1 anexo i
Anuncio

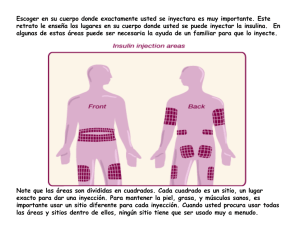
ANEXO I RESUMEN DE CARACTERÍSTICAS DEL PRODUCTO (Artículo 4a de la Directiva 65/65/CEE modificada) 1 RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO 1. NOMBRE DEL MEDICAMENTO BETAFERON 2. COMPOSICION CUALITATIVA Y CUANTITATIVA 1 ml de solución reconstituida para inyección contiene 0,25 mg (8 millones de UI) de Interferón beta-1b. La potencia se determina mediante un bioensayo basado en el efecto citopático (CPE) empleando el standard de referencia de la OMS del Interferón beta recombinante. Betaferon se ha formulado de forma que contenga 0,3 mg (9,6 millones UI) de Interferón beta-1b por vial, lo que incluye un excedente calculado del 20%. Interferón beta-1b es una proteína purificada, estéril, liofilizada, que consta de 165 aminoácidos. Se obtiene por técnicas de DNA recombinante a partir de una cepa de Escherichia coli portadora de un plásmido obtenido por ingeniería genética que contiene un gen para Interferón humano modificado, concretamente, para Interferón beta ser 17. Interferón beta-1b difiere estructuralmente del beta interferón natural humano por la presencia de serina en lugar de cisteína en posición 17, la falta de metionina en posición 1 y la ausencia de fracciones de hidrato de carbono. 3. FORMA FARMACEUTICA Polvo liofilizado estéril de color blanco o casi blanco apto para inyección. 4. DATOS CLINICOS 4.1. Indicaciones terapéuticas Betaferon está indicado para reducir la frecuencia y gravedad de las recaidas clínicas en pacientes capaces de andar (sin ayuda) que sufren esclerosis múltiple (EM) remitente recidivante, caracterizada por la aparición de, al menos, dos ataques de disfunción neurológica durante el período de los dos años anteriores, seguidos por recuperaciones completas o incompletas. Los pacientes tratados con Betaferon han mostrado una reducción en la frecuencia (30%) y gravedad de las exacerbaciones clínicas, así como una reducción en el número de hospitalizaciones relacionadas con la enfermedad. También se ha observado una prolongación de los períodos libres de recidivas. No existe evidencia del efecto de Betaferon sobre la duración de las exacerbaciones, sobre los síntomas entre exacerbaciones o sobre la progresión de la enfermedad. Tampoco hay evidencia del efecto de Betaferon en el desempeño de la actividad cotidiana o en el ámbito social. Betaferon no ha sido aún investigado en pacientes de esclerosis múltiple progresiva. No existe evidencia de efecto sobre la discapacidad. Los ensayos clínicos mostraron que no todos los pacientes responden al tratamiento con Betaferon. También se ha observado un empeoramiento de los ataques en algunos pacientes a pesar del tratamiento. No se dispone de criterios clínicos que permitan predecir la falta de respuesta o el empeoramiento en cada uno de los pacientes a tratar. 2 4.2. Posología y forma de administración El tratamiento con Betaferon deberá iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de esta enfermedad. La dosis recomendada de Betaferon es de 0,25 mg (8 MUI), correspondiente a 1 ml de solución reconstituida (ver 6.6 "Instrucciones de uso/manipulación), inyectada por vía subcutánea cada dos días. No está completamente establecida la dosis óptima. Actualmente no se conoce durante cuánto tiempo debe ser tratado el paciente. La eficacia del tratamiento durante más de dos años, no ha sido suficientemente demostrada. Debería efectuarse una evaluación clínica completa al cabo de dos años en todos los pacientes. La decisión de prolongar el tratamiento debe tomarla el médico caso por caso. No se dispone de datos de tratamientos de más de tres años. No se recomienda el tratamiento en pacientes que hayan sufrido menos de dos exacerbaciones en los dos años anteriores. El tratamiento con Betaferon se debe suspender si el paciente no responde al tratamiento y, por ejemplo, presenta progresión continua de la incapacidad durante 6 meses o requiere tratamiento adicional con ACTH o corticoides en tres ocasiones, como mínimo, durante un año. No se ha investigado la eficacia y seguridad de Betaferon en niños y adolescentes de menos de 18 años de edad. Por tanto, no deberá administrarse Betaferon en estas edades. 4.3. Contraindicaciones Betaferon está contraindicado en los siguientes casos: Embarazo (ver punto 4.6). Pacientes con historia de hipersensibilidad al interferón beta recombinante o natural, o a la albúmina humana. Pacientes con historia de alteraciones depresivas graves y/o ideación suicida Pacientes con hepatopatía descompensada. Pacientes con epilepsia no controlada adecuadamente mediante tratamiento. Pueden presentarse reacciones graves de hipersensibilidad (reacciones agudas, infrecuentes pero graves, tales como broncoespasmo, anafilaxia y urticaria). Ante reacciones graves, debe suspender Betaferon e instaurar el tratamiento médico adecuado. 3 4.4. Advertencias y precauciones especiales de empleo Antes del tratamiento con Betaferon los pacientes deben ser informados de que pueden presentarse alteraciones depresivas e ideación suicida como efecto secundario del tratamiento y de que estos síntomas deben comunicarse inmediatamente al médico que lo ha prescrito. Raramente estos síntomas pueden llevar a un intento de suicidio. Los pacientes con alteraciones depresivas e ideación suicida han de ser estrechamente vigilados y debe considerarse la interrupción del tratamiento. Betaferon debe administrarse con precaución a pacientes con historial de convulsiones y de alteraciones depresivas, así como a pacientes tratados con antiepilépticos (ver Interacciones). También debe usarse con precaución en pacientes con alteraciones depresivas y en aquellos que padezcan trastornos cardíacos preexistentes. También deberá tenerse precaución cuando se administre Betaferon a pacientes con supresión de la función medular; si se desarrolla neutropenia el paciente deberá vigilarse cuidadosamente por la posible aparición de fiebre o infección. Pueden presentarse reacciones graves de hipersensibilidad (reacciones agudas, infrecuentes pero graves, tales como broncoespasmo, anafilaxia y urticaria). Ante reacciones graves, debe discontinuarse Betaferon e instaurar el tratamiento médico adecuado. Es posible que reacciones adversas de otro tipo, de intensidad media a grave, requieran modificaciones del régimen de dosificación de Betaferon o incluso suspensión del fármaco. Antes de iniciar el tratamiento con Betaferon, y regularmente durante el mismo, debe determinarse la fórmula leucocitaria diferencial y los niveles de transaminasas (SGOT, SGPT). No se dispone de datos en pacientes con alteración de la función renal. La función renal deberá vigilarse cuidadosamente cuando tales pacientes reciban Betaferon. En los estudios sobre EM, el 45% de los pacientes ha desarrollado actividad neutralizante en suero contra el Interferon beta-1b, en al menos una ocasión. En un tercio de los pacientes se detectaron títulos positivos de actividad neutralizante en al menos dos ocasiones consecutivas. La aparición de actividad neutralizante se acompaña de una reducción de la eficacia clínica, que se hace evidente a los 18-24 meses. No se han asociado nuevos efectos adversos con la aparición de actividad neutralizante. No obstante, no se ha investigado la posibilidad de reactividad cruzada con el Interferón beta endógeno. Son escasos los datos de pacientes que habiendo desarrollado actividad neutralizante hayan completado el tratamiento con Betaferon. 4.5. Interacción con otros medicamentos y otras formas de interacción No se han realizado con Betaferon estudios específicos de interacción con medicamentos. No se conoce el efecto de la administración de 0,25 mg (8 MUI) de Betaferon, en días alternos, sobre el metabolismo de fármacos en pacientes de EM. El tratamiento de las recidivas con corticosteroides o ACTH durante hasta 28 días ha sido bien tolerado en pacientes que están recibiendo Betaferon. No se recomienda el empleo concomitante de Betaferon con otros inmunomoduladores, con excepción de corticoides o ACTH, por la falta de experiencia clínica en pacientes de EM. 4 Se ha comunicado que los Interferones originan una reducción de la actividad de enzimas dependientes del citocromo hepático P450, tanto en animales como en seres humanos. Por ello debe observarse precaución al administrar Betaferon en combinación con fármacos que tengan un estrecho índice terapéutico y dependan notablemente para su aclaramiento del sistema citocromo hepático P450, como por ejemplo los antiepilépticos. No se han realizado estudios de interacción con antiepilépticos. Influencia sobre resultados analíticos A la dosis recomendada puede presentarse leucopenia (linfopenia, neutropenia) o elevación de la SGPT. También se han observado niveles bajos de calcio, ácido úrico elevado o elevación de la SGOT, asociados al tratamiento con Betaferon. 4.6. Embarazo y lactancia Se desconoce si el Betaferon es inocuo para el feto cuando se administra a la mujer gestante, o si puede afectar la capacidad reproductora. Se han comunicado abortos espontáneos en pacientes de EM, en ensayos clínicos controlados. El Interferón beta-1b recombinante humano ha mostrado embriotoxicidad en monos rhesus causando la muerte de los fetos en el rango más alto de dosis. Por ello, está contraindicado durante el embarazo y las mujeres fértiles deben tomar medidas contraceptivas adecuadas. Si la paciente quedara embarazada, o tuviera intención de hacerlo durante el tratamiento con Betaferon, debe ser informada del riesgo potencial y recomendársele la interrupción del tratamiento (ver resultados preclínicos en la sección 5.3). No se sabe si Interferón beta-1b se excreta en la leche materna. A causa de la posible inducción por Betaferon de reacciones adversas serias en los lactantes, debe decidirse si interrumpir la lactancia o el tratamiento con el fármaco. 4.7. Efectos sobre la capacidad de conducir vehículos y utilizar maquinaria No ha sido investigado. Los efectos adversos sobre el SNC asociados al empleo de Betaferon podrían afectar la capacidad de conducir vehículos y utilizar maquinaria en pacientes susceptibles. 4.8. Reacciones adversas Es aún limitada la experiencia con Betaferon en pacientes con EM y, consecuentemente, no han podido observarse los efectos adversos de baja incidencia. Tras la administración de Betaferon se observaron frecuentemente reacciones en el lugar de inyección. El tratamiento con inyección de 0,25 mg (8 MUI) de Betaferon se ha asociado de manera significativa a manifestaciones de inflamación, dolor, hipersensibilidad, necrosis y a reacciones inespecíficas en el lugar de la inyección. La incidencia de todas estas reacciones disminuyó habitualmente con el tiempo. Frecuentemente se ha observado sintomatología gripal (fiebre, escalofríos, mialgia, malestar o sudoración). La incidencia de estos síntomas decreció con el tiempo de tratamiento. Pueden presentarse reacciones graves de hipersensibilidad (reacciones agudas, infrecuentes pero graves, tales como broncoespasmo, anafilaxia y urticaria). Ante reacciones graves, debe suspenderse Betaferon e instaurar el tratamiento médico adecuado. 5 En mujeres premenopáusicas pueden presentarse trastornos de la menstruación. Se han observado efectos adversos relacionados con el sistema nervioso central que incluían depresión, ansiedad, labilidad emocional, despersonalización, convulsiones, intentos de suicidio y confusión. 4.9. Sobredosificación Interferón beta-1b se ha administrado sin efectos adversos graves que comprometieran funciones vitales a pacientes adultos con cáncer, en dosis de hasta 5,5 mg (176 MUI) por vía i.v., tres veces/semana. 5. PROPIEDADES FARMACOLOGICAS 5.1. Propiedades farmacodinámicas Los Interferones pertenecen a la familia de las citoquinas, que son proteínas naturales. Los Interferones tienen pesos moleculares comprendidos entre 15.000 y 21.000 dalton. Se han identificado tres clases principales de Interferones denominados alfa, beta y gamma. Interferón alfa, Interferón beta e Interferón gamma tienen actividades biológicas diferentes, aunque se solapan en parte. Las actividades del Interferón beta-1b están restringidas a la especie y, por tanto, la información farmacológica de mayor interés es la que se deriva de los estudios realizados sobre cultivos de células humanas o los estudios in vivo en humanos. Interferón beta-1b ha demostrado poseer actividad antivírica y actividad inmunorreguladora. Los mecanismos mediante los cuales ejerce sus acciones en la esclerosis múltiple aún no están totalmente aclarados. Sin embargo, se sabe que las propiedades modificadoras de respuesta biológica de Interferón beta-1b están mediadas por sus interacciones con receptores celulares específicos que se hallan en la superficie de las células humanas. El enlace de Interferón beta-1b a estos receptores induce la expresión de un número de productos genéticos que se supone que son los mediadores de las acciones biológicas del Interferón beta-1b. Algunos de estos productos han sido determinados en el suero y en fracciones celulares de sangre recogida de pacientes tratados con Interferón beta-1b. Interferón beta-1b reduce la afinidad de enlace y aumenta la internalización y degradación del receptor de Interferón gamma. Interferón beta-1b también aumenta la actividad supresora de las células mononucleares de sangre periférica. No se han realizado ensayos específicos acerca de la influencia de Betaferon sobre el aparato cardiocirculatorio, aparato respiratorio ni sobre la función de órganos endocrinos. 6 5.2. Propiedades farmacocinéticas Los niveles séricos del fármaco se determinaron en pacientes y voluntarios sanos mediante un bioensayo no completamente específico, detectándose valores máximos en suero de aproximadamente 40 UI/ml en el periodo de 1-8 horas tras la inyección subcutánea de 0,5 mg (16,0 MUI) de Interferón beta-1b. Las tasas medias de aclaramiento sérico y los valores de la vida media de las fases de eliminación en suero, se han estimado a partir de varios estudios, en no más de 30 ml·min -1·kg-1 y de 5 horas, respectivamente. Las inyecciones del producto en días alternos no dan lugar a elevación de la concentración sérica del fármaco y la farmacocinética tampoco parece modificarse durante el tratamiento. La biodisponibilidad absoluta de Interferón beta-1b en administración subcutánea fue aproximadamente del 50 %. 5.3. Datos preclínicos sobre seguridad No se han realizado estudios de toxicidad aguda. Puesto que el Interferón beta humano no es activo en los roedores, los estudios de administración repetida se efectuaron en monos rhesus. Se observó hipertermia transitoria, así como un aumento significativo de los linfocitos y un descenso significativo de plaquetas y neutrófilos segmentados. No se han realizado estudios a largo plazo. Los estudios sobre reproducción en monos rhesus revelaron toxicidad materna y fetal, originando mortalidad prenatal. No se observaron malformaciones en los supervivientes. No se ha realizado investigación sobre la fertilidad. No se ha observado influencia alguna sobre el ciclo estral en monos. La experiencia con otros interferones sugiere un potencial deterioro de la fertilidad de machos y hembras. En un único estudio de genotoxicidad (test de Ames) no se observó efecto mutagénico. No se han realizado estudios de carcinogénesis. Un ensayo de transformación celular in vitro no dio indicio de potencial tumorigénico. Los estudios de tolerancia local tras administración subcutánea fueron negativos. Sin embargo, en estudios clínicos se han observado reacciones locales tras el empleo de Betaferon. 6. DATOS FARMACEUTICOS 6.1. Lista de excipientes Albúmina humana Dextrosa Ph. Eur. Ph. Eur. 6.2. Incompatibilidades Ninguna conocida 6.3. Periodo de validez del producto en el envase intacto 18 meses, a 2-8ºC, a partir de la fecha de la filtración estéril de la solución a granel después de la reconstitución según las instrucciones hasta 3 horas, a 2-8ºC 7 6.4. Precauciones especiales de conservación Conservar a temperatura entre 2 y 8ºC, tanto antes como después de reconstitución 6.5. Naturaleza y contenido del envase Vial de vidrio claro, de 3 ml, con tapón de butil-caucho de 13 mm y cierre de cápsula de aluminio. Cada vial de Betaferon va acompañado de otro vial de diluyente que contiene 2 ml de solución estéril de cloruro sódico (0,54% p/v). El diluyente está envasado en un vial de 3 ml con un tapón de butil-caucho de 13 mm y cierre de cápsula de aluminio. 6.6. Instrucciones de uso/manipulación Para reconstituir la solución a partir de Interferón beta-1b liofilizado, empléese una jeringa y aguja estériles; extraer 1,2 ml del diluyente suministrado (solución de cloruro sódico 0,54% p/v) e inyectarlo en el vial Betaferon. Disolver completamente el producto sin agitar. Observar visualmente la solución reconstituida, antes de su empleo, y desecharla si contiene partículas o está coloreada. La solución reconstituida contiene 0,25 mg (8 MUI) de Interferón beta-1b por ml. Almacenar todos los medicamentos de manera apropiada y mantenerlos fuera del alcance de los niños. 6.7. Nombre o razón social y domicilio permanente o sede social del titular de la autorización de comercialización Schering Aktiengesellschaft D-.13342 Berlin Alemania Tfno: (07) 49-30-468-0 7. NUMERO DE AUTORIZACION DE COMERCIALIZACION 8. FECHA DE APROBACION/REVISION DEL SPC 12 de Julio de 1995 8 ANEXO II AUTORIZACIONES DE FABRICATIÓN Y CONDICIONES DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN 9 ANEXO II AUTORIZACIONES DE FABRICACIÓN Y CONDICIONES DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN A. TITULAR(ES) DE LA AUTORIZACIÓN DE FABRICACIÓN (artículo 16 de la Directiva 75/319/CEE modificada) 1. Dr. Karl Thomae GmbH - Birkendorfer Straße 65, D-88397 Biberach an der Riß (Alemania). Certificado GMP expedido por el Regierungspräsidium, Tubinga, el 5 de marzo de 1992. Certificado GMP expedido por el Regierungspräsidium, Tubinga, el 3 de febrero de 1995. 2. Bender & Co. GmbH, Viena Autorización expedida por el Magistrat der Stadt Wien el 30 de mayo de 1985. Autorización expedida por el Bundesministerium für Gesundheit Konsumentenschutz, Viena, el 30 de enero de 1995. und Y Importador del medicamento (apartado 3 del artículo 16 de la Directiva 75/319/CEE modificada): Schering AG, D-13342 Berlín, Alemania, que importa de la Chiron Corporation, 4560 Horton St., Emeryville, California, EE.UU. La planta de fabricación de Chiron recibió la inspección de las autoridades europeas los días 11 y 12 de mayo de 1995. B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y UTILIZACIÓN (apartado 3 del artículo 13 del Reglamento (CEE) nº 2309/93 del Consejo, artículos 2 y 3 de la Directiva 92/26/CEE, y letra b) de la parte 4 G del anexo de la Directiva del Consejo 75/318/CEE) Especialidad farmacéutica sujeta a prescripción médica restringida no renovable. 10 C. OBLIGACIONES ESPECÍFICAS PARA EL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN (apartado 2 del artículo 13 del Reglamento (CEE) nº 2309/93 del Consejo, y letra a) de la parte 4 G del anexo de la Directiva del Consejo 75/318/CEE) La empresa presentará periódicamente informes actualizados de seguridad con arreglo a los requisitos legales (artículo 22 del Reglamento (CEE) nº 2309/93 del Consejo). La empresa, tras haber sido consultada (véase la carta de la empresa CPMP/357/95) cumplirá la obligación de completar el programa de estudios que se especifica a continuación. Los resultados de dichos estudios se someterán a la EMEA en el plazo fijado a partir de la concesión de la autorización, y constituirán la base de una reevaluación anual por la Agencia de las obligaciones del titular de la autorización de comercialización y de la relación beneficios/riesgos del medicamento Betaferon. 1. La empresa presentará un informe sobre el estudio realizado en Europa (a) y en los EE.UU./Canadá (b) sobre la forma progresiva secundaria de la esclerosis múltiple: a) estudio multicéntrico en doble ciego contra placebo para evaluar la seguridad y la eficacia de 8 millones de UI de interferón beta-1b frente a un placebo. El informe final se presentará antes del 31 de marzo de 1999; b) estudio multicéntrico en doble ciego contra placebo para evaluar la seguridad y la eficacia de dos dosis de interferón beta-1b (8 millones de UI por dosis; 4,9 millones de 2 UI/ m de superficie corporal). El informe final se presentará antes del 31 de diciembre de 2000. 2. Deberá diseñarse un ensayo sensible y fiable de la actividad de neutralización. La empresa presentará un informe sobre este ensayo, incluida la validación del mismo, antes del 31 de diciembre de 1995. 3. La empresa debe volver a analizar las muestras plasmáticas retenidas de todos los pacientes de los estudios piloto de actividad de neutralización mediante el nuevo ensayo. Los resultados se analizarán con relación a la eficacia y la seguridad, y se recogerán en un informe que se presentará antes del 31 de diciembre de 1996. 4. La empresa estudiará las reacciones cruzadas del Betaferon con el interferón beta in vitro, y presentará los resultados antes del 31 de diciembre de 1995. 5. La empresa presentará los datos combinados completos del estudio de seguimiento de 5 años que completa los estudios TB 01/3103 y TB 01/3104 antes del 31 de marzo de 1996. 6. La empresa deberá realizar y completar los estudios clínicos suplementarios detallados en el punto 3 de la sección II del informe de evaluación (CPMP/213/95), que se resumen así: a) estudio sobre la persistencia del efecto en la forma remitente-recurrente de la esclerosis múltiple: la empresa deberá presentar el proyecto detallado de protocolo de ensayo clínico antes del 31 de octubre de 1995. b) estudios farmacocinéticos: la investigación se iniciará el 1 de enero de 1996, para cuando se dispondrá de un ensayo sensible y validado de medición farmacocinética (véase el punto 2). 11 7. Las aclaraciones restantes sobre puntos de calidad que deberá presentar y el plazo previsto se indican en el punto 3.7 de la sección II del informe de evaluación (CPMP/213/95). 12 ANEXO III ETIQUETADO Y PROSPECTO 13 A - ETIQUETADO 14 Folding box Betaferon 0,25 mg (8 millones de UI) / ml Interferon beta-1b (pINN) Medicamento sujeto a prescripción médica. I. 1 vial con polvo liofilizado para inyección contiene: Interferon beta-1b 0,25 mg (8 millones de UI) / ml tras reconstitución* Albúmina humana, Dextrosa II. 1 vial de diluyente contiene: Solución al 0,54 % de cloruro sódico, 2 ml Para inyección subcutánea tras reconstitución con 1,2 ml de diluyente para Betaferon. *Betaferon se ha formulado de forma que contenga 0,3 mg (9,6 millones de UI) de interferon beta-1b por vial, lo que incluye una sobredosificación del 20%. 1 ml de solución reconstituida para inyección contiene 0,25 mg (8 millones de UI) de interferon beta-1b. Manténgase fuera del alcance de los niños Conservar a temperatura entre 2 - 8 °C 1 vial = 1 sólo uso Posologie: Véase el prospecto adjunto Schering AG, Alemania PVP IVA PVP Lote: Cad.: 0,25 mg 15 viales con polvo liofilizado para inyección 15 viales con diluyente s.c. Vial with powder for injection Betaferon Interferon beta-1b 1 vial contiene: Interferon beta-1b, 0,25 mg (8 millones de UI) / ml tras reconstitución Para inyección subcutánea tras reconstitución. Conservar a temperatura entre 2 - 8 °C Dosis única. SCHERING Lote: Caducidad: Diluent vial Diluyente para Betaferon Solución al 0,54 % de Cloruro sódico 2 ml Conservar a temperatura entre 2 - 8 °C Estéril. Dosis única. Para dilución del polvo liofilizado de Betaferon para inyección. SCHERING Lote: Caducidad: 15 B - PROSPECTO 16 Betaferon Información importante, léase con atención. Betaferon contiene las siguientes sustancias (Composición) Principios activos: 1 ml de solución reconstituida para inyección contiene 0,25 mg (8 millones de UI) de Interferón beta-1b. Betaferon se ha formulado de forma que contenga 0,3 mg (9,6 millones de UI) de Interferón beta-1b por vial, lo que incluye una sobredosificación del 20 %. Excipientes: Albúmina humana, dextrosa. ¿De qué presentaciones de Betaferon se dispone? Cada envase de Betaferon contiene 15 viales de Interferón beta-1b y 15 viales de solución de cloruro sódico al 0,54 %. ¿Cómo actúa Betaferon? (Propiedades del preparado) La esclerosis múltiple (EM) es una enfermedad del sistema nervioso central (SNC) (es decir, del cerebro y la médula espinal), de causa desconocida. Se supone que la existencia de una respuesta anormal del sistema inmunitario desempeña un importante papel en el proceso que lesiona el SNC. Se ha demostrado que el Interferón beta-1b modifica la respuesta del sistema inmunitario. Los interferones pertenecen a la familia de las citoquinas, que son proteínas naturales. ¿Quién es responsable de Betaferon? (Nombre y dirección del propietario de la autorización de comercialización, fabricante) Schering AG D-13342 Berlin Alemania ¿Con qué fin se utiliza Betaferon? (Indicaciones) Betaferon está indicado para el tratamiento de pacientes capaces de andar, que sufren esclerosis múltiple (EM) remitente recidivante, caracterizada por la aparición de al menos dos ataques de disfunción neurológica durante el periodo de los dos años precedentes, seguidos por recuperaciones completas o incompletas. En este colectivo de pacientes, Betaferon ha demostrado reducir la frecuencia y gravedad de las recaídas clínicas, disminuir el número de hospitalizaciones relacionadas con la EM y prolongar los periodos exentos de recaídas. No todos los pacientes responden al tratamiento con Betaferon. No hay evidencia del efecto de Betaferon sobre la duración de los ataques, síntomas entre ataques o sobre la progresión de la enfermedad. No se conoce el efecto de Betaferon sobre el desempeño de las actividades diarias ni sobre el ámbito social. 17 Betaferon no ha sido aún investigado en pacientes de esclerosis múltiple progresiva. No hay evidencia de efecto sobre la discapacidad. Los estudios clínicos mostraron que no todos los pacientes responden al tratamiento con Betaferon. A pesar del tratamiento, se ha observado un empeoramiento de los síntomas durante los ataques en algunos pacientes. No es posible predecir de antemano en qué pacientes no se obtendrá respuesta o en cuáles se producirá un empeoramiento a pesar del tratamiento. ¿Cuándo no debe usarse Betaferon? (Contraindicaciones) No deberá usarse Betaferon en caso de embarazo o antecedentes de hipersensibilidad al interferón beta, natural o recombinante, o a la albúmina humana. No deberá utilizarse Betaferon en menores de 18 años, ya que no se ha investigado en este grupo de edad. Tampoco deberá utilizarse Betaferon si existen antecedentes de enfermedad depresiva grave y/o ideación suicida, insuficiencia hepática descompensada o epilépsia tratada de forma inadecuada. Si se manifiestan reacciones graves de hipersensibilidad, el tratamiento con Betaferon deberá ser interrumpido. ¿Qué precauciones deben observarse durante el empleo de Betaferon? Algunos pacientes han comunicado depresión e ideación suicida. En raros casos éstas pueden conducir a un intento de suicidio. Si Vd. experimenta tales síntomas, contacte rápidamente con su médico. Debe administrarse con precaución en pacientes con historia de convulsiones o de depresión, o si sufre de trastornos cardíacos preexistentes. También deberán tomarse precauciones en pacientes sometidos a un tratamiento con antiepilépticos. Betaferon también deberá ser administrado con precaución si se padece de alguna alteración de la médula ósea. Si el número de glóbulos blancos desciende, el médico deberá prestar especial atención a la posible aparición de fiebre o infecciones. Se desconoce si Betaferon tiene un efecto negativo sobre la fertilidad en seres humanos, pero basándose en la experiencia con otros interferones, no puede ser descartada una reducción de la fertilidad en hombres o mujeres. No existe información sobre el uso de Betaferon en pacientes con problemas renales. Por lo tanto si se padece este tipo de problemas, deberá monitorizarse la función renal durante el tratamiento. Durante el tratamiento con Betaferon, el organismo puede producir sustancias que pueden reducir la eficacia del tratamiento. Se trata de los llamados "anticuerpos nuetralizantes", que sólo se producen en algunos pacientes, Sin embargo, no es posible saber de antemano si un determinado sujeto se contará o no entre los pacientes en los que el tratamiento tendrá eficacia reducida. 18 ¿Puede emplearse Betaferon durante el embarazo y la lactancia? Betaferon esta contraindicado en caso de embarazo o en mujeres que tienen intención de concebir. Si se desea un embarazo, debe discutirse primero con el médico. Mientras se utilice Betaferon, las mujeres en edad de concebir deberán adoptar medidas anticonceptivas adecuadas. Si la paciente quedara embarazada, deberá suspender el tratamiento y contactar con el médico inmediatamente. No se sabe si el Interferón beta-1b se excreta por la leche materna. Sin embargo, ya que teóricamente sería posible que ello originara reacciones serias en los lactantes, debe discutirse el asunto con el médico y optar por interrumpir la lactancia natural o la administración de Betaferon. ¿Qué debe Vd. tener en cuenta si está tomando otra medicación? (Interacciones) Betaferon no debería ser utilizado con sustancias que modifiquen la respuesta del sistema inmunitario, con la excepción de corticoides o ACTH. Debe observarse precaución al administrar Interferón beta-1b en combinación con otros fármacos que necesitan para su metabolismo un determinado sistema enzimático hepático (conocido como sistema citocromo P450). A este tipo de fármacos pertenecen algunos ampliamente utilizados contra la fiebre y el dolor, anticonceptivos orales y antiepilépticos. ¿Cómo se emplea Betaferon? (Dosificación y administración) El tratamiento con Betaferon debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la enfermedad. Por el momento se desconoce cuánto tiempo debe durar el tratamiento con Betaferon. No se ha establecido la eficacia de este tratamiento durante más de dos años. La duración del tratamiento debe decidirla el médico. No se recomienda el tratamiento en pacientes que hayan tenido menos de dos ataques durante los dos años precedentes. Antes de la administración, se prepara la solución para inyección de Betaferon, empleando para ello uno de los viales con el polvo de Betaferon, al que se añaden 1,2 ml del líquido de uno de los viales de disolvente. 1,0 ml de la solución así preparada se inyecta subcutáneamente (bajo la piel). Las inyecciones se repetirán en días alternos. 1,0 ml de la solución así preparada equivale a 0,25 mg (8 millones de UI). El procedimiento anteriormente descrito lo realizará su médico o practicante. También lo podrá efectuar Vd. mismo, una vez que se le haya instruido y adiestrado en la técnica de manera cuidadosa. Para ayudarle a poderse administrar Vd. mismo el Betaferon, en este prospecto se incluyen instrucciones detalladas, en las que también se explica cómo preparar la solución para inyección. Forma de actuar ante el olvido de una inyección Si se olvida la administración de una inyección en el horario previsto, deberá administrarse tan pronto como sea posible. La siguiente inyección deberá administrarse 48 h más tarde. 19 ¿Qué efecto podría tener una sobredosis de Betaferon? (Sobredosificación) La administración de dosis de Betaferon muy superiores a las recomendadas para la EM no ha dado lugar a situaciones que entrañasen riesgo para la vida. Sin embargo, en caso de sobredosis accidental, se debe consultar con el médico que ha prescrito el tratamiento con Betaferon. Se debe consultar también con el médico, si por error se administra la inyección con demasiada frecuencia (por ejemplo una inyección cada 24 h en lugar de una inyección cada 48 h). ¿Se presenta algún efecto secundario durante el empleo de Betaferon? Frecuentemente se observan reacciones en el lugar de inyección, incluyendo inflamación, dolor, hipersensibilidad, muerte de células cutáneas (necrosis), así como reacciones inespecíficas. La aparición de reacciones en el lugar de inyección suele decrecer con el tiempo de tratamiento. Se han observado frecuentemente síntomas gripales (fiebre, escalofríos, dolor muscular, sensación general de malestar o sudoración). También la presentación de estos síntomas decrece con el tiempo. En mujeres premenopáusicas pueden aparecer trastornos de la menstruación. Se han observado reacciones adversas relacionadas con el sistema nervioso central entre las que se cuentan depresión, ansiedad, inestabilidad emocional, pérdida de identidad o de la realidad (despersonalización), convulsiones, intentos de suicidio y confusión. Las reacciones graves de hipersensibilidad son raras, si se manifiesta una reacción grave consulte a su médico inmediatamente. El número de glóbulos blancos de la sangre puede disminuir y la actividad de enzimas estrechamente relacionadas con la función del hígado puede aumentar. Por ello, tanto antes del tratamiento como con regularidad a lo largo del mismo, deberá determinarse la fórmula leucocitaria diferencial y efectuarse un análisis bioquímico del suero (SGOT, SGPT). Comunique a su médico cualquier efecto secundario que observe, en especial si son graves o no figuran entre los citados anteriormente. ¿Cómo debe conservarse Betaferon? Betaferon debe conservarse a temperatura baja (2-8 C). Betaferon deberá estar guardado en la nevera (pero no en el congelador), de donde se sacará en el momento de ir a preparar la solución para inyección. Si la solución ya preparada de Betaferon no se inyecta inmediatamente, puede guardarse en la nevera (no en el congelador) durante un tiempo máximo de tres horas. La fecha de caducidad de este medicamento está impresa en el envase. ¡ No utilice el medicamento después de esa fecha en ningún caso !. Los medicamentos deben mantenerse fuera del alcance de los niños. Fecha de la última revisión del prospecto: 12 de julio de 1995. 20 Anexo: PROCEDIMIENTO PARA LA AUTO-INYECCIÓN Las instrucciones siguientes tienen por objeto explicar cómo debe preparar la solución de Betaferon y como debe proceder para inyectársela Vd. mismo. Por favor, lea cuidadosamente las instrucciones y sígalas paso a paso. Su médico o practicante le instruirá y adiestrará en el procedimiento y técnica de auto-administración. No intente la auto-administración hasta estar seguro de haber comprendido cómo ha de preparar la solución para inyección y cómo ha de inyectársela. Las instrucciones comprenden los siguientes pasos principales: I. Preparativos para la auto-inyección II. Extracción del disolvente (solución de cloruro sódico) con la jeringa III. Inyección del volumen requerido de disolvente (1,2 ml) en el vial de Betaferon (polvo) IV. Extracción del volumen de solución requerido para la inyección (1,0 ml) con la jeringa V. Elección y preparación del lugar de inyección y administración de la solución de Betaferon (1,0 ml) por vía subcutánea (bajo la piel) I. Preparativos para la auto-inyección 1. Reúna Vd. todo el equipo antes de comenzar el proceso. Vd. necesitará: - el vial con disolvente para Betaferon (solución de cloruro sódico al 0,54%) el vial de Betaferon (polvo) jeringa de 2 ml aguja del 21 aguja del 27 toallitas o torundas empapadas en alcohol contenedor para jeringas y agujas usadas 2. Lávese las manos cuidadosamente con agua y jabón. 3. Quite las cápsulas protectoras del vial de polvo y del de disolvente 4. Use torundas con alcohol para limpiar los tapones de los viales - frotando en una sola dirección y usando una torunda para cada vial. NOTA: Deje una torunda con alcohol sobre el tapón de cada vial hasta su empleo. II. Extracción del disolvente (solución de cloruro sódico) con la jeringa Para disolver el polvo blanco del vial de Betaferon sólo debe usarse el vial de disolvente incluido en el envase. 21 1. Con los brazos apoyados sobre una superficie estable, saque la jeringa de su envoltura. Tenga cuidado de no tocar la boquilla. 2. Saque de su envoltura la aguja del 21 y colóquela firmemente encajada en la boquilla de la jeringa. Retire la caperuza protectora de la aguja. No toque la aguja. 3. Ajuste el émbolo de la jeringa a la marca 1,2 ml. NOTA: Lea las etiquetas de los viales - tome el vial de disolvente y retire la torunda con alcohol que se halla sobre el tapón. 4. Con el vial de disolvente colocado sobre una superficie estable, introduzca lentamente la aguja, a través del tapón de goma, dejando la punta en la parte alta del vial. NOTA: Al introducir y sacar las agujas de los viales, asegúrese de no tocar con las manos ni las agujas ni los tapones de los viales. Si tocara el tapón, límpielo con una nueva torunda con alcohol. Si tocara la aguja o la boquilla de la jeringa, échela al contenedor y comience con una nueva. Si la aguja tocara cualquier superficie, échela al contenedor y comience con una nueva. 5. Empuje despacio el émbolo hasta el final para inyectar lentamente aire en el vial (deje la aguja en el vial de disolvente). 6. Vuelque el vial de disolvente. NOTA: Conserve la punta de la aguja dentro del líquido. 7. Con los brazos apoyados sobre una superficie estable, sostenga con una mano el vial y la jeringa y, con la otra mano, tire lentamente del émbolo de la jeringa, hasta alcanzar la marca 1,2 ml (para extraer esa cantidad de líquido). 8. Conservando el vial invertido, dé suaves golpecitos en la jeringa hasta que todas las burbujas de aire suban hasta el extremo superior del cuerpo de la jeringa. 9. Empuje el émbolo cuidadosamente para expulsar SOLO EL AIRE a través de la aguja. Asegúrese de que la jeringa contiene 1,2 ml de disolvente. 10. Retire la jeringa con su aguja del vial de disolvente. III. Inyección del volumen requerido de disolvente (1,2 ml) en el vial de Betaferon NOTA: Coja el vial de Betaferon y retire de su tapón la torunda con alcohol. 1. Manteniendo el vial de Betaferon sobre una superficie estable, introduzca lentamente la jeringa (que contiene 1,2 ml de líquido) con su aguja a través del tapón del vial. 2. Empuje lentamente el émbolo hacia abajo, dirigiendo la aguja hacia la pared del vial para que el líquido resbale por ésta (la inyección del disolvente directamente sobre el polvo produciría un exceso de espuma). 3. Asegúrese de que la aguja no entre en contacto con el polvo ni con la solución obtenida. 4. Después de haber inyectado completamente el disolvente de la jeringa en el vial de Betaferon, sostenga el vial entre los dedos pulgar, índice y corazón con la aguja, sin dejar de sujetar la jeringa. 5. Gire suavemente la mano para disolver completamente el polvo blanco de Betaferon. ¡NO AGITAR! 22 6. Observe la solución atentamente (debe ser transparente). NOTA: Si la solución contiene partículas o está coloreada, deséchela y comience de nuevo. IV. Extracción del volumen de solución requerido para la inyección ( 1,0 ml) 1. Incline ligeramente el vial de la solución de Betaferon manteniendo la punta de la aguja en el punto más bajo del vial. NOTA: Mantenga la punta de la aguja en el líquido. 2. Haga retroceder el émbolo para hacer pasar 1,0 ml de líquido a la jeringa. 3. Invierta el vial y mantenga la jeringa con la aguja dirigida hacia arriba. 4. Dé suaves golpecitos en la jeringa hasta que todas las burbujas de aire asciendan hasta el extremo superior del cuerpo de la jeringa. 5. Empuje cuidadosamente el émbolo para expulsar SOLO EL AIRE a través de la aguja. 6. Retire la jeringa, dejando la aguja en el vial. 7. Coloque la jeringa (sin aguja) sobre una superficie, asegurándose de que la boquilla no toque la superficie. 8. Tome la aguja del 27, sáquela de su envoltorio, sin quitar la caperuza protectora, e insértela firmemente en la boquilla de la jeringa. 9. Deseche el resto de solución que queda en el vial y la aguja que se encuentra en él. NOTA: La inyección debe administrarse inmediatamente después de preparar la solución. Si la inyección tuviera que retrasarse, deje la jeringa con la solución preparada en el frigorífico, sin congelarla. La solución para inyección ya preparada puede conservarse en la nevera durante 3 horas. La inyección deberá administrarse antes de transcurrido este plazo. V. Elección y preparación del lugar de inyección, e inyección de la solución de Betaferon (1,0 ml) por vía subcutánea (bajo la piel) 1. Elija el lugar de inyección (ver diagrama LUGARES DE INYECCIÓN en el "Calendario para la administración" que se acompaña). Sostenga la jeringa como un lápiz o un dardo. Use un lugar de inyección diferente cada día: - Brazos (zona superior trasera) Abdomen (excepto alrededor del ombligo y cintura) Nalgas Muslos (zona frontal y lateral, excepto rodilla e ingle) NOTA: No inyectar en zonas en las que Vd. perciba bultos, nódulos firmes, depresiones, dolor o cambio de coloración; informe a su médico o practicante de cualquier cosa que note. 2. Emplee una torunda con alcohol para limpiar la piel en el lugar de inyección; déjela secar al aire. 23 3. Deseche la torunda. 4. Tome la jeringa con la aguja del 27. Quite la caperuza protectora, asegurándose de no tocar la aguja. 5. Pellizque suavemente la piel que rodea al punto de inyección (para levantarla un poco). 6. Apoyando la muñeca en la zona próxima a la de inyección introduzca la aguja recta en la piel, con un movimiento rápido y sin titubeos. 7. Inyecte el medicamento empujando el émbolo con un movimiento lento y constante hasta el final, de modo que la jeringa quede vacía. 8. Apretando un algodón sobre el lugar de inyección, retire la jeringa con la aguja. 9. Masajee suavemente el lugar de inyección con una torunda seca de algodón o gasa. 10. Arroje la jeringa y la aguja en el contenedor. LUGAR DE INYECCIÓN Elección del lugar de inyección Betaferon (Interferón beta-1b) debe inyectarse en el tejido subcutáneo (bajo la piel). Las áreas más idóneas para la inyección son las flojas y blandas, alejadas de articulaciones y nervios. Cada día de tratamiento puede elegir un lugar de inyección entre los señalados en el diagrama. Es buena idea decidir el lugar de inyección antes de preparar la jeringa. Si le cuesta trabajo acceder a algunos de los posibles lugares de inyección, solicite ayuda de la persona que le atiende o de alguien entrenado en poner inyecciones. Rotación de los lugares de inyección El cambio de lugar de cada inyección ayuda a evitar reacciones locales, permitiendo que el punto de inyección tenga tiempo de recuperarse hasta que le vuelva a tocar una nueva inyección. La inyección del día no debe ponerse en la misma zona que la anterior. Conserve una anotación de dónde y cuándo se inyectó la última vez. Una forma de hacerlo es anotar esta información en el "Calendario para la administración" que se acompaña. Vd. puede volver a inyectarse en un lugar después de 1 semana. En el caso de que todas las zonas posibles estén doloridas, consulte con su médico acerca de la posibilidad de utilizar otras zonas. 24