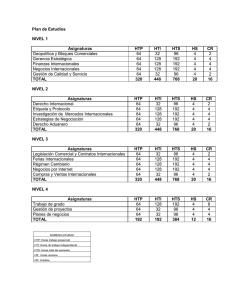
n por hormonas tiroideas de genes de se?alizaci?
Anuncio

UNIVERSIDAD DE BUENOS AIRES Facultad de Farmacia y Bioquímica Regulación por hormonas tiroideas de genes de señalización involucrados en vías de sobrevida y proliferación en células de linfoma T Lic. María Florencia Cayrol Directora: Dra. Graciela Cremaschi Co-Director: Dr. Leandro Cerchietti Lugar de Trabajo: Instituto de Investigaciones Biomédicas –CONICET. Facultad de Ciencias Médicas, Pontificia Universidad Católica Argentina 2014 1 Los trabajos publicados relacionados con este trabajo de tesis se detallan a continuación: Induction of apoptosis in T lymphoma cells by long-term treatment with thyroxine involves PKCz nitration by nitric oxide synthase. Barreiro Arcos ML, Sterle HA, Vercelli C, Valli E, Cayrol MF, Klecha AJ, Paulazo MA, Diaz Flaqué MC, Franchi AM, Cremaschi GA. Apoptosis. 2013, 18 (11): 1376-90. Thyroid status modulates T lymphoma growth via cell cycle regulatory proteins and angiogenesis. Sterle HA, Valli E, Cayrol F, Paulazo MA, Martinel Lamas DJ, Diaz Flaqué MC, Klecha AJ, Colombo L, Medina VA, Cremaschi GA, Barreiro Arcos ML. J Endocrinol. 2014, 222 (2): 243-55. Integrin αvβ3 acting as membrane receptor for thyroid hormones mediates angiogenesis in malignant T cells. Florencia Cayrol, María Celeste Díaz Flaqué, Tharu Fernando, Shao Ning Yang, Helena Andrea Sterle, Marcela Bolontrade, Mariana Amorós, Blanca Isse, Ricardo Norberto Farías, Haelee Ahn, Ye F. Tian, Fabrizio Tabbo, Ankur Singh, Giorgio Inghirami, Leandro Cerchietti, Graciela Alicia Cremaschi. Blood Journal. En prensa 2 Agradecimientos En primer lugar quisiera agradecer a mis compañeros de laboratorio que hicieron que la realización de este trabajo haya sido una de las experiencias más enriquecedoras de mi vida, no solo a nivel profesional sino también a nivel personal. De cada uno de ellos aprendí algo. A mi directora, Graciela, le agradezco haberme dado la oportunidad de formar parte de su grupo de trabajo en un momento donde se me hacía difìcil creer que uno se podía desarrollar en un lugar con obvias exigencias profesionales, pero rodeado de un ambiente de respeto, alegría y cariño. Por formarme y hacerme crecer tanto a nivel científico como personal, siempre con mucho amor y compañía, pero también mucha libertad. Gracias Gra por dejarme ser, quererme tal cual soy y acompañarme en todas mis locuras. A María Laura y Alicia por su preocupación y por siempre estar dispuestas a darme una mano cuando lo necesito. A Cele, Lenki, Ale y Edu por la compañía diaria con ricos mates, por las charlas sinceras cuando se plantean ciertos problemas, los consejos y por su colaboración en los experimentos realizados en este trabajo. También por todos los momentos divertidos y salidas que pasamos juntos. Quiero agradecer a mi co-director el Dr. Leandro Cerchietti el cual, en primer lugar, me abrió las puertas de su laboratorio en el Weill Cornell Medical College, y me permitió trabajar en un lugar de excelencia y conocer a personas increíbles. Gracias por tus enseñanzas que me formaron y me hicieron crecer tanto a nivel profesional como personal, por hacerme dar cuenta que se puede hacer buena ciencia básica pensando en una aplicación clínica y por todos los momentos increíbles vividos en NY. De esa experiencia conocí excelentes personas, de las cuales aprendí muchísimo y compartí muchisimos momentos de alegría e hicieron que mi estadía en NY sea una experiencia inolvidable. Quisiera agradecer a Lali, Maca, Carla, María, Kolko, Diego, Gabriel, Fabio, Cristian, Francisco y todos los compañeros del BIOMED, por siempre estar dispuestos a ayudar, por la buena onda en los almuerzos, las charlas, los chistes y todos los momentos compartidos. También quiero agradecer al Dr. Santa Coloma por ofrecernos y permitirnos trabajar en un hermoso instituto. 3 A toda la gente del CEFYBO, lugar donde comencé este trabajo. A Ana Genaro por su apoyo desde el inicio, a María, Miriam, María Rosa por su compañía. Especialmente a Ceci, Dami, Bere, Romi, Yami, Joy y Emi con quienes compartimos y seguiremos compartiendo momentos y experiencias tan lindas y divertidas. Quiero agradecer también a la Dra. Bolontrade y a Mariana Amorós con quienes realizamos ensayos in vivo y de migración. Al Dr. Diego Martinel, por su ayuda con los resultados de inmunohistoquímica y al Dr. Adrian Góngora por su ayuda en los ensayos donde se utilizaron los anticuerpos anti-VEGF. Al Dr. Ravinobich por siempre estar presente, especialmente, por haberme guíado en un momento profesional muy difícil y ayudarme a encontrar el camino que terminó llevandome a la realización de este trabajo. Agradecer a la Agencia Nacional de Promoción Científica y Tecnológica, y al CONICET, instituciones por las cuales a través de las becas otorgadas me permitieron desarrollar esta etapa de mi vida científica. A nivel personal, quiero agradecer a mis amigos de la UNQUI, Ana, Romi, Gustavo, Axel y Leti, que a pesar que algunos siguieron su camino y no nos vemos tan seguido, les agradezco tantos momentos lindos, de alegría y diversión. Gracias Ro querida por permitirme conocerte más y por compartir tantos momentos con Tizi. Gracias Anita por escucharme, por los consejos, por tu presencia incondicional, por alentarme y apoyarme en los momentos más difíciles, y por quererme tal cual soy. A la familia Montali, quiero agradecerle por dejarme formar parte de ella, particularmente a Lili por mimarme tanto, a David por todo su apoyo y a Fran por creer tanto en mí, y por todos los momentos compartidos junto con Mati. A mi familia, los Cayrol y los Molina-Schierenbeck, porque siempre me apoyaron y creyeron en mí. A mi mamá por inculcarme desde muy chica lo importante que es el estudio y la profesión para desarrollarse en la vida. A mi 4 papá, por estar siempre pendiente de mí a pesar de la distancia. Gracias por tu constante e increíble esfuerzo, el cual hizo realidad la posibilidad de estudiar fuera de Mar del Plata y formarme como profesional. Gracias por tus consejos y tu apoyo incondicional que hizo posible que hoy pueda concretar esta etapa de mí vida. Finalmente quisiera agradecerle a Mati, mi compañero desde hace ya 7 años. Por crecer y compartir la vida junto a mí, por estar tanto en los momentos más divertidos y alegres, como en los más tristes y difíciles. Por apoyarme en todas mis locuras, por soportar mis momentos de ansiedad y mal humor extremo, por enseñarme tantas cosas importantes que quizás no tengan que ver con la ciencia, pero sí con la vida misma. Gracias por enriquecer mi vida con tu conocimiento, tu arte y tu música, y por compartir conmigo tantas experiencias increíbles. 5 Resumen RESUMEN La proliferación, sobrevida y resistencia a drogas de las células T malignas son dependientes de una combinación de estímulos inducidos por el microambiente tumoral. En este trabajo se muestra que el receptor transmembrana para las hormonas tiroideas (HTs), la integrina αVβ3, juega un rol crítico entre la interacción de los linfomas de células T (LCT) y las señales externas del microambiente celular. Entre los ligandos de la integrina αVβ3 puede incluirse a proteínas extracelulares asociadas a matriz y factores solubles, como dichas hormonas. Demostramos que las HTs son capaces de estimular la proliferación de los LCT a través de vías de señalización intracelulares complementarias que involucran la activación del receptor de membrana para HTs (mTR). Por lo tanto, la hipótesis de este trabajo es que el bloqueo del mTR podría representar una nueva estrategia terapéutica para el tratamiento de pacientes con LCT. En este trabajo evaluamos el rol que ejercen las HTs en líneas celulares que representan los distintos subtipos de LCT humanos. Se encontró que las HTs a concentraciones fisiológicas inducen in vitro la proliferación y la transcripción de marcadores moleculares vinculados a la progresión del ciclo celular en células LCT. Entre ellos se induce la expresión del antígeno nuclear de células en proliferación, PCNA, y de las ciclinas D1, D2, D3, E2 y B1. En concordancia con lo mencionado se observó también, una disminución regulada por HTs del inhibidor de la quinasa dependiente de ciclina, p21, y del supresor tumoral, p53. Estos efectos se dan mayoritariamente por la acción de las HTs sobre el mTR. Es por ello que, particularmente, se focalizaron los estudios en los efectos mediados por HTs unidas a agarosa (HT-AG, impermeables a la célula) sobre el receptor de membrana en distintos subtipos de LCT, tanto de origen inmaduro como maduro. De estos estudios se pudo demostrar que la integrina αVβ3 es receptor de membrana para HTs en células T malignas. Utilizando técnicas de secuenciación del ARN y herramientas bioinformáticas, se identificaron los programas transcripcionales activados por HTs a través de su receptor de membrana. Se identificaron 118 y 5 transcriptos regulados de 6 Resumen manera positiva y negativa, respectivamente. Se encontró que a través de la activación de la integrina αvβ3, las HTs inducen vías de señalización que involucran la expresión de genes relacionados con la “traducción” (RPL13, RPL27A, RPL36), con la “cadena respiratoria oxidativa” NDUFA13, con UQCR11), la “angiogénesis” (NUDFB1/2, (VEGFB), con la proliferación/diferenciación de linfocitos y replicación del ADN (IL4, EDF1, DOK2). El factor de transcripción NF-κB está involucrado en la regulación transcripcional de los genes de proliferación y angiogénesis inducidos por la acción de las HTs a través del mTR. La regulación positiva mediada por HTs de IL-4, VEGFA y VEGFB, es disminuida de manera significativa por el inhibidor de NF-κB, demostrando la implicancia de este factor de transcripción sobre la inducción transcripcional mediada por HTs. Se ha descripto que la angiogénesis y la expresión de VEGF está asociada con la sobrevida y el pronóstico de pacientes con LCT; es por esto que la regulación mediada por HTs de genes vinculados a estos procesos podría contribuir al fenotipo maligno de estos linfomas. Por consiguiente, se analizó con mayor profundidad si la inducción transcripcional mediada por HTs se observa en los distintos subtipos de LCT. Se encontró que las HTs no sólo son capaces de inducir la expresión de VEGF en subtipos de LCT de origen inmaduro y maduro, sino que además, inducen la producción del ligando VEGF funcionalmente activo. La acción del VEGF inducido por HTs parecería influir a los LCT a través de mecanismos autócrinos y parácrinos. También se desarrollaron modelos in vivo donde se evaluó el crecimiento tumoral de células inmaduras, CUTLL1, en ratones inmunodeficientes NODSCID, y se encontró que la inhibición de la integrina αVβ3, utilizando ARN de silenciamiento, disminuye el crecimiento de los mismos. Más aún, se analizó el efecto de un inhibidor farmacológico de la integrina αVβ3, el cilengitide, sobre el crecimiento tumoral de la línea PTCL-NOS, OCI-Ly12, y sobre un modelo pre-clínico de linfoma T obtenido por el trasplante de tumores derivados de pacientes ALCL, ALK+ y ALK- en ratones humanizados. Se encontró que el tratamiento con cilengitide induce una disminución significativa del volumen 7 Resumen tumoral. Estos resultados nos proporcionan un marco de conocimiento para el desarrollo de nuevos regímenes terapéuticos, donde la inhibición selectiva de la integrina αVβ3, constituye una posible estrategia para el tratamiento de pacientes con linfomas de células T. 8 INDICE 1. INTRODUCCIÓN................................................................................ 12 1.1 HORMONAS TIROIDEAS ...................................................................... 13 12 1.1.1 Glándula tiroides........................................................................................................ 13 1.1.2 El eje Hipotalamo-Hipofiso (Pituitario)-Tiroideo (HPT).............................................. 15 1.1.3 Síntesis y metabolismo de las hormonas tiroideas ................................................... 17 1.1.4 Efectos biológicos de las hormonas tiroideas ........................................................... 21 1.1.5 Mecanismos de acción de las hormonas tiroideas.................................................... 23 1.1.5.1 Acciones clásicas de las HTs a través de los TRs ............................................ 24 1.1.5.2 Acciones no clásicas de las HTs........................................................................ 26 1.1.5.3 La integrina αVβ 3 como receptor de membrana para HTs (mTR) ................... 27 1.2 PROLIFERACIÓN Y CICLO CELULAR.................................................. 29 1.2.1 Ciclinas ...................................................................................................................... 30 1.2.2 Inhibidores de Cdks ................................................................................................... 32 1.2.3 Genes supresores tumorales .................................................................................... 33 1.3 PROCESOS NEOPLÁSICOS................................................................. 35 1.3.1 Características generales de los procesos neoplásicos ........................................... 35 1.3.2 Participación del eje tiroideo en procesos neoplásicos............................................. 38 1.3.3 Angiogénesis ............................................................................................................. 41 1.3.4 Participación del eje tiroideo en la regulación de la angiogénesis............................ 43 1.4 LINFOMAS.............................................................................................. 45 1.4.1 Características generales de los linfomas................................................................. 45 1.4.2 Linfomas T ................................................................................................................. 46 1.4.2.1 Linfomas Periféricos no especificados de otro modo (PTCL-NOS)................... 47 1.4.2.2 Linfomas Angioinmunoblasticos (AITL).............................................................. 48 1.4.2.3 Linfomas Anaplásicos de Células Grandes (ALCL)........................................... 49 1.4.2.4 Linfomas Cutáneos de células T (CTCL) ........................................................... 49 1.4.3 Rol de la angiogénesis en los linfomas ..................................................................... 50 1.4.4 Tratamientos de Linfomas T...................................................................................... 53 1.4.4.1 Anticuerpos monoclonales............................................................................ 54 1.4.4.2 Inhibidores de las Histonas Deacetilasas (HDAC).......................................... 56 1.4.4.3 Inhibidores de proteosomas .............................................................................. 57 1.4.4.4 Drogas citotóxicas convencionales y anti-metabolitos....................................... 57 1.4.4.5 Drogas inmunomoduladoras y no-citotóxicas .................................................... 58 1.4.4.6 Inhibidores de señales de transducción............................................................. 59 2. HIPÓTESIS Y OBJETIVOS................................................................. 60 2.1. HIPÓTESIS DE TRABAJO .................................................................... 60 61 2.2. OBJETIVOS........................................................................................... 61 2.2.1. Objetivo general ....................................................................................................... 61 2.2.2 Objetivos específicos................................................................................................. 61 3. MATERIALES Y MÉTODOS............................................................... 64 9 3.1 PREPARACIÓN DE LAS HORMONAS T3 Y T4 UNIDAS A AGAROSA 64 65 3.2 ENSAYOS IN-VITRO CON LINEAS CELULARES HUMANAS DE LCT .. 65 3.2.1 Líneas celulares de linfoma T.................................................................................... 65 3.2.2 Tratamientos in vitro de las líneas de LCT ................................................................ 66 3.2.3 Ensayos de proliferación celular................................................................................ 66 3.2.4 Crecimiento in vitro en cultivos en 3 dimensiones .................................................... 68 3.2.5 Transfecciones transientes con ARN de interferencia (siRNA) ................................ 69 3.2.6 Análisis por RT-PCR en tiempo real.......................................................................... 69 3.2.7 Análisis de expresión de proteínas por western blot................................................. 71 3.2.8 Análisis por citometría de flujo................................................................................... 72 3.2.9 Ensayos de migración ............................................................................................... 72 3.2.10 Ensayos de microscopía confocal........................................................................... 73 3.3 SECUENCIACIÓN DE ARNM Y MICROARREGLOS DE EXPRESIÓN . 74 3.4 ANÁLISIS DE LOS MODELOS IN-VIVO DE LCT .................................... 75 3.4.1 Ensayos in vivo de células CUTLL1 y OCI-Ly12....................................................... 75 3.4.2 Ensayos in vivo con tumores derivados de pacientes ALCL .................................... 76 3.4.3 Ensayos de Inmunohistoquímica............................................................................... 76 3.4.4 Evaluación de la apoptosis por la técnica de TUNEL ............................................... 77 3.4.5 Ensayo de unión de NF-κB activado a ADN ............................................................. 77 FIGURA 3.4: ENSAYO DE LA UNIÓN DE NF-κB ACTIVADO A ADN .......................... 78 3.5 ANÁLISIS ESTADÍSTICO ....................................................................... 78 4. RESULTADOS .................................................................................... 79 4.1 LAS HORMONAS TIROIDEAS INDUCEN LA PROLIFERACIÓN DE LOS LINFOMAS DE CÉLULAS T A TRAVÉS DE SUS RECEPTORES NUCLEAR 80 Y DE MEMBRANA........................................................................................ 79 4.1.1 Evaluación de la expresión del receptor nuclear y del de membrana de las hormonas tiroideas .............................................................................................................................. 80 4.1.2 Evaluación del efecto de las hormonas tiroideas sobre la proliferación y el ciclo celular de LCT. ................................................................................................................... 82 4.2 IDENTIFICACIÓN DEL RECEPTOR DE MEMBRANA PARA HTS EN LINFOMAS DE CÉLULAS T HUMANOS...................................................... 91 4.2.1 La activación de la integrina αVβ3 es la responsable de los efectos proliferativos mediados por HTs en linfomas de células T ...................................................................... 91 4.2.2 Evaluación del crecimiento regulado por HTs de células LCT en presencia del ligando biológico de la integrina αVβ3 ............................................................................... 97 4.3 PROGRAMAS TRANSCRIPCIONALES REGULADOS POR LAS HTS A TRAVÉS DE SU RECEPTOR DE MEMBRANA ......................................... 101 4.3.1. Las hormonas tiroideas regulan programas transcripcionales vinculados a proliferación y angiogénesis a través del mTR ................................................................ 101 4.3.2 Las hormonas tiroideas regulan genes involucrados en proliferación y angiogénesis a través de su receptor de membrana.............................................................................. 104 10 4.3.3 NF-κB está involucrado en la regulación transcripcional inducida por HTs sobre los genes de proliferación y angiogénesis ............................................................................. 110 4.4 LAS HTS INDUCEN LA EXPRESIÓN DE FACTORES ANGIOGÉNICOS FUNCIONALES .......................................................................................... 113 4.4.1 La activación de la integrina αvβ3 por HTs induce la expresión de VEGF en los distintos subtipos de linfomas de células T. .................................................................... 113 4.4.2 La expresión de VEGF mediada por HTs a través de la integrina αvβ3 induce la migración de células endoteliales..................................................................................... 116 4.4.3 La expresión de VEGF mediada por HTs cumple un rol autócrino sobre los LCT . 117 4.5 EVALUACIÓN DE LA INHIBICIÓN TRANSCRIPCIONAL O DEL BLOQUEO FARMACOLÓGICO DE LA INTEGRINA αVβ3 SOBRE EL CRECIMIENTO IN VIVO DE LOS LCT....................................................... 120 4.5.1 La regulación negativa de la integrina αvβ3 disminuye el crecimiento tumoral in vivo de las células CUTLL1. .................................................................................................... 120 4.5.2 La inhibición farmacológica de la integrina αvβ3 disminuye el crecimiento in vivo de las células OCI-Ly12......................................................................................................... 123 4.5.3 La inhibición farmacológica de la integrina αvβ3 disminuye el crecimiento in vivo de tumores ALCL derivados de pacientes............................................................................. 126 5. DISCUSIÓN....................................................................................... 129 5.1 Efectos de las hormonas tiroideas sobre la proliferación y ciclo celular de linfomas de células T humanos............................................................................................................ 131 5.2 Identificación del receptor de membrana para HTs en linfomas humanos de células T. .......................................................................................................................................... 135 5.3 Programas transcripcionales regulados por las hormonas tiroideas a través de su receptor de membrana ..................................................................................................... 138 5.3 Inducción de la expresión mediada por HTs de factores angiogénicos funcionales. 141 5.4 Modelos in vivo de LCT y efecto de la inhibición de la integrina αVβ3 sobre su crecimiento ....................................................................................................................... 145 6. CONCLUSIONES.............................................................................. 148 7. ABREVIATURAS .............................................................................. 154 8. BIBLIOGRAFÍA................................................................................. 157 11 1. INTRODUCCIÓN Introducción 1.1 HORMONAS TIROIDEAS 1.1.1 Glándula tiroides La tiroides es una glándula neuroéndocrina, encargada de la síntesis y secreción de las hormonas tiroideas (HTs) tiroxina y 3,3′-5 triyodotironina (T4 y T3 respectivamente; Figura 1.1). Figura 1.1: Estructura de las hormonas tiroideas La glándula tiroides se sitúa en la región infrahioidea de la parte frontal del cuello junto al cartílago tiroides y yace sobre la tráquea, a la que rodea hasta alcanzar posteriormente al esófago. Está compuesta por dos lóbulos, situados del lado izquierdo y derecho respecto de la tráquea, los cuales se encuentran conectados por el istmo. También existe un pequeño lóbulo, llamado piramidal, en contacto con el istmo (Figura 1.2) La tiroides está recubierta por una vaina aponeurótica que ayuda a mantener la glándula en su posición. La porción más externa de la cápsula de la tiroides se continúa con la aponeurosis cervical y hacia atrás con la vaina carotídea. Los músculos infrahioideos la recubren en su cara anterior; el músculo esternocleidomastoideo lo hace lateralmente; y por su cara posterior, está fijada a los cartílagos tiroides y traqueal y al músculo cricofaríngeo por medio de un engrosamiento de la aponeurosis (Dumont y col, 2008). 13 Introducción Esta glándula, tiene un flujo sanguíneo muy alto en relación a su tamaño. Está irrigada por las arterias tiroideas superiores, ramificaciones de la carótida externa, e inferiores que derivan de la subclavia. Hay tres venas que la drenan, que son las venas tiroideas superior, media e inferior y que desembocan en las yugulares internas. Los vasos linfáticos forman alrededor de la glándula un plexo paratiroideo. Los troncos que parten de él se dividen en linfáticos descendentes, que terminan en ganglios situados delante de la tráquea y encima del timo y en linfáticos ascendentes. La inervación es simpática, proveniente del simpático cervical, y parasimpática, proveniente de los nervios laríngeos que a su vez proceden del nervio vago. La inervación regula el sistema vasomotor y, a través de éste la irrigación de la glándula (Dumont y col, 2008). Figura 1.2: Localización y estructura de la glándula tiroidea Representación frontal de la glándula tiroides y sus límites. La tiroides, ubicada junto al cartílago tiroides, consta de un lóbulo derecho y otro izquierdo con respecto a la tráquea, unidos por el istmo, el cual presenta una prolongación a la que se denomina lóbulo piramidal. El istmo se encuentra justo debajo del nivel del cartílago cricoides. Se muestra además el ligamento cricotiroideo que une los cartílagos tiroides y cricoides. Adaptado de: www.highlands.edu 14 Introducción La tiroides está compuesta por folículos o acinos que pueden ser considerados, tanto desde el punto de vista estructural como funcional, como las unidades primarias o secretoras del órgano. Las células de los folículos son las productoras de las hormonas y el lumen es el depósito. Las paredes del folículo están formadas por un epitelio continuo de una célula de profundidad, el parénquima tiroideo. Dentro del folículo y contenido en el lumen se encuentra el coloide, constituido por una mezcla de proteínas, principalmente la tiroglobulina (Tg), y otras iodoproteinas de menor peso molecular y proteínas séricas, entre ellas la albúmina (Livolsi, 2001). Adicionalmente a las células foliculares, existen células individuales o pequeños grupos de células que se ubican entre los acinos. Estas células parafoliculares, o células C, secretan calcitonina (o tirocalcitonina) en respuesta al aumento del calcio sérico. Fuera de los folículos hay células endoteliales y fibroblastos (Livolsi, 2001; Koulouri y col, 2013). 1.1.2 El eje Hipotalamo-Hipofiso (Pituitario)-Tiroideo (HPT). La señalización neuroendócrina permite la integración de las funciones biológicas de distintos tejidos en organismos complejos, conduciendo una respuesta coordinada e incrementando la adaptación de ese organismo a una condición dada. El eje hipotálamo-pituitario-tiroideo (HPT) es un buen ejemplo de como el sistema neuroendócrino regula diferentes funciones de un organismo, tanto durante su desarrollo como en la vida adulta (Laudet, 2011; Patel y col, 2011). El eje hipotálamo-hipófisis-tiroideo se representa gráficamente en la Figura 1.3. La hormona hipotalámica involucrada en el eje HPT es la liberadora de tirotropina (TRH), y es secretada por un grupo específico de células en los núcleos arcuato y paraventricular del hipotálamo. La TRH es un tripéptido sintetizado a través del clivaje de una prohormona (pre-pro-TRH), cuyo procesamiento se inicia durante el transporte axonal luego de la remoción de un péptido señal. La TRH se secreta al sistema venoso portal hipofisario y al unirse específicamente a los receptores de la membrana hipofisaria, activa y estimula 15 Introducción la secreción de la hormona hipofisaria estimulante del tiroides (TSH), por exocitosis de los gránulos que la contienen. La unión del ligando a estos receptores acoplados a proteína G lleva también a la fosforilación y a cambios en la concentración de proteínas nucleares, las que interaccionan con los genes involucrados en la síntesis de TSH, incrementando de esta manera su transcripción. Figura 1.3: Esquema del eje hipotálamo-hipófisis-tiroideo Hipotálamo Hipófisis Glándula Tiroidea T3 y T4 ** Representación esquemática del eje HPT. La TRH viaja a través del sistema vascular porta hipofisario estimulando la síntesis y liberación de TSH. Esta molécula actúa en la glándula tiroides promoviendo la biosíntesis y liberación de T3 y T4. Estas son liberadas en la circulación general para cumplir su rol fisiológico sobre los distintos tejidos. Adicionalmente inducen la retroalimentación negativa en la hipófisis. Adaptado de Appleton y Lange, 1994. La TSH, hormona glicoproteica de 30 kDa, es un heterodímero formado por dos 16 Introducción subunidades, α y β, unidas de forma no covalente. La subunidad α es idéntica a la de las gonadotropinas LH y FSH, mientras que la subunidad β le confiere especificidad, siendo la que se une al receptor en la célula tiroidea. El receptor de TSH está acoplado a proteína G, tiene 7 dominios transmembrana y se expresa principalmente en la glándula tiroidea, aunque también fue identificado en varios tejidos incluyendo cerebro, testículo, riñón, corazón, hueso, timo, linfocitos, tejido adiposo y fibroblastos (Davies y col, 2002; de Lloyd y col. 2010). La TSH liberada de la hipófisis anterior, actúa a nivel de sus receptores en la glándula tiroidea. De esta forma, activa vías de señalización que involucran el sistema adenilatociclasa, la cual regula la síntesis y liberación a la circulación periférica de las HTs, T4 y T3. El 80 % de la T3 proviene de la conversión extra-tiroidea por monodeyodinación de la T4 en los tejidos periféricos; el resto se segrega por la glándula tiroides (Koulouri y col. 2013). Las hormonas tiroideas T3 y T4 están unidas reversiblemente en sangre a proteínas transportadoras, principalmente a globulinas fijadoras de tiroxina (TBG) y en menor proporción a prealbúmina fijadora de tiroxina (TBPA) y a albúmina. La T4 libre (FT4) y la T3 libre (FT3) son las formas metabólicas activas y son los mejores indicadores del estado de las hormonas tiroideas. La FT4 se aproxima al 0,03 % de la T4 total y la FT3 al 0,3 % de la T3 total. Las HTs que circulan en forma libre actúan a nivel de sus órganos diana en la modulación del metabolismo celular o participan en el mecanismo de retroalimentación negativa a nivel del hipotálamo y la hipófisis inhibiendo la síntesis y liberación de TRH y TSH, respectivamente. Este control tan preciso permite mantener constantes las concentraciones de T3 y T4 y así evitar tanto el exceso como la carencia de estas hormonas, ambas circunstancias sumamente adversas para el organismo, en especial en etapas tempranas de su desarrollo. 1.1.3 Síntesis y metabolismo de las hormonas tiroideas La síntesis de hormonas tiroideas, tiene lugar tanto en la glándula tiroides como en el tejido periférico; su síntesis dentro de la glándula tiroides requiere de 4 17 Introducción elementos esenciales: Iodo, Peróxido de Hidrogeno (H2O2), Tiroglobulina (Tg), Peroxidasa tiroidea (TPO de sus siglas en inglés). Las HTs controlan a través de una retroalimentación negativa, la liberación tanto de la hormona liberadora de tirotropina del hipotálamo (TRH); como de la TSH en la glándula pituitaria anterior (Lin X y col, 2004; Dietrich y col, 2012). El folículo tiroideo es la estructura funcional responsable de la biosíntesis, almacenamiento y secreción de las HTs. Las células tiroideas foliculares se encuentran polarizadas y especializadas en la producción de T4 y T3. La función celular depende, entonces, de la expresión de un set de proteínas bien caracterizadas que están involucradas en la biosíntesis de la hormona (Kopp P, 2013). Las HTs son sintetizadas dentro de una estructura de alto peso molecular, una proteína tiroidea específica, llamada tiroglobulina (Tg), y permanecen unidas covalentemente a la estructura primaria de esta molécula hasta que ocurre la degradación proteica y la hormona es secretada. Los pasos por los cuales estas células sintetizan y secretan las HTs se encuentran esquematizados en la Figura 1.4: El primer paso (A) es el transporte del ioduro por captación activa y mediante la proteína transportadora de iodo NIS (del inglés, Na/I symporter), la cual transporta dentro de la célula dos cationes de Sodio (Na+) por cada anión ioduro. El ioduro es transportado desde la membrana basal a la membrana apical, donde sale al coloide mediante una proteína transportadora del anión denominada pendrina, localizada en la membrana apical. En el siguiente paso (B), se produce la oxidación del ioduro a iodonio, proceso en el que participa la tiroperoxidasa (TPO), el que se incorpora a la Tg para producir iodotirosinas. La TPO nuevamente participa en el acoplamiento (C) de las iodotirosinas para formar las iodotironinas T3 y T4. Para poder disponer de las mismas para su secreción, la célula folicular capta gotitas del coloide por endocitosis (D). 18 Introducción Figura 1.4: Síntesis de las hormonas tiroideas LUMEN Membrana Apical B Tiroglobulina C D E F A Transporte ioduro Membrana Basal Se señalan los pasos que llevan a la síntesis de HTs, su almacenamiento en la Tg y finalmente su secreción. Adaptado de: fitsweb.uchc.edu Luego se produce la ruptura proteolítica de los enlaces que unen a las HTs a la Tg (E) y desiodación de la T4, de modo que tanto T4 como T3 pueden acceder al torrente sanguíneo. La liberación de las HTs (F) se da mayoritariamente en forma de T4, siendo su concentración en suero 40 veces mayor que la de T3; aunque, solo una pequeña proporción de las HTs circulan en forma libre con capacidad de entrar en la célula y ejercer los efectos biológicos. (Lester Reed y col, 2001; Dentice y Salvatore, 2011; Peying Fong, 2011). La mayor proporción de las HTs se encuentra unida a proteínas transportadoras. La T4 se une a la globulina transportadora de HTs (TBG) en un 70%, a la albumina en un 20% y a la transtiretina (TTR) en un 10%. La T3 se une principalmente a TBG (80%), y el resto a TTR y a albúmina. La 19 Introducción influencia de esta última, que tiene una afinidad más o menos baja por las HTs, se pone de manifiesto porque su concentración plasmática es elevada (Lester Reed, 2001). Por otra parte, la T3 puede producirse a través de la 5’-deiodinacion del anillo externo de la T4 por deiodinasas. La deiodinasa de tipo 1 (DIO1) se encuentra en tejidos periféricos como el hígado y el riñón y es responsable de la conversión de la mayor parte de T4 a T3 en circulación. La deiodinasa de tipo 2 (DIO2) tiene una distribución más amplia encontrándose en tiroides, cerebro, pituitaria, tejido adiposo, corazón y músculo esquelético, entre otros. Esta deiodinasa tiene como función la conversión de T4 a T3 para uso intracelular. Este paso metabólico constituye el camino principal para la provisión de T3 en diferentes tejidos (Gereben y col, 2008; Bianco, 2011). Por su parte, la deiodinasa tipo 3 (DIO3), puede inactivar a la T4 formando T3 reversa o a la T3, dando como resultado T2 y terminando así su acción hormonal. La localización celular de las deiodinasas individuales es una característica importante, ya que las HTs están en circulación, pero su mecanismo de acción tiene lugar en el interior celular mediante su interacción con receptores nucleares. Estudios realizados con modelos celulares donde esas enzimas son expresadas transitoria o endógenamente indican que DIO1 y DIO3 se encuentran en la membrana plasmática, mientras que DIO2 es una proteína residente en el retículo endoplásmico (Gereben y col, 2008). Por otra parte, las membranas celulares son relativamente impermeables a las HTs, es por esto, que son necesarios transportadores de membrana para que las mismas accedan al microambiente intracelular (Heuer, 2009). Una vez dentro de la célula, la pro-hormona T4 puede ser transformada por medio de la acción de DIO2 a T3, molécula biológicamente activa, la que difunde hacia el núcleo eventualmente uniéndose a sus receptores (TR) y dando como resultado la transcripción de sus genes blanco (Cheng, 2010). Por lo tanto, la señalización que inducen las HTs puede ser modificada por efecto de las deiodinasas, ya sea, potenciando su acción a través de la conversión de T4 a T3 por medio de la DIO2, o inactivando su acción a través de la expresión de la DIO3. Más aún, estos eventos suceden dentro de la célula sin que ocurran cambios relativos en los niveles plasmáticos de las HTs (Gereben y col, 2008). 20 Introducción Finalmente, las HTs se metabolizan periféricamente por diversas vías, esquematizadas en la Figura 1.5, a fin de transformarlas en sustancias más hidrosolubles para permitir su eliminación. Además de los mecanismos de inactivación ya mencionados, tiene lugar en el hígado la conjugación, que se puede dar con sulfato o la glucuronidación. Figura 1.5: Conversión metabólica periférica de las hormonas tiroideas Adaptado de slideplayer.es 1.1.4 Efectos biológicos de las hormonas tiroideas Las HTs modulan distintos procesos fisiológicos y son críticas para el crecimiento, desarrollo, diferenciación y mantenimiento de la homeostasis metabólica (Oetting y Yen, 2007). Las HTs ejercen su actividad en todos los sistemas del organismo, ya que sus receptores se expresan en casi todos los tejidos. Dado que, cada órgano posee diferente expresión de los mismos y de sus isoformas, y presentan distinta actividad de las deiodinasas, existen diferencias en la respuesta a las mismas. Las principales acciones sistémicas de estas hormonas abarcan el aumento del metabolismo basal, la estimulación 21 Introducción de la termogénesis y la regulación del consumo de oxígeno celular. Los cambios en la función tiroidea aumentan la captación de glucosa, la demanda de vitaminas y modifican las concentraciones de proteínas y enzimas séricas. Las HTs tienen la habilidad de inducir vías anabólicas y catabólicas, como la lipogénesis y la lipólisis, contribuyendo de esta manera a la inducción del consumo energético (Pucci y col, 2000; Brent, 2012). Las hormonas tiroideas ejercen una serie de acciones periféricas fundamentales para el correcto funcionamiento de varios órganos y sistemas. Suprimen la secreción de TSH por parte de las células tirotropas hipofisarias regulando negativamente la transcripción de sus genes por un mecanismo mediado por la deiodinación intracelular de T4 en T3. A nivel del sistema nervioso, tienen un papel fundamental en el desarrollo del cerebro fetal y durante el periodo neonatal. Además regulan proteínas y glúcidos que participan en la mielinización (Williams, 2008). En el hígado las HTs cumplen un importante rol en la hosmeostasis lipídica hepática (Malik y Hodgson, 2002) estimulando las enzimas que regulan la lipogénesis y la lipólisis, induciendo la síntesis de transaminasas y de proteínas plasmáticas y regulando la síntesis del colesterol. Son capaces de modular el número de βARs en el corazón aumentando la sensibilidad a catecolaminas. En el tejido adiposo, la T3 regula el consumo basal de oxígeno, el almacenamiento graso, la lipogénesis y la lipólisis. También induce la diferenciación del tejido adiposo y estimula la proliferación de los adipocitos (Hulbert, 2000; Brent, 2012). Los efectos hormonales se extienden al músculo esquelético, donde favorecen la acción contráctil, la biosíntesis de miosina y de enzimas lisosómicas, así como también la captación de glucosa. Un correcto funcionamiento tiroideo es fundamental para el desarrollo óseo (Gogakos y col, 2010). Por otro lado, una secreción inadecuada de estas hormonas se ha relacionado con irregularidades a nivel gonadal. 22 Introducción Si todo el eje endócrino funciona correctamente, el individuo se encuentra en estado eutiroideo. En el caso de existir patología tiroidea primaria, se pueden dar diferentes escenarios. Se denomina estado hipertiroideo a aquel donde los niveles hormonales están por encima de los normales. La consecuencia de este estado es la exacerbación, de acuerdo a la gravedad de la patología, de todas las funciones sistémicas y periféricas señaladas en este apartado (Potenza y col, 2009; Hörmann y Schumm-Draeger, 2010). Por el contrario, cuando la secreción hormonal es insuficiente y los niveles de las HTs se encuentran por debajo de los valores normales, se denomina estado de hipotiroidismo, estando los síntomas clínicos relacionados a una disminución del metabolismo. También se han definido estados leves y subclínicos de estos desórdenes, caracterizados por un aumento o reducción de la TSH circulante según se trate de hipo o hipertiroidismo subclínicos. Como sus nombres lo indican, en estas circunstancias los síntomas de la enfermedad son leves o inexistentes, pero pueden progresar a enfermedad manifiesta si no se realiza la intervención terapéutica (Karmisholt y col, 2011). También pueden darse casos de aumento o reducción de los niveles hormonales pero secundarios a una patología hipotalámica o hipofisaria. 1.1.5 Mecanismos de acción de las hormonas tiroideas La acción de las HTs se define como la modificación de la expresión de genes mediada por los receptores nucleares para HTs (TRs). Estos receptores están codificados por dos genes diferentes, el THRA localizado en el cromosoma 17 y el THRB en el cromosoma 3, que codifican para las proteínas TRα y TRβ respectivamente (Yen y col, 2001). Ambos genes comparten alta homología en su estructura proteica incluyendo el dominio de unión al ADN (DBD de su sigla en inglés) y la porción C-terminal del dominio de unión al ligando (LBD de su sigla en inglés). Una secuencia corta de aminoácidos entre el DBD media la unión al ADN e impone la especificidad de la secuencia. Dentro del DBD, los TRs reconocen elementos de respuesta a HTs (TREs) en los promotores de sus genes blanco. Los mismos actúan como potenciadores de la transcripción de aquellos genes que son regulados positivamente por las HTs. El LBD une 23 Introducción 10-15 veces con mayor afinidad a la T3 que a la T4 (Yen y col, 2006; Cheng y col. 2010). Existen varias isoformas de los TRs que se dan por empalme (splicing) alternativo. El gen THRA genera dos isoformas TRα1, TRα2, que difieren en la región carboxilo terminal donde está ubicado el dominio de unión a la hormona; y el gen THRB genera otras dos isoformas TRβ1 y TRβ2 a través de la elección de un promotor alternativo (Cheng y col, 2010). Los receptores para HTs (TRs) son factores de transcripción específicos. Los TRs se encuentran basalmente asociados al ADN, es decir aún en ausencia de hormona tiroidea, pero su capacidad de transcripción génica estaría impedida por unión a correpresores (ver más adelante). La expresión de los genes para estos receptores es variable, siendo el TRβ2 el que se encuentra involucrado principalmente en la regulación de la producción de la TSH. En el hipotálamo, esta isoforma es quizás la única responsable de la regulación negativa del gen de TRH por parte de la T3 (Oetting y Yen, 2007). 1.1.5.1 Acciones clásicas de las HTs a través de los TRs En el modelo clásico de inducción de la transcripción de genes regulada por HTs, los TRs actúan como factores de transcripción ligando-dependientes. Una vez que la T3 atraviesa la membrana basal se une a su receptor en el núcleo; generando un cambio conformacional, que permite que el TR dimerice formando homodímeros (TR/TR) o heterodimeros con el receptor de rexinoides (TR/RXR) que son los que tienen mayor actividad biológica (Lee y Privalsky, 2005; Moeller y Broecker-Preuss, 2011; Tata, 2013). Estos complejos hormonareceptor, se unen a los TREs en el promotor de los genes blanco. En ausencia de T3, el TR se une constitutivamente a un grupo de proteínas con actividad de histona deacetilasa (HDAC), denominadas correpresores nucleares, las cuales se caracterizan por reclutar otras proteínas accesorias que a su vez compactan a los nucleosomas y reprimen el proceso de transcripción (Figura 1.6-A). En presencia de T3, el receptor cambia de conformación y los correpresores son liberados y reemplazados por coactivadores, lo que lleva a que la cromatina se desenrolle, facilitándose por lo tanto la transcripción (Figura 1.6-B), (Aranda y 24 Introducción Pascual, 2001; Oetting y Yen, 2007; Moeller y Fuhrer, 2013). En el caso de los genes regulados negativamente el proceso es el inverso; estos pueden ser estimulados en ausencia de hormonas e inhibidos cuando la T3 se une a su receptor. De esta manera el TR estaría actuando como un factor de transcripción dependiente de ligando (Yen, 2001; You y col, 2010). Se han realizado estudios con ratones transgénicos que no expresan los genes del TR (knockout). Los mismos muestran una gran superposición de los patrones genéticos de expresión positiva de los genes THRA y THRB, sugiriendo que más que la isoforma, lo más importante es la cantidad de TR expresada (Yen y col, 2003). Sin embargo, las distintas isoformas del TR muestran diferentes afinidades de unión al ADN en un cierto promotor y, por lo tanto, la respuesta transcripcional a un cierto gen en un tejido particular, podría depender de la isoforma del TR predominante (Chan y Privalsky, 2009; Chiamolera y col, 2012). Figura 1.6: Mecanismo de acción clásico de las hormonas tiroideas T3 T4 Citoplasma T3 T4 D3 D1/D2 T3 rT3 Núcleo TRE TR Coactivador RXR TR RXR Corepresor T3 Transcripción genes blanco TRE A B Esquema que muestra la unión del TR al ADN. (A) La unión constitutiva del TR al correpresor provoca el reclutamiento de proteínas histidina acetilasas (HDAC), lo que causa la condensación de la cromatina y la represión de la transcripción. (B) Al unirse la T3 al TR provoca el intercambio del correpresor por un coactivador, se descondensa la cromatina y se facilita la transcripción. Adaptado Juvenal, 2009. 25 Introducción El estado de fosforilación del TR podría modular positivamente la unión del complejo de la HT a su receptor y a las secuencias TRE del ADN. Evidencias experimentales indican que las HTs podrían inducir la fosforilación de su receptor en sitios específicos. En tal sentido, Davis y col (2000) han demostrado que la T4 promueve la asociación entre la proteína quinasa activada por mitógeno (MAPK) y el TR, causando su fosforilación en residuos específicos de serina. 1.1.5.2 Acciones no clásicas de las HTs Existen evidencias que sugieren que las acciones no clásicas de las HTs, podrían ser mediadas por isoformas de los receptores nucleares asociados a la membrana plasmática, citoplasmáticos o bien por otros tipos de receptores de membrana de alta afinidad para HTs (Bassett y col, 2003). Davis y col. (2006), han postulado la existencia de efectos genómicos y no genómicos de las HTs que actuarían en forma cooperativa para la activación de la cascada de las MAPK. Entre los mecanismos no clásicos de las HTs, podemos mencionar la inducción de cambios en los patrones de fosforilación de proteínas efectoras de distintas vías de señalización (Shih y col, 2004, Cordeiro y col, 2013), la modulación de la disponibilidad de segundos mensajeros (Yamauchi y col, 2008) y la modificación de la estabilidad de ARN mensajeros (SerranoNascimento y col, 2010). Algunos años atrás y luego de varios estudios, resultó evidente que las HTs y un TRβ citoplasmático podían actuar a través de un mecanismo diferente al clásico: el TRβ unido al ligando puede activar, a tiempos muy cortos, la fosfoinositol 3-quinasa (PI3K), a través de la fosforilación de la vía de señalización Akt/PKB, la cual subsecuentemente termina modulando la expresión génica (Simoncini y col, 2000; Lei y col, 2004; Cao y col, 2005). Este mecanismo no clásico, es independiente de la unión del TRβ al ADN y TREs, pero también resulta en la inducción de la expresión génica. Resultados similares fueron luego encontrados en la acción del TRα. Por ejemplo, se demostró la activación de PI3K en células neuronales que sobreexpresan el TRα (Cao y col, 2009); y también se encontró una asociación entre el TRα y 26 Introducción p85a, seguido de la fosforilación de la vía Akt/PKB y la activación de la óxido nítrico sintasa endotelial (Hiroi y col, 2006). 1.1.5.3 La integrina αVβ3 como receptor de membrana para HTs (mTR) En 2005, Bergh y col. demostraron que las HTs pueden unirse a una proteína de membrana, la cual fue descripta en células fibroblásticas como la integrina αvβ3. Dicha integrina es el receptor de vitronectina y miembro de la superfamilia de glicoproteínas de moléculas de adhesión que median la unión de las células a la matriz extracelular por reconocer la secuencia conservada RGD (Arginina- Glicina- Aspartato) de varias proteínas plasmáticas y de matriz. Las acciones de la T3 y de la T4 sobre la integrina αvβ3 como receptor de membrana, fueron demostradas tanto en células normales, como células de vasos sanguíneos (Davis y col, 2011; Bergh y col, 2005), en células no cancerosas inmortalizadas, como células embriónicas de riñon; y en varios tipos de líneas celulares tumorales, como cáncer de próstata y carcinomas renales, entre otros (Kimbro y Simons, 2006). Utilizando modelados matemáticos de la cinética de unión de las hormonas a la integrina, se sugirió que el dominio de unión de HTs sobre la misma se encuentra cerca del sitio RGD (Arginina- Glicina- Aspartato) e incluye dos sitios de unión, denominados S1 y S2 (Figura 1.7) que transducen las señales inducidas por HTs diferencialmente (Davis y col 2011; Freindorf y col, 2012). El sitio S1 une exclusivamente a la T3 y activa la vía de señalización mediada por la fosfatidilinositol 3 kinasa (PI3K) activando el TRα y la posterior activación de genes blanco; este dominio puede ser inhibido por el análogo de la T4, el ácido tetraiodotiroacético (tetrac), y péptidos bloqueantes RGD. El sitio S2 une a T4, y en menor medida a T3, activando una vía de señalización que involucra a ERK1/2 y TRβ dando como resultado proliferación celular de células tumorales (Lin y col, 2009). Las HTs pueden, vía el sitio S2 de la integrina αvβ3, activar ERK1/2 e inducir la expresión del factor 2 de crecimiento fibroblástico, promoviendo así la angiogénesis (Davis, 2009). Estas formas de acción de las HTs sobre su receptor de membrana, tienen la potencialidad de promover el crecimiento tumoral. Por ejemplo, se ha descripto 27 Introducción que las HTs a través de una isoforma modificada del TRα estimulan programas proliferativos y de sobrevida, incrementando las concentraciones intracelulares de calcio, de óxido nítrico, y dando como resultado la activación de la proteína quinasa G II, Src, ERK y la señalización vía Akt (Kalyanaraman y col, 2014). Figura 1.7: Sitios de unión de las HTs a la integrina αvβ3 TRα HIF-1α TRβ1 Proliferación celular Representación esquemática propuesta, basada en modelos matemáticos, de la organización del dominio de unión de la T4 y T3 a la integrina αvβ3, y las consecuencias funcionales río abajo de la misma en las células humanas de glioma U87MG. El dominio de unión incluye un sitio exclusivo para T3 (sitio S1) que activa la fosfoinositol 3-quinasa (PI3K) y conlleva a la translocación del TRα al núcleo y la transcripción del factor de hipoxia 1α (HIF-1α). El sitio 2 une tanto T4 como T3, y traduce las señales inducidas por HTs mediadas por la activación de ERK1/2 que conducen a la proliferación celular a través de la acción de TRβ. Adaptado de: HungYun Lin y col, 2009. La vía oncogénica PI3K, también activada por HTs a través de su acción sobre su receptor de membrana (mTR), facilita el crecimiento celular y sobrevida e inhibe la apoptosis (Chalhoub y Baker, 2009). Vía la activación de PI3K, la T3 (a través del sitio S1 de la integrina αvβ3), induce la expresión de una subunidad del factor de transcripción inducible por hipoxia 1 (HIF1; Moeller y col, 2005). Los genes blanco de este factor de transcripción juegan roles claves 28 Introducción en la biología del cáncer, entre ellos la angiogénesis para la adaptación a la hipoxia, la cual se encuentra activada en tumores de crecimiento rápido, invasión y metástasis (Semenza, 2009). La expresión de HIF1 también es un marcador pronóstico en los carcinomas renales, cáncer de mama y de próstata, entre otros (Kimbro y Simons, 2006). 1.2 PROLIFERACIÓN Y CICLO CELULAR La célula puede encontrarse en dos estados claramente diferenciados: el estado de división o mitosis y el estado de no división o interfase. Cuando la célula se divide entra en el ciclo celular, proceso ordenado y repetitivo en el tiempo en el que la célula proliferante crece y se divide en dos células hijas. El ciclo celular consta de cuatro fases bien diferenciadas fase G1, fase S, fase G2 y fase M. (Figura 1.8) Figura 1.8: Fases del ciclo celular Esquema representativo de las fases del ciclo celular. Frente a estímulos proliferativos, las células atraviesan la interfase (I), que está compuesta por las fases Gap1 (G1), síntesis (S) y Gap2 (G2) y luego ingresan en la fase de mitosis (M), lo que lleva a la formación de dos células hijas. Adaptado de Galderisi y col, 2003. 29 Introducción La interfase es la fase más larga del ciclo, ocupando casi el 95% del mismo, y comprende tres etapas: Fase G1 (Gap 1): En esta primera fase del ciclo celular tiene lugar el crecimiento celular, dado que la célula sintetiza proteínas y ARN para su posterior división. Fase S: Fase en la cual ocurre la replicación del ADN. Fase G2 (Gap 2): Continúa el crecimiento celular y la duplicación de proteínas y ARN, observándose cambios en la estructura celular, que indican el principio de la división. Esta fase termina cuando los cromosomas empiezan a condensarse al inicio de la mitosis. La mitosis corresponde a la fase M del ciclo celular. En esta fase la célula progenitora se divide para formar dos células hijas. Para asegurar el correcto desarrollo del ciclo, es decir, que cada nueva célula hija reciba un genoma completo, tanto el inicio y progresión de la fase S como la mitosis deben ser controladas exhaustivamente. En este control del ciclo celular son muy importantes las ciclinas, las quinasas dependientes de ciclinas (Cdks) y sus inhibidores (CdkI) y los genes supresores tumorales. 1.2.1 Ciclinas Las ciclinas son proteínas reguladoras del ciclo celular, que forman complejos enzimáticos con quinasas dependientes de ciclinas (Cdks), encargadas de fosforilar residuos de serina y treonina de proteínas regulatorias específicas. Cada complejo ciclina-Cdk regula distintas partes del ciclo celular (Figura 1.9). Las ciclinas D conforman la subunidad regulatoria del complejo formado con Cdk4 o Cdk6 y son importantes durante la transición G0/G1. La ciclina D1 fosforila la proteína del retinoblastoma (Rb), bloqueando su efecto inhibitorio sobre el factor de transcripción E2F, involucrado en la expresión de un gran número de genes regulatorios. La ciclina D1 se ha visto sobreexpresada en varios tipos de cáncer, incluyendo el de paratiroides (Imanishi y col, 2001; Corbetta y col, 2007), mama (Casimiro y col, 2012), próstata, colon, linfomas y 30 Introducción melanomas (Siddon y col, 2012) entre otros. Las alteraciones de las otras ciclinas D en células tumorales son menos frecuentes; aunque existen trabajos donde muestran la sobreexpresión de la ciclina D2 en ciertos tumores gástricos y de colon (Leach y col, 1993; Takano y col, 2000) y de la ciclina D3 en algunos subtipos de carcinomas mamarios y tumores cerebrales (Bartkova y col, 1996; Buschges y col, 1999). Figura 1.9: Regulación de la expresión de ciclinas a lo largo del ciclo celular Esquema representativo de la regulación de la expresión de ciclinas a lo largo del ciclo celular. Adaptado de: Roberti y col, 2009 La ciclina E forma un complejo con Cdk2 en la fase G1 del ciclo celular, lo cual es necesario para la transición G1/S. Este complejo fosforila p27/Kip1, un inhibidor de las ciclinas D, que es degradado en el proteasoma y que además promueve la expresión de la ciclina A y, como consecuencia, la entrada y la progresión de la fase S. La ciclina A forma complejos enzimáticos con Cdk1 y Cdk2 y dependiendo con cual forma esta unión, favorece la progresión de la fase S o la transición de la fase G2 a la fase M, respectivamente. Esta ciclina se une a importantes reguladores del ciclo celular, como proteínas de la familia Rb, factores de transcripción E2F1 y a proteínas de la familia p21. Cdk1 también se une a la ciclina B, lo cual es necesario para la progresión de las células a lo largo de la 31 Introducción fase M. Las ciclinas A y B son consideradas ciclinas mitóticas y son rápidamente degradadas durante la mitosis. Se ha descripto que el estado tiroideo modula la proliferación mediante la regulación de proteínas del ciclo celular. En hepatocitos el hipertiroidismo aumenta la expresión de las ciclinas D1, E y A y disminuye la expresión de inhibidores de Cdks y proteínas proapoptóticas p53 y p27. Por el contrario, en condiciones de hipotiroidismo se encontró una disminución en los niveles de expresión de ciclinas y un incremento en la expresión de proteínas proapoptóticas (Alisi y col, 2005). En un trabajo reciente de nuestro grupo, se describió el aumento de las Ciclinas D1 y D3 en tumores de linfomas T crecidos en ratones hipertiroideos. Por el contrario los tumores crecidos en ratones hipotiroideos mostraron una disminución en la expresión de inhibidores del ciclo celular, como CDKN2A y CDKN1B, y de supresores tumorales como p53 (Sterle y col, 2013) 1.2.2 Inhibidores de Cdks El ciclo celular se interrumpe en respuesta a diversas señales antiproliferativas, como a la privación de factores de crecimiento y citoquinas, o al daño al ADN, principalmente por la inhibición de la fosforilación de la proteína de Rb. Esto lleva a la inhibición de factores de transcripción E2F, que son necesarios para la activación transcripcional de genes de síntesis del ADN. Los principales mediadores de este efecto son los inhibidores de Cdks. Los inhibidores de Cdks están divididos en dos familias: la familia CIP/KIP, que incluye a p21 y p27 y la familia INK4, que incluye las proteínas p15 y p16. Ambas familias difieren en su estructura, mecanismo de acción y especificidad. Los componentes de la familia INK4 interactúan previniendo la formación específica de complejos ciclina/Cdk e inhiben la actividad de Cdk4 y Cdk6, ambos involucrados en el control de la fase G1. Como consecuencia de esto, se inhibe la fosforilación de Rb, que lleva a la inhibición de la transcripción de 32 Introducción una variedad de proteínas necesarias para la progresión de G1 a S y para la replicación del ADN, incluyendo las ciclinas E y A. El componente más importante de la familia es p16, que se encuentra mutado en muchas neoplasias. Además de su rol como inhibidor del ciclo celular, participa en los procesos de angiogénesis, invasión tumoral, diseminación celular y apoptosis, mediante la regulación negativa de la transcripción de diversos genes implicados en estos procesos (Mirzayans y col, 2012a y 2012b). El silenciamiento transcripcional de genes supresores de tumores, asociado a la metilación del ADN, es un evento epigenético común en las neoplasias hematológicas. Cuando hay daño al ADN se genera un aumento en la concentración y actividad de la proteína p53, que promueve la transcripción de p21. Como consecuencia, p21 bloquea el ciclo celular en la transición G1/S. Este freno del ciclo celular permite a la célula reparar el ADN dañado antes de la replicación. A su vez, p21 también ejerce su efecto sobre el ciclo celular mediante la inhibición del antígeno nuclear de proliferación celular (PCNA) y la degradación de Rb (Mirzayans y col, 2012a). Asimismo, p21 inhibe también a la ciclina B1, regulando de esta manera la transición de G2 a S (Mirzayans y col, 2012a). Cabe mencionar que se ha descripto que la depleción in vitro de HTs inhibe la proliferación de células de glioblastoma mediante el arresto de las células en G1, como resultado de la inducción de p21 (Toms y col, 1998). 1.2.3 Genes supresores tumorales Los genes supresores tumorales son fundamentales en la regulación del ciclo celular. Su rol principal es inhibir el ciclo frente a condiciones celulares adversas e impedir la proliferación excesiva. Estos genes se encuentran frecuentemente mutados en diversos tipos de tumores. En linfomas los genes supresores tumorales más frecuentemente mutados son p53, Rb y PTEN (Murakami y Tanaka, 1991; Nagai y col, 2002). 33 Introducción Frente a situaciones como hipoxia, daño al ADN, acortamiento de telómeros, estrés oxidativo o activación de oncogenes, se activa p53, que a su vez activa otras proteínas efectoras que evitan el desarrollo de neoplasias. Como ya mencionamos anteriormente, p53 puede activar la transcripción de proteínas que frenan el ciclo celular, como p21 y regular la expresión de proteínas vinculadas a la apoptosis, como Bax (Chipuk y col, 2004). También regula negativamente proteínas involucradas en la angiogénesis, como VEGF (Ghahremani y col, 2013). Se demuestra claramente la importancia de esta proteína como supresora tumoral en distintos estudios clínicos, donde se observó que más del 50% de los tumores humanos en general y el 30% de los linfomas presentan mutaciones en el gen de p53 (Soussi, 2001). La proteína Rb también realiza un control no transcripcional del ciclo celular, mediante la estabilización de p27. Además del control del ciclo celular en G1, Rb tiene acciones sobre otras funciones celulares, como la diferenciación celular (Lipinski y Jacks, 1993), mediante la interacción con enzimas remodeladoras de la cromatina, con lo que regula la expresión global de genes (Harbour y Dean, 2000) y el control de la estabilidad genómica (Hernando y col, 2004). El PTEN es la fosfatasa del fosfatidilinositol (3, 4, 5)-trifosfato (PIP3). Regula negativamente los niveles intracelulares del PIP3 y la cascada de señalización Akt/PKB. Es un regulador de la homeostasis celular y también está relacionado con la regulación de la migración celular y la quimiotaxis (Sulis y Parsons, 2003). A PTEN se lo considera un gen supresor de tumores, ya que se ha reportado su mutación en un número significativo de cánceres de mama y de próstata, en carcinomas de endometrio, glioblastomas, algunos melanomas y en linfomas no-Hodgkin (Sakai y col, 1998). 34 Introducción 1.3 PROCESOS NEOPLÁSICOS 1.3.1 Características generales de los procesos neoplásicos Para estudiar en profundidad el efecto de las HTs sobre el desarrollo de neoplasias, es necesario conocer las características que distinguen a las células tumorales de las normales y que les otorgan la capacidad de proliferar descontroladamente y hacer metástasis en sitios distantes. A continuación se detallan las capacidades adquiridas por las células tumorales, descriptas por Hanahan y Weinberg en el año 2000, que se encuentran representadas en la Figura 1.10. Figura 1.10: Capacidades adquiridas en cáncer Capacidades adquiridas por la mayoría de las células tumorales durante su desarrollo a través de diversas estratégias mecanísticas. Adaptado de Hanahan y Weinberg, 2000. Autosuficiencia de señales de crecimiento: Para dividirse, las células normales requieren de señales de crecimiento como factores solubles, componentes de la matriz extracelular (ECM) o moléculas de adhesión célula- 35 Introducción célula, que se unen a sus receptores y activan la división celular. Las células tumorales adquieren la capacidad de dividirse independientemente de estos factores. Los mecanismos que utilizan para lograrlo incluyen la sobreexpresión de receptores que las hacen hiperreactivas, cambios en el patrón de expresión de integrinas hacia aquellas que generan señales que favorecen el crecimiento y alteraciones en las cascadas de señalización intracelular. Insensibilidad a señales anti-proliferativas: Las células tumorales pueden adquirir la capacidad de independizarse de factores solubles o de matriz extracelular que inhiben la división y llevan al arresto del ciclo celular. Lo hacen principalmente mediante la activación de proteínas que regulan la entrada a la fase G1 del ciclo celular o por la disminución de la expresión de los receptores de factores inhibitorios. Evasión de la apoptosis: Las células normales frente a la detección de anormalidades, tales como daño al ADN, desbalances en las señales intracelulares por activación de oncogenes e insuficiencia de factores de supervivencia o hipoxia, disparan procesos de apoptosis. Sin embargo, las células tumorales presentan mecanismos como la sobreexpresión de genes antiapoptóticos o la pérdida de reguladores proapoptóticos que impiden estos procesos. Potencial proliferativo ilimitado: Las células tumorales, tienen un potencial de crecimiento ilimitado, ya que no sufren de acortamiento de los telómeros y no entran en senescencia. Esto es posible porque sobreexpresan la enzima telomerasa, encargada de colocar las repeticiones de hexanucleótidos al final del ADN de los telómeros. Angiogénesis: Las células que conforman neoplasias incipientes en general no tienen capacidad angiogénica, pero para poder crecer en tamaño, hacen un “switch angiogénico” y comienzan a producir el factor de crecimiento del endotelio vascular (VEGF) y el factor de crecimiento de fibroblastos (FGF), disminuyen la expresión de factores anti-angiogénicos como trombospondina 1 36 Introducción o interferón (IFN) β o inducen cambios en la expresión de integrinas hacia aquellas que son pro-angiogénicas. Invasión tisular y metástasis: El asentamiento de las células tumorales en tejidos distantes es la principal causa de muerte por cáncer. Lo que permite la separación del tejido tumoral y la entrada en circulación, son los cambios en la expresión de integrinas y de proteínas de adhesión célula-célula, principalmente cadherinas, que son las que unen a las células con la matriz extracelular, y la activación de proteasas extracelulares que degradan la matriz. En el año 2011 Hanahan y Weinberg, han descripto otras dos características de las células tumorales y dos factores que facilitan el establecimiento de neoplasias. (Figura 1.11). Figura 1.11: Capacidades adicionales adquiridas en cáncer Capacidades adquiridas por la mayoría de las células tumorales y características tumorales que permiten el establecimiento de todas las capacidades descriptas. Adaptado de Hanahan y Weinberg, 2011. La desregulación del metabolismo celular, permite la supervivencia celular mediante la glicólisis anaeróbica, que afecta no sólo el crecimiento de las células tumorales, sino también la migración de las mismas para la generación 37 Introducción de metástasis. La otra, es la capacidad de evadir al sistema inmune responsable de la eliminación de la mayoría de las células tumorales incipientes, mediante la detección de antígenos extraños en las mismas. Los factores descriptos que facilitan el establecimiento de neoplasias son: (i) la aparición de mutaciones e inestabilidad genómica en las células; y (ii) el proceso inflamatorio, que podría aportar moléculas bioactivas al microambiente tumoral, tales como citoquinas y factores de crecimiento. En algunos tipos de cáncer, el proceso inflamatorio está presente antes de que ocurra la transformación maligna, mientras que, en otros casos, el cambio oncogénico induce un microambiente inflamatorio que promueve el desarrollo del tumor (Montovani y col, 2008). 1.3.2 Participación del eje tiroideo en procesos neoplásicos Como se mencionó anteriormente, las HTs son esenciales para la sobrevida y están involucradas en procesos fisiológicos como el desarrollo, el crecimiento y el metabolismo. Las enfermedades asociadas a las HTs están mayoritariamente relacionadas a la falta o exceso de las mismas (hipo- e hipertiroidismo) y a sus síntomas clínicos conocidos; pero las HTs también están asociadas al desarrollo y curso de otras patologías. El crecimiento y la progresión tumoral, están íntimamente vinculados con el entorno celular, como por ejemplo, la influencia del sistema inmunológico y del sistema endócrino. En este sentido existen trabajos donde asocian a las HTs con la transformación celular, tumorigénesis y metástasis (Hercbergs y col, 2010; Moeller y Führer, 2013) y proponen que las HTs podrían cumplir un rol importante en la inducción de angiogénesis (Pinto y col, 2011; Mondul y col, 2012). Teniendo en cuenta una perspectiva clínica, es importante preguntarse: ¿Las HTs pueden influenciar la incidencia y progreso tumoral y, en última instancia, el éxito de un tratamiento? Existen varios estudios epidemiológicos que sustentan la hipótesis que las HTs podrían influenciar el desarrollo tumoral; entre ellos podemos mencionar, trabajos retrospectivos donde asocian al 38 Introducción hipertiroidismo como una condición médica que incrementa en pacientes el riesgo de cáncer de ovario (Ness y col, 2000) y cáncer pancreático (Ko y col, 2007). También se ha descripto que los pacientes con cáncer prostático localizado, tienen mayores niveles de T3 en comparación a pacientes con hiperplasia prostática benigna; y asocian a los altos niveles de T3 con un mayor estadío clínico de la enfermedad y mayor riesgo de recurrencia (Lehrer y col, 2001 y 2002). Más aún, se han realizado estudios prospectivos en pacientes con cáncer de mama y se ha encontrado que las pacientes con hipotiroidismo eran diagnosticadas a una mayor edad, y tenían menores estadíos clínicos y tamaño tumoral (Cristofanilli y col, 2005; Tosovic y col, 2014), sugiriendo una relación entre el hipotiroidismo y la presencia de cáncer de mama menos invasivo. Por último existen también estudios más generales, donde encuentran conexión entre la función de las HTs y el factor de riesgo a distintos tipos de cáncer (Hellevik y col, 2009). Uno de los métodos experimentales que se ha utilizado para demostrar que las HTs están involucradas en el crecimiento tumoral fue el crecimiento in vivo de líneas tumorales murinas y humanas, en ratones singeneicos o inmunodeficientes, respectivamente, con distintos estados tiroideos. Una vez implantadas las células, se ha medido la tasa de crecimiento tumoral y la aparición de metástasis. Shoemaker y col (1976) realizaron uno de los primeros estudios donde se reportó una conexión entre la sobrevida y la inducción de hipotiroidismo en los ratones luego de la implantación de células de adenocarcinoma mamario. Resultados similares se encontraron utilizando otras líneas tumorales, como adenocarcinomas de próstata (Theodoussiou y col, 1999; Theodossiou y Schawrzener, 2000), hepatocarcinomas, células de cáncer de mama (Martinez-Iglesias y col, 2009) y líneas celulares de cáncer de pulmón (Mousa y col, 2012). Nuestro grupo de trabajo, ha demostrado que las HTs son capaces de inducir in vitro la proliferación de las células de linfoma T murinas BW5147 (Barreiro Arcos y col, 2006 y 2011); y que el crecimiento y volumen tumoral de las células de linfoma murino EL-4 es mayor en los ratones hipertiroideos (Sterle y col, 2014). 39 Introducción El efecto estimulante de las HTs sobre la proliferación celular y la angiogénesis vía su receptor de membrana, la integrina αVβ3, ha sido demostrado en diversas líneas de células tumorales. Por ejemplo, el estímulo con T4 y T3 induce la proliferación de células de cáncer de pulmón humanas de manera dosis dependiente (Mousa y col, 2012). Este efecto inductor fue inhibido tanto in vitro como in vivo, utilizando anticuerpos anti-integrina αVβ3, como también utilizando el análogo de la hormona T4, el ácido tetraiodotiroacético (Tetrac), (Mousa y col, 2012). El efecto antitumoral del Tetrac también ha sido demostrado en otros modelos de xenotrasplantes, como carcinoma medular tiroideo, carcinoma tiroideo folicular y carcinomas renales, entre otros (Yalcin y col, 2009 y 2010). Estas observaciones son prometedoras ya que podrían utilizarse a futuro para que surjan nuevos tratamientos. Por último cabe mencionar que las patologías neoplásicas pueden llevar a alteraciones en el eje tiroideo. En este sentido, se ha descripto la disminución de los niveles séricos de HTs en animales singeneicos transplantados con linfomas (Besedovsky y col, 1989). También existe una correlación positiva entre el cáncer de mama y el desarrollo de alteraciones tiroideas de origen autoinmune (Giani y col, 1996). Las alteraciones en el metabolismo de las HTs son dos veces más comunes en pacientes con cáncer que no han sido tratados por su enfermedad que en individuos sanos (Dişel y col, 2012). Más aún, es común el síndrome de T3 baja en pacientes de edad avanzada con injurias graves o diversas enfermedades terminales, siendo un indicador de mal pronóstico (Tognini y col, 2010). Sin embargo, también se ha detectado este síndrome en pacientes pediátricos con linfoma Hodgkin (Mohn y col, 2001). Teniendo en cuenta estas evidencias, se puede deducir la existencia de un fenómeno bidireccional en el que el estado tiroideo puede afectar el balance entre la proliferación y la muerte de las células tumorales y por otro lado, las neoplasias por sí mismas pueden afectar la función tiroidea. 40 Introducción 1.3.3 Angiogénesis La angiogénesis es el proceso por el cual se forman nuevos vasos sanguíneos a partir de la vasculatura normal preexistente. Este proceso ocurre en condiciones fisiológicas, como son la embriogénesis y la reparación de heridas, y también en condiciones patológicas, como las enfermedades inflamatorias y el cáncer. La angiogénesis está regulada por un balance de señales parácrinas, que incluyen factores de crecimiento y citoquinas proinflamatorias. Estas señales promueven y regulan la proliferación y migración de células endoteliales, remodelado de la matriz extracelular, reclutamiento de pericitos y la estabilización de los nuevos vasos. Entre las moléculas reguladoras de este proceso, se pueden mencionar las angiopoyetinas (Ang-1 y Ang-2), la familia del receptor de tirosina quinasas Tie-2, el factor de crecimiento del endotelio vascular (VEGF) y el factor de crecimiento de fibroblastos (FGF) (FigueroaVega y col, 2009). El crecimiento tumoral, la invasión y la formación de metástasis están fuertemente relacionados con la angiogénesis. Los nuevos vasos formados promueven el crecimiento tumoral ya que facilitan la llegada del oxígeno y nutrientes y permiten la remoción de catabolitos. El proceso de angiogénesis tumoral consiste en una serie de pasos regulados (Figura 1.12) donde, en primer lugar, las células tumorales secretan factores proangiogénicos (VEGF y FGF) activando a las células endoteliales que conforman capilares cercanos a la zona donde se generó la neoplasia. Como consecuencia, las células tumorales comienzan a secretar metaloproteasas de matriz (MMPs), con el fin de degradar la matriz extracelular y migrar hacia el estímulo. Allí, las células endoteliales proliferan y generan brotes vasculares. Estos dan lugar finalmente al desarrollo de una red circulatoria funcional con el posicionamiento perivascular de células de soporte, como los pericitos (Rundhaug y col, 2005). El proceso de neoangiogénesis en cáncer es influenciado de manera crítica por el microambiente tumoral (Carmeliet y col, 2000; Rafii y col, 2002). El mediador más importante de la angiogénesis es el factor de crecimiento vascular. Los miembros de la familia VEGF incluyen a VEGF-A, VEGF-B, VEGF-C, VEGF-D 41 Introducción y al factor de crecimiento placentario (PIGF de sus siglas en inglés) y regulan varios aspectos de la angiogénesis vascular a través de sus receptores, VEGFR1, VEGFR2 y VEGFR3. El VEGF-A es producido por una gran variedad de células tumorales como también por ciertas células asociadas al tumor (Fukumura y col, 1998; Kim y col, 1993) y puede ejercer su función a través de dos receptores con actividad de tirosina quinasa, VEGFR-1 y VEGFR-2 (Ferrara y col, 2003). VEGFR-2 es el principal receptor que transmite las señales mitogénicas de VEGF-A en células endoteliales a través de la activación de las vías de señalización Raf-Mek-Erk y PI3K-Akt (Takahashi y col, 1999). Figura 1.12: Proceso de angiogénesis tumoral 1 2 3 4 (1) Los tumores generan hipoxia, la cual lleva a la activación de genes proangiogénicos como el factor de crecimiento del endotelio vascular (VEFG) o el factor de crecimiento de fibroblastos (FGF). (2) La secreción de estos factores activa células endoteliales de capilares cercanos preexistentes, que producen metaloproteasas de matriz (MMPs) para romper la matriz extracelular y poder migrar hacia el estímulo. (3) Las células endoteliales proliferan y forman brotes vasculares. (4) El nuevo vaso sanguíneo está disponible para la oxigenación del tejido y la eliminación de desechos. Adaptado de: Weiss y col, 2012 42 Introducción Algunas células tumorales expresan VEGFR-1 y VEGFR-2 y se ha demostrado que promueven la sobrevida, la proliferación y las metástasis actuando a través de mecanismos autócrinos (Dias y col, 2001; Fragoso y col, 2006). La expresión de VEGF está regulada por la acción del factor inducible por hipoxia 1 o HIF-1 (del inglés hipoxia-induced factor) y por el gen supresor tumoral von Hippel-Lindau (VHL). En condiciones de niveles normales de oxígeno la proteína VHL marca a HIF-1 para su ubiquitinación y degradación proteosómica. En condiciones patológicas, la inactivación de VHL da como resultado la acumulación de HIF-1 y la regulación positiva de VEGF. Entre los reguladores positivos del eje HIF-1/VEGF se encuentran factores de crecimiento, como el factor de crecimiento fibroblástico básico (bFGF), los factores de crecimiento transformante α y β (TGF-α, TGF-β); citoquinas proinflamatorias como el factor de necrosis tumoral (TNF) y las interleuquinas 1-α y 6 (Yancopoulos y col, 2000); mutaciones inactivadoras de genes supresores como p53 y PTEN (Hamada y col, 2005; Kaelin y col, 2007) y mutaciones activadoras de proto-oncogenes como c-myc y ErbB/Her (Ferrara y col, 2005; Dews y col, 2006; Manning y col, 2007). La activación de AKT, una molécula de señalización río abajo de PI3K, PTEN y Ras, parece ser necesaria y suficiente para regular la expresión de VEGF y HIF-1. Aunque existen vías de señalización de VEGF complementarias, que involucran, entre otros factores, al factor de crecimiento derivado de plaquetas (PDGF) el cual, es producido por las células endoteliales y cumple un rol importante en el remodelamiento y maduración vascular (Abramsson y col, 2003). 1.3.4 Participación del eje tiroideo en la regulación de la angiogénesis Los estudios realizados sobre la modulación de la angiogénesis por el estado tiroideo arrojaron resultados muy diversos. Por un lado, la endostatina, un inhibidor natural de la angiogénesis, se vio aumentada en sueros de pacientes hipertiroideos y disminuida en pacientes hipotiroideos (Kucharz y col, 2003). Sin embargo, se han encontrado niveles séricos aumentados de moléculas proangiogénicas, como Tie-2 y Ang-2, en pacientes con la enfermedad de 43 Introducción Graves, lo cual fue revertido por el tratamiento con agentes antitiroideos (Figueroa-Vega y col, 2009). Los efectos pro-angiogénicos de las HTs han sido descriptos tanto a través de su receptor nuclear como el de membrana (Luidens y col, 2010). Se ha demostrado que la hormona T3 puede mediar la proliferación de capilares en el corazón a través del receptor TRβ, el cual actúa como factor de transcripción (Makino y col, 2009). También se ha demostrado la acumulación de Ang-2 en células endoteliales luego de la estimulación in vitro con análogos de las HTs (Mousa y col, 2008). El receptor de membrana para HTs parece estar involucrado en el efecto proliferativo de la hormona sobre las células de los vasos sanguíneos y las células tumorales (Cheng y col, 2010). Experimentos realizados in vitro demuestran un efecto positivo de la hormona T4 en la inducción de la angiogénesis, la cual sería mediada por la vía no genómica de acción de estas hormonas (Bergh y col, 2005). Varios trabajos han reportado la rápida inducción de la angiogénesis en el modelo de membrana corioalantoidea de embriones de pollo (Davis y col, 2004; Bergh y col, 2005). La integrina αvβ3, descripta como el receptor de membrana para HTs, induce la vía de transducción MAPK e interactúa con las vías del VEGF y FGF (Davis y col, 2004; Mousa y col, 2006; Mousa y col, 2008; Makino y col, 2009). Los pasos que siguen a la activación de las MAPKs y la consecuente neovascularización no se conocen completamente, pero si se sabe que es necesaria la traslocación de ERK1/2 activado al núcleo y la consecuente transcripción del gen para bFGF. Se ha descripto que la adición de una proteína anti-bFGF bloquea el efecto proangiogénico de las hormonas tiroideas (Davis y col, 2004). También se ha sugerido que la transcripción del gen de VEGF podría estar inducida en respuesta a la acción de las HTs sobre la integrina αvβ3 y su subsecuente activación. Es decir que las HTs podrían ser clasificadas como moduladores proangiogénicos “no clásicos” debido a estas acciones mediadas por su receptor de membrana (Pinto y col, 2011) 44 Introducción 1.4 LINFOMAS 1.4.1 Características generales de los linfomas Los linfomas son un grupo heterogéneo de células cancerosas que derivan del sistema inmune, el cual está conformado por un gran número de células especializadas, entre las que se encuentran los linfocitos B y T. Los linfomas conforman un grupo muy amplio de más de 40 desórdenes linfoproliferativos llamados tumores de los tejidos linfoides y hematopoyéticos (Vardiman et al, 2009). La Organización mundial de la Salud (OMS) clasifica estos desórdenes para definir entidades patológicas clínicamente significativas; para ello utiliza toda la información disponible y tiene en cuenta criterios morfológicos, inmunológicos, genéticos y clínicos (Jaffe, 2008; Vardiman y col, 2008). Según la OMS existen dos grupos principales de linfomas, (i) los linfomas Hodking (LH); y (ii) linfomas Non-Hodking (LNH) que junto con otros dos subtipos, los mielomas múltiples y las enfermedades inmunoproliferativas, conforman este grupo de enfermedades hemato- oncológicas (Jaffe, 2008). Los LH se caracterizan por la presencia de un tipo de célula característica llamada célula de Reed Sternberg. Entre los dos tipos principales de enfermedad de Hodgkin están el linfoma de Hodgkin clásico y linfoma de Hodgkin con predominio de linfocitos nodulares. Los LH tiene una distribución bimodal de ocurrencia, con el primer pico en adolescentes y jóvenes adultos (15-34 años) y el segundo en adultos mayores de 50 años. En general, los tratamientos para los LH son exitosos dado que el 80-90% de los pacientes luego de un tratamiento citotóxico quedan libres de enfermedad. Sin embargo, entre un 15-20% sufren una recaída dentro de los dos años post-tratamiento y tienen una media de sobrevida general de 3 años (Cuccaro y col, 2014; Romano y col, 2014). Los LNH son aproximadamente el 90% de todos los casos e incluyen un gran número de subtipos de neoplasias malignas heterogéneas con distintos 45 Introducción patrones de comportamiento y respuesta al tratamiento. Entre los LNH los linfomas de células B representan entre el 85-88% y los linfomas de células T alrededor del 15% del total de los LNH, aunque estos porcentajes tienen variaciones según las regiones geográficas. Entre los linfomas B podemos mencionar la incidencia de los distintos subtipos: 30% corresponden a linfomas difusos de células B grandes, 25 % a linfomas foliculares, 7% a linfomas de la zona marginal asociada a mucosas de tejido linfático, 7% a leucemias linfocíticas crónicas y 5 % a linfomas de las células del manto (Ribatti y col, 2013). Los tumores linfoides pueden clasificarse en dos categorias, indolentes o agresivos, de acuerdo a las características de la enfermedad en el momento de la ocurrencia y a la expectativa de vida de los pacientes en ausencia de tratamiento. Los linfomas de células T tienen, en general, un comportamiento clínico mucho más agresivo en comparación a los de células B (Ribatti ycol, 2013). 1.4.2 Linfomas T Los linfomas de células T (LCT) son un grupo heterogéneo de desórdenes clínicos linfoproliferativos agresivos de considerable variación clínica, morfológica, inmunofenotípica y genética, que incluyen aproximadamente entre el 10-15% del total de las neoplasias linfoides Non-Hodking (de Leval y Gaulard, 2011). Con una incidencia aproximada de 1.77 x 100.000 habitantes/año, su frecuencia varía geográficamente, siendo su prevalencia mayor en Asia (Dearden y col, 2011; Armitage, 2012). Los LCT se dividen en inmaduros y maduros dependiendo de su origen celular. Los linfomas de origen maduro, también llamados periféricos, son los más frecuentes, representan aproximadamente el 12% del total de las neoplasias linfoides y resultan de la proliferación clonal de linfocitos T maduros posttímicos. La última clasificación de neoplasias hematopoyéticas publicada por la OMS ha dividido a este grupo de desórdenes de acuerdo a su forma de presentación: predominantemente leucémica (diseminada), ganglionar, extraganglionar o cutánea (Jaffe y col, 2008; Dearden y col, 2011). Las 46 Introducción variedades más comunes incluyen: los linfomas de células T periféricas no especificados de otro modo (PTCL-NOS, del inglés Peripheral T Cell Lymphoma Non Otherwise Specified), los linfomas de células grandes anaplásicos (ALCL, del inglés Anaplastic Large Cell Lymphoma) y los linfomas de células T angioinmunoblásticos (AITL, del inglés Angioinmunoblastic T cell Lymphoma). Independientemente del subtipo, los linfomas T periféricos comparten ciertas características, como por ejemplo, un comportamiento clínico muy agresivo, refractariedad a los tratamientos quimioterapéuticos convencionales y un mal pronóstico en general. La excepción a estas características es el caso de los linfomas ALCL que expresan la enzima ALK (del inglés Anaplastic lymphoma Kinase), ya que en general responden mejor al tratamiento y tienen mejor pronóstico (Petrich y Rosen, 2013). 1.4.2.1 Linfomas Periféricos no especificados de otro modo (PTCL-NOS) Los linfomas PTCL-NOS representan entre el 40-60% del total de los linfomas de células T. Usualmente afectan a adultos de entre la quinta o sexta década de vida y son más frecuentes en hombres que en mujeres. Aunque algunos subtipos pueden seguir un curso prolongado más benigno, la gran mayoría de pacientes con PTCL-NOS tiene mal pronóstico, debido a la combinación de un curso clínico agresivo y a la falta de tratamientos específicos (Foss y col, 2011). En la mayoría de los casos, al tener caracteristicas tan heterogéneas, no pueden clasificarse según su morfología, fenotipo o biología molecular (Evens y Gartenhaus, 2004). Sin embargo, existen algunos casos donde se han podido distinguir variantes raras como, por ejemplo, subtipos foliculares o epiteloides (Rüdiger y col, 2000; Geissinger y col, 2004). A nivel clínico los PTCL-NOS son de los LNHs más agresivos debido a su muy baja respuesta a las terapias quimioterapéuticas convencionales, los pacientes tienen solo un 26% de posibilidades de no experimentar recaídas y un 20% de sobrevida general de 5 años (Kahl y col, 2002; Kojima y col, 2004; Gallaminiet y 47 Introducción col, 2004). La patobiología de los PTCL-NOS es, como para la mayoría de las neoplasias de células T, muy poco entendida. En particular, esto sucede porque existen muy pocos estudios que hayan explorado los perfiles de expresión genética de este tipo de patologías (Martinez-Delgado y col, 2004; Ballester y col, 2006; Cuadros y col, 2007; de Leval y col, 2007; Lamant y col, 2007; Piccaluga y col, 2007). Por lo mencionado anteriormente, sería muy importante profundizar en los estudios sobre los mecanismos moleculares que intervienen en la patogénesis de los LCT, ya que los mismos están basados en la evaluación de genes desregulados que resultan ser indispensables para la sobrevida de las células linfoides y aportan información vital para el diseño y desarrollo de regímenes terapéuticos específicos para los PTCL-NOS. 1.4.2.2 Linfomas Angioinmunoblasticos (AITL) Los linfomas angioinmunoblásticos de células T han emergido como un subtipo distintivo con características patobiológicas únicas que los diferencia de otros subtipos de linfomas periféricos. Este subtipo de LCT se observa principalmente en los adultos de edad avanzada, con una proporción parecida entre ambos sexos. No han sido descriptos en niños. En general los pacientes con AITL no tienen buen pronóstico aunque se los trate intensivamente. Sin embargo, no siempre es letal, el 30% de los pacientes tiene una sobrevida general de 7 años. Originalmente se pensaba que los AITL eran una respuesta anormal del sistema inmune, porque la mayoría de los pacientes presentan linfoadenopatía generalizada, hepatoesplenomegalia, erupciones en la piel entre otros síntomas clínicos marcados. La hipergammaglobulinemia policlonal es una de las características que se presenta en casi todos los casos asociada con fenómenos autoinmunes. Aunque los AITL son una malignidad de células T, presentan una característica expansión de células B y plasmáticas, que probablemente reflejan la función de las células neoplásicas como de células T 48 Introducción colaboradoras foliculares (TFH). Las células B pueden estar asociadas o no al virus de Epstein-Barr (Jaffe, 2006; Bajor-Dattilo y col, 2013). 1.4.2.3 Linfomas Anaplásicos de Células Grandes (ALCL) Los linfomas anaplásicos de células grandes constituyen uno de los subtipos de LCT periféricos más comunes. Se presentan más frecuentemente de forma nodal, pero pueden presentarse también en una gran variedad de sitios extranodales. Es más común en niños y jóvenes adultos, pero puede presentarse a cualquier edad. Una de las características patobiológicas más notorias es la alta expresión del marcador CD30. También es característica la expresión positiva de marcadores citotóxicos y, en general, hay una pérdida de marcadores pan-T, con un 75% de los casos que no presentan expresión de CD3 (Pulford y col, 1997; Jaffe, 2001; Bajor-Dattilo y col, 2013). Si bien la OMS provisionalmente ha incluido dos categorías de ALCL, ALK positivas y ALK negativas (Jaffe, 2008; Vardiman y col, 2008), se ha descripto que los ALCL-ALK negativos difieren en gran número de características con respecto a los ALCL-ALK +. Entre estas diferencias se puede mencionar que los ALCL-ALK- se presentan mayoritariamente en un grupo de edad más avanzada, tienen peor pronóstico y, generalmente, presentan un pleomorfismo nuclear mayor. Estas características sugieren que los ALCL-ALK- podrían, en realidad, pertenecer a un grupo independiente de patologías LCT (Jaffe, 2001; Bajor-Dattilo y col, 2013). 1.4.2.4 Linfomas Cutáneos de células T (CTCL) Los linfomas cutáneos incluyen una gama de neoplasias primarias de la piel y representan la segunda manifestación más común de linfomas extranodales (Dummer y col, 2012). Se clasifican en dos grandes grupos dependiendo de las células de las que derivan, células T o B. En muy pocos casos, también, pueden derivar de células NK (del inglés Natural Killer) o células dendríticas plasmocitoides (Klemke, 2014). Dos tercios de los pacientes con linfomas 49 Introducción cutáneos son CTCL. Los tumores malignos CTLC incluyen entre otros, la micosis Fungoide (MF), el síndrome de Sézary (SS) y el Síndrome Linfoproliferativo CD30+ cutáneo primario, los cuales representan el 90% del total de los CTCL. La mayoría de los casos de MF y SS, los cuales representan entre un 54-71% del total de los CTCL (Bradford y col, 2009), derivan de células T CD4+, presentan pérdida del marcador CD7 y muestran bajos niveles de marcadores de activación como CD25. Sin embargo, existen casos donde en efecto se observa expresión del marcador CD8. La MF se caracteriza por la proliferación de linfocitos T neoplásicos, epidermotróficos, de tamaño medio, con manifestaciones clínicas que varian entre la presencia de manchas, placas, tumores o eritrodermia. Por otro lado, el SS, una variante leucémica de CTCL, se caracteriza por la manifestación de eritrodermia, linfoadenopatía generalizada y la presencia de células T neoplásicas en la piel, nódulos linfáticos y sangre periférica. 1.4.3 Rol de la angiogénesis en los linfomas La importancia de la angiogénesis en desórdenes linfoproliferativos fue estudiada en relación a su impacto en la progresión de la enfermedad. Estos estudios sugieren una gran importancia de la angiogénesis en relación al pronóstico de los pacientes en distintos subtipos de linfomas (Kini y col, 2004; Koster y col, 2005; Ruan y col, 2009). El crecimiento y la progresión de los linfomas están potenciados por al menos dos mecanismos angiogénicos distintivos: 1- Estimulación autócrina de las células tumorales a través de la expresión de VEGF y VEGFR en las células de linfoma. 2- Influencias parácrinas del microambiente tumoral pro-angiogénico sobre la transformación neovascular local y el reclutamiento de progenitores derivados de médula ósea circulantes. 50 Introducción Existe evidencia donde el infiltrado de células asociado al linfoma, constituido por células derivadas de médula ósea, monocitos hematopoyéticos, células T y pericitos mesenquimales, fue relacionado con la patogénesis, la angiogénesis y el prónostico de los pacientes que presentan esta patología (Ruan, 2009). Los factores que afectan las propiedades pro-angiogénicas de un linfoma son: (i) los niveles de expresión de VEGF y VEGFR, (ii) la densidad microvascular del linfoma, (iii) el microambiente tumoral y (iv) los progenitores endoteliales circulantes. La expresión de VEGF en células neoplásicas fue demostrada en un amplio rango de subtipos de linfomas agresivos, que incluye, entre otros, a los linfomas T periféricos (PTCL), a los linfomas difusos de células B grandes (DLBCL) y al linfoma de células del manto (MCL), (Foss y col, 1997; DoussisAnagnostopoulou y col, 2002); así como también, en leucemias linfocíticas crónicas y en linfomas linfocíticos de células pequeñas (Chen y col, 2000; Kay y col, 2002). Como contraste, sólo una minoría de casos de linfomas foliculares indolentes muestra una expresión variable de VEGF (Koster y col, 2005; Jorgensen y col, 2007). Los niveles de expresión de VEGF han sido asociados con áreas de transformación de un linfoma B indolente a un DLBCL agresivo (Shipp y col, 2002). Más aún, los niveles séricos de VEGF correlacionan inversamente con la sobrevida de pacientes con LNH (Bertolini y col, 1999; Jorgensen y col, 2009). Se ha descripto que VEGF y sus receptores se expresan en un gran número de células de linfoma, influyendo sobre los mismos tanto por mecanismos autócrinos como parácrinos (Doussis-Anagnostopoulou y col, 2002; Wang y col, 2004; Jorgensen y col, 2009). El bloqueo de VEGFR-1 y VEGFR-2 redujo el crecimiento de linfomas humanos xeno-transplantados en ratones NODSCID (Wang y col, 2004). La señalización de VEGF a través de sus receptores confiere resistencia a la apoptosis y promueve la sobrevida de leucemias linfocíticas crónicas, pero este efecto es revertido con inhibidores de receptores con actividad de tirosina quinasa (Lee y col, 2004). En células de linfomas DLBCL se ha encontrado que entre el 42-60% expresan VEGF (Ganjoo y col, 51 Introducción 2006). Específicamente sobre los LCT, existen trabajos donde se ha observado una alta expresión de VEGF en linfomas AITL (Piccaluga y col, 2007). Por otro lado, se ha descripto una correlación positiva entre la expresión de VEGF y un peor pronóstico en pacientes con PTCL-NOS (Jorgensen y col, 2007 y 2009). La densidad microvascular (DMV) mide la neovascularización de los linfomas, la cual es generada en respuesta a las células tumorales, células proangiogénicas estromales, linfocitos T y B normales del infiltrado tumoral y células mieloides dentro del microambiente tumoral. La DMV varía mucho entre los distintos estudios debido a la gran heterogeneidad del estroma de los linfomas, al amplio rango de marcadores de membrana celular utilizados para la tinción y a las grandes diferencias en las metodologías para la clasificación de la marcación (score). En general los scores de DMV tienden a ser más elevados en los subtipos más agresivos de linfomas, incluyendo el linfoma de Burkitt´s y PTCL, en comparación a linfomas de agresividad intermedia, como DLBCL, o indolentes como los linfomas foliculares (Ribatti y col, 1996). Sin embargo, existen estudios donde no encuentran correlación entre la DMV y VEGF con el prónostico clínico (Ganjoo y col, 2006; Ruan y col, 2006). Estos estudios vislumbran la heterogeneidad y complejidad de los procesos angiogénicos en los linfomas. El microambiente tumoral ha sido reconocido como un factor que influencia la progresión y el crecimiento de los tumores. Se han realizado muchos estudios de perfiles genéticos a gran escala en distintos subtipos de linfomas. En estos estudios se ha demostrado que los perfiles genéticos que expresan las células estromales e inmunes del infiltrado tumoral definen grupos distintivos, de manera que esta información puede ser utilizada como una herramienta molecular pronóstica (Dave y col, 2004; Monti y col, 2005). Se ha descripto que grandes cantidades de macrófagos intratumorales correlacionan con mal pronóstico en pacientes con linfomas foliculares (Taskinen y col, 2007; Canioni y col, 2008) y que el aumento del infiltrado hematopoyético por progenitores mieloides y monocitos correlaciona con la progresión histológica de algunos subtipos de linfoma (Farinha y col, 2005; Ruan y col, 2006) 52 Introducción Las células endoteliales circulantes y los niveles de VEGF parecen correlacionar con los volúmenes tumorales de los linfomas humanos crecidos en ratones NOD-SCID (Monestiroli y col, 2001). Por otro lado, se ha descripto también, una disminución de los niveles de estas células en pacientes con una respuesta completa al tratamiento (Igreja y col, 2007), sugiriendo que la presencia de progenitores endoteliales circulantes en linfomas puede llegar a proporcionar un potencial biomarcador específico de angiogénesis para evaluar la respuesta luego de una terapia anti-angiogénica. Todos los mecanismos distintivos angiogénicos mencionados en esta sección, parecen ser importantes blancos terapéuticos o futuros biomarcadores del pronóstico de un paciente en ciertos subtipos de LNH. El conocimiento de estas vías podría dar como resultado la utilización de estrategias de tratamiento con drogas anti-angiogénicas para este tipo de patologías. 1.4.4 Tratamientos de Linfomas T Los LCT se incluyen entre el 13-15% de los 60000 LNH diagnosticados anualmente en Estados Unidos. Este resultado estima unos 8000-9000 casos nuevos anuales de LCT (Rodriguez-Abreu y col, 2008). A pesar de que cada subtipo de LCT tiene relativamente baja incidencia, la carga acumulada de estas patologías es muy significativa. La epidemiología de varios subtipos de LCT, particularmente aquellos asociados con factores ambientales tales como algunos virus, difiere substancialmente en distintos lugares del mundo. En algunas regiones de Asia, América Central, América del Sur y Medio Oriente, las neoplasias de células T pueden llegar a representar hasta un 35-40% del total de los LNH diagnosticados y, generalmente, están asociados con el Virus Epstain Barr (EBV) y el Virus linfotrópico de células T humanas-1 (HTLV-1) (Jaffe y col, 2006). Como la mayoría de los LCT son de origen maduro, en esta sección nos focalizaremos en los tratamientos disponibles para los mismos. 53 Introducción Con propósitos clínicos, los LCT pueden dividirse convenientemente en 2 grupos: 1- Los que involucran neoplasias primarias de piel, denominados genéricamente Linfomas Cutáneos de células T (CTCL), los cuales son patologías crónicas y con recaídas caracterizadas por erupciones, prurito, fatiga y susceptibilidad a infecciones debido a la ruptura de la protección y del sistema inmune que provee la piel. 2- Los que involucran nódulos linfáticos o sitios extra-nodales, los cuales en su mayoría son identificados como Linfomas Periféricos de Células T. Estos últimos, son patologías sistémicas agresivas, caracterizadas por el alto grado de avance de la enfermedad al momento de la aparición clínica, con un extensivo involucramiento extra-nodal y progresión clínica rápida, con baja respuesta completa a los tratamientos, aún cuando se utilicen protocolos con combinación de drogas quimioterapéuticas (Savage, 2007). Uno de los desafíos más importantes en el estudio de los LCT, es la rápida traslación a la clínica de los conocimientos básicos que se están obteniendo sobre la biología molecular y genética de las células T. Salvo casos excepcionales de LCT que tienen un pronóstico más favorable, la relevancia clínica de los marcadores de expresión, como CD8, CD4 y CD56, marcadores citotóxicos y una variedad de oncogenes, citoquinas y receptores de quimoquinas, no han sido estudiados en profundidad. El objetivo principal de esta sección es proveer una síntesis de las drogas que se han aprobado o que están en fases avanzadas de desarrollo para el tratamiento de CTCL y PTCL. 1.4.4.1 Anticuerpos monoclonales a. Zanolimumab: es un anticuerpo IgG1κ monoclonal dirigido contra el antígeno CD4 (Fishwild y col, 1996). Este marcador es específico de 54 Introducción células T y es expresado débilmente en monocitos y macrófagos. Originalmente fue testeado para la artritis reumatoidea y la psoriasis (Choy y col, 2000) y está siendo evaluado en estudios clínicos de fase II y III en CTCL y PTCL (Kim y col, 2007; Rider y col, 2007). b. Anticuerpos anti-CD30: El marcador CD30 es una glicoproteína de membrana miembro de la familia TNF (Horie y col, 1998) que se expresa en células B y T activadas y que en ciertas condiciones puede inducir muerte celular (Wahl y col, 2002; Younes y col, 2003). Se la ha descripto como una proteína con distribución restringida en ciertos tipos de leucemias maduras de células T y en PTCL (Pizzolo y col, 1994). En la actualidad, se están investigando varios anticuerpos contra CD30 que se encuentran en distintas fases de estudios clínicos, entre ellos podemos mencionar, el MDX-060 para linfomas foliculares y ALCL (Younes y col, 2003); el SGN-30 en modelos de linfomas Hodking (Wahl y col, 2002; Younes y col, 2008). c. Siplizumab (MEDI-507): Es un anticuerpo anti-CD2, un marcador celular expresado en células T maduras y en células NK, sobreexpresado en células T activadas en comparación con células en reposo. El Siplizumab fue estudiado inicialmente en modelos murinos de AITL y luego en estudios clínicos de fase I en leucemias/linfomas CTCL o PTCL CD2 positivos (Criscione y col, 2007; Rodriguez-Abreu y col, 2008). d. Alemtuzumab (Campath-1H): Es un anticuerpo IgG1κ humanizado dirigido contra el antígeno CD52, una glicoproteína expresada en todos los linfocitos B y T, monocitos, macrófagos, eosinófilos y células dendríticas (Frampton y Wagstaff, 2003). CD52 se encuentra expresado de manera variable en ciertos tipos de linfomas, incluyendo los LCT. Si bien existen algunos estudios que demuestran que el alemtuzumab es activo contra los LCT (Lundin y col, 2003; Zinzani y col, 2005; Dearden y col, 2006; Bernengo y col, 2007), los ensayos clínicos hasta el momento han sido con un número reducido de pacientes, por lo que la aprobación para su uso todavía no está garantizada. 55 Introducción 1.4.4.2 Inhibidores de las Histonas Deacetilasas (HDAC) Este grupo de drogas funciona a través de la modificación de las actividades epigenéticas que ocurren dentro de las células, es decir, la regulación de la transcripción de genes por alteraciones al ADN o en la estructura de la cromatina (Glaser, 2007). La efectividad de la modulación epigenética en enfermedades hemato-oncológicas, a través de la inhibición de las vías de las histonas deacetilasas, ya ha sido descripta en varios trabajos (Bhalla, 2007). a. Vorinostat (SAHA, Zolinza): Es un inhibidor de la actividad histona desacetilasa que se ha estudiado en estudios clínicos de fase I en una gran variedad de enfermedades hemato-oncológicas, donde se observó una dosis máxima tolerable con distintas respuestas clínicas (GarciaManero y col, 2008). La FDA aprobó al Vorinostat para su uso como droga única en el tratamiento de CTCL (Mann y col, 2007). b. Depsipeptide (Romidepsin): Se ha demostrado una respuesta parcial de un grupo pequeño de pacientes con CTCL y una respuesta completa en un paciente con PTCL (Piekarz y col, 2001). Este caso dio como resultado un estudio clínico de fase I en 37 pacientes con tumores sólidos (Sandor y col, 2002). Se ha publicado que la combinación de Romidepsin con doxorubicina, etopósidos, ciclofosfamida, entre otras drogas, genera un efecto aditivo en distintas líneas celulares (Kano y col, 2007). Estos estudios pre-clínicos ayudan a demostrar la sensibilidad general de los linfomas T a los inhibidores de histonas deacetilasas. c. LBH589 (Panobinostat): es un inhibidor histona deacetilasa nuevo, que se encuentra en estudios pre-clínicos contra células de leucemia aguda y de mieloma (George y col, 2005; Maiso y col, 2006). También se encuentran en desarrollo estudios clínicos de fase I y II en leucemias mieloides agudas y CTCL (Giles y col, 2006). 56 Introducción d. PXD-101 (Belinostat): Es una droga nueva que todavía no está estudiada en profundidad para su uso en desórdenes linfoproliferativos de células T, aunque ha mostrado un efecto sinérgico significativo con el bortezomib en líneas celulares de mieloma múltiple (Feng y col, 2007). 1.4.4.3 Inhibidores de proteosomas Bortezomib (Velcade, PS-341): Es un inhibidor de proteosomas reversible que muestra una marcada actividad inhibitoria sobre una gran variedad de enfermedades malignas hematológicas y sólidas. Los estudios clínicos se han realizado más profundamente en mielomas múltiples (Leonard y col, 2006), aunque también existe información sobre su efectividad en el tratamiento de distintos subtipos de LNH (Rajkumar y col, 2005). Se están desarrollando estudios clínicos de fase II en pacientes CTCL y PTCL donde se sugiere su uso como agente individual o en combinación con otras drogas (Hamamura y col, 2007). 1.4.4.4 Drogas citotóxicas convencionales y anti-metabolitos a. Pralatrexato: Es un análogo del folato, con efectos antitumorales efectivos bien descriptos, como los obtenidos en modelos murinos con el Metotrexato. Se han realizado estudios de fase clínica II/III en pacientes con linfomas B y linfomas T donde se ha observado una buena respuesta al antifolato (Zinzani y col, 1998). También existen otros estudios, con el anti-folato, PROPEL donde se ve que el 35% de los pacientes con PTCL-NOS responden a la terapia. b. Gencitabina: Es un análogo de pirimidinas muy estudiado como agente quimioterapéutico. Existen múltiples estudios de fases clínicas II y III que demostraron su uso en PTCL y CTCL (Marchi y col, 2005; Zinzani y col, 2000). 57 Introducción c. Doxorubicina liposomal pegilada (Doxil): La doxorubicina es una antraciclina que se ha utilizado frecuentemente para una gran variedad de LNH. La forma liposomal pegilada ha sido estudiada en varios ensayos clínicos de fases I y II en pacientes con CTCL (Pulini S y col, 2007; Wollina y col, 2003; Wollina y col, 2001). d. Pentostatina: Es un análogo de purinas y ha mostrado resultados promisorios iniciales en CTCL (Kurzrock y col, 1999). En base a los mismos, se han desarrollado estudios clínicos de fase I y II obteniendo resultados razonables con una toxicidad media (Tsimberidou y col, 2004). 1.4.4.5 Drogas inmunomoduladoras y no-citotóxicas a. Interferones (IFN): Son una familia de glicoproteínas que se producen y secretan en respuesta a variedad de inductores virales y no virales. Los IFNs inducen múltiples actividades biológicas y tienen actividad antiproliferativa, anti-viral, citotóxica y anti-angiogénica. El IFN-α recombinante se ha utilizado en el tratamiento de CTCL durante al menos dos décadas. En bajas dosis y en combinación con bexaroteno, el IFN-α ha demostrado una rápida mejoría en pacientes con CTCL y mielomas foliculares (McGinnis y col, 2004). Un aspecto negativo son sus efectos secundarios, los cuales son dosis dependientes. b. Agonistas de receptores Toll (TLR): Los patrones de expresión TLR 2, 4 y 9 fueron estudiados en la micosis fungoide y el síndrome de Sézary (Jarrousse y col, 2006). Un ejemplo de este tipo de drogas, es el Imiquimod, un agente inmunomodulador que activa el TLR7, aprobado por la FDA para el tratamiento de cánceres superficiales. El Imiquimod en ensayos in vivo, ha demostrado ser un potente inductor del TNFα, IFNγ e IFNα. El Imiquimod al 5% en crema fue evaluado en pacientes con micosis fungoide y ha demostrado mejorar la respuesta en la mayoría de las lesiones tratadas (Deeths y col, 2005). 58 Introducción 1.4.4.6 Inhibidores de señales de transducción El Enzastaurin es una molécula pequeña inhibidora de serina/treonina quinasas, la cual ha mostrado un efecto inhibitorio sobre las vías PKC β y PI3K/AKT. La experiencia clínica con esta droga en linfomas es todavía limitada, pero existe un estudio clínico de fase II en CTCL en progreso. El tratamiento efectivo del amplio espectro de los linfomas de células T todavía es dificultoso. Salvo en los estadíos tempranos de la Micosis Fungoides y los ALCL-ALK positivos, el pronóstico de las neoplasias de células T sigue siendo malo. El mayor desafío por delante, es focalizar y racionalizar la aplicación de los nuevos agentes terapéuticos y sus combinaciones para mejorar la eficacia de los tratamientos contra los LCT. 59 2. HIPÓTESIS Y OBJETIVOS Hipótesis y Objetivos 2.1. HIPÓTESIS DE TRABAJO En base a los antecedentes presentados, la hipótesis de este trabajo es que las hormonas tiroideas, a través de la regulación de programas genéticos, jugarían un rol importante en la proliferación y supervivencia de las células de linfomas T y, de esta manera, favorecerían al fenotipo maligno de los mismos. Adicionalmente consideramos que la caracterización de las redes de genes regulados por las HTs podría contribuir al conocimiento de la linfomagénesis T y servir como base, en un futuro, para la modificación terapéutica de estas vías. 2.2. OBJETIVOS 2.2.1. Objetivo general Para comprobar la hipótesis arriba mencionada, nos planteamos como objetivo general evaluar la acción in vitro de las HTs sobre la proliferación y sobrevida de linfomas de células T humanas y determinar que programas genéticos se regulan a partir de la acción de las mismas. Adicionalmente, mediante la intervención genética o farmacológica de estas vías in vivo evaluaremos su implicancia en el desarrollo tumoral. Para poder cumplir nuestro objetivo general, planteamos los objetivos específicos que se detallan a continuación. 2.2.2 Objetivos específicos A fin de analizar la acción in vitro de las HTs sobre LCT humanos, contamos con un panel de 9 líneas de linfomas T, las cuales representan distintos subtipos de LCT humanos, a saber: de origen inmaduro, Jurkat y CUTLL1; de origen maduro/periférico, OIC-LY12, OIC-Ly13.2, MJ, Mac2a, HuT 78, SUDHL1 y Karpas 299. En estas líneas nos propusimos: 1) Evaluar la expresión de los receptores de HTs, tanto nucleares como el de membrana, mediante: 61 Hipótesis y Objetivos a. RT-PCR en tiempo real para evaluar los niveles transcripcionales de THRA, THRB, ITGAV e ITGB3 b. Citometría de flujo, para evaluar los niveles de expresión proteica de los mismos. 2) Estudiar las acciones de las HTs sobre la proliferación y el ciclo celular, en particular: a. Evaluar el efecto de concentraciones fisiológicas de HTs libres o unidas a agarosa (HT-AG) sobre la proliferación celular. Dado que las HT-AG son impermeables a la membrana y no pueden ingresar a la célula, nos permiten distinguir el efecto total de las HTs vs el efecto inducido por su receptor de membrana (mTR). b. Evaluar el efecto de las HTs a distintos tiempos sobre la expresión de marcadores de proliferación y ciclo celular (como la expresión de PCNA, ciclinas y genes supresores tumorales, entre otros) 3) Identificar si la integrina αVβ3 es receptor de membrana para HTs en células de linfoma T. a. Utilizando inhibidores farmacológicos. b. Mediante el uso de ARN de interferencia contra las integrinas αV y β3 4) Determinar y comparar los programas transcripcionales inducidos por las HTs en los linfomas T. a. Por análisis de los genes regulados positiva y negativamente por las HTs mediante la técnica de secuenciación del ARN. b. Utilizando herramientas bioinformáticas de análisis como el software Ingenuity Pathway Analysis para la identificación de los genes regulados por HTs involucrados en programas biológicos relevantes para el desarrollo, proliferación y sobrevida de los LCT. 62 Hipótesis y Objetivos c. Validando la regulación por HTs de los genes de interés por RT-PCR en tiempo real. Por otra parte hemos desarrollado modelos tumorales mediante xenotrasplantes de las líneas de LCT en ratones inmunodeficientes. También contamos con muestras de pacientes de distintos subtipos de LCT de origen maduro. Con estas herramientas nos planteamos: 5) Analizar el efecto del bloqueo de la acción de HTs sobre el crecimiento in vivo de los tumores de LCT humanos desarrollados en ratones NODSCID. 6) Evaluar la expresión de los receptores para HTs y genes de interés regulados por las mismas en muestras primarias obtenidas de pacientes con LCT. 63 3. MATERIALES Y MÉTODOS Materiales y Métodos 3.1 PREPARACIÓN DE LAS HORMONAS T3 Y T4 UNIDAS A AGAROSA Las hormonas triyodotironina y tiroxina unidas a agarosa (T3-AG and T4-AG, respectivamente) se obtuvieron de la misma forma en la que se describió previamente en nuestros trabajos (Barreiro Arcos y col, 2011). Brevemente, las hormonas T3 y T4 adquiridas de Sigma Chemical Co. (St. Louis, MI) fueron utilizadas sin ninguna otra purificación extra. El brazo espaciador (entre la matriz y el grupo activado), “N-Hydroxysuccinimide (NHS)-activated Sepharose 4 Fast Flow 14-atom”, se obtuvo de la compañía GE Healthcare (Amersham Biosciences, Uppsala, Sweden). Los grupos amino primarios de las HTs fueron unidos directamente al grupo ester activo para formar una unión amida de acuerdo a las indicaciones del proveedor (Amersham Biosciences). La presencia de T3 y T4 libres, testeada por RIA, fue muy baja, aproximadamente menos de 1 mol de hormona libre cada 105 - 104 moles de HTs unidas a agarosa. La estabilidad del conjugado hormona-agarosa, se determinó en los sobrenadantes de cultivos de 24 horas con T3-AG o T4-AG y no se obtuvieron niveles detectables de hormonas libres en los mismos. 3.2 ENSAYOS IN-VITRO CON LINEAS CELULARES HUMANAS DE LCT 3.2.1 Líneas celulares de linfoma T Utilizamos 9 líneas celulares, las cuales representan los distintos subtipos de esta enfermedad. Este panel de líneas celulares incluye: células T de linfoma/leucemia de origen inmaduro Jurkat y CUTLL1 (cedidas por la Dra. Roxana Schillaci, IBYME-CONICET, Argentina, y por el Dr. Adolfo Ferrando, Columbia University, USA, respectivamente); linfomas T cutáneos representados por las líneas celulares MJ y HuT78 (ATCC) y linfomas T periféricos representados por las líneas celulares OIC-Ly12 y OIC.Ly13.2 (Ontario Cancer Institute, Canadá), Mac2a, Karpass 299 y SUDHL1 (ATCC). Las células fueron cultivadas en concentraciones óptimas para su crecimiento (1–5 x 105 cel/ml) en el medio RPMI 1640 (Invitrogen, Gibco, Grand Island, NY) 65 Materiales y Métodos suplementado con 10% de suero fetal bovino (SFB, Gibco BRL), 2mM de glutamina (Gibco BRL) y antibióticos (Gibco BRL). 3.2.2 Tratamientos in vitro de las líneas de LCT Para los distintos ensayos, las células LCT fueron tratadas a distintos tiempos con concentraciones fisiológicas de HTs libres (T3 = 1 nM; T4 = 100 nM, Sigma Aldrich Co) y con HTs unidas a agarosa (HT-AG) en concentraciones similares. El bloqueo de la acción de las HTs se evaluó pre-incubando 15 minutos antes del tratamiento hormonal a las células con el péptido RGD a una concentración de 10 µM. Antes de realizar los tratamientos, la células fueron sincronizadas, cultivándolas por 24 horas en RPMI con 1% SFB (para los ensayos de incorporación de timidina tritiada y los ensayos de migración, ver abajo) o con RPMI suplementado con 10% de un suero depletado de hormonas tiroideas (Sunny-Lab Co, UK). Para evaluar la inhibición de la acción del VEGF, 15 minutos antes del tratamiento con las HTs se pre-incubó con un anticuerpo anti-VEGF (Bevaczumab, 10 µg/ml) o con un inhibidor farmacológico del receptor VEGFR (Axitinib, 3 µM). También se evaluó el efecto del VEGF y del ligando Vitronectina (VN), utilizando, para ello 10 µg/ml de VEGF recombinante y 250 ng/ml de VN respectivamente. En los ensayos con VN, las placas fueron preincubadas durante 16 horas a 4°C, para luego descartar el exceso de la misma y realizar los estímulos con o sin HTs subsiguientes. Para evaluar el efecto de la inhibición del factor de transcripción NF-κB se pre-incubaron a las células durante 1 hora con el inhibidor farmacológico BAY 11-7082. 3.2.3 Ensayos de proliferación celular La proliferación celular fue evaluada utilizando, la técnica de incorporación de timidina tritiada ([3H]TdR), el conteo celular por marcación con Trypan Blue, o el ensayo fluorométrico Cell Titer Blue. La marcación convencional con [3H]TdR de las células en proliferación se realizó como se describió previamente 66 Materiales y Métodos (Barreiro Arcos 2006 y 2011). De manera resumida, 5 x 105 células/ml fueron colocadas en un volumen final de 0.2 ml por pocillo (placa de 96 pocillos) y fueron tratadas o no (control) con HTs libres o unidas a agarosa durante 24 horas. Se agregó un pulso de [3H]-TdR (Perkin Elmer, USA, 20 Ci/mmol) durante las últimas 16 horas de tratamiento. Todos los ensayos fueron realizados por cuadruplicado. También utilizamos el método fluorométrico de reducción de la resazurina (CellTiter-Blue; Promega, Figura 3.1) para medir proliferación celular. Se determinó la fluorescencia (560Ex/590Em) utilizando el Synergy4 microplate reader (BioTek). El porcentaje de células proliferativas se calculó utilizando la regresión lineal por cuadrados mínimos de la curva estándar. La fluorescencia fue determinada a partir de 6 replicados por cada tratamiento y la proliferación de las células tratadas con HTs fue normalizada a sus respectivos controles. Figura 3.1: Ensayo Cell Titer Blue para evaluar proliferación Célula Viable Reacciones de reducción Resazurina Resorufina Representación ilustrativa del método fluorométrico de reducción de la resazurina. Las células viables reducen la Resazurina a Resofurina, la cual emite a 590 nm. Adaptado de www.promega.com 67 Materiales y Métodos 3.2.4 Crecimiento in vitro en cultivos en 3 dimensiones La matriz tridimensional se obtuvo utilizando metoxi polietilenglicol maleimida (PEG-MAL, Laysan Bio, Inc) y un péptido, como entrecruzador de secuencia NH2-GCRDVPMSMRGGDRCG-COOH (Figura 3.2). Los hidrogeles fueron preparados como se describió anteriormente (Singh y col, 2011; Lee y col, 2014; Patel y col, 2014). Brevemente, 4 brazos espaciadores de PEG-MAL fueron funcionalizados con el ligando peptídico RGD (NH2-GRGDSPC-COOH) y entrecruzados con un péptido de colagenasa degradable (VPM) para formar el hidrogel. Cada 10 µl se encapsularon células OCI-Ly12 (PTCL-NOS) y células dendríticas HK foliculares derivadas de amigdalas en una proporción 2(PTCL-NOS):1 (HK). Los hidrogeles fueron incubados con o sin hormonas tiroideas (T3 y T4, 1 nM y 100 nM, respectivamente) y en presencia o ausencia del inhibidor farmacológico cilengitide (4 µM). Luego de 48 horas de cultivo, los hidrogeles fueron degradados enzimáticamente con la colegenasa tipo I durante 2 horas (Worthington, 50 U/mL), para luego analizar molecularmente las células cultivadas con los distintos tratamientos. Figura 3.2: Cultivo de células LCT en 3 dimensiones Hormonas tiroideas Droga RGD cíclica Hormonas Tiroideas Droga RGD cíclica Ligando RGD Células OCI-Ly12 Células estromales HK Representación ilustrativa del cultivo en 3 dimensiones de linfomas de células T. 68 Materiales y Métodos 3.2.5 Transfecciones transientes con ARN de interferencia (siRNA) Por cada transfección se utilizaron 1 x 107 células en 400 µl de medio RPMI 1640 y fueron mezcladas con una concentración final de 50 nM de los respectivos siRNA contra ITGAV y/o ITGB3, o contra una secuencia no codificante utilizada como control (L-004565, L-004124 and D-001810, respectivamente, ON-TARGETplus SMART pool, THERMO SCIENTIFIC). Luego fueron electroporadas a 250 V y por 13 ms en el electroporador BTX ECM 830 (Liu et al, 2007). Luego de la transfección, las células fueron cultivadas en medio RPMI suplementado con 10% de SFB por 24 horas. Posteriormente se empezó el protocolo ya mencionado para realizar los distintos tratamientos con las HTs. 3.2.6 Análisis por RT-PCR en tiempo real. Luego de los distintos tratamientos in vitro, aproximadamente 1-2.5 x106 células de cada condición fueron homogeneizadas a temperatura ambiente en 1 ml del reactivo Tri-Reagent (Genbiotech SRL) y el ARN total fue aislado siguiendo las instrucciones del fabricante. Los pellets de ARN se disolvieron en agua libre de ARNasas y guardados a -80°C. La concentración del ARN total fue cuantificada midiendo la absorbancia a 260 nm (Nanodrop ND-1000, UK). La pureza de las preparaciones de ARN se determinaron por espectrometría midiendo la relación de absorbancias a 260/280 nm y corriendo las muestras en un gel de agarosa 1% con Bromuro de Etidio. A partir de 2 µg de ARN por muestra, se sintetizó el ADN complementario (cADN) por transcripción reversa utilizando el kit Omniscript (Qiagen GMDH). La cuantificación del cADN presente en cada muestra fue determinada utilizando una mezcla comercial SYBR Green para PCR en tiempo real (qPCR) (Biodynamics SRL, Buenos Aires, Argentina). La reacción de qPCR fue llevada a cabo en el equipo Applied Biosystems 7500. Las secuencias de los primers (Biodynamics SRL, Buenos Aires, Argentina), las cuales se muestran en la Tabla 1, se diseñaron utilizando el Primer Express software version 3.0 (Applied Biosystems, California, USA) con una 69 Materiales y Métodos temperatura de melting de entre 58-60°C. Para verificar que el fluoróforo solo detecta un producto de PCR, todas las reacciones se sometieron al protocolo de disociación por calor luego del ciclo final de la PCR (Ririe y col., 1997). Tabla 3.1: Secuencia de primers utilizados para RT-PCR en tiempo real Gen B2M CCR8 CCNB1 CCND1 CCND2 CCND3 CCNE2 DBP DOK2 ELOVL4 IL2RA IL4 ITGAV ITGB3 PCNA THRA VEGFA VEGFB WNT5B Temperatura °C Secuencia AGATGAGTATGCCTGCCGTGTGAA TGCTGCTTACATGTCTCGATCCCA AAAGGTGAGGACGATCAGGA AATGGCGGTTAGCCATACTG CATGGTGCACTTTCCTCCTT TCTTAGCCAGGTGCTGCATA TCCTCTCCAAAATGCCAGAG TGAGGCGGTAGTAGGACAGG TGGGGAAGTTGAAGTGGAAC ATCATCGACGGTGGGTACAT AGGGATCACTGGCACTGAAG CTGTAGGAGTGCTGGTCTGG TACTGACTGCTGCTGCCTTG AAAAGTCTTCAGCTTCACTGGA CCTCGAAGACATCGCTTCTC GCCTCGTTGTTCTTGTACCG ACTGGCCCTACAGGTTTCTG CTCAAAGTTGCCCTCTCCAG GGCCCATGGATTCAGAAATA GGAAGGGGCAGTCAGTGTAA ACTTCCTGCCTCGTCACAAC GATCAGCAGGAAAACACAGC TGAACAGCCTCACAGAGCAG GCGAGTGTCCTTCTCATGGT TGCCAAGTTGGGAGATTAGA TGAAGCAGACGACTTCAGAGA TGAGGATGACTGTGTCGTCAG GTCTTGCTCTATCTTTCTTTGG ATCCTCAAGAAGGTGTTGGAGGCA ACGAGTCCATGCTCTGCAGGTTTA CTGATCCACATTGCCACAGA TTCCAGGTCCACCTTGTCTC CTTCCTACAGCACAACAAAT GTCTTGCTCTATCTTTCTTTGG GACAGTGCTGTGAAGCCAGA AGTGGGATGGGTGATGTCAG TTTGGGAGAGTCATGCAGAT TAGCCGTACTCCACGTTGTC F R F R F R F R F R F R F R F R F R F R F R F R F R F R F R F R F R F R F R 60 60 60 60 60 60 60 60 60 60 60 60 60 60 60 60 60 60 60 70 Materiales y Métodos La detección directa de los productos de PCR se monitoreó midiendo el incremento en la fluorescencia causada por la unión del colorante al ADN doble cadena. Todas las muestras fueron testeadas contra el gen de referencia de la proteína β2-microglobulina (“housekeeping gene”), usado como control interno para la normalización de los datos, procedimiento para la corrección de las variaciones en calidad y cantidad de cADN. La cuantificación de la expresión del gen blanco fue realizada mediante un análisis de ∆∆Ct para calcular las diferencias entre tratamientos (Livak y Schmittgen, 2001) 3.2.7 Análisis de expresión de proteínas por western blot Muestras de lisados de 5x106 células de LCT, incubados en ausencia o presencia de HT o HT-AG por distintos tiempos, fueron obtenidos por disolución de los pellets celulares en 100 µl de una solución con 2-βmercaptoetanol 10 mM, EDTA 2 mM, ácido etileglicol bis(β-aminetileter)N-N’ tetraacético (EGTA) 2 mM, fenilmetil sulfonil fluoruro (PMSF) 1 mM, leupeptina 10 µg/ml y ácido 4-(2-hidroxietil)-1-piperazina etanosulfónico (HEPES) 20 mM pH 7,4; y se centrifugaron para obtener las fracciones solubles en el sobrenadante. Se mezclaron 50 µg de proteínas de cada muestra en solución de carga (SDS 2%, glicerol 10%, Tris-HCl 62,5 mM pH 6,8; azul de bromofenol 0,2%, 2-β-mercaptoetanol 1%) y se sembraron en un gel de poliacrilamida al 10%, para separar dichas proteínas por SDS-PAGE y transferirlas a membranas de nitrocelulosa. Para corroborar la eficiencia de la carga y de la transferencia, dichas membranas fueron coloreadas con Ponceau S. Las membranas se bloquearon con leche descremada 5% en PBS-Tween-200 (0,1%), durante 1 hora. Luego, se incubaron con los respectivos anticuerpos primarios, anti-PCNA (Santa Cruz Biotechnology); ERK1/2 o ERK p42/44 (ab52245 y ab50011, respectivamente, Abcam), toda una noche a 4°C. Como control de carga se utilizó el anticuerpo anti-Actina (1:100, Santa Cruz Biotechnology). Tras realizar tres lavados, las membranas fueron incubadas con un anticuerpo policlonal anti-IgG de conejo o de ratón conjugado con peroxidasa, durante 1 hora (ab191866 y ab193651, respectivamente, Abcam). La quimioluminiscencia fue visualizada usando el kit comercial de Amersham 71 Materiales y Métodos ECL Western Blotting Analysis System (Amersham Biosciences), y utilizando el documentador de geles Image Quant (GE). Las densitometrías se cuantificaron mediante el software GelPro. 3.2.8 Análisis por citometría de flujo La expresión de los receptores de HTs en las líneas celulares de LCT fue analizada utilizando los anticuerpos primarios anti-TRαβ (sc-772, Santa Cruz Biotechnology), anti- integrina αvβ3 (ab78289, Abcam), y sus correspondientes anticuerpos anti-isotipo como control. Para evaluar el efecto proliferativo de las HTs utilizando como marcador el PCNA, se usó el anti-PCNA (sc- 25280, Santa Cruz Biotechnology) y su respectivo anti-isotipo control. La evaluación de la apoptosis se realizó mediante el marcado de las células con un anticuerpo antianexina conjugado a FITC y la marcación nuclear con Ioduro de Propidio. Para las muestras primarias de PTCL-NOS utilizamos los anticuerpos anti-integrina β3 y anti-integrina αv (ab7167 y ab16821, respectivamente, Abcam). En todos los casos, las muestras fueron fijadas en paraformaldehído al 1% y permeabilizadas en PBS 0,1% Tween, antes de incubarlas durante 1 hora a 4°C con los anticuerpos primarios. Luego las células fueron lavadas e incubadas con el anticuerpo secundario anti-IgG conjugado correspondiente anti-IgG de ratón-Cy5 o anti-IgG conejo-FITC (Abcam). En todos los casos se adquirieron al menos 10,000 eventos utilizando el citómetro de flujo BD Accuri TM C6 flow cytometer 3.2.9 Ensayos de migración La capacidad de inducción de respuesta quimiotáctica a medios condicionados fue evaluada utilizando un sistema de 2 dimensiones en una micro-cámara de Boyden modificada de 48 pocillos (#AP48 Neuropobe, Inc.) como se describió previamente (Richards et al, 1984). Brevemente, una alícuota de 28.4 µl de los medios condicionados de las células CUTLL1 tratadas durante 24 horas con T4-AG+T3-AG o Control (células incubadas sólo con agarosa) fue agregada en la parte inferior de la cámara. En algunos casos, se pre-incubó durante 1h a los 72 Materiales y Métodos medios condicionados con un anticuerpo anti-VEGF (10 µg/ml). En la parte superior de cada pocillo se agregaron 50 µl de una suspensión de 12.000 células endoteliales HMEC1 (Figura 3.3). Los dos compartimientos fueron separados utilizando un filtro de policarbonato con poros de 8 µm (Neuroprobe, Cabin John, MD). La cámara fue incubada por 4 horas a 37°C y 5% CO2. Se utilizó RPMI suplementado con 5% SFB como control positivo o RPMI solo como control negativo de la respuesta quimiotáctica. Luego del periodo de incubación, la membrana fue removida, fijada y teñida con el colorante Hoechst. El número de células migratorias fue contado con el software Cell profiler. Todos los experimentos fueron repetidos al menos 3 veces. Figura 3.3: Evaluación de la migración en cámara de Boyden modificada Células HMEC1 Medio condicionado Representación ilustrativa de un pocillo de la cámara de Boyden modificada. Las células endoteliales estimuladas por los factores quimioatractantes del medio condicionado proveniente de células LCT, migran a través de los poros de la membrana de policarbonato. 3.2.10 Ensayos de microscopía confocal Se analizó la localización celular del factor de transcripción NF-κB en las células CUTLL1. Para ello se incubó a las células con un anticuerpo primario anti-NF-κB (ab7970, Abcam). Antes de la incubación las células fueron fijadas en metanol frío por, al menos, 30 minutos, y luego permeabilizados con PBS- 73 Materiales y Métodos Tritón 0,1% durante 20 minutos. Luego se incubó a 4°C durante 16 horas con el anti-NF-κB. Al finalizar este paso, se lavaron las células y se incubaron durante 30 minutos con el anticuerpo secundario anti-conejo conjugado a FITC (ab6717, Abcam). Finalmente, para la marcación nuclear, se utilizó 0,02 mg/ml de Ioduro de Propidio (IP) durante 1 minuto. 3.3 SECUENCIACIÓN DE ARNm Y MICROARREGLOS DE EXPRESIÓN El ARN fue aislado de células CUTLL1 tratadas o no con T3+T4 durante 24 horas utilizando el reactivo TRI-reagent (Life Technologies). El ADN genómico fue eliminado y la concentración y pureza del ARN fue determinado en el equipo Nanodrop (Thermo Scientific). La integridad del mismo fue evaluada utilizando el Agilent 2100 Bioanalyzer (Agilent Technologies). Los valores de integridad fueron mayores a 8 en todas las muestras. Las bibliotecas de secuenciación fueron generadas con el oligo polyA+ ARN, usando el TruSeq RNA sample prep kit (Illumina). Brevemente, el ARNm fue aislado, utilizando 2 rondas de purificación con esferas magnéticas unidas al oligo Poly-T. El ARNm obtenido fue fragmentado y luego se realizó la síntesis de la primera cadena del cADN con primers aleatorios y transcripción reversa. La segunda cadena de cADN se sintetizó para remover el templado de ARN y generar cADN doble cadena. El cADN doble cadena generado fue reparado en sus extremos, convirtiéndolos de cohesivos a romos. Bases A fueron agregadas en los extremos 3´ de los fragmentos y múltiples adaptadores de indexación fueron agregados y ligados a los extremos del cADN doble cadena. Los fragmentos de ADN con las moléculas adaptadoras fueron amplificados por PCR utilizando 14 ciclos. Las bibliotecas fueron agrupadas, amplificadas y secuenciadas durante 50 ciclos utilizando el secuenciador Illumina HiSeq2000. Los resultados se obtuvieron en lecturas (del inglés reads). Estas fueron alineadas a la secuencia humana hg19 utilizando el software Tophat y la expresión diferencial de los genes fue evaluada utilizando el software Cuffdiff. Los microarreglos de expresión fueron reanalizados utilizando datos públicos de un trabajo anterior (GEO). Este estudio analizó la expresión de transcriptos 74 Materiales y Métodos por la técnica de microarreglos de ARN en muestras de pacientes de distintos subtipos de LCT (74 PTCL-NOS, 30 ALCL-ALK+, 24 ALCL-ALK-, y 41 AITL). 3.4 ANÁLISIS DE LOS MODELOS IN-VIVO DE LCT 3.4.1 Ensayos in vivo de células CUTLL1 y OCI-Ly12 Para los ensayos in-vivo, se utilizaron ratones NOD-SCID (NOD.CB17Prkdcscid/J) tanto hembras como machos, de 6-10 semanas, de las instalaciones para animales de la Fundación Instituto Leloir y de la empresa Taconic farms, NY. Todos los ensayos que involucraron animales fueron aprobados, tanto por el Comité para el uso y cuidado de animales de experimentación de la Fundación Instituto Leloir, como por el del Weill Cornell Medical College (WCMC). Un grupo de ratones fue inyectado subcutáneamente en el flanco derecho con 2 x 107 células CUTLL1 las cuales fueron previamente transfectadas con siRNA contra ITGAV, ITGB3 o contra una secuencia control no codificante (Control). Otro grupo de ratones fue inyectado subcutáneamente en el flanco izquierdo con 1 x 107 células OCI-Ly12. El volumen tumoral fue medido todos los días del periodo que duró el experimento y se calculó el área bajo la curva (ABC) del crecimiento tumoral. Cuando los tumores de células OCI-Ly12 llegaron a un tamaño palpable (aproximadamente 75-100 mm3), los ratones fueron divididos aleatoriamente en dos grupos y tratados intraperitonealmente; uno recibió el vehículo (PEG300 35%, Tween-80 5% y dextrosa 65% en 5% de agua) y el otro 125 mg/kg/día del inhibidor de la integrina αVβ3, cilengitide (MedKoo, Inc). Los ratones fueron pesados cada día del periodo de tratamiento. Todos los ratones fueron sacrificados bajo anestesia por dislocación cervical cuando al menos el 20% de los tumores alcanzaron un tamaño de 20 mm en todas sus dimensiones. Al final del ensayo, los tumores fueron recolectados, pesados y examinados macroscópicamente para evaluar signos de daño del tejido. 75 Materiales y Métodos 3.4.2 Ensayos in vivo con tumores derivados de pacientes ALCL Se utilizaron ratones humanizados (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) de 610 semanas provenientes de la empresa Taconic farms, USA. Todos los ensayos que involucraron animales fueron aprobados por el Comité para el uso y cuidado de animales de experimentación del Weill Cornell Medical College (WCMC). Se trasplantaron tumores derivados de 2 pacientes, uno ALCL-ALK positivo (n=16) y otro ALCL-ALK negativo (n=8), luego fueron divididos aleatoriamente en dos grupos de tratamiento. Los tratamientos se realizaron intraperitonealmente durante 10 días; un grupo fue tratado con vehículo (PEG300 35%, Tween-80 5% y dextrosa 65% en 5% de agua) y el otro con Cilengitide (125 mg/kg/día, MedKoo, Inc). Los ratones fueron pesados cada día del periodo de tratamiento. Todos los ratones fueron sacrificados bajo anestesia por dislocación cervical. Al final del ensayo, los tumores fueron recolectados, pesados y examinados macroscópicamente para evaluar signos de daño tisular. 3.4.3 Ensayos de Inmunohistoquímica Los tumores sólidos fueron extirpados y los tejidos tumorales se fijaron en paraformaldheído 4% durante 18 horas. Luego las muestras fueron incluídas en parafina y cortadas en secciones seriales de 4 µm de espesor utilizando un micrótomo. La recuperación antigénica se realizó en Buffer Citrato (10 mM, pH 6.0) a 95°C; y luego, se bloqueó la actividad de la peroxidada endógena con H2O2 al 3% en agua destilada. Después del bloqueo, se incubaron las muestras durante 18 horas a 4°C en una cámara húmeda con los anticuerpos primarios anti-integrina αV y anti-integrina β3 (1:25, ab16821 y ab7167, respectivamente, Abcam), anti-CD31 (1:30, ab28364, Abcam), anti-Caspasa 3 activa (1:30, ab2302, Abcam) y anti-VEGFA (1:30, Ab1m, cedido generosamente por el Dr. A. Baldi, IBYME-CONICET, Argentina). La inmunoreactividad fue detectada utilizando el sistema VECTASTAIN Elite ABC Kit (PK-6200) y fue visualizado con el DAB Substrate Kit (SK-4100, VECTOR). Para el control de la señal inespecífica, secciones seriales de las mismas muestras fueron tratadas bajo el 76 Materiales y Métodos mismo protocolo, pero reemplazando el anticuerpo primario por una IgG inespecífica de ratón o de conejo o con PBS. No se detectó señal en estos controles de marcación. 3.4.4 Evaluación de la apoptosis por la técnica de TUNEL La fragmentación del ADN ligada a la respuesta apoptótica fue detectada en los núcleos morfológicamente identificables y en los cuerpos apoptóticos presentes en los cortes de tumores fijados y embebidos en parafina utilizando el ensayo TUNEL (ApopTag, Chemicon) siguiendo las instrucciones del fabricante. Los portaobjetos con los tejidos fueron pre-tratados con tripsina al 0.5% por 15 minutos para aumentar la exposición del ADN. 3.4.5 Ensayo de unión de NF-κB activado a ADN La capacidad de unión al ADN de los miembros de la familia NF-κB fue determinada utilizando un ensayo basado en el método de ELISA (TransAM, Active Motif, Carlsbad, CA) siguiendo las instrucciones del fabricante (Figura 3.4). Brevemente, los tejidos de linfoma de los tumores de las células OCI-Ly12 tratados con el vehículo o con la droga Cilengitide, fueron disgregados y luego se extrajeron las fracciones nucleares de los mismos. Posteriormente se agregaron 10 µg de los lisados nucleares a pocillos pre-absorbidos con oligonucleótidos consenso para NF-κB (5’-GGGACTTTCC-3’). El ensayo fue realizado en ausencia o presencia de 20 pmol de oligonucleótidos competidores, los cuales contienen un sitio consenso de unión a NF-κB mutado o wild type. Extractos nucleares de células Raji (5 µg por well) fueron utilizados como control positivo para el ensayo. Luego de la incubación y lavado, se añadieron anticuerpos hechos en conejo anti-p65, -p50, -cRel, -p52 or –relB (1:1000, Active Motif) a sus correspondientes pocillos, seguido de un anticuerpo secundario anti-conejo conjugado a HRP (1:1000, Active Motif). Luego del agregado del sustrato HRP, se midió la absorbancia a 450 nm (Synergy4, Biotek, Winooski, VT). En este ensayo, las medidas de absorbancia son directamente proporcionales a la cantidad del factor de transcripción unido 77 Materiales y Métodos a ADN presente en la muestra. Los experimentos se realizaron por cuadruplicado y fueron expresados como el promedio de los valores de absorbancia ± error estándar. Figura 3.4: Ensayo de la unión de NF-κB activado a ADN Adición extracto nuclear Anticuerpo primario anti IgG conjugado a HRP solución de frenado de la reacción Extracto celular con factor de transcripción activo Placa cubierta con oligonucleótidos El factor de transcripción activado en los extractos celulares se une a los sitios de unión consenso en los wells con oligos inmovilizados. La incubación con los anticuerpos primarios y secundarios cuantifica específicamente la cantidad de factor de transcripción activo. Adaptado de http://www.activemotif.com/ 3.5 ANÁLISIS ESTADÍSTICO Para todos los experimentos se calcularon las medias, desviaciones estándares y errores estándares de los grupos. Las significancias estadísticas se evaluaron con el software GraphPad PRISM 4.0 Version para Windows (GraphPad Software Inc, La Jolla, California), utilizando la prueba estadística ANOVA de una vía seguido con contraste de Bonferroni o Tukey. Las diferencias entre las medias fueron consideradas significativas cuando p<0.05. Los resultados están expresados como la media ± error estándar (SEM). Los resultados de las correlaciones en las muestras de pacientes fueron analizadas utilizando el test de Pearson para poblaciones Gaussianas (dos colas), y el test de Spearman para las correlaciones no paramétricas. 78 4. RESULTADOS Resultados 4.1 LAS HORMONAS TIROIDEAS INDUCEN LA PROLIFERACIÓN DE LOS LINFOMAS DE CÉLULAS T A TRAVÉS DE SUS RECEPTORES NUCLEAR Y DE MEMBRANA 4.1.1 Evaluación de la expresión del receptor nuclear y del de membrana de las hormonas tiroideas Estudios recientes de nuestro laboratorio demostraron que las HTs inducen in vitro la activación de vías de señalización que llevan a la proliferación celular en la línea murina de linfoma T BW 5147, a través de la activación de su receptor nuclear, TR y del de membrana, mTR (Barreiro Arcos y col, 2011). Sin embargo, los programas genéticos que involucran estos efectos y en qué medida contribuyen ambos receptores a los mismos, no han sido estudiados hasta el momento. Para profundizar estos estudios y determinar la influencia de las HTs en la proliferación celular de LCT humanas, se caracterizó, en primer lugar, la expresión del TR y del posible mTR en un panel de 9 líneas celulares humanas de LCT, las cuales representan los distintos subtipos de esta enfermedad. Este panel incluye tanto linfomas T de origen inmaduro (Jurkat y CUTLL1), como de origen maduro (Hut78, Mac-2A, OCI-Ly12, OCI-Ly13.2, SU-DHL-1, Karpas299). Si bien el receptor de membrana para HTs no se ha descripto en células de linfoma, en otro tipos celulares, como fibroblastos (Bergh y col, 2005) y ciertos tipos de cáncer, como el de próstata y riñón (Kimbro y Simons, 2006), se demostró que la integrina αVβ3 es el mTR. Basados en este antecedente, se inició el estudio con la determinación de los niveles transcripcionales de los genes de la integrina αVβ3 (ITGAV e ITGB3) como posible mTR también en este tipo de células. Los resultados muestran que, en comparación con la expresión observada en células T periféricas, las líneas de LCT expresan niveles significativamente más elevados de THRA, ITGAV e ITGB3 (Figura 4.1.A). Los niveles proteicos de 80 Resultados esta integrina también fueron evaluados en algunas de las líneas de LCT analizadas (Figura 4.1.B). Figura 4.1: Niveles de expresión de los receptores para HTs en líneas celulares humanas de LCT 80 60 40 ** ** ** ** ** ** ** 12 ** ** ** 4 * * * * ITGAV ITGB3 THRA * * * 8 * * 50 H L1 29 9 SU D PA S M ac 2a KA R I-L Y1 3. 2 TR αβ 300 Cuentas 100 O C O C Integrina αVβ3 150 I-L Y1 2 M J 78 uT H U TL L1 Ju rk at C C C el -T -G 0 200 Cuentas ** ** ** ** 16 el -T -P Niveles mRNA relativo a B2M A 200 Isotipo CUTLL1 SUDHL1 OCI-Ly12 HuT 78 100 0 0 102.3 103 104.1 102.3 103.3 104.4 A: Niveles de expresión del ARNm de los genes ITGAV, ITGB3 y THRA en el panel completo de LCT en comparación a los linfocitos T periféricos (Cel-T-P). Se muestra el promedio de 3 experimentos independientes ± ES. B: Niveles proteicos de la integrina αVβ3 y del TRαβ evaluados por citometría de flujo, en las células CUTLL1, SUDHL1, OCI-Ly12 y HuT 78. Resultado representativo de 3 experimentos. *P<0.05; **P<0.01. 81 Resultados 4.1.2 Evaluación del efecto de las hormonas tiroideas sobre la proliferación y el ciclo celular de LCT. Para determinar las consecuencias funcionales de la activación hormonal de estos receptores para HTs, se evaluó la proliferación de la línea celular CUTLL1 luego de un estímulo de 24 horas con concentraciones fisiológicas de las hormonas T3 y T4 de forma libre y de las mismas unidas a agarosa (AG), T3-AG y T4-AG. Estas últimas se encuentran unidas covalentemente por sus grupos amino primarios a la mencionada matriz y por ende constituyen derivados impermeables a la membrana celular. De esta manera, se pueden evaluar de forma selectiva los efectos inducidos por la activación del mTR de manera independiente al efecto total (TR+mTR) inducido por las hormonas libres. Los resultados muestran que luego de 24 horas de estímulo, tanto las T3 y T4 libres como las unidas a agarosa, son capaces de inducir la proliferación de las células CUTLL1 de manera significativa en comparación con sus respectivos controles (Figura 4.2). Figura 4.2: Evaluación de los efectos de las HTs sobre la proliferación de las células CUTLL1 % Proliferación celular relativa al Control 200 160 ** * * T4 T3 120 ** * * 80 20 0 CT HT CT-AG T4-AG T3-AG HT-AG Evaluación de la proliferación por Cell Titer Blue de las células de origen inmaduro, CUTLL1, luego de 24 horas de tratamiento con concentraciones fisiológicas de las hormonas libres o acopladas a agarosa, T3 (1nM) y T4 (100nM), de manera individual o en combinación (HT). Los resultados representan el porcentaje de las células proliferativas con respecto al control y se muestra el promedio de 3 experimentos independientes ± ES. *P<0.05; **P<0.01. 82 Resultados Más aún, se observó una mayor inducción de la proliferación celular cuando las hormonas T3 y T4 fueron adicionadas en combinación, ya sea en su versión libre (HT) o acoplada a AG (TH-AG), en comparación con los efectos inducidos por cada una de las mismas por separado. Cabe mencionar, que en todos los casos las células se cultivaron en un medio RPMI con 10% de suero depletado de T3 y T4, de manera de poder analizar los efectos producidos solo por el agregado de las mismas. Luego se determinó si las HTs son capaces también de inducir la proliferación celular de los subtipos de LCT de origen maduro. Para ello, se decidió realizar los tratamientos con la combinación de las hormonas T3 y T4, ya que, como se mencionó anteriormente, en estas condiciones se observa un mayor efecto tanto de las HTs libres como unidas a AG (Figura 4.3). Figura 4.3: Evaluación del efecto de las HTs sobre la proliferación celular en el panel completo de líneas humanas de LCT % Proliferación celular relativa al control 180 160 * 140 HT * * * * * * * * * * HT-AG * * * * * * * 120 CT 100 80 20 L1 DH SU 29 9 2a Ka rp as M ac 2 y1 I-L O C -L CI 3. y1 2 J M O H uT 78 at rk Ju CU TL L1 0 Proliferación celular evaluada con el método Cell Titer Blue en el panel completo de líneas celulares humanas de LCT, luego de 24 horas de tratamiento con HT y HT-AG en las concentraciones antes especificadas. Los resultados representan el porcentaje de las células proliferativas con respecto al control (CT, línea discontinua roja) y se muestra el promedio de 3 experimentos independientes ± ES. *P<0.05. El tratamiento durante 24 horas con HT y HT-AG en el panel completo de líneas celulares de LCT mostró que las HTs son capaces de inducir, al igual que lo observado en las células CUTLL1, la proliferación celular de todo el 83 Resultados panel de LCT. Tanto las HTs libres como las HT-AG, aumentan entre un 3050% la proliferación, en comparación con sus respectivos controles. Para analizar en mayor profundidad el efecto proliferativo de las HTs sobre las células LCT, se decidió evaluar la síntesis de ADN por incorporación de [3H]TdR en las células CUTLL1, HuT 78 y OCI-Ly12, luego de 24 horas de estímulo con las HTs (Figura 4.4). En concordancia con los resultados mencionados anteriormente, se encontró que, tanto las HT libres como las HT-AG, aumentan entre un 80-120% la síntesis de ADN en las 3 líneas evaluadas (Figura 4.4, panel A). Figura 4.4: Efecto de las hormonas tiroideas sobre la síntesis de ADN de las células CUTLL1, HuT 78 y OCI-Ly12 Incorporación [3H]-TdR (dpm) A 3000 Control HT HT-AG ** 2000 ** ** * ** * 1000 0 CUTLL1 B CT TH HuT 78 OCI-Ly12 % Células apoptóticas CUTLL1 HuT 78 OCI-Ly12 5,2±0,8 4,6±0,6 3,4±1,1 4,2±0,6 3,4±0,9 3,1±0,7 A: Evaluación de la síntesis de ADN por incorporación de [3H]TdR en las líneas CUTLL1, HuT 78 y OCI-Ly12, luego de 24 horas de tratamiento con HT libres y HT-AG. B: Apoptosis celular evaluada por marcación de anexina-FITC por citometría de flujo en células LCT luego de 24 horas de tratamiento. Los resultados representan el promedio de 3 experimentos independientes ± ES. *P<0.05; **P<0.01 vs Control. 84 Resultados En estos experimentos, y de acuerdo a resultados previos de nuestro grupo (Barreiro Arcos y col, 2006), se realizó la sincronización celular por deprivación de SFB. En estas condiciones se puede inducir apotosis, por lo que podría ser que las HTs estuvieran impidiendo que este fenómeno tenga lugar. Se verificó, entonces, la apoptosis celular por marcación con Anexina-V en las células CUTLL1, HuT78 y OCI-Ly12 luego de 24 horas de tratamiento con HTs (Figura 4.4, panel B). No se observaron diferencias significativas entre ambos tratamientos en ninguna de las líneas celulares analizadas, lo que indicó que las HTs estarían regulando preferentemente la división celular. Con el objeto de profundizar el estudio en este aspecto, se evaluó el efecto de las HTs sobre la expresión de marcadores moleculares implicados en la progresión del ciclo celular, tales como las ciclinas. Para ello, se trataron células CUTLL1 con HTs por disitntos tiempos (entre 2 a 18 horas) tanto con HTs libres como con HT-AG (Figura 4.5, Panel A). Los resultados de la expresión de ciclinas, por RT-PCR en tiempo real, muestran un aumento inicial de los niveles de ARNm de las ciclinas D1, D2 y D3 (todas requeridas para la progresión de fase G0 del ciclo celular a G1) seguido por un aumento en la ciclina E2 (progresión de G1 a S, y fase S) y de la ciclina B1 (progresión de G1 a M). Es decir que el aumento de la proliferación celular de CUTLL1 está asociado a la inducción del ciclo celular dependiente de ciclinas. El efecto de las HTs sobre la expresión transcripcional de marcadores de progresión del ciclo celular también fue evaluado en células de linfomas T de origen maduro. Para ello, se realizó un tratamiento similar con HTs libres y HTAG en células de LCT cutáneas, HuT 78, y PTCL-NOS, OCI-Ly12; luego se evaluaron los niveles de expresión transcripcional de las ciclinas D1, E2 y B1 por RT-PCR en tiempo real (Figura 4.5, panel B). Los resultados muestran que tanto las HT libres como las HT-AG, inducen la expresión de estas ciclinas en las células HuT 78 y OCI-Ly12, en magnitudes similares a las observadas en las células CUTLL1. 85 Resultados Figura 4.5: Análisis de la progresión del ciclo celular por determinación de 5 4 HT vs CT ** * * 3 2 CT 1 0 2 6 6 18 h CCND1 OCI-Ly12 HuT78 ** ** ** 4 ** 2 CT 0 2 6 12 2 Niveles ARNm relativo a B2M Niveles ARNm relativo a B2M B 12 6 h 12 4 HT-AG vs CT 3 * * CT 1 0 2 6 12 18 CCNE2 3 OCI-Ly12 HuT78 ** ** 2 ** * CT 1 0 h 2 6 12 2 6 12 h CCNB1 3 OCI-Ly12 HuT78 2 ** * * HT HT-AG * CT 1 0 CCND1 CCND2 CCND3 CCNE2 CCNB1 2 Niveles ARNm relativo a B2M Niveles ARNm relativo a B2M A Niveles ARNm relativo a B2M los niveles de ARNm de ciclinas en células LCT tratadas con HTs 2 6 12 2 6 12 h Niveles de expresión de los genes de ciclinas analizados por RT-PCR en tiempo real, luego del tratamiento con HT libres y HT-AG (derecha e izquierda respectivamente) durante 2, 6, 12 y 18 horas en las células CUTLL1 (A). Similar tratamiento se realizó por 2, 6 y 12 horas en las LCT de origen maduro, HuT 78 y OCI-Ly12 (B). Los resultados representan los niveles relativos de ARNm con respecto al control (CT, línea discontinua roja). Se muestra el promedio de 3 experimentos independientes ± ES. *P<0.05; **P<0.01. 86 Resultados En concordancia con los resultados mencionados de los efectos de las HTs sobre el ciclo celular, se encontró que luego de 6 y 12 horas de estímulo con concentraciones fisiológicas de las mimas, disminuye la expresión del inhibidor de la quinasa dependiente de ciclina, p21; y del supresor tumoral, p53, en las células CUTLL1 y HuT 78 (Figura 4.6). Figura 4.6: Evaluación de los niveles transcripcionales de p21 y p53 en Niveles ARNm relativo a B2M 1.5 Niveles ARNm relativo a B2M células LCT tratadas con HTs 1.5 CUTLL1 p21 p53 1.0 CT * * 0.5 0.0 6 * * 12 6 12 HuT 78 p21 p53 CT 1.0 * * 0.5 0.0 h 6 * * 12 * * 6 HT HT-AG * * 12 h Niveles de expresión de los genes CDKN1A (p21) y TP53 (p53) analizados por RTPCR en tiempo real, luego del tratamiento con HT libres y HT-AG durante 6 y 12 horas en las células CUTLL1 y las células HuT 78 (Panel de arriba y abajo, respectivamente). Los resultados representan los niveles relativos de ARNm con respecto al control sin tratar (CT, línea discontinua roja). Se muestra el promedio de 3 experimentos independientes ± ES. *P<0.05. También se observó que los efectos mediados por las HTs sobre la inducción de la proliferación y progresión del ciclo celular en LCT, fue acompañada por un 87 Resultados aumento en los niveles de expresión del antígeno nuclear de proliferación (PCNA). Las células CUTLL1 fueron tratadas durante 12 y 24 horas con HT libres y HT-AG; luego se analizaron los niveles de expresión transcripcional y proteicos de PCNA por las técnicas de RT-PCR en tiempo real y citometría de flujo o western blot, respectivamente. Los resultados muestran una inducción de los niveles transcripcionales y proteicos del marcador evaluado (Figura 4.6, Panel A y B, respectivamente), luego de estímulo con HTs y HT-AG. Como señal intracelular de activación linfocitaria se evaluó la fosforilación de ERK1/2. Se observó que luego de 15 minutos de estímulo con HTs se ven aumentados los niveles de fosforilación del mismo (Figura 4.6, Panel C). Figura 4.6: Inducción por hormonas tiroideas de los niveles de PCNA en células LCT A ** ** 1 CT TH Isotipo Control TH TH-AG 150 2 0 CUTLL1 200 Cuentas Niveles ARNm de PCNA relativo a B2M 3 B CUTLL1 4 TH-AG 100 50 0 103.1 104 105.1 C CT TH TH-AG CT PCNA p-ERK (p42/44) ACTINA ERK total T3+T4 A: Niveles transcripcionales de PCNA luego de 12 horas de estímulo con HTs. Los resultados representan los niveles relativos de ARNm con respecto al control (CT, línea discontinua roja). Se muestra el promedio ± ES de 3 experimentos independientes. B: Niveles proteicos de PCNA evaluados por citometría de flujo luego de 24 horas de estímulo con HTs. C: Evaluación por Western Blot de los niveles proteicos de PCNA luego de 24 horas de estímulo y de la fosforilación de ERK1/2 luego de 15 minutos de tratamiento con HTs en células CUTLL1. Para los niveles proteicos se muestran resultados representativos de n=3 ensayos. **P<0.01. 88 Resultados El efecto de las HTs sobre la expresión de PCNA también se evaluó en líneas LCT de origen maduro. La figura 4.7 muestra que tanto las HT libres, como las HT-AG, son capaces de inducir la expresión de los niveles transcripcionales (Panel A) y proteicos (Panel B) de PCNA luego del tratamiento con HTs durante 12 y 24 horas, respectivamente, en las líneas LCT maduras, HuT 78 y OCILy12. Figura 4.7: Inducción por hormonas tiroideas de los niveles de PCNA en células LCT humanas de origen maduro Niveles ARNm relativo a B2M A B 3 2 HT HT-AG ** 200 100 * 1 0 CT HuT 78 OCI-Ly12 OCI-Ly12 400 Cuentas 300 ** * HuT 78 400 Cuentas PCNA 4 Control HT HT-AG 300 200 100 0 0 10 2.5 10 3 PCNA 10 4.5 10 2.8 10 4 10 4.7 PCNA Niveles del ARNm (A) y proteicos (B) de PCNA en las células HuT 78 y OCI-Ly12, a las 12 y 24 horas, respectivamente, de estímulo con HT libres y HT-AG. Los resultados representan los niveles relativos de ARNm con respecto al control (CT, línea discontinua roja). Se muestra el promedio ± ES de 3 experimentos independientes. Para los niveles proteicos de PCNA se muestra un gráfico de citometría de flujo representativo de 4 ensayos. *P<0.05; **P<0.01. 89 Resultados Teniendo en cuenta los resultados mencionados en esta sección, se puede inferir que las HTs, a través de la acción tanto sobre el TR como sobre el receptor de membrana, constituyen una señal proliferativa para las células T malignas, que podría ser necesaria para su crecimiento. Esto sugiere que los mecanismos genéticos activados por las HTs podrían contribuir al fenotipo maligno de los subtipos de linfomas de células T estudiados. 90 Resultados 4.2 IDENTIFICACIÓN DEL RECEPTOR DE MEMBRANA PARA HTs EN LINFOMAS DE CÉLULAS T HUMANOS 4.2.1 La activación de la integrina αVβ 3 es la responsable de los efectos proliferativos mediados por HTs en linfomas de células T Los resultados mencionados en la sección anterior sugieren que concentraciones fisiológicas de HTs inducen la proliferación de las células T malignas actuando tanto a través de su receptor nuclear, como del mTR. Por ende, el bloqueo de la acción de las HTs sobre las células de linfoma T resultaría de utilidad, como posible estrategia terapéutica para el tratamiento de los LCT. Sin embargo, la inhibición de la producción hormonal o el bloqueo del receptor canónico nuclear para HTs, no parecería ser la mejor estrategia terapéutica, ya que se generarían consecuencias negativas fisiológicas muy importantes. Por lo cual, el receptor de membrana para HTs, parecería ser un blanco terapéutico más atractivo para ser empleado en la terapia anti-linfoma. Se ha descripto a la integrina αVβ3 como mTR de las HTs en distintos tipos de células normales, como en fibroblastos y células vasculares (Bergh y col, 2005; Davis, 2004) y en varios tipos de líneas celulares tumorales, como cáncer de próstata y carcinomas renales, entre otros (Cohen y col, 2011; Kimbro y Simons, 2006). Sin embargo, hasta el momento no se ha descripto si esta integrina es el mTR y está involucrada en los efectos no genómicos de las HTs en células de linfoma T. El sitio de unión de las HTs en la integrina αVβ3 está muy próximo al sitio de reconocimiento para el péptido RGD (Arg-Gly-Asp) presente en los ligandos endógenos de este receptor (Bergh y col, 2005; Cheng et al, 2010). Por este motivo, se analizó si el péptido RGD podría actuar como competidor de las HTs y, de esta manera, bloquear el efecto proliferativo inducido por las mismas a través de su receptor de membrana en los LCT. Se evaluó la proliferación celular mediada por HTs luego de la pre-incubación con el péptido RGD en células CUTLL1. La Figura 4.8 muestra los resultados donde se observa que, 91 Resultados efectivamente, la pre-incubación con el péptido RGD inhibe el efecto inducido por las HT-AG en las células CUTLL1. Más aún, el péptido RGD también bloquea entre un 40 y 60 % el efecto proliferativo que inducen las HTs libres, resultado consistente con un bloqueo específico de la integrina αvβ3. Cabe señalar, que el péptido RGD por si sólo, no afecta de manera significativa la proliferación basal de las células CUTLL1 (Figura 4.8). Figura 4.8: Acción del péptido RGD sobre el efecto proliferativo de las HTs en células CUTLL1 % Proliferación celular relativa al Control 200 180 160 *** ** 140 # 120 # CT 100 80 20 0 D RG -A HT G HT AG GD +R HT D RG + HT Proliferación celular evaluada por Cell Titer Blue, luego de 24 horas de tratamiento con concentraciones fisiológicas de HT y HT-AG en células CUTLL1 pre-incubadas o no, 15 minutos antes del tratamiento hormonal con el péptido RGD (10 µM). Los resultados representan el porcentaje de células proliferativas con respecto al control (CT, línea discontinua roja) y se muestra el promedio de 4 experimentos independientes ± ES. ** P<0.01 *** P<0.001 vs control; # P<0.05 vs hormonas Con el objetivo de analizar en profundidad la utilización del bloqueo de la integrina αvβ3 como posible estrategia terapéutica en el tratamiento de LCT, se realizaron ensayos en células transfectadas de manera transiente con ARN de interferencia (siRNA, del inglés silencing RNA) contra la integrina αV (si-ITGAV) y/o la integrina β3 (si-ITGB3). Como control, se utilizó un siRNA contra una secuencia no codificante (si-CT). 92 Resultados En primer lugar, se evaluó como control, la efectividad de los siRNA, analizando la regulación negativa de los niveles transcripcionales de estas integrinas a distintos tiempos post-transfección. Los resultados obtenidos en las células CUTLL1 muestran que 24 y 48 horas post-transfección los si-ITGAV y si-ITGB3 regularon negativamente de manera efectiva los niveles transcripcionales y proteicos de la integrina correspondiente a cada siRNA (Figura 4.9). Figura 4.9: Regulación negativa de los niveles de ARNm y proteicos de la integrina αvβ3 en las células CUTLL1 B ITGAV 1.2 Integrina αVβ3 ITGB3 400 1.0 0.8 0.6 si-CT si-ITGAV si-ITGB3 siCT ** ** ** Cuentas Niveles ARNm relativo a B2M A ** 0.4 300 200 100 0.2 0 0.0 24 48 h 10 2.4 10 3 10 4.3 A: Niveles de expresión del ARNm de ITGAV e ITGB3 medidos por RT-PCR en tiempo real luego de 24 y 48 horas posteriores a la transfección por electroporación de células CUTLL1 con 20 µM de los si-RNA (si-ITGAV y si-ITGB3) vs una secuencia no codificante como control (si-CT). Los resultados son el promedio ± ES de 5 experimentos independientes. B: Niveles de expresión proteica de la integrina αVβ3 evaluados por citometría de flujo. El gráfico mostrado es representativo de 4 experimentos. **P<0.01. Como siguiente paso, se evaluó si la regulación negativa de la integrina αvβ3 afecta el crecimiento dependiente de HTs de las células CUTLL1 luego del tratamiento con las mismas por más de 120 horas (Figura 4.10). Los resultados muestran que luego de 96 y 120 horas de cultivo en medio completo con 10% de SFB, el crecimiento de las células que no expresan el receptor, disminuye. Este dato sugiere que el receptor heterodimérico es necesario para el normal crecimiento in vitro de las células CUTLL1. 93 Resultados Figura 4.10: Efecto de la regulación negativa de la integrina αvβ3 sobre el crecimiento de las células CUTLL1 n° de células (x 10 4) 24 * * 20 si-CT si-ITGAV si-ITGB3 16 12 8 0 24 48 72 96 120 144 h Crecimiento de las células CUTLL1 transfectadas con siRNA contra ITGAV (si-ITGAV), ITGB3 (si-ITGB3) o con una secuencia no codificante como control (si-CT), analizando el número de células por conteo microscópico con el colorante Trypan Blue. Los resultados mostrados son el promedio ± ES de 3 ensayos independientes. *P<0.05 Para determinar si la integrina αvβ3 está relacionada con los efectos proliferativos inducidos por las HTs sobre las células de LCT, analizamos la incorporación de [3H]-TdR sobre las células CUTLL1 transfectadas con siITGAV, si-ITGB3, si-ITGAV + si-ITGB3, o si-CT, luego de 24 horas de tratamiento con concentraciones fisiológicas de HTs libres y HT-AG. Los resultados muestran que, ninguna de las condiciones de transfección afecta de manera significativa la proliferación de las células CUTLL1; pero tanto el efecto proliferativo inducido por las HT libres como las HT-AG, se ve completamente inhibido en estas células (Figura 4.11). La inhibición producida por la combinación de ambos siRNA (si-ITGAV + si-ITGB3), no fue significativamente mayor a la inhibición producida por el si-ITGAV y el si-ITGB3 utilizados por separado. Este resultado sugiere que la integrina αvβ3 es requerida para los efectos proliferativos inducidos por HTs en células CUTTL1. 94 Resultados Figura 4.11: Evaluación del efecto de la regulación negativa de la integrina αvβ3 sobre la proliferación mediada por HTs en células CUTLL1 Control HT HT-AG 900 600 ** 300 ** ** ** ** ** 0 si C on s si i I trol IT TG G AV si AV I + TG si B3 IT G B3 si C o si ntro si IT l IT G G AV si AV I + TG si B3 IT G B3 si C o si ntro si IT l IT G G s AV i AV IT + G si B3 IT G B3 Incorporación [3H]-TdR (dpm) 1200 Proliferación celular evaluada por incorporación de [3H]TdR, en células CUTTL1 transfectadas con los siRNA si-ITGAV, si-ITGB3 o una secuencia no codificante (siCT), luego de 24 horas de tratamiento con HT y HT-AG. La regulación negativa de los genes ITGAV y/o ITGB3 inhibe el aumento de la proliferación inducido por HT libres y HT-AG. Los resultados mostrados son el promedio ± ES de n=3 ensayos independientes. **P<0.01. Luego se quiso determinar si la regulación negativa de la integrina αvβ3, afecta también la proliferación inducida por HTs en subtipos maduros de LCT. Para ello, se transfectaron las líneas celulares de LCT de origen maduro, HuT-78 y OCI-Ly12, con los siRNA contra los genes ITGAV e ITGB3 y se evaluó la expresión de los niveles transcripcionales de los mismos 24 y 48 horas posttransfección (Figura 4.12, panel A). Los resultados muestran una disminución de los niveles de ARNm de estos genes de forma similar a lo observado en las células CUTLL1. Como siguiente paso, se evaluó la proliferación luego del tratamiento durante 24 horas con concentraciones fisiológicas de HT y HT-AG. Los resultados muestran que, de manera similar a lo observado en las células inmaduras, la inducción de la proliferación mediada por HTs en las células HuT- 95 Resultados 78 y OCI-Ly12 se vio impedida en aquellas células que no expresan la integrina αvβ3 (Figura 4.12, panel B). Estos datos indican que el dímero αvβ3 es necesario para que la inducción de la proliferación mediada por HTs ocurra en los distintos subtipos de LCT. Figura 4.12: Efecto de la regulación negativa de la integrina αvβ3 sobre la si CT 1.0 0.8 0.6 ** 0.4 ** ** 0.2 0.0 24h B 48h HuT HuT 7878 180 TH-AG 140 120 * 100 * * * 80 20 1.0 si CT 0.8 0.6 CT ITGAV ** ** 0.4 ITGB3 ** ** 0.2 0.0 24h 48h OCI-Ly12 OCI-Ly12 180 TH 160 OCI-Ly12 OCI-Ly12 1.2 160 TH TH-AG 140 120 100 * * * * 80 20 B3 G IT si- G AV CT IT si- si- B3 G IT si- G AV CT IT si- si- si -C si -IT T G AV si - IT G B3 0 0 si -C si -IT T G AV si - IT G B3 % Proliferación celular relativa al Control ** Niveles ARNm relativo a B2M HuT 7878 HuT 1.2 % Proliferación celular relativa al Control A Niveles ARNm relativo a B2M proliferación mediada por HTs en células maduras LCT A: Niveles transcripcionales de ITGAV e ITGB3 medidos por RT-PCR en tiempo real luego de 24 y 48 horas post-transfección en las células HuT 78 y OCI-Ly12 con siITGAV, si-ITGB3, vs una secuencia no codificante como control (si-CT). Los resultados son el promedio ± ES de 3 experimentos independientes. B: Proliferación celular evaluada en las células HuT 78 y OCI-Ly12 transfectadas con los siRNA si-ITGAV, siITGB3 y si-CT, luego de 24 horas de tratamiento con HT y HT-AG. Los resultados representan el porcentaje de las células proliferativas con respecto al control (CT, línea discontinua roja) y se muestra el promedio de n=3 experimentos independientes ± ES. *P<0.05, **P<0.01. 96 CT Resultados 4.2.2 Evaluación del crecimiento regulado por HTs de células LCT en presencia del ligando biológico de la integrina αVβ 3 El contexto fisiológico en el que las células de linfoma T crecen y proliferan incluye un contexto biológico con distintos componentes de la matriz extracelular. Es por esto que se decidió evaluar la magnitud de la acción de las HTs mediada por la integrina αVβ3 sobre la proliferación de los LCT en presencia de la vitronectina (VN), el ligando biológico del receptor de membrana mencionado. Para ello se analizó, por incorporación de [3H]-TdR, la proliferación inducida por 24 horas de estímulo con HTs, por VN y por la combinación de ambos tratamientos, en las células CUTLL1, HuT 78 y OCILy12 (Figura 4.13) Figura 4.13: Evaluación del efecto del ligando vitronectina sobre la Incorporación [3H]-TdR (dpm) proliferación de células de LCT 8000 CUTLL1 HuT 78 ** 6000 ** ** OCI-Ly12 ** ** ** * 4000 2000 0 HT VN - + - + - - + + - + - + - - + + - + - + - - + + Proliferación celular evaluada por incorporación de [3H]TdR en las células CUTLL1, HuT 78 y OCI-Ly12, luego de 24 horas de tratamiento con HT libres en concentraciones fisiológicas y vitronectina (VN, 250 ng/ml). Los resultados representan el promedio ± ES de 3 ensayos independientes. *P<0.05 **P<0.01. 97 Resultados Los resultados muestran que la VN sola tiene una tendencia a incrementar la proliferación de las células de LCT, siendo significativo el aumento únicamente en las células HuT-78. Sin embargo este aumento fue menor que el observado con las HTs (Figura 4.13). Cuando se agregan juntas, VN y HTs, la VN no compitió con el efecto de las HTs, y para las células Hut-78 la combinación proporciona un mayor efecto proliferativo del observado con cada ligando solo. Para estudiar con mayor profundidad la proliferación mediada por HTs en un contexto biológico más real, se decidió evaluar el crecimiento de las células LCT en un modelo de cultivo en 3 dimensiones (3D). Para ello primero se desarrolló una matriz tridimensional que incluye al péptido RGD y a células estromales (ver Figura 3.2 en la sección de Materiales y Métodos). Recordemos que el péptido RGD forma parte de la estructura de la VN que se une al sitio de unión para su ligando de la integrina αVβ3. Una vez obtenida la matriz, se cultivaron en ella células OCI-Ly12 en presencia o ausencia de las HTs durante 48 horas. Los resultados muestran que existe un claro aumento en el número de células OCI-Ly12 cuando éstas son cultivadas con HTs en comparación con el control (Figura 4.14, panel A y B). También se estudió el efecto de un inhibidor farmacológico de la integrina αVβ3, el cilengitide, sobre el crecimiento de las células, mediado por HTs, en el cultivo 3D. El cilengitide es un pentapéptido cíclico con actividad anti-angiogénica que muestra actividad antagonista de la integrina αVβ3 en el orden subnanomolar y que se encuentra actualmente en estudio de fase clínica III en glioblastomas, y de fase II en otros tipos de tumores sólidos (Tabatabai y col, 2010; Nabors y col, 2009; Reardon y col, 2008; Friess y col, 2006). Como puede observarse, el cilengitide inhibió el crecimiento mediado por HTs de las células OCI-Ly12 (Figura 4.14, panel C) 98 Resultados Figura 4.14: Efecto de las HTs y de un inhididor de la integrina αVβ3 sobre el crecimiento in-vitro de células LCT en una matriz tridimensional Estroma RGD A C Estroma RGD + TH + cilengitide Estroma RGD + HT B Imágenes representativas de los distintos tratamientos realizados sobre un cultivo tridimensional de células OCI-Ly12. Se muestran imágenes del crecimiento control (A), tratado con HTs libres (B) y tratado con el inhibidor Cilengitide (4 uM, C). Aumento 100X, barra escala = 100 µm. 99 Resultados Estos resultados sustentan aún más la hipótesis de que la proliferación de células LCT mediada por HTs a través de la integrina αVβ3, podría contribuir al fenotipo maligno de las células de linfoma T y que esta vía proliferativa podría ser capitalizada para desarrollar una nueva estrategia terapéutica para este tipo de patologías. 100 Resultados 4.3 PROGRAMAS TRANSCRIPCIONALES REGULADOS POR LAS HTs A TRAVÉS DE SU RECEPTOR DE MEMBRANA 4.3.1. Las hormonas tiroideas regulan programas transcripcionales vinculados a proliferación y angiogénesis a través del mTR Como próximo objetivo se planteó analizar de manera exhaustiva los programas genéticos activados por las HTs en las células de LCT a través de su receptor de membrana (mTR), tratando de identificar aquellos involucrados en la proliferación de las células T malignas. Para ello, se analizaron los cambios transcripcionales inducidos por las HT-AG, luego de 24 horas de estímulo en células CUTLL1 utilizando una técnica de última generación como es la secuenciación de ARN (RNAseq). Los transcriptos fueron identificados utilizando el software edgeR (Robinson y col, 2010) y aquellos que mostraron un p-value <0.01, en comparación al control (agarosa sola), fueron considerados transcriptos expresados diferencialmente. Los resultados de este análisis muestran 118 y 5 genes regulados de manera positiva y negativa, respectivamente. Algunos de los transcriptos regulados diferencialmente se muestran en el mapa de calor de valores de expresión génica en el Panel A de la Figura 4.14. Utilizando herramientas informáticas que permiten analizar vías de señalización, como por ejemplo el software Ingenuity Pathway Analysis, se identificaron los programas genéticos mayormente regulados luego de 24 horas de estímulo con las HT-AG (Figura 4.15, Panel B). A través de la activación de la integrina αvβ3, las HTs inducen vías de señalización que involucran la expresión de genes relacionados con la “traducción proteica” (RPL13, RPL27A, RPL36), con la “cadena respiratoria oxidativa” (NUDFB1/2, NDUFA13, UQCR11), con la “angiogénesis” (VEGFB), con la proliferación/diferenciación de linfocitos y con la replicación del ADN (IL4, EDF1, DOK2). 101 Resultados Figura 4.15: Transcriptos regulados por las hormonas tiroideas a través de su receptor de membrana en células CUTLL1 TH-AG B T CT C A VEGFB RPL39L RPL38 LTB RPL36 AIF1 CCDC73 H2AFX EDF1 RPL13 NDUFB1 NME1 RPL37A NDUFB11 UQCR11 UQCRQ CD3D NDUFA13 NDUFB2 RPL27A FTH1 TCTEX1D2 IL4 FOXH1 DOK2 GPR97 DBP − 0 -3 log (FC) 3 2 A: Mapa de calor que representa los cambios significativos (p<0.01) de la expresión de los genes regulados por las HT-AG con respecto al control, luego de 24 horas de tratamiento en las células CUTLL1. Cada condición se realizó por triplicado en ensayos independientes B: Representación de las vías de señalización reguladas por las HT-AG respecto al control en las células CUTLL1 realizada con el programa informático Ingenuity pathway. En trabajos anteriores publicados por nuestro grupo, se identificó al factor de transcripción, NF-κB, como efector río abajo de la activación del mTR en linfomas T murinos (Barreiro Arcos y col, 2011). Los resultados que se obtuvieron utilizando herramientas bioinformáticas y por el análisis de los genes regulados por HT-AG, también sugieren una asociación con la molécula de señalización NF-κB en los LCT humanos (Figura 4.16). 102 Resultados Figura 4.16: Vías de señalización iniciadas por la acción de las HTs sobre su receptor de membrana en células LCT Vías y moléculas de señalización asociadas a los genes movilizados por las HT-AG (P<0.01) luego de 24 horas de tratamiento en las células CUTLL1. Gráfico realizado con el programa Ingenuity pathway. Luego de un análisis más exhaustivo, se encontró que los genes regulados por las HT-AG involucrados en angiogénesis, proliferación y replicación del ADN (DBP, VEGFB, IL4 y DOK2) presentan en sus regiones promotoras, sitios de unión a NF-κB1. Dada la implicancia de estos genes en la contribución al fenotipo maligno de los LCT, se decidió validar su regulación por HTs con la técnica RT-PCR en tiempo real. Para ello se evaluaron los niveles de los ARNm de los mismos, tanto en las células de origen inmaduro CUTLL1, como en las células LCT de origen maduro HuT 78 y OCI-Ly12 (Figura 4.17, Panel A y B, respectivamente). Los resultados de este ensayo muestran que la inducción de estos genes es regulada por las HT-AG en los subtipos de LCT tanto de origen maduro como inmaduro. 103 Resultados Figura 4.17: Validación de los transcriptos regulados diferencialmente por las HT-AG en distintos subtipos de LCT. Niveles ARNm relativo a B2M A CUTLL1 3.5 3.0 ** 2.5 ** 2.0 * * 1.5 1.0 CT 0.5 0.0 DBP Niveles ARNm relativo a B2M B 3 2 IL4 HuT 78 5 4 VEGFB DOK2 OCI-Ly12 * ** * * * * * * 1 CT 0 VEGFB IL4 DOK2 DBP VEGFB IL4 DOK2 DBP Niveles de expresión del ARNm de los genes DBP, VEGFB, IL4 y DOK2, evaluados por RT-PCR en tiempo real, en las células de origen inmaduro, CUTLL1 (A) y en las células de origen maduro HuT 78 y OCI-Ly12 (B), luego de 24 horas de estímulo con concentraciones fisiológicas de HT-AG. Los resultados representan los niveles relativos de ARNm con respecto al control (CT, línea discontinua roja). Se muestra el promedio ± ES de 3 experimentos independientes. *P<0.05 **P<0.01. 4.3.2 Las hormonas tiroideas regulan genes involucrados en proliferación y angiogénesis a través de su receptor de membrana. Para determinar si las HTs en su forma libre también pueden modular la expresión de los genes inducidos por las HT-AG, las células de LCT fueron tratadas con concentraciones fisiológicas de HTs libres durante distintos tiempos. Se observa que, luego de 2 horas de estímulo con las HTs, se induce 104 Resultados la expresión de los genes IL-4 y VEGFB en células CUTLL1. Para poder atribuir estos efectos a la acción de las HTs sobre su receptor de membrana, la integrina αvβ3, se analizó la expresión transcripcional de estos genes en células CUTLL1 que no expresan el receptor. Los resultados muestran que las HTs no son capaces de inducir la transcripción de IL-4 y VEGFB en las células CUTLL1 transfectadas con los ARN de interferencia si-ITGAV o si-ITGB3, en comparación con las células transfectadas con el si-CT (Figura 4.18). Figura 4.18: Efecto de la regulación negativa del mTR sobre la inducción VEGFB 3 IL4 ** ** 2 CT 1 AV si -IT G B3 G T si -IT si -C G si -IT si -C AV si -IT G B3 0 T Niveles ARNm relativo a B2M de la expresión de los genes IL4 y VEGFB mediada por HTs Expresión del ARNm de los genes VEGFB e IL-4, luego de 2 horas de tratamiento con concentraciones fisiológicas de HTs libres en células CUTLL1 transfectadas con los ARN de interferencia si-ITGAV o si-ITGB3 en comparación con el control (si-CT). Se muestra el promedio ± ES de 3 experimentos independientes. **P<0.01. Estos resultados indican que la regulación dependiente de HTs de estos genes, se debe a la activación del mTR. Como se mencionó anteriormente, VEGF está involucrado en la regulación de la angiogénesis y su expresión podría contribuir al fenotipo maligno de los LCT. De hecho, se ha demostrado una asociación entre la alta expresión de VEGF y la menor sobrevida y peor pronóstico de pacientes con LCT de los subtipos PTCL-NOS y AITL (Konstantinou y col, 2011; Konstantinou y col; 2009, 105 Resultados Jorgensen y col, 2009). El factor VEGFB es miembro de la familia de factores de crecimiento del endotelio vascular que incluye también a VEGFA, VEGFC, VEGFD y PLGF (Goel y col, 2013). Con el objetivo de evaluar con mayor profundidad la relación entre los genes de proliferación y angiogénesis inducidos por las HTs y el fenotipo maligno de los LCT, se analizó mediante microarreglos de ARN, la expresión de los miembros de la familia de VEGF en una serie de muestras de pacientes de LCT (74 PTCL-NOS, 30 ALCL-ALK+, 24 ALCL-ALK- y 41 AITL). Además del factor VEGFB, el único miembro que también se encuentra expresado en estas muestras es VEGFA (Figura 4.19). Figura 4.19: Niveles transcripcionales de VEGFB y VEGFA en muestras de pacientes con distintos subtipos de LCT VEGFB 15 Niveles ARNm relativo a GAPDH Niveles ARNm relativo a GAPDH 15 12 9 6 3 0 12 9 6 3 0 PTCL-NOS AITL ALK + ALK - VEGFA PTCL-NOS AITL ALK + ALK - Determinación de la abundancia de los transcriptos VEGFA (derecha) y VEGFB (izquierda) en el panel de muestras de pacientes de los distintos subtipos de LCT. Los resultados se obtuvieron por un analisis de microarreglos de ARN. Teniendo en cuenta los datos obtenidos de las muestras de pacientes de LCT, se decidió evaluar si la expresión de VEGFA se encuentra regulada por las HTs a través de la integrina αvβ3 en las células CUTLL1. De manera similar a IL-4 y VEGFB, la expresión de VEGFA es inducida luego de 2 horas de tratamiento con las HTs en comparación al control. Más aún, las células CUTLL1 transfectadas que no expresan el receptor, son incapaces de producir esta 106 Resultados inducción (Figura 4.20). De manera que este gen también forma parte de los programas genéticos regulados por HTs a través de la activación de mTR. Figura 4.20: Efecto de la regulación negativa del mTR sobre la inducción Niveles ARNm relativo a B2M de la expresión del gen VEGFA mediada por HTs VEGFA 2.5 2.0 Control HT ** 1.5 1.0 0.5 0.0 si-CT si-ITGAV si-ITGB3 Expresión del ARNm del gen de VEGFA, luego de 2 horas de tratamiento con HTs libres, en las células CUTLL1 transfectadas con los siRNA si-ITGAV o si-ITGB3 en comparación a la células control (transfectadas con si-CT). Se muestra el promedio ± ES de 3 experimentos independientes. **P<0.01 Con el fin de determinar si la expresión de VEGFA y VEGFB en el panel de muestras de pacientes de los distintos subtipos de LCT estudiado podría tener alguna asociación con la expresión de receptores para HTs en dichas muestras se decidió evaluar, en primer lugar, la expresión transcripcional de THRA, ITGAV e ITGB3, a través del análisis por microarreglos de ARN (Figura 4.21, Panel A). También se evaluaron los niveles proteicos de estos receptores en muestras de pacientes con PTCL-NOS del panel antes mencionado, mediante análisis por inmunohistoquímica en cortes de tejidos parafinados y por citometría de flujo en muestras frescas, respectivamente (Figura 4.21, Paneles B y C). 107 Resultados Figura 4.21: Expresión de receptores para HTs en muestras de pacientes con distintos subtipos de LCT Niveles ARNm relativo a GAPDH A 6 B THRA 5 TRα 4 3 2 Niveles ARNm relativo a GAPDH 0 12 PTCL-NOS AITL ALK + ALK - ITGAV 10 Integrina αV 1 8 4 2 8 7 6 5 4 3 2 1 0 PTCL-NOS AITL ALK + ALK - C ITGB3 Isotipo PTCL-1 PTCL-2 PTCL-3 PTCL-4 PTCL-5 PTCL-6 100 80 Cuentas Niveles ARNm relativo a GAPDH 0 Integrina β3 6 60 40 20 0 PTCL-NOS AITL ALK + ALK - Anti-integrina αVβ3 FITC A: Determinación de la abundancia de los transcriptos de THRA, ITGAV e ITGB3 en el panel de muestras de pacientes con los distintos subtipos de LCT. Los resultados se obtuvieron por analisis de microarreglos de ARN. B y C: Niveles proteicos del TRα y de las integrinas αV y β3 evaluada por inmunohistoquímica y citometría de flujo, respectivamente; en algunas muestras de pacientes PTCL-NOS. Aumento de las imágenes 630X (Barra de escala: 20 µm). 108 Resultados A fin de evaluar si existe una asociación entre los niveles del mTR y de VEGF en las muestras de pacientes con LCT, se analizó si había una correlación positiva entre los niveles transcripcionales de la integrina αVβ3 y de VEGF, en las muestras primarias de los distintos subtipos de LCT. Efectivamente, como puede observarse en las Figuras 4.22 y 4.23, existe una correlación positiva entre la expresión de ITGAV e ITGB3, y la expresión de VEGFA y VEGFB en la mayoría de los subtipos de LCT analizados. Estos resultados sugieren una posible asociación funcional entre el mTR y los factores angiogénicos VEGF. Figura 4.22: Correlación entre la abundancia de los transcriptos de ITGAV y los de VEGFA y VEGFB en muestras primarias de LCT PTCL-NOS PTCL-NOS 10 12 4 P<0.0001 pearson r = 0,5818 2 0 2 4 6 8 VEGFA 6 6 P<0.0001 pearson r = 0,7310 3 0 10 0 2 PTCL-NOS PTCL-NOS 8 0 10 4 P<0.001 pearson r = 0,3941 0 2 4 6 ITGAV 2 8 10 6 8 10 9 6 P<0.0001 pearson r = 0,7273 3 0 4 ALCL ALCL 12 VEGFB 6 2 0 ITGAV 9 VEGFB VEGFB 6 6 3 AITL AITL 12 8 0 4 9 ITGAV ITGAV 10 ALCL ALCL 15 9 VEGFA VEGFA 8 0 AITL AITL 12 0 2 4 6 ITGAV 8 10 6 3 0 P=0.0014 pearson r = 0,4251 0 2 4 6 8 ITGAV Correlación de los niveles de ARNm de VEGFA y VEGFB con los de la integrinas αV. Cada punto representa una muestra individual. Los resultados se analizaron utilizando el test de Pearson para poblaciones Gaussianas. 109 10 Resultados Figura 4.23: Correlación entre la abundancia de los transcriptos de ITGB3 con los de VEGFA y VEGFB en muestras primarias de LCT PTCL-NOS PTCL-NOS 10 4 2 0 2 4 6 8 ITGB3 ITGAB3 10 0 1 2 3 4 0 5 P<0.001 pearson r = 0,4201 6 3 2 0 0 2 ITGB3 ITGB3 4 6 1 2 3 ITGB3 ITGB3 4 5 ALCL ALCL 12 VEGFA VEGFB 6 0 15 9 4 6 3 AITL 12 8 VEGFB P<0.0001 pearson r= 0,7923 P<0.0001 pearson r = 0,6707 ITGB3 ITGB3 PTCL-NOS PTCL-NOS 10 0 6 3 P<0.001 pearson r = 0,3941 0 9 VEGFA 6 ALCL ALCL 12 9 VEGFA VEGFA 8 0 AITL AITL 12 P<0.0001 pearson r =0,5769 0 1 2 3 ITGB3 ITGB3 4 5 P<0.0001 pearson r = 0,5334 9 6 3 0 0 1 2 3 4 ITGB3 ITGB3 Correlación de los niveles transcripcionales de VEGFA y VEGFB con los de la integrina β3. Cada punto representa una muestra individual. Los resultados se analizaron utilizando el test de Pearson para poblaciones Gaussianas. 4.3.3 NF-κB está involucrado en la regulación transcripcional inducida por HTs sobre los genes de proliferación y angiogénesis Como ya se ha mencionado, nuestro grupo de trabajo demostró que el factor de transcripción NF-κB es activado por la acción de las HT-AG sobre el receptor de membrana en células de linfoma murinas (Barreiro Arcos y col, 2011). Más aún, los genes involucrados en procesos de proliferación y angiogénesis inducidos por las HTs a través de la integrina αVβ3, poseen en sus regiones promotoras, sitios de unión a este factor de transcripción. Es por eso que se decidió analizar si la acción de las HTs activa a NF-κB en células 110 5 Resultados humanas de LCT. Para ello, se evaluó la localización de NF-κB por microscopía confocal en células CUTLL1 luego del tratamiento con HTs libres durante 5, 15 y 30 minutos. Los resultados de estos ensayos muestran que luego de 15 minutos de tratamiento con HTs, el factor de transcripción NF-κB es traslocado del citosol al núcleo de las células LCT (Figura 4.24). Figura 4.24: Las HTs a través del mTR inducen la activación del factor de transcripción NF-κB en las células CUTLL1 HT Mezcla NF-κB IP Control Localización de NF-κB por microscopía confocal en las células CUTLL1 tratadas o no (Control) con concentraciones fisiológicas de HTs, durante 15 minutos. Se muestran imágenes representativas con un aumento X1000 (Barra de escala = 20 µm) Luego se evaluó la participación del factor de transcripción NF-κB en la regulación mediada por HTs de los genes IL-4, VEGFA y VEGFB, relacionados a procesos de proliferación y angiogénesis. Para ello, se analizó el efecto de las HTs sobre la expresión transcripcional de los mismos, en células CUTLL1, HuT 78 y OCI-Ly12, preincubadas o no con un inhibidor específico de NF-κB, 111 Resultados BAY-11-7082 (Figura 4.25). La regulación positiva de los genes IL-4, VEGFA y VEGFB, que se produce luego de 2 horas de estímulo con HTs, es significativamente disminuida por el inhibidor de NF-κB en las tres líneas celulares. Estos resultados, obtenidos en células humanas de linfoma T, demuestran que el factor NF-κB está involucrado en la inducción transcripcional mediada por HTs a través de su receptor de membrana. Figura 4.25: El factor de transcripción NF-κB participa en la inducción de ** 3 2 ** * # # 1 0 CT ## HT HT +BAY BAY 2 ** 3 2 ** * 1 0 # ## HT # HT +BAY CT BAY CUTLL1 HuT78 OCI-Ly12 * ** * # ## 1 0 VEGFA 4 IL4 4 3 Niveles ARNm relativo a B2M VEGFB 4 Niveles ARNm relativo a B2M Niveles ARNm relativo a B2M programas transcripcionales mediada por HTs en células LCT HT # HT +BAY CT BAY Efecto del inhibidor de NF-κB (1 µM), BAY 11-7082, sobre la regulación mediada por HTs de la expresión de los genes IL-4, VEGFB y VEGFA en las células CUTLL1, HuT 78 y OCI-Ly12. Se muestra el promedio ± ES de 3 experimentos independientes. * P<0.05, ** P<0.01 vs Control; # P<0.05, ## P<0.01 vs HT. 112 Resultados 4.4 LAS HTs INDUCEN LA EXPRESIÓN DE FACTORES ANGIOGÉNICOS FUNCIONALES 4.4.1 La activación de la integrina αvβ3 por HTs induce la expresión de VEGF en los distintos subtipos de linfomas de células T. Los factores de crecimiento endotelial vascular, VEGFA y VEGFB, pueden contribuir e influir sobre la proliferación de los LCT a través de dos mecanismos; el primero involucra un efecto autócrino a través de la expresión de los receptores de VEGF en las propias células de linfomas (Zhao y col, 2004; Piccaluga y col, 2007); el segundo puede darse por la inducción y mantenimiento de la angiogénesis a través de efectos parácrinos de estas moléculas sobre el microambiente celular (Goel y col, 2013). Esto sugiere que el incremento de VEGFA y VEGFB dependiente de HTs podría llegar a promover el crecimiento de los LCT a través de mecanismos autócrinos, induciendo la proliferación de las células malignas, o induciendo el crecimiento de los vasos sanguíneos que se encuentran en el microambiente tumoral. Para estudiar con mayor profundidad el efecto de las HTs sobre la expresión de estos factores angiogénicos, se determinó si la inducción de los mismos es un mecanismo generalizado que ocurre en los distintos subtipos de LCT, ya sean de origen inmaduros o maduros. Para ello, se trató durante 24 horas a todo el panel de líneas celulares de LCT con concentraciones fisiológicas de HT-AG y luego se midieron los niveles transcripcionales de VEGFB y VEGFA por RTPCR en tiempo real (Figura 4.26). De manera similar al efecto que se observa en las células CUTLL1, las HT-AG son capaces de inducir los niveles del ARNm de VEGFB en las células Jurkat, HuT-78, OCI-Ly12 y Karpas299, como también, los niveles de ARNm de VEGFA en las células Jurkat, HuT78, MJ y OCI-Ly12 113 Resultados Figura 4.26: Regulación de la expresión transcripcional mediada por HTs de VEGFB y VEGFA en distintos subtipos de LCT Expresión del ARNm de los genes VEGFA y VEGFB, luego de 24 horas de tratamiento con HT-AG en el panel completo de líneas celulares de LCT. Los resultados representan los niveles de ARNm en las células tratadas con HT-AG con respecto al control (CT, línea discontinua roja) y se muestra el promedio de 3 experimentos independientes ± ES. *P<0.05, **P<0.01. Para confirmar que los efectos sobre la inducción de VEGF son mediados por las HTs a través de la activación de la integrina αvβ3, células CUTLL1, HuT-78 y OCI-Ly12 fueron transfectadas con ARN de interferencia contra la integrina αVβ3 (si-ITGAV o si-ITGB3) o con una secuencia no codificante como control (si-CT), para luego tratarlas con concentraciones fisiológicas de HT-AG durante 24 horas. Los resultados muestran que la regulación negativa tanto de la integrina αV como de la β3, inhibe la inducción mediada por las HT-AG (Figura 4.27, Panel A). Estos datos validan nuestras observaciones anteriores, donde 114 Resultados se sugiere que en la mayoría de los subtipos de LCT la expresión de los ligandos VEGF depende de la actividad de las HTs sobre su receptor de membrana, la integrina αVβ3. Más aún, la inducción de la expresión de VEGFA y VEGFB no se observa cuando se trata a las células de LCT con el ligando biológico de la integrina, la vitronectina (VN) (Figura 4.27, Panel B). Con lo cual se puede inferir que, si bien la VN podría contribuir a la proliferación de las células, lo haría por programas genéticos diferentes. Figura 4.27: Efecto de la regulación negativa de la integrina αvβ3 y del ligando vitronectina sobre la expresión inducida por HTs de VEGF en células LCT A HuT 78 OCI-Ly12 # # # 2 ** 1 ** ** ** ** ** 0 CT Niveles ARNm relativo a B2M CUTLL1 3 CUTLL1 * HuT78 CUTLL1 # OCI-Ly12 * 1 CT HT VN HT VN HuT 78 # 2 # ** ** 1 OCI-Ly12 ** ** ** ** CT 0 VEGFA * 2 0 3 VEGFB VEGFB 4 HT VN Niveles ARNm relativo a B2M Niveles ARNm relativo a B2M B VEGFA VEGFA s si i-CT - IT G si- AV IT G B3 sisi CT - IT G si- AV IT G B3 sisi CT - IT G si AV - IT G B3 3 si si -CT -IT G si AV -IT G B3 si si -CT -IT G si AV -IT G B3 si si -CT -IT G si AV -IT G B3 Niveles ARNm relativo a B2M VEGFB VEGFB 3 CUTLL1 HuT78 * 2 * OCI-Ly12 * CT 1 0 HT VN HT VN HT VN A: Niveles de ARNm de VEGFA y VEGFB (derecha e izquierda, respectivamente) luego de 24 horas de tratamiento con HT-AG, en la células CUTLL1, HuT 78 y OCILy12 transfectadas con los siRNA; si-ITGAV, si-ITGB3 o si-CT. B: Niveles de ARNm de VEGFA y VEGFB (derecha e izquierda, respectivamente) luego de 24 horas de tratamiento con HT-AG y con el ligando VN (250 ng/ml) en líneas de LCT Se muestra el promedio de 3 experimentos independientes ± ES. **P<0.05 vs CT, **P<0.01 vs siCT 115 Resultados 4.4.2 La expresión de VEGF mediada por HTs a través de la integrina αvβ3 induce la migración de células endoteliales Con el objetivo de estudiar y caracterizar si existen consecuencias parácrinas funcionales de la inducción de la expresión de VEGF mediada por HTs a través de la integrina αvβ3, se evaluó la capacidad de inducción de respuesta quimiotáctica de medios condicionados provenientes de células LCT tratadas o no con hormonas. Utilizando un sistema de 2 dimensiones en una microcámara de quimiotaxis, se determinó la migración de células endoteliales HMEC1 en presencia de dichos medios condicionados. Para ello se compararon los resultados obtenidos con medios provenientes de células CUTLL1 tratadas con HT-AG durante 24 horas, con los correspondientes a medios similares generados en presencia de agarosa sola sin hormona (medio control). La cuantificación de los 3 ensayos realizados demostró que el número de células HMEC1 que migran es de 481 ± 118 con el medio proveniente de las CUTLL1 tratadas con HT-AG vs. 206 ± 82 con el medio control (P<0.001) (Figura 4.28). Estos resultados muestran una mayor migración de células endoteliales HMEC1 al ser cultivadas con un medio condicionado proveniente de células CUTLL1 tratadas con las HT-AG. Figura 4.28 Efecto de medios condicionados de células LCT tratadas con HT-AG sobre la migración de las células endoteliales HMEC1 Medio condicionado HT-AG Células HMEC1 Medio condicionado control 206 ± 82 481 ± 118 Migración de las células HMEC1 en presencia de medios condionados de células CUTLL1 tratadas o no con HT-AG. Fotos representativas de las células que migraron a través de la membrana de la microcámara (aumento 100x; barra escala= 60 µm). 116 Resultados El aumento del número de células HMEC1 migrantes también fue confirmado en las células LCT de origen maduro, HuT 78 y OCI-Ly12. Más aún, cuando los medios condicionados de células LCT tratadas con HT-AG durante 24 horas son luego pre-incubados con el anticuerpo anti-VEGF, Bevacizumab, la inducción de la migración de las células endoteliales se ve inhibida (Figura 4.29, Panel A). Estos resultados sugieren que la inducción de la expresión de los genes de VEGF mediada por HTs, lleva a la producción de VEGF funcional, presente en los medios condicionados. Más aún, la inducción de la migración de las células endoteliales se vio inhibida cuando las células LCT tratadas con HT-AG durante 24 horas, no expresan la integrina αVβ3 por su regulación negativa mediante siRNA contra ITGAV o ITGB3 (Figura 4.29, Panel B). Figura 4.29 Cuantificación de la migración de células endoteliales tratadas con medios condicionados de células LCT CUTLL1 600 HuT 78 OCI-Ly12 * 400 ** # # 200 * # 0 HT-AG - + - + - + - + - + - + Anti-VEGF - - + + - - + + - - + + Migración HMEC1 (n° células) B Migración HMEC1 (n° células) A 800 600 si-CT si-ITGAV si-ITGB3 ** 400 # # 200 0 CT HT-AG CT HT-AG CT HT-AG Cuantificación de la migración de las células HMEC1 luego de 4 horas de cultivo con: A: medios condicionados de distintos subtipos de LCT, control o HT-AG, preincubados o no con Becavizumab (10 µg/ml). B: medios condicionados de las células CUTLL1 transfectadas con siRNA contra ITGAV oITGB3 y tratadas o no con HT-AG. # P<0.05 vs si-CT, * P<0.05, ** P<0.01 vs Control. 4.4.3 La expresión de VEGF mediada por HTs cumple un rol autócrino sobre los LCT Se analizó el posible rol autócrino de la inducción de VEGF mediada por las HTs. Para ello, se evaluó el efecto del anticuerpo bloqueante para VEGF, 117 Resultados Bevacizumab, y de un inhibidor farmacológico del receptor de VEGF, Axitinib, sobre la inducción de la proliferación inducida por HTs en las células de linfomas T: CUTLL1, HuT 78 y OCI-Ly12. Tanto el Bevacizumab (anti-VEGF) como el Axitinib (inhibidor de la tirosina quinasa del VEGFR) disminuyen el efecto proliferativo de las HTs en todos los subtipos de LCT analizados (Figura 4.30), demostrando la contribución del VEGF y del incremento de su expresión inducido por HTs sobre la proliferación de estas células. Figura 4.30: Efecto de la inhibición del VEGF sobre la proliferación Incorporación [ 3 H]-TdR (dpm) regulada por HTs en LCT HuT 78 CUTLL1 8000 OCI-Ly12 * 6000 * * 4000 # # # # # # 2000 0 - - + + + - - - + + + - - Anti-VEGF - - + - + - - - + - + - - - + - + - Inhib VEGFR - - - + - + - - - + - + - - - + - + TH - + + + - Proliferación evaluada por incorporación de [3H]TdR en las células CUTLL1, HuT 78 y OCI-Ly12 preincubadas o no por una hora con Bevacizumab (anti-VEGF, 10 µg/ml) o Axitinib (inhib VEGFR, 3 µM) y luego tratadas por 24 horas con HTs. Los resultados representan el promedio ± ES de 3 ensayos independientes *P<0.05 vs CT; #P<0.05 vs HTs. Adicionalmente, se analizó el efecto de VEGF recombinante (VEGFr) sobre la proliferación de distintas líneas LCT. La adición de VEGFr fue capaz de inducir la proliferación de las células de los distintos subtipos de LCT de manera significativa en comparación al control (Figura 4.31). 118 Resultados Figura 4.31: Efecto de VEGF recombinante sobre la proliferación regulada Incorporación [3 H]-TdR (dpm) por HTs en LCT 8000 CUTLL1 HuT 78 OCI-Ly12 6000 * * * 4000 2000 0 VEGFr - + - + - + Proliferación evaluada por incorporación de [3H]TdR en las células CUTLL1, HuT 78 y OCILy12 tratadas por 24 horas con VEGF recombinante (VEGFr, 10ug/ml). Los resultados representan el promedio ± ES de 3 ensayos independientes. *P<0.05. Los resultados mencionados en esta sección sugieren que las HTs, a través de su receptor de membrana, la integrina αVβ3, inducen la producción del ligando VEGF funcionalmente activo. Los efectos funcionales de este factor de crecimiento, parecerían darse tanto por mecanismos autócrinos, induciendo la proliferación de las células de linfoma T, como parácrinos a través de la inducción de la migración de células endoteliales. 119 Resultados 4.5 EVALUACIÓN DE LA INHIBICIÓN TRANSCRIPCIONAL O DEL BLOQUEO FARMACOLÓGICO DE LA INTEGRINA αVβ3 SOBRE EL CRECIMIENTO IN VIVO DE LOS LCT 4.5.1 La regulación negativa de la integrina αvβ3 disminuye el crecimiento tumoral in vivo de las células CUTLL1. En las secciones anteriores se describió como, la acción de las HTs sobre su receptor de membrana, induce in vitro la proliferación de los distintos subtipos de LCT, a través de la activación de programas transcripcionales vinculados con la proliferación y la angiogénesis. Las vías de señalización activadas por HTs podrían utilizarse como blanco terapéutico para el tratamiento de esta patología. Para estudiar con mayor profundidad esta posible estrategia terapéutica se propuso como siguiente objetivo la evaluación del efecto que la inhibición de la integrina αvβ3 podría tener sobre el crecimiento in vivo de células humanas de linfoma T en animales de experimentación. Para ello, se desarrolló un modelo de xenotrasplante utilizando células CUTLL1 en ratones inmunodeficientes NOD-SCID (NOD.CB17-Prkdcscid/J). Se 7 inyectaron subcutáneamente 2 x 10 células CUTLL1 en cada ratón y se midió el volumen tumoral a lo largo del tiempo. Para evaluar el efecto de la inhibición de la integrina αvβ3 se transfectaron previamente las células CUTLL1 con los siRNA contra ITGAV (si-ITGAV), ITGB3 (si-ITGB3) o con una secuencia no codificante (si-CT), como control. Los resultados muestran que los tumores desarrollados a partir de las células transfectadas con los siRNA si-ITGAV o siITGB3, fueron significativamente menores que aquellos desarrollados con las CUTLL1 si-CT (Figura 4.32, Panel A). Como control, se analizaron los niveles proteicos de dichas integrinas por inmunohistoquímica en secciones de los tumores. Se observa claramente que, los tumores provenientes de células CUTLL1 transfectadas con si-ITGAV o si-ITGB3, expresan niveles proteicos mucho más bajos de la integrina correspondiente en comparación con los tumores control (Figura 4.32, Panel B). 120 Resultados Figura 4.32: Efecto de la regulación negativa de las integrinas αV y β3 sobre el crecimiento in vivo de los tumores de células CUTLL1 A Volumen tumoral día 4 a día 15 (ABC) 9000 P<0.05 P<0.01 6000 3000 si-CT si-CT si-ITGAV si-CT si-ITGB3 Integrina β3 Integrina αV B si-ITGAV si-ITGB3 A: Las células CUTLL1 fueron transfectadas con los siRNA, si-ITGAV, si-ITGB3 o siCT, y luego inyectadas subcutáneamente (2 x 107 cel) en ratones NOD.CB17Prkdcscid/J. El gráfico representa el área bajo la curva (ABC) del volumen tumoral. B: Expresión proteica de las integrinas αV y β 3 evaluada por inmunohistoquímica en secciones de los tumores obtenidas en A. Aumento de las fotografías 630X, barra de escala= 20 µm. También se analizó, por inmunohistoquímica, la expresión de VEGFA, CD31 y caspasa 3 activa en secciones de tejidos parafinados de dichos tumores (Figura 4.33). Aquellos tumores provenientes de células CUTLL1 transfectadas tanto con si-ITGAV como con si-ITGB3 mostraron una marcada disminución de 121 Resultados los niveles proteicos de VEGFA y un aumento en la expresión de caspasa 3 activa en comparación con los tumores controles. El aumento de esta última podría sugerir que la disminución en el volumen tumoral está relacionada con el incremento de la apoptosis celular. Figura 4.33: Efecto de la regulación negativa de las integrinas αV y β3 sobre la expresión de VEGFA y la apoptosis en los tumores CUTLL1. si-ITGAV si-ITGB3 Caspasa 3 activa VEGFA si-CT Niveles de VEGFA y de Caspasa 3 activa evaluados por inmunohistoquimica en secciones de tumores CUTLL1. Aumento de las fotografías 630X, barra de escala= 20 µm. Adicionalmente, se evaluó la vascularización intratumoral mediante la marcación con un anticuerpo contra el marcador endotelial CD31 y luego se cuantificó el área de los vasos visualizados por microscopía (Figura 4.34). En las imágenes obtenidas se observa que los tumores CUTLL1 si-ITGAV y siITGB3, desarrollan una vascularización anatómicamente defectuosa. También se encontró una disminución en el área de los vasos sanguíneos con respecto a los tumores si-CT (área en mm2: si-ITGAV= 0.40x10-3 ± 0.12x10-3, si-ITGB3= 0.34x10-3 ± 0.08 x10-3 vs. si-CT= 1.07x10-3 ± 0.25x10-3, p<0.01). Todos estos datos sugieren que podría existir una disminución en el potencial angiogénico en los tumores desarrollados a partir de las células CUTLL1 que no expresan la integrina αVβ3. 122 Resultados Figura 4.34: Efecto de la regulación negativa de las integrinas αV y β3 sobre la vascularización de los tumores CUTLL1. Área vasos sanguíneos (mm 2) 5.0×10 -3 3.0×10 -3 2.0×10 -3 ** ** 1.0×10 -3 0 si-CT si-ITGAV si-ITGB3 si-CT si-ITGAV si-ITGB3 CD31 Niveles de CD31 evaluados por inmunohistoquimica en secciones de tejidos de tumores CUTLL1 (izquierda). Aumento de las fotografías 630X, barra de escala=20 µm. Cuantificación del área de los vasos sanguíneos en los distintos grupos de tumores. **P<0.001 vs si-CT. 4.5.2 La inhibición farmacológica de la integrina αvβ3 disminuye el crecimiento in vivo de las células OCI-Ly12 Como siguiente objetivo se propuso determinar si la inhibición de la vía regulada por las HTs a través de su receptor de membrana, puede ser capitalizada terapéuticamente para el tratamiento de pacientes con linfoma de células T. Para ello, se evaluó el efecto del inhibidor farmacológico, cilengitide, sobre el crecimiento in vivo de células de LCT. Como se mencionó anteriormente, el cilengitide es un pentapéptido cíclico anti-angiogénico que muestra una actividad antagonista de la integrina αVβ3 en el orden 123 Resultados subnanomolar. Además se encuentra en estudios de fase clínica III en glioblastomas y en fase II en otros tipos de tumores sólidos. La expresión de VEGF se ha descripto como un marcador predictivo de sobrevida; su alta expresión está asociada a una mala respuesta a drogas quimoterapéuticas convencionales en pacientes con PTCL-NOS (Jorgensen y col, 2007, 2009). Por esto, se decidió desarrollar un modelo de xenotrasplante de células PTCL-NOS en ratones inmunodeficientes NOD-SCID utilizando células OCI-Ly12, para luego evaluar el efecto del cilengitide sobre su crecimiento in vivo. Se inyectaron subcutáneamente 1 x 107 células OCI-Ly12 en el flanco izquierdo de cada ratón. Una vez que los tumores llegaron a un volumen palpable de aproximadamente 75-100 mm3, se dividió de manera aleatoria a los ratones en dos grupos de tratamiento, el cuál se administró diariamente por 20 días. Uno de los grupos (n=9) recibió inyecciones intraperitoneales diarias de cilengitide (110 mg/kg) y el otro grupo (n=6) recibió el vehículo de la droga (PEG300 35%, Tween-80 5% y dextrosa 65% en 5% de agua) como control (Figura 4.35). Figura 4.35: Efecto del inhibidor farmacológico, cilengitide, sobre el crecimiento in vivo de las células PTCL-NOS OCI-Ly12 15000 Volumen tumoral día 1 a 20 (ABC) P < 0.001 10000 5000 0 CONTROL CILENGITIDE Crecimiento in vivo de células OCI-Ly12 en ratones NOD-SCID. Se muestra el gráfico del volumen tumoral (área bajo la curva, ABC) luego de 20 días de tratamiento diario por vía intraperitoneal con 110 mg/kg del inhibidor cilengitide (n=9) o con el vehículo como control (n=6). 124 Resultados Una vez finalizado el tratamiento, se observó que los ratones tratados con el inhibidor de la integrina αVβ3 desarrollaron tumores de tamaño significativamente menor a los generados en los ratones SCID tratados con el vehículo (Figura 4.35). El efecto inhibitorio del pentapéptido cíclico cilengitide sobre el crecimiento tumoral de las células OCI-Ly12 fue asociado a la disminución de la activación de la vía de señalización de NF-κB (Figura 4.36) Figura 4.36: Efecto del tratamiento con el inhibidor Cilengitide sobre la Actividad relativa de NFκB activación de las vías de señalización de NF-κB en los tumores OCI-Ly12 Vehículo Cilengitide Capacidad de unión al ADN de los distintos miembros de la familia NF-κB (ver Materiales y Métodos) en extractos nucleares de fracciones de tejido de linfoma obtenidos de los tumores OCI-Ly12. *P<0.05 y ** P<0.01 También se evaluó la apoptosis celular, mediante la técnica de TUNEL, en secciones tisulares de los tumores OCI-Ly12. Se encontró un aumento de células apoptóticas inducido por el tratamiento (7.3 ± 3.5% vs. 12 ± 4.2%, vehículo vs. cilengitide, respectivamente, Figura 4.37). Estos resultados sugieren que la reducción en el volumen tumoral podría estar relacionada con el aumento de la apoptosis celular. 125 Resultados Cabe mencionar que si bien la dosis de cilengitide utilizada en el ensayo es la concentración máxima tolerada descripta en pacientes, teniendo en cuenta el peso corporal y el análisis macroscópico tisular de los ratones tratados, se pudo determinar que no hay toxicidad asociada al tratamiento (datos no mostrados). Figura 4.37: Efecto del tratamiento con el inhibidor Cilengitide sobre la apoptosis en los tumores OCI-Ly12 Vehículo 7.3 ± 3.5 % Cilengitide 12 ± 4.2 % Apoptosis celular analizada por la técnica de TUNEL en secciones tisulares representativas de tumores OCI-Ly12 tratados con cilengitide o con vehículo como control. Aumento de las fotografías 200X, barra de escala = 100 µm. 4.5.3 La inhibición farmacológica de la integrina αvβ3 disminuye el crecimiento in vivo de tumores ALCL derivados de pacientes. Para determinar si la estrategia terapéutica descripta podría tener una traslación a la clínica, se evaluó el efecto del cilengitide en un modelo preclínico utilizando tumores derivados de pacientes. Los mismos se trasplantaron en ratones humanizados (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) que luego fueron tratados intraperitonealmente con el inhibidor farmacológico cilengitide como se indicó anteriormente. Se realizaron dos ensayos, uno con tumores derivados de un paciente ALCL-ALK+ y otro con tumores derivados un paciente ALCL-ALK– 126 Resultados (n=16 y n=8 respectivamente). Una vez que los tumores fueron trasplantados, se dividió aleatoriamente a los ratones en dos grupos de tratamiento (Cilengitide vs Vehículo). Los resultados muestran que el tratamiento con cilengitide durante 10 días indujo una disminución en el crecimiento in vivo de ambos subtipos de tumores ALCL, ALK- y ALK+, en comparación con los tumores tratados con el vehículo como control (Figura 4.38). Figura 4.38: Efecto del tratamiento con cilengitide sobre el crecimiento de tumores derivados de pacientes ALCL día 1 a10 (ABC) Volumen tumoral P =0.0104 VEHÍCULO CILENGITIDE día 1 a10 (ABC) Volumen tumoral P =0.0159 VEHÍCULO CILENGITIDE Evaluación del volumen tumoral (área bajo la curva, ABC) de los tumores derivados de un paciente ALCL-ALK+ y otro ALCL-ALK- (panel superior e inferior, respectivamente) crecidos en ratones NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ. Los mismos fueron divididos en dos grupos de tratamiento, un grupo fue tratado durante diez días con cilengitide (110 mg/kg) y el otro con el vehículo como control. 127 Resultados Teniendo en cuenta los resultados obtenidos en este trabajo podemos plantear que las HTs contribuirían al fenotipo maligno de los linfomas de células T a través de la regulación de programas genéticos vinculados con la proliferación y angiogénesis. Estos efectos se dan por la acción de las HTs sobre su receptor de membrana, la integrina aVβ3, dando como resultado la activación transcripcional de genes como VEGF, que favorece en última instancia la vascularización y el crecimiento tumoral. La inhibición selectiva de la integrina aVβ3, resulta en la inhibición de la proliferación y el crecimiento tumoral de los LCT. De esta manera, podríamos plantear la utilización de este receptor como nuevo blanco terapéutico para el tratamiento de esta patología. 128 5. DISCUSIÓN Discusión Las hormonas tiroideas modulan distintos procesos fisiológicos y son críticas para el crecimiento, desarrollo, diferenciación y mantenimiento de la homeostasis metabólica (Oetting y Yen, 2007). Las enfermedades asociadas a las HTs están relacionadas a la falta o exceso de las mismas (hipo e hipertiroidismo), pero también se las asocia al desarrollo y curso de otras patologías. El crecimiento y la progresión tumoral están íntimamente vinculados con el entorno celular. En el mismo juega un papel preponderante la influencia del sistema inmunológico y del sistema endócrino, mediante la producción de citoquinas y hormonas derivadas tanto del huésped como del tumor (Reiche y col, 2004; Hammacher y col, 2005). Los factores inmunológicos y endócrinos pueden orquestar la evolución y diseminación de una patología oncológica (Reiche y col, 2004; Hammacher y col, 2005). Las citoquinas derivadas de los linfomas de células T son capaces de modular la respuesta inmune antitumoral, favoreciendo la progresión tumoral (Shu y col, 2010; Gaulard y de Levald, 2014). Las células tumorales también se encuentran expuestas a un complejo microambiente parácrino y endócrino, compuesto por una variedad de factores de crecimiento y hormonas elaboradas por las células vecinas o distales (Singh y Singh, 2009). En este sentido existen trabajos donde asocian a las HTs con la transformación celular, tumorigénesis y metástasis (Hercbergs y col, 2010; Moeller y Führer, 2013) y proponen que las HTs podrían cumplir un rol importante en la inducción de la angiogénesis (Pinto y col, 2011; Mondul y col, 2012). Los LCT no son la excepción, también están influenciados por el entorno celular. Recientemente nuestro grupo de trabajo describió que las HTs se encuentran entre los factores que influencian, in vitro, la proliferación de células murinas de linfomas T (Barreiro Arcos y col, 2006; 2011 y 2013). Más aún, también se describió que el estado tiroideo afecta el crecimiento tumoral in vivo de la línea de linfoma T murino EL-4 creciendo en ratones singeneicos (Sterle y col, 2014). Es importante tener en cuenta que, salvo excepciones, los pacientes con LCT tienen mal pronóstico debido a la combinación de un curso clínico muy agresivo y a la falta de tratamientos específicos. Esto se pone en evidencia por la ausencia de protocolos estándares para el cuidado y tratamiento de los 130 Discusión pacientes y por la refractariedad a las quimioterapias convencionales. Por ello es tan importante focalizar esfuerzos en el estudio de la biología genética y tumoral de los LCT, para lograr el desarrollo de regímenes terapéuticos más específicos y efectivos para el tratamiento de esta patología. En este trabajo evaluamos el rol que ejercen las HTs a través de su acción sobre sus receptores, nuclear y de membrana (TR y mTR, respectivamente), sobre líneas de distintos subtipos de LCT humanos, demostrando por primera vez los programas genéticos activados por las HTs a través del mTR, que, en esta células, se identificó como la integrina αVβ3. Además, y en base a los resultados encontrados, extendimos nuestros estudios a modelos animales de xenotrasplante de células de LCT y de tumores de pacientes a fin de evaluar la potencialidad del blanco terapéutico que representa el receptor de membrana para las HTs. A continuación realizaremos un análisis de las acciones in vitro de las HTs sobre la proliferación de las células de LCT y la inducción de genes resultante de la activación del mTR, a fin de comprender mejor los mecanismos por los cuales las HTs podrían estar contribuyendo al desarrollo y diseminación de los LCT in vivo. Finalmente analizaremos la importancia de la inhibición farmacológica del mTR como potencial terapia para esta patología. 5.1 Efectos de las hormonas tiroideas sobre la proliferación y ciclo celular de linfomas de células T humanos. El estudio de las acciones de las HTs sobre el crecimiento tumoral no es reciente. Hace más de 30 años, se encontró que la remoción de las hormonas T3 y T4 del suero suprimía la transformación neoplásica inducida por rayos X en líneas celulares en cultivo, sin modificar la supervivencia celular (Guernsey y col, 1980). Más aún, la adición de T3 al medio depletado de hormonas restablecía la frecuencia de transformación esperada. También se ha descripto que la T3 facilita la carcinogénesis química (Borek y col, 1983). Adicionalmente, se demostró en ratas hipotiroideas una reducción de la inducción de cáncer de 131 Discusión mama luego de la exposición a 7,12-dimetilbenzantraceno (DMBA). El reemplazo hormonal incrementó un 78% su incidencia (Goodman y col, 1980). Por un largo periodo, los estudios mencionados no fueron tenidos en cuenta debido al tradicional concepto de la acción genómica de las HTs sobre la transcripción de genes relacionados con la homeostasis y el metabolismo de las células normales. Sin embargo más recientemente, luego de que se describiera un receptor para HTs ubicado en la membrana plasmática capaz de estimular señales intracelulares de activación, se pudo explicar la contribución de dichas hormonas a la proliferación celular de distintos tipos de neoplasias. Estudios realizados en líneas humanas de cáncer de mama (Tang y col, 2004), de tiroides papilar y folicular (Lin y col, 2006), de glioma U-87 MG (Lin y col, 2009) y de pulmón (Meng y col, 2011), demostraron que el tratamiento con concentraciones fisiológicas de T3 y T4 lleva a la activación de ERK1/2 y consecuentemente a la proliferación celular, evidenciada por una acumulación del antígeno de proliferación nuclear, PCNA. Pocos estudios se han centrado en el complejo rol que tienen los factores endócrinos presentes en el microambiente tumoral de LCT humanos. Entre ellos, se ha determinado la influencia de las HTs del microambiente tumoral en modelos experimentales desarrollados en animales con diferente estado tiroideo, demostrando que el mismo está asociado a la modificación significativa de la sensibilidad a la radioterapia, la que se ve favorecida por el estado hipotiroideo (Jordan y col, 2007). A fin de resaltar el rol de las HTs entre dichos factores, en este trabajo evaluamos, en primer lugar, su efecto sobre la proliferación celular en un panel de 9 líneas de LCT humanos, los cuales representan tanto subtipos de origen inmaduro (Jurkat y CUTLL1), como de origen maduro (Hut78, Mac-2A, OCI-Ly12, OCI-Ly13.2, SU-DHL-1, Karpas299). El tratamiento in vitro de las células LCT con concentraciones fisiológicas tanto de T3 y T4 libres (HT), como de las mismas acopladas a agarosa (TH-AG, impermeables a las células) indujo un aumento de la proliferación celular, de la síntesis del ADN, de la expresión del marcador proliferativo PCNA y de la fosforilación de ERK1/2. La magnitud del efecto proliferativo de las HTs fue similar en los distintos subtipos de LCT analizados; este resultado sugiere que 132 Discusión la proliferación mediada por HTs podría tratarse de un mecanismo generalizado que es aprovechado por las células de linfoma T para su supervivencia. Más aún, se encontró que el mayor efecto mediado por HTs se obtiene con la adición combinada de T3 y T4, o T3-AG y T4-AG. Asimismo, dicho efecto se observó con concentraciones similares a los niveles circulantes de ambas hormonas, acercando estos hallazgos in vitro a lo que tiene lugar fisiológicamente en el microambiente tumoral. Más aún, estudios recientes sugieren que los LCT podrían encontrarse en un microambiente enriquecido en HTs por un mecanismo que involucra el control epigenético transcripcional de la deiodinasa de tipo 3 (DIO3). El gen de esta enzima, que juega un rol esencial en regular la inactivación de las HTs en los tejidos, fue encontrado hipermetilado y reprimido en células T malignas (Martin-Subero et al, 2009). Existen evidencias clínicas que sugieren que el hipertiroidismo subclínico aumentaría la incidencia de ciertos tipos de tumores sólidos, y que el hipotiroidismo espontáneo o inducido podría estar asociado a un mejor pronóstico y mayor sobrevida de pacientes con distintos tipos de cáncer (Hercbergs et al, 2010; Hamnvik et al, 2011). Nuestro grupo de trabajo describió a las HTs como factores que inducen la proliferación de células murinas de linfomas T y capaces también de regular positivamente la activación de células T normales (Barreiro Arcos y col, 2006 y 2011). Adicionalmente comprobamos que el hipertiroidismo experimental aumenta el crecimiento in vivo de un linfoma T murino en ratones singeneicos (Sterle y col, 2014). La regulación del ciclo celular es el mecanismo más importante de control del crecimiento de los distintos tipos de células. Hay trabajos que describen ensayos in vitro con las HTs que demuestran que las mismas son capaces de modular la expresión de marcadores moleculares de la progresión del ciclo celular e inducir la proliferación y síntesis de ADN. En cardiomiocitos de rata, por ejemplo, se observó que el tratamiento con T3 induce la entrada al ciclo celular mediante un incremento de la expresión de la ciclina D1 (LeddaColimbano y col, 2006). Se decidió entonces evaluar marcadores del ciclo celular en las líneas LCT tratadas con HTs. 133 Discusión Luego del tratamiento por distintos tiempos con las HT o HT-AG, se encontró un aumento inicial de los niveles transcripcionales de las ciclinas D1, D2 y D3 (todas requeridas para la progresión de G0 a G1 del ciclo celular) seguido por un aumento de la ciclina E2 (progresión de G1 a S, y fase S) y de la ciclina B1 (progresión de G1 a M). También se encontró una disminución de los niveles transcripcionales del inhibidor de quinasas dependiente de ciclinas, p21, y del supresor tumoral, p53. Recientemente, efectos similares fueron observados por nuestro grupo en tumores sólidos de linfomas T murinos crecidos en ratones hipertiroideos, en los que encontramos un aumento de las ciclinas D1 y D3. Por el contrario los tumores crecidos en ratones hipotiroideos muestran una disminución en la expresión de inhibidores del ciclo celular, como CDKN2A y CDKN1B, y de supresores tumorales como p53 (Sterle et al, 2014). En concordancia con nuestros resultados también se describió la modulación por HTs de reguladores del ciclo celular en otros tipos tumorales. Así, en células de carcinoma papilar tiroideo se encontró que la T3 induce la expresión de la ciclina D1 a través de la activación y reclutamiento al núcleo del complejo transcripcional TRβ1/ Oct-1, promoviendo la proliferación celular (Perri y col, 2014). Resultados similares se describieron en una línea celular de carcinoma pancreático humano (Yalcin y col, 2013). La regulación de genes vinculados a la progresión del ciclo celular mediada por HTs es un punto muy importante, ya que se ha descripto a las ciclinas como marcadores moleculares del potencial oncogénico de los linfomas de células T y su regulación estaría involucrada con la linfomagénesis (Møller y col, 2001; Metcalf y col, 2010; Vaites y col, 2014). La inducción de la proliferación celular y de la transcripción de ciertas ciclinas, mediada por HTs a través del mTR en las líneas de LCT humanas, fue de magnitud similar al efecto total inducido por las HTs libres. Existen observaciones experimentales y clínicas que sugieren que tanto la T3 como la T4 son capaces de mantener la proliferación de células de distintos tipos de cáncer a través de su unión a un receptor de membrana. Se ha demostrado que la acción de las HTs sobre la integrina αVβ3 regula el crecimiento de células de mieloma (Cohen y col, 2011) y glioblastomas (Lin y col, 2009) a través de la activación de la vía de señalización MAPK y aumentan la actividad proliferativa sobre células humanas de carcinoma de pulmón y de 134 Discusión cáncer de ovario (Lin y col, 2013). También se ha descripto que el bloqueo de la integrina αVβ3 disminuye el crecimiento tumoral in vivo y la angiogénesis de células humanas de cáncer pancreático, acompañado por una disminución de la expresión de marcadores de progresión del ciclo celular (Yalcin y col, 2013). Como se mencionó anteriormente muchos trabajos asocian a las HTs con la transformación celular, la tumorigénesis y las metástasis (Hercbergs y col, 2010; Moeller y Führer, 2013). En este sentido y teniendo en cuenta los resultados mencionados, se describe por primera vez que los LCT humanos no son la excepción y que también son influidos por las HTs, induciendo su crecimiento y proliferación celular y la expresión de marcadores moleculares asociados a la progresión del ciclo celular. 5.2 Identificación del receptor de membrana para HTs en linfomas humanos de células T. En el año 2005 se demostró por primera vez que tanto la T3 como la T4 pueden unirse a una proteína de membrana en las células fibroblásticas CV-1. Este receptor fue descripto como la integrina αvβ3 (Bergh y col, 2005). Las integrinas son receptores de membrana αβ heterodiméricos que, luego de la unión de sus ligandos, inducen una serie de señales intracelulares que pueden regular la sobrevida, la proliferación, la migración y la diferenciación celular. La respuesta celular depende del tipo de ligando y de los componentes del receptor dimérico. La integrina αVβ3 es el receptor de proteínas de la matriz celular como la vitronectina (VN), Cyr61, eta-1/osteopontina, fibronectina, fibrinógeno y factores solubles, como las hormonas tiroideas. Las HTs se unen a una o varias isoformas de su receptor nuclear. El TR une preferencialmente a la T3; sin embargo, la integrina αVβ3 une tanto T3 como T4. Utilizando modelados matemáticos de la cinética de unión de las HTs a la integrina, se describió que el dominio de unión de HTs sobre la misma se encuentra cerca del sitio de unión a ligandos con secuencia RGD (Arginina- 135 Discusión Glicina- Aspartato) e incluye dos sitios de unión, que transducen señales mediadas por las HTs de manera diferencial (Davis y col 2011; Freindorf y col, 2012). Las acciones de las hormonas T3 y T4 sobre este receptor de membrana, fueron demostradas en células normales, en células de vasos sanguíneos (Davis, 2004; Bergh, 2005), en células no cancerosas inmortalizadas, en células embrionarias de riñón y en distintos tipos tumorales, tales como cáncer de próstata y carcinomas renales (Kimbro y Simons, 2006). Hasta el momento no se había descripto si esta integrina es también el receptor de membrana para las HTs en los LCT. En este trabajo se demuestra que, tanto el TRα como la integrina αVβ3, se encuentran expresados en todas las líneas celulares de linfomas T humanos evaluadas, indicando que estas células podrían ser afectadas por los niveles circulantes de T3 y T4 que se encuentran normalmente presentes en el microambiente tumoral. Más aún, la mayoría de las células de LCT evaluadas expresan mayores niveles de estos receptores en comparación a los linfocitos T periféricos normales, sugiriendo una mayor respuesta de los LCT a las HTs. Es importante mencionar que la integrina αVβ3 se encuentra altamente expresada en células endoteliales angiogénicas y en células tumorales cumpliendo un rol preponderante en procesos patológicos asociados a la supervivencia, al crecimiento tumoral y a las metástasis (Hood y Cheresh, 2002). Dicho receptor de membrana fue implicado en la patofisiología y progresión de melanomas (Dang y col, 2006), gliomas (Gingras y col, 1995) y en distintos tipos de cáncer como los de ovario (Hapke y col, 2003), próstata (Cooper y col, 2001) y mama (Harms y col, 2004; Vellon y col, 2006). Por ejemplo, la integrina αVβ3 está altamente expresada en prácticamente todos los cánceres de mama con metástasis óseas (Liapis y col, 2006). Estudios in vitro sobre cultivos de células tumorales mamarias describen la existencia de una correlación positiva entre la expresión de la integrina αVβ3 y la habilidad de las células tumorales de adherirse a la matriz extracelular, de migrar y de regular secreción de metaloproteasas, características que favorecen la diseminación metastásica (Felding-Habermann y col, 2001; Rolli y col, 2003; Gomes y col, 2004). También se ha descripto que las integrinas, ampliamente expresadas en tejidos hematopoyéticos y no hematopoyéticos, tienen una distribución diferencial en muestras primarias de Linfomas Non-Hodking, 136 Discusión encontrándose que la expresión de ciertas integrinas está relacionada con el subtipo histológico, influyendo en la difusión de la enfermedad y el pronóstico de los pacientes (Terol y col, 1999). En este estudio se analizó la expresión de las integrinas α2 a α6, αL, β1 y β2 en muestras primarias de LNH, pero no la expresión de las integrinas αV o β3, las cuales si se encuentran expresadas en nuestro panel de muestras primarias de los distintos subtipos de LCT. Se ha descripto que el efecto inductor de las HTs sobre la proliferación celular fue inhibido tanto in vitro como in vivo, utilizando anticuerpos monoclonales contra la integrina αVβ3, como también utilizando el análogo de la T4, ácido tetraiodotiroacético (Tetrac) (Mousa y col, 2012). El efecto antitumoral del Tetrac también fue demostrado en otros modelos de xenotrasplantes de células humanas de carcinomas medulares tiroideos, carcinomas tiroideos foliculares y carcinomas renales (Yalcin y col, 2009 y 2010). En este trabajo demostramos que la integrina αVβ3 es el receptor de membrana para las HTs en las células T malignas. En primer lugar, comprobamos que el mTR en células de LCT es una integrina con sitio de unión para ligandos con el motivo RGD, ya que el efecto proliferativo de las HTs fue inhibido in vitro de manera significativa cuando se preincuba a las células con el péptido RGD. Más aún, la regulación negativa de los niveles transcripcionales de las integrinas αV o β3 utilizando ARN de interferencia inhibió la inducción de la proliferación mediada por HTs tanto en las células LCT de origen inmaduro, CUTLL1, como en células de origen maduro, HuT 78 y OCI-Ly12. Estos estudios nos permitieron identificar a la integrina αVβ3 como receptor de membrana para las HTs en los LCT humanos. Se ha descripto que el ligando biológico de la integrina αVβ3, la VN, es capaz de estimular la proliferación de células tumorales de origen linfoide, incluyendo células T de leucemias linfoblásticas (Vacca y col, 2001). Es por ello que se evaluó la acción de la misma, como componente del microambiente tumoral, sobre la proliferación de los LCT. Para ello se trató a células de los distintos subtipos de LCT con VN, en presencia o ausencia de las HTs, de manera de poder analizar el impacto de las hormonas sobre la proliferación en presencia 137 Discusión del ligando natural de la integrina. Si bien la VN fue capaz de inducir la proliferación de los LCT en comparación al control, la magnitud de este efecto fue menor que la observada con HTs. Además el tratamiento con ambos factores no potenció el efecto proliferativo. Estos resultados fueron confirmados al evaluar la proliferación de células LCT mediada por HTs, en cultivos tridimensionales que incluyen en su matriz, células mesenquimales y el péptido RGD. De esta manera se pudieron analizar los efectos de las HTs en un contexto más cercano al fisiológico. Observamos no sólo que las HTs son capaces de inducir el crecimiento de las células LCT en los cultivos 3D, sino también que la inhibición farmacológica del mTR inhibió el efecto proliferativo de las HTs. Estos datos indicarían que las acciones mediadas por las HTs pueden darse de manera independiente y, aún, en presencia del ligando biológico de la integrina αVβ3. 5.3 Programas transcripcionales regulados por las hormonas tiroideas a través de su receptor de membrana El surgimiento de técnicas de última generación basadas en el análisis de blancos moleculares, como la expresión diferencial de genes, la secuenciación de ADN y ARN y los perfiles epigenéticos, han influido de manera significativa en la comprensión y el enfoque terapéutico de distintos subtipos de neoplasias, incluidas las linfoides (Tirado y col, 2012; Li y col, 2013). Estas nuevas tecnologías permiten el rápido análisis en forma global de los perfiles genéticos y de las vías de señalización activadas en estas patologías, ayudando de manera considerable a la clasificación de los distintos subtipos e identificando nuevas estrategias terapéuticas. Teniendo en cuenta lo mencionado anteriormente, se decidió evaluar los mecanismos genéticos involucrados en los efectos proliferativos mediados por HTs a través de su mTR. Para ello se analizaron los cambios transcripcionales inducidos por las HT-AG (vs. AG sola como control) en las células CUTTL1 por secuenciación de ARN. La activación mediada por las HT-AG a través de la 138 Discusión integrina αvβ3, estimula vías de señalización que inducen finalmente la expresión de genes relacionados con la “traducción proteica” (RPL13, RPL27A, RPL36), con la “cadena respiratoria oxidativa” (NUDFB1/2, NDUFA13, UQCR11), con la “angiogénesis” (VEGFB), con la proliferación / diferenciación de linfocitos y con la replicación del ADN (IL4, EDF1, DOK2). Dado que estos genes podrían contribuir al fenotipo maligno de los LCT, se validaron por RTPCR en tiempo real los genes involucrados en la angiogénesis, en la proliferación linfocitaria y en la replicación del ADN (DBP, VEGFB, IL4 y DOK2), no sólo en las células de origen inmaduro, CUTLL1, sino también, en las células de origen maduro HuT 78 y OCI-Ly12. Encontramos que las HT-AG incrementaron los niveles transcripcionales de IL4, DBP, DOK2 y VEGFB en todas las líneas celulares analizadas. Existen evidencias experimentales que demuestran que la IL-4 es capaz de estimular la proliferación de células T tumorales y podría participar de los efectos proliferativos de las HTs. Así, la producción de IL-4 por células T CD4+ y CD8+ de pacientes con diferentes LCT periféricos puede inducir in vitro la proliferación de células T malignas (Raziuddin y col, 1998). También se ha demostrado que esta citoquina induce la proliferación de células T de leucemia linfoblástica aguda, a través de la regulación de la progresión del ciclo celular dependiente de mTOR (Cardoso y col, 2009). Por su parte DOK2 es una proteína adaptadora, expresada principalmente en tejidos hematopoyéticos, que actúa como sustrato común para varias tirosina quinasas y que regula negativamente la señalización intracelular de inmuno-receptores (Mashima y col, 2009). Más aún, DOK2 ha sido involucrado en la regulación positiva de la migración celular por interactuar con un amplio rango de proteínas de señalización, incluyendo la cola citoplasmática de la integrina β3 (Calderwood y col, 2003), y se ha demostrado su sobreexpresión en linfomas anaplásicos de células grandes (ALCL) (Lamant y col, 2007). El DBP es un factor de transcripción involucrado en la regulación de ciertos genes metabólicos, como el de la albúmina, el CYP2A4 y el CYP2A5 y se ha descripto su regulación positiva por la acción de la T3 en el proceso de diferenciación de oligodendrocitos y de regeneración mielínica (Dugas y col, 2012). Con respecto a VEGF, cabe mencionar que su expresión ha sido relacionada con la sobrevida y el pronóstico de pacientes con distintos subtipos 139 Discusión de linfomas de células T (Jorgensen y col, 2009), lo que resalta la importancia de este factor angiogénico en la progresión de esta patología y cuyo estudio hemos profundizado tal como se detalla más adelante. También demostramos que las HTs libres a través de su acción sobre la integrina αVβ3 son capaces de inducir la expresión de los genes evaluados. Esta inducción se vio inhibida cuando la expresión transcripcional de dicho receptor es disminuida utilizando ARN de interferencia. Estos resultados confirman que la acción de las hormonas sobre su mTR y su posterior activación es responsable de la regulación observada en los genes de proliferación y angiogénesis. En trabajos anteriores publicados por nuestro grupo, se identificó al factor de transcripción NF-κB, como un efector río abajo de la activación de un putativo mTR en linfomas T murinos (Barreiro Arcos y col, 2011). En este trabajo se observó que dicho factor de transcripción es activado y translocado al núcleo de células de LCT humanas luego del tratamiento por tiempos cortos con HTs, lo que concuerda con los resultados observados en las células de linfoma T murinas. De hecho, del análisis realizado con herramientas bioinformáticas surge una asociación entre los genes regulados por HT-AG y el factor de transcripción NF-κB: estos genes presentan en sus regiones promotoras sitios de unión a NF-κB. Más aún, cuando se preincuba a los distintos subtipos de LCT con un inhibidor específico de este factor de transcripción, la inducción de los genes IL-4 y VEGF mediada por HTs se vio inhibida. Este resultado sustenta aún más la implicancia de NF-κB en la regulación transcripcional mediada por HTs de IL4 y VEGF. NF-κB regula la expresión de genes involucrados en muchos procesos que juegan un rol central en el desarrollo y progresión de las neoplasias, como la proliferación, la migración y la apoptosis (Dolcet y col, 2005). Se ha encontrado expresado y activado constitutivamente de forma aberrante en distintos tipos de cáncer, entre los que podemos mencionar el cáncer de mama (Khan y col, 2013), linfomas de Hodgkin (Carbona y col, 2011) y linfomas cutáneos de 140 Discusión células T (Thakur y col, 1994, Döbbeling, 2007). También se ha vinculado su sobre activación con el desarrollo de distintos tipos de linfomas de células T en ratones transgénicos Notch-/- (Bellavia y col, 2000). Más aún la expresión y activación de este factor de transcripción ha sido relacionada de manera directa con la expresión de VEGF y la angiogénesis. Así por ejemplo, en células de glioma se ha descripto que el bloqueo de la señalización de NF-κB disminuye el crecimiento tumoral in vivo acompañado por la disminución de factores angiogénicos como el VEGF (Xie y col, 2010). Resultados similares han sido descriptos en células de cáncer de próstata (Huang y col, 2001) ¿Podrían también estos mecanismos estar involucrados en la patobiología de los LCT? De acuerdo a nuestros datos, la activación de NF-κB induciría la expresión de VEGF y precisamente el elemento desencadenante de estas vías serían las HTs presentes en el microambiente tumoral al menos en concentraciones semejantes a sus niveles plasmáticos. Es por ello que cabe preguntarse si es a través de estos mecanismos de acción que las HTs contribuyen al fenotipo maligno de los LCT. Nuestros resultados parecerían indicarlo. Por este motivo se profundizó el estudio de la participación del VEGF en las acciones de las HTs. 5.3 Inducción de la expresión mediada por HTs de factores angiogénicos funcionales. El factor de crecimiento del endotelio vascular, VEGF, está involucrado en la regulación de la angiogénesis y su expresión podría contribuir al fenotipo maligno de los LCT. De hecho, existen trabajos donde asocian la expresión de VEGF y la angiogénesis, con la sobrevida y el pronóstico de pacientes con PTCL-NOS y AITL (Konstantinou y col; 2009, Jorgensen y col, 2009; Konstantinou y col, 2011; Geskin y col, 2014). VEGFB es miembro de una gran familia de factores de crecimiento del endotelio vascular que incluye también a VEGFA, VEGFC, VEGFD y PLGF (Goel y col, 2013). Por análisis de microarreglos de ARN, se analizó la expresión transcripcional de los miembros de la familia de VEGF en una serie de muestras provenientes de pacientes con distintos subtipos de linfomas T. Además de VEGFB, el único miembro que 141 Discusión también se encuentra expresado en estas muestras es VEGFA. Más aún, el tratamiento de las líneas celulares de los distintos subtipos de LCT con HT-AG indujo la expresión transcripcional de ambos factores en la mayoría de las mismas; efecto que se vio inhibido en las células cuyo mTR fue regulado negativamente. Precisamente este es el hallazgo más importante de nuestros estudios, ya que señala a la integrina αvβ3 como un potencial blanco terapéutico. Está reportado que las células T malignas producen VEGF y expresan sus receptores; sustentando la idea de la existencia de vías autócrinas y parácrinas mediadas por VEGF en la linfomagénesis. Estudios realizados en muestras provenientes de pacientes con linfoma T angioinmunoblástico (AILT), muestran un aumento en la expresión de VEGF tanto en las células de linfoma como en las células endoteliales (Zhao y col, 2004; Piccaluga y col, 2007). El grupo de Zhang y col, en el año 2001, encontró que la expresión de VEGF está íntimamente relacionada con el estadio tumoral, la invasión de la médula ósea y el pronóstico en muestras de pacientes con PTCL-NOS. En estos estudios se sugiere que VEGF podría estar involucrado en la tumorigénesis, la progresión, la invasión y la angiogénesis en esta patología. Resultados similares fueron descriptos en pacientes con otros subtipos de LCT, y en todos los casos se encontró una correlación entre la expresión de VEGF, la sobrevida y el pronóstico de la enfermedad (Konstantinou y col, 2009; Jorgensen y col, 2009; Geskin y col, 2014). Más aún, la relación entre la expresión de la integrina αvβ3 y del VEGF y la promoción tumoral y crecimiento metastásico fue descripta en otros tipos de neoplasias (Lorger y col, 2009; Chetty y col, 2010; Zhao y col, 2014) Para evaluar la relación entre la integrina αVβ3 y el VEGF descripta en otras neoplasias y que nosotros observamos en las líneas de LCT, analizamos la expresión transcripcional de las integrinas en las muestras primarias de pacientes de LCT y estudiamos su posible asociación con los niveles de VEGF. Encontramos una correlación positiva entre los niveles de VEGFA/B y los de ITGAV e ITGB3 en todos los subtipos analizados, lo que reforzaría la existencia 142 Discusión de una asociación funcional entre los mismos en la patología humana. Es de destacar, que la inducción de la expresión de VEGFA y VEGFB parecería ser exclusiva de la acción de las HTs sobre la integrina, ya que no se observó este efecto cuando se trata a las células con el ligando natural de dicha integrina, la VN. Si bien la VN podría contribuir a la proliferación de las células LCT a través de la integrina, este efecto se daría por la activación de programas genéticos diferenciales a los inducidos por HTs. Como ya hemos referido, las células de LCT pueden encontrarse en un microambiente “enriquecido” en HTs por inactivación epigenética del gen DIO3. Se podría pensar entonces que este mecanismo prioriza la acción de las HTs por encima de la ejercida por la VN. Sin embargo el análisis de transcriptos de nuestro panel de muestras primarias de LCT mostró niveles indetectables de DIO3 y DIO2 y bajos niveles de DIO1 (datos no mostrados). A pesar de esto, usando niveles fisiológicos de T3 y T4 hemos demostrado que las HTs son factores necesarios para el crecimiento de los LCT. Se ha descripto que la VN vía la integrina αvβ3 es capaz de activar al factor de transcripción NF-κB en células de melanoma y de cáncer de páncreas (Yebra y col, 1995; Das y col, 2005). Sin embargo, la relación entre dicha molécula de la ECM y la expresión de VEGF ha sido muy poco estudiada. Recientemente se describió que la acción de la VN jugaría un rol en la inducción de la angiogénesis a través de la activación del receptor del VEGF en células endoteliales. Más aún, se ha demostrado que la estimulación de células endoteliales con VEGF resulta en la formación de un complejo entre VEGFR-2 y la integrina αvβ3 (Serini y col, 2008 a y b); la consecuencia funcional de esta interacción es el aumento de la actividad de dicha integrina (Payaningal y col, 2009). Esto sugiere que las HTs y la VN podrían estar ejerciendo una función complementaria que favorecería la angiogénesis. El crecimiento y progresión de los linfomas está potenciado por, al menos, dos mecanismos angiogénicos distintivos: (i) influencias parácrinas del microambiente tumoral pro-angiogénico sobre la transformación neovascular local y el reclutamiento de progenitores derivados de médula ósea circulantes; y por (ii) la estimulación autócrina de las células tumorales a través de la 143 Discusión expresión de VEGF y VEGFR en las células de linfoma (Ruan y col, 2009). La expresión de VEGF en células neoplásicas fue demostrada en un amplio rango de subtipos de linfomas agresivos, entre los que se incluyen linfomas T periféricos, linfomas difusos de células B grandes, linfoma de células del manto (Foss y col, 1997; Doussis-Anagnostopoulou y col, 2002), como también en leucemias linfocíticas crónicas y linfomas linfocíticos (Chen y col, 2000; Kay y col, 2002). En células de linfoma, el factor VEGF y sus receptores fueron involucrados tanto en mecanismos autócrinos como parácrinos (DoussisAnagnostopoulou y col, 2002; Wang y col, 2004). La señalización de VEGF a través de sus receptores confiere resistencia a la apoptosis y promueve la sobrevida de leucemias linfocíticas crónicas, pero este efecto es revertido con inhibidores de tirosina quinasas que impiden la activación del VEGFR (Lee y col, 2004; Ruan y col, 2009; Pedersen y col, 2013). En este sentido, se realizaron estudios para analizar si existen consecuencias parácrinas y autócrinas funcionales de la expresión de VEGF inducida por las HTs a través de la integrina αvβ3 en las células LCT. La evaluación de la capacidad de inducción de la respuesta quimiotáctica con medios condicionados de LCT mostró que un mayor número de células endoteliales migran cuando éstas son cultivadas con los medios condicionados provenientes de células tratadas con las HT-AG. Estos resultados se observaron tanto cuando se utilizaron líneas celulares de LCT de origen inmaduro como de origen maduro. Más aún, cuando los medios condicionados son pre-incubados con un anticuerpo anti-VEGF, Bevacizumab, o provienen de líneas de LCT que no expresan la integrina αVβ3, la inducción de la migración de las células endoteliales se vio inhibida. Con respecto a los efectos autócrinos de VEGF, se encontró que tanto el anticuerpo bloqueante para VEGF como un inhibidor farmacológico de su receptor, el Axitinib, disminuyen el efecto proliferativo de las HTs sobre las células de LCT. Más aún, la adición de VEGF humano recombinante también fue capaz de inducir la proliferación de las líneas de LCT. 144 Discusión Estos resultados refuerzan el importante rol que juega el VEGF en el fenotipo maligno de los LCT. De hecho, se ha descripto que el Bevacizumab tiene efectos anti-linfoma y anti-angiogénicos que potencian la acción de drogas quimioterapéuticas sobre linfomas/leucemias de células T (Wang y col, 2011; Mori y col, 2014) 5.4 Modelos in vivo de LCT y efecto de la inhibición de la integrina αVβ3 sobre su crecimiento Uno de los desafíos más importantes en el estudio de los linfomas de células T es la rápida traslación a la clínica de los conocimientos que se van obteniendo sobre la biología molecular y genética de los mismos. Como se mencionó anteriormente, los pacientes que padecen distintos subtipos de linfomas T, salvo escasas excepciones, tienen mal pronóstico debido a la combinación de un curso clínico muy agresivo y la falta de tratamientos específicos. Es por ello, que es tan importante profundizar el estudio de esta patología, de manera de poder desarrollar nuevos regímenes terapéuticos con mayor especificidad y efectividad. Como último objetivo, se propuso determinar si la inhibición de la vía regulada por las HTs a través de su receptor de membrana, la integrina αVβ3, puede ser capitalizada terapéuticamente para el tratamiento de pacientes con LCT. En los últimos años se ha logrado un gran progreso en el estudio, tanto a nivel preclínico como clínico, del bloqueo de las integrinas en patologías oncológicas. Estos estudios muestran cierta efectividad de los inhibidores de distintas integrinas para tratar de frenar la progresión tumoral de algunos tipos de cáncer (Desgrosellier et al, 2010). En primer lugar, se evaluó el efecto de la regulación negativa de la transcripción de las integrinas αV o β3, utilizando ARN de silenciamiento, sobre el crecimiento tumoral de células CUTLL1 en ratones inmunodeficientes NODSCID. Se observó en comparación a los tumores control, una reducción del volumen tumoral, de la expresión de VEGF y del área de los vasos sanguíneos. 145 Discusión También se observó un aumento del marcador de apoptosis caspasa 3 activa, sugiriendo una asociación entre la inducción de la apoptosis y la disminución del volumen tumoral. Estos datos sugieren que existiría una disminución en el potencial angiogénico en los tumores que provienen de las células CUTLL1 que no expresan tanto la integrina αV como la β3. Como enfoque terapéutico y para un análisis más exhaustivo del efecto de la inhibición de la integrina αVβ3 sobre el crecimiento tumoral in vivo de los LCT, se evaluó el efecto del inhibidor farmacológico cilengitide, que muestra actividad anti-angiogénica. Esta droga se encuentra en estudios de fase clínica III en glioblastomas y en estudios de fase II en otros tipos de tumores sólidos (Friess y col, 2006; Reardon y col, 2008; Nabors y col, 2009; Tabatabai y col, 2010). Como ya se mencionó, la alta expresión de VEGF está asociada a una mala respuesta a drogas quimoterapéuticas convencionales en pacientes con PTCL-NOS (Jorgensen y col, 2007, 2009). Es por esto que se desarrolló un modelo de xenotrasplante de células de PTCL-NOS, OCI -Ly12, para evaluar el efecto del cilengitide sobre su crecimiento in vivo. Se encontró que el tratamiento diario con el inhibidor redujo significativamente el crecimiento tumoral. El efecto inhibitorio del cilengitide, fue asociado a una disminución de la activación de la vía de señalización de NF-κB y al aumento de la apoptosis celular. Se ha descripto que el cilengitide induce la apoptosis celular en distintos modelos experimentales; en xenotrasplantes de células de cáncer de mama, por ejemplo, tiene una acción sinérgica con la radioterapia a través de la inducción de la apoptosis (Burke y col, 2002); resultados similares se observaron en células de glioblastoma (Oliveira-Ferrer y col, 2008) El receptor de membrana para HTs constituye un blanco terapéutico atractivo para utilizar como tratamiento anti-linfoma. Para determinar si el impacto de la estrategia terapéutica descripta podría tener una traslación a la clínica, se evaluó el efecto del cilengitide en un modelo pre-clínico utilizando tumores derivados de pacientes ALCL xenotrasplantados en ratones humanizados. El tratamiento diario con el inhibidor durante 10 días, indujo una disminución del crecimiento tumoral, sin toxicidad asociada al tratamiento, posicionando al 146 Discusión cilengitide como potencial herramienta terapéutica para el tratamiento de los LCT humanos. Figura 5.1 Mecanismo activados por las HTs vía la integrinaαVβ3 en la células de linfomas T humanos ´ Finalmente podemos concluir que los resultados más salientes de este trabajo señalan a las HTs como componentes reguladores fundamentales de la patobiología de los LCT. A través de la activación del mTR, la integrina αVβ3, las HTs estimulan vías de señalización relacionadas con la proliferación y sobrevida en los distintos subtipos de LCT. Como consecuencia de dichas vías, que incluyen a ERK 1/2 y al NF-κB, tiene lugar la activación transcripcional de genes como el de VEGF y la producción de dicho factor vascular, favoreciendo en última instancia el crecimiento tumoral y la vascularización (ver Figura 7.1). Más aún, el bloqueo farmacológico selectivo de la integrina αVβ3, resulta en la inhibición de la proliferación y del crecimiento tumoral in vivo de distintos subtipos de LCT. Estos resultados constituyen un marco teórico para la utilización de la integrina αVβ 3 como posible blanco terapéutico para el tratamiento de pacientes con linfomas de células T. 147 6. CONCLUSIONES Conclusiones En este trabajo de Tesis pudimos demostrar que las HTs, en concentraciones fisiológicas y a través de la integrina αVβ3, inducen la angiogénesis y la proliferación de células de LCT inmaduras y maduras. Demostramos que la integrina αVβ3 es el receptor de membrana para las HTs en células de LCT y caracterizamos el programa transcripcional activado por las HTs vía dicho receptor. Este programa está ligado a la inducción de genes de proliferación celular y angiogénesis, tales como el VEGF. Más aún, hemos encontrado que la inhibición de la integrina αVβ3 reduce la proliferación de células de linfoma T en modelos pre-clínicos de LCT incluyendo xenotrasplantes de tumores ALCL derivados de pacientes. Los estudios que nos han permitido demostrar lo antedicho incluyen experimentos realizados tanto in vitro como in vivo. Primeramente hemos visto el efecto del tratamiento con HTs sobre 9 líneas celulares representativas de los distintos subtipos de linfomas de células T humanos. En base a los resultados obtenidos in vitro podemos concluir que: 1- Las HTs inducen la proliferación celular de distintos subtipos de LCT: El tratamiento in vitro con concentraciones fisiológicas de T3 y T4 aumenta la proliferación celular, la expresión de marcadores proliferativos, como PCNA, y la síntesis de ADN en los distintos subtipos de LCT. El tratamiento con HT-AG tiene casi el mismo efecto sobre la proliferación de los LCT que las HTs libres, sugiriendo que los efectos proliferativos mediados por HTs se dan también a través de su receptor de membrana. El aumento de la proliferación mediada por HTs está asociado a la inducción de la expresión de marcadores del ciclo celular como las ciclinas D1, D2, D3, E2 y B1; y a la disminución de la expresión del inhibidor de ciclinas, p21 y del supresor tumoral, p53. 2- La integrina αVβ 3 es receptor de membrana para HTs en LCT: 149 Conclusiones Las líneas celulares de LCT expresan mayores niveles transcripcionales de THRA, ITGAV e ITGB3 en comparación a los linfocitos T periféricos, sugiriendo una mayor respuesta de las células T malignas a los efectos de las HTs. El efecto proliferativo mediado por las HTs en células de LCT es inhibido cuando se pre-incuba con un péptido bloqueante RGD, lo que sugiere que el mTR en células de LCT es una integrina con sitio de unión para ligandos con dicho motivo RGD. Esto fue confirmado por la inhibición de los efectos proliferativos de las HTs mediante técnicas de ARN de silenciamiento e inhibidores farmacológicos selectivos para la integrina αVβ3, lo que señaló a dicha integrina como el mTR en células de LCT. La unión del ligando biólogico de ECM, la vitronectina (VN), a la integrina αVβ3, no disminuye el efecto de la proliferación de HT. En cultivos, la VN es capaz de inducir la proliferación de los LCT, pero en menor proporción que las HTs y algo similar ocurre en un cultivo 3D por activación simultánea con ligandos que contienen RGD y HTs, en los que el inhibidor de la integrina, el Cilengitide impide estos efectos. Esto indicaría que las acciones sobre la proliferación mediadas por las HTs se dan de manera independiente a las acciones de los otros componentes del microambiente tumoral. 3- Las HTs regulan, a través del mTR, programas transcripcionales vinculados a proliferación y angiogénesis Los programas genéticos mayormente regulados por HT-AG a través de la activación de la integrina αvβ3, en las células CUTLL1, involucran la expresión de genes relacionados con la “transducción” (RPL13, RPL27A, RPL36), con la “cadena respiratoria oxidativa” (NUDFB1/2, NDUFA13, UQCR11), con la “angiogénesis” (VEGFB) y con la proliferación/diferenciación de linfocitos y replicación del ADN (IL4, EDF1, DOK2). 150 Conclusiones La inducción de los genes VEGFB, IL4, DOK2 y DBP fue validada en estas células y también en líneas celulares de LCT de origen maduro. Dada la función de estos genes, puede sugerirse que los mismos pueden contribuir al fenotipo maligno de los linfomas de células T. El NF-κB está involucrado en las acciones de las HTs a través de su receptor de membrana, ya que las HTs inducen la translocación de este factor de transcripción al núcleo celular y el inhibidor irreversible de su activación disminuye significativamente la regulación positiva de los genes de IL-4, VEGFA y VEGFB mediada por HTs. 4- Existe una asociación entre la expresión de la integrina αvβ3 y la expresión de VEGF en muestras de pacientes con LCT El análisis de la expresión transcripcional demostró la presencia tanto de las integrinas αV y β3 como de VEGFA y VEGFB en muestras de pacientes con distintos subtipos LCT. Encontramos que los niveles de expresión de la integrina αV y β3 correlacionan positivamente con los de VEGF, sugiriendo una asociación entre la activación de la integrina αvβ3 y la expresión de VEGFA y VEGFB en el panel de muestras primarias de pacientes estudiados. 5- Las HTs inducen la expresión de factores angiogénicos funcionales que actúan de forma parácrina y autócrina La expresión transcripcional de los factores de crecimiento del endotelio vascular VEGFA y VEGFB, aumenta luego del tratamiento con HT-AG en las líneas celulares de distintos subtipos de LCT. Esta inducción es mediada por la integrina αVβ3, ya el efecto inductor de las HTs es inhibido cuando los ensayos se realizan sobre líneas celulares en las que se silenció la expresión del mTR. 151 Conclusiones Los medios condicionados provenientes de líneas LCT inmaduras y maduras tratadas con HT-AG, son capaces de inducir un incremento de la migración in vitro de células endoteliales HMEC1. Ese efecto es mediado por VEGF ya que la pre-incubación de dichos medios condicionados con un anticuerpo anti-VEGF, inhibe la inducción de la migración de las células endoteliales, sugiriendo la presencia de VEGF en los mismos. La expresión de VEGF mediada por HTs tiene un rol autócrino sobre los LCT ya que la inducción de la proliferación mediada por HTs se ve inhibida por la acción del anticuerpo bloqueante para VEGF, Bevacizumab y del inhibidor farmacológico del receptor de VEGF, Axitinib. Por otra parte se realizaron xenotrasplantes de células humanas de LCT en ratones inmunodeficientes y se evaluó el efecto de la inhibición de la integrina αvβ3 sobre el crecimiento tumoral. De los resultados de estos estudios in vivo podemos concluir que: La regulación negativa de la integrina αV o β3 por ARN de interferencia en células CUTLL1, reduce el volumen tumoral, disminuye la expresión de VEGF y el área de los vasos sanguíneos; sugiriendo una disminución en el potencial angiogénico de los tumores que provienen de las células CUTLL1 que no expresan el mTR. El tratamiento diario con el inhibidor farmacológico de la integrina, el cilengitide, redujo significativamente el crecimiento tumoral in vivo de las células OCI-Ly12 de origen PTCL-NOS. El efecto inhibitorio del cilengitide, fue asociado con la disminución de la activación de la vía de señalización de NF- κB y con el aumento de la apoptosis celular. El tratamiento diario durante 10 días con cilengitide indujo una disminución en el crecimiento de tumores derivados de pacientes ALCL-ALK- y ALCL-ALK+, sin toxicidad asociada al tratamiento. 152 Conclusiones Los resultados obtenidos en este trabajo indican que las hormonas tiroideas regulan vías de señalización relacionadas con la proliferación y sobrevida de los distintos subtipos de linfomas de células T. Estos efectos mediados por HTs se dan por la acción de las mismas sobre la integrina αVβ3, la cual da como resultado la activación transcripcional de genes como el VEGF, favoreciendo en última instancia el crecimiento tumoral y la vascularización. Más aún, la inhibición selectiva de la integrina αVβ3, resulta en la inhibición de la proliferación y el crecimiento tumoral in vivo de distintos subtipos LCT (Figura 6.1). Todos los resultados obtenidos en este trabajo y mencionados anteriormente, sugieren que el receptor de membrana para HTs, la integrina αVβ3, constituye un blanco terapéutico atractivo para la terapia de esta patología. Figura 6.1: Esquema representativo de la acción de las HTs a través de su receptor de membrana en células de linfoma T T3 T4 αv β3 NFkB Células LCT Proliferación VEGF Angiogénesis 153 7. ABREVIATURAS Abreviaturas ADN, Ácido desoxiribonucleico histona desacetilasa AITL, linfoma de células T HIF1, factor de transcripción inducible angioinmunoblástico por hipoxia 1 AKR1C1–3, aldo-keto-reductasas 1-3 HPT, hipotálamo-pituitario-tiroideo ALCL, linfoma de células grandes IFN, interferón anaplásico IL, interleuquinas ALK, Kinasa del linfoma anaplástico IP, ioduro de propidio Ang, angiopoyetinas LCT, linfomas de células T ARN, Ácido ribonulceico LH, linfomas Hodking ARNm, Ácido ribonulceico LNH, linfomas Non-Hodking mensajero MAPK, proteína quinasa activada ARNasa, Ribonucleasa por mitógeno ARs, Receptores adrenérgicos MCL, linfoma de células del manto bFGF, factor de crecimiento de MF, micosis Fungoide el fibroblastos básico MMPs, metaloproteasas de matriz cADN, Ácido desoxiribonucleico mTR, Receptor de membrana de complementario hormonas tiroideas Cdk, quinasa dependiente de ciclinas NIS, proteína transportadora de yodo CdkI, inhibidor de quinasas OMS, Organización mundial de la dependientes de ciclinas Salud CR, respuesta completa PCNA, antígeno nuclear de CTCL, Linfomas Cutáneos de proliferación celular células T PDGF, factor de crecimiento derivado DBD, dominio de unión a DNA de plaquetas DIO, deiodinasa PI3K, fosfoinositol 3-quinasa DLBCL, linfomas difusos de células B PIGF, factor de crecimiento placentario grandes PTCL, linfoma de células T periféricas DMV, densidad micovascular PTCL-NOS, linfoma células T ECM, matriz extracelular periféricas ERK1/2, quinasa reguladora de de otro modo no especificado señales extracelulares ½ PTEN,fosfatasa del fosfatidilinositol FGF, factor de crecimiento de (3, 4, 5)-trifosfato fibroblastos Rb, proteína del retinoblastoma FT4, tiroxina libre RGD, péptido FT3, 3,3′-5 triyodotironina libre Arginina- Glicina- Aspartato HDAC, proteínas con actividad RIA, radioinmunoensayo 155 Abreviaturas RXR, receptor de rexinoides TR, Receptor nuclear de S1, Sitio de unión 1 hormonas tiroideas S2, Sitio de unión 2 TREs, elementos de respuesta SFB, suero fetal bovino a hormonas tiroideas SS, síndrome de Sézary TRH, hormona hipotalámica T3, 3,3′-5 triyodotironina liberadora de tirotropina T3r, 3,3′-5 triyodotironina TSH, hormona hipofisaria reversa estimulante del tiroides T4, tiroxina TPO, peroxidasa tiroidea Tetrac, ácido tetraiodotiroacético TTR, transtiretina Tg, tiroglobulina VEGF, factor de crecimiento TBG, globulina transportadora endotelial vascular de hormonas tiroideas VEGFR, Receptor del factor de TNF, factor de necrosis tumoral crecimiento endotelial vascular 156 8. BIBLIOGRAFÍA Bibliografía Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003; 112(8):1142-51. Alisi A, Demori I, Spagnuolo S, Pierantozzi E, Fugassa E, Leoni S. Thyroid status affects rat liver regeneration after partial hepatectomy by regulating cell cycle and apoptosis. Cell Physiol Biochem. 2005; 15(1-4):69-76. Ballester B, Ramuz O, Gisselbrecht C, Doucet G, Loï L, Loriod B, Bertucci F, Bouabdallah R, Devilard E, Carbuccia N, Mozziconacci MJ, Birnbaum D, Brousset P, Berger F, Salles G, Briére J, Houlgatte R, Gaulard P, Xerri L. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas. Oncogene. 2006; 25(10):1560-70. Barreiro Arcos ML, Sterle HA, Paulazo MA, Valli E, Klecha AJ, Isse B, Pellizas CG, Farias RN, Cremaschi GA. Cooperative nongenomic and genomic actions on thyroid hormone mediatedmodulation of T cell proliferation involve up-regulation of thyroid hormone receptor and inducible nitric oxide synthase expression. J Cell Physiol. 2011; 226(12):3208-18 Bartkova J, Zemanova M, Bartek J. Abundance and subcellular localisation of cyclin D3 in human tumors. Int J Cancer. 1996; 1996;65:323–327. Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, Stoppacciaro A, Tiveron C, Tatangelo L, Giovarelli M, Gaetano C, Ruco L, Hoffman ES, Hayday AC, Lendahl U, Frati L, Gulino A, Screpanti I. Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J. 2000; 3;19(13):3337-48. Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. Integrin avb3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogenactivated protein kinase and induction of angiogenesis. Endocrinology, 2005; 146: 2864-2871. Bernengo MG, Quaglino P, Comessatti A, Ortoncelli M, Novelli M, Lisa F, Fierro MT. Low-dose intermittent alemtuzumab in the treatment of Sézary syndrome: clinical and immunologic findings in 14 patients. Haematologica. 2007; 92:784–94. Bertolini F, Paolucci M, Peccatori F, Cinieri S, Agazzi A, Ferrucci PF, Cocorocchio E, Goldhirsch A, Martinelli G. Angiogenic growth factors and endostatin in non-Hodgkin's lymphoma. Br J Haematol. 1999; 106(2):504-9. Besedovsky H, del Rey A, Normann SJ. Host endocrine responses during tumor growth. Prog Clin Biol Res. 1989; 288:203-13. Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005; 23:3971–93. Bianco AC. Cracking the Metabolic Code for Thyroid Hormone Signaling. Endocrinology. 2011; 152(9): 3306–3311. Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2001;1;62(15):4263-72. Buschges R, Weber RG, Actor B, Lichter P, Collins VP, Reifenberger G. Amplification and expression of cyclin D genes (cyclin D1, CCND2 and cyclin D3) in human malignant gliomas. Brain Pathol. 1999; 9:435–442. Calderwood DA, Fujioka Y, de Pereda JM, Garcia-Alvarez B, Nakamoto T, Margolis B, et al. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2272-7. 158 Bibliografía Canioni D, Salles G, Mounier N, Brousse N, Keuppens M, Morchhauser F, Lamy T, Sonet A, Rousselet MC, Foussard C, Xerri L. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J Clin Oncol. 2008; 26(3):440-6. Cao X, Kambe F, Moeller LC, Refetoff S and Seo H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamcyn-p70S6K cascade througth phosphatidylinositol 3-kinase in human fibroblast. Mol Endocrinol. 2005;19(1):102-12. Cao X, Kambe F, Yamauchi M and Seo H. Thyroid-hormone-dependent activation of the phosphoinositide 3-kinase/ Akt cascade requires Src and enhances neuronal survival. Biochem J. 2009; 11;424(2):201-9. Carbone A, Spina M, Gloghini A, Tirelli U. Classical Hodgkin's lymphoma arising in different host's conditions: pathobiology parameters, therapeutic options, and outcome. Am J Hematol. 2011;86(2):170-9. Cardoso BA, Martins LR, Santos CI, Nadler LM, Boussiotis VA, Cardoso AA, et al. Interleukin-4 stimulates proliferation and growth of T-cell acute lymphoblastic leukemia cells by activating mTOR signaling. Leukemia. 2009; 23:206-8. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000; 407(6801):249-57. Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget. 2012; 3(3):224-225. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009; 4:127-50. Chan IH and Privalsky ML. Isoform-specific transcriptional activity of overlapping target genes that respond to thryroid hormones receptors α1 and β1. Mol Endocrinol. 2009; 23(11):1758-75. Chen H, Treweeke AT, West DC et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood 2000; 96:3181–3187. Cheng SY1, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31(2):139-70. Chetty C, Lakka SS, Bhoopathi P, Rao JS. MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated PI3K/AKT signaling in A549 lung cancer cells. International journal of cancer Journal international du cancer. 2010;127:1081-95. Chiamolera MI, Sidhaye, Matsumoto S, He Q, Hashimoto K, Ortiga-Cavalho TM and Wondisford FE. Fundamentally distinct rolesof thyroid hormones receptor isoforms in a thyrotroph cell line are due todifferential DNA binding. Mol Endocrinol. 2012; 26(6):926-39. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membranepermeabilization and apoptosis. Science. 2004; 303(5660):1010-4. Choy EH, Connolly DJ, Rapson N, Jeal S, Brown JC, Kingsley GH, Panayi GS, Johnston JM. Pharmacokinetic, pharmacodynamic and clinical effects of a humanized IgG1 anti-CD4 monoclonal antibody in the peripheral blood and synovial fluid of rheumatoid arthritis patients. Rheumatology (Oxford). 2000; 39:1139–46. Cohen K, Ellis M, Khoury S, Davis PJ, Hercbergs A, Ashur-Fabian O. Thyroid hormone is a MAPK-dependent growth factor for human myeloma cells acting via alphavbeta3 integrin. Molecular cancer research : MCR. 2011;9:1385-94. 159 Bibliografía Cooper CR, Chay CH, Pienta KJ: The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia 2002, 4:191-194 Corbetta S, Eller-Vainicher C, Vicentini L, Lania A, Mantovani G, Beck-Peccoz P, Spada A. Modulation of cyclin D1 expression in human tumoral parathyroid cells: effects of growth factors and calcium sensing receptor activation. Cancer Lett. 2007; 255(1):34-41. Cordeiro A, Lopes Souza L, Einicker-Lamas M, Pazos-Moura CC. Non-classic thyroid hormone signaling involved in hepatic lipid metabolism. Journal of endocrinology, 2013; 216,R47-R57. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007; 143:854–59. Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M, Hsu L, Krishnamurthy S, Theriault RL, Hortobagyi GN. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005 Mar 15;103(6):1122-8 Cuadros M, Dave SS, Jaffe ES, Honrado E, Milne R, Alves J, Rodríguez J, Zajac M, Benitez J, Staudt LM, Martinez-Delgado B. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas. J Clin Oncol. 2007; 25(22):3321-9. Cuccaro A, Bartolomei F, Cupelli E, Galli E, Giachelia M, Hohaus S. Prognostic factors in hodgkin lymphoma. Mediterr J Hematol Infect Dis. 2014; 6(1):e2014053. Dang D, Bamburg JR, Ramos DM: Alphavbeta3 integrin and cofilin modulate K1735 melanoma cell invasion. Exp Cell Res 2006, 312:468-477 Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, Miller TP, LeBlanc M, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Connors JM, Lansdorp PM, Ouyang Q, Lister TA, Davies AJ, Norton AJ, Muller-Hermelink HK, Ott G, Campo E, Montserrat E, Wilson WH, Jaffe ES, Simon R, Yang L, Powell J, Zhao H, Goldschmidt N, Chiorazzi M, Staudt LM. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004 Nov 18;351(21):2159-69. Davies T, Marians R & Latif R. The TSH receptor reveals itself. Journal of Clinical Investigation 2002; 110 161–164. Davis FB, Mousa SA, O´Connor L, Mohamed S, Lin HY, Cao HJ, Davis PJ. Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Cir Res 2004; 94: 1500-1506. Davis FB, Tang H, Shih A, Keating T, Lansing L, Hercbergs A, Fenstermaker R, Mousa A, Mousa S, Davis P, Lin H. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Res. 2006; 66(14): 7270-5. Davis PJ, Davis FB, Lin HY, Mousa SM & Luidens MK. Translational implications of nongenomic actions of thyroid hormones initiated at its integrin receptor. Am J Physiol Endocrinol Metab, 2009; 297: E1238-E1246. Davis PJ, Davis FB, Mousa S, Luidens M, Lin H. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol. 2011; 51:99-115. Davis PJ, Shih A, Lin H, Martino L, Davis F. Thyroxine promotes association of mitogenactivated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J Biol Chem. 2000; 275 (48): 38032-9. Davis SL. Environmental modulation of the immune system via the endocrine system. Domestic Animal Endocrinology. 1998; 15: 283-289. 160 Bibliografía de Leval L, Gaulard P. Tricky and terrible T-cell tumors: these are thrilling times for testing: molecular pathology of peripheral T-cell lymphomas. Hematology Am Soc Hematol Educ Program. 2011;2011:336-43. de Leval L, Rickman DS, Thielen C, Reynies Ad, Huang YL, Delsol G, Lamant L, Leroy K, Brière J, Molina T, Berger F, Gisselbrecht C, Xerri L, Gaulard P. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007; 109(11):4952-63. Dearden CE, Johnson R, Pettengell R, Devereux S, Cwynarski K, Whittaker S, McMillan A; British Committee for Standards in Haematology. Guidelines for the management of mature Tcell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma). Br J Haematol. 2011; 153(4):451-85. Dearden CE, Matutes E. Alemtuzumab in T-cell lymphoproliferative disorders. Best Pract Res Clin Haematol. 2006; 19:795–810. Deeths M, Chapman J, Dellavalle R, Zeng C, Aeling JL. Treatment of patch and plaque stage mycosis fungoides with imiquimod 5% cream. J Am Acad Dermatol. 2005; 52:275–80. Dentice M1, Salvatore D. Deiodinases: the balance of thyroid hormone: local impact of thyroid hormone inactivation. J Endocrinol. 2011; 209(3):273-82. doi: 10.1530/JOE-11-0002. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature reviews Cancer. 2010;10:9-22. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Mycactivated microRNA cluster. Nat Genet. 2006; 38(9):1060-5. Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA, Rafii S. Inhibition of both paracrine and autocrine VEGF/ VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A. 2001; 98(19):10857-62 Dietrich, J.W.; Landgrafe, G.; Fotiadou, E.H. TSH and thyrotropic agonists: Key actors in thyroid homeostasis. J. Thyroid Res. 2012; 2012, 351864:1–51864:29. Dişel U, Beşen A, Karadeniz C, Mertsoylu H, Sezer A, Köse F, TanerSümbül A, Gürkut O, Muallaoğlu S, Abali H, Ozyilkan O. Prevalence of thyroid dysfunction in untreated cancer patients: a cross-sectional study. Med Oncol. 2012; 29(5):3608-13. Döbbeling U.Transcription factor profiling shows new ways towards new treatment options of cutaneous T cell lymphomas. Curr Drug Discov Technol. 2007;4(1):24-30. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005 May;446(5):475-82. Doussis-Anagnostopoulou IA, Talks KL, Turley H, Debnam P, Tan DC, Mariatos G, Gorgoulis V, Kittas C, Gatter KC. Vascular endothelial growth factor (VEGF) is expressed by neoplastic Hodgkin-Reed-Sternberg cells in Hodgkin's disease. J Pathol. 2002; 197(5):677-83. Dugas JC1, Ibrahim A, Barres BA. The T3-induced gene KLF9 regulates oligodendrocyte differentiation and myelin regeneration. Mol Cell Neurosci. 2012;50(1):45-57. Dummer R, Goldinger SM, Cozzio A, French LE, Karpova MB. Cutaneous lymphomas: molecular pathways leading to new drugs. J Invest Dermatol. 2012; 132(3 Pt 1):517-25. 161 Bibliografía Dumont J., Opitz R., Christophe D., Vassart G., Roger P., Maenhaut C. Chapter 1 – The Phylogeny, Ontogeny, Anatomy and Regulation of the Iodine Metabolizing Thyroid. En Groot L. Thyroid Disease Manager. 2008. Evens AM, Gartenhaus RB. Treatment of T-cell non-Hodgkin's lymphoma. Curr Treat Options Oncol. 2004; 5(4):289-303 Farinha P, Masoudi H, Skinnider BF, Shumansky K, Spinelli JJ, Gill K, Klasa R, Voss N, Connors JM, Gascoyne RD. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL). Blood. 2005; 106(6):2169-74. Felding-Habermann B, O'Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, Mueller BM: Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA. 2001, 98:1853-1858. Feng R, Oton A, Mapara MY, Anderson G, Belani C, Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007; 139:385–97 Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003; 9(6):669-76. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature.2005; 438(7070):967-74. Fishwild DM, O'Donnell SL, Bengoechea T, et al. High-avidity human IgG kappa monoclonal antibodies from a novel strain of minilocus transgenic mice. Nat Biotechnol. 1996; 14:845–51. Fon P. Thyroid iodide efflux: a team effort?. J Physiol. 2011;15;589(Pt 24):5929-39. Foss HD, Araujo I, Demel G et al. Expression of vascular endothelial growth factor in lymphomas and Castleman’s disease. J Pathol 1997; 183: 44–50. Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011; 117(25):6756-67. Fragoso R1, Pereira T, Wu Y, Zhu Z, Cabeçadas J, Dias S. VEGFR-1 (FLT-1) activation modulates acute lymphoblastic leukemia localization and survival within the bone marrow, determining the onset of extramedullary disease. Blood. 2006; 107(4):1608-16. Epub 2005 Oct 25. Frampton JE, Wagstaff AJ. Alemtuzumab. Drugs. 2003; 63:1229–43. Freindorf M1, Furlani TR, Kong J, Cody V, Davis FB, Davis PJ. Combined QM/MM study of thyroid and steroid hormone analogue interactions with αvβ3 integrin. Journal of Biomedicine Biotechnology. 2012;2012:959057. Fukumura D1, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK, Seed B. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998; 94(6):71525. Gallamini A, Stelitano C, Calvi R, Bellei M, Mattei D, Vitolo U, Morabito F, Martelli M, Brusamolino E, Iannitto E, Zaja F, Cortelazzo S, Rigacci L, Devizzi L, Todeschini G, Santini G, Brugiatelli M, Federico M; Intergruppo Italiano Linfomi. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood. 2004; 103(7):2474-9. Ganjoo KN, An CS, Robertson MJ, Gordon LI, Sen JA, Weisenbach J, Li S, Weller EA, Orazi A, Horning SJ. Rituximab, bevacizumab and CHOP (RA-CHOP) in untreated diffuse large B-cell 162 Bibliografía lymphoma: safety, biomarker and pharmacokinetic analysis. Leuk Lymphoma. 2006; 47(6):9981005. Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM. Phase 1 study of the histone deacetylase inhibitor vorinostat in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008; 111:1060–66. Gaulard P, de Leval L.The microenvironment in T-cell lymphomas: emerging themes. Semin Cancer Biol. 2014;24:49-60. Geissinger E, Odenwald T, Lee SS, Bonzheim I, Roth S, Reimer P, Wilhelm M, MüllerHermelink HK, Rüdiger T. Nodal peripheral T-cell lymphomas and, in particular, their lymphoepithelioid (Lennert's) variant are often derived from CD8(+) cytotoxic T-cells. Virchows Arch. 2004; 445(4):334-43. George P, Bali P, Annavarapu S, Scuto A, Fiskus W, Guo F, Sigua C, Sondarva G, Moscinski L, Atadja P, Bhalla K. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005; 105:1768–76 Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeöld A, Bianco AC.. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr 2008; Rev 29:898–938. Geskin LJ, Akilov OE, Lin Y, Lokshin AE. Distinct age-matched serum biomarker profiles in patients with cutaneous T-cell lymphoma. Exp Dermatol. 2014;23(8):598-600 Ghahremani MF, Goossens S, Nittner D, Bisteau X, Bartunkova S, Zwolinska A, Hulpiau P, Haigh K, Haenebalcke L, Drogat B,Jochemsen A, Roger PP, Marine JC, Haigh JJ. p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ. 2013; 20(7):888-97. Giani C, Fierabracci P, Bonacci R, Gigliotti A, Campani D, De Negri F, Cecchetti D, Martino E, Pinchera A. Relationship between breast cancer and thyroid disease: relevance of autoimmune thyroid disorders in breast malignancy. J Clin Endocrinol Metab. 1996; 81(3):990-4. Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006; 12:4628–35 Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP: Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol 1995, 57:143-53. Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol. 2007; 74:659–71. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nature reviews Cancer. 2013;13:871-82. Gomes N, Vassy J, Lebos C, Arbeille B, Legrand C, Fauvel-Lafeve F: Breast adenocarcinoma cell adhesion to the vascular subendothelium in whole blood and under flow conditions: effects of alphavbeta3 and alphaIIbbeta3 antagonists. Clin Exp Metastasis 2004, 21:553-561. Graves PN, Davies TF. New insights into the thyroid-stimulating hormone receptor. The major antigen of Graves' disease. Endocrinol Metab Clin North Am. 2000 Jun;29(2):267-86 163 Bibliografía Gregory A. Brent. Mechanisms of thyroid hormone action. J Clin Invest. 2012, 122(9): 3035– 3043. Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, Miyazaki J, Suda T, Itoh H, Nakao K, Mak TW, Nakano T, Suzuki A. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005; 19(17):2054-65. Hamamura RS, Ohyashiki JH, Kurashina R, Kobayashi C, Zhang Y, Takaku T, Ohyashiki K. Induction of heme oxygenase-1 by cobalt rotoporphyrin enhances the antitumour effect of bortezomib in adult T-cell leukaemia cells. Br J Cancer. 2007; 97:1099–1105. Hamnvik OP, Larsen PR, Marqusee E. Thyroid dysfunction from antineoplastic agents. Journal of the National Cancer Institute. 2011;103:1572-87. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-74. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100: 57-70. Hapke S, Kessler H, Luber B, Benge A, Hutzler P, Hofler H, Schmitt M, Reuning U: Ovarian cancer cell proliferation and motility is induced by engagement of integrin alpha(v)beta3/vitronectin interaction. Biol Chem 2003, 384:1073-1083 Harbour JW, Dean DC. Chromatin remodeling and Rb activity. Curr Opin Cell Biol. 2000; 12(6):685-9. Harms JF, Welch DR, Samant RS, Shevde LA, Miele ME, Babu GR, Goldberg SF, Gilman VR, Sosnowski DM, Campo DA, Gay CV, Budgeon LR, Mercer R, Jewell J, Mastro AM, Donahue HJ, Erin N, Debies MT, Meehan WJ, Jones AL, Mbalaviele G, Nickols A, Christensen ND, Melly R, Beck LN, Kent J, Rader RK, Kotyk JJ, Pagel MD, Westlin WF, Griggs DW. A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metástasis. Clin Exp Metastasis 2004, 21:119-128; Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84(5):1361-92 Hellevik AI1, Asvold BO, Bjøro T, Romundstad PR, Nilsen TI, Vatten LJ. Thyroid function and cancer risk: a prospective population study. Cancer Epidemiol Biomarkers Prev. 2009 Feb;18(2):570-4 Hercbergs AH, Ashur-Fabian O, Garfield D. Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Current opinion in endocrinology, diabetes, and obesity. 2010;17:432-6. Hernando E, Nahlé Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004; 430(7001):797-802. Heuer H, Visser TJ. Minireview: pathophysiological importance of thyroid hormone transporters. Endocrinology 2009; 150:1078–1083. Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Norma K,Ueki K, Nguyen NH, Scanlan TS. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006; 103(38):14104-9. Hood JD and Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer 2002; 2: 91-100. 164 Bibliografía Horie R, Watanabe T. CD30: expression and function in health and disease. Semin Immunol. 1998;10:457–70. Hörmann R, Schumm-Draeger PM. Organ manifestations of hyperthyroidism. Internist (Berl). 2010; 51(5):596-602. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 200;12;20(31):4188-97. Hulbert AJ. Thyroid hormones and their effects: a new perspective. Biol Rev Camb Philos Soc. 2000 Nov;75(4):519-631. Hung-Yun Lin , Mingzeng Sun , Heng-Yuan Tang , Cassie Lin , Mary K. Luidens , Shaker A. Mousa , Sandra Incerpi , George L. Drusano , Faith B. Davis , Paul J. Davis. l-Thyroxine vs. 3,5,3′-triiodo-l-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. American Journal of Physiology 2009; 296:5, C980-C991 Igreja C, Courinha M, Cachaço AS, Pereira T, Cabeçadas J, da Silva MG, Dias S. Characterization and clinical relevance of circulating and biopsy-derived endothelial progenitor cells in lymphoma patients. Haematologica. 2007; 92(4):469-77. Imanishi Y, Hosokawa Y, Yoshimoto K, Schipani E, Mallya S, Papanikolaou A, Kifor O, Tokura T, Sablosky M, Ledgard F, Gronowicz G, Wang TC, Schmidt EV, Hall C, Brown EM, Bronson R, Arnold A. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J Clin Invest. 2001; 107(9):1093-102 Jaffe ES. Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod Pathol. 2001; 14(3):219-28 Jaffe ES. Pathobiology of peripheral T-cell lymphomas. Hematology Am Soc Hematol Educ Program. 2006: 317-22 Jaffe ES. The 2008 WHO classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program. 2009:523-31. Jarrousse V, Quereux G, Marques-Briand S, Knol AC, Khammari A, Dreno B. Toll-like receptors 2, 4 and 9 expression in cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome). Eur J Dermatol. 2006; 16:636–41. Jordan BF, Christian N, Crokart N, Grégoire V, Feron O, Gallez B. Thyroid status is a key modulator of tumor oxygenation: implication for radiation therapy. Radiat Res. 2007;168(4):42832). Jorgensen JM, Sorensen FB, Bendix K, Nielsen JL, Funder A, Karkkainen MJ, et al. Expression level, tissue distribution pattern, and prognostic impact of vascular endothelial growth factors VEGF and VEGF-C and their receptors Flt-1, KDR, and Flt-4 in different subtypes of nonHodgkin lymphomas. Leukemia & lymphoma. 2009;50:1647-60. Jørgensen JM, Sørensen FB, Bendix K, Nielsen JL, Olsen ML, Funder AM, d'Amore F. Angiogenesis in non-Hodgkin's lymphoma: clinico-pathological correlations and prognostic significance in specific subtypes. Leuk Lymphoma. 2007; 48(3):584-95. Kaelin WG Jr. The von hippel-lindau tumor suppressor protein: an update. Methods Enzymol. 2007;435:371-83. Kahl C, Leithäuser M, Wolff D, Steiner B, Hartung G, Casper J, Freund M. Treatment of peripheral T-cell lymphomas (PTCL) with high-dose chemotherapy and autologous or allogeneic hematopoietic transplantation. Ann Hematol. 2002; 81(11):646-50. 165 Bibliografía Kano Y, Akutsu M, Tsunoda S, Izumi T, Kobayashi H, Mano H, Furukawa Y. Cytotoxic effects of histone deacetylase inhibitor FK228 (depsipeptide, formally named FR901228) in combination with conventional anti-leukemia/ lymphoma agents against human leukemia/lymphoma cell lines. Invest New Drugs. 2007; 25:31–40. Kay NE, Bone ND, Tschumper RC et al. B-CLL cells are capable of synthesis and secretio n of both pro- and anti-angiogenic molecules. Leukemia 2002; 16:911–919. Khan S, Lopez-Dee Z, Kumar R, Ling J. Activation of NFkB is a novel mechanism of prosurvival activity of glucocorticoids in breast cancer cells. Cancer Lett. 2013 28;337(1):90-5 Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993; 362(6423):841-4. Kim YH, Duvic M, Obitz E, et al. Clinical efficacy of zanolimumab (HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood. 2007; 109:4655–62. Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006 Sep;13(3):739-49. Kini AR. Angiogenesis in leukemia and lymphoma. Cancer Treat Res. 2004;121:221-38. Klemke CD. Cutaneous lymphomas. J Dtsch Dermatol Ges. 2014 Jan;12(1):7-28; quiz 29-30. Kojima H, Hasegawa Y, Suzukawa K, Mukai HY, Kaneko S, Kobayashi T, Kamoshita M, Shinagawa A, Komeno T, Komatsu T, Mitsuhashi S, Kawachi Y, Yamashita Y, Mori N, Nagasawa T. Clinicopathological features and prognostic factors of Japanese patients with "peripheral T-cell lymphoma, unspecified" diagnosed according to the WHO classification. Leuk Res. 2004; 28(12):1287-92. Konstantinou K, Yamamoto K, Ishibashi F, Mizoguchi Y, Kurata M, Nakagawa Y, et al. Angiogenic mediators of the angiopoietin system are highly expressed by CD10-positive lymphoma cells in angioimmunoblastic T-cell lymphoma. British journal of haematology. 2009;144:696-704. Kopp P. Thyroid hormone synthesis; in Braverman LE, Cooper D: Werner and Ingbar´s The Thyroid. A fundamental and Clinical Text, ed20. Philadelphia, Lippincott Williams & Wilkins, 2013, pp 48-74. Koster A y Raemaekers JMM. Angiogenesis in malignant lymphoma. Curr Opin Oncol. 2005; 17: 611-616. Koster A, van Krieken JH, Mackenzie MA, Schraders M, Borm GF, van der Laak JA, Leenders W, Hebeda K, Raemaekers JM. Increased vascularization predicts favorable outcome in follicular lymphoma. Clin Cancer Res. 2005; 11(1):154-61. Koulouri, Moran, Halsall, Chatterjee, and Gurnella. Pitfalls in the measurement and interpretation of thyroid function tests. Best Pract Res Clin Endocrinol Metab. Dec 2013; 27(6): 745–762. Kurzrock R, Pilat S, Duvic M. Pentostatin therapy of T-cell lymphomas with cutaneous manifestations. J Clin Oncol. 1999; 17:3117–21. [PubMed: 10506607] Lamant L, de Reyniès A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S, Brugières L, Gaulard P, Espinos E, Delsol G. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007; 109(5):2156-64 166 Bibliografía Laudet V. The origins and evolution of vertebrate metamorphosis.Current Biology, 2011, 21:R726–R737 Leach FS, Elledge SJ, Sherr CJ, Willson JKV, Markowitz S. Amplification of cyclin genes in colorectal carcinomas. Cancer Res. 1993; 53:1986–1989. Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004; 104(3):788-94. Lehrer S, Diamond EJ, Bajwa AM, Kornreich R, Stagger S, Stone NN, Droller MJ, Stock RG. Association between serum triiodothyronine (t3) level and risk of disease recurrence in men with localized prostate cancer. Prostate Cancer Prostatic Dis. 2001;4(4):232-234. Lehrer S, Diamond EJ, Stone NN, Droller MJ, Stock RG. Serum triiodothyronine is increased in men with prostate cancer and benign prostatic hyperplasia. J Urol. 2002;168(6):2431-3. Leonard JP, Furman RR, Coleman M. Proteasome inhibition with bortezomib: a new therapeutic strategy for non-Hodgkin's lymphoma. Int J Cancer. 2006; 119:971–79. Lester Reed H. Thyroid Physiology: Síntesis and release, iodine metabolism, binding and transport. En Becker K. Principles and practice of endocrinology and metabolism. 2001; 3rd Edition. Wartofsky L. (Ed). Part III. The thyroid gland. Chapter 30. Lippincott Williams y Wilkins Publishers. pp 314-318. Li T, Kung HJ, Mack PC, Gandara DR. J Clin Oncol. 2013 10;31(8):1039-49. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. Liapis H, Flath A, Kitazawa S: Integrin alpha V beta 3 expression by bone-residing breast cancer metastases. Diagn Mol Pathol 1996, 5:127-35. Lin HY, Su YF, Hsieh MT, Lin S, Meng R, London D, et al. Nuclear monomeric integrin alphav in cancer cells is a coactivator regulated by thyroid hormone. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:3209-16. Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, et al. L-Thyroxine vs. 3,5,3'-triiodo-Lthyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. American journal of physiology Cell physiology. 2009;296:C98091. Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene.1999; 18(55):7873-82. Liu Y, Kruhlak MJ, Hao JJ, Shaw S. Rapid T cell receptor-mediated SHP-1 S591 phosphorylation regulates SHP-1 cellular localization and phosphatase activity. Journal of leukocyte biology. 2007;82:742-51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4):402-8. Livolsi V. Morphology of the thyroid gland. En Becker K. (Ed). Principles and practice of endocrinology and metabolism, 2001; 3rd Edition. Wartofsky L. (Ed). Part III. The thyroid gland. Chapter 29. Lippincott Williams y Wilkins Publishers. pp 311-313. Lorger M, Krueger JS, O'Neal M, Staflin K, Felding-Habermann B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10666-71. Luidens MK, Mousa SA, Davis FB, Lin HY, Davis PJ. Thyroid hormone and angiogenesis. Vascul Pharmacol. 2010; 52 (3-4):142-145. 167 Bibliografía Lundin J, Hagberg H, Repp R, Cavallin-Ståhl E, Fredén S, Juliusson G, Rosenblad E, Tjønnfjord G, Wiklund T, Osterborg A. Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome. Blood. 2003; 101:4267– 72. Maiso P, Carvajal-Vergara X, Ocio EM, López-Pérez R, Mateo G, Gutiérrez N, Atadja P, Pandiella A, San Miguel JF. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006; 66:5781–89. Makino A, Suarez J, Wang H, et al. Thyroid hormone receptor-beta is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinology. 2009; 150:2008–15. Malik R & Hodgson H. The relationship between the thryroidglans and the liver. QJM: Monthly Journal ofthe Association of Physicians, 2002; 95 559-569. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007; 12:1247–52. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129(7):126174. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454(7203):436-44. Marchi E, Alinari L, Tani M, et al. Gemcitabine as frontline treatment for cutaneous T-cell lymphoma: phase II study of 32 patients. Cancer. 2005; 104:2437–41 Martinez-Delgado B, Meléndez B, Cuadros M, Alvarez J, Castrillo JM, Ruiz De La Parte A, Mollejo M, Bellas C, Diaz R, Lombardía L, Al-Shahrour F, Domínguez O, Cascon A, Robledo M, Rivas C, Benitez J. Expression profiling of T-cell lymphomas differentiates peripheral and lymphoblastic lymphomas and defines survival related genes. Clin Cancer Res. 2004; 10(15):4971-82. Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, et al. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PloS one. 2009;4:e6986. Mashima R, Hishida Y, Tezuka T, Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunological reviews. 2009;232:273-85. McGinnis KS, Junkins-Hopkins JM, Crawford G, Shapiro M, Rook AH, Vittorio CC. Low-dose oral bexarotene in combination with low-dose interferon alfa in the treatment of cutaneous T-cell lymphoma: clinical synergism and possible immunologic mechanisms. J Am Acad Dermatol. 2004; 50:375–79. Metcalf RA, Zhao S, Anderson MW, Lu ZS, Galperin I, Marinelli RJ, Cherry AM, Lossos IS, Natkunam Y. Characterization of D-cyclin proteins in hematolymphoid neoplasms: lack of specificity of cyclin-D2 and D3 expression in lymphoma subtypes. Mod Pathol. 2010; 23(3):420433. Mirzayans R, Andrais B, Hansen G, Murray D. Role of p16 (INK4A) in Replicative Senescence and DNA Damage-Induced Premature Senescence in p53-Deficient Human Cells. Biochem Res Int. 2012a:951574. Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol. 2012b:170325. 168 Bibliografía Moeller LC y Broecker-Preuss M. Transcriptional regulation by nonclassical action thyroid homrone. Thyroid Research, 2011; 4 (Suppl 1) S6. Moeller LC, Dumitrescu AM, Refetoff S. Cytosolic action of thyroid hormone leads to induction of hypoxia-inducible factor-1alpha and glycolytic genes. Mol Endocrinol. 2005;19(12):2955-63. Moeller LC, Führer. Thyroid hormone, thyroid hormone receptors, and cancer: a clinical perspective. D.Endocrine Related Cancer. 2013; 22;20(2):R19-29. Mohn A, Di Marzio A, Cerruto M, Angrilli F, Fioritoni C, Chiarelli F. Euthyroid sick syndrome in children with Hodgkin disease. Pediatr Hematol Oncol. 2001; 18(3):211-5. Møller MB, Nielsen O, Pedersen NT. Cyclin D3 Expression in Non-Hodgkin Lymphoma Correlation With Other Cell Cycle Regulators and Clinical Features. Am J Clin Pathol 2001;115:404-412 Mondul AM, Weinstein SJ, Bosworth T, Remaley AT, Virtamo J, Albanes D. Circulating thyroxine, thyroid-stimulating hormone, and hypothyroid status and the risk of prostate cancer. PLoS One. 2012;7(10):e47730 Monestiroli S, Mancuso P, Burlini A, Pruneri G, Dell'Agnola C, Gobbi A, Martinelli G, Bertolini F. Kinetics and viability of circulating endothelial cells as surrogate angiogenesis marker in an animal model of human lymphoma. Cancer Res. 2001; 61(11):4341-4. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, Dal Cin P, Ladd C, Pinkus GS, Salles G, Harris NL, Dalla-Favera R, Habermann TM, Aster JC, Golub TR, Shipp MA. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105(5):1851-61. Mori F, Ishida T, Ito A, Sato F, Masaki A, Narita T, Suzuki S, Yamada T, Takino H, Ri M, Kusumoto S, Komatsu H, Hishizawa M, Imada K, Takaori-Kondo A, Niimi A, Ueda R, Inagaki H, Iida S. Antitumor effects of bevacizumab in a microenvironment-dependent human adult T-cell leukemia/lymphoma mouse model. Eur J Haematol. 2014;92(3):219-28. Mousa SA, Bergh JJ,Dier E. Tetraiodothyroacetic acid a samall molecule integrin ligand, blocks angiogenesis induced by vascular endothelialgrowth factora and basic fibroblast factor. Angiogénesis 2008;11: 183-190. Mousa SA, O´Connor L, Davis FB. Proangiogenesis action of the thyroid hormone analog 3,5diiodothyropropionic (DITPA) is initiated at the cell surface and is integrin mediated. Endocrinology. 2006;147:1602-1607. Mousa SA, Yalcin M, Bharali DJ, Meng R, Tang HY, Lin HY, Davis FB, Davis PJ.Tetraiodothyroacetic acid and its nanoformulation inhibit thyroid hormone stimulation of nonsmall cell lung cancer cells in vitro and its growth in xenografts. Lung Cancer. 2012; 76(1):39-45 Murakami A, Tanaka H. Identification of RB and p53 mutations in mouse lymphoma cell lines. Mol Carcinog. 1991; 4(5):354-7. Nagai H, Kinoshita T, Ichikawa A, Murate T. Malignant Lymphoma and Tumor Suppressor Genes. J. Clin. Exp. Hemathopatol. 2002; 42(1):11-24. Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab, 2007; 21(2): 193-208. Oliveira-Ferrer L, Hauschild J, Fiedler W, Bokemeyer C, Nippgen J, Celik I, Schuch G. Cilengitide induces cellular detachment and apoptosis in endothelial and glioma cells mediated by inhibition of FAK/src/AKT pathway. J Exp Clin Cancer Res. 2008; 29;27:86 169 Bibliografía Patel J, Landers K, Li H, Mortimer RH, Richard K. Thyroid hormones and fetal neurological development. J Endocrinol. 2011; 209:1–8 Payaningal R. Somanath, Alieta Ciocea, and Tatiana V. ByzovaJoseph J. Jacobs. Integrin and Growth Factor Receptor Alliance in Angiogenesis. Cell Biochem Biophys. 2009; 53(2): 53-64 Pedersen IH, Willerslev-Olsen A, Vetter-Kauczok C, Krejsgaard T, Lauenborg B, Kopp KL, Geisler C, Bonefeld CM, Zhang Q, Wasik MA, Dabelsteen S, Woetmann A, Becker JC, Odum N. Vascular endothelial growth factor receptor-3 expression in mycosis fungoides. Leuk Lymphoma. 2013;54(4):819-26 Penning TM1, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33-42. Perri A, Catalano S, Bonofiglio D, Vizza D, Rovito D, Qi H, Aquila S, Panza S, Rizza P, Lanzino M, Andò S. T3 enhances thyroid cancer cell proliferation through TRβ1/Oct-1-mediated cyclin D1 activation. Mol Cell Endocrinol. 2014 25;382(1):205-17 Petrich AM, Rosen ST. Peripheral T-cell lymphoma: new therapeutic strategies. Oncology (Williston Park). 2013; 27(9):878-84. Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer research. 2007;67:10703-10. Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, Went P, Klein U, Zinzani PL, Baccarani M, Dalla Favera R, Pileri SA. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007; 117(3):823-34 Piekarz RL1, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001; 98:2865– 68 Pinto M, Soares P, Ribatti D. Thyroid hormone as a regulator of tumor induced angiogenesis. Cancer Lett. 2011;301: 119-126. Pizzolo G, Vinante F, Chilosi M, Romagnani S, Del Prete G. CD30 antigen and cellular biology of Reed-Sternberg cells. Blood. 1994; 84:3983–84. Potenza M, Via M, Yanagisawa R. Excess thyroid hormone and carbohydrate metabolism Organ Endocr Pract, 2009; 15(3): 254-62. Pucci E, Chiovato L, Pichera A. Thyroid and lipid metabolism. International Journal of Obesity and Related Metabolic Disorders. 2000; 24 (Suppl 2) S109-S112. Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997;89:1394 – 404. Pulini S, Rupoli S, Goteri G, et al. Pegylated liposomal doxorubicin in the treatment of primary cutaneous T-cell lymphomas. Haematologica. 2007; 92:686–89. Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002; 2(11):826-35Kim 1993 Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005; 23:630–39. 170 Bibliografía Raziuddin S, Sheikha A, Abu-Eshy S, Al-Janadi M. Regulation of interleukin-4 production and cytokine-induced growth potential in peripheral T-cell non-Hodgkin's lymphomas. British journal of haematology. 1998;100:310-6. Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A. The role of angiogenesis in human nonHodgkin lymphomas. Neoplasia. 2013; 15(3):231-8 Ribatti D, Vacca A, Nico B, Fanelli M, Roncali L, Dammacco F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin's lymphomas. An immunohistochemical and ultrastructural study. Eur J Haematol. 1996; 56(1-2):45-53. Richards KL, McCullough J. A modified microchamber method for chemotaxis and chemokinesis. Immunological communications. 1984;13:49-62. Rider DA, Havenith CE, de Ridder R, Schuurman J, Favre C, Cooper JC, Walker S, Baadsgaard O, Marschner S, vandeWinkel JG, Cambier J, Parren PW, Alexander DR. A human CD4 monoclonal antibody for the treatment of T-cell lymphoma combines inhibition of T-cell signaling by a dual mechanism with potent Fc-dependent effector activity. Cancer Res. 2007; 67(20):9945-53. Roberti A, Macaluso M, Giordano A. Alterations in Cell Cycle Regulatory Genes in Breast Cancer. Breast Cancer in the Post-Genomic Era. Current Clinical Oncology. 2009; 55-77 Rodriguez-Abreu D, Filho VB, Zucca E. Peripheral T-cell lymphomas, unspecified (or not otherwise specified): a review. Hematol Oncol. 2008; 26:8–20. Rolli M, Fransvea E, Pilch J, Saven A, Felding-Habermann B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc Natl Acad Sci USA 2003, 100:9482-9487. Romano A, Vetro C, Caocci G, Greco M, Parrinello NL, Di Raimondo F, La Nasa G. Immunological deregulation in classic hodgkin lymphoma. Mediterr J Hematol Infect Dis. 2014; 6(1):e2014039. Ruan J, Hajjar K, Rafii S and Leonard JP. Angiogenesis and antiangiogenic theraphy in nonHodking´s lymphoma. Ann Oncol. 2009; 20: 413-424. Ruan J, Hyjek E, Kermani P, Christos PJ, Hooper AT, Coleman M, Hempstead B, Leonard JP, Chadburn A, Rafii S. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin's lymphoma. Clin Cancer Res. 2006; 1;12(19):5622-31. Rüdiger T, Ichinohasama R, Ott MM, Müller-Deubert S, Miura I, Ott G, Müller- Hermelink HK. Peripheral T-cell lymphoma with distinct perifollicular growth pattern: a distinct subtype of T-cell lymphoma? Am J Surg Pathol. 2000; 24(1):117-22. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005; 9(2):267-85. Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M. PTEN gene alterations in lymphoid neoplasms. Blood. 1998; 92(9):3410-5. Salati M, Cesaretti M, Macchia M, Mistiri ME, Federico M. Epidemiological Overview of Hodgkin Lymphoma across the Mediterranean Basin. Mediterr J Hematol Infect Dis. 2014; 6(1):e2014048. Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002; 8:718–28. Savage KJ. Peripheral T-cell lymphomas. Blood Rev. 2007; 21(4):201-16 171 Bibliografía Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer research. 2012;32:4213-23. Semenza GL. Regulation of vascularization by hypoxia-inducible factor 1. Ann N Y Acad Sci. 2009 ;1177:2-8 Serini G, Napione L, et al. Besides adhesion: New perspectives of integrin functions in angiogenesis.Cardiovascular Research 2008a;78(2):213-22220. Serini G, Napione L, et al. Integrins team up with tyrosine kinase receptors and plexins to controlangiogenesis. Current Opinion in Hematology 2008b;15(3):235-242.; Serrano-Nascimento C, Calilis-SilveiraJ & Nunes MT. Posttranscripcional regulation ofsodiumiodide symporter mRNA expression in the thyrod gland by acute iodide administration. Am J Physiol Cell Physiol. 2010; 298(4):C893-9. Shih A, Zhang S, Cao HJ,Tang HY, Davis FB, Davis PJ & Lin HY. Disparate effects of thyoid hormone on actions of epidermal growth factor and transforming growth factor-α are mediated by 3´5´- cyclic adenosine 5´monophosphate-dependent protein kinase II. Endocrinology. 2004; 145(4):1708-17. Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR. Diffuse large B-cell lymphoma outcome prediction by geneexpression profiling and supervised machine learning. Nat Med. 2002; 8(1):68-74. Shoemaker JP, Bradley RL, Hoffman RV. Increased Survival and Inhibition of Mammary Tumors in Hypothyroid Mice. J Surg Res. 1976; 21(3):151-4. Siddon AJ, Torres R, Rinder HM, Smith BR, Howe JG, Tormey CA. Normalized CCND1 expression has prognostic value in mantle cell lymphoma. Br J Haematol. 2012; 158(4):551-3. Simoncini T, Hafezi-Moghadam A,Brazil DP, Ley K, Chin WW and Liao JK. Interaction of estrogen receptor with regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000; 407(6803):538-41. Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, Anderson RL: Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res 2006, 8:R20 Soussi T. p53 protein, biological and clinical aspects. Encyclopedic Reference of Cancer. 2001; 669-675. Sterle HA, Valli E, Cayrol F, Paulazo MA, Martinel Lamas DJ, Diaz Flaqué MC, Klecha AJ, Colombo L, Medina VA, Cremaschi GA, Barreiro Arcos ML. Thyroid status modulates T lymphoma growth via cell cycle regulatory proteins and angiogenesis. J Endocrinol. 2014; 222(2):243-55 Sulis ML, Parsons R.PTEN: from pathology to biology. Trends Cell Biol.2003; 13(9):478-83. Takahashi T1, Ueno H, Shibuya M.VEGF activates protein kinase C-dependent, but Rasindependent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999; 18(13):2221-30. Takano Y, Kato Y, van Diest PJ, Masuda M, Mitomi H, Okayasu I. Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am J Pathol. 2000; 156:585-594. 172 Bibliografía Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppä S. A high tumorassociated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res. 2007;13(19):5784-9. Tata JR. The road to nuclear receptors of thyroid hormone. Biochim Biophys Acta. 2013; 1830(7):3860-6 The Lymphoma Guide Information for Patients and Caregivers. Leukemia and Lymphoma Society. 2013; Retrieved 2014. Thakur S, Lin HC, Tseng WT, Kumar S, Bravo R, Foss F, Gélinas C, Rabson AB. Rearrangement and altered expression of the NFKB-2 gene in human cutaneous T-lymphoma cells. Oncogene. 1994;9(8):2335-44. Theodossiou C, Schwarzenberger P. Propylthiouracil reduces xenograft tumor growth in an athymic nude mouse prostate cancer model. Am J Med Sci. 2000; 319(2):96-9. Theodossiou C, Skrepnik N, Robert EG, Prasad C, Axelrad TW, Schapira DV, Hunt JD. Propylthiouracil-induced hypothyroidism reduces xenograft tumor growth in athymic nude mice. Cancer. 1999 Oct 15;86(8):1596-601. Tirado CA, Chen W, García R, Kohlman KA, Rao N. Genomic profiling using array comparative genomic hybridization define distinct subtypes of diffuse large B-cell lymphoma: a review of the literature. J Hematol Oncol. 2012; 11;5:54. Tognini S, Marchini F, Dardano A, Polini A, Ferdeghini M, Castiglioni M, Monzani F. Nonthyroidal illness syndrome and short-term survival in a hospitalised older population. Age Ageing. 2010; 39(1):46-50. Toms SA, Hercbergs A, Liu J, Kondo S, Haqqi T, Casey G, Iwasaki K, Barnett GH, Barna BP. Antagonist effect of insulin-like growth factor I on protein kinase inhibitor-mediated apoptosis in human glioblastoma cells in association with bcl-2 and bcl-xL. J Neurosurg. 1998; 88(5):884-9. Tosovic A, Bondeson AG, Bondeson L, Ericsson UB, Manjer J. T3 levels in relation to prognostic factors in breast cancer: a population-based prospective cohort study. BMC Cancer. 2014 Jul 24;14:536. Tsimberidou AM, Giles F, Duvic M, Fayad L, Kurzrock R. Phase II study of pentostatin in advanced T-cell lymphoid malignancies: update of an M.D. Anderson Cancer Center series. Cancer. 2004; 100:342–49. Vaites LP, Lian Z, Lee EK, Yin B, DeMicco A, Bassing CH, Diehl JA ATM deficiency augments constitutively nuclear cyclin D1-driven genomic instability and lymphomagenesis. Oncogene. 2014; 33(1):129-33 Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002; 100(7):2292-302. Vardiman, JW; Thiele, J; Arber, DA; Brunning, RD; Borowitz, MJ; Porwit, A; Harris, NL; Le Beau, MM; Hellström-Lindberg, E; Tefferi, A; Bloomfield, CD (Jul 30, 2009). "The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes.". Blood 114 (5): 937–51. Vellon L, Menendez JA, Lupu R: A bidirectional "alpha(v)beta(3) integrin-ERK1/ERK2 MAPK" connection regulates the proliferation of breast cancer cells. Mol Carcinog 2006, 45:795-804. Wahl AF, Klussman K, Thompson JD, et al. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin's disease. Cancer Res. 2002; 62:3736–42. 173 Bibliografía Wang ES, Teruya-Feldstein J, Wu Y, Zhu Z, Hicklin DJ, Moore MA. Targeting autocrine and paracrine VEGF receptor pathways inhibits human lymphoma xenografts in vivo. Blood. 2004; 104(9):2893-902. Wang L, Shi WY, Yang F, Tang W, Gapihan G, Varna M, Shen ZX, Chen SJ, Leboeuf C, Janin A, Zhao WL. Bevacizumab potentiates chemotherapeutic effect on T-leukemia/lymphoma cells by direct action on tumor endothelial cells. Haematologica. 2011;96(6):927-31. Weiss A, den Bergh Hv, Griffioen AW, Nowak-Sliwinska P. Angiogenesis inhibition for the improvement of photodynamic therapy: the revival of apromising idea. Biochim Biophys Acta. 2012; 1826(1):53-70. Williams G. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol. 2008; 20(6):784-94. Wollina U, Dummer R, Brockmeyer NH, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer. 2003; 98:993–1001. Wollina U, Graefe T, Kaatz M. Pegylated doxorubicin for primary cutaneous T cell lymphoma: a report on ten patients with follow-up. Ann NY Acad Sci. 2001; 941:214–16 Xie TX, Xia Z, Zhang N, Gong W, Huang S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol Rep. 2010;23(3):725-32. Yalcin M, Dyskin E, Lansing L, Bharali DJ, Mousa SS, Bridoux A, Hercbergs AH, Lin HY, Davis FB, Glinsky GV, Glinskii A, Ma J, Davis PJ, Mousa SA. Tetraiodothyroacetic acid (tetrac) and nanoparticulate tetrac arrest growth of medullary carcinoma of the thyroid. J Clin Endocrinol Metab. 2010; 95(4):1972-80 Yalcin M, Lin HY, Sudha T, Bharali DJ, Meng R, Tang HY, Davis FB, Stain SC, Davis PJ, Mousa SA. Response of human pancreatic cancer cell xenografts to tetraiodothyroacetic acid nanoparticles. Horm Cancer. 2013;4(3):176-85. Yamauchi M, Kambe F, Cao X, Lu X, Kozaki Y,Oiso Y & Seo H. Thyroid hormone activates adenosine 5´-monophosphate-activated protein kinase vía intracelular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase-β. Mol Endocrinol. 2008; 22(4):893903. Yancopoulos GD1, Davis S, Gale NW, Rudge JS, Wiegand SJ, HolashJ. Vascular-specific growth factors and blood vessel formation. Nature. 2000; 407(6801):242-8. Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81(3):1097-142. Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol. 2006; 246 (1-2): 121-7. Yen PM, Feng X, Flamant F, Chen Y,Walker R1,Weiss, Chassande O, Samarut J, Refetoff S and Meltzer PS.Effects of ligand and thyroid hormone recpetor isoforms on hepatic gene expression profiles of thyroid hormone receptor knowkout mice EMBO Rep. 2003;4(6):581-7. You SH, Liao X, Weiss RE, Lazar MA. The interaction between nuclear receptor corepressor and histone deacetylase 3 regulates both positive and negative thyroid hormone action in vivo. Mol Endocrinol. 2010; 24(7):1359-67. Younes A, Forrero-Torres A, Bartlett NL, et al. Objective responses in a phase I dose-escalation study of SGN-35, a novel antibody-drug conjugate (ADC) targeting CD30, in patients with relapsed or refractory Hodgkin lymphoma. ASCO. 2008 meeting (abstract # 8526). 174 Bibliografía Younes A, Kadin ME. Emerging applications of the tumor necrosis factor family of ligands and receptors in cancer therapy. J Clin Oncol. 2003; 21:3526–34. Zhang W, Wang L, Zhou D, Cui Q, Zhao D, Wu Y. Expression of tumor-associated macrophages and vascular endothelial growth factor correlates with poor prognosis of peripheral T-cell lymphoma, not otherwise specified. Leukemia & lymphoma. 2011;52:46-52. Zhao F, Li L, Guan L, Yang H, Wu C, Liu Y. Roles for GP IIb/IIIa and αvβ3 integrins in MDA-MB231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014; 1;344(1):62-73 Zhao WL, Mourah S, Mounier N, Leboeuf C, Daneshpouy ME, Legres L, et al. Vascular endothelial growth factor-A is expressed both on lymphoma cells and endothelial cells in angioimmunoblastic T-cell lymphoma and related to lymphoma progression. Lab Invest. 2004 Nov;84(11):1512-9. Zinzani PL, Alinari L, Tani M, Fina M, Pileri S, Baccarani M. Preliminary observations of a phase II study of reduced-dose alemtuzumab treatment in patients with pretreated T-cell lymphoma. Haematologica. 2005; 90:702–3. Zinzani PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated cutaneous T-cell lymphoma: experience in 44 patients. J Clin Oncol. 2000; 18:2603–06. Zinzani PL, Magagnoli M, Bendandi M, et al. Therapy with gemcitabine in pretreated peripheral T-cell lymphoma patients. Ann Oncol. 1998; 9:1351–53 175