OXCARBAZEPINA TRILEPTAL®® (Novartis)
Anuncio
")
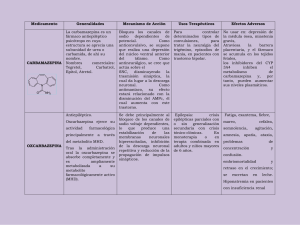
OXCARBAZEPINA TRILEPTAL (Novartis) GRUPO TERAPÉUTICO - Grupo anatómico: (N) SISTEMA NERVIOSO. - Grupo específico: N03AF. ANTIEPILÉPTICOS. Carboxamidas INDICACIÓN AUTORIZADA Tratamiento de las crisis epilépticas parciales con o sin generalización secundaria con crisis tónico-clónicas. ANTECEDENTES La epilepsia es una alteración o, más propiamente, un conjunto de alteraciones que tienen como manifestación común la aparición con carácter recurrente de alteraciones electroquímicas, las cuales conducen a movimientos involuntarios y/o experiencias sensitivas anómalas, acompañadas eventualmente por crisis convulsivas y/o pérdida del conocimiento. En términos generales, se estima que la prevalencia de la enfermedad oscila entre un 0,5% y un 3% de la población general, aunque se ha evaluado en un 6% el porcentaje de la población que experimentarán a lo largo de su vida al menos una crisis convulsiva de tipo no febril. Las formas clínicas en que se pueden manifestar las crisis epilépticas varían extraordinariamente. Por este motivo, se ha intentado en numerosas ocasiones realizar clasificaciones que agrupasen de una forma más o menos racional tales presentaciones clínicas. A) Crisis parciales (locales o focales): De comienzo localizado en una parte de uno de los hemisferios cerebrales. Una crisis parcial puede terminar como tal o evolucionar hacia una crisis generalizada (generalización secundaria). Puede ser simples o complejas: - Simples: El paciente no pierde la consciencia durante la crisis epiléptica. - Con signos motores: Pueden afectar a cualquier parte del organismo. Los síntomas pueden ser locales o extenderse a áreas corticales vecinas, produciendo el movimiento secuencial de varias partes del cuerpo ("marcha epiléptica" o "crisis jacksoniana"). Tras experimentar la crisis, la zona afectada puede quedar paralizada durante varios minutos o incluso horas. - Con síntomas somatosensoriales: Se suelen manifestar como pinchazos o entumecimiento y pueden seguir una marcha similar a las crisis jacksonianas. Puede ser visuales (destellos luminosos, alucinaciones visuales), auditivas (incluyendo sonidos musicales), olfativas (generalmente en forma de olores desagradables), gustativas (frecuentemente, bajo la forma de sabor metálico). - Con síntomas vegetativos: Vómitos, palidez, sudación profusa, piloerección, dilatación de la pupila (midriasis), incontinencia urinaria o fecal, etc. - Con síntomas psíquicos: Suelen evolucionar a formas complejas (con pérdida de la consciencia) e incluyen alteraciones de las funciones superiores. Son frecuentes las alteraciones del lenguaje, la distorsión de la memoria y del sentido del tiempo. Los síntomas afectivos suelen manifestarse en forma extrema, tanto si son de tipo agradable como desagradable. Son frecuentes los episodios de miedo intenso y de depresión, que duran apenas unos minutos, aunque dado que aparecen de forma brusca, pueden provocar una fuga del paciente. - Complejas: Se produce una alteración de la consciencia desde el inicio de la crisis. Pueden comenzar como crisis parcial simple, progresando posteriormente a compleja. La alteración de la consciencia puede ser la única manifestación de la crisis epiléptica, o bien pueden venir acompañada de automatismos, consistentes en actividades motoras involuntarias más o menos coordinadas. Son típicos los automatismos alime ntarios (masticación o deglución), los mímicos y los verbales. B) Crisis generalizadas: Tienen un origen bilateral y simétrico (ambos hemisferios), sin localización precisa del inicio. - Ausencias (Petit mal): Implican una interrupción brusca de la actividad, con mirada extraviada y, en algunos casos, con rotación de los ojos (crisis oculógiras), que comúnmente duran menos de 30 segundos. Suelen finalizar repentinamente, sin dejar ningún tipo de secuela ni pérdida de la consciencia. En algunas ocasiones, las crisis de ausencia pueden ir acompañadas de sacudidas clónicas de los párpados o de los labios, así como de automatismos o de otros signos y síntomas vegetativos (cambios en los ritmos cardíaco y respiratorio, incontinencia urinaria, etc). - Crisis mioclónicas: Consisten en contracciones súbitas y breves, que pueden ser generalizadas o afectar sólo a cara, tronco y/o extremidades, o incluso a grupos musculares o músculos aislados. - Crisis clónicas: Suele tratarse de contracciones repetitivas muy bruscas. En ocasiones pueden ir enlazadas. - Crisis tónicas: Implican la contracción generalizada de la musculatura de los mie mbros, generalmente acompañada de rotación de los ojos o incluso del cuerpo hacia un lado. - Crisis tonicoclónicas (Grand mal): La crisis suele iniciarse con una pérdida brusca de la consciencia, produciéndose a continuación una contracción muscular tónica intensa, acompañada de dificultades respiratorias y un grito o quejido. El paciente suele caer al suelo, completamente rígido. Tras unos segundos, se inicia la fase clónica, con sacudidas repetidas, respiración estertórea, cianosis e hipersalivación. Finalmente, el paciente se relaja y la respiración se hace profunda, pudiendo permanecer el paciente inconsciente durante períodos que van desde unos segundos a varias horas. Al despertar, el paciente no suele recordar lo sucedido (amnesia) y experimentará dolor muscular difuso, cefalea e incluso agarrotamiento general. - Crisis atónicas: Se caracterizan por una pérdida brusca del tono corporal, que puede ser fragmentaria o generalizada, lo cual puede conducir desde la flexión de la cabeza y la caída del maxilar, hasta la caída súbita del paciente. La pérdida de la consciencia suele ser momentánea, recuperándose el paciente de forma rápida. De acuerdo con los tipos de crisis y su posible origen, se ha establecido un amplio abanico de síndromes epilépticos. Cada una de las formas de epilepsia o de síndromes epilépticos combina uno o más tipos de crisis epilépticas, con otros signos y síntomas. Algunos de los más comunes son la epilepsia benigna infantil con puntas centrotemporales (forma más común de epilepsia benigna infantil), la epilepsia mioclónica benigna de la infancia, la epilepsia ausencia infantil (típica de niños en edad escolar, especialmente entre los 6 y los 7 años de edad), la epilepsia ausencia juvenil (más infrecuente que la variedad infantil), la epilepsia con crisis generalizadas tonicoclónicas del despertar (aparece de forma casi exclusiva durante la primera media hora tras el despertar del paciente y supone entre el 20% y el 50% de todas las epilepsias con crisis convulsivas generalizadas tonicoclónicas), la epilepsia mioclónica juvenil (representa en torno al 6% de todas las epilepsias), el síndrome de West (un síndrome epiléptico infantil, con crisis espasmódicas, grave retraso mental y otras secuelas neurológicas) y el síndrome de Lennox- Gastaut (afecta al 3-10% de los niños con epilepsia y cursa con crisis muy frecuentes y de diverso tipo, siendo también frecuente el desarrollo de un estado de mal epiléptico o status epilepticus y se suele asociar a retraso mental). Un síndrome bastante especial es el estado de mal epiléptico (Status epilepticus) referido como cualquier crisis epiléptica que dure más de 30 minutos o la reiteración de crisis sin restablecimiento de la consciencia entre ellas. Salvo ciertas formas muy peculiares (y muy infrecuentes, por otro lado), la epilepsia no debe ser enfocada como una entidad patológica concreta en sí misma. De hecho, se tiende a considerar la epilepsia como un grupo de enfermedades cuya única característica en común es la aparición con carácter recurrente (repetitivo) de fenómenos electroquímicos anómalos en el sistema nervioso central. En general, todos los ataques epilépticos son debidos a una descarga anómala, de frecuencia muy elevada, en uno o varios pequeños grupos de neuronas, que pueden difundir a otras áreas o incluso generalizarse. Estas descargas son producidas por la activación sincrónica (al mismo tiempo) de estos pequeños grupos celulares, formados por neuronas intrínsecamente hiperexcitables (epileptógenas), capaces de desencadenar una actividad sináptica excitatoria excesiva, a través de descargas de potenciales de acción de frecuencia muy alta. Las neuronas "epileptógenas" presentan despolarizaciones anormales, siendo lentas, muy amplias y prolongadas (más de 100 milisegundos), que son las responsables de la génesis de los potenciales de acción de alta frecuencia. Estas despolarizaciones son conocidas como PDS (del inglés, Paroxysmal Depolarization Shift). La dispersión de las descargas epilépticas está regulada por equilibrio de dos tipos de aminoácidos neurotransmisores de acción muy rápida, uno de tipo inhibitorio y otro de tipo excitatorio. El principal neurotransmisor inhibitorio es el GABA (ácido gammaaminobutírico), aunque existen otros, como la glicina y la taurina. Por contra, el ácido aspártico y, especialmente, el ácido glutámico actúan como mediadores en la transmisión excitat oria sináptica rápida. Se han descrito numerosos receptores celulares de aminoácidos neuroexcitatorios, casi todos en el sistema nervioso central. Cuatro de ellos han sido estudiados con cierto detalle. Reciben el nombre del sustrato específico al que se fijan: - NMDA: Acido N- Metil-D- Aspártico. - AMPA (también llamado QUIS/AMPA): Acido α-amino-3-hidroxi-4-isoxazolpropiónico y ácido quiscuálico. - KAIN: Acido kaínico. - AP4: Acido L-2-amino-4-fosfonobutanoico. De todos ellos, el más estudiado, a mucha diferencia, es el primero (NMDA). La activación de este receptor por el ácido glutámico es capaz de producir una intensa despolarización de la membrana, suficiente como para permitir la apertura de canales iónicos de calcio (Ca2 +) dependientes del voltaje. Esto conduce a una entrada (influjo) de calcio en el interior neuronal, lo cual, a su vez, conduce a la liberación de neurotransmisores exc itatorios capaces de estimular otras neuronas. Una excesiva entrada de calcio puede acabar con la muerte neuronal. Asimismo, la activación de los receptores NMDA conduce a la formación de brotes axónicos en varias áreas cerebrales, como consecuencia de la estimulación de ciertos genes de "respuesta inmediata". La expresión de estos genes conduce a la síntesis de Factor de Crecimiento Neural, responsable último de la aparición de los brotes axónicos, que facilitan la expansión de las descargas epilépticas en posteriores ataques. Por su parte, el GABA es el más estudiado de aminoácidos neuroinhibitorios. Se han descrito dos tipos de receptores para el GABA: - GABA A : Se trata de un receptor postsináptico. Su estímulo por el GABA conduce a una hiperpolarización de la membrana neuronal, que impide a la neurona excitarse. Está ac oplado a un canal iónico de cloruro (Cl- ), cuya apertura regula, tanto en tiempo como en amplitud. A este canal de cloruro están acopladas, en asociación al receptor del GABA, otras estructras receptoras de diversas sustancias, algunas de ellas fármacos (ignorándose hasta el momento cuál será el ligando endógeno). Entre ellas, se han descrito los receptores de benzodiazepinas y de barbitúricos, ambos diferenciables entre sí. Posiblemente, el receptor de barbitúricos lo sea también de otras productos con estructura ureídica o carbámica, aunque esto último está por confirmar. Otras sustancias que se fijan sobre receptores más o menos específicos acoplados a los canales de cloruro neuronales son la avermectina y la picrotoxina. La activación de estos receptores "adicionales" es capaz de amplificar la respuesta del canal de cloruro a la acción agonista del GABA sobre el receptor GABAA , magnificando la hiperpolarización de la membarana neuronal y con ello, su insensibilidad a los estímulos externos. - GABA B : Es un receptor presináptico. Actúa facilitando la liberación de GABA. Se conoce mucho menos acerca de la bioquímica de este receptor, aunque se sabe que sobre él actúa el baclofeno (un miorrelajante de acción central). Existen dos enfoques terapéuticos básicos frente a los cuadros epilépticos: - Impedir la génesis de la descarga epiléptica. - Impedir o dificultar la difusión de la descarga epiléptica. Hasta ahora no se ha encontrado ninguna forma farmacológica eficaz de prevenir la génesis de las descargas de las neuronas hiperxcitables epileptógenas. Sólo la neurocirugía es capaz de solucionar algunos casos muy seleccionados, casi siempre restringidos a cuadros de epilepsia parcial. Si no puede impedirse farmacológicamente que las neuronas epilépticas se exciten, en cambio sí se puede impedir o, al menos, moderar la propagación de la descarga. Para ello se puede actuar a varios niveles: - Modular el funcionamiento de canales iónicos dependientes del voltaje, implicados en la producción y propagación del potencial de acción: - a) Canales de sodio (Na+): El fármaco de referencia es la fenitoína, aunque otros muchos agentes antiepilépticos tienen efecto a este nivel, como carbamazepina, lamotrigina, ácido valproico o felbamato - b) Canales de calcio (Ca2 +): Etosuximida, ácido valproico. - Modular la neurotransmisión aminoacidérgica: - a) Favoreciendo la acción de los aminoácidos neuroinhibitorios, especialmente del GABA: - a1 : Aporte exógeno de GABA: Progabida. - a2 : Frenar la degradación del GABA endógeno: Vigabatrina, ácido valproico. - a3 : Potenciar la respuesta del GABA sobre el canal de cloruro: Benzodiazepinas, barbitúricos. - A4 : Inhibir la recaptación presináptica de GABA: Tiagabina. - b) Inhibiendo la acción de los aminoácidos neuroexcitatorios, especialmente del ácido glutámico: - b1 : Reducir la liberación de ácido glutámico: Lamotrigina. - b2 : Bloqueando los receptores NMDA del ácido glutámico: Felbamato. Un aspecto farmacológico importante es que, como se ha visto, una parte de los fármacos indicados son capaces de actuar a través de dos o más mecanismos a un tiempo. Se estima que entre un 30 y un 70% de los pacientes no presentan recaídas tras una primera crisis. Por tanto, como norma general, las crisis epilépticas aisladas no deben ser tratadas, salvo que el neurólogo identifique algún factor de riesgo específico. Sólo cuando se haya confirmado la existencia de dos crisis en menos de dos años se sugiere iniciar un tratamiento farmacológico preventivo. Aproximadamente un 80% de los casos de epilepsia se controlan con un solo medic amento. De acuerdo con esto, lo normal es comenzar un tratamiento con un solo medic amento. Si las crisis persisten con las dosis máximas del fármaco seleccionado, se cambia a otro considerado como de primera línea en el tipo de epilepsia considerado. Sólo cuando se plantee una resistencia generalizada es aceptable recurrir a la politerapia, aunque siempre valorando rigurosamente la posibilidad de interacciones, muy alta por otra parte. No existe un acuerdo unánime sobre la selección de fármacos antiepilépticos, pero son generalmente aceptadas las siguientes líneas terapéuticas: - Crisis genralizadas tonicoclónicas: 1º: Acido valproico, carbamazepina. 2º: Lamotrigina, fenitoína. - Crisis generalizadas de ausencia: 1º: Acido valproico, etosuximida. 2º: Clonazepam, lamotrigina. - Crisis generalizadas mioclónicas: 1º: Acido valproico. 2º: Clonazepam 3º: Lamotrigina, fenobarbital. - Crisis parciales: 1º: Acido valproico, carbamazepina. 2º: Vigabatrina, lamotrigina, fenitoína, fenobarbital. Los síndromes epilépticos especialies, como el de Lennox- Gastaut, los fármacos preferidos son topiramato, felbamato, lamotrigina y vigabatrina; esta última también está indicada en el síndrome de West. ACCIÓN Y MECANISMO DE LA OXCARBAZEPINA La oxacarbazepina es un derivado de la carbamazepina, que debe buena parte de su actividad a su principal metabolito (MHD). Su espectro y mecanismo de acción es similar al la carbamazepina. El bloqueo de uno o varios tipos de canales iónicos voltaje-dependientes de la membrana neuronal constituye el mecanismo de acción antiepiléptica más conocido de la carbamazepina, aunque no es el único. Esta dependencia del voltaje indica que el grado de inhibición del canal iónico es proporcional al voltaje (se produce más fácilmente en las células despolarizadas) y a la frecuencia (los canales más afectados son aquellos con más alto ritmo de cierre y apertura). Esto supone un cierto grado de selectividad frente a las neuronas hiperexcitables ("epilépticas"). Actúa fundamentalmente sobre canales de sodio (Na+) voltaje-dependientes, produciendo el bloqueo de éstos. Ello se traduce en una estabilización de las membranas neuronales hiperexcitadas, así como en la inhibición de la descarga neuronal repetitiva y reducción de la propagación de impulsos sinápticos. Se ha indicado que la acción sobre otros canales iónicos de este tipo de fármacos sobre otros canales iónicos, tales como los de potasio o los de calcio, también podría contribuir a la acción antiepiléptica. ASPECTOS MOLECULARES DEL NUEVO FÁRMACO La oxacarbazepina es un derivado de la carbamazepina y, como ésta, presenta una estructura tricíclica de tipo iminoestilbénico, que recuerda abiertamtente la de algunos antidepresivos tricíclicos, como la imipramina y sus derivados. La oxcarbazepina es la 10,11dihidro-10-oxocarbamazepina. Por consiguiente, se trata del cetoderivado de la carbamazepina, similar desde el punto de vista quí mico y farmacológico pero con diferencias farmacocinéticas, en especial en lo que se refiere a las vías de metabolización. La oxacarbazepina es transformada rápidamente y casi por completo en el derivado monohidroxilado, conocido como MHD (10,11-dihidro-10hidroxicarbamazepina), sin formación del epóxido en la posición 1011. Este hecho es clínicamente relevante, ya que este epoxi-derivado es considerado como responsable de algunos de los efectos colatera- les neurotóxicos más graves de la carbamazepina. Una cantidad relativamente pequeña de MHD es transformada posteriormente en el derivado dihidroxilado, el cual es farmacológicamente inactivo. EFICACIA CLÍNICA La eficacia clínica de la oxcarbazepina en el control de pacientes con crisis epilépticas de inicio parcial ha sido adecuadamente demostrada en ensayos clínicos controlados con placebo, así como en estudios comparativos con carbamazepina y fenitoína, incluyendo un cierto porcentaje de pacientes refractarios a estos últimos fármacos (en torno al 25%, en estudios a corto plazo). Los datos clínicos disponibles indican que el fármaco es eficaz tanto en monoterapia como asociado a otros medicamentos antiepilépticos. En este sentido, la utilización en monoterapia sobre pacientes recientemente diagnosticados y no tratados con anterioridad, permite mantener a un 60% de los pacientes sin padecer crisis convulsivas durante al menos un año de tratamiento. Por otro lado, la oxacarbazepina ha demostrado tener un buen perfil toxicológico y de interacciones con otros medicamentos antiepilépticos considerados como de primera lí nea. En los ensayos clínicos realizados no se ha observado diferencias con el placebo en lo que se refiere a efectos adversos frecuentemente relacionados con los fármacos antiepilépticos, tales como hiperplasia gingival, hirsutismo, dificultades en la concentración mental, problemas de memoria, descoordinación psicomotriz, aumento de peso, temblor, alopecia o erupciones cutáneas. En el caso de la oxcarbazepina, los más comúnmente descritos son vértigo, náuseas, vómitos, cefalea, diarrea o estreñimiento, entre otros. Las crisis epilépticas de inicio parcial suponen la forma más común de epilepsia (hasta un 70% de los casos). El objetivo terapéutico es conseguir el control permanente de tales crisis mediante la utilización de un único medicamento, a fin de mejorar el nivel de cumplimiento terapéutico y experimentar una menor incidencia de efectos adversos. Por ello, es interesante disponer de nuevos fármacos que resulten útiles en monoterapia y que, además, puedan ser combinados con otros agentes antiepilépticos sin alterar drástic amente la posología de estos últimos (por las interacciones). Además de lo indicado anteriormente, entre un 30% y un 50% de los pacientes epilépticos siguen experimentando alguna crisis epiléptica a pesar de estar bajo tratamiento con alguno de los diversos fármacos actualmente disponibles, y en aquellos en los que se consigue controlar tales crisis lo es a costa de provocar una elevada tasa de efectos adversos. Parece claro que la oxcarbazepina es un fármaco comparable con otros antiepilépticos de primera línea, al menos con respecto de su eficacia. Por lo general es bien tolerada, y los efectos adversos más graves son relativamente raros. Fue desarrollada a partir de la carbamazepina y ha demostrado ser tan efectiva como ésta y muestra una tolerabilidad superior en la mismas indicaciones terapéuticas. Una ventaja adicional de la oxcarbazepina consiste en que es metabolizada principalmente mediante enzimas de tipo reductasa, lo que limita los posibles efectos inductores o inhibidores sobre los enzimas de tipo oxidativo (como el sistema P450) y, en general, la tendencia para provocar interacciones farmacológicamente importantes, al menos en comparación con muchos otros anticonvulsivantes, incluida la propia carbamazepina. En general, la instauración del tratamiento con oxcarbazepina es mucho más rápida y fácil que con la carbamazepina, debido a su toxicidad menos pronunciada en relación con la dosis. La autorización de comercialización de la oxcarbazepina fue concedida en Estados Unidos en enero de 2000. Asimismo, el procedimiento de reconocimiento mutuo europeo finalizó en noviembre de 1999. OTROS FÁRMACOS SIMILARES REGISTRADOS ANTERIORMENTE EN ESPAÑA Fármaco Carbamazepina Especialidad Tegretol Laboratorio Novartis COSTES DIRECTOS DEL TRATAMIE NTO Indicación: Monoterapia de crisis epilépticas parciales (dosis de mantenimiento). Dosis diarias y coste Dosis adulto Coste diario adultos Coste anual adultos OXCARBAZEPINA 1200-2400 mg/día 319-639 Ptas. 116.691-233.381 Ptas. CARBAMAZEPINA 800-1600 mg/día 44-89 Ptas. 16.184-32.368 Ptas. VALORACIÓN OXCARBAZEPINA TRILEPTAL (Novartis) Grupo Terapéutico (ATC): N03AF. ANTIEPILÉPTICOS. Carboxamidas Indicaciones autorizadas: Tratamiento de las crisis epilépticas parciales con o sin generalización secundaria con crisis tónico-clónicas. VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, ♣♣ pero no implica cambios sustanciales en la terapéutica estándar. Reduce la incidencia o la frecuencia de efectos adversos de la terapia farmaco⇑ lógica estándar. Mejora las características farmacocinéticas (posología más cómoda y eficaz, au⇑ sencia de interacciones, etc) BIBLIOGRAFÍA - Barcs G, Walker EB, Elger CE, et al. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia 2000; 41(12): 1597-607. Beydoun A, Sachdeo RC, Rosenfeld WE, Krauss GL, Sessler N, Mesenbrink P, Kramer L, D'Souza J. Oxcarbazepine monotherapy for partial-onset seizures: a multicenter, double-blind, clinical trial. Neurology 2000; 54(12): 2245-51. Beydoun A. Safety and efficacy of oxcarbazepine: results of randomized, double-blind trials. Pharmacotherapy 2000; 20(8 Pt 2): 152S-158S. Bill PA, Vigonius U, Pohlmann H, Guerreiro CA, et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in adults with previously untreated epilepsy. Epilepsy Res 1997; 27(3): 195204. Christe W, Kramer G, Vigonius U, et al. A double-blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Res 1997; 26(3): 451-60. Cramer JA. Oxcarbazepine in a monotherapy trial for partial seizures--placebo-controlled studies in neurology: where do they stop? Neurology 1999; 53(9): 2212-3. Glauser TA. Expanding first-line therapy options for children with partial seizures. Neurology 2000; 55(11 Suppl 3): S30-7. Glauser TA, Nigro M, Sachdeo R, et al. Adjunctive therapy with oxcarbazepine in children with partial seizures. The Oxcarbazepine Pediatric Study Group. Neurology 2000; 54(12): 2237-44. Guerreiro MM, Vigonius U, Pohlmann H, et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Res 1997; 27(3): 205-13. McCabe PH. New anti-epileptic drugs for the 21st century. Expert Opin Pharmacother 2000; 1(4): 633-74. Pellock JM. Treatment of epilepsy in the new millennium. Pharmacotherapy 2000; 20(8 Pt 2): 129S-138S. Sabers A, Gram L. Newer anticonvulsants: comparative review of drug interactions and adverse effects. Drugs 2000; 60(1): 23-33. Schachter SC, Vazquez B, Fisher RS, et al. Oxcarbazepine in a monotherapy trial for partial seizures-placebo-controlled studies in neurology: where do they stop? Neurology 1999; 53(9): 2211-2. Schachter SC, Vazquez B, Fisher RS, et al. Oxcarbazepine: double-blind, randomized, placebo-control, monotherapy trial for partial seizures. Neurology 1999; 52(4): 732-7. Willmore LJ. Choice and use of newer anticonvulsant drugs in older patients. Drugs Aging 2000; 17(6): 441-52.