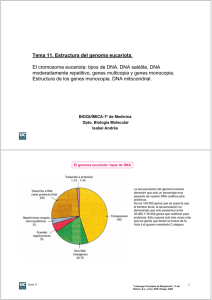
Descargar
Anuncio

1. INTRODUCCIÓN A LA BM DE MICROORGANISMOS -Conceptos básicosMicroorganismo: conjunto de seres vivos que se caracterizan por tener un tamaño pequeño de modo que la mayoría de ellos no son visibles a simple vista, teniendo gran sencillez en su estructura y organización. Su importancia radica en que han dado origen a una rama de la biología dedicada a su estudio: la microbiología. -Diferencias entre eucariotas y procariotas- -Diferencias entre los Phylum de Archaea y BacteriaLas Archaea son parecidas a Bacteria en la mayoría de los detalles de la estructura y del metabolismo de la célula, pero difiere sobre todo en la composición de los lípidos de la membrana celular, estructura de flagelos y en los aspectos de la maquinaria genética: En cuanto a la transcripción y traducción genéticas. La maquinaria de transcripción de Archaea es generalmente como en Bacteria y Eukaria, con 70S ribosomas. Los genes se colocan en racimos co-transcritos llamados operones (los genes de Eukaryas se transcriben generalmente de forma separada en lugar de en racimos). También como en las Bacterias, se unen transcripción y transcripción (es decir, ocurren simultáneamente), y el fracaso de un mRNA al ser traducido causa que la ARN polimerasa aborte la transcripción. Sin embargo, en muchos sentidos la traducción en Archaea es como en Eukaria. La traducción se comienza con metionina y es inhibida por la toxina de la difteria, como en los ribosomas de los eukarya, pero no es afectado por la mayoría de antibióticos inhibidores de la traducción bacteriana (Estreptomicina y Cloramfenicol). La ARN polimerasa es una enzima crucial requerida para la síntesis de nuevas moléculas de ARN. Las Eubacterias tienen un solo tipo de ARN polimerasa para la transcripción de todos los genes de la célula y utilizan Formil-metionina como iniciador para la síntesis de proteína. Archaea, como las Bacterias, tienen una sola ARN polimerasa que transcribe todos los genes. Sin embargo, las ARN polimerasas de Archaea son como los de los eucariotas (contienen 3 o 4 subunidades grandes y muchas pequeños Sólo algunos genes de Archaea contienen intrones en su DNA, en contraste con la falta completa de intrones en Eubacteria, y la presencia de intrones en todos los genes de los eucariotas. Muchos GENES ARNr y ARNt de las Archaea poseen INTRONES únicos que no se encuentran ni en eucariontes ni en bacterias. En cuanto a los lípidos: las Archaea poseen FOSFOLÍPIDOS basados en GLICEROL, pero con 3 características que los hacen únicos: 1) ESTEREOQUÍMICA del GLICEROL: Los lípidos de las Archaeas son únicos porque la estereoquímica del glicerol es opuesta a la encontrada en bacterias y eucariontes. Esto evidencia fuertemente un camino biosintético independiente. 2) ENLACES ÉTER: La mayoría de BACTERIAS y eucariontes tienen membranas compuestas principalmente por GLICEROL que se une a las cadenas laterales mediante ENLACES ÉSTER, mientras que en las Archaea la unión es por ENLACES ÉTER. Aun cuando alguna bacteria tiene lípidos ligados por éter, la estereoquímica del glicerol sigue siendo de tipo bacteriano. Estas diferencias pueden ser debidos por una adaptación de las Archaea a los ambientes hipertermófilos. Sin embargo, hay que hacer notar que incluso las Archaea mesófilas tienen lípidos ligados por éter. 3) CADENAS ISOPRENOIDES: Las cadenas laterales de las membranas celulares también tiene una composición distintiva en Archaea, pues son cadenas ISOPRENOIDES (compuestas de 20 o incluso 40 átomos de carbono), mientras que en Bacteria y Eukarya están compuestas por ÁCIDOS GRASOS (usualmente de 16 a 18 átomos de carbono). Las cadenas isoprenoides son comunes en la industria del caucho y como componente de algunas vitaminas comunes en bacterias y eucariontes, sin embargo, solo las Archaea incorporan estos compuestos a sus lípidos celulares. En algunas Archaea la cadena de isoprenoide C-40 es lo suficientemente larga para atravesar el espesor de la membrana, uniendo el fosfato de glicerol de ambos extremos y formando una monocapa (en lugar de la bicapa habitual). Esta adaptación hace más estable y resistente la membrana, siendo habitual, por lo tanto, en las Archaea hipertemófilas. Las Archaea no tienen paredes de PEPTIDOGLUCANOS como en las BACTERIAS, aunque en un grupo de metanógenos contiene PSEUDOPEPTIDOGLUCANOS (se diferencia en la composición y en los enlaces). Tampoco tienen paredes de CELULOSA como las plantas ni de QUITINA como los hongos. Las paredes celulares de otras Archaea carecen tanto de peptidoglicano como de seudopeptidoglicano y se componen de POLISACÁRIDOS, GLUCOPROTEÍNAS o PROTEÍNAS. El tipo más común de pared es la capa superficial paracristalina (CAPA S) que está formada por proteína o glucoproteína, generalmente de simetría hexagonal. Las Archaea también tienen flagelos que son diferentes en composición y desarrollo de los flagelos de las bacterias. El flagelo bacteriano es un sistema de secreción de tipo III modificado, mientras que los flagelos de las Archaea se asemejan a los pilis de tipo IV, los cuales utilizan un sistema de secreción algo similar pero no idéntico al sistema de secreción de tipo II 2. BIOLOGÍA MOLECULAR DE LEVADURAS -Introducción. HistoriaLas levaduras son hongos no filamentosos, seres unicelulares que viven de manera independiente (individualmente o formando colonias, lo cual no es estrictamente una organización pluricelular). La levadura fue el primer microorganismo doméstico: hace más de 6000 años, durante la edad de los sumerios, era empleada para obtener bebidas alcohólicas. En Roma, intentaron explicar la fermentación desde la mitología, al carecer del conocimiento necesario para descubrir sus mecanismos. Aun así, realizaron una aproximación bastante cierta: según la leyenda, el dios del vino Baco nació de una protuberancia de la pierna de Zeus (sería una ancestral “gemación”). Ya en la edad moderna, Pasteur hizo los primeros estudios sobre levadura, que constituyeron la “ciencia básica”: estableció el sistema bioquímico de metabolismo de la fermentación. En las últimas décadas, el mayor avance en el estudio de levaduras ha sido la capacidad para introducir DNA de otros organismos en las levaduras. Tanto bacterias como las propias levaduras poseen pequeños ADN-circulares llamados plásmidos. Este ADN extracromosómico le confiere a las levaduras propiedades especiales (como resistencia a antibióticos o ventajas sobre otras cepas que compiten con ellas). Otro hito importante en la evolución de la investigación en levaduras fue la secuenciación del genoma de S. cerevisiae a partir de este momento se estableció como organismo modelo en la Biología Molecular. -Aplicaciones de la Biología Molecular de levadurasLa biotecnología de levaduras abarca variados campos: industrias de la fermentación (alcohólica), tecnología química (como la obtención de metabolitos secundarios, por ejemplo los isoprenoides que dan lugar a pigmentos o aromas), industria alimentaria, producción de fármacos y vacunas, investigación biológica (función como organismo modelo por ejemplo en el estudio de la respuesta transitoria a estímulos) y biomédica (efectos de drogas sobre levaduras, estudio de enfermedades genéticas que afecten al desarrollo neuronal…) Algunos ejemplo de productos que se purifican a partir de levaduras (normalmente con una producción con mayor rendimiento que en bacterias): productos procariotas (fragmentos de toxina del tétanos, streptoquinasa), antígenos de superficie de virus, antígeno contra la malaria, productos animales (uridina, interferón porcino, interleucina), hormonas humanas (insulina, hormonas de crecimiento), factores de crecimiento humanos, proteínas de la sangre humana (hemoglobina, factores VIII y XIII), enzimas humanos…. -Tipos de levaduras que funcionan como organismo modeloLas principales levaduras que sirven como organismo modelo son Candida albicans, Saccharomyces cerevisiae y Schizosaccharomyces pombe: Saccharomyces cerevisiae: levadura de gemación (budding yeast). Poseen formas apepinadas, con una yema que poco a poco generará una célula hija. En S. cerevisiae puedo saber cuantas hijas ha tenido una célula contando sus cicatrices de gemación. La más utilizada en investigaciones, con características mejores que S. pombe (podemos diferencia célula madre de la hija por las cicatrices y diferente tamaño celular) y además con una ventaja sobre Candida, ya que tiene rasgos similares a ella, pero sin ser patógena. Se reproducen de forma asexual por gemación. En condiciones muy determinadas la forma diploide es capaz de reproducirse sexualmente. En estos casos se produce la meiosis en la célula formándose un asca que contiene cuatro ascosporas haploides. S. cerevisiae es uno de los modelos más adecuados para el estudio de problemas biológicos. Es un sistema eucariota, con una complejidad sólo ligeramente superior a la de la bacteria pero que comparte con ella muchas de sus ventajas técnicas. Además de su rápido crecimiento, la dispersión de las células y la facilidad con que se replican cultivos y aíslan mutantes, destaca por un sencillo y versátil sistema de transformación de ADN. Por otro lado, la ausencia de patogenicidad permite su manipulación con las mínimas precauciones. Características del genoma de S. cerevisiae: DNA genómico -12.8 MB en 16 cromosomas. -6000 ORF (cada 2kb un gen) -120 rRNA (casi todos en el cromosoma 12) -274 tRNA DNA extracromosómico -Plasmido 2micras (6Kb). -Killer plasmid. -Mitocondrial (75kb) Schizosaccharomyces pombe: Levadura de fisión (fission yeast). Forma más alargada, para su división sufren un progresivo alargamiento y forman un septum que separará la célula madre de la hija, estando la madre más envejecida que su descendiente. En el estudio del envejecimiento celular, por ejemplo, no conviene utilizar S. pombe, ya que al no presentar cicatrices consecuencia de anteriores divisiones, no podemos diferenciar la célula madre de la hija. Levadura como organismo modelo en Biología Molecular -Es un organismo eucariota unicelular, fácil de manipular genéticamente. -Es muy fácil trabajar con ella, económica. -Existen numerosas mutaciones y cepas que permiten estudiar variabilidades. -Fue el primer organismo secuenciado (1996). -Se emplea en el estudio de procesos básicos (transcripción, traducción, replicación, ect…). Rogert Konberg, premio Nobel 2006 por su trabajo en transcripcion -Se emplea en el estudio de procesos complejos conservados (envejecimiento celular, ciclo celular, apoptosis, ect..). Lee Hartwell, Paul Nurse, y Tim Hunt premio Nobel 2001 por su trabajo en la identificacion de componentes del ciclo celular (identificaron parámetros del ciclo celular importantes en el tratamiento del cáncer) -Se emplea como modelo para la biología de sistemas. Generación -ómica y post -ómica -Ciclo vital de las levaduras- Diferenciarlo del concepto de ciclo celular. Las levaduras viven como organismos diploides. En condiciones de estrés pueden sufrir meiosis y formar individuos haploides con el objetivo de luego poder cruzarse y por recombinación obtener al azar posibles ventajas frente a dicho estrés. Este hecho constituye una ventaja en su uso para investigación (se trabaja siempre con individuos en forma haploide), donde dependiendo del laboratorio se emplean levaduras de un sexo (math α) u otro (math β). -Ciclo celular de las levadurasLa división celular sólo representa una pequeña fracción del ciclo vital de una célula (en general una hora). La mayor parte de su vida la célula permanece en interfase, período en el que como mencionamos se duplica la masa y se replica el ADN. -En S. pombe: G1 se alcanza el tamaño de célula madura, S síntesis del ADN, durante G2 la célula se alarga y toma su forma característica y en la fase M se produce la mitosis, con la formación del septum. S. Pombe se utiliza más en procesos de estudio de la diferenciación celular y ciclo celular aplicado a mamíferos. -En S. cerevisiae la G2 desaparece, y la síntesis del ADN está solapada con la mitosis, dando lugar a una célula hija pequeña y una madre grande. Se emplea en el estudio de los mecanismos moleculares del funcionamiento eucariota. Puntos de control del ciclo celular: cuando las células tienen un problema como falta de nutrientes o calor y frío extremos (estrés ambiental), estos puntos de control indican donde tiene que parar el ciclo celular. Favorecen la sincronización del ciclo celular en la misma fase. Para estudios del ciclo celular se utiliza S. pombe, que en ese aspecto es muy similar a los humanos. Screaning de mutantes en levadura permitió identificar los componentes del ciclo celular -Arquitectura celular de levadurasImagen de tomografía con electrones muestra una célula completa de levadura: podemos distinguir la membrana celular, microtúbulos y las vacuolas ligeras, el núcleo, vacuolas más grandes y vesículas diversas, mitocondrias… Macromoléculas presentes en levaduras: Cicatriz de célula hija o Pared celular: Levaduras tienen pared celular, por eso se utilizan como modelo en plantas, sobre todo en temas de ósmosis (estrés salino). Constituye un 20-30% del peso celular. - Protege de la deshidratación y de los cambios de osmolaridad. - Se puede digerir con liticasa para formar protoplastos (purificaciones) - La composición altera la capacidad de flocular. - La distribución de los componentes no es homogénea Uniones entre la membrana y la pared celular hacen que sea más estable y no se pierda. Si degradamos la pared celular obtenemos el protoplasto (muy sensibles a cambios osmóticos) o Membrana celular: -Entre la pared celular y la membrana tenemos el periplasma. -Composición de lípidos de membrana en levaduras: destaca el ergosterol (de la familia del colesterol, único en levaduras). Al ser específico de este grupo de microorganismos, se utiliza como blanco en las drogas contra el patógeno de tipo levadura Candida albicans. Estas drogas son en su mayoría azoles que atacan al ergosterol drogas capaces de eliminar de forma selectiva la levadura. -Las hebras de beta glucano y quitina, además de las manoproteínas de la pared celular les da la propiedad de floculación: se puede producir bajo determinadas condiciones, se produce una unión entre células y requiere calcio (reversible con EDTA). Si por ejemplo, centrifugamos, algunas células decantan. Los iones de calcio mantienen estable la unión entre la pared y la membrana celular. Hay cepas que floculan solas. Si añadimos EDTA y quitamos calcio vuelven al aspecto normal. Determinadas cepas que presentan mutaciones en sus manoproteínas, si floculan se sedimentan lo cual no interesa en muchas ocasiones para su uso industrial. -Bud scar: cicatrices que quedan en las levaduras en los puntos donde ha habido gemación de células hijas, se pueden contar. Manipulando S. cerevisiae somos capaces de ver qué genes podemos manipular para acortar el envejecimiento replicativo. o Peroxisomas: Realizan la beta-oxidación de los ácidos grasos. Como consecuencia dan un efecto negativo: traspaso del H del NADPH al NADH, lo cual causa un envejecimiento celular acelerado. Si bajamos concentración del NADPH quitamos un cofactor importante que detoxifica la célula, sin él, las proteínas de estrés oxidativo no pueden actuar. o Mitocondrias: -Contiene unas 500 proteínas, la mayoría codificadas en el núcleo. -El genoma contiene muchos intrones, lo cual no coincide con la teoría endosimbionte. -DNA mitocondrial se encuentra organizado formando los nucleoides, que cambian de conformación. -Es una estructura dinámica. Son muy plásticas, van fusionándose y separándose. -Su morfología y biogénesis depende de las condiciones ambientales. -Presencia de mutantes rho, que pierden DNA mitocondrial y al final se van degradando: La ventaja de trabajar con mitocondrias de levaduras es que cuando crecen en un medio con la suficiente glucosa reprimen la respiración y son prácticamente inactivas. Si eliminas la glucosa del medio empiezan a respirar. Muchas enfermedades se producen por problemas en la respiración, y el envejecimiento celular de la respiración. Si se muere el DNA mitocondrial, el resto de la célula queda inutilizada. -Metabolismo de las levadurasLas levaduras del tipo S. cerevisiae son, por norma general fermentadores aerobios facultativos, es decir, presentan una respiración limitada, fermentación aeróbica y anaeróbica y su crecimiento anaerobio es facultativo (pueden dividirse en anaerobiosis: si yo pongo glucosa en el medio, van a evitar siempre el realizar la respiración, lo cual evitará un prematuro envejecimiento). S. cerevisiae prefiere fermentar, aun así, si la concentración de glucosa es baja, utiliza el alcohol de la fermentación como fuente de carbono para la respiración. Otras levaduras, como S. pombe, no crecen en anaerobiosis, y si eliminamos el oxígeno no son capaces de vivir. Como ya hemos dicho, a levadura Saccharomyces cerevisiae en presencia de glucosa y oxigeno, primero fermenta la glucosa y produce etanol, hasta que los niveles de glucosa bajan. Muchos de los genes de respiración están reprimidos en presencia de glucosa (ruta de transmisión de señal snf1 y rtg). Se pierde represión y se activan los genes de la respiración (ruta retrógrada). Rgt1 es muy importante, y se encuentra conservado tanto en mamíferos como en levaduras. Aunque la acumulación de etanol en determinadas concentraciones puede llegar a ser tóxica, para algunas cepas. Mediante manipulación genética se ha buscado alterar la sensibilidad y el componente a fermentar (producción de bioetanol). Otra parte importante del metabolismo de levaduras es la Gluconeogénesis: en algunas ocasiones se requiere la síntesis de polisacaridos complejos. Con la gluconeogénesis se consigue la síntesis de glucosa a partir de piruvato, lo que requiere ATP y el poder reductor de NADH. Se requiere para: -Síntesis de polisacaridos estructurales de la pared celular. -Síntesis de carbohidratos de reserva (glucógeno y trealosa) Por último, cabe destacar la biosíntesis de aminoácidos en levaduras, en los que los metabolitos de las otras rutas de síntesis y degradación (de glúcidos o lípidos) se emplean para formar parte de la síntesis de los aminoácidos que darán lugar a proteínas. Las levaduras se emplean para producir aminoácidos de interés industrial en alimentación (glutamato), fármacos… -BIOTECNOLOGÍA DE LEVADURA Clases de vectores para Biología molecular de levadura S. cerevisiae Las levaduras contienen plásmidos (moléculas circulares de DNA, de múltiples copias o solo una). Estos plásmidos tienen capacidad de replicación, y no aparecen en el resto de eucariotas superiores. Hay plásmidos que podemos introducir para producir proteína purificada de forma eficiente, o vectores integrativos que se introducen por recombinación homóloga en los plásmidos. Aquí se presentan algunos de los plásmidos y vectores más importantes en levaduras (no aprender detalles): YIp-Yeast integrative plasmid YEp-Yeast episomal plasmid (YRp-Yeast replicative plasmid) YCp-Yeast centromeric plasmid MCS = Multiple Cloning Site, sitios de restricción para subclonar fragmentos de DNA. 2m = secuencia del plásmido natural 2 micron de levadura, necesario para mantener el plásmido en varias copias. ARS = autonomously replicating sequence. CEN = secuencia de centrómero, sirve para mantener el plásmido como minicromosoma (1 o pocas copias).URA3, LEU2, TRP1 son marcadores de levadura, complementan auxotrofias en la cepa transformada.Todos los plásmidos contienen secuencias de E. coli (amp) para mantenerlos en bacterias. Para saber si un plásmido se ha introducido correctamente en la célula de interés utilizamos los marcadores de selección: - Complementan auxotrofías como LEU2, TRP1 etc. - Son detectables en ensayos enzimáticos (lacZ, luciferasa) - Pueden ser resistencias a antibióticos (KAN, cloramfenicol, etc) Por ejemplo, la Kanamicina (KAN) es un marcador de selección negativa de resistencia a un antibiótico. LEU2 es un marcador de selección negativa. Se elimina uno de los genes para la síntesis de leucina. Como la levadura a la que introduzco el plásmido no tiene el gen LEU2 y el plásmido de interés lleva acompañado el LEU2, haciendo crecer las levaduras en un medio sin leucina, solo podrán sobrevivir aquellas que incorporan el plásmido de interés mutantes auxótrofos. Promotores constitutivos e inducibles. Un promotor constitutivo es un conjunto de secuencias de ADN que permiten que los genes bajo su control estén activos en todos los tejidos y en todo momento, a diferencia de "promotores regulados", que permiten la activación de genes únicamente en ciertas condiciones y en determinados órganos.El uso de promotores constitutivos e inducibles permite la expresión (sobreexpresión) controlada de proteínas en levadura. Promotores de otros organismos son funcionales (Plantas, humanos etc.). Normalmente son promotores de enzimas glicolíticos, que situamos antes del DNA poli-linker (en el que vamos a introducir el gen de interés). Silenciado siempre que no pongamos la galactosa o el mineral cobre útil en la producción de proteínas tóxicas. Los promotores fuertes se utilizan para conseguir una producción elevadas de proteínas concretas, aunque una fabricación masiva puede llegar a ser negativa (toxicidad) Transformación de levadura. La transformación nos permite introducir el plásmido de una forma bastante eficiente. 1) Transformación de levadura por el método químico Principio: Permeabilizar temporalmente las células por tratamiento con Acetato de litio (LiAc) y un choque térmico (42oC). En la transformación ayuda un alto porcentaje de poli etilenglicol (PEG) y la presencia de “carrier DNA” (DNA inespecífico). Cultivo de levadura Tratar células con 0.1M LiAc Mezclar con DNA (plasmídico y “carrier”) Añadir PEG Choque térmico (20min 42oC) 2) Transformación de levadura por el método de electroporación Principio: Aplicación de un campo eléctrico en pulsos permeabiliza temporalmente a las células y facilita la entrada de moléculas de DNA, ventaja del método: alta eficiencia. Cultivo de levadura Plásmido Identificación de transformantes en placas selectivas Vectores para fusiones GFP en levadura Expresión de proteínas recombinantes de una variedad de organismos de acogida se ha convertido en una práctica común. Sin embargo, la producción de proteínas debidamente dobladas con alto rendimiento y pureza no siempre se logra. Cuestiones como probabilidad, solubilidad, la estabilidad de la proteína, transcripción y traducción de eficiencia, postraduccionales procesamiento, la secreción, la carga metabólica y otras respuestas de estrés resultante de la producción de proteínas recombinantes, así como la purificación de proteínas, hay que abordarlas con el fin de obtener las proteínas biológicamente activas recombinante con alta pureza y rendimiento. En este sentido, fluorescentes reporteros genéticamente codificados ofrecen nuevas oportunidades para enfrentar estas cuestiones. Fusiones de proteínas con péptidos fluorescentes permite la detección de la proteína in vivo. Por microscopía fluorescente se puede localizar la proteína en la célula viva. Existen sistemas de plásmidos de levadura para clonar cualquier gen en fusión con GFP (green fluorescent protein). La GFP constituye una herramienta para visualizar proteínas en células vivas La expresión puede ser regulada o de forma constitutiva en copia única o multicopia. Además permiten fusionar la proteína en el extremo N- o C-terminal. Análisis global de la localización de proteínas por GFP Rox3 nuclear Nic96 membrana nuclear Pho86 ER Hof1 bud neck Ilv6 mitocondria Erg6 lipid particle Zonas de recombinación homóloga en los extremos de la zona del gen (azul y rojo). Fusión de genes con epítopos en levadura Empleado para introducir epítopos, con TAP detrás: pequeño péptido que nos va a servir para inmunoprecipitar, permite captar la proteína que nos interesa en mezclas complejas. El sistema del doble híbrido de levadura El sistema del doble híbrido permite identificar la interacción de dos proteínas en la célula viva.Aprovecha el caracter modular de los factores específicos de transcripción como Gal4 Utilizando lacZ o HIS3 como reporteros. Tenemos un dominio de activación que va a activar la transcripción aprovechar Gal-BD que tiene tanto dominio de unión como de activación, se separan los dominios en dos plásmidos distintos Disrupción de genes en la levadura Saccharomyces cerevisiae Integración de un marcador por recombinación homóloga Reciclaje del marcador posible con el sistema Cre-loxP Colecciones de cepas de S. cerevisiae La combinación de la manipulación genética exacta con el conocimiento de la secuencia entera del genoma nuclear permite construir colecciones de cepas que contienen representantes para (casi) cada gen de la levadura: Delecciones de todos los genes -Haploides: genes no esenciales -Diploides: todos los genes Fusiones con GFP Fusiones con epítopos (myc, HA, TAP) Gen X HA Análisis de complejos KAN protéicos al nivel Análisis funcionalde los fenotipos Análisis genómico de los mutantes de levadura genómico localisación del GFP del proteoma Principio: Cuantificación del proteoma crecimiento de cada mutante en distintas condiciones ambientales, comparación con la cepa silvestre. Las condiciones pueden ser estreses: calor, drogas, ayuno de nutrientes, estrés salino, pH, oxidativo etc. El análisis permite encontrar funciones de genes al nivel genómico. Banco de datos: Prophecy (profiling of phenotypic characteristics in yeast. Análisis global de la expresión génica en levadura (microarray) Condición A Condición B Purificar mRNA Síntesis de cDNA por transcriptasa reversa Marcaje del DNA con fluorescentes Cy3 Cy5 La comparación de genomas evolutivamente cercanos: Evolución de genomas: Mecanismos y dinámica Identificación de los genes que son específicos para una especie Identificación de secuencias conservadas evolutivamente: Dominios funcionales en proteínas y en promotores 3. PRINCIPIOS GENERALES DE GENÉTICA MICROBIANA -Organización de la información genéticaCaracterísticas: o o o Una única molécula circular en el citoplasma, es decir, un solo cromosoma situado en un lugar del citoplasma que recibe el nombre de nucleoide. Aun así hay excepciones como cromosoma lineal o presencia de 3 cromosomas en algunas bacterias DNA desnudo Superenrollamiento: en un cromosoma podemos encontrar zonas relajadas y superenrolladas. Dos tipos: o Negativo: realizado por la DNA girasa, enzima no presente en eucariotas (equivalente a la topoisomerasa de tipo II de eucariotas). Es importante por ejemplo en fabricación de antibióticos que son inhibidores de la actividad DNA girasa. Divide al cromosoma en varios dominios superenrollados. La relajación la llevará a cabo la topoisomerasa I. Este fenómeno de superenrollamiento afecta a la expresión génica Positivo: de signo contrario, catalizado por el enzima girasa inversa, propia de la Archaea hipertermófilas. Se cree que ayuda a evitar la desnaturalización del DNA cuando está sometido a altas temperaturas. Es un cromosoma que carece de intrones. Los intrones son fragmentos de DNA que no codifican para proteínas, rRNA o tRNA. Así en eucariotas siempre aparecen intrones y exones siendo DNA monocistrónico mientras que en procariotas el DNA se transcribe sin intrones en forma policistrónica (secuencias que van a originar más de una proteína cuando se traducen). Por tanto, la densidad codificadora del cromosoma procariota es mucho mayor que en eucariotas. En los casos en que en procariotas existen intrones, se produce un autoprocesamiento por ribozimas. o o o o Escasas o única copia de la mayoría de los genes (excepción: RNA ribosómico puede tener más de una copia). Secuencias repetitivas: secuencias muy cortas altamente repetidas. Por ejemplo en E. coli hay una secuencia de 38 pb que se repite más de 500 veces. Presencia de elementos transponibles (replicación como parte de alguna otra molécula de DNA) secuencias de inserción, transposones o virus (Mu) Presencia de pequeños DNA circular llamados plásmidos. -Replicación del DNA en procariotas – DNA circular, por ello necesaria la acción de las topoisomerasas para relajar el superenrollamiento. Un único origen de replicación (OriC). La apertura de la doble hélice se produce por la acción de la enzima helicasa. Se forma una horquilla de replicación, en la que se realiza una replicación bidireccional muy rápida estructuras theta. El resultado final son dos cromosomas circulares hijos. -Transcripción de DNAUna única RNA polimerasa procariota, es una holoenzima enorme cuyas secuencias están muy conservadas. Aun así, la RNA pol I de los eucariotas es más similar a la RNA pol de Archaeas que a la de las Eubacterias. La RNA pol Eubacteria presenta una estructura simple: El factor sigma es el responsable de reconocer el sitio donde se tiene que unir en el DNA la RNApol. Este factor es múltiple en procariotas y específico para la mayoría de los genes. Factor sigma realiza un reconocimiento holoenzimático, no exige que esté el enzima al completo al ser un factor independiente del resto del enzima. El factor sigma reconoce tanto secuencias consenso (altamente conservadas como la caja Pribnow) como promotores fuertes (los más efectivos, mayor parecido a la secuencia consenso le da mayor afinidad, lo cual es de enorme interés en biotecnología). Para que una secuencia se considere conservada deben haber 3 coincidencias de entre 3 a 5 nucleótidos como mínimo. A mayor coincidencia con secuencias consenso mayor afinidad entre el factor sigma y secuencia de DNA Terminación de la transcripción: En Bacteria: 1) Secuencias con repeticiones invertidas con un segmento central no repetido: Hacen que el RNA se cierre en bucle Bucle seguido de varias U es reconocido como zona de terminación por la RNApol, que detiene su avance cuando llega a él 2) Secuencias ricas en GC seguidas de otras ricas en AT 3) Secuencias específicas dependientes de proteínas (Rho): los enzimas Rho reconocen la secuencia específica y disocian la unión del DNA con la RNApol. En Archaea: grupos filogenéticamente muy distintos, teorías muy divergentes. Por lo general la terminación es poco conocida. Bucle seguido de una secuencia rica en AT que actúa como secuencia de terminación de tipo intrínseco. También aparecen secuencias de nucleótidos con fragmentos repetidos (poliT). Resultado de la transcripción en procariotas La transcripción da lugar a mRNA policistrónicos (sin procesamiento a RNA maduro), que contiene información para traducir proteínas relacionadas normalmente en la misma ruta metabólica concepto de operón. Operón: unidad de expresión génica completa. A menudo implica genes que codifican varios péptidos en un RNA policistrónico. Con frecuencia, la transcripción está regulada por un operador: región del DNA adyacente a la región codificante del primer gen del operón. -Traducción: síntesis de proteínas1) INICIACIÓN Codón de inicio: AUG (fundamental) o GUG (menos común). Codifican para N-formilmetionina en Bacteria, a diferencia de en eucariotas que es para metionina. Códigos alternativos ligeramente diferentes a los del código genético universal, codones que codifican aminoácidos no esperados (Micoplasma). Asignan sentido a codones habitualmente sin sentido (por ejemplo UGA, en algunas bacterias como E. coli codifican para el “aminoácido 21”, la selenocisteína. Dependiendo del contexto de los genes de alrededor se traduce en forma de selenocisteína o no) Ribosomas procariota 70S: compuestos de dos subunidades. No todas las subunidades igual de unidas o afines a este ribosoma. Complejo de iniciación: 30SrRNA + mRNA + formilmetionina tRNA + factores de iniciación (proteínas específicas) + energía en forma de GTP se forma el ribosoma 70S activo Unión 50S Secuencia de Shine-Dalgarno: de 3-9 nucleótidos inmediatamente anteriores al codón de iniciación. Presente en el extremo 5’ del mRNA y complementario al extremo 3’ del RNA 16S. Permite la traducción del mRNA policistrónico y el uso de otros codones de inicio (pero con menos frecuencia) 2) ELONGACIÓN En la elongación participan diversos factores de elongación y como fuente energética el GTP. Se produce la traslocación al sitio peptidílico P: factor de elongación específico + 1 GTP. 3) TERMINACIÓN La terminación sucede cuando se llega a un codón sin sentido o de parada (stop) que marca el fin de la traducción. Hay veces, en procariotas, que algunos otros codones que no son de terminación pueden ser reconocidos en determinadas condiciones como codón de terminación, por ejemplo el GUG que codifica para valina (incluso a veces hace de iniciación). Son procesos muy plásticos. También aparecen los factores de liberación, que causan la hidrólisis del complejo polipéptido-tRNA Terminal, y finalmente se produce la disociación del ribosoma. -Secreción de proteínasModificación post-traduccional de la proteína (se conocen menos mecanismos de modificación que en eucariotas). El más importante: Implica la participación de las SRP (Signal Recognition Particle), de 20-25 aminoácidos, reconoce la secuencia señal. Participa en el transporte a complejos de la membrana para su secreción en proteínas que tienen que ser secretadas, cuando reconocen esta secuencia, se unen a la proteína y la llevan a los complejos membranosos donde serán hidrolizados de la secuencia señal y finalmente secretados. En Bacteria: una única proteína y un RNA 4,5S. Importancia en Biotecnología manipulación de la secuencia señal en microorganismos modificados para producir proteínas heterólogas. 4. REGULACIÓN DE LA EXPRESIÓN GÉNICA. -Tipos generales de regulación de la expresión génicaEl estudio de la regulación de la expresión génica se perfila como útil en el tratamiento de infecciones en un futuro próximo (por ejemplo desarrollo de enzimas inhibidoras de la resistencia a antibióticos). En microorganismos, los enzimas no son constitutivos, sino adaptativos, es decir, solo se expresan en determinadas condiciones activando vías metabólicas complejas (el operón también ayuda a regularlas). Podemos encontrar dos modos de regulación: De la actividad enzimática: no es el mecanismo más común, la enzima es procesada (se pasa de un precursor a moldearse para dar lugar a su conformación definitiva). Se modifica la actividad de una enzima ya producida y existente. Es por lo general un proceso postraduccional que se realiza de manera muy rápida ya que obedece a mecanismos moleculares; ocurre en pocos segundos. Generalemente la modificación de la actividad se realiza por procesos reversibles; son por procesos de retroalimentación. De la cantidad (determina cuanta enzima aparece en la célula). La modificación en la producción de la enzima se produce tanto en la transcripción (modelos de E. Coli) como en la traducción. Es un proceso más lento, ocurre en minutos ya que hace falta que se alcance una concentración adecuada. -Regulación de la actividad enzimáticaSíntesis de precursores que luego se activan. Se da gracias a la existencia de inestabilidad en las enzimas, que tienen una vida media muy curta y acaban degradándose. Es posible tanto inducir, como hacer que no se produzca (traducción), como hacer que no se transcriba. 1) Activación de una molécula precursora (“procesamiento” de la enzima o modificación): esto es para enzimas que se mantienen de forma inactiva, como las enzimas (irreversible). A nivel bacteriano La Girasa: DNA Gyr 2) Degradación de la molécula de enzima: Al degradar la enzima la molécula desaparece. (irreversible también) 3) Inhibición de la actividad enzimática: (mecanismo reversible) Por retroalimentación. La aparición de una molécula en el medio ambiente inhibe la producción de esa molécula o alguno de sus derivados. Pueden ser metabolitos intermedios, aunque es menos frecuente. Se basa en: -Se trata de enzimas alostéricas, a las que se le une el metabolito y bloquean centros activos (presentan varios de ellos). -Necesitan para su funcionamiento un efector. Posibilidad de que en una ruta metabólica haya varias isoenzimas: enzimas que regulan la misma función pero que son inducidas por distintos efectores. Importancia en Microbiología Industrial: Uno de los avances es conseguir mutantes que han eliminado la retroalimentación para que la producción del producto de interés (por ejemplo aminoácidos) sea independiente de la contaminación por presencia de dicho producto en el medio. Por ejemplo el enzima DHAP tiene 3 formas análogas diferentes, cada una inhibida por diferentes aminoácidos. Se disminuirá la velocidad de la ruta pero no la llegará a inhibir. Los fenómenos de retroalimentación son comunes en rutas complejas. El producto final o un intermediario de las rutas es capaz de inhibir la enzima que acabará dando metabolismo (retroalimentación negativa). La enzima reguladora ha de ser una enzima alostérica (con dos sitios de unión; uno para el sustrato y otro para el efector). En este caso (derecha) cada isoenzima es un inhibidor de un aminoácido distinto. Modificación covalente o EPIGENÉTICA: en algunas enzimas se produce una unión de una molécula que bloquea la acción de la enzima. A la enzima se le añade alguna molécula que modifica sustancialmente su acción. Un ejemplo de este tipo de modificación es la Adenilación de Glutamino Sintasa, donde el GS está compuesto por doce subunidades y depende de la concentración de AMP-c. -Regulación de la cantidad de la enzima – Regulación de la transcripción; es un mecanismo pre-traduccional. Se debe a la interacción de proteínas reguladoras-DNA: las proteínas reguladoras están codificadas por los genes reguladores que forman parte de los operones. La regulación se produce por la combinación específica de proteínas reguladoras al DNA bloqueando (control negativo) o activando (control positivo) la transcripción. Determinado por sustancias metabólicas presentes en el ambiente. En bacterias existen diversos mecanismos, y todos ellos muy influenciados por el medio ambiente (un cambio en el medio ambiente se traduce en un cambio en la expresión de los genes); estando la mayoría de los genes regulados por más de un mecanismo. Importancia en biotecnología: modificando el medio de cultivo podemos obtener distintos productos. A. Regulación de la transcripción: 1. Represión enzimática: Las enzimas que catalizan la síntesis de un producto específico no se sintetizan si el producto está presente en el medio; tampoco lo hacen o incluso desaparecen sin un sustrato determinado. La molécula efectora (determina la acción de la represión), que es el producto final, actúa de CORREPRESOR. Es un proceso específico, como la síntesis de aminoácidos (el caso más típico la arginina), de purinas y pirimidinas, por lo que no afecta al resto del metabolismo celular (el resto de procesos metabólicos siguen a la misma velocidad). 2. Inducción enzimática: La síntesis de una enzima sólo se produce cuando su sustrato está presente, se usa para degradar dicho sustrato. Este tipo de regulación se produce en rutas catabólicas, es decir, en rutas de degradación. La célula sólo sintetiza las enzimas necesarias para la degradación de un sustrato, sólo si este sustrato está en el medio. Catabolismo de fuentes de carbono y energía. En este caso la molécula efectora, que es el producto final, actúa de INDUCTOR (suele ser el sustrato de las enzimas). Los mecanismos moleculares por los cuales se regula la transcripción a un nivel molecular son el control negativo y el control positivo. 1.Control negativo: Los efectores se combinan con proteínas alostéricas (reprsores) que actúan a nivel de la región operadora. Existen dos métodos distintos de control negativo. A-Si la proteína represora o represor no está unido al operador, la transcripción continúa. Los represores se van a unir a la región operadora impidiendo la transcripción de un gen causando represión enzimática. Sin embargo, cuando el correpresor interactúa con el represor, este cambia de conformación y se une al operador; bloqueando la transcripción. Por ejemplo, en vez de alolactosa podemos utilizar análogos sintéticos. B- Si al represor está unido al operador, impide la transcripción, con la unión de un inductor al represor, éste cambia su conformación. Dejando libre al operador, permitiendo la transcripición. 2.Control positivo: los efectores se combinan con las proteínas alostéricas (activadores), que se unen al sitio de unión del activador, sólo si el efector está presente implica un aumento en la síntesis de mRNA Solo produce procesos de inducción enzimática (no reprime). Por ejemplo, en la maltosa, gracias a un inductor se pueden transcribir mejor sus genes, y aumentar su concentración. La unión al promotor de la ARN plimerasa no es demasiado efectiva (al contrario que la DNA pol) por lo que se requiere la unión de la proteína reguladora, que altera el plegamiento del ARN. Cuando en el medio aparece en inductor, este induce un cambio conformacional en la proteína reguladora, facilitando la unión de la RNA pol al promotor. Concepto de regulón: un regulón son varios operones que codifican genes implicados en una misma vía metabólica, regulados por una sola proteína reguladora. Ejemplos: Regulón maltosa (control positivo) Regulón arginina (control negativo) En bacterias hay mucha densidad genética; nos encontramos varios operones que codifican genes implicados en una ruta metabólica. Dichos operones pueden estar regulados por una única proteína; el regulón. B. Sistema de regulación global Existe un mecanismo de regulación de la expresión de diversos genes como respuesta a cambios ambientales, antibióticos, desinfectantes, temperaturas… Permiten la adaptación de los microorganismos. Mecanismos reguladores que responden a señales ambientales mediante la regulación de la expresión de muchos genes: Represión catabólica (efecto glucosa) Inhibición de la síntesis de enzimas no relacionadas cuando en el medio hay una fuente de energía preferida; cuando una bacteria es capaz de usar varías fuentes de carbono, se inhiben aquellas enzimas que no interesan (las enzimas que se encargan de asimilar el carbono de fuentes de asimilación más compleja). Se da cuando hay efectos más “bruscos” que influyen en la síntesis. El efecto glucosa (su nombre se debe a que la glucosa fue el primer sistema de este tipo estudiado) es consiste en que si en el medio hay varias fuentes de carbono (glucosa, sacarosa, lactosa), la bacteria es capaz de reconocer la fuente de carbono de mayor rendimiento (glucosa). Mientras esté presente la glucosa como fuente de carbono, las enzimas para utilizar otras fuentes de carbono se inactivan. Crecimiento diaúxico: aparece cuando en el medio glucosa y otras fuentes de carbono. Al principio se utiliza la glucosa hasta agotarla, y se empieza con otra fuente de carbono Llegados al punto en el que la glucosa se termina, se tendrá que usa otra fuente de carbono, como la lactosa, y para ello será necesario que se sintetice la β-galactosidasa. Dos niveles de actuación: Utiliza glucosa Síntesis de enzimas degradadotas de lactosa 1. Implica mecanismos de control positivo: Al disminuir la glucosa, aumenta en AMP cíclico, que, al unirse al CAP, sintetiza la β-galactosidasa. La proteína activada por catabolito (CAP) es una proteína alostérica que para activarse necesita que se una a ella el AMPc. A su vez, la síntesis de AMPc está inhibida por la presencia de glucosa. Si hay glucosa en el medio, no habrá AMPc, que no podrá unirse a CAP y por tanto la galactosidasa permanecerá Utilización de la lactosa inactividad. La proteína CAP actúa en varios operones y cada uno tiene sus proteínas reguladoras. 2. Operón lactosa: síntesis de β-galactosidasa. Con altos niveles de AMPc el CAP se une a su sitio, si hay lactosa presente (inductor) impide que el represor bloquee la transcripción. En resumen; solo se produce β-galactosidasa si, además de haber lactosa, no hay glucosa. Ejemplos de sistemas de regulación global Atenuación: En procariotas, la transcripción y traducción están acoplados determina que se completen menos transcritos de los que se inician. Control posterior al inicio de la síntesis del mRNA, pero antes de su terminación. El número de transcritos que se completan se reduce, aunque el número de los que se inician no varía; la transcripción se detiene antes de terminar. Son frecuentes en vías de síntesis de aminoácidos (operón triptófano, histidina o fenilalanina), y especialmente frecuentes en Gram negativas. Este control se da para variaciones más pequeñas, es un control más “fino”. Operón triptófano de E. coli: implica la traducción de un péptido líder (rico en el aminoácido regulado Trp). Para que se pueda reducir es necesario que en el medio haya mucho Trp; si existe el suficiente Trp se puede sintetizar el péptido líder que bloquea la transcripción del resto de genes estructurales y provoca el plegamiento del mRNA en forma de lazo de parada para la rRNA polimerasa. Se va traduciendo un péptido líder rico en el aminoácido que se está sintetizando (trp). La transcripción se detiene cuando se forma algún bucle u horquilla. El RNAm para el operón trp tiene 4 regiones: cuando el péptido líder puede ser sintetizado directamente se induce la terminación con la formación de una horquilla En Gram positivas (Bacillus) también existen mecanismos de atenuación, pero en este caso hay proteínas que unen a región del mRNA induciendo la formación de un bucle de terminación: Unión de proteínas de atenuación que controlan estructuras alternativas del mRNA a la región líder del mRNA (cajaT), para terminar la transcripción (Interruptores o riboswitchers) Biosíntesis de pirimidinas. En Gram positivas está menos estudiado, aunque se sabe que existen interruptores de RNA como la riboflavina: si en el medio hay metabolitos, la riboflavina se una al RNA, le cambia su conformación y detiene la transcripción. Atenuación traduccional: la traducción del péptido líder previene la traducción del siguiente gen en el mRNA policistrónico, mediante modificación de la accesibilidad de la secuencia de Shine-Dalgarno (ejemplo: en genes de resistencias a antibióticos en gram positivas). “Interruptores de RNA” (síntesis de tiamina en bacterias y Archaeas) • Factores sigma (σ) alternativos: RNA pol emplea un factor σ que no es el habitual. Se sintetizan solo en casos determinados. Si está presente en el medio ese factor σ concreto, prevalece y se produce la transcripción de gener concretos. Ejemplo 1: formación de endosporas como respuesta a estrés ambiental en Bacillus Ejemplo 2: Producción de proteínas de choque térmico (DNA-K), son proteínas chaperonas (pliegan proteínas) y degradadoras del factor σ32 (en condiciones normales eliminan los factores σ32). Si hay choque térmico las DNA-K plegarán las proteínas anti choque térmico y no podrán degradar los factores sigma 32, por lo que aumenta la concentración de σ 32 y se transcriben genes específicos. Quorum sensing: Expresión de un gran número de genes diferentes cuando la densidad de población llega a ser suficientemente alta, es decir, cuando hay muchos microorganismos. Dichos microorganismos producen proteínas específicas de especie, lo que da la señal. Esta proteína se detecta en grandes cantidades (cuando hay factores de virulencia). Producción de proteínas difundibles al medio, que actúan sólo como indicadores cuando se encuentran en la célula a altas concentraciones. Esta expresión se produce por información química: hasta que no hay suficientes células, no se alcanza la concentración de proteína que puede actuar. Ejemplo 1: producción de AHL (lactona homoserina acilada) en Gram negativas (siendo específica de cada especie). Ejemplo 2: estudios en Vibrio han demostrado que si se juntan, al aumentar su concentración, adquieren fluorescencia. Ejemplo 3: la Pseudomona se puede encontrar latente en el pulmón, pero cuando alcanzan una densidad de población determinada, producen a la vez factores de virulencia. Transducción de señales, Transducción: detección de la señal por un sensor y transmisión al sistema regulado. Son sistemas de detección de señales ambientales. Con frecuencia, se trata de sistemas de dos componentes: -Proteína detectora en la membrana (quinasa): autofosforilación en un residuo histidina de la parte citoplasmática. Quinasa: proteína reguladora fosforilada puede actuar de inhibidor, se tratará de un proceso reversible porque también hay fosfatasas que pueden realizar el proceso inverso. -Proteína reguladora de la transcripción Ejemplos: Asimilación del fosfato en E.coli, fijación del Nitrógeno en Rhizobium, respuesta al estrés osmótico en E. coli (útil en investigación con plantas) -Diferencias en la expresión génica entre procariotas y eucariotasEn eucariotas: No hay RNA policistrónicos No hay acoplamiento transcripción-traducción Los operones ejercen su control sólo sobre una única enzima Escasos mecanismos de control negativo Abundantes sistemas de control positivo Modificación post-transcripcional en proteínas muy frecuentes (esta es muy poco frecuente en procariotas) Procesamiento del mRNA, que no existe en procariotas modificación La modificación de la información genética en microorganismos viene determinada por mutaciones, que afectan a pequeñas secuencias del mismo genoma (T.3) y por recombinaciones, que afectan a grandes secuencias de distintos genomas (T.4). 5. MUTAGÉNESIS. -Introducción. Conceptos generalesMutación: es un cambio estable y heredable de la secuencia de nucleótidos del DNA, interesan las mutaciones que son detectables. Si el cambio en la secuencia de DNA no se hereda, no se considera mutación. Son modificaciones de la información genética en los microorganismos, muy importantes en biotecnología microbiana. Las mutaciones vienen generalmente acompañadas de alteraciones fenotípicas del tipo: 1. Morfológicas: cambio del color, del aspecto, etc ya sea de la colonia o del microorganismo en concreto, siendo esta última menos frecuente. 2. Bioquímicas (auxotrofismo): cambios en las rutas metabólicas. Los auxótrofos no son capaces de sintetizar un metabolito concreto que recibirá el nombre de factor de crecimiento (metabolito para el que se es deficiente; se es auxótrofo para un factor concreto) y hay que añadírselos al medio para que el microorganismo crezca. Por el contrario, un protrótrofo es un m.o capaz de sintetiza un factor de crecimiento, es decir, que no depende de su presencia en el medio para su desarrollo. Por ejemplo, un His- es un auxótrofo que no puede sintetizar histidina, y para su crecimiento será necesario añadirla al medio. La His sería el factor de crecimiento, que la variedad salvaje (protrótrofo) sí que es capaz de sintetizar. 3. Resistencias: tanto a agresión química, biológica como al estrés ambiental. Por ejemplo a determinados antibióticos o a agentes letales para la bacteria silvestre. Para ver que las clonaciones han sido correctas o para ver que las transformaciones han sido correctas. Las mutaciones también determinan la variabilidad poblacional, ya que es el mecanismo evolutivo que le permite al microorganismo colonizar prácticamente en cualquier nicho ecológico y adaptarse a las hostiles variaciones del medio; y además, una importante aplicación biotecnológica, ya que podemos utilizar la mutagénesis para crear mutantes para el aprovechamiento industrial (mutantes in vitro). Los mecanismos moleculares por los que se produce la mutagénesis son: 1. Daños directos en el DNA 2. Errores en la replicación, como desplazamiento del marco de lectura ó sustituciones de pares de bases. 3. Secuencias de inserción (transposones). Tipos de mutaciones: o Mutaciones puntuales como sustituciones de pares de bases o microinserciones / microdelecciones. Afectan a muy pocas pares de bases o a solo una de ellas. Se utilizan para mejora de cepas en ingeniería genética, cambios mínimos. o Mutaciones en un gran número de bases: inserciones, delecciones, inversiones y translocaciones. Cambios mayores, aumentan la tasa de letalidad. -Tipos de mutaciones- Mutagénesis natural o espontánea en ausencia de exposición voluntaria a agentes externos es aleatoria aunque en ocasiones parece tener un aparente carácter adaptativo y parecen ser inducidas por condiciones ambientales (“Evolución forzada”). Este término es incorrecto, realmente lo que ocurre es la selección de mutantes en ambientes hostiles, favoreciendo la supervivencia de aquellas mutaciones que han aparecido aleatoriamente y las hace más resistentes en esas condiciones. Es decir, se producen por selección de mutantes (hipermutación: mecanismos SOS, polimerasas IV-V…). Aplicación industrial rama de la biotecnología llamada evolutiva (término no muy correcto). Presenta una frecuencia muy baja; 1 / 10 7–106por gen y por generación. Al principio la biotecnología era un proceso empírico que se basaba en este tipo de mutaciones, seleccionando las especies con mejores mutaciones naturales para fabricación de alimentos.; La mutagénesis durante la reparación: Las mutaciones en los genes que codifican para las DNA polimerasas provocan lo que se conoce como fenotipo hipermutador. Este fenotipo se da bajo situaciones de estrés y aumentan la tasa de mutación considerablemente: Sistemas de reparación con propensión a los errores, como los sistemas SOS o las DNA polimerasas IV y V (DNA mutasas). Estos son sistemas que evitan la muerte celular a cualquier precio (una alta tasa de mutaciones). Hot spots: que son secuencias cortas con alta frecuencia de errores de la DNA polimerasa. Mutagénesis inducida (dirigida o forzada) se basa en someter a los microorganismos a factores (adicionando al medio dichos factores) que aumentan la probabilidad de mutaciones, aparentemente adaptativas como puede ser aprovechar determinadas fuentes de carbono o poder vivir sin algún tipo de nutriente. Las mutaciones inducidas son aparentemente adaptativas frente al stress ambiental, la deprivación de nutrientes y la adición de sustratos (biodegradación). Este tipo de mutagénesis presenta, por tanto, una frecuencia de mutación mucho mayor que la espontánea: 1 / 10 5-101 aumento de la tasa de mutación. Hay agentes mutágenos de dos tipos: físicos y químicos. 1. Físicos: radiaciones -Radiaciones no ionizantes (260 nm): Este tipo de radiación provoca la formación de dímeros de timina (del tipo ciclobutano) y el cambio en el marco de lectura, transversiones o delecciones. -Radiaciones ionizantes: muy agresivas, provocan la formación de radicales libres y rupturas tanto simples como dobles, lo que causan mutaciones de grandes fragmentos, por lo que este tipo de radiación es poco utilizada en microorganismos (no se utilizan desde el punto de vista industrial). 2. Químicos: - Análogos de bases (en vez de utilizar bases los microorganismos utilizan compuestos que se asemejan estructuralmente a las bases) o agentes alquilantes. Causan apareamiento específico incorrecto - Agentes intercalantes: como por ejemplo el bromuro de etidio, que se intercala en la doble hebra de ADN deformándola desfase - Ausencia de replicación por daño directo del DNA; compuestos acetilados oxidan el DNA dañándolo y evitando que se produzca la replicación. Normalmente son agentes muy carcinógenos. A parte de los agentes mutágenos físicos o químicos también hay otras posibilidades para inducir la mutación, como la deprivación de nutrientes o la adición de sustratos para que se produzca la biodegradación. -Efecto de las mutacionesUna lesión del DNA (pre-mutacional) puede desembocar en: 1. Muerte del microorganismo 2. Reparación por diversos mecanismos y solución de la lesión 3. Dar lugar a una mutación que puede ser: -Inaparente (sin cambio fenotípico) -Letal (causa la muerte del microorganismo) -Condicionalmente letal: el microorganismo es capaz de crecer en unas determinadas condiciones (“permisivas”) y en otras condiciones (llamadas “no permisivas”) no crecen y mueren, cuando la cepa silvestre sí que crecería y sobreviviría. Son mutaciones que afectan sobretodo a la resistencia a diferentes temperaturas, sobre todo al calor -Cambio fenotípico -Expresión de las mutaciones – Si aparece en la zona de lectura que codifica para una proteína : En los genes estructurales: -Mutaciones silenciosas: cambia una base pero codifica el mismo aminoácido, por lo que ocurren en el último triplete del codón. No se reflejan en la proteína resultante. -Mutaciones con sentido erróneo: el cambio de un aminoácido por otro, y dicho cambio conlleva un cambio de codón y alteración de la proteína. Este tipo de mutaciones puede ocasionar desde proteínas inactivas (proteínas con actividad pero sensibles a determinados factores) a mutaciones letales (provocan la muerte) o condicionalmente letales (provocan la muerte ante determinadas condiciones). -Mutaciones sin sentido: la mutación da lugar a un codón de terminación; provocan proteínas inactivas a no ser que la mutación se encuentre al final del gen. Son poco frecuentes porque hay pocos codones de parada, son casi todas letales. -Desplazamiento del marco de lectura: se originan grandes alteraciones; también da lugar a proteínas inactivas. Si aparecen en los genes reguladores : puede ocurrir dos cosas, que se produzca el producto si la secuencia del operador no es reconocida por el represor (producirá una transcripción continua), o que no llegue a producirse el producto (ausencia del mismo), esto ocurre si la secuencia del promotor no es funcional. Mutaciones en los genes que codifican para las DNA polimerasas (DNA pol III: fenotipo hipermutador la polimerasa sigue funcionando pero con mucha frecuencia de error, lo cual aumenta la tasa de mutación). Mutaciones en los genes del rRNA y tRNA: se produce una alteración de la síntesis proteica (expresión fenotípica frecuente). -Mutaciones hacia atrás (Reversiones)Un mutante revertiente es aquél que ha recuperado el fenotipo silvestre tras dos mutaciones consecutivas (segunda mutación restaura el fenotipo original), sea cual sea el mecanismo molecular que las haya originado. Si las dos mutaciones ocurren en el mismo sitio, la mutación puede ser: - Restauradora: da lugar a un “verdadero revertiente” se recupera justamente la misma secuencia (codón) original, de manera exacta. - Equivalente (restaura un codón que codifica para el mismo aminoácido que el original aunque no tiene la misma secuencia de codón que el anterior. Mientras que si las dos mutaciones han tenido lugar en sitios distintos puede ocurrir que: -Sean en el mismo gen (intargénicas) -Se produzcan en distintos genes (extragénicas), como es el caso de la supresión fisiológica, la nueva proteína anula el efecto de la primera mutación. En este caso, una segunda enzima, por mutación de reversión, es capaz de realizar la función de una primera enzima que ha sido inactivada por una primera mutación. En general, la tasa de reversión es mucho menor que la tasa de mutación: es mucho más fácil probabilísticamente que se produzca una sola mutación, que se produzcan 2 y que precisamente la segunda restaure el fenotipo de la otra. -Detección de mutantesCuanto mayor sea el porcentaje o volumen de mutación, más fácil será la detección de los mutantes, y por tanto, más fácil será localizar mutaciones puntuales. La tasa de mutación se ve reflejada únicamente en cambios fenotípicos detectables. La detección de los mutantes depende de la velocidad de mutación: Errores replicación: Alta frecuencia, se detectan con relativa facilidad. Transposición: Muy alta frecuencia. Mutaciones sin sentido: Baja frecuencia, dan lugar a fenotipos no viables. Según su carácter encontramos: Mutaciones no seleccionables: los mutantes no seleccionables son aquellos que no ofrecen ninguna ventaja ecológica al mutante, y los métodos para su detección son el rastreo o screening, que son métodos muy tediosos. Por ejemplo los auxótrofos. Dentro del rastreo se pueden encontrar distintos métodos: 1. Observación de cambios morfológicos, de color, aspecto de las colonias, etc. buscar diferencias morfológicas 2. Selección negativa con penicilina: los microorganismos protrótrofos crecen a pesar de que no estén presentes en el medio todos los factores de crecimiento, aun así, como la penicilina solo afecta a los microorganismos en fase exponencial (que estarán en crecimiento protrótrofos), al añadir penicilina, ésta matará a los protrótrofos, mientras que los auxótrofos, al no poder crecer por la ausencia del factor de crecimiento, sobrevivirán al tratamiento con penicilina (serán inmunes a ella). Por ejemplo, tenemos en primer lugar un cultivo sin Histidina en el que introducimos mutantes auxótrofos para Histidina con resistencia a penicilina (His-) y también protrótrofos His+. Si añadimos la penicilina al medio, se destruirán casi todos los que estén creciendo en fase exponencial (los His+), mientras que los auxótrofos, con mutación en penicilina, sobrevivirán al tratamiento con penicilina. Luego cambiamos el cultivo a un medio con Histidina y en él crecerán en su mayoría auxótrofos His- inmunes a penicilina y algunos His+ supervivientes al tratamiento. Este método nos ayudará a obtener con altos rendimientos (aunque no del 100%) únicamente mutantes auxótrofos selección negativa. 3. Método de la réplica en placa. Este método sólo sirve para detectar auxótrofos para un determinado factor de crecimiento, como por ejemplo la Lisina. Se siembra una alícuota del medio en una placa con lisina, y una vez han crecido los microorganismos se coge una esponja de terciopelo; como en el terciopelo se quedan fijas las posiciones de las colonias y se adhiere una parte de la colonia, estor permite “copiar” la posición de las colonias. Ahora se junta la esponja de terciopelo en un medio con lisina y en uno sin, quedándose los m.o en el mismo lugar que en el original. Al crecer las colonias, en el medio con lisina crecerán todos, mientras que en el medio sin lisina crecerán todos menos los auxótrofos para lisina. Por simple comparación de placas se puede saber cuáles son los auxótrofos y cuáles los protrótrofos y luego seleccionarlos. Mutaciones seleccionables: Este tipo de mutaciones confieren ventajas ecológicas al mutante, de ahí que se pueda seleccionar. El método de detección de este tipo de mutaciones es la selección: colocaremos al mutante en condiciones que tan sólo el mutante podrá aguantar. Permite separar el microorganismo que es mejor competidor, que se seleccionará sobre el resto. El crecimiento en condiciones ambientales adecuadas da lugar a mutantes de resistencia; es decir, m.o que crecen incluso en presencia de antibióticos, antimetabolitos, fagos, temperaturas limitantes. En el caso de los metabolitos: antimetabolito es una sustancia que se puede acoplar a la ruta metabólica pero del cual no se puede extraer energía directamente. En el medio con presencia de antimetabolitos crecerán solamente aquellos m.o que produzcan metabolitos en cantidades más altas de los normales, o el que, al haber mutado, haya desarrollado otra ruta metabólica y sea capaz de sintetizar el metabolito de interés, ya que a nivel de fenotipo se parecía lo mismo. Crecimiento en ausencia de nutrientes: se elimina el nutriente que nosotros estamos estudiando, se utiliza para la selección de protótrofos revertientes (microorganismos que al sufrir una mutación se han vuelto auxótrofos, y que al volver a mutar se han vuelto otra vez protótrofos). -Prueba de mutagenicidad. Test de AmesSe utiliza en toxicología para valorar si una determinada sustancia tiene capacidad mutagénica o no. Empleado sobre procariotas, puede llegar a ser bastante sofisticado. En general, compara la probabilidad de que aparezcan protrótrofos revertidos con mutágeno y sin mutágeno. Este test de carcinogeneicidad se realiza cuando se prueban nuevas sustancias químicas. Procedimiento: Se usa una cepa con una mutación puntual y cuya probabilidad de mutación sea muy alta en sistemas de reparación de errores de DNA (normalmente en E. coli o Salmonella). Los auxótrofos para un determinado factor de crecimiento (histidina por ejemplo) se cultivan en dos medios: a uno se le ha añadido la sustancia química a estudiar y al otro no. En ambos se pone un poco de histidina para el periodo de adaptación (para que el medio sea lo menos estresante posible). Cuando termina la histidina, los auxótrofos dejarán de crecer. Tras dejar pasar un tiempo, en ambos cultivos se verá crecimiento, ya que siempre habrá un pequeño número de mutantes revertientes espontáneos. Si la sustancia no es mutagénica se verá más o menos el mismo número de mutantes revertientes en el control que en la prueba, mientras que si la sustancia es mutagénica se verán muchos más mutantes revertientes en la prueba. Es decir, dependiendo de los mutantes revertidos que aparezcan en la placa con producto químico de prueba concluiremos que si han revertido muchos, es carcinógeno, y si hay pocos revertidos, su uso no es peligroso por ser agente mutagénico agresivo. Normalmente en la zona periférica del disco de aplicación de la sustancia química a estudiar no hay crecimiento, porque la sustancia está en tal concentración que es letal para los microorganismos. 6. RECOMBINACIÓN GENÉTICA -IntroducciónLa recombinación genética es un proceso de intercambio físico de material genético y es la causante de modificaciones en la información genética en los microorganismos. Produce unión de elementos genéticos de dos genomas y cambios mayores que la mutación. Encontramos dos tipos principales de recombinación: homóloga y heteróloga. La recombinación origina generalmente reordenamiento genético que se expresa fenotípicamente. -Recombinación homólogaEl intercambio de material genético es produce entre secuencias de DNA largas y muy parecidas (han de ser suficientemente homólogas en composición nucleotídica como para que se apareen) pero de origen distinto, es decir, entre secuencias similares en nucleótidos de dos orígenes diferentes. En procariotas se produce por transferencia horizontal (entre poblaciones) y se basa en que un microorganismo cede el material genético intercambiado a otro habrá un elemento donador (virus, bacterias) y uno aceptor (bacterias). Este tipo de recombinación se produce tanto de forma natural como dirigida (en ingeniería genética), y se realiza, igual que cualquier clonación de un microorganismo, para producir un m.o con características de interés. Es la más frecuente en procariotas. En la recombinación homóloga diferenciamos un exogenote o elemento dador y un endogenote, que será el receptor del material genético. Se trata de una recombinación no recíproca: el DNA de la bacteria receptora ha cambiado, pero el de la bacteria donadora se mantiene intacto. Se asocia el exogenote con el endogenote y a continuación se produce la invasión de las ramas por acción de las enzimas RecA. Finalmente obtenemos un DNA heterodúplex recombinante Si los cortes y empalmes afectan a las cadenas de ADN que antes no participaron en el entrecruzamiento, el resultado será dos moléculas recombinantes recíprocas, donde cada una de las 4 cadenas son recombinantes (ha habido intercambio de marcadores entre donador y receptor) Si los cortes y empalmes afectan a las mismas cadenas que ya habían participado en el primer entrecruzamiento, el resultado consistirá en dos dobles hélices que presentan solamente sendas porciones de ADN heterodúplex -Recombinación heterólogaEs específica del sitio, como por ejemplo ocurre en infección con virus. Secuencias completamente diferentes. En la recombinación heteróloga se introducen genes nuevos a un replicón, mientras que en la recombinación homologa se modifica la posición de los genes ya existentes dentro del replicó -Transferencia genéticaEl fenómeno de transferencia genética se produce por tres mecanismos distintos: Transformación: el hospedador integra DNA exogenote (libre en el medio) Transducción: proceso de transferencia genética mediada por virus. Conjugación: en este proceso es necesario el contacto físico entre los 2 microorganismos para transferir el material genético de uno al otro y la presencia de un plásmido conjugativo en la célula donadora. -TransformaciónConsiste en la incorporación de DNA liberado al medio, ya sea: - Cromosómico (tras una suave lisis de las bacterias) - Plasmídico (puede haberse liberado al medio tras lisis): es el más frecuente. - De un virus bacteriano (se considera transfección) Este proceso se produce en la naturaleza de forma aleatoria ya que depende del azar, y sólo ocurre en algunos géneros de bacterias y arqueas, a estos géneros se les conoce como especies naturalmente transformables. DNA del plásmido: el plásmido de DNA entra en la bacteria y se produce una transformación estable, que se reproducirán en las células hijas. Sin recombinación DNA libre en el medio (incorporación de DNA monocatenario): se incorpora ADN libre del medio. Para que sea estable, ha de producirse necesariamente la recombinación (derecha). Presenta una baja frecuencia La recombinación no recíproca entre distintas especies o géneros es más difícil porque es más difícil que tengan secuencias homólogas largas. Siempre se incorpora una hebra monocatenaria, apareándose esta con la de la célula. Lo más normal es que se degrade el ADN del medio y que fracase la transformación. La transformación implica la competencia de las células receptoras, que no es más que la capacidad de una célula para poder transformarse, en la naturaleza sólo se transforman las que entran en competencia. La competencia es una característica determinada genéticamente. No tiene por que ser estable en el tiempo, es decir, puede no ser competente en parte de su ciclo y por una señal, pasar a competencia. Las proteínas específicas de la competencia incluyen: Proteínas de membrana, de unión al DNA: específica y propia de las células competentes. Protegen el DNA extraño de la acción de nucleasas, impiden la restricción que implicaría el fracaso de la transformación Autolisinas de la pared: para romper la pared. Nucleasas: pasan a estado monocatenario las hebras; en gram negativas se encuentran en el espacio del periplasma y en gram positivas parece que es externo. Proteína RecA: una vez el exogenote se ha unido, la proteína RecA comienza el proceso de recombinación. Pasos: 1) Incorporación de DNA La unión comienza siendo reversible, pero pasa a ser irreversible. Tras esto se producen poros en la pared, con lo que puede entrar en DNA. Si es bicatenario, será degradado a monocatenario en el espacio periplásmico (G-), o puede ser que se degrade el ADN a monocatenario en la propia membrana por acción de nucleasas (G+), pero el DNA que entra al citoplasma es siempre monocatenario. Para que el ADN monocatenario no se degrade se le unen proteínas que la protegen de las nucleasas (proteínas de unión al ADN específicas de competencia). Es más eficaz con plásmidos y requiere la inactivación de exonucleasas, además de un exceso de DNA transformante (las células van a competir por el DNA). A pesar del exceso de DNA, la probabilidad de que una célula incorpore el DNA con el marcador genético adecuado es pequeña (no pasa de una eficiencia máxima del 20%, y lo lo normal es entre 0,1 y 1%). La concentración mínima de DNA que produce transformantes detectables es de 10 -5 µg/ml, que es tan baja que resulta químicamente indetectable. El fenómeno de transformación tiene una frecuencia baja (10-3 población bacteriana), ya que la probabilidad de que al inducir una transformación el gen deseado haya mejorado es muy reducida. 2) Integración del DNA transformante Tras la incorporación, el DNA se asocia con una proteína específica de la competencia que permanece unida al DNA hasta que alcanza el cromosoma, donde es sustituida por la RecA. El DNA se integra luego en el genoma del receptor por procesos de recombinación. Durante la replicación de este DNA heterodúplex, se forma una molécula de DNA parental y otra de DNA recombinante. Tras la segregación por división celular, la última estña presente en cada célula transformada, que está genéticamente alterada en comparación con el tipo parental. La competencia puede ser natural o artificial: Natural, sobretodo en organismos como: o Bacillus: solo un porcentaje entra en competencia, cuando hay señales químicas o de quórum sensing o Streptococcus pneumoniae: solo entra en competencia cuando el cultivo llega a un número deerminado, y entonces todas entran en competencia. o Haemophilus: no participa ningún factor de competencia; solo incorpora ácidos nucleicos muy similares al suyo. Solamente incorpora competencia de especies muy relacionadas. o Azotobacter o Pseudomonas o Acinobacterium Artificial: se puede inducir mediante un tratamiento de calcio y frío, pero es un tratamiento poco eficaz para el que sólo sirven las Gram negativas (como E. coli) y los plásmidos. Actualmente el modo de inducir la competencia artificialmente de Ca y frío se está sustituyendo por la electroporación, que es el método más eficaz para transformar, se utiliza para bacterias, arqueas e incluso eucariotas. Este método consiste en la aplicación de pulsos eléctricos a las células para la apertura de los poros en las membranas, permitiendo tanto la entrada como la salida de DNA en las células. -TransducciónEs el proceso de recombinación más frecuente para procariotas y es prácticamente específico, consiste en una transferencia de DNA entre células por la acción de un virus. Se da en Escherichia, Pseudomonas, Rhodococcus, Archaea, hipertermófilas, metanógenas… Cuando un fago infecta una célula, se fusionan su cápside con la membrana de la célula y se introduce su DNA, es aquí donde pueden darse dos ciclos distintos: - Ciclo lítico. El DNA vírico utiliza la maquinaria de la célula para replicarse, generar más material genético y las proteínas de la envuelta (sintetizar sus cápsides). Las cápsides empaquetan el material genético del virus y una vez fabricados, se liberan rompiendo la célula (provocan lisis celular) - Ciclo lisogénico: (lisogenia, bacteria lisogénica) en ocasiones el DNA del fago se introduce y se incorpora en el DNA de la bacteria, convirtiéndose en un profago o fago atemperado (bacteriófago cuyo genoma se ha incorporado a la bacteria huésped). La bacteria está lisogenizada, y cuando la bacteria se dividida, lo hará con el fago. El profago (DNA del fago en el DNA de la bacteria) se multiplicará dependiendo del DNA bacteriano. Además, el fago induce cambios fenotípicos en la bacteria conversión fágica: las bacterias se hacen inmunes a otro tipo de fagos, por ejemplo, en Salmonella cambian polisacáridos de membrana al lisogenizarse y la hacen más virulenta. En cualquier momento se puede producir inducción lítica, es decir, que el profago se separe del material genético de la bacteria y comience un ciclo lítico que desembocará en la lisis bacteriana. Esta inducción se puede dar por ejemplo, por exposición a luz UV o a agentes tóxicos. Ensamblaje de cabezas y DNA del fago para formar nuevas partículas víricas Distinguimos dos tipos de transducción: Generalizada: el DNA foráneo se empaqueta en la partícula vírica en lugar de su genoma. Si los genes del donador no sufren recombinación homóloga con el cromosoma de la bacteria receptora se perderán, ya que no pueden replicarse independientemente, y por tanto, el DNA del donador no se transmitirá a la descendencia. Se da lugar siempre en ciclo lítico, por inducción de un lisógeno y posterior infección lítica. En este proceso se puede transferir prácticamente cualquier marcador genético desde el donador al receptor. Durante una infección lítica (lleva a la lisis) las enzimas responsables del empaquetamiento del DNA vírico en el bacteriófago a veces introducen DNA del hospedador (bacteriano) de forma accidental, creando así una partícula transductora, que será liberada al medio tras la lisis con el resto de viriones normales. Las partículas transductoras son defectivas, porque no tienen DNA vírico, no tienen capacidad de replicación, pero sí de infección, aunque no pueden empezar una infección vírica. Si el lisado se usa para infectar una población de células, la mayoría se infectará con el virus normal, pero una pequeña proporción recibirá las partículas transductantes, que, aunque no pueda replicar el material genético, puede sufrir procesos de recombinación del DNA del nuevo hospedador. Es un fenómenos de baja probabilidad (prob. de tener una partícula transductante es baja, y como sólo tiene un fragmento pequeño de DNA, la prob. total de que en dicha partícula haya un gen es muy pequeña: 1 / 10 6 -108). Las partículas transductantes son causadas o bien por fagos atemperados o bien por fagos virulentos Especializada: el DNA foráneo de una zona específica sustituye a algunos genes virales y se integra en el genoma viral. Se trata de un fenómeno de bajísima frecuencia (tienen que darse muchos sucesos de baja probabilidad a la vez) que permite una transferencia muy eficiente y que una pequeña región del cromosoma bacteriano se replique independientemente del resto. Un ejemplo muy representativo es E. coli con gen de galactosidasa, muy útil en biotecnología. Partimos siempre de un profago en ciclo lisogénico: cuando el fago se integra en el genoma bacteriano la bacteria es la responsable de la replicación del material del fago. Mediante inducción del ciclo lítico (luz UV es un agente externo inductor que provoca la separación de los dos genomas, la recirculación y que se vuelva a introducir en el genoma) se separa el DNA viral del DNA del huésped por un proceso inverso de integración, ocurriendo a veces que el DNA del fago se escinda de forma incorrecta (suceso raro), escindiéndose con parte del DNA del hospedador. Cuando se produce la inducción, lo normal es que se restablezcan las secuencias originales, sin embargo, en muy pocas ocasiones, el fago se lleva parte del DNA bacteriano. Si el DNA del fago conserva la capacidad de replicación, éste se replicará y cuando sean liberados al medios tras la lisis, llevarán parte del genoma de la bacteria fagos defectivos, ya que les faltan algunos genes. Para que el fago sea viable hay un máximo de DNA que se puede intercambiar, ya que debe conservar un mínimo del fago para suministrar información para la producción de las proteínas de la cubierta del fago y otras proteínas causantes de la lisis. Es posible la participación de un fago auxiliar. A veces, quedan tan pocos genes propios del fago tras la inducción que no son capaces de replicarse. Por otra parte, la bacteria estará infectada por dos tipos de fagos: defectivos y auxiliares. Gracias a los auxiliares, se podrán replicar los defectivos, requiriendo menos información propia, necesitándose únicamente la región att (attachment), el sitio cos (extremos cohesivos para el empaquetamiento) y el origen de replicación. En biotecnología siempre empleamos fagos auxiliares para asegurarnos de que ocurre la transducción. Diferencia entre especializada y generalizada: la forma de producir el lisado transductor. En al especializada debe ocurrir por inducción de un lisógeno, mientras que en la generalizada puede ocurrir por este método o por infección de una bacteria no lisogénica. -ConjugaciónAntes de hablar de la conjugación es necesario hablar de los plásmidos. Un plásmido es un elemento genético capaz de replicarse independientemente del cromosoma del hospedador. No tiene forma extracelular (no pueden vivir fuera de la célula) y se encuentran dentro de la célula como moléculas de ácido nucleico. Características físicas de los plásmidos: poseen casi todo su DNA bicatenario y la mayoría se encuentra en forma circular. Son de tamaño variable, su tamaño oscila entre 1 y 1000 kilopares de bases. Generalmente en configuración superenrollada. En cuanto al número de copias, aunque estén controladas por los genes del plásmido, lo normal es que tengan solo una. Replicación Intervención de enzimas celulares, por lo que los genes del plásmido dedicados a la replicación se limitan a controlar temporalmente el proceso de iniciación y el repartote los plásmidos entre las células hijas. Número de copias generalmente entre 1 y 3, pero pueden llegar a más de 100. En Gram negativas: la mayoría se replican de forma similar a la cromosómica, lo que implica la iniciación y replicación en un origen bidireccional, algunos de forma unidireccional. En Gram positivas: la mayoría por el método del círculo rodante. La replicación empieza en el punto de origen por rotura de una cadena de DNA. La hebra interior comienza a girar a la vez que se va copiando la hebra externa. Después de sintetizar la nueva cadena (una vuelta del círculo), la proteína del gen A corta la cadena nueva y liga sus dos extremos. Este método genera un intermediario monocatenario, por eso a los plásmidos que utilizan este método también se les conoce como plásmidos con DNA monocatenario. En una célula pueden coexistir varios tipos de plásmidos si son compatibles (bacterias hospitalarias pueden tener 3-4 plásmidos de resistencia). Incompatibilidad de plásmidos: hay plásmidos que tiene un mecanismo común para regular su replicación, por lo que la existencia de uno imposibilitará la existencia de otro, ya que el segundo no puede mantenerse y se pierde durante la replicación celular subsiguiente (no permite incorporación de nuevos plásmidos). Dichos grupos son conocidos como plásmidos de incompatibilidad (Inc). Dos plásmidos del mismo grupo no pueden coexistir, pero plásmidos de distintos grupos sí. - Significación biológica de los plásmidos: Todos poseen genes para su propia replicación Muchos tienen genes que modifican el fenotipo celular y codifican para sustancias de: o Rutas metabólicas: menos frecuente, dar capacidad de sintetizar o utilizar productos metabólicos o Plásmidos de resistencia (plásmidos R) o Producción de toxinas (toxinas de Vibrio, por ejemplo). o Síntesis de bacteriocinas: sustancias antibacterianas de competencia ecológica Los plásmidos son interesantes, entre otras cosas, porque permiten destruir sustancias tóxicas (Pseudomonas el tolueno, por ejemplo), lo cual nos puede resultar útil en biorremediación si insertamos estos plásmidos en otras bacterias. La CONJUGACIÓN es la transferencia de material genético plasmídico (plásmidos) entre células. Este proceso se asemeja a una infección, es más frecuente entre bacterias, pero también se produce entre bacterias y arqueas, hongos o eucariotas. Es necesario que los plásmidos sean conjugativos, y para ello requieren tener región tra, que propicia la transferencia (mínimo 28 genes en esta zona). La región Tra del plásmido se va a encargar de codificar los componentes necesarios para que se produzca el contacto específico entre las células, retracción, fusión de membranas externas y finalmente la transferencia. - En gram negativas, la conjugación se produce a partir de la emisión de un puente citoplasmático, un pelo llamado “pili sexual”, encargado de la retracción y fusión de las membranas plasmáticas. - En gram positivas, no existe el pelo sexual, y se cree que el proceso se basa en la liberación de péptidos cortos que actúan como señal química para la activación de los genes de transferencia. La unión entre células también se produce por interacción con proteínas codificadas por el plásmido. Más complicado de estudiar. Es un proceso replicativo (infectivo, cuando acaba el proceso, ambas bacterias poseen el plásmido); cada bacteria se apropia de una hebra, y ésta hebra se replica y pasa a estado bicatenario dentro de cada bacteria. La transmisibilidad por conjugación está controlada por genes de la región tra, que codifican proteínas que funcionan la transmisión de DNA y su replicación. Si la región tra se integra en el cromosoma (episomas los que lo pueden hacer) puede modificar la transferencia de DNA cromosómico de una célula a otra. La conjugación es específica entre bacterias que tienen proteínas que se reconocen mutuamente. Transmisión del plásmido F: La conjugación es la conversión de cepas F- en F+ (plásmidos de fertilidad). El plasmido F de una célula F+ pasará a la célula F-. El plásmido F codifica genes que aportan: - Capacidad para formar el “pelo sexual” (cepa F+) - Movilización del DNA para su transferencia - Alteraciones de la membrana celular (impermeabilización): incapacidad para que se vuelva a fusionar con otras bacterias F+ con el mismo tipo de plásmidos; no podrán conjugarse de nuevo con la célula. Si es otro tipo de sí se puede incorporar, lo que no estará permitido es el mismo plásmido que acaba de ser conjugado. Durante la conjugación, lo último que se transferirá es la región tra. Para que una célula pase de F- a F+ tiene que completarse totalmente la conjugación. Si se interrumpe, no se transfiere la zona tra (que es la última) y no podrá conjugar otras bacterias, ya que no dispone de la maquinaria para la conjugación. Se convertiría en una bacteria F’ no conjugativa, con parte del plásmido, pero que no ha llegado a adquirir la zona tra. Es posible que se produzca curación: la curación es un proceso por el cual se eliminan los plásmidos de las células hospedadoras, sea por algún tipo de tratamiento o por presión selectiva. Parece ser que es el resultado de inhibir la replicación del plásmido sin inhibir la replicación del cromosoma. La curación puede ocurrir de forma espontánea, pero se incrementa con colorantes de acridina que se insertan en el DNA, aunque también se puede utilizar la electroporación Si se produce una pérdida de presión selectiva, se puede perder el plásmido. Sin embargo, los plásmidos de resistencia a antibióticos, incluso sin presión selectiva se transfieren perjudicial para tratamientos médicos, y aumento progresivo de la resistencia a antibióticos por parte de bacterias. Actualmente se busca que las bacterias puedan perder el plásmido si no hay presión selectiva. Que una cepa sea F+ no implica que se vaya a conjugar siempre, tiene que haber una serie de condiciones concretas. Al final del proceso de conjugación del plásmido F, las dos cepas son F+ Integración del plásmido F (episoma): un episoma es un plásmido capaz de integrarse en el cromosoma, en zonas con secuencias de inserción homólogas. Cuando integran el plásmido F, reciben el nombre de cepas Hfr. Los plásmidos que transmiten mucho DNA cromosomal son los Hfr (High frequency of recombination). Existen diferentes cepas Hfr según el lugar de inserción del plásmido (una misma cepa puede conjugar utilizando recombinantes muy diferentes). El plásmido pierde la capacidad de replicarse autónomamente, pero la región trap sigue siendo activa y puede provocar la conjugación. Muy poco probable la transferencia si el plásmido está integrado; frecuente transferencia de DNA bacteriano, aunque difícil conversión de F- a F+. Pregunta examen: diferencia entre cepas F+ y cepas Hfr En los dos casos se puede producir conjugación, pero en cepas F+ el plásmido está separado del cromosoma bacteriano y se transferirá a F- individualmente. En cepas Hfr, sin embargo, el plásmido F está integrado en el genoma bacteriano (episoma), que inducirá la transferencia a cepas F- junto con parte del material genético no plasmídico (parte de cromosoma). Sexoducción: plásmido F’ escisión incorrecta que arrastra y transfiere material cromosómico. Frecuente mecanismo de intercambio genético entre bacterias. El plásmido se recircularías y arrastra una porción del genoma bacteriano. Ya no será un plásmido F, será F’. La cepa F’ ha originado otra bacteria F’ con el mismo plásmido que ha arrastrado el fragmento bacteriano A. Podrá generar otras cepas F’ que puedan conjugar, ya que la nueva F’ sigue conservando las zonas oriT y trap. Aplicaciones de la conjugación: elaboración de mapas genéticos Se basa en la elaboración de experimentos de cruce interrumpido. Se utiliza una cepa Hfr y otra cepa aceptora. El método es observar cuantos genes de la cepa dadora hay en la cepa aceptora cada cierto tiempo. Los genes que aparezcan más pronto, serán los que estén más cerca del oriT. Esta referencia nos permite construir un mapa relativo viendo el tiempo en el que los genes tardan en aparecer en los recombinantes. Los últimos genes en aparecer serán los de la región trap. Con los datos, realizamos una curva, en la que se vea el tiempo desde que mezclas hasta que rompes el puente citoplasmático. Para que E. coli transfiriera todo su material, harían falta 100 minutos. Si para la realización del mapa genético usamos una sola cepa Hfr, obtendremos un mapa muy deficitario, y cuanto más nos alejemos de la zona oriT más inexacto nos saldrán los datos; por ello realizamos el estudio con varias cepas Hfr y el mismo plásmido. -Recombinación in VitroGracias a la recombinación in Vitro, hacemos que algunas cepas adquieran genes que a mi me interesan. Producción de cepas recombinantes de aplicación biotecnológica: - Pseudomonas, E. coli: conjugación, transducción. Técnicas efectivas en Gram negativas. Bacillus: transformación, transducción (fagos PBS1, SP10, se manipulan muy bien y realizan una recombinación muy efectiva). Gram positivas Etapas: 1. Secuencia de DNA que se desea insertar en la cepa silvestre. 2. Incorporación de la secuencia en un vector 3. Transformación: introducción del vector en la célula hospedadora y replicación estable del inserto 4. Selección del clon transformante (Selección de recombinantes buscamos obtener cultivos puros de recombinantes de interés) a) Identificación de células recombinantes: aislar los recombinantes. Solo son detectables si se induce un cambio fenotípico estable entre el recombinante y el progenitor: - Uso de progenitores auxótrofos: incorporo en la secuencia que quiero transferir genes que les permitan tener protrotrofía. Para que no se produzca la reversión con facilidad se utilizan varias auxotrofías a la vez, lo cual reduce la probabilidad de reversión. Es una técnica muy restrictiva: para cuando quiero que se transfiera toda la secuencia. Al final del proceso se eliminan todos los progenitores y nos quedamos solo con recombinantes (los únicos que sobreviven). Inconvenientes del método: pierdo algunos recombinantes. Añado estreptomicina para quitarme a los donadores. Dos medios finales que lleven antibiótico siempre; luego adicionas los factores de crecimiento u otros para seleccionar los distintos tipos de recombinantes. - Inactivación del marcador: método complicado. Uno de los marcadores se sitúa en el sitio de reconocimiento de la enzima de restricción que se utiliza para clonar la secuencia: INACTIVACIÓN al insertarse en el genoma hospedador. Los marcadores contienen secuencias de restricción y resistencia a antibióticos; uno de los marcadores secuencia que le concedía resistencia a la ampicilina para progenitores hacer cepas sensibles. Selecciono por rastreo (método de rastreo por placa), ver qué colonia crece en una y cuál en otra b) Detección del DNA foráneo en los recombinantes -Hibridación con sondas específicas, PCR, secuenciación c) Detección de la expresión del producto: cuando el recombinante es para producir proteínas heterólogas (como la insulina humana) -Detección de mRNA -Expresión de proteínas en bacterias: bajo peso molecular, pocos puentes disulfuro, no glicosilación -Las bacterias como factorías de proteínas heterólogas tienen un escaso rendimiento y una purificación final muy compleja. Pero como podemos tener billones de ellas en poco espacio, compensan por la producción de ingentes cantidades de proteína y se consideran útiles en industria.