ed. 2004 R…GIMEN DE DOSIFICACI”N Se administra por vía IM, IV ó SC. La dosis recomendada para el tratamiento del carcinoma de células escamosas, del linfoma Hodgkin y no-Hodgkin y del carcinoma testicular es de 0,250,50 U/kg (10-20 U/m2) 1-2 veces por semana. Una vez lograda una respuesta del 50%, se administra una dosis de mantenimiento IV ó IM de 1U/día o 5 U/semana. La mejoría clínica en la enfermedad de Hodgkin y en los tumores testiculares se observa en 2 semanas o menos. Si no hay respuesta en ese tiempo, es poco probable que ocurra. El cáncer de células escamosas responde más lentamente; a veces son necesarias 3 semanas para observar una mejoría. mos (bacterias o virus) vivos. El cisplatino altera la eliminación renal de la bleomicina, aumentando las concentraciones séricas y el potencial tóxico del antineoplásico. Durante la anestesia, puede aumentar los requerimientos de oxígeno. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y en el embarazo. No debe administrarse en personas que hayan presentado reacciones idiosincrásicas. Debe usarse con extrema precaución en pacientes con falla renal o con compromiso de la función pulmonar. Se debe evaluar frecuentemente la función pulmonar mediante radiografía de tórax (ca- L da 1-2 semanas) ya que la detección REACCIONES ADVERSAS Cardiovasculares: Fenómeno de Ray- de la toxicidad pulmonar es difícil pornaud, hipotensión. Dermatológicas: que carece de un síndrome clínico Hiperpigmentación de la piel, acné, específico. Si hay cambios radiológiestrías, sensibilidad de la piel, prurito, cos se debe descontinuar la terapia alopecia reversible, hiperqueratosis, hasta determinar si están relacionacambios en las uñas. Gastrointesti- dos con el medicamento. Dado que los nales: Estomatitis, anorexia, náuseas, pacientes en tratamiento con bleomivómitos, diarrea. Neurológicas: Agre- cina presentan sensibilización del sividad, debilidad. Reacciones de hi- tejido pulmonar y tienen alto riesgo de persensibilidad: Reacciones anafilác- desarrollar toxicidad pulmonar por ticas. Renales: Hematuria. Respirato- exposición al O2 en la cirugía, se rias: Toxicidad respiratoria tal como recomienda mantener la fracción neumonitis intersticial, fibrosis pul- inspiratoria de O2 (FiO2) en concenmonar. Otras: Escalofrío, pérdida de traciones del 25% durante la peso, fiebre. operación y en el período postoperatorio, así como una observación 303 2ed. 2004 * Formulario TerapÈutico Nacional de volumen, agentes presores, antihistamínicos y esteroides) si fuese necesario. Todos los pacientes con linfoma deben recibir una dosis de prueba antes de iniciar la terapia. No se conoce la seguridad del uso en niños. Se recomienda suspender la lactancia durante la terapia. SOBREDOSIS Manejo sintomático y terapia de soporte. Evaluación de la función pulmonar. BUSULFANO FARMACOLOGÍA Agente antineoplásico alquilante bifuncional, con dos grupos metanosulfonato lábiles. En medio acuoso, el busulfano se hidroliza y se liberan los grupos metanosulfonato que forman iones carbonio reactivos que atacan la molécula de ADN, alterando su estructura e impidiendo su replicación. También afecta la transcripción del ARN. Es activo durante todas las fases del ciclo celular. El daño al ADN es responsable por gran parte de la citotoxicidad del busulfano. FARMACOCINÉTICA Después de la administración oral, se absorbe rápida y casi completamente en el tracto gastrointestinal alcanzando niveles séricos pico en 0,5-2 horas. 304 en un 10-50% en la orina en 48 horas. La vida media de eliminación es de 2,3-3,4 horas. INDICACIONES Tratamiento paliativo de la leucemia mielógena crónica. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Para la inducción de la remisión en adultos y niños iniciar con dosis de 0,6 mg/kg/día o 1,8 mg/m2/día a un máximo de 4 mg. Si el contaje leucocitario cae por debajo de 15.00020.000/mm3, suspender el tratamiento. Si la remisión no se mantiene por más de 3 meses, usar dosis de mantenimiento de 1-3 mg/día. Si durante la remisión el contaje leucocitario alcanza valores de 50.000/mm 3 , reiniciar la terapia con la misma dosis de la inducción. REACCIONES ADVERSAS Cardiovasculares: Taponamiento cardiaco (en pacientes con talasemia y fibrosis endocardiaca) (rara). Dermatológicas: Hiperpigmentación y alopecia. Gastrointestinales: Náuseas, vómito, estomatitis, diarrea o constipación, anorexia, dolor abdominal, boca seca, dispepsia, pancreatitis y esofagitis. Hematológicas: Mielosupresión (leucopenia, trombocitopenia, anemia o pancitopenia). Hepáticas: Enfermedad Formulario Terapéutico Nacional * 2ed. 2004 tiga, hipotensión, anorexia, náusea, vómitos, pérdida de peso y melanoderma). Neurológicas: Mareo, cefalea, depresión, pérdida de la conciencia, insomnio, ansiedad y convulsiones. Reacciones de hipersensibilidad: Erupción, urticaria, eritema multiforme y eritema nodoso. Renales: Aumentos del BUN y de la creatinina sérica, insuficiencia renal aguda y hematuria. Otras: Fibrosis pulmonar intersticial, visión borrosa, cataratas, hiperpigmentación de la piel, excesiva resequedad y sensibilidad de la piel, mialgia, artralgia, miastenia gravis, ginecomastia y porfiria cutánea tardía. INTERACCIONES En combinación con otros agentes mielosupresores, incrementa el riesgo de dicho efecto. Con medicamentos citotóxicos aumenta la posibilidad de toxicidad pulmonar. Con tioguanina puede causar hepatotoxicidad, várices esofágicas o hipertensión portal. El itraconazol y el acetaminofén pueden disminuir la depuración del busulfano e incrementar su toxicidad. La fenitoína, por el contrario, aumenta su depuración. La warfarina y la aspirina aumentan el riesgo de sangramiento. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en el embarazo. Usar con precaución en pa- debe suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Advertir al paciente sobre la posibilidad de alopecia, hiperpigmentación, infecciones y hemorragia. Mantener una adecuada hidratación (2-3 L/día) durante el tratamiento. Evitar el contacto con multitudes y personas con infecciones o enfermedades contagiosas. Practicar medidas anticonceptivas durante el tratamiento. Notificar de inmediato al médico si se presenta alguna reacción o sintomaL tología inusual durante la terapia, en especial: sangramiento, infección, CARBOPLATINO FARMACOLOGÕA Antineoplásico del grupo de los agentes alquilantes, análogo del cisplatino y con actividad antineoplásica similar (ver: CISPLATINO). FARMACOCIN…TICA Después de la administración IV, exhibe una eliminación plasmática bifásica, con una vida media terminal de 3-6 horas. No se une en forma apreciable a las proteínas 2ed. 2004 * Formulario TerapÈutico Nacional 305 excretándose después con una vida media de 5 días o más. La evidencia disponible revela que el platino se distribuye predominantemente al riñón, hígado, piel y tejido tumoral. Se elimina casi completamente como fármaco intacto en la orina (70% de la dosis en 24 horas). Su eliminación se prolonga en pacientes con falla renal. INDICACIONES Solo, o en combinación con otros antineoplásicos, en el tratamiento del carcinoma avanzado de ovario, de células pequeñas de pulmón y en el cáncer de cabeza y cuello. R…GIMEN DE DOSIFICACI”N Se administra por vía IV. La dosis usual es de 360 mg/m2 como dosis única, cada 4 semanas. REACCIONES ADVERSAS Ver: CISPLATINO. INTERACCIONES Ver: CISPLATINO. CONTRAINDICACIONES/ PRECAUCIONES Ver: CISPLATINO. CICLOFOSFAMIDA FARMACOLOGÍA Antineoplásico del grupo de los agentes alquilantes, químicamente 306 relacionada con las mostazas nitrogenadas. Es metabolizado a metabolitos alquilantes por el sistema de la oxidasa de función mixta. Produce entrecruzamiento de las bandas del ADN de las células tumorales e interfiere con el crecimiento de las células malignas. Posee, además, propiedades inmunosu-presoras. FARMACOCINÉTICA Se absorbe bien por vía oral, con una biodisponibilidad superior a 75%, alcanzando niveles plasmáticos pico en 60 minutos. Se distribuye ampliamente a los tejidos y se une a las proteínas plasmáticas en escasa proporción (menos del 10%). Se metaboliza en el hígado, transformándose en metabolitos con actividad citostática que alcanzan concentraciones séricas máximas en 2-3 horas y se unen a las proteínas plasmáticas en un 60%. Se excreta como fármaco intacto (5-25%) y metabolitos en 48 horas, por vía renal. La vida media de eliminación es de 3-12 horas. INDICACIONES Sola o en combinación con otros agentes antineoplásicos en el tratamiento de linfomas malignos, mieloma múltiple, leucemias, micosis fúngica, neuroblastoma, adenocarcinoma de ovario, retinoblastoma y carcinoma de mama. Formulario Terapéutico Nacional * 2ed. 2004 dosis en 1/3 ó 1/2. La dosis inicial en niños es de 2-8 mg/kg por día. Las dosis de mantenimiento se establece con base en la respuesta clínica y hematológica a la terapia y considerando la tolerancia del paciente. Se han usado diferentes regímenes de dosificación: 1-5 mg/kg/día por vía oral, 10-15 mg/kg IV cada 7-10 días o 3-5 mg/kg IV 2 veces por semana. En niños, se recomienda dosis de mantenimiento de 2-8 mg/kg por vía oral, 2 veces por semana. A menos que exista alguna limitación o impedimento, es recomendable administrar la mayor dosis que pueda tolerarse, usando como guía el contaje absoluto de granulocitos, intentando mantener la cuenta por encima de 1000 células/mm3. Si se requiere un efecto terapéutico en presencia de cifras menores, se puede utilizar factores estimuladores de colonias. pulmonar intersticial. Otras: Caída del cabello, hiperpigmentación y engrosamiento de la piel y las uñas, mixedema, dermatitis, disminución de las concentraciones séricas de colinesterasa. INTERACCIONES La ciclofosfamida sola o en combinación con otros antineoplásicos, disminuye las concentraciones séricas de digoxina hasta niveles subterapéu-ticos. Dado que la ciclofosfamida disminuye los niveles plasmáticos de la colinesterasa plasmática, puede potenciar el bloqueo neuromuscular de la succinilcolina. Los barbitúricos y otros fármacos que inducen el sistema microsomal hepático pueden aumen- L tar la toxicidad de la ciclofosfamida al favorecer su conversión en metabolitos activos. Por el contrario, los medicamentos que inhiben el sistema microsomal hepático pueden interferir REACCIONES ADVERSAS con el metabolismo y la respuesta Cardiovasculares: Hemorragia farmacológica de la ciclofosfamida. La transmural, cardiotoxicidad y vasculitis administración conjunta de diuréticos de las arterias coronarias. Gastrointiazídicos puede incrementar la testinales: Anorexia, náusea, vómitos, toxicidad sobre la médula ósea. La diarrea, estomatitis, colitis hemorráciclofosfamida incrementa el riesgo de gica, ulceración de la mucosa oral y infección por vacunas de organismos alteración de la función hepática. (bacterias o virus) vivos. El alopurinol Geni-tourinarias: Cistitis hemorrágica aumenta la mielosupresión. La asagu-da, amenorrea, azoospermia, pirina aumenta el riesgo de sangrado. fibrosis del ovario y formación de Los medicamentos cardiotóxicos coágulos en la pelvis renal. incrementan los efectos adversos 307 2ed. 2004 * Formulario TerapÈutico Nacional CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y en el embarazo. No se debe administrar a pacientes con aplasia medular, cistitis hemorrágica e infección sistémica o urinaria aguda. Usar con precaución en pacientes con diabetes mellitus, trombocitopenia, leucopenia, infiltración tumoral de médula ósea, radioterapia previa, terapia citotóxica previa, alteración de la función hepática y/o renal y en pacientes geriátricos o debilitados. Durante el tratamiento se debe realizar la evaluación semanal de la función hematopoyética. Como posee propiedades inmunosupresoras, se debe considerar la posibilidad de reducción de la dosis o interrupción de la terapia en pacientes con infecciones bacterianas, por hongos o por virus, especialmente en quienes reciben o han recibido recientemente tratamiento con esteroides. Durante el tratamiento se debe suspender la lactancia. La ciclofosfamida puede interferir con la cicatrización de las heridas. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. médico si se presentan reacciones inusuales durante la terapia, en especial: Fiebre, escalofríos, irritación bucal, retardos en la menstruación, sangre en la orina, dolor al orinar, tos, dificultad para respirar, taquicardia, inflamación de los pies o de las extremidades inferiores, dolor estomacal o articular. Advertir al paciente que el tratamiento puede ocasionar oscurecimiento de la piel y de las CISPLATINO FARMACOLOGÕA Antineoplásico del grupo de los agentes alquilantes, que contiene en su estructura átomos de platino que forman un complejo de coordinación. Se intercala entre las bandas y bases del ADN, generando enlaces cruzados que distorsionan la molécula, impidiendo así el proceso de síntesis. Posee también propiedades inmunosupresoras y radiosensibilizantes. FARMACOCIN…TICA Después de la administración IV, sufre una transformación no enzimática dando lugar a uno o más metabolitos. El platino del cisplatino, pero no el cisplatino mismo, se une a varias proteínas plasmáticas incluyendo la albúmina, la transferrina y la gamma-globulina en un 90%. La fracción libre (no unida a proteínas) INFORMACIÓN AL PACIENTE 308 Formulario Terapéutico Nacional * 2ed. 2004 abandona el plasma de una manera bifásica, con una vida media inicial de 25-49 minutos y de 58-73 horas de vida media terminal. El platino se excreta principalmente por la orina, aunque en forma incompleta y prolongada; se ha reportado una eliminación urinaria de 25-45% en 5 días e, incluso, ha sido posible detectar el platino en los tejidos meses después de finalizada la terapia. INDICACIONES Se usa como medicamento único en el tratamiento del carcinoma avanzado de vejiga y, en combinación con otros antineoplásicos, en el tratamiento de tumores metastásicos de testículo y de ovario. R…GIMEN DE DOSIFICACI”N Se administra por vía IV. Las dosis se ajustan en función de la respuesta clínica, efectos hematológicos, renales y la tolerancia del paciente. - Tumor metastásico de testículo: 20 mg/m2/día por 5 días consecutivos (un ciclo) cada 3 semanas, por 3 ciclos. - Tumor metastásico de ovario: 75100 mg/m2/día como dosis única, cada 4 semanas. - Cáncer de vejiga: 50-70 mg/m2/día como dosis única, cada 3-4 semanas. dosis 18 a 23, con recuperación para el día 39. Reacciones de hipersensibili-dad: Reacciones anafilactoides y anafilácticas (edema facial, sibilan-cias, taquicardia e hipotensión a pocos minutos de la administración). Renales: Nefrotoxicidad severa y prolongada (con cursos repetidos de terapia). Neurológicas: Neuritis pe-riférica, convulsiones. Otras: Tinitus, pérdida de la audición, toxicidad vestibular, neuritis óptica, papiledema, ceguera central, visión borrosa, hipomagnesemia, hipokalemia, hipocalcemia, hiponatremia, hipofosfatemia, hiperuricemia. INTERACCIONES El cisplatino puede disminuir los ni- L veles plasmáticos de los anticonvulsivantes. Usado en combinación con aminoglicósidos, amfotericina B, diuréticos de asa y otros agentes con potencial ototóxico y/o nefrotóxico, incrementa el riesgo de tales efectos tóxicos. La aspirina aumenta el riesgo de sangramiento. El uso concomitante de agentes mielosupresores aumenta el riesgo de mielosupresión. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a los compuestos que contienen platino, en pacientes REACCIONES ADVERSAS con insuficiencia renal, mielosupreGastrointestinales: Náuseas, pérdida 309 2ed. 2004 * Formulario TerapÈutico Nacional auditiva. Para minimizar el riesgo de nefrotoxicidad se debe hidratar adecuadamente al paciente con 1-2 L de fluidos 8-12 horas antes de la administración. Para la administración se debe evitar el uso de agujas, jeringas, catéteres y equipos que contengan aluminio, ya que ocurre una reacción de precipitación del platino que compromete la eficacia de la droga. No se conoce la seguridad de su empleo en niños. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico sobre CITARABINA FARMACOLOGÕA Agente antineoplásico de tipo antimetabolito. La citarabina es convertida intracelularmente a trifosfato de citarabina. Este metabolito inhibe la síntesis del ADN. Produce daño al ADN por varios mecanismos, incluyendo la inhibición de la polimerasa alfa del ADN, inhibición de la reparación del ADN a través de un efecto sobre la polimerasa beta del ADN y la inhibición de la transformación de la citidina en desoxicitidina. La 310 FARMACOCIN…TICA Después de la inyección IM ó SC alcanza niveles plasmáticos en 20-60 minutos y concentraciones relativamente estables en 8-12 horas. Una vez absorbida, se distribuye rápida y ampliamente a los tejidos y fluidos corporales, incluido el líquido cefalorraquídeo, donde alcanza concentraciones superiores a las obtenidas en el plasma. Se une a las proteínas plasmáticas en aproximadamente un 13%. Se metaboliza principalmente en el hígado y en menor proporción en el riñón, en la mucosa gastrointestinal, los granulocitos y otros tejidos donde es transformada en metabolitos inactivos. Es convertida intracelularmente a trifosfato de citarabina, metabolito activo que es posteriormente inactivado por la acción de enzimas desaminasas. Se excreta en un 7080% por la orina en 24 horas, un 90% como metabolitos y el resto como fármaco. Después de la administración IV, tiene una vida media de eliminación de 1-3 horas. Después de la administración intratecal, 2 horas. INDICACIONES En combinación con otros agentes antineoplásicos para la inducción de la remisión de la leucemia linfocítica y no linfocítica aguda y por vía intratecal en la profilaxis y tratamiento de la leuce-mia meníngea. Formulario Terapéutico Nacional * 2ed. 2004 100 mg/m2 IV cada 12 horas. Se administra por 7 días y se repite cada 2 semanas. Para el mantenimiento, 1 mg/kg SC una o dos veces a la semana. En leucemia linfocítica aguda: 200 mg/m 2 /día por infusión IV contínua por 5 días a intervalos de 2 semanas. En el tratamiento de la leucemia meníngea por vía intratecal, la dosis se ubica en el rango de 5-75 mg/m2 o 30-70 mg cada 2-7 días o una vez al día por 4-5 días. El régimen de dosifi-cación debe ajustarse según el tipo y severidad de las manifestaciones centrales y la respuesta del paciente a la terapia previa. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómito, diarrea, anorexia, dolor abdominal, inflamación o ulceración oral y/o anal, esofagitis y hemorragia gastrointestinal. Hematológicas: Supresión de la médula ósea con manifestaciones de megaloblastosis, leucopenia, reticulocitopenia, trombocitopenia y anemia. Hepáticas: Alteración de la función hepática. Neurológicas: Mareos, somnolencia, neuritis. Reacciones de hipersensibilidad: Anafilaxis. Renales: Retención urinaria, alteración de la función renal, hiperuricemia. Otras: Fiebre, sepsis, erupción, alopecia, ulceración cutánea, conjuntivitis. Se ha descrito un síndrome específico INTERACCIONES La citarabina puede alterar la actividad antimicótica de la fluocitosina. Puede incrementar el riesgo de infección por vacunas de organismos vivos. El uso en combinación con medicamentos potencialmente mielosupresores incrementa el riesgo de mielosupresión. La administración concomitante con clozapina, tiene el potencial de causar agranulocitosis. No se recomienda la administración de filgrastim o lenograstim entre 24 horas antes y 24 horas después de la quimioterapia. Reduce la absorción de la fenitoína. CONTRAINDICACIONES/ PRECACIONES Contraindicada en pacientes con hipersensibilidad al medicamento y en L mujeres embarazadas. Usar con extrema precaución en pacientes con alteración de la función renal y hepática. Durante la terapia se debe practicar evaluaciones periódicas de la función hematológica, hepática y renal. En pacientes sometidos a tratamiento con dosis elevadas, es recomendable realizar con frecuencia exámenes de la función neurológica. Dado que el medicamento puede producir hiperuricemia, es conveniente determinar los niveles de ácido úrico y establecer medidas farmacológicas en caso necesario. Durante el tratamiento se debe suspender la 2ed. 2004 * Formulario TerapÈutico Nacional 311 presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial manifestaciones que sugieran la posibilidad de una infección, como: Fiebre, dolor de CLORMETINA (MOSTAZA NITROGENADA) FARMACOLOGÕA Antineoplásico del grupo de los agentes alquilantes. Interfiere con la replicación del ADN y la transcripción del ARN, resultando en una alteración de la función de los ácidos nucléicos. FARMACOCIN…TICA Después de la administración IV, sufre una rápida transformación química al reaccionar con los fluidos corporales y componentes celulares de manera tal que resulta indetectable como fármaco intacto después de algunos minutos. Se absorbe muy poco tras la admi-nistración intracavitaria, probable-mente debido a la rápida inactivación por contacto con los fluidos corporales. Menos del 0,01% de la dosis adminis-trada es recuperada intacta en la orina. INDICACIONES En combinación con otros antineoplásicos para el tratamiento de la enfermedad de Hodgkin (estadios III y IV), linfosarcoma, leucemia linfocítica 312 R…GIMEN DE DOSIFICACI”N Se administra por vía IV e intracavitaria. La dosis usual por vía IV es de 0,4 mg/kg en cada curso de tratamiento, administrada como dosis única o dividida en 2-4 dosis. Los cursos subsecuentes se administran a intervalos de 3-6 semanas, dependiendo del estado hematológico del paciente. Cuando se emplea como parte del esquema MOPP (mecloretamina, vincristina, procarbazina y prednisona) se usa en dosis de 6 mg/kg los días 1, 8 y 28 del ciclo. Generalmente, los pacientes reciben, como mínimo, 6 ciclos del esquema, o los necesarios para producir remisión completa, más 2-3 ciclos adicionales para consolidar la quimioterapia. La dosis usual para la administración intracavitaria (intrapleural, intraperitoneal o intracardíaca) es de 0,2-0,4 mg/kg. Después de la ad-ministración se debe cambiar de posi-ción al paciente cada 5-10 minutos durante 1 hora, a fin de lograr una dis-tribución uniforme del medicamento dentro de la cavidad. REACCIONES ADVERSAS Cardiovasculares: Trombosis, tromboflebitis, disritmias, taquicardia, hipertrofia ventricular. Gastrointestinales: Náuseas, vómitos, diarrea, anorexia y hematemesis. Hematológicas: Linfopenia, granulocitopenia y trombocitopenia. Neurológicas: Debilidad, cefalea, vértigo, parálisis muscular, oto- Formulario Terapéutico Nacional * 2ed. 2004 herpes zoster (común en pacientes con linfoma), trastornos de la menstruación y de la espermatogénesis, fiebre, sordera, aumento de la presión del líquido cefalorraquídeo, hiperurice-mia. INTERACCIONES Con anticoagulantes y aspirina aumenta el riesgo de sangramiento. Con agentes mielosupresores, mielosupresión aditiva. infiltración de la médula ósea por células malignas. Los pacientes con leucemia linfocítica crónica resultan especialmente sensibles a la toxicidad hematológica del medicamento. Durante la terapia es recomendable suspender la lactancia. No se ha establecido la seguridad y la eficacia en pacientes pediátricos. SOBREDOSIS Dosis superiores a la recomendada pueden producir leucopenia severa, anemia, trombocitopenia y diátesis hemorrágica. Se ha reportado muertes por esta causa. Tratamiento sintomático y de soporte con hemoderivados, antibióticos y medidas generales, según el caso. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, enfermedades infecciosas, inflamación supurativa aguda o crónica (ya que el medicamento contribuye al desarrollo L de la amiloidosis) y en el embarazo. INFORMACIÓN AL PACIENTE Las mujeres con potencial de ges- Informar de inmediato al médico sobre tación deben evitar el embarazo. Los pacientes sometidos a tratamiento DACARBAZINA presentan un riesgo elevado de complicaciones infecciosas (bacterianas, FARMACOLOGÍA virales y/o por hongos), por lo que es Antineoplásico del grupo de los fundamental mantener las medidas agentes alquilantes. Forma iones profilácticas que dicha circunstancia metilcarbonio reactivos que atacan a amerita. Si durante el tratamiento se los grupos nucleofílicos de la desarrolla infección por herpes zoster, molécula del ADN. Inhibe también la se debe interrumpir la administración síntesis del ADN actuando como para evitar su diseminación. Se debe antimetabolito análogo de las purinas. evaluar la función hematológica antes Interactúa, además, con los grupos de iniciar la terapia y frecuentemente sulfhidrilo de las proteínas. Es activo durante su desarrollo. Los ancianos y durante todas las fases del ciclo las personas debilitadas son parti- celular. cularmente susceptibles2ed. a los efectos 313 2004 * Formulario TerapÈutico Nacional produce una respuesta inicial en el melanoma maligno que se evidencia a los 18-24 días. Se distribuye rápidamente en el organismo y se une a las proteínas plasmáticas en un porcentaje inferior al 5%. Se metaboliza extensamente en el hígado, transformándose en productos que se cree poseen alguna actividad farmacológica. Un 40% de la dosis es excretada intacta por secreción tubular en la orina y un 9-18% como meta-bolitos. También se ha descrito excreción por vía biliar. La vida media de eliminación es bifásica, con una vida media inicial de 19 minutos y una vida media final de 5 horas. INDICACIONES Melanoma metastásico maligno y, en combinación con otros antineoplásicos, para el tratamiento de la enfermedad de Hodgkin. tológicas: Leucopenia, trombocitopenia, eosinofilia, mielosupresión. Hepáticas: Elevación de los niveles de las aminotransferasas, oclusión de la vena hepática, necrosis hepatocelular. Neurológicas: Confusión, dolor de cabeza, parestesia, letargia, convulsiones, visión borrosa. Reacciones de hipersensibilidad: Eritema, erupción máculopapular, fotosensibilidad, anafilaxis. Otras: Síndrome tipo influenza, elevación del BUN, alopecia y dolor intenso en el sitio de la inyección. INTERACCIONES El uso en combinación con fármacos mielosupresores incrementa el riesgo de mielosupresión. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con falla hepática y/o renal. La extravasación durante la administración IV puede causar lesiones tisulares serias. Durante el tratamiento se debe vigilar frecuentemente la función hematológica y hepática. Su uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales para la madre superen al posible riesgo para el feto. Se recomienda suspender la lactancia durante el tratamiento. RÉGIMEN DE DOSIFICACIÓN Se administra por infusión IV en 15-30 minutos o en bolo en 1-2 minutos. - Melanoma: 2-4,5 mg/kg/día por 10 días continuos, con repeticiones a intervalos de 4 semanas. Alternativamente, 250 mg/m2/día por 5 días, con repeticiones cada 3 semanas. - Enfermedad de Hodgkin: 150 mg/m2/día por 5 días, en combinación con otros antineoplásicos, y repeticiones cada 4 semanas. AlSOBREDOSIS 314 Formulario Terapéutico Nacional * 2ed. 2004 síndrome tipo influenza e infecciones. Evitar la exposición excesiva a la luz solar. Evitar contacto con multitudes y con personas con enfermedades infecciosas. Notificar de inmediato al médico si se presenta alguna DACTINOMICINA (ACTINOMICINA D) mediante técnica de perfusión aislada regional, como terapia paliativa o coadyuvante de la cirugía en el tratamiento de sarcomas, carcinomas y adenocarcinomas que no responden a los tratamientos tradicionales. R…GIMEN DE DOSIFICACI”N Se administra por vía IV (excepto en la FARMACOLOGÕA técnica de perfusión regional). La Antineoplásico del tipo de los anti- dosis debe ser basada en la respuesta bióticos. Se postula que el medi- clínica y hematológica y en la camento se intercala entre los tolerancia del paciente. La dosis usual residuos de guanina de la molécula de cada curso de tratamiento en del ADN, formando un complejo que adultos es de 500 mcg/día IV por un im-pide la síntesis del ARN. Posee, máximo de 5 días. El régimen puede a d e - m á s , p r o p i e d a d e s repetirse después de 3 o más inmunosupresoras y actividad semanas si no se evidencian signos de hipocalcémica. efectos tóxicos residuales. La dosis diaria no debe exceder de 15 L FARMACOCIN…TICA mcg/Kg/día o 400-600 mg/m2/día por Después de la administración IV, se 5 días. Se pueden administrar dosis distribuye rápidamente a los tejidos, inferiores cuando se combina la dactialcanzando mayores concentracio- nomicina con otros antineoplásicos o nes en la médula ósea y en las con radioterapia. La dosis usual en células nucleadas, incluyendo niños es de 15 mcg/kg/día por 5 días granulocitos y linfocitos. No atraviesa o un total de 2,5 mg/m2 administrados la barrera hematoencefálica. Se en dosis divididas en un periodo de 7 metaboliza en muy escasa proporción, días. En adultos y niños, los cursos por lo que se excreta casi suce-sivos deben administrarse a completamente intacta por la orina y intervalos de 2-4 semanas, una vez las heces (30%) en una semana. La desapa-recidos los signos de vida media de eliminación es de t o x i c i d a d . L a s d o s i s p a r a l a aproximadamente 36 horas. administración locali-zada, mediante la técnica de perfusión aislada INDICACIONES regional, es de 50 mcg/kg para las Sola o en combinación con otros an- extremidades inferiores y de 5 mcg/kg 315 2ed. 2004 * Formulario TerapÈutico Nacional terapia produce eritema cutáneo y de las mucosas oral y faríngea que puede progresar a hiperpigmentación, ede-ma, descamación y vesiculación. Gastrointestinales: Náuseas, vómito, queilitis, esofagitis, estomatitis, faringitis, anorexia, dolor abdominal, diarrea, ulceración gastrointestinal y proctitis. Hematológicas: Anemia aplásica, pancitopenia, reticulocitopenia, agranulocitosis y trombocitopenia. Reacciones de hipersensibilidad: Erupción maculopapular prurítica, reacciones anafilactoides. Otras: Malestar, fatiga, letargo, fiebre, mialgia, hipocalcemia y reacciones anafilactoides. La administración por técnica de perfusión puede generar mielosupresión, absorción de productos tóxicos por destrucción masiva de tejido, aumento de la susceptibilidad a infecciones y edema del área perfundida. Es extremadamente corrosiva para los tejidos blandos. Produce dolor y daño severo, necrosis, celulitis, flebitis e inflamación cuando ocurre extravasación durante la administración. medicamento puede producir mielosupresión, los pacientes presentan riesgo de sufrir complicaciones infecciosas y hemorrágicas. Se debe realizar evaluación hematológica com-pleta diariamente durante la terapia. Se recomienda evaluar frecuentemen-te la función hepática y la función renal. Si se presentan estomatitis o diarrea severa durante la terapia, el medica-mento debe suspenderse hasta la recuperación. Debe tenerse precau-ción en pacientes que han recibido radioterapia poco tiempo antes del tratamiento, especialmente en pacientes con tumor de Wilms debido a que se puede presentar hepatomegalia. Dado que el medicamento es altamente tóxico, debe ser manipulado y administrado con mucho cuidado. Evitar la inhalación del polvo o los vapores y el contacto con los ojos y las mucosas. En caso de ocurrir, se debe lavar inmediatamente la zona expues-ta con abundante agua por 15 minutos. Los niños son más susceptibles a los efectos adversos del fármaco. Se debe suspender la lactancia durante el tratamiento. INTERACCIONES El uso concurrente o secuencial con doxorubicina incrementa la cardioto- DAUNORUBICINA xicidad de ésta. La dactinomicina incrementa el riesgo de infección por FARMACOLOGÍA Antineoplásico de tipo antibiótico vacunas de organismos (bacterias o glicósido antraciclínico cuyo mecavirus) vivos. Disminuye la efectividad 316 Formulario Terapéutico Nacional * 2ed. 2004 nismo de acción no ha sido completamente elucidado. La evidencia experimental sugiere que inhibe la síntesis del ADN al intercalarse entre los pares de bases de la molécula y producir distorsiones estructurales, a la vez que inhibe la actividad de la enzima topoisomerasa II que interviene en los procesos de reparación. FARMACOCINÉTICA Después de la administración IV, se distribuye rápida y ampliamente en el organismo, alcanzando concentraciones elevadas en riñón, bazo, hígado, pulmón y corazón. Se metaboliza en el hígado y otros tejidos por la acción de enzimas aldo-ceto-reductasas transformándose en daunorubicinol, metabolito principal con actividad antineoplásica, y otros productos inactivos. Un 13-25% de la dosis se excreta por la orina como fármaco intacto y como metabolito activo y un 40% por la bilis. La vida media de eliminación de la daunorubicina es de 18,5 horas y la del daunorubicinol de 26,7 horas. Estos valores se incrementan en pacientes con insuficiencia hepática y/o renal. INDICACIONES En combinación con otros agentes antineoplásicos para la inducción de remisión de la leucemia no linfocítica aguda (mielógena, monocítica, eri- - InducciÛn de la remisiÛn en casos de leucemia no linfocÌtica aguda en adultos menores de 60 aÒos: 30-45 mg/m2/dÌa por 3 dÌas consecutivos en el primer curso y por 2 dÌas en los cursos subsiguientes. En adultos mayores de 60 aÒos: 30 mg/m2/dÌa por 3 dÌas consecutivos en el primer curso y por 2 dÌas en los cursos subsiguientes. - InducciÛn de la remisión en casos de leucemia linfocítica aguda en adultos: 45 mg/m2/día por 3 días consecutivos. En niños: 25 mg/m2/día 1 vez por semana por 46 semanas. REACCIONES ADVERSAS Cardiovasculares: Cardiotoxicidad (al- L teraciones en el ECG, arritmias, trastornos de conducción, insuficiencia cardiaca congestiva). Gastrointestinales: Náuseas, vómitos, diarrea, dolor abdominal. Hematológicas: Supresión de la médula ósea (con eventuales consecuencias hemorrágicas e infecciosas). Reacciones de hipersensibilidad: Anafilaxis, erupción, urticaria. Otras: Hiperuricemia, alo-pecia y reacciones en el sitio de la inyección (necrosis tisular, celulitis severa, tromboflebitis e induración dolorosa) generalmente debidas a extravasación. INTERACCIONES 2ed. 2004 * Formulario TerapÈutico Nacional 317 norubicina. Lo mismo es válido para los medicamentos nefrotóxicos. La administración de vacunas de virus vivos a pacientes inmunosuprimidos por antineoplásicos pueden generar infecciones severas e, inclusive, fatales. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento, en pacientes con terapia previa con doxorubicina y en el embarazo. Usar con extrema precaución en pacientes con enfermedad cardiaca preexistente. En pacientes con falla renal y/o hepática se debe administrar en dosis reducidas. Se recomienda administrar en los vasos sanguíneos de gran calibre. La extravasación durante la administración puede causar lesiones tisulares serias. Se debe realizar evaluación de la función cardiaca, hematológica, renal y hepática antes de iniciar el tratamiento, y a intervalos frecuentes durante su desarrollo. La vigilancia de la función cardiaca, en particular, debe mantenerse por varios meses después de finalizada la terapia. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE 318 tudes y con personas con enfermedades infecciosas. Practicar medidas anDOXORUBICINA FARMACOLOGÕA Antineoplásico de tipo antibiótico glicósido antraciclínico cuyo mecanismo de acción no ha sido completamente dilucidado. La evidencia experimental sugiere que se intercala entre los pares de bases de la molécula de ADN formando un complejo que impide la síntesis de ácidos nucléicos y la acción de las ADN y ARN polimerasas. El medicamento es activo durante todo el ciclo celular, incluyendo durante la interfase. Además, se une a los lípidos de la membrana. FARMACOCIN…TICA Como sufre degradación por acción de los jugos gastrointestinales y, además, resulta extremadamente irritante para los tejidos, sólo puede ser adminis-trada por vía IV. Se distribuye amplia-mente en los tejidos, alcanzando ni-veles apreciables en el hígado, el pulmón, el corazón y los riñones en sólo 30 segundos. No atraviesa la barrera hematoencefálica. Penetra a la célula, donde se une a diversos componentes celulares y, en especial, a los ácidos nucléicos. Se metaboliza en el hígado transformándose en doxorubicinol Formulario Terapéutico Nacional * 2ed. 2004 Sigue una distribución multifásica. La vida media distributiva inicial de 5 minutos sugiere una captación tisular rápida, pero la lenta eliminación de los tejidos se refleja en la vida media terminal de 20-48 horas. INDICACIONES Para inducir la regresión en condiciones neoplásicas diseminadas como la leucemia linfoblástica aguda, leucemia mieloblástica aguda, tumor de Wilms, neuroblastoma, sarcoma de hueso y tejido blando, linfoma Hodgkin y no Hodgkin y carcinoma de mama, estómago, ovario, vejiga, tiroides y broncogénico (especialmente de célu-las pequeñas). R…GIMEN DE DOSIFICACI”N Para obtener el máximo beneficio con un mínimo de efectos adversos, la dosis debe basarse en la respuesta clínica, hepática, renal y hematológica, en la tolerancia del paciente y en el uso simultáneo de otros antineoplá-sicos. La dosis usual es de 60-75 mg/m2 en una sola dosis a intervalos de 21 días. Otros regímenes proponen 30 mg/m2/día por 3 días continuos cada 4 semanas o 20 mg/m2 una vez/semana. Este último régimen de dosificación puede producir menor incidencia de insuficiencia cardiaca congestiva. La dosis total no debe exceder 550 mg/m2 dado el riesgo de cardiotoxicidad REACCIONES ADVERSAS Cardiovasculares: Cardiotoxicidad aguda que se manifiesta por cambios en el ECG, taquicardia sinusal y extrasístoles; cardiotoxicidad crónica que se manifiesta con insuficiencia cardiaca congestiva y descompensación cardiorespiratoria que puede ocurrir semanas o inclusive meses después de descontinuada la terapia. Puede ocurrir fleboesclerosis y enrojecimiento facial por administración rápida y/o en venas pequeñas. Gastrointestinales: Náuseas y vómitos severos, estomatitis, esofagitis, ulceración y necrosis del colon. Hematológicas: Leucopenia (principalmente granulocitopenia), trombocitopenia y anemia. Reacciones de hipersensibilidad: Urticaria y anafilaxis. Otras: L Fiebre, escalofrío, alopecia completa y reversible, hiperpigmentación de las uñas y de los pliegues de la piel. Si hay extravasación puede ocurrir necrosis tisular local, celulitis, vesicación, trom-boflebitis e induración dolorosa. INTERACCIONES Puede disminuir la concentración plas-mática de digoxina. Potencia la cistitis hemorrágica producida por la ciclofos-famida y aumenta la hepatotoxicidad de la 6mercaptopurina. Puede reducir la eficacia anticonvulsivante de la fenitoína. Incrementa el riesgo de infección por vacunas de organismos 319 2ed. 2004 * Formulario TerapÈutico Nacional mar un precipitado. Con los bloqueantes de los canales de calcio puede potenciar los efectos cardiotóxicos. séricos de ácido úrico durante el tratamiento y establecer el manejo respectivo en caso necesario. El medicamento produce una coloración roja en la orina sin importancia clínica. Debe tenerse la precaución de evitar la extravasación. Se recomienda la administración del medicamento en venas de gran calibre. Debe evitarse la lactancia durante el tratamiento. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, insuficiencia cardiaca, en pacientes que han recibido previamente tratamiento completo con dosis acumulativas de daunorubicina y en el embarazo. No debe iniciarse tratamiento en pacien- SOBREDOSIS tes con mielosupresión severa induci- Tratamiento sintomático y de soporte, da por quimioterapia o radioterapia especialmente orientado al manejo de previa. El melanoma maligno, el carci- los efectos hematológicos, cardíacos noma de hígado, el carcinoma de in- y gastrointestinales. testino grueso, los tumores cerebrales y la metástasis en sistema nervioso INFORMACIÓN AL PACIENTE central no responden bien al medi- Informar de inmediato al médico si se c a m e n t o . S e r e c o m i e n d a l a presenta alguna reacción o sintomaevaluación de la función hepática, tología inusual durante el tratamiento, hematológica y cardiaca antes de en especial cualquier manifestación iniciar la terapia y a intervalos frecuentes durante su desarrollo. ETOPÓSIDO Puede ser necesario disminuir la dosis o descontinuar el medicamento en FARMACOLOGÕA Antineoplásico semisintético cuyo pacientes con mielosupresión severa mecanismo de acción no se conoce o en aquellos que presenten con precisión. Se cree que provoca infiltración de células malignas en la alteraciones estructurales en la momédula ósea. Si se presentan lécula de ADN, generando trastornos manifestaciones de infec-ción y/o sangramiento debe conside-rarse la o inhibición del proceso de síntesis. terapia con antibióticos y/o derivados FARMACOCIN…TICA sanguíneos. La cardiotoxi-cidad La absorción por vía oral se ha puede ser potenciada por la estimado en un 50%, con concenciclofosfamida o por la daunorubicina. 320 Formulario Terapéutico Nacional * 2ed. 2004 intraindividual e interindividual. Con la administración IV también se observan variaciones en los niveles plasmáticos y en la cantidad de fármaco absorbido, pero menos marcadas que con la dosificación oral. Una vez absorbido se distribuye rápidamente a los tejidos y fluidos corporales, pero no atraviesa la barrera hematoencefálica. Se distribuye a la bilis en escasa proporción. Se une en un 97% a las proteínas plasmáticas. El metabolismo no está bien determinado; se sabe que se transforma en diversos derivados sin actividad antineoplásica. Un 40-60% del fármaco intacto y sus metabolitos son excretados en 72 horas por la orina, mientras un 2-16% lo hace por las heces. La eliminación parece ser bifásica, con una vida media inicial de 0,6-2 horas y de 5,3-10,8 horas en la fase terminal. INDICACIONES En combinación con otros antineoplásicos para el tratamiento de tumores testiculares refractarios y del carcinoma de células pequeñas de pulmón. R…GIMEN DE DOSIFICACI”N Se administra por vía oral o por infusión IV en 30-60 minutos. La dosis dependerá del estado clínico, la respuesta hematológica, la tolerancia terapia. En el carcinoma de células pequeñas de pulmón la dosis usual varía desde 35 mg/m2/día IV por 4 días seguidos hasta 50 mg/m2/día IV por 5 días consecutivos cada 3-4 semanas. La dosis por vía oral es el doble de la dosis IV. REACCIONES ADVERSAS Cardiovasculares: Hipotensión (en la administración IV rápida), taquicardia. Dermatológicas: Alopecia, pigmentación de la piel, erupción y prurito. Gastrointestinales: Náusea y vómitos, anorexia, dolor abdominal, diarrea y estomatitis. Hematológicas: Leucopenia, trombocitopenia y anemia. La mielosupresión no es acumulativa pero puede ser más severa en pacientes que han recibido previamente L radioterapia o tratamiento con otros antineoplásicos. Neurológicas: Neuropatía periférica, fatiga, somnolencia y dolor de cabeza. Reacciones de hipersensibilidad: Reacciones anafilactoides que se manifiestan con fiebre, escalofríos, disnea, broncoespasmo, taquicardia y alteraciones de la presión arterial (hipotensión o hipertensión); anafilaxis. INTERACCIONES Pacientes tratados previamente con cisplatino pueden manifestar alteraciones en la eliminación del etopósido. El etopósido incrementa el riesgo de infección por vacunas de organismos 321 2ed. 2004 * Formulario TerapÈutico Nacional CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes hipersensibles al medicamento y durante el embarazo. Debido a la elevada incidencia y severidad, se debe prestar atención especial a la aparición precoz de reacciones anafilactoides y a la hipotensión. Se debe practicar evalua-ciones hematológicas previas al inicio de la terapia, frecuentemente durante su desarrollo y después de finalizada. Si el contaje plaquetario resulta menor que 50.000/mm3 o el de neutrófilos menor que 500/mm3, debe suspen-derse el tratamiento hasta la recu-peración de la médula ósea. La efica-cia y la seguridad en niños no han sido totalmente establecidas. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico si se FLUOROURACILO (5-FLUOROURACILO) FARMACOLOGÕA Antineoplásico fluorado que actúa como antimetabolito inhibiendo a la enzima timidilato sintetasa, interfiriendo con la síntesis de ADN. El fármaco, así mismo, se incorpora al ARN 322 terado que no es reconocido por el ARN de transferencia al momento de su lectura para la síntesis de proteínas. Adicionalmente, el fluorouracilo blo-quea la enzima fosfatasa de uracilo impidiendo la utilización del uracilo en la síntesis de ARN. FARMACOCIN…TICA Después de la administración IV, el medicamento es distribuido a los tumores, médula ósea, mucosa intestinal, hígado, sistema nervioso central y otros tejidos. Algunos estudios han demostrado que el fármaco y sus metabolitos logran concentraciones más elevadas y persistentes en el tejido tumoral que en el tejido sano circundante, lo que sugiere una especificidad del fluorouracilo contra ciertos tumores en comparación con el tejido normal. Una pequeña porción del fármaco se transforma en los tejidos en un metabolito activo y el resto se metaboliza en el hígado. Un 10-15% del fármaco se excreta intacto en la orina, mientras los metabolitos (6080%) se eliminan como dióxido de carbono por vía respiratoria y como urea por la orina. La vida media de eliminación es de 16 minutos. INDICACIONES Tratamiento paliativo del carcinoma de Formulario Terapéutico Nacional * 2ed. 2004 paciente; si es obeso o está edematizado se utiliza el peso ideal. No obstante, la dosis diaria no debe exceder, en ningún caso, 800 mg totales. La dosis inicial usual es de 12 mg/kg una vez al día por 4 días consecutivos. Si la toxicidad no impide continuar el tratamiento, administrar 6 mg/kg los días 6, 8, 10 y 12, a menos que ocurran reacciones tóxicas antes. En pacientes con estado nutricional inadecuado se recomienda una dosis inicial de 6 mg/kg por 3 días consecutivos, seguidos por 3 mg/kg los días 5, 7 y 9, a menos que ocurran reacciones tóxicas previamente. En pacientes sin problemas de toxicidad, repetir el curso de tratamiento 30 días después de la última dosis. Cuando la toxicidad del curso inicial ha mejorado, se puede proceder al próximo ciclo con una dosis de mantenimiento de 10-15 mg/kg una vez por semana, sin exceder de 1 g/semana. REACCIONES ADVERSAS Cardiovasculares: Isquemia miocárdica, angina y tromboflebitis. Dermatológicas: Alopecia, fotosensibilidad, dermatitis, hiperpigmentación, cambios en las uñas. Gastrointestinales: Anorexia, náuseas, vómito, estomatitis, faringitis, esofagitis, gastritis, duodenitis, diarrea y úlcera duodenal. Hematológicas: Leucopenia (especialmente granulo- epistaxis. INTERACCIONES El fluorouracilo incrementa el efecto anticoagulante de la warfarina y puede aumentar la posibilidad de infección por vacunas de organismos (bacterias o virus) vivos. La cimetidina incrementa las concentraciones séricas de fluorouracilo. El ácido folínico aumenta la toxicidad del fluorouracilo. Los diuréticos tiazídicos pueden incremen-tar el potencial mielosupresor del fluorouracilo. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento y durante el embarazo. También, en paL cientes con estado nutricional pobre, depresión de la médula ósea o infecciones potencialmente serias o que hayan sido sometidos recientemente a una cirugía mayor. Usar con extrema precaución en pacientes que hayan recibido altas dosis de radiación pélvica o agentes alquilantes, pacientes con insuficiencia hepática o renal, pacientes con metástasis generalizada que involucre la médula ósea y pacientes con enfermedad coronaria preexistente. Si se presentan vómitos intratables, estomatitis severa con ulceración, esofagitis o faringitis con dolor de garganta o disfagia, diarrea severa, úlceras gastrointestinales, he- 2ed. 2004 * Formulario TerapÈutico Nacional 323 debajo de 100.000/mm3, el tratamiento debe descontinuarse. Si los glóbulos blancos están por debajo de 2000/mm3 el paciente debe aislarse para evitar infecciones. No se ha establecido la eficacia y la seguridad del medicamento en niños. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACI”N AL PACIENTE Se debe informar al médico de inmediato sobre cualquier manifestación de GEMCITABINA FARMACOLOGÍA Antineoplásico tipo antimetabolito específico de fase. Provoca la muerte de la célula durante la fase S (síntesis de ADN) del ciclo celular y se cree, además, que bloquea la progresión de la célula de la fase G1 a la fase S. Luego de la administración sistémica es metabolizada intracelularmente por enzimas quinasas de nucleósido y convertida en los nucleósidos difosfato y trifosfato activos que interfieren con los mecanismos que conducen a la síntesis del ADN. buye más ampliamente, lo que parece resultar de un lento equilibrio en el compartimiento tisular. La unión a las proteínas plasmáticas es insignificante. Luego de la activación intracelular, es posteriormente desaminada en el hígado, el riñón y el plasma, dando lugar a un producto inactivo derivado de la uridina. Un 92-98% de la dosis administrada es recuperada en la orina en aproximadamente una semana, 5-10% como fármaco intacto y el resto como metabolitos inactivos. El tiempo de eliminación varía con la edad y el género del paciente, lo cual, sumado a la variabilidad de la distribución como resultado de la duración de la infusión, da lugar a un amplio rango de vida media de eliminación de 32-638 minutos. INDICACIONES Cáncer de páncreas y cáncer de células no-pequeñas de pulmón. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV. - C·ncer de páncreas: 1000 mg/m2 por infusión IV en 30 minutos, semanalmente por 7 semanas continuas, seguido por 1 semana de descanso y reinicio con ciclos subsiguientes de 3 semanas continuas cada 4 semanas. - Cáncer de células no-pequeñas de pulmón: Se han descrito 2 esquemas en combinación con cisplatino: FARMACOCINÉTICA Después de la administración IV, la distribución al organismo depende de la duración de la infusión; con Nacional infu324 Formulario Terapéutico * 2ed. 2004 después de la infusión de la gemcitabina. - Esquema de 3 semanas: 1250 mg/m2 por infusión IV en 30 minutos los días 1 y 8 de un ciclo de 21 días (3 semanas), en combinación con cisplatino 100 mg/m2 IV el día 1 de cada ciclo, inmediatamente después de la infusión de la gemcitabina. REACCIONES ADVERSAS Cardiovasculares: Hipertensión arterial, infarto al miocardio, arritmias, accidente cerebro-vascular. Gastrointestinales: Náusea, vómito, diarrea, constipación, estomatitis. Hematológicas: Anemia, neutropenia, leucopenia, trombocitopenia, mielosupresión. Hepáticas: Elevación de los niveles de las enzimas hepáticas. Neurológicas: Cefalea, parestesia. Reacciones de hipersensibilidad: Erupción, broncoespasmo y reacciones anafilactoides, anafilaxis. Renales: Síndrome urémico hemolítico, proteinuria, hematuria, falla renal. Respiratorias: Disnea, broncoespasmo. Otras: Fiebre, ede-ma, síndrome tipo gripe, infección, alopecia. INTERACCIONES Las vacunas de virus vivos en pacientes inmunosuprimidos por antineoplásicos pueden dar origen a infecciones severas e, inclusive, fatales. El uso de la gemcitabina en embarazo. Se debe realizar una evaluación de la función cardiaca, hematológica, renal y hepática antes de iniciar el tratamiento y a intervalos frecuentes durante su desarrollo. Usar con precaución en pacientes con falla renal y/o hepática, historia de enfermedad cardiovascular, en ancianos y durante la radioterapia (la gemcitabina es radiosensibilizante). No se conoce la eficacia y seguridad de su uso en niños. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Posible mielosupresión. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Advertir al paciente sobre la posiL bilidad de sangramiento e infecciones, y sobre la necesidad de notificar de inmediato al médico la ocurrencia de manifestaciones de tal naturaleza o de HIDROXICARBAMIDA (HIDROXIUREA) FARMACOLOGÕA Antineoplásico derivado de la urea. El mecanismo de acción no se conoce con precisión. Se cree que produce una inhibición inmediata de la síntesis de ADN sin interferir con la síntesis de ARN y de las proteínas. Existe evidencia que sugiere que el medicamento inhibe la incorporación de timidina en 2ed. 2004 * Formulario TerapÈutico Nacional 325 FARMACOCIN…TICA Se absorbe bien en el tracto gastrointestinal, alcanzando concentraciones séricas pico en aproximadamente 1-4 horas. Se distribuye ampliamente a los tejidos y fluidos corporales ge-nerando concentraciones máximas en el líquido cefalorraquídeo en 3 horas. Se metaboliza parcialmente en el hígado y se excreta en un 80% en la orina en 12 horas. Una pequeña fracción se elimina como dióxido de carbono por vía respiratoria. La vida media de eliminación es de 3 horas. INDICACIONES Tratamiento del melanoma, leucemia mielocítica crónica y carcinoma de ovario inoperable, recurrente o metastásico. También es usada en combi-nación con radioterapia para el control local de carcinoma primario de células escamosas de cabeza y cuello. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. La dosis usual en tumores sólidos es de 80 mg/kg cada 3 días o 20-30 mg/kg/día. En la leucemia mielocítica crónica: 20-30 mg/kg/día durante el tiempo necesario para mantener remisión. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, anorexia, constipación, diarrea, mu326 INTERACCIONES No se conocen casos de interacción entre hidroxicarbamida y otros medicamentos en humanos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al fármaco, mielosupresión y en el embarazo. Usar con extrema precaución en pacientes con falla renal y/o hepática y en aquellos sometidos recientemente a quimioterapia o a radioterapia. Se debe evaluar la función hematológica, hepática y renal antes de iniciar el tratamiento y frecuentemente durante su desarrollo. Dado que los pacientes geriátricos resultan particularmente sensibles a los efectos del medicamento, en ellos se deben emplear dosis menores. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y medidas de soporte. IFOSFAMIDA FARMACOLOGÍA Antineoplásico del grupo de los agentes alquilantes, análogo de la ciclofosfamida, químicamente relacio-nado con la mostaza Formulario Terapéutico Nacional * 2ed. 2004 Debido a que produce entrecruzamiento de las bandas, interfiere con la replicación del ADN y la transcripción del ARN, alterando la función de los ácidos nucléicos, llevando a la muerte celular. FARMACOCINÉTICA Luego de su administración IV la respuesta alquilante se inicia en 1-6 horas. La distribución en el organismo no ha sido bien establecida. Se une a las proteínas plasmáticas en un 20%. Se metaboliza extensamente en el hígado dando lugar a metabolitos activos e inactivos. Se ha demostrado que los metabolitos alquilados de la ifosfamida interactúan con el ADN. También se ha encontrado que produce acroleína. La eliminación después de dosis altas (3,8-5 g/m2), exhibe un comportamiento bifásico con una vida media de eliminación de 15 horas, mientras que con dosis bajas (1,6-2,4 g/m2) la eliminación es monoexponencial, con una vida media de 7 horas. La ifosfamida y sus metabolitos se excretan por vía renal en un 12-18% con dosis bajas y en un 70-86% con dosis altas. INDICACIONES En combinación con otros antineoplásicos para el tratamiento del cáncer testicular de células germinales, cáncer de cabeza y cuello, mama, cervix, ovarios, pulmón, sarcoma de RÉGIMEN DE DOSIFICACIÓN Se usa por vía IV en dosis de 1,2 g/m≈/día por 5 días consecutivos, cada 3 semanas o después de la recu-peración de valores hematológicos deprimidos (plaquetas sobre 100.000/mm3 y glóbulos blancos sobre 4.000/mm 3 ). Se usa en combinación con mesna para prevenir la cistitis hemorrágica y abundante hidratación (2 L/día de fluidos por vía oral o IV). Ver: MESNA en la sección VARIOS. REACCIONES ADVERSAS Cardiovasculares: Arritmias supraventriculares y cambios en el ECG, flebitis. Gastrointestinales: Náusea y vómitos, anorexia, diarrea y constipación. Hematológicas: Mielosupresión L (leucopenia y/o trombocitopenia). Hepáticas: Alteración de la función hepática. Neurológicas: Somnolencia, psicosis depresiva, confusión, alucinaciones, mareo, desorientación, disfunción de nervios craneales, convulsiones y coma. Reacciones de hipersensibilidad. Renales: Cistitis hemorrágica, disuria, urgencia urinaria, falla renal aguda y síndrome de Fanconi. Otras: alopecia, fiebre. INTERACCIONES En combinación con warfarina incrementa el riesgo de hemorragia. Aumenta el riesgo de infección por vacunas de bacterias o virus vivos. Los 2ed. 2004 * Formulario TerapÈutico Nacional 327 hipersensibilidad al medicamento, con depresión severa de la médula ósea y durante el embarazo. Debe usarse con precaución en pacientes con insuficiencia renal o con insuficiencia hepática, así como en aquellos con alteraciones del sistema nervioso central. Se debe realizar evaluación hematológica antes de iniciar la terapia y periódicamente durante su desa-rrollo. Asimismo, se recomienda la vigilancia frecuente de la función renal y atención particular al desarrollo de cistitis hemorrágica. Se debe asociar a mesna y a una hidratación adecuada durante el tratamiento. La eficacia y seguridad de su uso en niños no ha sido bien establecida. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Tratamiento sintomático y de soporte (Ver: MESNA en la sección VARIOS). INFORMACIÓN AL PACIENTE Durante el tratamiento se debe MELFALÁN FARMACOLOGÕA Antineoplásico alquilante del tipo de la bis-cloroetilamina, que produce entrecruzamiento de las bandas de ADN e interfiere con la replicación del mismo y con la transcripción del ARN. También posee propiedades inmu328 FARMACOCIN…TICA Se absorbe en forma errática e incompleta en el tracto gastrointestinal, alcanzando concentraciones plasmáticas variables. Se distribuye rápidamente en el agua corporal total y se une a las proteínas plasmáticas en un 50-60% inicialmente y en 80-90% a las 4-12 horas. Aproximadamente 20% se une irreversiblemente a las proteínas plasmáticas. Penetra muy poco a través de la barrera hematoencefálica. Sufre hidrólisis en la sangre, trans-formándose en derivados mono y dihidroxilados que son eliminados, junto con el fármaco intacto, en las heces y en menor proporción en la orina. La vida media de eliminación plasmática del melfalán es de 1,5 horas y la de sus derivados hi-droxilados 2-3 veces mayor. INDICACIONES En combinación con otros antineoplásicos para el tratamiento paliativo del mieloma múltiple y del carcinoma epitelial ovárico no operable. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. La dosis debe fundamentarse en la respuesta clínica y hematológica, y en la tolerancia del paciente. A veces puede ser necesario mantener cierto grado de mielosupresión; un contaje leucocitario entre 3000-4000/mm 3 es Formulario Terapéutico Nacional * 2ed. 2004 elementos sanguíneos y reiniciar tratamiento con dosis de mantenimiento de 2 mg/día. Otro esquema propone dosis de 0,15 mg/kg/día por 7 días o 0,25 mg/kg/día por 4 días, a intervalos de 4-6 semanas, usualmente con prednisona. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, taquicardia, edema. Gastrointestinales: Náusea, vómitos, diarrea, ulceración de la mucosa oral, estomatitis. Hematológicas: Leucopenia, trombocitopenia, anemia hemolítica, pancitopenia, agranulocitosis. Hepáticas: Hepatotoxicidad. Reacciones de hipersensibilidad: Anafilaxis, urticaria. Respiratorias: Neumonitis, fibrosis pulmonar, disnea, broncoespasmo. Otras: Alopecia, trastornos menstruales, ulceración en el sitio de inyección, hiperuricemia. INTERACCIONES La administración conjunta de dosis IV elevadas de melfalán y ácido nalidíxico en niños ha producido enterocolitis he-morrágica severa. Aumento del riesgo de sangramiento con anticoagulantes y aspirina. Disminuye la eficacia de los agentes antigotosos. Efecto aditivo con los supresores de la médula ósea. Disminuye la eficacia de las vacunas de virus muertos y aumenta el riesgo de toxicidad con las vacunas de virus con funciÛn medular comprometida o en recuperaciÛn despuÈs de radioterapia o de quimioterapia. Observar cuidadosamente a los pacientes con azoemia para reducir las dosis, si es necesario. Es recomendable realizar una evaluaciÛn hematolÛgica completa antes de iniciar la terapia y frecuentemente durante la misma. Se debe descontinuar temporalmente la terapia o reducir la dosis al primer signo de depresiÛn de la mÈdula Ûsea (leucocitos por debajo de 3000/mm3 y plaquetas por debajo de 100.000/mm3). No se conoce bien la eficacia y seguridad del uso del melfal·n en niÒos. Debe suspenderse la lactancia durante el tratamiento. No administrar vacunas hasta por lo menos 3 meses despuÈs de la ˙ltima L dosis de melfal·n. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE MERCAPTOPURINA FARMACOLOGÕA Antineoplásico que actúa como antimetabolito al convertirse intracelularmente en un ribonucleótido antagonista de las purinas, y que interfiere con la síntesis de ADN y ARN. Posee, además, propiedades inmunosupresoras. 2ed. 2004 * Formulario TerapÈutico Nacional 329 FARMACOCIN…TICA Se absorbe en forma errática e incompleta por vía oral alcanzando concentraciones plasmáticas variables que llegan a su valor máximo en aproximadamente 2 horas. Se cree que sufre metabolismo en la mucosa gastrointestinal y/o metabolismo hepático de primer paso. Se distribuye rápidamente en los líquidos corporales, pero penetra poco a través de la barrera hematoencefálica. Se une a las proteínas plasmáticas en un 19%. Es oxidado parcialmente en el hígado a ácido 6-tioúrico por la acción de las enzimas oxidasas de la xantina y de las 6-metiltiopurinas, excretándose después, junto con el fármaco intacto, en la orina. La vida media plasmática es de 21 minutos en los niños y de 47 minutos en los adultos. INDICACIONES En combinación con otros antineoplásicos en el tratamiento de leucemias (linfáticas y mielógenas) agudas. REACCIONES ADVERSAS Dermatológicas: Hiperpigmentación de la piel. Gastrointestinales: Ulceración intestinal, náusea, vómito, anorexia y diarrea. Hematológicas: Leucopenia, trombocitopenia, anemia, agranulocitosis y pancitopenia. Hepáticas: Ictericia colestástica, ascitis, encefalopatía hepática y/o elevación de los niveles de las enzimas hepáticas. Reacciones de hipersensibilidad: Urticaria. Otras: Hiperuricemia, fiebre. INTERACCIONES El alopurinol inhibe el metabolismo oxidativo de la mercaptopurina incrementando su toxicidad. La administración con fármacos potencialmente hepatotóxicos, incrementa el riesgo de reacciones hepáticas adversas. Antagoniza el efecto de los relajantes musculares no despolarizantes. Trimetoprima/sulfametoxazol aumenta la supresión de la médula ósea. Aumento o disminución de los efectos de la warfarina. CONTRAINDICACIONES/ REGIMEN DOSIFICACI”N Se administra por vía oral. La dosis PRECAUCIONES debe ser individualizada considerando Contraindicada en pacientes con la respuesta hematológica y la hipersensibilidad al medicamento y en tolerancia del paciente. La dosis usual el embarazo. No debe usarse para el para la terapia de inducción en niños tratamiento de la leucemia meníngea, es de 2,5 mg/kg/día o 75 mg/m2 y en leucemias linfáticas crónicas, linfomas adultos 2,5 mg/kg/día o 80-100 y tumores sólidos. Debe realizarse mg/m2, como dosis única diaria. Si no u n a e v a l u a c i ó n h e m a t o l ó g i c a 330 Formulario Terapéutico Nacional * 2ed. 2004 binación con alopurinol, se debe reducir la dosis a un cuarto o a un tercio de la dosis usual. Se debe considerar la posibilidad de infecciones o neoplasia subsiguiente como consecuencia del efecto inmunosupresor. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico sobre cualquier manifestación de infección METOTREXATO FARMACOLOGÍA Antineoplásico. Antagonista del ácido fólico que actúa como antimetabolito inhibiendo la enzima dihidrofolato reductasa que reduce al ácido fólico a ácido tetrahidrofólico. Al impedir la formación del tetrahidrofolato, limita la disponibilidad de fragmentos monocarbonados necesarios para la síntesis de purinas y la conversión del desoxiuridilato en timidilato, interfiriendo así con la síntesis de ADN y con la replicación celular. FARMACOCINÉTICA Se absorbe bien en el tracto gastrointestinal alcanzando concentraciones plasmáticas pico en 1-2 horas. La administración IM ó IV produce vesícula, el bazo, el hígado y la piel. Para lograr concentraciones terapéuticas en el SNC, debe administrarse por vía intratecal. Se une a las proteínas plasmáticas en un 50%. Sufre metabolismo hepático a poliglutamatos que pueden ser reconvertidos a metrotexato. Estos poliglutamatos actúan como inhibidores de la dihidrofolato reductasa y de la timidilato sintetasa. También es metabolizado por la flora intestinal. Se elimina principalmente por vía renal y en menor proporción por las heces. Con la administración IV, 80-90% de la dosis es excretada intacta en la orina dentro de 24 horas. La eliminación es trifásica con una vida media inicial (alfa) de menos de 1 hora, vida media intermedia (beta) de 2-3 horas y una L vida media terminal (gamma) de 8-12 horas. La fase de eliminación terminal prolongada parece ser debida a una combinación de factores como: Liberación del fármaco de compartimientos profundos, recirculación enterohepática y reabsorción tubular renal. La administración de dosis repetidas produce concentraciones plasmáticas sostenidas y acumulación en los tejidos. Puede ser retenido por semanas en el riñón y hasta por meses en el hígado. INDICACIONES En el tratamiento de neoplasias trofoblásticas en mujeres con 331 2ed. 2004 * Formulario TerapÈutico Nacional en niños. En dosis elevadas, seguida de rescate con leucovorina, en el tratamiento del osteosarcoma no metastásico (combinada con otros medicamentos y/o cirugía), cáncer de mama, cáncer epidermoide de cabeza y cuello, tratamiento de linfoma de Burkitt, estados avanzados de linfosarcoma y en la micosis fungoide. - (máximo 15 mg) intratecalmente, a intervalos de 2-5 días. Repetir hasta que el contaje de células en el líquido cefalorraquídeo sea normal y luego administrar una dosis adicional. La dosis para la profilaxis es igual que para el tratamiento, pero con intervalos diferentes. Cáncer de mama: 40 mg/m2 por vía IV los días 1 y 8 de un ciclo de 14 días, en combinación con ciclofosfamida 100 mg/m2 los días 1 a 14 y fluorouracilo 600 mg/m2 los días 1 y 8 del mismo ciclo, por 6-12 ciclos y períodos de descanso de 2 semanas entre cada ciclo. Osteosarcoma no metastásico: En combinación con otros antineoplásicos dentro de diversos esquemas de quimioterapia en dosis de 12 g/m2 por infusión IV lenta (4 horas) por ciclo. Cáncer de cabeza y cuello: 30 mg/m2 los días 1, 15 y 22 de un ciclo de 22 días, en combinación con otros antineoplásicos, con períodos de descanso de 4 semanas entre cada ciclo. Linfoma de Burkitt grado I y II: 10-25 mg/día por vía oral por 4-8 días con intervalos de reposo de una semana; en estados avanzados se usa en combinación con otros antineoplásicos. Linfosarcoma grado III: 0,625-2,5 mg/kg/día, generalmente asociada RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM, IV e intratecal. - Enfermedades trofoblásticas: La dosis usual es de 15-30 mg/kg/día por vía oral o IM por 5 días. Repetir el curso 3-5 veces, según sea necesario, con intervalos de una o más semanas. Evaluar la efectividad de la terapia mediante la determinación de las gonadotropinas coriónicas en orina de 24 horas. Este valor debe retornar a lo normal o a menos de 50 UI/24 horas después del tercer o cuarto curso de terapia, seguido de una completa resolución de las lesiones, medible en 4-6 semanas. Se recomienda 1-2 cursos de metotrexato después de la normalización de los valores de gonadotropinas coriónicas. El número máximo de cursos es de 3 a 5. - Leucemia linfocítica aguda: La dosis usual para inducción de la remisión es de 3,3 mg/m2/día combinado con prednisona 40-60 332 Formulario Terapéutico Nacional * 2ed. 2004 REACCIONES ADVERSAS Dermatológicas: Erupción, foliculitis, vasculitis, alopecia, fotosensibilidad, despigmentación, equimosis, acné y furunculosis (tienden a agravarse con la exposición a la luz solar). Gastrointestinales: Gingivitis, estomatitis, faringitis, glositis, enteritis, ulceración y sangramiento de la mucosa oral y otras zonas del tracto gastrointestinal, dolor abdominal, náusea, vómitos, hematemesis, diarrea y melena. Genitourinarias: Espermatogénesis y oogénesis defectuosas, oligospermia, insuficiencia ovárica, trastornos menstruales e infertilidad, aborto. Hematológicas: Leucopenia, trombocitopenia, anemia y hemorragias. Hepáticas: Atrofia hepática aguda, necrosis, metamorfosis grasa, fibrosis periportal y cirrosis hepática. Neurológicas: Cefalea, mareos, visión borrosa, tinitus, fatiga, hemiparesis, paresis, convulsiones, aracnoiditis (a las horas del uso intratecal), neurotoxicidad subaguda, leucoencefalopatía, desmielinización. Reacciones de hipersensibilidad: Anafilaxis, erupción eritematosa, prurito, urticaria. Renales: Insuficiencia renal, azotemia, cistitis, hematuria, nefropatía severa. Respiratorias: Neumonitis y fibrosis pulmonar (se ha reportado muertes por toxicidad pulmonar), infiltrados intersticiales pulmonares, tos no productiva. Otras: Conjuntivitis, tras- INTERACCIONES Fármacos con elevada unión a las proteínas plasmáticas (como: salicilatos, sulfonamidas, fenitoína, sulfonilúreas, fenilbutazona, tetraciclinas y cloranfenicol) pueden desplazar al metotrexato de su unión a las proteínas plasmáticas e incrementar su toxicidad. Los ácidos débiles pueden retardar la excreción renal del metotrexato y favorecer su acumulación. Los antiinflamatorios no esteroideos incrementan los niveles séricos del metotrexato y la posibilidad de sus efectos adversos. El meto-trexato puede incrementar el riesgo de infección por vacunas de organismos (bacterias o virus) vivos. El meto-trexato puede disminuir la depuración renal de la teofilina. El L ácido fólico o sus derivados pueden disminuir la respuesta al metotrexato. El uso concurrente de aciclovir con meto-trexato intratecal puede causar anormalidades neurológicas. Disminuye los niveles séricos de la digoxina. Los fármacos hepatotóxicos y el alcohol aumentan el riesgo de hepa-totoxicidad. Los antibióticos orales disminuyen la absorción del meto-trexato. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al fármaco, con insuficiencia renal y/o hepática severa 2ed. 2004 * Formulario TerapÈutico Nacional 333 terapia, y frecuentemente durante su desarrollo, se debe practicar evaluaciones de la función hematológica, renal, hepática y pulmonar, junto con radiografías de tórax. En caso de infecciones o hemorragias debidas a mielosupresión, se debe instituir tratamiento con antibióticos y/o derivados sanguíneos, según el caso. Puede ser necesario realizar biopsias hepáticas y aspirados de médula ósea en pacientes sometidos a terapia con dosis elevadas y/o por tiempo prolongado. Debe prestarse especial cuidado a pacientes con efusión pleural o ascitis, ya que la eliminación del fármaco puede alterarse. Para la administración por vía intratecal, no debe emplearse formulaciones que contengan preservativos. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Manejo sintomático y terapia de soporte. Administrar ácido folínico (leucovorina) por infusión IV en dosis hasta de 75 mg dentro de las primeras 12 horas, seguidos por 12 mg IM cada 6 horas por 4 dosis. La dosis de leucovorina debe ser igual o mayor que la del metotrexato y debe ser administrada, preferiblemente, dentro de la primera hora. La hemoperfusión sobre carbón activado ha sido usada para disminuir los niveles séricos de 334 garganta, sangramiento inusual, tos, dificultad para respirar, pérdida del apetito, dolor de cabeza, náusea incontrolable, vómitos, coloración amarillenta de los ojos o la piel, orina oscura o sanguinolenta y disminución en la cantidad diaria de orina. Se debe evitar la exposición prolongada a la luz del sol, así como el consumo de alcohol. Antes de tomar algún medicamento no prescrito se debe PACLITAXEL FARMACOLOGÍA Agente antineoplásico. Promueve el ensamblaje y estabilización de los microtúbulos en oposición a la organización dinámica normal de éstos, previniendo la despolimerización y provocando, en consecuencia, una interferencia con la interfase vital y la función mitótica, lo que resulta en la inhibición de la multiplicación celular. Además, promueve la for-mación de arreglos anormales de microtúbulos a través del ciclo celular. FARMACOCINÉTICA Después de la administración IV, se distribuye ampliamente en el organismo. Se une a las proteínas plasmáticas en un 89-98%. Se metaboliza en el hígado a través del sistema microsomal del citocromo P450, dando origen a productos inactivos Formulario Terapéutico Nacional * 2ed. 2004 INDICACIONES Para el tratamiento del carcinoma metastásico de ovario y de mama en combinación con otros agentes antineoplásicos. RÉGIMEN DE DOSIFICACIÓN Se administra por infusión IV. - Carcinoma de ovario: - Pacientes sin tratamiento previo: 175 mg/m 2 por infusión IV durante 3 horas, seguido por cisplatino 75 mg/m 2 . Alternativamente, 135 mg/m2 por infusión IV en 24 horas, seguido por cisplatino 75 mg/m2. En cualquiera de los casos, repetir cada 3 semanas. - Pacientes previamente tratados: 135 ó 175 mg/m2 por infusión IV durante 3 horas, cada 3 semanas. - Carcinoma de mama: 175 mg/m2 por infusión IV durante 3 horas y repetido cada 3 semanas. Todos los pacientes deben ser premedicados a fin de evitar las severas reacciones de hipersensibilidad. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, taquicardia, hipertensión, anormalidades en el ECG, arritmias, trastornos de conducción, insuficiencia cardiaca congestiva y edema, dolor severo en el pecho. Dermatológicas: Alopecia y alteraciones en las uñas. Gastrointes- periférica, ataxia y convulsiones. Reacciones de hipersensibilidad: Erupciones, erupción máculopapular, broncoespasmo, rubor y anafilaxis. Renales: Falla renal. Respiratorias: Neumonitis intersticial. Otras: Infecciones (tracto urinario, respiratorias y sepsis), artralgia, mialgia, pérdida de la agudeza visual y dolor en el sitio de la inyección. INTERACCIONES Cuando se usa a continuación de una terapia con cisplatino, se incrementa la incidencia y severidad de la mielosupresión; si se administra previo al cisplatino, el efecto es menor. Los medicamentos que inducen o inhiben el citocromo P450 pueden alterar la disposición del paclitaxel y ocasionar L fallas en la respuesta terapéutica o toxicidad, según el caso. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, en pacientes con contaje de neutrófilos inferior a 1500/mm3 y en el embarazo. Durante el tratamiento se debe vigilar periódicamente la función cardiaca y realizar evaluaciones de hematología completa con frecuencia. Usar con precaución en pacientes con falla hepática severa. No se conoce la eficacia y la seguridad del uso del paclitaxel en niños. Se recomienda suspender la 2ed. 2004 * Formulario TerapÈutico Nacional 335 INFORMACIÓN AL PACIENTE Advertir al paciente sobre la posibilidad de pérdida del cabello, vómitos, debilidad generalizada, neuropatías periféricas e infecciones. Mantener una hidratación adecuada (2-3 li-tros/día) a lo largo de todo el trata-miento. Evitar el contacto con multitu-des y personas con enfermedades infecciosas. Tomar medidas para la prevención del embarazo. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial: Entumecimiento u hormigueo en los dedos de las manos o de los pies PREDNISONA (Antineopl·sico) FARMACOLOGÍA Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. FARMACOCINÉTICA Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. INDICACIONES En combinación con agentes antineoplásicos, dentro de diversos esquemas o protocolos, para el tratamiento de leucemia y linfomas en adultos y leucemia linfocítica aguda en niños. 336 según la condición a tratar y los agentes involucrados en el protocolo. La dosis, según el caso, se ubica entre 5 a 60 mg/día en adultos en 2-4 dosis divididas y en niños, 0,14 a 2 mg/kg/día en 4 dosis divididas. REACCIONES ADVERSAS Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. INTERACCIONES Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. CONTRAINDICACIONES/ PRECAUCIONES Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. SOBREDOSIS Ver: PREDNISONA en la sección PREPARADOS HORMONALES SISTÉMICOS. PROCARBAZINA FARMACOLOGÍA Agente antineoplásico. Aunque se desconoce su mecanismo de acción, la evidencia experimental sugiere una interferencia en la transmetilación de los grupos metilo de la metionina en la molécula del ARN-t que resulta en la inhibición de la síntesis de ARN y Formulario Terapéutico Nacional * 2ed. 2004 mente, el fármaco parece actuar directamente sobre el ADN, produciendo alteraciones estructurales. El peróxido de hidrógeno formado durante la autooxidación de la procarbazina se une a los grupos sulfhidrilos de la proteína residual fuertemente unida al ADN. FARMACOCINÉTICA Después de la administración oral, la procarbazina se absorbe rápida y completamente a la circulación sistémica, logrando niveles séricos pico en aproximadamente 1 hora. Se distribuye rápidamente alcanzando concentraciones elevadas en el hígado, el riñón, la pared intestinal, la piel y el SNC. Se metaboliza principalmente en el hígado y en los riñones transformándose en metabolitos inactivos que se excretan en un 70%, junto a una pequeña proporción del fármaco intacto, en la orina en 24 horas. El resto se elimina en las heces y a través de la vía respiratoria. La vida media plasmática es de 1 hora. Por vía intravenosa es de 10 minutos. INDICACIONES Sola o en combinación con otros agentes antineoplásicos para el tratamiento de la enfermedad de Hodgkin. RÉGIMEN DE DOSIFICACIÓN - (leucocitos por debajo de 4000/mm3 ) o trombocitopenia (plaquetas por debajo de 100.000/mm3), en cuyo caso se debe suspender la terapia. Una vez obtenida la respuesta deseada o lograda la recuperación hematológica, mantener o reiniciar (según el caso) la terapia con 1-2 mg/kg/día. - En niños: Iniciar con 50 mg/m2/día la primera semana, seguido por 100 mg/m2/día hasta lograr la res-puesta deseada o evidencia clínica de leucopenia y/o trombo-citopenia. Una vez obtenida la respuesta deseada o lograda la recuperación hematológica, man-tener o reiniciar (según el caso) la terapia L con 50 mg/m2/día. Cuando se usa en combinación con otros agentes antineoplásicos, la dosis usual en adultos es de 100 mg/m2/día durante los días 1-14 de un ciclo de 28 días. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, taquicardia, síncope, crisis hipertensivas y edema. Dermatológicas: Alopecia reversible, dermatitis, hiperpigmentación, enrojecimiento de la piel, herpes. Gastrointestinales: Náuseas, vómito, anorexia, dolor abdominal, hematemesis, melena, estomatitis, boca seca, disfagia, constipación y diarrea. He- 2ed. 2004 * Formulario TerapÈutico Nacional 337 coma y convulsiones. Oculares: Hemorragia retiniana, nistagmo, papiledema, fotofobia, diplopía y dificultad para enfocar. Reacciones de hipersensibilidad: Erupción, dermatitis, prurito, fotosensibilidad y anafilaxis. Respiratorias: Efusión pleural, tos, neumonitis. Renales: Hematuria, frecuencia urina-ria y nocturia. Otras: Infecciones, gine-comastia, sudoración, ronquera y pér-dida de la audición. INTERACCIONES Como la procarbazina posee actividad inhibitoria de la MAO, puede desencadenar crisis hipertensivas si se usa en combinación con medicamentos simpaticomiméticas, antidepresivos tricíclicos y ciertos alimentos o bebidas ricos en tiramina (consultar fuentes especializadas para conocer el contenido de tiramina en alimentos y bebidas). Con alcohol puede provocar reacción tipo antabuse. La procarbazina puede disminuir la absorción gastrointestinal de la digoxina. Los barbitúricos, opiáceos, antihistamínicos y fenotiazinas pueden potenciar la depresión inducida por la procarbazina en el SNC. Usada con meperidina (petidina) puede producirse excitación, sudoración, rigidez e hipertensión severa. Usada concurrentemente con fluoxetina puede producir confusión, agitación, intran338 embarazo. Usar con precaución en pacientes con falla renal y/o hepática y en niños. Se debe realizar evaluación de la función hematológica, renal y hepática antes de iniciar el tratamiento y a intervalos frecuentes durante el mismo. Se debe suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Debido al potencial de la procarbazina para producir toxicidad hepática y hematológica, se debe practicar pruebas de función hepática y hematología completa frecuentemente durante la recuperación y hasta 2-4 semanas después. INFORMACIÓN AL PACIENTE Advertir al paciente sobre la posibilidad de sangramiento e infecciones y sobre la necesidad de notificar de inmediato al médico la ocurrencia de cualquier manifestación de tal naturaleza o de algún otro tipo. Informar al paciente cuáles alimentos y bebidas (ricos en tiramina) debe evitar mientras dure el tratamiento. Evitar VINBLASTINA FARMACOLOGÍA Antineoplásico alcaloide derivado de la Vinca rosea. Se cree que altera las Formulario Terapéutico Nacional * 2ed. 2004 mitóticos impidiendo su polimeración e interrumpiendo así la división celular en la etapa de metafase. Posee también cierta actividad inmunosu-presora. FARMACOCINÉTICA Luego de la administración IV, el fármaco es rápidamente distribuido a los tejidos corporales, aunque no penetra bien la barrera hematoencefálica ni otros tejidos grasos. Se metaboliza parcialmente en el hígado transformándose en un producto (desacetilvinblastina) con actividad farmacológica superior a la de su precursora, además de otros metabolitos inactivos. El fármaco y sus metabolitos se excretan por vía biliar. La eliminación sigue un patrón cinético tricompartamental con una vida media terminal (gamma) de aproximadamente 25 horas. INDICACIONES En combinación con otros antineoplásicos para el tratamiento paliativo de: Linfoma de Hodgkin (estadios III y IV), linfoma no-Hodgkin incluyendo linfoma linfocítico (nodular y difuso) y linfoma histiocítico, micosis fungoide (estados avanzados), carcinoma de testículo, sarcoma de Kaposi, enfermedad de Letterer-Siwe, coriocarcinoma refractario a otros agentes y en el carcinoma de mama que no responde a cirugía y/o terapia hor- con daño hepático la dosis debe reducirse en un 50%. Las dosis subsiguientes se establecen en función de la respuesta y tolerancia del paciente (las dosis pueden variar, de la primera a la quinta, entre 3,7 y 11,1 mg/m2). No debe administrarse una nueva dosis si el contaje de leucocitos es menor a 4000/mm3, aún habiendo transcurrido los 7 días. Usualmente las dosis se incrementan cada semana en aproximadamente 1,8 mg/m2 en adultos y 1,25 mg/m2 en niños, hasta obtener la respuesta terapéutica deseada (disminución de leucocitos a 3000/mm3), pero sin exceder de 18 mg/m2 en adultos y de 12,5 mg/m2 en niños. Una vez que se ha determinado la dosis requerida para producir un contaje de leucocitos de 3000/mm3 se inicia la L terapia de mantenimiento restando a 2 dicha dosis 1,8 mg/m (en el caso de los adultos) o 1,25 mg/m2 (en el caso de niños) y se administra semanalmente durante 4-6 semanas. REACCIONES ADVERSAS Cardiovasculares: Flebitis, hipertensión, infarto del miocardio. Dermatológicas: Alopecia, vesiculación de la piel, dermatitis y fotosensibilidad. Gastrointestinales: Náuseas, vómitos, diarrea, anorexia, constipación, dolor abdominal, íleo paralítico, faringitis, estomatitis, enterocolitis hemorrágica y sangramiento rectal. Hematológicas: Leucopenia, trombocitopenia, anemia. 2ed. 2004 * Formulario TerapÈutico Nacional 339 nervioso autónomo que se manifiesta con retención urinaria, hipotensión ortostática, taquicardia sinusal, sequedad de la boca. La neurotoxicidad puede ser incapacitante. Respiratorias: Dificultad para respirar, broncoespasmo. Otras: irritación tisular, dolor y debilidad muscular, celulitis y necrosis (por extravasación), osteopenia, amenorrea. INTERACCIONES El uso de medicamentos que inhiben el sistema microsomal del citocromo P450 puede incrementar de manera significativa la toxicidad de la vinblastina. Se ha reportado casos de broncoespasmo severo y dificultad respiratoria en pacientes que reciben vinblastina simultáneamente con o después de la administración de mitomicina. Regímenes de quimioterapia que contienen vinblastina pueden reducir los niveles plasmáticos de fenitoína. Los medicamentos ototóxicos pueden potenciar la pérdida de la audición. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, leucopenia severa o con procesos infecciosos, insuficiencia hepática severa y en el embarazo. Se debe realizar una evaluación hematológica completa antes de iniciar el tratamiento y semanalmente durante su 340 ancianos y en aquellos con compromiso de la función hepática. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Notificar de inmediato al médico sobre cualquier manifestación de infección o sangramiento inusual que se VINCRISTINA FARMACOLOGÍA Antineoplásico tipo alcaloide derivado de la Vinca rosea, con mecanismo de acción similar al de la vinblastina (ver: VINBLASTINA). FARMACOCINÉTICA Luego de la administración IV, el fármaco es rápidamente distribuido a los tejidos corporales (90% en 15-30 minutos), aunque no penetra bien la barrera hematoencefálica ni al tejido adiposo. Sufre descomposición in vivo y metabolismo hepático. Los metabolitos y productos de la degradación se excretan por las heces en un 80% y en un 10-20% por la orina. La cinética de eliminación sigue un patrón tricompartamental con una vida media terminal de aproximadamente 85 horas. Formulario Terapéutico Nacional * 2ed. 2004 Hodgkin diseminado, linfoma noHodgkin avanzado, rabdomiosarcoma, neuroblastoma, tumor de Wilms, púrpura trombocitopénica idiopática, sarcoma de Kaposi, carcinoma de mama, carcinoma de vesícula, sarcoma osteogénico, linfoma de Burkitt y mieloma múltiple. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV a intervalos de 1 semana. La dosis usual en adultos es de 0,4 -1,4 mg/m2 y en niños con peso mayor de 10 kg: 1,5-2 mg/m2. En niños con peso menor o igual a 10 kg o con superficie corporal menor a 1 m 2 , administrar 0,05 mg/kg. En pacientes con concentración sérica de bilirrubina superior a 3 mg/dl se sugiere la reducción de la dosis en un 50% (0,05-1 mg/m2). REACCIONES ADVERSAS Cardiovasculares: Hipotensión e hipertensión, flebitis. Dermatológicas: Alopecia reversible. Gastrointestinales: Náusea, vómito, constipación, íleo paralítico, anorexia, necrosis y per-foración intestinal. Genitourinarias: Atonía vesical, incontinencia o reten-ción urinaria, secreción inapropiada de hormona antidiurética, oliguria, disuria y poliuria. Hematológicas: Leucopenia y trombocitopenia. Neurológicas: Neuropatía periférica, pérdida de los reflejos tendinosos profundos, pa- visuales, ceguera, diplopía, neuropatía óptica y extraocular, ptosis palpebral. Reacciones de hipersensibilidad: Erupciones, reacciones con extravasación de líquido. Renales: Hiponatremia, hiperuricemia, nefropatía por ácido úrico. Otras: Fiebre, debilidad muscular, pérdida de peso, mialgia y celulitis en el sitio de inyección. INTERACCIONES El uso de medicamentos que inhiben el sistema microsomal del citocromo P450 puede incrementar, en forma significativa, la toxicidad de la vincristina. Se han reportado casos de broncoespasmo severo y dificultad para respirar en pacientes que reciben vincristina simultáneamente con o L después de mitomicina. En regímenes de quimioterapia que contienen vin-cristina se ha observado disminución de los niveles plasmáticos de fe-nitoína. La administración de L-as-parginasa antes de la vincristina puede reducir la depuración hepática del fármaco. Los bloqueantes de los canales de calcio aumentan la acu-mulación de la vincristina en las células. Disminuye los efectos de la digoxina. Los medicamentos ototóxi-cos pueden potenciar la pérdida de la audición. CONTRAINDICACIONES/ PRECAUCIONES 2ed. 2004 * Formulario TerapÈutico Nacional 341 tóxicos y en pacientes geriátricos, debido a que son más susceptibles a los efectos neurotóxicos del medicamento. Se debe realizar evaluación hematológica completa antes de iniciar la terapia y frecuentemente durante su desarrollo. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Tratamiento sintomático y de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico sobre cualquier manifestación de infección o sangramiento inusual que se FLUTAMIDA FARMACOLOGÕA Agente no esteroideo y no hormonal con potente actividad antiandrogénica. Interfiere con la unión de los an-drógenos (dihidrotestosterona y tes-tosterona) a los receptores nucleares en las células del tejido prostático, suprimiendo así la influencia de dichas hormonas sobre el cáncer de próstata. Se ha observado elevación de los niveles plasmáticos de testosterona y estradiol después de la administración de flutamida. FARMACOCIN…TICA Se absorbe rápida y completamente 342 un producto alfa-hidroxilado (hidroxiflutamida) es farmacológicamente activo, y cuyas concentraciones plasmáticas pico a las 2 horas superan a las de la flutamida. A los 60 minutos de la administración sólo es posible detectar un 2,5% del fármaco intacto. La flutamida y sus metabolitos se distribuyen principalmente a la próstata sin alcanzar concentraciones apreciables en otros tejidos. Se une a las proteínas plasmáticas en un 9496%, mientras que el metabolito hidroxilado lo hace en un 92-94%. El fármaco y sus metabolitos son excretados, en su mayoría, por la orina en 72 horas y sólo un 4,2% por las heces. La vida media de eliminación del metabolito hidroxilado es de 8 horas. INDICACIONES En combinación con agonistas de la hormona liberadora de gonadotropinas (como la leuprolida) para el tratamiento del carcinoma prostático avanzado. R…GIMEN DE DOSIFICACI”N Se usa por vía oral en dosis diarias de 250 mg cada 8 horas. Se han usado dosis hasta de 1500 mg/día sin evidencia de toxicidad. La terapia se debe mantener hasta lograr la remisión completa (usualmente 3-30 meses). Formulario Terapéutico Nacional * 2ed. 2004 nación con leuprolida: Cardiovasculares: Edema periférico, hipertensión. Gastrointestinales: Diarrea, náusea, vómito, anorexia. Hematológicas: Anemia, leucopenia, trombocitopenia. Hepáticas: Elevación de los niveles de las transaminasas, ictericia, hepatitis. Neurológicas: Confusión, decaimiento, depresión, ansiedad, nerviosismo, cefalea, insomnio. Reacciones de hipersensibilidad: Fotosensibilidad. Otras: Oleadas de calor, pérdida de la libido, impotencia, ginecomastia, visión borrosa. INTERACCIONES En pacientes que reciben flutamida y warfarina se ha observado incrementos en el efecto del anticoagulante. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento. Usar con precaución en pacientes con disfunción hepática. Durante el tratamiento se debe evaluar periódicamente la función hepática y realizar determinaciones del antígeno prostáti-co específico. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y medidas de soporte. MEDROXIPROGESTERONA (MEDROXIPROGESTERONA ACETATO) (Antineopl·sico) FARMACOLOGÍA Progestágeno sintético cuyo mecanismo de acción antineoplásico no se conoce con precisión. Se postula que el fármaco difunde libremente al núcleo de las células blanco (ubicadas en los órganos del sistema reproductor femenino) para unirse a los receptores de progesterona, alterando la trans-cripción de los genes involucrados en la síntesis de proteínas. Inhibe la liberación de la hormona liberadora de las gonadotropinas, además de la inhibición de la proliferación celular. Este último efecto puede ser máximo en la fase G del ciclo celular, aunque L no es específico de fase. FARMACOCINÉTICA Después de la administración IM, se absorbe lentamente, alcanzando la concentración plasmática pico después de 3 semanas. Una vez en la sangre, se une a las proteínas plasmáticas en un 90%. Se metaboliza en el hígado dando lugar a, por lo menos, 16 productos diferentes que se excretan en su mayoría por la orina (20-40%) y en menor proporción por las heces (5-13%). La vida media de eliminación, tras la administración IM, es de 50 días. 2ed. 2004 * Formulario TerapÈutico Nacional 343 RÉGIMEN DE DOSIFICACIÓN Iniciar con 400-1000 mg IM una vez por semana. Luego de lograda la mejoría clínica y la estabilización del cuadro, mantener la misma dosis inicial, pero una vez al mes. REACCIONES ADVERSAS Cardiovasculares: Tromboflebitis, embolismo pulmonar, edema, tromboembolismo, accidente cerebrovascular. Dermatológicas: Abscesos estériles, acné, melasma, prurito, alopecia, hirsutismo. Gastrointestinales: Náuseas, vómito. Genitourinarias: Sangramiento vaginal, dismenorrea, amenorrea, erosión cervical, secreciones anormales, dolor abdominal. Hepáticas: Ictericia colestática. Neurológicas: Depresión, nerviosismo, insomnio, somnolencia, cefalea, fatiga, mareos. Reacciones de hipersensibilidad: Erupciones, urticaria, anafilaxis. Otras: Intolerancia a la glucosa, ganancia o pérdida de peso, lesiones oculares; sensibilidad, aumento de tamaño o secreción de las mamas. INTERACCIONES La rifampicina, la carbamazepina, el fenobarbital y la fenitoína pueden inducir el metabolismo de la medroxiprogesterona y comprometer su eficacia terapéutica. La medroxiprogesterona puede disminuir el efecto hipoglicémico de la gliben344 CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, hemorragia uterina no diagnosticada, antecedentes tromboembólicos, tromboflebitis, apoplejía, disfunción hepática, carcinoma genital o de mama y durante el embarazo. Durante el tratamiento se debe evaluar frecuentemente la función oftalmológica. Dado que en los pacientes diabéticos puede producir intolerancia a la glucosa, se debe practicar determinaciones periódicas de la glicemia. Usar con precaución en pacientes con diabetes mellitus, historia de depresión mental y enfermedades que pueden agravarse por la retención de líquido como la epilepsia, la migraña, la hipertensión arterial, el asma y la disfunción cardiaca o renal. Se debe suspender la lactancia durante la terapia. SOBREDOSIS No es factible la intoxicación, aún con la administración de dosis elevadas. INFORMACIÓN AL PACIENTE Durante el tratamiento se recomienda usar métodos anticonceptivos no hormonales. Ante la sospecha o la certeza de un embarazo, suspender el tratamiento y consultar al médico. Evitar el consumo de café y el hábito del tabaquismo. Informar de inmediato al médico si se presenta alguna Formulario Terapéutico Nacional * 2ed. 2004 TAMOXIFENO FARMACOLOGÍA Agente no esteroideo y no hormonal con actividad antiestrogénica. Aunque su mecanismo de acción no ha sido totalmente aclarado, se postula que el fármaco y algunos de sus metabolitos compiten con el estradiol por los receptores citoplasmáticos en las células de los órganos blanco, impidiendo su influencia en el crecimiento de tumores dependientes de estrógenos. La unión a la cromatina nuclear es atípica y persiste por mayor tiempo que la unión tamoxifeno-receptor. FARMACOCINÉTICA Se absorbe lentamente en el tracto gastrointestinal alcanzando concentraciones plasmáticas máximas en 3-6 horas. La evidencia disponible sugiere que el fármaco sufre un extenso ciclo enterohepático. El metabolismo resulta en varios productos, siendo el principal de ellos el N-desmetiltamoxifeno, metabolito con actividad farmacológica similar a la del fármaco padre y que, con la administración continua, llega a alcanzar niveles séricos en el estado estable que igualan o duplican a las del tamoxifeno. Las concentraciones de tamoxifeno en el estado estable se alcanzan a las 3-4 semanas de iniciada la terapia, mientras que la de su metabolito lo hace a las 3-8 se- semanas; el tamoxifeno intacto y los metabolitos no conjugados cuentan con un 30% de la excreción fecal. La vida media del metabolito N-desmetiltamoxifeno es de 9-14 días. INDICACIONES Tratamiento del carcinoma avanzado de mama y como coadyuvante de la cirugía en el tratamiento del carcinoma de mama en mujeres con nódulos linfáticos axilares negativos y en mu-jeres posmenopáusicas con nódulos linfáticos axilares positivos. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral en dosis diarias de 10-20 mg hasta por 5 años. Cantidades superiores a los 20 mg/día deben administrarse en dosis L di-vididas. REACCIONES ADVERSAS Cardiovasculares: Edema periférico, vasculitis, trastornos tromboembólicos. Gastrointestinales: Náuseas, vómitos, cólicos abdominales, anorexia. Genitourinarias: Trastornos menstruales, sangramiento vaginal, menstruación irregular, amenorrea, prurito vulvar, quistes ováricos. Hematológicas: Leucopenia, trombocitopenia. Hepáticas: Elevación de los niveles de las aminotransferasas, hepatitis, colestasis, ictericia, necrosis hepática, hígado graso. Neurológicas: Cefalea, depresión 2ed. 2004 * Formulario TerapÈutico Nacional 345 caída del cabello, hipercalcemia, dolor óseo. INTERACCIONES La combinación con otros agentes antineoplásicos aumenta el riesgo de fenómenos tromboembólicos. El medicamento puede potenciar la acción de los anticoagulantes cumarínicos. Los antiácidos pueden afectar la absorción del tamoxifeno. La bromocriptina puede aumentar los niveles sanguíneos del tamoxifeno. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, con antecedentes de trombosis venosa o de embolismo pulmonar y en el embarazo. Usar con precaución en pacientes con leucopenia y trombocitopenia. Se debe realizar evaluación hematológica periódica durante el tratamiento. Debido al efecto antiestrogénico, puede causar daño fetal y/o abortos espontáneos, por lo que se debe evitar su uso en el embarazo. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y medidas de soporte. el consumo de sal. Aunque se debe evitar el embarazo, no es prudente hacerlo mediante el uso de anticonceptivos orales. Debe usarse un FILGRASTIM (G-CSF) FARMACOLOGÍA Factor estimulante de colonias de granulocitos obtenido por tecnología de ADN recombinante en cultivos de Escherichia coli. Induce la proliferación y diferenciación de las células precursoras de neutrófilos en la médula ósea, así como la producción y activación funcional de los mismos. FARMACOCINÉTICA Se absorbe rápida y completamente después de la administración SC, alcanzando concentraciones plasmáticas pico a las 2-8 horas. En animales, se distribuye y concentra principalmente en la médula ósea, glándulas adrenales, riñón e hígado. No se conoce con precisión su metabolismo y rutas de excreción. La vida media de eliminación es de aproximadamente 3,5 horas. INDICACIONES Prevención y tratamiento de la neutropenia en pacientes con neoplasias no mieloides sometidos a quimioterapia antineoplásica mielosupresora. Para reducir el período de neutropenia en pacientes con trans- INFORMACIÓN AL PACIENTE 346 Formulario Terapéutico Nacional * 2ed. 2004 RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV en infusión corta (15-30 minutos) o continua, y por vía SC en bolo o infusión continua. El régimen debe mantenerse hasta obtener un contaje de neutrófilos de 1.500-10.000/mm3. - En pacientes con quimioterapia mielosupresora: 5 mcg/kg/día con incrementos, en caso necesario, de 5 mcg/kg/ciclo según la duración y severidad de la neutropenia. - En pacientes con transplante de médula ósea: 10 mcg/kg/día con incrementos, en caso necesario, de 5 mcg/kg/ciclo según la duración y severidad de la neutropenia. - Para la movilización de células precursoras en sangre periférica en donantes sanos: 10 mcg/kg/día SC, como un bolo o por infusión continua por lo menos 4 días antes de la leucoparesis y se continúa hasta la última leucoparesis. - En pacientes con neutropenia crónica: 6 mcg/kg, 2 veces al día (congénita) o 5 mcg/kg/día (idiopática), por vía SC. REACCIONES ADVERSAS Cardiovasculares: Dolor en el pecho, hipotensión, arritmia supraventricular transitoria, depresión del segmento ST del ECG, tromboflebitis. Gastrointestinales: Náusea, vómito, diarrea, mucositis, esplenomegalia, anorexia, constipación, estomatitis. He- sitio de la inyección. INTERACCIONES Aunque no se han descrito interacciones con filgrastim, se recomienda precaución en pacientes sometidos a terapia con medicamentos que disminuyen el contaje plaquetario o que potencian la liberación de neutrófilos, tales como el litio. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento. No debe usarse dentro de las 24 horas previas o siguientes a la quimioterapia antineoplásica o a la radioterapia. Antes de iniciar la terapia, y periódicamente (mínimo 2 veces/semana) durante su desarrollo, se debe realizar el contaje L de células sanguíneas, incluyendo las plaquetas. Cuando se usa en pacientes con neutropenia congénita existe el riesgo del síndrome mielodisplásico o de leucemia mieloide aguda. Usar con precaución en pacientes con leucocitosis. Si el contaje de glóbulos blancos alcanza 100.000/mm3 o más, puede requerirse un ajuste de dosis. Contraindicado en pacientes con hipersensibilidad a las proteínas derivadas de la Escherichia coli, al medicamento o a sus componentes. El uso en el embarazo debe restringirse a situaciones de verdadera necesidad en las que el beneficio 2ed. 2004 * Formulario TerapÈutico Nacional 347 INFORMACIÓN AL PACIENTE (sólo en caso de pacientes ambulatorios) Durante la terapia, evitar el contacto con multitudes y personas que padezcan enfermedades infecciosas. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. En el caso de pacientes que se autoadministran el medicamento, se les debe enseñar cómo hacerlo y INTERFERÓN ALFA-2B FARMACOLOGÍA Proteína hidrosoluble altamente purificada de interferón, sub-tipo alfa2b, obtenida por tecnología de ADN recombinante cuyo mecanismo de acción, aunque no ha sido completamente elucidado, se cree que sus efectos resultan de una compleja secuencia de eventos farmacológicos y de modulación biológica interrelacionados, que se traducen en una actividad antiviral, antineoplásica e inmunomoduladora. Los interferones ejercen su actividad celular mediante la unión a receptores específicos de membrana en la superficie celular. Luego, se inicia una compleja secuencia de eventos intracelulares como: Inducción de ciertas enzimas, supresión de la proliferación celular, inhibición de la replicación viral en células infectadas, incremento de la actividad fagocítica de los 348 FARMACOCINÉTICA Luego de la administración IM ó SC se absorbe en un 80% o más a la circulación sistémica alcanzando niveles plasmáticos pico en 3-12 horas, mientras que con la administración IV se producen concentraciones máximas a los 15-60 minutos. Tras la administración IM ó SC el fármaco es detectable en el plasma hasta por 16-30 horas y por 4-8 horas después de la administración IV. Un 90-96% de la dosis es metabolizada en los túbulos renales por degradación proteolítica y posteriormente excretada con la orina. Se cree que una pequeña fracción es metabolizada también en el hígado y excretada después por la bilis. La vida media de eliminación terminal es de 2-3 horas. INDICACIONES Leucemia de células velludas, melanoma maligno, linfoma no-Hodgkin, sarcoma de Kaposi relacionado con el SIDA, hepatitis B crónica, hepatitis C crónica e infección por papilomavirus humano. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM, SC, IV o intralesión. - Leucemia de células velludas: 2 millones de UI/m2 por vía IM ó SC 3 veces a la semana, por 2-6 meses. - Melanoma maligno: Inducción con Formulario Terapéutico Nacional * 2ed. 2004 - - - - UI/m2 por vía SC 3 veces semanales por 18 meses, en combinación con antineoplásicos antracíclicos. Sarcoma de Kaposi: 30 millones de UI/m2 por vía IM ó SC 3 veces a la semana. Hepatitis B: En adultos 5 millones de UI/m2 /día o 10 millones de UI/m2 por vía IM ó SC 3 veces a la semana, por 16 semanas. En niños, se han recomendado dosis de 3 millones de UI/m2 por vía SC, 3 veces durante la primera semana, seguido por dosis crecientes hasta los 6 millones de UI/m2 3 veces a la semana, por 16-24 semanas. Hepatitis C: 3 millones de UI/m2 por vía SC por vía IM ó SC 3 veces a la semana, por 18-24 semanas. Papilomavirus humano: 1 millón de UI/m 2 intralesión (máximo 5 lesiones por tratamiento) 3 veces a la semana, por 3 semanas. REACCIONES ADVERSAS Cardiovasculares: Arritmias, taquicardia, bradicardia, hipertensión, hipotensión, ortostasis, angina, cardiomegalia, trastornos coronarios, trastornos valvulares, embolismo pulmonar y vasculitis. Dermatológicas: Alopecia, acné, fotosensibilidad, eczema, quistes sebáceos y despigmentación cutánea. Endocrinas: Ginecomastia, hipotiroidismo, hipertiroidismo, hiper- tológicas: Mielosupresión, anemia, granulocitopenia, linfocitosis, anemia hemolítica, leucopenia, neutropenia y trombocitopenia. Hepáticas: Elevación de los niveles de las enzimas hepá-ticas, bilirrubinemia y falla hepática. Músculo-esqueléticas: Mialgia, artritis, dolor óseo, síndrome de túnel carpal, calambres y debilidad. Neurológicas: Somnolencia, confusión, mareo, fati-ga, depresión, parestesia, dolor de cabeza, dificultad para concentrarse, vértigo, amnesia, irritabilidad, ansie-dad, nerviosismo, trastornos auditivos, trastornos extrapiramidales, neuropa-tía periférica, psicosis, ideas suicidas, convulsiones y coma. Reacciones de hipersensibilidad: Erupción eritematosa y maculopapular, prurito, psoria- L sis, necrólisis epidérmica, angioedema y anafilaxis. Renales: Incrementos del BUN y de la creatinina sérica, falla renal aguda, hematuria, proteinuria, incontinencia, cistitis e infecciones del tracto urinario. Respiratorias: Disnea, sibilancias, tos, faringitis, sinusitis, congestión nasal, neumonía, bronqui-tis, broncoespasmo, insuficiencia res-piratoria, hemoptisis, efusión pleural y fibrosis pulmonar. Otras: Síndrome similar a la gripe (fiebre, escalofríos, cefalea, malestar general, mialgia, sudoración y fatiga), deshidratación, dolor y ardor en el sitio de inyección, dolor de oído, mastitis, 2ed. 2004 * Formulario TerapÈutico Nacional 349 mielosupresión. Los alcaloides de la vinca pueden incrementar la toxicidad hematológica, hepática y neurológica del interferón. La zidovudina potencia la toxicidad hematológica. El interferón inhibe el metabolismo hepático de la teofilina y posiblemente del fenobar-bital, incrementando así la factibilidad de sus reacciones adversas. En pacientes que reciben interferón, la radioterapia puede producir mucositis severa, obstrucción de las vías respiratorias y hemorragias. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, trastornos neuropsiquiátricos, trastornos tiroideos, historia de convulsiones, insuficiencia renal, hepática o pancreática, enfermedades autoinmunes, miocardiopatía grave y en el embarazo. Usar con extrema precaución en pacientes con enfermedad pulmonar, discrasias sanguíneas, diabetes mellitus y en ancianos. Antes de iniciar el tratamiento, y periódicamente durante su desarrollo, se deben practicar evaluaciones de la función hematopoyética, cardiaca, pulmonar, hepática, renal y neurológica, así como determinaciones de glicemia y de lípidos sanguíneos. Se debe procurar mantener la misma marca del 350 soporte. INFORMACIÓN AL PACIENTE El paciente debe ser informado de los efectos adversos más frecuentes e importantes asociados con la terapia, así como de la importancia de informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial infecciones, sangramiento, trastornos cardíacos, respiratorios o neurológicos. Administrar preferible-mente por las noches, antes de dormir. Se debe mantener una adecuada hidratación (2-3 litros/día), sobretodo al inicio del tratamiento. El producto puede alterar la capacidad para realizar actividades que requieren concentración, alerta MOLGRAMOSTIM (GM-CSF) FARMACOLOGÍA Factor estimulante de colonias de granulocitos y macrófagos obtenido por tecnología de ADN recombinante en cultivos de Escherichia coli que contiene un plásmido portador de un gen de GM-CSF humano. Induce la división y diferenciación de las células progenitoras comprometidas en la línea granulocito-macrófago, así como la proliferación y activación funcional de los granulocitos y macrófagos ma-duros. El efecto se traduce en un incremento del contaje Formulario Terapéutico Nacional * 2ed. 2004 cia del G-CSF (filgrastim) que sólo genera incrementos en el contaje de neutrófilos. FARMACOCINÉTICA Se absorbe rápida y completamente después de la administración SC, alcanzando concentraciones plasmáticas pico a las 4-6 horas y una respuesta clínica (aumento del contaje de glóbulos blancos) a los 2-10 días de terapia continua. No se conoce con precisión su metabolismo y rutas de excreción. La vida media de eliminación tras la administración SC es de aproximadamente 2-3 horas y de 1-2 horas con la administración IV. INDICACIONES Prevención y tratamiento de la neutropenia en pacientes sometidos a quimioterapia mielosupresora. Estimulación de la regeneración mieloide posterior al transplante de médula ósea. RÉGIMEN DE DOSIFICACIÓN Se administra por vía SC e IV. - En pacientes con terapia mielosupresora: 5-10 mcg/kg/día por vía SC por 7-10 días, comenzando 24 horas después de la última dosis del antineoplásico. - En pacientes con transplante de médula ósea: 10 mcg/kg/día por in-fusión IV durante 4-6 horas, REACCIONES ADVERSAS Cardiovasculares: Hipotensión, arritmias, falla cardiaca, pericarditis, aumento de la presión intracraneana. Gastrointestinales: Náusea, vómito, diarrea, anorexia, estomatitis, dolor abdominal. Hematológicas: Linfocitosis, trombocitosis, trombocitopenia, disminución de la hemoglobina, eosinofilia. Hepáticas: Elevación de los niveles de las aminotransferasas, hiperbilirrubinemia. Neurológicas: Dolor de cabeza, mareo, fatiga, depresión paranoide, debilidad. Reacciones de hipersensibilidad: Disnea, broncoespasmo, erupción, prurito, angioedema y anafilaxis. Renales: Insuficiencia renal. Respiratorias: Efusión pleural, pneumonitis. Otras: Dolor óseo, hipoalbu- L minemia, edema, aumento de peso, sudoración, precipitación o agravamiento de enfermedades autoinmunes pre-existentes. En algunos pacientes se ha presentado un síndrome que ocurre, particularmente, después de la primera dosis de molgramostim y que se caracteriza por hipotensión, taquicardia, fiebre, rubor, náusea, vómito, diaforesis, dolor de espalda, espasmos en las piernas, disnea e hipoxia. Parece más común con el tratamiento IV y no recurre con dosis subsiguientes, aunque puede repetir con un nuevo tratamiento después de un 2ed. 2004 * Formulario TerapÈutico Nacional 351 tratamiento. INTERACCIONES Aunque no se conocen interacciones con el molgramostim, se recomienda precaución en pacientes sometidos a terapia con medicamentos que potencian la liberación de los neutrófilos (como el litio). Como la administración del molgramostim ha sido asociada con disminución de la albúmina sérica, se debe tener cuidado con la ad-ministración concomitante con fárma-cos con alta unión a las proteínas. soporte. No se ha reportado sobredosis accidental con molgramostin, pero a dosis de 20 a 30 mcg/kg/día se ha observado taquicardia, hipotensión y un síndrome tipo gripe, efectos que son tratados sintomáticamente. INFORMACIÓN AL PACIENTE (sólo en caso de pacientes ambulatorios) Durante la terapia evitar el contacto con multitudes y personas que padezcan enfermedades infecciosas. Notificar de inmediato al médico si se ÁCIDO MICOFENÓLICO (MICOFENOLATO MOFETIL) CONTRAINDICACIONES/ FARMACOLOGÍA PRECAUCIONES Agente inmunosupresor. Ejerce un Contraindicado en casos de hipersenefecto citostático (antiproliferativo) sosibilidad al medicamento. No debe ser bre los linfocitos T y B como resultado usado en malignidades mieloides. No de la inhibición de la enzima inosina debe usarse dentro de las 24 horas monofosfato deshidrogenasa y conseprevias o siguientes a la quimioterapia cuente bloqueo de la síntesis de puriantineoplásica. Antes de iniciar la nas. terapia, y periódicamente durante su desarrollo, se debe realizar un análisis FARMACOCINÉTICA d e h e m a t o l o g í a c o m p l e t a y , Se absorbe rápida y extensamente asimismo, vigilar el peso corporal, la (94%) por vía oral, aunque sufre un temperatura, la función cardiaca y la intenso metabolismo pre-sistémico. función respiratoria. Usar con Después de la administración oral e precaución en pacientes con falla IV, prácticamente todo el micofenolato hepática y/o renal, trastornos mofetil es transformado en el hígado respiratorios y enferme-dades en ácido micofenólico, metabolito actiautoinmunes. No se conoce la vo que es metabolizado a un glucuseguridad del uso del molgramostin en rónido de ácido micofenólico inactivo. niños, en el embarazo y durante la Este metabolito sufre ciclo enterohelactancia. Debe ser usado bajo la 352 Formulario Terapéutico Nacional * 2ed. 2004 minutos de su administración y otra similar a las 6-12 horas. El ácido micofenólico se une a las proteínas plasmáticas en un 97% y el metabolito glucurónido en un 82%. Un 93% de la dosis de micofenolato mofetil administrada es excretada en la orina por filtración glomerular y secreción tubular (87% como glucurónido y menos del 1% como ácido micofenólico) y el resto (6%) por las heces. La vida media de eliminación del ácido micofenólico es de 16-18 horas. INDICACIONES En combinación con ciclosporina y corticosteroides para la prevención del rechazo post-transplante alogénico de corazón, hígado o riñón. adultos. PrevenciÛn del rechazo posttransplante cardíaco en adultos: 1,5 g por vía oral o IV, 2 veces al día. - PrevenciÛn del rechazo posttransplante hepático en adultos: 1,5 g por vía oral o 1 g IV, 2 veces al día en ambos casos. Independientemente del tipo de transplante, la administración debe iniciarse tan pronto como sea posible después de la cirugía, siempre dentro de las primeras 24 horas. - REACCIONES ADVERSAS Los efectos más comunes incluyen: Cardiovasculares: Hipertensión, arritmias, edema y dolor en el pecho. Dermatológicas: Acné, erupción. L Gas-trointestinales: Náusea, vómito, RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral e IV. diarrea, constipación, gingivitis, hiperCuando se usa la vía IV el producto plasia gingival, moniliasis bucal, debe diluirse a concentración de 6 dispepsia, anorexia, dolor abdominal, mg/ml en dextrosa al 5% y pancreatitis, hemorragia y perforación administrarse por infusión lenta en un gastrointestinal. Hematológicas: perÌodo no me-nor de 2 horas. La Leucopenia, neutropenia severa, anevía IV no debe emplearse por más de mia hipocrómica, leucocitosis y trom14 días con-tinuos. Se debe procurar bocitopenia. Hepáticas: Alteración de cambiar al paciente de la terapia por las pruebas de función hepática, vía IV a la terapia oral tan pronto como hepatitis e ictericia colestásica. Neula tole-rancia al medicamento por esa rológicas: Temblor, dolor de cabeza, debilidad, mareo e insomnio. Renales: vía lo permita. - PrevenciÛn del rechazo post- Necrosis tubular y hematuria. Restransplante renal en adultos: 1 g por piratorias: Disnea y tos. Otras: Infecvía oral o IV, 2 veces al día en am- ción, sepsis, fiebre, edema periférico, bos casos. En niños: 600 mg/m2 hipercolesterolemia, hiperglicemia, 353 2ed. 2004 * Formulario TerapÈutico Nacional antiácidos reducen la absorción gastrointestinal del micofenolato. La colestiramina puede interferir con el ciclo enterohepático del ácido micofenólico y disminuir sus niveles plasmáticos. Los medicamentos que afectan la flora bacteriana gastrointestinal pueden también ejercer un efecto similar. El probenecid puede interferir con la secreción tubular de los metabolitos del micofenolato. Dosis elevadas de salicilados pueden incrementar la fracción libre de ácido micofenólico. El aciclovir y el ganciclovir, en pacientes con falla renal, pueden competir con el glucurónido del ácido micofenólico por la secreción tubular e incrementar sus niveles séricos. Puede afectar la eficacia de los anticonceptivos orales. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento o a los constituyentes de la formulación. Como no se conoce la seguridad en el embarazo, el uso en tales circunstancias debe limitarse a situaciones de verdadera necesidad, en las que los beneficios potenciales superen al riesgo para el feto. De no ser ese el caso, se debe practicar medidas orientadas a evitar el embarazo antes de iniciar la terapia, durante la misma y hasta 6 meses después de finalizada. 354 resta del año. De igual manera, se debe vigilar periódicamente la función renal y la función hepática. Usar con precaución en pacientes con enfermedad gastrointestinal activa. La terapia puede provocar mielosupresión y neutropenia, incrementando el riesgo de infecciones. Asimismo, se ha descrito un incremento del riesgo para el desarrollo de linfomas y otras neoplasias. Durante el tratamiento se debe evitar el uso de vacunas de virus vivos. SOBREDOSIS No existe experiencia en humanos que revelen los efectos de una sobredosificación con micofenolato mofetil. En caso de ingestión accidental de dosis elevadas, si es reciente, se recomienda vaciamiento gástrico, carbón activado y un catártico salino, manejo sintomático y tratamiento de soporte. Los niveles plasmáticos del ácido micofenólico podrían reducirse interrumpiendo la circulación enterohepática del metabolito glucurónido con colestiramina. INFORMACIÓN AL PACIENTE Administrar con el estómago vacío (1 hora antes o 2 horas después de las comidas), procurando tomar la dosis siempre a la misma hora del día o de la noche. No morder ni triturar la cápsula. No alterar la dosis o la frecuencia de administración, ni Formulario Terapéutico Nacional * 2ed. 2004 adecuada y permanente higiene bucal y dental. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial cualquier malestar que sugiera la posibilidad de una infección. Debe usar un método anticonceptivo eficaz AZATIOPRINA FARMACOLOGÕA Derivado imidazólico de la 6-mercaptopurina (6-MP) con propiedades inmunosupresoras. Aunque no se conoce bien su mecanismo de acción, se cree que podría involucrar la inhibición de la síntesis de ARN y ADN, la incorporación de los análogos de la tiopurina que produce daño del ADN, bloqueo de los grupos SH por alquilación, y la 6-MP que actúa como antimetabolito de la purina. Hay la consecuente alteración de los cromosomas, producción de proteínas fraudulentas, trastornos en el metabolismo celular, inhibición de la mitosis, supresión de la hipersensibilidad mediada por células y alteración de la producción de anticuerpos. mándose en diversos productos, de los cuales resulta más abundante el ácido 6-tioúrico. Se excreta como metabolitos y en muy escasa proporción como fármaco intacto en la orina. Los efectos pueden persistir aún después de suspendida la terapia. Para la azatioprina, se estima una vida media de 6-28 minutos y para la 6-MP de 38-114 minutos. INDICACIONES Prevención del rechazo de órgano en transplante alogénico de riñón, en combinación con otros inmunosupresores. R…GIMEN DE DOSIFICACI”N Iniciar con una dosis única de 3-5 mg/kg/día por vía oral el mismo día L del transplante o 1-3 días antes del transplante. En pacientes incapaces de tomar la medicación oral, se inicia el tratamiento por vía IV con el uso subsiguiente de tabletas (a la misma dosis) después del período postoperatorio. La dosis de mantenimiento es de 1-3 mg/kg/día. REACCIONES ADVERSAS Cardiovasculares: Hipotensión. GasFARMACOCIN…TICA trointestinales: Náusea, vómitos, panDespués de la administración oral, se creatitis, esteatorrea, diarrea, dolor absorbe bien en el tracto gastroin- abdominal, úlceras bucales. Hematotestinal. Se une a las proteínas plas- lógicas: Leucopenia, mielosupresión, máticas en un 30% y se distribuye anemia, trombocitopenia, inmunosupresión (posiblemente marcada), eo355 2ed. 2004 * Formulario TerapÈutico Nacional piel y de hígado, linfoma no-Hodgkin). INTERACCIONES La administración con inhibidores de la enzima convertidora de angiotensina puede causar anemia y leucopenia severa. El alopurinol puede inhibir el metabolismo de la azatioprina e incre-mentar el riesgo de toxicidad. La azatioprina incrementa el riesgo de infecciones cuando se administran vacunas de organismos vivos: Disminuye la eficacia de los anticoagulantes orales y revierte el bloqueo neuromuscular de los relajantes musculares no despolarizantes. Los aminosalicilatos aumentan el riesgo de leucopenia. Con trimetropima/sulfametoxazol aumenta el riesgo de toxicidad hematológica. El efecto anticoagulante de la warfarina puede ser reducido. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al fármaco, en el embarazo y en individuos tratados previamente con agentes alquilantes (ciclofosfamida, clorambucilo o melfalán) debido al elevado riesgo de desarrollo de neoplasias. Se debe realizar una hematología completa semanalmente durante el primer mes de tratamiento, 2 veces al mes durante el segundo y tercer mes, y luego una vez al mes (o más frecuentemente si hay aumento 356 dicamente la función hepática. Reducir la dosis o suspender la terapia en caso de una caída rápida del contaje leu-cocitario o de alguna otra manifesta-ción que sugiera la posibilidad de mielodepresión. Tomar las medidas necesarias a objeto de evitar las infec-ciones que, en caso de presentarse, pueden resultar fatales. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Tratamiento sintomático y terapia de soporte. INFORMACI”N AL PACIENTE En caso de molestias gastrointestinales, administrar el medicamento en dosis divididas o tomarlo con las comidas. Se recomienda practicar medidas anticonceptivas durante el tratamiento. Evitar el contacto con multitudes y personas con enfermedades infecciosas. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología CICLOSPORINA FARMACOLOGÕA Polipéptido cíclico con propiedades in-munosupresoras. Suprime la respues-ta inmune mediada por Formulario Terapéutico Nacional * 2ed. 2004 que no se conoce bien su mecanismo de acción se cree que podría involucrar una inhibición de la proliferación y funciones de los linfocitos T, aparentemente debido a la inhibición específica y reversible de las fases G0 ó G1 del ciclo celular. La ciclosporina también inhibe la producción y liberación de linfoquinas, incluyendo la interleukina-2 o el factor de crecimiento celular-1 (TCGF). después de la cirugía por 1-2 semanas y disminuir un 5% por semana hasta niveles de mantenimiento de 5-10 mg/kg/día. Se recomienda la deter-minación de los niveles sanguíneos de ciclosporina. REACCIONES ADVERSAS Cardiovasculares: Hipertensión, edema, falla cardiaca, arritmias y dolor anginoso. Gastrointestinales: Náuseas, vómito, anorexia, dolor abdominal, constipación, disfagia, úlcera FARMACOCIN…TICA La absorción desde el tracto gas- gástrica, esofagitis e hiperplasia trointestinal es variable e incompleta. gingival. Hematológicas: Leucopenia, Después de la administración oral, linfadenopatía, anemia y complicaalcanza niveles séricos pico a las 3,5 ciones tromboembólicas. Hepáticas: horas. Los alimentos alteran su ab- Aumento de los niveles de las enzimas sorción. Se distribuye en un 33-47% hepáticas y de la bilirrubina. Neuen plasma, 4-9% en los linfocitos, 5- rológicas: Temblor, convulsiones, L 12% en los granulocitos y 41-58% en cefalea, parestesia, confusión, los eritrocitos. A concentraciones ele- neuropatías, vértigo, mareos, ansievadas se satura la captación por los dad, nerviosismo y somnolencia. leucocitos y los eritrocitos. Se une a las Reacciones de hipersensibilidad: proteínas plasmáticas en un 90%, Hiperpigmentación, erupción, prurito, primariamente a las lipoproteínas. Se urticaria, dermatitis, eczema, acné, metaboliza extensamente en el foliculitis, angioedema y anafilaxis. hígado. Se elimina principalmente por Renales: Aumentos del BUN y de la la bilis y en escasa proporción (6%) creatinina sérica, disuria, hematuria, por la orina, menos de 1% como falla renal, urgencia e incontinencia fármaco intacto. La vida media de urinaria. Respiratorias: Infecciones, eliminación en fase terminal es de 8- disnea, faringitis, tos, bronquitis y broncoespasmo. Otras: Alteraciones 19 horas. de la glicemia, sudoración, trastornos INDICACIONES oculares y auditivos, artralgia, sínEn combinación con otros inmuno- drome gripal, infecciones bacterianas, virales y micóticas, fiebre, rubor, 357 2ed. 2004 * Formulario TerapÈutico Nacional niveles sanguíneos y la eficacia de la ciclosporina, dando lugar a la posibilidad de rechazo del órgano transplantado. El diltiazem, la eritromicina, la nicardipina, el verapamilo, el danazol, la bromocriptina, los antimicóticos imidazoles, la metiltestosterona, los anticonceptivos orales, el imipenem-cilastatina, el jugo de toronja y la metoclopramida pueden aumentar los niveles séricos de ciclosporina y el riesgo de reacciones adversas. La asociación con amfotericina B, aminoglicósidos, trimetoprima/sulfametoxazol, AINEs, cimetidina, ranitidina y melfalán, aumenta el riesgo de nefrotoxicidad. El uso con azatioprina, corticosteroides, verapamilo y ciclofosfamida incrementa la inmunosupresión y la posibilidad de infección. La combinación con metilprednisolona puede conducir a convulsiones. La ciclosporina puede reducir la depuración de la prednisolona, la digoxina y la lovastatina. Con los diuréticos ahorradores de potasio puede provocar hiperpotasemia. Puede disminuir la respuesta inmunológica a las vacunas de organismos vivos. Ha ocurrido miositis cuando se administra con lovastatina; hipertrofia gingival frecuente con nifedipino. la función renal, hepática y hematológica, así como la presión arterial. Se debe tomar medidas orientadas a evitar infecciones. Debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente (debido a su lenta absorción se puede practicar el vaciamiento gástrico, aún después de 2 horas de su ingestión). Tratamiento sintomático y terapia de soporte. La ciclosporina no es dializable. INFORMACI”N AL PACIENTE No alterar la dosis ni suspender el tratamiento sin el consentimiento del médico. Tomar la dosis a intervalos regulares, siempre a la misma hora cada día. En caso de usar la solución oral, se puede diluir con jugo de naranja o de manzana a temperatura ambiente y enjuagar después el vaso con el mismo vehículo, a fin de ingerir la dosis completa. Evitar el jugo de toronja. Durante el tratamiento es de vital importancia asistir al control periódico. Evitar el contacto con multitudes y personas con enfermedades infeccio-sas. Consultar al médico antes de usar algún otro medicamento durante la terapia. Se recomienda practicar medi-das anticonceptivas durante el tratamiento. Notificar de inmediato al mé- CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hiper358 Formulario Terapéutico Nacional * 2ed. 2004 M03 MIORRELAJANTES Bromuro de Vecuronio Suxametonio (Succinilcolina) Tiocolchicósido M04 PREPARADOS ANTIGOTOSOS Alopurinol Colchicina M05 DROGAS PARA EL TRATAMIENTO DE ENFERMEDADES ”SEAS Ácido Alendrónico (Alendronato Sódico) M SISTEMA M⁄SCULO-ESQUEL…TICO M01 PRODUCTOS ANTIINFLAMATORIOS Y ANTIRREUM¡TICOS Celecoxib Cloroquina* (Antiinflamatorio y Antirreumático) Diclofenaco (Diclofenac Sódico) (Sistémico) Ibuprofeno Indometacina Ketoprofeno Nimesulida 2ed. 2004 * Formulario TerapÈutico Nacional 359 M CELECOXIB FARMACOLOGÍA Antiinflamatorio no esteroideo derivado del pirazol, que posee un grupo sulfonamida y una sustitución diaril en su estructura. Ejerce una actividad antiinflamatoria, analgésica y antipirética que parece ser debida a la inhibición selectiva de la isoenzima ciclooxigenasa 2 (COX-2) que interviene en la síntesis de prostaglandinas. No inhibe la ciclooxigenasa 1 (COX-1). FARMACOCINÉTICA Genera concentraciones séricas pico a las 3 horas de la administración por vía oral de una dosis única y una concentración al estado estable a los 5 días con la administración de dosis múltiples. Se une a las proteínas plasmáticas en un 97%. Se metaboliza en el hígado a través del sistema del citocromo P450 y se elimina como metabolitos inactivos, y menos del 3% como fármaco intacto, en un 57% en las heces y en 27% en la orina. La vida media de eliminación es de 11 horas. INDICACIONES Tratamiento sintomático de condiciones inflamatorias como osteoartritis y artritis reumatoidea y alivio del dolor de intensidad leve a moderada. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis REACCIONES ADVERSAS Cardiovasculares: Taquicardia, agravamiento de la hipertensión, palpitaciones. Gastrointestinales: Dolor abdominal, diarrea, dispepsia, flatulencia, náusea, reflujo gastroesofágico, sangrado gastrointestinal. Hematológicas: Anemia, epistaxis, trombocitopenia, equimosis. Hepáticas: Aumento de los niveles de las aminotransferasas, hepatitis, insuficiencia hepática. Neurológicas: Mareos, cefalea, insomnio, vértigo, neuralgia, neuropatía. Reacciones de hipersensibilidad: Reacciones alérgicas. Respiratorias: Rinitis, sinusitis, infección del tracto respiratorio superior. Otras: Dolor de espalda, faringitis, edema periférico, erupciones de la piel, calambres musculares, fotosensibilidad, pérdida de la audición, alopecia. M INTERACCIONES Ver: DICLOFENACO. El fluconazol puede incrementar los niveles plasmáticos del celecoxib. CONTRAINDICACIONES/ PRECAUCIONES Ver: DICLOFENACO. CLOROQUINA (Antiinflamatorio y Antirreum·tico) FARMACOLOGÍA El mecanismo de acción en la terapia de la artritis reumatoidea es desco- 2ed. 2004 * Formulario TerapÈutico Nacional 361 nocido. La evidencia experimental revela que el medicamento tiene propiedades antiinflamatorias. Estudios con cÈlulas de mamÌferos demuestran que posee actividad antihistamÌnica, antiserotonÌnica e inhibidora del efecto de las prostaglandinas, presumiblemente por inhibiciÛn de la conversiÛn de ·cido araquidÛnico en prostaglandina F2. Estudios in vitro indican que inhibe la quimiotaxis en leucocitos polimorfonucleares, macrÛ-fagos y eosinÛfilos. FARMACOCINÉTICA Ver: CLOROQUINA en la sección de PRODUCTOS ANTIPARASITARIOS (antimaláricos). INDICACIONES Como alternativa a las sales de oro o a la penicilamina en el tratamiento de la artritis reumatoidea en pacientes que no responden a los AINEs. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Las dosis se expresan en términos de cloroquina base. La dosis recomendada en adultos es de 150 mg/día, hasta control de la patología según criterio médico. Por lo general, genera una respuesta terapéutica a las 4-6 semanas. REACCIONES ADVERSAS 362 CONTRAINDICACIONES/ PRECAUCIONES Ver: CLOROQUINA en la sección de PRODUCTOS ANTIPARASITARIOS. SOBREDOSIS Ver: CLOROQUINA en la sección de PRODUCTOS ANTIPARASITARIOS. INFORMACIÓN AL PACIENTE Ver: CLOROQUINA en la sección de DICLOFENACO (DICLOFENAC SÓDICO) (SistÈmico) FARMACOLOGÍA Antiinflamatorio no esteroideo derivado del ácido fenilacético, con actividad analgésica, antiinflamatoria y antipirética. Su acción parece debida, al menos en parte, a la inhibición de la síntesis de prostaglandinas por inac-tivación de las isoenzimas ciclooxi-genasa-1 (COX-1) y ciclooxigenasa-2 (COX-2). FARMACOCINÉTICA Se absorbe rápida y completamente en el tracto gastrointestinal, aunque la biodisponibilidad se reduce a un 5060% por el efecto metabólico de primer pasaje. Por vía oral, genera niveles plasmáticos pico en aproximadamente 1 hora, mientras que por inyección IM, las concentraciones máximas se logran en 10-20 minutos. Se une a las proteínas plasmáticas en un 99% y se Formulario Terapéutico Nacional * 2ed. 2004 mándose en productos con actividad farmacológica débil algunos e inactivos otros, que se excretan en la orina en un 65% y en un 35% por las heces. La vida media de eliminación es de 1,2-2 horas y se prolonga en pacientes con insuficiencia renal severa. INDICACIONES Tratamiento sintomático de condiciones inflamatorias como osteoartritis y artritis reumatoidea y alivio del dolor de intensidad leve a moderada. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM e IV. La dosis usual en adultos para el manejo de la artritis reumatoidea es 150-200 mg/día por vía oral divididos en 2-3 dosis. En la osteoartritis: 100-150 mg/día por vía oral, divididos en 2-3 dosis. Como analgésico en condiciones dolorosas no articulares: 100 mg por vía oral, seguido por 50 mg cada 8 horas, o 75 mg IM dos veces al día, o 1 mg/kg administrado por bolo IV o por infusión lenta. REACCIONES ADVERSAS Cardiovasculares: Edema periférico, dolor anginoso, hipertensión e insuficiencia cardiaca congestiva. Gastrointestinales: Náuseas, vómito, diarrea, faringitis, constipación, dispepsia, dolor abdominal, gastritis, úlcera péptica, perforación y hemorragia aminotransferasas, ictericia y hepatitis. Neurológicas: Cefalea, mareo, fatiga, desvanecimiento, depresión, insomnio, ansiedad e irritabilidad. Reacciones de hipersensibilidad: Erupción, prurito, dermatitis, fotosensibilidad, eritema multiforme, síndrome de Stevens-Johnson, angioedema, broncoespasmo, reacciones anafilactoides y anafilaxis. Renales: Aumento del BUN y de la creatinina sérica, falla renal aguda, proteinuria, síndrome nefrótico, infección del tracto urinario, cálculos renales, oliguria e hiponatremia. Respiratorias: Asma, neumonía y bronquitis. Otras: Alopecia, acné, disminución de la agudeza visual, visión borrosa, epistaxis, cambios en la visión de colores, tinitus y pérdida de la audición. INTERACCIONES Los AINEs pueden aumentar los M niveles plasmáticos y la consecuente toxicidad del metotrexato, del litio y de la digoxina. Puede disminuir la actividad de los inhibidores de la enzima convertidora de angiotensina y de los bloqueantes de los receptores betaadrenérgicos, aumentar la nefrotoxicidad de la ciclosporina y reducir el efecto natriurético de los diuréticos de asa. La combinación con otros AINEs aumenta el riesgo de sangramiento y de falla renal. Los corticosteroides y el consumo elevado de alcohol 2ed. 2004 * Formulario TerapÈutico Nacional 363 la insulina e hipoglicemiantes orales. Puede aumentar los niveles plasmáticos de la fenitoína. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otros AINEs y en pacientes con úlcera péptica activa, enfermedad hepática y/o renal severa. Usar con precaución en pacientes con historia de úlcera péptica, trastornos de coagulación, alteración de la función renal y/o hepática leve a moderada, asma, insuficiencia cardiaca congestiva, hipertensión y, en general, en aquellas condiciones que pueden agravarse por retención o sobrecarga de fluidos. Durante el tratamiento se debe vigilar periódicamente la función hepática, renal y hematológica. Los pacientes geriátricos resultan muy susceptibles a los efectos adversos de los AINEs y, por lo general, padecen enfermedades que pueden ser afectadas o agravadas por el uso de estos medicamentos. En el embarazo, se recomienda limitar su empleo a situaciones de verdadera necesidad en las que los beneficios potenciales a la madre superen a los riesgos para el feto. Se debe evitar en embarazos a término dado que podría inhibir las contracciones uterinas, inducir complicaciones hemorrágicas y provocar el cierre prematuro del ducto arterioso en el 364 tratamiento de soporte. INFORMACI”N AL PACIENTE Evitar el consumo de alcohol durante el tratamiento. Informar al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial: Malestar gastrointestinal, sangrado anormal y/o heces negras. En caso de erupción generalizada, dificultad respiratoria e inflamación de los párpados, de los labios o de la nariz, suspender el tratamiento y notificar al médico. Instruir al paciente acerca de la importancia de notificar de inmediato la ocurrencia de náuseas, IBUPROFENO FARMACOLOGÍA Antiinflamatorio no esteroideo derivado del ácido propiónico. El mecanismo de acción es similar al descrito para el diclofenaco sódico (ver: DICLOFENACO). FARMACOCINÉTICA Se absorbe en un 80% en el tracto gastrointestinal alcanzando concentraciones séricas pico en 1-2 horas. En niños, la respuesta antipirética se evidencia a los 60 minutos y se mantiene por 6-8 horas. Se une en un 99% a las proteínas plasmáticas y se distribuye ampliamente a los tejidos y a los fluidos corporales. Se metaboliza en el hígado transformándose en Formulario Terapéutico Nacional * 2ed. 2004 la orina. La excreción es casi completa en 24 horas. Tiene una curva bifásica de eliminación plasmática con una vida media de aproximadamente 2 horas. INDICACIONES Tratamiento sintomático de condiciones inflamatorias como osteoartritis, artritis reumatoidea y artritis juvenil, como analgésico para el alivio del dolor de intensidad leve a moderada y como antipirético para el manejo de la fiebre en niños de 6 meses a 12 años de edad. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - Artritis reumatoidea y osteoartritis en adultos: 300-800 mg 3-4 veces al día. No exceder 3,2 g diarios. - Artritis reumatoidea juvenil: 30-40 mg/kg/día divididos en 3-4 dosis. - Dolor de intensidad leve a moderada: 200-400 mg cada 4-6 horas, según sea necesario. - Fiebre en niños: 5-10 mg/kg cada 68 horas, según sea necesario. REACCIONES ADVERSAS Ver: DICLOFENACO. INTERACCIONES Ver: DICLOFENACO. CONTRAINDICACIONES/ PRECAUCIONES Ver: DICLOFENACO. INDOMETACINA FARMACOLOGÍA Antiinflamatorio no esteroideo derivado del ácido indolacético. El mecanismo de acción es similar al descrito para el diclofenaco sódico (ver: DICLOFENACO). FARMACOCINÉTICA Se absorbe rápida y completamente en el tracto gastrointestinal, alcanzando una concentración sérica pico en 2 horas. Se une en un 99% a las proteínas plasmáticas y se distribuye ampliamente a los tejidos y fluidos corporales, lográndose una concentración en líquido sinovial equivalente a un 20% de la plasmática. Se metaboliza en el hígado a productos inactivos que se excretan, junto a un 1,5% del fármaco intacto, en un 60% por la orina y 33% por las heces. La M vida media de eliminación es de 4,5 horas. INDICACIONES Tratamiento sintomático de condiciones inflamatorias como la artritis reumatoidea, osteoartritis y espondilitis anquilosante y para el alivio del dolor de intensidad leve a moderada. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos es de 25 mg 2-3 veces al día, con comida o con antiácido. En caso necesario, se puede incrementar 2ed. 2004 * Formulario TerapÈutico Nacional 365 INTERACCIONES Ver: DICLOFENACO. CONTRAINDICACIONES/ PRECAUCIONES Ver: DICLOFENACO. SOBREDOSIS Ver: DICLOFENACO. INFORMACI”N AL PACIENTE KETOPROFENO FARMACOLOGÍA Antiinflamatorio no esteroideo derivado del ácido propiónico. El mecanismo de acción es similar al descrito para el diclofenaco sódico (ver: DICLOFENACO). FARMACOCINÉTICA Se absorbe en un 90% en el tracto gastrointestinal alcanzando niveles plasmáticos pico en 0,5 a 2 horas y produciendo una respuesta analgésica en 30 minutos, la cual persiste por 4-6 horas. Se une en un 99% a las proteínas plasmáticas y se distribuye ampliamente a los tejidos y fluidos corporales. Se metaboliza en el hígado transformándose en productos inactivos que se excretan en un 50-90% por la orina y 1-8% por las heces. La vida media de eliminación es de 1-4 horas y se prolonga en pacientes con alteración de la función renal. 366 RÉGIMEN DE DOSIFICACIÓN La dosis usual por vía oral en adultos es: 75 mg 3 veces al día o 50 mg 4 veces al día. Dosis máxima: 300 mg/día. Por vía IV: 100-300 mg/día en dosis divididas y administradas cada 8-12 horas por un máximo de 48 horas. REACCIONES ADVERSAS Ver: DICLOFENACO. INTERACCIONES Ver: DICLOFENACO. CONTRAINDICACIONES/ PRECAUCIONES Ver: DICLOFENACO. SOBREDOSIS NIMESULIDA FARMACOLOGÍA Antiinflamatorio no esteroideo con un grupo sulfonanilida en su estructura. Ejerce una actividad antiinflamatoria, analgésica y antipirética que parece ser debida, al menos en parte, a la inhibición selectiva de la isoenzima ciclooxigenasa 2 (COX-2) que interviene en la síntesis de prostaglandinas. Además, inhibe la liberación de histamina in vivo e in vitro. FARMACOCINÉTICA Después de la administración oral, se absorbe a la circulación sistémica alcanzando concentraciones séricas Formulario Terapéutico Nacional * 2ed. 2004 metaboliza en el hígado casi en su totalidad y se excreta como metabolitos en un 50-60% en la orina y en un 18-36% en las heces. La vida media de eliminación es de 1,8-5,2 horas. INDICACIONES Tratamiento sintomático de condiciones inflamatorias, del dolor de intensidad leve a moderada y de la fiebre. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos es de 100-200 mg 2 veces al día. REACCIONES ADVERSAS Gastrointestinales: Pirosis, dolor epigástrico, náusea, vómito, diarrea. Hepáticas: Hepatitis aguda. Neurológicas: Vértigo, hiperexcitabilidad, decaimiento. Reacciones de hipersensibilidad: Erupciones, prurito, eritema, lesiones purpúricas. Otras: Diaforesis. INTERACCIONES Ver: DICLOFENACO. CONTRAINDICACIONES/ PRECAUCIONES Ver: DICLOFENACO. BROMURO DE VECURONIO FARMACOLOGÕA Bloqueante neuromuscular no des- polarizante. Compite con la acetilcolina por sus sitios receptores en la unión neuromuscular de la placa motora, impidiendo la interacción de la acetilcolina con los receptores nicotínicos y, en consecuencia, la actividad colinérgica sobre el músculo, provocando una parálisis transitoria. No ejerce actividad agonista sobre los receptores ni altera el potencial de reposo de la placa motora. FARMACOCIN…TICA DespuÈs de la administraciÛn IV produce un efecto inicial en 1 minuto, que se hace máximo a los 3-5 minutos. La duración del bloqueo bajo anestesia con tiopental, óxido nitroso y fentanilo es de 25-30 minutos y de 30-40 minutos con halotano. La administración repetida de dosis de mantenimiento no tiene un efecto acumulativo apreciable M en la duración del bloqueo neuromuscular. Se une a las proteínas plasmáticas en un 60-90% y se distribuye rápidamente al espacio extracelular. Se metaboliza por desacetilación transformándose en metabolitos hidroxilados, activos e inactivos que se excretan principalmente por la bilis y en escasa proporción por la orina. Su eliminación es bifásica, con una vida media en fase terminal de 30-80 minutos. 2ed. 2004 * Formulario TerapÈutico Nacional 367 en adultos de 0,08-0,1 mg/kg produce condiciones adecuadas de intubación en 2,5-3 minutos. Bajo anestesia balanceada el bloqueo neuromuscular clínicamente requerido dura entre 2530 minutos, con recuperación del 25% en 25-40 minutos y del 95% en 45-65 minutos. Cuando se usa en combinación con anestésicos generales que potencian su acción bloqueante (enflurano, halotano o isoflurano), la dosis inicial debe reducirse en alrededor de un 15% (0,06-0,085 mg/kg), si es administrado más de 5 minutos después de los anestésicos. Cuando se usa después de la succinilcolina, debe administrarse en dosis inicial de 0,005-0,06 mg/kg con anestesia balanceada y de 0,04-0,06 mg/kg con anestesia por inhalación. La dosis usual de mantenimiento en procedimientos prolongados con anestesia balanceada es de 0,010,015 mg/kg y en pacientes con anestesia por inhalación 0,008-0,012 mg/kg. La primera dosis de mantenimiento se requiere a los 25-40 minutos y las dosis subsiguientes a intervalos de 12-15 minutos con anestesia balanceada, o más prolongados con la anestesia por inhalación. El bloqueo también puede mantenerse con la infusión continua de 0,8-1,2 mcg/kg/minuto iniciada a los 20-40 minutos de la dosis inicial. En niños mayores de 10 años, administrar la 368 la recuperación; aunque reciben dosis similares a las de los adultos, posiblemente la frecuencia de las dosis de mantenimiento es menor. REACCIONES ADVERSAS Las reacciones más comunes son extensión del efecto farmacológico e incluyen: Debilidad o parálisis de músculos esqueléticos, insuficiencia respiratoria y apnea; esta última más factible con el uso simultáneo de agentes depresores del SNC. Se han reportado casos poco frecuentes de broncoespasmo, hipotensión, taquicardia, eritema y urticaria. INTERACCIONES El uso concurrente de anestésicos halogenados, succinilcolina, aminoglicósidos, polimixina B, clindamicina, lincomicina, quinina, quinidina, procainamida, bloqueantes de los receptores beta-adrenérgicos, lidocaína (en dosis altas), litio, diuréticos tiazida, furosemida, tetraciclinas, amfotericina B, inhibidores de la anhidrasa carbónica, sulfato de magnesio y corticosteroides puede incrementar y/o prolongar el bloqueo neuromuscular y dificultar la recuperación del paciente. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y/o a Formulario Terapéutico Nacional * 2ed. 2004 hepática y/o renal, enfermedad cardiovascular, obesidad, edema, miastenia gravis u otras enfermedades neuromusculares, trastornos hidroelectrolíticos y debilidad. Los pacientes quemados pueden desarrollar resistencia a los bloqueantes neuromusculares no despolarizantes. El uso en el embarazo debe limitarse a circunstancias en las que resulte estrictamente necesario. Durante su uso se debe suspender la lactancia. SOBREDOSIS Manejo sintomático y terapia de soporte. Mantener permeable la vía aérea con ventilación asistida o controlada. Soporte cardiovascular con posición Trendelenburg, fluidoterapia IV y, en caso necesario, fármacos vasopresores. La reversión del bloqueo puede requerir el uso de inhibidores de colinesterasa como neostigmina, piridostigmina o edroSUXAMETONIO (SUCCINILCOLINA) FARMACOLOGÕA Bloqueante despolarizante de la unión neuromuscular de acción ultracorta. Compite con la acetilcolina por los sitios receptores nicotínicos en la unión neuromuscular produciendo despolarización persistente (por resistencia a la acción de la acetilcolinesterasa) y la consecuente relajación muscular. FARMACOCINÉTICA Después de la administración IV, produce relajación muscular completa en 0,5-1 minuto, la cual persiste por 23 minutos y desaparece por completo a los 10 minutos. Con la administración IM se inicia el efecto a los 2-3 minutos y perdura por 10-30 minutos. Es metabolizada rápidamente por la enzima pseudocolinestarasa a succinilmonocolina y colina. Una fracción de la succinilmonocolina es excretada en la orina y el resto transformado por hidrólisis alcalina en succinato y colina. Un 10% de la dosis se elimina en la orina como fármaco intacto y el resto como metabolitos. INDICACIONES Como relajante muscular en procedimientos quirúrgicos de corta duraM ción. R…GIMEN DE DOSIFICACI”N Se administra por vía IV. - Para procedimientos cortos en adultos: 0,3-1,1 mg/kg en 10-30 segundos. En niños: 1-2 mg/kg IV. En caso necesario administrar dosis adicionales, según la respuesta del paciente. - Para procedimientos prolongados en adultos: Infusión IV de 0,5-10 mg/min. La infusión continua no es recomendable en niños. 2ed. 2004 * Formulario TerapÈutico Nacional 369 intragástrica, salivación excesiva. Reacciones de hipersensibilidad: Erupciones, anafilaxis. Respiratorias: Apnea o depresión respiratoria, especialmente en pacientes con actividad deficiente de la pseudocolinesterasa. Otras: Aumento de la presión intraocular, hipertermia maligna, fasciculaciones musculares, dolor muscular post-operatorio, rabdomiolisis (con posible falla renal aguda mioglobinúrica, salivación excesiva e hiperpotasemia). nación con glicósidos digitálicos puede provocar arritmia. El atracurio y el mivacurio pueden producir antagonismo de la actividad bloqueante del suxametonio. La furosemida puede causar alteraciones del bloqueo neuromuscular. La melatonina potencia los efectos bloqueantes de la succinilcolina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al fármaco, trastornos genéticos de la pseudocolinesterasa, glaucoma de ángulo cerrado, historia personal o familiar de hipertermia maligna e historia de miopatías asociadas a niveles séricos elevados de la creatina quinasa. Usar con precaución en pacientes con enfermedad hepatocelular, deshi-dratación, anemia severa, quema-duras, cáncer, enfermedades del colágeno y mixedema, ya que estas condiciones se asocian con reduccio-nes de la concentración y/o actividad de pseudocolinesterasa. También se debe usar con cautela en pacientes que se recuperan de traumas severos o con desbalances electrolíticos, hiperpotasemia, paraplejia, desnervación músculo-esquelética, enfermedad neuromuscular distrófica o degenerativa y durante la cirugía ocular. El uso en el embarazo debe INTERACCIONES La administración con agentes que reducen la actividad de la pseudocolinestarasa plasmática como: neostigmina, piridostigmina, edrofonio, ciclofosfamida, anticonceptivos orales, prednisona, prednisolona, trimetafán, fenelzina, fenotiazinas, tiotepa, fisostigmina e inhibidores de la MAO puede potenciar el efecto del suxametonio y causar apnea e, inclusive, la muerte. La procaína compite con el suxametonio por la pseudocolinesterasa y su uso conjunto puede provocar apnea prolongada. Agentes como la promazina, aprotinina, oxitocina, aminoglicósidos, vancomicina, colistina, polimixina B, lincomicina, clindamicina, opiáceos, procainamida, quinidina, quinina, terbutalina, lidocaína (dosis elevadas), bloqueantes de los receptores beta-adrenérgicos, litio, 370 Formulario Terapéutico Nacional * 2ed. 2004 SOBREDOSIS La sobredosificación causa bloqueo neuromuscular más allá del tiempo necesario para la cirugía y la anestesia, que puede conducir a debilidad músculo-esquelética severa, disminución de la reserva respiratoria y apnea. El tratamiento consiste en establecer y/o mantener funcional la vía aérea y suministrar respiración artificial con oxígeno hasta recuperación de la respiración espontánea. No se debe intentar revertir la intoxicación con neostigmina o me-dicamentos TIOCOLCHICÓSIDO FARMACOLOGÕA Análogo glucosulfurado de la colchicina con propiedades relajantes de la musculatura esquelética. Posee afinidad selectiva por los receptores del ácido gamma-aminobutírico (GABA). Produce acciones similares a las del GABA y a las de la glicina. Aunque no se conoce con precisión su mecanismo de acción, se postula que podría ser el resultado de una activación directa del receptor del GABA a nivel espinal. FARMACOCIN…TICA Luego de su administración por vía oral o IM se absorbe rápidamente hacia la circulación sistémica. ambos casos) por 24 horas. Una vez en la sangre se une a las proteínas plasmáticas en un 13% y se distribuye a los tejidos. No se conoce bien su metabolismo. Se elimina intacto y como metabolitos principalmente por la bilis y en un 20% por vía renal. La vida media de eliminación es de 3-5 horas. INDICACIONES Estados de espasticidad o contractura de la musculatura esquelética. REGIMEN DE DOSIFICACION Se administra por vía oral e IM. La dosis por vía oral es de 4 mg 4 veces al día y por vía IM 4 mg 2 veces diarias. REACCIONES ADVERSAS Son por lo general leves en intensidad y asociadas, la mayoría de las veces, M a la inyección IM. Se han descrito casos poco frecuentes de hipotensión, agitación, decaimiento, gastralgia, náuseas, diarrea, pirosis y eritema. INTERACCIONES No se describen en la literatura. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al producto y en la miastenia gravis. Usar con precaución en pacientes con hipotonía muscular 2ed. 2004 * Formulario TerapÈutico Nacional 371 de soporte. INFORMACIÓN AL PACIENTE Informar al médico si se presenta alguna reacción o sintomatología ALOPURINOL FARMACOLOGÕA El alopurinol actúa sobre el catabolismo de las purinas sin alterar la biosíntesis. Siendo un análogo estruc-tural de hipoxantina, disminuye las concentraciones séricas y urinarias de ácido úrico por inhibición de la xantino-oxidasa, enzima que cataliza la conversión de hipoxantina en xantina y ésta en ácido úrico. FARMACOCIN…TICA Se absorbe en un 80-90% en el tracto gastrointestinal alcanzando concentraciones plasmáticas pico en 2-6 horas. El efecto reductor de los niveles séricos de urato se observa a las 2448 horas y se hace máximo después de 1-3 semanas. Se distribuye ampliamente en los líquidos tisulares, excepto en el cerebro donde las c o n c e n t r a c i o n e s s o n aproximadamente el 50 % de las alcanzadas en otros tejidos. No se une a las proteínas plasmáticas. Es metabolizado por acción de la xantinooxidasa a oxipurinol y posteriormente conjugado, junto con alopurinol intacto, a sus respectivas formas de 372 fármaco se elimina intacto por las heces en 48-72 horas. La vida media de eliminación del alopurinol es de 1-3 horas y la del oxipurinol 18-30 horas. INDICACIONES Tratamiento de la gota. Prevención de la hiperuricemia, de los depósitos tisulares de uratos y de la nefropatía por ácido úrico asociada a trastornos neoplásicos mieloproliferativos. Cálculos renales de oxalato de calcio y de ácido úrico recurrentes. R…GIMEN DE DOSIFICACI”N Se administra por vía oral, usualmente como dosis única después de las comidas. Si la cantidad a administrar diariamente excede los 300 mg, administrar en dosis divididas. - Gota: Iniciar con 100 mg/día e incrementar 100 mg/semana hasta lograr una concentración sérica de uratos inferior a 6 mg/dl o alcanzar una dosis máxima de 800 mg/día. La dosis usual en caso de gota moderada es de 200-300 mg/día y, en casos severos, 400600 mg/día. Una vez alcanzada la concen-tración sérica deseada de uratos, se debe reducir la dosis a 300 mg/día. - Trastornos neoplásicos mieloproliferativos: En adultos 600-800 mg/día 1-2 días antes de iniciar la Formulario Terapéutico Nacional * 2ed. 2004 - C·lculos renales: Iniciar con 200300 mg/día y ajustar posteriormente en función de los valores de ácido úrico en orina de 24 horas. REACCIONES ADVERSAS Cardiovasculares: Angeítis necrotizante. Gastrointestinales: Náusea, vómitos, diarrea, dolor abdominal, gastritis, pérdida o perversión del gusto. Hematológicas: Agranulocitosis, anemia, anemia aplásica, trombocitopenia, leucopenia, leucocitosis, eosinofilia. Hepáticas: Aumento de los niveles de la fosfatasa alcalina, ALT y AST; hepatitis, necrosis hepática, hepatomegalia, ictericia colestásica. Reacciones de hipersensibilidad: Erupción (usualmente maculopapular), dermatitis exfoliativa, urticaria y lesiones purpúricas, eritema multiforme, vasculitis por hipersensibilidad. Renales: Insuficiencia renal, uremia, obstrucción ureteral bilateral. Neurológicas: Somnolencia, cefalea, parestesia, neuropatía periférica, neuritis. Otras: Epistaxis, furunculosis severa de la nariz, ictiosis, alopecia, necrólisis epidérmica tóxica, artralgia, equimosis, fiebre, miopatía, escalofrío. INTERACCIONES Dosis elevadas de alopurinol inhiben el metabolismo oxidativo de la azatioprina y la mercaptopurina, aumentando la posibilidad de sus efectos probenecid y la ciclosporina, incrementando el riesgo de toxicidad. El uso en combinación con ampicilina o amoxicilina aumenta la incidencia de urticaria. Los diuréticos tiazídicos y el ácido etacrínico aumentan la concentración sérica de oxipurinol, principal metabolito del alopurinol. La combinación con trimetoprima/sulfametoxazol puede provocar trombocitopenia. Puede aumentar el efecto del dicumarol. Los diuréticos tiazídicos aumentan la toxicidad del alopurinol. El diazóxido y la pirazinamida au-mentan los niveles sanguíneos del ácido úrico. Aumenta el efecto hipoglicemiante de la cloropropamida. Los agentes acidificantes de la orina (cloruro de amonio, ácido ascórbico, fosfato de potasio o de sodio) aumentan la formación de cálculos. El alcohol M disminuye la eficacia del alopurinol ya que aumenta los niveles de ácido úrico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. Dado que el alopurinol puede aumentar la frecuencia de los ataques de gota durante los primeros 6-12 meses de la terapia, se debe combinar con dosis profilácticas de colchicina durante los 3-6 primeros meses de 2ed. 2004 * Formulario TerapÈutico Nacional 373 para asegurar una diuresis adecuada. Se debe procurar lograr una orina neutra o ligeramente alcalina. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Tanto el medicamento como su metabolito son removibles por diálisis peritoneal y por hemodiálisis. INFORMACI”N AL PACIENTE El medicamento puede alterar la capacidad del paciente para realizar actividades que requieren alerta mental, concentración y destreza motriz. Es mejor tolerada si se ingiere después de las comidas. Ingerir COLCHICINA FARMACOLOGÕA Agente con actividad antigotosa y efecto antiinflamatorio débil. No tiene acción analgésica ni afecta la excreción urinaria o la concentración sérica de ácido úrico. Aunque no se conoce con precisión su mecanismo de acción, se postula que reduce la respuesta inflamatoria a la deposición de cristales de urato de sodio en los tejidos articulares, probablemente por inhibición del metabolismo, la movilidad, la quimiotaxis y otras funciones 374 Tiene un efecto profiláctico supresor que ayuda a reducir la incidencia de los ataques agudos y a aliviar el dolor residual y la incomodidad que a veces sufren los pacientes con gota. FARMACOCIN…TICA Después de la administración oral, se absorbe bien en el tracto gastrointestinal. Se distribuye rápidamente a diversos tejidos como el riñón, el bazo, los leucocitos (donde se concentra) y el hígado. En el hígado es parcial-mente metabolizada (por desace-tilación) y de allí, por vía biliar, reingresa nuevamente al intestino como fármaco intacto y como me-tabolitos, desde donde se reabsorbe pasando otra vez a la circulación sistémica; lo cual explica que las concentraciones plasmáticas del fármaco y sus metabolitos declinen 1-2 horas después de la administración oral y luego vuelvan a aumentar. El fármaco intacto y sus metabolitos se excretan, en su mayoría, en las heces y, en menor proporción, en la orina. La vida media de eliminación plasmática es de 20 minutos, mientras que en los leucocitos es de 60 horas. INDICACIONES Alivio del ataque de artritis gotosa aguda y manejo profiláctico de la artritis gotosa recurrente. Formulario Terapéutico Nacional * 2ed. 2004 La cantidad total diaria se ubica en el rango de 4-8 mg y el ataque debe desaparecer en 48-72 horas. - Tratamiento profiláctico de la artritis gotosa recurrente en pacientes con al menos 1 ataque por año: 0,5-0,60 mg/día 1-4 veces por semana, en forma contínua. En pacientes con más de 1 ataque por año: 0,5-0,60 mg/día, en forma contínua. Dado que los procedimientos quirúrgicos pueden desencadenar ataques, pacientes próximos a una cirugía deben recibir 0,5-0,65 mg 3 veces/día por 3 días antes y 3 días después de la intervención. REACCIONES ADVERSAS Gastrointestinales: Náuseas, vómitos, diarrea, dolor abdominal, espasmo vesicales, estomatitis, íleo paralítico. Hematológicas: En terapias prolongadas se han descrito casos de agranulocitosis, leucopenia, trombocitopenia y anemia aplásica. Reacciones de hipersensibilidad: Urticaria. Otras: Hipotiroidismo, postración, púrpura no trombocitopénica, alopecia, neuritis periférica, dermatitis vesicular, miopatía, falla renal, hematuria y aumentos de la fosfatasa alcalina. INTERACCIONES Altera la absorción oral de la vitamina B12. Estudios realizados en animales, sugieren que la colchicina aumenta la la profilaxis con la colchicina. La fenilbutazona aumenta el riesgo de leucopenia y trombocitopenia. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con trastornos severos de la función cardiaca, gastrointestinal, renal, con discrasias sanguíneas, en casos de hipersensibilidad al medicamento y durante el embarazo. Usar con precaución en pacientes con alteración de la función hepática, en ancianos y en pacientes debilitados. La terapia debe suspenderse de inmediato si se presentan manifestaciones de toxicidad gastrointestinal y no debe reiniciarse hasta la desaparición completa de la sintomatología. La seguridad y la eficacia en niños no han sido establecidas. Se recomienda suspender la lactancia M durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. ÁCIDO ALENDRÓNICO (ALENDRONATO S”DICO) FARMACOLOGÍA El alendronato sódico es un 2ed. 2004 * Formulario TerapÈutico Nacional 375 aminobifosfonato sintético que actúa como inhibidor específico de la resorción ósea por los osteoclastos. Los bifosfonatos son análogos sintéticos del pirofosfato que se une a la hidroxiapatita en el hueso. FARMACOCINÉTICA La biodisponibilidad oral es inferior al 1%. Los alimentos y la presencia de calcio reducen su absorción. Se une a las proteínas plasmáticas en un 78% y se distribuye rápidamente a los huesos adhiriéndose a los sitios en los que ocurre la resorción, donde ejerce su acción antiosteoclástica. Posteriormente, se incorpora a la matriz ósea donde cesa su actividad farmacológica y se acumula por largo tiempo. La porción no unida al hueso (aproxima-damente un 50% de la dosis) se excreta en 72 horas como fármaco intacto por la orina. No sufre me-tabolismo. La vida media de eliminación terminal se estima que excede los 10 años, reflejando, probablemente, la liberación del alendronato del esqueleto. INDICACIONES Condiciones que cursan con un aumento de la resorción ósea, como la osteoporosis y la enfermedad de Paget. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis 376 REACCIONES ADVERSAS Gastrointestinales: Dolor abdominal, distensión abdominal, gastritis, disfagia, náusea, vómitos, úlcera esofági-ca, regurgitación, flatulencia, diarrea, constipación, dispepsia. Neurológicas: Cefalea, mareos. Reacciones de hipersensibilidad: Eritema y erupción (raras veces). Otras: Dolor musculo-esquelético, calambres musculares, perversión del gusto. INTERACCIONES Los antiácidos contienen cationes multivalentes (calcio, magnesio, aluminio) que se unen al ácido alendrónico y los suplementos de calcio comprometen su absorción gastrointestinal. Los alimentos y las bebidas lácteas actúan de manera similar. Se ha observado reducción de la absorción de ácido alendrónico con café, jugo de naranja y alimentos en general. El consumo diario de aspirina puede incrementar el riesgo de efectos gastrointestinales adversos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, trastornos esofágicos, hipocalcemia y en pacientes con incapacidad para mantenerse de pie o en posición erguida durante los 30-60 minutos siguientes a la administración del medicamento. Usar con precaución en Formulario Terapéutico Nacional * 2ed. 2004 vitamina D. No se conoce la seguridad de su uso en niños, en el embarazo, durante la lactancia y en pacientes con depuración de creatinina menor de 35 ml/minuto. SOBREDOSIS Debido al riesgo de lesión esofágica, no es recomendable inducir el vómito en casos de sobredosis. Se debe mantener al paciente en posición erguida y administrar leche o antiácidos para impedir o minimizar la absorción del ácido alendrónico. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Tomar siempre por las mañanas al levantarse, 30-60 minutos antes de la primera comida y evitar, durante ese tiempo, la ingestión de bebidas, alimentos u otros medicamentos. Administrar ˙nicamente con 240 ml de agua y mantenerse de pie o en posición erguida por no menos de 3060 minutos. La tableta no debe triturarse o masticarse. Notificar al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial cualquier molestia a nivel del esófago. 2ed. 2004 * Formulario TerapÈutico Nacional M 377 N01 ANESTÉSICOS Bupivacaína Enflurano Fentanilo (Anestésico) Isoflurano Ketamina Lidocaína (Anestésico) Lidocaína / Epinefrina* (Anestésico) Propofol Tiopental N03 ANTIEPILÉPTICOS Ácido Valpróico (Valproato Sódico) Carbamazepina Fenitoina Fenobarbital Gabapentina Sulfato de Magnesio* (Anticonvulsivante en Toxemia) N SISTEMA NERVIOSO N02 ANALGÉSICOS Ácido Acetilsalicílico (Analgésico y Antipirético) Ergotamina / Cafeína / Paracetamol (Ergotamina / Cafeína / Acetaminofen) Fentanilo (Analgésico) Metamizol Sódico (Dipirona) Morfina (Liberación Continua y Solución Inyectable) Oxicodona Paracetamol (Acetaminofen) Paracetamol / Codeína (Acetaminofen / Codeína) Petidina (Meperidina) Tramadol N N04 DROGAS ANTIPARKINSONIANAS Biperideno (Biperideno Clorhidrato y Biperideno Lactato) Bromocriptina Levodopa / Carbidopa N05 PSICOLÉPTICOS Bromazepam 2ed. 2004 * Formulario TerapÈutico Nacional 379 Midazolam Olanzapina Risperidona Tioridazina Trifluoperazina N06 PSICOANALÉPTICOS Fluoxetina Imipramina Metilfenidato N07 OTRAS DROGAS QUE ACT⁄AN SOBRE EL SISTEMA NERVIOSO Bromuro de Neostigmina (Neostigmina) 380 Formulario Terapéutico Nacional * 2ed. 2004 BUPIVACAÍNA FARMACOLOGÍA Ver: LIDOCAINA / EPINEFRINA. FARMACOCINÉTICA La bupivacaína es más liposoluble que la lidocaína. Después de la administración produce una respuesta anestésica en 4-10 minutos, la cual persiste por 3,5-8,5 horas. Una vez absorbida, se une a las proteínas plasmáticas en un 95% y se distribuye ampliamente a los tejidos penetrando con facilidad la barrera hematoencefálica. Se metaboliza en el hígado por conjugación con ácido glucurónico y se excreta como metabolitos, junto con 4-10% del fármaco intacto, en la orina. La vida media de eliminación tras la administración IV es de 2-3 horas y de 2,7-5,5 horas después de la administración peridural. INDICACIONES Anestesia local o regional y analgesia en procedimientos quirúrgicos, dentales, obstétricos y de diagnóstico. - Bloqueo parcial a moderado: 2550 mg. - Anestesia caudal: - Bloqueo moderado a completo: 75-150 mg. - Bloqueo parcial a moderado: 37,5-75 mg. - Anestesia retrobulbar: - Bloqueo completo: 15-30 mg. - Bloqueo nervioso periférico: 25175 mg. - Infiltración local: hasta 175 mg máximo. - Anestesia dental: 9 mg. - Anestesia simpática (sin epinefrina): 50-125 mg. - Como dosis de prueba antes de la administración epidural (sin epinefrina): 10-15 mg. REACCIONES ADVERSAS Ver: LIDOCAINA / EPINEFRINA. INTERACCIONES Ver: LIDOCAINA / EPINEFRINA. CONTRAINDICACIONES/ PRECAUCIONES Además de lo descrito para la lidocaína (Ver: LIDOCAINA / EPINERÉGIMEN DE DOSIFICACIÓN FRINA), la bupivacaína está contraLas dosis deben individualizarse en indicada en el bloqueo paracervical y función del tipo y la duración del en la anestesia regional IV. Para la procedimiento clínico o quirúrgico, la analgesia obstÈtrica no se debe profundidad y la duración de la usar la soluciÛn al 0,75%. La anestesia requerida, la concentración dosificación debe reducirse en del producto y la condición del pa- pacientes debilitados, en ancianos y ciente. La dosis única máxima no debe en presencia de enfermedad cardiaca 381 2ed. 2004 * Formulario TerapÈutico Nacional N INFORMACIÓN AL PACIENTE ENFLURANO FARMACOLOGÕA Anestésico volátil halogenado. Aunque no se conoce con precisión el mecanismo por el cual los anestésicos inhalatorios producen pérdida de la percepción de las sensaciones e inconsciencia, existe evidencia que demuestra una correlación directa entre la potencia de los anestésicos y su solubilidad en aceite. Con base en ello, se ha sugerido que el efecto anestésico podría ser el resultado de una acción sobre la matriz lipídica de las membranas neuronales en el cerebro que produce una alteración en su funcionamiento fisiológico. Hay evidencia de que algunos anestésicos inhalados inhiben la liberación de neurotransmisores y que, por otra parte, actúan a nivel post-sináptico, impidiendo la actividad del neurotransmisor a nivel del receptor. FARMACOCIN…TICA Después de la inhalación, es absorbido rápidamente a la circulación sistémica produciendo el inicio de la respuesta anestésica a los 7-10 minutos, la cual se hace máxima a los 20 minutos. Tiene un coeficiente de partición sangre/gas de 1,91 y aceite/gas de 98,5. Un 2,5-10% del 382 (2,4% de la dosis). INDICACIONES Inducción y mantenimiento de la anestesia general. R…GIMEN DE DOSIFICACI”N La concentración alveolar mínima de enflurano es de 1,68% en oxígeno y de 0,57% en 70% de óxido nitroso. Tales valores; sin embargo, tienden a disminuir en presencia de hipotermia, embarazo, hipotensión, con el uso concurrente de otros depresores del SNC y mientras mayor sea la edad del paciente. La concentración de enflurano para la inducción de anestesia con oxígeno o en combinación con óxido nitroso/oxígeno, en adultos, es 2- 4,5% y para mantenimiento 0,5-3%. Para la cirugía ocular se emplea usualmente al 1%. Como suplemento en la anestesia obstétrica (sección cesárea) 0,5-1% y para la analgesia en el parto vaginal 0,25-1%. En niños de 5-14 años se usa una concentración de 2%; en infantes o en pacientes aprehensivos podría ser necesaria una concentración hasta de 3-4%. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, cambios en el ECG. Gastrointestinales: Náuseas, vómito. Hepáticas: Hepatotoxicidad. Neurológicas: Excitación, agitación, delirio, convulsiones. Renales: Insuficiencia renal. Respiratorias: Formulario Terapéutico Nacional * 2ed. 2004 crementa los requerimientos de anestesia; la metildopa los disminuye. Los depresores del SNC potencian el efecto de los anestésicos. La adrenalina, la levodopa y las xantinas aumentan el riesgo de arritmias cardíacas. La succinilcolina incrementa la posibilidad de hipertermia maligna y bradicardia. Los anestésicos inhalatorios prolongan la vida media de la ketamina y el tiempo de recu-peración de los pacientes; interfieren con la actividad de los medicamentos usados en el tratamiento de la miastenia gravis (neostigmina y piri-dostigmina) reduciendo su efectividad; aumenta el efecto de los anticoa-gulantes cumarínicos; sensibiliza al miocardio a la acción de las catecolaminas y otros simpato-miméticos; disminuye la respuesta uterina a los oxitócicos y el uso concurrente con aminoglicósidos, lincomicina, relajantes musculares no despolarizantes o con polimixina B, puede provocar bloqueo neuromuscular aditivo. Posible potenciación de la hepatotoxicidad de la isoniazida. La isoniazida incrementa la formación de metabolitos fluorados inorgánicos potencialmente nefrotóxicos. Aumenta el efecto de los agentes hipotensores. Reduce del efecto de la oxitocina pero aumenta el efecto hipotensor y el riesgo de arritmias. sivos. Utilizar con precaución en pacientes con insuficiencia hepática y/o renal, durante el embarazo y en combinación con bloqueantes neuromusculares. El olor ácido y picante del enflurano puede provocar tos y laringoespasmo. SOBREDOSIS Manejo sintomático y terapia de soporte. Ventilación asistida y administración de oxígeno 100%. Corrección del equilibrio ácido-base e hidro-electrolítico. En caso de bradicardia severa, usar atropina. Si se presenta hipotensión marcada, colocar el paciente en posición Trendelenburg y administrar fluidoterapia IV; no usar medicamentos vasopresores. En caso de hipertermia maligna, usar FENTANILO (AnestÈsico) FARMACOLOGÍA N Analgésico narcótico. Actúa como agonista de los receptores de opiáceos (principalmente los receptores mu [ ]) cerebrales, espinales y de otros tejidos, generando efectos dependientes de la dosis que incluyen: Analgesia, sopor, miosis, disminución de la motilidad intestinal, supresión del reflejo de la tos y depresión respiratoria. A dosis terapéuticas, no afecta el sistema 2ed. 2004 * Formulario TerapÈutico Nacional 383 la acción analgésica se cree que involucra una alteración de la percepción del dolor y de la respuesta emocional al estímulo doloroso. FARMACOCINÉTICA Después de la administración IV produce, casi de inmediato, un efecto analgésico que se hace máximo en pocos minutos y persiste por 30-60 minutos. Se distribuye rápidamente desde la sangre a los pulmones, al tejido músculo-esquelético y más lentamente al tejido adiposo, desde donde se redistribuye en forma gradual y sostenida nuevamente a la circulación sistémica. Se une a las proteínas plasmáticas en un 80-85% pero la unión disminuye a medida que el fármaco se ioniza. Es metabolizado principalmente en el hígado por Ndesalquilación a productos farmacológicamente inactivos. Demuestra una alta depuración de primer pasaje. El 75% de una dosis IV es eliminado en 72 horas por la orina como metabolitos y 10% como fármaco intacto, mientras un 9% se excreta por las heces, principalmente como metabolitos. peso, la condición clínica y el uso de otros medicamentos, así como también en función del tipo de anestesia y del procedimiento quirúrgico involucrado. - Premedicación: 50-100 mcg IM, 3060 minutos antes de la cirugía. - Como adjunto en la anestesia general: Se puede usar en regímenes de dosis bajas de 2 mcg/kg IV, en regímenes de dosis moderadas de 2-20 mcg/kg IV (seguido por dosis adicionales de 25-100 mcg, en caso necesario) y en regímenes de dosis altas de 20-50 mcg/kg (seguido por dosis adicionales que se ubican entre 25 mcg y 50% de la dosis inicial, IV, según sea necesario). - Como adjunto en anestesia regional y durante la recuperación postoperatoria: 50-100 mcg IM ó IV lentamente, durante 1 a 2 minutos, según sea necesario. REACCIONES ADVERSAS Ver: MORFINA. INTERACCIONES Ver: MORFINA. INDICACIONES Analgesia de corta duración en la CONTRAINDICACIONES/ premedicación, inducción y manteni- PRECAUCIONES miento de la anestesia y en el período Ver: MORFINA. post-operatorio durante la recuperación del paciente. Como suplemento analgésico narcótico en la anestesia 384 Formulario Terapéutico Nacional * 2ed. 2004 ISOFLURANO FARMACOLOGÕA Anestésico halogenado inhalatorio con mecanismo de acción similar al descrito para el enflurano (ver: ENFLURANO). FARMACOCIN…TICA Después de la administración por vía inhalatoria se absorbe rápidamente a la circulación sistémica y se distribuye de manera predominante a órganos altamente irrigados como el cerebro, riñón, corazón e hígado. Se observa un efecto anestésico en aproximadamente 7-10 minutos. Tiene un coeficiente de partición sangre/gas de 1,43 y aceite/gas de 97,8. Se metaboliza en el hígado en muy escasa proporción (0,17%), por lo que se excreta prácticamente intacto, casi en su totalidad, por vía respiratoria. INDICACIONES Inducción y mantenimiento de la anestesia general. R…GIMEN DE DOSIFICACI”N La concentración alveolar mínima de isoflurano es de 1,15% en oxígeno y de 0,5% en óxido nitroso. Tales valores; sin embargo, tienden a disminuir en presencia de hipotermia, embarazo, hipotensión, con el uso concurrente de otros depresores del SNC y con la edad del paciente. La se usa con oxígeno sólo, se debe adicionar 0,5-1%. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, arritmias cardíacas. Gastrointestinales: Náusea, vómito. Hepáticas: Hepatotoxicidad. Neurológicas: Convulsiones, alucinaciones, excitación, delirio. Renales: Nefrotoxicidad. Respiratorias: Depresión respiratoria. Otras: Rabdomiolisis y síndrome neuroléptico maligno. En el período post-operatorio: Temblores, náuseas, vómitos, íleo paralítico. INTERACCIONES Ver: ENFLURANO. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al isoflurano o a otros anestésicos halogenados y en individuos con sospecha o certeza de predisposición genética al desarrollo N de hipertermia maligna. Utilizar con precaución en pacientes con presión intracraneana alta, en pacientes diabéticos (el isoflurano incrementa la glicemia durante la cirugía), en niños, en el embarazo y en combinación con bloqueantes neuromusculares. Es importante evitar la isquemia miocárdica en pacientes con enfermedad de las arterias coronarias. La seguridad en niños menores de 2 años no ha 2ed. 2004 * Formulario TerapÈutico Nacional 385 hospitalario. KETAMINA FARMACOLOGÍA Anestésico general de acción corta, que produce un estado anestésico caracterizado por una analgesia profunda, reflejos laríngeos y tono muscular normales, estimulación cardiovascular y respiratoria y, ocasionalmente, una leve depresión respiratoria. Aunque no se conoce bien su mecanismo de acción, la evidencia disponible sugiere que podría resultar del bloqueo de los impulsos aferentes asociados al componente afectivoemocional de la percepción dolorosa en la formación reticular, supresión de la actividad medular o interacción con diversos neurotransmisores en el SNC. Al estado anestésico inducido por la ketamina se le ha denominado “anestesia disociativa”. FARMACOCINÉTICA Después de la administración IV produce una respuesta anestésica a los 4-10 minutos, la cual persiste por 5-10 minutos, mientras que por vía IM el efecto se evidencia a los 3-4 minutos y se mantiene por 12-25 minutos. Sufre metabolismo hepático transformándose en norketamina, con actividad farmacológica menor, que se excreta, junto con un 4% del fármaco intacto, en un 90% por la orina y en un 386 duración y que no requieren de relajación muscular. Inducción de anestesia general y para anestesia suplementaria en procedimientos con óxido nitroso. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV e IM. Las dosis para adultos y niños son similares. - Inducción de anestesia: 1-2 mg/kg IV como dosis única o por infusión a una velocidad de 0,5 mg/kg/minuto, o por vía IM en dosis de 5-10 mg/kg. - Mantenimiento de la anestesia: 0,1 a 0,5 mg/minuto durante el tiempo que sea necesario. - SedaciÛn y analgesia: Iniciar con 200-750 mcg/kg IV en 2-3 minutos o 2-4 mg/kg IM, seguido en ambos casos por infusión IV de 5-20 mcg/kg/minuto. REACCIONES ADVERSAS Cardiovasculares: Aumento de la presión arterial y la frecuencia del pulso. También se ha observado hipotensión y bradicardia. Arritmia. Gastrointestinales: Anorexia, náuseas, vómitos. Oculares: Diplopía, nistagmo, aumento de la presión intraocular, lagrimeo. Reacciones de hipersen-sibilidad: Eritema transitorio y/o erupción multiforme. Respiratorias: Depresión respiratoria severa (con la inyección muy rápida) y laringoes-pasmo, aumento de la Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Los anestésicos halogenados pueden prolongar la vida media de la ketamina y el tiempo de recuperación del paciente. Los barbitúricos y narcóticos usados concurrentemente, prolongan el período de recuperación. Los antihipertensivos y los depresores del SNC (incluidos los agentes para premedicación, inducción y mantenimiento de la anestesia) pueden incrementar el riesgo de hipotensión y depresión respiratoria. En pacientes que reciben hormonas tiroideas incrementa el riesgo de hipertensión y taquicardia. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento, en pacientes en quienes un aumento de la presión arterial podría resultar peligrosa y en personas con trastornos psiquiátricos como esquizofrenia o psicosis aguda. Usar con precaución en pacientes con enfermedad cardiaca o en aquellos sometidos a terapia con hormonas tiroideas. Dado que la ketamina incrementa la secreción salival y traqueobronquial, se debe usar previamente atropina para evitar dicha eventualidad y usar con precaución en procesos quirúrgicos de faringe, laringe, tráquea y árbol bronquial. La administración IV rápida produce hipertensión y taquicardia. El SOBREDOSIS Manejo sintomático y terapia de soporte. Si ocurre depresión respiratoria, se prefiere el soporte mecánico al uso de analépticos. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso hospitalario. Sin embargo, si el paciente es dado de alta en un tiempo breve (procedimientos diagnósticos), se le debe advertir que no puede LIDOCAÍNA (AnestÈsico) FARMACOLOGÍA AnestÈsico local. Bloquea reversiblemente la generación y conducción de los impulsos nerviosos a través de las neuronas sensitivas, motoras y autonómicas mediante una disminu-ción de la permeabilidad de la mem-brana a los iones sodio, que se traduce en aumento del umbral de excita-bilidad eléctrica, reducción de N la ca-pacidad de despolarización y preven-ción de la propagación del potencial de acción a lo largo de la fibra. FARMACOCIN…TICA Ver: LIDOCAÍNA / EPINEFRINA. INDICACIONES Ver: LIDOCAÍNA / EPINEFRINA. RÉGIMEN DE DOSIFICACIÓN 2ed. 2004 * Formulario TerapÈutico Nacional 387 INTERACCIONES Ver: LIDOCAÍNA / EPINEFRINA. CONTRAINDICACIONES/ PRECAUCIONES Ver: LIDOCAÍNA / EPINEFRINA. SOBREDOSIS Ver: LIDOCAÍNA / EPINEFRINA. INFORMACI”N AL PACIENTE LIDOCAÍNA / EPINEFRINA (AnestÈsico) FARMACOLOGÍA Anestésico local. Bloquea reversiblemente la generación y conducción de los impulsos nerviosos a través de las neuronas sensitivas, motoras y autonómicas mediante una disminución de la permeabilidad de la membrana a los iones sodio, que se traduce en un aumento del umbral de excitabilidad eléctrica, reducción de la capacidad de despolarización y prevención de la propagación del potencial de acción a lo largo de la fibra. La incorporación de una amina simpaticomimética, la epinefrina, a la formulación provoca una vasoconstricción local debida a su acción sobre los receptores alfa-adrenérgicos vasculares que impide la absorción sistémica del anestésico, prolongando así la duración de su efecto en la zona de aplicación. duración depende del tiempo de contacto con el tejido nervioso y de la concentración del anestésico. Se ha descrito tiempos de duración de la anestesia de 0,5-1 hora (sin epinefrina) y 2-6 horas (con epinefrina). Una vez absorbida, la lidocaína se une a las proteínas plasmáticas en un 80% y se distribuye ampliamente a los tejidos. Se metaboliza extensamente (90%) en el hígado generando metabolitos activos que se excretan, junto con menos de 10% del fármaco intacto, por la orina. La vida media de eliminación terminal es de 1,5-2 horas. INDICACIONES Anestesia superficial, bloqueo nervioso central o periférico y anestesia regional IV. RÉGIMEN DE DOSIFICACIÓN Las dosis deben individualizarse en función del tipo y la duración del procedimiento clínico o quirúrgico, la profundidad y la duración de anestesia requerida, la concentración del producto y la condición del paciente. Las siguientes son dosis referenciales (con y sin epinefrina): - Infiltración percutánea: 5-300 mg. - Bloqueo nervioso central (epidural o caudal): 200-300 mg. - Bloqueo nervioso braquial: 225-300 mg. - Anestesia dental: 20-100 mg. - Bloqueo nervioso intercostal: 30 mg. FARMACOCINÉTICA 388 Formulario Terapéutico Nacional * 2ed. 2004 - Anestesia regional IV: 50-300 mg. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, hipotensión y colapso cardiovascular que puede conducir a un paro cardíaco, arritmias. Neurológicas: Cefalea posicional y dolor de espalda; escalofrío, síntomas nerviosos periféricos, diplopía, ansiedad. Reacciones de hipersensibilidad: Prurito, eritema, urticaria, edema angioneurótico, congestión nasal, elevación de la temperatura, sudoración, síncope, broncoconstricción, lesiones cutáneas, reacciones anafilactoides y anafilaxis. Respira-torias: Paro respiratorio, status asmaticus. Otras: Visión borrosa, tinitus. Algunas de estas reacciones pueden ser debidas a la técnica anestésica usada, con o sin con-tribución del anestésico local. Cuando se trata de bloqueo caudal o epidural lumbar, por penetración del espacio subaracnoideo: Bloqueo epidural, hipotensión secundaria a bloqueo epidural, pérdida de control de la vejiga y del intestino, pérdida de la sensación perineal y de la función sexual. Déficit motor, sensorial y/o autonómico persistente de algunos segmentos espinales inferiores (recuperación lenta). Reacciones debidas a la presencia de epinefrina: Palpi-taciones, taquicardia, cefalea, angina, hipertensión y, en casos extremos, edema pulmonar y reciben antidepresivos tricíclicos o inhibidores de la MAO pueden provocar hipertensión; con alcaloides del ergot puede ocasionar hipertensión severa y persistente y, en casos graves, accidente cerebrovascular. Las fenotiazinas pueden reducir o revertir el efecto presor de la epinefrina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento o a otros anestésicos del grupo de las amidas. Usar con precaución en pacientes con falla hepática y/o renal, enfermedad neurológica preexistente, niños, ancianos, pacientes debilitados, hipertensión severa, septicemia y en el embarazo. Durante su uso se debe vigilar frecuentemente el ECG, la función respiratoria y el estado de conciencia del paciente. Las solu-ciones con epinefrina deben usarse con precaución en áreas corporales irrigadas por arterias N terminales o con irrigación comprometida, en pacientes con hipertensión, enfermedad vas-cular periférica, ateroesclerosis, bloqueo cardíaco, tirotoxicosis y diabetes mellitus. Se debe suspender temporalmente la lactancia. SOBREDOSIS Las emergencias son debidas a niveles elevados del anestésico durante la administración o a la inyección 389 2ed. 2004 * Formulario TerapÈutico Nacional hipotensión severa, administrar fluidos IV y agentes vasopresores. INFORMACIÓN AL PACIENTE No aplicable; medicamento para uso hospitalario. Sin embargo, cuando sea apropiado, debe advertirse al paciente que puede experimentar una disminución temporal de la sensación y la actividad motora después de la PROPOFOL FARMACOLOGÍA Hipnótico-sedante de acción rápida, con propiedades de anestésico general. FARMACOCINÉTICA Después de la administración en bolo IV, produce un efecto anestésico en 30-40 segundos el cual se mantiene por 3-10 minutos, según la dosis. Se une a las proteínas plasmáticas en un 97-99%. Por su naturaleza lipofílica se distribuye ampliamente en el organismo, alcanzando concentraciones efectivas a nivel del SNC en muy pocos segundos. La vida media del equilibrio hematocerebral es 1 a 3 minutos. La corta duración de sus efectos es debida a una rápida redistribución desde el SNC hacia otros tejidos, sumado a un rápido y extenso metabolismo hepático que da lugar a metabolitos inactivos que se excretan en un 88% por vía renal y 390 fases previas. La eliminación total tiene una mayor duración en caso de terapia prolongada debido, probablemente, a un efecto de saturación tisular. INDICACIONES Inducción y mantenimiento de anestesia general, sedación en pacientes conscientes durante procedimientos quirúrgicos o de diagnóstico y sedación en pacientes con intubación y ventilación mecánica en cuidados intensivos. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV en bolo o en infusión continua. Las dosis deben individualizarse cuidadosamente en función de la respuesta y las condiciones clínicas de cada paciente. - Inducción y mantenimiento de la anestesia general: - Adultos menores de 55 años: Iniciar con 40 mg cada 10 segundos hasta el comienzo de la inducción (2-2,5 mg/kg) y mantener con 100-200 mcg/kg/minuto (6-12 mg/kg/hora). Bolo intermitente: Incrementos de 20-50 mg según sea necesario. - Ancianos, pacientes debilitados o pacientes ASA (Sociedad Americana de Anestesiólogos) III ó IV: Para la inducción: 20 mg cada 10 segundos hasta el Formulario Terapéutico Nacional * 2ed. 2004 300 mcg/kg/minuto (7,5-18 mg/kg/hora). - Sedación en pacientes conscientes durante procedimientos quirúrgicos o de diagnóstico: - Adultos menores de 55 años: Iniciar con 100-150 mcg/kg/minuto (6-9 mg/kg/hora) por 3-5 minutos y mantener con 25-75 mcg/kg/minuto (1,5-4,5 mg/kg/hora) o dosis crecientes en bolos IV de 10-20 mg. - Ancianos, debilitados o pacientes ASA III ó IV: 80% de las dosis anteriores. - Sedación para pacientes adultos con intubación y ventilación mecánica en cuidados intensivos: - Iniciar con 5 mcg/kg/minuto (0,3 mg/kg/hora) durante 5 minutos como mínimo con incrementos subsecuentes de 5-10 mcg/kg/minuto (0,3-0,6 mg/kg/hora) por 5-10 minutos adicionales hasta lograr el nivel de sedación deseado y mantener con 5-50 mcg/kg/minuto (0,3-3 mg/kg/hora) o más, según sea necesario. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, hipotensión, hipertensión, disminución de gasto cardíaco, contracciones auriculares prematuras, síncope. Gastrointestinales: Sialorrea. Neurológicas: Movimientos involuntarios, Otras: Sensación de quemadura en el sitio de inyección, hiperlipemia (en pacientes en unidad de cuidados intensivos), ambliopía, orina turbia u orina de color verdoso, hipo. INTERACCIONES La premedicación con opiáceos como fentanilo, alfentanil, morfina o meperidina, o la combinación de opiáceos y sedantes como benzodiazepinas, barbitúricos, hidrato de cloral o droperidol, incrementa los efectos anestésicos del propofol, lo cual puede resultar en descensos más marcados de la presión arterial y el gasto cardíaco. La combinación con anes-tésicos inhalatorios como el óxido nitroso, el halotano, el isoflurano o el enflurano, aumenta el riesgo de de-presión cardio-respiratoria. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a sus N componentes (lecitina de huevo, aceite de soya y glicerina). Cuando la anestesia general o la sedación están contraindicadas. No es recomendable su empleo en obstetricia. Usar con precaución en pacientes con epilepsia y/o historia de convulsiones, enfermedad cardiaca o respiratoria e insuficiencia hepática. No se conoce la seguridad de su uso para inducción de la anestesia en niños menores de 3 2ed. 2004 * Formulario TerapÈutico Nacional 391 etilendiamino-tetracético), un fuerte quelante de metales; considerar un suplemento de zinc durante la terapia prolongada. Durante el uso se debe suspender la lactancia. El medicamento debe ser protegido de la luz. SOBREDOSIS Suspender inmediatamente la administración del medicamento. La sobredosificación puede causar depresión cardio-respiratoria. Manejar con ventilación artificial con oxígeno, cambiar de posición al paciente, elevando las piernas, aumentando la tasa de flujo de los fluidos intravenosos y administración de agentes vasopresores y/o anticolinérgicos, según el caso. TIOPENTAL FARMACOLOGÕA Anestésico tiobarbitúrico de acción ultracorta que deprime el sistema nervioso central produciendo sedación, hipnosis y anestesia sin analgesia. Se cree que actúa aumentando la respuesta al GABA, en virtud de la interacción con el sitio de unión de los barbitúricos en el complejo del receptor GABA A , disminuyendo la respuesta al glutamato y aumentando la conductancia de la membrana, generando una disminución neta de la excitabilidad neuronal y, en consecuencia, la 392 conducción en la formación reticular ascendente, interfiriendo la transmisión de impulsos a la corteza. FARMACOCIN…TICA Después de la administración IV, produce un efecto hipnótico en 10-40 segundos y uno anestésico en 30-60 segundos, el cual se mantiene por 2030 minutos. El rápido inicio y breve duración de su efecto es consecuencia de su elevada liposolubilidad. Atra-viesa fácilmente la barrera hematoen-cefálica pero es rápidamente re-distribuido de allí a otros tejidos; primero a los altamente perfundidos (hígado, riñón, corazón), luego a los músculos y por último al tejido adiposo. La administración continua causa acumulación en el tejido adiposo, de donde se libera gradualmente para producir un efecto sostenido. Se une a las proteínas plasmáticas en un 80-85% y se metaboliza principalmente en el hígado, aunque ocurre alguna transformación en otros tejidos, como el cerebro y los riñones. Se excreta parcialmente por vía renal como metabolitos inactivos. La vida media de eliminación es de 3-8 horas. INDICACIONES Inducción de la anestesia. Suplemento de otros agentes anestésicos. Anestesia IV para Formulario Terapéutico Nacional * 2ed. 2004 - - respuesta deseada. En niños: Neonatos, 0-14 días: 3,4 mg/kg; Niños, 1-6 meses: 5-8 mg/kg, 112 años: 5-6 mg/kg, cada 30 segundos hasta lograr la respuesta deseada. Para el mantenimiento: Dosis adicionales de 25-50 mg con la frecuencia que la circunstancia demande. Se aconseja la administración IV lenta para minimizar la posibilidad de depresión respiratoria. Estados convulsivos: 75-125 mg tan pronto como sea posible, una vez iniciada la convulsión. Para convulsiones secundarias a la anestesia local, podría requerirse dosis de hasta 125-250 mg en períodos de 10 minutos. REACCIONES ADVERSAS Cardiovasculares: Depresión circulatoria, depresión miocárdica, tromboflebitis, arritmias, hipotensión. Gastrointestinales: Náuseas, vómito, sialorrea. Neurológicas: Confusión, delirio, dolor de cabeza, ansiedad, somnolencia, excitación, déficit de funciones psicomotoras. Reacciones de hipersensibilidad: Eritema, prurito, urticaria y anafilaxis. Respiratorias: Depresión respiratoria, apnea, rinitis, disnea, laringoespasmo, broncoespasmo, tos. Otras: Dolor en el sitio de la inyección, hipo. tiopental en pacientes sometidos a terapia con hipotensores, incluidos los diuréticos, incrementa el riesgo de hipotensión ortostática. El tiopental reduce las concentraciones plasmáticas de los bloqueantes de los receptores beta-adrenérgicos. La ketamina antagoniza los efectos del tiopental. La administración concomitante con fármacos adrenérgicos puede inducir disritmia cardiaca. No debe mezclarse con soluciones de carácter acídico como la de los relajantes musculares (succinilcolina) y otros fármacos, debido a que pueden formarse agregados. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en individuos hipersensibles al medicamento o a otros barbitúricos, en pacientes con porfiria latente o manifiesta, en la enfermedad cardiovascular severa, en el estado asmático y en condiciones inflamaN torias de la boca, la mandíbula y el cuello. Usar con precaución en condiciones en las que el efecto hipnótico podría prolongarse o potenciarse (premedicación excesiva, enfermedad de Addison, falla hepática y/o renal, mixedema, uremia, anemia o miastenia gravis), en la hipotensión, shock, presión intracraneana aumentada, trastornos circulatorios y durante el embarazo. El uso repetido y/o pro- 2ed. 2004 * Formulario TerapÈutico Nacional 393 ministración del medicamento, establecer o mantener las vías aéreas permeables y, en caso necesario, administrar oxígeno por ventilación asistida. INFORMACI”N AL PACIENTE ÁCIDO ACETILSALICÍLICO (AnalgÈsico y AntipirÈtico) FARMACOLOGÍA Derivado salicilado con actividad analgésica, antiinflamatoria y antipirética. Su acción parece debida, al menos en parte, a la inhibición de la síntesis de prostaglandinas por la inactivación irreversible de las isoenzimas ciclooxigenasa-1 (COX-1) y ciclooxigenasa-2 (COX-2). Produce analgesia por un efecto periférico y central. Periféricamente inhibe la síntesis de prostaglandinas; centralmente parece que actúa a nivel hipotalámico, pero el mecanismo se desconoce. También actúa en el hipotálamo para producir antipiresis. Aumenta la disipación de calor y el flujo sanguíneo periférico. El efecto antipirético puede estar relacionado con la inhibición de la síntesis de prostaglandinas. FARMACOCINÉTICA El ácido acetilsalicílico se absorbe con rapidez en parte en el estómago, 394 hidrólisis que tiene lugar primariamente a nivel del estómago y del hígado. El ácido salicílico se conjuga con la glicina, formando el ácido salicilúrico, y también con el ácido glucurónico; ambos tipos de metabolitos son excretados en la orina. El ácido acetilsalicílico y el ácido salicílico se unen a las proteínas plasmáticas, particularmente a la albúmina, en un 30% y 90%, respectivamente. Los metabolitos se excretan en la orina en un 75%, conjuntamente con 10% de ácido salicílico y 1% de ácido acetilsalicílico. La vida media de eliminación del ácido acetilsalicílico es de 20 minutos y la del ácido salicílico es de 1,7 horas, incrementándose ambas en pacientes con insuficiencia hepática y renal. INDICACIONES Analgésico y antipirético. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos: 500-1000 mg cada 46 horas, sin exceder 4 g en 24 horas. En niños de 7-12 años: 200-300 mg cada 4-6 horas y en niños de 2-6 años: 100-150 mg cada 4-6 horas. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, taquicardia. Gastrointestinales: Náuseas, vómito, dispepsia, gastritis, Formulario Terapéutico Nacional * 2ed. 2004 hipersensibilidad: Angioedema, broncoespasmo, urticaria, edema de la laringe, anafilaxis. Renales: Falla renal. Respiratorias: Edema pulmonar, taquipnea, alcalosis respiratoria. Otras: Edema cerebral, acidosis metabólica, alteraciones de la glicemia e hiperpotasemia, problemas de la audición. INTERACCIONES El ácido acetilsalicílico puede desplazar de su unión a las proteínas a las sulfonilúreas, warfarina, ácido valpróico, fenitoína y sulfonamidas e incrementar la posibilidad de sus efectos adversos. Puede aumentar los niveles plasmáticos, con la consecuente toxicidad, del metotrexato y la acetazolamida, al competir por su secreción en los túbulos renales. También, puede incrementar el efecto anticoagulante de la warfarina y la heparina, disminuir la actividad de los inhibidores de la enzima convertidora de angiotensina, antagonizar la acción uricosúrica del probenecid y reducir el efecto natriurético de la espironolactona. La combinación con AINEs aumenta el riesgo de sangramiento y de falla renal. Los corticosteroides y el elevado consumo de alcohol predisponen a la ulceración gastrointestinal. El fenobarbital disminuye la eficacia del ácido acetilsalicílico por inducción del metabolismo. También puede interferir con el efecto de los agentes (AINEs), úlcera péptica activa, trastornos hemorrágicos, dengue, varicela (lechina), tratamiento simultáneo con anticoagulantes y en pacientes con alteración de la función hepática y/o renal severa. El uso en niños o adolescentes con infección viral o enfermedades eruptivas ha sido asociado con el síndrome de Reye. Si ocurre zumbido en los oídos o pérdida de la audición se debe consultar al médico antes de tomar dosis adi-cionales del medicamento. Usar con precaución en pacientes con a n t e - c e d e n t e s d e hipoprotrombinemia, deficiencias de vitamina K, enferme-dad renal y/o hepática leve a moderada, asma, insuficiencia cardia-ca congestiva, hipertensión, edema y, en general, aquellas condiciones que pueden agravarse por la retención de fluidos. Se debe evitar el uso en el embarazo a término. No es recomen-dable el uso durante la lactancia. N SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Debe determinarse los niveles plasmáticos de salicilato para establecer la severidad de la intoxicación. Manejo sintomático y tratamiento de soporte. Corrección del balance hidro-electrolítico y del equilibrio ácido-base. La diuresis alcalina favorece la eliminación. En 395 2ed. 2004 * Formulario TerapÈutico Nacional gastrointestinales. Evitar el consumo de alcohol. Si el paciente es alérgico a la tartrazina, debe evitar el uso del ácido acetilsalicílico. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial molestias gastrointestinales, zumbidos en los oídos o pérdida de la niveles plasmáticos pico en 0,5-3 horas. El inicio del efecto; sin embargo, depende de cuán pronto se administre el medicamento después que se presenta el dolor. Por vía oral se logran concentraciones sistémicas muy ba-jas, debido al extenso metabolismo de primer pasaje. Se dice que la ad-ministración con cafeína aumenta la biodisponibilidad pero no ERGOTAMINA / CAFEÍNA / PARACETAMOL se logra más de 1%. Se metaboliza (ERGOTAMINA / CAFEÕNA / ACETAMINOFEN) extensamente en el hígado a productos activos e inactivos que se FARMACOLOGÍA La formulación combina el efecto excretan, en su mayoría (90%), por la analgésico del paracetamol (vía inhi- bilis y en escasa proporción por la bición de la síntesis de prostaglan- orina. La vida media de eliminación es dinas) con la acción vasopresora bifásica con un valor inicial de 2-7 central de la ergotamina. La ergo- horas y de 21 horas en su fase tamina produce una vasoconstricción terminal. La administración oral de selectiva del lecho vascular cerebral y cafeína produce concentraciones una disminución de la amplitud de las séricas pico en 50-75 minutos. Se pulsaciones arteriales como resultado metaboliza en el hígado y los mede una actividad agonista parcial y tabolitos se excretan en la orina. La antagonista sobre los receptores alfa- vida media de eliminación es de 3-5 adrenérgicos, serotoninérgicos y horas. dopaminérgicos y, aparentemente, de INDICACIONES un efecto inhibidor de la recaptación Tratamiento sintomático de dolores de de noradrenalina que contribuye a la cabeza de tipo vascular, como acción vasoconstrictora. Aunque la migraña y cefaleas histamínicas. cafeína también posee propiedades constrictoras de la vasculatura ceRÉGIMEN DE DOSIFICACIÓN rebral, la evidencia reciente sugiere La tableta para la administración oral que su participación en el efecto final contiene 450 mg de paracetamol, 1 mg de la combinación es debida a la de ergotamina y 40 mg de cafeína. En inducción de un incremento en la adultos iniciar con 1-2 tabletas. Si al velocidad y magnitud de la absorción cabo de 20-30 minutos no se logra el 396 Formulario Terapéutico Nacional * 2ed. 2004 prolongada puede provocar dolor precordial, valvulopatías, isquemia miocárdica e infarto. Gastrointestinales: Náuseas, vómito, dolor abdominal, diarrea. Neurológicas: Fatiga, depresión, debilidad en las piernas, hormigueo en las manos y los pies. Otras: Mialgia, erupción, prurito y rigidez en las extremidades, cuello y hombros. INTERACCIONES Con eritromicina y otros macrólidos pueden causar síntomas de toxicidad del ergot (vasoespasmo periférico severo con posible isquemia), cianosis y adormecimiento. El uso de bloqueantes beta-adrenérgicos puede resultar en vasoconstricción excesiva. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a alguno de los constituyentes de la formulación o a derivados del ergot y en pacientes con insuficiencia hepática y/o renal, enfermedad vascular periférica o coronaria, sepsis, hipertensión severa, en niños, en el embarazo y durante la lactancia. Usar con precaución en ancianos. No debe usarse para la profilaxis de la migraña. SOBREDOSIS Vaciamiento gástrico, carbón activado mas no de calor, y de protección de los miembros isquémicos. Puede usarse vasodilatadores con beneficio, pero debe ejercerse precaución para no agravar una hipotensión ya existente. Ante la posibilidad de daño hepático inducido por el paracetamol, proceder según se indica en la monografía respectiva (ver: PARACETAMOL). INFORMACIÓN AL PACIENTE No usar por más de 10 días continuos. Tomar con los alimentos. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. El FENTANILO (AnalgÈsico) FARMACOLOGÍA V e r : F E N T A N I L O ANEST…SICOS. e n FARMACOCINÉTICA Después de la colocación del parche N (sistema transdérmico de fentanilo), el fentanilo, se libera hacia la capa superficial de la piel formando un depósito desde el cual es absorbido en forma lenta y sostenida a la circulación sistémica. Produce una respuesta analgésica a las 12-24 horas, la cual se mantiene por 72 horas. Una vez en la sangre, el patrón farmacocinético es similar al descrito en la monografía del fentanilo como anestésico (ver: 2ed. 2004 * Formulario TerapÈutico Nacional 397 media de eliminación puede alcanzar las 17 horas y hasta 22 horas con la adminisración contínua. INDICACIONES Tratamiento de condiciones que cursan con dolor de intensidad moderada a severa. RÉGIMEN DE DOSIFICACIÓN La dosis debe individualizarse según las necesidades particulares de cada paciente. La dosis inicial usual en adultos sin tolerancia a opiáceos es como sigue: Un parche de 10 cm2 (que libera 25 mcg/hora) cada 72 horas. En caso necesario ajustar las dosis subsiguientes hasta lograr la respuesta deseada. Debido al lento inicio de acción con el primer parche, podría ser necesaria una analgesia suplementaria por vía oral o parenteral. REACCIONES ADVERSAS Ver: FENTANILO en ANESTÉSICOS. El uso de parches de fentanilo puede provocar aumento de la sudoración, prurito, erupción, eritema, edema en la zona de aplicación, dermatitis exfoliativa y pápulas. El fentanilo puede causar dependencia física. Se ha producido muertes por hipoventilación por mal uso del sistema transdérmico de fentanilo. operatorio. No iniciar la terapia con dosis mayores de 25 mcg/hora. Reducir la dosis inicial en pacientes debilitados y en ancianos. En pacientes con fiebre aumenta la liberación de fentanilo del parche y se incrementa la permeabilidad cutánea. No se conoce la seguridad del uso de los parches de fentanilo en niños. El uso prolongado puede conducir a tolerancia y dependencia. SOBREDOSIS Ver: FENTANILO en ANESTÉSICOS. INFORMACIÓN AL PACIENTE (cuando se trate de pacientes ambulatorios) Previo a la colocación del parche, debe lavarse con agua y secarse bien el sitio de aplicación. No se debe usar jabón, aceites, lociones o alcohol para limpiar la piel. Aplicar el parche en zonas libres de vello, costras, irritación o lesiones. Se debe elegir áreas planas como la espalda, el torso, el pecho o la parte superior del brazo. Al colocar el parche, se debe presionar el mismo con la palma de la mano durante 30 segundos para garantizar un ade-cuado contacto con la superficie cutá-nea. No se debe cortar o alterar en forma alguna la integridad del parche. Es recomendable cambiar periódica-mente el sitio de colocación del parche. No se debe alterar la dosificación, frecuencia o duración del INTERACCIONES Ver: FENTANILO en ANESTÉSICOS. 398 Formulario Terapéutico Nacional * 2ed. 2004 la terapia. Notificar al médico, de inmediato, si se presenta alguna reacción o sintomatología inusual METAMIZOL SÓDICO (DIPIRONA) FARMACOLOGÕA Agente derivado de la pirazolona que posee marcada actividad como analgésico, antipirético y antiespasmódico y, en menor grado, posee propiedades antiinflamatorias. El modo de acción como analgésico y antipirético no está claro, aunque parece que hay un componente periférico y uno central involucrados. El efecto antiespasmódico ha sido atribuido a la disminución de la excitabilidad del músculo liso. Su acción parece debida, al menos en parte, a la inhibición de la síntesis de prostaglandinas por inhibición de las isoenzimas ciclooxigenasa-1 (COX-1) y ciclooxigenasa-2 (COX-2). FARMACOCIN…TICA Luego de la administración oral se metaboliza extensamente en la pared intestinal, dando lugar al metabolito activo 4-metilaminoantipirina (4-MAA) que se absorbe e induce una respuesta antipirética inicial a los 3060 minutos, que persiste por 4-6 horas. El 4-MAA se une a las proteínas plasmáticas en un 58% y se distribuye ampliamente en el organismo. Al llegar INDICACIONES Antipirético y analgésico. R…GIMEN DE DOSIFICACI”N Se administra por vía oral, IM e IV siendo las dosis, en cada caso, similares. La dosis usual en adultos es de 0,5-1 g cada 6-8 horas, según sea necesario. En niños, se recomienda 10-12 mg/kg/dosis cada 6-8 horas, según sea necesario, sin exceder 2 g/día en niños de 6-12 años y 1 g/día en niños de 2-6 años. Se debe procurar que la duración del tratamiento sea lo más breve posible. Dosis máxima: 3 g/día (6 tabletas). REACCIONES ADVERSAS Cardiovasculares: Hipotensión, que puede ser marcada si se administra en forma rápida por vía intravenosa, flebitis. Hematológicas: Raras veces leucopenia, agranulocitosis y trombocitopenia. Reacciones de hipersensibilidad: Anafilácticas y anafilacN toides, aunque raras, pueden aparecer después de usar dipirona en varias ocasiones sin complicaciones. También pueden ser agudas o leves: Prurito, ardor, enrojecimiento de la piel, urticaria, edema, disnea. Severas: Urticaria generalizada, angioedema, broncoespasmo, arritmias cardíacas, hipotensión y shock circulatorio, asma, síndrome de Stevens-Johnson o síndrome de 2ed. 2004 * Formulario TerapÈutico Nacional 399 plasmáticos de la ciclosporina. No se debe mezclar con otros medicamentos en la inyectadora. Evitar el consumo de alcohol. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, a los excipientes o a otros analgésicos no narcóticos, enfermedad hepática y/o renal severa, en el embarazo y durante la lactancia. Usar con precaución en pacientes con historia de úlcera péptica, trastornos de la coagulación, alteración de la función renal y/o hepática leve a moderada, asma, insuficiencia cardiaca congestiva, hipertensión y, en general, aquellas condiciones que pueden agravarse por retención o sobrecarga de fluidos. Contraindicado en la porfiria hepática intermitente aguda, deficiencia de glucosa-6-fosfato deshidrogenasa, deterioro de la función de la médula ósea, niños menores de 3 meses de edad. Se debe interrumpir el tratamiento si aparecen signos de posible agranulocitosis o de trombocitopenia. INFORMACIÓN AL PACIENTE Informar al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial: Malestar gastrointestinal, sangrado anormal y/o heces negras. En caso de erupción generalizada, dificultad respiratoria e inflamación de los párpados, de los labios o la nariz, suspender el tratamiento y notificar al médico. Instruir al paciente acerca de la importancia de notificar de inmediato la ocurrencia de fiebre elevada, escalofríos, debilidad e inflamación, o ulceración de la boca y/o la garganta, dado que podrían constituir la fase prodrómica de una agranulocitosis inducida por el medicamento. Cuando se usan dosis altas debe evitarse realizar tareas que requieran estar alerta como conducir MORFINA (LiberaciÛn Continua y SoluciÛn Inyectable) FARMACOLOGÍA Analgésico narcótico. Actúa como agonista de los receptores opiáceos (mu [ ] y kappa [ ] principalmente) cerebrales, espinales y de otros tejidos, generando efectos dependientes de la dosis que incluyen: Analgesia, sopor, miosis, disminución de la motilidad intestinal, supresión del reflejo de la tos y depresión respiratoria. El mecanismo de la acción analgésica se cree que involucra una SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. No usar estimulantes respiratorios, controlar 400 Formulario Terapéutico Nacional * 2ed. 2004 doloroso. FARMACOCINÉTICA Se absorbe en forma variable por vía oral. Debido a su rápida eliminación sistémica (por metabolismo en la mucosa del tracto gastrointestinal durante la absorción y por el efecto de primer pasaje) sólo un 40-50% de la dosis alcanza la circulación sistémica como fármaco intacto. Se observa un efecto analgésico máximo en aproximadamente 60 minutos. Por vía IM se evidencia el efecto pico a los 3060 minutos y a los 20 minutos con la administración IV, manteniéndose, en ambos casos, hasta por 7 horas. Se distribuye a numerosos tejidos y se une a las proteínas plasmáticas en 30-40%. Se metaboliza principalmente en el hígado, transformándose en productos activos como el 3-glucurónido de morfina y el 6-glucurónido de morfina, que tiene la mayor actividad analgésica de los dos metabolitos, indistinguible del de la morfina. También se producen metabolitos inactivos como el 3,6-glucurónido. Los meta-bolitos se excretan, junto con 212% de fármaco intacto, en un 90% por la orina y 7-10% por las heces. La vida media de eliminación es de 1,5-4,5 horas y se incrementa en pacientes con alteración de la función renal. cada 4 horas, según sea necesario. La dosis con formas de liberación prolongada es: 15-30 mg cada 12 horas, aunque en algunos pacientes podría ser necesaria la administración cada 8 horas. La dosis por vía IM en adultos: 10 mg cada 4 horas, según sea necesario y en niños: 0,1-0,2 mg/kg, también cada 4 horas. La dosis por vía IV en adultos: 2,5-15 mg diluidos en 4-5 ml de agua estéril para inyección, administrados en 4-5 minutos y en niños: 0,05-0,1 mg/kg. Para la infusión IV continua se debe iniciar con 0,8-10 mg/hora, con ajustes posteriores hasta lograr la respuesta deseada. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, bradicardia, shock, paro cardíaco, taquicardia. Gastrointestinales: Náuseas, vómitos, constipación, íleo paralítico, boca seca, espasmo del tracto biliar, anorexia. Hematológicas: N Trombocito-penia. Neurológicas: Sedación, som-nolencia, problemas sensoriales, con-vulsiones (con grandes dosis), ma-reos, pesadillas (con las formas de liberación prolongada), aturdimiento, alucinaciones, nerviosismo, depresión, síncope. Reacciones de hipersensibilidad: Prurito y enrojecimiento de la piel, erupciones, urticaria. INDICACIONES Respiratorias: Depresión respiratoria, Manejo del dolor agudo o crónico, de apnea, paro respiratorio. Otras: 401 2ed. 2004 * Formulario TerapÈutico Nacional hipnóticos, antihistamínicos y alcohol, potencian el efecto de los opiáceos. El uso de neurolépticos con la morfina oral puede aumentar el riesgo de depresión respiratoria, hipotensión, sedación profunda o coma. Los opiá-ceos pueden incrementar el bloqueo neuromuscular de los relajantes mus-culares, aumentando así el riesgo de depresión respiratoria. Se ha descrito casos de potenciación del efecto de los anticoagulantes cumarínicos. depresión respiratoria, estupor, coma, flacidez muscular, hipotensión, bradicardia, colapso circulatorio, paro cardio-respiratorio y muerte. Como la formulación oral es de liberación prolongada, puede esperarse que la absorción de morfina continúe por varias horas. El antídoto indicado es la naloxona, asociada a medidas de resucitación respiratoria con ventilación asistida. Dado que la duración del efecto del opiáceo excede al de la naloxona, el paciente debe mantenerse bajo observación continua y repetirse la dosificación de naloxona, si fuese necesario, para mantener una respiración adecuada. Cuando está indicado, puede inducirse el vómito y usar carbón activado. Debe usarse medidas de soporte. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento o a otros opiáceos, depresión central severa, íleo paralítico, insuficiencia cardiaca congestiva e insuficiencia respiratoria. Usar con precaución en pacientes con INFORMACIÓN AL PACIENTE alteración de la función hepática y/o No aplicable a las formas de renal, hipertensión, hipotiroidismo, administración parenteral por ser de hipertrofia prostática, constricción ure- uso hospitalario. Con formas de tral, enfermedad pulmonar obstructiva dosificación oral de liberación procrónica, arritmias cardíacas, en an- longada, se debe advertir al paciente cianos y durante el embarazo. La que no debe partirla, fraccionarla o morfina oral debe ser usada con masticarla. No consumir alcohol. No extrema precaución en pacientes con alterar la dosificación, la frecuencia o aumento de la presión intracraneana o la duración del tratamiento, ni suspencon lesiones de la cabeza. No debe ser der el mismo sin el conocimiento del usada inmediatamente antes o médico. Informar al paciente que el durante el parto. No se recomienda el producto puede alterar la capacidad uso en pacientes pediátricos. Durante para realizar actividades que el tratamiento se debe suspender la requieren concentración, alerta y lactancia. El uso prolongado puede habilidad motora. Notificar al médico 402 Formulario Terapéutico Nacional * 2ed. 2004 OXICODONA liberación controlada, 10 mg cada 12 horas. FARMACOLOGÍA Ver: MORFINA. FARMACOCINÉTICA Administrada por vía oral, se absorbe en un 60-87%. Los alimentos con elevado contenido de grasa mejoran su absorción. Luego de la administración oral de una forma de dosificación de liberación convencional de oxicodona, el efecto analgésico se inicia en 10-15 minutos, se hace máximo a los 30-60 minutos y se mantiene por 3-6 horas. Cuando se usan formas de liberación controlada la analgesia comienza a los 60 minutos y se alcanza el estado estable, con la administración continua, a las 24-36 horas. Se une en un 45% a las proteínas plasmáticas. Se metaboliza en el hígado transformándose en productos con baja actividad farmacológica, noroxicodona y oximorfona (ésta mediante la CYP2D6) y sus glucurónidos, que se excretan, junto con un 19% del fármaco intacto, por la orina. La vida media de eliminación con formas de liberación convencional es de 3,2 horas y de 4,5 horas con formas de liberación controlada. Estos valores se incrementan en pacientes con insuficiencia hepática y renal. INDICACIONES Manejo del dolor agudo o crónico de REACCIONES ADVERSAS Ver: MORFINA. INTERACCIONES Ver: MORFINA. La oxicodona puede aumentar los efectos de los anticoagulantes. Debe controlarse el tiempo de protrombina. CONTRAINDICACIONES/ PRECAUCIONES Ver: MORFINA. SOBREDOSIS Ver: MORFINA. INFORMACIÓN AL PACIENTE Cuando se trata de formas de dosificación oral de liberación controlada, se debe advertir al paciente que no debe partirlas, fraccionarlas o masticarlas. No debe consumir alcohol durante el trataN miento. No debe alterar la dosificación, la frecuencia, la duración del tra-tamiento ni suspender el mismo sin el conocimiento del médico. Informar al paciente que el producto puede alterar la capacidad para realizar actividades que requieren PARACETAMOL (ACETAMINOFEN) FARMACOLOGÍA Ejerce un efecto analgésico y anti- 2ed. 2004 * Formulario TerapÈutico Nacional 403 pirético a través de la inhibición de la síntesis de prostaglandinas en el encéfalo y de una acción sobre el centro termorregulador hipotalámico de manera similar a como lo hace el ácido acetilsalicílico (ver: ÁCIDO ACE-TILSALICÍLICO). Sin embargo, y a diferencia de éste, el paracetamol no posee una actividad antiinflamatoria significativa, ni inhibe la agregación plaquetaria. No inhibe la activación de los neutrófilos. Se ha demostrado la existencia de una isoenzima, la COX3, susceptible de inhibición por el acetaminofén. INDICACIONES Manejo sintomático del dolor de intensidad leve a moderada y de la fiebre. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral y rectal (en supositorios) cada 4-6 horas, según sea necesario. Las dosis según la edad son las siguientes: Adultos y niños mayores de 12 años: 325-600 mg (en algunos casos puede ser necesario hasta 1000 mg). - Niños: 11 años: 480 mg; 9-10 años: 400 mg; 6-8 años: 320 mg; 4-5 años: 240 mg; 2-3 años: 160 mg; 12 años: 120 mg; 4-11 meses: 80 mg; niños menores de 3 meses: 40 mg. La dosis máxima diaria para adultos: 4 g y para niños: 2 g. FARMACOCINÉTICA Se absorbe rápida y casi completamente en el tracto gastrointestinal alcanzando concentraciones plasmáti-cas pico a los 30-60 minutos. Se une a las proteínas plasmáticas en un 25% y se distribuye rápida y REACCIONES ADVERSAS Hematológicas: Anemia hemolítica, uniformemente a los tejidos neutropenia, leucopenia, pancitopecorporales. Se metaboliza casi nia, trombocitopenia (rara). Hepáticas: totalmente en el hígado, dando lugar a Daño hepático severo (con dosis conjugados glucurónicos (60%), tóxicas), ictericia. Reacciones de sulfatos (35%) y mercaptatos (3%) que se excretan, junto con un 2% del hipersensibilidad: Erupción, urticaria. fármaco intacto, en la orina. Los niños Otras: Hipoglicemia. muestran menor capacidad de INTERACCIONES glucuronidación, pero producen una En pacientes sometidos a terapia con mayor proporción de sulfatos. El uso fenotiazinas se puede presentar de dosis elevadas y/o por períodos hipotermia. El consumo excesivo de prolongados puede saturar las rutas alcohol aumenta el riesgo de metabólicas de conjugación con ácido hepatotoxicidad, al igual que la glucurónico y ácido sulfúrico y 404 Formulario Terapéutico Nacional * 2ed. 2004 isoniazida. El uso concomitante con zidovudina puede aumentar la incidencia de mielosupresión por alteración del metabolismo de la zidovudina. El berro (Nasturtium officinale) puede inhibir el metabolismo oxidativo del paracetamol. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con insuficiencia hepática, cirrosis o alcoholismo. No debe usarse por más de 10 días continuos en adultos, ni por más de 5 días en niños. No usar por más de 3 días seguidos en caso de fiebre superior a 39,5º C. El uso crónico puede conducir a la acumulación de metabolitos hepato-tóxicos por agotamiento del sistema metabólico que interviene en la depuración de los mismos. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. La toxicidad hepática se presenta con dosis superiores a 7,5 g en 24 horas. Los signos más factibles incluyen: Náusea, vómito y dolor abdominal (usualmente a las 2-3 horas) y metahemoglobinemia. El antídoto es la N-acetilcisteína y debe ser administrado tan pronto como sea posible, preferiblemente dentro de las 16 horas caso de adultos y 2 g en el caso de los niños. No usar por más de 10 días continuos en adultos, ni por más de 5 días en niños. Informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Si la condición tratada con paracetamol persiste por más de 48 horas a pesar de su uso, o PARACETAMOL / CODEÍNA (ACETAMINOFEN / CODEÕNA) FARMACOLOGÍA Combina el efecto de un analgésico de acción central, la codeína, con el de otro de acción periférica, el paracetamol, para producir una analgesia superior en intensidad y calidad que la que se obtiene con ambos compuestos por separado. La codeína es un opiáceo con actividad similar a la descrita para la morfina (ver: MORFINA) aunque de menor potencia y el paracetamol actúa, en parte, por inhibición de la síntesis de N prostaglan-dinas (ver: PARACETAMOL). FARMACOCINÉTICA El comportamiento cinético de la combinación es el que cabe esperar de cada uno de los constituyentes por separado (ver monografía de CODEÍNA en la sección SISTEMA RESPIRATORIO y monografía de PARACETAMOL). 2ed. 2004 * Formulario TerapÈutico Nacional 405 RÉGIMEN DE DOSIFICACIÓN 1 tableta (500 mg de paracetamol + 25 mg de codeína) cada 4-6 horas, según sea necesario. REACCIONES ADVERSAS Ver la monografía de CODEÍNA en la sección SISTEMA RESPIRATORIO y la monografía de PARACETAMOL en esta misma sección. INTERACCIONES Ver la monografía de CODEÍNA en la sección SISTEMA RESPIRATORIO y la monografía de PARACETAMOL en esta misma sección. CONTRAINDICACIONES/ PRECAUCIONES Ver la monografía de CODEÍNA en la sección SISTEMA RESPIRATORIO y la monografía de PARACETAMOL en esta misma sección. SOBREDOSIS Ver la monografía de CODEÍNA en la sección SISTEMA RESPIRATORIO y la monografía de PARACETAMOL en esta misma sección. PETIDINA (MEPERIDINA) FARMACOLOGÍA Analgésico narcótico sintético que interactúa con los receptores de opiáceos y posee actividad farmacológica similar a la descrita para la 406 FARMACOCINÉTICA Después de la administración SC ó IM produce un efecto analgésico inicial en aproximadamente 10-15 minutos que se hace máximo a los 30-60 minutos y persiste por 2-4 horas. Se une a las proteínas plasmáticas en un 60-80%. Se metaboliza en el hígado por N-desmetilación dando lugar a la normeperidina, metabolito con un 50% de la actividad farmacológica de la petidina, y que es posteriormente hidrolizado y conjugado con ácido glucurónico y finalmente excretado, junto con un 5% del fármaco intacto, en la orina. La vida media de eliminación de la petidina es de 3-5 horas y la del metabolito activo 8-21 horas, incrementándose ambas en pacientes con insuficiencia hepática y/o renal. INDICACIONES Alivio del dolor moderado a severo y en la medicación preoperatoria. RÉGIMEN DE DOSIFICACIÓN Se administra por vía SC, IM e IV. Las dosis deben individualizarse en función de las necesidades particulares de cada paciente y de acuerdo a la respuesta clínica. - Dolor moderado a severo: Adultos: 50-150 mg por vía SC ó IM cada 3-4 horas, según sea necesario, o 15-35 mg/hora en infusión IV lenta; niños: 1,1-1,8 mg/kg por vía SC ó IM cada 3-4 horas, según Formulario Terapéutico Nacional * 2ed. 2004 REACCIONES ADVERSAS Ver: MORFINA. INTERACCIONES Ver: MORFINA. CONTRAINDICACIONES/ PRECAUCIONES Ver: MORFINA. SOBREDOSIS Ver: MORFINA. INFORMACIÓN AL PACIENTE TRAMADOL FARMACOLOGÍA Analgésico de acción central. Es un análogo sintético de la codeína. El tramadol y su metabolito M-1 se unen a los receptores mu [ ] de los opioides, y posee una leve actividad inhibidora de la recaptación de la serotonina y la noradrenalina. FARMACOCINÉTICA El tramadol racémico se absorbe en un 75% por vía oral produciendo un efecto analgésico máximo a los 30-60 minutos, el cual persiste por 4-6 horas. Cuando se administra por vía IM, la biodisponibilidad es de 100%. La afinidad por los receptores mu de opioides es 1/6000 la de la morfina. Se une a las proteínas plasmáticas en un 20% y se distribuye ampliamente en el do del tramadol es 2-4 veces más potente que el compuesto padre y puede explicar parte del efecto analgésico. Los metabolitos se excretan, junto con un 30% del fármaco intacto, en la orina. La vida media de eliminación es de aproximadamente 6,3 y 7,4 horas, respectivamente, para el tramadol y su metabolito o-desmetilado. INDICACIONES Tratamiento del dolor de intensidad moderado a severo. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM e IV. La dosis oral usual en adultos es 50-100 mg cada 4-6 horas. La dosis IM usual es 50-100 mg cada 6-8 horas, sin exceder los 400 mg/día. La dosis IV es 50-100 mg en infusión o en bolo administrado en 2-3 minutos cada 4-6 horas. La dosificación máxima es 400 mg por día. En pacientes de más de N 75 años de edad, la dosis máxima es 300 mg/día en dosis divididas. La dosis debe ser ajustada en pacientes con insuficiencia hepática y/o renal. REACCIONES ADVERSAS Cardiovasculares: Vasodilatación. Gastrointestinales: Náuseas, constipación, vómitos, dispepsia, boca seca, dolor abdominal, anorexia, flatulencia. Genitourinarias: Retención urinaria, frecuencia urina-ria, 2ed. 2004 * Formulario TerapÈutico Nacional 407 Respiratorias: Depresión respiratoria, disnea. Reacciones de hipersensibilidad: Prurito, diaforesis, erupción, síndrome de Stevens-Johnson, necrólisis epidérmica, anafilaxis. Otras: Malestar, hipertonía, trastornos visuales, diaforesis, pérdida de peso. INTERACCIONES Puede potenciar la acción de otros depresores del SNC como opiáceos, barbitúricos, fenotiazinas, anestésicos, benzodiazepinas y alcohol. La combinación con warfarina puede incrementar el tiempo de protrombina. El uso con inhibidores de la MAO o con inhibidores selectivos de la recaptación de serotonina incrementa el riesgo de reacciones adversas, síndrome serotoninérgico y convulsiones. Los neurolépticos pueden favorecer la ocurrencia de convulsiones. La car-bamazepina acelera el metabolismo hepático del tramadol, por inducción del citocromo P450. La quinidina, la cimetidina, el ritonavir y los inhibidores de la CYP2D6 (fluoxetina, paroxetina y amitriptilina) inhiben el metabolismo del tramadol. Puede incrementar la toxicidad de la digoxina (raro). CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otros opiáceos y en adictos o personas con 408 toria de convulsiones, traumatismo craneano, presión intracraneal elevada, enfermedad hepática y/o renal, mixedema, hipotiroidismo, hipoadrenalismo, ancianos, pacientes debilitados y durante el embarazo. No se ha establecido la seguridad de su uso en niños. Se recomienda suspender la lactancia durante la administración del medicamento. El uso crónico puede generar tolerancia y dependencia física. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Vigilancia constante de la función respiratoria y atención a la posibilidad de convulsiones. La naloxona no revierte en su totalidad las manifestaciones tóxicas de la sobredosificación pero sí la depresión respiratoria y, además, incrementa el riesgo de convulsiones. INFORMACIÓN AL PACIENTE Se debe evitar el consumo de alcohol durante el tratamiento. El tramadol puede alterar la capacidad para realizar actividades que requieren concentración, alerta mental y destreza motora. No se debe modificar la dosis o la frecuencia de administración ni suspender el medicamento sin el conocimiento del médico. Debe usarse con precaución cuando Formulario Terapéutico Nacional * 2ed. 2004 dependencia física. ÁCIDO VALPRÓICO (VALPROATO SÓDICO) FARMACOLOGÕA Agente anticonvulsivante. Muestra efectos semejantes a los de otros anticonvulsivantes. Inhibe la activación repetitiva sostenida, inducida por la despolarización de las neuronas corticales o de la médula espinal. Este efecto parece estar mediado por la recuperación prolongada de los canales de sodio activados por voltaje. La actividad parece estar relacionada con un incremento en los niveles cerebrales del ácido gamma-aminobutírico (GABA), por activar la descarboxilasa del ácido glutámico. FARMACOCIN…TICA Después de la administración oral se absorbe rápida y completamente en el tracto gastrointestinal, alcanzando niveles plasmáticos pico en 1-4 horas. La concentración plasmática requerida para el efecto terapéutico es 50100 mcg/ml y la dosis para lograrla es 1,2-1,5 g/día. Para el inicio del efecto anticonvulsivante son necesarios entre 7 y 15 días de terapia continua. Se distribuye rápidamente a los tejidos y se une a las proteínas plasmáticas en un 90%. Se metaboliza casi completamente (95%) en el hígado por oxidación y glucuronidación, y se excreta en la orina como productos media de eliminación es de 9-16 horas, pero se reduce en pacientes que toman otros medicamentos antiepilépticos. INDICACIONES Solo o en combinación con otros anticonvulsivantes para el manejo profiláctico de los ataques simples (pequeño mal) y complejos de ausencia. RÉGIMEN DE DOSIFICACIÓN Adultos y niños: Dosis inicial de 15 mg/kg/día e incrementos semanales de 5-10 mg/kg/día hasta el control de la condición, o hasta la aparición de efectos colaterales que limiten la posibilidad de incrementos adicionales. La dosis máxima diaria no debe exceder de 60 mg/kg. En caso de que la dosis diaria supere 250 mg, debe dividirse en 2 o más dosis iguales a objeto de evitar las molestias gastrointestinales. REACCIONES ADVERSAS Dado que el ácido valpróico, por lo general, es usado en combinación con otros anticonvulsivantes, es difícil discriminar si los eventos adversos que se reportan en tales terapias son exclusivamente atribuibles al ácido valpróico o a la combinación de fármacos. Dermatológicas: Alopecia, fotosensibilidad. Gastrointestinales: Náuseas, vómitos, cólicos abdo409 2ed. 2004 * Formulario TerapÈutico Nacional N hematomas, hemorragias, linfocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia y supresión de la médula ósea. Hepáticas: Elevación de las aminotransferasas, la bilirrubina y de otras pruebas de funcionamiento hepático, hepatitis tóxica. Neurológicas: Sedación, dolor de cabeza, temblor, ataxia, somnolencia (particularmente en ancianos), incoordinación, visión borrosa, diplopía, ambliopía, labilidad emocional, amnesia, ideas anormales. Psiquiátricas: Trastornos emocionales, psicosis, depresión, agresión, hiperactividad y alteraciones de conducta. Reacciones de hipersensibilidad: Erupción, eritema multiforme, síndrome de Stevens-Johnson y prurito generalizado. Otras: Disartria, debilidad, galactorrea, ginecomastia, hiperamonemia, hiperglicemia, aumento del apetito y edema de las extremidades. ción con la fenitoína puede ocasionar tanto disminución de los niveles plasmáticos de fenitoína como aumento de la frecuencia de ataques, asi como incrementos de la concentración de fenitoína libre e intoxicación por ésta. La rifampicina aumenta la depuración del valproato en un 40%. La combinación con agentes que afectan la coagulación puede aumentar el riesgo de sangramiento. Los salicilados inhiben el metabolismo del valproato y elevan los niveles séricos de ácido valproico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, con alteración de la función hepática y en el embarazo. Se ha reportado insuficiencia hepática resultante en muerte en pacientes que reciben ácido valpróico. Los niños menores de dos años están en riesgo considerable de desarrollar hepatotoxicidad fatal, particularmente aquellos que reciben varios anticonvulsivantes, sufren retardo mental, trastornos metabólicos congénitos o enfermedad cerebral orgánica. También se han descrito casos de pancreatitis con riesgo de la vida en adultos y niños. En caso de pancreatitis, el tratamiento debe ser suspendido de inmediato. Debido a la posibilidad de interacción con otros INTERACCIONES El valproato inhibe el metabolismo de los fármacos metabolizados por la CYP2C9, entre los cuales se encuentran la fenitoína y el fenobarbital, aumentando las concentraciones plasmáticas de estos medicamentos; igualmente, inhibe la UDPglucuronil-transferasa (UGT) y aumenta los niveles plasmáticos de lamotrigina y lorazepam. Potencia los efectos del alcohol, benzodiazepinas y de otros agentes depresores del SNC. 410 Formulario Terapéutico Nacional * 2ed. 2004 Durante la terapia se debe realizar evaluaciones frecuentes de la función hepática, hematológica y de la coagulación; descontinuar el tratamiento a la menor evidencia de alteración. Se debe suspender la lactancia durante el tratamiento. canales de sodio activados por voltaje. El metabolito 10,11-epoxicarbamazepina tiene un efecto similar. Adicionalmente, alivia el dolor de la neuralgia del trigémino por reducción de la transmisión sináptica entre los núcleos trigeminales. SOBREDOSIS Lavado gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. FARMACOCIN…TICA Luego de la administración oral se absorbe lentamente, en forma errática, alcanzando niveles séricos pico en 28 horas, aunque son necesarios hasta 4 días de terapia continua para lograr el estado estable. La concentración plas-mática terapéutica (como anticon-vulsivante y para el alivio de la neuralgia del trigémino) se ubica en el rango de 3-14 mcg/ml. Se distribuye ampliamente en todo el organismo. Se metaboliza en el hígado a compuestos con actividad farmacológica, princi-palmente el 10,11-epóxido, que se excretan, junto a un 1-3% del fármaco intacto, por vía N renal. La vida media de eliminación es de 8-72 horas. Dicha amplitud parece ser debida a que el fármaco induce su propio metabo-lismo; mientras su vida media al inicio de la terapia es de 2565 horas, en la medida en que ésta progresa se re-duce a 12-17 horas. Inhibe las enzimas CYP3A, CYP3C y la transferasa del UDP con los consiguientes efectos so-bre el metabolismo de otros fármacos. INFORMACI”N AL PACIENTE No modificar la dosificación ni suspender la terapia sin consultar al médico. No consumir alcohol u otros depresores del sistema nervioso sin conocimiento del médico. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, particularmente dolor abdominal, náusea, vómitos y anorexia, considerados síntomas de pancreatitis. Como el CARBAMAZEPINA FARMACOLOGÕA Agente anticonvulsivante. Limita la activación repetitiva de los potenciales de acción inducida por la despolarización sostenida de la médula espinal o de las neuronas corticales. Bloquea la potenciación postetánica. Aparentemente hay un descenso en la 2ed. 2004 * Formulario TerapÈutico Nacional 411 R…GIMEN DE DOSIFICACI”N tromboflebitis, edema, agravamiento La dosificación debe ser indivi- de enfermedad coronaria, arritmias y dualizada y ajustada lentamente a los bloqueo AV. Gastrointestinales: Náurequerimientos de cada paciente. Se seas, vómitos, malestar abdominal, debe iniciar con dosis bajas e ir diarrea, constipación, anorexia, boca incrementando gradualmente hasta seca, glositis y estomatitis. Genitoualcanzar el control, para luego dis- rinarias: Micción frecuente, retención minuir hasta la mínima dosis efectiva urinaria, oliguria con hipertensión, falla posible. renal, azotemia, impotencia, protei- Como anticonvulsivante en adultos nuria, glicosuria y aumento del BUN. y niños con más de 12 años: Iniciar Hematológicas: Anemia aplásica, con 200 mg/día, dividido en 4 leucopenia, agranulocitosis, eosinodosis, e incrementar 200 mg a filia, leucocitosis, trombocitopenia y intervalos semanales hasta lograr porfiria intermitente aguda. Hepáticas: la respuesta óptima, sin exceder la Alteración de las pruebas de función dosis máxima de 1,2 mg/día en hepática, ictericia colestásica y adultos y niños mayores de 15 hepatocelular, hepatitis fatal y necrosis años, o de 1 g en niños de 13-15 hepatocelular masiva fatal (con años. Una vez logrado el control, pérdida total del tejido hepático). ajustar la dosis a su mínimo nivel Metabólicas: Escalofríos, fiebre, sínefectivo, el cual se ubica en el rango drome de excreción inadecuada de de 800-1200 mg/día. hormona antidiurética, intoxicación - En niños de 6-12 años, iniciar el hídrica, hiponatremia e hipocalcemia. tratamiento con 100 mg 2 ve- Músculo-esqueléticas: Mialgia, arces/día, e incrementar semanal- tralgia, calambres de las piernas y mente 100 mg hasta el control osteomalacia. Neurológicas: Incooradecuado de la condición, sin dinación motora, mareos, confusión, exceder 1 g/día. Luego de logrado dolor de cabeza, visión borrosa, el control, ajustar al mínimo nivel t i n i t u s , f a t i g a , a l u c i n a c i o n e s , efectivo, que se ubica en el rango trastornos del habla, movimientos de 400-800 mg/día. involuntarios anormales, neuritis - Neuralgia del trigémino: Iniciar con periférica y pares-tesia, depresión 100 mg 2 veces al día e con agitación, cam-bios de conducta incrementar 200 mg a intervalos (niños), parálisis y síntomas de diarios hasta el control de la insuficiencia arterial cerebral. sintomatología, sin exceder 1,2 Oftalmológicas: Diplopía transitoria, g/día. Una vez logrado el control, 412 Formulario Terapéutico Nacional * 2ed. 2004 forme y eritema nodoso, necrólisis epidérmica tóxica y agravamiento del lupus eritematoso. Respiratorias: Hipersensibilidad pulmonar caracterizada por fiebre, disnea, pneumonitis o neumonía. Otras: Casos aislados de lupus eritematoso y aumento de los lípidos sanguíneos. INTERACCIONES La carbamazepina puede disminuir las concentraciones plasmáticas y/o la respuesta terapéutica de: fenitoína, fenobarbital, ácido valpróico, primidona, doxiciclina, haloperidol, warfarina y anticonceptivos orales. El verapamilo, la eritromicina y la fluoxetina pueden incrementar los niveles séricos de carbamazepina y aumentar con ello el riesgo de toxicidad. Con la teofilina puede ocurrir (por inducción metabólica recíproca) una disminución de las concentraciones sanguíneas de ambos medicamentos. Se han repor-tado alteraciones de la función tiroidea con los anticonvulsivantes. Se puede producir sangrado en pacientes que reciben anticonceptivos orales. los primeros 3 meses y luego mensual durante los 2-3 años siguientes, si fuera el caso). Suspender la terapia a la menor señal de mielosupresión: Eritrocitos <4 millones, hematocrito <32%, hemoglobina <11 g/100 ml, leucocitos <400/mm3, reticulocitos <0,3% (20.000/mm3) y hierro sérico >150 mcg/100 ml. Usar con precaución en pacientes con glaucoma. En ancianos, el medicamento puede activar una psicosis latente o producir cuadros de confusión o agitación. Durante el tratamiento se debe vigilar frecuentemente la función hepática, renal y oftalmológica. No se conoce la eficacia y seguridad del uso en niños menores de 6 años. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales para la madre superen al posible riesgo para el feto. Se debe suspender la lactancia durante la terapia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. El lavado gástrico puede realizarse aún después de transcuCONTRAINDICACIONES/ rridas 4 horas de la ingestión. Además, PRECAUCIONES se puede inducir el vómito, o tratar de Contraindicada en pacientes con reducir la absorción. Considerar la hipersensibilidad al medicamento, posibilidad de diálisis en caso de falla historia de depresión de la médula renal. Manejo sintomático y terapia ósea, discrasias sanguíneas, tratade soporte. Evaluar la función respiramiento con inhibidores de la monoatoria, el ECG, la presión arterial, la 413 2ed. 2004 * Formulario TerapÈutico Nacional N médico. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante la terapia, en especial: Sangramiento, petequias, dolor abdominal, coloración amarillenta de la piel o los ojos, fiebre, oscurecimiento de la orina, trastornos neurológicos, trastornos de la visión y úlceras en la boca. Como los mareos y la FENITOÍNA FARMACOLOGÍA Agente derivado de la hidantoína con propiedades anticonvulsivantes. El sitio primario de acción es la corteza motora. Se ha postulado que impide la propagación de la onda convulsiva por bloqueo de la potenciación posttetánica de la transmisión sináptica, aparentemente por reducción del influjo pasivo de iones sodio o por incremento en la eficiencia de la bomba de sodio evitando, en ambos casos, la acumulación intraneuronal excesiva de sodio durante la estimulación tetánica. FARMACOCINÉTICA Después de la administración oral, se absorbe lentamente a la circulación sistémica. La velocidad y magnitud de la absorción dependen de la formulación que se use. Las formas orales de liberación inmediata 414 el rango de 10-20 mcg/ml. Se une a las proteínas plasmáticas en un 95%. Se metaboliza en el hígado por oxidación dando lugar a productos inactivos. Dado que la ruta metabólica de la fenitoína es saturable, pequeños incrementos en la dosificación pueden producir aumentos considerables en la concentración plasmática conduciendo a toxicidad (un incremento de 10% en la dosis puede traducirse en una duplicación o triplicación del nivel sérico). La mayoría del fármaco se excreta por la bilis como metabolitos inactivos que son reabsorbidos en el tracto gastrointestinal y excretados en la orina por filtración glomerular y secreción tubular, y 1% como fármaco intacto. La vida media de eliminación es de aproximadamente 22 horas (rango de 7-42 horas). El estado estable se alcanza en 7 a 10 días. INDICACIONES Control y prevención de desórdenes convulsivos tipo gran mal y convulsiones psicomotoras. RÉGIMEN DE DOSIFICACIÓN La dosificación debe individualizarse y ajustarse en cada paciente en función del nivel plasmático (que en el estado estable debe ubicarse entre 10 y 20 mcg/ml) y la respuesta terapéutica. Se administra por vía oral e IV. - Por vía oral: En adultos, iniciar con 100 mg 3 veces diarias por 5-10 Formulario Terapéutico Nacional * 2ed. 2004 niños, iniciar con 5 mg/kg/día o 250 mg/m2 2-3 veces al día, sin exceder 300 mg/día, y ajustar posteriormente según la respuesta del paciente. Por lo general, la dosis de mantenimiento se ubica en el rango de 4-8 mg/kg. - Por vía IV (status epilepticus): En adultos, iniciar con 10-15 mg/kg lentamente, seguido por dosis de mantenimiento de 100 mg por vía oral o IV cada 6-8 horas. En niños: 15-20 mg/kg en dosis divididas a una tasa que no exceda 1-3 mg/kg/min. Las dosis de mantenimiento deben ser individualizadas. Los ancianos pueden requerir menores dosis. depresión, temblor y dolor de cabeza. Niveles séricos superiores al rango terapéutico pueden causar delirio, psicosis y encefalopatía. Reacciones de hipersensibilidad: Erupción morbiliforme o escarlatiforme; dermatitis bulosa, exfoliativa o purpúrica; necrólisis epidérmica tóxica, síndrome de Stevens-Johnson. Respiratorias: Neumonía, faringitis, disnea, asma, bronquitis, aumento de secreción bronquial, tos, epistaxis y fibrosis pul-monar. Otras: Osteomalacia, nistag-mo, diplopía, tinitus, sordera, trastornos oculares, fotofobia, disar-tria, hiperglicemia, ganancia de peso, porfiria y ginecomastia. REACCIONES ADVERSAS Cardiovasculares: Periarteritis nodosa, dolor en el pecho. Dermatológicas: Lupus eritematoso, hipertricosis, fotosensibilidad; dolor, fiebre e inflamación en el sitio de inyección, coloración de la piel. Gastrointestinales: Náuseas, vómitos, diarrea, constipación, dolor epigástrico, disfagia, pérdida del gusto, anorexia e hiperplasia gingival (especialmente en niños). Hematológicas: Trombocitopenia, agranulocitosis, leucopenia, pancitopenia con o sin supresión de la médula ósea, anemia megaloblástica, monocitosis, macrocitosis, anemia hemolítica, eosinofilia, leucocitosis, INTERACCIONES El efecto farmacológico de la fenitoína es incrementado por la administración concurrente de inhibidores del citocromo P 4 5 0 , alopurinol, cloran-fenicol, cimetidina, N diazepam, etanol (ingestión aguda), isoniazida, miconazol, fenilbutazona, ibuprofeno, sulfonamidas, trimetoprima, ácido valpróico, clorfeniramina e imipramina. El efecto es disminuido por inductores del citocromo P450 como los barbi-túricos, carbamazepina, etanol (ingestión crónica), diazóxido y por ácido fólico, teofilina, antiácidos, antineoplásicos, calcio, alimentación enteral, 2ed. 2004 * Formulario TerapÈutico Nacional 415 nutrición enteral. La administración enteral debe suspenderse dos horas antes o dos horas después de la administración de fenitoína. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, en el embarazo, bradicardia sinusal, bloqueo SA, bloqueo AV de segundo grado y tercer grado o síndrome de Stoke-Adams. Su uso por períodos prolongados ha sido asociado a degeneración cerebelosa. La suspensión repentina o abrupta de la fenitoína puede ocasionar convulsiones. Usar con precaución en pacientes con hipotensión, diabetes mellitus, insu-ficiencia cardiaca severa, porfiria, falla hepática, historia de discrasias sanguíneas y durante la lactancia. Si durante el tratamiento se desarrollan trastornos hepáticos, hematológicos, efectos dermatológicos o linfadeno-patía, suspender de inmediato la tera-pia. Debido a sus múltiples posi-bilidades de interacción se recomien-da consultar fuentes especializadas antes de asociar la fenitoína con cualquier otro medicamento. Se debe realizar, periódicamente, determinaciones del nivel plasmático de fenitoína. SOBREDOSIS 416 sión total en intoxicaciones severas en niños. INFORMACIÓN AL PACIENTE Evitar el consumo de alcohol. No alterar la dosificación ni suspender el tratamiento sin consultar al médico. No iniciar terapia con otros medicamentos sin el conocimiento y la aprobación del médico. Notificar de inmediato al médico si se presenta alguna reacción o síntoma inusual durante la terapia, en especial: Fiebre, FENOBARBITAL FARMACOLOGÍA Ejerce su acción anticonvulsivante disminuyendo la excitabilidad de la fibra neuronal como resultado de un efecto a nivel de la sinapsis, el cual se traduce en una reducción de la transmisión del impulso nervioso, presumiblemente debido a un incremento o a la mimetización de la acción inhibitoria del ácido gamma-aminobutírico (GABA), en virtud de que su sitio de unión se encuentra en el complejo del receptor GABAA. Adicionalmente, el fenobarbital parece elevar el umbral de la corteza motora a la estimulación eléctrica. FARMACOCINÉTICA Luego de su administración por vía oral se absorbe en un 70-90%, alcanzando concentraciones Formulario Terapéutico Nacional * 2ed. 2004 sivante es de 10-40 mcg/ml, que se alcanza por lo general después de 2-3 semanas de terapia continua con dosis de 100-200 mg/día. Cuando el fenobarbital se administra por vía IV el inicio del efecto ocurre en 5 minutos y la acción máxima en 30 minutos, persistiendo por 4-6 horas. Se une a las proteínas plasmáticas en un 20-45% y se metaboliza en el hígado a productos inactivos que se excretan, junto a un 25% del fármaco intacto, por la orina. La vida media de eliminación es de 2-6 días (promedio 96 horas). INDICACIONES Control y prevención de convulsiones tónico-clónicas características del gran mal y en convulsiones parciales. RÉGIMEN DE DOSIFICACIÓN La dosificación debe individualizarse y ajustarse en cada paciente en función del nivel plasmático (10-40 mcg/ml) y la respuesta terapéutica. Se administra por vía oral e IV. La dosis diaria por vía oral en adultos: 100-300 mg y en niños: 3-5 mg/kg. Debido a la prolongada vida media, no es necesario dividir la dosificación. La dosis IV en adultos: 200-600 mg y en niños: 100-400 mg. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, hipotensión, síncope. Gastrointestinales: niños. Reacciones de hipersensibilidad: Erupciones cutáneas y angioedema. Respiratorias: Cuando se administra por vía IV en forma rápida puede ocurrir depresión respiratoria severa. Apnea, hipoventilación. INTERACCIONES El fenobarbital reduce la eficacia terapéutica de los anticonceptivos orales, glucocorticoides, glicósidos cardioactivos, anticoagulantes orales, ciclosporina, dacarbazina, fenoprofeno, quinidina, levotiroxina, doxiciclina, griseofulvina, metronidazol, carbamazepina, hidantoínas, vitamina D y del ácido ascórbico. Potencia los efectos de otros depresores del SNC. Incrementa la toxicidad de la ciclofosfamida, gua-netidina, antipiréticos e inhibidores de la anhidrasa carbónica. El ácido valpróico, los inhibidores de la MAO, la fenitoína y el felbamato inhiben el N metabolismo hepático e incrementan las concentraciones plasmáticas del fenobarbital. Los antidepresivos tricíclicos antagonizan la acción anticonvulsivante. La furosemida aumenta los niveles séricos del fenobarbital; la piridoxina los reduce. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad a la droga o a otros 2ed. 2004 * Formulario TerapÈutico Nacional 417 nos y pacientes debilitados. Durante el tratamiento se debe practicar determinaciones periódicas de los niveles plasmáticos, cuando ello sea posible. La suspensión brusca del tratamiento puede desencadenar convulsiones. El uso crónico puede generar tolerancia y dependencia física. Se recomienda suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Las concentraciones tóxicas se ubican sobre los 50 mcg/ml y las letales alrededor de los 80 mcg/ml. Tratamiento de soporte con adecuado mantenimiento de la función respiratoria (administración de oxígeno en caso necesario). Si la función renal es normal, forzar diuresis. La alcalinización de la orina facilita la eliminación renal. INFORMACIÓN AL PACIENTE No alterar la dosis ni suspender el tratamiento sin el conocimiento del médico. Evitar el consumo de alcohol y de otros depresores del SNC durante la terapia. Informar de inmediato al médico si se presenta alguna GABAPENTINA FARMACOLOGÍA Anticonvulsivante relacionado estruc418 turalmente con el ácido gammaaminobutírico (GABA), pero no interactúa con sus receptores, no es convertido a GABA ni interfiere con procesos relacionados con el mismo. Puede favorecer la liberación no vesicular del GABA, por un mecanismo aún poco claro. Se ha postulado la existencia de sitios de unión para la gabapentina en la neocorteza y el hipocampo. En general, el mecanismo de acción no se conoce bien. FARMACOCINÉTICA La biodisponibilidad oral es de 60%. La absorción no es afectada por los alimentos. Se une en menos de un 3% a las proteínas plasmáticas. No sufre metabolismo apreciable ni induce la actividad de las enzimas hepáticas. Se excreta intacto por la orina. La vida media de eliminación es de 5-7 horas y ésta se incrementa en pacientes con insuficiencia renal. INDICACIONES En combinación con otros antiepilépticos para el tratamiento de ataques parciales, con y sin generalización. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos y niños mayores de 12 años: Iniciar con 300 mg/día e incrementar la dosis en forma gradual hasta alcanzar 900- Formulario Terapéutico Nacional * 2ed. 2004 (dividido en 3 dosis). REACCIONES ADVERSAS Cardiovasculares: Edema periférico, vasodilatación. Dermatológicas: Prurito, abrasión. Gastrointestinales: Náu-seas, vómitos, dispepsia, garganta seca, boca seca, constipación. Hema-tológicas: Leucopenia. Neurológicas: Fatiga, somnolencia, mareo, ataxia, nistagmo, temblor, nerviosismo, disar-tria, amnesia, depresión, ideas anormales, contracciones, incoordinación. Otros: Diplopía, impotencia, rinitis, faringitis, tos, ambliopía, anormalidades dentales, aumento del apetito, aumento de peso, dolor de espalda, mialgia, fracturas. INTERACCIONES Los antiácidos reducen la biodisponibilidad oral de la gabapentina. Deben administrarse separados por un intervalo de dos horas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Para la evaluación del régimen no se requiere la determinación de los niveles séricos. Dado que la suspensión brusca de la terapia puede desencadenar convulsiones, se recomienda hacerlo (en caso necesario) de manera gradual en un período no y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. El medicamento es removible por hemodiálisis. INFORMACIÓN AL PACIENTE No alterar la dosificación ni suspender de manera abrupta la terapia. Respetar los horarios de administración. El tiempo transcurrido entre dos dosis nunca debe superar las 12 horas. La terapia puede alterar la capacidad para realizar actividades que requieren concentración, alerta mental y coordinación motora. Notificar de SULFATO DE MAGNESIO (Anticonvulsivante en Toxemia) FARMACOLOGÍA Cuando se administra en dosis suficientes para producir hipermagnesemia (niveles séricos superiores a 2,5 mEq/L), el magnesio deprime el SNC y bloquea la transmisión neuromuscular periférica produciendo N un efecto anticonvulsivante. El mecanismo por el cual se produce el efecto depresor, se cree que es debido a una disminución en la cantidad de acetilcolina liberada por el impulso nervioso motor. A mayores concentraciones se produce depresión de los reflejos tendinosos profundos, desaparición de dichos reflejos y parálisis respiratoria. También puede producirse bloqueo cardiaco, apa- 2ed. 2004 * Formulario TerapÈutico Nacional 419 FARMACOCINÉTICA Luego de su administración IV, se une a las proteínas plasmáticas en 2535% y se distribuye ampliamente a todos los tejidos y fluidos corporales. Se excreta, en su mayoría, por filtración glomerular y, en escasa proporción, por las heces. INDICACIONES Prevención y control de convulsiones en la toxemia (preeclampsia y eclampsia) del embarazo. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IV y por vía IM. Se usa como sal heptahidratada y la dosis se expresa en términos de magnesio, teniendo en cuenta que 1 g de sulfato de magnesio heptahidratado contiene 8,1 mEq o 4,1 mmoles de magnesio. Iniciar con 4-5 g (32,4-40,5 mEq) diluidos en 250 ml de dextrosa al 5% o solución salina de NaCl al 0,9% por infusión IV en 30 minutos, simultáneamente con una inyección IM de 4-5 g (solución al 50%) en cada glúteo. Alternativamente, luego de la inyección IV inicial, se puede administrar 1-3 g/hora por infu-sión IV continua. Otro esquema propone una dosis inicial de 4 g (32,4 mEq) diluidos en una solución al 10-20% por infusión IV en 3-4 minutos, seguidos por inyección IM de 4-5 g (solución al 50%) en cada glúteo, cada 4 horas 420 total diaria no debe exceder 30-40 g. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, rubor, colapso circulatorio, depresión de la función cardiaca. Neurológicas: Somnolencia, hiporreflexia, parálisis flácida, hipotermia. Otras: Diaforesis, parálisis respiratoria, hipocalcemia; hipocalcemia con signos de tetania. INTERACCIONES Puede potenciar el efecto de los bloqueantes neuromusculares y la depresión central de los barbitúricos, opiáceos y otros depresores del sistema nervioso central. El uso concomitante con digitálicos puede exacerbar las arritmias y conducir a bloqueo cardíaco. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con bloqueo cardíaco, lesión miocárdica e insuficiencia renal severa. Usar con precaución en pacientes con falla renal leve a moderada y con miastenia gravis. Durante el tratamiento se debe vigilar las concentraciones séricas de magnesio a objeto de evitar la toxicidad (niveles superiores a 4 mEq/L). Debe usarse con extrema precaución en pacientes digitalizadas. Se debe evitar el uso del sulfato de magnesio antes del parto (al menos 24 horas) para evitar la hipermagnesemia Formulario Terapéutico Nacional * 2ed. 2004 10 mEq de gluconato de calcio por vía IV (solución al 10%) para revertir la depresión respiratoria y el bloqueo cardíaco y forzar diuresis con manitol. En casos graves practicar diálisis peritoneal o hemodiálisis. 33%. Después de la administración oral, alcanza concentraciones séricas pico en 1-1,5 horas. Se distribuye a todos los tejidos. Su metabolismo y excreción no se conocen bien, pero involucra hidroxilación. La vida media de eliminación es de 18-24 horas. INFORMACI”N AL PACIENTE BIPERIDENO (BIPERIDENO CLORHIDRATO Y BIPERIDENO LACTATO) FARMACOLOGÕA Amina terciaria con propiedades de antagonista de los receptores muscarínicos que tiene efectos en el sistema nervioso central y periférico. Ha sido usado en el parkinsonismo idiopático, arterioesclerótico y postencefalítico. Aunque su mecanismo como antiparkinsoniano no ha sido bien aclarado, se postula que el biperideno antagoniza la acción de la acetilcolina sobre los receptores colinérgicos en el cuerpo estriado, restituyendo así el balance entre la actividad del sistema excitatorio (colinérgico) y el inhibitorio (dopaminérgico) que, según se cree, está vinculado al origen de la enfermedad. Por vía parenteral resulta efectivo para el control de las reacciones extrapira-midales asociadas al uso terapéutico o a la sobredosificación por algunos medicamentos. Debido a sus efectos anticolinérgicos INDICACIONES Coadyuvante en el tratamiento de todas las formas de parkinsonismo. Manejo de las reacciones extrapiramidales inducidas por medicamentos. R…GIMEN DE DOSIFICACI”N La dosis debe ajustarse a los requerimientos individuales y tolerancia de cada paciente. Se emplea por vía oral como clorhidrato y por vía parenteral como lactato. - Tratamiento de la enfermedad de Parkinson: 2 mg por vía oral 3-4 veces al día. Si la respuesta no es adecuada, o si se desarrolla tolerancia durante la terapia N prolongada, puede incrementarse la dosis sin exceder 16 mg/día. - Tratamiento de las reacciones extrapiramidales inducidas por medicamentos: 2 mg por vía oral 1-3 veces por día dependiendo de la severidad. En casos severos (intoxicaciones) se usa 2 mg por vía IM cada 30 minutos, hasta un má-ximo de 8 mg en 24 horas. No se ha establecido la dosis para 2ed. 2004 * Formulario TerapÈutico Nacional 421 de otras pruebas de funcionamiento hepático, hepatitis tóxica. Neurológicas: Trastornos de coordinación (cuando se administra por vía parenteral). Renales: Retención urinaria y hematuria. Otras: Visión borrosa, midriasis. Estos efectos disminuyen con la administración lenta. Las reacciones adversas se relacionan con la dosis y se asemejan a las de la toxicidad de la atropina. INTERACCIONES Disminuye los niveles plasmáticos de la clorpromazina por incremento de su metabolismo. Aumenta la biodisponibilidad de los digitálicos. Puede potenciar los efectos del alcohol y de otros depresores del SNC. En combinación con quinidina, antidepresivos tricíclicos, agonistas opiáceos (como la meperidina), antihistamínicos, inhibidores de la MAO, fenotiazinas y otros agentes antipsicóticos puede potenciar los efectos adversos anticolinérgicos. Los antiácidos y los antidiarreicos disminuyen su absorción. enfermedad pulmonar crónica, trastornos psicóticos, insuficiencia renal y/o hepática, en niños, ancianos y en climas muy cálidos. El uso durante el embarazo debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales a la madre superen al posible riesgo para el feto. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS La sobredosis con biperideno produce síntomas típicos de la intoxicación atropínica (síndrome anticolinérgico central). Vaciamiento gástrico, carbón activado y catártico salino, aún cuando hayan transcurrido más de 2 horas de la ingestión. Manejo sintomático y terapia de soporte. Si se observa excitación del sistema nervioso central, puede administrarse una pequeña dosis de diazepam o de un barbitúrico de acción corta. La hiperpirexia debe ser revertida, el volumen de fluido reemplazado y el balance ácido-básico mantenido. En casos de intoxicación severa administrar fisostigmina como antídoto, aunque el uso de rutina es controversial. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, glaucoma de ángulo cerrado, esteno- INFORMACI”N AL PACIENTE sis pilórica-duodenal, úlcera péptica, Administrar con las comidas para atonía intestinal, colitis ulcerativa o evitar las molestias gástricas. El megacolon tóxico, hipertrofia prostáti- medicamento puede alterar la ca, estenosis del cuello vesical, capacidad para realizar actividades 422 Formulario Terapéutico Nacional * 2ed. 2004 BROMOCRIPTINA FARMACOLOGÕA Es un derivado del ergot con potente actividad agonista de los receptores dopaminérgicos post-sinápticos D2. Ejerce un efecto terapéutico en la enfermedad de Parkinson mediante la estimulación directa de los receptores dopaminérgicos en el cuerpo estriado del SNC. FARMACOCIN…TICA Después de la administración oral, se absorbe solamente un 6% como fármaco intacto debido al efecto del metabolismo de primer pasaje. Produce una mejoría de los síntomas del síndrome parkinsoniano a los 3090 minutos de la administración, la cual se hace máxima a las 2 horas. Se une a las proteínas plasmáticas en un 90-96%. Se metaboliza completamente en el hígado transformándose en productos inactivos que se excretan principalmente por vía biliar, y por ende, en las heces, y sólo un 2-5% por la orina. La vida media es de 7 +- 5 horas y la vida media de eliminación en fase terminal es de 45-50 horas. INDICACIONES Manejo del síndrome parkinsoniano idiopático o post-encefálico. R…GIMEN DE DOSIFICACI”N Iniciar con 1,25 mg 2 veces al día con puesta óptima. No se ha demostrado la seguridad de la bromocriptina en dosis que exceden 100 mg/día. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, signos y síntomas de ergotismo, o exa-cerbación del síndrome de Raynaud, edema de los pies y de los tobillos. Dermatológicas: Erupciones, moteado de la piel. Gastrointestinales: Náu-seas, vómitos, cólicos abdominales, constipación, diarrea, anorexia, disfagia, dolor epigástrico, indigestión, constipación. Genitourinarias: Reten-ción urinaria, poliuria. Hepáticas: Aumento transitorio de las amino-transferasas y de la fosfatasa alcalina. Neurológicas: Movimientos involun-tarios anormales, alucinaciones, con-fusión, mareos, fatiga, somnolencia, síncope, insomnio, depresión, vértigo, frialdad y palidez de dedos, manos y pies, pesadillas. N Respiratorias: Con-gestión nasal, infiltrados pulmonares. Otras: Ardor de los ojos, trastornos visuales. INTERACCIONES En dosis elevadas puede disminuir la tolerancia al alcohol. Su uso en combinación con medicamentos antihipertensivos puede ocasionar un efecto hipotensor aditivo. El uso concomitante de alcaloides del ergot puede 2ed. 2004 * Formulario TerapÈutico Nacional 423 eritromicina aumenta los niveles de la bromocriptina y las reacciones adversas potenciales. Los anticonceptivos orales interfieren con los efectos de la bromocriptina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, en tratamiento con alcaloides del ergot, hipertensión no controlada, enfermedad vascular periférica, isquemia cardiaca y en la preeclampsia. Dado que el medicamento suprime la secreción láctea, se debe evitar su uso durante la lactancia. Se debe usar con precaución en pacientes con enfermedad pulmonar, infarto del miocardio, angina severa, enfermedad psiquiátrica e insuficiencia hepática. En terapias prolongadas, se debe evaluar periódicamente la función hepática, renal, pulmonar y cardiovascular. No se conoce la seguridad del uso en la terapia de la enfermedad de Parkinson por períodos mayores de 2 años. La bromocriptina, sola o en combinación con levodopa, ha ocasionado alucinaciones visuales y auditivas. No se recomienda su uso con otros alcaloides del ergot. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es 424 INFORMACIÓN AL PACIENTE Administrar con las comidas para minimizar las molestias gástricas. Durante el tratamiento se debe evitar el consumo de alcohol. El medicamento puede alterar la capacidad para realizar actividades que requieren alerta mental, concentración y destreza motriz. Informar de inmediato al médico si se presenta alguna LEVODOPA / CARBIDOPA FARMACOLOGÕA Agente antiparkinsoniano. El uso de la combinación permite aumentar los niveles de dopamina en el cuerpo estriado. La carbidopa inhibe a la enzima descarboxilasa de aminoácidos aromáticos que transforma la levodopa en dopamina en la periferia. La dopamina como tal, no atraviesa la barrera hematoencefálica, aunque sí lo hace la levodopa. Una vez en el cerebro, la levodopa es transformada en dopamina por la enzima cerebral, pudiendo la dopamina ocupar los receptores dopaminérgicos en el cuer-po estriado. De esta manera, el uso de la combinación permite reducir la dosificación de levodopa en un 75% y, con ello, la incidencia de efectos colaterales debidos a la levodopa. FARMACOCIN…TICA Después de la administración oral, la levodopa y la carbidopa se absorben Formulario Terapéutico Nacional * 2ed. 2004 proteico, afectan la absorción de la levodopa y reducen su biodisponibilidad. Luego de absorbida, en ausencia de la carbidopa, la levodopa es casi totalmente convertida en dopamina por acción de la enzima descarboxilasa de aminoácidos aromáticos y sólo una pequeña fracción del fármaco logra penetrar al SNC. La carbidopa, luego de absorbida, se distribuye también ampliamente pero no penetra la barrera hematoencefálica. En presencia de la carbidopa, una mayor cantidad de levodopa pasa al SNC. Una vez en el cerebro, la levodopa es transformada en dopamina y una pequeña cantidad metabolizada a norepinefrina, epinefrina, metoxitiramina y metildopa. La dopamina, por su parte, es degradada a productos sin actividad farmacológica que, junto con los anteriores, son excretados todos por la orina. Cerca de un 80-85% de la dosis de levodopa se excreta en 24 horas, 6% como fármaco intacto. La carbidopa no sufre grandes cambios y se excreta casi completamente en 24 horas por la orina. La vida media de eliminación de la levodopa es de aproximadamente 1 hora. INDICACIONES Tratamiento sintomático de la enfermedad de Parkinson. R…GIMEN DE DOSIFICACI”N - incrementar 1/2 tableta cada 1-2 días hasta lograr una respuesta óptima, sin exceder la cantidad de 8 tabletas por día. La mayoría de los pacientes exhibe una respuesta adecuada con una dosis de 3-6 tabletas por día. En pacientes que reciben terapia con levodopa sola, se debe descontinuar la medicación 12 horas antes de iniciar tratamiento con la combinación. Hecho esto, iniciar la terapia con una dosis equivalente al 20% de la usada en el tratamiento con levodopa sola. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, flebitis, irregularidades cardíacas. Gastrointestinales: Boca seca, sabor amargo, sialorrea, náuseas, vómitos, anorexia, pérdida de peso, constipación, flatulencia, diarrea, dolor abdominal. Genitourinarias: Frecuen-cia urinaria, N retención urinaria, in-continencia urinaria, orina oscura, priapismo. Hematológicas: Anemia hemolítica, trombocitopenia, leuco-penia, agranulocitosis. Hepáticas: Hepatotoxicidad. Neurológicas: Movimientos coreiformes, distónicos, discinéticos; muecas involuntarias, movimientos de la cabeza, movimientos mioclónicos, ataxia, temblor, contracciones musculares, episodios 2ed. 2004 * Formulario TerapÈutico Nacional 425 crisis oculogíricas. Otras: Sudoración oscura, hiperventilación, hipo, activación de melanoma. INTERACCIONES La administración conjunta con ciclopropano o con halotano provoca arritmias cardíacas. Los anticonvulsivantes, las fenotiazinas, clonidina, benzodiazepinas, droperidol, sales de hierro, butirofenonas, antidepresivos tricíclicos, isoniazida, clozapina, papaverina, risperidona, fenitoína y la piridoxina disminuyen la eficacia de la levodopa; la amantadina, el trihexifenidilo y la bromocriptina la aumentan. El uso de la levodopa en combinación con cisaprida, metildopa o con metoclopramida puede incrementar la incidencia de efectos adversos de la levodopa. Los inhibidores de la MAO aumentan los niveles de dopamina y norepinefrina y, en consecuencia, el riesgo de hipertensión. Efecto aditivo con los agentes antihipertensivos. Los alimentos ricos en proteínas disminuyen la absorción de la levodopa. antihipertensiva y en el embarazo. Durante la terapia se debe practicar evaluaciones periódicas de la función hepática, hematopoyética, renal y cardiovascular, así como de la respuesta del paciente al tratamiento. Cuando se interrumpe repentinamente la medicación se debe considerar la posibilidad de aparición de un cuadro parecido al síndrome neuroléptico maligno. La seguridad del uso en menores de 18 años no ha sido establecida. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Control electrocardiográfico y observación cuidadosa del paciente por arritmias. INFORMACIÓN AL PACIENTE Administrar una hora antes o dos horas después de las comidas. No alterar la dosificación ni suspender el tratamiento, sin consultar al médico. Durante el tratamiento evitar el consumo de vitaminas del complejo B. El medicamento puede alterar la capacidad para realizar tareas que PRECAUCIONES/ CONTRAINDICACIONES Contraindicada en individuos con hipersensibilidad al medicamento, en pacientes sometidos a terapia con BROMAZEPAM inhibidores de la MAO, glaucoma de ángulo cerrado, lesiones cutáneas no FARMACOLOGÍA diagnosticadas y en personas con Ver: DIAZEPAM. 426 Formulario Terapéutico Nacional * 2ed. 2004 FARMACOLOGÍA Ver: DIAZEPAM. CONTRAINDICACIONES/ PRECAUCIONES Ver: DIAZEPAM. FARMACOCINÉTICA Después de la administración oral se absorbe bien y rápidamente en el tracto gastrointestinal (biodisponibilidad 84%). Alcanza la concentración plasmática pico en 1-2 horas. Se une a las proteínas plasmáticas en un 70% y se distribuye ampliamente en el organismo. Se metaboliza en el hígado por hidroxilación y conjugación con ácido glucurónico originando meta-bolitos inactivos que se excretan, junto con un 2-3% del fármaco intacto, en un 70% por orina y en un 2-6% por las heces. INDICACIONES Tratamiento de corta y mediana duración de los trastornos de ansiedad. RÉGIMEN DE DOSIFICACIÓN La dosis usual por vía oral en adultos es de 6-30 mg/día, como dosis única o en dosis divididas. REACCIONES ADVERSAS Ver: DIAZEPAM. INTERACCIONES Las mismas del diazepam (ver: DIAZEPAM), excepto lo relacionado con los antiácidos, ya que parece que éstos no afectan la absorción del SOBREDOSIS Ver: DIAZEPAM. INFORMACIÓN AL PACIENTE BUSPIRONA FARMACOLOGÍA Agente con propiedades ansiolíticas no relacionado con las benzodiazepinas. Se ha sugerido que el mecanismo de acción se debe a la interacción con los receptores 5-HT1A de la serotonina, actuando como agonista parcial. El efecto ansiolítico no es inmediato, sino que tarda varios días en aparecer. No tiene efectos anticon-vulsivantes ni relajantes musculares. No tiene el marcado efecto sedante de los otros ansiolíticos. Su efecto no es antagonizado por el flumazenil. TamN bién se postula que la buspirona interactúa con los receptores dopaminérgicos, con los correspondientes efectos asociados a ello. FARMACOCINÉTICA Aunque se absorbe rápida y casi completamente por vía oral, sólo un 4% de la dosis alcanza la circulación sistémica como fármaco intacto debido al efecto del metabolismo de primer pasaje. La administración con 2ed. 2004 * Formulario TerapÈutico Nacional 427 conoce bien su distribución. Se metaboliza en el hígado por oxidación produciendo metabolitos que se eliminan en un 29-63% por la orina y 18-36% por las heces en 24 horas. Se excreta en un porcentaje inferior al 1% como fármaco intacto. La vida media de eliminación es de 2-3 horas y se prolonga en pacientes con insuficiencia renal. INDICACIONES Tratamiento de la ansiedad. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos iniciar con 15 mg/día, divididos en 2-3 dosis, e incrementar 5 mg cada 2-4 días, según la respuesta, hasta ubicar la dosis en el rango de 20-30 mg/día. REACCIONES ADVERSAS Gastrointestinales: Sequedad de la boca, náuseas, diarrea, trastornos abdominales, constipación. Genitourinarias: Aumento de la frecuencia urinaria. Neurológicas: Mareos, somnolencia, nerviosismo, cefalea, fatiga, adormecimiento, trastornos del sueño, tinitus. Otras: Visión borrosa, dolor de garganta, congestión nasal, hiper-prolactinemia y alteraciones tiroideas (raras veces). INTERACCIONES La combinación con inhibidores de la MAO puede producir aumentos de la 428 ciones adversas. La buspirona puede aumentar los niveles séricos de haloperidol. Los depresores del sistema nervioso central y el alcohol aumentan la depresión central. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con trastornos renales y/o hepáticos, durante el embarazo y la lactancia. El medicamento no debe ser usado dentro de los 14 días siguientes de una terapia con un inhibidor de la MAO. Aunque la buspirona no tiene efectos sedantes, se debe alertar a los pacientes acerca de manejar automóviles o maquinaria peligrosa. Debido a que la buspirona no presenta tolerancia cruzada con las benzodiazepinas, la buspirona no es un sustituto cuando se suspende una benzodiazepina u otros hipnóticos y sedantes ya que el medicamento no protege contra el síndrome de abstinencia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Se debe evitar el consumo de alcohol Formulario Terapéutico Nacional * 2ed. 2004 diato si se presenta alguna reacción o sintomatología inusual durante el tratamiento, o si está embarazada o CLORPROMAZINA FARMACOLOGÍA Antipsicótico derivado de la fenotiazina. Aunque no se conoce con precisión el mecanismo de acción, su actividad parece vinculada al bloqueo post-sináptico de los receptores D2 de dopamina en el sistema nervioso central. La evidencia disponible indica que las fenotiazinas antagonizan la neurotransmisión sináptica mediada por la dopamina y a la vez se comportan como antagonistas de los receptores alfa-adrenérgicos, serotoninérgicos, histamínicos (H 1 ) y musca-rínicos centrales y periféricos, lo cual podría explicar algunos de los efectos adversos como la hipotensión. Ejerce efectos antieméticos, sedantes y ansiolíticos. La clorpromazina, en par-ticular, posee una marcada acción sedante, antiemética, anticolinérgica y bloqueante de los receptores alfaadrenérgicos. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal y desde los sitios de administración parenteral; sin embargo, tras la administración por vía oral, sólo un 32% de la dosis alcanza la circulación sistémica como fármaco sistentemente. Con la administración IM se logran mayores concentraciones plasmáticas (4-10 veces mayores). El inicio de la actividad después de la administración IM es de 15-30 minutos. Se une a las proteínas plasmáticas en un 92-97% y se distribuye amplia-mente a los tejidos corporales. Atra-viesa la barrera hematoencefálica, la placenta y se distribuye a la leche materna. Se metaboliza en el hígado y el riñón y se excreta casi totalmente por la orina como metabolitos y menos del 1% como fármaco intacto. La vida media de eliminación es de 6 horas. INDICACIONES Tratamiento de las manifestaciones de los trastornos psicóticos. Control de náuseas y vómitos. Porfiria intermitente aguda. Coadyuvante en el tratamiento del tétanos. Hipo intratable. Alivio de la ansiedad previa a la cirugía. Control de las N manifestaciones maníacas de la enfermedad bipolar. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM e IV. - Para el manejo sintomático de los trastornos psicóticos leves, la dosis inicial en pacientes adultos ambulatorios por vía oral, es de 2575 mg diarios divididos en 2-4 dosis. Para el control de síntomas 2ed. 2004 * Formulario TerapÈutico Nacional 429 rápido de la sintomatología se veces diarias o de 25 mg IM 3-4 recomienda la administración IM de veces diarias. 25 mg y repetir, en caso necesario, - Hipo: Dosis oral en adultos: 25-50 1 hora después. Una vez logrado el mg 3-4 veces al día; y 25-50 mg IM, control, pasar a la vía oral con 25si los síntomas persisten. 50 mg 3 veces al día. - Tétanos: Dosis oral en adultos: 25- Para el manejo de síntomas 50 mg 3-4 veces al día, en severos en pacientes hospitalicombinación con barbitúricos. En zados, iniciar con 25 mg IM y, en niños de 6 meses o más: 0,55 caso necesario, administrar 25-50 mg/kg IM ó IV. La máxima dosis mg adicionales 1 hora después. parenteral para niños con peso Podría ser necesario incrementos menor de 22 kg es 40 mg diarios, y graduales en los días para aquellos con peso de 22-45 kg: subsiguientes (máximo 400 mg 75 mg excepto en los casos cada 4-6 horas) hasta el control severos. total de las manifestaciones. Tan - Alivio de la ansiedad preoperatoria pronto como ello se logre, cambiar a en adultos: 25-50 mg por vía oral 2la vía oral titulando la dosis. La 3 horas antes de la cirugía o 12,5dosis usual de mantenimiento en la 25 mg IM 2 horas antes de cirugía; mayoría de los pacientes es de 500 en niños: 0,55 mg/kg por vía oral 2mg. Para el control de los síntomas 3 horas antes de la cirugía o IM 1-2 en niños mayores de 6 meses, la horas antes de la cirugía. dosis oral usual es de 0,55 mg/kg cada 4-6 horas o 0,55 mg/kg IM REACCIONES ADVERSAS cada 6-8 horas. La dosis máxima IM Cardiovasculares: Hipotensión ortosen niños de menos de 5 años es de tática, taquicardia, arritmias y 40 mg/día y en niños de 5-12 alteraciones del electrocardiograma (ECG). Gastrointestinales: Anorexia, años, 75 mg/día. - Para la prevención y tratamiento de sequedad de la boca, náuseas, las náuseas y los vómitos en adul- constipación, íleo paralítico y tos, la dosis usual por vía oral es d i s p e p - s i a . H e m a t o l ó g i c a s : de 10-25 mg cada 4-6 horas, tantas A g r a n u l o c i t o s i s , l e u c o p e n i a , veces como sea necesario. La t r o m b o c i t o p e n i a , e o s i n o - f i l i a , dosis IM inicial es de 25 mg, con granulocitopenia, anemia hemo-lítica, dosis adicionales de 25-50 mg cada anemia aplásica y pancitopenia. 3-4 horas hasta el control del Neurológicas: Reacciones extrapiracuadro. En niños mayores de 6 midales (enfermedad de Parkinson, 430 Formulario Terapéutico Nacional * 2ed. 2004 CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, depresión de la médula ósea o historia de discrasias sanguíneas, depresión severa del SNC, coma, daño cerebral subcortical y durante el primer tri-mestre del embarazo. Usar con cautela en la insuficiencia hepática y/o renal, trastornos cardiovasculares, hipertensión o hipotensión severa, alcoholismo, hipertrofia prostática, glaucoma, hipocalcemia, enfisema, asma, INTERACCIONES Los antidiarreicos, los adsorbentes y epilepsia, infecciones agudas del los antiácidos disminuyen la absorción tracto respiratorio, en pacientes de la clorpromazina. El litio disminuye expuestos a altas o bajas temperalos efectos de la clorpromazina. La turas, en ancianos o en personas cafeína contrarresta el efecto anti- debilitadas y en niños. El medicapsicótico. La clorpromazina, al igual mento puede provocar fotosensibilizaque otras fenotiazinas, puede inhibir el ción. Se debe usar con precaución, y metabolismo hepático de los antide- en dosis reducidas, en pacientes que presivos tricíclicos y de la fenitoína, han presentado reacciones severas a disminuir la eficacia de la levodopa y la insulina o a la terapia electroconN de la guanetidina, potenciar los efectos vulsiva. En tratamientos prolongados hipotensores de los nitratos y la acción se debe practicar evaluaciones sedante de los barbitúricos, narcóti- periódicas de la función hematológica cos, anestésicos, alcohol y otros de- y oftálmica. El uso durante el empresores centrales. También, puede barazo (segundo y tercer trimestre) b l o q u e a r l o s e f e c t o s a l f a - debe limitarse a situaciones de adrenérgicos de la epinefrina verdadera necesidad en las que los favoreciendo el predo-minio de los potenciales beneficios para la madre efectos beta-adrenérgi-cos. La superen el posible riesgo para el feto. administración simultánea con Durante la terapia se debe suspender propranolol produce incremento en los la lactancia. Las ampollas de clorpromazina deben ser protegidas 431 2ed. 2004 * Formulario TerapÈutico Nacional Reacciones de hipersensibilidad: Erupción, urticaria, eritema, fotosensibilidad, angioedema y anafilaxis. Hepáticas: Ictericia colestásica, anormalidades en las pruebas de la función hepática. Otras: Mastalgia, hiperprolactinemia, ginecomastia, galactorrea, alteraciones de la glicemia, trastornos de la termorregulación, midriasis, congestión nasal, retención urinaria e impotencia, pigmentación de la piel y de la córnea, síndrome neuroléptico maligno. aéreas permeables. No se debe inducir la émesis. En caso de hipotensión, no se debe administrar epinefrina; manejar con norepinefrina o fenilefrina. Para el control de los trastornos extrapiramidales se puede administrar agentes antiparkinsonianos y difenhidramina. Controlar las convulsiones con diazepam, seguido de fenitoína. Vigilar la función cardiaca por lo menos durante 5 días. INFORMACIÓN AL PACIENTE El medicamento puede alterar la capacidad para la realización de actividades que requieran atención, concentración y destreza motriz. Durante el tratamiento se debe evitar el consumo de alcohol u otros depresores centrales y la exposición prolongada a la luz solar. Tener cuidado al efectuar cambios bruscos de posición o subir escaleras. Notificar de inmediato al médico si se presenta DIAZEPAM FARMACOLOGÍA Benzodiazepina. Las benzodiazepinas actúan a nivel del sistema límbico, talámico e hipotalámico del SNC produciendo efectos ansiolíticos, hipnóticos, sedantes, anticonvulsi-vantes y relajantes musculares. Los efectos son mediados por la inter-acción de las benzodiazepinas con los receptores (GABA), mediador inhibitorio del SNC. Dependiendo de la dosis pueden llegar a producir todos los niveles de depresión del SNC, desde una sedación leve hasta hipnosis y coma. FARMACOCINÉTICA Se absorbe bien y rápidamente por vía oral y en forma errática por vía IM. Alcanza niveles plasmáticos pico a las 0,5-2 horas de la administración oral, a los 60 minutos de la inyección IM y a los 8 minutos por vía IV. Con la administración oral produce un inicio del efecto sedante en aproximadamente 30 minutos. Se une a las proteínas plasmáticas en un 94-99% y se distribuye a todo el organismo, especialmente al hígado y al cerebro. Las concentraciones alcanzadas en el SNC son similares a las plasmáticas. Se metaboliza en el hígado por las enzimas CYP3A4 y CYP2C19, transformándose en productos activos e inactivos que se excretan, junto con un porcentaje inferior al 25% del fármaco intacto, por la orina. Los principales metabolitos activos son el nordiazepam y el oxazepam. El primero tiene una vida media mayor de 48 horas y el segundo de 6-24 horas. La vida media de eliminación es bifásica, con un tiempo de 7-10 horas en su fase inicial y de 20-80 horas en su fase terminal. INDICACIONES 432 Formulario Terapéutico Nacional * 2ed. 2004 RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral, IM e IV. - Tratamiento de la ansiedad leve en adultos: 2-10 mg por vía oral 3-4 veces al día. En la ansiedad moderada: 2-5 mg IV y en ansiedad severa: 5-10 mg IV con repeticiones, en ambos casos, cada 2-4 horas de ser necesario. En niños: 0,04-0,2 mg/kg IV, con repeticiones cada 3-4 horas en caso necesario, sin exceder 0,6 mg/kg en 8 horas. - Manejo de las convulsiones en adultos: 5-10 mg IV con repeticiones a intervalos de 10-15 minutos hasta el control del cuadro, sin exceder 30 mg. Luego de controladas las convulsiones, repetir la dosis cada 2-4 horas, en caso necesario. Niños de 1 mes a 5 años: 0,2-0,5 IV a intervalos de 2-5 minutos, sin exceder 5 mg. Niños de 5 años o mayores: 1 mg IV a intervalos de 2-5 minutos, sin exceder 10 mg. Luego de controladas las convulsiones, repetir la dosis cada 2-4 horas en caso necesario. - Sedación preoperatoria en adultos: 10 mg IM 1-2 horas antes de la cirugía. En niños mayores de 2 años: 0,4 mg/kg IM 1-2 horas antes de la cirugía. - Coadyuvante en el alivio de la espasticidad y condiciones músculo-esqueléticas dolorosas en adultos: 2-10 mg por vía oral 3-4 por vía oral 3-4 veces al día durante el primer día, seguido por 5 mg 3-4 veces al día, de acuerdo a las necesidades. En casos severos: 10 mg IV, seguido por 5-10 mg cada 3-4 horas, según sea necesario. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, taquicardia, colapso, hipotensión, hipertensión y edema. Gastrointestinales: Náuseas, vómito, boca seca, diarrea, constipación, sialorrea y gastritis. Hematológicas: Agranulocitosis, leucopenia, neutropenia, anemia hemolítica, anemia aplásica, eosinofilia y disminución del hematocrito. Hepáticas: Trastornos hepáticos, ictericia. Genitourinarias: Irregularidades menstruales, retención urinaria. Neurológicas: Sedación excesiva, depresión, letargia, apatía, fatiga, desmayos, alucinaciones, trastornos de la memoria, desorientación, amnesia, cansancio, dolor de cabeza, ataxia, N confusión, vértigo, dificultad para la concentración, temblor, llanto, pesadillas, psicosis e histeria. Reacciones de hipersensibilidad: Erupción, prurito, urticaria, dermatitis, alopecia, edema facial. Renales: Insuficiencia renal. Respiratorias: Depresión respiratoria. Otras: Trastornos visuales, fotosensibilidad, nistagmo, sudoración, hipo, disartria, alteraciones de la libido, hirsutismo, calambres, artralgia, reac- 2ed. 2004 * Formulario TerapÈutico Nacional 433 macrólidos, propofol, ritonavir, ácido valpróico, fluoxetina, digoxina, fluvoxamina, isoniazida, itraconazol, ketoconazol, omeprazol y propoxifeno inhiben el metabolismo del diazepam, incrementando el riesgo de toxicidad. El alcohol y demás depresores del SNC incrementan el efecto depresor. La rifampicina incrementa el metabolismo del diazepam disminuyendo su eficacia. El diazepam inhibe la depuración de la zidovudina incrementando el riesgo de toxicidad. El diazepam puede disminuir los efectos de la levodopa. La administración conjunta con fentanilo puede disminuir las concentraciones plasmáticas de las catecolaminas con una consecuente reducción de la presión arterial. La teofilina y la cafeína disminuyen el efecto ansiolítico y sedante del diazepam. Los agentes hipotensores pueden potenciar el efecto hipotensor del diazepam. Puede incrementar los niveles séricos de la digoxina y el riesgo de toxicidad. El cigarrillo puede disminuir la eficacia del diazepam. debilitados. En casos de hipoalbuminemia o desnutrición se debe disminuir la dosis. Durante la terapia se debe evitar el alcohol o la combinación con otros agentes depresores del SNC. El uso en el embarazo (segundo y tercer trimestre) debe limitarse a situaciones de verdadera necesidad en las que los beneficios potenciales para la madre superen el posible riesgo para el feto. Se debe suspender la lactancia durante la terapia. El uso prolongado puede generar dependencia física. En pacientes con dependencia física, la supresión del medicamento puede inducir un síndrome de abstinencia. La eficacia y la seguridad del diazepam en neonatos no se han establecido. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Cuadros de toxicidad moderada a severa se presentan con la ingestión de dosis superiores a los 1200 mg. Vigilar la función cardiaca y la respiratoria. Si hay hipotensión administrar fluidos IV; en casos severos dopamina o norepinefrina. Si hay depresión respiratoria grave, usar flumazenil como antídoto (ver: FLUMAZENIL en la sección VARIOS). CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes hipersensibles al medicamento o a otras benzodiazepinas, en el glaucoma de ángulo cerrado, hipotonía muscular, miastenia gravis, coma, enfermedad respiratoria severa, apnea del sueño y INFORMACIÓN AL PACIENTE 434 Formulario Terapéutico Nacional * 2ed. 2004 depresoras del SNC. El uso prolongado puede generar dependencia física. En estos casos, debe consultar al médico antes de suspender el HALOPERIDOL FARMACOLOGÍA Agente antipsicótico derivado de la butirofenona. Aunque el mecanismo de acción no está totalmente claro, su acción fundamental parece ser debida al bloqueo de los receptores D≤ de dopamina en el sistema nervioso central y, en menor grado, de los Dπ. FARMACOCINÉTICA Aunque se absorbe bien en el tracto gastrointestinal, sólo un 60% de la dosis oral alcanza la circulación sistémica como fármaco intacto debido al efecto de metabolismo de primer pasaje. Alcanza concentraciones séricas pico a las 2-6 horas de su administración oral. Por vía IM se absorbe un 75%, alcanzando niveles pico a los 10-20 minutos y efectos farmacológicos a los 30-45 minutos. Se une a las proteínas plasmáticas en un 92%. No se conoce bien su distribución. Se metaboliza en el hígado transformándose en productos débilmente activos e inactivos que se excretan lentamente, junto con el fármaco intacto, por la orina en un 40% y 15% en la bilis. La eliminación completa del organismo hiperactividad con actividad motora excesiva y acompañada de impulsividad, dificultad de concentración, agresividad, malhumor y poca tolerancia a la frustración) y tratamiento del síndrome de Tourette. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral e IM. - Tratamiento de trastornos psicóticos y síndrome de Tourette en adultos, ancianos y pacientes debilitados: Iniciar con una dosis oral de 0,5-2 mg 2-3 veces al día, con ajustes posteriores en función de la respuesta y tolerancia del paciente. En presencia de síntomas severos y/o crónicos: 3-5 mg 2-3 veces al día. Pacientes con sintomatología severa pueden requerir hasta 100 mg/día. Por vía IM, iniciar con 2-5 mg, con repeticiones cada hora o a intervalos de 4-6 horas, según la respuesta. Una vez controlado el cuadro se debe pasar a vía oral tan N pronto como sea posible. La primera dosis oral debe ser administrada a las 12-24 horas de la última dosis IM. La dosis máxima es 100 mg diarios por VO. - En niños de 3-12 años con trastornos psicóticos, trastornos de conducta no psicóticos o con síndrome de Tourette: Iniciar con 0,05-0,15 mg/kg/día divididos en 23 dosis, con incrementos de 0,5 2ed. 2004 * Formulario TerapÈutico Nacional 435 REACCIONES ADVERSAS Cardiovasculares: Taquicardia, hipotensión, hipertensión, cambios electrocardiográficos, incluyendo prolongación del intervalo QT. Gastrointestinales: Boca seca, anorexia, constipación, diarrea, náuseas, vómitos, dispepsia. Genitourinarias: Retención urinaria, irregularidades menstruales, ginecomastia, priapismo. Hematológicas: Leucopenia, leucocitosis. Hepáticas: Alteración de la función hepática, ictericia. Neurológicas: Reacciones extrapiramidales severas, discinesia tardía, sedación, somnolencia, letargia, cefalea, insomnio, confusión, vértigo, convulsiones, síndrome neuroléptico maligno. Reacciones de hipersensibilidad: Erupciones. Otras: Visión borrosa, diaforesis. INTERACCIONES Los agentes antimuscarínicos pueden intensificar los efectos anticolinérgicos del haloperidol. La administración conjunta de litio puede causar toxi-cidad neurológica (síndrome encefalo-pático). Potencia el efecto depresivo del alcohol y otros depresores del SNC y las reacciones extrapiramidales de las fenotiazinas. Antagoniza el efecto presor de la dopamina y la epinefrina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hi436 o respiratoria severa, glaucoma, en ancianos, en el embarazo y durante la lactancia. Extremar la precaución cuando se use en combinación con otros antipsicóticos. No se conoce la seguridad del uso por vía oral en menores de 3 años. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. Los efectos extrapiramidales se pueden controlar con medicamentos antiparkinsonianos. Si se presenta hipotensión severa, emplear fluidoterapia IV; no usar epinefrina. Vigilar los signos vitales y el ECG. INFORMACIÓN AL PACIENTE Tomar con alimentos o leche para contrarrestar la irritación gástrica; no tomar con té o café. Evitar el consumo de alcohol y otros depresores del SNC. Durante la terapia, evitar la exposición excesiva a la luz solar. El haloperidol puede alterar la capacidad para LEVOMEPROMAZINA FARMACOLOGÕA Medicamento derivado de la fenotiazina con potente acción depresora del SNC. Deprime áreas subcorticales del cerebro a nivel del tálamo, hipotálamo y los sistemas Formulario Terapéutico Nacional * 2ed. 2004 supresión de los impulsos sensoriales, reducción de la actividad motora, sedación, alteración de la regulación de la temperatura y un efecto tranquilizante. Aumenta el umbral del dolor, produce amnesia y ejerce un marcado efecto hipotensor. Tiene actividad antiemética, antihistamínica, antiprurítica, anestésica local y anticolinérgica. FARMACOCIN…TICA Después de la administración oral, se absorbe en un 50% en el tracto gastrointestinal y alcanza concentraciones séricas efectivas después de 1-4 horas. Tras la administración IM, genera un efecto analgésico máximo a los 20-40 minutos, el cual persiste por 4 horas. Se metaboliza en el hígado transformándose en productos inac-tivos que se excretan, junto con un 1% del fármaco intacto, por la orina y las heces. La vida media de eliminación es de 15-30 horas. INDICACIONES Manejo del dolor de moderado a severo. En la sedación y analgesia obstétrica cuando se requiere evitar la depresión respiratoria. Como medicación pre-anestésica para producir sedación, somnolencia y alivio de la aprehensión y ansiedad previas a la cirugía. utilizado dosis hasta de 100-200 mg/día. Por vía IM: - Analgesia: 10-20 mg cada 4-6 horas. En pacientes geriátricos: Iniciar con 5-10 mg, con incrementos graduales, en caso necesario. - Analgesia en trabajo de parto: 15-20 mg. - MedicaciÛn pre-anestésica: 10 mg 1-3 horas antes de la cirugía. - Analgesia post-operatoria: Se recomienda dosis bajas de 2,5-7,5 mg debido a que los efectos residuales de los anestésicos pueden resultar aditivos. Repetir cada 4-6 horas, según sea necesario. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, palpitaciones, taquicardia o bradicardia. Gastrointestinales: Boca seca, náuseas, vómito, dolor abdominal. Hematológicas: Agranulocitosis N (terapia prolongada). Hepáticas: Ictericia (terapia prolongada). Neurológicas: Debilidad, mareo, desorientación, somnolencia, amnesia, sedación excesiva, euforia, cefalea, reacciones extrapiramidales y convulsiones. Otras: Congestión nasal, dificultad para orinar, escalofríos. INTERACCIONES Los depresores del SNC potencian la acción depresora de la levomepro437 2ed. 2004 * Formulario TerapÈutico Nacional presivos tricíclicos. Administrado concomitantemente con amiodarona, disopiramida, procainamida, quinolonas y tioridazina, incrementa el riesgo de arritmias ventriculares. Disminuye la eficacia de los anticonvulsivantes. Incrementa el riesgo de toxicidad de la sibutramina sobre el SNC. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al medicamento, en asociación con agentes antihipertensivos o con inhibidores de la MAO, en pacientes con depresión central, coma, epilepsia, enfermedad hepática, cardiovascular o renal, hipotensión y en menores de 12 años de edad. No se debe usar por más de 30 días, excepto en pacientes terminales o cuando los analgésicos narcóticos están contraindicados. En caso de tratamiento prolongado, se debe practicar el control periódico de la función hepática y hematológica. Usar con precaución en pacientes geriátricos o debilitados, en el embarazo y durante la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es 438 LITIO (CARBONATO DE LITIO) FARMACOLOGÍA Agente con actividad antimaníaca cuyo mecanismo de acción no ha sido completamente aclarado. Se cree que compite en el SNC con otros cationes monovalentes y divalentes (sodio, calcio, potasio, magnesio) por los sitios de transporte transmembranal, alterando la respuesta neuronal a la influencia de algunos neurotransmisores. Tiene un pequeño gradiente de distribución a través de las membranas; no es capaz de mantener potenciales de membrana porque no es un buen sustrato para la bomba de Na + , tendiendo a permanecer intracelularmente. Existe evidencia que sugiere que el litio inhibe la liberación de la norepinefrina y la dopamina (neurotransmisores involucrados en la patogénesis de los trastornos afectivos). Incrementa la recaptación de los neurotransmisores en los terminales nerviosos. El litio, además, inhibe el sistema de transducción de señales mediado por el fosfatidil inositol al inhibir diferentes enzimas en el sistema. Aún no se ha establecido la relación entre este efecto y el efecto terapéutico. FARMACOCINÉTICA Se absorbe rápidamente en el tracto gastrointestinal alcanzando concentraciones plasmáticas máximas en Formulario Terapéutico Nacional * 2ed. 2004 Concentraciones de 1,5-2 mEq/L por lo general generan reacciones adversas; niveles superiores a 2 mEq/L resultan tóxicos. Se distribuye ampliamente en el organismo y alcanza concentra-ciones en el SNC equivalentes al 40-50% de la concentración plasmática. No se une a las proteínas plasmáticas. Se excreta en su totalidad como fármaco intacto por la orina. La vida media de eliminación es de 20-27 horas y se incrementa en pacientes con alteración de la función renal. mg (24-32 mEq) diarios divididos en 2-4 dosis, sin exceder 2,4 g/día (65 mEq). - En niños: Iniciar con 15-60 mg/kg (0,4-1,6 mEq/kg) o 0,5-1,5 g/m2/día (13,5-41,1 mEq/m2). La dosis usual de mantenimiento es 15-60 mg/kg/día (0,4-1,6 mEq/kg) o 0,5-1,5 g/m2 /día (13,5-41,1 mEq/m2) en dosis divididas. No exceder la dosis de adultos. REACCIONES ADVERSAS Están relacionadas con los niveles plasmáticos de litio. Reacciones de intensidad leve a moderada suelen INDICACIONES Tratamiento de las fases maníacas y ocurrir con niveles plasmáticos de 1,5depresivas del trastorno bipolar. 2,5 mEq/L y reacciones más severas se observan con niveles de 2,5-3,5 RÉGIMEN DE DOSIFICACIÓN mEq/L. Las reacciones más comunes Se administra solamente por vía oral. incluyen: Cardiovasculares: Arritmias, Las dosis se deben individualizar en cambios en el ECG, hipotensión y función de los niveles séricos y la trastornos de conducción, colapso respuesta del paciente. Durante los circulatorio periférico, bradicardia que episodios agudos se requieren puede ser severa y originar un N concentraciones séricas de 1-1,5 síncope. Dermatológicas: Prurito, mEq/L. Una vez controlada la con- erupciones acneiformes, foliculitis, dición, se recomienda disminuir la sequedad y adelgazamiento del dosis hasta lograr niveles ubicados en cabello, alopecia, dermatitis exfoliael rango de 0,6-1,2 mEq/L. No se debe tiva, exacerbación de la psoriasis, exceder 2 mEq/L durante la fase agu- úlceras cutáneas, foliculitis crónica y da del tratamiento; y si esto sucede se xerosis cutis. Endocrinas: Hipotiroidebe suspender la terapia por 24 dismo y bocio eutiroideo y/o hipotiroihoras. deo. Gastrointestinales: Náuseas, vó- En adultos: Iniciar con 1,8 g/día o mito, anorexia, diarrea, dolor abdo20-30 mg/kg/día, divididos en 2-3 minal, sed y boca seca, edema de las dosis. En pacientes geriátricos glándulas salivales, sialorrea, 439 2ed. 2004 * Formulario TerapÈutico Nacional convulsiones, retardo psicomotor, dificultad para hablar, incoordinación, alucinaciones, empeoramiento del síndrome orgánico cerebral. Renales: Diabetes insípida nefrogénica que se manifiesta con polidipsia y poliuria; glucosuria, proteinuria, disminución de la depuración de creatinina, oliguria. Otras: Disfunción eréctil, visión borrosa, pérdida o aumento de peso, hiperglicemia transitoria, hipercal-cemia, artralgia, malestar general, caries dental, hiperparatiroidismo, alteración del gusto, debilidad muscular, fasciculaciones, movimien-tos clónicos de los miembros. Se han reportado casos de pseudotumores cerebrales. discinesia tardía. Con los yoduros puede ocurrir un efecto aditivo sobre el hipotiroidismo. La sibutramina, administrada conjuntamente con litio, puede incrementar el riesgo de síndrome serotoninérgico. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con insuficiencia renal y/o cardiaca, deshidratación severa, depleción de sodio o en pacientes debilitados y en el embarazo. Usar con precaución en pacientes con enfermedad cardiovascular o tiroidea, enfermedad de Parkinson, epilepsia, deshidratación leve, hiponatremia, debilidad, infecciones severas, daño cerebral orgánico, ancianos y en asociación con diuréticos o AINEs. En infecciones con manifestaciones febriles se debe reducir o suspender la terapia. Debido a que el litio puede disminuir la reabsorción renal de sodio, se debe mantener una dieta que incluya sal y al menos 2,5-3 litros diarios de agua durante el período de estabilización del tratamiento. Se debe vigilar periódicamente la concentración sérica del litio y el estado clínico del paciente. Antes de iniciar el tratamiento se debe realizar una evaluación de la función renal y vigilar la creatinina sérica durante los primeros 6 meses de tratamiento. Realizar un uroanálisis cada 1-2 INTERACCIONES El ácido etacrínico, diuréticos tiazídicos, antiinflamatorios no esteroideos, metildopa, inhibidores de la enzima convertidora de angiotensina, losartán, espironolactona, meclofenamato, metronidazol, fluoxetina, probenecid y tetraciclinas aumentan los niveles séricos del litio y pueden conducir a toxicidad. La acetazolamida, diuréticos osmóticos, cloruro de sodio, teofilina, cafeína, verapamilo y el bicarbonato de sodio disminuyen los niveles de litio y, por ende, sus efectos farmacológicos. La fenitoína, carbamazepina, haloperidol y las fenotiazinas incrementan los efectos 440 Formulario Terapéutico Nacional * 2ed. 2004 SOBREDOSIS Los síntomas tempranos de la toxicidad del litio pueden ser tratados con la reducción o la suspensión de la dosis y reiniciar el tratamiento a dosis más bajas a las 24-48 horas. En casos de intoxicación severa, se debe proceder al lavado gástrico, corrección del desbalance hidro-electrolítico y regulación de la función renal. Manejo sintomático y terapia de soporte. La hemodiálisis por 8-12 horas es recomendable cuando hay niveles séricos de litio que exceden 3 mEq/L, o cuando los niveles están en 2-3 mEq/L y las condiciones del paciente están deterioradas, o cuando no se logra reducir la concentración sérica de litio al menos en un 20% en 6 horas. INFORMACIÓN AL PACIENTE Tomar el medicamento con las comidas. Mantener una ingesta adecuada de sal y líquidos (2,5-3 L/día) a objeto de evitar la deshidratación. Notificar al médico de inmediato si se presenta alguna reacción o sintomaMIDAZOLAM FARMACOLOGÍA Benzodiazepina de acción corta con mecanismo de acción similar al descrito para el diazepam (ver: DIAZEPAM). FARMACOCINÉTICA Se absorbe bien por vía oral y por vía IM. La respuesta (sedación preoperatoria) se inicia a los 10-20 minutos después de la administración oral. Después de la administración intramuscular en adultos, los efectos sedantes se inician a los 15 minutos, ocurriendo la sedación pico 30 a 60 minutos después de la inyección. Después de la administración intravenosa, en pacientes adultos y pediátricos, la sedación se alcanza a los de 3-5 minutos. Cuando se administra por vía IV para la inducción anestésica, la inducción ocurre en 2 a 2,5 minutos (cuando no se administra otra premedicación sedante). Cuando se administra por vía IM, la biodisponibilidad es de 90%. Se une en un 97% a las proteínas plasmáticas. Se metaboliza en el hígado (CYP3A4) a metabolitos hidroxilados que son conjugados y excretados en la orina. La vida media N de eliminación es de 2-2,5 horas y se prolonga en pacientes con insuficiencia renal, insuficiencia hepática, insuficiencia cardiaca congestiva, en ancianos y en obesos. INDICACIONES Sedación preoperatoria, sedación previa a procedimientos diagnósticos e inducción de la anestesia. RÉGIMEN DE DOSIFICACIÓN 2ed. 2004 * Formulario TerapÈutico Nacional 441 usual IM en adultos menores de 60 sedación preoperatoria. años es de 70-80 mcg/kg 30-60 - Inducción de la anestesia: En minutos antes de la cirugía. En adultos no premedicados, iniciar pacientes mayores de 60 años, o con dosis de 300-350 mcg/kg por debilitados, o con enfermedad infusión IV en 20-30 segundos; pulmonar obstructiva crónica se esperar 2 minutos para evaluar la debe reducir la dosis a 20-50 respuesta y completar la inducción, mcg/kg. En niños, la dosis IM en caso necesario, con dosis recomendada es de 100-150 equivalente al 25% de la dosis mcg/kg, sin exceder 10 mg totales. inicial. En adultos premedicados La dosis IV en niños depende de la (fundamentalmente con opiáceos) edad, debe administrarse como iniciar con 150-350 mcg/kg y infusión en 2-3 minutos y esperar proceder de manera similar. En un lapso similar para evaluar la niños la dosis se ubica entre 50 y respuesta antes de iniciar el 200 mcg/kg. procedimiento o repetir la dosis. En niños de 6 meses a 5 años: Iniciar REACCIONES ADVERSAS con 50-100 mcg/kg, sin exceder 6 Cardiovasculares: Variaciones en la mg totales. En niños de 6-12 años: presión arterial y en la frecuencia del Iniciar con 25-50 mcg/kg, sin pulso. Gastrointestinales: Náuseas, exceder 10 mg totales. En niños de vómitos. Neurológicas: Cefalea, so12-16 años, usar la dosis de bresedación, somnolencia, amnesia, movimientos involuntarios, nistagmo, adultos. - Sedación previa a procedimientos conducta paradójica o excitación. diagnósticos: En adultos menores Respiratorias: Disminución de la de 60 años iniciar con dosis de 1- frecuencia respiratoria, apnea. Otras: 2,5 mg por infusión IV durante 2 Dolor en el sitio de la inyección, hipo. minutos; esperar 2 minutos para INTERACCIONES evaluar la respuesta y, en caso Puede potenciar la acción de otros necesario, para el mantenimiento depresores del SNC como opiáceos, repetir con el equivalente al 25% de barbitúricos, anestésicos y alcohol. La la dosis inicial. Puede usarse una eritromicina, ketoconazol, fluconazol, dosis total de 5 mg. En pacientes itraconazol, verapamilo, diltiazem, mayores de 60 años, o debilitados, ritonavir y el nelfinavir pueden inhibir el o con enfermedad pulmonar metabolismo hepático del midazolam obstructiva crónica, iniciar con 1- incrementando las concentraciones 1,5 mg por infusión IV durante 2 442 Formulario Terapéutico Nacional * 2ed. 2004 de toronja aumentan significativamente la biodisponibilidad oral del midazolam. Los anticonceptivos orales prolongan la vida media y la teofilina puede antagonizar los efectos del midazolam. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento o a otras benzodiazepinas, glaucoma de ángulo cerrado y en el embarazo. Usar con precaución en pacientes con enfermedad pulmonar, disfunción renal y/o hepática, alteraciones hidro-electrolíticas, insuficiencia cardiaca congestiva, en ancianos y en niños. Se recomienda una reducción de la dosis en pacientes ancianos o debilitados. Se debe evitar la administración en bolo. SOBREDOSIS Manejo sintomático y tratamiento de soporte. En casos graves usar flumazenil para revertir la depresión inducida por la sobredosis. INFORMACIÓN AL PACIENTE Informar al médico si ha consumido alcohol y acerca de otros medicamentos que esté tomando, especialmente medicamentos para la presión arterial, antibióticos o medicamentos de uso sin prescripción. Además, se debe informar al médico si la paciente está embarazada o planea estarlo, o si OLANZAPINA FARMACOLOGÍA Agente antipsicótico que es un antagonista selectivo de los receptores monoaminérgicos. Tiene alta afinidad por los receptores de serotonina 5-HT2A/2C, dopamina DA1-4, histamina H1, muscarínicos M1-5 y 1adrenérgicos. Se une con baja afinidad a los receptores del GABA y a los -adrenérgicos. Sin embargo, el sugiere que ejerce su acciÛn a travÈs de los receptores dopaminÈrgicos y 5HT2. Su afinidad por los receptores muscarÌnicos M1-5, explica sus efectos anticolinÈrgicos y aquella por los receptores H1 explica la somnolencia y el bloqueo de los receptores adrenÈrgicos, explica la hipotensiÛn ortost·tica observada con este f·rmaco. FARMACOCINÉTICA Aunque se absorbe bien por vía oral, el fármaco sufre metabolismo de N primer pasaje en un 40%. Luego de iniciado el tratamiento produce una respuesta terapéutica, en la esquizofrenia, que se evidencia en aproximadamente 7 días. Se une a las proteínas plasmáticas en un 93% y se distribuye ampliamente en el organismo. Se metaboliza en el hígado transfor-mándose en productos inactivos que se excretan, junto con el fármaco intacto, en un 57% 2ed. 2004 * Formulario TerapÈutico Nacional 443 trastornos psicóticos. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En adultos se debe iniciar el tratamiento con 5-10 mg/día con ajustes, en caso necesario, de 5 mg/día a intervalos semanales hasta lograr la respuesta deseada. La mayoría de los pacientes responde a dosis de 10 mg/día. No es reco-mendable exceder la dosis de 20 mg/día. REACCIONES ADVERSAS Cardiovasculares: Hipotensión ortostática, taquicardia, dolor en el pecho, hipotensión, edema. Gastrointestinales: Constipación, boca seca, dolor abdominal, aumento del apetito, sialorrea, náuseas, vómitos, sed. Genitourinarias: Síndrome premenstrual, metrorragia, hematuria, incontinencia urinaria, infección del tracto urinario. Neurológicas: Somnolencia, agitación, insomnio, cefalea, nerviosismo, hostilidad, parkinsonismo, mareos, ansiedad, trastornos de personalidad, acatisia, hipertonía, temblor, amnesia, dificultad para articular palabras, euforia, discinesia tardía, síndrome neuroléptico maligno. Reacciones de hipersensibilidad: Erupción vesículobulosa. Respira-torias: Tos, rinitis, faringitis, disnea. Otras: Ganancia o pérdida de peso, fiebre, lesiones intencionales, am-bliopía, 444 efectos de la levodopa y de los agonistas dopaminÈrgicos. La combinaciÛn con agentes anticolinÈrgicos aumenta las reacciones adversas de Èstos. La rifampicina y la carbamazepina incrementan el metabolismo de la olanzapina por inducciÛn del citocromo P450 y disminuyen su eficacia terapÈutica. El omeprazol, la fluvoxamina, la fluoxetina y la ciprofloxacina, por el contrario, pueden inhibir el metabolismo de la olanzapina e incrementar asÌ el riesgo de toxicidad. La administraciÛn con litio puede generar trastornos encefalop·ticos. Los agentes antihipertensivos pueden aumentar los efectos hipotensores de la olanzapina. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Usar con precaución en pacientes con enfermedad hepática, trastornos convulsivos, hipertrofia prostática, glaucoma de ángulo cerrado, íleo paralítico, tendencia a la hipotensión, diabetes, discinesia tardía, historia de síndrome neuroléptico maligno, en el embarazo y durante la lactancia; también en pacientes con riesgo de neumonía por broncoaspiración. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es Formulario Terapéutico Nacional * 2ed. 2004 hipotensión ortostática, especialmente durante el período inicial y cuando se administran medicamentos que pueden potenciar el efecto hipotensor. Deben evitar el exceso de calor y la deshidratación. Notificar de inmediato al médico si se RISPERIDONA FARMACOLOGÍA Agente antipsicótico cuyo mecanismo de acción, aunque no ha sido completamente aclarado, se cree que involucra el antagonismo, a nivel central, de receptores dopaminérgicos D2 y serotoninérgicos 5-HT2. Adicionalmente, existe evidencia de su afinidad por los receptores alfa1- y alfa2-adrenérgicos y H1 de histamina. FARMACOCINÉTICA Se absorbe bien en el tracto gastrointestinal (85%). Se une a las proteínas plasmáticas en un 90%. Se metaboliza en el hígado a través del sistema microsomal del citocromo P450, transformándose en 9-hidroxirisperidona, producto con actividad farmacológica similar a la de la risperidona. En consecuencia, el efecto clínico del fármaco resulta de la combinación de las concentraciones del fármaco y su metabolito. Se excreta en un 70% en la orina y 15% en las heces. La vida media es de 20 RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos es de 1 mg 2 veces diarias, con incrementos de 1 mg 2 veces al día en el segundo y tercer día, según la tolerancia, hasta alcanzar la dosis de 3 mg dos veces al día en el tercer día. En caso de requerir ajustes subsiguientes, se puede incrementar o disminuir la dosis en 1-2 mg/día, pero a intervalos de una semana. La res-puesta terapéutica óptima se observa, en la mayoría de los pacientes, con la dosis en el rango de 4-6 mg/día. REACCIONES ADVERSAS Cardiovasculares: Taquicardia, dolor torácico, hipotensión ortostática, prolongación del intervalo QT. Dermatológicas: Erupciones, sequedad de la piel, fotosensibilidad, seborrea. Gastrointestinales: Constipación, náuseas, vómitos, dispepsia, dolor abdominal, sialorrea, disfagia. N Neurológicas: Síntomas extrapiramidales, somnolencia, insomnio, cefalea, agitación, ansiedad, discinesia tardía, agresividad. Respiratorias: Rinitis, tos, sinusitis, faringitis, disnea, infección del tracto respiratorio superior. Otras: Visión anormal, dolor de espalda, fiebre, hiperprolactinemia, síndrome neuroléptico maligno (raro). INTERACCIONES 2ed. 2004 * Formulario TerapÈutico Nacional 445 clozapina disminuye la depuración. La fluoxetina aumenta las concentraciones plasmáticas de la risperidona por inhibición del metabolismo. consumo de alcohol durante el tratamiento. Debe tomar precauciones antes de la exposición al sol. Notificar de inmediato al médico si se presenta alguna reacción o sintomatología CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en casos de hipersensibilidad al medicamento. Se debe usar con precaución en pacientes con alteración de la función renal y/o hepática, en ancianos y en individuos con historia de convulsiones y/o síndrome neuroléptico maligno. Se desconoce la seguridad de su uso en niños. Debe ser usada con precaución en pacientes con riesgo de neumonía por aspiración y en pacientes con problemas cardíacos. El uso en el embarazo debe limitarse a situaciones de verdadera necesidad, en las que el beneficio para la madre supere al riesgo potencial para el feto. Durante el tratamiento se debe suspender la lactancia. TIORIDAZINA SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Mantener las vías aéreas permeables, vigilar el ECG y prestar atención a la posibilidad de reacciones distónicas o convulsiones. FARMACOLOGÕA Antipsicótico del grupo de las fenotiazinas, con potencia y efectos farmacológicos similares a los de la clorpromazina (ver: CLORPROMAZINA). A diferencia de otras fenotiazinas, posee una débil acción antiemética y produce una mínima estimulación del sistema extrapiramidal. Además, muestra una marcada actividad sedante y anticolinérgica. FARMACOCIN…TICA Se absorbe bien en el tracto gastrointestinal. Alcanza niveles plasmáticos pico en 60 minutos (rango de 2-4 horas). Una vez en la sangre, se une a las proteínas plasmáticas en un 99% y se distribuye ampliamente en el organismo. Se metaboliza en el hígado originando metabolitos como la mesoridazina y la sulforidazina, los cuales poseen actividad farmacodinámica semejante a la de la tioridazina. La mesoridazina se usa como medicamento per se. Se excreta principalmente por las heces (50%) pero también por el riñón (menos del INFORMACIÓN AL PACIENTE Advertir al paciente que el medicamento puede alterar la capacidad 446 Formulario Terapéutico Nacional * 2ed. 2004 INDICACIONES Manejo sintomático de trastornos psicóticos en pacientes que no toleran o no logran una respuesta satisfactoria con otros medicamentos antipsicóticos. Tratamiento de corta duración de los estados de depresión asociada a diferentes grados de ansiedad. R…GIMEN DE DOSIFICACI”N Se administra solamente por vía oral. Para el manejo sintomático de trastornos psicóticos en adultos: Iniciar con 50-100 mg 3 veces diarias con incrementos graduales, en caso necesario, hasta el control de las manifestaciones. En casos graves, la dosis podría superar los 300 mg/día; en pacientes hospitalizados se ha usado hasta 800 mg/día en 2 ó 4 dosis divididas. Para el tratamiento de corta duración de la depresión acompañada de ansiedad, iniciar con 25 mg 3 veces al día y ajustar gradualmente según la respuesta del paciente. La dosis usual se ubica, por lo general, en el rango de los 20-200 mg/día, según la severidad del cuadro. Para el manejo sintomático de trastornos psicóticos: En niños de 212 años, se recomienda una dosis de 0,5-3 mg/kg/día. REACCIONES ADVERSAS Los efectos adversos de la tioridazina son similares a los de otras fenotiazinas, pero, además, con este INTERACCIONES Ver: CLORPROMAZINA. CONTRAINDICACIONES/ PRECAUCIONES Ver: CLORPROMAZINA. SOBREDOSIS Ver: CLORPROMAZINA. INFORMACI”N AL PACIENTE TRIFLUOPERAZINA FARMACOLOGÍA Antipsicótico del grupo de las fenotiazinas con efectos farmacológicos similares a las de la clorpromazina (ver: CLORPROMAZINA), aunque más potentes y de mayor duración. Posee una débil actividad sedante y antimuscarínica, y una marcada acción antiemética y extrapiramidal. FARMACOCINÉTICA Es rápidamente absorbida en el tracto N gastrointestinal y desde los sitios de administración parenteral. Alcanza niveles plasmáticos pico a las 2-4 horas de su administración. Una vez absorbida se une a las proteínas plasmáticas en un 90-95% y se distribuye a todos los tejidos. Por la naturaleza lipofílica de la molécula, las concentraciones en el SNC resultan superiores a los niveles plasmáticos. Se metaboliza en el 2ed. 2004 * Formulario TerapÈutico Nacional 447 INDICACIONES En el tratamiento sintomático de los trastornos psicóticos y como alternativa en terapias de corta duración de la ansiedad no psicótica en pacientes que no toleran o no responden adecuadamente a otro tratamiento. 12 semanas. REACCIONES ADVERSAS Ver: CLORPROMAZINA. INTERACCIONES Ver: CLORPROMAZINA. CONTRAINDICACIONES/ RÉGIMEN DE DOSIFICACIÓN Para el tratamiento de trastornos PRECAUCIONES Ver: CLORPROMAZINA. psicóticos: - En adultos: 1-2 mg por vía oral, 2 SOBREDOSIS veces al día aumentando graVer: CLORPROMAZINA. dualmente hasta obtener la respuesta terapéutica, la cual se logra por lo general con dosis de FLUOXETINA 15-20 mg/día. Los niveles terapéuticos óptimos se alcanzan FARMACOLOGÍA Agente antidepresivo, inhibidor en unas 3 semanas. selectivo de la recaptación neuronal - Para pacientes hospitalizados y de serotonina (5-HT). También se ha bajo supervisión: 2-5 mg por vía demostrado que bloquea la captación oral 2 veces al día. de 5-HT en las plaquetas. Bloquea - Para el control rápido de síntomas débilmente la recaptación de noraseveros: 1-2 mg IM (inyección drenalina. profunda) cada 4-6 horas según sea necesario. La mayoría de los FARMACOCINÉTICA pacientes responden a 15 a 20 mg Se absorbe bien y completamente por diarios. vía oral, alcanzando concentraciones - En niños de 6-12 años hospitaséricas pico a las 6-8 horas y una lizados: Iniciar con una dosis oral respuesta antidepresiva inicial en 1-2 de 1 mg 1-2 veces al día, con semanas, que se hace máxima a las 4 incrementos graduales hasta semanas de iniciado en tratamiento. control de los síntomas o hasta Se une a las proteínas plasmáticas en que los efectos adversos se un 95% y se distribuye ampliamente en conviertan en un problema. En el organismo. Se metaboliza extenniños, gene-ralmente la dosis samente en el hígado transformáxima requerida es 15 mg diarios. 448 Formulario Terapéutico Nacional * 2ed. 2004 60% por la orina y en un 12% por las heces. La vida media de eliminación, con la administración de una dosis única, es de 1-3 días y con la administración crónica es de 4-6 días. La vida media del metabolito norfluoxetina es de 4 a 16 días. Estos tiempos se prolongan en pacientes con insuficiencia hepática. INDICACIONES Tratamiento de la depresión, trastorno obsesivo-compulsivo, bulimia y disforia premenstrual. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La dosis usual en adultos para la depresión, el trastorno obsesivo-compulsivo y la disforia premenstrual es de 20 mg/día, en la mañana. En caso de respuesta insuficiente al cabo de varias semanas, se puede incrementar la dosis de manera gradual hasta un máximo de 80 mg/día, en dosis divididas. La dosis usual para el tratamiento de la bulimia es de 60 mg/día. REACCIONES ADVERSAS Cardiovasculares: Angina, variaciones de la presión arterial y la frecuencia cardiaca, hipertensión pulmonar y prolongación del intervalo QT. Gastrointestinales: Náusea, vómito, constipación, dispepsia, dolor abdominal, flatulencia, boca seca y Dolor de cabeza, mareo, insomnio temblor, nerviosismo, psicosis, astenia, trastornos extrapiramidales, trastornos psicomotores y convulsiones. Reacciones de hipersensibilidad: Urticaria, erupción, diaforesis, angioedema, broncoespasmo y anafilaxis. Otras: Hipopotasemia, hiponatremia, síndrome de secreción inapropiada de la hormona antidiurética, hipertrigliceridemia, galactorrea e hipoglicemia, trastornos visuales, pérdida de peso. INTERACCIONES La combinación con inhibidores de la monoaminooxidasa (IMAO) o con agentes con actividad serotoninérgica puede provocar el síndrome serotonínico. La fluoxetina y su metabolito norfluoxetina pueden inhibir el metabolismo hepático e incrementar los niveles séricos y el riesgo de toxicidad de los siguientes medicamentos: terfenadina, astemizol, antidepresivos tricíclicos, antiarrítmicos clase IC, N fenotiazinas, cisaprida, vinblastina, diazepam, alprazolam, clozapina, haloperidol, insulina, antidiabéticos, carbamazepina, fenitoína, bloqueantes de los receptores betaadrenérgicos y warfarina. Se ha reportado cambios en los niveles de litio, incluyendo casos de toxicidad por litio. CONTRAINDICACIONES/ PRECAUCIONES 2ed. 2004 * Formulario TerapÈutico Nacional 449 trastornos renales y/o hepáticos, tendencias suicidas, historia de convulsiones, diabetes mellitus y durante el embarazo. No se conoce la seguridad de su uso en niños. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Mantener las vías aéreas permeables, vigilar el ECG y los signos vitales y prestar atención a la posibilidad de convulsiones. INFORMACIÓN AL PACIENTE Advertir al paciente que el medicamento puede alterar la capacidad para realizar actividades que requieren concentración, alerta mental y destreza motora. No se debe modificar la dosis ni la frecuencia de administración sin el consentimiento del médico. También debe informar al IMIPRAMINA FARMACOLOGÍA Antidepresivo tricíclico, inhibidor de la recaptación neuronal de la norepinefrina y la serotonina, aunque principalmente de la noradrenalina. Exhibe, además, una marcada actividad anticolinérgica y bloqueante de 450 FARMACOCINÉTICA Se absorbe rápida y completamente en el tracto gastrointestinal, aunque sufre un intenso efecto de metabolismo de primer pasaje. Alcanza concentraciones plasmáticas pico a los 60-120 minutos de su administración oral, y produce una respuesta antidepresiva a las 2-4 semanas de iniciado el tratamiento. Se distribuye ampliamente en el organismo. Se metaboliza en el hígado transformándose en productos activos como la desipramina, la cual es inhibidora selectiva de la recaptación de noradrenalina, y metabolitos inactivos que se excretan principalmente en la orina y en escasa proporción en las heces. La vida media de eliminación es de 8-16 horas. INDICACIONES Tratamiento de trastornos depresivos en adultos y enuresis nocturna funcional en niños mayores de 6 años. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. Para el tratamiento de la depresión en adultos iniciar con 25-75 mg/día y ajustar gradualmente hasta lograr la respuesta deseada, sin exceder la dosis total diaria de 200 mg para pacientes ambulatorios; hasta 300 mg para pacientes hospitalizados. Para ancianos y adolescentes: Inicialmente, 30 a 40 mg/día. Usualmente no es Formulario Terapéutico Nacional * 2ed. 2004 75 mg/día en mayores de 12 años. La dosis total diaria no debe exceder de 2,5 mg/kg/día. REACCIONES ADVERSAS Cardiovasculares: Alteraciones en el ECG, arritmias, hipotensión postural y trastornos de la conducción, taquicardia, bloqueo cardíaco, precipitación de la insuficiencia cardiaca, accidente cerebrovascular, infarto del miocardio. Endocrinas: Secreción inapropiada de hormona antidiurética, alteraciones de la glicemia, galactorrea, ginecomastia y alteración de la libido. Gastrointestinales: Náusea, vómito, boca seca, reflujo gastroesofágico, constipación, íleo paralítico, elevación de aminotransferasas, hepatitis, lengua negra y cólicos abdominales. Hematológicas: Agranulocitosis, trombocitopenia, eosinofilia, leucopenia y púrpura. Neurológicas: Sedación, confusión, mareo, fatiga, agitación, agresión, ataxia, alucinaciones, neuropatía periférica, pesadillas, exacerbación de la psicosis, manía, trastornos extrapiramidales, trastornos psicomotores, convulsiones y cambios en el EEG. Oculares: Visión borrosa, aumento de la presión intraocular y midriasis. Reacciones de hipersensibilidad: Erupción, eritema, urticaria y anafilaxis. Otras: Tinitus, hipertermia, falla renal, retención urinaria, aumento o pérdida de peso, simpatomiméticos, aumentando el riesgo de arritmias e hipertensión. La combinación con inhibidores de la MAO aumenta el riesgo de crisis hipertensivas y convulsiones. Por su efecto enlentecedor del vaciamiento gástrico, la imipramina puede alterar la absorción gastrointestinal de numerosos fármacos, como la levodopa y fenilbutazona. La cimetidina, los inhibidores selectivos de la recaptación de serotonina, el metilfenidato, la quinidina, las fenotiazinas y el haloperidol inhiben el metabolismo de la imipramina por la enzima CYP2D6, incrementando las concentraciones séricas y el riesgo de toxicidad. Los medicamentos tiroideos aumentan la incidencia de arritmias cardíacas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y durante la fase aguda de recuperación N posterior a un infarto al miocardio. Usar con precaución en pacientes con hipertrofia prostática, retención urinaria, presión intraocular elevada o glaucoma de ángulo cerrado, hipertiroidismo o tratamiento con medicamentos tiroideos, enfermedad respiratoria, estreñimiento, trastornos convulsivos, enfermedad cardiovascular, discrasias sanguíneas, falla renal y/o hepática, en ancianos y en pacientes alcohólicos. Durante el tra451 2ed. 2004 * Formulario TerapÈutico Nacional prolongada, puede producir náuseas, cefalea y malestar. El uso en el embarazo debe restringirse a situaciones de verdadera necesidad en las que los beneficios de la terapia superen los riesgos potenciales para el feto. Se recomienda suspender la lactancia durante el tratamiento. en la noche, se le debe alertar sobre la posibilidad de hipotensión postural en la mañana. No se debe suspender abruptamente el medicamento. El fármaco puede alterar la capacidad para realizar actividades que requieran concentración, estado de alerta y coordinación motora. Notificar de inmediato al médico si se presenta SOBREDOSIS Vaciamiento gástrico, carbón activado METILFENIDATO y catártico salino, si la ingestión es reciente. Manejo sintomático y FARMACOLOGÍA Agente estimulante respiratorio y del tratamiento de soporte. Vigilar contiSNC, con propiedades nuamente la función cardiaca (ECG) y farmacológicas similares a las de la respiratoria, la presión arterial, el anfetamina y una débil actividad equilibrio hidro-electrolítico y la simpaticomimética. No se conoce con temperatura. Mantener las aéreas exactitud su meca-nismo de acción, permeables. En caso de convulsiones pero se cree que actúa a nivel cortical y usar diazepam (evitar los barbitúricos, sobre algunas estructuras excepto en casos refractarios) o subcorticales, incluido el tálamo, para fenitoína. Las arritmias severas pueproducir una estimulación que se den responder a fenitoína, lidocaína traduce en incremento de la actividad o propranolol (no usar quinidina, motora y de la alerta mental, euforia procainamida o disopiramida). La hipotensión debe ser tratada con leve, disminución de la fatiga y fluidoterapia y se debe colocar al pérdida del apetito. Se postula que paciente en posición Trendelenburg promueve la transmisión del impulso (no usar fármacos simpatomiméticos nervioso mediante la liberación de o vasopresores). La administración IV noradrenalina de los terminales de bicarbonato para alcanzar un pH nerviosos. No hay evidencia que sistémico de 7,4-7,5 puede resultar de explique los efectos mentales y utilidad para controlar los trastornos de conductuales en niños, ni evidencia conducción cardiaca. Los pacientes conclusiva con respecto a cómo se con bloqueo AV severo pueden relacionan estos efectos con la requerir un marcapaso. La diálisis y la condición del sistema nervioso central. diuresis forzada son inefectivas. 452 Formulario Terapéutico Nacional * 2ed. 2004 3-6 horas. Se une a las proteínas plasmáticas en un 15%. Se metaboliza extensamente en el hígado, dando lugar a productos que se excretan, junto a menos de un 1% del fármaco intacto, por vía renal. Debido al metabolismo de primer pasaje, tiene una biodisponibilidad de 30%. El principal metabolito es el ácido ritalínico. La vida media de eliminación es de 2-7 horas. INDICACIONES Trastornos por déficit de atención con hiperactividad en niños y en la narcolepsia. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. En niños de 6 años y más, con déficit de atención asociado a hiperactividad, iniciar con 5 mg dos veces/día antes del desayuno y el almuerzo, e incrementar, en caso necesario, en 510 mg/día a intervalos semanales, sin exceder 60 mg/día. En el tratamiento de la narcolepsia la dosis usual en adultos es de 10 mg 2-3 veces/día, 30-45 minutos antes de las comidas. En algunos casos puede requerirse hasta 40-60 mg/día. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones, angina, taquicardia, cambios en la presión arterial y en la presión de pulso, arritmias. Gastrointestinales: de Tourette, mareos, cefalea, acatisia, discinesia, convulsiones, somnolencia y psicosis tóxica. Reacciones de hipersensibilidad: Erupción, urticaria, dermatitis exfoliativa, eritema multiforme. INTERACCIONES Los inhibidores de la MAO pueden potenciar los efectos presores del metilfenidato y provocar hipertensión severa. El metilfenidato puede inhibir el metabolismo hepático del fenobarbital, la fenitoína, la primidona, la fenilbutazona y de los anticoa-gulantes cumarínicos. También puede incrementar los niveles plasmáticos de los antidepresivos tricíclicos, particularmente la imipramina y la desipramina. Antagoniza los efectos de los agentes antihipertensivos. Las bebidas que contienen cafeína pueden aumentar los efectos del metilfenidato. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento, estados de ansiedad, tensión o agitación, hipertiroidismo, glaucoma, tics motores e historia familiar o personal del síndrome de Tourette. Usar con precaución en pacientes con hipertensión arterial, psicosis, trastornos convulsivos, anormalidades en el ECG 2ed. 2004 * Formulario TerapÈutico Nacional 453 N dependencia psíquica. Se requiere supervisión cuidadosa de la suspensión del medicamento ya que puede causar depresión. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Debe proveerse cuidado intensivo para mantener una circulación e intercambio respiratorio adecuado. Procedimientos de enfriamiento externo pueden ser requeridos para la hiperpirexia. INFORMACIÓN AL PACIENTE A objeto de evitar el insomnio, la última dosis del día debe tomarse horas antes de dormir. Evitar el consumo de BROMURO DE NEOSTIGMINA (NEOSTIGMINA) FARMACOLOGÕA Agente anticolinesterásico. Inhibe la hidrólisis de la acetilcolina por inhibición de la enzima acetilcolinesterasa, aumentando las concentraciones sinápticas del neurotransmisor y, consecuentemente, la intensidad y duración del efecto colinérgico. FARMACOCIN…TICA Después de la administración parenteral, se observa actividad peristáltica a los 10-30 minutos y un 454 persiste por 2,5-4 horas. Una vez en la sangre, se une a las proteínas plasmáticas en un 15-25%. Debido a su estructura amonio cuaternario no atraviesa la barrera hematoencefálica. Se hidroliza por la acción de la colinesterasa y también sufre metabolismo hepático. Se excreta en la orina en un 50% como fármaco intacto y el resto como metabolitos. La vida media de eliminación es de 42-60 minutos. INDICACIONES Tratamiento de la miastenia gravis, cuando la terapia oral no es factible. Reversión del bloqueo neuromuscular causado por relajantes musculares no despolarizantes. Prevención y tratamiento de la distensión y retención urinaria post-operatoria. R…GIMEN DE DOSIFICACI”N Se administra por vía parenteral (SC, IM e IV) como sal metilsulfato. - Tratamiento de miastenia gravis: Iniciar con 0,5-2,5 mg SC, IM ó IV cada 1-3 horas e individualizar las dosis subsiguientes según la respuesta clínica. - Reversión del bloqueo neuromuscular en adultos: 0,5-2,5 mg IV lenta, precedidos de 0,6-1,2 mg de atropina IV unos minutos antes. En caso necesario repetir la neostigmina para un total de 5 mg. En los Formulario Terapéutico Nacional * 2ed. 2004 - Tratamiento de la distensión y retención urinaria post-operatoria: 0,5-1 mg SC ó IM. Si no hay respuesta en 1 hora se debe cateterizar al paciente. Una vez que la vejiga ha sido vaciada repetir la dosis cada 3 horas por 5 dosis. REACIONES ADVERSAS Cardiovasculares: Hipotensión, bradicardia, taquicardia, síncope, paro cardíaco, bloqueo AV. Gastrointestinales: Excesiva secreción salival, hipersecreción gastrointestinal, náuseas, vómito, aumento del peristaltismo, diarrea, cólicos, flatulencia. Genitourinarias: Incontinencia urinaria. Neurológicas: Cefalea, debilidad muscular, somnolencia, pérdida de la conciencia, convulsiones. Reacciones de hipersensibilidad: Erupción, urticaria y anafilaxis. Respiratorias: Aumento de la secreción bronquial, broncoespasmo, disnea, depresión respiratoria, laringoespasmo, parálisis de los músculos respiratorios, parálisis respiratoria central. Otras: Calambres y fasciculaciones musculares, miosis, lagrimeo, artralgia, diaforesis. INTERACCIONES La atropina y otros agentes anticolinérgicos antagonizan el efecto de la neostigmina. La neostigmina puede disminuir el metabolismo de la succinilcolina e incrementar el bloqueo incrementando sus requerimientos. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, peritonitis y obstrucción urinaria o intestinal. Usar con cautela en pacientes con asma bronquial, epilepsia, oclusión coronaria reciente, vagotonía, bradicardia, hipertiroidismo, arritmias cardíacas, úlcera péptica y en el embarazo. Debe evitarse la administración de dosis elevadas en pacientes que presentan megacolon o disminución de la motilidad intestinal. Se recomienda suspender la lactancia durante el tratamiento. SOBREDOSIS Manejo sintomático y terapia de soporte. Vigilar la función respiratoria. Puede requerirse aspiración bronquial o drenaje postural para mantener la vía aérea permeable. Controlar la N crisis colinérgica con atropina 1-4 mg IV y repetir cada 5-30 minutos, según la respuesta del paciente. INFORMACI”N AL PACIENTE No aplicable; medicamento para uso hospitalario. 2ed. 2004 * Formulario TerapÈutico Nacional 455 P02 ANTIHELMÕNTICOS Albendazol Ivermectina Piperazina Prazicuantel P03 ECTOPARASITICIDAS, INCLUYE ESCABICIDAS, INSECTICIDAS Y REPELENTES Benzoato de Benzilo Crotamitón* Lindano 2ed. 2004 * Formulario TerapÈutico Nacional P PRODUCTOS ANTIPARASITARIOS, INSECTICIDAS Y REPELENTES P01 ANTIPROTOZOARIOS Antimoniato de Meglumina (N-Metil-Glucamina) Artemetero Artesunato Benznidazol Cloroquina (Cloroquina Base y Cloroquina Clorhidrato) (Antimalárico) Doxiciclina* (Antimalárico) Mefloquina (Mefloquina Clorhidrato) Metronidazol (Metronidazol y Metronidazol-Benzoil) (Antiprotozoario) Pirimetamina Pirimetamina / Sulfadoxina Primaquina Quinina (Quinina Diclorhidrato y Quinina Sulfato) Tinidazol 457 P ANTIMONIATO DE MEGLUMINA (N-METIL-GLUCAMINA) FARMACOLOGÍA Compuesto de antimonio pentavalente con propiedades anti-leishmaniásicas. Aunque no se conoce con precisión su mecanismo de acción, se ha propuesto que los antimoniales pentavalentes inhiben la actividad de diversas enzimas citoplasmáticas vitales del parásito, tales como la fosfofructoquinasa, llevando a una disminución de la fructosa difosfato y de la glicólisis. FARMACOCINÉTICA Después de la administración IM, se absorbe rápidamente alcanzando niveles plasmáticos pico en aproximadamente 2 horas, aunque la respuesta clínica se observa a las 2 semanas en el caso de la leishmaniasis visceral y mucocutánea y a las 8 semanas en la leishmaniasis cutánea. Una cantidad no determinada se metaboliza a antimonio trivalente en el hígado (lo cual podría contribuir a la toxicidad observada en las terapias prolongadas). Se excreta por vía renal en más de 90%. Exhibe un patrón de eliminación bifásico caracterizado por una vida media de eliminación rápida de 2 horas y una vida media de eliminación lenta de 76 horas (probablemente debida a su conversión hepática en antimonio trivalente). INDICACIONES (hasta un máximo de 850 mg) por 20 días o hasta 2 semanas después de evidenciar una respuesta clínica favorable o ausencia del parásito en el aspirado esplácnico. - Leishmaniasis mucocut·nea: 20 mg/kg/día (hasta un máximo de 850 mg) por 30 días. En caso de reacción severa o respuesta clínica insufi-ciente, modificar la dosificación a 10-15 mg/kg cada 12 horas. - Leishmaniasis cut·nea: En la fase inicial, tratar con infiltración de 1-3 ml de solución 100 mg/ml (100-300 mg), pudiendo repetirse 1-2 veces, en caso necesario, a intervalos de 12 días. Si las lesiones persisten o se agravan, usar terapia sistémica con 20 mg/kg/día hasta 2 semanas después de evidenciar una respuesta clínica favorable o la ausencia del parásito en el aspirado esplácnico. REACCIONES ADVERSAS Cardiovasculares: Arritmias cardíacas, prolongación del intervalo QT y aplanamiento o inversión de la onda P T. Dermatológicas: Erupción, edema facial. Gastrointestinales: Náuseas, vómitos, dolor abdominal y anorexia. Hematológicas: Anemia, leucopenia y agranulocitosis. Hepáticas: Elevación de las enzimas hepáticas, ictericia y degeneración grasa del hígado. Neurológicas: Cefalea, letargia y polineuritis. Reacciones de hipersensibilidad: 459 2ed. 2004 * Formulario TerapÈutico Nacional cansancio, mialgias, artralgia. INFORMACIÓN AL PACIENTE Antes de iniciar la terapia, se requiere corregir los niveles de hierro en sangre y, durante el tratamiento, la dieta debe ser hiperproteica. Evitar el consumo de alcohol. Informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento, en especial INTERACCIONES Los fármacos que prolongan el intervalo QT y los agentes con propiedades arritmogénicas pueden incrementar el riesgo de arritmias cardíacas. El alcohol aumenta el potencial hepatotóxico del antimoniato de meglumina. Los antiarrítmicos, los antidepresivos tricíclicos (amitriptilina, amoxapina, ARTEMETERO clomipramina, desipramina, doxepina, imipramina, nortriptilina, protriptilina, FARMACOLOGÍA Agente derivado de la artemisinina, trimipramina) pueden incrementar los con actividad antimalárica similar a efectos cardiacos. aquella descrita para el artesunato. Ver: ARTESUNATO. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hiper- FARMACOCINÉTICA sensibilidad al medicamento, en Después de la administración IM, pacientes con insuficiencia cardiaca, alcanza niveles séricos pico a las 6 renal y hepática y en el embarazo. horas y produce una respuesta teraUsar con precaución en pacientes con péutica (reducción de la fiebre y disantecedentes de pancreatitis, discra- minución de la parasitemia) a las 28sias sanguíneas y durante la 48 horas. Una vez en la sangre se une lactancia. Durante la terapia se debe a las proteínas plasmáticas en un vigilar periódicamente la función 95%. Se metaboliza rápidamente en el c a r d i a c a , h e p á t i c a , r e n a l y hígado, transformándose en la forma hematológica. Este producto contiene farmacológicamente activa, la dihisulfitos que pueden provocar o agravar droartemisinina, la cual se distribuye a reacciones anafilácticas existentes. los eritrocitos parasitados donde ejerce su actividad. La vida media de SOBREDOSIS eliminación es de aproximadamente 2 Manejo sintomático y terapia de horas y se prolonga de manera sigsoporte. Los signos observados, en nificativa en pacientes con disfunción casos de intoxicación son: Ictericia renal. severa, insuficiencia renal aguda, 460 Formulario Terapéutico Nacional * 2ed. 2004 combinación con la mefloquina para evitar o minimizar las recaídas. RÉGIMEN DE DOSIFICACIÓN Se administra por vía IM. La dosis inicial en adultos y niños es de 3,2 mg/kg el primer día, seguidos por 1,6 mg/kg/día por 4 días (5 días de terapia) o, alternativamente, 200 mg como dosis inicial, seguidos por 100 mg cada 12 horas, hasta alcanzar un total de 600 mg (3 días de terapia). En ambos casos, el artemetero es seguido por una dosis oral única de 750-1000 mg de mefloquina 12 horas después. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, anormalidades en el ECG. Hematológicas: Disminución del contaje de neutrófilos y reticulocitos. Hepáticas: Elevación de los niveles de las enzimas hepáticas. Reacciones de hipersensibilidad: Erupción y otras reacciones de hipersensibilidad. Otras: Mareos, fiebre medicamentosa, dolor en el sitio de la inyección. INTERACCIONES Ver: ARTESUNATO. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a los agentes derivados de la artemisinina y durante el primer trimestre del embarazo, malaria regulares en pacientes con prolongación preexistente del intervalo QT, historia de arritmias cardiacas sintomáticas, trastornos del balance hidro-electrolítico (hipokalemia, hipomagnesemia), bradicardia clínicamente relevante, insuficiencia cardiaca congestiva o pacientes que reciban medicamentos que prolonguen el intervalo QT. Disfunción renal o hepática severa. Controlar los niveles de potasio. Este medicamento no está indicado para profilaxis de la malaria. Usar con precaución en pacientes con disfunción hepática y/o renal, enfermedad cardiaca (en especial trastornos de conducción), mielosupresión y en el embarazo (segundo y tercer trimestre). Durante el tratamiento se debe vigilar periódicamente la función hepática, renal, cardiaca y hematológica. No se conoce si el artemetero se excreta en la leche materna; por tanto, debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS P Manejo sintomático y tratamiento de soporte. Prestar atención a la posibilidad de alteraciones cardíacas, disfunción hepática y discrasias sanARTESUNATO FARMACOLOGÍA Agente derivado de la artemisinina con 2ed. 2004 * Formulario TerapÈutico Nacional 461 actividad antimal·rica. Se cree que su efecto esquizonticida involucra una acciÛn directa sobre los fosfolÌpidos de la membrana del par·sito, o una alteraciÛn estructural de la molÈcula de ADN que impide la sÌntesis de proteÌnas, o una reacciÛn con el HEME en las vacuolas alimenticias del plasmodium. Dado que no es activo frente a las formas exoeritrocÌticas del par·sito, resulta inefectivo contra el P. vivax y el P. ovale. FARMACOCINÉTICA Después de la administración oral, alcanza niveles plasmáticos pico en 15-30 minutos y se observa una respuesta terapéutica (reducción de la fiebre y disminución de la parasitemia) a las 27-40 horas. Una vez absorbido, el artesunato es rápidamente meta-bolizado por la colinesterasa plas-mática y tisular, transformándose en su forma farmacológicamente activa, la dihidroartemisinina, que alcanza concentraciones séricas pico a los 60 minutos de la administración oral y que persisten por 4 horas. La experimentación in vitro revela que dicho metabolito se acumula en una magnitud significativa y en forma selectiva en los eritrocitos parasitados. La vida media de eliminación del artesunato es de 3-29 minutos y la de 462 RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. La Organización Mundial de la Salud recomienda, para adultos y niños, una dosis total de 10 mg/kg en dosis divididas en 3 días, combinadas con una dosis única oral de mefloquina de 25 mg/kg administrados en el segundo día del tratamiento. La dosis de mefloquina también puede ser administrada 12 horas después de la última dosis de artesunato. REACCIONES ADVERSAS Cardiovasculares: Bradicardia, anormalidades en el ECG (que incluyen prolongación del intervalo QT). Gastrointestinales: Náusea, vómito, diarrea. Hematológicas: Neutropenia, disminución del contaje de reticulocitos. Hepáticas: Elevación de los niveles de las aminotransferasas. Neurológicas: Cefalea, mareos, tinitus, disfunción cerebelar, convulsiones. Reacciones de hipersensibilidad: Erupción, urticaria y anafilaxis. Otras: Dolor generalizado. INTERACCIONES El uso de aurotioglucosa con agentes antimaláricos incrementa el riesgo de discrasias sanguíneas. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad a los agentes derivados de la artemisinina y durante el primer Formulario Terapéutico Nacional * 2ed. 2004 trimestres). Durante el tratamiento se debe vigilar periódicamente la función hepática, hematológica, cardiaca y neurológica. No se conoce si el artesunato se excreta en la leche materna; por lo tanto, debe suspenderse la lactancia durante el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. Prestar atención a la posibilidad de alteraciones cardíacas, disfunción hepática, trastornos neurológicos y discrasias sanguíneas. INFORMACIÓN AL PACIENTE BENZNIDAZOL FARMACOLOGÕA Agente derivado del nitroimidazol con actividad inhibitoria de la síntesis de proteínas y del ácido ribonucléico en las células del Tripanosoma cruzi. En estudios experimentales, el benznidazol ha mostrado que incrementa la actividad fagocitaria en los macrófagos, la liberación de citoquinas y la producción de intermediarios nitrogenados reactivos, generando la destrucción del parásito. FARMACOCIN…TICA séricas pico en 3-4 horas. Se une a las proteínas plasmáticas en un 44%. Se elimina en un 60-67% por vía renal dentro de 4 días y en 22-28% por las heces. Exhibe una vida media de eliminación promedio de 12 horas. INDICACIONES Tratamiento de la tripanosomiasis (enfermedad de Chagas) durante la fase aguda e intermedia. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. En adultos se ha usado dosis de 5-7 mg/kg/día divididos en 2 dosis por 30-60 días, 3 mg/kg/día por 90 días, o 10 mg/kg/día por 10 días, seguido de 3 mg/kg/día por 50 días. En niños menores de 12 años: 5-7,5 mg/kg/día (divididos en 2 dosis) por 30-60 días. En algunos casos, se recomienda el tratamiento por 120 días. REACCIONES ADVERSAS Dermatológicas: Dermatitis purpúrica progresiva. Gastrointestinales: Dolor abdominal o estomacal, náusea, P vómitos. Hematológicas: Discrasias sanguíneas (raras), específicamente leucopenia y trombocitopenia. Reacciones de hipersensibilidad: Erupciones en la piel (raras). Neurol ó g i c a s : N e u r o p a t í a (adormecimiento, dolor escociante, debilidad en las manos y en los pies), convulsiones. 2ed. 2004 * Formulario TerapÈutico Nacional 463 efectos de los anticoagulantes orales (warfarina) por inhibición del metabolismo. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento. No se recomienda su uso durante el primer trimestre del embarazo. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Notificar de inmediato al médico si se presenta alguna manifestación o sintomatología inusual durante el tratamiento (fiebre o escalofríos, sangrado inusual, hematomas, heces de CLOROQUINA (CLOROQUINA BASE Y CLOROQUINA CLORHIDRATO) (Antimal·rico) FARMACOLOGÍA Agente derivado de la 4-aminoquinolina, con propiedades antimaláricas. Interfiere con la síntesis de proteínas en especies susceptibles, mediante la unión a la molécula de ADN. También inhibe a las polimerasas del ADN y del ARN. El fármaco se concentra en las vacuolas digestivas del Plasmodium 464 par·sito para metabolizar y utilizar la hemoglobina. De allÌ que, las formas exoeritrocÌticas del plasmodium, carentes de vacuolas y que no utilizan hemoglobina, no sean afectadas por la cloroquina. Es por ello que no es efectiva como f·rmaco ˙nico para la curaciÛn radical de infecciones agudas por Plasmodium vivax o por Plasmodium ovale (formas exoeritrocÌticas). En tales casos, se debe combinar con un agente activo contra esas formas, como la primaquina. La resistencia del P. falciparum a la cloroquina es amplia y se ha reportado casos de resistencia del P. vivax. FARMACOCINÉTICA Se absorbe rápida y casi completamente en el tracto gastrointestinal, alcanzando niveles plasmáticos pico en 1-2 horas. Se distribuye ampliamente en el organismo alcanzando concentraciones elevadas en el hígado, el bazo, el riñón, el corazón, los ojos, el pulmón, los eritrocitos, las plaquetas y los granulocitos. Se une a las proteínas plasmáticas en un 5065%. Se deposita en los tejidos en cantidades considerables. Se acumula en la retina y otros tejidos que contienen melanina, en el hígado, el bazo, los riñones, los pulmones, los leucocitos, el cerebro y la médula espinal. Se metaboliza parcialmente en el hígado, transformándose en Formulario Terapéutico Nacional * 2ed. 2004 REACCIONES ADVERSAS Auditivas: Sordera tipo nervioso; INDICACIONES tinitus, reducción de la audición en Tratamiento supresivo y profiláctico de pacientes con daño auditivo previo. la malaria producida por cepas sus- Cardiovasculares: Hipotensión, camceptibles de Plasmodium falciparum. bios en el ECG y miocardiopatía. Dermatológicas: Cambios en la RÉGIMEN DE DOSIFICACIÓN pigmentación en piel y mucosas, Se administra por vía oral como prurito y erupción que empeora con la cloroquina base y por vía parenteral exposición a la luz solar, pérdida del como clorhidrato. Las dosis se excabello y de la pigmentación del presan en términos de cloroquina mismo. Gastrointestinales: Náusea, base. vómito, dolor epigástrico, anorexia, - Tratamiento de la malaria no diarrea y cólicos abdominales. complicada: En adultos: Iniciar con Hematológicas: Neutropenia, agra600 mg por vía oral y continuar con nulocitosis, trombocitopenia, anemia 300 mg a las 6, 24 y 48 horas de la aplásica (rara) y hemólisis. Neurodosis inicial. En niños: Iniciar con lógicas: Cefalea, fatiga, ansiedad, 10 mg/kg y continuar con 5 mg/kg a nerviosismo, apatía, irritabilidad, las 6, 24 y 48 horas de la dosis agresividad, confusión, cambios en la inicial. Cuando no es factible iniciar personalidad, neuropatía periférica, el tratamiento por vía oral, en convulsiones, conducción nerviosa adultos: Administrar 200 mg por anormal. Oculares: Visión borrosa y vía IM cada 6 horas y en niños: 5 dificultad para enfocar, nictalopía, mg/kg cada 12 horas. En niños no visión escotomatosa con defectos de debe excederse la dosis de 10 mg diferentes campos, dificultad para leer. (base)/kg/día. Se debe procurar La terapia prolongada y a altas dosis cambiar a la terapia oral tan pronto puede provocar queratopatía rever- P como sea posible. sible al descontinuar el medicamento, - Profilaxis en individuos que viajan a y retinopatía que puede progresar zonas endémicas: Iniciar la terapia aún después de suspendido el 1-2 semanas antes del ingreso a la tratamiento. Otras: Dolor e induración zona endémica, mantenerla en el sitio de inyección, durante la estadía y continuarla hipersensibilidad, miopatía o hasta 4-6 semanas después de neuromiopatía, que puede estar haber abandonado dicha zona. La asociada con cambios sensoriales, dosis para adultos es 300 depresión de los reflejos osteoten465 2ed. 2004 * Formulario TerapÈutico Nacional finalizada la terapia. succinilcolina, interfiere con la respuesta inmune a la vacuna antirrábica. Puede inducir el metabolismo de la levotiroxina, reduciendo su eficacia terapéutica. La combinación con aurotioglucosa aumenta el riesgo de discrasias sanguíneas. Los antiácidos interfieren con la absorción gastroin-testinal de la cloroquina. La cimetidina reduce el metabolismo hepático de la cloroquina predisponiendo al paciente a la toxicidad. La cloroquina aumenta los niveles plasmáticos de la ciclosporina. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Para que el carbón activado sea efectivo, debe darse una dosis por lo menos 5 veces la dosis estimada de cloroquina. Si se presentan convulsiones, deben ser controladas antes del lavado gástrico. Manejo sintomático y tratamiento de soporte. INFORMACI”N AL PACIENTE Se recomienda tomar con las comidas para minimizar las molestias gastrointestinales. No alterar la dosis ni suspender el tratamiento sin consultar al médico. Informar de inmediato al médico si se presenta alguna reacción o sintomatología CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes con trastornos de la retina o DOXICICLINA cambios en el campo visual. Usar con (Antimal·rico) precaución en pacientes con FARMACOLOGÍA psoriasis, porfiria, deficiencia de Ver: DOXICICLINA en la sección de glucosa-6-fosfato deshidrogenasa, ANTIINFECCIOSOS SISTÉMICOS. insuficiencia hepática y alcoholismo. Antes de iniciar la terapia, y FARMACOCINÉTICA periódicamente durante su desarrollo, Ver: DOXICICLINA en la sección de s e d e b e p r a c t i c a r e x á m e n e s ANTIINFECCIOSOS SISTÉMICOS. oftalmológicos, hematológicos y de la función auditiva. Dado que los niños INDICACIONES son particular-mente susceptibles a Profilaxis y tratamiento de malaria por los efectos tóxicos de la cloroquina, en Plasmodium falciparum resistente a la ellos no es recomendable el uso por cloroquina. vía parenteral, a menos que sea esRÉGIMEN DE DOSIFICACIÓN trictamente necesario. No se conoce la Se administra por vía oral. seguridad de su uso en el embarazo; - Profilaxis de la malaria en personas por ello, debe evitarse el uso de la 466 Formulario Terapéutico Nacional * 2ed. 2004 - 1-2 días antes del ingreso al área de riesgo y manteniendo la administración diaria hasta 4 semanas después de abandonar dicha zona. En niños, se aplica el mismo régimen pero con dosis de 2 mg/kg. Tratamiento de la malaria: En adultos: 100 mg 2 veces diarias por 7 días, en combinación con 650 mg de sulfato de quinina 3 veces al día por 3-7 días. En niños: 2 mg/kg/día por 7 días, en combinación con 25 mg/kg/día de sulfato de quinina divididos en 3 dosis por 3-7 días. REACCIONES ADVERSAS Ver: DOXICICLINA en la sección de ANTIINFECCIOSOS SISTÉMICOS. INTERACCIONES Ver: DOXICICLINA en la sección de ANTIINFECCIOSOS SISTÉMICOS. CONTRAINDICACIONES/ PRECAUCIONES Ver: DOXICICLINA en la sección de ANTIINFECCIOSOS SISTÉMICOS. MEFLOQUINA (MEFLOQUINA CLORHIDRATO) FARMACOLOGÍA Agente derivado de la 4-metanolquinolina, con propiedades antimaláricas, efectivo frente a todas las especies de Plasmodium, incluyendo al Plasmodium falciparum resistente a la cloroquina. Aunque el mecanismo de acciÛn no se conoce con exactitud, se postula que la actividad antimal·rica depende de la formaciÛn de complejos con la hemina que daÒan las membranas del par·sito e interact˙an con otros componentes del plasmodio. PodrÌa actuar aumentando el pH de las vesÌculas acÌdicas del par·sito. Adicionalmente, la evidencia experimental revela que el medica-mento produce cambios morfolÛgicos en las etapas tempranas del desarrollo del P. falciparum y P. vivax. Dado que el f·rmaco no es activo contra las formas exoeritrocíticas del P. vivax, puede ocurrir que los pacientes trata-dos tengan recaídas, por lo que se recomienda iniciar un curso de tratamiento con primaquina al finalizar la terapia. FARMACOCINÉTICA Después de la administración por vía oral, se absorbe casi por completo P (85%) aunque lentamente, alcanzando concentraciones séricas máximas en 6-24 horas. Los alimentos aumentan la biodisponibilidad en un 40%. Se une a las proteínas plasmáticas en un 98% y se distribuye ampliamente en los tejidos corporales, alcanzando concentraciones elevadas en los 2ed. 2004 * Formulario TerapÈutico Nacional 467 INDICACIONES Prevención y tratamiento de la malaria producida por Plasmodium vivax y Plasmodium falciparum, incluida la especie resistente a la cloroquina. RÉGIMEN DE DOSIFICACIÓN Se presenta como tabletas con 250 mg de mefloquina base. - Profilaxis en viajeros a zonas endémicas: En adultos, iniciar el tratamiento con 1 tableta, una semana antes del ingreso a la zona de riesgo, continuando con dosis subsiguientes semanales (el mismo día de cada semana, preferiblemente después de la comida principal) durante la permanencia en el área y hasta 4 semanas después de abandonar la zona. La dosis semanal en niños de 31-45 kg es de 3/4 de tableta; en niños de 20-30 kg media tableta y en niños de 15-19 kg 1/4 de tableta. - Tratamiento de la malaria (dosis ˙nica con un vaso lleno de agua (240 ml) con la comida): - Adultos:: 1250 mg. - Niños menores de 5 años: 15-25 mg/kg. - Niños de 5-12 años: 20-30 mg/kg . REACCIONES ADVERSAS Cardiovasculares: Extrasístoles, bradicardia y cambios en la presión arterial. Dermatológicas: Erupción, 468 dolor de cabeza, fatiga, astenia, problemas emocionales, confusión, alucinaciones, ansiedad, depresión, psicosis y convulsiones. Otras: Trastornos oculares, hepatitis, mialgia, fiebre medicamentosa, escalofrío, hipersensibilidad, tinitus. INTERACCIONES La mefloquina disminuye la eficacia anticonvulsivante del ácido valproico. La quinina puede incrementar el riesgo de convulsiones. La ampicilina altera el patrón farmacocinético de la mefloquina. La aurotioglucosa incrementa el riesgo de discrasias sanguíneas. Con los bloqueantes de los receptores beta-adrenérgicos, la quinidina y la quinina, se han descrito casos de alteraciones del ECG y paro cardiaco. CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al producto, falla hepática severa, trastornos convulsivos, trastornos psiquiátricos y durante el primer trimestre del embarazo. Antes y durante la terapia se recomienda una evaluación oftalmológica y de la función hepática. El uso en el segundo y tercer trimestres del embarazo debe limitarse a circunstancias de verdadera necesidad en las que los potenciales beneficios a la madre superen al riesgo potencial para el feto. Durante el tratamiento se debe Formulario Terapéutico Nacional * 2ed. 2004 y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte, particularmente para los trastornos cardiovasculares. INDICACIONES Amibiasis (intestinal y abscesos hepáticos), giardiasis y tricomoniasis. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral como tabletas y suspensión (benzoil metronidazol) y por infusión IV lenta. - Amibiasis intestinal: En adultos, por vía oral: 750 mg 3 veces al día por METRONIDAZOL (METRONIDAZOL Y 5-10 días. En niños: 35-50 METRONIDAZOL- BENZOIL) mg/kg/día (dividido en 3 dosis) por (Antiprotozoario) 10 días. FARMACOLOGÍA - Absceso hepático: En adultos, por Antiinfeccioso derivado del nitroimivía oral: 500-750 mg 3 veces al día dazol con actividad antibacteriana y por 5-10 días. Si la terapia oral no antiprotozoaria. Aunque el mecanises factible, administrar por infusión mo de acción no ha sido completaIV lenta (1 hora) 500 mg cada 6 mente aclarado, se cree que al horas por 10 días. En niños, por penetrar la célula del patógeno el vía oral: 30-50 mg/kg/día (dividido fármaco sufre una reducción que da en 3 dosis) por 10 días. lugar a compuestos reactivos que - Giardiasis: En adultos, por vía oral: alteran la estructura del ADN e impiden 250 mg 3 veces diarias por 5-7 días. En niños, por vía oral: 15 la síntesis de los ácidos nucléicos. mg/kg/día (dividido en 3 dosis) por FARMACOCINÉTICA 5-7 días. Después de la administración oral, se - Tricomoniasis: En adultos, por vía absorbe casi en su totalidad, aloral: dosis única de 2 g o dividida en P canzando concentraciones séricas 2 dosis de 1 g cada una; alterpico en 1-2 horas. Se une a las nativamente 250 mg 3 veces al día proteínas plasmáticas en un 10-20% y por 7 días. En la tricomoniasis se distribuye ampliamente en el refractaria, la dosis para adultos es organismo. Se metaboliza en el 250 mg, 2 veces al día por 10 días hígado transformándose en o, alternativamente, 500 mg 2 productos activos e inactivos que se veces al día por 7 días. En niños, eliminan, junto con el fármaco intacto, por vía oral: 15 mg/kg/día (dividido en un 60-80% por la orina y el resto por en 3 dosis) por 7-10 días. 469 2ed. 2004 * Formulario TerapÈutico Nacional INFORMACIÓN AL PACIENTE Tomar con abundante agua y con las comidas. Notificar de inmediato al médico si se presenta alguna cistitis, oscurecimiento de la orina. Hematológicas: Leucopenia, eosinofilia, agranulocitosis, neutropenia, trombocitopenia y anemia aplásica. Neurológicas: Dolor de cabeza, mareo, decaimiento, vértigo, ataxia, irritabilidad, depresión, debilidad, insomnio, confusión, neuropatía periférica y convulsiones. Respiratorias: Infecciones del tracto respiratorio superior, rinitis, sinusitis y faringitis. Otras: Trastornos visuales, disminución de la libido, sobrecrecimiento de organismos susceptibles, especialmente C. albicans; fiebre, dolor articular tipo enfermedad del suero. INTERACCIONES El metronidazol potencia el efecto de los anticoagulantes cumarínicos. El fenobarbital y la fenitoína pueden incrementar el metabolismo hepático del metronidazol y comprometer su eficacia terapéutica. La cimetidina, por el contrario, disminuye el metabolismo del metronidazol y aumenta sus niveles séricos. El metronidazol puede reducir la eliminación del litio, la fenitoína y la carbamazepina, aumentando el riesgo de reacciones adversas. Con disulfiram, induce psicosis aguda y confusión. Con alcohol, efecto antabuse (náusea, vómitos, cefalea, cólicos, enrojecimiento de la cara). Sanguíneas, disfunción hepática y/o renal y durante el segundo y tercer trimestres del embarazo. El tratamiento puede causar candidiasis oral, vaginal o intestinal. No se debe usar hasta 2 semanas después de la última dosis de disulfiram. No se debe usar alcohol hasta 3 días después de la última dosis. Durante el tratamiento se debe suspender la lactancia y no reiniciarla hasta después de 48 horas de finalizado el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. El metronidazol es removible por hemodiálisis. INFORMACIÓN AL PACIENTE Administrar con los alimentos para disminuir las molestias gastrointestinales. No alterar la dosificación ni interrumpir la terapia antes del tiempo establecido, aunque hayan desaparecido los signos y síntomas de la infección. Evitar el consumo de PIRIMETAMINA FARMACOLOGÍA Antiparasitario derivado de la aminopirimidina, con propiedades de antagonista de ácido fólico. Inhibe la reductasa del ácido dihidrofólico impidiendo la transformación en ácido CONTRAINDICACIONES/ 470 Formulario Terapéutico Nacional * 2ed. 2004 esencial para la biosíntesis celular de purinas, pirimidinas y algunos aminoácidos en el patógeno. FARMACOCINÉTICA Se absorbe casi completamente en el tracto gastrointestinal alcanzando niveles séricos pico en 2-6 horas. Se une a las proteínas plasmáticas en un 80-87% y se distribuye ampliamente en el organismo, alcanzando concentraciones elevadas en hígado, pulmón, riñón y bazo. Se metaboliza en el hígado y se excreta por vía renal. También se excreta por la leche materna. La vida media de eliminación es de 54-148 horas (promedio 111 horas). INDICACIONES Tratamiento de la toxoplasmosis (en combinación con una sulfonamida) y de la malaria por Plasmodium falciparum resistente a la cloroquina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral. - Toxoplasmosis: En adultos iniciar con 50-75 mg/día (combinado con sulfonamida en dosis usuales) por 1-3 semanas, según la respuesta y tolerancia del paciente, seguido por mantenimiento con la mitad de la dosis inicial por 4-5 semanas adicionales. En niños iniciar con 1 mg/kg/día (dividido en 2 dosis) por 2-4 días, reducir la dosis a la mitad y mantener por 30 días náuseas, vómito, boca seca, diarrea, glositis. Hematológicas: Anemia megaloblástica, leucopenia, agranulocitosis, trombocitopenia, pancitopenia. Neurológicas: Ataxia, temblor, convulsiones, dolor de cabeza, depre-sión, fatiga. Reacciones de hipersen-sibilidad: Erupción, dermatitis y fotosensibilidad. Respiratorias: Eosinofilia pulmonar, falla respiratoria. Otras: Hematuria, fiebre medicamentosa, hiperpigmentación de la piel, hiperfenilalaninemia. INTERACCIONES En algunos pacientes la administración conjunta de pirimetamina y lorazepam ha generado hepatotoxicidad moderada. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al fármaco y en pacientes con anemia megaloblástica por deficiencia de folatos. Dado que su uso en toxoplasmosis implica la admi- P nistración de dosis elevadas (cercanas al rango tóxico) resulta altamente recomendable combinarla con ácido folínico (ácido tetrahidrofólico o leucovorina) como terapia de rescate. Usar con precaución en pacientes con falla hepática y/o renal, síndrome de 2ed. 2004 * Formulario TerapÈutico Nacional 471 necesidad en las que otras alternativas terapéuticas resulten ineficaces o contraindicadas. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tratamiento de soporte. El ácido folínico (ácido tetrahidrofólico o leucovorina), administrado dentro de las 2 horas siguientes a la ingestión, previene las manifestaciones hematológicas. Debido a la prolongada vida media de la pirimetamina, los pacientes deben ser evaluados por varios días hasta el restablecimiento completo de los valores hematológicos. INFORMACIÓN AL PACIENTE No alterar la dosis, ni interrumpir la terapia antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. Tomar con las comidas para minimizar las molestias gástricas. Informar de PIRIMETAMINA / SULFADOXINA FARMACOLOGÍA La sulfadoxina inhibe competitivamente la síntesis del ácido dihidrofólico, mientras que la pirimetamina (por inhibición de la enzima dihidrofolato reductasa) evita la conversión 472 tetrahidrofólico. De esta manera, la asociación de ambos agentes actúa de manera sinérgica para impedir la síntesis de ácido tetrahidrofólico, cofactor involucrado en reacciones de transferencia de unidades de átomos de carbono para la biosíntesis de ácidos nucléicos. FARMACOCINÉTICA: Ambos agentes se absorben bien en el tracto gastrointestinal. La pirimetamina alcanza niveles séricos pico en 1,5-8 horas y la sulfadoxina en 2,5-6 horas. Se unen a las proteínas plasmáticas en un 80-87% y 90-95%, respectivamente, y se distribuyen ampliamente en el organismo. Se metabolizan en el hígado y se excretan por vía renal. La vida media de eliminación de la pirimetamina es de 54-148 horas (promedio 111 horas) y la de la sulfadoxina 100-231 horas (pro-medio 169 horas). INDICACIONES Tratamiento de la malaria causada por Plasmodium falciparum resistente a la cloroquina. RÉGIMEN DE DOSIFICACIÓN La combinación se presenta comercialmente como tabletas de 500 mg de sulfadoxina y 25 mg de pirimetamina. Se administra el último día de un curso de quinina de 3-7 Formulario Terapéutico Nacional * 2ed. 2004 de tableta. REACCIONES ADVERSAS Cardiovasculares: Arritmias (con grandes dosis), miocarditis alérgica. Gastrointestinales: Anorexia, vómitos, glositis atrófica, estomatitis, dolor abdominal. Hematológicas: Agranulocitosis, anemia aplásica, anemia megaloblástica, anemia hemolítica, leucopenia, trombocitopenia, pancitopenia. Hepáticas: Aumento de las pruebas de función hepática, ictericia. Neurológicas: Cefalea, neuritis periférica, depresión, convulsiones, ataxia, alucinaciones, fatiga. Reacciones de hipersensibilidad: Síndrome de Stevens-Johnson, erupciones generalizadas de la piel, urticaria, edema periorbital, necrólisis epidérmica, dermatitis exfoliativa, enfermedad del suero, fiebre medicamentosa, anafilaxis. Renales: nefrosis tóxica con oliguria y anemia, cristaluria, hematuria, aumento de la creatinina sérica (efectos debidos a la sulfonamida). Otras: Fotosensibilidad, irritación de la esclerótica. megaloblástica por deficiencia de folatos, en niños menores de 2 meses y en el embarazo a término. Usar con precaución en pacientes con falla hepática y/o renal, asma bronquial y deficiencia de glucosa-6-fosfato deshidrogenasa. Durante la terapia se debe practicar, periódicamente, evaluaciones hematológicas y uroaná-lisis. El paciente debe mantener una adecuada hidratación durante todo el tratamiento a objeto de reducir la posibilidad de cristaluria inducida por la sulfadoxina. El tratamiento debe interrumpirse si se presenta erupción y/o infección bacteriana o micótica. Su uso en el embarazo debe restringirse a situaciones de verdadera necesidad en las que otras alternativas terapéuticas resulten ineficaces o contraindicadas. Durante el tratamiento se debe suspender la lactancia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y tra- P tamiento de soporte. Mantener una INTERACCIONES El uso de agentes mielosupresores y adecuada hidratación IV para prevenir otras sulfonamidas puede aumentar el el daño renal. El ácido folínico (ácido riesgo de discrasias sanguíneas y de tetrahidrofólico o leucovorina), admidepresión de la médula ósea. En nistrado dentro de las 2 horas algunos pacientes, la administración siguientes a la ingestión, previene las conjunta de pirimetamina y lorazepam manifestaciones hematológicas. Se ha producido hepatotoxicidad mo- puede usar con diazepam para controlar las convulsiones. Debido a la 473 2ed. 2004 * Formulario TerapÈutico Nacional INFORMACI”N AL PACIENTE No alterar la dosis, ni interrumpir la terapia antes del tiempo previsto, aunque hayan desaparecido los signos y síntomas de la infección. Administrar con los alimentos para minimizar las molestias gástricas. Si ocurre anorexia y vómitos, el medicamento debe administrarse con la comida. Se debe ingerir abundantes líquidos durante la terapia para evitar la cristaluria. Informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Suspender la adminis-tración en caso dolor de PRIMAQUINA tracto gastrointestinal, alcanzando niveles plasmáticos pico en aproximadamente 6 horas. La biodisponibilidad es de alrededor de 96%. Se distribuye ampliamente en el organismo aunque, a diferencia de otros derivados aminoquinolina, no se acumula en los eritrocitos. Se metaboliza rápidamente en el hígado a carboxiprimaquina. Se excreta por vía renal (menos de 1% como fármaco intacto). La vida media de eliminación es de 3,7-7,4 horas y la de la carboxiprimaquina es de 22-30 horas. INDICACIONES Tratamiento supresivo de la malaria causada por cepas susceptibles de Plasmodium vivax y Plasmodium ovale. Erradicación de las formas gametocíticas del Plasmodium falciparum. FARMACOLOGÍA Agente derivado de la 8-aminoquinolina que posee propiedades antimaláricas. Se cree que su acción es debida a su capacidad para unirse a RÉGIMEN DE DOSIFICACIÓN la molécula de ADN del parásito, Se administra por vía oral como produciendo alteraciones en la es- fosfato de primaquina. Las dosis se tructura que impiden su reproducción expresan en términos de primaquina y p r o v o c a n l a m u e r t e . E s base. Cada 26,3 mg de fosfato de especialmente activa contra las primaquina equivale a 15 mg de formas exoeritro-cíticas del parásito y primaquina base. particularmente letal contra las formas - Tratamiento supresivo de la malaria no complicada provocada por gametocíticas (sexuales) de todas las especies susceptibles: En adultos: especies de Plasmodium. Dosis única diaria de 15 mg por 14 Aparentemente, actúa sobre las días; en niños: 0,2-0,3 mg/kg/día formas hepáticas del parásito, por 14 días. Alternativamente, en afectando la función mitocondrial. 474 Formulario Terapéutico Nacional * 2ed. 2004 de 45 mg. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, malestar epigástrico, cólicos abdominales. Hematológicas: Leucopenia, anemia hemolítica en pacientes con deficiencia de glucosa-6-fosfato deshidrogenasa, metahemoglobinemia en pacientes con deficiencia de la NADH-metahemoglobina reductasa. INTERACCIONES La combinación con agentes potencialmente mielosupresores o inductores de anemia hemolítica incrementa el riesgo en tal sentido. La quinacrina potencia la toxicidad de la primaquina. Las sales de aluminio y de magnesio disminuyen la absorción. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en pacientes sometidos a terapia con fármacos potencialmente mielosupresores o inductores de anemia hemolítica. Usar con precaución en pacientes con deficiencia de glucosa6-fosfato deshidrogenasa. Los pacientes con deficiencias de la enzima reductasa de la NADH-metahemoglobina resultan particularmente propensos al desarrollo de metahemoglobinemia. Durante la terapia se debe realizar una evaluación y catártico salino, si la ingestión es reciente. Manejo sintomático y Tratamiento de soporte. INFORMACIÓN AL PACIENTE Se recomienda tomar el medicamento con las comidas para minimizar las molestias gastrointestinales. No alterar la dosis ni suspender el tratamiento sin consultar al médico. Informar de inmediato al médico si se presenta alguna reacción o sintomaQUININA (QUININA DICLORHIDRATO Y QUININA SULFATO) FARMACOLOGÍA Agente derivado de la 4-metanolquinolina con propiedades antimaláricas. Se cree que interfiere con la funciones del ADN del parásito. Deprime la captación de oxígeno y el metabolismo de carbohidratos del plasmodio. Dado que la quinina es activa sólo contra las formas eritrocíticas del Plasmodium, no previene las recaídas en infecciones por Plasmodium ovale o Plasmodium P vivax debido a que dichas especies presentan formas de desarrollo exoeritrocíticas. Por ello, cuando se usa quinina en la profilaxis o el tratamiento de la malaria por P. ovale o P. vivax se debe combinar con primaquina. FARMACOCINÉTICA 2ed. 2004 * Formulario TerapÈutico Nacional 475 horas. Se distribuye ampliamente a los tejidos y en los eritrocitos alcanza concentraciones equivalentes al 40% de la concentración plasmática. No atraviesa la barrera hematoencefálica. Se une a las proteínas plasmáticas en un 94% y se metaboliza parcialmente en el hígado dando origen a productos hidroxilados que se excretan, junto con parte del fármaco intacto, en un 95% por la orina y en una pequeña proporción por las heces. En pacientes adultos con malaria, la vida media de eliminación es de 8-21 horas y en niños 11-12 horas. INDICACIONES Prevención y tratamiento de malaria por Plasmodium falciparum resistente a la cloroquina en asociación con pirimetamina/sulfadoxina o con doxiciclina. RÉGIMEN DE DOSIFICACIÓN Se administra por vía oral como sulfato de quinina y por vía IV como diclorhidrato de quinina. Se usa en combinación con sulfadoxina/pirimetamina o con doxiciclina (ver las monografías respectivas). La administración por vía IM no es recomendable debido a que puede producir abscesos estériles y causar tétanos. - En la supresión o profilaxis de la malaria: Dosis oral de 350 mg 2 476 tratamiento en niños, también cada 8 horas por 7 días, establece 75 mg para niños menores de 2 años o menos de 15 kg, 150 mg para niños de 2-4 años o 15-19 kg, 300 mg para niños de 5-8 años o 20-30 kg y 450 mg para niños de 915 años o 31-45 kg. - Para el manejo de la malaria severa: Usar la sal diclorhidrato de quinina, iniciando con 20 mg/kg por infusión IV en 4 horas, seguido de 10 mg/kg en 2-8 horas. Se debe cambiar la administración del medicamento a la vía oral tan pronto como sea posible y la evolución del paciente lo permita. REACCIONES ADVERSAS Cardiovasculares: Angina, arritmia y vasculitis. Dermatológicas: Eritema multiforme, prurito, fotosensibilidad e hiperpigmentación. Endocrinas: Hipoglicemia y porfiria. Gastrointestinales: Náuseas, vómitos, diarrea y dolor epigástrico. Hematológicas: Hemólisis, trombocitopenia, leucopenia, agranulocitosis, hipoprotrombinemia y pancitopenia. Hepáticas: Trastornos hepáticos. Neurológicas: Cinconismo (tinitus, disminución de la agudeza auditiva, mareos, vértigo, cefalea, alteración visual), vértigo, dolor de cabeza, cansancio, confusión, aprehensión, delirio e intranquilidad. Oftalmológicas: Alteración de la visión de colores y percepción de la luz, Formulario Terapéutico Nacional * 2ed. 2004 Respiratorias: Disnea, asma. Otras: Tinitus, sordera y miastenia gravis. INTERACCIONES Puede aumentar las concentraciones plasmáticas de los digitálicos. Potencia los efectos bloqueantes neuromusculares del pancuronio, la succinilcolina y la d-tubocurarina. Los antiácidos interfieren con la absorción de la quinina. Los alcalinizantes urinarios (acetazolamida y bicarbonato) aumentan los niveles plasmáticos de la quinina y el riesgo de toxicidad. La quinina puede incrementar la acción de la warfarina y los niveles plasmáticos de astemizol y terfenadina. La cimetidina puede inhibir el metabolismo hepático de la quinina y aumentar sus niveles séricos. La aurotioglucosa incrementa el riesgo de reacciones hematológicas y la mefloquina la posibilidad de efectos cardiovasculares. Tiene efectos vagolíticos. Los anticonvulsivantes, los barbitúricos y la rifampicina disminuyen la eficacia de la quinina por inducción del metabolismo. luaciones hematológicas y oftalmológicas. Debe suspenderse la lactancia durante la terapia. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Tomar medidas para evitar el embarazo. Informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual TINIDAZOL FARMACOLOGÕA Agente derivado del nitroimidazol con propiedades antiprotozoarias y antimicrobianas similares a las del metronidazol (ver: METRONIDAZOL). FARMACOCIN…TICA Después de la administración por vía oral se absorbe casi por completo en el tracto gastrointestinal alcanzando P niveles séricos pico en menos de 2 horas. Se une a las proteínas CONTRAINDICACIONES/ plasmáticas en un 12% y se distribuye PRECAUCIONES con rapidez y amplitud en todo el Contraindicada en pacientes con organismo. Se metaboliza parcialhipersensibilidad al medicamento, mente en el hígado y se excreta como deficiencia de glucosa-6-fosfato fármaco intacto y metabolitos, deshidrogenasa, miastenia gravis y principalmente en la orina y en menor durante el embarazo. Usar con grado por las heces. La vida media de 477 2ed. 2004 * Formulario TerapÈutico Nacional INDICACIONES Tratamiento de infecciones causadas por Entamoeba histolytica, Giardia lamblia, Trichomonas vaginalis y bacterias anaeróbicas susceptibles. ZOL). R…GIMEN DE DOSIFICACI”N Se administra sólo por vía oral. - Amibiasis en adultos: Dosis única diaria de 2 g por 2-3 días. En abscesos hepáticos: Dosis única diaria de 2 g por 2-6 días; niños de 4 meses-11 años: Dosis única diaria de 50-63 mg/kg por 3 días. - Giardiasis en adultos: Dosis única de 2 g; niños hasta 1 año: Dosis única de 500 mg; niños hasta 7 años: Dosis única de 1 g y niños hasta 12 años: Dosis única de 1,5 g. - Tricomoniasis: E n adultos: Dosis única de 2 g; en niños de 6-13 años: Dosis única de 1 g. - Vaginosis bacteriana inespecífica: 2 g/día por 2 días. SOBREDOSIS Ver: METRONIDAZOL. CONTRAINDICACIONES/ PRECAUCIONES Ver: METRONIDAZOL. ALBENDAZOL FARMACOLOGÕA Antihelmíntico de amplio espectro derivado del benzimidazol, activo contra todas las etapas del desarrollo del helminto (huevo, larva y estado adulto). Causa degeneración selectiva de los microtúbulos citoplasmáticos de las células tegumentales e intestinales del parásito; disminuye la captación de glucosa y desacopla la fosforilación oxidativa, produciendo una dismi-nución en la producción intracelular de ATP y, en consecuencia, la inmoviliza-ción y muerte del helminto. Además, reduce la reductasa del fumarato mitocondrial. Sin embargo, se ha de-mostrado que la acción primaria corresponde a la inhibición de la polimerización de los microtúbulos al unirse a la betatubulina. REACCIONES ADVERSAS Cardiovasculares: Palpitaciones. Gastrointestinales: Náusea, vómitos, pérdida del apetito, diarrea, trastornos del gusto, constipación. Hematológicas: Leucopenia. Hepáticas: Insuficiencia hepática. Reacciones de hipersensibilidad: Erupción, prurito. Neurológicas: Cefalea, debilidad, FARMACOCIN…TICA agitación, confusión, mareos, decai- Se absorbe escasamente a través del tracto gastrointestinal, metabolizánmiento. Otras: Decoloración de la 478 Formulario Terapéutico Nacional * 2ed. 2004 incrementa significativamente (de 4 a 5 veces) con comidas ricas en grasa. Se une a las proteínas plasmáticas en un 70% y se distribuye ampliamente en el organismo. Es metabolizado nuevamente en el hígado y convertido en productos inactivos que se excretan en su mayoría por las heces y en escasa proporción por la orina (1%). La vida media de eliminación es de 812 horas. INDICACIONES Tratamiento de parasitosis causadas por Ascaris lumbricoides, Ancylostoma duodenale, Necator americanus, larva migrans cutánea, Enterobius vermicularis, Strongyloides stercoralis, Trichuris trichiura, Hymenolepis nana y especies de Taenia. R…GIMEN DE DOSIFICACI”N Se administra por vía oral. - Enterobiasis, ascaridiasis, estrongiloidiasis, teniasis, trichuriasis y anquilostomiasis, en adultos y niños mayores de 2 años: Dosis única de 400 mg. Niños menores de 2 años: Dosis única de 200 mg. Si es necesario, repetir el tratamiento después de 3 semanas. - Larva migrans cutánea: Administrar entre 400-800 mg diarios por 3-5 días. REACCIONES ADVERSAS Dermatológicas: Erupción y prurito. Gastrointestinales: Dolor abdominal, náusea, vómito, anorexia, constipación, sequedad de la mucosa oral, dolor epigástrico. Hematológicas: Eosinofilia, trombocitopenia, granulocitopenia y pancitopenia. Hepáticas: Elevación de los niveles de las aminotransferasas e ictericia. Neurológicas: Cefalea, mareos y vértigo, aumento de la presión intracraneana, signos de irritación meníngea. Reacciones de hipersensibilidad: Erupción, urticaria y prurito. Renales: Insuficiencia renal aguda. Otras: Alopecia, fiebre, neuritis óptica y pérdida de la visión. INTERACCIONES La cimetidina, la dexametasona y el prazicuantel incrementan significativamente las concentraciones séricas del metabolito sulfóxido de albendazol, aumentando el riesgo de reacciones adversas. CONTRAINDICACIONES/ P PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento y durante el embarazo o cuando se sospeche su existencia. Usar con precaución en pacientes con insuficiencia renal y/o hepática, particularmente en terapias prolongadas. No se conoce la seguridad del uso durante la 2ed. 2004 * Formulario TerapÈutico Nacional 479 SOBREDOSIS Manejo sintomático y tratamiento de soporte. INFORMACIÓN AL PACIENTE Informar de inmediato al médico si se presenta alguna reacción o sintomatología inusual durante el tratamiento. Este medicamento debe ser tomado con la comida. Puede causar daño fetal; por lo tanto, las mujeres en edad de gestación deben iniciar el tratamiento después de haber obtenido una prueba de embarazo negativa. La mujer no debe quedar embarazada mientras recibe el tratamiento y hasta un mes después IVERMECTINA FARMACOLOGÍA Se postula que actúa como agonista de los receptores del ácido gammaaminobutírico (GABA), produciendo un intenso efecto inhibidor de la neurotransmisión en las neuronas motoras del parásito, lo que conduce a la parálisis y muerte del mismo. El hecho que los humanos no resulten susceptibles a la acción farmacológica de la ivermectina es debido a que, en los mamíferos, el GABA está limitado al SNC y el fármaco no atraviesa la barrera hematoencefálica. debe transcurrir varias semanas para observar una respuesta clínica satisfactoria. Se metaboliza en el hígado y se elimina principalmente por vía biliar. La vida media de eliminación se ubica en el rango de 1635 horas. INDICACIONES Infecciones causadas por Onchocerca volvulus, Strongyloides stercolaris y Larva migratoria cutanea. RÉGIMEN DE DOSIFICACIÓN Se administra solamente por vía oral. Para tratamiento de oncocercosis en adultos y en niños con peso superior a 15 kg, la dosis recomendada es de 150 mcg/kg como dosis única. En caso de recurrencia, repetir el tratamiento después de 6-12 meses. Para larva migratoria cutánea y estrongiloidiasis en adultos y niños con peso superior a 15 kg, la dosis recomendada es de 200 mg/kg/día por 2 días consecutivos. REACCIONES ADVERSAS Cardiovasculares: Hipotensión, alteraciones del electrocardiograma, taquicardia. Neurológicas: Cefalea, mareos, somnolencia, insomnio, vértigo, fatiga y temblor. Reacciones de hipersensibilidad: Erupción y prurito. Otras: Mialgia, fiebre. INTERACCIONES FARMACOCINÉTICA Después de la administración oral, se No se describen en la literatura. 480 Formulario Terapéutico Nacional * 2ed. 2004 no reiniciarla hasta pasados 3 días de terminado el tratamiento. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Administrar la dosis con un vaso grande de agua 1 hora antes del desayuno. Notificar al médico si se PIPERAZINA FARMACOLOGÕA Se postula que la piperazina altera la permeabilidad de la membrana celular del parásito a los iones, dando lugar a una hiperpolarización, con la consecuente supresión de los potenciales en espiga. Esto ocasiona una parálisis flácida de la musculatura del helminto que hace posible su fácil expulsión del tracto gastrointestinal por simple peristalsis. FARMACOCIN…TICA Después de la administración por vía oral, se absorbe en un 20-40%, alcanzando niveles plasmáticos pico en aproximadamente 60 minutos. Se metaboliza parcialmente en el hígado y se excreta en un 60-90% como fármaco intacto en la orina. Parece que hay una gran diferencia interindividual R…GIMEN DE DOSIFICACI”N Se administra solamente por vía oral. - Enterobiasis en adultos y niños: 65 mg/kg/día (máximo 2,5 g) por 7 días. Alternativamente, se ha propuesto para niños hasta 7 kg: 250 mg/día; niños de 7-14 kg: 500 mg/día; niños de 14- 27 kg: 1 g/día; niños con peso superior a 27 kg y adultos: 2 g/día, en todos los casos por 7 días. En casos severos, re-petir la terapia después de 1 semana. - Ascaridiasis: E n adultos: 3-5 g/día por 2 días; en niños: 75 mg/kg/día (hasta un máximo de 3,5) por 2 días. En casos severos, repetir la terapia después de 1 semana. REACCIONES ADVERSAS Gastrointestinales: Náusea, vómitos, cólicos, diarrea. Hematológicas: Hemólisis. Reacciones de hipersensibilidad: Urticaria, broncoespasmo, síndrome de StevensJohnson, angioedema. Neurológicas: Cefalea, mareos, somnolencia, nistagmo, vértigo, confusión y con- P tracciones clónicas en pacientes con trastornos neurológicos o con anormalidades renales, alteraciones del EEG, trastornos de la memoria. Otras: Incoordinación muscular, visión borrosa, artralgia, lagrimeo, rinorrea. INTERACCIONES 2ed. 2004 * Formulario TerapÈutico Nacional 481 CONTRAINDICACIONES/ PRECAUCIONES Contraindicada en pacientes con hipersensibilidad al fármaco, insuficiencia hepática y/o renal, trastornos convulsivos y en el embarazo. Usar con precaución en pacientes con desnutrición o anemia. Debido al potencial neurotóxico, se debe evitar los tratamientos prolongados o que sobrepasen los lapsos recomendados. Si ocurren reacciones severas, sobre todo del SNC, debe suspenderse el tratamiento de inmediato. No se ha establecido la seguridad de su uso durante la lactancia. FARMACOCINÉTICA Después de la administración oral, se absorbe rápidamente en un 80% generando niveles séricos máximos en 1-3 horas. Las concentraciones plasmáticas se incrementan cuando se administra con comidas ricas en carbohidratos. Se une a las proteínas plasmáticas en un 80% y se distribuye ampliamente en el organismo. Sufre metabolismo de primer pasaje. Se metaboliza en el hígado generando metabolitos inactivos que se excretan en un 80% por vía renal en 24 horas. La vida media de eliminación es de 4-5 horas. SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INDICACIONES Tratamiento de infecciones causadas por tremátodos, céstodos y por todas las especies de Schistosoma que parasitan al ser humano. INFORMACIÓN AL PACIENTE: Administrar 1 hora antes o 2 horas REGIMEN DE DOSIFICACIÓN Se administra solamente por vía oral. - Schistosomiasis en adultos y niños mayores de 4 años: 50-60 mg/kg por 1 día de tratamiento, divididos en 3 dosis a intervalos de 4-6 horas. - Cestodiasis: Para adultos y niños: Dosis única de 5-10 mg/kg. - Neurocisticercosis: Para adultos y niños: 50-60 mg/kg/día divididos en 3 dosis iguales, por 15 días, en combinación con corticosteroides para minimizar la incidencia y PRAZICUANTEL FARMACOLOGÍA Antihelmíntico. Altera la permeabilidad de la membrana celular del parásito a los iones calcio dando origen a una contractura muscular sostenida que conduce a la parálisis y muerte del helminto. También se ha descrito un efecto de vacuolización, desintegración del tegumento y 482 Formulario Terapéutico Nacional * 2ed. 2004 vómito, cólicos, urgencia defecatoria y diarrea. Hematológicas: Eosinofilia. Hepáticas: Elevación de los niveles de las aminotransferasas. Neurológicas: Mareos, dolor de cabeza y fatiga. En pacientes con neurocisticercosis puede ocurrir un síndrome neurológico severo caracterizado por fiebre, meningitis, aracnoiditis, hipertensión intracraneal y convulsiones, que se cree es debido a una reacción infla-matoria como resultado de las larvas muertas. Reacciones de hipersen-sibilidad: Erupciones, prurito, urticaria. Otras: Mialgia, artralgia y sudoración. debe ser tratada con este compuesto. INTERACCIONES El prazicuantel puede aumentar los niveles plasmáticos del albendazol, con el consecuente riesgo de reacciones adversas. La carbamazepina, la fenitoína, la cloroquina y la dexametasona reducen las concentraciones plasmáticas del prazicuantel. La cimetidina, por el contrario, incrementa los niveles plasmáticos del prazicuantel y la vida media de eliminación. BENZOATO DE BENCILO CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al medicamento, historia de convulsiones y en niños menores de 4 años o peso inferior a 20 kg. No se conoce la seguridad de su SOBREDOSIS Vaciamiento gástrico, carbón activado y catártico salino, si la ingestión es reciente. Manejo sintomático y terapia de soporte. INFORMACIÓN AL PACIENTE Administrar con un vaso grande de agua y con las comidas. El medicamento puede alterar la capacidad para realizar actividades que requieren alerta, concentración y destreza motora; por tanto, el paciente no debe manejar ni operar maquinaria durante el día del tratamiento y el día FARMACOLOGÕA Ectoparasiticida sintético que ejerce un efecto tóxico contra el ácaro que produce la sarna (Sarcoptes scabiei) y, en menor grado, contra el piojo de la cabeza (Pediculus capitis). No se conoce con precisión su mecanismo de acción, pero se piensa que puede P actuar sobre el sistema nervioso del parásito, resultando en su muerte. FARMACOCINÉTICA No se dispone de información sobre la absorción sistémica del benzoato de bencilo posterior a su aplicación tópica. En caso de ser absorbido, es rápidamente hidrolizado a ácido 2ed. 2004 * Formulario TerapÈutico Nacional 483 hipúrico, el cual es excretado en la orina. INDICACIONES Tratamiento tópico de la escabiosis y de la pediculosis. RÉGIMEN DE DOSIFICACIÓN Se emplea tópicamente como loción al 25%. - Escabiosis: El paciente debe bañarse con agua y jabón antes de aplicar el producto, frotando bien para remover escamas y costras, y después secar la piel con una toalla limpia. Aplicar el producto con masaje suave hasta cubrir con una capa fina y uniforme toda la superficie corporal desde el cuello hasta los pies, evitando el contacto con la cara, los ojos, membranas mucosas y el meato uretral. Una vez que se ha secado la loción, aplicar una segunda capa y mantenerla, sin bañarse, por 24-48 horas. Transcurrido ese tiempo, remover el producto con abundante agua y jabón. En caso de nuevas lesiones repetir el tratamiento 7-10 días después. - Pediculosis: Aplicar la loción en el área afectada evitando el contacto con los ojos y remover 12-24 horas después con abundante agua y jabón. Posterior al secado, se debe usar un peine de dientes finos para remover cualquier resto del 484 cutánea. INTERACCIONES No aplicable. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en casos de inflamación cutánea severa o de agrietamiento de la piel o cuero cabelludo. En caso de hipersensibilidad al producto, debe descontinuarse y ser removido inmediatamente con agua y jabón. Toda la ropa y lencería usadas por el paciente deben lavarse con agua caliente para evitar la reinfección. Usar con precaución en el embarazo. SOBREDOSIS No existen reportes de toxicidad por sobredosis del medicamento. En caso de ingestión accidental de cantidades elevadas del fármaco, practicar medidas orientadas a evitar su absorción gastrointestinal y, en caso necesario, manejo sintomático. Uso de anticonvulsivantes en caso de convulsiones. INFORMACI”N AL PACIENTE No ingerir. Informar detalladamente al paciente la técnica adecuada para el Formulario Terapéutico Nacional * 2ed. 2004 CROTAMITÓN FARMACOLOGÕA Ectoparasiticida sintético que ejerce un efecto tóxico contra el ácaro que produce la sarna (Sarcoptes scabiei). No se conoce con precisión su mecanismo de acción. FARMACOCINÉTICA No se dispone de información sobre la absorción sistémica del crotamitón después de la aplicación tópica. INDICACIONES Tratamiento tópico de la escabiosis (sarna). RÉGIMEN DE DOSIFICACIÓN El crotamitón se emplea tópicamente en crema y loción al 10%. El paciente debe bañarse con agua y jabón antes de aplicar el producto, frotando bien para remover escamas y costras, y después secar la piel con una toalla limpia. Aplicar el producto con masaje suave hasta cubrir con una capa fina y uniforme toda la superficie corporal desde el cuello hasta los pies, evitando el contacto con la cara, los ojos, membranas mucosas y el meato uretral. Aplicar una segunda capa del producto al día siguiente y mantenerla, sin bañarse, por 48 horas. Transcu-rrido ese tiempo bañarse con abun-dante agua y jabón para remover el medicamento. En caso de nuevas lesiones repetir el tratamiento reacciones de hipersensibilidad cutánea. INTERACCIONES No aplicable. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en casos de hipersensibilidad al medicamento y en casos de inflamación cutánea severa o piel agrietada. En caso de hipersensibilidad, el medicamento debe descontinuarse y ser removido inmediatamente con abundante agua y jabón. Toda la ropa y lencería usadas por el paciente deben lavarse con agua caliente para evitar la reinfección. El prurito causado por la infección, que en ocasiones persiste por varios días aún después de usado el producto, no debe ser interpretado como una falla del tratamiento o como una circunstancia que justifique repetir su aplicación. Usar en el embarazo solamente si es claramente necesario. No se ha establecido la seguridad del uso del P crotamitón en niños. SOBREDOSIS No existen reportes de toxicidad por sobredosis del producto. En caso de ingestión accidental de cantidades elevadas, practicar medidas orientadas a evitar la absorción gastrointestinal y, en caso necesario, tratamiento sintomático. 485 2ed. 2004 * Formulario TerapÈutico Nacional alergia, suspender la aplicación, remover con agua y jabón y notificar al LINDANO FARMACOLOGÍA: Hidrocarburo cíclico halogenado con actividad ectoparasiticida y ovicida específica contra el ácaro productor de la sarna (Sarcoptes scabiei) y contra los piojos que parasitan al hombre (Pediculus humanus capitis, Pediculus corporis y Phthirus pubis). El fármaco penetra el exoesqueleto del parásito y ejerce un efecto estimulante sobre su sistema nervioso que genera convulsiones y produce la muerte. FARMACOCINÉTICA: Después de la aplicación tópica se absorbe parcialmente y en forma lenta a través de la piel intacta, aproximadamente un 9%; la absorción puede ser mayor cuando se aplica sobre la cara, cabeza, axilas, cuello, escroto y piel lesionada. Por ser liposoluble, después de absorbido puede concentrarse en el tejido adiposo y en el cerebro. Se metaboliza en el hígado y se excreta en la orina y en las heces. La vida media de eliminación es de 1821 horas. INDICACIONES Tratamiento tópico de la escabiosis y la pediculosis capitis. 486 secarse con una toalla limpia. - Escabiosis: Aplicar una capa fina y uniforme con masaje firme en todo el cuerpo, desde el cuello a los pies, evitando el contacto con la cara, los ojos, membranas mucosas y el meato urinario; mantener por 8-12 horas y remover después con agua y jabón. - Pediculosis: Lavar primero la cabeza con un champú ordinario, enjuagar y secar bien; aplicar entonces con masaje firme y mantener por 4 minutos (evitando, en lo posible, el contacto con los ojos, la nariz y la boca) y quitar después con abundante agua. Posterior al secado, se debe usar un peine de dientes finos para remover cualquier resto del parásito, sus crías y sus huevos. Aunque, por lo general, el uso como escabicida o pediculicida resulta efectivo con una sola aplicación, podría repetirse después de 1 semana en caso de nuevas lesiones o evidencia de reinfección. REACCIONES ADVERSAS Cuando se emplea en forma apropiada el producto resulta bien tolerado y, por lo general, seguro. Sin embargo, se ha descrito reacciones de irritación, erupciones, prurito y sensibilidad. Existen reportes aislados que refieren convulsiones aún con la aplicación de las dosis recomendadas. La Formulario Terapéutico Nacional * 2ed. 2004 INTERACCIONES Los aceites pueden aumentar la absorción; por lo tanto, debe evitarse el uso simultáneo de cremas, ungüentos o aceites. CONTRAINDICACIONES/ PRECAUCIONES Contraindicado en pacientes con hipersensibilidad al producto, en niños menores de 2 años, en el embarazo y durante la lactancia (o suspender temporalmente la lactancia hasta 4 días después del tratamiento). Toda la ropa y lencería usadas por el paciente deben lavarse con agua caliente para evitar la posibilidad de reinfección. En caso de hipersensibilidad, el medi-camento debe descontinuarse y ser removido inmediatamente de la piel con agua y jabón. No debe usarse sobre áreas lesionadas, inflamadas o escoriadas. Usar con precaución en ancianos y en personas con ante-cedentes convulsivos. contacto recomendado. Explicar las medidas de higiene asociadas al tratamiento, así como la importancia de notificar al médico si se presenta alguna reacción o sintomatología inusual durante o después del SOBREDOSIS La aplicación inapropiada, en cantidades excesivas, o por períodos mayores al recomendado puede generar toxicidad del SNC y riesgo de convulsiones. Lo mismo aplica cuando se ingiere de manera accidental o intencional. En tales casos, proceder al vaciamiento gástrico, administración de carbón activado y catártico 2ed. 2004 * Formulario TerapÈutico Nacional P 487