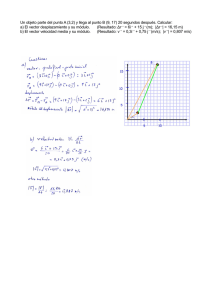
UNIVERSIDAD AUTÓNOMA DE BARCELONA
Anuncio

UNIVERSIDAD AUTÓNOMA DE BARCELONA FACULTAD DE BIOCIENCIAS MÁSTER EN GENÉTICA AVANZADA “EVALUACIÓN DEL FUNCIONAMIENTO DE UN VECTOR BICISTRÓNICO EN EL ANÁLISIS DE LA ACTIVIDAD DE SECUENCIAS IRES EN DIFERENTES LÍNEAS CELULARES” 2011 – 2012 MARÍA JOSÉ MUÑOZ 1 LABORATORIO DE ANATOMÍA PATOLÓGICA HOSPITAL VALL D’HEBRON Certifico que el trabajo para disertación de Máster en Genética Avanzada de la candidata María José Muñoz Guijarro ha sido concluido de conformidad con las normas establecidas; por lo tanto, puede ser presentado para la calificación correspondiente. Dr. Trond Aasen Director Barcelona, 11 de Julio 2012 2 Contenido 1 RESUMEN ............................................................................................................................ 5 2 INTRODUCCIÓN ................................................................................................................ 7 2.1 Mecanismos de inicio de traducción ...................................................................................... 7 2.1.1 Características del inicio de la traducción cap-depediente .......................................... 7 2.1.2 Características de la iniciación de la traducción cap-independiente ........................... 8 2.2 Revisión de los sitios de entrada internos del ribosoma (IRES) y las proteínas de unión al ARN ....................................................................................................................................... 9 2.2.1 Elementos IRES .......................................................................................................... 9 2.2.2 Factores trans-activadores IRES (ITAFs) ................................................................... 9 2.3 IRES celulares ........................................................................................................................ 9 2.3.1 Identificación de elementos IRES en mARN celular .................................................. 9 2.3.2 Datos funcionales e ITAFs importantes para la actividad celular de secuencias IRES ………………………………………………………………………………………………………………………………10 2.4 Vectores bicistrónicos .......................................................................................................... 11 3 OBJETIVOS ....................................................................................................................... 13 3.1 Objetivos generales .............................................................................................................. 13 3.2 Objetivos específicos ............................................................................................................ 13 4 MATERIALES Y MÉTODOS .......................................................................................... 14 4.1 Descripción de la metodología ............................................................................................. 14 4.2 Reacción en cadena de la polimerasa (PCR) ........................................................................ 14 4.3 Purificación de los fragmentos de PCR usando el kit Wizard® SV gel and PCR clean-up system. .................................................................................................................................. 16 4.4 Digestión con enzimas de restricción ................................................................................... 16 4.5 Formación de moléculas de ADN recombinante.................................................................. 16 4.5.1 Desfosforilación del vector ....................................................................................... 17 4.5.2 Ligación..................................................................................................................... 17 4.5.3 Transformación ......................................................................................................... 18 4.6 Extracción de ADN plasmídico (mini y midipreps) ............................................................. 18 4.7 Análisis del ADN extraído ................................................................................................... 19 4.8 Test de mycoplasma ............................................................................................................. 19 4.9 Transfección a líneas celulares ............................................................................................. 20 3 4.9.1 Preparación y contaje de células para transfectar ...................................................... 21 4.9.2 Transfección celular usando fosfato de calcio .......................................................... 21 4.9.3 Mecanismo de transfección celular con jetPEITM (polímero catiónico) .................... 22 4.10 Western blot ......................................................................................................................... 22 4.10.1 Lisado celular ............................................................................................................ 22 4.10.2 Cuantificación de proteínas mediante el kit bio-rad protein assay ............................ 23 4.10.3 Electroforesis en gel de poliacrilamida SDS-PAGE. ................................................ 24 4.10.4 Revelado de la membrana de nitrocelulosa ............................................................... 25 4.11 Cultivo de líneas celulares bajo condiciones de estrés ......................................................... 25 4.12 Construcción de partículas infeccionas con el plásmido pPLCX ......................................... 26 4.12.1 Construcción del plásmido pLPC-SRp20.................................................................. 27 4.12.2 Construcción del plásmido pPLC-MFR .................................................................... 27 4.13 Producción de partículas virales usando células Phoenix .................................................... 27 5 RESULTADOS ................................................................................................................... 29 5.1 Amplificación mediante PCR de secuencias IRES .............................................................. 29 5.2 Clonación de secuencias IRES en el plásmido reportero eucariota pMFR .......................... 29 5.3 Extracción y análisis de ADN plasmídico (mini y midipreps) ............................................. 30 5.4 Cultivo de líneas celulares en medio DMEM y test de mycoplasma ................................... 31 5.5 Transfección del vector pMFR – IRES en células 293T ...................................................... 32 5.5.1 Análisis de la fluorescencia de las proteínas Cherry y GFP ...................................... 32 5.6 Western blot cuantificación de la expresión de Cherry y GFP ............................................. 33 5.6.1 Cuantificación de proteínas mediante el kit bio-rad protein assay ............................ 33 5.7 Producción viral en células Phoenix..................................................................................... 37 5.8 Establecimiento de líneas celulares productoras de virus .................................................... 37 5.9 Cultivo de líneas celulares bajo condiciones de estrés ......................................................... 39 5.10 Evaluación de la expresión del plásmido pLPC-SRp20 ....................................................... 40 6 DISCUSIÓN ........................................................................................................................ 42 7 CONCLUSIONES Y FUTURO ........................................................................................ 46 8 BIBLIOGRAFIA ................................................................................................................ 48 4 1 RESUMEN La traducción del mARN en células eucariotas típicamente ocurre mediante el mecanismo “cap-dependiente”. Un modelo alternativo “mediado por IRES” es empleado por varios virus y, recientemente este mecanismo de traducción cap-independiente también se ha visto que es utilizado por ciertos mARNs eucariotas. La traducción IRES-dependiente es frecuentemente activada bajo ciertas circunstancias incluyendo el estrés celular como: la hipoxia, la falta de nutrientes, o durante la mitosis. Sin embargo, es poco conocido el número de mARNs eucariotas que contienen elementos IRES funcionales, o como estos son regulados por varios estímulos celulares (estrés), y la función de las denominadas proteínas ITAF, las cuales modulan su represión traduccional, o activación. El objetivo de este proyecto, por lo tanto, fue probar un reportero bicistrónico fluorescente y su utilidad para analizar elementos IRES y como estos pueden ser influenciados por estrés celular, modulación de las vías de señalización y su regulación por factores ITAF específicos conocidos. Se realizó amplificación por PCR de varias secuencias IRES conocidas, las mismas que fueron clonadas para el análisis de la expresión cap-dependiente (visualizada por la expresión de la proteína mCherry) y la mediada por IRES (que activa la traducción de GFP). Una vez obtenidos los constructos, se analizó la expresión de Cherry/EGFP mediante western blot, microscopía de fluorescencia y con un lector de fluorescencia múltiple. Los constructos cherry-IRES-GFP fueron clonados en un vector retroviral y transfectados en células Phoenix. Con este vector se produjeron partículas virales que se utilizaron para infectar una variedad de líneas celulares como HaCaT, HeLa, A549 y MDA-MB-231. Además, se evaluó el funcionamiento de los ITAF SRp20 y YB-1 mediante transfección en líneas celulares que expresaban cherry-IRES-EGFP. Se observó una fuerte activación del IRES de C-myc en la línea celular de cáncer mamario MDA-MB-468 determinada por SRp20. En resumen, en este estudio de prueba, nos hemos concentrado en el uso de un vector fluorescente para estudiar elementos IRES, conocidos y desconocidos, se ha demostrado la utilidad del vector, además se utilizó retrovirus para la generación de líneas celulares 5 reporteras estables, y la identificación de cómo el estrés celular, las vías de señalización y los factores ITAFs, pueden modular la traducción cap-independiente de proteínas. 6 2 INTRODUCCIÓN La biosíntesis de proteínas es un proceso complejo que consta de tres fases principales: iniciación, elongación y terminación (Lodish, et al., 2000; Strachan & Read., 1999). Los organismos eucariotas, tienen varias estrategias para regular la traducción del mARN (Fitzgerald & Semler, 2009), siendo la iniciación la fase más compleja de la traducción, la cual puede continuar por diferentes rutas (Sonenberg & Hinnebusch, 2009; Jackson, et al., 2010). Además del modelo canónico cap-dependiente de reconocimiento y escaneo del ribosoma, un método alternativo, cap-independiente, de traducción fue descrito por primera vez en los picornavirus y, posteriormente en un subconjunto creciente de mARNs celulares. En este mecanismo 5’-independiente de inicio de la traducción, los ribosomas son reclutados hacia el mARN en un sitio de entrada interna del ribosoma (IRES). Los IRES’s son elementos estructurales del ARN que funcionan en la unión de los ribosomas al mARN y modulan la traducción de mARNs celulares especialmente durante el desarrollo, el estrés (por ejemplo hypoxia en tumores) y la enfermedad, lo que sugiere que estas estructuras del ARN son importantes para la dirección de la traducción en contextos celulares en los que la traducción cap-dependiente esta down-regulado (Fitzgerald & Semler, 2009). 2.1 Mecanismos de inicio de traducción 2.1.1 Características del inicio de la traducción cap-depediente La mayor parte de regulación traduccional se produce durante la fase de iniciación, por lo tanto se ha puesto mayor énfasis en el estudio de los detalles moleculares de los mecanismos convencionales y los mecanismos alternativos de inicio (Sonenberg & Hinnebusch, 2009; Jackson, 2005). La traducción de los mARNs celulares ocurre con mayor frecuencia a través del mecanismo de iniciación cap-dependiente. Los mARNs celulares contienen un capuchón de 7-metil guanosina en el extremo 5’del ARN y esta estructura es reconocida por el factor de inicio eucariota 4F (eIF4F) del complejo de unión cap. El complejo consta de los factores de iniciación 4A, 4G y 4E, que reclutan al ribosoma al sitio de inicio. El factor eIF4E es el componente del complejo de unión, mientras eIF4G es una proteína de andamiaje que se une a eIF4E, eIF4A y al mARN. eIF4A es una helicasa dependiente de ATP responsable de la relajación de la estructura secundaria y terciaria del ARN durante el proceso. Esta actividad helicasa es estimulada por el factor asociado, 7 eIF4B. La subunidad 40S ribosomal se une a un complejo proteico que consta de eIF1, eIF2-GTP-Met-tRNA (complejo ternario), eIF3, y eIF5. El ensamblaje del complejo proteico, denominado 43S complejo de pre-inicio, se une al mARN en la estructura del cap mediante la interacción de un dominio central de eIF4G con eIF3. El límite del complejo de pre-inicio entonces examina la molécula de mARN hasta encontrar un codón de inicio AUG que es reconocido en un contexto favorable (Fitzgerald & Semler, 2009). Cuando se encuentra un codón de inicio adecuado, el GTP se hidroliza a GDP en presencia de eIF5, y varios de los factores de iniciación se disocian. La subunidad ribosómica grande entonces, se une a la subunidad pequeña para generar la competente elongación 80S del ribosoma. En este punto se inicia la síntesis de proteínas, y los factores de inicio se reciclan para posteriores rondas de iniciación. Algunas condiciones celulares, tales como el estrés o una infección viral puede resultar en una baja regulación de la traducción cap-dependiente, frecuentemente al interferir con los factores de iniciación (Fitzgerald & Semler, 2009). 2.1.2 Características de la iniciación de la traducción cap-independiente En el mecanismo de inicio de traducción cap-independiente, los ribosomas son reclutados al ARN mediante un mecanismo desconocido. La subunidad 40S ribosomal reconoce una secuencia de ARN, una estructura o un complejo de ribonucleoproteína dentro de la región 5’ no codificante (5’ UTR), y el sitio de inicio en el codón de inicio auténtico puede producirse varios cientos de nucleótidos downstream del extremo 5’. Esta forma alternativa de inicio de traducción no requiere un cap en el extremo 5’ como un sitio de reunión para los factores de iniciación, y el reconocimiento del cap de la subunidad ribosomal 40S a través de la formación del complejo de unión formado por el factor eIF4F intacto no se produce. El mecanismo de inicio cap-independiente, por lo tanto, involucra características que se distinguen de la unión al cap canónica, se lleva a cabo por el modelo de análisis ribosomal (Fitzgerald & Semler, 2009). 8 2.2 Revisión de los sitios de entrada internos del ribosoma (IRES) y las proteínas de unión al ARN 2.2.1 Elementos IRES Los poliovirus (PV) y el virus de encefalomiocarditis (EMCV) presentan genomas virales de ARN sin capuchón y se ha visto que contienen secuencias en sus extremos 5’UTR que están mediando la traducción eficiente en células eucariotas a través de la unión interna de los ribosomas (Pelletier & Sonenberg, 1988; Pelletier, et al., 1988; Jang, et al., 1988). En casi todas las infecciones por picornavirus, la traducción cap-dependiente se apaga y el ARN viral utiliza la traducción mediada por IRES para dirigir la síntesis de proteínas virales (Fitzgerald & Semler, 2009). 2.2.2 Factores trans-activadores IRES (ITAFs) Los elementos IRES típicamente consisten en largas y altamente estructuradas secuencias de ARN que funcionan para reclutar ribosomas, aunque el mecanismo de reclutamiento no ha sido totalmente definido y puede variar entre los diferentes elementos IRES. Algunos, por ejemplo, requieren por lo menos un subconjunto de eIFs así como ciertas proteínas de unión al ARN para facilitar la traducción mediada por IRES (CostaMattioli, et al., 2004; Walter, et al., 2002). Estos factores no canónicos se denominan factores trans-activadores IRES, o ITAFs. Varios ITAFs han demostrado, previamente, que se unen a los elementos IRES y actúan en la traducción mediada por IRES (Mitchell, et al., 2005; Holcik, et al., 2003; Schepens, et al., 2007; Paek, et al., 2008). 2.3 IRES celulares 2.3.1 Identificación de elementos IRES en el mARN celular Ha sido difícil identificar las clases funcionales de IRES celulares porque existe una secuencia primaria pequeña principal o porque presentan similitudes estructurales entre ellos, lo que provoca variaciones en su eficiencia. Mientras que algunos IRES virales pueden tener contextos comunes de síntesis y traducción en el citoplasma de células cíclicas (dando lugar a clasificaciones estructurales y funcionales), un gran número de contextos celulares que requieren regulación diferencial podría dar lugar a un número muy 9 grande de clases IRES con secuencia diferente y muchos componentes estructurales (Baird, et al., 2006). Por lo tanto, los IRES celulares han sido a menudo discutidos en términos de las condiciones que inducen su actividad o de las condiciones que resultan de su actividad. Estructuras secundarias se han derivado de varios IRES celulares utilizando la estructura enzimática y química obtenida de los transcritos, incluyendo c-Myc, Apaf-1, Kv1.4, y FGF2, entre otros (Le Quesne, et al., 2001; Mitchell, et al., 2003; Jang, et al., 2004; Bonnal, et al., 2003). 2.3.2 Datos funcionales e ITAFs importantes para la actividad celular de secuencias IRES La identificación de muchos elementos IRES celulares se ha basado en la demostración de la función de estos elementos y su capacidad para dirigir el inicio interno de la traducción. El test más común para la actividad IRES es un ensayo de células de mamíferos utilizando la transfección con vectores bicistrónicos reporteros. La expresión de un segundo marco de lectura downstream, depende de secuencias IRES que dirigen la entrada interna del ribosoma y el inicio de la traducción. Aunque es un primer paso importante en la determinación de si una secuencia de ARN tiene actividad IRES, este enfoque ha sido criticado debido a que los mARNs generados a partir de células transfectadas con vectores plasmídicos bicistrónicos de ADN generalmente tienen una actividad IRES mucho mayor que cuando se transfecta los ARNs correspondientes (Jimenez, et al., 2005). La diferencia en los niveles de expresión se puede interpretar como un artefacto de la transfección con ADN que conduce a la expresión reportera que es independiente de la función IRES (Van Eden, et al., 2004; Kozak, 2005; Bert, et al., 2006). A pesar de esta deficiencia, la traducción mediada por secuencias IRES se produce por un subconjunto de mARN celulares. Un IRES muy estudiado es el elemento IRES de c-myc (Nanbru, et al., 1997). Las proteínas que pertenecen a la familia Myc tienen un papel en la proliferación y la apoptosis celular, y la sobre expresión de Myc ha sido asociado con la tumorigénesis (Pelengaris, et al., 2002). El ARN mensajero de c-myc tiene un cap en el extremo 5’, y puede ser traducido ya sea mediante la vía cap dependiente o independiente (Stoneley, et al., 2000). Por lo tanto, es probable que la variación en la abundancia de ITAFs específicos contribuya 10 a esta diferencia en la eficiencia de traducción. Se han identificado varios ITAFs que estimulan la traducción de c-myc, incluyendo: PCBP2, IRP, unr, DAP5, YB-1, GRSF-1, PSF, p54nrb (Cobbold, et al., 2008; Evans, et al., 2003; Martínez-Salas, et al., 2001). Todo esto sugiere que los elementos IRES están regulados diferencialmente en diversas condiciones fisiológicas. Mientras que las secuencias IRES celulares continúan siendo identificadas y son añadidas a la creciente lista de mARN que poseen IRES, existen factores ITAFs que también están siendo investigados por estos IRES. Además, hay pruebas de que muchos ITAFs que regulan la traducción de secuencias celulares IRES son proteínas que se encuentran tanto en el núcleo como en el citoplasma (Schepens, et al., 2007; Graber & Holcik, 2007). 2.4 Vectores bicistrónicos La expresión de dos secuencias codificantes diferentes bajo el control de un único promotor permite la identificación del producto codificado por el segundo cistrón, lo que asegura que el primer cistrón también se ha expresado. Algunos vectores bicistrónicos se encuentran comercialmente disponibles basados en secuencias IRES a partir del virus EMCV, en los cuales el codón AUG ha sido modificado (Pelletier, et al., 1988). Además, los vectores bicistrónicos proveen la posibilidad de infectar células transfectadas tomando en cuenta la base de la fluorescencia de las proteínas controladas por IRES, así como por la expresión de la proteína de interés, fusionada a una proteína fluorescente separable espectralmente (Bonnal, et al., 2003). Además de revelar el insólito mecanismo de traducción, las secuencias IRES han sido exitosamente introducidas entre 2 cistrones para la construcción de vectores bicistrónicos, por ejemplo se ha demostrado la co-expresión simultánea de 2 proteínas, siendo una un marcador de selección o un gen reportero y la otra el gen de interés (Martin, et al., 2006). Por lo tanto, la identificación y el desarrollo de elementos IRES útiles para los vectores de expresión se han vuelto una herramienta muy útil en la biotecnología. En este estudio, evaluamos varias secuencias IRES que podrían permitir la expresión génica bicistrónica en cultivos celulares mediante la transfección del plásmido pMFR, el cual ha sido mejorado en el laboratorio de Anatomía Patológica, por Dr. Trond Aasen 11 (Grupo de Patología Molecular, VHIR). Se trata de una construcción bicistrónica fluorescente que contiene la expresión de mCherry bajo una regulación cap-dependiente, un sitio de clonación múltiple para insertar una secuencia IRES de interés, seguida por la expresión de la proteína EGFP que depende directamente de la actividad de la secuencia IRES clonada (Figura 1). Después de la clonación de las secuencias IRES candidatas en este vector, se procederá a evaluar la activación de la traducción a través de estas secuencias. Figura 1. Estructura del vector pMFR Construcción bicistrónica fluorescente, en la cual mCherry está regulado por la vía normal de traducción, un MSC para insertar secuencias IRES de interés, seguida por la expresión de la proteína EGFP 12 3 3.1 OBJETIVOS Objetivos generales Evaluar el comportamiento de un vector bicistrónico en el análisis de secuencias IRES y factores ITAFs, mediante la transfección en líneas celulares. Generación de partículas virales, portadoras de las secuencias IRES, para la infección de varias líneas celulares y selección de clones estables. Evaluar el efecto de varios ITAFs y el estrés celular como la falta de suero en la eficiencia de traducción cap-dependiente Cherry e IRES-dependiente EGFP. 3.2 Objetivos específicos Introducir secuencias IRES en el en MCS del vector pMFR. Evaluar la funcionalidad de los constructos mediante la transfección en líneas celulares. Clonación del inserto Cherry-MCS-GFP en un plásmido retroviral pLPCX, para generar partículas virales, e infectar líneas celulares y seleccionar con puromicina para establecer líneas celulares estables. Comparar los niveles de expresión de las secuencias IRES al someter a las células a diferentes mecanismos de estrés o con la modulación de los niveles de ITAFs (como YB-1 y SRp20). Evaluar el efecto del ITAF SRp20, en líneas celulares. 13 4 MATERIALES Y MÉTODOS 4.1 Descripción de la metodología Se analizaron varias secuencias IRES y (posibles ITAFs) de cMyc, Connexin 26, Connexin 43, EMCV, 19kD (un IRES putativo dentro de la región codificante de connexin 43) relacionados con el proceso de traducción cap-independiente mediante el uso del vector de expresión pMFR. Para este estudio se determinó las diferencias de expresión de estos genes utilizando líneas celulares sometidas a situaciones de estrés. Se realizó la Reacción en Cadena de la Polimerasa (PCR), con el fin de amplificar las secuencias de los genes a analizar en el estudio. Los fragmentos amplificados fueron identificados en geles de agarosa al 1%. A continuación, se procedió a realizar el corte de la banda del gel para poder obtener el ADN amplificado y purificarlo para posteriormente evaluar su concentración utilizando el Nanodrop 2000. Cada uno de los productos obtenidos, así como el vector, fueron sometidos a digestión con las enzimas de restricción KpnI y BamHI; los productos resultantes fueron purificados y ligados. Se procedió a la transformación bacteriana de la cepa de E. coli DH5, y a la obtención de ADN plasmídico de alta calidad. Finalmente, se transfectó líneas celulares y se evalúo la expresión proteica mediante Western Blot. Cuando se obtuvieron los resultados de los tratamientos y se recopilaron todos los datos de las líneas celulares analizadas. 4.2 Reacción en cadena de la polimerasa (PCR) La Reacción en Cadena de la Polimerasa es una técnica in vitro que permite la amplificación exponencial de una región específica del ADN, situada entre dos regiones de secuencia conocida. Su eficiencia y especificidad son determinadas por muchos factores como: calidad del ADN templado, diseño de los primers, concentración del MgCl2 y de la ADN polimerasa (Newton y Graham, 1994). En este estudio las condiciones de la PCR se estandarizaron tomando en cuenta la capacidad de síntesis de la ADN polimerasa. Cada tubo de reacción para la amplificación de la muestra contenía: 32µl de H2O Mili-Q, 5µl de buffer de PCR 10X, 3µl de MgCl2 25 mM, 5µl de dNTPs (dideoxinucleótidos trifosfato) a una concentración de 2 mM, 1,5µl de cada uno de los primers a una concentración de 10µM, 1µl del ADN templado en una 14 concentración de 50ng/µl, 1µl de KOD Hot Start DNA Polymerase (Novagen®), en un volumen final de 50µl. Todas las reacciones de amplificación se llevaron a cabo en un termociclador Veriti® 96-Well Thermal Cycler (Life Technologies, Applied Biosystems Inc., Van Allen Way, Carlsbad, CA., USA). Los primes que se utilizaron es este estudio fueron diseñados para la amplificación de fragmentos de 250, 450, 600 y 700 pares de bases. Las secuencias se muestran en la tabla 1. Tabla 1. Secuencias de los genes analizados y primers utilizados para su amplificación Genes rCx43 Cx26 Secuencia (forward y reverse) Fw: 5’- GCGACCACGAGGTATCAGCACTTTTC -3’ R: 5’-GCGGGATCCGTCTGGGCACCTCTCTTTCAC -3 Fw: 5’- GCGGGTACCCAGGTGTTGCGCCCCGCGCG-3’ R: 5’-GCGGGATCCCTTCTACTCTGGGCGGTTT -3’ 19Kd Fw: 5’- GCGGGTACCGGTGACTGGAGCGCCTTAGGC-3’ Rv: 5’- GCGGGATCCGAAGATGATGAAGATGGTTT- 3 Cmyc Fw: 5’- GCGCGTACCAATTCCAGCGAGAGGCAAGG-3’ Rv: 5’- GCGCGTACCAATTCCAGCGAGAGGCAGAGG-3 Fw: 5’- CGGGTACCACGAGGTATCAGCACTTTTC -3’ Rv: 5’- GCGGGATCCGTCTGGGCACCTCTCTTTCAC -3’ hCx43 Las condiciones utilizadas para la amplificación de todos los fragmentos fue: desnaturalización inicial a 95°C por 2 minutos; seguido de 29 ciclos a 95°C por 20 segundos, 60°C por 30 segundos, con una disminución de 0,5°C por cada ciclo y 70°C por 15 segundos. Finalmente hubo un ciclo de extensión final a 72°C por 10 minutos, estas condiciones se usaron para amplificar las secuencias de todos los genes descritos. Después de la PCR, se realizó una electroforesis horizontal en geles de agarosa en una concentración de 1%, y de este modo se determinó la presencia de los fragmentos amplificados para cada uno de los genes estudiados. Los geles se prepararon con SYBR Safe® (Invitrogen) como colorante para su observación en el transiluminador. Estos geles fueron corridos en una cámara de electroforesis, sumergidos en solución de buffer TAE 1X, a 70 voltios durante una hora. Para poder determinar el tamaño de los fragmentos amplificados se utilizó un marcador de peso molecular de 100 pares de bases (pb), de la casa comercial Fermentas. 15 4.3 Purificación de los fragmentos de PCR usando el kit Wizard® SV gel and PCR clean-up system. Se extrajo el fragmento de ADN de interés a partir de un gel de agarosa, usando un bisturí. El mecanismo de purificación se llevo a cabo siguiendo las indicaciones del fabricante, de acuerdo al protocolo de la casa comercial. 4.4 Digestión con enzimas de restricción Los productos de amplificación por PCR, de los diferentes genes, así como del vector fueron sometidos a la digestión mediante enzimas de restricción; en todos los casos las enzimas empleadas fueron KpnI y BamHI, las mismas que dan como resultado la formación de los sitios de reconocimiento para la inserción en el vector. Cada tubo eppendorf utilizado para la digestión de los productos de PCR y del vector contenía: 22µl de H2O Mili-Q, 3µl de fast digest buffer 10X, 1,5µl de cada una de las enzimas (KpnI y BamHI) (Fermentas®), 1µl del ADN (purificado de PCR y del vector) en una concentración de 1g/µl, en un volumen final de 30µl. Estos tubos fueron incubados a 37°C durante 1 hora. La visualización de los productos de las digestiones, fueron llevadas a cabo mediante una electroforesis horizontal en geles de agarosa en una concentración del 1%. El vector linealizado y los productos de PCR, obtenidos mediante la digestión con enzimas de restricción, fueron purificados a partir del corte del gel de agarosa utilizando el mecanismo descrito previamente. 4.5 Formación de moléculas de ADN recombinante Los cuatro pasos esenciales de la clonación de ADN basado en células son: 1. Construcción de moléculas de ADN recombinantes mediante una unión covalente in vitro (ligación) de los fragmentos de ADN deseado (ADN diana) a un replicón (cualquier secuencia capaz de una replicación independiente del ADN). Este paso es facilitado por el corte del ADN diana y las moléculas del replicón con 16 endonucleasas de restricción específicas antes de la unión de los diferentes fragmentos de ADN utilizando la enzima ADN ligasa. 2. Transformación, las moléculas de ADN recombinante son transferidas hacia el interior de las células hospederas (frecuentemente bacterianas o células de levadura) en la que el replicón elegido puede ser sometido a la replicación del ADN independientemente del cromosoma de la célula hospedadora. 3. Propagación selectiva de los clones, que se da en dos pasos. Inicialmente las células transformadas son cultivadas en placas con agar con el fin de promover el crecimiento de colonias aisladas. Estos son los clones (poblaciones de células idénticas, todas descendientes de una sola célula). Subsecuentemente, las colonias individuales pueden seleccionadas de una placa de cultivo para ampliar su crecimiento en medio líquido. 4. Aislamiento de clones de ADN recombinantes mediante la cosecha de los cultivos bacterianos y el aislamiento selectivo del ADN recombinante (Strachan & Read, 1999). 4.5.1 Desfosforilación del vector El proceso de desfosforilación de extremos 5’ con la enzima FastAPTM Thermosensitive Alkaline Phosphatase (Fermentas®), se realizó en un volumen final de 20µl de H2O Mili-Q, 2µl de FastAPTM buffer 10x (100 mM Tris-HCl (pH 8.0 at 37°C), 50 mM MgCl2, 1 MKCl, 0.2% Triton X-100 and 1 mg/ml BSA), 1l de enzima, 1l de vector lineal en una concentración de 80ng/µl. Los tubos fueron incubados a 37°C durante 10 minutos. 4.5.2 Ligación Los productos de amplificación por PCR digeridos con las enzimas de restricción, así como el vector linealizado y desfosforilado, fueron ligados utilizando la enzima T4 DNA ligasa, la misma que requiere ATP como cofactor. La reacción de ligación se llevo a cabo a 22oC, durante toda la noche. Cada tubo eppendorf contenía, en un volumen total de 20µl: 2µl de T4 DNA ligase buffer 10X (400mM Tris-HCl, 100mM MgCl2, 100mM DTT, 5mM 17 ATP (pH 7.8 at 25°C)), 1l de enzima, 80ng/l de vector lineal, la concentración de trabajo del inserto se calculó con la siguiente fórmula: inserto ng.vector * bp.inserto bp.vector 4.5.3 Transformación Para la transformación se utilizaron células competentes de la cepa de bacterias E. coli DH5, las mismas que se conservan a -80oC, y se mantienen en hielo hasta que se encuentren totalmente descongeladas. El mecanismo de transformación se llevó a cabo de la siguiente manera: se colocó 50l de bacterias en un eppendorf y se agregó 5l de producto de ligación, se mantuvo la mezcla en hielo durante 30 minutos. Se realizó un choque térmico a 42oC, durante 42 segundos. Posteriormente, se colocaron los tubos en hielo durante 1 minuto. Y finalmente, se colocó 1ml de medio LB líquido sin antibiótico y se dejaron las muestras en agitación a 37oC durante 1 hora. Se procedió a retirar las muestras, se las centrifugó durante 3 minutos a 3000 rpm y se sembró 100l de pellet en placas de agar LB con el antibiótico kanamicina. Por último, las placas fueron incubadas toda la noche a 37oC. Se prepararon además dos controles: 1) Negativo: sólo las bacterias para verificar la resistencia al antibiótico y 2) Positivo: plásmido sin digerir transformado. Una vez obtenidas las colonias se procedió a ampliar el crecimiento bacteriano sembrando colonias individuales en 3 ml de medio LB con kanamicina en concentración 1:200 ml. Los cultivos se colocaron en incubación con agitación a 37oC, durante toda la noche; se trabajó con 3-4 repeticiones. Al siguiente día se realizó la extracción de ADN. 4.6 Extracción de ADN plasmídico (mini y midipreps) La extracción de ADN de miniprep se realizó a partir de 3ml medio de cultivo líquido LB, incubado durante toda la noche a 37oC y con agitación de colonias individuales que fueron seleccionadas con antibiótico siguiendo las indicaciones del fabricante (Promega®). 18 Así mismo, para la extracción de ADN plasmídico midiprep de gran rendimiento y pureza se utilizó el kit: Extracción de ADN midiprep con el kit Pure link™ hipure plasmid filter purification kit (Invitrogen). En este caso se dejó incubando 50 ml de medio líquido LB con antibiótico y la colonia de bacteria respectiva. Se siguió el protocolo descrito por el fabricante. 4.7 Análisis del ADN extraído Un ADN de alto peso molecular principalmente libre de contaminantes es muy importante para la aplicación de técnicas moleculares, como la clonación de secuencias de ADN que es la base para el resto del análisis (Manak, 1993; Ausbel et al., 1995). Para determinar la calidad y cantidad del ADN, se utilizó el espectrofotómetro Thermo Scientific NanoDrop™ 2000. Para lo cual se calibró el equipo utilizando 1µl del agua con la cual se diluyó el ADN. Posteriormente se midió la concentración de cada ADN colocando 1µl de muestra sobre el lector. Luego de haber determinado la calidad y cantidad de ADN, este material fue diluido a una concentración final de 1µg/µl. 4.8 Test de mycoplasma Los cultivos celulares frecuentemente se contaminan con diferentes especies de mycoplasmas, lo cual altera el crecimiento celular, los patrones enzimáticos, la composición de la membrana e induce anormalidades cromosómicas. Dichas alteraciones influyen considerablemente en la toma de decisiones de los resultados obtenidos. Los mycoplasmas son un grupo de microrganismos que pertenecen a la clase Mollicutes y se caracterizan por ser los procariotes más pequeños de vida libre hasta ahora descritos. Carecen de pared celular, el tamaño de su genoma oscila entre 577 y 2220 kb, son exigentes desde el punto de vista nutricional y difíciles de cultivar (Rivera-Tapia, et al., 2002). Los métodos para detección de contaminación por mycoplasmas son el cultivo microbiológico, técnicas de microscopia, métodos bioquímicos y uso de técnicas de biología molecular. El protocolo usado para la detección de mycoplasma se describe a continuación: 19 Se tomó 1 ml de medio de cultivo de cada línea celular a analizar. Se centrifugó la muestra durante 1 minuto a 250 rpm. Se retiró el sobrenadante y se volvió a centrifugar a máxima velocidad durante 10 minutos. Se aspiró el sobrenadante y se eliminó. Se resuspendió el pellet en 10 l de buffer solution (kit). Se sometió a la muestra a choque térmico durante 3 minutos a 95oC. La muestra fue preparada en tubos de 0,2 ml. La reacción contenía 7 l de agua MiliQ estéril, 2 l de reaction mix (kit), 1 l de la muestra con buffer solution. Se preparó bajo las mismas condiciones un control positivo, incluido en el kit, y un control negativo que únicamente contenía agua. Las condiciones en el termociclador fueron: denaturación inicial a 94oC durante 30 segundos, seguido de 36 ciclos a 94oC 30 segundos, annealing 60oC durante 2 minutos y extensión a 72oC durante 1 minuto. Finalmente se realizó una extensión final a 72oC durante 5 minutos. Las muestras fueron mantenidas a 16oC. La comprobación se realizó mediante electroforesis en gel de agarosa al 2%. 4.9 Transfección a líneas celulares Es un mecanismo mediante el cual se introduce un material genético externo en células eucariotas mediante plásmidos, vectores víricos (en este caso también se habla de transducción) u otras herramientas para la transferencia. La transfección de células animales generalmente se lleva a cabo abriendo poros o "agujeros" transitorios en la membrana plasmática de las células, lo que permite el paso del material genético. Hay varios métodos para introducir DNA en una célula eucariota. Se usan muchos materiales como vehículos para la transfección, divisibles en tres tipos: polímeros (catiónicos), liposomas y nanopartículas. Uno de los métodos más baratos y óptimos es la transfección mediante fosfato de calcio (Graham & Van der, 1973). En este mecanismo se utiliza una solución salina tamponada con HEPES buffer que contiene iones fosfato y se combina con una solución de cloruro de calcio que contiene el ADN a transfectar. Cuando ambas se combinan, se forma un precipitado fino formado por el calcio cargado positivamente y el fosfato cargado negativamente. Esta suspensión se añade a las células que se quieren transfectar (normalmente un cultivo celular en monocapa). Mediante un proceso no 20 comprendido completamente, las células toman parte del precipitado, y junto con él, el ADN. 4.9.1 Preparación y contaje de células para transfectar Se tomó células en cultivo de la línea celular humana HEK-293, las cuales fueron preparadas de la siguiente manera: Se realizaron 2 lavados con 10 ml de PBS 1X estéril. Se añadió 1.5ml de tripsina y se colocó las placas a 37oC en la incubadora durante 2 minutos. Se añadió 3 ml de medio de cultivo DMEN (Dulbecco‘s modified Minimum Essential Medium) Gibco® suplementado (5.5 ml de penicilina-estreptomicina, 10% BSA, 5 ml L-Glutamina) para inactivar la tripsina. Se re suspendió las células y se las transfirió a un tubo falcon de 15ml. Se centrifugó las células a 1200 rpm durante 5 minutos a temperatura ambiente. Se retiró el medio y se colocó nuevamente 5 ml de medio y se resuspendió las células. A continuación, se contó las células para sembrar un número determinado de las mismas para transfectar. Se colocó 1ml de las células resuspendidas en un tubo eppendorf de 1,5ml. De esta alícuota, se tomó 10l de células y se colocó sobre la placa Neubauer. Una vez obtenido el total de células se procedió a sembrar 3 millones de células en placas p100, y se agregó 10ml de medio. Las placas se colocaron en la incubadora con 5% de CO2 a 37oC. 4.9.2 Transfección celular usando fosfato de calcio Para la transfección se utilizó el mecanismo del fosfato de calcio, para lo cual se prepararon dos tubos falcon de 15ml por cada muestra a transfectar. Un tubo contenía 50l de CaCl 2.5M, 10g de ADN de cada vector en un volumen total de 500l. En el otro tubo se colocó 500l de HBS. Una vez obtenida la mezcla del vector, se colocó gota a gota sobre el HBS para permitir la formación de los cristales de ADN. Se mezcló 10 segundos en el vortex y se dejó reposar durante 15 minutos a temperatura ambiente. Finalmente, la mezcla se añadió en las células sembradas el día anterior, a las cuales, previamente, se les cambió el medio. Se dejó incubando durante 24 horas a 37oC con CO2. A las 24 horas de transfección, se realizó un control de las células y se procedió a cambiar el medio. 21 4.9.3 Mecanismo de transfección celular con jetPEITM (polímero catiónico) JetPEI™ es un reactivo de transfección de gran alcance que asegura una efectiva y reproducible transfección con una baja toxicidad. El reactivo, permite una transfección superior in vitro en comparación con varios lípidos catiónicos y polímeros. JetPEI ™ ha demostrado una buena entrega de material genético a diversas líneas celulares establecidas, así como células primarias (Horbinski, et al., 2001). JetPEI™ compacta el ADN en partículas con carga positiva, capaces de interactuar con proteoglicanos aniónicos en la superficie celular, de esta manera ingresa en las por endocitosis; además posee la propiedad única de actuar como una "esponja de protones" que amortigua el pH endosomal y protege el ADN de la degradación (jetPEITM Polypus Transfecction, manual). 4.10 Western blot Es una técnica analítica que permite detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis, electroblotting, en gel de SDS-poliacarilamida se separan las proteínas de acuerdo al peso molecular, estructura, hidrofobicidad, etc. Posteriormente, las proteínas son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella. Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. Debido al marcaje, el anticuerpo produce una banda oscura sobre un film de rayos X al momento de revelado (autorradiograma). De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas (Sambrook, et al., 1989; Berg, et al., 2002). 4.10.1 Lisado celular Los cultivos celulares mantenidos en incubación con CO2 y a 37oC durante 48-72 horas, fueron retirados de la incubadora y mantenidos en hielo para su procesamiento. Las placas de células primeramente fueron lavadas 2 veces con 5ml de PBS 1x. Posteriormente se preparó 1ml de buffer NP 40 de lisis (hepes 1M, NaCl 2,5M; NP 40, glicerol, MgCl2 1.5M, EDTA 0.5M), al mismo que se le adicionó 1l de DTT, 10l de 22 PMSE, 20l de Naf, 5l de NaPPi (0.1M), 1l de Na3VO4 y 10l de -glicerofosfato (1M). Una vez preparado en buffer se colocó entre 30-150l de buffer en cada una de las placas. Las placas fueron raspadas para obtener el lisado celular, se colocó en eppendorfs estériles de 1.5ml y se mantuvo en hielo durante 15 minutos. Finalmente se centrifugó durante 20 minutos a 14.000 rpm a 4oC. 4.10.2 Cuantificación de proteínas mediante el kit bio-rad protein assay El kit de Bio-Rad, se basa en el método Bradford, el mismo que es un método simple y preciso para determinar la concentración de proteínas solubilizadas. Requiere la adición de un tinte acídico a la solución proteica, y la subsecuente medición a 595 nm con un espectrofotómetro o un lector de microplacas. El ensayo se basa en la fijación de colorante, en el cual un cambio diferencial de color se produce en respuesta a varias concentraciones proteína. La absorbancia máxima de la solución acídica del tinte azul brillante de Coomassie® G-250 cambia de 465 nm a 595 nm cuando la unión a proteínas ocurre. El colorante azul de Coomassie se une a principalmente residuos de aminoácidos básicos y ácidos aromáticos, especialmente arginina. Para medir la concentración de las proteínas, se preparó el reactivo de Bradford diluyendo 1 volumen de reactivo concentrado (5x) con 4 partes de agua miliQ (1x) y se mezcló con el vortex. En una placa de 96-well, en cada pocillo, se colocó 200 μl del reactivo de Bradford, diluido, de acuerdo al número de muestras y a la escalera patrón. Por último se colocó 1l de muestra, tanto de proteína como de recta patrón y se mezcló por pipeteo. Se incubó durante 5 minutos a temperatura ambiente y se midió la absorbancia a 595 nm. La regla patrón para la medición de la concentración de proteína se preparó como se indica el fabricante. Una vez obtenida la medición de las proteínas, se calculó la concentración de cada una, y las muestras fueron mantenidas a una concentración de 2g/l, para lo cual se agregó la cantidad de buffer de lisis adecuada a cada una. 23 4.10.3 Electroforesis en gel de poliacrilamida SDS-PAGE. Una molécula con una carga neta puede moverse en un campo eléctrico, mediante electroforesis, lo cual permite la separación de proteínas y otras macromoléculas como el ADN y el ARN. La velocidad de migración de una proteína (o cualquier molécula) en un campo eléctrico depende de la intensidad del campo eléctrico, la carga neta de la proteína y un coeficiente de fricción. Aquellas moléculas que sean más pequeñas que el tamaño del poro del gel se moverán más rápido a través del gel, mientras que las moléculas mucho más grandes que los poros son casi inmóviles. Las moléculas de tamaño intermedio se mueven a través del gel con diferentes grados de facilidad (Sambrook, et al., 1989; Berg, et al., 2002). Las proteínas se pueden separar en base a su masa por electroforesis bajo condiciones denaturantes. La mezcla de proteínas es primeramente disuelta en una solución de dodecil de sodio (SDS), un detergente aniónico que rompe caso todas las interacciones no covalentes en las proteínas nativas. Mercaptoetanol (2-tioetanol) o ditiotreitol (DTT) también es añadido para reducir los enlaces disulfuro. Este complejo SDS con una proteína denaturada posee una gran carga negativa neta que es aproximadamente proporcional a la masa de la proteína. La carga negativa adquirida con la unión del SDS es usualmente mucho mayor que la carga de la proteína original, esta carga nativa se vuelve entonces insignificante. Una vez completada la electroforesis, las proteínas en el gel se pueden visualizar mediante tinción con plata o con un tinte como el azul de Coomassie, el cual revela una serie de bandas. El marcaje radiactivo puede ser detectado colocando una hoja de film de rayos X sobre el gel, mediante auto radiografía (Sambrook, et al., 1989; Berg, et al., 2002). Para la electroforesis se preparó un gel de electroforesis cuya concentración fue de 12%, en un volumen de 10m, utilizando 3.3 ml de H2O miliQ, 4 ml de mix de acrilamida 30%, 2.5 ml de Tris 1.5M (pH 8.8), 100 l de SDS 10%, 100 l de persulfato de amonio 10% y 4 l de TEMED. Para su polimerización se cubrieron los vidrios hasta el borde con Isopropanol. Una vez polimerizado el gel, se retiro el isopropanol y se preparó el gel Stacking fue a una concentración del 5% en un volumen final de 4 ml, para lo cual e mezcló 2,7 ml de H2O miliQ, 670 l de mix de acrilamida 30%, 500 l de Tris 1.0 M (pH 6.8), 40 l de SDS 10%, 40 l de persulfato de amonio 10% y 4 l de TEMED (Sambrook, 24 et al., 1989). Una vez polimerizado el gel se procedió a preparar las muestras para la electroforesis. Todas las muestras fueron preparadas en un volumen total de 20 l, se adicionó 2,5 l de buffer de carga 5x y 0,2 l de DTT. Se desnaturalizó las muestras previamente durante 5 minutos a 96oC. La electroforesis se llevo a cabo en una cubeta Bio Rad, durante 3 horas a 80V. El buffer de corrida fue preparado a una concentración 1x. Una vez terminada la electroforesis se procedió a la transferencia del gel a la membrana de nitrocelulosa, para lo cual primeramente se activó la membrana colocándola durante 1 minuto en metanol 100%, se lavó con agua destilada y finalmente se la mantuvo en buffer de transferencia 1x que contenía metanol. La electroforesis de trasferencia se llevó a cabo durante una hora y media a 300 mA. 4.10.4 Revelado de la membrana de nitrocelulosa Una vez terminada la electroforesis de transferencia, la membrana fue bloqueada durante 30 minutos utilizando el buffer de leche (DifcoTM Skim Milk) (TBS 1x y 0.01% Tween 20). Posteriormente se procedió a lavar la membrana 3 veces con el buffer TBST durante 10 minutos cada una. Por último, la membrana fue incubada con agitación durante una hora con los anticuerpos -His y -GFP a temperatura ambiente. Una vez transcurrido el tiempo de incubación la membrana fue lavada, nuevamente, 3 veces y cada lavado durante 10 minutos. A continuación, se preparo 1 ml de solución de revelado, se colocó la membrana sobre el casete y se adicionó la solución sobre la membrana. Las condiciones de revelado dependieron de la intensidad de la proteína pero se mantuvo el film de rayos X entre 10 y 15 segundos para una exposición mínima y de 1 a 3 minutos para obtener una máxima exposición. 4.11 Cultivo de líneas celulares bajo condiciones de estrés El uso de sistemas de cultivos celulares para evaluar los efectos de la toxicidad principalmente, de agentes anticancerígenos empezó 50 años atrás. Existe una gran variedad de ensayos para evaluar la sensibilidad de las células a situaciones de estrés 25 provocado por cambios en la expresión génica de las mismas ó mediante la adición de sustancias químicas que pueden alterar el metabolismo celular (Langdon, 2010). En este ensayo se utilizaron las líneas celulares SW620 y HaCat para probar diferentes tipos de estrés celular. Para ello se transfectaron células en placas de 96-well con, aproximadamente, 10.000 células por tratamiento y por línea celular. Los tratamientos se realizaron por triplicado y las condiciones fueron: medio de cultivo DMEM con 10% FBS (control), medio DMEM con 0% FBS, PP242 2.5 M, CoCl2 250 μM y CGP 40M. Las células fueron incubadas a 37oC durante 24 horas con 5% de CO2. Para la evaluación de la expresión de la proteína Cherry y GFP, transcurridas las 24 horas de incubación, se realizó la medición de la absorbancia de las proteínas. Las mediciones se realizaron en los intervalos de 485/535 nm para GFP y entre 544/590 nm para Cherry. Las mediciones fueron realizadas por duplicado, una primera vez con el medio de cultivo. Para la segunda medición de fluorescencia, primero se realizó 2 lavados de las placas con 30 l de PBS 1x, posteriormente se colocó 30 l de buffer de lisis NP40 sobre cada tratamiento y se dejó reposar durante 10 minutos. La medición de la absorbancia se realizó bajo las mismas condiciones anteriores. 4.12 Construcción de partículas infeccionas con el plásmido pPLCX Es posible construir vectores retrovirales de replicación competente añadiendo secuencias a virus existentes, pero más comúnmente el diseño involucra el remplazo de las secuencias retrovirales para la creación de vectores deficientes de replicación. Además, la cantidad de ADN externo que debe ser acomodado en los vectores de replicación competente es mucho más pequeña que la que se debe colocar en los vectores de replicación defectiva. La expresión de proteínas retrovirales en la mayor parte de retrovirus oncogénicos que se producen naturalmente es dirigida por un solo promotor en las regiones 5’ LTR (Long Terminal Repeats), y la expresión de múltiples regiones virales codificantes se alcanza mediante el splicing alternativo. Sin embargo, el diseño del vector no se limita al uso de un único promotor retroviral con splicing alternativo. Otras estrategias incluyen el uso de múltiples promotores, inserción de genes en orientación reversa, y la utilización de secuencias IRES (Coffin, et al., 1997). 26 4.12.1 Construcción del plásmido pLPC-SRp20 A partir del vector pEGFPC1-SRp20 se realizó la extracción de la secuencia SRp20, que es un factor de splicing, mediante la digestión con las enzimas de restricción EcoRI y NotI. Al mismo tiempo se trató al vector pLPCX con las mismas endonucleasas como se ha descrito anteriormente. Por último los fragmentos fueron ligados y transformados para obtener el vector para transfectar células de carcinoma mamario MDA-MB-468. 4.12.2 Construcción del plásmido pPLC-MFR A partir del vector pMFR se realizó la extracción de las secuencias IRES EMCV, 19kD (secuencia interna en la región codificante del gen connexin 43) y c-Myc mediante la digestión con las enzimas de restricción BglII y NotI. El vector pLPCX fue digerido con las mismas enzimas como se ha descrito. Los fragmentos fueron ligados y transformados para obtener el vector retroviral para infectar células Phoenix (derivadas a partir de las células HEK-293). 4.13 Producción de partículas virales usando células Phoenix Durante las últimas dos décadas, los vectores retrovirales han surgido exclusivamente como un sistema de transferencia de genes de gran alcance. Las características que dotan a los retrovirus con su potencial patogénico también los hacen atractivos para la ingeniería de vectores seguros para transferencia genética. Por ejemplo, los retrovirus se integran establemente en muchos tipos celulares. Además, la expresión de los retrovirus puede ser constitutiva o inducible usando una variedad de configuraciones del promotor (Pear, et al., 1993; Hitoshi, et al., 1998). Para el empaquetamiento del vector se utilizó líneas celulares Phoenix (empaquetadores de retrovirus), mediante la transfección de las mismas con fosfato cálcico; para la recolección de las partículas virales se siguió el protocolo descrito a continuación: A las 24 horas de la transfección en células Phoenix, se aspiró el medio de cultivo DMEM (suplementado) que contenía las partículas virales, el mismo que fue pasado a través de filtro de 0,2 m. Sobre las células Phoenix se añadió 10 ml de medio DMEM y se las dejó en la incubadora a 37°C. El medio filtrado, que contenía los virus, fue colocado en tubos falcon de 15 ml y se añadió 10 Polybrene (0.8mg/ml) por cada ml de sobrenadante. 27 La mezcla viral y el polybrene fueron distribuidos sobre las líneas celulares HOP-62, MDA-MB-231, 293T y A549 para su infección y producción estable del virus. El proceso de infección sobre líneas celulares se repitió a las 24 horas. Finalmente, sobre las células infectadas se colocó medio de selección y se las mantuvo en paso normal de selección durante una semana. 28 5 RESULTADOS 5.1 Amplificación mediante PCR de secuencias IRES Para amplificar los correspondientes IRES de 19kD, rCx43, hCx43 (Cx43 humano) y Cx26, se utilizó como base un plásmido que ya tenía el la secuencia clonada. Usando primers específicos diseñados para introducir las dianas KpnI - BamHI se amplificó los fragmentos por PCR y se comprobó mediante electroforesis en gel de agarosa (figura 2). Figura 2. Amplificación mediante PCR de los genes Cx26, eCx43, C-myc y hCx43, gel de agarosa 1%. Mw 1kb Cx26 rCx26 Cmyc 19kD MW 100pb 750pb 350pb 200pb 1 2 3 4 5 6 7 8 9 10 11 12 Productos de PCR correspondientes a los fragmentos de los genes Cx26, rCx43 (filas 2, 35, 6) con un tamaño de 200 pares de bases. En la fila 8, el producto de amplificación del gen c-Myc con un tamaño de 350 pares de bases. Las filas 10 y 11 el producto de amplificación del gen 19kD con un peso de 750 pares de bases. Además los marcadores de peso molecular de 1kb, en las fila 1 y de 100 pares de bases en la fila 12. Los fragmentos de PCR fueron amplificados usando ADN plasmídico. 5.2 Clonación de secuencias IRES en el plásmido reportero eucariota pMFR Una vez obtenidos los fragmentos amplificados mediante PCR de las secuencias IRES, se cortaron las bandas correspondientes del gel y se procedió a purificarlas con el kit de promega. A continuación, el vector pMFR y los productos de PCR purificados, fueron digeridos con las enzimas de restricción KpnI y BamHI, lo cual permitió lineralizar el plásmido y generar los sitios de inserción del producto de PCR. Finalmente se realizó la transformación bacteriana para la obtención de colonias. 29 De cada una de las placas transformadas con las secuencias IRES, en agar con kanamicina, se escogieron 2-3 colonias y se hizo el cultivo más grande miniprep. Se extrajo el ADN plasmídico usando el kit de extracción de miniprep y se verificó la presencia del inserto mediante la digestión con las enzimas KpnI – BamHI, obteniéndose así las bandas del vector lineal, sin inserto, y los fragmentos de cada uno de los genes amplificados mediante PCR como se indica en la figura 3. Figura 3. Verificación mediante digestión con las enzimas de restricción KpnI y BamHI de la transformación bacteriana, gel de agarosa 1%. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 MW 1kb 15 MW 100 pb Cx26 rCx26 Cmyc 19kD hCx43 750pb 200pb En la primera fila se observa el marcador de peso molecular de 1kb, en las filas 2-3, 5-6 y 13-14 se observa un fragmento de 200 pares de bases de los fragmentos Cx26, rCx26 y hCx43; en la fila 8 un producto de 350 pares de bases, que corresponden al fragmento cMyc. La banda de 6000 pares de bases corresponde al vector pMFR lineal. 5.3 Extracción y análisis de ADN plasmídico (mini y midipreps) El ADN obtenido mediante extracción de miniprep a partir de 3 ml de cultivos bacterianos fue de alto peso molecular, así como también el ADN obtenido a partir del protocolo de Midiprep a partir de 50 ml de medio de cultivo. La concentración obtenida y la calidad del ADN se muestran en la figura 4, obtenida a partir de la medición de concentración con el Nanodrop. 30 Figura 4. Curva de absorbancia de la concentración del ADN obtenido a partir de midiprep. La figura muestra la medición de una muestra de ADN obtenida mediante la extracción con el kit de midipreps. La relación OD 260/OD280 es de 1.87, lo cual demuestra que es un material genético con una alta pureza, y cuya concentración fue 4g/l. 5.4 Cultivo de líneas celulares en medio DMEM y test de mycoplasma Para los ensayos de expresión génica se utilizó el modelo del cultivo celular. Para ello primero, todas las líneas celulares utilizadas fueron analizadas para detectar mycoplasma y, en algunas ocasiones, se encontró que fueron positivas como se indica en la figura 5. En estos casos se descongeló y usó nuevos clones que fueron negativos para mycoplasma. Figura 5. Análisis de mycoplasma en líneas celulares mediante PCR, gel de agarosa 2%. 1 2 MW 231 100pb 3 4 5 6 7 8 HeLa HeLa A549 A549 HOP 293-T 62 En la primera fila se observa el marcador de peso molecular de 100 pb, en las filas 2-7 (líneas celulares 231, HeLa, A549, HOP-62 y 293T), la amplificación del fragmento que corresponde a la banda de mycoplasma de aproximadamente 300 pares de bases. 31 5.5 Transfección del vector pMFR – IRES en células 293T 5.5.1 Análisis de la fluorescencia de las proteínas Cherry y GFP Una vez obtenidas todas las construcciones deseadas, se transfectó en líneas celulares 293T mediante la técnica del fosfato cálcico, para comprobar que fueron clonadas correctamente y que los vectores expresaban adecuadamente GFP y Cherry. Las construcciones transfectadas fueron: pMFR-Cx43, pMFR-19kD, pMFR-cMyc, pMFRCx26 y pMFR-EMCV como control. Los resultados se analizaron 48 horas después de la transfección en el microscopio de fluorescencia. Las fotografías muestran los niveles de expresión proteica GFP/Cherry. Todos los tratamientos son positivos para la expresión de Cherry,; sin embargo se puede observar un leve background debido a la fluorescencia emitida por GFP, ya que GFP se expresa mucho en la condición de pMFR-EMCV, que era lo que se esperaba ya que es nuestro control positivo. La figura 6 muestra los resultados obtenidos. Las imágenes fueron adquiridas con el microscopio de fluorescencia Olympus FSX-BSW 100. Figura 6. Niveles de expresión proteica de Cherry/GFP en células 293 de los genes transfectadas con pMFR-IRES GFP/mCherry mCherry GFP a) b) c) 32 GFP/mCherry mCherry GFP d) e) f) Ensayo de transfección transitoria de la actividad de GFP/Cherry en ausencia de infección retroviral. Células 293T fueron transfectadas con 1g de plásmido pMFR-IRES y observadas 48 horas después de la transfección bajo microscopio de fluorescencia. a) expresión de la secuencia 19kD. b) secuencia Cx26. c) Expresión de EMCV como control. Los tratamientos a) y b) revelan un background de fluorescencia verde a diferencia del tratamiento c) en el cual la expresión de GFP es más fuerte. d) expresión de la secuencia Cx43. e) secuencia c-Myc. f) Expresión rCx43. Los tratamientos a), b), d) e) y f) revelan un background de fluorescencia verde. La fluorescencia roja debida a la expresión de mCherry se observa en todos los tratamientos. La fotografías fueron adquiridas al mismo tiempo, con exposición de 1/3 ms y en un campo con un canal para rodamina (rojo) y un filtro de 510/560 nm y un canal FITC (verde) con un filtro de 450/490. 5.6 Western blot cuantificación de la expresión de Cherry y GFP 5.6.1 Cuantificación de proteínas mediante el kit bio-rad protein assay Por otra parte, se analizaron estas transfecciones por western blot. Para ello se trasnfectaron las células como se ha descrito antes y se procedió a realizar un lisado celular para correr estas muestras en un gel de acrilamida. Una vez obtenidos todos los lisados pMFR-IRES, las proteínas obtenidas fueron mantenidas a una concentración final de 2µg/µl. La concentración final que se utilizó para llevar a cabo la electroforesis fue de 33 20µg/µl de cada lisado proteico. Para el cálculo de las concentraciones se utilizó una regresión, en la cual la curva y la ecuación de la curva nos permitió calcular la cantidad exacta de buffer de lisis que se debía añadir a los lisados para obtener la concentración deseada, figura 7. Figura 7. Recta de regresión para calcular la concentración de proteínas. Recta de regresión 1 Abs (690) 0,8 0,6 y = 0,1425x + 0,5352 R2 = 0,8464 0,4 0,2 0 0 0,2 0,4 0,6 0,8 1 [mg/ml] 1,2 1,4 1,6 1,8 2 En el eje de las X se indica la concentración de la proteína y en el eje Y la absorbancia a 690nm. La recta nos permite identificar una relación entre las proteína obtenidas en cada línea celular, ya que la cantidad utilizada para la medición de cada una fue 1µl. La ecuación de la recta nos permitió calcular la concentración de proteína en mg/ml. Una vez obtenidas las concentraciones adecuadas de los lisados proteicos de las células que expresaban mCherry-His y EGFP-His bajo la activación de los IRES conrrespondientes de las proteínas 19kD, Cx26, rCx43, C-myc, Cx43 y EMCV, se cargó 20 g/l de proteína para realizar una electroforesis SDS-PAGE. Una vez terminada la electroforesis y realizada la transferencia, la membrana fue sometida al anticuerpo -His. Las células que contenían las construcciones de 19kD, c-Myc y rCx43 casi no expresaban GFP, es decir, casi no mostraron actividad IRES, mientras que pMFR-EMCV expresó una mayor cantidad de GFP-His, lo que quiere decir que el IRES está activo figura 8. En contraste, la expresión de mCherry fue muy similar entre todos los vectores, indicando que el primer cistrón se expresa de manera similar en todos los constructos. 34 Figura 8. Western Blot del vector pMFR y las secuencias de 5 genes IRES. 1 2 3 4 5 6 La figura indica la expresión de las proteínas Cherry y GFP por análisis de western blot. Cantidades equivalentes de lisado de proteína fueron cargados. En el carril 6 se muestra el vector control pMFR-EMCV en el cual se observa claramente la fuerte expresión de GFP. En los carriles 1, 4 y 5 se distinguen las bandas de 29kD y 27kD correspondientes a Cherry y GFP, respectivamente. Por otro lado, también se transfectó los vectores PMFR que contenían las secuencias de los genes IRES en dos líneas celulares HeLa y MDA-MB-231. Los tratamientos aplicados en las células fueron: como control el vector pMFR; se realizó dos cotransfecciones 1) pMFR-cMyc con el plásmido control pLPCX y 2) pMFR-cMyc con un vector que expresa el ITAF YB-1. pMFR-19kD y el disolvente DMSO como control; pMFR-19kD con la droga CGP (un inhibidor de Mnk1 kinasa que previene la fosforilación de eIF4E) y el vector pMFR-EMCV como control. En este caso, con todos los vectores transfectados y las co-transfecciones se realizó el análisis de western blot. Las membranas fueron sometidas, igualmente, al anticuerpo His. Sorprendentemente, ningún constructo mostró expresión de eGFP y solamente se pudo detectar la proteína Cherry (figura 9). 35 Figura 9. Western Blot para el análisis de co-transfección en células HeLa. 1 2 3 4 5 6 Efecto de la co-transfección del vector pMFR-IRES. Corrida electroforética SDS-PAGE y analizada con el anticuerpo -His. Debido a que mediante western blot no se pudo detectar la proteína GFP en células HeLa, se realizó mediciones en el lector de fluorescencia Thermo Scientific Appliskan para determinar la relación de expresión proteica de GFP/mCherry. La figura 10 muestra que GFP aumenta en pMFR-EMCV (control positivo) tanto en la línea celular HeLa como en 231, pero en las otras construcciones no se observa una expresión transitoria. Figura 10. Ratio de expresión de las proteínas Cherry-GFP en dos líneas celulares HeLa y 231. 16 14 12 10 8 6 4 2 0 -2 HeLa 231 En la figura se muestra la proporción de expresión de las proteínas GFP/Cherry. La expresión del vector pMFR se toma como inicial, a partir de la cual se hace un ratio para cada tratamiento. Se puede observar que la proporción de la expresión en el vector que posee el gen EMCV es mayor en las dos líneas celulares; así mismo la expresión de c-Myc se ve incrementada únicamente en la línea celular 231 en el tratamiento con el vector viral. Sin embargo, al realizar la co-transfección cMyc-Yb1 se observa una mayor expresión en las células 231. Igualmente sucede con la expresión del gen 19kD, en el tratamiento con DMSO hay una mayor expresión en las células HeLa, en cambio con el tratamiento de CGP se incrementa la proporción GFP/Cherry en las células 231. 36 Podemos ver que en la línea celular MDA-MB-231 hay una mayor expresión de los vectores (cMyc+pPLCX, pMFR-EMCV y pMFR-19kd (CGP)) y no en las células HeLa excepto por pMFR-EMCV cuya expresión es fuerte en los dos casos. Los resultados fueron obtenidos con la relación de GFP IRES-dependiente dividido por la expresión de Cherry cap-dependiente. 5.7 Producción viral en células Phoenix A partir del vector pMFR se realizó la extracción del fragmento Cherry-MCS-GFP el cual contiene el MCS de diferentes secuencias IRES, EMCV, 19kD (secuencia interna en la región codificante del gen connexin 43) y c-Myc mediante la digestión con las enzimas de restricción BglII y NotI. El vector pLPCX fue digerido con las mismas enzimas como se ha descrito. Por último los fragmentos fueron ligados usando la ligasa T4 (FERMENTAS®). Para la transformación, con los productos ligados, se utilizó bacterias competentes de E. coli de la cepa DH5; las mismas que fueron plaqueadas en agar LB con ampicilina como antibiótico de selección. Finalmente, se obtuvieron colonias positivas, se realizó la extracción de ADN plasmídico el mismo que fue utilizado para infectar células Phoenix (derivadas a partir de las células HEK-293). 5.8 Establecimiento de líneas celulares productoras de virus Obtenidas las líneas celulares Phoenix transfectadas con los vectores pLPCX: pLPCXCherry-EMCV-GFP, pLPCX-Cherry-c-Myc-GFP, pLPCX-Cherry-19kD-GFP, se recogió los virus y se los filtró. Se adicionó polibreno al medio y se infectó 4 líneas celulares: 1) HeLa (cáncer de cérvix), 2) Hop-62 (cáncer de pulmón), 3) MDA-MB-231 (cáncer de mama) y 4) A549 (línea celular de cáncer de pulmón) para la producción de virus de manera continua. El proceso de infección se repitió a las 24 y 48 horas de producción viral. Una vez obtenidas las células infectadas se realizó el cambio de medio de cultivo y se adicionó puromicina para seleccionar clones positivos. Se mantuvo las células en selección con antibiótico durante una semana cambiando el medio y añadiendo puromicina cada 48 horas. Posteriormente, se procedió a cambiar las células a medio de cultivo normal, sin antibiótico) para que se recuperen al tratamiento y así aislar las colonias positivas. Sin embargo, el tratamiento con el antibiótico afecto la morfología de las células, y la línea 37 celular Hop-62 no sobrevivió ni al tratamiento con virus ni a la selección con puromicina, confirmado así que la puromicina elimina eficazmente las células no infectadas. Se logró obtener clones en las las líneas celulares A549, HeLa, y MDA-MB-231 a partir de la construcción que contiene el IRES de c-Myc figura 11. Además se obtuvieron clones positivos de las secuencias de los genes EMCV y 19kD en la línea celular 231, además se obtuvo el vector pMFR como control figura 12. Figura 11. Clones del gen c-Myc y la relación entre la expresión de las proteínas Cherry-GFP en líneas celulares HeLa, A549 y 231 Contraste de fases GFP mCherry a) HeLa – c-Myc b) A549 – c-Myc c) 231 – c-Myc Detección de clones positivos de las secuencias del gen c-Myc en 3 líneas celulares mediante la expresión de eGFP-mCherry. a) se muestra una colonia positiva para la expresión de mcherry y GFP. b) se muestra una colonia positiva para mCherry y muy pocas células positivas para GFP. c) En la línea celular 231 únicamente se distinguen células positivas para mCherry. Todas la fotografías fueron tomadas en el mismo campo con un canal para rodamina (rojo) con un filtro de 510/560 nm y un canal FITC (verde) con un filtro de 450/490. Todas las fotografías fueron tomadas con el mismo tiempo de exposición 1/3 260 ms. 38 Figura 12. Clones estables en la línea celular 231 de EMCV, 19kD y del vector control pMFR. GFP/Cherry Cherry GFP Contraste de fases a) 231 pMFR b) 231 EMCV c) 231 – 19kD Detección de clones positivos en la línea celular 231 mediante la evaluación de la expresión de eGFP-mCherry. a) Se muestran colonias positivas para la expresión de mcherry y GFP control. b) colonias positivas para la expresión de EMCV IRES c) Del fragmento 19kD únicamente se distinguen colonias positivas para la expresión de mcherry. 5.9 Cultivo de líneas celulares bajo condiciones de estrés Se utilizaron dos líneas celulares estables, obtenidas a través de la infección viral, SW620 (línea celular humana de cáncer de colon metastático) y HaCat (línea celular huma de keratinocitos inmortalizados), para identificar como ciertas sustancias afectan la traducción de GFP iniciada a partir de cMyc-IRES. En los tratamientos llevados a cabo, se eliminó el suero fetal bovino del medio de cultivo (FCS 0%). También, se inhibió la proteína reguladora mTOR mediante el uso de PP242 (muy similar a rapamicina), además se sometió a las células a estrés por la ausencia de oxígeno usando cloruro de cobalto (como se ha visto en ciertos tumores). Al someter a las células, en cultivo, a diferentes condiciones de estrés, se provocó una sobre regulación de la actividad traduccional de MycIRES. La condición de 0% FCS provoca, en las células, estrés debido a la ausencia de la señal mitogénica del crecimiento; CPG tiene un menor efecto. Se observa que los 39 tratamientos aplicados en las líneas celulares incrementan el inicio de la traducción IRESdependiente, como se indica en la figura 13. Figura 13. Condiciones de estrés en células HaCat. Fold Change EGFP / mCherry 5 4 3 2 1 0 10% FBS0% FBS PP242 CoCl2 CGP Comparación de la expresión de GFP y mCherry en células HaCat sometidas a 5 tratamientos de estrés celular. Las condiciones 0 y 10% suero fetal bovino (FBS), PP242 (2.5 M), CoCl2 (250 M) y CGP (40M). Las células HaCat utilizadas eran células estables que expresan el vector pLPCX-Cherry-c-Myc-GFP. A las 24 horas de la transfección, se evalúo la fluorescencia emitida. Todos los datos fueron normalizados de acuerdo a la eficiencia de transfección efectuando la proporción GFP/mcherry y presentada como medidas promedio de los 5 tratamientos. Se puede observar que bajo las condiciones de estrés hay un incremento en la expresión del cMyc-IRES (tanto en el tratamiento con 0% FBS como en el del PP242); este incremento en la expresión puede deberse a que los elementos IRES celulares están presentes en mARNs que codifican proteínas bajo ciertas condiciones de estrés. 5.10 Evaluación de la expresión del plásmido pLPC-SRp20 Se trabajo en células MDA-MB-468, estables, con los vectores pMFR-EMCV y pMFR-cMyc. Los cultivos fueron inducidos con diferentes tipos de estrés: 1) 10% FBS, 2) 0% FBS, 3) CoCl2 250 M 3) CoCl2 250 M 4) PP242 2.5 M y 5) rapamicina. Además se realizó la transfección de la construcción pLPC-SRp20 que es un conocido ITAF, utilizando el reactivo JetPei como se ha descrito anteriormente. Transcurridas 24 horas de la transfección se trató las células con 0 y 10% de FBS; 24 horas después del tratamiento se realizó la medición de la fluorescencia de GFP con respecto a 40 Cherry como se indica en la figura 14. Los resultados muestran que el ITAF SRp20 está activando la expresión mediada por los IRES EMCV y c-Myc, siendo mayor en cMyc en ausencia de FBS. No obstante, la simulación del estado de hipoxia, tratamiento con CoCl2, no activa IRES en estas células ya que podría estar provocando en las células apoptosis. Figura 14. Evaluación de la expresión del vector pLPC-SRp20 y condiciones de estrés en líneas celulares MDA-MB-468 7 6 5 4 3 2 1 0 -1 MDA-MB-468 EMCV MDA-MB-468 c-myc Comparación de la expresión de GFP y mCherry debida expresión mediada por los IRES c-Myc y EMCV en células MDA-MB-468 sometidas a varios tratamientos de estrés celular. En las condiciones de 0 y 10% de FBS y PP242 no se observa una marcada diferencia en cuanto a la expresión de GFP y Cherry mediada por los IRES c-Myc y EMCV. Por el contrario, el tratamiento de CoCl2, muestra que no hay expresión del IRES c-Myc. En el tratamiento con rapamicina se ve un incremento en la expresión de EMCV. Sin embargo al realizar el tratamiento con SRp20 se muestra una gran activación de la expresión del IRES-cMyc. - 41 6 DISCUSIÓN El control de la traducción de proteínas juega un papel esencial en el crecimiento celular y es un proceso altamente regulado en la progresión tumoral. En particular, se ha visto que la traducción mediada por secuencias IRES, aunque aún sea considerada como una vía “alternativa”, se utiliza con relativa frecuencia tanto en condiciones fisiológicas normales como en condiciones patológicas o de estrés, y el mecanismo de escaneo capdependiente está comprometido (Komar & Hatzoglou; 2011). Por lo tanto, se piensa que la vía de traducción cap-independiente podría ser estar contribuyendo a la transformación tumoral. Con el fin de evaluar la activación de la traducción mediada por IRES, en este trabajo se ha puesto a punto el clonaje de diferentes secuencias IRES ya conocidas en un vector bicistrónico fluorescente denominado pMFR, previamente generado en el laboratorio, cuyos insertos dan como resultado la expresión constante de Cherry y la expresión alternativa de GFP como dos especies de proteínas distintas. Para analizar la posibilidad de que varias proteínas presenten secuencias IRES, se clonaron las secuencias Connexin 26, Connexin 43, EMCV (el cual no es traducido, pero funciona como una secuencia que permite el inicio de la traducción downstream del ARN, en este caso hacia GFP), la secuencia IRES ya conocida de c-Myc y 19kD (un IRES putativo dentro de la región codificante de connexin 43, ya que las secuencias putativas pueden actuar como promotores crípticos, sitios de splicing que modulan el fraccionamiento por RNAasa) (Van Eden, et al., 2004), en un vector bicistrónico, de modo que si las secuencias IRES se activan, se impulsaría la expresión de GFP en este vector. Así, en la primera parte de los resultados podemos concluir que hemos clonado con éxito las todas las secuencias IRES, ya que la posterior comprobación de los clonajes, así lo demuestra. En una segunda fase, se valoró la actividad mediada por las secuencias IRES y se evalúo la activación de las mismas mediante transfecciones (figura 9) y cotransfecciones (figura 10), de las construcciones generadas, en líneas celulares. El posterior análisis indica que el experimento está bien ejecutado, ya que el control positivo, el IRES viral EMCV está activando la expresión de GFP. Éste resultado se ha 42 observado con los tres métodos utilizados: microscopía de fluorescencia (figura 6), cuantificación de la fluorescencia con el fluorímetro (figura 10) y también se confirmó mediante el análisis de western blot, en el cual se observó una banda correspondiente a la proteína quimérica GFP-His (figura 10). Los otros IRES no son muy efectivos o en realidad no son secuencias IRES ya que no se observó una expresión significativa de GFP con respecto a Cherry, esto fue evaluado en condiciones normales de cultivo celular con 10% FBS. Otro mecanismo de evaluación fue a través de infecciones virales, se trabajó con líneas celulares estables con capacidad de traducir la fluorescencia roja de mCherry a partir de la traducción cap-dependiente y fluorescencia verde correspondiente a la proteína EGFP mediada por secuencias IRES (figuras 11 -12). Una vez obtenidas las líneas celulares estables, que contenían el vector bicistrónico, se comprobó si las secuencias IRES funcionaba y se indujo la expresión de GFP (figuras 13-14). Se vio que, para estudiar la activación de IRES es mejor tener líneas estables que transfecciones transitorias, ya que en estas, a veces se observan muy pocas células transfectadas y no podemos ver el efecto. Por el contrario, en líneas estables la mayoría ó todas las células, al menos deberían estar expresando la proteína cherry. Posteriormente, para determinar si el IRES de c-Myc es activo en nuestro sistema, planteamos provocar condiciones de estrés en las líneas celulares para activar esta secuencia. Se observó que con falta de nutrientes se incrementa la fluorescencia cuantificada con fluorímetro en células no tumorales HaCat (figura 14), pero no sucede lo mismo en células MDA-MB-468 (carcinoma mamario) (figura 15). Es conocido que con falta de nutrientes, se inactiva el mecanismo de traducción cap-dependiente y se activa el sistema mediado por IRES en el 5’UTR de los mRNAs (Fitzgerald & Semler, 2009). El hecho de que en células tumorales no se active el IRES de c-Myc puede deberse a que estas células sobre expresan Myc para permitir la proliferación normal de células. En cuanto al tratamiento con PP242, inhibidor de mTOR, se inhibió la vía cap-dependiente y consecuentemente se observó vía cap-independiente, mediada por el IRES de c-Myc y la expresión de GFP en células HaCat (figura 14) y también en células MDA-MB-468 (figura 15). El funcionamiento de mTOR, es un mecanismo crucial para el crecimiento tumoral, 43 por lo que se ha vuelto un blanco popular para los tratamientos terapéuticos (Hoang, et al., 2012). Con estos resultados, podríamos pensar que se ha identificado una nueva vía de activación de la sobre regulación del oncogén Myc. Así mismo, la inducción de hipoxia afectó sólo a las células no tumorales HaCat y se vio que se activa el IRES de c-Myc, pero no se activó en células MDA-MB-468 posiblemente porque podría estar induciendo apoptosis y no estar favoreciendo el efecto deseado. De esta manera se puede relacionar al estrés producido con un incremento endógeno de la proteína Myc (la cual contiene la secuencia myc-IRES en su región 5’UTR). A pesar de que las células responden al estrés fisiológico (hipoxia) y patológico mediante la fosforilación de la subunidad eIF2 la cual da como resultado la atenuación global de la síntesis de proteínas (Joachims, et al., 1999). Además, para analizar el funcionamiento de las secuencias de los genes tranfectados junto con el vector pMFR-c-Myc se añadió el plásmido YB-1 para determinar si el factor YB-1 ITAF podría afectar la traducción de GFP desde el sitio del IRES-cMyc. De acuerdo a los datos obtenidos parece que estaría reprimiendo la traducción en las dos líneas celulares, HeLa y 231 (figura 11), lo cual es inconsistente con datos reportados previamente, en los cuales se indica que YB1 se une y regula promotores que no pertenecen sólo a la caja-Y, entre ellos el receptor del factor de crecimiento epidermal (Stratford, et al., 2007), aunque YB-1 puede regular positiva o negativemente la transcripción y la traducción; además activa genes asociados con la porliferacion celular y el cáncer, y reprime genes asociados con la muerte celular (Wang, & Hirschberg, 2011). Finalmente, se observó que la expresión de un ITAF (SRp20) en líneas celulares estables transfectadas con los vectores bicistrónicos de los IRES EMCV y Myc provocan una activación de la expresión de GFP (figura 15). Probablemente este factor ha sido implicado en funciones alternativas de expresión génica, posiblemente proporcionando un vínculo entre el procesamiento y exportación del mARN y la traducción. Sin embargo, SRp20 ha sido detectado en fracciones de la subunidad ribosomal, pero la sobre expresión de SRp20 no tiene un efecto estimulatorio en la traducción cap-dependiente (Bedard, et al., 2007). 44 En definitiva, el sistema combinado de expresión de la proteína GFP y mCherry podría ser una alternativa interesante en varias circunstancias: 1) una herramienta para la elaboración de líneas celulares estables (incluyendo el uso para la selección de células mediante citometría de flujo), 2) creación de ratones transgénicos que expresen genes reporteros fluorescentes, 3) como un instrumento para la revisión de librerías de shARNi o la investigación de químicos usando lecturas evaluaciones fluorescentes de intensidad, ya sea manualmente o mediante robots. 45 7 CONCLUSIONES Y FUTURO 1. Algunas secuencias IRES conocidas (EMCV viral, c-Myc eucariota, Cx26 y Cx43) así como un IRES putativo no probado (denominado 19kD) fueron exitosamente amplificados por PCR. Por lo tanto, el enfoque de los estudios futuros en el campo deben centrarse en el significado fisiológico celular de la traducción mediada por IRES; ya que esta vía puede permitir el entendimiento de varias hipótesis sobre los mecanismos de traducción cap-independiente que permitan descubrir nuevos mARNs candidatos, cuyo mecanismo de traducción cap-independiente mediada por IRES sea facilitada/impulsado por las secuencias reguladoras ITAFs. 2. Las secuencias IRES fueron clonadas con éxito dentro del sitio de clonaje múltiple entre Cherry y EGFP para permitir el análisis de la expresión de cherry capdependiente y la expresión de GFP mediada por IRES. 3. Estudios de expresión transitoria confirmaron, como se esperaba, que el IRES viral EMCV presenta la más fuerte activación traduccional de EGFP. El c-Myc-IRES posee una actividad más baja, lo cual depende de la línea celular que haya sido utilizada y, sorprendentemente, parece ser reprimido cuando co-expresa el factor ITAF YB-1. El IRES putativo 19kDa, aparentemente no presenta actividad o esta es limitada. 4. La transfección de células Phoenix con este vector permitió la producción de partículas virales y la infección de una variedad de líneas celulares como HaCaT, HeLa, A549 y MDA-MB-231. Usando la resistencia a la puromicina, presente en el vector, estas líneas celulares fueron seleccionadas adecuadamente por una expresión de fluorescencia estable y actualmente están siendo expandidas para su uso en una variedad de proyectos. 5. Las líneas celulares han sido, hasta ahora, utilizadas con éxito para investigar la actividad IRES en células sujetas ya sea a través de estrés o por la manipulación de las vías de señalización por fármacos. Usando células HaCaT, una fuerte activación de la actividad del IRES de Myc fue descubierta ya sea a través de la tensión o la manipulación de las vías de señalización por fármacos, la privación de suero y la simulación de hipoxia con cloruro de cobalto. Adicionalmente, la represión de la vía 46 de mTOR por PP242, permitió una elevada activación del IRES de cMyc, mientras que Mnk1, inhibidor CGP, tuvo poco efecto. 6. Finalmente, el conocido ITAF SRp20 fue clonado en un vector de expresión y transfectado en líneas celulares que ya expresaban establemente cherry-EGFP conteniendo varios elementos IRES. Una fuerte activación de la actividad del IRES de myc se observó en la línea celular de cáncer mamario MDA-MB-468 determinada por SRp20. Esta es la primera vez que se observa una asociación entre SRp20 y la activación de Myc. El análisis de muchos otros ITAFs mediante sobre expresión de ARNi es actualmente en marcha. 7. En el futuro, se obtendrá una línea celular estable con IRES específicos (por ejemplo HCV IRES); el screening de ARN (una librería de siARN cubriendo la mayor parte del genoma. Se podrán realizar transfecciones robóticas en 2 días, y lecturas automatizada de la proporción de GFP para que bioinformáticos puedan identificar todos los genes que regulan específicamente la actividad IRES (pero no la traducción normal de cherry), así mismo se podrá realizar el screening químico de bibliotecas mediante robots. 47 8 BIBLIOGRAFIA Ausubel, F., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K. 1995. Short Protocols in Molecular Biology. John Wiley & Sons, Inc., USA. Baird, S.D., Turcotte, M., Korneluk, R.G., Holcik, M. 2006. Searching for IRES. RNA; 12(10): 1755-1785. Bedard, K.M., Daijogo, S., Semler, B.L. 2007. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J; 26(2): 459 - 67. Berg, J.J., Tymoczko, J.L., Stryer, L. 2002. Biochemistry. 5th edition. New York: W H Freeman. Bert, A.G., Grepin, R., Vadas, M.A., Goodall, G.J. 2006. Assessing IRES activity in the HIF-1alpha and other cellular 5′ UTRs. RNA; 12(6): 1074-1083. Bonnal, S., Schaeffer, C., Creancier, L., Clamens, S., Moine, H., Prats, A.C., Vagner, S. 2003. A single internal ribosome entry site containing a G quartet RNA structure drives fibroblast growth factor 2 gene expression at four alternative translation initiation codons. J. Biol. Chem; 278(41): 39330-39336. Cobbold, L.C., Spriggs, K.A., Haines, S.J., Dobbyn, H.C., Hayes, C., de Moor, C.H., Lilley, K.S., Bushell, M., Willis, A.E. 2008. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol. Cell Biol; 28(1): 40-49. Coffin, J.M., Hughes, S.H., Varmus, H.E. 1997. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. Costa-Mattioli, M., Svitkin, Y., Sonenberg, N. 2004. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol. Cell Biol; 24(15): 6861-6870. Evans, J.R., Mitchell, S.A., Spriggs, K.A., Ostrowski, J., Bomsztyk, K., Ostarek, D., Willis, A.E. 2003. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene; 22(39): 80128020. Fitzgerald, K.D. & Semler, B.L. 2009. Bridging IRES elements in mRNAs to the eukaryotic translationapparatus. Biochim Biophys Acta; 1789(9-10): 518–528. Graber, T.E. &Holcik, M. 2007. Cap-independent regulation of gene expression in apoptosis. Mol. Biosyst; 3(12): 825-834. Graham, F.L., Van der Eb A.J., 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology; 52(2): 456-467. 48 Hitoshi, Y., Lorens, J., Kitada, S.I., Fisher, J., La-Barge, M., Ring, H., Francke, U., Reed, J.C., Kinoshita, S., Nolan, G.P. 1998. Toso, a cell surface, specific regulator of Fasinduced apoptosis in T cells. Immunity; 8(4): 461-471. Hoang, B., Benavides, A., Shi, Y., Yang, Y., Frost, P., Gera, J., Lichtenstein, A. 2012. J Biol Chem; 287(26): 21796 – 21805. Holcik, M., Gordon, B.W., Korneluk, R.G. 2003. The internal ribosome entry sitemediated translation ofantiapoptotic protein XIAP is modulated by the heterogeneous nuclear ribonucleoproteins C1 andC2. Mol. Cell Biol; 23(1): 280-288. Horbinski, C.M., Stachowiak, K., Higgins, D., Finnegan, S.G. 2001 Polyethylenimine mediated transfection of cultured postmitotic neurons from rat sympathic ganglia and adult human retina. BMC Neuroscience; 2: 2. Jackson, R.J., Hellen, C.U., Pestova, T.V. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol; 11(2): 113-127. Jackson, R.J. 2005. Alternative mechanisms of initiating translation of mammalian mRNAs. Biochem. Soc.Trans; 33(6): 1231-1241. Jang, G.M., Leong, L.E., Hoang, L.T., Wang, P.H., Gutman, G.A., Semler, B.L, 2004. Structurally distinct elements mediate internal ribosome entry within the 5′-noncoding region of a voltage-gated potassium cannel mRNA. J. Biol. Chem; 279(46): 47419-47430. Jang, S.K., Kräusslich, H.G., Nicklin, M.J., Duke, G.M., Palmenberg, A.C., Wimmer, E. 1988. A segment of the 5’ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol; 62(8): 2636-2643. Jimenez, J., Jang, G.M., Semler, B.L., Waterman, M.L. 2005. An internal ribosome entry site mediates translation of lymphoid enhancer factor-1. RNA; 11(9): 1385-1399. Joachims, M., Van Breugel, P.C., Lloyd, R.E. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol; 73(1): 718-727. Kozak, M. 2005. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res; 33(20): 6593-6602. Komar, A.A & Hatzoglou, M. 2011. Cellular IRES-mediated translation The war of ITAFs in pathophysiological states. Cell Cycle; 10(2): 229-240. Langdon, S.P. 2010. Cancer Cell Culture. Methods and Protocols. Humana Press. Totowa, New Jersey. Le Quesne, J.P., Stoneley, M., Fraser, G.A., Willis, A.E. 2001. Derivation of a structural model for the c-myc IRES. J. Mol. Biol; 310(1): 111-126. Lodish, H., Berk, A., Zipursky, S.L., Matsudaira, P., Baltimore, D. Darnell, J. 2000. Molecular Cell Biology, 4th edition. New York: W. H. Freeman. 49 Manak, M.M. 1993. Sample Preparation. En: DNA probes (G.H. Keller y M.M. Manak, eds) pp. 27 - 68. Stockton Press, New York, USA. Martin, P., Albagli, O., Poggi, M.C., Boulukos, K.E., Pognonec, P. 2006. Development of a new bicistronic retroviral vector with strong IRES activity. BMC Biotechnol: 12; 6:4. Martínez-Salas, E., Ramos, R., Lafuente, E., López, de Q.S. 2001. Functional interactions in internal translation initiation directed by viral and cellular IRES elements. J. Gen. Virol; 82(5): 973-984. Mitchell, S.A., Spriggs, K.A., Bushell, M., Evans, J.R., Stoneley, M., Le Quesne, J.P., Spriggs, R.V., Willis, A.E. 2005. Identification of a motif that mediates polypyrimidine tract-binding protein-dependent internal ribosome entry. Genes Dev; 19(13): 1556-1571. Mitchell, S.A., Spriggs, K.A., Coldwell, M.J., Jackson, R.J., Willis, A.E. 2003. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol. Cell; 11(3): 757-771. Nanbru, C., Lafon, I., Audigier, S., Gensac, M.C., Vagner, S., Huez, G., Prats, A.C. 1997. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J. Biol. Chem; 272(51): 32061-32066. Newton, C.R., Graham, A. 1994. PCR. Bios Scientific Publishers, Oxford, UK. Paek, K.Y., Kim, C.S., Park, S.M., Kim, J.H., Jang, S.K. 2008. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J. Virol; 82(24): 12082-12093. Pear, W.S., Nolan, G.P., Scott, M.L., Baltimore,D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl.Acad.Sci. USA; 90(18): 83928396. Pelengaris S, Khan, M., Evan, G. 2002. c-MYC: more than just a matter of life and death. Nat. Rev. Cancer; 2(10): 764-776. Pelletier, J., Sonenberg, N. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature; 334(6180): 320-325. Pelletier, J., Kaplan, G., Racaniello, V.R., Sonenberg, N. 1988. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5′ noncoding region. Mol. Cell Biol; 8(3): 1103-1112. Rivera-Tapia, J.A., Cedillo, R.L., Gil, J.C. 2002. Some biological featuresof Mollicutes. Rev Latinoam Microbiol; 44(2); 53-57. Sambrook, J., Fritsch, E.F., Maniatis, T. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York, USA. 50 Schepens, B., Tinton, S.A., Bruynooghe, Y., Parthoens, E., Haegman, M., Beyaert, R., Cornelis, S. 2007. A role forhnRNP C1/C2 and Unr in internal initiation of translation during mitosis. EMBO J; 26(1); 158-169. Sonenberg, N., Hinnebusch, A.G. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell; 136(4):731-745. Strachan, T., Read, A.P., 1999. Human Molecular Genetics, 2nd edition. New York: Wiley-Liss. Garland Science. Stoneley, M., Subkhankulova, T., Le Quesne, J.P., Coldwell, M.J., Jopling, C.L., Belsham, G.J., Willis, A.E. 2000. Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res; 28(3); 687694. Stratford, A.L., Habibi, G., Astanehe, A., Jiang, H., Hu, K., Park, E., Shadeo, A., Buys, T.P.H., Lam, W., Pugh, T., Marra, M., Nielsen, T.O., Klinge, U., Mertens, P.R., Aparicio, S., Dunn, S.E. 2007. Breast Cancer Res; 9(5): R61.1. Van Eden, M.E., Byrd, M.P., Sherrill, K.W., Lloyd, R.E. 2004. Demonstrating internal ribosome entry sites in eukaryotic mRNAs using stringent RNA test procedures. RNA; 10(4): 720-730. Walter, B.L., Parsley, T.B., Ehrenfeld, E., Semler, B.L. 2002. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J. Virol; 76(23): 12008-12022. Wang, S., Hirschberg, R. 2011. Y-Box Protein-1 is a Transcriptional Regulator of BMP7. Journal of Cellular Biochemistry; 112(4):1130-1137. 51