R e v i s i ó n
Anuncio

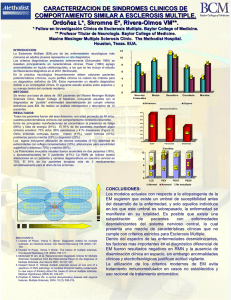
Revisión VOL. 19 / N ÚM. 2 Y 3 / 2000 2000; PP 81-89 INMUNOLOGÍA, Cuestiones sobre la patogenia de la esclerosis múltiple E.M. MARTÍNEZ-CÁCERES, X. M ONTALBÁN Unitat Neuroinmunología Clínica. Hospital Vall d’Hebron. Barcelona ISSUES ON THE PATHOGENESIS OF MULTIPLE SCLEROSIS RESUMEN La esclerosis múltiple (EM) es una enfermedad crónica inflamatoria y desmielinizante del sistema nervioso central de etiología desconocida. Estudios clínicos y en el modelo animal de la enfermedad, la encefalitis autoinmune experimental, sugieren que se trata de una enfermedad autoinmune órgano-específica mediada por linfocitos T autorreactivos CD4 + Th1. Sin embargo, todavía existe un gran número de interrogantes sobre los mecanismos implicados en el desarrollo de la misma. Aunque parece existir una cierta predisposición genética, aún no se han identificado los genes responsables; no se han determinado el/los antígeno/s diana y todavía se desconoce dónde se inicia la reacción autoinmune. Como en otras enfermedades autoinmunes, un buen conocimiento de su etiología y patogenia mejorará la posibilidad de desarrollar estrategias terapéuticas específicas. ABSTRACT Multiple sclerosis is a inflammatory demyelinating chronic disease of the central nervous system. The aethiology of MS is still unknown. Clinical results and experimental evidence in the animal model of the disease, experimental autoimmune encephalomyelitis, support that MS is an organ-specific autoimmune disease mediated by T helper 1 CD4+ autoreactive T cells. However, a number of issues on the mechanisms involved in the pathogenesis of the disease are still to be disclosed. It seems to exist a genetic predisposition, although responsible genes have not yet been identified; the antigen/s triggered have not yet been defined and it is still unknown where the autoimmune response starts. As in other autoimmune diseases, a thorough understanding of disease aetiology and pathogenesis of MS will improve the chances for a more differentiated therapeutic approach. PALABRAS CLAVE: Esclerosis múltiple. Encefalitis autoinmune experimental. Patogenia. KEY WORDS: Multiple sclerosis. Experimental autoimmune encephalomyelitis. Pathogenesis. INTRODUCCIÓN apuntan que el riesgo a sufrir la enfermedad estaría determinado por la combinación de factores genéticos -en los que se verían implicados múltiples genes- (1) y ambientales. A nivel anatomopatológico, los infiltrados inflamatorios en el SNC de estos pacientes están constituídos principalmente por linfocitos T y macrófagos, con destrucción selectiva de las láminas de mielina, aunque la lesión axonal puede ser evidente desde fases tempranas de la enfermedad (2-4). La mayoría de estudios sobre la patogenia de la EM se han realizado en el modelo animal de enfermedad, la encefalitis autoinmune experimental (EAE), y apoyan la hipótesis de que la EM es una L a esclerosis múltiple (EM) es una enfermedad crónica inflamatoria y desmielinizante del Sistema Nervioso Central (SNC). En nuestro medio tiene una prevalencia relativamente alta (60/100.000 habitantes), siendo una de las principales causas de discapacidad en adultos jóvenes. El espectro clínico de la enfermedad es variable. A grandes rasgos se han definido tres formas de evolución: remitenterecurrente, secundariamente progresiva y primariamente progresiva. La etiología de la EM es desconocida y estudios epidemiológicos recientes 81 CUESTIONES SOBRE LA PATOGENIA DE LA ESCLEROSIS MÚLTIPLE VOL. enfermedad autoinmune mediada por linfocitos T CD4+ de tipo Th1 específicos frente a antígenos de la mielina (5,6). La interacción de su TCR con el/los antígeno/s presentado/s por las células presentadoras de antígeno (macrófagos y posiblemente también astrocitos y microglía del SNC) a través de las moléculas HLA-II, induciría su activación, proliferación y secreción de citocinas, las cuales activarían a su vez a otros macrófagos y linfocitos B. Como consecuencia de esta reacción inmunológica se produciría la destrucción de la mielina con la ulterior aparición de manifestaciones clínicas en los pacientes (Fig. 1). Existen muchos interrogantes en relación a los puntos clave de esta reacción del sistema inmune. agente infeccioso era el responsable de causar la enfermedad. Se han sugerido varios mecanismos por los que los virus desencadenarían la enfermedad: —Infección aguda, durante los primeros años de la vida, que se continuaría con una inflamación crónica responsable de la afectación neurológica posterior. —Infecciones virales recurrentes que inducirían destrucción de la mielina de forma inespecífica. —Persistencia de virus en el SNC. Se han identificado secuencias víricas en células del SNC de gran número de pacientes con EM así como en controles sanos. Virus relacionados con la patogenia de la EM han sido, entre otros, el EBV, virus de la hepatitis B, Coronavirus, Paramyxovirus, JC virus o measles. Últimamente los estudios se han centrado en dos grupos de virus, los virus Herpes, concretamente el HHV-6 (7-10) (Tabla I) y los Retrovirus (11-15) (Tabla II). Sin embargo, hasta el momento no existen evidencias de que su presencia indique patogenicidad (16). ¿QUÉ DESENCADENA LA REACCIÓN AUTOINMUNE EN EM? Existe gran controversia sobre si la EM tiene una etiología vírica y el ataque al SNC es una consecuencia de la respuesta inmune hacia el patógeno o si lo que ocurre es una reacción autoinmune desencadenada por diversos factores (tales como reactividad cruzada frente a antígenos víricos o bacterianos, proteínas de estrés o activación por superantígenos) que tendría también como consecuencia el ataque al SNC. Reactividad cruzada con antígenos víricos o bacterianos en individuos genéticamente susceptibles Las reacciones cruzadas entre patógenos y componentes propios se han propuesto como una vía por la que se puede iniciar la respuesta autoinmune frente a antígenos de la mielina. De hecho, existen datos epidemiológicos relacionando las fases de exacerbación de la EM con infecciones virales. Hipótesis vírica La hipótesis vírica parte de muy antiguo. Pierre Marie, en 1884, fue el primero en proponer que un Blood-brain barrier Blood Central nervous system Activated myelin-reactive CD4+ T cell HelperT cell T Activated CD4+ T cell Th Microglia astrocyte (APC) T IL-2 Antigen TGFβ Th1 IFNγ TGFβ T IL-4, 5, 6 IL-4, 10 MO Th2 IL-2 LT TGFα NO SupressorT cell Activated microglia/MO B B TGFβ Antibodies TGFα OH NO C’ C5b-9 T SupressorT cell Antibodies Figura 1. Patogenia de la esclerosis múltiple (según Hartung et al., 1995) (5). 82 19 NÚM. 2 Y 3 / 2000 Myelin INMUNOLOGÍA E.M. MARTÍNEZ-CÁCERES ET AL Tabla I HHV-6 y esclerosis múltiple Challoner PB et al (7) Soldan SS et al (8) Sanders VJ et al (9) Mayne M et al (10) Aparece HHV-6 en el 70% de EM y controles. En los OLD sólo aparece en EM. Relaciona EM con HHV-6 y destrucción de los OLD Demuestran incremento de respuesta IgM anti p41/38 de HHV-6 en EM remitente-recurrente. Detectan DNA de HHV-6 en suero de pacientes. Detectan la presencia de virus HHV-6 en cerebros de pacientes con EM y controles Falta de correlación entre la infección de PBMC por HHV-6 y el desarrollo de EM Wucherpfenning y Strominger (17) identificaron 129 péptidos víricos y bacterianos capaces de simular estructuralmente al péptido PMB 85-99 de la proteína mielínica básica (PMB). De estos péptidos candidatos, siete virales y uno bacteriano activaron de forma eficiente linfocitos T específicos frente a la PMB. Martin y cols., realizando sustituciones de aminoácidos individuales en cada posición de la secuencia del péptido inmunodominante PMB 83-99 observaron que algunas sustituciones no cambiaban la respuesta funcional de los clones testados, mientras que otras causaban una reducción o abolición de la respuesta (18) o incluso se obtenían péptidos "superagonistas", estimuladores más potentes que el péptido nativo de la PMB (19). Además, el efecto de sustituciones "negativas" pudo ser compensado por sustituciones "positivas" en otras posiciones (20,21) e incluso se diseñaron péptidos que diferían en todas las posiciones de la secuencia antigénica nativa y eran capaces de estimular los clones específicos (20), sustentando la hipótesis de que no se requiere necesariamente una homología de secuencia para la reactividad cruzada (22). A pesar de los datos in vitro, hasta el momento hay muy pocas evidencias in vivo de inducción de EAE basadas en reactividad cruzada (23,24). Activación de células T específicas frente a antígenos propios por superantígenos Los superantígenos son antígenos virales o bacterianos capaces de estimular poblaciones com- pletas de células T por unión a la cadena Vβ del TCR y a moléculas HLA-II de las APC y que inducen la activación de ambos tipos celulares (25). Se ha sugerido que los superantígenos podrían estar involucrados en la patogenia de las enfermedades autoinmunes al activar linfocitos T autorreactivos que expresen una cadena Vβ determinada. En EAE se ha demostrado que superantígenos de estafilococo son capaces de producir exacerbaciones de la enfermedad (26), pero no son suficientes para inducirla. Según estos resultados, los superantígenos no estarían implicados en el inicio de la respuesta sino en fases posteriores. Por otro lado, hay autores que defienden que podrían actuar eliminando linfocitos T reguladores y, por tanto, favoreciendo la reacción autoinmune. Reactividad cruzada con proteínas del shock térmico (heat shock proteins) Se ha demostrado la presencia de proteínas de estrés (hsp) en lesiones de EM (27). También se ha encontrado respuesta T y B específica frente a hsp (28). Existe una familia particular de hsp de bajo peso molecular, denominada hsp25 que parece ser única de células eucariotas. En humanos se han descrito dos miembros, la hsp27 y la αB-cristallin (29). Una hipótesis acerca del posible papel de la hsp de bajo peso molecular αB-cristallin en la patogenia de la EM ha sido recientemente postulada por Van Noort y cols. (30). Según estos autores, una infección vírica en la infancia provocaría Tabla II Retrovirus y esclerosis múltiple Ehrlich GD et al (11) Rassmussen HB et al (12) Hackett J Jr et al (13) Perron H et al (14) Brahic M et al (15) No encuentra relación entre HTLV-I , HTLV-II u otros agentes con EM Analizan el retrovirus endógeno ERV3. No encuentran diferencias entre EM y controles. No encuentran evidencia de retrovirus en suero, PBMC o LCR de pacientes con EM. Caracterizan parcialmente un retrovirus (LM7) asociado a EM. Se le denomina MSRV (MS related virus). Enfatizan que secuencias casi idénticas a MSRV se encuentran tanto en EM como en controles. 83 CUESTIONES SOBRE LA PATOGENIA DE LA ESCLEROSIS MÚLTIPLE la inducción de αB-cristallin en las células infectadas (i.e. oligodendrocitos) junto con la aparición de una respuesta T específica frente a ella. En un momento posterior de la vida, cuando se desencadenase una reacción similar con expresión de esta molécula (en presencia o ausencia de patógeno), estos linfocitos específicos que ya formarían parte del repertorio T podrían desencadenar la reacción autoinmune. Desequilibrio en los mecanismos de control de la respuesta inmune, favoreciendo la pérdida de la tolerancia a antígenos propios En condiciones normales existen en periferia precursores de células potencialmente autorreactivas que probablemente han escapado a los procesos de selección tímica por presentar TCR de baja afinidad. Además, experimentalmente, en el modelo de EAE, Lafaille y cols. (31) han demostrado que la mayoría de ratones transgénicos con TCR específico frente a la PMB no desarrollan la enfermedad espontáneamente a pesar de la presencia de numerosas células T autorreactivas. Todos estos datos apoyan la existencia de mecanismos a nivel periférico capaces de controlar la respuesta autoinmune. En este sentido, se ha demostrado recientemente en EAE que una subpoblación de linfocitos T CD4 + reguladores, previene el desarrollo de la enfermedad ya que su ausencia desencadena la reacción autoinmune (31-33). Por otro lado, la liberación de citocinas y otros factores solubles en el curso de determinadas infecciones podrían favorecer de forma inespecífica la activación de células potencialmente autorreactivas (34,35) produciendo una respuesta autoinmune. ¿DÓNDE SE INICIA LA RESPUESTA AUTOINMUNE? Unos autores defienden que la respuesta autoinmune se iniciaría dentro del SNC. Otros, sin embargo, consideran que la activación inicial ocurre fuera del SNC. Dentro del SNC Neumann y cols. (36) defienden que debe haber una alteración primaria de la neurona para que se desarrolle EM. En un sistema experimental de cultivo mixto de neuronas/glía, observaron que la glía sólo expresaba moléculas MHC-II tras estimulación con IFN-γ si se encontraba físicamente distante de neuronas eléctricamente activas. Al añadir tetrodotoxina (TTX) a los cultivos para inhibir 84 VOL. 19 NÚM. 2 Y 3 / 2000 el potencial de acción de las neuronas, las células gliales expresaron MHC-II en todo el cultivo. Al mismo tiempo, Demerens y cols. (37) demostraron que la producción de PMB por los oligodendrocitos (OLD) depende de la actividad eléctrica neuronal, ya que el tratamiento con TTX inhibía la mielinización. Otros autores postulan que el primer acontecimiento que ocurre en la EM es la interferencia funcional y/o el daño sobre la capacidad de mielinización del OLD (38). Inicialmente, este hecho no se asociaría a alteraciones morfológicas, pero posteriormente se produciría la degeneración de la mielina. Como consecuencia de ello, epítopos antigénicos que no eran accesibles al sistema inmune (epítopos crípticos) podrían ser presentados por la microglía, astrocitos o células endoteliales en el contexto de determinadas moléculas HLA. Tras la entrada en el SNC de linfocitos T capaces de reconocer esos antígenos crípticos, se iniciaría la reacción autoinmune. Los linfocitos T citotóxicos, macrófagos, citocinas y complemento aumentarían el proceso patológico de daño al OLD y/o inhibición de la función de los axones desnudos. Fuera del SNC El SNC ha sido considerado un sistema "privilegiado" desde el punto de vista inmunológico, ya que tiende a reducir y limitar las reacciones inmunitarias locales. Esto es debido a que una reacción inflamatoria importante en el SNC podría producir secundariamente una destrucción irreversible de las células nerviosas. Este efecto lo consigue de varias formas: por una expresión muy baja de moléculas HLA-I y II, poca eficiencia -en condiciones normales- en la presentación antigénica y existencia de factores solubles en el LCR capaces de neutralizar o contrarrestar el efecto de determinadas citocinas pro-inflamatorias. En este contexto, el SNC no sería un lugar adecuado para iniciar una respuesta autoinmune que requiere de gran eficacia, por lo que gran número de autores sugieren que el proceso de desmielinización se iniciaría tras la entrada en el SNC de los linfocitos activados (2-6, 39). En este sentido, se han encontrado linfocitos activados en sangre periférica de pacientes con EM en brote con características fenotípicas similares a linfocitos encefalitogénicos descritos en EAE (40) (Barrau y cols. pendiente de publicación). Como conciliación de ambas hipótesis, se postula que como consecuencia de una afectación inicial dentro del SNC se produciría una salida de antígeno a través de ciertos nervios craneales (primariamente olfatorios) hacia los ganglios linfáticos cervicales (41), donde se realizaría una presentación antigénica eficiente. Los linfocitos activados específicos para esos antígenos migrarían al INMUNOLOGÍA SNC donde iniciarían la reacción autoinmune. Detractores de esta hipótesis argumentan que resultaría poco eficaz desde el punto de vista inmunológico, ya que las células, una vez activadas, tendrían que realizar un largo recorrido y atravesar la barrera hematoencefálica (BHE) antes de llegar al lugar de actuación. ¿QUÉ ELEMENTOS SON IMPORTANTES EN EL DESARROLLO DE LA RESPUESTA AUTOINMUNE EN EL SNC? Una vez desencadenada la respuesta autoinmune la cadena de acontecimientos y los elementos que intervienen son básicamente los mismos que en cualquier tipo de respuesta no autoinmune, aunque con las peculiaridades propias del órgano diana, el SNC, y la existencia de la BHE. Muchos investigadores han tratado de analizar exhaustivamente cada uno de los elementos de esta cadena para poder evaluar la importancia relativa de determinados tipos celulares o determinadas citocinas y así bloquear la respuesta lo más específicamente posible. De hecho, se ha demostrado en EAE la eficacia de tratamientos con anticuerpos monoclonales anti-citocinas, antimoléculas de adhesión y anti-moléculas coestimuladoras, así como la administración de citocinas con un posible efecto inmunomodulador, como el interferón-ß, el único tratamiento para la EM aceptado hasta el momento en nuestro país. Siguiendo estrategias similares, en la actualidad se están realizando diversos ensayos clínicos en pacientes. Por razones prácticas sólo discutiremos brevemente el papel de los linfocitos T CD4 +, las moléculas HLA y los antígenos candidatos. E.M. M ARTÍNEZ-CÁCERES ET AL la aparición de brotes, detener la progresión de la enfermedad o prevenir la formación de nuevas lesiones (45,46). Es por ello, por lo que se comenzó a pensar en otros tipos celulares que pudieran ser importantes en la EM. Hasta el momento, se sabe muy poco de la contribución de los linfocitos CD8+, linfocitos T γδ y células NK a la patogenia de la enfermedad (47-50). Restricción del repertorio T en EM Para demostrar si los linfocitos T de pacientes con EM muestran una especificidad antigénica limitada, se han estudiado las regiones variables (V) del TCR. En la EAE se ha demostrado que un número restringido de genes Vß puede ser importante en el desarrollo de la enfermedad ya que la administración de anticuerpos monoclonales frente a estas regiones previene la EAE (51,52). Sin embargo, en EM existen controversias sobre si el repertorio de los genes del TCR es restringido. Se han realizado estudios con linfocitos T específicos frente a la PMB, tanto de sangre periférica, LCR de pacientes (53,54) como en placas de esclerosis (55) y se ha encontrado restricción. Sin embargo, otros autores demostraron un amplio repertorio de genes Vα y Vß en las lesiones (56). Meinl y cols. (57) demostraron la presencia de diversos genes Vß en paneles de líneas T obtenidas de la misma persona y específicas frente al mismo epítopo de la PMB. Otros autores sugieren que en un mismo paciente el repertorio sería restringido al principio de la enfermedad, pero posteriormente, se produciría un incremento de reactividad frente a nuevos determinantes ("determinant spreading") y el repertorio se ampliaría (58-60). HLA y esclerosis múltiple Linfocitos T CD4+ en EM En base a las similitudes entre EAE y EM, se ha sugerido que la EM es también una enfermedad autoinmune mediada por linfocitos T CD4 +. Numerosos autores se han centrado en el estudio de la reactividad de estas células frente a antígenos de la mielina. Se ha demostrado un incremento en la frecuencia de precursores reactivos frente a PMB y proteína proteolipídica (PLP) en pacientes con EM (42,43). Sin embargo, otros autores defienden que podría tratarse de linfocitos expandidos secundariamente a la afectación de la mielina (44). Además, está ampliamente demostrado que existen linfocitos T frente a antígenos de la mielina en el repertorio de linfocitos T de individuos sanos (44). Por otra parte, el tratamiento con anticuerpos anti-CD4 inhibe la EAE en ratones y primates. En la EM, la utilización de estos anticuerpos no evitó Por medio de técnicas de tipificación de "alta resolución" se ha encontrado asociación entre el haplotipo HLA DR2 y más especificamente el DRB1*1501 DQA1*0102 DQB1*0602 y la EM (61). Otros estudios apuntan que las formas primariamente progresivas de la enfermedad estarían más asociadas al DR4. Sin embargo, investigaciones recientes dirigidas a identificar regiones genómicas que confieran predisposición a EM muestran que no existe ningún locus concreto que contribuya significativamente al riesgo familiar a padecer la enfermedad (62,63), aunque existe una asociación débil/moderada con ciertos loci. Haines y cols. (64) han identificado 19 regiones; los datos confirman que la región situada en la posición q21.1-21.3 del cromosoma 6 y que contiene los genes codificantes del sistema HLA parece hallarse directamente implicada en la etiología de la EM. Sin embargo, Kuokkanen y cols. (65), en un estu- 85 CUESTIONES SOBRE LA PATOGENIA DE LA ESCLEROSIS MÚLTIPLE dio realizado en la población finesa relacionan la EM con un locus situado en la región 5p14-p12, mientras que Sawcer y cols. (66) encuentran dos regiones relacionadas con la EM: 17q22 y 6p21 (HLA). Existen, pues, divergencias respecto a la importancia relativa de los loci que contienen los genes que codifican el HLA, según los grupos, pero en conjunto parece admitirse que existe una asociación moderada. ¿Cuáles son los antígenos diana en la EM? Se han descrito gran número de posibles antígenos implicados en la patogenia de la EAE/EM. Hasta muy recientemente, los estudios se centraron en dos antígenos: PMB y PLP, las dos proteínas más abundantes de la mielina del SNC. Sin embargo, a pesar del gran número de trabajos, no se ha podido demostrar claramente su función como antígenos diana de la enfermedad. Esto ha dado lugar a que en los últimos años se hayan investigado otros posibles candidatos. Los más estudiados son los antígenos proteicos de la mielina. Entre ellos, aparte de la PMB y la PLP (67), se incluyen la transaldolasa 2´3´cyclic nucleotide 3´phosphodiesterase (CNPase) (68), dos miembros de la superfamilia de las inmunoglobulinas -la glicoproteína mielínica oligodendroglial (MOG) y la glicoproteína asociada a la mielina (MAG) (69) - y la hsp αB-cristallin, antígeno no específico de la mielina (29). Otro antígeno candidato es la proteína S100-ß (70), ampliamente expresada en astroglía pero no en oligodendrocitos. Antígenos mielínicos no proteicos, como los glucocerebrósidos (71) y los gangliósidos (72,73) también han sido considerados como candidatos en EM /EAE. Cada antígeno induce un curso clínico, una topografía y una composición de las lesiones diferente y se han encontrado diferencias en la susceptibilidad a estos antígenos en diferentes modelos de EAE (74,75). Antígenos muy abundantes en la mielina como PMB o PLP se localizan en las capas gruesas de mielina; MOG, antígeno localizado en la superficie de la mielina, se localiza preferentemente en zonas donde las capas de mielina son muy finas; la transferencia pasiva de células específicas frente a S100-ß induce lesiones en el córtex cerebral, retina y úvea, además de en la sustancia blanca. La composición celular de los infiltrados también varía. Por ejemplo, en lesiones inducidas tras inmunización con PMB predominan los macrófagos y en las inducidas por S100-ß, los linfocitos T. Hay antígenos que inducen una elevada respuesta inflamatoria (S100-ß) mientras que otros inducen gran desmielinización (MOG). La inmunización con PMB en rata Lewis induce un curso clínico agudo, el péptido 135-155 de la PLP un curso remitente-recurrente en ratón SJL mientras que la inmunización con MOG en rato- 86 VOL. 19 NÚM. 2 Y 3 / 2000 nes susceptibles H-2b induce una enfermedad de curso crónico no-remitente. Por otro lado, S-100ß, aún induciendo gran inflamación a nivel del SNC, no induce clínica de EAE. Trasladadas a la patogenia de la EM, estas observaciones podrían ayudar a explicar la heterogeneidad de la enfermedad (76). Como en la EAE, esta variabilidad podría reflejar patrones individuales en respuesta al mismo antígeno, una respuesta variable a diferentes antígenos, o ambas cosas. Además, como se ha comentado anteriormente, diferentes autoantígenos podrían estar implicados en diversos estadios de la enfermedad, por ampliación del número de determinantes antigénicos ("determinant spreading"). En este sentido, trabajos recientes tanto a nivel clínico, como radiológico (resonancia magnética) (77), inmunológico (78) o anatomo-patológico (79), comienzan a ilustrar la existencia de diferentes "subgrupos" dentro de la EM. CONTROVERSIA Th1/Th2. LINFOCITOS B Y ANTICUERPOS. PAPEL QUE JUEGAN LOS AUTOANTICUERPOS EN LA DESMIELINIZACIÓN-REMIELINIZACIÓN Como ya se ha comentado anteriormente, basándose en el modelo de EAE, se ha considerado la EM como una enfermedad autoinmune de tipo "Th1". Sin embargo, estudios recientes han mostrado que los linfocitos B y los anticuerpos producidos por ellos son de gran importancia, especialmente en el proceso de desmielinización (70,80-83). Los linfocitos Th2 juegan un papel importante en la activación de los linfocitos B. Es por ello por lo que últimamente se está replanteando por algunos grupos de investigación el concepto de EM como enfermedad "Th1" y por tanto, se está poniendo en tela de juicio la utilización de tratamientos que induzcan "desviación inmune" hacia Th2 (82). Se postula que en la EM los linfocitos Th1 específicos frente al/los autoantígeno/s diana iniciarían el proceso inflamatorio y la apertura de la BHE y posteriormente se produciría la activación de los linfocitos B productores de autoanticuerpos que tendrían un papel preponderante en la desmielinización (83). No obstante, existe una gran controversia en relación al papel de los autoanticuerpos en la EM y EAE (84,85). De hecho, ha sido postulado por algunos (86) que inmunoglobulinas dirigidas frente a componentes del SNC pueden promover la proliferación de los oligodendrocitos. En el modelo de EAE inducida por virus Theiler, se ha demostrado que una serie de anticuerpos monoclonales frente a antígenos del OLD promueven la remielinización (87-89). En vista de estos resultados en la actualidad se están llevando a cabo ensayos clínicos multicéntricos para evaluar el poten- INMUNOLOGÍA cial de un "pool" de inmunoglobulina policlonal humana para promover la remielinización en la EM (90). E.M. M ARTÍNEZ-CÁCERES ET AL 2. 3. RESUMEN Y PERSPECTIVAS DE FUTURO La EM es considerada una enfermedad autoinmune del SNC. Sin embargo, de los puntos anteriormente expuestos se desprende que aún quedan muchas cuestiones pendientes sobre su patogenia. No se ha demostrado una predisposición genética clara; no se ha/n establecido el/los antígenos diana y se desconoce qué desencadena y dónde se inicia la reacción autoinmune. Además, existen referencias contradictorias sobre el papel de los autoanticuerpos en la desmielinización y/o remielinización. Por otro lado, los resultados obtenidos de los diferentes modelos de EAE y en pacientes comienzan a revelar una gran heterogeneidad dentro de la enfermedad que hoy denominamos esclerosis múltiple y que puede explicar en parte los datos controvertidos entre los diferentes autores. Lógicamente, existe un gran interés en encontrar tratamientos efectivos para esta enfermedad. El único tratamiento (no sintomático) aceptado hasta el momento en nuestro país es el IFN-β, que ha demostrado ser efectivo en un subgrupo de pacientes, disminuyendo la frecuencia e intensidad de los brotes, por un mecanismo de acción no bien caracterizado. Sin embargo, como en otras enfermedades, un tratamiento racional es dependiente de un extenso conocimiento de la patogenia. Una clasificación más detallada de la enfermedad, basada tanto en criterios clínicos, radiológicos como inmunológicos puede ser de gran utilidad para poder entender mejor los mecanismos implicados en la misma y mejorar la probabilidad de desarrollar aproximaciones terapéuticas más específicas. CORRESPONDENCIA Eva Mª Martínez-Cáceres Unitat de Neuroinmunología Clínica Hospitals Vall d’Hebron Pg. Vall d’Hebron 119-129 08035 Barcelona Tel.: 93 274 62 02 Fax: 93 274 60 84 e-mail: emart@hg.vhebron.es 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. Bibliografía 1. Compston A. Genetic susceptibility to multiple sclerosis. In: A. Compston(ed) McAlpine´s Multiple Sclerosis; 3rd Edition; Churchill Livingstone, London, 1998; 101-144. 20. Raine CS. Demyelinating diseases. In Textbook of N e u ro p a t h o l o g y. RL Davis and DM Robertson, editors. Williams and Wilkins, Baltimore, MD, 1992; 535-552. Wekerle H. T-cell autoimmunity in the central nervous system. Intervirology 1993; 35: 95-100. Lassmann H. Pathology of Multiple sclerosis. In: A. Compston(ed) McAlpine´s Multiple Sclerosis; 3rd Edition; Churchill Livingstone, London, 1998; 323358. Hartung HP. Pathogenesis of inflammatory demyelination: implications for therapy. Curr Op Neurol 1995; 8: 191-199. Martin R, McFarland HF, McFarlin D. Immunological aspects of demyelinating diseases. Annu Rev Immunol 1992; 10: 153-187. Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A 1995; 92: 7440-7444. Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, et al. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response to HHV-6 early antigen and detection of serum HHV6 DNA. Nat Med 1997; 3: 1394-1397. Sanders VJ, Felisan S, Waddell A, Tourtellotte WW. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol 1996 Aug; 2(4): 249-258. Mayne M, Krishnan J, Metz L, Nath A, Auty A, Sahai BM, et al. Infrequent detection of human herpesvirus 6 DNA in peripheral blood mononuclear cells from multiple sclerosis patients. Ann Neurol 1998 Sep; 44(3): 391-394. Ehrlich GD, Glaser JB, Bryz-Gornia V, Maese J, Waldmann TA, Poiesz BJ, et al. Multiple sclerosis, retroviruses, and PCR. The HTLV-MS Working Group. Neurology 1991; 41: 335-343. Rasmussen HB, Heltberg A, Lisby G, Clausen J. Three allelic forms of the human endogenous retrovirus, ERV3, and their frequencies in multiple sclerosis patients and healthy individuals. Autoimmunity 1996; 23: 111-117 Hackett J Jr, Swanson P, Leahy D, Anderson EL, Sato S, Roos RP, et al. Search for retrovirus in patients with multiple sclerosis. Ann Neurol 1996; 40: 805-809. Perron H, Garson JA, Bedin F, Beseme F, ParanhosBaccala G, Komurian-Pradel F, et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci U S A 1997; 94: 7583-7588. Brahic M, Bureau JF. Multiple sclerosis and retroviruses. Ann Neurol 1997; 42: 984-985. Monteyne P, Bureau JF, Brahic M. Viruses and multiple sclerosis. Curr Opin Neurol 1998 Aug; 11(4): 287-91. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell mediated autopimmunity: viral peptides activate human T cell clones specific for MBP. Cell 1995; 80: 695-705. Vergelli M, Hemmer B, Utz U, et al. Differential activation of human autoreactive T cell clones by altered peptide ligands derived from myelin basic protein peptide (87-99). Eur J Immunol 1996; 26: 2624-2634. Vergelli M, Hemmer B, Kalbus M, Vogt AB, Ling N, Conlon P, et al. Modifications of peptide ligands enhancing T cell responsiveness implylarge numbers of stimulatory ligands for autoreactive T cells. J Immunol 1997 Apr 15; 158(8): 3746-3752. Hemmer B, Vergelli M, Gran B, Ling N, Conlon P, Pinilla C, et al. Predictable TCR antigen recognition based on peptide scans leads to the identification of agonist ligands with no sequence homology. J Immunol 1998; 160: 3631-3636. 87 CUESTIONES SOBRE LA PATOGENIA DE LA ESCLEROSIS 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 88 VOL. MÚLTIPLE Ausubel LJ, Kwan CK, Sette A, Kuchroo V, Hafler DA. Complementary mutations in an antigenic peptide allow for crossreactivity of autoreactive T-cell clones. Proc Natl Acad Sci U S A 1996; 93: 15317-15322. Gran B, Hemmer B, Vergelli M, McFarland HF, Martin R. Molecular mimicry and multiple sclerosis: degenerate T-cell recognition and the induction of autoimmunity. Ann Neurol 1999; 45: 559-567. Fujinami RS, Oldstone MBA. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanisms for autoimmunity. Science 1995; 230: 1043-4105 Gautam AM, Liblau R, Chelvanayagam G, et al. A viral peptide with limited homology to a self peptide can induce clinical signs of experimental autoimmune encephalomyelitis. J Immunol 1998; 161: 60-64. Fleischer, B. Superantigens (review). APMIS 1994; 102: 3-12 Brocke S, Gaur A, Piercy C, et al. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigens . Nature 1993; 365: 642-4. Bajramovic JJ, Lassmann H, Van Noort JM. Expression of alpha B-cristallin during lesional development in multiple sclerosis. J. Neuroimmunol. 1997; 78: 143-151. Fisher HP, Sharrock CE, Panayi GS. High frequency of cord blood lymphocytes against the mycobacterial 65kDa heat-shock protein. Eur J Immunol 1992; 1667-1672. Van Noort JM, van Sechel AC, Bajramovic JJ, El Ouagmiri M, Polman CH, Lassmann H, et al. The small heat shock protein alpha-B cristallin as a candidate autoantigen in multiple sclerosis. Nature 1995; 375: 798-801. Van Noort JM, Amor S. Cell biology of autoimmune diseases. Int Rew Cytology 1998; 178: 127-206. Lafaille JJ, Nagashima K, Katsuki M , Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell 1994; 78: 399-408. Van de Keere F, Tonegawa S. CD4+ T cell prevent spontaneous experimental autoimmune encephalomyelitis in anti-myelin basic protein T cell receptor transgenic mice. J Exp Med 1998; 188: 1875-1882. Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med 1998; 188: 1883-1894. Sun D, Coleclough C, Cao L, Hu X, Sun S, Whitaker JN. Reciprocal stimulation between TNF-alpha and nitric oxide may exacerbate CNS inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol 1998; 89: 122-130. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type 1 IFN in vivo. Science 1996; 272: 1947-1950. Neumann H, Boucraut J, Hahnel C, Misgeld T, Wekerle H. Neuronal control of MHC class II inducibility in rat astrocytes and microglia. Eur J Neurosci 1996; 8: 25822590. Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, et al. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci U S A 1996; 93: 9887-9892. Rodriguez M, Scheithauer BW, Forbes G, Kelly PJ. O l i g o d e n d rocyte injury is an early event in lesions of multiple sclerosis. Mayo Clin Proc 1993; 68: 627636. Prineas JW, Raine CS. Electron microscopy and immunoperoxidase studies of early multiple sclerosis lesions. Neurology (1976); 26: 29-32 Qin Y, Van den Noort S, Kurt J, Gupta S. Dual expression of CD45RA and CD45RO isoforms on myelin basic protein-specific CD4+ T-cell lines in multiple sclerosis. J Clin Invest 1993; 13: 152-161. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 19 NÚM. 2 Y 3 / 2000 Cserr HF, Harling-Berg CJ, Knopf FM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol 1992; 2: 269-276. Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cell responsive to mielin basic protein in patients with multiple sclerosis . Science 1990; 247: 718-721. Lodge PA, Johnson C, Sriram S. Frequency of MBP and MBP peptide-reactiva T cells in the HPRT mutant T cell population of MS patients. Neurology 1996; 1410-1415. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature 1990; 346: 183-187. Lindsey JW, Hodgkinson S, Metha R , Mitchell D, Enzmann D, Steinmann L. Repeated treatment with chimaeric anti-CD4 antibody in multiple sclerosis. Ann Neurol 1994; 36: 183-189. Van Oosten BW, Lai M, Hodkingson S, Barkhof F, Miller DH, et al. Treatment of MS with the monoclonal antiCD4 antibody cM-T412: results of a randomized, double blind, placebo-controlled, MR-monitored phase II trial. Neurology 1997; 49: 351-357. Biddison WE, Taub DD, Cruikshank WW, Center SM, Connor EW, Honma K. Chemokine and matrix metalloproteinase secretion by mielin proteolipid protein-specific CD8+ T cells. Potential roles in inflammation. J Immunol 1997; 158: 3046-3053. Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol 1998; 160: 5955-5962. Zeine R, Pon R, Ladiwala U, Antel JP, Filion LG, F reedman MS. Mechanism of gammadelta T cellinduced human oligodendrocyte cytotoxicity: re l evance to multiple sclerosis. J Neuroimmunol 1998; 87: 49-61. Kastrukoff LF, Morgan NG, Zecchini D, White R, Petkau AJ, Satoh J, et al. A role for natural killer cells in the immunopathogenesis of multiple sclerosis. J Neuroimmunol 1998; 86: 123-133. Acha-Orbea H, Mitchell DJ, Timmermann L, et al. Limited heterogeneity of T cell receptors fro m l y mphocytes mediating autoimmune encephalomyelitis allows specific immune intervention. Cell 1988; 54: 263-273. Owhashi M, Heber-Katz E. protection from EAE conferred by a monoclonal antibody directed against a shared idiotype on rat T cell receptors specific for MBP. J Exp Med 1988; 168: 2153-2164. Kotzin BL, Karuturi S, Chou YK, et al. Preferential T cell receptor beta chain variable gene use in MBP -reactive T cell clones from patients with multiple sclerosis. Proc Natl Acad Sci USA 1991; 88: 9161-9165. Zang YC, Kozovska M, Aebischer I, Li S, Boehme S, Crowe P, et al. Restricted TCR Valpha gene rearrangements in T cells recognizing animmunodominant peptide of myelin basic protein in DR2 patients with multiple sclerosis. Int Immunol 1998; 10: 991-998. Oksenberg JR, Stuart S, Begovich AB, et al. Limited heterogeneity of rearranged T cell receptor V alpha transcripts in brains of multiple sclerosis patients (published erratum appear in Nature 1991; 353-94). Nature 1990; 345: 344-346. Wucherpfennig KW, Newcombe J, Li H, et al. T cell receptor Valpha-Vbeta repertorie and citokine gene expression in active multiple sclerosis lesions. J Exp Med 1992; 175: 993-1002. Meinh E, Weber F, Drexler K, et al. Mielin basic protein -specific T lymphocyte repertorie in multiple sclerosis: complexity of the response and dominance of nested epitopes due to recruitment of multiple T cell clones. J Clin Invest 1993; 92: 2633-2643. INMUNOLOGÍA 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. Yu M, Johnson JM, Tuohy VK. A predictable sequential determinant spreading cascade invariable accompanies progression of EAE: a basis for peptide-specific therapy after onset of clin disease. J Exp Med 1996; 183: 17771788. McFarland HI, Lobito AA, Johnson MM, Nyswaner JT, Frank JA, Palardy GR, et al. Determinant spreading associated with demyelination in a nonhuman primate model of multiple sclerosis. J Immunol 1999; 162: 2384-2390. Tuohy VK, Yu M, Yin L, Kawczak JA, Johnson JM, Mathisen PM, et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev 1998 Aug; 164: 93-100. Ghabanbasani MZ, Gu XX, Spaepen M, Vandevyver C, Raus J, Marynen P, et al. Importance of HLA-DRB1 and DQA1 genes and of the amino acid polymorphisms in the functional domain of DRbeta 1 chain in multiple sclerosis. J. Neuroimmunol. 1995; 59: 77-82. Ebers GC, Dyment DA. Genetics of multiple sclerosis. Semin Neurol 1998; 18: 295-299. Chataway J, Feakes R, Coraddu F, Gray J, Deans J, Fraser M, et al. The genetics of multiple sclerosis: principles, background and updated results of the United Kingdom systematic genome screen. Brain 1998; 121: 1869-1887. Haines JL, Ter-Minassian M, Bazyk A, Gusella JF, Kim DJ, Terwedow H, et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat Genet 1996; 13: 469-471. Kuokkanen S, Sundvall M, Terwilliger JD, Tienari PJ, Wikstrom J, Holmdahl R, et al. A putative vulnerability locus to multiple sclerosis maps to 5p14-p12 in a region syntenic to the murine locus Eae2. Nat Genet 1996; 13 : 477-480. Sawcer S, Jones HB, Feakes R, Gray J, Smaldon N, Chataway J, Robertson N, Clayton D, Goodfellow PN, Compston A. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet 1996; 13: 464-468. McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol 1992; 38: 229-240. Rosener M, Muraro PA, Riethmuller A, Kalbus M, Sappler G, Thompson RJ, et al. 2',3'-cyclic nucleotide 3'-phosphodiesterase: a novel candidate autoantigen in demyelinating diseases. J Neuroimmunol 1997; 75: 2834. Weerth S, Berger T, Lassmann H, Linington C. Encephalitogenic and neuritogenic T cell responses to the myelin-associated glycoprotein (MAG) in the Lewis rat. J Neuroimmunol 1999; 95: 157-164. Kojima K, Berger T, Lassmann A, et al. Experimental autoimmune panencephalitis and uveoretinitis transferred to the Lewis rat by T lymphocytes specific for the S100-beta molecule, a calcium binding protein of astroglia. J Exp Med 1994; 180: 817-829 Krzalic LJ, Nedeljkovic DR, Cvetkovic DH, Skender MK. Serum lipids and demyelination in experimental a l l e rgic encephalomyelitis induced by the incre a s e d encephalitogenicity of myelin basic protein given w ith galactocerebroside. J Neuroimmunol 1989; 22: 193-199. E.M. M ARTÍNEZ-CÁCERES ET AL 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. Cohen O, Sela BA, Schwartz M, Eshhar N, Cohen IR. Multiple sclerosis-like disease induced in rabbits by immunization with brain gangliosides. Isr J Med Sci 1981; 17: 711-714. Sáez-Torres I, Díaz-Villoslada P, Martínez-Cáceres EM, Ferrer I, Montalban X. J Neuroimmunol 1998; 84: 24-29. Wekerle H, Kojima K, Lannes-Vieira J, et al. Animal models. Ann Neurol 1994; 36 Suppl. S47-53. Lassmann H, Vass K. Are current immunological concepts of multiple sclerosis reflected by the immunopathology of its lesions?. Springer Seminar Immunopathol 1995; 17: 77-87 Luchinetti CF, Brück W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity in pathogenesis. Brain Pathol 1996; 6: 259-274. Thompson AJ, Polman CH, Miller DH, McDonald WI, Brochet B, Filippi M, et al. Primary progressive multiple sclerosis. Brain 1997; 120: 1085-1096. Immunological profile of patients with primary progressive multiple sclerosis. Expression of adhesion molecules Durán I, Martínez-Cáceres EM, Río J, Barberà N, Marzo ME, Montalban X. Brain 1999 (in press). Lucchinetti CF, Brück W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity in pathogenesis. Brain Pathol 1996; 6: 259-274. Linington C, Bradl M, Lassmann H, et al. Aumengtation of demielinination in rat acute EAE by circulating mouse monoclonal antibodies directed against MOG. Am J Pathol 1988; 130: 443-454. Linington C, Berger T, Perry L, et al. T cells specific for the MOG mediate an unusual autoimmune inflammatory response in the CNS. Eur J Immunol 1993; 23: 1364-1372. Genain CP, Roberts T, Davis RL, et al. Late complications of immune deviation therapy in a nonhuman primate. Science 1996; 274: 2054-2057. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple. Nat Med 1999; 5: 170-175. Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH. B-celldeficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol. 1998; 161: 4480-4483. Karni A, Bakimer-Kleiner R, Abramsky O, Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch Neurol 1999; 56: 311-315. Rodriguez M, Scheithauer B. ultraestructure of multiple sclerosis. Ultraestruc pathol 1994; 18: 3-13. Rodriguez M, Lennon VA. Immunglobulins promote remyelination in the CNS. Ann Neurol 1990; 27: 12-17. Miller DJ, Sanborn KS Katzmann, Rodríguez M. Monoclonal autoantibodies promote CNS repair in an animal model of MS. J Neurosci 1994; 14: 6230-6238. Asakura K, Miller DJ, Murray K, et al. Monoclonal autoantibody SCH94.03 , which promotes CNS remyelination, recognices an antigen on the surface of oligodendrocytes. J Neurosci Res 1996; 43: 273-281. Noseworthy JH, O’Brien PC, van Engelen BGM, Rodriguez M. Intravenous immunoglobulin therapy in multiple sclerosis: progress from remyelination in the Theiler´s virus model to a randomised, double-blind, placebo-controlled clinical trial. J Neurol Neurosurg Psychiatry 1994; 57 (suppl): 11-14. 89