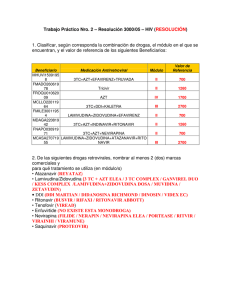
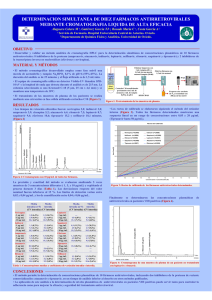
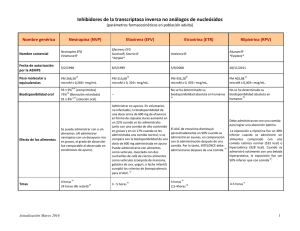
Viramune, INN-Nevirapine
Anuncio

RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO 1/34 1. DENOMINACIÓN DEL MEDICAMENTO VIRAMUNE 200 mg comprimidos 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Cada comprimido contiene 200 mg de nevirapina anhidra (principio activo). Lista de excipientes, en 6.1. 3. FORMA FARMACÉUTICA Comprimido VIRAMUNE 200 mg comprimidos son comprimidos blancos, ovalados y biconvexos. Una cara tiene grabado el código “54 193”, con una única ranura que separa el “54” y el “193”. La cara opuesta está marcada con el símbolo de la compañía. 4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas VIRAMUNE está indicado como parte de la terapia combinada para el tratamiento antiviral de los pacientes infectados por VIH-1 con inmunodeficiencia avanzada o progresiva. La mayor parte de la experiencia con VIRAMUNE es en combinación con inhibidores nucleósidos de la transcriptasa inversa. No se dispone, en la actualidad, de datos suficientes sobre la eficacia del uso de la triple combinación incluyendo inhibidores de la proteasa, después del tratamiento con VIRAMUNE. Ver apartado 5.1 Propiedades farmacodinámicas. 4.2 Posología y forma de administración Pacientes mayores de 16 años La dosis recomendada de VIRAMUNE es de un comprimido diario de 200 mg durante los primeros 14 días (es preciso seguir este período inicial, ya que se ha demostrado que reduce la frecuencia de exantema), seguido de un comprimido de 200 mg dos veces al día, en asociación con al menos dos agentes antirretrovirales adicionales a los cuales el paciente no haya sido expuesto previamente. Los virus resistentes aparecen rápida y uniformemente cuando VIRAMUNE se administra como monoterapia; por lo tanto, VIRAMUNE siempre debe administrarse en terapia combinada. Por lo que respecta a la terapia antirretroviral concomitante, deben seguirse la dosificación y monitorización recomendadas. Pacientes pediátricos (adolescentes) Los comprimidos de 200 mg de VIRAMUNE, siguiendo el esquema de dosificación anteriormente descrito, son adecuados para niños mayores, particularmente adolescentes, menores de 16 años, de peso igual o superior a 50 kg. Para niños de este grupo de edad, de peso inferior a 50 kg está disponible una forma de dosificación en suspensión oral, que puede dosificarse según el peso corporal (ver Ficha Técnica de VIRAMUNE suspensión oral). 2/34 Consideraciones para el manejo de la dosis Antes de iniciar el tratamiento con VIRAMUNE y a intervalos adecuados durante el mismo, deben realizarse pruebas químico-clínicas, entre las que se incluyan pruebas de la función hepática. Por lo que respecta a la toxicidad que requiera la interrupción del tratamiento con VIRAMUNE, ver apartado 4.4 Advertencias y precauciones especiales de empleo. Los pacientes que presenten exantema durante el período inicial de 14 días con 200 mg/día, no deben aumentar su dosis de VIRAMUNE hasta que se haya resuelto el exantema. El exantema aislado debe ser estrechamente monitorizado (ver apartado 4.4. Advertencias y precauciones especiales de empleo). Los pacientes que interrumpan la administración de VIRAMUNE durante más de 7 días, deben reiniciar el tratamiento con la dosis inicial recomendada, tomando un comprimido diario de 200 mg durante los primeros 14 días, seguido de un comprimido de 200 mg dos veces al día. VIRAMUNE debe ser administrado por médicos expertos en el tratamiento de la infección por VIH. 4.3 Contraindicaciones Hipersensibilidad al principio activo o a alguno de los excipientes. VIRAMUNE no debe readministrarse a pacientes que hayan requerido una interrupción permanente por exantema grave, exantema acompañado de sintomatología general, reacciones de hipersensibilidad o hepatitis clínica debida a nevirapina. VIRAMUNE no debe administrarse a pacientes con daño hepático grave. VIRAMUNE no debe readministrarse en pacientes que hayan presentado anteriormente valores de SGOT o SGPT > 5 veces el LSN durante el tratamiento con VIRAMUNE y tuvieran una rápida recurrencia de las anomalías de la función hepática durante la readministración de VIRAMUNE, (ver apartado 4.4 Advertencias y precauciones especiales de empleo). Los preparados a base de plantas medicinales que contengan Hipérico (Hypericum perforatum) no deben ser utilizados durante la administración de VIRAMUNE debido al riesgo de disminución de las concentraciones plasmáticas y del efecto clínico de la nevirapina (ver apartado 4.5 Interacciones con otros medicamentos y otras formas de interacción). Los datos farmacocinéticos disponibles sugieren que el uso concomitante de la rifampicina y VIRAMUNE no está recomendado (ver apartado 4.5 Interacciones con otros medicamentos y otras formas de interacción). 4.4 Advertencias y precauciones especiales de empleo En base a los datos farmacodinámicos VIRAMUNE sólo debe utilizarse en asociación con otros dos agentes antirretrovirales como mínimo (ver apartado 5.1 Propiedades farmacodinámicas). Las primeras 8 semanas de tratamiento con VIRAMUNE constituyen un periodo crítico, que requiere una monitorización estrecha de los pacientes para revelar la potencial aparición de reacciones cutáneas graves y que supongan un riesgo para la vida (incluyendo casos de síndrome de Stevens-Johnson y necrolisis epidérmica tóxica) o hepatitis grave/insuficiencia hepática. Además, debe respetarse estrictamente la pauta de dosificación, especialmente durante los 14 días del período inicial (ver apartado 4.2 Posología y forma de administración). 3/34 Reacciones cutáneas Se han producido reacciones cutáneas graves y que suponen un riesgo para la vida, incluyendo casos fatales, en pacientes tratados con VIRAMUNE, principalmente durante las primeras 6 semanas de tratamiento. Estas han incluido casos de síndrome de Stevens-Johnson, necrolisis epidérmica tóxica y reacciones de hipersensibilidad, caracterizadas por exantema, sintomatología general y afectación visceral. Los pacientes deben ser estrechamente monitorizados durante las primeras 8 semanas de tratamiento. Los pacientes deben ser estrechamente monitorizados si se produce un exantema aislado. VIRAMUNE debe interrumpirse permanentemente en pacientes que presenten exantema grave o exantema acompañado de sintomatología general (como fiebre, formación de vesículas, lesiones orales, conjuntivitis, hinchazón, dolores musculares o articulares, o malestar general), incluyendo síndrome de Stevens-Johnson o necrolisis epidérmica tóxica. VIRAMUNE debe interrumpirse permanentemente en cualquier paciente que presente reacciones de hipersensibilidad (caracterizadas por exantema con sintomatología general además de afectación visceral, tal como hepatitis, eosinofilia, granulocitopenia y disfunción renal) ver apartado 4.4 Advertencias y precauciones especiales de empleo. La administración de VIRAMUNE por encima de la dosis recomendada puede incrementar la frecuencia y la gravedad de las reacciones cutáneas, tales como el síndrome de Stevens-Johnson y la necrolisis epidérmica tóxica. La utilización simultánea de prednisona (40 mg/día durante los primeros 14 días de administración de VIRAMUNE) ha demostrado no disminuir la incidencia del exantema asociado de VIRAMUNE y puede asociarse con un aumento en la incidencia y gravedad del exantema durante las primeras 6 semanas de tratamiento con VIRAMUNE. Se han identificado algunos factores de riesgo en el desarrollo de reacciones cutáneas graves, que incluyen errores en el seguimiento de la pauta de dosificación inicial de 200 mg diarios durante el período inicial y un retraso prolongado entre la aparición de los síntomas iniciales y la consulta al médico. Debe indicarse a los pacientes que la principal toxicidad de VIRAMUNE es el exantema. Debe aconsejárseles que comuniquen de inmediato a su médico la aparición de cualquier exantema y que eviten el retraso entre la aparición de los síntomas iniciales y la consulta al médico. La mayoría de los casos de exantemas asociados a VIRAMUNE aparecen durante las primeras 6 semanas desde el inicio del tratamiento. Por lo tanto, debe monitorizarse cuidadosamente la aparición de exantemas en los pacientes durante este periodo. Debe indicarse a los pacientes que si aparece algún tipo de exantema durante el periodo inicial de dos semanas no se procederá al aumento de dosis hasta que éste desaparezca. Todo paciente que presente exantema grave o exantema acompañado de sintomatología general, tal como fiebre, formación de vesículas, lesiones orales, conjuntivitis, hinchazón, dolores musculares, dolores articulares o malestar general, debe interrumpir la medicación y consultar al médico. En estos pacientes VIRAMUNE no debe reiniciarse. Si se producen reacciones de hipersensibilidad, caracterizadas por exantema con sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral, tal como hepatitis, eosinofilia, granulocitopenia y disfunción renal, VIRAMUNE debe interrumpirse permanentemente y no volver a administrarse. Reacciones hepáticas En pacientes tratados con VIRAMUNE se ha producido hepatotoxicidad grave y que supone un riesgo para la vida, incluyendo hepatitis fulminante fatal. En pacientes tratados con VIRAMUNE, los casos 4/34 de hepatitis graves e insuficiencia hepática se producen con mayor frecuencia durante las primeras 8 semanas de tratamiento. Un tercio de los casos, se han descrito después de este período crítico. En individuos no infectados por el VIH que recibieron dosis múltiples de VIRAMUNE como profilaxis post-exposición, un uso no autorizado, se ha descrito hepatotoxicidad grave, incluyendo casos de fallo hepático que requirieron transplante. La administración de VIRAMUNE no ha sido evaluada en un estudio específico sobre profilaxis post-exposición, especialmente en términos de duración de tratamiento. El aumento de los valores de SGOT o SGPT y/o historia de infección por hepatitis crónica (B y C) antes del inicio del tratamiento con regímenes que contengan VIRAMUNE, se asocia con un mayor riesgo de reacciones adversas hepáticas. Debe indicarse a los pacientes que las reacciones hepáticas constituyen la principal toxicidad de VIRAMUNE, requiriéndose una estrecha monitorización durante los primeros 2 meses. Se les debe indicar que la aparición de cualquier síntoma que sugiera hepatitis, debe llevarles a contactar inmediatamente con su médico. Monitorización hepática Se han descrito anomalías de las pruebas de función hepática en el tratamiento con VIRAMUNE, algunas en las primeras semanas de tratamiento. Se describen frecuentemente elevaciones asintomáticas de las enzimas hepáticas que no constituyen necesariamente una contraindicación para usar VIRAMUNE. Elevaciones asintomáticas de la GGT no constituyen una contraindicación para la continuación del tratamiento. Debería realizarse una monitorización de las pruebas hepáticas cada dos semanas durante los 2 primeros meses de tratamiento, al 3er mes y a partir de entonces cada 3-6 meses. Debe realizarse una monitorización de las pruebas hepáticas si los pacientes presentan signos o síntomas que sugieran hepatitis y/o hipersensibilidad. Si los valores de SGOT o SGPT > 2 veces el LSN, las pruebas hepáticas deben monitorizarse más frecuentemente durante las visitas clínicas regulares. Los médicos y los pacientes deben estar alerta ante signos prodrómicos o hallazgos de hepatitis, tales como anorexia, náuseas, ictericia, bilirrubinuria, heces acólicas, hepatomegalia o dolor a la palpación hepática. Debe indicarse a los pacientes que soliciten rápidamente atención médica si esto se produce. Si los valores de SGOT o SGPT aumentan a > 5 veces el LSN, la administración de VIRAMUNE debe interrumpirse inmediatamente. Si los valores de SGOT o SGPT regresan a los valores basales, se puede reintroducir VIRAMUNE, según cada caso particular, a la dosis inicial de 200 mg/día durante 14 días seguida de 400 mg/día. En estos casos, se requiere una monitorización hepática más frecuente. Si vuelven a aparecer rápidamente las anomalías de la función hepática, VIRAMUNE debe interrumpirse permanentemente. Si se produce hepatitis clínica, caracterizada por anorexia, náuseas, vómitos, ictericia Y hallazgos de laboratorio (tales como anomalías de las pruebas de función hepática moderadas o graves (excluyendo GGT)), VIRAMUNE debe interrumpirse permanentemente y no volver a administrarse. VIRAMUNE no debe ser readministrado a pacientes que hayan requerido una interrupción permanente, por hepatitis clínica debida a la nevirapina. 5/34 Otras advertencias El tratamiento combinado con VIRAMUNE no es un tratamiento curativo de los pacientes infectados por VIH-1; los pacientes pueden seguir sufriendo enfermedades asociadas a la infección avanzada por VIH-1, incluyendo infecciones oportunistas. En la actualidad, los efectos a largo plazo de la nevirapina son desconocidos. El tratamiento combinado con VIRAMUNE no ha demostrado reducir el riesgo de transmisión del VIH-1 a terceros a través de contacto sexual o sangre contaminada. Se han descrito también los siguientes acontecimientos cuando VIRAMUNE se ha utilizado en asociación con otros agentes antirretrovirales: pancreatitis, neuropatía periférica y trombocitopenia. Estos acontecimientos se asocian generalmente con otros agentes antirretrovirales y puede esperarse que se produzcan cuando VIRAMUNE se utiliza en asociación con otros agentes; sin embargo, es improbable que estos acontecimientos sean debidos al tratamiento con VIRAMUNE. Se han registrado raramente síndromes hepatorrenales. La terapia antirretroviral combinada se ha asociado con una redistribución de la grasa corporal (lipodistrofia) en pacientes con infección por VIH. Actualmente se desconocen las consecuencias de estos acontecimientos a largo plazo. El conocimiento sobre el mecanismo es incompleto. Se han propuesto como hipótesis una posible conexión entre lipomatosis visceral y el tratamiento con inhibidores de la proteasa y entre lipoatrofia y el tratamiento con inhibidores de la transcriptasa inversa análogos de nucleósidos. Se ha relacionado un mayor riesgo de lipodistrofia con factores del individuo, tales como la edad avanzada, y con factores relacionados con el fármaco, tales como una larga duración del tratamiento antirretroviral, y trastornos metabólicos asociados. El examen clínico debe incluir una evaluación de los signos físicos de redistribución de la grasa. Se deben tener en cuenta los niveles de lípidos en suero y de glucosa en sangre, en condiciones de ayuno. Los trastornos lipídicos deben tratarse como se considere clínicamente apropiado (véase apartado 4.8 Reacciones adversas). La nevirapina puede interaccionar con algunos medicamentos; por consiguiente, debe advertirse a los pacientes que informen a su médico del uso de cualquier otra medicación. En mujeres en tratamiento con VIRAMUNE, no deben utilizarse anticonceptivos orales ni otros tratamientos hormonales de control de la natalidad como único método de anticoncepción, ya que la nevirapina podría disminuir las concentraciones plasmáticas de estos medicamentos. Por este motivo, y con el fin de reducir el riesgo de transmisión del VIH, se recomiendan los métodos anticonceptivos de barrera (por ej., preservativos). Además, cuando se utilicen anticonceptivos orales para la regulación hormonal durante la administración de VIRAMUNE, debe monitorizarse el efecto terapéutico. Los resultados farmacocinéticos sugieren que debe tenerse precaución cuando VIRAMUNE es administrado a pacientes con disfunción hepática moderada y que no debe administrarse a pacientes con disfunción hepática grave. En general, los resultados sugieren que los pacientes con disfunción hepática de leve a moderada, con una puntuación ≤ 7 de acuerdo con la clasificación Child-Pugh, no requieren un ajuste de la dosis de VIRAMUNE. En pacientes con disfunción renal, que estén siendo sometidos a diálisis, los resultados farmacocinéticos sugieren que el tratamiento suplementario con una dosis adicional de 200 mg de VIRAMUNE después de cada diálisis, ayudaría a contrarrestar los efectos de la diálisis sobre el aclaramiento de la nevirapina. Por otra parte los pacientes con CLcr ≥ 20 ml/min no requieren un ajuste de la dosis de VIRAMUNE (ver 5.2 Propiedades farmacocinéticas). 4.5 Interacción con otros medicamentos y otras formas de interacción Análogos nucleósidos: No se requieren ajustes de dosis cuando VIRAMUNE se toma en asociación con zidovudina, didanosina o zalcitabina. Al agrupar los datos de zidovudina procedentes de dos estudios (n = 33), en los cuales pacientes infectados por VIH-1 recibieron 400 mg diarios de VIRAMUNE, solos o en combinación con 200-300 mg/día de didanosina o a 0,375 a 0,75 mg/día de 6/34 zalcitabina con un tratamiento de fondo con zidovudina, la nevirapina produjo un descenso no significativo de un 13 % del área bajo la curva (AUC) de la zidovudina y un aumento no significativo del 5,8 % de la Cmax de la zidovudina. En un subgrupo de pacientes (n = 6), a los cuales se administraron 400 mg/día de VIRAMUNE y didanosina con un tratamiento de fondo con zidovudina, la nevirapina dio lugar a un descenso significativo del 32 % de la AUC de la zidovudina y a un descenso no significativo del 27 % de la Cmax de la zidovudina. Los datos pareados sugieren que la zidovudina careció de efecto sobre la farmacocinética de la nevirapina. En un estudio cruzado, la nevirapina careció de efecto sobre la farmacocinética en estado de equilibrio de la didanosina (n = 18) o la zalcitabina (n = 6). Los resultados de un estudio de 36 días en pacientes infectados por VIH (n = 25), a los que se administró VIRAMUNE, nelfinavir (750 mg tres veces al día) y estavudina (30-40 mg dos veces al día), no mostraron cambios estadísticamente significativos en la AUC ni en la Cmax de la estavudina. Por otra parte, un estudio farmacocinético realizado en una población de 90 pacientes, a la que se administró lamivudina con VIRAMUNE o placebo, no reveló cambios en el aclaramiento aparente ni en el volumen de distribución de la lamivudina, lo que sugiere que la nevirapina no ejerce efecto de inducción sobre el aclaramiento de la lamivudina. Análogos no nucleósidos: Los resultados de un ensayo clínico (n=14) mostraron que en el estado de equilibrio los parámetros farmacocinéticos de la nevirapina no fueron afectados por la administración concomitante del efavirenz. Sin embargo, los niveles de efavirenz se redujeron significativamente en presencia de nevirapina. La AUC del efavirenz disminuyó en un 22 % y la Cmin en un 36%. Cuando se administra de manera conjunta con la nevirapina debe asegurarse que la dosis de efavirenz se incremente a 800 mg una vez al día. Inhibidores de la proteasa: La nevirapina es un inductor ligero a moderado del enzima hepático CYP3A; por lo tanto, es posible que la administración conjunta con inhibidores de la proteasa (también metabolizados por CYP3A) produzca una modificación de las concentraciones plasmáticas de cada uno de ellos. Los resultados de un ensayo clínico (n = 31) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE y saquinavir (cápsulas de gelatina dura, 600 mg 3 veces al día), indicaron que su administración simultánea produce una reducción media del 24 % (p = 0,041) de la AUC del saquinavir y no produce cambios significativos de los niveles plasmáticos de nevirapina. La reducción de los niveles de saquinavir, debidos a esta interacción, pueden reducir adicionalmente los niveles plasmáticos marginales del saquinavir que se alcanzan con la formulación en cápsulas de gelatina dura. Otro estudio (n= 20) evaluó la administración una vez al día de saquinavir cápsula de gelatina blanda con una dosis de 100 mg de ritonavir. Todos los pacientes recibieron VIRAMUNE concomitantemente. El estudio mostró que la asociación de saquinavir cápsula de gelatina blanda y 100 mg de ritonavir no tuvo un efecto medible sobre los parámetros farmacocinéticos de la nevirapina, comparado con controles históricos. El efecto de la nevirapina sobre la farmacocinética del saquinavir cápsula de gelatina blanda, en presencia de 100 mg de ritonavir, fue discreto y no significativo clínicamente. Los resultados de un ensayo clínico (n = 25) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE e indinavir (800 mg cada 8 horas), indicaron que su administración simultánea produce un descenso medio del 28 % (p < 0,01) de la AUC del indinavir y no produce cambios significativos de los niveles plasmáticos de nevirapina. No se ha llegado a conclusiones clínicas definitivas, respecto al impacto potencial de la administración simultánea de nevirapina e indinavir. Debe considerarse un aumento de la dosis de indinavir a 1.000 mg cada 8 horas si el indinavir se administra junto con nevirapina, 200 mg dos veces al día; sin embargo, actualmente no se dispone de datos que permitan afirmar que la actividad antiviral a corto o a largo plazo de 1.000 mg de indinavir cada 8 horas con 200 mg de nevirapina dos veces al día, diferirá de la de 800 mg de indinavir cada 8 horas con 200 mg de nevirapina dos veces al día. 7/34 Los resultados de un ensayo clínico (n = 25) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE y ritonavir (600 mg dos veces al día [utilizando un régimen de escalado de dosis gradual]), indicaron que su administración conjunta no da lugar a cambios significativos en los niveles plasmáticos de ritonavir o nevirapina. Los resultados de un estudio de 36 días en pacientes infectados por VIH (n = 25), a los que se administró VIRAMUNE, nelfinavir (750 mg tres veces al día) y estavudina (30-40 mg dos veces al día), no demostraron cambios estadísticamente significativos en los parámetros farmacocinéticos del nelfinavir después de la adición de nevirapina (AUC + 4 %, Cmax +14 % y Cmin - 2 %). En comparación con los controles históricos, los niveles de nevirapina se mostraron inalterados. No se observó un aumento en el riesgo en relacion a la seguridad en la administración simultánea de VIRAMUNE con cualquiera de estos los inhibidores de la proteasa cuando se prescriben en combinaci ón. No hubo cambio aparente en la farmacocinética del lopinavir cuando se utilizó concomitantemente con VIRAMUNE en voluntarios sanos. En pacientes tratados con un único inhibidor de la proteasa, la nevirapina, utilizada en combinación con lopinavir/ritonavir 400/100 mg (3 cápsulas) dos veces al día y análogos nucleósidos, proporcionaron una tasa de respuesta virológica muy buena. Los resultados de un estudio farmacocinético en pacientes pediátricos revelaron una disminución de las concentraciones de lopinavir durante la administración conjunta con nevirapina. Se desconoce la significación clínica de esta interacción. Sin embargo, debe considerarse un aumento de la dosis de lopinavir / ritonavir de 533/133 mg (4 cápsulas o 6,5 ml) cuando se utilizan asociados con la nevirapina en pacientes donde se sospecha clínicamente (por la historia del tratamiento o la evidencia de laboratorio) una reducción de la sensibilidad a lopinavir / ritonavir. Ketoconazol: En un estudio, la administración de 200 mg de nevirapina dos veces al día con ketoconazol 400 mg cuatro veces al día produjo una reducción significativa (63 % de reducción media de la AUC del ketoconazol y un 40 % de reducción media de la Cmax del ketoconazol). En el mismo estudio, la administración de ketoconazol produjo un aumento del 15-28 % de los niveles plasmáticos de nevirapina en comparación con los controles históricos. El ketoconazol y VIRAMUNE no deben administrarse simultáneamente. No se conocen los efectos de la nevirapina sobre el itraconazol. Fluconazol: La administración simultánea de fluconazol y VIRAMUNE dio como resultado un aumento de la exposición a la nevirapina de aproximadamente el 100%, en comparación con los datos históricos en los que VIRAMUNE se administró solo. Debido al riesgo de una exposición aumentada a la nevirapina, debe procederse con precaución si los medicamentos se administran concomitantemente y los pacientes deben ser estrechamente controlados. No hubo efectos clínicamente relevantes de la nevirapina sobre el fluconazol. Anticonceptivos orales: Como los anticonceptivos orales no deben utilizarse como único método anticonceptivo en pacientes infectados por VIH-1, se recomiendan otros métodos anticonceptivos (tales como métodos de barrera) en pacientes tratados con VIRAMUNE. Se ha identificado además una interacción farmacocinética. Se administró 200 mg de nevirapina dos veces al día, conjuntamente con una dosis única de un anticonceptivo oral que contenía 0,035 mg de etinil-estradiol (EE) y 1,0 mg de noretindrona (NET). Comparando las concentraciones plasmáticas observadas antes de la administración de la nevirapina, la AUC media del 17α-EE disminuyó significativamente en un 29 %, a los 28 días de la administración de nevirapina. Hubo una reducción significativa en el tiempo medio de residencia y en la vida media del EE. Hubo una reducción significativa (18%) en la AUC media de la NET, sin cambios en el tiempo medio de residencia o vida media. La magnitud de este efecto sugiere que la dosis del anticonceptivo oral debe ser ajustada para permitir un tratamiento adecuado de indicaciones distintas a la anticoncepción (p.ej., endometriosis), si se utiliza con la nevirapina. Otros medicamentos metabolizados por CYP3A: La nevirapina es un inductor del CYP3A y potencialmente del CYP2B6, produciendo una inducción máxima al cabo de 2-4 semanas de iniciar la terapia a dosis múltiples. En base al metabolismo conocido de la metadona, la nevirapina puede disminuir las concentraciones plasmáticas de metadona por aumento de su metabolismo hepático. Se 8/34 ha descrito síndrome de abstinencia a los narcóticos en pacientes tratados con VIRAMUNE y metadona simultáneamente. Los pacientes tratados con metadona, que inicien una terapia con VIRAMUNE, deben ser monitorizados en relación a la aparición de síndrome de abstinencia y debe ajustarse la dosis de metadona adecuadamente. Otros compuestos que son sustratos de CYP3A y CYP2B6 pueden experimentar la disminución de sus concentraciones plasmáticas cuando se administran simultáneamente con VIRAMUNE. Por lo tanto, se recomienda una monitorización cuidadosa de la eficacia terapéutica de los medicamentos metabolizados por P450 cuando se administren en asociación con VIRAMUNE. Inhibidores del isoenzima CYP: Los resultados de un estudio de interacción de nevirapinaclaritromicina (n = 18) indicaron una reducción significativa de la AUC (30 %) y de la Cmax (- 21 %) de la claritromicina, con un incremento significativo de la AUC (58 %) y la Cmax (62 %) del metabolito activo 14-OH claritromicina. La nevirapina presentó un incremento significativo de la Cmin (28 %) y un incremento no significativo de la AUC (26 %) y de la Cmax (24 %). Estos resultados sugieren que no son necesarios ajustes de dosis ni de claritromicina ni de VIRAMUNE cuando se administran conjuntamente ambos medicamentos. No obstante, se recomienda una estricta monitorización de las anomalías hepáticas y de la actividad frente al complejo intracelular del Myobacterium avium (CMA). La monitorización de las concentraciones plasmáticas valle de nevirapina, en pacientes que recibían un tratamiento con VIRAMUNE a largo plazo, reveló que las concentraciones valle de nevirapina estaban elevadas en pacientes que recibían cimetidina (+ 7 %, n = 13). Inductores del isoenzima CYP: Un estudio abierto (n = 14) para determinar los efectos de la nevirapina sobre la farmacocinética en estado de equilibrio de la rifampicina, no reveló cambios significativos en la Cmax ni en la AUC de la rifampicina. Por el contrario, la rifampicina produjo una disminución significativa de la AUC (-58 %), Cmax (- 50 %) y Cmin (- 68 %) de la nevirapina, en comparación con los datos históricos. Los datos farmacocinéticos disponibles sugieren que el uso concomitante de rifampicina y VIRAMUNE no está recomendado. Por lo tanto, estos medicamentos no deben utilizarse en combinación. Los médicos que necesiten tratar pacientes co-infectados con tuberculosis y que utilicen un régimen que contenga VIRAMUNE, deben en su lugar, considerar la utilización de rifabutina. La rifabutina y VIRAMUNE pueden administrarse conjuntamente sin ajustes de dosis (ver más abajo). Alternativamente los médicos podrían considerar el cambio a una asociación triple de análogos nucleósidos durante un periodo variable de tiempo, dependiendo del régimen de tratamiento de la tuberculosis (ver apartado 4.3 Contraindicaciones). En un estudio farmacocinético la administración simultánea de VIRAMUNE con rifabutina, reveló un aumento no significativo del 12 % (media) en la AUC en el estado de equilibrio, una reducción no significativa del 3% en la Cminee y un aumento significativo del 20 % en la Cmaxee. No se detectaron cambios significativos en la AUC, Cminee o Cmaxee de la 25-O-desacetil-rifabutina (metabolito activo de la rifabutina). Se reveló un aumento estadísticamente significativo en el aclaramiento aparente de la nevirapina (9 %) comparado con datos farmacocinéticos históricos. Este estudio sugiere que no hay interacciones clínicamente relevantes entre la nevirapina y la rifabutina. Por lo tanto, los dos fármacos pueden ser administrados simultáneamente sin necesidad de ajuste de la dosis, siempre que se lleve a cabo una monitorización cuidadosa de las reacciones adversas. Warfarina: La interacción entre la nevirapina y el agente antitrombótico warfarina es compleja, con la posibilidad de producirse tanto aumentos como descensos en el tiempo de coagulación cuando se usan concomitantemente. El efecto neto de la interacción puede variar durante las primeras semanas de administración simultánea o en la discontinuación de VIRAMUNE, y por lo tanto, debe garantizarse una estrecha monitorización de los niveles anticoagulación. Hypericum perforatum: Los niveles séricos de la nevirapina pueden reducirse por la utilización concomitante de preparaciones a base de plantas medicinales que contengan Hipérico (Hypericum perforatum). Esto es debido a la inducción de enzimas del metabolismo del fármaco y/o proteínas de 9/34 transporte producida por el Hipérico. Por lo tanto, las preparaciones a base de plantas medicinales que contengan Hipérico, no deben asociarse con VIRAMUNE. Si el paciente está tomando Hipérico, deben comprobarse los niveles de nevirapina y si es posible la carga viral e interrumpir la administración de Hipérico. Los niveles de nevirapina pueden aumentar al interrumpir la administración de Hipérico. Puede ser necesario un ajuste de la dosis de VIRAMUNE. El efecto inductor puede continuar durante al menos 2 semanas después de la interrupción del tratamiento con Hipérico. Otra información: Los estudios en microsomas hepáticos humanos indicaron que la formación de metabolitos hidroxilados de nevirapina no se vio afectada por la presencia de dapsona, rifabutina, rifampicina y trimetoprim/sulfametoxazol. El ketoconazol y la eritromicina inhibieron significativamente la formación de metabolitos hidroxilados de nevirapina. 4.6 Embarazo y lactancia En estudios sobre la reproducción realizados en conejos y ratas en estado de gestación no se detectó teratogenicidad observable. No se dispone de estudios adecuados y bien controlados en mujeres embarazadas. Por tanto, VIRAMUNE sólo debe utilizarse durante el embarazo si el beneficio esperado justifica el posible riesgo para el niño y debe tenerse precaución cuando se prescriba VIRAMUNE a mujeres embarazadas. Los resultados de un estudio farmacocinético(ACTG 250) en 10 mujeres embarazadas infectadas por VIH-1, a las cuales se administró una dosis oral única de 100 ó 200 mg de VIRAMUNE 5,8 horas antes del parto por término medio, han demostrado que la nevirapina atraviesa fácilmente la placenta y aparece en la leche materna. Se recomienda que madres con infección por VIH no amamanten a sus hijos para evitar el riesgo de transmisión postnatal del VIH y que interrumpan la lactancia si reciben tratamiento con VIRAMUNE. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas No se han realizado estudios específicos sobre la capacidad para conducir vehículos y utilizar máquinas. 4.8 Reacciones adversas Adultos Los acontecimientos adversos relacionados con el tratamiento con VIRAMUNE descritos con mayor frecuencia durante los ensayos clínicos fueron exantema, náuseas, fatiga, fiebre, cefalea, vómitos, diarrea, dolor abdominal y mialgia. En circunstancias muy raras, pueden asociarse casos de anemia y neutropenia al tratamiento con VIRAMUNE. En raras circunstancias, se ha descrito artralgia como un acontecimiento aislado en pacientes que reciben regímenes conteniendo VIRAMUNE. La experiencia post-comercialización ha demostrado que las reacciones adversas más graves son el síndrome de Stevens-Johnson y la necrolisis epidérmica tóxica y la hepatitis grave/insuficiencia hepática y reacciones de hipersensibilidad, caracterizados por exantema con sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral como hepatitis, eosinofilia, granulocitopenia y disfunción renal. Las 8 primeras semanas de tratamiento, constituyen un período crítico que requiere una estrecha monitorización (ver apartado 4.4 Advertencias y precauciones especiales de empleo). La terapia antirretroviral combinada se ha asociado con una redistribución de la grasa corporal (lipodistrofia) en pacientes VIH, que incluye pérdida de grasa subcutánea periférica y facial, aumento 10/34 de la grasa intra-abdominal y visceral, hipertrofia de las mamas y acumulación de la grasa dorsocervical (joroba de búfalo). La terapia antirretroviral combinada se ha asociado con anomalías metabólicas tales como hipertrigliceridemia, hipercolesterolemia, resistencia a la insulina, hiperglucemia e hiperlactacidemia (véase apartado 4.4 Advertencias y precauciones especiales de empleo). Piel y tejido subcutáneos La toxicidad clínica más frecuente de VIRAMUNE es el exantema, con la aparición de exantema atribuible a VIRAMUNE en el 16% de los pacientes con regímenes combinados en estudios controlados de Fase II/III. En estos ensayos clínicos el 35% de los pacientes tratados con VIRAMUNE experimentaron exantema en comparación con el 19% de los pacientes en los grupos control tratados con zidovudina + didanosina o zidovudina sola. En un 6,6% de los pacientes tratados con VIRAMUNE aparecieron reacciones cutáneas graves o que suponen un riesgo para la vida en comparación con el 1,3% de los pacientes tratados en los grupos control. En conjunto, el 7% de los pacientes interrumpieron el tratamiento con VIRAMUNE a causa del exantema. Por lo general, los exantemas son ligeros a moderados, erupciones cutáneas eritematosas maculopapulares, con o sin prurito, localizados en el tronco, cara y extremidades. Se han descrito reacciones alérgicas (anafilaxis, angioedema y urticaria). Los exantemas se producen en forma aislada o en el contexto de reacciones de hipersensibilidad, caracterizadas por exantema asociado a sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral como hepatitis, eosinofilia, granulocitopenia y disfunción renal. Se han producido reacciones cutáneas graves y que suponen riesgo para la vida en pacientes tratados con VIRAMUNE, incluyendo síndrome de Stevens-Johnson (SSJ) y necrolisis epidérmica tóxica (NET). Se han descrito casos fatales de SSJ, NET y reacciones de hipersensibilidad. La mayoría de los exantemas graves se produjeron durante las primeras 6 semanas de tratamiento y algunos necesitaron hospitalización, precisando un paciente intervención quirúrgica. Hepato-biliares Las anomalías observadas con mayor frecuencia en las pruebas de laboratorio fueron elevaciones en las pruebas de la función hepática (PFHs), incluyendo SGPT, SGOT, GGT, bilirrubina total y fosfatasa alcalina. Las elevaciones asintomáticas de los niveles de GGT son las más frecuentes. Se han descrito casos de ictericia. En pacientes tratados con VIRAMUNE se han producido casos de hepatitis (hepatotoxicidad grave y que supone un riesgo para la vida, incluyendo hepatitis fulminante fatal). En un ensayo clínico extenso, el riesgo de acontecimientos hepáticos graves entre 1.121 pacientes que recibieron VIRAMUNE durante una duración media de más de un año, fue del 1,2 % (frente al 0,6 % en el grupo placebo). El mejor indicio de un acontecimiento hepático grave, fue una elevación de las pruebas de función hepática basales. Las primeras 8 semanas de tratamiento constituyen un período crítico que requiere una monitorización estrecha (ver apartado 4.4 Advertencias y precauciones especiales empleo). Pacientes pediátricos En base a la experiencia de 361 pacientes pediátricos tratados en ensayos clínicos, los acontecimientos adversos relacionados con VIRAMUNE descritos con mayor frecuencia fueron similares a los observados en adultos, a excepción de la granulocitopenia que se presentó más frecuentemente en niños. En esta población se han descrito casos aislados de síndrome de Stevens-Johnson o síndrome de Stevens-Johnson/transición a necrolisis epidérmica tóxica. 4.9 Sobredosis No se conoce un antídoto para la sobredosificación de VIRAMUNE. Se han descrito casos de sobredosificación con VIRAMUNE, a dosis entre 800 y 1.800 mg por día, durante un período de hasta 11/34 15 días. Los pacientes presentaron edema, eritema nudoso, fatiga, fiebre, cefalea, insomnio, náuseas, infiltraciones pulmonares, exantema, vértigo, vómitos y disminución de peso. Todos estos efectos disminuyeron al interrumpir la administración de VIRAMUNE. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Grupo farmacoterapéutico: agente antiviral, código ATC J05A G01. Mecanismo de acción La nevirapina es un inhibidor no nucleósido de la transcriptasa inversa (NNRTI) del VIH-1. Se une directamente a la transcriptasa inversa y bloquea las actividades ARN-dependientes y ADNdependientes de la ADN-polimerasa, produciendo una rotura del punto catalítico del enzima. La actividad de la nevirapina no compite con el patrón ni con los trifosfatos nucleósidos. La transcriptasa inversa del VIH-2 y las ADN polimerasas eucariotas (tales como las ADN polimerasas humanas α, β, γ o δ) no son inhibidas por la nevirapina. Resistencia In vitro aparecen aislados de VIH con sensibilidad reducida (100 a 250 veces) a la nevirapina. Se producen cambios fenotípicos y genotípicos en aislados de VIH de pacientes tratados con VIRAMUNE o VIRAMUNE + zidovudina al cabo de 1 a 12 semanas. A la semana 8 de la monoterapia con VIRAMUNE, el 100 % de los pacientes ensayados presentaban aislados de VIH con una disminución de la sensibilidad a la nevirapina > 100 veces, independientemente de la dosis. La terapia de combinación de VIRAMUNE + zidovudina no alteró el grado de aparición de virus resistentes a la nevirapina. En el estudio INCAS se investigó la resistencia genotípica y fenotípica en pacientes que recibieron VIRAMUNE en el tratamiento combinado en terapia doble o triple y en el grupo comparativo que no recibió VIRAMUNE. Los pacientes sin tratamiento previo con antirretrovirales, con recuentos de células CD4 de 200-600/mm3 se trataron con VIRAMUNE + zidovudina (n = 46), zidovudina + didanosina (n = 51) o VIRAMUNE + zidovudina + didanosina (n = 51) y fueron controlados durante 52 semanas o más de tratamiento. Se realizaron evaluaciones virológicas basales, a los 6 meses y a los 12 meses. La prueba de resistencia fenotípica realizada requirió, un mínimo de 1000 copias/ml de ARN VIH con el fin de amplificar el virus. De los 3 grupos en estudio, 16, 19 y 28 pacientes respectivamente, presentaron aislados basales evaluables y posteriormente permanecieron en el estudio durante al menos 24 semanas. Por lo que respecta a los valores basales, hubo cinco casos de resistencia fenotípica a la nevirapina; los valores de CI50 fueron de 5 a 6,5 veces superiores en 3 de los casos y > 100 veces en los otros dos. A las 24 semanas, todos los aislados disponibles recuperables de pacientes que recibieron nevirapina fueron resistentes a este agente, mientras 18/21 (86 %) de los pacientes presentaron estos aislados a las 30 - 60 semanas. En 16 de los pacientes la eliminación viral estuvo por debajo de los límites de detección (< 20 copias/ml = 14, < 400 copias/ml = 2). Asumiendo que esta eliminación por debajo de < 20 copias/ml, implica sensibilidad del virus a nevirapina, el 45 % (17/38) de los pacientes presentaron virus medibles o con sensibilidad atribuible a nevirapina. Todos los 11 pacientes que recibieron VIRAMUNE + zidovudina en los cuales se ensayó la resistencia fenotípica, fueron resistentes a la nevirapina durante los seis meses. Durante el periodo completo de observación, se observó un caso de resistencia a la didanosina. La resistencia a la zidovudina apareció más frecuentemente a las 30 - 60 semanas, especialmente en pacientes que recibieron doble terapia combinada. Basándose en el aumento en la CI50, la resistencia a la zidovudina fue inferior en el grupo de VIRAMUNE + zidovudina + didanosina que en los otros grupos de tratamiento. Por lo que respecta a la resistencia a la nevirapina, todos los aislados secuenciados, presentaron al menos una mutación asociada a resistencia, siendo los cambios simples más comunes el K103N y el Y181C. En nueve de los 12 pacientes observados, se detectaron combinaciones de mutaciones. Estos datos del estudio INCAS reflejan que el uso de tratamientos muy activos está asociado con un retraso en el desarrollo de resistencia a los fármacos antirretrovirales. 12/34 La relevancia clínica de los cambios fenotípicos y genotípicos asociados al tratamiento con VIRAMUNE no ha sido establecida. Además de los datos presentados anteriormente, existe el riesgo de rápida aparición de resistencia a los NNRTIs en caso de fallo virológico. Resistencia cruzada Se ha observado in vitro una rápida aparición de cepas de VIH que presentan resistencia cruzada a los NNRTIs. Los datos sobre resistencia cruzada entre el NNRTI nevirapina y los análogos nucleósidos inhibidores de la transcriptasa inversa son muy limitados. En cuatro pacientes, los aislados resistentes a la zidovudina ensayados in vitro mantenían la sensibilidad a la nevirapina y, en seis pacientes, los aislados resistentes a la nevirapina eran sensibles a la zidovudina y didanosina. La resistencia cruzada entre nevirapina e inhibidores de la proteasa VIH es improbable, debido a que los enzimas diana involucrados son diferentes. La resistencia cruzada entre los NNRTIs actualmente registrados es extensa. Algunos datos de resistencia genotípica indican que en la mayoría de los pacientes que fracasan con NNRTI , las cepas virales expresan resistencia cruzada a los otros NNRTIs. En la actualidad, los datos disponibles no respaldan el uso secuencial de los NNRTIs. Efectos farmacodinámicos VIRAMUNE ha sido evaluado tanto en pacientes sin tratamiento previo como en pacientes tratados. Los resultados de un ensayo (ACTG 241) evaluaron el tratamiento triple con VIRAMUNE, zidovudina y didanosina en comparación con zidovudina + didanosina, en 398 pacientes infectados por VIH-1 (valores basales medios 153 células CD4+/mm3, ARN VIH-1 en plasma 4,59 log10 copias/ml), los cuales habían recibido, como mínimo, seis meses de tratamiento con análogos nucleósidos antes de la incorporación (media 115 semanas). Estos pacientes, extensamente tratados, demostraron una mejoría significativa en el grupo de terapia triple, en relación con el grupo de terapia doble durante un año, tanto en relación al ARN viral como al recuento de células CD4+. En un ensayo (INCAS), se documentó una respuesta duradera de como mínimo un año para el grupo de terapia triple con VIRAMUNE, zidovudina y didanosina, en comparación con zidovudina + didanosina o VIRAMUNE + zidovudina, en 151 pacientes infectados por VIH-1 sin tratamiento previo, con recuentos de células CD4+ de 200-600 células/mm3 (media 376 células/mm3) y una concentración basal media de ARN VIH-1 en plasma de 4,41 log10 copias/ml (25.704 copias/ml). Las dosis de tratamiento fueron VIRAMUNE, 200 mg diarios durante dos semanas seguidos de 200 mg dos veces al día, o placebo; zidovudina, 200 mg tres veces al día; didanosina, 125 ó 200 mg dos veces al día (dependiendo del peso). VIRAMUNE ha sido también estudiado en asociación con otros agentes antirretrovirales, por ej., zalcitabina, estavudina, lamivudina, indinavir, ritonavir, nelfinavir, saquinavir y lopinavir. No se han descrito problemas de seguridad nuevos y evidentes para estas asociaciones. Se están llevando a cabo estudios para evaluar la eficacia y seguridad de los tratamientos combinados con VIRAMUNE en pacientes en los cuales la terapia con inhibidores de la proteasa no es eficaz. Transmisión perinatal Dos estudios evaluaron la eficacia de VIRAMUNE para prevenir la transmisión vertical de la infección por VIH-1. Durante estos ensayos, las madres recibieron únicamente el tratamiento antirretroviral en estudio. En el estudio HIVNET 012 en Kampala (Uganda) se randomizaron pares madre-recién nacido para la administración de VIRAMUNE por vía oral (madre: 200 mg al principio del parto; recién nacido: 2 mg/kg durante las 72 horas posteriores al nacimiento) o un tratamiento ultracorto de zidovudina por vía oral (madre: 600 mg al principio del parto y 300 mg cada 3 horas hasta que éste finaliza; recién nacido: 4 mg/kg dos veces al día durante 7 días). Las tasas de infección por VIH-1 acumuladas en el 13/34 recién nacido a las 14 - 16 semanas fueron del 13,1 % (n = 310) en el grupo de VIRAMUNE, frente al 25, 1 % (n = 308) en el grupo de tratamiento ultracorto de la zidovudina (p = 0.00063). En el estudio SAINT realizado en Sudáfrica, se randomizaron pares madre-recién nacido para la administración de VIRAMUNE por vía oral (madre: 200 mg durante el parto y 200 mg entre 24 y 48 horas después del parto; recién nacido: 6 mg entre 24 y 48 horas después del parto); o un tratamiento corto de zidovudina y lamivudina por vía oral (madre: 600 mg de zidovudina, luego 300 mg cada 3 horas durante el parto, seguido de 300 mg dos veces al día durante 7 días después del parto más 150 mg de lamivudina dos veces al día durante el parto y durante 7 días después del mismo; recién nacido: 12 mg de zidovudina dos veces al día más 6 mg de lamivudina dos veces al día durante 7 días [si el peso del recién nacido es <2 kg, 4 mg/kg de zidovudina dos veces al día más 2 mg/kg de lamivudina dos veces al día durante 7 días]). No hubo diferencias significativas en la tasa de transmisión del VIH1 a las 6-8 semanas entre el grupo tratado con VIRAMUNE (5,7%, n = 652) y el grupo tratado con zidovudina más lamivudina (3,6%, n = 649). Hubo un mayor riesgo de transmisión del VIH-1 a recién nacidos cuyas madres habían recibido las dosis de VIRAMUNE o zidovudina más lamivudina menos de 2 horas antes del parto. En el estudio SAINT el 68% de las madres expuestas a nevirapina tuvo cepas resistentes, aproximadamente 4 semanas después del parto. No se ha establecido la relevancia clínica de estos datos en la población Europea. Además, en el caso de que VIRAMUNE se utilice en dosis única para prevenir la transmisión vertical de la infección por VIH-1, el riesgo de hepatotoxicidad en la madre y en el niño no puede ser descartado. Un ensayo clínico ciego, randomizado, en mujeres que ya tomaban tratamiento antirretroviral durante el embarazo (PACTG 316) no demostró una reducción adicional de la transmisión vertical del VIH-1 cuando se administró una dosis única de VIRAMUNE a la madre y al recién nacido, durante el parto y después del nacimiento, respectivamente. Los índices de transmisión VIH-1 fueron similarmente bajos en ambos grupos de tratamiento (1,3% en el grupo de VIRAMUNE, 1,4% en el grupo del placebo). La transmisión vertical tampoco disminuyó en mujeres con ARN VIH-1 por debajo del límite de cuantificación ni en mujeres con ARN VIH-1 por encima del límite de cuantificación antes del parto. De las 95 mujeres que recibieron VIRAMUNE intra-parto, un 15% desarrollaron mutaciones de resistencia a la nevirapina a las 6 semanas después del parto. 5.2 Propiedades farmacocinéticas Adultos La nevirapina se absorbe fácilmente (> 90 %) después de la administración oral en voluntarios sanos y en adultos con infección por VIH-1. La biodisponibilidad absoluta en 12 adultos sanos después de administración de una dosis única fue del 93 ± 9 % (media ± DE) para un comprimido de 50 mg y del 91 ± 8 % para una solución oral. Las concentraciones pico en plasma de nevirapina, de 2 ± 0,4 µg/ml (7,5 µM), se alcanzaron a las 4 horas después de una dosis única de 200 mg. Después de dosis múltiples, las concentraciones pico de nevirapina parecieron aumentar linealmente dentro del rango de dosis de 200 hasta 400 mg/día. Las concentraciones valle de nevirapina en estado de equilibrio de 4,5 ± 1,9 µg/ml (17 ± 7 µM), (n = 242) se alcanzaron con 400 mg/día. Los comprimidos y la suspensión oral de VIRAMUNE han demostrado ser comparables en cuanto a su biodisponibilidad e intercambiables a dosis de hasta 200 mg. La absorción de la nevirapina no se ve afectada por el alimento, antiácidos o medicamentos formulados con un agente tamponador alcalino (por ej., didanosina). La nevirapina es lipofílica y se encuentra esencialmente no ionizada a pH fisiológico. Después de la administración intravenosa a adultos sanos, el volumen de distribución (Vdss) de la nevirapina fue de 1,21 ± 0,09 l/kg, sugiriendo una amplia distribución en el ser humano. La nevirapina atraviesa fácilmente la placenta y se detecta en la leche materna. La unión a proteínas plasmáticas es de alrededor del 60 % en el intervalo de concentración en plasma de 1-10 µg/ml. Las concentraciones de nevirapina en líquido cefalorraquídeo humano (n = 6) fueron del 45% (± 5 %) de las concentraciones en plasma; esta proporción es aproximadamente igual a la fracción no ligada a proteínas plasmáticas. 14/34 Los estudios in vivo realizados en el ser humano y los estudios in vitro en microsomas de hígado humano han demostrado que la nevirapina experimenta una amplia biotransformación por metabolismo vía citocromo P450 (oxidativo) a diversos metabolitos hidroxilados. Los estudios in vitro en microsomas de hígado humano sugieren que el metabolismo oxidativo de la nevirapina es mediado esencialmente por isozimas de citocromo P450 de la familia CYP3A, si bien es posible que otros isozimas jueguen un papel secundario. En un estudio de ajuste de peso/excreción realizado en ocho voluntarios sanos del sexo masculino, tratados hasta el estado de equilibrio con 200 mg de nevirapina dos veces al día, seguidos de una dosis única de 50 mg de 14C-nevirapina, se recuperó aproximadamente el 91,4 ± 10,5 % de la dosis marcada radioactivamente, siendo la orina (81,3 ± 11,1 %) la vía principal de excreción en comparación con las heces (10,1 ± 1,5 %). Más del 80 % de la radioactividad en orina estaba constituida por conjugados glucurónidos de metabolitos hidroxilados. Por consiguiente, el metabolismo por citocromo P450, la conjugación glucurónida y la excreción urinaria de metabolitos glucurónidos representan la vía principal de biotransformación y eliminación de nevirapina en el ser humano. Solamente una pequeña fracción (< 5 %) de la radioactividad en orina (lo que representa < 3 % de la dosis total) correspondió al compuesto original; por lo tanto, la excreción renal juega un papel secundario en la eliminación del compuesto original. La nevirapina ha demostrado ser un inductor de enzimas metabólicos citocromo P450 hepáticos. La farmacocinética de la autoinducción se caracteriza por un aumento de aproximadamente 1,5 a 2 veces del aclaramiento oral aparente de la nevirapina cuando al tratamiento con una dosis única siguen 2-4 semanas de tratamiento con 200-400 mg/día. Asimismo, la autoinducción determina un descenso correspondiente de la vida media de la fase terminal de la nevirapina en plasma desde aproximadamente 45 horas (dosis única) hasta aproximadamente 25-30 horas después de la dosificación múltiple con 200-400 mg/día. Disfunción renal: La farmacocinética de dosis única de nevirapina ha sido comparada en 23 pacientes con disfunción renal leve (50 ≤ CLcr < 80 ml/min), moderada (30 ≤ CLcr < 50 ml/min) o grave (CLcr < 30 ml/min), fallo renal o enfermedad renal en estadío terminal que requiere diálisis, y 8 pacientes con función renal normal (CLcr > 80 ml/min). El fallo renal (leve, moderado y grave) no produce un cambio significativo en la farmacocinética de nevirapina. Sin embargo, los pacientes con enfermedad renal en estadío terminal que requerían diálisis mostraron una reducción del 43,5 % en la AUC de nevirapina después de un periodo de exposición de una semana. Hubo también acumulación de hidroximetabolitos de nevirapina en plasma. Los resultados sugieren que el tratamiento suplementario con una dosis adicional de 200 mg de VIRAMUNE después de cada diálisis, ayudaría a contrarrestar los efectos de la diálisis sobre el aclaramiento de nevirapina. Por otra parte los pacientes con CLcr ≥ 20 ml/min no requieren un ajuste de la dosis de VIRAMUNE. Disfunción hepática: La farmacocinética de dosis única de nevirapina ha sido comparada en 10 pacientes con disfunción hepática y 8 pacientes con función hepática normal. En general, los resultados sugieren que los pacientes con disfunción hepática de leve a moderada, con una puntuación ≤ 7 de acuerdo con la clasificación Child-Pugh, no requieren un ajuste de la dosis de VIRAMUNE. Sin embargo, la farmacocinética de nevirapina en un paciente con una puntuación de 8, de acuerdo con la clasificación Child-Pugh y ascitis de moderada a grave, sugiere que los pacientes con empeoramiento de la función hepática pueden tener el riesgo de acumulación de nevirapina en la circulación sistémica. A pesar de que se detectó un volumen de distribución ajustado al peso de la nevirapina, ligeramente superior en las mujeres en comparación con los hombres, no se observaron diferencias significativas ligadas al sexo, en las concentraciones plasmáticas de nevirapina después de la administración de dosis únicas o múltiples. La farmacocinética de la nevirapina en adultos infectados por VIH-1 no parece modificarse con la edad (rango 19-68 años) o raza (negros, hispanos o caucásicos). VIRAMUNE no ha sido investigado específicamente en pacientes de edad superior a 65 años. 15/34 Pacientes pediátricos Se ha estudiado la farmacocinética de la nevirapina en dos estudios abiertos en niños con infección por VIH-1. En uno de los estudios, fue administrada una dosis única (7,5 mg, 30 mg ó 120 mg por m2; n = 3 por dosis) de VIRAMUNE suspensión oral a nueve niños infectados con VIH, de edades entre 9 meses y 14 años, después de una noche de ayuno. La AUC y la concentración máxima de nevirapina aumentaron en proporción a la dosis. Después de la absorción, las concentraciones plasmáticas medias de nevirapina disminuyeron de forma linealmente logarítmica con respecto al tiempo. La vida media en la fase terminal de la nevirapina, después de una dosis única, fue de 30,6 ± 10,2 horas. En un segundo estudio de dosis múltiple, se administró VIRAMUNE en forma de suspensión oral o de comprimidos (240 a 400 mg/m2/día) como monoterapia o en asociación con zidovudina o zidovudina y didanosina a 37 pacientes pediátricos infectados por VIH-1, con las siguientes características demográficas: varones (54 %), grupos de minorías raciales (73 %), edad media de 11 meses (intervalo: 2 meses - 15 años). Estos pacientes recibieron 120 mg/m2/día de nevirapina, durante 4 semanas aproximadamente, seguidos de 120 mg/m2/dos veces al día (pacientes > 9 años de edad) ó 200 mg/m2/dos veces al día (pacientes ≤ 9 años de edad). El aclaramiento de la nevirapina, ajustado al peso corporal, alcanzó valores máximos hasta la edad de 1 a 2 años y a partir de entonces disminuyó a medida que aumentaba la edad. El aclaramiento aparente de la nevirapina fue aproximadamente dos veces mayor en los niños menores de 8 años, en comparación con los adultos. La vida media de la nevirapina para el grupo estudiado en su conjunto, desde la administración hasta el estado estacionario, fue de 25,9 ± 9,6 horas. Con la administración del fármaco a largo plazo, los valores medios para la vida media terminal de la nevirapina variaron con la edad del modo siguiente: 2 meses a 1 año (32 horas), 1 a 4 años (21 horas), 4 a 8 años (18 horas) y mayores de 8 años (28 horas). 5.3 Datos preclínicos sobre seguridad Los datos preclínicos no mostraron riesgos especiales para los seres humanos, aparte de los observados en los ensayos clínicos, según los estudios convencionales sobre seguridad, farmacología, toxicidad de dosis repetidas y genotoxicidad. En los estudios de toxicidad para la reproducción, se observó una fertilidad disminuida en ratas. En estudios de carcinogénesis, la nevirapina induce tumores hepáticos en ratas y ratones. En ratas estos hallazgos están, mayoritariamente, relacionados con la fuerte actividad inductora de enzimas hepáticos de la nevirapina y no son debidos a un mecanismo de acción genotóxico. El mecanismo de los tumores en ratones no está aún clarificado, de modo que está aún por determinar su implicación en el ser humano. 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Celulosa microcristalina, lactosa monohidratada, povidona K26/28 ó K25, glicolato de almidón sódico, dióxido de sílice coloidal y estearato de magnesio 6.2 Incompatibilidades No aplicable 6.3 Período de validez 2 años 6.4 Precauciones especiales de conservación No se precisan condiciones especiales de conservación 16/34 6.5 Naturaleza y contenido del recipiente Blisters que se abren por presión de cloruro de polivinilo (PVC)/folio de aluminio (tira blister de 10 comprimidos, 6 tiras blister por caja). 6.6 Instrucciones de uso y manipulación, y eliminación Ninguna especial 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN Boehringer Ingelheim International GmbH Binger Strasse 173 55216 Ingelheim am Rhein, Alemania 8. NÚMERO DEL REGISTRO COMUNITARIO DE MEDICAMENTOS EU/1/97/055/001 9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN 17.02.2003 10. FECHA DE LA REVISIÓN DEL TEXTO 17/34 1. DENOMINACIÓN DEL MEDICAMENTO VIRAMUNE 50 mg/5 ml suspensión oral 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Suspensión oral que contiene 10 mg/ml de nevirapina (principio activo), equivalentes a 10,35 mg/ml de nevirapina hemihidrato. Lista de excipientes, en 6.1. 3. FORMA FARMACÉUTICA Suspensión oral VIRAMUNE 50 mg/ml suspensión oral es una suspensión homogénea, blanca o casi blanca. 4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas VIRAMUNE está indicado como parte de la terapia combinada para el tratamiento antiviral de pacientes infectados por VIH-1 con inmunodeficiencia avanzada o progresiva. La mayor parte de la experiencia con VIRAMUNE es en combinación con análogos nucleósidos inhibidores de la transcriptasa inversa. No se dispone en la actualidad de datos suficientes, sobre la eficacia del uso de la triple combinación incluyendo inhibidores de la proteasa después del tratamiento con VIRAMUNE. Ver apartado 5.1 Propiedades farmacodinámicas. 4.2 Posología y forma de administración Pacientes de 2 meses a 8 años La dosis recomendada para los pacientes de 2 meses hasta 8 años es de 4 mg/kg una vez al día, durante dos semanas, seguida de 7 mg/kg dos veces al día a continuación. La dosis diaria total no debe exceder los 400 mg para ningún paciente. Pacientes de 8 a 16 años La dosis recomendada para los pacientes de 8 a 16 años es de 4 mg/kg una vez al día durante dos semanas, seguida de 4 mg/kg dos veces al día a continuación. La dosis diaria total no debe exceder los 400 mg para ningún paciente. Pacientes mayores de 16 años La dosis recomendada de VIRAMUNE es de 20 ml (200 mg) de suspensión oral una vez al día, durante los primeros 14 días (es preciso seguir este período inicial, ya que se ha demostrado que reduce la frecuencia de exantema), seguidos de 20 ml (200 mg) de suspensión oral dos veces al día, en asociación con al menos dos agentes antirretrovirales adicionales a los cuales el paciente no haya sido expuesto previamente. Los virus resistentes aparecen rápida y uniformemente cuando VIRAMUNE se administra como monoterapia; por lo tanto, VIRAMUNE siempre debe administrarse en terapia 18/34 combinada. Por lo que respecta a la terapia antirretroviral concomitante, deben seguirse la dosificación y monitorización recomendadas. Es importante que se administre la dosis total medida de la suspensión oral de VIRAMUNE. Ello se facilita mediante el uso de la jeringa dispensadora que se suministra. Si se utiliza un dispositivo medidor alternativo (por ej. un vasito o cuchara dosificadores para dosis mayores), es importante aclarar el dispositivo para asegurar la administración de la suspensión oral residual. VIRAMUNE se presenta también en forma de comprimidos de 200 mg para pacientes mayores de 16 años o para niños mayores, particularmente adolescentes, de peso igual o superior a 50 kg. Todos los pacientes menores de 16 años que reciban Viramune suspensión oral, deberán someterse con frecuencia a control de peso, para evaluar si son necesarios ajustes de dosis. La dosis máxima diaria para todos los pacientes no deberá rebasar los 400 mg. Consideraciones para el manejo de la dosis Antes de iniciar el tratamiento con VIRAMUNE y a intervalos adecuados durante el mismo, deben realizarse pruebas químico-clínicas, entre las que se incluyan pruebas de la función hepática. Por lo que respecta a la toxicidad que requiera la interrupción del tratamiento de VIRAMUNE, ver apartado 4.4 Advertencias y precauciones especiales de empleo. Los pacientes que presenten exantema durante el período inicial de 14 días con 200 mg/día (4 mg/kg/día para pacientes pediátricos), no deben aumentar su dosis de VIRAMUNE hasta que se haya resuelto el exantema. El exantema aislado debe ser estrechamente monitorizado (ver apartado 4.4 Advertencias y precauciones especiales de empleo). Los pacientes que interrumpan la administración de VIRAMUNE durante más de 7 días, deben reiniciar el tratamiento con la dosis inicial recomendada, tomando 200 mg diarios (4 mg/kg/día para pacientes pediátricos) durante los primeros 14 días, seguidos de 200 mg dos veces al día (4 ó 7 mg/kg dos veces al día para pacientes pediátricos, dependiendo de la edad; ver lo mencionado anteriormente). VIRAMUNE debe ser administrado por médicos expertos en el tratamiento de la infección por VIH. 4.3 Contraindicaciones Hipersensibilidad al principio activo o a alguno de los excipientes. VIRAMUNE no debe readministrarse a pacientes que hayan requerido una interrupción permanente por exantema grave, exantema acompañado de sintomatología general, reacciones de hipersensibilidad o hepatitis clínica debida a nevirapina. VIRAMUNE no debe administrarse a pacientes con daño hepático grave. VIRAMUNE no debe readministrarse en pacientes que hayan presentado anteriormente valores de SGOT o SGPT > 5 veces el LSN durante el tratamiento con VIRAMUNE y tuvieran una rápida recurrencia de las anomalías de la función hepática durante la readministración de VIRAMUNE, (ver apartado 4.4 Advertencias y precauciones especiales de empleo). Los preparados a base de plantas medicinales que contengan Hipérico (Hypericum perforatum) no deben ser utilizados durante la administración de VIRAMUNE debido al riesgo de disminución de las concentraciones plasmáticas y del efecto clínico de la nevirapina (ver 4.5 Interacciones con otros medicamentos y otras formas de interacción). 19/34 Los datos farmacocinéticos disponibles sugieren que el uso concomitante de la rifampicina y VIRAMUNE no se recomienda (ver apartado 4.5 Interacciones con otros medicamentos y otras formas de interacción). 4.4 Advertencias y precauciones especiales de empleo En base a los datos farmacodinámicos VIRAMUNE sólo debe utilizarse en asociación con otros dos agentes antirretrovirales como mínimo (ver apartado 5.1 Propiedades farmacodinámicas). Las primeras 8 semanas de tratamiento con VIRAMUNE constituyen un periodo crítico, que requiere una monitorización estrecha de los pacientes para revelar la potencial aparición de reacciones cutáneas graves y que supongan un riesgo para la vida (incluyendo casos de síndrome de Stevens-Johnson y necrolisis epidérmica tóxica) o hepatitis grave/insuficiencia hepática. Además, debe respetarse estrictamente la pauta de dosificación, especialmente durante los 14 días del período inicial (ver apartado 4.2 Posología y forma de administración). Reacciones cutáneas Se han producido reacciones cutáneas graves y que suponen un riesgo para la vida, incluyendo casos fatales, en pacientes tratados con VIRAMUNE, principalmente durante las primeras 6 semanas de tratamiento. Estas han incluido casos de síndrome de Stevens-Johnson, necrolisis epidérmica tóxica y reacciones de hipersensibilidad, caracterizadas por exantema, sintomatología general y afectación visceral. Los pacientes deben ser estrictamente monitorizados durante las primeras 8 semanas de tratamiento. Los pacientes deben ser estrechamente monitorizados si se produce un exantema aislado. VIRAMUNE debe interrumpirse permanentemente en pacientes que presenten exantema grave o exantema acompañado de sintomatología general (como fiebre, formación de vesículas, lesiones orales, conjuntivitis, hinchazón, dolores musculares o articulares o malestar general), incluyendo síndrome de Stevens-Johnson o necrolisis epidérmica tóxica. VIRAMUNE debe interrumpirse permanentemente en cualquier paciente que presente reacciones de hipersensibilidad (caracterizadas por exantema con sintomatología general además de afectación visceral, tal como hepatitis, eosinofilia, granulocitopenia y disfunción renal) ver apartado 4.4 Advertencias y precauciones especiales de empleo. La administración de VIRAMUNE por encima de la dosis recomendada puede incrementar la frecuencia y la gravedad de las reacciones cutáneas, tales como el síndrome de Stevens-Johnson y la necrolisis epidérmica tóxica. La utilización simultánea de prednisona (40 mg/día durante los primeros 14 días de administración de VIRAMUNE) ha demostrado no disminuir la incidencia del exantema asociado de VIRAMUNE y puede asociarse con un aumento en la incidencia y gravedad del exantema durante las primeras 6 semanas de tratamiento con VIRAMUNE. Se han identificado algunos factores de riesgo en el desarrollo de reacciones cutáneas graves, que incluyen errores en el seguimiento de la pauta de dosificación inicial de 200 mg diarios (4 mg/kg/día para pacientes pediátricos) durante el período inicial y un retraso prolongado entre la aparición de los síntomas iniciales y la consulta al médico. Debe indicarse a los pacientes que la principal toxicidad de VIRAMUNE es el exantema. Debe aconsejárseles que comuniquen de inmediato a su médico la aparición de cualquier exantema y que eviten el retraso entre la aparición de los síntomas iniciales y la consulta al médico. La mayoría de los casos de exantemas asociados a VIRAMUNE aparecen durante las primeras 6 semanas desde el inicio del tratamiento. Por lo tanto, debe monitorizarse cuidadosamente la aparición de exantemas en los pacientes durante este período. Debe indicarse a los pacientes que si aparece algún tipo de exantema durante el período inicial de dos semanas no se procederá al aumento de dosis hasta que éste desaparezca. Se debe garantizar una monitorización cuidadosa especialmente en los pacientes 20/34 pediátricos, particularmente durante las 8 primeras semanas de tratamiento, ya que estos pacientes es menos probable que comuniquen o informen de reacciones cutáneas, que los adultos. Todo paciente que presente exantema grave o exantema acompañado de sintomatología general, tal como fiebre, formación de vesículas, lesiones orales, conjuntivitis, hinchazón, dolores musculares, dolores articulares o malestar general, debe interrumpir la medicación y consultar al médico. En estos pacientes VIRAMUNE no debe reiniciarse. Si se producen reacciones de hipersensibilidad, caracterizadas por exantema con sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral, tal como hepatitis, eosinofilia, granulocitopenia y disfunción renal, VIRAMUNE debe interrumpirse permanentemente y no volver a administrarse. Reacciones hepáticas En pacientes tratados con VIRAMUNE se ha producido hepatotoxicidad grave y que supone un riesgo para la vida, incluyendo hepatitis fulminante fatal. En pacientes tratados con VIRAMUNE, los casos de hepatitis graves e insuficiencia hepática se producen con mayor frecuencia en las primeras 8 semanas de tratamiento. Un tercio de los casos, se han descrito después de este período crítico. En individuos no infectados por el VIH que recibieron dosis múltiples de VIRAMUNE como profilaxis post-exposición, un uso no autorizado, se ha descrito hepatotoxicidad grave, incluyendo casos de fallo hepático que requirieron transplante. La administración de VIRAMUNE no ha sido evaluada en un estudio específico sobre profilaxis post-exposición, especialmente en términos de duración de tratamiento. El aumento de los valores de SGOT o SGPT y/o historia de infección por hepatitis crónica (B y C) antes del inicio del tratamiento con regímenes que contengan VIRAMUNE, se asocia con un mayor riesgo de reacciones adversas hepáticas. Debe indicarse a los pacientes que las reacciones hepáticas constituyen la principal toxicidad de VIRAMUNE, requiriéndose una estrecha monitorización durante los primeros 2 meses. Se les debe indicar que la aparición de cualquier síntoma que sugiera hepatitis, debe llevarles a contactar inmediatamente con su médico. Monitorización hepática Se han descrito anomalías de las pruebas de función hepática en el tratamiento con VIRAMUNE, algunas en las primeras semanas de tratamiento. Se describen frecuentemente elevaciones asintomáticas de las enzimas hepáticas que no constituyen necesariamente una contraindicación para usar VIRAMUNE. Elevaciones asintomáticas de la GGT no constituyen una contraindicación para la continuación del tratamiento. Debería realizarse una monitorización de las pruebas hepáticas cada dos semanas durante los 2 primeros meses de tratamiento, al 3er mes y a partir de entonces cada 3-6 meses. Debe realizarse una monitorización de las pruebas hepáticas si los pacientes presentan signos o síntomas que sugieran hepatitis y/o hipersensibilidad. Si los valores de SGOT o SGPT > 2 veces el LSN, las pruebas hepáticas deben monitorizarse más frecuentemente durante las visitas clínicas regulares. Los médicos y los pacientes deben estar alerta ante signos prodrómicos o hallazgos de hepatitis, tales como anorexia, náuseas, ictericia, bilirrubinuria, heces acólicas, hepatomegalia o dolor a la palpación hepática. Debe indicarse a los pacientes que soliciten rápidamente atención médica si esto se produce. 21/34 Si los valores de SGOT o SGPT aumentan a > 5 veces el LSN, la administración de VIRAMUNE debe interrumpirse inmediatamente. Si los valores de SGOT o SGPT regresan a los valores basales, se puede reintroducir VIRAMUNE, según cada caso particular, a la dosis inicial de 200 mg/día durante 14 días seguida de 400 mg/día. En estos casos, se requiere una monitorización hepática más frecuente. Si vuelven a aparecer rápidamente las anomalías de la función hepática, VIRAMUNE debe interrumpirse permanentemente. Si se produce hepatitis clínica, caracterizada por anorexia, náuseas, vómitos, ictericia Y hallazgos de laboratorio (tales como anomalías de las pruebas de función hepática moderadas o graves (excluyendo GGT)), VIRAMUNE debe interrumpirse permanentemente y no volver a administrarse. VIRAMUNE no debe ser readministrado a pacientes que hayan requerido una interrupción permanente, por hepatitis clínica debida a la nevirapina. Otras advertencias El tratamiento combinado con VIRAMUNE no es un tratamiento curativo de los pacientes infectados por VIH-1; los pacientes pueden seguir sufriendo enfermedades asociadas a la infección avanzada por VIH-1, incluyendo infecciones oportunistas. En la actualidad, los efectos a largo plazo de la nevirapina son desconocidos. El tratamiento combinado con VIRAMUNE no ha demostrado reducir el riesgo de transmisión del VIH-1 a terceros a través de contacto sexual o sangre contaminada. Se han descrito también los siguientes acontecimientos cuando VIRAMUNE se ha utilizado en asociación con otros agentes antirretrovirales: pancreatitis, neuropatía periférica y trombocitopenia. Estos acontecimientos se asocian, generalmente, con otros agentes antirretrovirales y puede esperarse que se produzcan cuando VIRAMUNE se utiliza en asociación con otros agentes; sin embargo, es improbable que estos acontecimientos sean debidos al tratamiento con VIRAMUNE. Se han registrado raramente síndromes hepatorrenales. La terapia antirretroviral combinada se ha asociado con una redistribución de la grasa corporal (lipodistrofia) en pacientes con infección por VIH. Actualmente se desconocen las consecuencias de estos acontecimientos a largo plazo. El conocimiento sobre el mecanismo es incompleto. Se han propuesto como hipótesis una posible conexión entre lipomatosis visceral y el tratamiento con inhibidores de la proteasa y entre lipoatrofia y el tratamiento con inhibidores de la transcriptasa inversa análogos de nucleósidos. Se ha relacionado un mayor riesgo de lipodistrofia con factores del individuo, tales como la edad avanzada, y con factores relacionados con el fármaco, tales como una larga duración del tratamiento antirretroviral, y trastornos metabólicos asociados. El examen clínico debe incluir una evaluación de los signos físicos de redistribución de la grasa. Se deben tener en cuenta los niveles de lípidos en suero y de glucosa en sangre, en condiciones de ayuno. Los trastornos lipídicos deben tratarse como se considere clínicamente apropiado (véase apartado 4.8 Reacciones adversas). La nevirapina puede interaccionar con algunos medicamentos; por consiguiente, debe advertirse a los pacientes que informen a su médico del uso de cualquier otra medicación. En mujeres en tratamiento con nevirapina, no deben utilizarse anticonceptivos orales ni otros tratamientos hormonales de control de la natalidad como único método de anticoncepción, ya que VIRAMUNE podría disminuir las concentraciones plasmáticas de estos medicamentos. Por este motivo, y con el fin de reducir el riesgo de transmisión del VIH, se recomiendan los métodos anticonceptivos de barrera (por ej., preservativos). Además, cuando se utilicen anticonceptivos orales para la regulación hormonal durante la administración de VIRAMUNE, debe monitorizarse el efecto terapéutico. 22/34 Los resultados farmacocinéticos sugieren que debe tenerse precaución cuando VIRAMUNE es administrado a pacientes con disfunción hepática moderada y que no debe administrarse a pacientes con insuficiencia hepática grave. En general, los resultados sugieren que los pacientes con disfunción hepática de leve a moderada, con una puntuación ≤ 7 de acuerdo con la clasificación Child-Pugh, no requieren un ajuste de la dosis de VIRAMUNE. En pacientes con disfunción renal, que estén siendo sometidos a diálisis, los resultados farmacocinéticos sugieren que el tratamiento suplementario con una dosis adicional de 200 mg de VIRAMUNE después de cada diálisis, ayudaría a contrarrestar los efectos de la diálisis sobre el aclaramiento de la nevirapina. Por otra parte los pacientes con CLcr ≥ 20 ml/min no requieren un ajuste de la dosis de VIRAMUNE (ver 5.2 Propiedades farmacocinéticas ). 4.5 Interacción con otros medicamentos y otras formas de interacción Análogos nucleósidos: No se requieren ajustes de dosis cuando VIRAMUNE se toma en asociación con zidovudina, didanosina o zalcitabina. Al agrupar los datos de zidovudina procedentes de dos estudios (n = 33), en los cuales pacientes infectados por VIH-1 recibieron 400 mg diarios de VIRAMUNE, solos o en combinación con 200-300 mg/día de didanosina o a 0,375 a 0,75 mg/día de zalcitabina con un tratamiento de fondo con zidovudina, la nevirapina produjo un descenso no significativo de un 13 % del área bajo la curva (AUC) de la zidovudina y un aumento no significativo del 5,8 % de la Cmax de la zidovudina. En un subgrupo de pacientes (n = 6), a los cuales se administraron 400 mg/día de VIRAMUNE y didanosina con un tratamiento de fondo con zidovudina, la nevirapina dio lugar a un descenso significativo del 32 % de la AUC de la zidovudina y a un descenso no significativo del 27 % de la Cmax de la zidovudina. Los datos pareados sugieren que la zidovudina careció de efecto sobre la farmacocinética de la nevirapina. En un estudio cruzado, la nevirapina careció de efecto sobre la farmacocinética en estado de equilibrio de la didanosina (n = 18) o la zalcitabina (n = 6). Los resultados de un estudio de 36 días en pacientes infectados por VIH (n = 25), a los que se administró VIRAMUNE, nelfinavir (750 mg tres veces al día) y estavudina (30-40 mg dos veces al día), no mostraron cambios estadísticamente significativos en la AUC ni en la Cmax de la estavudina. Por otra parte, un estudio farmacocinético realizado en una población de 90 pacientes, a la que se administró lamivudina con VIRAMUNE o placebo, no reveló cambios en el aclaramiento aparente ni en el volumen de distribución de la lamivudina, lo que sugiere que la nevirapina no ejerce efecto de inducción sobre el aclaramiento de la lamivudina. Análogos no nucleósidos: Los resultados de un ensayo clínico (n=14) mostraron que en el estado de equilibrio los parámetros farmacocinéticos de la nevirapina no fueron afectados por la administración concomitante del efavirenz. Sin embargo, los niveles de efavirenz se redujeron significativamente en presencia de nevirapina. La AUC del efavirenz disminuyó en un 22 % y la Cmin en un 36%. Cuando se administra de manera conjunta con la nevirapina debe asegurarse que la dosis de efavirenz se incremente a 800 mg una vez al día. Inhibidores de la proteasa: La nevirapina es un inductor ligero a moderado del enzima hepático CYP3A; por lo tanto, es posible que la administración conjunta con inhibidores de la proteasa (también metabolizados por CYP3A) produzca una modificación de las concentraciones plasmáticas de cada uno de ellos. Los resultados de un ensayo clínico (n = 31) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE y saquinavir (cápsulas de gelatina dura, 600 mg 3 veces al día), indicaron que su administración simultánea produce una reducción media del 24 % (p = 0,041) de la AUC del saquinavir y no produce cambios significativos de los niveles plasmáticos de nevirapina. La reducción de los niveles de saquinavir, debidos a esta interacción, pueden reducir adicionalmente los niveles plasmáticos marginales del saquinavir que se alcanzan con la formulación en cápsulas de gelatina dura. Otro estudio (n= 20) evaluó la administración una vez al día de saquinavir cápsula de gelatina blanda con una dosis de 100 mg de ritonavir. Todos los pacientes recibieron VIRAMUNE concomitantemente. El estudio mostró que la asociación de saquinavir cápsula de gelatina blanda y 23/34 100 mg de ritonavir no tuvo un efecto medible sobre los parámetros farmacocinéticos de la nevirapina, comparado con controles históricos. El efecto de la nevirapina sobre la farmacocinética del saquinavir cápsula de gelatina blanda, en presencia de 100 mg de ritonavir, fue discreto y no significativo clínicamente. Los resultados de un ensayo clínico (n = 25) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE e indinavir (800 mg cada 8 horas), indicaron que su administración simultánea produce un descenso medio del 28 % (p < 0,01) de la AUC del indinavir y no produce cambios significativos de los niveles plasmáticos de nevirapina. No se ha llegado a conclusiones clínicas definitivas, respecto al impacto potencial de la administración simultánea de nevirapina e indinavir. Debe considerarse un aumento de la dosis de indinavir a 1.000 mg cada 8 horas si el indinavir se administra junto con nevirapina, 200 mg dos veces al día; sin embargo, actualmente no se dispone de datos que permitan afirmar que la actividad antiviral a corto o a largo plazo de 1.000 mg de indinavir cada 8 horas con 200 mg de nevirapina dos veces al día, diferirá de la de 800 mg de indinavir cada 8 horas con 200 mg de nevirapina dos veces al día. Los resultados de un ensayo clínico (n = 25) en pacientes infectados por VIH, a los cuales se había administrado VIRAMUNE y ritonavir (600 mg dos veces al día [utilizando un régimen de escalado de dosis gradual]), indicaron que su administración conjunta no da lugar a cambios significativos en los niveles plasmáticos de ritonavir o nevirapina. Los resultados de un estudio de 36 días en pacientes infectados por VIH (n = 25), a los que se administró VIRAMUNE, nelfinavir (750 mg tres veces al día) y estavudina (30-40 mg dos veces al día), no demostraron cambios estadísticamente significativos en los parámetros farmacocinéticos del nelfinavir después de la adición de nevirapina (AUC + 4 %, Cmax +14 % y Cmin -2 %). En comparación con los controles históricos, los niveles de nevirapina se mostraron inalterados. No se observó un aumento en el riesgo en relacion a la seguridad en la administración simultánea de VIRAMUNE con cualquiera de estos los inhibidores de la proteasa cuando se prescriben en combinaci ón. No hubo cambio aparente en la farmacocinética del lopinavir cuando se utilizó concomitantemente con VIRAMUNE en voluntarios sanos. En pacientes tratados con un único inhibidor de la proteasa, la nevirapina, utilizada en combinación con lopinavir/ritonavir 400/100 mg (3 cápsulas) dos veces al día y análogos nucleósidos, proporcionaron una tasa de respuesta virológica muy buena. Los resultados de un estudio farmacocinético en pacientes pediátricos revelaron una disminución de las concentraciones de lopinavir durante la administración conjunta con nevirapina. Se desconoce la significación clínica de esta interacción. Sin embargo, debe considerarse un aumento de la dosis de lopinavir / ritonavir de 533/133 mg (4 cápsulas o 6,5 ml) cuando se utilizan asociados con la nevirapina en pacientes donde se sospecha clínicamente (por la historia del tratamiento o la evidencia de laboratorio) una reducción de la sensibilidad a lopinavir / ritonavir. Ketoconazol: En un estudio, la administración de 200 mg de nevirapina dos veces al día con ketoconazol 400 mg cuatro veces al día produjo una reducción significativa (63 % de reducción media de la AUC del ketoconazol y un 40 % de reducción media de la Cmax del ketoconazol). En el mismo estudio, la administración de ketoconazol produjo un aumento del 15-28 % de los niveles plasmáticos de nevirapina en comparación con los controles históricos. El ketoconazol y VIRAMUNE no deben administrarse simultáneamente. No se conocen los efectos de la nevirapina sobre el itraconazol. Fluconazol: La administración simultánea de fluconazol y VIRAMUNE dio como resultado un aumento de la exposición a la nevirapina de aproximadamente el 100%, en comparación con los datos históricos en los que VIRAMUNE se administró solo. Debido al riesgo de una exposición aumentada a la nevirapina, debe procederse con precaución si los medicamentos se administran concomitantemente y los pacientes deben ser estrechamente controlados. No hubo efectos clínicamente relevantes de la nevirapina sobre el fluconazol. 24/34 Anticonceptivos orales: Como los anticonceptivos orales no deben utilizarse como único método anticonceptivo en pacientes infectados por VIH-1, se recomiendan otros métodos anticonceptivos (tales como métodos de barrera) en pacientes tratados con VIRAMUNE. Se ha identificado además una interacción farmacocinética. Se administró 200 mg de nevirapina dos veces al día, conjuntamente con una dosis única de un anticonceptivo oral que contenía 0,035 mg de etinil-estradiol (EE) y 1,0 mg de noretindrona (NET). Comparando las concentraciones plasmáticas observadas antes de la administración de la nevirapina, la AUC media del 17α-EE disminuyó significativamente en un 29 %, a los 28 días de la administración de nevirapina. Hubo una reducción significativa en el tiempo medio de residencia y en la vida media del EE. Hubo una reducción significativa (18 %) en la AUC media de la NET, sin cambios en el tiempo medio de residencia o vida media. La magnitud de este efecto sugiere que la dosis del anticonceptivo oral debe ser ajustada para permitir un tratamiento adecuado de indicaciones distintas a la anticoncepción (p.ej., endometriosis), si se utiliza con la nevirapina. Otros medicamentos metabolizados por CYP3A: La nevirapina es un inductor del CYP3A y potencialmente del CYP2B6, produciendo una inducción máxima al cabo de 2-4 semanas de iniciar la terapia a dosis múltiples. En base al metabolismo conocido de la metadona, la nevirapina puede disminuir las concentraciones plasmáticas de metadona por aumento de su metabolismo hepático. Se ha descrito síndrome de abstinencia a los narcóticos en pacientes tratados con VIRAMUNE y metadona simultáneamente. Los pacientes tratados con metadona, que inicien una terapia con VIRAMUNE, deben ser monitorizados en relación a la aparición de síndrome de abstinencia y debe ajustarse la dosis de metadona adecuadamente. Otros compuestos que son sustratos de CYP3A y CYP2B6 pueden experimentar la disminución de sus concentraciones plasmáticas cuando se administran simultáneamente con VIRAMUNE. Por lo tanto, se recomienda una monitorización cuidadosa de la eficacia terapéutica de los medicamentos metabolizados por P450 cuando se administren en asociación con VIRAMUNE. Inhibidores del isoenzima CYP: Los resultados de un estudio de interacción de nevirapinaclaritromicina (n = 18) indicaron una reducción significativa de la AUC (30 %) y de la Cmax (- 21 %) de la claritromicina, con un incremento significativo de la AUC (58 %) y la Cmax (62 %) del metabolito activo 14-OH claritromicina. La nevirapina presentó un incremento significativo de la Cmin (28 %) y un incremento no significativo de la AUC (26 %) y de la Cmax (24 %). Estos resultados sugieren que no son necesarios ajustes de dosis ni de claritromicina ni de VIRAMUNE cuando se administran conjuntamente ambos medicamentos. No obstante, se recomienda una estricta monitorización de las anomalías hepáticas y de la actividad frente al complejo intracelular del Myobacterium avium (CMA). La monitorización de las concentraciones plasmáticas valle de nevirapina, en pacientes que recibían un tratamiento con VIRAMUNE a largo plazo, reveló que las concentraciones valle de nevirapina estaban elevadas en pacientes que recibían cimetidina (+ 7 %, n = 13). Inductores del isoenzima CYP: Un estudio abierto (n = 14) para determinar los efectos de la nevirapina sobre la farmacocinética en estado de equilibrio de la rifampicina, no reveló cambios significativos en la Cmax ni en la AUC de la rifampicina. Por el contrario, la rifampicina produjo una disminución significativa de la AUC (- 58 %), Cmax (- 50 %) y Cmin (- 68 %) de la nevirapina, en comparación con los datos históricos. Los datos farmacocinéticos disponibles sugieren que el uso concomitante de rifampicina y VIRAMUNE no está recomendado. Por lo tanto, estos medicamentos no deben utilizarse en combinación. Los médicos que necesiten tratar pacientes co-infectados con tuberculosis y que utilicen un régimen que contenga VIRAMUNE, deben en su lugar, considerar la utilización de rifabutina. La rifabutina y VIRAMUNE pueden administrarse conjuntamente sin ajustes de dosis (ver más abajo). Alternativamente los médicos podrían considerar el cambio a una asociación triple de análogos nucleósidos durante un periodo variable de tiempo, dependiendo del régimen de tratamiento de la tuberculosis (ver apartado 4.3 Contraindicaciones). En un estudio farmacocinético la administración simultánea de VIRAMUNE con rifabutina, reveló un aumento no significativo del 12% (media) en la AUC en el estado de equilibrio, una reducción no 25/34 significativa del 3 % en la Cminee y un aumento significativo del 20 % en la Cmaxee. No se detectaron cambios significativos en la AUC, Cminee o Cmaxee de la 25-O-desacetil-rifabutina (metabolito activo de la rifabutina). Se reveló un aumento estadísticamente significativo en el aclaramiento aparente de la nevirapina (9 %) comparado con datos farmacocinéticos históricos. Este estudio sugiere que no hay interacciones clínicamente relevantes entre la nevirapina y la rifabutina. Por lo tanto, los dos fármacos pueden ser administrados simultáneamente sin necesidad de ajuste de la dosis, siempre que se lleve a cabo una monitorización cuidadosa de las reacciones adversas. Warfarina: La interacción entre la nevirapina y el agente antitrombótico warfarina es compleja, con la posibilidad de producirse tanto aumentos como descensos en el tiempo de coagulación cuando se usan concomitantemente. El efecto neto de la interacción puede variar durante las primeras semanas de administración simultánea o en la discontinuación de VIRAMUNE, y por lo tanto, debe garantizarse una estrecha monitorización de los niveles anticoagulación. Hypericum perforatum: Los niveles séricos de la nevirapina pueden reducirse por la utilización concomitante de preparaciones a base de plantas medicinales que contengan Hipérico (Hypericum perforatum). Esto es debido a la inducción de enzimas del metabolismo del fármaco y/o proteínas de transporte producida por la Hipérico. Por lo tanto, las preparaciones a base de plantas medicinales que contengan Hipérico, no deben asociarse con VIRAMUNE. Si el paciente está tomando Hipérico, deben comprobarse los niveles de nevirapina y si es posible la carga viral e interrumpir la administración de Hipérico. Los niveles de nevirapina pueden aumentar al interrumpir la administración de Hipérico. Puede ser necesario un ajuste de la dosis de VIRAMUNE. El efecto inductor puede continuar durante al menos 2 semanas después de la interrupción del tratamiento con Hipérico. Otra información: Los estudios en microsomas hepáticos humanos indicaron que la formación de metabolitos hidroxilados de nevirapina no se vio afectada por la presencia de dapsona, rifabutina, rifampicina y trimetoprim/sulfametoxazol. El ketoconazol y la eritromicina inhibieron significativamente la formación de metabolitos hidroxilados de nevirapina. 4.6 Embarazo y lactancia En estudios sobre la reproducción realizados en conejos y ratas en estado de gestación, no se detectó teratogenicidad observable. No se dispone de estudios adecuados y bien controlados en mujeres embarazadas. Por tanto, VIRAMUNE sólo debe utilizarse durante el embarazo si el beneficio esperado justifica el posible riesgo para el niño y debe tenerse precaución cuando se prescriba VIRAMUNE a mujeres embarazadas. Los resultados de un estudio farmacocinético (ACTG 250) en 10 mujeres embarazadas infectadas por VIH-1, a las cuales se administró una dosis oral única de 100 ó 200 mg de nevirapina 5,8 horas antes del parto por término medio, han demostrado que la nevirapina atraviesa fácilmente la placenta y aparece en la leche materna. Se recomienda que madres con infección por VIH no amamanten a sus hijos para evitar el riesgo de transmisión postnatal del VIH y que interrumpan la lactancia si reciben tratamiento con nevirapina. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas No se han realizado estudios específicos sobre la capacidad para conducir vehículos y utilizar máquinas. 4.8 Reacciones adversas Adultos Los acontecimientos adversos relacionados con el tratamiento con VIRAMUNE descritos con mayor frecuencia durante los ensayos clínicos fueron exantema, náuseas, fatiga, fiebre, cefalea, vómitos, 26/34 diarrea, dolor abdominal y mialgia. En circunstancias muy raras, pueden asociarse casos de anemia y neutropenia al tratamiento con VIRAMUNE. En raras circunstancias, se ha descrito artralgia como un acontecimiento aislado en pacientes que reciben regímenes conteniendo VIRAMUNE. La experiencia post-comercialización ha demostrado que las reacciones adversas más graves son el síndrome de Stevens-Johnson y la necrolisis epidérmica tóxica y la hepatitis grave/insuficiencia hepática y reacciones de hipersensibilidad, caracterizados por exantema con sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral como hepatitis, eosinofilia, granulocitopenia y disfunción renal. Las 8 primeras semanas de tratamiento, constituyen un período crítico que requiere una estrecha monitorización (ver apartado 4.4 Advertencias y precauciones especiales de empleo). La terapia antirretroviral combinada se ha asociado con una redistribución de la grasa corporal (lipodistrofia) en pacientes VIH, que incluye pérdida de grasa subcutánea periférica y facial, aumento de la grasa intra-abdominal y visceral, hipertrofia de las mamas y acumulación de la grasa dorsocervical (joroba de búfalo). La terapia antirretroviral combinada se ha asociado con anomalías metabólicas tales como hipertrigliceridemia, hipercolesterolemia, resistencia a la insulina, hiperglucemia e hiperlactacidemia (véase apartado 4.4 Advertencias y precauciones especiales de empleo). Piel y tejido subcutáneos La toxicidad clínica más frecuente de VIRAMUNE es el exantema, con la aparición de exantema atribuible a VIRAMUNE en el 16% de los pacientes con regímenes combinados en estudios controlados de Fase II/III. En estos ensayos clínicos el 35% de los pacientes tratados con VIRAMUNE experimentaron exantema en comparación con el 19% de los pacientes en los grupos control tratados con zidovudina + didanosina o zidovudina sola. En un 6,6% de los pacientes tratados con VIRAMUNE aparecieron reacciones cutáneas graves o que suponen un riesgo para la vida en comparación con el 1,3% de los pacientes tratados en los grupos control. En conjunto, el 7% de los pacientes interrumpieron el tratamiento con VIRAMUNE a causa del exantema. Por lo general, los exantemas son ligeros a moderados, erupciones cutáneas eritematosas maculopapulares, con o sin prurito, localizados en el tronco, cara y extremidades. Se han descrito reacciones alérgicas (anafilaxis, angioedema y urticaria). Los exantemas se producen en forma aislada o en el contexto de reacciones de hipersensibilidad, caracterizadas por exantema asociado a sintomatología general, como fiebre, artralgia, mialgia y linfadenopatía, además de afectación visceral como hepatitis, eosinofilia, granulocitopenia y disfunción renal. Se han producido reacciones cutáneas graves y que suponen riesgo para la vida en pacientes tratados con VIRAMUNE, incluyendo síndrome de Stevens-Johnson (SSJ) y necrolisis epidérmica tóxica (NET). Se han descrito casos fatales de SSJ, NET y reacciones de hipersensibilidad. La mayoría de los exantemas graves se produjeron durante las primeras 6 semanas de tratamiento y algunos necesitaron hospitalización, precisando un paciente intervención quirúrgica. Hepato-biliares Las anomalías observadas con mayor frecuencia en las pruebas de laboratorio fueron elevaciones en las pruebas de la función hepática (PFHs), incluyendo SGPT, SGOT, GGT, bilirrubina total y fosfatasa alcalina. Las elevaciones asintomáticas de los niveles de GGT son las más frecuentes. Se han descrito casos de ictericia. En pacientes tratados con VIRAMUNE se han producido casos de hepatitis (hepatotoxicidad grave y que supone un riesgo para la vida, incluyendo hepatitis fulminante fatal). En un ensayo clínico extenso, el riesgo de acontecimientos hepáticos graves entre 1.121 pacientes que recibieron VIRAMUNE durante una duración media de más de un año, fue del 1,2 % (frente al 0,6 % en el grupo placebo). El mejor indicio de un acontecimiento hepático grave, fue una elevación de las 27/34 pruebas de función hepática basales. Las primeras 8 semanas de tratamiento constituyen un período crítico que requiere una monitorización estrecha (ver apartado 4.4 Advertencias y precauciones especiales empleo). Pacientes pediátricos En base a la experiencia de 361 pacientes pediátricos tratados en ensayos clínicos, los acontecimientos adversos relacionados con VIRAMUNE descritos con mayor frecuencia fueron similares a los observados en adultos, a excepción de la granulocitopenia que se presentó más frecuentemente en niños. En esta población se han descrito casos aislados de síndrome de Stevens-Johnson o síndrome de Stevens-Johnson/transición a necrolisis epidérmica tóxica. 4.9 Sobredosis No se conoce un antídoto para la sobredosificación de VIRAMUNE. Se han descrito casos de sobredosificación con VIRAMUNE, a dosis entre 800 y 1.800 mg por día, durante un período de hasta 15 días. Los pacientes presentaron edema, eritema nudoso, fatiga, fiebre, cefalea, insomnio, náuseas, infiltraciones pulmonares, exantema, vértigo, vómitos y disminución de peso. Todos estos efectos disminuyeron al interrumpir la administración de VIRAMUNE. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Grupo farmacoterapéutico: agente antiviral, código ATC J05A G01. Mecanismo de acción La nevirapina es un inhibidor no nucleósido de la transcriptasa inversa (NNRTI) del VIH-1. Se une directamente a la transcriptasa inversa y bloquea las actividades ARN-dependientes y ADNdependientes de la ADN polimerasa, produciendo una rotura del punto catalítico del enzima. La actividad de la nevirapina no compite con el patrón ni con los trifosfatos nucleósidos. La transcriptasa inversa del VIH-2 y las ADN polimerasas eucariotas (tales como las ADN polimerasas humanas α, β, γ o δ) no son inhibidas por la nevirapina. Resistencia In vitro aparecen aislados de VIH con sensibilidad reducida (100 a 250 veces) a la nevirapina. Se producen cambios fenotípicos y genotípicos en aislados de VIH de pacientes tratados con VIRAMUNE o VIRAMUNE + zidovudina al cabo de 1 a 12 semanas. A la semana 8 de la monoterapia con VIRAMUNE, el 100 % de los pacientes ensayados presentaban aislados de VIH con una disminución de la sensibilidad a la nevirapina > 100 veces, independientemente de la dosis. La terapia de combinación de VIRAMUNE + zidovudina no alteró el grado de aparición de virus resistentes a la nevirapina. En el estudio INCAS se investigó la resistencia genotípica y fenotípica en pacientes que recibieron VIRAMUNE en el tratamiento combinado en terapia doble o triple y en el grupo comparativo que no recibió VIRAMUNE. Los pacientes sin tratamiento previo con antirretrovirales, con recuentos de células CD4 de 200-600/mm3 se trataron con VIRAMUNE + zidovudina (n = 46), zidovudina + didanosina (n = 51) o VIRAMUNE + zidovudina + didanosina (n = 51) y fueron controlados durante 52 semanas o más de tratamiento. Se realizaron evaluaciones virológicas basales, a los 6 meses y a los 12 meses. La prueba de resistencia fenotípica realizada requirió un mínimo de 1000 copias/ml de ARN VIH con el fin de amplificar el virus. De los 3 grupos en estudio, 16, 19 y 28 pacientes respectivamente, presentaron aislados basales evaluables y posteriormente permanecieron en el estudio durante al menos 24 semanas. Por lo que respecta a los valores basales, hubo cinco casos de resistencia fenotípica a la nevirapina; los valores de CI50 fueron de 5 a 6,5 veces superiores en 3 de los casos y > 100 veces en los otros dos. A las 24 semanas, todos los aislados disponibles recuperables de pacientes que recibieron nevirapina fueron resistentes a este agente, mientras 18/21 (86 %) de los pacientes presentaron estos aislados a las 30-60 semanas. En 16 de los pacientes la eliminación viral estuvo por debajo de los límites de detección (<20 copias/ml = 14, <400 copias/ml = 2). Asumiendo que esta eliminación por debajo de <20 copias/ml, implica 28/34 sensibilidad del virus a nevirapina, el 45 % (17/38) de los pacientes presentaron virus medibles o con sensibilidad atribuible a nevirapina. Todos los 11 pacientes que recibieron VIRAMUNE + zidovudina, en los cuales se ensayó la resistencia fenotípica, fueron resistentes a nevirapina durante los seis meses. Durante el periodo completo de observación, se observó un caso de resistencia a la didanosina. La resistencia a la zidovudina apareció más frecuentemente a las 30 - 60 semanas, especialmente en pacientes que recibieron doble terapia combinada. Basándose en el aumento en la CI50, la resistencia a la zidovudina fue inferior en el grupo de VIRAMUNE + zidovudina + didanosina que en los otros grupos de tratamiento. Por lo que respecta a la resistencia a la nevirapina, todos los aislados secuenciados, presentaron al menos una mutación asociada a resistencia, siendo los cambios simples más comunes el K103N y el Y181C. En 9 de los 12 pacientes observados, se detectaron combinaciones de mutaciones. Estos datos del estudio INCAS reflejan que el uso de tratamientos muy activos está asociado con un retraso en el desarrollo de resistencia a los fármacos antirretrovirales. La relevancia clínica de los cambios fenotípicos y genotípicos asociados al tratamiento con VIRAMUNE no ha sido establecida. Además de los datos presentados anteriormente, existe el riesgo de rápida aparición de resistencia a los NNRTIs en caso de fallo virológico. Resistencia cruzada Se ha observado in vitro una rápida aparición de cepas de VIH que presentan resistencia cruzada a los NNRTIs. Los datos sobre resistencia cruzada entre el NNRTI nevirapina y los análogos nucleósidos inhibidores de la transcriptasa inversa son muy limitados. En cuatro pacientes, los aislados resistentes a la zidovudina ensayados in vitro mantenían la sensibilidad a la nevirapina y, en seis pacientes, los aislados resistentes a la nevirapina eran sensibles a la zidovudina y didanosina. La resistencia cruzada entre nevirapina e inhibidores de la proteasa VIH es improbable, debido a que los enzimas diana involucrados son diferentes. La resistencia cruzada entre los NNRTIs actualmente registrados es extensa. Algunos datos de resistencia genotípica indican que en la mayoría de los pacientes que fracasan con NNRTI , las cepas virales expresan resistencia cruzada a los otros NNRTIs. En la actualidad, los datos disponibles no respaldan el uso secuencial de los NNRTIs. Efectos farmacodinámicos VIRAMUNE ha sido evaluado tanto en pacientes sin tratamiento previo como en pacientes tratados. Los resultados de un ensayo (ACTG 241) evaluaron el tratamiento triple con VIRAMUNE, zidovudina y didanosina en comparación con zidovudina + didanosina, en 398 pacientes infectados por VIH-1 (valores basales medios 153 células CD4+/mm3, ARN VIH-1 en plasma 4,59 log10 copias/ml), los cuales habían recibido, como mínimo, seis meses de tratamiento con análogos nucleósidos antes de la incorporación (media 115 semanas). Estos pacientes, extensamente tratados, demostraron una mejoría significativa en el grupo de terapia triple, en relación con el grupo de terapia doble durante un año, tanto en relación al ARN viral como al recuento de células CD4+. En un ensayo (INCAS), se documentó una respuesta duradera de como mínimo un año para el grupo de terapia triple con VIRAMUNE, zidovudina y didanosina, en comparación con zidovudina + didanosina o VIRAMUNE + zidovudina en 151 pacientes infectados por VIH-1 sin tratamiento previo con recuentos de células CD4+ de 200-600 células/mm3 (media 376 células/mm3) y una concentración basal media de ARN VIH-1 en plasma de 4,41 log10 copias/ml (25.704 copias/ml). Las dosis de tratamiento fueron VIRAMUNE, 200 mg diarios durante dos semanas seguidos de 200 mg dos veces al día, o placebo; zidovudina, 200 mg tres veces al día; didanosina, 125 ó 200 mg dos veces al día (dependiendo del peso). 29/34 VIRAMUNE ha sido también estudiado en asociación con otros agentes antirretrovirales, por ej., zalcitabina, estavudina, lamivudina, indinavir, ritonavir, nelfinavir, saquinavir y lopinavir. No se han descrito problemas de seguridad nuevos y evidentes para estas asociaciones. Se están llevando a cabo estudios para evaluar la eficacia y seguridad de los tratamientos combinados con VIRAMUNE en pacientes en los cuales la terapia con inhibidores de la proteasa no es eficaz. Transmisión perinatal Dos estudios evaluaron la eficacia de VIRAMUNE para prevenir la transmisión vertical de la infección por VIH-1. Durante estos ensayos, las madres recibieron ùnicamente el tratamiento antirretroviral en estudio. En el estudio HIVNET 012 en Kampala (Uganda) se randomizaron pares madre-recién nacido para la administración de VIRAMUNE por vía oral (madre: 200 mg al principio del parto; recién nacido: 2 mg/kg durante las 72 horas posteriores al nacimiento) o un tratamiento ultracorto de zidovudina por vía oral (madre: 600 mg al principio del parto y 300 mg cada 3 horas hasta que éste finaliza; recién nacido: 4 mg/kg dos veces al día durante 7 días). Las tasas de infección por VIH-1 acumuladas en el recién nacido a las 14 - 16 semanas fueron del 13,1 % (n = 310) en el grupo de VIRAMUNE, frente al 25, 1 % (n = 308) en el grupo de tratamiento ultracorto de la zidovudina (p = 0.00063). En el estudio SAINT realizado en Sudáfrica, se randomizaron pares madre-recién nacido para la administración de VIRAMUNE por vía oral (madre: 200 mg durante el parto y 200 mg entre 24 y 48 horas después del parto; recién nacido: 6 mg entre 24 y 48 horas después del parto); o un tratamiento corto de zidovudina y lamivudina por vía oral (madre: 600 mg de zidovudina, luego 300 mg cada 3 horas durante el parto, seguido de 300 mg dos veces al día durante 7 días después del parto, más 150 mg de lamivudina dos veces al día durante el parto y durante 7 días después del mismo; recién nacido: 12 mg de zidovudina dos veces al día más 6 mg de lamivudina dos veces al día durante 7 días [si el peso del recién nacido es <2 kg, 4 mg/kg de zidovudina dos veces al día más 2 mg/kg de lamivudina dos veces al día durante 7 días]). No hubo diferencias significativas en la tasa de transmisión del VIH1 a las 6-8 semanas entre el grupo tratado con VIRAMUNE (5,7%, n = 652) y el grupo tratado con zidovudina más lamivudina (3,6%, n = 649). Hubo un mayor riesgo de transmisión del VIH-1 a recién nacidos cuyas madres habían recibido las dosis de VIRAMUNE o zidovudina más lamivudina menos de 2 horas antes del parto. En el estudio SAINT el 68% de las madres expuestas a nevirapina tuvo cepas resistentes, aproximadamente 4 semanas después del parto. No se ha establecido la relevancia clínica de estos datos en la población Europea. Además, en el caso de que VIRAMUNE se utilice en dosis única para prevenir la transmisión vertical de la infección por VIH-1, el riesgo de hepatotoxicidad en la madre y en el niño no puede ser descartado. Un ensayo clínico ciego, randomizado, en mujeres que ya tomaban tratamiento antirretroviral durante el embarazo (PACTG 316) no demostró una reducción adicional de la transmisión vertical del VIH-1 cuando se administró una dosis única de VIRAMUNE a la madre y al recién nacido, durante el parto y después del nacimiento, respectivamente. Los índices de transmisión VIH-1 fueron similarmente bajos en ambos grupos de tratamiento (1,3% en el grupo de VIRAMUNE, 1,4% en el grupo del placebo). La transmisión vertical tampoco disminuyó en mujeres con ARN VIH-1 por debajo del límite de cuantificación ni en mujeres con ARN VIH-1 por encima del límite de cuantificación antes del parto. De las 95 mujeres que recibieron VIRAMUNE intra-parto, un 15% desarrollaron mutaciones de resistencia a la nevirapina a las 6 semanas después del parto. 5.2 Propiedades farmacocinéticas Adultos La nevirapina se absorbe fácilmente (> 90 %) después de la administración oral en voluntarios sanos y en adultos con infección por VIH-1. La biodisponibilidad absoluta en 12 adultos sanos después de administración de una dosis única fue del 93 ± 9 % (media ± DE) para un comprimido de 50 mg y del 30/34 91 ± 8 % para una solución oral. Las concentraciones pico en plasma de nevirapina, de 2 ± 0,4 µg/ml (7,5 µM), se alcanzaron a las 4 horas después de una dosis única de 200 mg. Después de dosis múltiples, las concentraciones pico de nevirapina parecieron aumentar linealmente dentro del rango de dosis de 200 hasta 400 mg/día. Las concentraciones valle de nevirapina en estado de equilibrio de 4,5 ± 1,9 µg/ml (17 ± 7 µM), (n = 242), se alcanzaron con 400 mg/día. Los comprimidos y la suspensión oral de VIRAMUNE han demostrado ser comparables en cuanto a su biodisponibilidad e intercambiables a dosis de hasta 200 mg. La absorción de la nevirapina no se ve afectada por el alimento, antiácidos o medicamentos formulados con un agente tamponador alcalino (por ej., didanosina). La nevirapina es lipofílica y se encuentra esencialmente no ionizada a pH fisiológico. Después de la administración intravenosa a adultos sanos, el volumen de distribución (Vdss) de la nevirapina fue de 1,21 ± 0,09 l/kg, sugiriendo una amplia distribución en el ser humano. La nevirapina atraviesa fácilmente la placenta y se detecta en la leche materna. La unión a proteínas plasmáticas es de alrededor del 60 % en el intervalo de concentración en plasma de 1-10 µg/ml. Las concentraciones de nevirapina en líquido cefalorraquídeo humano (n = 6) fueron del 45 % (± 5 %) de las concentraciones en plasma; esta proporción es aproximadamente igual a la fracción no ligada a proteínas plasmáticas. Los estudios in vivo realizados en el ser humano y los estudios in vitro en microsomas de hígado humano han demostrado que la nevirapina experimenta una amplia biotransformación por metabolismo vía citocromo P450 (oxidativo) a diversos metabolitos hidroxilados. Los estudios in vitro en microsomas de hígado humano sugieren que el metabolismo oxidativo de la nevirapina es mediado esencialmente por isozimas de citocromo P450 de la familia CYP3A, si bien es posible que otros isozimas jueguen un papel secundario. En un estudio de ajuste de peso/excreción realizado en ocho voluntarios sanos del sexo masculino tratados hasta el estado de equilibrio con 200 mg de nevirapina dos veces al día, seguidos de una dosis única de 50 mg de 14 C-nevirapina, se recuperó aproximadamente el 91,4 ± 10,5 % de la dosis marcada radioactivamente, siendo la orina (81,3 ± 11,1 %) la vía principal de excreción en comparación con las heces (10,1 ± 1,5 %). Más del 80 % de la radioactividad en orina estaba constituida por conjugados glucurónidos de metabolitos hidroxilados. Por consiguiente, el metabolismo por citocromo P450, la conjugación glucurónida y la excreción urinaria de metabolitos glucurónidos representan la vía principal de biotransformación y eliminación de nevirapina en el ser humano. Solamente una pequeña fracción (< 5 %) de la radioactividad en orina (lo que representa < 3 % de la dosis total) correspondió al compuesto original; por lo tanto, la excreción renal juega un papel secundario en la eliminación del compuesto original. La nevirapina ha demostrado ser un inductor de enzimas metabólicos citocromo P450 hepáticos. La farmacocinética de la autoinducción se caracteriza por un aumento de aproximadamente 1,5 a 2 veces del aclaramiento oral aparente de la nevirapina cuando al tratamiento con una dosis única siguen 2-4 semanas de tratamiento con 200-400 mg/día. Asimismo, la autoinducción determina un descenso correspondiente de la vida media de la fase terminal de la nevirapina en plasma desde aproximadamente 45 horas (dosis única) hasta aproximadamente 25-30 horas después de la dosificación múltiple con 200-400 mg/día. Disfunción renal: La farmacocinética de dosis única de nevirapina ha sido comparada en 23 pacientes con disfunción renal leve (50 ≤ CLcr < 80 ml/min), moderada (30 ≤ CLcr < 50 ml/min) o grave (CLcr < 30 ml/min), fallo renal o enfermedad renal en estadío terminal que requiere diálisis, y 8 pacientes con función renal normal (CLcr > 80 ml/min). El fallo renal (leve, moderado y grave) no produce un cambio significativo en la farmacocinética de nevirapina. Sin embargo, los pacientes con enfermedad renal en estadío terminal que requerían diálisis mostraron una reducción del 43,5 % en la AUC de nevirapina después de un periodo de exposición de una semana. Hubo también acumulación de hidroximetabolitos de nevirapina en plasma. Los resultados sugieren que el tratamiento suplementario con una dosis adicional de 200 mg de VIRAMUNE después de cada diálisis, ayudaría a contrarrestar los efectos de la diálisis sobre el aclaramiento de nevirapina. Por otra parte los pacientes con CLcr ≥ 20 ml/min no requieren un ajuste de la dosis de VIRAMUNE. 31/34 Disfunción hepática: La farmacocinética de dosis única de nevirapina ha sido comparada en 10 pacientes con disfunción hepática y 8 pacientes con función hepática normal. En general, los resultados sugieren que los pacientes con disfunción hepática de leve a moderada, con una puntuación ≤ 7 de acuerdo con la clasificación Child-Pugh, no requieren un ajuste de la dosis de VIRAMUNE. Sin embargo, la farmacocinética de nevirapina en un paciente con una puntuación de 8, de acuerdo con la clasificación Child-Pugh y ascitis de moderada a grave, sugiere que los pacientes con empeoramiento de la función hepática pueden tener el riesgo de acumulación de nevirapina en la circulación sistémica. A pesar de que se detectó un volumen de distribución ajustado al peso de la nevirapina, ligeramente superior en las mujeres en comparación con los hombres, no se observaron diferencias significativas ligadas al sexo, en las concentraciones plasmáticas de nevirapina después de la administración de dosis únicas o múltiples. La farmacocinética de la nevirapina en adultos infectados por VIH-1 no parece modificarse con la edad (rango 19-68 años) o raza (negros, hispanos o caucásicos). VIRAMUNE no ha sido investigado específicamente en pacientes de edad superior a 65 años. Pacientes pediátricos Se ha estudiado la farmacocinética de la nevirapina en dos estudios abiertos en niños con infección por VIH-1. En uno de los estudios, fue administrada una dosis única (7,5 mg, 30 mg ó 120 mg por m2; n = 3 por dosis) de VIRAMUNE suspensión oral a nueve niños infectados con VIH, de edades entre 9 meses y 14 años, después de una noche de ayuno. La AUC y la concentración máxima de nevirapina aumentaron en proporción a la dosis. Después de la absorción, las concentraciones plasmáticas medias de nevirapina disminuyeron de forma linealmente logarítmica con respecto al tiempo. La vida media en la fase terminal de la nevirapina, después de una dosis única, fue de 30,6 ± 10,2 horas. En un segundo estudio de dosis múltiple, se administró VIRAMUNE en forma de suspensión o de comprimidos (240 a 400 mg/m2/día) como monoterapia o en asociación con zidovudina o zidovudina y didanosina a 37 pacientes pediátricos infectados por VIH-1, con las siguientes características demográficas: varones (54 %), grupos de minorías raciales (73 %), edad media de 11 meses (intervalo: 2 meses - 15 años). Estos pacientes recibieron 120 mg/m2/día de nevirapina, durante 4 semanas aproximadamente, seguidos de 120 mg/m2/dos veces al día (pacientes > 9 años de edad) ó 200 mg/m2/dos veces al día (pacientes ≤ 9 años de edad). El aclaramiento de la nevirapina, ajustado al peso corporal, alcanzó valores máximos hasta la edad de 1 a 2 años y a partir de entonces disminuyó a medida que aumentaba la edad. El aclaramiento aparente de la nevirapina fue aproximadamente dos veces mayor en los niños menores de 8 años, en comparación con los adultos. La vida media de la nevirapina para el grupo estudiado en su conjunto, desde la administración hasta el estado estacionario, fue de 25,9 ± 9,6 horas. Con la administración del fármaco a largo plazo, los valores medios para la vida media terminal de la nevirapina variaron con la edad del modo siguiente: 2 meses a 1 año (32 horas), 1 a 4 años (21 horas), 4 a 8 años (18 horas) y mayores de 8 años (28 horas). 5.3 Datos preclínicos sobre seguridad Los datos preclínicos no mostraron riesgos especiales para los seres humanos, aparte de los observados en los ensayos clínicos, según los estudios convencionales de seguridad, farmacología, toxicidad de dosis repetidas y genotoxicidad. En los estudios de toxicidad para la reproducción, se observó una fertilidad disminuida en ratas. En estudios de carcinogénesis, la nevirapina induce tumores hepáticos en ratas y ratones. En ratas estos hallazgos están, mayoritariamente, relacionados con la fuerte actividad inductora de enzimas hepáticos de la nevirapina y no son debidos a un mecanismo de acción genotóxico. El mecanismo de los tumores en ratones no está aún clarificado, de modo que está aún por determinar su implicación en el ser humano. 32/34 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Carbómero, parahidroxibenzoato de metilo, parahidroxibenzoato de propilo, sorbitol, sacarosa, polisorbato 80, hidróxido sódico y agua purificada 6.2 Incompatibilidades No aplicable 6.3 Periodo de validez 2 años El medicamento debe ser utilizado dentro de los 2 meses después de abierto. 6.4 Precauciones especiales de conservación No se precisan condiciones especiales de conservación 6.5 Naturaleza y contenido del recipiente Frasco de polietileno de alta densidad (HDPE) blanco, con cierre de dos piezas resistente a los niños (cubierta exterior de polietileno blanco de alta densidad, cubierta interior de polipropileno natural) con obturador de espuma de polietileno de baja densidad (LDPE). Cada frasco contiene 240 ml de suspensión oral. Jeringa dosificadora de 5 ml, de polipropileno transparente, con émbolo de cierre de goma de silicona. Adaptador frasco-jeringa de polietileno de baja densidad transparente. 6.6 Instrucciones de uso y manipulación, y eliminación Los volúmenes de dosificación necesarios deben ser medidos utilizando la jeringa dosificadora y el adaptador, como se describe a continuación en las etapas 1 a 5. El volumen máximo que puede ser medido con la jeringa dosificadora es de 5 ml; por lo tanto, las etapas 3-5 deben ser repetidas para volúmenes de dosificación superiores a 5 ml. 1. Agitar el frasco suavemente 2. Ajustar (presionando primero y después enroscando) el adaptador al cuello del frasco abierto 3. Insertar la jeringa en el adaptador 4. Invertir el frasco 5. Extraer el volumen de dosificación requerido El frasco puede mantenerse cerrado con la tapa flexible del adaptador. La suspensión oral de VIRAMUNE debe ser utilizada dentro de los 2 meses después de abrir el frasco por primera vez. 33/34 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN Boehringer Ingelheim International GmbH Binger Strasse 173 55216 Ingelheim am Rhein, Alemania 8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN EU/1/97/055/002 9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN 17.02.2003 10. FECHA DE LA REVISIÓN DEL TEXTO 34/34