Enfermedad de Pompe - Asociación Distrofia Muscular para las
Anuncio

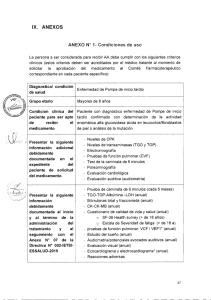
This page was exported from - Asociación Distrofia Muscular para las Enfermedades Neuromusculares Export date: Sat Nov 26 17:36:27 2016 / +0000 GMT Enfermedad de Pompe ¿Qué es la Enfermedad de Pompe? La Enfermedad de Pompe es una enfermedad neuromuscular que como la gran mayoría produce pérdida progresiva de la fuerza muscular y función motora Fue descripta en 1932 y pasaron muchos años hasta que se descubrió que la causa de la misma es la falta de una enzima (llamada alfa glucosidasa ácida) en las fibras musculares que es capaz de desdoblar el glicógeno, una de las fuentes principales de energía para su funcionamiento. La acumulación de glicógeno en el músculo produce un daño irreversible en el mismo provocando la degeneración de las fibras musculares. Su incidencia global (cantidad de nacidos que llevan la enfermedad en sus genes) se estima en 1/40.000. La imagen representa una célula muscular normal, mientras que la imagen de la derecha ilustra lo que puede suceder en una célula afectada con la enfermedad de Pompe. A medida que el glucógeno se acumula en las células afectadas, los lisosomas se pueden agrandar, lo que eventualmente deteriora la función muscular. Diferentes formas de aparición La enfermedad puede aparecer en la infancia (Enfermedad de Pompe Infantil), antes del año de vida y se la conoce como forma infantil. Los niños que la presentan sufren de una cardiomegalia (agrandamiento cardíaco) y gran debilidad de los músculos respiratorios por lo que la enfermedad es fatal antes del año de vida si no es reconocida y tratada. Existe otra forma, que es la más común y de comienzo en la vida adulta (Enfermedad de Pompe de comienzo tardío), que puede aparecer en la primera o segunda infancia, la adolescencia o frecuentemente en la vida adulta (hasta los 50 o mas años de edad). Su aparición es muy insidiosa con síntomas variados lo que complica mucho su reconocimiento y diagnóstico. La gran simuladora La enfermedad puede comenzar simulando un cansancio fácil, intolerancia al ejercicio, debilidad en las piernas, dolor lumbar (de espalda) o hasta fatiga y agitación por falta de aire. En los niños y adolescentes como una falta de crecimiento o ganancia de peso. A veces sólo la elevación de enzimas hepáticas o de la CPK que se pesquisan en exámenes de laboratorio de rutina es la única manifestación de su presencia. Con esta variedad de síntomas de presentación, el diagnóstico temprano es muy infrecuente ya que ni los médicos clínicos generales ni los pediatras, ortopedistas u otros especialistas la sospechan. Varios estudios efectuados en todo el mundo demuestran que existe una demora de varios años (hasta 10 o más) en llegar al diagnóstico correcto desde la aparición de los primeros síntomas. De hecho hasta hace muy pocos años atrás la EP era (y lo sigue siendo hoy) considerada muy rara, ya casi nadie pensaba en ella. La investigación y un cambio en la Historia Sin embargo un grupo de investigadores en la Universidad de Duke en los Estados Unidos investigaba y trabajaba activamente en la Output as PDF file has been powered by [ Universal Post Manager ] plugin from www.ProfProjects.com | Page 1/2 | This page was exported from - Asociación Distrofia Muscular para las Enfermedades Neuromusculares Export date: Sat Nov 26 17:36:33 2016 / +0000 GMT búsqueda de una solución. Este gran esfuerzo en conjunto con el Laboratorio Genzyme produjo el gran cambio en la historia y esa historia fue muy bien relatada en una película protagonizada por el actor Harrison Ford hace un par de años atrás que se llamó ?Medidas Extraordinarias?. En ella se reflejó cómo se llevó adelante el esfuerzo hasta lograr un tratamiento efectivo para la enfermedad. 1. El niño hereda dos copias normales del gen, una de cada padre. 2. El niño hereda un gen mutante de la madre, un gen normal del padre. 3. El niño hereda un gen mutante del padre, un gen normal de la madre. 4. El niño hereda dos copias mutantes del gen, una de cada padre. Nota: La herencia de la enfermedad de Pompe no está relacionada con el género, ya que tanto los hombres como las mujeres resultan igualmente afectados. ¿Cómo evoluciona un paciente con EP? Lamentablemente, una vez iniciada la enfermedad ya no se detiene y con el paso del tiempo los pacientes comienzan a experimentar franca debilidad muscular, dificultad para caminar y subir escaleras o realizar actividades de la vida diaria. Es en este período en que la enfermedad puede ser indistinguible entre muchas enfermedades neuromusculares como las distrofias musculares, las enfermedades de motoneurona, la miastenia o la polimiositis. Pero mucho más dramático que eso es el avance silencioso de la pérdida de fuerza en los músculos respiratorios que, sin avisar con síntomas, avanza más rápido que la pérdida de la función motora general. Lamentablemente, los síntomas de falta de aire aparecen muy tarde cuando ya para entonces se debe recurrir a mecanismos de ayuda respiratoria (sistemas de respiración asistida como el BIPAP). Muchos pacientes pueden caminar con mayor o menor dificultad pero deben dormir o pasar muchas horas del día con un respirador artificial. ] ¿Cómo se hace el diagnóstico? Todos los estudios efectuados demuestran que existe una demora de varios años (hasta 10 o más) en llegar al diagnóstico correcto desde la aparición de los primeros síntomas. Sin embargo, el diagnóstico es sencillo ya que se puede hacer con una gota de sangre seca en papel de filtro. Con este material tan simple se puede determinar el nivel de actividad enzimática. Este método se utiliza en todo el mundo y fue desarrollado aquí en la Argentina por el Dr. Néstor Chamoles. Programa de Diagnóstico de Enfermedad de Pompe - ADM] ¿Cuál es el Tratamiento? La EP cuenta hoy con tratamiento específico a través de la administración de Enzima Recombinante (lo que se conoce como Terapia de Reemplazo Enzimático), lo que ha cambiado de manera radical el pronóstico de los pacientes. Ésta es una enzima que reemplaza la que el organismo no produce y se ha convertido en el primer tratamiento específico para una enfermedad muscular. ¿Qué es entonces lo importante? Como se comprende, el poder sospechar de la enfermedad en pacientes que presentan debilidad muscular progresiva puede significar el tratamiento temprano y evitar el avance de esta grave afección. Nuevamente volvemos a la importancia de un diagnóstico correcto y precoz toda vez que sea posible. El difundir esta enfermedad y sus múltiples formas de presentación ayudará a reconocerla y evitar entonces la progresión de la misma. © Copyright 2010-All Rights Reserved · ADM Argentina Adaptación del Artículo ?La Distrofia Facioescapulohumeral?. Revista Seguir Andando No12. Diciembre 2012. Asociación Distrofia Muscular Output as PDF file has been powered by [ Universal Post Manager ] plugin from www.ProfProjects.com | Page 2/2 |