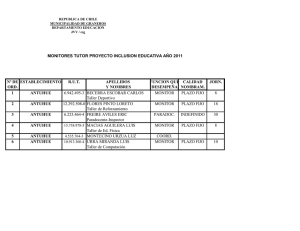
clínica de occidente - Repositorio Institucional UAO
Anuncio

DISEÑO DE UN PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA PARA LA CLÍNICA DE OCCIDENTE JAIRO ALBERTO SARRIA VARGAS UNIVERSIDAD AUTÓNOMA DE OCCIDENTE FACULTAD DE INGENIERÍA DEPARTAMENTO DE AUTOMÁTICA Y ELECTRÓNICA PROGRAMA INGENIERÍA BIOMÉDICA SANTIAGO DE CALI 2012 1 DISEÑO DE UN PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA PARA LA CLÍNICA DE OCCIDENTE JAIRO ALBERTO SARRIA VARGAS Pasantía institucional para optar al título de Ingeniero Biomédico Directora FABIOLA MARGOTH OBANDO REINA Ingeniera Electricista UNIVERSIDAD AUTÓNOMA DE OCCIDENTE FACULTAD DE INGENIERÍA DEPARTAMENTO DE AUTOMÁTICA Y ELECTRÓNICA PROGRAMA INGENIERÍA BIOMÉDICA SANTIAGO DE CALI 2012 2 Nota de aceptación: Aprobado por el Comité de Grado en cumplimiento de los requisitos exigidos por la Universidad Autónoma de Occidente para optar al título de Ingeniero Biomédico Ana Maria Sanchez Benavides Jurado Julian David Quintero Ospina Jurado Santiago de Cali, 19 de Junio de 2012 3 AGRADECIMIENTOS A mis Padres, Hermana y familia por su colaboración y apoyo para optar por el título de Ingeniero Biomédico. A mis docentes y personal de la Clínica de Occidente por abrir sus puertas, aportar conocimiento y recomendaciones que permitieron finalizar con éxito mi práctica en el área Biomédica. 4 CONTENIDO pág. GLOSARIO 13 SIGLAS 15 RESUMEN 16 INTRODUCCION 17 1. PRESENTACION DE LA EMPRESA 21 1.2. SEGURIDAD DEL PACIENTE 24 2. ESTRUCTURA ORGANIZACIONAL 29 2.1 MARCO LEGAL 29 2.2 NORMATIVA A NIVEL INTERNACIONAL 29 2.3 NORMATIVA A NIVEL NACIONAL 30 2.3.1 MINISTERIO DE LA PROTECCIÓN SOCIAL 31 2.3.2 INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS (INVIMA) 31 2.3.3 INSTITUTO COLOMBIANO DE NORMAS TÉCNICAS Y CERTIFICACIÓN (ICONTEC) 31 2.3.4 DECRETO 4725 DE 2005 32 2.3.5 RESOLUCIÓN 004816 DE 2008 32 3. TECNOVIGILANCIA 36 3.1 REPORTES DE TECNOVIGILANCIA 41 3.1.1 Reportes inmediatos 41 3.1.2 Reportes periódicos 41 3.1.3 Reportes de retiro de productos o lotes de producto 41 5 3.1.4 Reporte de alertas internacionales 41 3.1.5 Información para el reporte 43 3.1.6 Información del reporte 44 3.2 ANÁLISIS Y EVALUACIÓN DE EVENTOS E INCIDENTES ADVERSOS 44 3.2.1 Multicausalidad 44 3.2.2 Modelo SHELL 45 3.2.3 Protocolo de Londres 46 3.3 51 VIGILANCIA ACTIVA 4. DISEÑO DE UN PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA PARA LA CLÍNICA DE OCCIDENTE 55 4.1 ASIGNACIÓN DEL RESPONSABLE DEL PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA 57 4.2 INVENTARIO TÉCNICO FUNCIONAL 58 4.2.1 Definición del tipo de dispositivos médicos objeto de vigilancia 60 4.3 GUÍAS RÁPIDAS DE EQUIPOS BIOMÉDICOS 64 4.4 DISEÑO DE UN SISTEMA SENSIBLE 67 4.4.1 Diseño del formato y medios de reporte de eventos e incidentes adversos 67 4.4.2 Formato para el reporte del visitante 68 4.4.3 Formato para el reporte del empleado 70 4.5 73 GESTIÓN DEL REPORTE 4.5.1 Análisis y valoración de los reportes 75 4.5.2 Reporte al fabricante y autoridad sanitaria 75 4.6 75 DISEÑO DE FOLLETOS Y MATERIAL INFORMATIVO 6 4.6.1 DISEÑO DEL MANUAL DE TECNOVIGILANCIA 78 4.7 DISEÑO DE ESTRATEGIAS DE VIGILANCIA DE EQUIPOS BIOMÉDICOS 78 4.7.1 Seguimiento a acciones y responsables 91 5. CONCLUSIÓN 94 6. RECOMENDACIONES 95 BIBLIOGRAFÍA 96 ANEXOS 98 7 LISTA DE CUADROS pág. Cuadro 1. Agencias Internacionales relacionadas con la Tecnovigilancia 29 Cuadro 2. Normatividad del Sistema Obligatorio de Garantía de Calidad en Salud relacionado con Tecnovigilancia 34 Cuadro 3. Responsabilidad de los actores institucionales para la gestión del riesgo de equipos biomédicos 39 Cuadro 4. Tiempo establecido para reportar ante el INVIMA 42 Cuadro 5. Factores contributivos que influencian la práctica clínica 47 Cuadro 6. Principales riesgos de la tecnología en salud 52 Cuadro 7. Diagnóstico del estado actual de Tecnovigilancia en la Clínica de Occidente 56 Cuadro 8. Inventario técnico funcional de la UCI Coronaria 1 58 Cuadro 9. Equipos biomédicos objeto de vigilancia 60 Cuadro 10. Formato de guías rápidas 63 Cuadro 11. Pasos para el reporte de eventos e incidentes adversos 71 Cuadro 12. Sistematizacion de respuestas 75 Cuadro 13. Medida cualitativa de la probabilidad del evento 79 Cuadro 14. Medida cualitativa de la severidad del evento 79 Cuadro 15. Matriz de riesgo de la Clínica de Occidente 80 Cuadro 16. Matriz de riesgo máquina de anestesia 82 Cuadro 17. Matriz de riesgo desfibrilador 84 Cuadro 18. Matriz de riesgo ventilador 86 8 Cuadro 19. Matriz de riesgo monitor de signos vitales 88 Cuadro 20. Barreras 89 9 LISTA DE FIGURAS pág. Figura 1. Seguridad del paciente en siete pasos 25 Figura 2. Sistema integrado de vigilancia en salud 27 Figura 3. Niveles de operación de los Programas de Tecnovigilancia en Colombia 38 Figura 4. Tecnovigilancia en el ciclo de vida del dispositivo médico 39 Figura 5. Modelo del queso suizo 45 Figura 6. Modelo organizacional del Protocolo de Londres 46 Figura 7. Secuencia de investigación y análisis del Protocolo de Londres 48 Figura 8. Diagrama de espina 50 Figura 9. Ciclo PHVA de tecnovigilancia 55 Figura 10. Diagrama de barreras resultado de la jornada de capacitación al personal de la Clínica de Occidente 75 Figura 11. Pasos para el diseño de una matriz de riesgo 78 10 LISTA DE IMÁGENES pág. Imagen 1. Formato para el reporte del visitante 66 Imagen 2. Formato para el reporte del empleado 68 Imagen 3. Pasos para el reporte de eventos e incidentes adversos 72 11 LISTA DE ANEXOS pág. Anexo A. Hojas de vida equipos biomédicos 96 Anexo B. Inventario técnico funcional 97 Anexo C. Guías rápidas de equipos biomédicos 136 Anexo D. Folleto informativo 142 Anexo E. Evaluación 144 Anexo F. Registro fotográfico de la capacitación al personal 146 Anexo G. Formato de asistencia 147 Anexo H. Sistema de indicadores del programa de tecnovigilancia 161 Anexo I. Manual de tecnovigilancia 162 12 GLOSARIO La información que a continuación se relaciona, fue obtenida de la Resolución 004816 de 20081 y del Decreto 4725 de 20052 EQUIPO BIOMÉDICO: dispositivo medico operacional y funcional que reúne sistemas y subsistemas eléctricos, electrónicos o hidráulicos, incluidos los programas informáticos que intervengan en su buen funcionamiento, destinado por el fabricante a ser usado en seres humanos con fines de prevención, diagnóstico, tratamiento o rehabilitación. No constituyen equipo biomédico, aquellos dispositivos médicos implantados en el ser humano o aquellos para un solo uso. EVENTO ADVERSO: daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. EVENTO ADVERSO NO SERIO: evento no intencionado, diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, operador o todo aquel que se vea implicado directa o indirectamente, como consecuencia de la utilización de un dispositivo o aparato de uso médico. EVENTO ADVERSO SERIO: evento no intencionado que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, operador o todo aquel que se vea implicado directa o indirectamente, como consecuencia de la utilización de un dispositivo médico. FORMATO DE REPORTE: es el medio por el cual un reportante notifica a la institución hospitalaria, al fabricante y/o a la autoridad sanitaria, sobre un evento o incidente adverso asociado a un dispositivo médico. INCIDENTE ADVERSO: potencial daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. 1 COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 4816 de 2008 (Noviembre 27). por la cual se reglamenta el Programa Nacional de Tecnovigilancia. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2008. [Consultado 20 se septiembre de 2011]. Disponible en internet: http://www.alcaldiabogota.gov.co 2 COLOMBIA, MINISTERIO DE LA PROTECCION SOCIAL. Decreto 4725 de2005 (Diciembre 26). Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2005. [Consultado Octubre 01 de 2011]. Disponible en internet: http://www.presidencia.gov.co 13 INCIDENTE ADVERSO NO SERIO: potencial riesgo de daño no intencionado diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. INCIDENTE ADVERSO SERIO: potencial riesgo de daño no intencionado que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. TECNOVIGILANCIA: es el conjunto de actividades que tienen por objetivo la identificación y la cualificación de efectos adversos serios e indeseados producidos por los dispositivos Biomédicos, así como la identificación de los factores de riesgo asociados a estos efectos o características, con base en la notificación, registro y evaluación sistemática de los efectos adversos de los dispositivos Biomédicos, con el fin de determinar la frecuencia, gravedad e incidencia de los mismos para prevenir su aparición. 14 SIGLAS CDO: Clínica de Occidente DANE: Departamento Administrativo Nacional de Estadística DM: Dispositivos Médicos EA: Evento Adverso ECRI: Emergency Care and Research Institute FDA: Food and Droug Administration GHTF: Global Harmonization Task Force IA: Incidente Adverso INVIMA: Instituto Nacional de Vigilancia de Medicamentos y Alimentos IPS: Institución Prestadora de Salud JCAHO: Joint Commission on Accreditation of Healthcare Organizations MHRA: Medicines and Healthcare products Regulatory Agency OMS: Organización Mundial de la Salud OPS: Organización Panamericana de la Salud SP: Seguridad del Paciente SOGC: Sistema Obligatorio de Garantía de Calidad de la Atención en Salud UCI: Unidad de Cuidados Intensivos 15 RESUMEN La inclusión de la seguridad de los pacientes en el ámbito clínico es una importante iniciativa que ha sido fuertemente impulsada por el gobierno nacional, de allí, que ha propuesto la Política de Seguridad del Paciente, liderada por el Sistema Obligatorio de Garantía de Calidad de la Atención en Salud SOGC, con la que se pretende prevenir la ocurrencia de situaciones que afecten la seguridad del paciente, reducir y de ser posible eliminar la ocurrencia de eventos adversos para contar con instituciones seguras y competitivas internacionalmente. Este proceso de seguridad del paciente es un paso inicial que debe evolucionar e incorporar experiencias importantes de instituciones de salud, la evolución del conocimiento científico en el tema, incluyendo nuevas prácticas y relaciones con otros sistemas que le son afines como la Tecnovigilancia3. En consecuencia con los requerimientos normativos nacionales, este proyecto de grado, en cumplimiento a la ley presenta el diseño de un Programa Institucional de Tecnovigilancia para la Clínica de Occidente del municipio de Santiago de Cali, en el cual se desarrolló el Inventario Técnico Funcional de los equipos biomédicos con que cuenta la Institución, el diseño de formatos de reporte de eventos e incidentes adversos desde dos enfoques: (i) Un enfoque interno para el área administrativa y el cuerpo médico y (ii) Un enfoque externo para los visitantes, pacientes y acompañantes que interactúan con la Clínica de Occidente, la entrega, capacitación e implementación de los formatos de reporte para la notificación de los eventos e incidentes adversos, el diseño del manual de tecnovigilancia, y el diseño de guías rápidas de los equipos biomédicos de la institución, teniendo en cuenta el Inventario Técnico Funcional actualizado y sus respectivas precauciones del manejo de los equipos. Finalmente, presenta los resultados administrativos y procedimentales llevados a cabo por el Comité de Seguridad al Paciente de la clínica, quienes ejecutaron la codificación y evaluación de los formatos diligenciados para permitir dar inicio al Programa Institucional de Tecnovigilancia de la Clínica de Occidente. Palabras claves: Evento adverso, Incidente adverso, Tecnovigilancia. 3 COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Dirección General de Calidad de Servicios. Unidad sectorial de Normalización. Guía técnica: Buenas prácticas para la seguridad del paciente en la atención en salud. Bogotá, 2010. 16 INTRODUCCIÓN El desarrollo tecnológico ha alcanzado altos niveles de progreso y la industria biomédica no ha sido ajena a esos cambios. En la actualidad se encuentran dispositivo biomédicos sofisticados que facilitan los procedimientos clínicos, haciéndolos menos invasivos y en el menor tiempo posible. Así mismo, como se ha venido incrementado apreciablemente la oferta y la demanda de los dispositivos biomédicos en el mercado, también lo han hecho las técnicas de seguridad en éstos equipos, durante los últimos 20 años. Sin embargo la garantía de que ninguna falla se presente, no es absoluta, ya que técnica y médicamente el diseño del equipo por sí solo no garantiza la protección al paciente, y se hace necesario considerar otras variantes como lo son las medidas de seguridad en su instalación, mantenimiento y manejo que le proporciona el personal técnico y/o médico al equipo4. En la actualidad las grandes clínicas alrededor del mundo que han lamentado miles de muertes por errores en la atención médica están preocupadas por analizar los riesgos, mejorar procesos, obtener tecnología de punta que aleje la posibilidad de error del factor humano, adoptar prácticas seguras, en fin, hacer todo aquello que conlleve a no cometer errores que causen daño a los pacientes. Sin embargo, debido a lo altamente complejo que es la atención en salud, desafortunadamente los errores y daños se siguen presentando, lo anterior no se puede adoptar como un sinónimo de seguridad al paciente. A pesar de ello, se reconoce que la tecnología biomédica contribuye a la prevención de enfermedades y es la principal herramienta de diagnóstico y tratamiento, que permite mitigar el impacto de éstas sobre las personas, así como también acortar el período de convalecencia o la recuperación de los individuos, permitiendo agilizar su reincorporación a la sociedad. Como es de suponer los pacientes acuden a un servicio médico con el objetivo de hacerse exámenes de rutina o de encontrar, según sea el caso de cada paciente, una cura al malestar o padecimiento que les aqueja y no son conscientes de los riesgos a los que podrían estar sometidos; encontrándose indefensos ante una eventual falla durante el procedimiento clínico que reciben. 4 Seguridad Eléctrica en Equipos e Instalaciones Médicas. [En línea]. Capítulo V. [Consultado Septiembre 20 de 2011]. Disponible en internet: http://micronet.cujae.edu.cu/Docencia 17 Con respecto al tema, el panorama en Latinoamérica es diferente en cuanto a la implementación de programas de vigilancia, mientras en algunas instituciones de salud se tienen programas efectivos para prevenir estos eventos, en otras no las hay, debido a que no existe una cultura de gestión de riesgos, no se cuenta internamente con un departamento que se responsabilice de las gestiones de vigilancia o no se dispone de los recursos necesarios para su implementación. Esta problemática ha sido materia de estudio de gran cantidad de organismos de salud a nivel mundial, dado el reporte de cifras alarmantes de pacientes que mueren como consecuencia de un mal procedimiento clínico, o el porcentaje de pacientes que sufren alguna lesión física o prolongación de su tratamiento u hospitalización; además de la elevación de los costos de la clínica por indemnizaciones por muerte o manutención del paciente afectado. Países como Australia, Estados Unidos, Gran Bretaña, Canadá, Dinamarca, los países Bajos, Suecia, Nueva Zelanda y más países miembros de la Organización para la Cooperación y el Desarrollo Económico (OCDE), estudian seriamente el problema y cuentan con programas de vigilancia de eventos adversos. Sin embargo la situación en los países en desarrollo es muy diferente, ya que en muchos casos no existe o no se le presta la debida atención a los programas de vigilancia. En éstos países, el mal estado de las infraestructuras, los equipos, la irregularidad del suministro y de la calidad de los medicamentos, las deficiencias en la gestión de desechos y en la lucha contra las infecciones, una actuación deficiente del personal, por falta de motivación o insuficiencia de sus conocimientos técnicos, y la grave escasez de recursos para financiar los costos de funcionamiento esenciales de los servicios de salud hacen que la probabilidad de que se produzcan eventos adversos sea mucho más alta que en las naciones industrializadas5. Es así como en países como Argentina se está elaborando la regulación de equipos y dispositivos médicos nuevos y fijan las directrices de los productos importados que deben cumplir con guías especificadas, incluida la rotulación en español y la provisión de información relativa al importador. En Brasil, la regulación también se está actualizando y se trabaja en los procedimientos de registro requeridos a los fabricantes no brasileños y actualmente planean la aplicación de una guía de buenas prácticas de fabricación, en Chile, se cuenta con 5 ORGANIZACIÓN MUNDIAL DE LA SALUD. [En línea]. [Consultado Septiembre 20 de 2011]. Disponible en internet http://www.who.int/gb/ebwha/pdf_files/WHA55/sa5513.pdf 18 una regulación nueva de equipos y dispositivos médicos que incluye un esquema de clasificación del riesgo y promueven el establecimiento de una entidad autorizadas que realice las pruebas, conforme a las guías de su país u otras, solo por citar algunos ejemplos. En Colombia, según el Ministerio de Protección Social6, la vigilancia de eventos adversos constituye una de las estrategias a través de las cuales el Sistema Obligatorio de Garantía de Calidad (SOGC), busca garantizar que las instituciones de atención en salud sean instituciones seguras para el paciente y los mismos profesionales que laboran en ellas. Esta entidad identifica cinco aspectos que pueden derivar accidentes o eventos adversos ocurridos durante procedimientos clínicos como: Fallas en el equipo o en la estructura de la prestación del servicio. Ausencia de monitoreo, vigilancia, control. Problemas de planificación de la atención. Fallas en la definición de procedimientos. Negligencia o descuido por parte del personal médico al no tener en cuenta las normas de seguridad. En el país, se reglamenta el régimen de registros sanitarios, el permiso de comercialización y vigilancia sanitaria de los Dispositivos Biomédicos para uso humano, en el decreto 4725 del 2005 del Ministerio de la Protección Social7, y se reglamenta el Programa Nacional de Tecnovigilancia en la Resolución 004816 del 2008 de dicho ministerio. Dentro de los compromisos establecidos en dicho decreto y resolución se encuentra la generación de los mecanismos para recolección, evaluación y gestión de la información sobre eventos o incidente adversos asociados a los dispositivos biomédicos usados en la prestación de servicio de salud; aspecto que también se explica en la Norma Técnica Colombiana NTC 5736 que presenta los requisitos para una estructura de codificación cuyo fin sea describir los eventos adversos relacionados con dispositivos biomédicos. 6 COLOMBIA, MINISTERIO DE PROTECCIÓN SOCIAL. Lineamientos para la implementación de la Política de Seguridad del Paciente [En línea]. [Consultado Septiembre 20 de 2011]. Disponible en internet: http://www.minproteccionsocial.gov.co/Documentos%20y%20Publicaciones/LINEAMIENTOS%20S EGURIDAD%20DEL%20PACIENTE.pdf 7 Ibíd., p. 13 19 En consecuencia, la Clínica de Occidente de Santiago de Cali, cuenta con el área de electromedicina que se encarga de realizar la revisión, mantenimiento y reposición de los equipos biomédicos. Sin embargo, es importante resaltar que aunque se cuente con el personal capacitado para realizar la gestión de los equipos biomédicos, estos dispositivos médicos son sometidos a diferentes controles durante su desarrollo, que por lo general no son suficientes para garantizar que durante su uso, se presenten problemas o incidentes que pueden desencadenar daños o potenciales daños para la salud de los pacientes que los utilizan pues “todos los dispositivos biomédicos poseen un cierto grado de riesgo el cual podría causar problemas en circunstancias específicas”8. Es por esto, que la importancia de estructurar un sistema de vigilancia de dispositivos biomédicos parte de la necesidad de garantizar la seguridad y efectividad de estos productos una vez salen al mercado y son usados individual y colectivamente, lo que se conoce como control post-mercado de la tecnología biomédica. Partiendo de lo anterior y en consideración con la normatividad nacional, se formula este proyecto cuyo objetivo general es diseñar un programa de tecnovigilancia para la clínica de occidente que permita identificar, registrar, evaluar y gestionar los reportes de eventos adversos en equipos biomédicos. Para el logro de este proyecto se plantearon cuatro objetivos específicos cuyo cumplimiento va a permitir contar con información actualizada de los equipos biomédicos con que cuenta la clínica de occidente por medio de un inventario técnico funcional., el diseño de los formatos que permita reportar los eventos adversos asociados al uso de los dispositivos biomédicos, la capacitación al personal de la clínica en la identificación y cultura del reporte de eventos adversos, relacionados con dispositivos médicos, y la elaboración de guías rápidas para equipos biomédicos utilizados en las unidades de cuidados intensivos y salas de cirugía. 8 COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Dispositivos médicos y que es la tecnovigilancia [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2005 [consultado el 01 de octubre 2011]. Disponible en internet: http://issuu.com/leadyrs/docs/revista_decreto_4725 20 1. PRESENTACION DE LA EMPRESA La Clínica de Occidente, se ubica al norte de la ciudad de Santiago de Cali. Creada desde 1938 y fundada nuevamente en 1943 como Clínica de Occidente S.A. Actualmente cuenta con aproximadamente 800 empleados. Su visión se basa en el deseo de consolidarse al año 2010 como una de las mejores clínicas nivel IV del País, enfocada a la venta de productos de alta complejidad y tecnología de punta. Para este propósito cuenta con certificación ISO 9001 y su política de calidad se enfoca en la minimización del riesgo a los pacientes de sufrir Eventos adversos en el proceso de atención; mediante la aplicación de una política de mejoramiento del personal que labora directa o indirectamente en la organización; capacitándolos también en procesos efectivos y mejorando los equipos y técnicas, con el fin de reducir el riesgo de contaminación ambiental; generando excelentes relaciones comerciales con los proveedores, donde se destaque el cumplimiento de lo pactado. Todos estos aspectos contribuirán a la generación de niveles de rentabilidad necesarios para la permanencia de la organización en el tiempo 9. La clínica ofrece los siguientes servicios los cuales pueden ser consultados en su página web http://clinicadeoccidente.com.co: Urgencias: El servicio está conformado por: Sala de cuidado crítico. Sala para colocación y retiro de yesos. Sala de observación. Sala de procedimientos y pequeña Cirugía. Sala de espera. Hospitalización: La Clínica cuenta con diferentes tipos de habitación de acuerdo a las necesidades y recursos del paciente. 9 CLÍNICA DE OCCIDENTE. Información Corporativa. Santiago de Cali, 2011, [En línea] [consultado el 01 de octubre 2011]. Disponible en internet: http://clinicadeoccidente.com.co/FRONT/texto.php?Id=20 21 Laboratorio Clínico: El Laboratorio Clínico cuenta con equipos automatizados y con el recurso humano que garantiza ciertos estándares de calidad para brindar resultados confiables. En él se realizan procedimientos y exámenes completamente sistematizados, con el manejo de tecnología de punta en las áreas de: Hematología Coagulación Bioquímica Sanguínea Gases Arteriales Hormonas e Infecciosas Uroanálisis Hemocultivos Microbiología Departamento de Rehabilitación: La unidad de Rehabilitación presta servicios como: Fisioterapia (Fisioterapeutas) Manejo de patologías Terapia Respiratoria (Terapeutas respiratorios) Espirometria Rehabilitación Cardíaca Terapia del lenguaje (Fonoaudiólogas) Acondicionamiento físico Unidad de Cuidados Intensivos: La Clínica cuenta con 62 camas dotadas con tecnología moderna para el monitoreo invasivo y soporte ventilatorio: Cardiovasculares Coronarias I y II Convenio General Neurovasculares Intermedios 22 U.C.I Neonatal y Pediátrico: Cuenta con un grupo conformado por Médicos Pediatras, subespecialistas y Neonatologos, terapeutas respiratorios, enfermeras con amplia experiencia. La unidad está dotada con equipos para el cuidado integral del menor: Ventiladores Monitores Incubadoras Mesas de calor radiante Cámara de Fototerapia Cirugía General y Especializada: La Unidad de Cirugía cuenta con 9 quirófanos completamente dotados con los equipos necesarios para procedimientos quirúrgicos de Nivel II, III y IV de complejidad. Cirugía Cardiovascular: La Clínica de Occidente cuenta con dos salas con equipos para realizar Cirugía Extracorpórea, Recuperación Celular, Balón Intraórtico de Contrapulsación y equipos de Asistencia Ventricular. Unidad de imágenes diagnósticas: La Unidad de Imágenes diagnósticas cuenta con equipos que garantizan diagnósticos oportunos y confiables para realizar: Radiología convencional Radiología especializada: Ecografía convencional Ecografía endocavitaria Ecografía 3 D Drenaje percutane o guiado por Ecografía Mamografía Tomografía Axial Computarizada Además cuenta con servicios de atención a Maternidad, Unidad de Urología, Unidad de Angiografía, Unidad de Endoscopia digestiva. 23 1.2 SEGURIDAD DEL PACIENTE La seguridad del paciente es el conjunto de elementos estructurales, procesos, instrumentos, análisis y prevención de las fallas de la atención en salud que con frecuencia son causas de eventos adversos. Esta seguridad al paciente implica la evaluación permanente de los riesgos asociados a la atención en salud para diseñar y minimizar el riesgo de un evento o incidente adverso. Lo anterior de acuerdo con el observatorio de la calidad en salud del Ministerio de la Protección Social, que manifiesta que la seguridad del paciente es el conjunto de elementos estructurales, procesos, instrumentos y metodologías basadas en evidencias científicamente probadas que propenden por minimizar el riesgo de sufrir un evento adverso en el proceso de atención en salud o de mitigar sus consecuencias10. La seguridad del paciente es la prioridad de los sistemas sanitarios en todo el mundo, especialmente desde que diversos estudios epidemiológicos han manifestado que la misma atención sanitaria destinada a mejorar la salud de las personas es una fuente importante de daños, siendo los errores de medicación una de las principales causas de daño prevenible. Un ejemplo claro para mejorar la seguridad del paciente es la guía “Seguridad del paciente en siete pasos” de la Agencia Nacional para la Seguridad del Paciente (NPSA) perteneciente al Sistema Nacional de Salud (NHS) del Reino Unido. Esta guía describe los pasos claves de actividades en las organizaciones y equipos de atención para salvaguardar la seguridad de los pacientes que atienden. Estos sietes pasos proporcionan una sencilla lista de control para ayudar a planificar la actividad y evaluar la actuación en seguridad al paciente. El seguimiento de estos pasos ayuda a planificar la actividad, evaluar la actuación del paciente, alcanzar los objetivos de gestión clínica que tienen como principio incrementar la eficiencia y la calidad de las prestaciones sanitarias, y la gestión de riesgos de cada organización. Esta guía constituye una referencia para la planificación y seguimiento de las actividades ligadas a la Seguridad del Paciente11. En la figura 1 se pueden observar. 10 Observatorio de calidad en la atención en salud [En línea]: Seguridad del paciente. Bogotá D.C.: Ministerio de la Protección Social [consultado 13 de Marzo de 2011] Disponible en Internet:http://201.234.78.38/ocs/public/seg_paciente/Default.aspx 11 REINO UNIDO. Ministerio de Sanidad y Consumo. Agencia Nacional para Seguridad del Paciente NPSA. Sistema Nacional de Salud NHS. La seguridad del paciente en siete pasos. 24 Figura 1. Seguridad del paciente en siete pasos. Construir una cultura de seguridad. Lideraz go de equipo de persona s. Integrar las tareas de gestión de riesgos Aprender y compartir lecciones de seguridad (RCA) Involucrar y comunicars e con pacientes y público Promove r que se informe Implementar soluciones para prevenir daños Fuente: REINO UNIDO. Ministerio de Sanidad y Consumo. Agencia Nacional para Seguridad del Paciente NPSA. Sistema Nacional de Salud NHS. La seguridad del paciente en siete pasos. La cultura de seguridad hace referencia a la conciencia de la probabilidad que pueda ocurrir un evento, la capacidad de reconocer los errores, aprender de ellos y actuar para el mejoramiento continuo; lo cual puede ser analizado desde el punto de vista de la planeación estratégica mediante el planear, el hacer, el verificar y el actuar. Es por ello que esta cultura de seguridad aporta un impacto positivo para el funcionamiento de los establecimientos de salud y debe ser abierta e imparcial que permita compartir información para tratar los eventos adversos. El liderazgo del recurso humano abarca un proceso que inicia desde el propio reconocimiento de los errores y las deficiencias del trabajo en equipo hasta la declaración de políticas claras, de motivación y de fomentar un entorno abierto a cambios, comunicación, aprendizaje y mejora continua. La integración de las tareas de gestión de riesgos hace referencia a cómo las organizaciones pueden incorporar la gestión de riesgo a su sistema de gestión, teniendo en cuenta los tipos de riesgo existentes, herramientas de análisis y valoración de riesgos. Existen dos tipos de riesgo. El riesgo clínico, el cual está ligado directamente a los pacientes. 25 El riesgo no clínico relacionado con los aspectos tecnológicos, administrativos, seguridad, finanzas, recursos humanos, entre otros. A nivel mundial, la Organización Mundial de la Salud (OMS) es una de las entidades que ha ejercido un liderazgo para el mejoramiento de las funciones de las instituciones de salud para alcanzar la seguridad de los pacientes. En este sentido, Colombia impulsó la Política de Seguridad del Paciente, que es liderada por el Sistema Obligatorio de Garantía de Calidad de la Atención en Salud. Esta Política tiene por objetivo prevenir la ocurrencia de situaciones que afecten la seguridad del paciente, reducir y de ser posible eliminar la ocurrencia de eventos adversos para contar con instituciones seguras y competitivas. El Ministerio de la Protección Social expidió los “Lineamientos para la implementación de la Política de Seguridad del Paciente”; el propósito de esta Norma Técnica Sectorial es brindar a las instituciones directrices técnicas para la operación e implementación practica de los mencionados lineamientos en sus procesos asistenciales. La política de seguridad al paciente del Ministerio de la Protección Social, tiene siete principios orientadores, que se describen a continuación, enfocados hacia la atención segura, abarcando desde el establecimiento de normas hasta la sensibilización, la promoción y la coordinación. Enfoque de atención centrado en el usuario. Significa que lo más importante son los resultados obtenidos en él y su seguridad, lo cual es el eje principal en donde giran todas las acciones del paciente. Cultura de Seguridad. El desarrollo de las actividades de seguridad del paciente deben darse en un entorno de familiaridad y de respeto entre pacientes, profesionales, aseguradores y la comunidad. Es obligación del personal facilitar las condiciones que permitan dicho desarrollo. Integración con el Sistema Obligatorio de Garantía de Calidad de la Atención en Salud. La política de seguridad del paciente es parte total del Sistema Obligatorio de Garantía de Calidad de la Atención en Salud, y es transversal a todos sus componentes. Multicausalidad. El problema de la seguridad del paciente es un problema sistémico y multicausal en el cual deben involucrarse las áreas organizacionales y el personal de la institución. Validez. Para impactarlo, soportadas con evidencias. se 26 requiere implementar metodologías, Alianza con el paciente y su familia. La política de seguridad debe contar con los pacientes y sus familiares e integrarlos en las acciones de mejora. Alianza con el profesional de la salud. La política de seguridad parte del reconocimiento de la atención brindada por el profesional de la salud y de la complejidad de estos procesos por lo cual apoyara con la participación de ellos y procurará defenderlo de indicaciones injustas. La figura 2 muestra los aspectos que se deben tener en cuenta a la hora de implementar un sistema integrado de vigilancia en salud, todos enfocados en mejorar la seguridad del paciente. Figura 2. Sistema integrado de vigilancia en salud. Farmaco vigilancia Sistema de vigilancia ambiental Tecno vigilancia Gestion de riesgo Hemo vigilancia Vigilancia epidemiologica Reactivo vigilancia 27 Este trabajo está enfocado en la vigilancia de equipos biomédicos (tecnovigilancia), uno de los factores que se constituye como un pilar fundamental en la evaluación de la efectividad y seguridad real de los dispositivos biomédicos y una herramienta para la evaluación razonada de los beneficios y riesgos que su utilización representa para la salud de un paciente12. 12 Información general: Tecnovigilancia. [En línea] Bogotá D.C.: Instituto Nacional de Vigilancia de Medicamentos y Alimentos INVIMA [C onsultado 13 de Marzo de 2011]. Disponible en Internet:http://www.invima.gov.co/Invima///tecnovigilancia/tecnovigila.jsp?codigo=489 28 2. ESTRUCTURA ORGANIZACIONAL 2.1 MARCO LEGAL A nivel internacional el tema de la Tecnovigilancia se encuentra regulado por un amplio número de agencias conformadas desde hace más de diez años cuyas funciones se describen a continuación en el cuadro 1. 2.2 NORMATIVIDAD A NIVEL INTERNACIONAL Cuadro 1. Agencias Internacionales relacionadas con la Tecnovigilancia NOMBRE PAÍS AÑO DE ORIGEN Administración Nacional de Medicamentos, Alimentos y Tecnología Médica Argentina 1992 AFSSAPS Francia 1998 Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) España 1999 Agencia Nacional de Vigilancia Sanitaria (ANVISA) Brasil 1999 Emergency Care and Research Institute (ECRI) EEUU 1955 DESCRIPCIÓN Organismo que colabora en la protección de la salud humana, asegurando la calidad de los productos de su competencia: medicamentos, alimentos, productos médicos, reactivos de diagnóstico, cosméticos, suplementos dietarios y productos de uso doméstico. Su misión es esencial para evaluar los beneficios y los riesgos asociados con el uso de productos de salud y así contribuir a la intervención, identificación, análisis y control de riesgos inherentes a cada producto cada vez que sea posible, teniendo en cuenta las necesidades terapéuticas y la necesidad de continuidad de la atención. Tiene como misión garantizar a la sociedad, desde la perspectiva de servicio público, la calidad, seguridad, eficacia y correcta información de los medicamentos y productos sanitarios y la protección y promoción de la salud de las personas y de los animales. La finalidad institucional de la Agencia es promover la protección de la salud de la población por intermedio del control sanitario de la producción y de la comercialización de productos y servicios sometidos a la vigilancia sanitaria, incluso de los ambientes, de los procesos, de los insumos y de las tecnologías relacionados con ellos. Corporación dedicada a la evaluación y perfeccionamiento continúo de la tecnología del cuidado de la salud. 29 Cuadro 1. (continuación) Global Harmonization Task Force (GHTF) Unión Europea EEUU Canadá Gran Bretaña Japón 1992 Heads of Medicines Agencies (HMA) Países de la Unión Europea 2004 HealthCanada Canadá 1996 Joint Commission EEUU 1951 Medicines and healthcare products regulatory agency (MHRA) Reino Unido 2003 Food and Drug Administration (FDA) EEUU 1906 Grupo voluntario de representantes de autoridades nacionales reguladoras de dispositivos médicos y de la industria regulada. El propósito del GHTF es para fomentar la convergencia en las prácticas reglamentarias relacionadas con garantizar la seguridad, eficacia y rendimiento y la calidad de los productos sanitarios, la promoción de la innovación tecnológica, entre otros. La HMA coopera con la Agencia Europea del Medicamento y la Comisión Europea (CE) en el funcionamiento de la Red Europea Reguladora de Medicamentos y tiene como visión proteger la salud pública y animal en Europa. Health Canadá es un departamento federal responsable de ayudar a los canadienses a mantener y mejorar su salud, respetando las opciones individuales y las circunstancias. Es una organización independiente y sin ánimo de lucro que acredita y certifica organizaciones de la salud en los EUA. Se dedica a mejorar en forma continua la seguridad y calidad de la atención ofrecida al público. Es la agencia del gobierno de los Estados Unidos responsable de la regulación de alimentos, vacunas, suplementos alimenticios, medicamentos, cosméticos, aparatos médicos, productos biológicos y productos que emiten radiaciones. Es la agencia del gobierno de los Estados Unidos responsable de la regulación de alimentos, suplementos alimenticios, medicamentos, cosméticos, aparatos médicos, productos biológicos y derivados sanguíneos. Fuente: Adaptado de FRANCO, Catalina Vásquez, TRUJILLO, Mauricio Pérez. Guía para la implementación del Programa Nacional de Tecnovigilancia en el Hospital General de Medellín, 2011. 2.3 NORMATIVIDAD A NIVEL NACIONAL La normativa de Tecnovigilancia en Colombia, está definida fundamentalmente por tres entidades: el Ministerio de Protección Social, el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) y el Instituto Colombiano de Normas Técnicas y Certificación (ICONTEC). 30 2.3.1 Ministerio de la protección social. El Ministerio de la Protección Social tiene como misión, orientar el Sistema de Protección Social y el Sistema de Seguridad Social hacia su integración y consolidación, mediante la aplicación de los principios básicos de universalidad, solidaridad, calidad, eficiencia y equidad, con el objeto de tener un manejo integral del riesgo y brindar asistencia social a la población colombiana13. El Artículo 9 del Decreto 4725 de 2005, determina las funciones del Ministerio de la Protección Social frente al tema de la Tecnovigilancia de Equipos Biomédicos: Art. 9. El Ministerio de la Protección Social con el apoyo del INVIMA diseñara el Programa Nacional de Tecnovigilancia que permita identificar los incidentes adversos no descritos, cuantificar el riesgo, proponer y realizar medidas de salud pública para reducir la incidencia y mantener informados a los usuarios, a otros profesionales de la salud, a las autoridades sanitarias a nivel nacional y a la población general.14 2.3.2 Instituto nacional de vigilancia de medicamentos (INVIMA). El INVIMA se encarga de la vigilancia y control de carácter técnico científico, para proteger la salud individual y colectiva de los colombianos mediante la aplicación de las normas sanitarias relacionadas con los productos de su competencia. Este instituto se rige bajo varios decretos y resoluciones. Para las competencias definidas en este proyecto, se tiene en cuenta el decreto 4725 de 2005 por el cual se reglamenta el régimen de registros sanitarios, permisos de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano, resolución 004816 del 2008 por el cual se reglamenta el programa nacional de Tecnovigilancia. 2.3.3 Instituto colombiano de normas técnicas y certificación (ICONTEC). El Instituto Colombiano de Normas Técnicas y Certificación (ICONTEC) es un organismo multinacional de carácter privado, sin ánimo de lucro, que trabaja para fomentar la normalización, la certificación, la metrología y la gestión de la calidad en Colombia. 13 COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 3133 del 14 de septiembre de 2005. Por la cual se ajusta el Manual Específico de Funciones, Requisitos y de Competencias Laborales para los empleos de la Planta de Personal del Ministerio de la Protección Social [En línea] Bogotá d.c.2005. [consultado el 01 de octubre 2011]. Disponible en internet: http://www.minproteccionsocial.gov.co. 14 Ibíd., p. 12 31 En el campo de la normalización, la misión del Instituto es promover, desarrollar y guiar la aplicación de Normas Técnicas Colombianas (NTC) y otros documentos normativos, con el fin de alcanzar una economía óptima de conjunto, el mejoramiento de la calidad y también facilitar las relaciones cliente-proveedor, en el ámbito empresarial nacional o internacional. Para el tema de la Tecnovigilancia, el ICONTEC, promueve la aplicación de la Norma Técnica Colombiana 5736, sobre Dispositivos Médicos, su estructura de codificación para tipos de eventos adversos y sus causas, la NTC 5460, sobre Dispositivos Médicos, su aplicación del manejo del riesgo a los Dispositivos Medico. 2.3.4 Decreto 4725 de 2005. El Decreto 4725 de 2005 por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano e imparte responsabilidades al Ministerio de la Protección Social e INVIMA frente al tema. Así mismo, en su artículo 5, presenta una clasificación de los dispositivos médicos y los relaciona con los riesgos potenciales de los mismos y clasifica dichos riesgos según tres clases15 que a continuación se presenta: Clase I. Son los dispositivos médicos de bajo riesgo, no destinados para mantener la vida y que no signifiquen un riesgo potencial para la salud. Clase IIa. Son los dispositivos médicos de riesgo bajo, sujetos a controles especiales en la fase de fabricación, no destinados para mantener la vida. Clase Ilb. Son los dispositivos médicos de riesgo moderado alto, sujetos a controles especiales en el diseño y fabricación para demostrar su seguridad y efectividad. Clase III. Son los dispositivos médicos de muy alto riesgo, destinados a proteger o mantener la vida para la prevención de la salud humana. 2.3.5 Resolución 004816 de 2008. Es la resolución por la cual se reglamenta el Programa Nacional de Tecnovigilancia16, en su artículo 5, clasifica los eventos e incidentes adversos que pueden presentar los equipos biomédicos y fija la responsabilidad de los actores del nivel nacional, departamental, local e institucional frente al tema. 15 16 Ibid., p. 13 Ibid., p. 13 32 1. Evento adverso serio: Daño no intencionado al paciente u operador o medio ambiente que pudo haber llevado a la muerte o al deterioro serio de la salud. 2. Evento adverso no serio: Daño no intencionado, diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, operador. 3. Incidente adverso serio: Daño no intencionado al paciente u operador, que pudo haber llevado a la muerte o al deterioro serio de la salud pero que por la intervención de un profesional en salud, operario, o persona no genero un desenlace adverso. 4. Incidente adverso no serio: Potencial riesgo de daño no intencionado diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente. Con respecto a la responsabilidad de los actores del nivel local, la resolución 004816 del 2008 según articulo 9 fija los siguientes aspectos a tener en cuenta: Estar vigilantes y alertas del desempeño, calidad y seguridad de los dispositivos médicos previos a su uso. Comunicar, divulgar y aplicar las prácticas adecuadas de utilización de dispositivos médicos. Diseñar e implementar un Programa Institucional de Tecnovigilancia, que permita identificar, registrar, evaluar y gestionar los reportes de eventos e incidentes adversos con los dispositivos médicos. Designar como mínimo un profesional competente en el tema, responsable del Programa Institucional de Tecnovigilancia en la institución que a sus ves será la persona encargada de realizar los reportes ante el Gobierno. Tomar las acciones preventivas o correctivas que sean del caso y las que le sean exigidas por el Instituto Nacional de Medicamentos y Alimentos, INVIMA, de forma inmediata. En el cuadro 2 se sintetizan algunas otras leyes, decretos y resoluciones del Sistema Obligatorio de Garantía de Calidad en Salud que tienen alguna relación con Tecnovigilancia. 33 Cuadro 2. Normatividad del Sistema Obligatorio de Garantía de Calidad en Salud relacionado con Tecnovigilancia. NORMATIVA LEY 100 DE 1993 OBJETIVO “Por la cual se crea el sistema de seguridad social integral y se dictan otras disposiciones” “Por la cual se dictan normas para la evaluación de importación de tecnologías biomédicas, se definen las de importación controlada y se dictan otras disposiciones” RESOLUCIÓN 434 DE 2001 DECRETO 4725 DEL 2005 DECRETO 1011 DE 2006 RESOLUCIÓN 1043 DE 2006 “Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano‟‟ “Por el cual se establece el Sistema Obligatorio de Garantía de Calidad de la Atención de Salud del Sistema General de Seguridad Social en Salud” „‟Por la cual se establecen las condiciones que deben cumplir los Prestadores de Servicios de Salud para habilitar sus servicios e implementar el componente de auditoria para el mejoramiento de la calidad de la atención y se dictan otras disposiciones‟‟. 34 CONTENIDO Los artículos 190 y 245 de la Ley 100 de 1993, establecen algunos aspectos para la evaluación, vigilancia y control de la tecnología. Objetivo de la resolución, relacionado en el Artículo 2 que menciona la necesidad de establecer metodologías y procedimientos de evaluación técnica y económica así como aquellos que permitan determinar la más eficiente localización, de tecnología biomédica y determinar los criterios para su importación o adquisición y adecuada incorporación a las Instituciones Prestadoras de Servicios de Salud. El capítulo IX del Decreto 4725 contiene los artículos 59 (Obligación de informar a la autoridad sanitaria), 60 (Notificación) y 61 (Del programa nacional de Tecnovigilancia). En el artículo 4 se definen los cuatro componentes del SOGCS: El Sistema Único de Habilitación, La Auditoria para el Mejoramiento de la Calidad de la Atención de Salud, El Sistema Único de Acreditación, El Sistema de Información para la Calidad. En el numeral 4.2 (que hace parte de los Estándares de Habilitación en Gestión de Medicamentos y Dispositivos): „‟Los procedimientos de adquisición de medicamentos y dispositivos médicos, incluyen la verificación del registro expedido por el INVIMA y el programa de Farmacovigilancia y Tecnovigilancia‟‟. Esto aplica para todos los servicios. Cuadro 2. (continuación) RESOLUCIÓN 1445 DE 2006 RESOLUCIÓN 1446 DE 2006 RESOLUCIÓN 4002 DE 2007 RESOLUCIÓN 004816 DE 2008 “Por la cual se definen las funciones de la Entidad Acreditadora y se adoptan otras disposiciones” “Por la cual se define el Sistema de Información para la Calidad y se adoptan los indicadores de monitoreo del Sistema Obligatorio de Garantía de Calidad de la Atención en Salud” “Por la cual se adopta el Manual de Requisitos de Capacidad de Almacenamiento y Acondicionamiento para Dispositivos Médicos” “Por la cual se reglamenta el Programa Nacional de Tecnovigilancia” “Código Eléctrico Colombiano” Norma Técnica Colombiana – NTC 2050 Norma Técnica Colombiana – NTC 5736 Norma Técnica Colombiana – NTC 5460 “Estructura de codificación para tipos de eventos adversos y sus causas “Dispositivos Médicos, su aplicación del manejo del riesgo a los Dispositivos Medico” En el numeral 12 de los estándares de Gestión de la tecnología dice lo siguiente: „‟La organización garantiza que existe un sistema de reporte de eventos adversos asociados con el uso de la tecnología. El indicador I.3.3 hace referencia a la proporción de vigilancia de eventos adversos, el cual tiene como numerador el número total de eventos adversos detectados y gestionados, y como denominador, el número total de eventos adverso detectados. Hace parte del dominio de la gerencia del riesgo. Contiene las prácticas y procedimientos para el almacenamiento y/o acondicionamiento que se debe aplicar a los dispositivos médicos y así mantener la calidad de los mismos, durante todo el proceso de almacenamiento. Artículo 1°. Objeto y ámbito de aplicación. El objeto de la presente resolución es reglamentar el Programa Nacional de Tecnovigilancia Capítulo 5: Ambientes especiales, sobre las instituciones de asistencia médica. Instalaciones eléctricas de los Equipos Biomédicos en Clínicas y Hospitales (Unidades de Cuidados Intensivos Esta norma específica los requisitos para una estructura de codificación cuyo fin sea describir los eventos adversos relacionados con los dispositivos médicos. Esta es una norma para la aplicación del manejo de riesgo a dispositivos médicos. Fuente: Adaptado de FRANCO, Catalina Vásquez, TRUJILLO, Mauricio Pérez. Guía para la implementación del Programa Nacional de Tecnovigilancia en el Hospital General de Medellín, 2011. 35 3. TECNOVIGILANCIA La Vigilancia de dispositivos biomédicos o Tecnovigilancia es, en su definición más amplia, el proceso de obtención de información sobre incidentes ocurridos con dispositivos médicos durante su utilización en seres humanos con fines de prevención, diagnóstico y tratamiento de enfermedades. La detección de la ocurrencia de un evento o incidente adverso relacionado con un dispositivo biomédico permite alertar a toda la comunidad médica, fabricantes sobre el mismo y tomar las correspondientes medidas correctivas para minimizar sus efectos, después de un proceso de investigación que determine las causas que lo originaron. La Tecnovigilancia es considerada por el INVIMA, como el “conjunto de actividades que tienen por objeto la identificación y la cualificación de efectos indeseados producidos por los dispositivos médicos, así como la identificación de los factores de riesgo asociados a éstos efectos o características relacionados con éste riesgo, con base en la notificación, registro y evaluación sistemática de los problemas relacionados con los dispositivos médicos, con el fin de determinar la frecuencia, gravedad e incidencia de los mismos para prevenir su aparición”17. Según el INVIMA en su documento Guía de reporte de incidentes adversos a dispositivos médicos, “la importancia de estructurar un sistema de vigilancia de dispositivos médicos parte de la necesidad de garantizar la seguridad y efectividad de estos productos una vez salen al mercado y son usados individual y colectivamente”18. 17 Lineamientos para al desarrollo de la Tecnovigilancia en Colombia. Documento Marco. INVIMA, Subdirección de Insumos para la Salud y Productos Varios .2005. Citado en INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS. Programa Nacional de Tecnovigilancia. Guía de reporte de incidentes adversos a dispositivos médicos. [En línea] Bogotá d.c.2005. [consultado el 01 de octubre 2011]. Disponible en internet: http://www.consultorsalud.com/biblioteca/ 18 INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS. Subdirección de insumos para la salud y productos varios. Programa Nacional de Tecnovigilancia. Guía de reporte de incidentes adversos a dispositivos médicos. [En línea] Bogotá d.c.2005. [consultado el 01 de octubre 2011]. Disponible en internet: http://www.consultorsalud.com/biblioteca/ 36 Por su parte, el Ministerio de la Protección Social corrobora la información anterior al decir que la Tecnovigilancia es un “sistema de vigilancia postmercado”19 que lo constituyen normas a nivel nacional, mecanismos, procesos, instituciones, que requiere la asignación de recursos económicos técnicos y humanos y que debe estar acompañado por procesos y funciones para su correcto funcionamiento. La Tecnovigilancia de dispositivos biomédicos, es muy importante pues permite Identificar, recolectar, evaluar, gestionar y divulgar los eventos e incidentes adversos no descritos, que presentan los dispositivos médicos durante su uso. Esta, como actividad valiosa dentro de la gestión de la evaluación y regulación sanitaria y como integridad de un sistema de calidad en salud, requiere de la ratificación de sistemas o programas que le permitan su adecuado desarrollo, y es por esto, que tanto las entidades sanitarias nacionales y regionales de salud, como de los prestadores de salud, deben movilizar esfuerzos por generar los elementos necesarios para su buen desarrollo. Lo anterior, se hace necesario para cuantificar el riesgo y la realización de medidas que propendan por el mejoramiento de la protección de la salud y la seguridad del cuerpo médico, pacientes, usuarios y todo aquel que se vea implicado directa o indirectamente con la utilización del dispositivos biomédico. Como se mencionó, el funcionamiento de estos programas requiere de la articulación interinstitucional para su funcionamiento. Los niveles de operación del Plan de Tecnovigilancia a nivel nacional están compuestos de la siguiente manera tal como se muestra en la figura 3: 19 COLOMBIA. MINISTERIO DE PROTECCIÓN SOCIAL, INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS. [En línea] Bogotá d.c.2010. [consultado el 01 de octubre 2011]. Disponible en internet: http://www.valledelcauca.gov.co 37 Figura 3. Niveles de operación de los Programas de Tecnovigilancia en Colombia Fuente: Elaboración propia. Tomado de INVIMA, 2005 La Tecnovigilancia hace parte de la fase postmercado de la vigilancia sanitaria de los dispositivos médicos, según Antonio Hernández20, los elementos esenciales que debe tener el sistema de vigilancia son: la clasificación, la detección notificación, la investigación, el reporte, la información y la difusión. Igualmente se debe realizar un seguimiento de los EA ocurridos tanto en la propia institución como en otras, para evitar la ocurrencia y/o repetición de estos, lo que constituye un valioso punto de partida para una eficaz gestión de riesgo. 20 Hernández, Antonio. Asesor regional en infraestructura física y tecnología de servicios de salud OPS/OMS, 2007. En presentación “Conceptos de Tecnovigilancia, estatus en Latinoamérica y el Mar Caribe prospectiva de la OPS”. [En línea]. [consultado el 08 de agosto 2011]. Disponible en internet: http://www.cenetec.salud.gob.mx/descargas/presentacionesforo2007/17sep/clinica/antonio_hernandez.pdf. 38 Figura 4. Tecnovigilancia en el ciclo de vida del dispositivo médico Fuente: Hernández, Antonio. Asesor regional en infraestructura física y tecnología de servicios de salud OPS/OMS, 2007. En presentación “Conceptos de Tecnovigilancia, estatus en Latinoamérica y el Mar Caribe prospectiva de la OPS”. [En línea] [Disponible en Internet]: http://www.cenetec.salud.gob.mx/descargas/presentacionesforo2007/17sep/clinica/antonio_hernandez.pdf La responsabilidad de la formulación e implementación de los Programas Institucionales de Tecnovigilancia recae en los prestadores de salud y profesionales independientes, fabricantes e importadores de equipos biomédicos, Secretarias de Salud Departamentales y Locales, el Instituto Nacional de Vigilancia de Medicamentos y Alimentos - INVIMA y el Ministerio de la Protección Social. A continuación, se presenta en el cuadro 3 las responsabilidades de estos actores institucionales para la implementación de programas de Tecnovigilancia para la gestión del riesgo en equipos biomédicos, en Colombia. Cuadro 3. Responsabilidad de los actores institucionales para la gestión del riesgo de equipos biomédicos ENTIDAD PRESTADORES DE SALUD Y PROFESIONALES INDEPENDIENTES FABRICANTES E IMPORTADORES RESPONSABILIDAD Creación y desarrollo del Programa Institucional de Tecnovigilancia (PITV), verificar la calidad y seguridad de DM, promover prácticas adecuadas de uso y tomar acciones preventivas y correctivas. Creación y desarrollo del PITV, garantizar la calidad y seguridad de DM, informar sobre prácticas adecuadas de uso y tomar acciones preventivas y correctivas. 39 Cuadro 3. (continuación) SECRETARIAS DE SALUD DEPARTAMENTALES Y LOCALES INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS INVIMA MINISTERIO DE PROTECCIÓN SOCIAL Promover el desarrollo y articulación de los PITV (trabajo en red), capacitar a los actores institucionales, comunicación y direccionamiento hacia el PNTV, proponer y aplicar reglamentación y acciones de IVC. Fortalecer el PNTV, capacitar a todos los actores del PNTV, fortalecer y promover el trabajo en red, clasificar y registrar ordenadamente los reportes de las diferentes fuentes, proponer y aplicar reglamentación y acciones de IVC. Reglamentación, evaluar y valorar, establecer e Informar y desarrollar relaciones. Fuente: Elaboración propia. Tomado de INVIMA, 2005 Toda institución donde se desarrolle el programa de Tecnovigilancia debe tener claras las actividades y la asignación de una persona responsable de su gestión. Estas actividades y responsabilidades se describen a continuación: Definir y proponer estudios sobre: Tipos de dispositivos médicos, servicios o grupos de pacientes objeto de vigilancia. Protocolizar: Estrategia de vigilancia y recolección de reportes, análisis y valoración de los resultados, reporte al fabricante y autoridad sanitaria. Realizar el análisis: De acuerdo a la gravedad, frecuencia, impacto a pacientes que hacen el uso de estos dispositivos médicos implicados en evento o incidente adverso. Documentar: Las funciones y actividades del Programa Institucional de Tecnovigilancia mediante procedimientos que deben ser aprobados por los responsables del programa. Desarrollar / adquirir un programa de administración y gestión de datos: Debe permitir exactitud, fiabilidad, consistencia, confidencialidad y seguimiento en el tiempo. Elaborar y enviar: De manera inmediata al INVIMA, todo reporte de evento o Incidente adverso y trimestralmente a las Secretarías Departamentales y Distritales de Salud todo reporte serio junto con el resultado del análisis realizado. Para llevar a cabo lo anterior, el INVIMA estableció los mecanismos para recolectar, evaluar y gestionar la información relacionada con la seguridad de los dispositivos médicos, y de esta manera, propender por la toma de las medidas necesarias para preservar la salud y protección de los usuarios de este tipo de tecnología. 40 3.1 REPORTES DE TECNOVIGILANCIA La Tecnovigilancia identifica, evalúa y sirve para hacer el seguimiento de las situaciones relacionadas con dispositivos médicos que puedan ocasionar daño a los pacientes, usuarios o personas inherentes a los establecimientos médicos. Estas situaciones se consideran eventos adversos o potencialmente adversos, si se genera daño al paciente o pudo haber sido un evento adverso, respectivamente. El reporte de estos eventos tiene como objetivo brindar información al INVIMA, al fabricante o la institución hospitalaria sobre la generación de uno de esto, relacionado con un dispositivo médico durante su uso. Según la Guía de Reporte de Eventos Adversos a Dispositivos Médicos del Programa Nacional de Tecnovigilancia, los reportes considerados por el Programa Nacional son: 3.1.1 Reportes inmediatos. Reportes de Tecnovigilancia que relacionan eventos adversos serios asociados a dispositivos médicos para uso en humanos. Estos reportes deben realizarse al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA dentro de las setenta y dos horas (72) horas siguientes al conocimiento del eventos. 3.1.2 Reportes periódicos. Estos reportes deben ser presentados trimestralmente y en forma consolidada al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA o a las Direcciones Distritales o Departamentales de Salud según sea el caso, junto con las posibles medidas preventivas tomadas. 3.1.3 Reportes de retiro de productos o lotes de producto. Reportes de Tecnovigilancia mediante los cuales un fabricante o importador, informa a la autoridad sanitaria sobre el retiro de un producto o lote de producto del mercado, cuando estos supongan un riesgo para la salud de los pacientes en que van a ser usados. 3.1.4 Reporte de alertas internacionales. Reporte de Tecnovigilancia mediante el cual un importador autorizado para comercializar dispositivos médicos en Colombia informa al INVIMA de la generación de una alerta internacional por parte de la casa fabricante en el país de origen o por una agencia sanitaria a nivel mundial en la que se vea involucrado un dispositivo médico comercializado en Colombia. Existen 4 tipos de reporte de seguridad estos se describen en el cuadro 4. 41 Cuadro 4. Tiempo establecido para reportar ante el INVIMA TIPO DE REPORTE PROFESIONALES DE LA SALUD E INSTITUCIONES FABRICANTE E IMPORTADORES DE DISPOSITIVOS MÉDICOS REPORTES INMEDIATOS 72 horas siguientes a la ocurrencia del incidente adverso serio o potencialmente serio 72 horas siguientes al conocimiento de la ocurrencia del incidente adverso no serio o potencialmente no serio REPORTES PERIÓDICOS Trimestral Trimestral REPORTE DE RETIRO DE PRODUCTOS No aplica Cuando el fabricante o importador decida iniciar el retiro del producto en el país REPORTE DE ALERTAS INTERNACIONALES No aplica 72 horas siguiente en que se obtuvo conocimiento de la generación de una alerta internacional por parte de la casa fabricante en el país de origen o por una agencia sanitaria a nivel mundial en la que se vea involucrado un dispositivo médico comercializado en Colombia Fuente: Instituto Nacional de Vigilancia de Medicamentos y Alimentos INVIMA, subdirección de insumos para la salud y productos varios. Programa Nacional de Tecnovigilancia. Guía de reporte eventos adversos a Dispositivos médicos. En el INVIMA el Grupo de Tecnovigilancia, dentro de su gestión de alertas sanitarias, realiza una búsqueda de alertas emitidas por Organismos Reguladores Internacionales mediante monitoreo de las páginas web oficiales de las Agencias Sanitarias que se mencionan a continuación: • Agencia Nacional de seguridad de medicamentos y productos de salud “ANSM” de Francia • Agencia regulatoria para productos de medicina y salud “MHRA” de Reino Unido • Agencia de Administración de Alimentos y Drogas “FDA” de Estados Unidos • Agencia Española de medicamentos y productos sanitarios “AEMPS” • Agencia Salud de Canadá “HS-SC” • Agencia Nacional de Vigilancia Sanitaria “ANVISA” de Brasil y • Agencia Sanitaria de Australia “TGA” 42 En esta información se encuentran recomendaciones, información general, alertas sobre productos como también los retiros de producto del mercado notificado por los fabricantes de dispositivos médicos en cumplimiento de sus responsabilidades respecto a la seguridad de los productos que comercializa. El Objetivo de estos reportes es conocer de manera directa la información relacionada con la Seguridad y Desempeño de los productos notificados por los actores, para la toma de medidas sanitarias a las que haya lugar en función de la protección de Salud Pública del país.21. La información sobre alertas aportada por el INVIMA se registra desde 1999 a mayo de 2012 y aporta los enlaces para verificar la información directamente desde las páginas oficiales. 3.1.5 Información para el reporte22. La información para el reporte debe ser información clara, veraz y confiable sobre el riesgo de generación de un incidente adverso (IA) o generación de un evento adverso (EA) relacionado con un dispositivo médico durante su uso. En concordancia con el artículo 60 del decreto 4725 de 2005, el reporte debe realizarse ante cualquier evento o circunstancias relacionadas con cualquier funcionamiento defectuoso, efecto adverso o alteración de las características o de las funciones, también se debe reportar un etiquetado inadecuado o instrucciones de uso que puedan o haya causado la muerte o un deterioro grave del estado de salud de un paciente o de un usuario. Así mismo se debe reportar cualquier razón de carácter técnico o sanitario que haya inducido al fabricante a retirar del mercado los productos que pertenezcan al mismo tipo. Los objetivos de reportar, según el INVIMA, son: Obtener información para la evaluación de los riesgos asociados con el uso de dispositivos médicos. Tomar acciones necesarias para la reducción o eliminación del riesgo de generación de eventos e incidentes adversos. Contribuir en el mejoramiento de los sistemas de información en salud en el país referentes a la vigilancia epidemiológica. 21 Inspección, vigilancia y control. [en línea]. Instituto Nacional de Vigilancia de Medicamentos y Alimentos INVIMA [consultado 17 de junio de 2012].Disponible en internet http://web.invima.gov.co/portal/faces/index.jsp?id=4592&page=2 22 Preguntas frecuentes sobre el reporte de incidentes adversos [en línea]. Bogotá D.C.: Instituto Nacional de Vigilancia de Medicamentos y Alimentos INVIMA [consultado 17 de junio de 2011]. Disponible en Internet: http://web.invima.gov.co/portal/faces/index.jsp?id=5081 43 3.1.6 Información del reporte. Según el artículo 17 de la resolución 4816 del 2008, los reportes periódicos deben contener 5 aspectos: Identificación del paciente. Descripción del evento. Información del DM involucrado. Otras informaciones adicionales, como la gestión realizada y las acciones correctivas y/o preventivas. Identificación de la persona que reporta. 3.2 ANÁLISIS Y EVALUACIÓN DE EVENTOS E INCIDENTES ADVERSOS Según la Guía Técnica para las Buenas Prácticas para la Seguridad del Paciente en atención en Salud23, para que el reporte sea útil es necesario que se desarrolle un análisis de las causas que favorecieron la ocurrencia del evento adverso. Se debe precisar cuál será el manejo dado a la lección aprendida, como se establecerán barreras de seguridad, cuales procesos inseguros deberán ser rediseñados, y el apoyo institucional a las acciones de mejoramiento. Si bien es cierto, el “Protocolo de Londres” a través del modelo de Reason es la metodología utilizada en Colombia y recomendada para el análisis de los eventos adverso (EA), existen también otros aspectos que deben ser tenidos en cuenta, los cuales se describen a continuación: 3.2.1 Multicausalidad. Pocas veces los eventos son consecuencia de un solo error, por esto importante entender los errores multicausales, los cuales son reconocidos universalmente en los procesos de seguridad de todos los sectores. La multicausalidad hace referencia a que un evento adverso es el resultado de una serie de circunstancias concatenadas interactuantes; este concepto es conocido también como el modelo del queso suizo24, que se entiende como un 23 COLOMBIA, Ministerio de la Protección Social. Sistema Obligatorio de Garantía de Calidad en Salud SOGC. Dirección General de Calidad de Servicios. Unidad Sectorial de Normalización. Guía Técnica “Buenas Practicas para la seguridad del paciente en atención en salud”. 24 AMAYA, Luengas Sergio. Centro de Gestión Hospitalaria. Centro Médico imbanaco. Seguridad del Paciente. Conceptos y análisis de eventos adversos. Disponible en http://www.cgh.org.co/imagenes/calidad1.pdf 44 conjunto de fallas latentes que se activa simultáneamente durante la atención de un paciente y se presenta por los agujeros tal como se ilustra en el figura 5, el cual explica gráficamente los resultados que se obtienen cuando existen grietas en las barreras de seguridad del sistema. Figura 5. Modelo del queso suizo Fuente: ESPAÑA. Ministerio de Sanidad y Consumo. Dirección General de la Agencia de Calidad del Sistema Nacional de Salud. [En línea] Seguridad del Paciente, una prioridad del sistema nacional de salud. XV Congreso Nacional de Hospitales Roquetas de mar Almería. 17 de Mayo 2007. [Consultado el 18 febrero 2012]. Disponible en http://www.seguridaddelpaciente.es/opsc/boletin9/seguridad_del_paciente_priorida d_SNS.pdf. 3.2.2 Modelo SHELL. Es un modelo conceptual de explicación de la realidad que se centra en la importancia que se le da al ser humano como núcleo central de dicha realidad. Reconoce que la persona posee una serie de debilidades que inciden directamente en su desempeño laboral. El nombre SHELL proviene de las siguientes palabras en inglés. Software: Corresponde al soporte lógico, como por ejemplo los procedimientos para llevar a cabo una tarea, los manuales, las listas de chequeos, las reuniones de coordinación, las instrucciones, etc. Hardware: Corresponde al soporte físico, como puede ser cualquier tipo de máquina o equipos. Envirorment: Corresponde al soporte físico, como puede ser cualquier tipo de máquina o equipos. Liveware: Corresponde al elemento humano (hombre en el puesto de trabajo). 45 3.2.3 Protocolo de Londres. El protocolo de Londres25 es considerado como una guía práctica para los administradores de riesgo y tiene como propósito facilitar la investigación clara y objetiva de los incidentes clínicos, con una visión global de la situación y no centrándose en la simple identificación de la falla o responsables. En este modelo, las decisiones para la gestión del riesgo, son tomadas desde los niveles directivo y gerencial de la organización y luego son transmitidas hacia el resto del equipo de profesionales. Esta dinámica permite crear las condiciones que puedan generar conductas inseguras de diversa índole. Durante el análisis de un incidente cada uno de estos elementos se considera detalladamente y por separado, comenzando por las acciones inseguras y las barreras que fallaron, hasta llegar a la cultura y procesos organizaciones, como se muestra en la figura 6. Figura 6. Modelo organizacional del Protocolo de Londres Fuente: AMAYA, Luengas Sergio. Centro de Gestión Hospitalaria. Centro Médico imbanaco. [En línea] Seguridad del Paciente. Conceptos y análisis de eventos adversos. [Consultado el 12 de Diciembre de 2011]. Disponible en http://www.cgh.org.co/imagenes/calidad1.pdf. 25 TAYLOR Sally, VINCENT Adams y VINCENT Charles. Proceso sugerido para la investigación y análisis de eventos adversos (Protocolo de Londres). [En línea]. Londres Clinical Safety Research Unit. Imperial College Lodon, UK. [consultado el 14 de julio de 2011]. Disponible en Internet.http://www.seguridadclinica.tk/descargas/pdf/Protocolo_de_Londres.pdf 46 El paso siguiente es considerar el contexto institucional general y las circunstancias en que se cometieron los errores, las cuales son conocidas como factores contributivos tal como se observa en el cuadro 5. Cuadro 5. Factores contributivos que influencian la práctica clínica ORIGEN FACTOR CONTRIBUTIVO Complejidad y gravedad Lenguaje y comunicación Personalidad y factores sociales Diseño de la tarea y claridad de la estructura Disponibilidad y uso de protocolos Disponibilidad y confiabilidad de las pruebas diagnósticas Ayudas para toma de decisiones INDIVIDUO Conocimiento, habilidades y competencia Salud física y mental EQUIPO DE TRABAJO Comunicación verbal y escrita Supervisión y disponibilidad de soporte Estructura del equipo (consistencia, congruencia, etc.) Personal suficiente; mezcla de habilidades; carga de trabajo; patrón de turnos; diseño, d disponibilidad y mantenimiento de equipos; soporte Administrativo y gerencial; clima laboral; ambiente físico (luz, espacio, ruido) PACIENTE TAREA Y TECNOLOGÍA AMBIENTE ORGANIZACIÓN Y GERENCIA Recursos y limitaciones financieras; estructura organizacional; Políticas, estándares y metas; prioridades organización y gerencia y cultura organizacional CONTEXTO Económico y regulatorio Contactos externos INSTITUCIONAL Fuente: Servicios integrales de salud. Clínica Colsanitas S.A. [En línea]. Protocolo de Londres [Consultado el 18 de Febrero 2012].Disponible en: http://200.47.156.201/pacienteseguro/docs/Lectura_protocolodelondres.pdf En la figura 7 se ilustra la sucesión de pasos a seguir para investigar y analizar un incidente clínico, es decir, tanto un error como un evento adverso. 47 El proceso básico de investigación y análisis está bastante estandarizado. Fue diseñado pensando en que sea fácil y que se pueda utilizar tanto en eventos o incidentes menores hasta eventos graves, depende de cada institución lo extenso y profundo de la investigación del evento dependiendo de la gravedad, de los recursos y de la institución. Figura 7. Secuencia de investigación y análisis del Protocolo de Londres Fuente: TAYLOR Sally, VINCENT Adams y VINCENT Charles. Proceso sugerido para la investigación y análisis de eventos adversos (Protocolo de Londres). [En línea]. Londres: Clinical Safety Research Unit. Imperial College London, UK. [Consultado el 14 de julio de 2011]. Disponible en Internet.http://www.seguridadclinica.tk/descargas/pdf/Protocolo_de_Londres.pdf A continuación se detallan cada uno de los pasos de la investigación de incidentes: Investigar evento adverso (identificación y decisión de investigar). Esta decisión se toma por el impacto y la frecuencia que tiene el evento adverso en la institución. Selección del equipo investigador. Debe integrar al personal asistencial involucrado en el evento, liderado por un experto de investigación y análisis de incidentes clínicos. 48 Experto en investigación y análisis de incidentes clínicos. Miembro de junta directiva sin conocimiento médico del caso. Autoridad administrativa como director médico o jefe de enfermería. Autoridad clínica como jefe de departamento o especialista reconocido. Miembro de la unidad asistencial donde ocurrió el incidente. El equipo de investigación deberá obtener y organizar toda la información relacionada con el evento adverso. Esta acción recoge todos los hechos, conocimientos y elementos involucrados en el evento. Historia clínica completa. Guías clínicas y protocolos de atención asociados a pacientes con cuadros clínicos. Declaraciones y observaciones inmediatas. Entrevista con los involucrados. Hojas de vida de los equipos involucrados. Otros aspectos relevantes tales como índice de rotación del personal y disponibilidad de personal capacitado. Cronología del incidente. Detallar el paso o pasos del incidente con el fin de hallar las causas que lo provocaron, mediante los pasos anteriores (entrevistas, declaraciones, formatos, historia clínica, entre otros) se puede establecer qué y cuándo ocurrió. Hay dos metodologías para precisar la cronología. La narrativa. El método diagrama. El método que se usará será el método diagrama en el cual los movimientos de personas, materiales, documentos e información pueden representarse mediante un dibujo esquemático. Puede ser útil la secuencia de hechos como deberían haber ocurrido de acuerdo con las políticas, protocolos y procedimientos, y compararla con lo que verdaderamente ocurrió cuando se presentó el incidente. 49 Identificación de acciones inseguras. Estas son las fallas activas en el proceso de atención en salud. Identificar las condiciones asociadas con cada acción insegura (factores contributivos). Estos predisponen la aparición de las acciones inseguras. El paciente. Tarea y tecnología ej: falta de claridad del protocolo de uso y reusó de mascarillas en urgencias, escases de suministros de servicios, falta de investigación de los síntomas. Los individuos ej: toma de decisión sin comunicarle a los jefes de área, tomar una conducta sin realizar exámenes más rigurosos. Equipo de trabajo ej: la falta de supervisión de un profesional del servicio. El ambiente ej: estrés de servicio de urgencias. La organización y gerencia ej: escases de suministros en la organización, políticas de uso y reuso de dispositivos médicos. Contexto institucional. Se puede hacer un diagrama de espina donde se asocia una acción insegura con los factores contributivos. Como se muestra en la figura 8. Figura 8. Diagrama de espina Fuente: TAYLOR Sally, VINCENT Adams y VINCENT Charles. Proceso sugerido para la investigación y análisis de eventos adversos (Protocolo de Londres). [En línea]. Londres, Clinical Safety Research Unit. Imperial College Lodon, UK. [Consultado el 18 de Febrero de 2012]. Disponible en Internet.http://www.seguridadclinica.tk/descargas/pdf/Protocolo_de_Londres.pdf Recomendaciones y planes de acción. Diseño de planes de acción que ayuden a prevenir o mitigar los eventos adversos. 50 Definir políticas del uso de adquisición biomédica. Revisar y ajustar el proceso de adquisición de equipos biomédicos. Evaluar la adherencia del personal. Establecer las necesidades de insumos en la Clínica de Occidente. Sensibilizar al personal sobre la importancia de la utilización de equipos biomédicos. Gestión y calificación del riesgo de la atención en salud en equipos biomédicos. Diseño de guías rápidas de los equipos biomédicos. Capacitación al personal asistencial. 3.3 VIGILANCIA ACTIVA La mayoría de los incidentes adversos, en los que se encuentran implicadas las tecnologías de la salud, se pueden prevenir con un mantenimiento adecuado y asegurándose de la pertinencia y buen uso de las mismas 26. Por lo anterior, los sistemas de Tecnovigilancia deben tener incluido el componente de sistema de vigilancia activa. El sistema de vigilancia activa enfoca sus esfuerzos en vigilar los dispositivos médicos que presentan un riesgo alto para la salud de la población, caso particular son los equipos catalogados como riesgo III, equipos de nueva tecnología, de soporte de vida, y equipos de tecnología de importación controlada. La vigilancia también se puede centrar en aquellas áreas donde existe un alto riesgo para la salud del paciente a causa del uso de los dispositivos médicos, tales como las unidades de cuidados intensivos, salas de cirugía, servicios de neonatología, neurocirugía, entre otros27. Dentro de la vigilancia activa también se contemplan los programas de muestreo de dispositivos con el fin de determinar la calidad con que fueron elaborados, almacenados y suministrados a los pacientes, así mismo se realizan matrices de riesgo de los dispositivos. 26 La mayoría de los incidentes adversos con tecnologías de la salud se pueden prevenir [En línea]. Madrid: Europapress, 2010 [consultado 10 de julio de 2011]. Disponible en Internet: http://www.europapress.es/salud/noticia-mayoria-incidentes-adversos-tecnologias-salud-puedenprevenir-20101214152712.html 27 Vigilancia activa [En línea]. Bogotá D.C.: INVIMA, 2008 [consultado 10 de julio de 2011]. Disponible en Internet: http://web.invima.gov.co/portal/faces/index.jsp?id=4583 51 A continuación, se presenta en el cuadro 6 el resumen de algunos de los principales riesgos de la tecnología en salud, presentados por el Instituto ECRI28 (Emergency Care and Research Institute). Cuadro 6. Principales riesgos de la tecnología en salud FACTORES DE RIESGO PREVENCIÓN SOBREDOSIS DE RADIACIÓN Y OTROS ERRORES DE DOSIS DURANTE RADIOTERAPIA Margen de error cada vez más estrecho. Potencial de errores que se repiten. Transferencia de datos entre el tratamiento planeado y los dispositivos terapéuticos. Dotación y clasificaciones del personal. Sistema de instalación e integración. Protocolos locales y aseguramiento de la calidad. Manejo de alarmas. PELIGROS DE ALARMAS Falsa alarma. Alarmas mal configuradas. Alarmas inaudibles. Dispositivos/diseños similares. Evaluación de áreas de atención al paciente. Protocolos y permisos de usuarios definidos. Normalización y estandarización. CONTAMINACIÓN CRUZADA DE LOS ENDOSCOPIOS FLEXIBLES Variación en el diseño del endoscopio. Mala técnica de limpieza o secado. Cambios en el equipo o proceso. Modelo específico de protocolos de desinfección. Asegurarse de conocer y comunicar los nuevos modelos. Revisión periódica de los protocolos y del entrenamiento. Considerar los accesorios. DOSIS DE RADIACIÓN DE TOMOGRAFÍA COMPUTARIZADA (TC) Uso creciente de TC. Dosis: 1 TC = 3 años de estudio de fondo. 29.000 casos de cáncer en EE.UU. cada año 28 Promover la conciencia entre los médicos. Exámenes justificados. Revisar y optimizar los protocolos de exploración. Entrenamiento de los tecnólogos. Control de calidad Sacks, Eric. Top 10 technology hazards for 2011. En: Health devices ECRI. Noviembre de 2010, vol 39, no. 11, p. 386 – 400. 52 Cuadro 6. (continuación) COMPLICACIONES EN LA INFORMACIÓN TECNOLÓGICA EN SALUD Cambios en las interfaces del equipo médico de la red. Proyectos a gran escala Planificación eficaz entre las partes interesadas. Especificación de las necesidades de interoperabilidad en los contratos de compra. Coordinación de mantenimiento y actualizaciones. Pruebas y verificación. JERINGAS LUER INCORRECTAS Administración intravenosa de otros fluidos. Embolia de gas. Equipamiento no intravenoso con accesorios Luer. Adaptadores. Consideraciones de compra. Etiquetado de la clave de acceso de los catéteres. Capacitación del personal. SEDACIÓN DURANTE EL USO DE BOMBAS DE INFUSIÓN PCA Orden y administración errónea de medicamentos. Medicamentos potentes. Manipulación. Organizar protocolos. Plan para monitorear pacientes. Revisar los protocolos de evaluación del paciente. Doble control, de las órdenes y configuración de la bomba PINCHAZOS CON AGUJAS Y LESIONES CON OBJETOS CORTOPUNZANTES Técnica pobre Evaluación continua. Análisis y ajuste de la meta. Plan de acción para cada meta. Aplicación coordinada y formación. Repetición. INCENDIOS QUIRÚRGICOS Medio ambiente quirúrgico. Conocimiento. Respuesta. 53 Control. Control de la fuente de oxígeno. Control de la fuente de ignición. Control de la fuente de combustible. Protocolos de respuesta. Cuadro 6. (continuación) FALLAS DEL DESFIBRILADOR EN LOS INTENTOS DE REANIMACIÓN DE EMERGENCIA Baterías dañadas. Conexión incorrecta. Malinterpretación de los códigos de error. Control diario. Verificación de la carga de la batería. Reacción a fallos en las pruebas de autorevisión. Fuente: VARGAS, Flórez Angélica, ARISTIZÁBAL, Polanco María Alejandra. Diseño de un programa de tecnovigilancia para la clínica Rey David – Cali. Santiago de Cali, 2011. 54 4. DISEÑO DE UN PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA PARA LA CLÍNICA DE OCCIDENTE El Programa Institucional de tecnovigilancia de la Clínica de Occidente se diseñó con base en la normatividad nacional que fija las directrices para su formulación. Este programa dependerá del área de electromedicina de la Clínica que tiene por objetivo mantener en buen estado físico y funcional, dentro de las especificaciones del fabricante, los equipos médicos de la institución, facilitando los requerimientos tanto para su instalación como para su desempeño en las áreas; de igual forma, asesorar en la adquisición de tecnología a fin de obtener la satisfacción y el bienestar del paciente. El programa Institucional de Tecnovigilancia fue incluido en dicho proceso ya que esta área maneja los recursos económicos y humanos para su implementación. Este proceso cuenta con un ciclo PHVA (planear, hacer, verificar y actuar) en donde se incluyeron los aspectos propios del programa de tecnovigilancia lo que permitirá monitorear el proceso de planeación de manera efectiva con el objetivo de constituirlo en una estrategia de mejora continua en donde se planee, se tomen acciones, se verifiquen si los resultados fueron los esperados y se actúe sobre dichos resultados para reiniciar el ciclo. Para el caso específico del programa institucional de tecnovigilancia el planear consiste en el diseño del programa, el hacer en la implementación de este que comprende la asignación de recursos, responsables y el desarrollo de la estrategia, el verificar en el seguimiento, la aplicación de indicadores y el análisis de datos para la medición de resultados y el actuar en la implementación de las estrategias o acciones para solucionar las dificultades problemas encontrados en el verificar. 55 Figura 9. Ciclo PHVA de Tecnovigilancia •Diseñar programa de tecnovigilancia •Implementar programa de tecnovigilancia (asignacion de rescursos y responsables) Planificar Hacer Actuar Verificar •Solucionar problemas encontrados en la verificación •Seguimiento, medir resultados, Aplicar los indicadores y analisis de datos Trabajar con la filosofía de mejoramiento continuo permite obtener beneficios como: mejoramiento en la calidad, alta productividad, mejor disponibilidad y confiabilidad de cada uno de los equipos, estandarización, servicios de preventa a los clientes y competitividad en un futuro29. Para este trabajo, se partió de un diagnóstico de reconocimiento del estado actual de la tecnovigilancia en la clínica. Como se observa en el cuadro 7, en este diagnóstico, se evaluó la existencia de los siguientes aspectos: 29 Área o departamento encargado de la tecnovigilancia en la clínica. Inventario técnico funcional. Formatos de reporte de eventos e incidentes adversos. Guías rápidas. Manual de tecnovigilancia. PEREZ, Carlos Mario. Soporte y Cia LTDA. Los indicadores de Gestión. 56 Cuadro 7. Diagnóstico del estado actual de Tecnovigilancia en la Clínica de Occidente ASPECTO EXISTENCIA SI NO Área o personal encargado de Tecnovigilancia en la Clínica Inventario Técnico Funcional x x Formatos de reporte de Eventos e incidentes adversos Manual de Tecnovigilancia OBSERVACIONES Si bien la Clínica cuenta con la Oficina de electromedicina, no cuenta con un programa de vigilancia estructurado, ni con una persona designada para este fin. La oficina de electromedicina cuenta un coordinador y un analista. A pesar de existir un Inventario Técnico Funcional que se actualiza una vez al año. Se procedió a actualizar el inventario encontrando equipos sin marcación de activos fijos No existe x x Guías rápidas x Protocolo de manejo y espacio seguro de los Equipos Biomédicos x No existe Se encontraron algunas guías rápidas incluidas en el manual de procesos de electromedicina y cuentan con copia controlada del área de calidad. Sin embargo se identificaron equipos sin guías rápidas y durante la ejecución del proyecto ingresaron a la Clínica nuevos equipos. A estos se les realizó la guía rápida. Se consideró importante reportar en este diagnóstico la existencia del Protocolo de manejo y espacio seguro de los Equipos Biomédicos que está dirigido al personal asistencial de la Clínica. El resultado de este diagnóstico permitió establecer las estrategias para el diseño del Programa Institucional de Tecnovigilancia, teniendo en cuenta las falencias y las necesidades de mejora de los aspectos evaluados. De esta manera, se procedió a ejecutar las siguientes actividades requeridas para el diseño del programa en la Clínica de Occidente. 4.1 ASIGNACIÓN DEL RESPONSABLE DEL PROGRAMA INSTITUCIONAL DE TECNOVIGILANCIA Dando cumplimiento a los requerimientos legales, se presentó al Comité de Seguridad del Paciente la normatividad que indica la necesidad de nombrar y asignar el personal y área de la clínica responsable del Programa Institucional de Tecnovigilancia. Teniendo en cuenta que la Institución ya contaba con la oficina de Electromedicina, el Comité de Seguridad del Paciente aprobó y asigno la responsabilidad del Programa Institucional de Tecnovigilancia al Coordinadora de dicha área. 57 Esta persona será la encargada de reportar los eventos o incidentes adversos que se presenten en la Clínica de Occidente. Para el caso de los incidentes se registrará, evaluará, informará, y los dará a conocer en el comité de Seguridad al Paciente quien a su vez valorará los casos más relevantes ocasionados con los equipos biomédicos, se darán a conocer junto con las posibles medidas preventivas tomadas al INVIMA con un reporte trimestralmente y en forma consolidada. Para el caso de los eventos el reporte se hará dentro de las 72 horas siguientes de evento por medio de la página oficial del INVIMA. Estos reportes estarán a cargo del coordinador de área de electromedicina. 4.2 INVENTARIO TÉCNICO FUNCIONAL Se considera el Inventario Técnico Funcional como una herramienta útil que permite desarrollar un registro descriptivo y permanente de las principales características de los equipos biomédicos, que permite la planeación, programación, adquisición de equipos, entre otros procesos. Además brinda la posibilidad de realizar la ejecución de otras acciones operativas, propias del servicio de mantenimiento dentro de la institución. El inventario técnico funcional de la clínica de occidente está compuesto por las hojas de vida de los equipos biomédicos la cual brinda información sobre las características principales de dichos equipos como la serie modelo, activo, nombre del equipo, fecha de adquisición del equipo, ubicación, área, descripción del equipo, clasificación de riesgo, protección contra descargas, fuente de alimentación mantenimientos preventivos, correctivos y metrología; la cual se puede observar en el anexo A Con base en lo anterior y reconociendo que la clínica ya cuenta con el inventario, se realizó su revisión encontrando algunas deficiencias en cuanto al contenido e información actualizada. Se encontró información que no correspondía con la generada anteriormente (activos fijos de los equipos, modelo, serie), y así mismo que el número de activo fijo de algunos equipos eran erróneos, algunos equipos se habían dado de baja, y aparecían en el inventario; por lo tanto, se optó por su actualización para conocer la ubicación del equipo y así saber el funcionamiento de cada uno de ellos y poder dar la clasificación de riesgo acorde al decreto 4725 de 2005. Formato para la actualización de inventario: Para la actualización del inventario, se utilizó el formato establecido por la Clínica de Occidente para realizar el seguimiento al mantenimiento de los equipos. Se diligenciaron los 58 campos correspondientes a: nombre del equipo, marca, modelo, activo fijo, serie, ubicación y clasificación de equipos biomédicos, a continuación se muestra en el cuadro 8 un ejemplo del inventario Técnico Funcional del Área de la Unidad de Cuidado Intensivo Coronaria 1 y el inventario completo realizado en todas las áreas de la clínica (UCI‟s, urgencias, rayos-x, laboratorio, cirugía, partos, hospitalización rehabilitación) se puede observar en el anexo B Cuadro 8. Inventario Técnico Funcional de la UCI Coronaria 1 INVENTARIO TECNICO FUNCIONAL DE LAS UNIDADES DE CUIDADOS INTENSIVOS Clinica de Occidente Área Unidad de Cuidado Intensivo Coronaria 1 Equipo Desfibrilador Electrocardiografo Balanza Flujometro Flujometro Flujometro Flujometro Monitor de Signos Vitales Monitor de Signos Vitales Monitor de Signos Vitales Monitor de Signos Vitales Ventilador Mecánico Ventilador Mecánico Ventilador Mecánico Ventilador Mecánico Vacuometro Vacuometro Vacuometro Marca Primedic Burdick Seca Chemetrom Chemetrom Chemetrom Chemetrom Space Lab Nihon Khoden Nihon Khoden Nihon Khoden Puritan Bennett Siemens Siemens Puritan Bennett Chemetrom Chemetrom Chemetrom Modelo DefiMonitor Eclipse LE N/A Timeter Timeter Timeter Timeter N/A Life Escope 9 Life Escope 9 Life Escope 9 740 Servo 900C Servo 900C 740 Continuos Continuos Continuos Activo Fijo 019150 004004 003908 077909 S/A 76520 097488 003921 003939 004018 005312 004646 003859 005320 004034 34079 34908 004681 Serie 73505001850 944621 8492 1077909 1021628 0404 1097488 309-011095 00040 00399 01145 3501982235 168936 161803 3501982219 S/S S/S S/S Clasificacion de equipos Biomedicos III III I I I I I III III III III III III III III I I I Visitas para la revisión de equipos y actualización de inventario: Para la actualización del inventario, se ejecutaron visitas a todas las áreas de la clínica (UCI‟s, urgencias, rayos-x, laboratorio, cirugía, partos, hospitalización rehabilitación), realizando entrevista con los coordinadores de estas áreas y la coordinadora del área de electromedicina. Revisión y análisis del inventario actualizado: Como etapa final del proceso, se procedió a revisar el inventario actualizado con el inventario 59 anterior con el área administrativa de la Clínica, específicamente, el área de activos fijos. Se realizó la socialización de lo encontrado en el nuevo inventario para corroborar la información en la base de datos y la actualización del listado de activos fijos de la Clínica. En la Clínica de Occidente se cuenta con más de 500 equipos biomédicos, 8 Unidades de Cuidados Intensivos (UCI‟s General, Coronaria 1, Cardiovascular, Coronaria 2, Nuevo Convenio, Neurovascular, Neonatal, Intermedios) y 9 Salas de Cirugía. Las UCI‟s y salas de Cirugía están equipadas con monitores de signos vitales PM9000, ventiladores Siemens, máquinas de anestesia Ohmeda, desfibriladores Primedic, electrocardiógrafos Edan, fuente de luz Storz, entre otros. 4.2.1 Definición del tipo de dispositivos médicos objeto de vigilancia. Posterior al Inventario Técnico Funcional, se realizó la descripción de los elementos conceptuales de los eventos e incidentes adversos teniendo en cuenta la resolución 004816 de 2008. A continuación, se presenta en el cuadro 9 la descripción de los equipos biomédicos, su función, su ubicación en la clínica, algunas causas de eventos o incidente adverso que pueda ocasionar, algunos riesgos que se han presentado con mayor frecuencia en estos equipos y la fotografía del mismo; considerando relevante aclarar que estos no son los únicos riesgos que pueden presentar y la información que se referencia es a manera de ejemplo. Esta información se dio a conocer a todo el personal asistencial y administrativo de la Clínica de Occidente por medio del Boletín de Seguridad al Paciente NO 15 del mes de Febrero del 2012” 60 Cuadro 9. Equipos biomédicos objeto de vigilancia EQUIPO BIOMÉDICO Monitor de signos vitales FUNCIÓN Dispositivo que permite detectar, procesar funciones fisiológicas del paciente UBICACIÓN UCI‟s, hospitalización, Salas de Cirugía CAUSAS DE EVENTO ADVERSO Mal funcionamiento intermitente. Falla en la fuente de poder. Falla en los accesorios del dispositivo. Falla en el Software. RIESGOS El monitor de signos vitales se queda en blanco periódicamente y luego reaparece la imagen. El equipo da una toma de presión errónea del paciente El equipo se recalienta ya que presenta una falla en la fuente de poder. EQUIPO BIOMÉDICO Desfibrilador FUNCIÓN Dispositivo medico electrónico que diagnostica y descarga un choque eléctrico al corazón para restablecer el ritmo cardiaco UBICACIÓN UCI‟s hospitalización, salas de Cirugía , CAUSAS DE EVENTO ADVERSO Eléctrico. Falla en la fuente de poder. Humo, fuego, explosión. Función imprevista. 61 RIESGOS El desfibrilador realiza su máxima descarga y se recalienta y se incendia El desfibrilador no entrega la descarga seleccionada. No hay una buena impedancia entre el paciente y las palas del desfibrilador provocando quemaduras al paciente. Cuadro 9. (continuación) EQUIPO BIOMÉDICO Electrocardiógrafo FUNCIÓN Dispositivo médico que registra y amplía la actividad eléctrica del corazón a través de electrodos. UBICACIÓN UCI‟s, hospitalización. CAUSAS DE EVENTO ADVERSO Mal funcionamiento del equipo. Conexión. Desconexión. EQUIPO BIOMÉDICO Ventilador mecánico 62 RIESGOS El electrocardiógrafo registra una lectura errónea aplicando medicamentos innecesarios. No se realiza una buena lectura debido a que las chupas se encuentran sucias provocando repetitividad del examen. Cuadro 9. (continuación) FUNCIÓN Dispositivo médico que suple o colabora con la función respiratoria de una persona. UBICACIÓN CAUSAS DE EVENTO ADVERSO Medidas de protección, software UCI‟s. RIESGOS No se activa la alarma del ventilador cuando un paciente presenta obstrucción en la vía aérea que reduce el flujo de aire. Paciente conectado al ventilador mecánico y se apaga constantemente, provocando la muerte al paciente. EQUIPO BIOMÉDICO Máquina de anestesia FUNCIÓN Dispositivo médico que permite ventilar al paciente y administrar los gases necesarios para el soporte de la anestesia. UBICACIÓN Salas de Cirugía CAUSAS DE EVENTO ADVERSO Medidas de protección, software, desconexión. 63 RIESGOS Se apaga constantemente la maquina cuando se produce un movimiento de ella, provocando la descompensación del paciente. El paciente se despierta en medio de la Cirugía los vaporizadores están mal puestos. Cuadro 9. (continuación) EQUIPO BIOMÉDICO Electrobisturi FUNCIÓN Dispositivo capaz de producir una serie de ondas electromagnéticas con el fin de cortar o eliminar tejido blando UBICACIÓN CAUSAS DE EVENTO ADVERSO Falla en la fuente de poder. Eléctrico. Salas de Cirugía RIESGOS El dispositivo recalienta provocando quemaduras al paciente se 4.3 GUÍAS RÁPIDAS DE EQUIPOS BIOMÉDICOS Se elaboraron las guías rápidas de acuerdo al cuadro 10, de los equipos utilizados en las UCI‟s y salas de Cirugía, con el fin de facilitarle al cuerpo médico, la lectura de las características de estos equipos; como su manejo, limpieza, frecuencia de mantenimientos, precauciones de manejo, modo de empleo del equipo y aspectos técnicos de los mismos que se muestran a continuación. Vale la pena aclarar, que las guías rápidas no sustituyen al manual de usuario. 64 Cuadro 10. Formato de Guías Rápidas MODO DE EMPLEO 1 3 2 4 5 6 7 8 65 Cuadro 10. (continuación) MODO DE EMPLEO 9 10 11 PARAMETRO DE ALARMA PRECAUCIONES LIMPIEZA DEL EQUIPO FRECUENCIA DE MANTENIMIENTO 66 Se realizaron guías rápidas para los siguientes equipos, los cuales se presentan en el anexo C: Monitor Mindray pm9000 Ventilador 840 Ventilador 740 5.4 DISEÑO DE UN SISTEMA SENSIBLE Todo programa de Tecnovigilancia requiere la implementación de estrategias que generen sensibilización. Cada persona debe ser consciente de su responsabilidad dentro del programa y debe cumplirla. En la etapa de diseño del sistema sensible se deben tener en cuenta los siguientes aspectos: El diseño del formato y medios de reporte El diseño de folletos y material informativo El diseño del manual de tecnovigilancia El diseño de estrategias de vigilancia 4.4.1 Diseño del formato y medios de reporte de eventos e incidentes adversos. Para la realización de los formatos de reporte de eventos e incidentes adversos, se revisó la resolución 4816 de 2008, que permitió encontrar información clave para el desarrollo del proyecto y el diseño de los formatos. Después de realizar el diseño de los formatos, se procedió a realizar la presentación de los mismos ante el comité de Seguridad al Paciente, que permitió incluir información propia de la institución y aclarar puntos necesarios para el formato del reporte. Con base en la información encontrada y concertada con el comité, finalmente se diseñaron y aprobaron los formatos de reporte de eventos e incidentes adversos desde dos enfoques: (i) Un enfoque interno para el área administrativa y el cuerpo médico y (ii) Un enfoque externo para los visitantes, pacientes y acompañantes que interactúan con la Clínica de Occidente. Los formatos de reporte se manejan de esta manera teniendo en cuenta que los visitantes, acompañantes, pacientes no manejan información técnica de los equipos lo cual puede generar confusión al manejar un formato unificado del reporte; es por esto que se aprobó el manejo de dos formatos. 67 4.4.2 Formato para el reporte del visitante. Para el diseño del formato de eventos e incidentes adversos de dispositivos médicos, se tomaron en cuenta las recomendaciones de la resolución 4816 de noviembre del 2008. Ver imagen 1 Imagen 1. Formato para el reporte del visitante Nº de Consecutivo Fecha de actualizacion Version Pagina Formato Visitante CLÍNICA DE OCCIDENTE ¡ Cada día Mejor ! FORMATO DE REPORTE DE EVENTO O INCIDENTE ADVERSO DE DISPOSITIVOS BIOMEDICOS 1. INSTITUCION REPORTANTE Fecha: Dia Hora: Mes Año 2. IDENTIFICACION DEL PACIENTE Nombre del paciente: Numero de Historia Clinica: Edad: Sexo: M F Ubicación del paciente: Cama Nº : Entidad: 3. DESCRIPCION DEL EVENTO ADVERSO EL EVENTO FUE A Paciente: Acompañante: Visitante: Otro: Cual: DESCRIPCION DEL EVENTO INFORMACION QUE CONSIDERE IMPORTANTE USTED PARA ESTE REPORTE NOTA: Si es necesario adicione hojas 68 El formato fue diseñado teniendo en cuenta el propuesto por el INVIMA. Posteriormente fue llevado al comité de seguridad al paciente donde se incluyeron algunos aspectos propios de la clínica. Este formato cuenta con 4 aspectos principales: 1. Institución reportante: Se debe mencionar la fecha, hora y área donde se presentó el evento. Fecha: Se establece la fecha cuando se produjo el evento. Hora: Cuando se presentó el evento (hora estimada). 2. Identificación del paciente: Se debe reportar la información del paciente / usuario que sufrió los efectos del evento adverso o riesgo de evento adverso. Nombre: Nombre del paciente o persona involucrada en el evento. Numero de historia Clínica: Número asignado al paciente durante la estadía en la Clínica. Edad: Indique la edad del paciente, si el paciente es menor de 1 año indique los meses cumplidos. Sexo: Seleccione con una (X) si el paciente es de sexo femenino (F) o masculino (M). Ubicación del paciente: Es el área donde se encuentra el paciente, hospitalizado en la Clínica, o el lugar donde se hallen los visitantes, los familiares. N° Cama: Es el número asignado al paciente dependiendo del área donde se encuentre el paciente. Entidad: Es el nombre de la IPS o EPS que presta el servicio de salud al paciente. 3. Descripción del evento adverso: Este ítem del formato de reporte cuenta con tres (3) sub ítems: El evento fue a: Corresponde a la identificación del paciente o persona involucrada en el evento que puede ser paciente, acompañante, visitante, otro, cual. Descripción del evento: En este espacio, el paciente o persona afectada debe describir con sus palabras el evento ocurrido. Información que considere importante usted para este reporte: Alguna información del paciente que se considere relevante. 69 4.4.3 Formato para el reporte del empleado Para el diseño del formato de eventos e incidentes adversos de equipos biomédicos, se tomaron en cuenta las recomendaciones de la resolución 004816 de noviembre del 2008. Ver imagen 2 Imagen 2. Formato para el reporte del empleado Nº de Consecutivo Fecha de actualizacion Version Pagina Formato Empleado CLÍNICA DE OCCIDENTE ¡ Cada día Mejor ! FORMATO DE REPORTE DE EVENTO O INCIDENTE ADVERSO DE DISPOSITIVOS BIOMEDICOS 1. INSTITUCION REPORTANTE Fecha: Dia Mes Año Hora: AREA REPORTANTE 3. INFORMACION DEL DISPOSITIVO MEDICO NOMBRE DEL EQUIPO: SERIE: MODELO: ACTIVO: UBICACIÓN: SI SE REPORTO AL FABRICANTE: NO Cirugia: Sala: 2. IDENTIFICACION DEL PACIENTE Nombre del paciente: Numero de Historia Clinica: Edad: Sexo: M F EL EVENTO FUE A Paciente: Acompañante: Visitante: Otro: 4. DESCRIPCION DEL EVENTO ADVERSO DESCRIPCION DEL EVENTO O INCIDENTE ADVERSO Tipo de reporte Fecha del evento o incidente adverso Primera vez Seguimiento ¿SE RESOLVIÓ EL PROBLEMA? SI [ ] NO [ ] Medidas que se tomaron: SEÑALE SEGÚN EL(LOS) DESENLACE(S) QUE APLIQUE(N) [ ] No hubo consecuencia(s) [ ] Daño de una Función o Estructura Corporal [ ] Enfermedad o Daño que Amenace la Vida [ ] Hospitalización: Inicial o Prolongada [ ] Intervención Médica o Quirúrgica [ ] Muerte [ ] Otros: ¿Cuál? DESCRIPCION DEL EVENTO ADVERSO INCIDENTE ADVERSO NOTA: Si es necesario adicione hojas 70 Posteriormente fue llevado al comité de seguridad al paciente donde se incluyeron algunos aspectos propios de la clínica. Este formato cuenta con 5 aspectos principales: 1. Información de la institución reportante Se debe incluir la información sobre la institución donde se presentó el evento o incidente adverso, especificando su ubicación y nivel de atención. Fecha: Se establece la fecha cuando se produjo el evento. Hora: Registrar la hora en que se produjo el evento (hora estimada). Área reportante: Registrar el área donde se produjo el evento. 2. Identificación del paciente Se debe reportar la información del paciente / usuario que sufrió las consecuencias del evento o incidente adverso. Nombre: Nombre del paciente o iniciales. Numero de historia Clínica: Número asignado al paciente durante la estadía en la clínica. Edad: Indique la edad del paciente, si el paciente es menor de 1 año indique los meses cumplidos. Sexo: Seleccione con una (X) si el paciente es de sexo Femenino (F) o Masculino (M). 3. Información del dispositivo medico Nombre del equipo: Nombre genérico del dispositivo medico Ej.: Monitor de Signos Vitales, Ventiladores, Electrocardiógrafos, Desfibriladores. Serie: Numero designado por la compañía de fabricación, la serie de los equipos se encuentra casi siempre en la parte trasera del dispositivo. Activo fijo: Numero designado por la institución prestadora de salud para un control de los equipos biomédicos, se encuentra a los laterales del equipo o en la parte trasera. Ubicación: Es la ubicación del equipo en el área donde ocurrió el evento. 4. Descripción del evento adverso El evento fue a: Corresponde a la identificación del paciente o persona involucrada en el evento que puede ser paciente, acompañante, visitante, otro, cual. Descripción del evento o incidente adverso: En este espacio, el cuerpo médico debe describir con sus palabras el evento ocurrido. Se establece la 71 fecha en que ocurrió el evento y se selecciona si el evento es por primera vez o si fue un evento recurrente. Los desenlaces: Son desenlaces que pueden ocasionar la muerte o deterioro de la salud del paciente, familiares o cuerpo médico, por consecuencia del dispositivo. De igual forma en este cuadro se debe señalar la opción con una X cualquiera de las siguientes opciones: No hubo consecuencias. Enfermedad o daño que amenace la vida. Hospitalización: inicial o prolongada. Intervención médica o quirúrgica. Muerte. Medidas tomadas: Esta sección de formato la diligenciara el coordinador de electromedicina quien describirá las medidas tomadas durante y después de ocurrido el evento. Esta información es importante pues aportara la elaboración del informe trimestral de reporte periódico. 72 4.5 GESTIÓN DEL REPORTE El reporte de los eventos e incidentes adversos debe agotar unos pasos previos a la toma de decisiones para el reporte a la autoridad sanitaria, en este caso, el INVIMA. A continuación, se detalla en el cuadro 11. Cuadro 11. Pasos para el reporte de eventos e incidentes adversos PASO 1 ACTIVIDADES Estar atentos, vigilantes ante algún evento o incidente adverso con los dispositivos médicos, asegurar el bienestar del paciente, realizar un seguimiento en las instalaciones de la clínica (UCI‟s, Hospitalización, Urgencias, Cirugía, etc.). RESPONSABLE Coordinador de Electromedicina Coordinador de UCI‟s Coordinador de salas de Cirugía Coordinador de rayos-x Coordinador de urgencias Coordinador de laboratorio Coordinador de partos Coordinador de hospitalización 2 Tomar medidas para realizar el reporte de los eventos e incidentes adversos serios y no serios. Recolectar los formatos de reportes de los eventos e incidentes adversos definidos por la Clínica de Occidente. Los formatos de tipo uno y de tipo dos pueden ser depositados en los buzones de sugerencia.. Divulgación del evento o incidente adverso en el Comité de Seguridad al Paciente Enviar el reporte de los eventos e incidentes adversos al INVIMA o el informe consolidado trimestral. Personal asistencial al Comité de Seguridad al Paciente 3 4 5 Coordinador Electromedicina de Coordinador Electromedicina de Coordinador Electromedicina de A continuación, la imagen 3 complementa el paso a paso de la gestión que se debe realizar con el reporte de los eventos e incidentes adversos. 73 Imagen 3. Pasos para el reporte de eventos e incidentes adversos Para la recolección de los reportes se realizaron visitas a las Unidades de Cuidados Intensivos, salas de Cirugía, Hospitalización, Urgencias, rayos-x, etc, dos veces al día para obtener un informe inmediato de los eventos. Estos eran analizados y en caso de presentarse un evento o incidente adverso serio se reportaba al INVIMA dentro de las (72) horas siguientes, de lo contrario se realizaba un informe que era registrado y archivado y reportado al INVIMA posteriormente dentro del informe trimestral con las posibles medidas preventivas tomadas. 74 4.5.1 Análisis y valoración de los reportes. Los reportes de los eventos e incidentes adversos son evaluados por el Coordinador del área de electromedicina de la clínica cuya función es dar claridad al evento reportado, siguiendo un procedimiento de investigación del evento o incidente el cual requiere de un trabajo coordinado con el cuerpo médico para determinar el suceso y llegar a la conclusión de la situación que genero el evento. Una vez se cuenta con los resultados se hace una reunión con el Comité de Seguridad al Paciente aclarando el incidente con el paciente involucrado y el equipo biomédico que presento falla. 4.5.2 Reporte al fabricante y autoridad sanitaria. Una vez agotados los pasos anteriores, se envía comunicación al fabricante del equipo biomédico detallando el equipo que presento falla, tipo de falla, daño ocasionado a la paciente y demás información relevante. 4.6 DISEÑO DE FOLLETOS Y MATERIAL INFORMATIVO Esta etapa incluye la capacitación al personal de la clínica sobre la importancia del Programa Institucional de Tecnovigilancia y poner al tanto a los miembros de la institución acerca de los riesgos que pueden presentar los equipos biomédicos. Con el objetivo de informar al cuerpo médico y la comunidad que interactúa con la clínica, se diseñó un folleto ver anexo D, que cuenta con información de fácil comprensión sobre el Programa Institucional de Tecnovigilancia y especialmente las acciones a realizar en caso de presentarse un evento o incidente adverso y por ende, el uso de los formatos de reporte. Para el caso del cuerpo médico, la entrega de este folleto estuvo acompañada de jornadas de capacitación y socialización realizadas en la clínica. Estas jornadas de capacitación se encuentran planeadas anualmente desde el 2011 cuando se dio inicio al diseño del Programa de Tecnovigilancia. En consecuencia, en ese año se hicieron las jornadas respectivas a manera de exposición en el auditorio de la Clínica de Occidente en la semana del 11 al 15 en el mes de abril del 2011 donde se dio a conocer el detalle del formato, ejemplos ilustrativos de eventos e incidentes adversos, la normativa que regula la Tecnovigilancia, ejemplos de cómo diligenciar los formatos de reporte, etc. La capacitación tuvo una duración de dos horas (2) por día, para un total de diez horas (10) y fue dirigida al cuerpo médico, administrativo y de servicios generales de la clínica. 75 En el 2012 se continuó con la estrategia de capacitación en donde se han realizado ejercicios de casos puntuales que han permitido evaluar el personal. En el caso de que las evaluaciones no sean satisfactorias o que no cumplan con el umbral establecido por el área de calidad, el cual debe ser superior al 80% de comprensión, se discutirán los resultados con el departamento de recursos humanos para tomar las medidas necesarias que logren fortalezcan los conceptos y conocimientos del personal. Para la evaluación de esta jornada se diseñó un formato con casos de eventos que pueden presentarse en donde los asistentes debían relacionarlos con la clasificación de los eventos (Evento o Incidente), ver evaluación en el anexo E. Lo anterior con el objetivo de determinar el nivel de entendimiento de las personas. Esta evaluación fue aplicada a 80 asistentes del cuerpo administrativo y médico de la clínica los cuales asistieron a la jornada. Ver registro fotográfico en el anexo F y formato de asistencia en el anexo G Los casos evaluados fueron los siguientes: Caso A: Una bomba de infusión se detiene y no da alarma de esta situación. El paciente no recibe la cantidad suficiente de fluidos. Caso B: Una bomba de infusión que se detiene, debido a un mal funcionamiento, pero entrega una adecuada señal de alarma y no provoca daño al paciente. Caso C: Un profesional de la salud informó que durante el implante de una válvula cardíaca, se descubre que viene con defectos de fabricación. Esta válvula no se utilizó y se tuvo que utilizar una nueva, por lo tanto, el tiempo de bombeo durante la cirugía se extendió. Caso D: El envase de un dispositivo médico estéril, de un solo uso, señala la advertencia “no usar si el envase está abierto o dañado”. Previo al uso, el usuario observó que el envase estaba dañado y, obviamente, el dispositivo médico no fue usado Caso E: Un paciente muere después de una diálisis. El paciente tenía una enfermedad renal terminal y murió de una falla renal. Las investigaciones demostraron que el dispositivo médico funcionaba adecuadamente y el evento no fue atribuido al funcionamiento de éste. Caso F: Durante el uso de un desfibrilador externo en un paciente, el equipo no entrega el nivel de energía programado debido a un mal funcionamiento. El paciente falleció. 76 Los resultados fueron evaluados con base en el acierto o desacierto de las respuestas tal como se detalla a continuación: Cuadro 12. Sistematización de respuestas RESPUESTA CASO % RESPUESTAS CORRECTAS CORRECTO INCORRECTO CASO 1 54 26 68 % CASO 2 78 2 98 % CASO 3 78 2 98 % CASO 4 80 0 100 % CASO 5 78 2 98 % CASO 6 72 8 90 % Como se puede observar en el cuadro anterior y en la figura 10 que se presenta a continuación, en la mayoría de los casos evaluados la respuesta fue correcta. Solamente en el caso 1 el porcentaje de respuesta correcta fue del 68% mientras que en el resto supero el 90%. Figura 10. Diagrama de barras resultado de la jornada de capacitación al personal de la Clínica de Occidente. Personas capacitadas = 80 Evaluacion Capacitacion a personal de la Clinica de Occidente 90 80 70 60 50 40 30 20 10 0 CASO 1 CASO 2 CASO 3 CASO 4 CASO 5 CASO 6 CORRECTO 54 78 78 80 78 72 INCORRECTO 26 2 2 0 2 8 77 Para fortalecer el reconocimiento del Programa Institucional de Tecnovigilancia de la clínica se utilizaron estrategias divulgativas como el uso de avisos con lecturas rápidas, información en carteleras institucionales y la entrega del folleto diseñado a los visitantes de la clínica. 4.6.1 DISEÑO DEL MANUAL DE TECNOVIGILANCIA Uno de los pasos más importantes para el diseño e implementación del Programa de Tecnovigilancia es el diseño de un manual, entendiendo como tal, el documento institucional que define el tipo de equipos médicos objeto de vigilancia, elementos conceptuales de los eventos e incidentes adversos, estrategia de vigilancia y recolección de reportes, análisis y valoración de los resultados, reporte al fabricante y autoridad sanitaria. El objetivo del manual es el de orientar a todo el personal asistencial de la Clínica de Occidente – CDO sobre el Programa Institucional de Tecnovigilancia generando el conocimiento y las herramientas necesarias para realizar reportes de eventos e incidentes adversos a equipos biomédicos. Este manual detalla el marco legal del programa de Tecnovigilancia, la descripción de equipos médicos, el detalle de la clasificación de eventos de acuerdo al riesgo, detalle del reporte de Tecnovigilancia, los objetivos de reporte, los eventos, la información que debe contener el reporte, el análisis de evaluación del reporte, entre otros aspectos de interés que dan las directrices a la Clínica de Occidente de cómo implementar el programa. Manual institucional de tecnovigilancia, está indicado en el anexo I. 4.7 DISEÑO DE ESTRATEGIAS DE VIGILANCIA DE EQUIPOS BIOMÉDICOS Con el objetivo de disminuir la generación de eventos e incidentes adversos se implementaron estrategias de vigilancia, chequeo visual, mantenimiento preventivo, mantenimientos predictivos, metrología, listas de chequeo o check list en las Unidades de Cuidados Intensivos, Cirugía y Hospitalización, urgencias, rayos-x, laboratorio, etc. 78 Estas actividades se implementaron tres veces a la semana donde el analista de electromedicina registró la información y documentó el funcionamiento del equipo. Al momento de encontrar fallas en equipos biomédicos, las cuales también se pueden generar por condiciones del área, se notifica la situación al funcionario a cargo del área, que a su vez notifica al coordinador de electromedicina para su conocimiento y acciones pertinentes. La realización de reuniones mensuales del comité de tecnovigilancia involucrado en el comité de seguridad al paciente donde asisten los Coordinadores de Electromedicina, UCI‟s, salas de Cirugía, rayos-x, urgencias, laboratorio, partos, hospitalización. Con los datos e información obtenida a través de las alertas, se deben generar planes de acción para los eventos adversos más comunes, de esta manera ir organizando una base de datos con planes de acción y de prevención aplicables para que en caso de que el evento adverso relacionado ocurra en la clínica, estén claras las medidas que se deben tomar. Además se deben implementar los planes de prevención cuando las alertas están relacionadas a equipos con los que cuenta la institución. El diseño de barreras es importante, debido a que mediante estas es posible la prevención de algún evento o incidente adverso y actuar de manera oportuna cuando este suceda y disminuir al máximo el daño al paciente. Para el diseño de las barreras es necesario realizarla por medio de matrices de riesgo las cuales los priorizan según la ocurrencia en cada uno de los equipos biomédicos generando una base para el diseño de las barreras, se debe hacer énfasis principalmente en los entornos donde se encuentra la tecnología biomédica, los focos de peligro de los equipos biomédicos y los riesgo que pueden ocurrir con estos equipos. Los pasos a seguir para realizar la matriz de riesgo se muestran en la figura 11. 79 Figura 11. Pasos para el diseño de una matriz de riesgo SELECCION DEL AREA LISTA DE LOS EQUIPOS BIOMEDICOS DE ESTA AREA IDENTIFICAR LOS RIESGOS DISTRIBUCION DE LOS RIESGOS IDENTIFICADOS EN LA MATRIZ IDENTIFICAR LAS VARIABLES DE LA MATRIZ La norma NTC 5254 aporta un marco genérico para establecer el contexto y los elementos del proceso de gestión del riesgo, sin embargo, es presentada como una propuesta que puede ser adaptada según las necesidades de cada establecimiento; pues, de acuerdo con la misma, el diseño e implementación del sistema de gestión del riesgo se verá influenciado por las necesidades variables de una organización, sus objetivos particulares, sus productos y servicios y los procesos y practicas especificas empleadas. En consecuencia con lo anterior, en la Clínica de Occidente se estudió esta propuesta y otras surgidas del proceso de acreditación para consolidar la matriz institucional de riesgo según el contexto institucional. La matriz de riesgo de la Clínica de Occidente presenta, entonces, unas probabilidades y unas categorías de severidad del evento. En este caso, el análisis del riesgo se realiza a través de un análisis cualitativo que emplea palabras y escalas descriptivas que permiten identificar aspectos como las consecuencias, potencialidades y posibilidades de que esto ocurra. Para el caso de las probabilidades, las cataloga en frecuente, ocasional, remoto, improbable y extremadamente improbable; esta probabilidad se valora de 1 a 5 en donde 5 es una probabilidad frecuente y 1 es extremadamente improbable, como se observa en el cuadro 13. 80 Cuadro 13. Medida cualitativa de la probabilidad del evento PROBABILIDAD DEL EVENTO DEFINICIÓN CUALITATIVA FRECUENCIA OCASIONAL REMOTO IMPROBABLE EXTREMADAMENTE IMPROBABLE SIGNIFICADO Probable que ocurra muchas veces (ha ocurrido frecuentemente) Probable que ocurra algunas veces (ha ocurrido infrecuentemente) Improbable, pero es posible que ocurra (ocurre raramente) Muy improbable que ocurra (no se conoce que haya ocurrido) Casi inconcebible que el evento ocurra VALOR 5 4 3 2 1 Para el caso de las categorías de severidad, se presentan unos valores de la A a la E en donde A es un evento catastrófico y E es un evento insignificante, como se observa en el cuadro 14. Cuadro 14. Medida cualitativa de la severidad del evento SEVERIDAD DEL EVENTO DEFINICIÓN SIGNIFICADO Muertes múltiples, Muerte del Paciente, invalidez de CATASTRÓFICO más de 50%, Destrucción o daños severos en infraestructura o equipos VALOR A Lesiones o secuelas permanentes PELIGROSO MAYOR Daños mayores a pacientes Equipos o infraestructura Una reducción en la habilidad del operador en responder a condiciones operativas adversas como resultado del incremento de la carga de trabajo, o como resultado de condiciones que impiden su eficiencia B C Incidente serio Lesiones temporales a las personas Paciente insatisfecho MENOR Aumento de trámites administrativos D Incidentes menores INSIGNIFICANTE Consecuencias leves E 81 Con base en lo anterior, se diseñó entonces la matriz que se observa en el cuadro 15 en donde se confronta la probabilidad y la severidad del evento y se asignan los valores correspondientes para cada caso. Cuadro 15. Matriz de riesgo de la Clínica de Occidente PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante 5A 5B 5C 5D 5E 4A 4B 4C 4D 4E 3A 3B 3C 3D 3E 2A 2B 2C 2D 2E 1A 1B 1C 1D 1E Fuente. Clínica de Occidente Esta matriz, permite catalogar tres tipos de riesgo identificados como riesgo extremo, medio y bajo, los cuales se explican a continuación: RIESGO EXTREMO: Cuando la situación requiere acción inmediata. La dirección debe conocer esta situación y deben implementarse acciones correctivas urgentes. Si ocurre el Evento Adverso o Incidente, el mismo debe ser reportado dentro de las 72 horas y se deben analizar causas raíces. RIESGO MEDIO: Cuando la situación requiere acciones lo antes posible y no más allá de los 6 meses. La alta dirección y el director médico deben conocer esta situación. RIESGO BAJO: Manejo por procedimientos de rutina. Implementar medidas fáciles y rápidas y soluciones de fondo cuando los recursos lo permitan. A continuación se presentan cuatro ejemplos relacionados con los riesgos que pueden ocasionar algunos equipos biomédicos disponibles en la Clínica de Occidente, detallando el área, el equipo y las fuentes de peligro. Estas fuentes de peligro fueron tomadas de casos reales presentados en la Clínica de Occidente los cuales fueron identificados y por lo tanto se considera pertinente traerlos a colación en este documento. 82 Para diseñar los ejemplos que se presentan a continuación, se seleccionaron dos casos del área de cirugía y otros dos casos de las áreas de UCI´s ya que son las áreas que presenta mayor riesgo dentro de la institución. EJEMPLO 1 ÁREA: cirugía EQUIPOS: máquina de anestesia, desfibrilador, monitor de signos vitales, lámpara cielítica, máquina de circulación extracorpórea, electrobisturí, entre otros. EQUIPO SELECCIONADO: Máquina de anestesia FUENTE DE PELIGRO DE LA MÁQUINA DE ANESTESIA: Válvula de inspiración dañada. Circuito de respiración en mal estado. Monitor dañado no se registran parámetros. Alarma de sensor de oxigeno deshabilitado. Vaporizadores mal ajustados. Fuga en absorvedor. DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS A LA MÁQUINA DE ANESTESIA: A continuación, se presentan algunos casos que pueden generar riesgos asociados a la máquina de anestesia, los cuales son numerados del 1 al 8; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Válvula de inspiración dañada: Al tener esta válvula dañada o en mal estado el absorvedor de la máquina de anestesia no realiza su función quedando pegado y no envía flujo al paciente provocando la muerte. Caso 2 (C2). Circuito de respiración en mal estado: El paciente se despierta en medio de la cirugía ya que hay fuga en la manguera faltando gas anestésico al paciente. Caso 3 (C3). Al tener una fuga en el circuito el anestesiólogo se ve obligado a suministrar más oxigeno o aire y gas anestésico provocando una recuperación prolongada. Caso 4 (C4). Monitor dañado no se registran parámetros: El anestesiólogo no identifica en la monitoria de la máquina de anestesia los flujos de aire, oxigeno, entregados al paciente provocando daños cerebrales. 83 Caso 5 (C5). No se registran señales de alerta de apnea o de concentración de flujo, alta presión de flujo de aire. Caso 6 (C6). Alarma de sensor de oxigeno deshabilitada: No se registra la alarma de para baja y alta concentración de flujo. Caso 7 (C7). Vaporizadores mal ajustados: No entregan el suficiente gas anestésico al paciente provocando que el paciente se despierte en medio de la cirugía. Caso 8 (C8). Fuga en el absorvedor: Al tener esta fuga hay que suministrarle más aire o oxígeno al paciente y por ende más gas anestésico. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. Cuadro 16. Matriz de riesgo máquina de anestesia PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C2 C3 C7 C6 C8 C4-C5-C1 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar barreras para los casos 2, 3, 5 7 y 8 Es indispensable realizar un chequeo diario del circuito paciente antes de que inicie una cirugía debido a que puede ocasionar daños al paciente mientras se encuentra anestesiado. ACCIONES INMEDIATAS: Realizar un chequeo en las salas de cirugía diariamente en las horas de la mañana antes que inicien las cirugías. No esterilizar más de cinco veces los circuitos pacientes 84 Verificación del buen estado del equipo antes de las cirugías preferiblemente por las mañanas Tener a la mano el repuesto de lo que está fallando u otro circuito respiratorio. EJEMPLO 2 EQUIPO SELECCIONADO: Desfibrilador FUENTE DE PELIGRO DEL DESFIBRILADOR: Daño en el botón de descarga Entrega de energía más alta de lo que se indica Batería dañada Equipo desconectado DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS AL DESFIBRILADOR A continuación, se presentan algunos casos que pueden generar riesgos asociados al desfibrilador, los cuales son numerados del 1 al 5; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Daño en el botón de descarga: No se puede hacer la descarga al paciente ya que está dañado el botón provocando la muerte al paciente ó daño mientras se consigue otro equipo de urgencia. Caso 2 (C2). Entrega de energía más alta de los que se indica: Se puede producir quemaduras al paciente. Caso 3. (C3). Descarga no adecuada para patología del paciente. Caso 4 (C4). Batería dañada: No se puede hacer la carga ni la descarga al paciente provocando la muerte o daño. Caso 5. (C5). Equipo desconectado: No se visualiza el led de carga del desfibrilador confiándose que se encuentra conectado a la red eléctrica, al utilizarlo no prende el equipo y no se puede hacer la descarga al paciente. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 85 Cuadro 17. Matriz de riesgo desfibrilador PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C4-C5 C2 C3 C1 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 4, 5, 2 y 3. Es indispensable realizar un cheque diariamente del estado del desfibrilador cerciorarse que se estén realizando las descargas y verificar que este encendido el led de carga. ACCIONES INMEDIATAS: Realizar un chequeo diario del funcionamiento del desfibrilador cerciorarse que se esté realizando las descargas. Realizar todos los días el test interno que tiene el desfibrilador y llevar un reporte del test. Limpiar las palas cada vez que se usan para evitar que haya material que este generando resistencia o baja impedancia y no permita entregar la energía necesaria. Verificar que se encuentre conectado a la red. 86 EJEMPLO 3 ÁREA: UCI‟s EQUIPOS: desfibrilador, monitor de signos electrocardiografos, ventiladores, entre otros. vitales multiparametricos, EQUIPO SELECCIONADO: ventilador FUENTE DE PELIGRO DE UN VENTILADOR: Filtro espiratorio con fugas. Circuito paciente con fugas. Alarma no audibles. Falla en la batería. Desajuste del peep DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS AL VENTILADOR: A continuación, se presentan algunos casos que pueden generar riesgos asociados al ventilador, los cuales son numerados del 1 al 4; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Filtro espiratorio con fugas: Al tener fugas no hay una buena compensación del paciente y se ve obligado el fisioterapeuta a dar respiración manual “ambu” al paciente. Caso 2 (C2). Circuito paciente con fuga: Al tener una fuga en el circuito el fisioterapeuta se ve obligado a suministrar 100% de oxígeno al paciente provocando lesiones pulmonares. Caso 3 (C3). Desajuste del peep: Puede producir lesiones pulmonares por alta presión en las vías respiratorias. Caso 4 (C4). Falla en la batería: Paciente se puede morir si no hay planta de emergencia eléctrica. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 87 Cuadro 18. Matriz de riesgo ventilador PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C1-C4 C2 C3 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 1 y 4. Es indispensable realizar la metrología de estos equipos ya que son soporte de vida, hay que cerciorarse que los circuitos no se esterilicen más de cinco veces, realizar un chequeo visual de los accesorios (filtros inspiratorios, espiratorio, circuitos). ACCIONES INMEDIATAS: Antes de que llegue un paciente a la UCI‟s se pone a ciclar el ventilador por 30 minutos. Verificar el estado de los filtros, cerciorarse que están en buen estado que no estén partidos. Algunos ventiladores tiene un test interno donde se realizan pruebas de compensación de fugas, hay que realizar este test cada vez que cambian de paciente. Verificar que se encuentre conectado a la red. 88 EJEMPLO 4 EQUIPO SELECCIONADO: monitor de signos vitales multiparamétrico FUENTE DE PELIGRO DE UN MONITOR: Bomba de presión arterial en mal estado Cable de saturación de oxígeno en mal estado Calibración de presión invasiva desajustada Batería interna en mal estado Alarmas audibles y visibles en ,mal estado DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS A LOS MONITORES: A continuación, se presentan algunos casos que pueden generar riesgos asociados a los monitores, los cuales son numerados del 1 al 5; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Bomba de presión arterial en mal estado: Toma de presiones erróneas no acordes a la patología del paciente suministrando medicamentos innecesarios, provocando la muerte, o prolongando la estadía. Caso 2 (C2). Cable de saturación de oxígeno en mal estado: No se registra la saturación de oxigeno del paciente ya que el accesorio está mal ubicado o se encuentra malo, dando un mal diagnóstico a la enfermera o a la fisioterapeuta si hay que suministrar más oxígeno al paciente. Caso 3 (C3). Calibración de presión invasiva desajustada: Toma de presiones erróneas no acordes a la patología del paciente suministrando medicamentos innecesarios, provocando la muerte, estadía prolongada. Caso 4 (C4). Batería interna en mal estado: Paciente se puede morir si no hay planta de emergencia eléctrica. Caso 5 (C5). Alarmas audibles y visibles en mal estado: Al tener las alarmas en nivel bajo o dañado no se podría registrar los cambios fisiológicos del paciente provocando desenlaces graves como la muerte o deterioro de la salud. Cada uno de estos casos se evalúan mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 89 Cuadro 19. Matriz de riesgo Monitor de signos vitales PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C1 C4-C2 C5 C3 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 1, 4, 2 y 5. Es indispensable realizar la metrología de estos equipos ya que son soporte de vida, hay que cerciorarse que los accesorios de los equipos estén funcionando correctamente, realizar un chequeo visual de los accesorios (cable de ECG, saturador de oxígeno, brazalete, cable de invasiva). ACCIONES INMEDIATAS: Antes de que llegue un paciente a la UCI‟s se verifica que los accesorios estén correctamente funcional. Realizar un chequeo visual y funcional de los accesorios. Realizar la metrología de los equipos biomédicos. Verificar que se encuentre conectado a la red el equipo. A nivel internacional existen organizaciones encargadas de recopilar y brindar alertas de casos ocurridos en dispositivos biomédicos. Una de estas agencias es la FDA (Food and Drugs Administration) que presenta unos antecedentes importantes de alertas tempranas donde se reportan regularmente aquellos incidentes con dispositivos biomédicos ocurridos en Estados Unidos Teniendo en cuenta que muchos de los equipos manejados en este país son similares a los de Estados Unidos, es necesario considerar esta información pues permite hacer un monitoreo permanente para estar enterados de los casos ocurridos y de esta manera revisar si hay coincidencias con eventos presentados por equipos médicos que se manejen en la institución. Esto genero la necesidad de que en la Clínica se maneje esta información adecuadamente para prevenir estos sucesos. 90 4.7.1 Seguimiento a acciones y responsables. Para todos los casos, el seguimiento de los planes de acción está a cargo del área de electromedicina que es el área responsable de llevar a cabo estas acciones inmediatas. El seguimiento se hace por medio de un formato de chequeo que se aplica semanalmente para el área de las UCI´s y diario para el área de cirugía. Basados en la matriz de riesgo como se mostró en los ejemplos anteriores, las barreras se diseñan al identificar los riesgos que presentan los equipos biomédicos, estas barreras se deben implementar para evitar la ocurrencia de los eventos o para actuar de manera oportuna cuando ocurra un evento o incidente adverso. Estas barreras aplican para todos los equipos. A continuación en el cuadro 20 se presentan las barreras. Cuadro 20. Barreras BARRERAS RECURSO HUMANO INFRAESTRUCTURA CAPACITACIÓN RED ELÉCTRICA VERIFICACIÓN DE LA IDONEIDAD DEL RED DE GASES PERSONAL GESTIÓN MANTENIMIENTO LISTA DE CHEQUEO INDICADORES CALIDAD METROLOGÍA DE RECURSO HUMANO Capacitación al personal: Se realiza capacitación y evaluación al personal asistencial de la clínica cada vez que se requiera o se vean falencias en algunas áreas; estas falencias son vistas en los llamados inoficiosos o en procedimientos, se realiza cada mes una capacitación en el auditorio a todo el personal donde se explica el buen manejo de los equipos biomédicos. Verificación de la idoneidad del personal: Verificar por el área de recurso humano las hojas de vida del personal que va a ingresar a la Clínica de Occidente que cumpla con los requerimientos establecidos por las normas reglamentarias de la empresa. 91 INFRAESTRUCTURA Red eléctrica: La clínica cuenta con un sistema de contingencia de dos plantas eléctricas de emergencia en caso que falle el servicio eléctrico. Estas plantas de energía suministran energía a las áreas más críticas como las UCI´s, cirugía, rayos-x, alarmas contra incendio, e iluminación, tal como lo exige la norma NTC2050 en la sección 517. Red de gases: La clínica cuenta con una planta de generación de oxígeno, aire, vacío para el suministro a la Clínica de Occidente. Se cuenta con sistemas de alarma de ausencia de gases en cada área crítica como las UCI‟s, Cirugía, Rayos-x, etc. Para el oxígeno se cuenta con un sistema de contingencia de un tanque de criogénico de 5 toneladas y un sistema manual con cilindros de oxigeno que respalda el suministro de la planta, para el sistema de aire se cuenta solamente con la técnica manual. Para el sistema de vacío se cuenta con dos compresores para el suministro a la clínica, además hay una planta de emergencia para suplir la energía al sistema de vacío en caso de fallo del suministro eléctrico. GESTIÓN Plan de mantenimiento preventivo: A los equipos biomédicos que se encuentran en la Clínica de Occidente se les realiza cada 6 meses un mantenimiento preventivo, de acuerdo a la criticidad de los equipos, de su uso constante y lugar donde esté ubicado el equipo, los informes quedan registrados en las hojas de vida de cada equipo. Plan de mantenimiento predictivo: El mantenimiento predictivo se realiza todos los días en las salas de Cirugía a las 6:30 am antes de iniciar procedimientos, en las Unidades de Cuidados Intensivos se realiza cada viernes. Aseguramiento metrológico: Se realiza cada año con una entidad externa, se utilizan equipos patrones para realizar la metrología, los informes quedan registrados en las hojas de vida de cada equipo. En la clínica de Occidente, se realiza la calibración posterior al mantenimiento preventivo. 92 Acompañamientos en procedimientos quirúrgicos: Se realiza este acompañamiento cada vez que un médico, o jefe lo requiera, si es para el manejo de algún equipo en especial como los siguientes: Equipo de Laser, Microscopio, Shaver, entre otros. Indicadores de calidad: Verificar por medio de indicadores el cumplimiento de los mantenimientos preventivos, correctivos, de los equipos biomédicos de la Clínica de Occidente. 93 5. CONCLUSIÓN Se diseñó el programa institucional de tecnovigilancia para la Clínica de Occidente, se elaboró los formatos para el reporte de eventos o incidentes adversos, se designó el responsable del programa de tecnovigilancia quien será el encargado de reportar ante el gobierno, y se diseñó el manual de tecnovigilancia para la Clínica de Occidente. Se realizaron capacitaciones con el fin de dar a conocer el programa de tecnovigilancia y de fomentar la cultura del reporte, inicialmente al personal médico y asistencial. La metodología empleada generó un acercamiento y confianza al personal asistencia. La actualización del inventario técnico funcional de los equipos biomédicos con los que cuenta la institución es una etapa importante para el desarrollo del programa de tecnovigilancia, ya que permite conocer el funcionamiento de cada uno de los equipos, e identificar los riesgos asociados a esa tecnología. Actualmente la Clínica de Occidente maneja el método del protocolo de Londres, indicado para el análisis y evaluación de eventos adversos, porque además de ser uno de los más usados, es un método de fácil implementación y facilita la investigación clara y objetiva de los incidentes clínicos, así mismo se pueden detectar las causas que llevaron a la ocurrencia de efecto adverso, permitiendo prevenir la ocurrencia de estos daños no intencionados al paciente u operación. Se logró que la clínica iniciara el proceso de culminación del Programa Institucional de tecnovigilancia al acceder al comité de Seguridad del Paciente que permite relacionar todos los aspectos que buscan mejorar la gestión de riesgo de la institución. El Comité de Tecnovigilancia es el encargado de identificar y analizar los efectos adversos producidos por los dispositivos médicos en la institución, se establecieron reuniones cada 15 días para el análisis y discusión de los efectos adversos que se presente, si son eventos adversos serios se realiza una reunión con el coordinador del área y se reportara inmediatamente al INVIMA. 94 6. RECOMENDACIONES Dada la importancia que tiene la Tecnovigilancia en los procesos misionales de la Clínica de Occidente, se recomienda incorporar el programa Institucional de tecnovigilancia en la política de calidad de la institución. Incorporar al plan de gestión integral de residuos hospitalarios, el procedimiento de disposición final adecuada de los equipos biomédicos dados de baja. Se recomienda que las directivas de la Clínica de Occidente den estricto cumplimiento a la normatividad que exige que exista un funcionario con dedicación exclusiva para el manejo del programa institucional de tecnovigilancia de la Clínica de Occidente. Buscar un mayor y mejor acompañamiento de las entidades encargadas de velar por la tecnovigilancia en el Valle del Cauca y Colombia. Institucionalizar mediante la implementación, capacitación y manejo al personal de la clínica el correcto y oportuno diligenciamiento de los formatos del reporte de eventos e incidentes adversos. Implementar una cartelera por piso, donde se expliquen los tipos de eventos e incidentes adversos que pudiesen presentarse durante la estadía del paciente en la clínica, con el fin de reforzar al cuerpo médico y al cuerpo administrativo la importancia de tomar las medidas preventivas para que estos eventos no sucedan. Se recomienda que la clínica adopte o implemente un sistema de indicadores del programa de tecnovigilancia que permita realizar un constante seguimiento de estos eventos y medir la efectividad del Programa, como se propone en el anexo H de este documento. 95 BIBLIOGRAFÍA CLÍNICA DE OCCIDENTE. Información Corporativa.[En línea] Santiago de Cali, 2011. [Consultado el 01 de octubre 2011]. Disponible en internet: http://clinicadeoccidente.com.co/FRONT/texto.php?Id=20 COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 4816 de 2008 (Noviembre 27). por la cual se reglamenta el Programa Nacional de Tecnovigilancia. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2008. [Consultado 20 se septiembre de 2011]. Disponible en internet: http://www.alcaldiabogota.gov.co COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Dispositivos médicos y que es la Tecnovigilancia [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2005 [consultado el 01 de octubre 2011]. Disponible en internet: http://issuu.com/leadyrs/docs/revista_decreto_4725 COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 3133 del 14 de septiembre de 2005. Por la cual se ajusta el Manual Específico de Funciones, Requisitos y de Competencias Laborales para los empleos de la Planta de Personal del Ministerio de la Protección Social [En linea] Bogotá d.c.2005. [Consultado el 01 de octubre 2011]. Disponible en internet: http://www.minproteccionsocial.gov.co. COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Decreto 4725 de2005 (Diciembre 26). Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2005. [Consultado Octubre 01 de 2011]. Disponible en internet: http://www.presidencia.gov.co COLOMBIA, MINISTERIO DE PROTECCIÓN SOCIAL. Lineamientos para la implementación de la Politica de Seguridad del Paciente [En línea]. [Consultado Septiembre 20 de 2011]. Disponible en internet: http://www.minproteccionsocial.gov.co/Documentos%20y%20Publicaciones/LINEA MIENTOS%20SEGURIDAD%20DEL%20PACIENTE.pdf INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS. Subdirección de insumos para la salud y productos varios. Programa Nacional de Tecnovigilancia. Guía de reporte de incidentes adversos a dispositivos médicos. [En línea] Bogotá d.c.2005. [Consultado el 01 de octubre 2011]. Disponible en internet: http://www.consultorsalud.com/biblioteca/ 96 Lineamientos para al desarrollo de la Tecnovigilancia en Colombia. Documento Marco. INVIMA, Subdirección de Insumos para la Salud y Productos Varios .2005. Citado en INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS. Subdirección de insumos para la salud y productos varios. Programa Nacional de Tecnovigilancia. Guía de reporte de incidentes adversos a dispositivos médicos. [En línea] Bogotá d.c.2005. [Consultado el 01 de octubre 2011]. Disponible en internet: http://www.consultorsalud.com/biblioteca/ ORGANIZACIÓN PANAMERICANA DE LA SALUD. ORGANIZACIÓN MUNDIAL DE LA SALUD. 34a sesión del subcomité de planificación y programación del comité ejecutivo equipos y dispositivos médicos. [En línea] Washington, D.C., 29 al 31 de marzo de 2000 [consultado el 01 de octubre 2011]. Disponible en internet: http://www.paho.org/spanish/gov/ce/spp/spp34_7.pdf ORGANIZACIÓN MUNDIAL DE LA SALUD. [En línea]. [Consultado Septiembre 20 de 2011]. Disponible en internet http://www.who.int/gb/ebwha/pdf_files/WHA55/sa5513.pdf Seguridad Eléctrica en Equipos e Instalaciones Médicas. [En línea]. Capítulo V. [Consultado Septiembre 20 de 2011]. Disponible en internet: http://micronet.cujae.edu.cu/Docencia 97 ANEXOS ANEXO A. HOJA DE VIDA EQUIPOS MÉDICOS HOJA DE VIDA EQUIPOS MÉDICOS Identificación del Equipo Fecha de Ingreso Equipo Marca Modelo Activo Fijo Serie Ubicación Descripción del equipo: Características del Equipo Tecnología Predominante Fuentes Alimentación Características Técnicas/Dimensiones Eléctrica Agua Tensión Presión Electrónica Aire/O2 Corriente Vel. (RPM) Mecánica Gas Frecuencia Peso (Kg) Neumática Vapor Potencia Alto Hidráulica Electricidad Temperatura Ancho Otra Otros Humedad Largo Clasificación del Equipo Clasificación Biomédica Riesgo Protección contra Descargas Soporte Vital Clase III B Diagnóstico Clase IIB BF Terapia Clase IIA CF Rehabilitación Clase I Prevención Análisis de Laboratorio Información Adicional Frecuencia Mantenimiento Manuales 4 meses Servicio 6 meses Usuario 12 meses Componentes Recomendaciones/Observaciones Despiece Código: MTEFM025 Original: Electromedicina Última modificación: Abril 2012 98 ANEXO B. INVENTARIO TÉCNICO FUNCIONAL Área: Unidad de Cuidado Intensivo Neonatal Equipo Marca Modelo Activo Fijo Serie Clasificaci ón de Equipos biomédic os Cuna de calor radiante David medical HKN-93B 02501 24101001001 2 Clase IIa Desfibrilador Burdick Medic 5 00367 33095 0 Clase III Flujometro Chemetrom Timeter 01963 1069520 Clase I Flujometro Chemetrom Timeter S/A Flujometro Chemetrom Timeter Flujometro Chemetrom Timeter Flujometro Amvex Flujometro Doble Chemetrom Incubadora Cerrada 229021 Clase I 0605 S/S Clase I 0605 S/S Clase I 9353-2 3100 Clase I Timeter 00885 S/S 49 Clase I Ohmeda OHIO 00237 HAFU00559 4 Incubadora Cerrada Medics NATALCA 00368 20405 RE 5 Clase IIb Incubadora Cerrada Medics NATALCA 02229 649-06 RE 3 Clase IIb Incubadora Cerrada Ohmeda Care Plus 00369 HBHX00672 4 Clase IIb Incubadora Cerrada Fanem Vision 2186 03802 CJ3129 2 Clase IIb Incubadora Cerrada Medics NATALCA 02294 1174-08RE 3 1065097 Manta Termica Nellcor Warmtou 01935 CI01101261 99 Clase IIb Clase IIb Clase I ch 5 Monitor de NIBP General Electric Dinamap 00370 AAW0607009 7 1SA Clase IIb Monitor de NIBP Welch Allyn 53NT0 025078 JA092548 Clase IIb Monitor de Signos Vitales Nellcor N5500 00368 0200-05128 0120 Clase IIb Monitor de Signos Vitales Nellcor N5600 01935 7 Clase IIa Monitor de Signos Vitales Nellcor N5500 00368 0200-05121 0026 Clase IIb Monitor de Signos Vitales Siemens CS9000 00367 5220568171 5 Clase IIb Monitor de Signos Vitales Siemens CS9000 00368 5220561873 3 Clase IIb Monitor de Signos Vitales Siemens CS9000 00371 5220570275 1 Clase IIb Monitor de Signos Vitales Siemens CS9000 00366 5220567270 7 Clase IIb Monitor de Signos Vitales Mindray Mec 1200 02257 CC73 2053 8 Clase IIb Monitor de Signos Vitales Mindray Beneview 01974 CM3 9C110073 Clase IIb Monitor de Signos Vitales Penlon M5 02426 CM0B114685 8 Clase IIb Monitor de Signos Vitales Penlon M5 024269 Clase IIb Monitor de Signos Vitales Penlon M5 24690 Monitor de Signos Vitales Penlon M5 24689 Monitor de Signos Vitales Mindray Mec 1200 025076 Monitor de Signos Vitales Mindray Mec 1200 025077 Neopuff Fisher & Paykel Pulsoximetro Portátil Criticare 02331003000 4 CM0B114683 CM-13116645 CM-13116640 CC-14117325 CC-14117326 101104003578 503 100 00434 193162844 Clase IIb Clase IIb Clase IIb Clase IIb Clase IIb Clase IIa 1 Vacuometro Puritan Bennett Vacutro m 3682 S/S Clase I Vacuometro Puritan Bennett Vacutro m 00317 S/S 08 Clase I Vacuometro Chemetrom Continuo s 00935 003695 23 Clase I Vacuometro Chemetrom Vacutro m 00317 S/S 06 Clase I Vacuometro Chemetrom Vacutro m 00317 S/S 05 Clase I Vacuometro ONT Vacutro m 00366 8838-2 6 Clase I Vacuometro Chemetrom Vacuometro Puritan Bennett N/A 7856 Ventilador Neonatal Sechrist IV200 00236 22420 7 Ventilador Neonatal Sechrist IV100 S/A 8728 Clase III Ventilador Neonatal Sechrist IV100 8045 1458 Clase III Ventilador Neonatal Infant Start 500 00373 17671-076 6 Clase III Ventilador Neonatal Bear cup 750 00236 53224118 8 Clase III Ventilador Neonatal Puritan Bennett 840 01924 3510095504 5 Clase III Ventilador Neonatal Sechrist IV100 00371 7590 7 Clase III Ventilador Neonatal Puritan Bennett 840 01936 3510094624 5 Clase III 101 S/A 31704 Clase I S/S Clase I Clase III Área: Unidad de Partos Equipo Marca Modelo Activo Fijo 004497 2012850 Serie Clasificacio n de Equipos biomedico s Cuna de Calor Radiante Atom N/A Doppler Fetal Edan Sonotrax 01974 Ecografo Mindray PP6600 019546 BE-87100866 Flujometro Chemetron Timeter S/A Flujometro Doble Chemetron Timeter 31925 Clase I Flujometro Doble Chemetron Timeter 31923 Clase I Lámpara Cielítica Skylux 005211 Clase I Monitor de Signos Vitales Criticare 503 004140 19720498 Clase IIA Monitor de Signos Vitales Mediana M20M1 A 022328 10G708080002 Clase IIA Monitor de Signos Vitales Mindray Mec1200 024335 CC-0C116152 Clase IIA Monitor de Signos Vitales Mindray Mec1200 024901 CC-14117280 Clase IIA Monitor Fetal Edan Cadence 004552 Monitor Fetal Edan Cadence Monitor Fetal Edan Cadence CB3031139809E 025087 II H Neopuff Fisher & Packel Unidad de Calor David Medical 103122426 4291 CADA303075154 8B 002289 30308B540FDS 6 022262 070713001895 HKN-93B 025011 24110103010 102 Clase IIA Clase IIA Clase IIA Clase I Clase IIA Clase IIA Clase IIA Clase IIB Clase IIA Radiante Regulador de Vacio Vacutron Succionador Pulmonec Pulsioximetro Nellcor Purittan Bennett ONT NPB-40 103 S/A 9352-1 Clase I 25058 00060 Clase I S/A HRD00088 Clase IIA Área: Unidad de Cuidado Intensivo Coronaria 1 Equipo Activ Modelo o Fijo Marca Serie Clasifica cion de Equipos biomedic os Desfibrilador Primedic DefiMon 0191 73505001850 itor 50 Clase III Electrocardiografo Edan SE3 1974 SE3A323098 4 96481 Clase IIa Flujometro Chemetro m Timeter 0779 1077909 09 Clase I Flujometro Chemetro m Timeter 0974 1097488 88 Clase I Monitor de signos vitales Penlon SMP5 2496 CM-13116648 2 Clase IIb Monitor de signos vitales Penlon SMP5 2496 CM-13116649 3 Clase IIb Monitor de signos vitales Penlon SMP5 2497 CM-13116647 7 Clase IIb Monitor de signos vitales Penlon SMP5 2497 CM-13116643 6 Clase IIb Ventilador Mecánico Siemens Servo 900C S/A 165746 Clase III Ventilador Mecánico Siemens Servo 900C 0053 161803 20 Clase III Ventilador Mecánico Puritan Bennett 740 0040 3501982219 34 Clase III Ventilador Mecánico VIASYS VELA 0194 AKT02823 53 Clase III Ventilador Mecánico Newport E360 3537 N0736121183 1 6 Clase III Vacuometro Chemetro m Continu os 0046 S/S 81 Clase I 104 Área: Unidad de Cuidado Intensivo Coronario 2 Equipo Marca Modelo Activ o Fijo Desfibrilador Primedic Defimoni 0194 tor 98 Electrocardiografo WelchAllyn cp50 Flujometro Chemetr om Flujometro Serie Clasificaci on de Equipos biomedic os 73675000110 Clase III 0248 99 109200621011 Clase IIa Amdex 3566 3 FMA011898BJ Clase I Chemetr om Amdex 3566 2 FMA012310BJ Clase I Flujometro Chemetr om Amdex 3566 5 FMA011926BJ Clase I Flujometro Chemetr om Amdex 3566 0 FMA011789BJ Clase I Flujometro Chemetr om Amdex 1021722 Clase I Flujometro Chemetr om Amdex 3566 4 FMA011767BJ Clase I Manta Termica Nellcor Warm Touch 2513 9 CI1110J115 Clase I Monitor de Signos Vitales Mindray PM-9000 0195 04 W-05105993 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0195 06 W-05105988 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0195 05 W-05105995 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0194 77 W-03105746 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0195 01 W-03105775 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0195 00 W-03105777 Clase IIb Monitor de Signos Vitales Edan M8 0197 M83031042818 Clase IIb 105 46 LBCJ Vacuometro Chemetr om Amdex 3565 6 N/A Clase I Vacuometro Chemetr om Amdex 3565 9 N/A Clase I Vacuometro Chemetr om Amdex 3565 5 N/A Clase I Vacuometro Chemetr om Amdex 3565 8 N/A Clase I Ventilador Mecánico Siemens Servo 900C 0197 02 164375 Clase III Ventilador Mecánico VIASYS VELA 0194 52 AKT02749 Clase III Ventilador Mecánico Siemens Servo 900C 0038 08 159124 Clase III Ventilador Mecánico Viasys Vela 0194 54 AKT02818 Clase III Ventilador Mecánico Viasys Vela 0192 51 ACT01023 Clase III Ventilador Mecánico Viasys Vela 0192 55 AET05201 Clase III Ventilador Mecánico Viasys vela 1945 1 AKT02795 Clase III Ventilador Mecánico Viasys vela 0192 54 ADT03210 Clase III 106 Área: Unidad de Cuidado Intensivo Neuro Vascular Convenio Equipo Marca Modelo Activ o Fijo Serie Clasificaci on de Equipos biomedic os Desfibrilador Hewlett Packard Code Master 0041 23 US00108855 Compresor vascular Kendal 9525 0042 9 v0839483 Clase IIb Compresor vascular Kendal 9525 0042 8 v0839449 Clase IIb Electrocardiografo Edan SE-3 0198 25 SE3B32310511 6771 Flujometro AGA N/A 2173 9 S/S Clase I Flujometro Chemetrom Timeter 7651 4 S/S Clase I Flujometro AGA N/A 2143 8 S/S Clase I Flujometro Chemetrom Timeter S/A 1041091 Clase I Flujometro Chemetrom Timeter S/A 1087829 Clase I Flujometro AGA N/A 2144 1 S/S Clase I Manta Termica Nellcor Warm Touch 2513 8 CI1110J193 Clase I Monitor de Signos Vitales Mindray PM-9000 0221 80 W79101266 Clase IIb Monitor de Signos Vitales Criticare 508 S/A 294221684 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0068 6 W-0C106905 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0068 7 W-0C106904 Clase IIb 107 Clase III Clase IIa Monitor de Signos Vitales Mindray PM-9000 0068 8 W-0C106901 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 4 W-0C106866 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 5 W-0C106863 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 6 W-0C106906 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 7 W-0C106872 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 8 W-0C106891 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0069 9 W-0C106881 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0070 0 W-05105852 Clase IIb Ventilador Mecánico Puritan Bennett 840 0053 16 3510074527 Clase III Ventilador Mecánico Puritan Bennett 840 2499 6 3512101945 Clase III Ventilador Mecánico Puritan Bennett 7200 0038 99 9770308 Clase III Ventilador Mecánico Siemens Servo 900C 0053 18 161152 Clase III Ventilador Mecánico Siemens Servo 900C 0038 29 159118 Clase III Ventilador Mecánico Siemens Servo 900C 0038 59 168936 Clase III Ventilador Mecánico Siemens Servo 900C S/A 162978 Clase III Ventilador Mecánico Siemens Servo 900C 0038 81 182223 Clase III Ventilador Mecánico Siemens Servo 900C 0028 71 182208 Clase III Vacuometro Chemetrom Timeter 0041 97 S/S Clase I Vacuometro Chemetrom Continuos 0041 76 S/S Clase I 108 Vacuometro Chemetrom Continuos 0042 16 S/S Clase I Vacuometro Chemetrom Continuos 0041 92 S/S Clase I Vacuometro Chemetrom 3433 5 S/S Clase I 3433 6 S/S Clase I Vacuometro Chemetrom 109 Área: Unidad de Cuidado Intensivo CONVENIO Equipo Marca Modelo Activ o Fijo Serie Clasifica cion de Equipos biomedi cos Desfibrilador HEWELLET PACKARD CODE MASTER XL 0193 330499874 67 Clase III Electrocardiografo Edan SE-3A 0057 SE3A323098 2 96101 Clase IIa Flujometro Chemetrom Timeter 3489 FMA057559A 5 I Clase I Flujometro Chemetrom Timeter 3490 S/A 2 Clase I Flujometro Chemetrom Timeter 3488 FMA055776A 3 I Clase I Flujometro Chemetrom Timeter 3489 FMA055759A 3 I Clase I Flujometro Chemetrom Timeter 3489 FMA057559A 5 I Clase I Flujometro Chemetrom Timeter 3489 FMA055758A 2 I Clase I Flujometro Chemetrom Timeter 3488 FMA057536A 6 I Clase I Flujometro Chemetrom Timeter 3489 FMA057514A 6 I Clase I Marcapaso externo Transitorio Medtronic 5330 0058 EH1011704R 9 Clase III Marcapaso externo Transitorio Medtronic 5330 0058 EH0006700R 8 Clase III Monitor de Signos Vitales Mindray PM9000 0057 BX-96120727 Clase IIb 7 Monitor de Signos Vitales Mindray PM-9000 0055 W-93104573 8 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0055 W-93104578 9 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0056 W-95104729 Clase IIb 110 1 Monitor de Signos Vitales Mindray PM-9000 0056 w-92104345 3 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0055 W-93104579 6 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0056 W-93104571 5 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0057 W-93104567 1 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0057 W-93104575 4 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0056 W-92104349 9 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0056 W-95104727 7 Clase IIb Monitor de Signos Vitales Mindray PM-9000 0057 W-93104574 5 Clase IIb Vacuometro Chemetron Vacutron 3490 S/A 2 Clase I Vacuometro Chemetron Vacutron 3490 S/A 8 Clase I Vacuometro Chemetron Vacutron 3490 S/A 5 Clase I Vacuometro Chemetron Vacutron 3490 S/A 6 Clase I Vacuometro Chemetron Vacutron 3490 S/A 4 Clase I Vacuometro Chemetron Vacutron 3490 S/A 1 Clase I Vacuometro Chemetron Vacutron S/A Clase I Ventilador Mecanico Puritan Bennet 760 0056 3502990131 2 Clase III Ventilador Mecanico Puritan Bennet 760 0055 3402990254 7 Clase III Ventilador Mecanico Puritan Bennet 760 0055 3500990199 4 Clase III Ventilador Mecanico Puritan Bennet 760 0055 3501990045 Clase III 111 5 Ventilador Mecanico Puritan Bennet 760 0056 3502005487 6 Clase III Ventilador Mecanico Puritan Bennet 760 0056 3502990007 4 Clase III Ventilador Mecanico Puritan Bennet 760 0057 3502992467 0 Clase III Ventilador Mecanico Puritan Bennet 760 0057 3511006598 3 Clase III Ventilador Mecanico Newport E360 0057 N093610147 6 70 Clase III Ventilador Mecanico Newport E360 0056 N103603159 0 99 Clase III Ventilador Mecanico Newport E360 0056 N103663160 8 51 Clase III Ventilador Mecanico Bird Avian 0057 B058792262 8 0 Clase III 112 Área: Unidad de Cuidado Intensivo Quirurgica Equipo Marca Modelo Activ o Fijo Serie Clasifica cion de Equipos biomedi cos Desfibrilador Hewlett Packard Code Master M1722B 0225 2527A00224 80 Clase III Electrocardiografo Edan SE3 0198 SE3A3231061 39 17791 Clase IIa Flujometro Chemetrom Timeter 0899 S/S Clase I Flujometro Chemetrom Timeter 7651 S/S 1 Clase I Flujometro Chemetrom Timeter S/A 1097542 Clase I Flujometro Chemetrom Timeter S/A 6690 Clase I Flujometro Chemetrom Timeter 7651 S/S 3 Manta termica Nellcor Warmtouch Monitor de Signos Vitales Penlon M5 2433 CM-0C115114 Clase IIb 8 Monitor de Signos Vitales Penlon M5 2433 CM-0C115116 Clase IIb 9 Monitor de Signos Vitales Penlon M5 Monitor de Signos Vitales Penlon M5 Monitor de Signos Vitales Penlon M5 2427 CM-0B114686 Clase IIb 2 Monitor de Signos Vitales Penlon M5 2427 CM-08113099 1 Clase IIb Monitor de Signos Vitales Penlon M5 2428 CM-08113114 Clase IIb 113 CI0110J249 Clase I Clase I 2427 Clase IIb 0 CM-0B114682 2427 CM-0B114684 Clase IIb 3 0 Monitor de Signos Vitales Penlon M5 2467 CM-13116641 Clase IIb 2 Monitor de Signos Vitales Transporte Mindray PM-9000 0221 W79101274 81 Vacuometro Chemetrom Continuos 2144 S/S 8 Clase I Vacuometro Chemetrom Continuos S/A Clase I Vacuometro Chemetrom Continuos 0044 S/S 73 Clase I Vacuometro Chemetrom Continuos 0046 S/S 94 Clase I Vacuometro Chemetrom Continuos 0040 S/S 20 Clase I Vacuometro Chemetrom Continuos 0046 S/S 38 Clase I Ventilador de Transporte Airox N/A 0053 40G6200K039 26 909 Clase IIb Ventilador Mecánico Puritan Bennett 840 0193 3510101287 63 Clase III Ventilador Mecánico Puritan Bennett 840 2499 3512102113 7 Clase III 840 2499 3512100756 4 Clase III Ventilador Mecánico Puritan Bennett 8385 Clase IIb Ventilador Mecánico Puritan Bennett 840 2499 3512100774 5 Clase III Ventilador Mecánico Puritan Bennett 840 3914 7 3121110630 Clase III Ventilador Mecánico Puritan Bennett 840 3914 8 3121108910 Clase III Ventilador Mecánico Puritan Bennett 840 0193 3510095264 64 Clase III 840 0193 3510101301 62 Clase III Ventilador Mecánico Puritan Bennett 114 Área: Unidad de Cuidado Intensivo CardioVascular Convenio Serie Clasificaci on de Equipos biomedic os 0037 99 s72777-F5 Clase IIb 9525 0193 60 908808 Clase IIa KENDALL 9525 0193 61 908807 Clase IIa Desfibrilador Hewlett Packard Code Master 0037 96 3304A14036 Clase III Electrocardiografo Edan SE-3 0191 54 SE3B32309897 221 Clase IIa Flujometro chemetrom Timeter S/A 76516 Clase I Flujometro chemetrom Timeter S/A 1087375 Clase I Flujometro chemetrom Timeter S/A 1021722 Clase I Flujometro chemetrom Timeter S/A 1063961 Clase I Flujometro chemetrom Timeter 2144 0 S/S Clase I Flujometro chemetrom Timeter S/A 5070 Clase I Flujometro chemetrom Timeter S/A 1112627 Clase I Flujometro chemetrom Continuos S/A 1087374 Clase I Manta termica Nellcor Warmtou ch CI0110J260 Clase I Monitor de Signos Vitales Mindray PM9000 3623 8 W-08106291 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3624 W-08106292 ClaseIIb Marca Modelo Activ o Fijo Consola de Contrapulsación Datascope System97 Compresor vascular KENDALL Compresor vascular Equipo 115 0 Monitor de Signos Vitales Mindray PM9000 3623 6 W-08106293 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3623 5 W-08106295 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3624 1 W-08106296 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3624 2 W-08106298 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3623 7 W-08106299 ClaseIIb Monitor de Signos Vitales Mindray PM9000 3623 9 W-08106300 ClaseIIb Vacuometro Vacutrom Continuos 0038 18 S/S Clase I Vacuometro Vacutrom Continuos 0343 37 S/S Clase I Vacuometro Vacutrom Continuos 0343 38 S/S Clase I Vacuometro Vacutrom ONT 0038 33 8385 Clase I Vacuometro Vacutrom Continuos 0194 84 S/S Clase I Vacuometro Vacutrom Continuos 0053 22 S/S Clase I Vacuometro chemetrom Continuos 0038 65 31685 Clase I Vacuometro chemetrom Continuos 0038 55 S/S Clase I Ventilador Mecánico Siemens Servo 900C 005319 110195 Clase III Ventilador Mecánico Siemens Servo 900C 0023 73 165889 Clase III Ventilador Mecánico Siemens Servo 900C 0323 38 157316 Clase III Ventilador Mecánico Siemens Servo 900C 0053 21 152316 Clase III Ventilador Mecánico Siemens Servo 0038 161813 Clase III 116 900C 70 Ventilador Mecánico Siemens Servo 900C 0044 74 183252 Clase III Ventilador Mecánico Siemens Servo 900C 0046 82 164161 Clase III Ventilador Mecánico Siemens Servo 900C 0046 76 182221 Clase III 117 Área: Urgencias Equipo Marca Modelo Activo Fijo Serie Clasificaci on de Equipos biomedico s Desfibrilador Primedic XD10Xe 24906 73505002301 Clase III Electrocardiografo Edan SE3 02266 SE33A030846055 5 2B Clase IIa Electrocauterio Yfrecator 796 00443 HU081C11073 7 Clase III Flujometro Amvex N/A 34179 S/S Clase I Flujometro Chemetrom Timeter 21085 0605 Clase I Flujometro Chemetrom Timeter 21436 Clase I Flujometro Chemetrom Timeter 01110 33993 2 Clase I Flujometro Chemetrom Timeter 9353 3100 Clase I Flujometro Amvex N/A 03418 4050 0 Clase I Flujometro Chemetrom Timeter 32791 0308 Clase I Flujometro Chemetrom Timeter 33992 FMA012808FHI Clase I Flujometro Chemetrom Timeter NO Clase I Flujometro Chemetrom Classic 0-3 34888 S/S Clase I Flujometro Chemetrom Timeter 07830 011097 1 Clase I Flujometro Chemetrom classic 0-3 35256 FMA057537AI Clase I Flujometro Amvex N/A 24197 Clase I 118 1206 FMA009735BJ Flujometro Chemetrom Timeter 35255 FMA057545AI Flujometro Doble Chemetrom Timeter 00945 962414 7 Clase I Flujometro Doble Chemetrom Timeter 6226 Clase I Flujometro Doble Amvex N/A 33564 FMA012010FH Clase I Monitor de Signos Vitales Mindray PM900 Monitor de Signos Vitales Welch allyn 45NTO-SI SPOT LXI 02489 20100704355 7 Clase IIa Monitor de Signos Vitales Welch allyn 45NTO-SI SPOT LXI 02498 20101208059 3 Clase IIa Monitor de Signos Vitales Mindray IMEC10 39260 EX22000534 Clase IIb Monitor de Signos Vitales Mindray IMEC10 39272 EX22000530 Clase IIb Monitor de Signos Vitales Mindray PM900 01931 W-8C104154 7 Clase IIb Mec 1200 02402 CC-0A115286 0 Clase IIa W-08106263 Clase I Clase IIb 24911 Monitor de Signos Vitales Mindray Pulsioximetro Edan H100B 02465 H100B103107713 2 759 Clase IIa Pulsioximetro Edan H100B 02484 H100B103113237 5 58 Clase IIa Vacuometro Chemetrom Continuos 00432 31684 1 Clase I Vacuometro Chemetrom Continuos 00432 21449 5 Clase I Vacuometro Vacutrom ONT S/A Clase I Vacuometro Puritan Bennett N/A 00443 S/S 4 Clase I Vacuometro Vacutrom Continuos 63110 22-13-1108-0307 Clase I Vacuometro Vacutrom ONT 00443 S/S Clase I 119 004414 9 Ventilador Mecánico Puritan Bennett 00464 3501982235 6 740 120 Clase III Área: Imagnenes Diagnósticas Equipo Marca Modelo Serie Clasificaci on de Equipos biomedic os 019058 73675000 108 Clase III 036500 6620 Clase III 10172850 4 Clase IIa 51087315 4 Clase IIa 60167 Clase IIb Activo Fijo Desfibrilador Primedic Desfibrilador Zolld PD1400 Ecografo General Electric 5264008 (Logiq P5) N/A Ecografo Philips HD11 36667 Escanografo Siemens Higt Definition 24014 Escanografo Siemens Somaton Spirit Flujometro Amvex Flujometro Chemetron Timeter Inyector de Medio de Contraste LIEBERFLRASHEIM Inyector de Medio de Contraste XD10Xe 79578 25188 Clase IIb 24638 Clase I N/A 9327-6 Clase I MALLINCKRO DT 00002 CI0306B0 25 covidien optivantage 36491 SI0810B58 Clase IIb 7 Inyector de Medio de Contraste Medrad H5M 001337 648101 Clase IIb Mamografo Siemens Mammomat 3000 N/A 02244511 Clase IIb Máquina de anestesia Ohmeda Excel 210 005213 AMAW004 83 Clase III Monitor de Signos Vitales Mindray Mec-1200 019857 CC07114135 Clase IIa Monitor de Signos Vitales Nellcor N5500 023310030 019358 007 Multix Pro Siemens 1175640 001359 Neumoinsuflador Smith + Nephew Dyonics 121 025132 02844950 1 14799EJA Clase IIb Clase IIa Clase IIb Clase IIa Seriografo Siemens 4466053RHD3 2 001335 013905A4 Clase IIb Polymobil Plus Siemens Plus 24082 21325 Clase IIb Vacuometro Chemetrom Continuos 261001 9224-5 Clase I Vacuometro Chemetrom 001418 122 Clase I Área: Hospitalización Equipo Marca Modelo Activo Fijo Serie Clasificaci on de Equipos biomedico s Electrocardiografo Cardioline Delta Plus 00433 3 MDL101927 Clase IIa Electrocardiografo welch-allyn cp50 02489 8 109200651011 Clase IIa Desfibrilador Mindray Benehear D3 EL.19001146 Clase III Desfibrilador Primedic XD10Xe 01987 5 735005002216 Clase III Desfibrilador Primedic XD10Xe 39259 73505002496 Clase III Desfibrilador Mindray Benehear D3 EL.19001147 Clase III Desfibrilador Mindray Benehear D3 25170 EL.19001148 Clase III Desfibrilador Primedic XD10Xe 24516 73505002306 Clase III Desfibrilador Primedic XD10Xe 01982 2 736715000107 Doppler Oxford Instruments SonicaidOne 10954 Flujometro chemetron timeter 32792 No Clase I Flujometro chemetron timeter 31811 No Clase I Flujometro Chemetron No 07792 4 1077924 Clase I Flujometro Allied Timeter 31803 0307 Clase I flujometro doble chemetrom No 33498 No Clase I No 21622 No Clase I No 96241 No Clase I Flujometro Flujometro doble Precision medical Chemetron 123 25168 25169 007347 Clase III Clase IIa 7 Flujometro doble Chemetron No 10813 37 No Clase I Flujometro Doble Amvex Corp 4050 No 31927 Clase I Flujometro doble chemetrom No 33555 No Clase I Flujometro Chemetrom Timeter No No Clase I Flujometro AGAFANO No 33563 No Clase I Flujometro AGAFANO No 32790 No Clase I Flujometro AGAFANO No 21624 No Clase I Flujometro Chemetrom Timeter 21085 No Clase I Flujometro Chemetrom Timeter No No Clase I Flujometro doble AGA No 33562 No Clase I Flujometro Chemetron Timeter 02243 0 No Clase I Flujometro Chemetron Timeter 31804 Clase I Flujometro Chemetron Timeter 1057911 Clase I Flujometro Doble Amvex Corp No FMA011923FH Clase I Flujometro Chemetron Timeter 33560 Clase I Flujometro Chemetrom Timeter 23002 0 1075022 Clase I Flujometro Chemetrom Timeter 31928 No Clase I Flujometro Chemetrom Timeter No Clase I Flujometro Chemetrom Timeter No Clase I Flujometro Chemetrom Timeter No Clase I 33559 124 96241 5 Flujometro Doble Allied Timeter 96242 0 No Clase I Flujometro Chemetrom Timeter 76517 S/S Clase I Flujometro AGA No 21623 No Clase I Flujometro Doble Flowmeter No 31926 4050 Clase I Flujometro AGAFANO No 33499 No Clase I Flujometro doble AGA No No 33558 Clase I Flujometro Chemetron Timeter 33556 No Clase I Flujometro Chemetron Timeter 23003 6 1081257 Clase I Flujometro Chemetrom No 22901 7 No Clase I Flujometro Chemetron Timeter 230025 Clase I Flujometro Chemetron Timeter No 31924 Clase I Flujometro doble chemetrom timeter 33500 No Clase I Flujometro doble chemetrom timeter 31807 No Clase I Flujometro AGA No 9353-1 No Clase I Flujometro AGA No 33557 No Clase I Flujometro AGA No No Clase I Flujometro AGA No 93275 No Clase I Flujometro AGA No 21435 No Clase I Flujometro AGA No 76512 No Clase I Flujometro AGA No 35260 No Clase I 125 Flujometro Allied Timeter 33353 No Clase I Flujometro Allied Timeter 31810 No Clase I Flujometro Allied Timeter 2008 No Clase I Flujometro Allied Timeter 24661 FMAO0799AKJ Clase I Flujometro Allied Timeter 24662 FMAO010565 DJ Clase I Flujometro Allied Timeter 24663 FMAO10565DJ Clase I Flujometro Allied Timeter 24664 FMA012286BJ Clase I Flujometro Allied Timeter 24665 FMAO07975KJ Clase I Flujometro Allied Timeter 24666 FMA018268BJ Clase I Flujometro Allied Timeter 24667 FMAO5560AI Clase I Flujometro Allied Timeter 24668 FMAO18142BJ Clase I Flujometro Doble Allied Timeter 24669 FMAO19964KJ Clase I Flujometro Doble Allied Timeter 24670 Nellcor N5500 00366 0200-05124 0112 Clase IIb Mindray Mec-1200 24513 CC-0C116158 Clase IIa Monitor de Signos Vitales Mindray Mec-1200 24515 CC-0C116164 Clase IIa Monitor de Signos Vitales Nellcor N5600 01935 6 023310030015 Clase IIa Monitor de Signos Vitales Nellcor N5600 01924 7 023310010009 Clase IIa Mindray Mec-1200 24900 CC-14117260 Clase IIa Mindray Mec-1200 24514 CC-0C116173 Clase IIa Monitor de Signos Vitales Monitor de Signos Vitales Monitor de Signos Vitales Monitor de Signos 126 Clase I Vitales Monitor de Signos Vitales Nellcor N5600 Monitor de Signos Vitales Mindray Mec-1200 Monitor de signos vitales Mindray 01935 023310030012 9 Clase IIa 02474 5 CC-0C116174 Clase IIa Mec 1200 25018 CC-14117351 Clase IIa Monitor NIBP GE Dinamap procare 200 02217 5 AAW0706024 2SA Clase IIa Monitor NIBP GE Dinamap procare 200 02217 3 AAW0706024 6SA Clase IIa Regulador de vacío Chemetron Vacutron 01948 6 No Clase I Regulador de vacío Chemetron Vacutron 34185 No Clase I Regulador de vacío Chemetrom Vacutrom 30214 No Clase I Regulador de vacío Chemetron Vacutron 20035No 1 Clase I Regulador de vacío Chemetron Vacutron 8838-3 No Clase I Regulador de vacío Chemetron Vacutron 34176 No Clase I Regulador de vacío Chemetron Vacutron 01942 No Clase I Regulador de vacío Puritan-bennett 92043 No 7598 Clase I Regulador de vacío Chemetron Vacutron No 37188 Clase I Regulador de vacío Chemetron Vacutron 01948 7 VRA05094BJ Clase I Regulador de vacío Chemetron Vacutron 01948 5 No Clase I Regulador de vacío Chemetron Vacutron 01948 8 No Clase I Regulador de vacío Chemetron Vacutron No VRA08879LI Clase I Regulador de vacío Chemetron Vacutron 34187 No Clase I Regulador de vacío Chemetron Vacutron 34188 127 Clase I Regulador de vacío Chemetron Vacutron 30213 No Clase I Regulador de vacío Chemetron Vacutron 01948 9 No Clase I Regulador de vacío Chemetron Vacutron 21451 No Clase I Regulador de vacío Chemetron Vacutron 21451 No Clase I Regulador de vacío Chemetron Vacutron 30215 No Clase I Regulador de vacío Chemetron Vacutron 31815 No Clase I Regulador de vacío Chemetron Vacutron 35261 No Clase I Regulador de vacío Chemetron Vacutron No 35260 Clase I 128 Área: Cirugía Equipo Marca Modelo Activ o Fijo Serie Clasificac ion de Equipos biomedic os 0051 06 22351 Clase IIb 3776494 0051 08 02017507 Clase IIb Stockert Scpc 0059 1 60S10505 Clase III Coagulador Jhonson y Jhonson 801170 HP2086A Clase III Desfibrilador HP M1222B 0051 74 US00104337 Clase III Desfibrilador HP M1722A 0048 68 3601A17963 Clase III Desfibrilador Primedic XD10Xe 2490 5 73505002204 Clase III Arco en C Philips Arco en C Siemens Bomba centrifuga Electrobisturí Daiwha DT300 0052 22 DT3P03007003 Clase III Electrobisturí Valleylab Force FX 0051 98 F6D46043A Clase III Electrobisturí Valleylab Force 2 2043 7 F5D0192T Clase III Electrobisturí Valleylab Force 2 0051 67 F4D43607T Clase III Electrobisturí Valleylab Force 2 0052 38 F4B43478T Clase III Electrobisturí Valleylab Force FX No F5CI41804A Clase III Electrobisturi Valleylab Force 2 0048 07 FAL44487T Clase III Electrobisturi Valleylab Force FX 0391 42 SF0L08795A Clase III Electrobisturí Bovie IDS - 200 2490 7 BV1511010 Clase III 129 Electrobisturí Erbe VIO300 3946 0 11333234 Clase III Electrocardiógrafo Edan instruments SE-3 0221 02 Se33A0307A455 4ZB Clase III Equipo para Litotripsia Nidhi Lith Nidhi Lith Digital No NLD-240 Clase IIa Flujometro Doble Allied Timeter 9624 22 No Clase I Flujometro Doble Allied Timeter 9624 16 No Clase I Flujometro Doble Allied Timeter 9624 21 No Clase I Flujometro Doble Allied Timeter No 1014965 Clase I Flujometro Amvex Timeter 3417 8 4090 Clase I Flujometro Amvex Timeter 3418 2 No Clase I Flujometro Amvex Timeter 3417 7 No Clase I Fuente de luz Storz 615C 0049 43 3249 Clase I Fuente de luz Stryker X8000 0195 52 A004514 Clase I Fuente de luz Stryker X6000 0228 06 04E026704 Clase I Fuente de luz Stryker X6000 0228 23 02A002844 Clase I Fuente de luz Stryker X6000 0198 21 04D038594 Clase I Fuente de luz Stryker X6000 0198 23 04E014234 Clase I Fuente de luz Sunoptics XLS-300 No 300-00241 Clase I Fuente de luz Olympus CLK4 8419689 Clase I Generador armónico USSC Autosonix 0226 17 As3363 Clase III Generador Force Triad Valleylab Force Triad 0391 43 T1K26275EX Clase III 130 Intercambiador de Calor 3M Sarns No 2963B Clase I Intercambiador de Calor 3M Sarns TCM II 0051 70 2963A Clase I Lámpara cielítica Trident Medical ELD-85H Co 0052 30 E018-G3-0003 Clase I Lámpara cielítica de pie Amsco 0052 28 No Clase I Lámpara cielítica Trident Medical ELD-85H Co 0048 59 E018-G3-0002 Clase I Lámpara cielítica System one ss2s1120 0508 18 1109041 Clase I Lámpara cielítica System one ss2s1120 0227 43 0609162 Clase I Lámpara cielítica Trident Medical ELD-85H Co 0048 06 E018-FA-0007 Clase I Lámpara cielítica Vamada Shadowless 0048 72 AC-2035 Clase I Lámpara cielítica (Satélite) Trident Medical ELD-85H Co 0048 58 E018-G3-0003 Clase I Lámpara cielítica (Satélite) Trident Medical ELD-85H Co 0048 05 E018-FA-0007 Clase I Lámpara cielítica (Satélite) Trident Medical ELD-85H Co 0052 31 E018-G3-0002 Clase I Lámpara cielítica (Satélite) Vamada Shadowless 9656 0048 73 AC-2035 Clase I Lámpara cielítica (Satélite) SKYLUX EXCEL 0053 25 3AY-78 Clase I Lámpara cielítica (Satélite) SKYLUX EXCEL 0051 38 3AY-78 Clase I Lámpara cielítica de pie SKYLUX Excel Minor 0021 61 50-453 Clase I Lámpara cielítica de pie SKYLUX A5 Stand 0051 09 Lampara Cielitica Mindray Hylite 6700 0250 15 G5-0A000117 Clase I Láser Surgilase SL60 0049 16 SL0-00-93-0624-01-D Clase III Manta térmica Nellcor Warm touch 2225 3 CI0107J840 Clase I Examiner 10 9656 131 Clase I Máquina CEC 3M Sarns 9000 0051 94 4194 Clase III Maquina de ACT Medtronic ACTPlus 0225 12 ACT1002638 Clase I Maquina de Anestesia Penlon SP2 0229 85 SP2100831 Clase III Máquina de anestesia Ohmeda Modulus II 0048 03 ABDF00331 Clase III Máquina de anestesia Penlon SP2 0242 79 SP2100962 Clase III Máquina de anestesia Penlon SP2 0194 50 SP2100736 Clase III Máquina de anestesia Penlon SP2 2434 0 SP2100964 Clase III Máquina de anestesia Ohmeda Modulus II No ABQN03235 Clase III Maquina de Anestesia ohmeda Modulus II 0051 41 ABQQ00870 Clase III Máquina de anestesia Ohmeda Modulus II No ABDG02074 Clase III Máquina de anestesia Ohmeda Modulus II 0048 65 ABDM00488 Clase III Máquina de anestesia Ohmeda Modulus II 0051 85 ABQR01812 Clase III Marcapasos externo transitorio APC Medical EV4543 No MVBS 3056 Clase III Marcapasos externo transitorio APC Medical EV4543 No MVBX 4072 Clase III Microscopio Cx Carl Zeiss Universal S3 No 252803 Clase I Microscopio Cx Carl Zeiss Superlux 300 0225 90 304953 Clase I M20M1A 0223 27 10G708080001 Clase IIa Monitor de Signos Vitales Mediana Monitor de signos vitales Criticare 508 0049 56 195249339 Clase IIa Monitor de signos vitales Criticare 508 0053 13 195249340 Clase IIa Monitor de signos vitales Criticare 508 0049 53 294221603 Clase IIa 132 Monitor de signos vitales Nellcor N5500 0049 03 020005120123 Clase IIa Monitor de signos vitales Mindray PM-9000 0197 72 W-05105989 Clase IIb Monitor de signos vitales Mindray PM-9000 0197 74 W-05105992 Clase IIb Monitor de signos vitales Mindray Beneview 2402 CM-9C110068 6 Clase IIb Monitor de signos vitales Transporte Mindray PM-9000 0197 73 W-05105990 Clase IIb Monitor de signos vitales Mindray PM-9000 0229 36 W-93104572 Clase IIb Monitor de signos vitales Mindray Beneview 2402 7 CM-9C110083 Clase IIb 2433 7 Clase IIb Monitor de signos vitales Penlon M5 Monitor de signos vitales Mindray T5 2499 8 CM-14116955 Monitor de signos vitales Mindray T5 2499 9 CM-14116961 5150400 CC-14117270 Monitor de signos vitales Datex-ohmeda IPX1 0047 99 Monitor de signos vitales Mindray Mec 1200 0249 03 Monitor de signos vitales Mindray Monitor de signos vitales Mindray Monitor de Video CM-0C115115 Clase IIb Clase IIb Clase IIb Clase IIa Mec 1200 2501 7 CC-14117335 Mec 1200 2490 2 CC-14117316 Sony PVM1953MD 0051 01 2007099 Clase I Monitor de Video Medical Display SC-SX19A1A11 0195 50 05-55907 Clase I Monitor de Video Sony PVN120M2M 2282 1 2001873 Clase I Monitor de Video Sony LMD-214MD 0250 73 Monitor de Video Striker 240-030-920 0251 40 SV219510355 Monitor para Saturacion Venosa Terumo CDI 101 0198 45 1781 133 Clase IIa Clase IIa Clase I Clase I Clase IIb Monitor NIBP Critikon Inc. 8100 No 8100J6784 Clase IIa Motor eléctrico Bbraun Aesculap Microspeed 0225 64 003069 Clase III Motor eléctrico Bbraun Aesculap Torre aesculap N/A N/A Clase III Motor eléctrico Bbraun Aesculap Elan E 0051 66 FABR2611 Clase III Neumoinsuflador Striker 40lhigh flow 0195 53 0207ce127 Clase IIa Neumoinsuflador Smith & Nephew Dyonics Access 40 0049 42 16655KMF Clase IIa Neumoinsuflador Striker 40lhigh flow 0250 96 040LCE149 Clase IIa Procesador cámara de video Storz 20221120 Procesador Camara de video Storz 20221120 Procesador Dgital de video Storz 20202020 Procesador cámara de video Striker 9883chip Procesador cámara de video Storz 202221-20 Procesador camara de video Striker 1088hd Procesador camara de video Storz 20222120 Pulsoximetro Criticare 503 Pulsoximetro Criticare Regulador de vacío 0198 24 0228 26 0222 94 IEI52834 Clase I IEE48825 Clase I IFH60530 Clase I 04k010714 Clase I dip031605 Clase I 0195 51 Clase I dh038694-p Clase I 0048 99 291105329 Clase I 503 0049 00 491121642 Clase I Chemetron Vacutron 0049 07 229451 Clase I Regulador de vacío Chemetron Vacutron 0049 51 229010 Clase I Regulador de vacío Chemetron Vacutron 30209 Clase I Regulador de vacío Chemetron Vacutron 229008 Clase I 134 0051 88 0049 55 Regulador de vacío Chemetron Vacutron 3417 3 No Clase I Regulador de vacío Chemetron Vacutron 3417 8 No Clase I Regulador de vacío Chemetron Vacutron 3417 5 No Clase I Regulador de vacío Chemetron Vacutron 0048 63 No Clase I Regulador de vacío Chemetron Vacutron 0051 37 Regulador de vacío Chemetron Vacutron 0047 96 No Clase I Regulador de vacío Chemetron Vacutron 5227 No Clase I Regulador de vacío Chemetron Vacutron No No Clase I Regulador de vacío Chemetron Vacutron 0052 34 No Clase I Regulador de vacío Chemetron Vacutron 0047 81 No Clase I Reproductor/Grabador DVD Toshiba D-KR10 No PC114028529 Clase I Shaver Striker 150 0228 28 51001 Clase III Shaver Striker Core 0249 20 07431887 Clase III Televisor Sony KV-21FS120 0050 98 4248559 Clase I Televisor Sony KV-21FS140 No 4011123 Clase I Televisor Samsung LCD 32" 2473 9 Z3743CGB3006 24T Clase I Televisor Samsung LCD 32" 2471 7 21ZC3CPB3003 3J Clase I 135 Clase I Área: Unidad de Cuidado Intensivo Intermedio Equipo Marca Modelo Activo Fijo Serie Inventari oy Program a de Mtto Preventiv o 2011 Desfibrilador Primedic XD10xc 01946 7367500011 e 7 2 Clase III Electrocardiografo Welch Allyn CP50 25136 1092001010 11 Clase IIa Monitor de Signos Vitales Mindray PM9000 01949 W-04105819 7 Clase IIb Monitor de Signos Vitales Mindray PM9000 01947 W-04105820 8 Clase IIb Monitor de Signos Vitales Mindray PM9000 01948 W-03105752 0 Clase IIb Monitor de Signos Vitales Mindray PM9000 01947 W-03105749 9 Clase IIb Monitor de Signos Vitales Mindray PM9000 01948 W-03105785 1 Clase IIb Monitor de Signos Vitales Mindray PM9000 01948 W-04105818 2 Clase IIb Monitor de Signos Vitales Transporte Mindray PM9000 01977 W-05105994 6 Clase IIb Flujometro Chemetro Timeter m FMA055737 AI Clase I Flujometro Chemetro Timeter S/A m 1087621 Clase I Flujometro Chemetro Timeter m Clase I Flujometro Chemetro Timeter m Clase I Flujometro Doble Chemetro Timeter S/A m Flujometro Regulador de vacío 0899 Chemetro Timeter 1087633 m Chemetro Vacutro 00405 No 136 Clase I Clase I Clase I n n 6 Regulador de vacío Chemetro Vacutro n n No Clase I Regulador de vacío Chemetro Vacutro 00393 No n n 2 Clase I Regulador de vacío Chemetro Vacutro 00531 No n n 5 Clase I Regulador de vacío Chemetro Vacutro 01949 No n n 0 Clase I Regulador de vacío Chemetro Vacutro 01949 No n n 1 Clase I 137 ANEXO C. GUÍAS RÁPIDAS DE EQUIPOS BIOMÉDICOS 138 139 140 141 142 143 ANEXO D. FOLLETO INFORMATIVO 144 145 ANEXO E. EVALUACIÓN ¿De los siguientes enunciados cuál es su respuesta? A: Una bomba de infusión se detiene y no da alarma de esta situación. El paciente no recibe la cantidad suficiente de fluidos. B: Una bomba de infusión que se detiene, debido a un mal funcionamiento, pero entrega una adecuada señal de alarma y no provoca daño al paciente. C: Un profesional de la salud informó que durante el implante de una válvula cardíaca, se descubre que viene con defectos de fabricación. Esta válvula no se utilizó y se tuvo que utilizar una nueva, por lo tanto, el tiempo de bombeo durante la cirugía se extendió. D: El envase de un dispositivo médico estéril, de un solo uso, señala la advertencia “no usar si el envase está abierto o dañado”. Previo al uso, el usuario observó que el envase estaba dañado y, obviamente, el dispositivo médico no fue usado E: Un paciente muere después de una diálisis. El paciente tenía una enfermedad renal terminal y murió de una falla renal. Las investigaciones demostraron que el dispositivo médico funcionaba adecuadamente y el evento no fue atribuido al funcionamiento de éste. F: Durante el uso de un desfibrilador externo en un paciente, el equipo no entrega el nivel de energía programado debido a un mal funcionamiento. El paciente falleció. Respuesta cuales debemos reportar A, C, F porque son eventos adversos serios que ocasionaron deterioro serio de la salud del paciente, o muerte, en estos casos el paciente necesita una hospitalización prolongada, una intervención quirúrgica prolongada para prevenir un daño al paciente. Respuesta que no debemos reportar B, D, E, B: El evento que no conduce a un deterioro del estado de salud o daño grave, porque el dispositivo presenta un diseño que alerta las fallas que pudieran presentar algún peligro, no requiere notificarse. D: Si en las instrucciones de uso del fabricante se indican advertencias que el usuario detecta fácilmente previo al uso, no es necesario notificarlas, sin perjuicio que el usuario le informe al fabricante/distribuidor de la deficiencia detectada. 146 E: Cuando la causa del evento se debe a una condición de salud del paciente, el evento no requiere ser notificado, ya que esta condición podría ser una preexistencia u ocurrir durante el uso del dispositivo médico. 147 ANEXO F. REGISTRÓ FOTOGRÁFICO DE LA CAPACITACIÓN AL PERSONAL 148 ANEXO G. FORMATO DE ASISTENCIA 149 150 151 152 153 154 155 156 157 158 159 160 161 162 ANEXO H. PROPUESTA BÁSICA DE INDICADORES DEL PROGRAMA INSTITUCIONAL DE LA CLÍNICA DE OCCIDENTE N. NOMBRE DEL INDICADOR Cumplimiento del programa de mantenimiento preventivo 1 Cumplimiento de órdenes de servicio 2 FORMULA Numero de orden de mantenimiento preventivo Total de órdenes de mantenimiento preventivo programadas Número de órdenes de servicio gestionadas Número de órdenes de servicio tramitadas Numero de eventos adversos tramitados Reporte de eventos adversos Numero de eventos adversos sucedidos 3 Número de incidentes adversos tramitados Reporte de incidentes adversos Número de incidentes adversos sucedidos 4 UNIDAD PERIODO Porcentaje (%) Mensual Porcentaje (%) Mensual Porcentaje (%) Mensual Porcentaje (%) Mensual Porcentaje (%) Mensual Porcentaje (%) Mensual Porcentaje (%) Mensual Jornadas realizadas 5 Capacitación a personal médico y administrativo de la clínica Jornadas programadas Reuniones realizadas 6 Operatividad comité de seguridad al paciente Reuniones programadas 7 Socialización y divulgación del programa institucional de tecnovigilancia Jornadas de socialización realizadas Jornadas de socialización programadas 163 ANEXO I. MANUAL DE TECNOVIGILANCIA 164 2012 MANUAL DE TECNOVIGILANCIA 165 JAIRO ALBERTO SARRIA CLÍNICA DE OCCIDENTE 01/01/2012 CONTENIDO pág. INTRODUCCIÓN 172 1. OBJETIVO DEL MANUAL 173 2. ALCANCE DEL MANUAL 174 3. DESCRIPCION DEL COMITÉ DE TECNOVIGILANCIA 173 4. NORMATIVA 174 5. DEFINICIONES 177 6. CLASIFICACIÓN DE ACUERDO AL RIESGO 179 7. CLASIFICACIÓN DEL EVENTO E INCIDENTE ADVERSOS (SEGÚN RESOLUCIÓN 4816 DE 2008) 180 8. REPORTE DE TECNOVIGILANCIA 181 9. TIPOS DE REPORTE 183 9.1 REPORTES INMEDIATOS 183 9.2 REPORTES PERIÓDICOS 183 9.3 REPORTES DE RETIRO DE PRODUCTOS O LOTES DE PRODUCTO 183 9.4 REPORTE DE ALERTAS INTERNACIONALES 183 10. INFORMACIÓN QUE DEBE CONTENER LOS REPORTES DE EVENTOS ADVERSOS 184 166 10.1 CONFIDENCIALIDAD DE LA INFORMACIÓN 184 11 ANÁLISIS Y EVALUACIÓN DEL REPORTE 186 12 DISEÑO DE ESTRATEGIAS DE VIGILANCIA DE EQUIPOS BIOMÉDICOS 189 12.1 SEGUIMIENTO A ACCIONES Y RESPONSABLE 201 13 PROPUESTA PARA LA EVALUACIÓN DEL CUMPLIMIENTO DE ACTIVIDADES 204 BIBLIOGRAFIA 207 ANEXOS 208 167 LISTA DE CUADROS pág. Cuadro 1. Marco Normativo 174 Cuadro 2. Cuando hacer el reporte 183 Cuadro 3. Medida cualitativa de la probabilidad del evento 191 Cuadro 4. Medida cualitativa de la severidad del evento 191 Cuadro 5. Matriz de riesgo 192 de la Clínica de Occidente Cuadro 6. Matriz de riesgo máquina de anestesia 194 Cuadro 7. Matriz de riesgo desfibrilador 196 Cuadro 8. Matriz de riesgo ventilador 198 Cuadro 9. Matriz de riesgo monitor de signos vitales 200 Cuadro 10. Barreras 201 Cuadro 11. Propuesta para la evaluación básica de actividades del Programa Institucional de Tecnovigilancia de la Clínica de Occidente Barreras 204 168 LISTADO DE IMÁGENES pág. Imagen 1. Descripcion del comité de tecnovigilancia 173 Imagen 2. Modelo organizacional del protocolo de Londres 185 169 LISTADO DE FIGURAS pág. Figura 1. Investigación de eventos o incidentes adversos 186 Figura 2. Pasos para el diseño de una matriz de riesgo 190 170 ANEXOS pág. ANEXO A. Formato de reporte establecido por el Ministerio de la Protección Social 208 ANEXO B. Formato para el reporte del visitante 209 ANEXO C. Formato para el reporte del empleado 210 171 INTRODUCCIÓN Uno de los pasos más importantes para el diseño e implementación del Programa de Tecnovigilancia es la constitución de un manual, entendiendo como tal, el documento institucional que define el tipo de dispositivos biomédicos objeto de vigilancia, elementos conceptuales de los eventos e incidentes adversos, estrategia de vigilancia y recolección de reportes, análisis y valoración de los resultados, reporte al fabricante y autoridad sanitaria. El objetivo del manual es el de orientar a todo el personal asistencial de la Clínica de Occidente – CDO sobre el Programa Institucional de Tecnovigilancia generando el conocimiento y las herramientas necesarias para realizar reportes de eventos e incidentes adversos a dispositivos biomédicos. Este manual detalla el marco legal del programa de Tecnovigilancia, la descripción de equipos médicos, el detalle de la clasificación de eventos de acuerdo al riesgo, detalle del reporte de tecnovigilancia, los objetivos de reporte los eventos, la información que debe contener el reporte, el análisis de evaluación del reporte, entre otros aspectos de interés que dan las directrices a la Clínica de Occidente de cómo implementar el programa. 172 1. OBJETIVO DEL MANUAL Orientar a todo el personal de la Clínica de Occidente – CDO sobre el Programa Institucional de Tecnovigilancia generando el conocimiento y las herramientas necesarias para realizar reportes de eventos e incidentes adversos a dispositivos biomédicos al responsable de tecnovigilancia de la institución, fabricantes, importadores y al INVIMA. 173 2. ALCANCE DEL MANUAL Desde el proceso general de electromedicina, adquisición de tecnología biomédica, seguimiento de inspección para el ingreso de tecnología, y montaje respectivo, puesta en marcha, y demás procesos que estén relacionados con el desarrollo de los mismo. Este manual está dirigido al cuerpo médico, administrativo y personal de la Clínica de Occidente. 174 3. DESCRIPCIÓN DEL COMITÉ DE TECNOVIGILANCIA Imagen 1. Descripción del comité de tecnovigilancia DESCRIPCIÓN DEL COMITÉ Clínica de Occidente S.A. TECNOVIGILANCIA OBJETIVO: Orientar a todo el personal de la Clínica de Occidente – CDO sobre el Programa Institucional de Tecnovigilancia generando el conocimiento y las herramientas necesarias para realizar reportes de eventos e incidentes adversos a dispositivos biomedicos al responsable de tecnovigilancia de la institución, fabricantes, importadores y al INVIMA LÍDER: Coordinador de electromedicina PARTICIPANTES: Directora Médica, Supervisora Hospitalaria, Coordinadora de Comité de Infecciones, Coordinadora de laboratorio, Coordinadora de Imágenes Dx, Cordinador de Enfermería UCI ADULTOS, Coordinador de Enfermeria Urgencias, Coordinadora de Central de Esterilizacion, Analistas de Calidad, Coordinador de calidad, Jefe de servicios farmaceuticos, Coordinador de cirugia, etc. PERIODICIDAD DE REUNIÓN: Quincenalmente Viernes 8:00 a.m, 10 minutos en el comité de seguridad al paciente FUNCIONES: 1) Tomar las acciones preventivas o correctivas que sean del caso y las que le sean exigidas por el Instituto Nacional de Medicamentos y Alimentos, Invima, de forma inmediata. 2) Comunicar al fabricante o importador del dispositivo médico correspondiente, la ocurrencia del evento e incidente adverso, si se estima pertinente. 3) Comunicar al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, Invima, o a las Secretarías Departamentales y Distritales de Salud, la ocurrencia de eventos e incidentes adversos, en los términos de la presente Resolución. 4) Desarrollar actividades de promoción y formación con los profesionales de la salud de la institución, en relación al desarrollo e implementación del Programa Nacional de Tecnovigilancia y la gestión de eventos o incidentes adversos con dispositivos médicos. 5) Registrar, analizar y gestionar todo evento o incidente adverso susceptible de ser causado por un dispositivo médico. 6) Recomendar medidas preventivas para tomar acciones inmediatamente ocurrido el evento. 7) Orientar a los informantes en el correcto diligenciamiento del formato de reporte. 8) Estar atentos y vigilantes del desempeño, calidad y seguridad de los dispositivos médicos previos a su uso. 9) Informar de manera inmediata al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, Invima, todo reporte de evento o incidente adverso serio dentro de las 72 horas siguientes a la ocurrencia del evento o incidente. 10) Enviar trimestralmente los informes periódicos al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, Invima, o a las Secretarías Departamentales y Distritales de Salud, de todo reporte de evento o incidente adverso no serio, los reportes periodicos seran de caracter voluntario durante los primeros 48 meses de expedida la presente resolucion. Posterior a estos 48 meses, los reportes periodicos seran de caracter obligatorio. 175 4. NORMATIVA La normativa que rige el programa de tecnovigilancia, indica aspectos importantes en cuanto a los pasos que se deben seguir para implantar un programa de tecnovigilancia donde se debe enunciar primordialmente que son los dispositivos médicos, su clasificación, definición, entre otros. Por tal motivo para la reglamentación e implementación del programa de tecnovigilancia en la Clínica de Occidente – CDO, es de suma importancia para el desarrollo y cumplimiento del programa de tecnovigilancia. Cuadro 1. Marco Normativo. DECRETO 4725 DEL 2005 “Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano‟‟ El capítulo IX del Decreto 4725 contiene los artículos 59 (Obligación de informar a la autoridad sanitaria), 60 (Notificación) y 61 (Del programa nacional de Tecnovigilancia). “Por la cual se reglamenta Artículo 1°. Objeto y ámbito el Programa Nacional de de aplicación. El objeto de la RESOLUCIÓN Tecnovigilancia” presente resolución es 004816 DE 2008 reglamentar el Programa Nacional de Tecnovigilancia “Estructura de codificación Esta norma específica los para tipos de Eventos requisitos para una Norma Técnica adversos y sus causas estructura de codificación Colombiana – NTC cuyo fin sea describir los 5736 Eventos adversos relacionados con los dispositivos médicos. Fuente. Adaptado de FRANCO, Catalina Vásquez, TRUJILLO, Mauricio Pérez. Guía para la implementación del Programa Nacional de Tecnovigilancia en el Hospital General de Medellín, 2011 176 5. DEFINICIONES La información que a continuación se relaciona, fue obtenida de la Resolución 004816 de 200830 y del Decreto 4725 de 200531 EQUIPO BIOMÉDICO: dispositivo medico operacional y funcional que reúne sistemas y subsistemas eléctricos, electrónicos o hidráulicos, incluidos los programas informáticos que intervengan en su buen funcionamiento, destinado por el fabricante a ser usado en seres humanos con fines de prevención, diagnóstico, tratamiento o rehabilitación. No constituyen equipo biomédico, aquellos dispositivos médicos implantados en el ser humano o aquellos para un solo uso. EVENTO ADVERSO: daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. EVENTO ADVERSO SERIO: evento no intencionado que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, operador o todo aquel que se vea implicado directa o indirectamente, como consecuencia de la utilización de un dispositivo médico. EVENTO ADVERSO NO SERIO: evento no intencionado, diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, operador o todo aquel que se vea implicado directa o indirectamente, como consecuencia de la utilización de un dispositivo o aparato de uso médico. INCIDENTE ADVERSO: potencial daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. INCIDENTE ADVERSO SERIO: potencial riesgo de daño no intencionado que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. 30 COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 4816 de 2008 (Noviembre 27). por la cual se reglamenta el Programa Nacional de Tecnovigilancia. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2008. [Consultado 20 se septiembre de 2011]. Disponible en internet: http://www.alcaldiabogota.gov.co 31 COLOMBIA, MINISTERIO DE LA PROTECCION SOCIAL. Decreto 4725 de2005 (Diciembre 26). Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano. [En línea]. Bogotá D.C. Ministerio de la Protección Social, 2005. [Consultado Octubre 01 de 2011]. Disponible en internet: http://www.presidencia.gov.co 177 INCIDENTE ADVERSO NO SERIO: potencial riesgo de daño no intencionado diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. RIESGO: posibilidad o probabilidad de que pueda producirse un daño, para el paciente y para el personal que manipula el equipo. REPORTES INMEDIATOS DE TECNOVIGILANCIA: reportes de tecnovigilancia que relacionan un evento adverso serio o un incidente adverso serio con un dispositivo médico en particular. REPORTES PERIÓDICOS DE TECNOVIGILANCIA: conjunto de reportes de tecnovigilancia que relacionan la ocurrencia de eventos adversos no serios e información sobre la seguridad de un dispositivo médico o grupos de dispositivos médicos en un período definido. TECNOVIGILANCIA: es el conjunto de actividades que tienen por objeto la identificación y la cualificación de efectos adversos serios e indeseados producidos por los dispositivos médicos, así como la identificación de los factores de riesgo asociados a estos efectos o características, con base en la notificación, registro y evaluación sistemática de los efectos adversos de los dispositivos médicos, con el fin de determinar la frecuencia, gravedad e incidencia de los mismos para prevenir su aparición. 178 6. CLASIFICACIÓN DE ACUERDO AL RIESGO La clasificación de los dispositivos médicos realizada por el fabricante, se fundamenta en los riesgos potenciales relacionados con el uso y el posible fracaso de los dispositivos con base en la combinación de varios criterios tales como, duración del contacto con el cuerpo, grado de invasión y efecto local contra efecto sistémico. Los dispositivos médicos, según lo establecido en el artículo 5 del Decreto 4725 del 2005 expedido por el Ministerio de la Protección Social, clasifica los dispositivos médicos en 4 categorías según su riesgo: I, IIA, IIB y III, teniendo en cuenta 18 reglas descritas en el artículo 7. Clase I. Son los dispositivos médicos de bajo riesgo, no destinados para mantener la vida y que no signifiquen un riesgo potencial para la salud. Clase IIa. Son los dispositivos médicos de riesgo bajo, sujetos a controles especiales en la fase de fabricación, no destinados para mantener la vida. Clase Ilb. Son los dispositivos médicos de riesgo moderado alto, sujetos a controles especiales en el diseño y fabricación para demostrar su seguridad y efectividad. Clase III. Son los dispositivos médicos de muy alto riesgo, destinados a proteger o mantener la vida para la prevención de la salud humana. 179 7. CLASIFICACIÓN DEL EVENTO E INCIDENTE ADVERSOS (SEGÚN RESOLUCIÓN 4816) Evento adverso serio: Daño no intencionado al paciente u operador o medio ambiente que pudo haber llevado a la muerte o al deterioro serio de la salud. Evento adverso no serio: Daño no intencionado, diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, operador. Incidente adverso serio: Daño no intencionado al paciente u operador, que pudo haber llevado a la muerte o al deterioro serio de la salud pero que por la intervención de un profesional en salud, operario, o persona no genero un desenlace adverso. Incidente adverso no serio: Potencial riesgo de daño no intencionado diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente. Con respecto a la responsabilidad de los actores del nivel local, la resolución 004816 del 2008 según articulo 9 fija los siguientes aspectos a tener en cuenta: Estar vigilantes y alertas del desempeño, calidad y seguridad de los dispositivos médicos previos a su uso. Comunicar, divulgar y aplicar las prácticas adecuadas de utilización de dispositivos médicos. Diseñar e implementar un Programa Institucional de Tecnovigilancia, que permita identificar, registrar, evaluar y gestionar los reportes de eventos e incidentes adversos con los dispositivos médicos. Designar como mínimo un profesional competente en el tema, responsable del Programa Institucional de Tecnovigilancia en la institución que a sus ves será la persona encargada de realizar los reportes ante el Gobierno. Tomar las acciones preventivas o correctivas que sean del caso y las que le sean exigidas por el Instituto Nacional de Medicamentos y Alimentos, INVIMA, de forma inmediata. 180 8. REPORTE DE TECNOVIGILANCIA La Tecnovigilancia realiza la identificación, evaluación y seguimiento permanente de cualquier situación relacionada con el dispositivo médico que pueda llevar a un daño en un paciente, operario u otro; estas situaciones son consideradas como problemas de seguridad, los cuales pueden ser reportados o notificados a la Clínica de Occidente, fabricante, o al INVIMA, sobre le evento o incidente adverso asociado a dispositivos médicos. El formato de reporte es el medio por el cual un reportante notifica a la institución hospitalaria, al fabricante y/o a la autoridad sanitaria, sobre un evento o incidente adverso asociado a un dispositivo médico. El formato de reporte establecido por el Ministerio de la Protección Social mediante el cual se realizan los reportes periódicos está en el anexo A de este manual. El formato de reporte interno esta en el anexo B y C de este manual. ¿Quién reporta? : Todo ciudadano colombiano que sea víctima o testigo, de un evento adverso producido por el uso de la tecnología biomédica. Persona natural o jurídica responsable del diseño, fabricación, acondicionamiento y etiquetado de un dispositivo médico para ser comercializado en su propio nombre o de un titular autorizado, independientemente de que algunas de estas operaciones sean realizadas por esta misma persona o por un tercero contratado. ¿Qué se reporta? : Todo evento o incidente que perjudique o pueda perjudicar la integridad del paciente u operario, cualquiera por insignificante que parezca. ¿A quién se reporta? : Al responsable de la tecnovigilancia de la Clínica de Occidente, el cual se encargará de hacer a su vez el reporte al ente gubernamental o al fabricante según lo amerite el caso. ¿Qué se debe reportar?: Cualquier tipo de evento e incidente que ocurra o pueda ocurrir al interior de la Clínica de Occidente. Está relacionado directamente con tres factores in excluibles: Paciente: Todo aquel evento e incidente que pudiera afectar la seguridad e integridad del paciente, directa o indirectamente; debe ser considerado un riesgo latente para él, y debe reportarse. 181 Dispositivo: Toda aquella falla o interferencia de cualquier dispositivo médico, que pueda interferir con el normal desarrollo del procedimiento, y que pueda afectar finalmente la seguridad e integridad del paciente, debe ser reportado. Entorno: Todo aquel evento relacionado con el entorno del paciente, puede significar un riesgo inminente, ya sea en la red eléctrica, en la red de gases, red de agua, etc. Por eso toda falla presente en el entorno y que pueda afectar, directa o indirectamente la seguridad e integridad del paciente, debe reportarse. Recuerde que es importante nombrar cualquier tipo de evento o incidente por insignificante que parezca su riesgo, o por su poca y casi nula frecuencia de existencia. ¿Cómo realizar el reporte? Identificar el evento o incidente adverso. Ubicar el formato de notificación oficial del INVIMA o el diseñado por la institución. Diligenciar el formato haciendo particular énfasis en los datos del paciente, evento adverso, datos del dispositivo médico y reportante y las acciones tomadas cuando aplique. Entregar el formato de notificación al jefe de cada área El jefe de cada área llamara al encargado del programa de tecnovigilancia que le notificara del evento o incidente adverso. Tomar las medidas pertinentes para mejorar la situación clínica del paciente en caso que la salud de éste haya sido afectada por el evento. ¿Quién analiza el reporte? Una vez identificados los riesgos y notificados, los reportes los analizan el grupo de tecnovigilancia que está conformado por el comité de seguridad al paciente el cual se tratan las posibles causas y los métodos de control a utilizar. Cada responsable de proceso está informado de los incidentes adversos que se presentan en las diferentes áreas de la institución y será responsable junto con el personal técnico de la institución de divulgar y vigilar las medidas de control que se determinaron. Esto es tomado según los artículos 59, 60, 62, 63 y 64 del Decreto 4725 de 2005. 182 9. TIPOS DE REPORTE 9.1 REPORTES INMEDIATOS: Reportes de Tecnovigilancia que relacionan eventos adversos serios asociados a dispositivos médicos. Estos reportes deben realizarse al INVIMA dentro de las siguientes setenta y dos horas (72) horas de tener conocimiento del evento. 9.2 REPORTES PERIÓDICOS: Conjunto de reportes de Tecnovigilancia que relacionan la ocurrencia de eventos e incidentes adversos no serios e información sobre la seguridad de un dispositivo médico o grupos de dispositivos médicos en un periodo definido y se ha realizado un proceso de gestión interna eficiente por parte del reportante. Estos reportes deben ser presentados trimestralmente y en forma consolidada al INVIMA o a las Secretarias Departamentales de Salud según sea el caso, junto con las posibles medidas preventivas tomadas. 9.3 REPORTES DE RETIRO DE PRODUCTOS O LOTES DE PRODUCTO: Reportes de Tecnovigilancia mediante los cuales un fabricante o importador informa a la autoridad sanitaria sobre el retiro de un producto o lote de producto del mercado cuando estos supongan un riesgo para la salud de los pacientes en que van a ser usados. Aunque estos reportes no los debe hacer la clínica, es importante tenerlos en cuenta dado que alguno de estos dispositivos puede hacer parte del inventario de equipos de la clínica. 9.4 REPORTE DE ALERTAS INTERNACIONALES: Reporte de Tecnovigilancia mediante el cual un importador autorizado para comercializar dispositivos médicos en Colombia informa al INVIMA de la generación de una alerta internacional por parte de la casa fabricante en el país de origen o por una agencia sanitaria a nivel mundial en la que se vea involucrado un dispositivo médico comercializado en Colombia. 183 10. INFORMACIÓN QUE DEBE CONTENER LOS REPORTES DE EVENTOS ADVERSOS Los reportes deben contener como mínimo la siguiente información: 1. Datos de identificación de la Institución (sede) donde se generó el evento adverso. Nombre, nivel de atención y Ciudad/ Municipio / Departamento. 2. Datos de identificación del paciente afectado. Edad, sexo, identificación (documento de identidad) 3. Descripción detallada del evento e incidente adverso y su desenlace. Información relevante del evento ocurrido que describa las circunstancias en las que se presentó el evento adverso serio, estado del dispositivo médico, estado del paciente y desenlace final del evento (muerte, daño irreversible o temporal, etc). Para el caso de incidentes adversos se debe describir la situación que pudo haber llevado a un desenlace adverso. 4. Descripción del dispositivo médico asociado al evento adverso. Nombre, marca, modelo, serie o referencia, lote, versión de software, fabricante e importador. 5. Datos del reportante. Nombre, cargo en la institución y datos de ubicación (Esta última información aunque no es de carácter obligatorio es muy importante). 10.1 Confidencialidad de la Información: La información que contenga los diferentes reportes del Programa Institucional de Tecnovigilancia será de total confidencialidad y únicamente será usada con fines de vigilancia sanitaria. La información será de carácter reservado de acuerdo a la Ley 57 de 1985. 184 Cuadro 2. Cuando hacer el reporte TIPO DE REPORTE PROFESIONALES DE LA SALUD EN INSTITUCIONES HOSPITALARIAS FABRICANTE E IMPORTADORES DE DISPOSITIVOS MÉDICOS REPORTES INMEDIATOS Dentro de las 72 horas siguientes a la ocurrencia de eventos o incidentes adversos serios Dentro de las 72 horas siguientes al conocimiento de la ocurrencia de eventos o incidentes adversos serios. REPORTES PERIÓDICOS REPORTES DE RETIRO DE PRODUCTOS TRIMESTRALMENTE NO APLICA Al momento en que el fabricante o importador decida iniciar el retiro del producto en el país. REPORTE DE NO APLICA ALERTAS INTERNACIONALES Dentro de las 72 horas siguientes en que se tuvo conocimiento de la generación de una alerta internacional por parte de la casa fabricante en el país de origen o por una agencia sanitaria a nivel mundial en la que se vea involucrado un dispositivo médico comercializado en Colombia 185 11. ANÁLISIS Y EVALUACIÓN DEL REPORTE Una vez se diligencia el formato de reporte, el grupo responsable del programa de tecnovigilancia de la Clínica de Occidente, debe proceder a analizar si lo que se registra es un evento adverso serio o no serio, y establecer, cómo, donde y cuando ocurrió el evento. En caso de no tener la información correspondiente es necesario que se investigue más a fondo, ya sea en el Comité de Seguridad al Paciente o preguntar al jefe del área donde ocurrió el evento o incidente adversos. Cuando se ha determinado que lo que se reporta sí es un evento adverso, se realiza la evaluación del evento. Existen diversos métodos tales como, causa-raíz, causa-efecto, protocolo de Londres. En este caso se evaluará el reporte mediante el protocolo de Londres, ya que la Clínica de Occidente maneja y utiliza el protocolo de Londres. Su propósito es facilitar la investigación clara y objetiva de los incidentes clínicos, lo cual implica ir mucho más allá de simplemente identificar la falla o de establecer las responsabilidades. Este protocolo se basa en el modelo organizacional de accidente de James Reason como se muestra en la imagen 2. 186 Imagen 2. Modelo organizacional del Protocolo de Londres Fuente: AMAYA, Luengas Sergio. Centro de Gestión Hospitalaria. Centro Médico imbanaco. [en línea] Seguridad del Paciente. Conceptos y análisis de eventos adversos. [Consultado el 12 de Diciembre de 2011]. Disponible en http://www.cgh.org.co/imagenes/calidad1.pdf 187 La investigación y análisis de eventos adversos tiene una secuencia la cual se explica a continuación y se muestra en la siguiente figura 1. Figura 1. Investigación de eventos o incidentes adversos Indentificación y decisión de investigar Establecer cronología del incidente Seleccion del equipo investigador Identificar las acciones inseguras Obtención y organización de información Identificar factores contributivos Recomendacione s y plan de accion Investigar evento adverso (identificación y decisión de investigar): Esta decisión se toma por el impacto y la frecuencia que tiene el evento adverso en la institución. Selección del equipo investigador: Debe integrar al personal asistencial involucrado en el evento, liderado por un experto de investigación y análisis de incidentes clínicos. Experto en investigación y análisis de incidentes clínicos Miembro de junta directiva sin conocimiento médico del caso Autoridad administrativa como director médico o jefe de enfermería Autoridad clínica como jefe de departamento o especialista reconocido Miembro de la unidad asistencial donde ocurrió el incidente El equipo de investigación deberá obtener y organizar toda la información relacionada con el evento adverso: Esta acción recoge todos los hechos, conocimientos y elementos involucrados en el evento como: Historia clínica completa Guías clínicas y protocolos de atención asociados a pacientes con cuadros clínicos. 188 Declaraciones y observaciones inmediatas Entrevista con los involucrados Hojas de vida de los equipos involucrados Otros aspectos relevantes tales como índice de rotación del personal y disponibilidad de personal capacitado Cronología del incidente: Detallar el pasos a paso del incidente con el fin de hallar las causas que lo provocaron, mediante los pasos anteriores (entrevistas, declaraciones, formatos, historia clínica entre otros) se puede establecer qué y cuándo ocurrió. Hay dos metodologías para precisar la cronología la narrativa y el método diagrama. El método que se usará será el método diagrama en el cual los movimientos de personas, materiales, documentos e información pueden representarse mediante un dibujo esquemático. Puede ser útil la secuencia de hechos como deberían haber ocurrido de acuerdo con las políticas, protocolos y procedimientos, y compararla con lo que verdaderamente ocurrió cuando se presentó el incidente. Identificación de acciones inseguras: Estas son las fallas activas en el proceso de atención en salud Identificar las condiciones asociadas con cada acción insegura (factores contributivos): Estos predisponen la aparición de las acciones inseguras. El paciente Tarea y tecnología ej: falta de claridad del protocolo de uso y reusó de mascarillas en urgencias, escases de suministros de servicios, falta de investigación de los síntomas Los individuos ej: toma de decisión sin comunicarle a los jefes de área, tomar una conducta sin realizar exámenes más rigurosos Equipo de trabajo ej: la falta de supervisión de un profesional del servicio El ambiente ej: estrés de servicio de urgencias La organización y gerencia ej: escases de suministros en la organización, políticas de uso y reuso de dispositivos médicos Contexto institucional Recomendaciones y planes de acción. Diseño de planes de acción que ayuden a prevenir o mitigar los eventos adversos. Definir políticas del uso de adquisición biomédica. Revisar y ajustar el proceso de adquisición de equipos biomédicos. Evaluar la adherencia del personal. Establecer las necesidades de insumos en la Clínica de Occidente. 189 Sensibilizar al personal sobre la importancia de la utilización de equipos biomédicos. Gestión y calificación del riesgo de la atención en salud en equipos biomédicos. Diseño de guías rápidas de los equipos biomédicos. Capacitación al personal asistencial. 190 12. DISEÑO DE ESTRATEGIAS DE VIGILANCIA DE EQUIPOS BIOMÉDICOS Con el objetivo de disminuir la generación de eventos e incidentes adversos se implementaron estrategias de vigilancia, chequeo visual, mantenimiento preventivo, mantenimientos predictivos, metrología, listas de chequeo o check list en las Unidades de Cuidados Intensivos, Cirugía y Hospitalización, urgencias, rayos-x, laboratorio, etc. Estas actividades se implementaron tres veces a la semana donde el analista de electromedicina registró la información y documentó el funcionamiento del equipo. Al momento de encontrar fallas en equipos biomédicos, las cuales también se pueden generar por condiciones del área, se notifica la situación al funcionario a cargo del área, que a su vez notifica al coordinador de electromedicina para su conocimiento y acciones pertinentes. La realización de reuniones mensuales del comité de tecnovigilancia involucrado en el comité de seguridad al paciente donde asisten los Coordinadores de Electromedicina, UCI‟s, salas de Cirugía, rayos-x, urgencias, laboratorio, partos, hospitalización. Con los datos e información obtenida a través de las alertas, se deben generar planes de acción para los eventos adversos más comunes, de esta manera ir organizando una base de datos con planes de acción y de prevención aplicables para que en caso de que el evento adverso relacionado ocurra en la clínica, estén claras las medidas que se deben tomar. Además se deben implementar los planes de prevención cuando las alertas están relacionadas a equipos con los que cuenta la institución. El diseño de barreras es importante, debido a que mediante estas es posible la prevención de algún evento o incidente adverso y actuar de manera oportuna cuando este suceda y disminuir al máximo el daño al paciente. Para el diseño de las barreras es necesario realizarla por medio de matrices de riesgo las cuales los priorizan según la ocurrencia en cada uno de los equipos biomédicos generando una base para el diseño de las barreras, se debe hacer énfasis principalmente en los entornos donde se encuentra la tecnología biomédica, los focos de peligro de los equipos biomédicos y los riesgo que pueden ocurrir con estos equipos. Los pasos a seguir para realizar la matriz de riesgo se muestran en la figura 2. 191 Figura 2. Pasos para el diseño de una matriz de riesgo SELECCION DEL AREA LISTA DE LOS EQUIPOS BIOMEDICOS DE ESTA AREA IDENTIFICAR LOS RIESGOS DISTRIBUCION DE LOS RIESGOS IDENTIFICADOS EN LA MATRIZ IDENTIFICAR LAS VARIABLES DE LA MATRIZ La norma NTC 5254 aporta un marco genérico para establecer el contexto y los elementos del proceso de gestión del riesgo, sin embargo, es presentada como una propuesta que puede ser adaptada según las necesidades de cada establecimiento; pues, de acuerdo con la misma, el diseño e implementación del sistema de gestión del riesgo se verá influenciado por las necesidades variables de una organización, sus objetivos particulares, sus productos y servicios y los procesos y practicas especificas empleadas. En consecuencia con lo anterior, en la Clínica de Occidente se estudió esta propuesta y otras surgidas del proceso de acreditación para consolidar la matriz institucional de riesgo según el contexto institucional. La matriz de riesgo de la Clínica de Occidente presenta, entonces, unas probabilidades y unas categorías de severidad del evento. En este caso, el análisis del riesgo se realiza a través de un análisis cualitativo que emplea palabras y escalas descriptivas que permiten identificar aspectos como las consecuencias, potencialidades y posibilidades de que esto ocurra. Para el caso de las probabilidades, las cataloga en frecuente, ocasional, remoto, improbable y extremadamente improbable; esta probabilidad se valora de 1 a 5 en donde 5 es una probabilidad frecuente y 1 es extremadamente improbable, como se observa en el cuadro 3. 192 Cuadro 3. Medida cualitativa de la probabilidad del evento PROBABILIDAD DEL EVENTO DEFINICIÓN CUALITATIVA FRECUENCIA OCASIONAL REMOTO IMPROBABLE EXTREMADAMENTE IMPROBABLE SIGNIFICADO Probable que ocurra muchas veces (ha ocurrido frecuentemente) Probable que ocurra algunas veces (ha ocurrido infrecuentemente) Improbable, pero es posible que ocurra (ocurre raramente) Muy improbable que ocurra (no se conoce que haya ocurrido) Casi inconcebible que el evento ocurra VALOR 5 4 3 2 1 Para el caso de las categorías de severidad, se presentan unos valores de la A a la E en donde A es un evento catastrófico y E es un evento insignificante, como se observa en el cuadro 4. Cuadro 4. Medida cualitativa de la severidad del evento SEVERIDAD DEL EVENTO DEFINICIÓN SIGNIFICADO Muertes múltiples, Muerte del Paciente, invalidez de CATASTRÓFICO más de 50%, Destrucción o daños severos en infraestructura o equipos VALOR A Lesiones o secuelas permanentes PELIGROSO MAYOR Daños mayores a pacientes Equipos o infraestructura Una reducción en la habilidad del operador en responder a condiciones operativas adversas como resultado del incremento de la carga de trabajo, o como resultado de condiciones que impiden su eficiencia B C Incidente serio Lesiones temporales a las personas Paciente insatisfecho MENOR Aumento de trámites administrativos D Incidentes menores INSIGNIFICANTE Consecuencias leves E 193 Con base en lo anterior, se diseñó entonces la matriz que se observa en el cuadro 5 en donde se confronta la probabilidad y la severidad del evento y se asignan los valores correspondientes para cada caso. Cuadro 5. Matriz de riesgo de la Clínica de Occidente PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante 5A 5B 5C 5D 5E 4A 4B 4C 4D 4E 3A 3B 3C 3D 3E 2A 2B 2C 2D 2E 1A 1B 1C 1D 1E Fuente. Clínica de Occidente Esta matriz, permite catalogar tres tipos de riesgo identificados como riesgo extremo, medio y bajo, los cuales se explican a continuación: RIESGO EXTREMO: Cuando la situación requiere acción inmediata. La dirección debe conocer esta situación y deben implementarse acciones correctivas urgentes. Si ocurre el Evento Adverso o Incidente, el mismo debe ser reportado dentro de las 72 horas y se deben analizar causas raíces. RIESGO MEDIO: Cuando la situación requiere acciones lo antes posible y no más allá de los 6 meses. La alta dirección y el director médico deben conocer esta situación. RIESGO BAJO: Manejo por procedimientos de rutina. Implementar medidas fáciles y rápidas y soluciones de fondo cuando los recursos lo permitan. A continuación se presentan cuatro ejemplos relacionados con los riesgos que pueden ocasionar algunos equipos biomédicos disponibles en la Clínica de Occidente, detallando el área, el equipo y las fuentes de peligro. Estas fuentes de peligro fueron tomadas de casos reales presentados en la Clínica de Occidente los cuales fueron identificados y por lo tanto se considera pertinente traerlos a colación en este documento. 194 Para diseñar los ejemplos que se presentan a continuación, se seleccionaron dos casos del área de cirugía y otros dos casos de las áreas de UCI´s ya que son las áreas que presenta mayor riesgo dentro de la institución. EJEMPLO 1 ÁREA: cirugía EQUIPOS: máquina de anestesia, desfibrilador, monitor de signos vitales, lámpara cielítica, máquina de circulación extracorpórea, electrobisturí, entre otros. EQUIPO SELECCIONADO: Máquina de anestesia FUENTE DE PELIGRO DE LA MÁQUINA DE ANESTESIA: Válvula de inspiración dañada. Circuito de respiración en mal estado. Monitor dañado no se registran parámetros. Alarma de sensor de oxigeno deshabilitado. Vaporizadores mal ajustados. Fuga en absorvedor. DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS A LA MÁQUINA DE ANESTESIA: A continuación, se presentan algunos casos que pueden generar riesgos asociados a la máquina de anestesia, los cuales son numerados del 1 al 8; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Válvula de inspiración dañada: Al tener esta válvula dañada o en mal estado el absorvedor de la máquina de anestesia no realiza su función quedando pegado y no envía flujo al paciente provocando la muerte. Caso 2 (C2). Circuito de respiración en mal estado: El paciente se despierta en medio de la cirugía ya que hay fuga en la manguera faltando gas anestésico al paciente. Caso 3 (C3). Al tener una fuga en el circuito el anestesiólogo se ve obligado a suministrar más oxigeno o aire y gas anestésico provocando una recuperación prolongada. Caso 4 (C4). Monitor dañado no se registran parámetros: El anestesiólogo no identifica en la monitoria de la máquina de anestesia los flujos de aire, oxigeno, entregados al paciente provocando daños cerebrales. 195 Caso 5 (C5). No se registran señales de alerta de apnea o de concentración de flujo, alta presión de flujo de aire. Caso 6 (C6). Alarma de sensor de oxigeno deshabilitada: No se registra la alarma de para baja y alta concentración de flujo. Caso 7 (C7). Vaporizadores mal ajustados: No entregan el suficiente gas anestésico al paciente provocando que el paciente se despierte en medio de la cirugía. Caso 8 (C8). Fuga en el absorvedor: Al tener esta fuga hay que suministrarle más aire o oxígeno al paciente y por ende más gas anestésico. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. Cuadro 6. Matriz de riesgo máquina de anestesia PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C2 C3 C7 C6 C8 C4-C5-C1 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar barreras para los casos 2, 3, 5 7 y 8 Es indispensable realizar un chequeo diario del circuito paciente antes de que inicie una cirugía debido a que puede ocasionar daños al paciente mientras se encuentra anestesiado. ACCIONES INMEDIATAS: Realizar un chequeo en las salas de cirugía diariamente en las horas de la mañana antes que inicien las cirugías. No esterilizar más de cinco veces los circuitos pacientes 196 Verificación del buen estado del equipo antes de las cirugías preferiblemente por las mañanas Tener a la mano el repuesto de lo que está fallando u otro circuito respiratorio. EJEMPLO 2 EQUIPO SELECCIONADO: Desfibrilador FUENTE DE PELIGRO DEL DESFIBRILADOR: Daño en el botón de descarga Entrega de energía más alta de lo que se indica Batería dañada Equipo desconectado DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS AL DESFIBRILADOR A continuación, se presentan algunos casos que pueden generar riesgos asociados al desfibrilador, los cuales son numerados del 1 al 5; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Daño en el botón de descarga: No se puede hacer la descarga al paciente ya que está dañado el botón provocando la muerte al paciente ó daño mientras se consigue otro equipo de urgencia. Caso 2 (C2). Entrega de energía más alta de los que se indica: Se puede producir quemaduras al paciente. Caso 3. (C3). Descarga no adecuada para patología del paciente. Caso 4 (C4). Batería dañada: No se puede hacer la carga ni la descarga al paciente provocando la muerte o daño. Caso 5. (C5). Equipo desconectado: No se visualiza el led de carga del desfibrilador confiándose que se encuentra conectado a la red eléctrica, al utilizarlo no prende el equipo y no se puede hacer la descarga al paciente. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 197 Cuadro 7. Matriz de riesgo desfibrilador PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C4-C5 C2 C3 C1 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 4, 5, 2 y 3. Es indispensable realizar un cheque diariamente del estado del desfibrilador cerciorarse que se estén realizando las descargas y verificar que este encendido el led de carga. ACCIONES INMEDIATAS: Realizar un chequeo diario del funcionamiento del desfibrilador cerciorarse que se esté realizando las descargas. Realizar todos los días el test interno que tiene el desfibrilador y llevar un reporte del test. Limpiar las palas cada vez que se usan para evitar que haya material que este generando resistencia o baja impedancia y no permita entregar la energía necesaria. Verificar que se encuentre conectado a la red. 198 EJEMPLO 3 ÁREA: UCI‟s EQUIPOS: desfibrilador, monitor de signos electrocardiografos, ventiladores, entre otros. vitales multiparametricos, EQUIPO SELECCIONADO: ventilador FUENTE DE PELIGRO DE UN VENTILADOR: Filtro espiratorio con fugas. Circuito paciente con fugas. Alarma no audibles. Falla en la batería. Desajuste del peep DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS AL VENTILADOR: A continuación, se presentan algunos casos que pueden generar riesgos asociados al ventilador, los cuales son numerados del 1 al 4; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Filtro espiratorio con fugas: Al tener fugas no hay una buena compensación del paciente y se ve obligado el fisioterapeuta a dar respiración manual “ambu” al paciente. Caso 2 (C2). Circuito paciente con fuga: Al tener una fuga en el circuito el fisioterapeuta se ve obligado a suministrar 100% de oxígeno al paciente provocando lesiones pulmonares. Caso 3 (C3). Desajuste del peep: Puede producir lesiones pulmonares por alta presión en las vías respiratorias. Caso 4 (C4). Falla en la batería: Paciente se puede morir si no hay planta de emergencia eléctrica. Cada uno de estos casos se evalúa mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 199 Cuadro 8. Matriz de riesgo ventilador PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C1-C4 C2 C3 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 1 y 4. Es indispensable realizar la metrología de estos equipos ya que son soporte de vida, hay que cerciorarse que los circuitos no se esterilicen más de cinco veces, realizar un chequeo visual de los accesorios (filtros inspiratorios, espiratorio, circuitos). ACCIONES INMEDIATAS: Antes de que llegue un paciente a la UCI‟s se pone a ciclar el ventilador por 30 minutos. Verificar el estado de los filtros, cerciorarse que están en buen estado que no estén partidos. Algunos ventiladores tiene un test interno donde se realizan pruebas de compensación de fugas, hay que realizar este test cada vez que cambian de paciente. Verificar que se encuentre conectado a la red. 200 EJEMPLO 4 EQUIPO SELECCIONADO: monitor de signos vitales multiparamétrico FUENTE DE PELIGRO DE UN MONITOR: Bomba de presión arterial en mal estado Cable de saturación de oxígeno en mal estado Calibración de presión invasiva desajustada Batería interna en mal estado Alarmas audibles y visibles en ,mal estado DESCRIPCIÓN DE LOS RIESGOS ASOCIADOS A LOS MONITORES: A continuación, se presentan algunos casos que pueden generar riesgos asociados a los monitores, los cuales son numerados del 1 al 5; sin que esta numeración tenga correspondencia en la evaluación del riesgo. Caso 1 (C1). Bomba de presión arterial en mal estado: Toma de presiones erróneas no acordes a la patología del paciente suministrando medicamentos innecesarios, provocando la muerte, o prolongando la estadía. Caso 2 (C2). Cable de saturación de oxígeno en mal estado: No se registra la saturación de oxigeno del paciente ya que el accesorio está mal ubicado o se encuentra malo, dando un mal diagnóstico a la enfermera o a la fisioterapeuta si hay que suministrar más oxígeno al paciente. Caso 3 (C3). Calibración de presión invasiva desajustada: Toma de presiones erróneas no acordes a la patología del paciente suministrando medicamentos innecesarios, provocando la muerte, estadía prolongada. Caso 4 (C4). Batería interna en mal estado: Paciente se puede morir si no hay planta de emergencia eléctrica. Caso 5 (C5). Alarmas audibles y visibles en mal estado: Al tener las alarmas en nivel bajo o dañado no se podría registrar los cambios fisiológicos del paciente provocando desenlaces graves como la muerte o deterioro de la salud. Cada uno de estos casos se evalúan mediante el cruce de la probabilidad y la severidad y se ubican en la matriz, lo que arroja el nivel de riesgo de cada caso, según el color, tal como se observa a continuación. 201 Cuadro 9. Matriz de riesgo Monitor de signos vitales PROBABILIDAD DEL EVENTO Frecuente Ocasional Remoto Improbable Extremadamente improbable SEVERIDAD DEL RIESGO Catastrófico Peligroso Mayor Menor Insignificante C1 C4-C2 C5 C3 Teniendo en cuenta los resultados de la matriz de riesgo se deben implementar las siguientes barreras para los casos 1, 4, 2 y 5. Es indispensable realizar la metrología de estos equipos ya que son soporte de vida, hay que cerciorarse que los accesorios de los equipos estén funcionando correctamente, realizar un chequeo visual de los accesorios (cable de ECG, saturador de oxígeno, brazalete, cable de invasiva). ACCIONES INMEDIATAS: Antes de que llegue un paciente a la UCI‟s se verifica que los accesorios estén correctamente funcional. Realizar un chequeo visual y funcional de los accesorios. Realizar la metrología de los equipos biomédicos. Verificar que se encuentre conectado a la red el equipo. A nivel internacional existen organizaciones encargadas de recopilar y brindar alertas de casos ocurridos en dispositivos biomédicos. Una de estas agencias es la FDA (Food and Drugs Administration) que presenta unos antecedentes importantes de alertas tempranas donde se reportan regularmente aquellos incidentes con dispositivos biomédicos ocurridos en Estados Unidos Teniendo en cuenta que muchos de los equipos manejados en este país son similares a los de Estados Unidos, es necesario considerar esta información pues permite hacer un monitoreo permanente para estar enterados de los casos ocurridos y de esta manera revisar si hay coincidencias con eventos presentados por equipos médicos que se manejen en la institución. Esto genero la necesidad de que en la Clínica se maneje esta información adecuadamente para prevenir estos sucesos. 202 12.1 Seguimiento a acciones y responsables. Para todos los casos, el seguimiento de los planes de acción está a cargo del área de electromedicina que es el área responsable de llevar a cabo estas acciones inmediatas. El seguimiento se hace por medio de un formato de chequeo que se aplica semanalmente para el área de las UCI´s y diario para el área de cirugía. Basados en la matriz de riesgo como se mostró en los ejemplos anteriores, las barreras se diseñan al identificar los riesgos que presentan los equipos biomédicos, estas barreras se deben implementar para evitar la ocurrencia de los eventos o para actuar de manera oportuna cuando ocurra un evento o incidente adverso. Estas barreras aplican para todos los equipos. A continuación en el cuadro 20 se presentan las barreras. Cuadro 10. Barreras BARRERAS RECURSO HUMANO INFRAESTRUCTURA CAPACITACIÓN RED ELÉCTRICA VERIFICACIÓN DE LA IDONEIDAD DEL RED DE GASES PERSONAL GESTIÓN MANTENIMIENTO LISTA DE CHEQUEO INDICADORES CALIDAD METROLOGÍA DE RECURSO HUMANO Capacitación al personal: Se realiza capacitación y evaluación al personal asistencial de la clínica cada vez que se requiera o se vean falencias en algunas áreas; estas falencias son vistas en los llamados inoficiosos o en procedimientos, se realiza cada mes una capacitación en el auditorio a todo el personal donde se explica el buen manejo de los equipos biomédicos. Verificación de la idoneidad del personal: Verificar por el área de recurso humano las hojas de vida del personal que va a ingresar a la Clínica de Occidente que cumpla con los requerimientos establecidos por las normas reglamentarias de la empresa. 203 INFRAESTRUCTURA Red eléctrica: La clínica cuenta con un sistema de contingencia de dos plantas eléctricas de emergencia en caso que falle el servicio eléctrico. Estas plantas de energía suministran energía a las áreas más críticas como las UCI´s, cirugía, rayos-x, alarmas contra incendio, e iluminación, tal como lo exige la norma NTC2050 en la sección 517. Red de gases: La clínica cuenta con una planta de generación de oxígeno, aire, vacío para el suministro a la Clínica de Occidente. Se cuenta con sistemas de alarma de ausencia de gases en cada área crítica como las UCI‟s, Cirugía, Rayos-x, etc. Para el oxígeno se cuenta con un sistema de contingencia de un tanque de criogénico de 5 toneladas y un sistema manual con cilindros de oxigeno que respalda el suministro de la planta, para el sistema de aire se cuenta solamente con la técnica manual. Para el sistema de vacío se cuenta con dos compresores para el suministro a la clínica, además hay una planta de emergencia para suplir la energía al sistema de vacío en caso de fallo del suministro eléctrico. GESTIÓN Plan de mantenimiento preventivo: A los equipos biomédicos que se encuentran en la Clínica de Occidente se les realiza cada 6 meses un mantenimiento preventivo, de acuerdo a la criticidad de los equipos, de su uso constante y lugar donde esté ubicado el equipo, los informes quedan registrados en las hojas de vida de cada equipo. Plan de mantenimiento predictivo: El mantenimiento predictivo se realiza todos los días en las salas de Cirugía a las 6:30 am antes de iniciar procedimientos, en las Unidades de Cuidados Intensivos se realiza cada viernes. Aseguramiento metrológico: Se realiza cada año con una entidad externa, se utilizan equipos patrones para realizar la metrología, los informes quedan registrados en las hojas de vida de cada equipo. En la clínica de Occidente, se realiza la calibración posterior al mantenimiento preventivo. 204 Acompañamientos en procedimientos quirúrgicos: Se realiza este acompañamiento cada vez que un médico, o jefe lo requiera, si es para el manejo de algún equipo en especial como los siguientes: Equipo de Laser, Microscopio, Shaver, entre otros. Indicadores de calidad: Verificar por medio de indicadores el cumplimiento de los mantenimientos preventivos, correctivos, de los equipos biomédicos de la Clínica de Occidente. 205 13 PROPUESTA PARA LA EVALUACIÓN DEL CUMPLIMIENTO DE ACTIVIDADES Como complemento al proyecto, a continuación se propone que el Programa Institucional de Tecnovigilancia que cuente con un sistema de seguimiento y evaluación de los eventos o incidentes adversos ocasionados por equipos biomédicos. Cuadro 11. Propuesta para la evaluación básica de actividades del Programa Institucional de Tecnovigilancia de la Clínica de Occidente ACTIVIDAD RESPONSABLE CRITERIO Estar atentos y Área de Manteamiento preventivo y vigilantes del Electromedicina, correctivo a equipos biomédicos. desempeño, calidad y usuarios de Metrología a equipos biomédicos. seguridad de los dispositivos Vigilancia de alertas equipos biomédicos. médicos, internacionales Coordinador de la Guías rápidas de los equipos UCI‟s, todo biomédicos. personal Visitas diarias a los diferentes asistencial del niveles de la Clínica de Occidente. cuerpo médico. Evaluación de tecnología. Renovación de tecnología. Hojas de vidas actualizadas. Capacitación al personal del manejo de los equipos biomédicos. Chequeo a los dispositivos biomédicos en el área de Cirugía. Informar, divulgar, y Área de Capacitación al personal del aplicar las practicas Electromedicina, manejo de los equipos biomédicos. adecuadas de usuarios de Elaboración de guías rápidas para utilización de los dispositivos el manejo de los equipos dispositivos biomédicos médicos, biomédicos. Coordinador de la Elaboración de folletos de UCI‟s, todo tecnovigilancia. personal asistencial del cuerpo médico. Diseñar e implementar Coordinador de Formato de reporte de eventos e un Programa Electromedicina incidentes adversos. Institucional de Comité de seguridad al paciente Tecnovigilancia, que asegure un permanente seguimiento de los 206 eventos e incidentes adversos que puedan causar los dispositivos médicos durante su uso y que permitan identificar, registrar, evaluar y gestionar los reportes de eventos e incidentes adversos con los dispositivos médicos que use. Designar como mínimo un profesional competente en el tema, responsable del Programa Institucional de Tecnovigilancia quien también será el corresponsal ante el gobierno. Tomar las acciones preventivas o correctivas que sean del caso y las que le sean exigidas por el Instituto Nacional de Medicamentos y Alimentos, INVIMA, de forma inmediata. Comunicar al fabricante o importador del dispositivo médico correspondiente, la ocurrencia del evento o incidente adverso, si se estima pertinente. Comunicar al Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA, o a las Secretarías Departamentales y Distritales de Salud, la ocurrencia de eventos e Coordinador de Persona encargada del Programa Electromedicina Institucional de Tecnovigilancia. Coordinador de Revisión periódica a los Electromedicina. comunicados expedidos por el Comité de INVIMA. Seguridad al Paciente. Coordinador de Formato de eventos e incidentes Electromedicina. adversos para los fabricantes o importadores Comunicación al fabricante o importador Coordinador de Reporte al INVIMA de reporte se Electromedicina. eventos e incidentes adversos. 207 incidentes adversos, en los términos de la Resolución 4816 de 2008. Desarrollar actividades de promoción y formación con los profesionales de la salud de la institución, en relación al desarrollo e implementación del Programa Nacional de Tecnovigilancia y la gestión de eventos o incidentes adversos con dispositivos médicos. Coordinador de Actividades de promoción y Electromedicina. formación en relación al Programa Integrantes del Institucional de Tecnovigilancia. Comité de Seguridad al Paciente. Oficina de Comunicaciones 208 BIBLIOGRAFÍA COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Decreto 4725 de 2005. Por el cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de los dispositivos médicos para uso humano [en línea] Bogotá DC. Ministerio de la protección social, 2005. [Consultado 25 de Agosto de 2010]. Disponible en Internet: http://www.presidencia.gov.co/prensa_new/decretoslinea/2005/diciembre/26/dec47 25261205.pdf COLOMBIA, MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 4816 de 2008 (noviembre 27). Por la cual reglamenta el Programa Nacional de Tecnovigilancia. [en línea] Bogotá DC.: Ministerio de la Protección Social, 2008. [Consultado 25 de Agosto de 2010]. Disponible en Internet: http://web.invima.gov.co/portal/documents/portal/documents/root//resolucion_0048 16_nov2008.pdf VINCENT, Charles; TAYLOR – ADAMS, Sally.Protocolo de Londres [en línea] Londres, Clinical Safety ResearchUnit, Imperial College. [Consultado 13 de Enero de 2011]. Disponible en Internet: https://mailattachment.googleusercontent.com/attachment?ui=2&ik=4b64b1fd40&view=att&th =12ab073ca83b1c4b&attid=0.11&disp=inline&safe=1&zw&saduie=AG9B_P_bDwJ pMg1OOPYdH3lNpNC0&sadet=1301586834752&sads=3Bize4HCvspQzOl9yJGzd OxujV8&sadssc=1 209 ANEXOS ANEXO A. Formato de reporte establecido por el Ministerio de la Protección Social 210 ANEXO B. Formato para el reporte del visitante Nº de Consecutivo Fecha de actualizacion Version Pagina Formato Visitante CLÍNICA DE OCCIDENTE ¡ Cada día Mejor ! FORMATO DE REPORTE DE EVENTO O INCIDENTE ADVERSO DE DISPOSITIVOS BIOMEDICOS 1. INSTITUCION REPORTANTE Fecha: Dia Hora: Mes Año 2. IDENTIFICACION DEL PACIENTE Nombre del paciente: Numero de Historia Clinica: Edad: Sexo: M F Ubicación del paciente: Cama Nº : Entidad: 3. DESCRIPCION DEL EVENTO ADVERSO EL EVENTO FUE A Paciente: Acompañante: Visitante: Otro: Cual: DESCRIPCION DEL EVENTO INFORMACION QUE CONSIDERE IMPORTANTE USTED PARA ESTE REPORTE NOTA: Si es necesario adicione hojas 211 ANEXO C. Formato para el reporte del empleado Nº de Consecutivo Fecha de actualizacion Version Pagina Formato Empleado CLÍNICA DE OCCIDENTE ¡ Cada día Mejor ! FORMATO DE REPORTE DE EVENTO O INCIDENTE ADVERSO DE DISPOSITIVOS BIOMEDICOS 1. INSTITUCION REPORTANTE Fecha: Dia Mes Año Hora: AREA REPORTANTE 3. INFORMACION DEL DISPOSITIVO MEDICO NOMBRE DEL EQUIPO: SERIE: MODELO: ACTIVO: UBICACIÓN: SI SE REPORTO AL FABRICANTE: NO Cirugia: Sala: 2. IDENTIFICACION DEL PACIENTE Nombre del paciente: Numero de Historia Clinica: Edad: Sexo: M F EL EVENTO FUE A Paciente: Acompañante: Visitante: Otro: 4. DESCRIPCION DEL EVENTO ADVERSO DESCRIPCION DEL EVENTO O INCIDENTE ADVERSO Tipo de reporte Fecha del evento o incidente adverso Primera vez Seguimiento ¿SE RESOLVIÓ EL PROBLEMA? SI [ ] NO [ ] Medidas que se tomaron: SEÑALE SEGÚN EL(LOS) DESENLACE(S) QUE APLIQUE(N) [ ] No hubo consecuencia(s) [ ] Daño de una Función o Estructura Corporal [ ] Enfermedad o Daño que Amenace la Vida [ ] Hospitalización: Inicial o Prolongada [ ] Intervención Médica o Quirúrgica [ ] Muerte [ ] Otros: ¿Cuál? DESCRIPCION DEL EVENTO ADVERSO INCIDENTE ADVERSO NOTA: Si es necesario adicione hojas 212