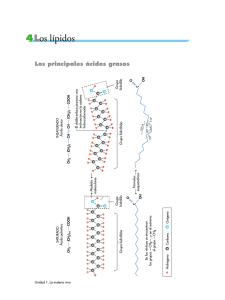
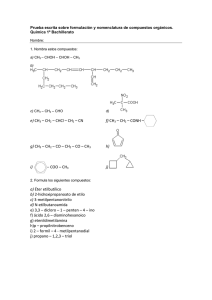
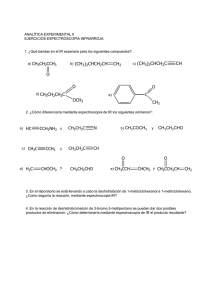
Ver/Abrir
Anuncio

Centro de Estudios de Productos Naturales Facultad de Química Universidad de La Habana Síntesis de híbridos péptido-esteroidales mediante la reacción de Ugi-4C Tesis de Diploma Autor: Radell Echemendía Pérez Tutor: Dr. Daniel García Rivera MSc. Odette Concepción González Ciudad de La Habana Mayo/ 2010 Agradecimientos Quisiera comenzar agradeciendo a toda mi familia, en especial a mis padres, mi hermana y a mi gordita querida, mi madre del alma. También quisiera agradecer al Dr Roidel Pérez Pérez mi segundo padre y amigo que siempre me ha brindado su apoyo y amor durante estos 5 años de batallas pírricas y por supuesto a su esposa que ha sido incondicional conmigo. Quisiera agradecer a Daniel, mi tutor por todo el conocimiento que me ha brindado, a la dulce Vivian (literalmente), a Odett , Karel, el Racho por ser un excelente amigo y hermano, a todo el laboratorio por la ayuda brindada, a Patri por su empeño en las columnas, al Micjel por los consejos brindados, a Nancy por su cariño desinteresado, a Rusela por su ayuda constante, a Roxana que si no fuese por ella esta tesis nunca estuviese impresa, al Juli por su análisis metodológico, a Shari, el Cane y a todo aquel que de una u otra forma ha contribuido con la realización de esta Tesis, a todos ellos que están y a los que no, un millón de gracias por todo… Abreviaturas AcOH Ácido acético AcOEt Acetato de etilo Bn Bencilo CCD Cromatografía de Capa Delgada CH2Cl2 Diclorometano CC Cromatografía de Columna DCC Diciclohexilcabodiimida DMF Dimetilformamida EM Espectrometría de Masas ESI-MS Espectrometría de Masa con Ionización Electrospray Et3N Trietilamina EtOH Etanol ESI-FT-ICR Electrospray Ionization Fourier Transform Ion Cyclotron Resonance HOBt 1-hidroxibenzotriazol HRMS Espectrometría de Masa de Alta Resolución (siglas en inglés) Me Metilo MeOH Metanol Ph Fenilo Py Piridina RMC Reacción Multicomponente RMCI Reacción Multicomponente basada en Isocianuros RMN Resonancia Magnética Nuclear SN Sustitución Nucleofílica SN2 Sustitución Nucleofílica Bimolecular TsCl Cloruro de tosilo THF Tetrahidrofurano TFA Ácido trifluoracético Ugi-4C Reacción de Ugi-4 componentes 1 Resumen Resumen En este trabajo se desarrolla un nuevo procedimiento de acoplamiento de péptidos a aminoesteroides basado en la reacción de Ugi-4 componentes. La reacción de Ugi-4 componentes consiste en la condensación de una amina primaria, un aldehído ó una cetona, un ácido carboxílico y un isocianuro o isonitrilo para dar lugar a un dipéptido N-sustituido con formación de cuatro nuevos enlaces covalentes. La metodología empleada consiste en el empleo de péptidos N-protegidos como componente ácido, amino esteroides como el componente amina y paraformaldehído e isocianoacetato de metilo como componente oxo e isonitrilo respectivamente. De esta manera los productos obtenidos de la conjugación son oligopéptidos N-sustituidos con esqueletos esteroidales que presentan dos residuos glicina más que los péptidos de origen. Los híbridos péptido-esteroidales obtenidos muestran una gran diversidad estructural como resultado de la variación tanto de la secuencia peptídica como de la posición de la conjugación en el esqueleto esteroidal. Todos los nuevos compuestos obtenidos fueron caracterizados mediante el empleo de técnicas espectroscópicas (RMN y espectrometría de masa). Abstract This work describes the development of a novel coupling procedure of peptides to amino-steroids. The process is based on the use of the Ugi four-component reaction, i.e., the condensation of a primary amine, an aldehyde or ketone, a carboxylic acid and an isocianide or isonitrile, to afford a N-substituted dipeptide with formation of a four new covalent bonds in one pot. The methodology consists on the use of N-protected oligopeptides as the acid component, amino-steroids as the amine component, as well as paraformaldehyde and methyl isocyanoacetate as the oxo and isonitrile components, respectively. Consequently, the conjugation products are branched oligopeptides N-substituted with steroidal skeletons bearing two additional glycines compared to the original peptides. The resulting peptido-steroidal hybrids show a wide structural diversity owing to the variation of both the peptidic sequence and the conjugation position at the steroidal core. All new compounds were appropriately characterized by spectroscopics techniques. 2 Índice Introducción 1. Revisión Bibliográfica 1.1 Péptidos en la naturaleza. Características e importancia biológica 1.2 Esteroides. Características e importancia biológica 1.3 Aplicaciones químico biológicas de los conjugados Péptidos-esteroidales 1.4 Métodos de síntesis 1.4.1 Reacciones multicomponentes 1.4.2 La química de los isonitrilos 1.4.3 Reacción de Ugi-4 componente 1.4.4 Síntesis de péptidos en disolución 1.4.5 Funcionalización de esteroides con grupos amino 2. Parte experimental 2.1 Equipos Materiales y Métodos 2.1.1 Procedimientos generales 2.1.2 Cromatografía, reactivos y solvente 2.2 Síntesis de aminas esteroidales 2.2.1 Síntesis del (25R)-3α-azido-5β-espirostan-5,6β-diol (20) 2.2.2 Síntesis del (25R)-3α-amino-5α-espirostan-5,6β-diol (21) 2.2.3 Síntesis de la 3α-azido-5α-androstan-6-ona (24) 2.2.4 Síntesis de la 3α-amino-5α-androstan-6-ona (25) 2.2.5 Síntesis del 3β-azido-5β-colano-24-ato de metilo (28) 2.2.6 Síntesis del 3β-amino-5β-colano-24-ato de metilo (29) 2.2.7 Síntesis del (25R)-12E-oximino-5α-espirostan-3β-ol (30) 2.2.8 Síntesis del (25R)-12α-amino-5α-espirostan-3β-ol (31) 2.2.9 Síntesis del 17α-amino-5α-androstan-3β-ol (33) 2.2.10 Síntesis del (25R)-6E-oximino-5α-espirostan-3β-ol (35) 2.2.11 Síntesis del (25R)-6α-amino (Boc)-5α-espirostan-3β-ol (37) 2.3 Síntesis de péptido-esteroides conjugados 2.3.1 Procedimiento General 2.3.2 Síntesis del conjugado 42 2.3.3 Síntesis del conjugado 43 2.3.4 Síntesis del conjugado 44 2.3.5 Síntesis del conjugado 45 2.3.6 Síntesis del conjugado 46 2.3.7 Síntesis del conjugado 47 3. Discusión de los resultados 3.1 Metodología de trabajo 3.2 Síntesis de aminas esteroidales 3.3 Síntesis de los híbridos péptido-esteroidales Conclusiones Recomendaciones Anexos Bibliografía 4 6 6 7 9 14 14 15 16 17 19 23 23 23 23 24 24 25 25 26 26 27 27 28 28 29 29 31 31 32 33 34 35 36 37 38 39 40 46 55 56 57 62 3 Introducción Introducción Se conoce que la actividad biológica de un importante número de polipéptidos y proteínas se debe a la disposición espacial que presentan determinadas secuencias de aminoácidos a lo largo de la cadena peptídica, factor que determina el tipo y alcance de las interacciones no-covalentes que tienen lugar durante dicho efecto biológico. Estas secuencias, conocidas como “secuencias activas” pueden llegar a ser tan cortas como de 3-5 residuos de amino-ácidos, aunque frecuentemente llegan a tener de 10 a 20, dependiendo del peso molecular y las propiedades del péptido o la proteína. La preorganización conformacional de dicha “secuencia activa” influye en una serie de factores estructurales, como: direccionalidad de la densidad electrónica, presencia de interacciones electrostáticas repulsivas o atractivas y de puentes de hidrógeno, que determinan las propiedades de la biomolécula. La conjugación de cadenas peptídicas “activas” a esqueletos rígidos se ha convertido en los últimos años en una metodología muy exitosa en el desarrollo de híbridos moleculares capaces de mimetizar el efecto biológico de determinadas proteínas y péptidos naturales. El esqueleto esteroidal ha sido una de las plataformas moleculares más empleadas con esta finalidad, debido a que permite imponer determinada preorganización conformacional a las cadenas peptídicas enlazadas a su sistema de anillos fusionados. El esqueleto esteroidal combina, en una única estructura, rigidez, quiralidad y gran diversidad de sitios de funcionalización ya sea con disposición axial o ecuatorial. Desde hace algún tiempo se han diseñado metodologías sintéticas para obtener híbridos péptidoesteroidales con importantes aplicaciones químico-biológicas. El procedimiento general ha consistido en la conjugación de la secuencia peptídica responsable del efecto biológico a dichos esqueletos rígidos, para mimetizar la preorganización conformacional de la secuencia nativa y lograr el efecto deseado sin necesidad de acceder a la biomolécula natural. Así, se han reportado conjugados péptido-esteroidales con diferentes aplicaciones, como enzimas artificiales del tipo serina-proteasa, reconocimiento selectivo de oligopéptidos endógenos del sistema neurológico, miméticos de los péptidos catiónicos naturales con potente actividad antibacteriana, inhibidores del proceso de adhesión celular y de reproducción descontrolada de las células cancerígenas, entre otras. No obstante, a pesar del progreso alcanzado en este campo, el único procedimiento químico de conjugación reportado en la literatura ha sido el clásico acoplamiento peptídico entre péptidos y amino- esteroides. Esta poca diversidad de procedimientos de conjugación ha limitado el desarrollo de nuevas aplicaciones biológicas de esta 4 Introducción importante familia de híbridos moleculares, debido fundamentalmente a la escasa novedad estructural a la que se tiene acceso mediante el empleo de un único tipo de reacción química. En este sentido, el problema científico que presenta esta importante línea de investigación es la poca disponibilidad de métodos de conjugación de péptidos a esqueletos esteroidales. En consecuencia, parte del esfuerzo por desarrollar nuevas familias de híbridos péptido-esteroidales está dirigido a la búsqueda de nuevos procedimientos sintéticos que permitan la conjugación eficiente de péptidos a esteroides funcionalizados. Entre los posibles procedimientos sintéticos disponibles en la literatura, las reacciones multicomponentes con isonitrilos ofrecen un gran potencial debido a la elevada eficiencia química con que transcurren, fácil implementación y amplia diversidad estructural que proporcionan con bajo costo sintético. En el presente trabajo se propone como hipótesis de trabajo que la reacción de Ugi 4-componentes es un procedimiento sintético versátil y eficiente para la conjugación de oligopéptidos a amino-esteroides. Por lo tanto nos proponemos como Objetivo General: Desarrollar un nuevo procedimiento de conjugación de péptidos a amino-esteroides basado en el empleo de la reacción de Ugi 4componentes (Ugi-4C). Para cumplir este objetivo general, nos trazamos los siguientes Objetivos Específicos: • Funcionalizar esteroides con grupos amino a través de la reducción de oximas y azidas. • Evaluar la eficiencia y versatilidad química del método de conjugación. • Caracterizar los compuestos obtenidos por técnicas espectroscópicas de RMN y EM. 5 Revisión Bibliográfica 1. Revisión Bibliográfica 1.1 Péptidos en la naturaleza. Características e importancia biológica Los péptidos son un grupo de biomoléculas que se definen estructuralmente como polímeros de α-aminoácidos naturales, los cuales se encuentran conectados covalentemente a través de enlaces amida ó enlaces peptídicos. Los péptidos naturales están formados por una familia que posee más de 20 α-aminoácidos diferentes (Anexo 1), todos pertenecientes a la serie L y diferenciados entre sí por las cadenas laterales situadas en el carbono α. 1 Estas cadenas laterales no sólo intervienen en la actividad biológica de dichas moléculas, sino que determinan la estructura secundaria y terciaria de los polipéptidos y proteínas. A finales de la década de los ´50 e inicios de los ´60 del siglo pasado con el perfeccionamiento de los métodos analíticos de aislamiento y separación de compuestos naturales, comienza una nueva era en este campo de investigación. Desde ese entonces se han aislado péptidos de diferentes fuentes naturales, entre las que se encuentran bacterias, hongos, plantas y diversas especies de invertebrados. De estos organismos mencionados se aíslan frecuentemente péptidos que contienen tanto β- como D-aminoácidos, estructuras cíclicas con anillos de diferente tamaño cuya obtención por vía química constituye un reto para los químicos sintéticos (Figura 1). Figura 1. Representación estructural de dos ciclopéptidos naturales y del glutatión. Los péptidos de organismos superiores modulan un gran número de funciones fisiológicas y bioquímicas, entre las que se encuentra como enzimas, hormonas, antígenos, neurotransmisores, transportadores de sustancias y reguladores de la expresión genética. En mamíferos son conocidos más de 100 péptidos con funciones en el sistema nervioso central y periférico, en procesos que involucran el sistema inmunológico, cardiovascular e intestinal, 1 entre otros. También actúan como receptores en el intercambio de información intercelular y participan en una variedad de procesos vinculados con el metabolismo, como receptores del dolor, en la reproducción y en la respuesta inmune de los organismos. 6 Revisión Bibliográfica Por otra parte, los péptidos endógenos de plantas, hongos, bacterias e invertebrados son una parte importante del sistema inmunológico innato de estos organismos, por lo que presentan un rol importante en los diversos mecanismos de defensa química. Al igual que otras familias de metabolitos secundarios, estos compuestos han sido producidos para actuar con gran especificidad contra agentes patógenos microbianos, depredadores o competidores. En consecuencia, son de gran importancia para el hombre al tener gran potencial como agentes terapéuticos antibióticos, antivirales, antiparasitarios o antitumorales. Los péptidos catiónicos endógenos presentan una marcada actividad antibacteriana. Que viene dada porque dichas estructuras son moléculas catiónicas facialmente anfifílicas (una cara de la molécula contiene grupos hidrofílicos y la otra, grupos hidrofóbicos) que favorecen un rápido tiempo de aniquilación y selectividad bacteriana sobre la célula huésped, debido fundamentalmente a su capacidad de permeabilidad o alteración de las membranas bacterianas. Un ejemplo de ello lo constituye la Magainina l, antibiótico aislado de un anfibio, cuya estructura de -hélice exhibe la separación entre las caras de residuos catiónicos e hidrofóbicos. De igual manera ocurre en una proteína humana con actividad antibacteriana, cuya estructura secundaria tipo hoja-β antiparalela muestra en caras separadas las porciones hidrofílicas e hidrofóbicas. Fig 2 Esta anfifilicidad es típica de aquellos péptidos que forman α-hélices u hojas-β. 2 Un ejemplo de ello lo constituye la Magainina I, antibiótico aislado de un anfibio, cuya estructura de α-hélice exhibe la separación entre las caras de residuos catiónicos e hidrofóbicos. De igual manera ocurre en una proteína humana con actividad antibacteriana, cuya estructura secundaria tipo hoja-β antiparalela muestra en caras separadas las porciones hidrofílicas e hidrofóbicas, como se muestra en la figura 2. Figura 2. Porciones del antibiotico Magainina I y de una proteína humana con características anfipáticas. 7 Revisión Bibliográfica 1.2 Esteroides. Características estructurales e importancia biológica Los esteroides representan una gran familia de productos naturales que se encuentran ampliamente distribuidos en el reino animal y vegetal. Esta importante familia de compuestos, desde el punto de vista estructural, posee un esqueleto carbonado del tipo 2’,3’ciclopentanoperhidrofenantreno, a lo que se denomina, núcleo esteroidal. Los esteroides que tienen en el núcleo esteroidal un metilo en el C-13 se les denominan estranos, mientras que si poseen además, otro metilo en C-10, se le llaman androstanos (Figura 3A). Los esteroides naturales pueden tener sustituyentes hidrocarbonados en C-17, estos sustituyentes pueden ser de 2, 5, 8, 9 ó 10 átomos de carbono, para dar lugar a familias de esteroides conocidas como: pregnanos, colanos, colestanos, ergostanos y estigmastanos, respectivamente. Figura 3. A) Representación esquemática y numeración del anillo esteroidal de la serie 5α y 5β. B) Ejemplos de esteroides naturales con diversas propiedades biológicas. El esqueleto esteroidal presenta características estructurales que lo convierten en una estructura química única. En primer lugar, se caracteriza por ser un tipo de molécula donde la naturaleza logra combinar rigidez, quiralidad y lipofilicidad en una única plataforma estructural; y lo más importante, sin hacer uso de la aromaticidad para conseguir este objetivo. 3 Esta característica se debe al distintivo sistema de anillos fusionados que forma un esqueleto policíclico muy rígido y generalmente lipofílico. Dicho esqueleto puede presentar un gran número de centros estereogénicos, y puede ser funcionalizado con dos tipos diferentes de orientación espacial en cada carbono no-cabeza de puente (Figura 3A). Tanto la gran rigidez de estos sistemas como la presencia de los metilos 18 y 19 que introducen impedimento estérico en la cara β, influyen en la demostrada estereoselectividad de las reacciones químicas que se realizan en el esqueleto 8 Revisión Bibliográfica esteroidal. Esta característica ha sido fundamental en la amplia utilización de los esteroides como materiales de partida para la obtención de moléculas bioactivas. La conjugación de los mismos a biomoléculas tales como los carbohidratos, ha sido una metodología ampliamente utilizada por los químicos sintéticos en la búsqueda de nuevas propiedades químico-biológicas. La 4 mayoría de los esteroides naturales presentan funciones oxigenadas y olefínicas en su estructura, las cuales permiten la funcionalización de dicho esqueleto con el objetivo de buscar nuevas moléculas con un amplio espectro de propiedades químico-biológicas.3 Un grupo funcional ampliamente encontrado en los esteroides naturales es el OH-3, el cual puede ser transformado a funciones cetona por oxidación como reemplazado por otros grupos funcionales a través de activación y sustitución nucleofílica. Otras funciones químicas de gran utilidad, son las cetonas en los carbonos 6 y 12 de sapogeninas esteroidales, pues pueden ser reducidas a alcoholes, transformadas a grupos amino a través de formación de oxima y posterior reducción, entre otras. 5 Un caso muy especial es el doble enlace entre los carbonos 5 y 6, – nombrado Δ5 en nomenclatura esteroidal –que puede ser transformado a una serie de grupos funcionales por oxidación o simplemente reducido por hidrogenación catalítica. Como se podrá apreciar en el epígrafe 1.4, en nuestro trabajo se hace un amplio uso de estos grupos funcionales para la introducción de grupos amino, los que son posteriormente utilizados en la conjugación de los esteroides a oligopéptidos. Las importantes aplicaciones biológicas de los esteroides se deben principalmente a su función como hormonas sexuales en mamíferos 6 (ej. compuestos 5 y 6), de crecimiento y de control antiestrés en plantas 7 (ej. brasinoesteroide 3), así como de regulación de otros procesos metabólicos como los mediados por el colesterol (1), la vitamina D, y los ácidos biliares (2). La diversidad de agentes terapéuticos y agroquímicos de naturaleza esteroidal que se comercializa en la actualidad se debe en gran medida a la disponibilidad de esteroides naturales utilizables como materias primas en la industria químico-farmacéutica.3 Estos pueden obtenerse con facilidad tanto de desechos de la producción animal (ej. colesterol y ácidos biliares), como de fuentes vegetales (ej. fitosteroles y sapogeninas como la hecogenina 4) y de hongos (ej. ergosterol), por lo que se dispone de una amplia variedad de esqueletos esteroidales para su empleo en química sintética. 1.3 Aplicaciones químico-biológicas de los conjugados péptido-esteroidales La conjugación de cadenas peptídicas bioactivas a esteroides se ha convertido en los últimos años en una metodología exitosa para reproducir el efecto biológico de determinadas proteínas y péptidos naturales. Es conocido que la actividad biológica de un importante número de oligopéptidos y proteínas se debe, entre otros requisitos, a la estructura primaria completa de 9 Revisión Bibliográfica estas biomoléculas. Sin embargo, en algunos casos sólo depende de la disposición espacial que presentan determinadas secuencias de amino-ácidos a lo largo de la misma. Estas secuencias, conocidas como “secuencias activas” pueden llegar a ser tan cortas como de 3-5 residuos, aunque en ocasiones pueden tener de 15-20 dependiendo del peso molecular de la biomolécula. 8 La conformación activa de dicha secuencia peptídica, o sea, la disposición espacial en la cual se pone de manifiesto el efecto biológico, viene determinada por una serie de factores estructurales tales como la rigidez conformacional, densidad electrónica, presencia de interacciones electrostáticas y por puentes de hidrógeno, que sí están determinados por las estructuras primaria y secundaria del oligopéptido o proteína que presentan determinados requerimientos espaciales. La conjugación de oligopéptidos a esqueletos rígidos ha sido una de las metodologías más empleadas para imponer rigidez conformacional en cadenas peptídicas diseñadas para obtener determinados efectos biológicos, como: interactuar con otra biomolécula o ejercer una acción enzimática. En general, dichos esqueletos rígidos presentan estructuras cóncavas (Figura 4) capaces de alinear o dirigir las cadenas peptídicas hacia la misma cara, con lo que se logra producir arquitecturas moleculares tipo podantes que favorecen la ocurrencia de múltiples interacciones no-covalentes durante el proceso de reconocimiento molecular. De esta forma, se han diseñado receptores sintéticos para el reconocimiento selectivo de aminoácidos, péptidos y glicoproteínas, 9 pero siempre utilizando esqueletos caracterizados por la presencia de anillos aromáticos, del tipo calixarenos, 10, 11 , 12 , 13 , 14 ciclotriveratrilenos 15, 16 , 17 , 18 y trialquilbencenos. 19, 20 , 21 Figura 4. Receptores sintéticos basados en esqueletos tipo A) calix[4]arenos, B) ciclotriveratrilenos y C) trialquilbencenos. 10 Revisión Bibliográfica Sin embargo, ha sido el esqueleto esteroidal la plataforma molecular más empleada para imponer preorganización conformacional a cadenas peptídicas, ya sea a través de la formación de estructuras rígidas macrocíclicas, 22 como acíclicas en las que las cadenas peptídicas quedan alineadas en disposición convergente. 23 En los últimos años se han reportado diferentes tipos de conjugados péptido-esteroidales a partir de esteroides funcionalizados por la cara α del esqueleto23, lo que permite alinear las cadenas peptídicas con disposición convergente para propiciar interacciones multivalentes. Dicha disposición espacial de elevada preorganización conformacional ha permitido obtener conjugados péptido-esteroidales con diversos tipos de aplicaciones biológicas. Still y col. reportaron la síntesis de la quimioteca combinatoria de conjugados péptido-esteroidales 10 (Figura 5) para su utilización en el reconocimiento selectivo de péptidos opioides en presencia de otras familias de péptidos.23g, h Por otra parte, De Clercq y col. produjeron la quimioteca de conjugados 11 en las que incorporaron por métodos combinatorios variantes de las secuencias peptídicas encontradas en el centro activo de uno de los tipos de enzimas serina-proteasa.23d Como en casos anteriores, el empleo del esqueleto esteroidal tiene como objetivo mimetizar la preorganización conformacional de la secuencia peptídica nativa. La evaluación biológica de dicha quimioteca propició la obtención de varios conjugados con elevada actividad catalítica, similar a la de la serina-proteasa nativa, por lo que dichos conjugados son considerados como enzimas artificiales.23c Empleando también métodos combinatorios, Savage y col. sintetizaron una quimioteca de conjugados péptido-esteroidales 12 (Figura 5), mediante la introducción de tres cadenas peptídicas con diferentes secuencias que contienen múltiples aminoácidos Lisina.23b Figura 5. Quimiotecas combinatorias de conjugados péptido-esteroidales. El objetivo de producir dichas cadenas peptídicas policatiónicas es mimetizar las propiedades anfipáticas del péptido antibacteriano Polimixina B2. Este producto natural presenta un esqueleto ciclopeptídico que alterna aminoácidos hidrofóbicos y aminoácidos catiónicos, formando así una 11 Revisión Bibliográfica estructura de elevada anfifilicidad facial, capaz de permeabilizar las membranas de bacterias y causar la muerte de las mismas. El tamizaje antibacteriano de dicha quimioteca produjo varios conjugados con elevada actividad bactericida, similar a la del ciclopéptido natural. Este reporte constituye un resultado trascendental, ya que demuestra la utilidad del esqueleto esteroidal, para alinear preorganizadamente cadenas peptídicas. Así, debido a la lipofilicidad de la cara β esteroidal, este puede ser empleado para la construcción de arquitecturas moleculares anfipáticas, mediante la funcionalización de la cara α con grupos polares. El diseño racional de péptido-esteroides capaces de mimetizar las propiedades químico-biológicas de oligopéptidos y proteínas, ha sido un área de importantes resultados. Recientemente Banerjee y col. obtuvieron conjugados péptido-esteroidales con elevada actividad antagonista de la ανβ3 integrinas (Figura 6). 24 La metodología seguida fue la introducción de análogos del tripéptido Arg-Gly-Asp (RGD) a esteroides reconocidos por membranas celulares. Dicha triada peptídica es la ‘secuencia activa’ de la familia de proteínas ανβ3 integrinas, responsables del proceso de adhesión celular y que tiene un rol fundamental en el proceso de reproducción descontrolada de las células cancerígenas. 25 Figura 6. Conjugados péptido-esteroidales antagonistas de las integrinas. Por tanto la inhibición de dicho proceso es un importante reto científico en la búsqueda de terapias anticancerígenas. Anteriormente se habían reportado varios peptidomiméticos antagonistas de las integrinas, mediante la incorporación de la secuencia RGD en esqueletos ciclopeptídicos rígidos, los que están en fase de evaluación por ser utilizados como agentes terapéuticos para el tratamiento del cáncer, la osteoporosis y otras enfermedades. 26 En este sentido, la conjugación a esqueletos esteroidales demostró una vez más ser una vía alternativa a la ciclización para la imposición de rigidez conformacional en cadenas peptídicas bioactivas, pues 12 Revisión Bibliográfica los conjugados obtenidos presentaron actividad antagonista de las ανβ3 integrinas similar a la de otros peptidomiméticos antes reportados. Una estrategia aun más exitosa para la obtención de cadenas peptídicas con alta preorganización conformacional, es la unificación del concepto de conjugación a un esqueleto rígido y ciclización en una única estructura. En este sentido, varios grupos de investigación se han enfocado en diseñar y sintetizar nuevas familias de conjugados péptido-esteroidales con estructuras macrocíclicas.22 En estos péptido-esteroides cíclicos la cadena peptídica se alterna con el esqueleto esteroidal para cerrar la cavidad macrocíclica, lo que resulta en una mayor preorganización estructural de la secuencia peptídica al combinarse la rigidez proporcionada por el esteroide con la pérdida de libertad conformacional debido a la ciclización. El primer reporte de un conjugado péptido-esteroidal macrocíclico lo realizó Wess y col. en 1996,22g al sintetizar el conjugado 15 como parte de un amplio programa de estudio del efecto de la conjugación de péptidos a ácidos biliares. 27 A pesar de que en este compuesto las cadenas peptídicas no están directamente unidas al esqueleto esteroidal, estudios estructurales demostraron que la cadena dipeptídica se encuentra en una conformación del tipo giro-β-II’ similar a las encontradas en ciclohexapéptidos, por lo que se demostró la posibilidad de mimetizar la conformación de motivos peptídicos biológicamente relevantes. Posteriormente, Feigel y col. reportaron una serie de estudios estructurales sobre ciclopéptido-esteroides análogos de 16.22e, f Figura 7. Macrociclos péptido- esteroidales. De este compuesto se obtuvo una estructura cristalina, lo que demostró la gran pre-organización conformacional que adquieren dichas cadenas dipeptídicas al alternarse dentro de una cavidad macrocíclica con el esteroide colánico, a pesar de que el residuo N-terminal se encuentra unido a la cadena lateral del esteroide que es altamente flexible. Recientemente, Rivera y col. reportaron el conjugado ciclotripéptido-esteroidal 17 (Figura 7), en el que se introduce nuevas restricciones conformacionales a la cadena peptídica.22h Este nuevo aporte se enfoca en el empleo de un esteroide de la serie 5α (fusión trans de anillos A/B), el que presentan una estructura más plana y alargada que los esteroides colánicos de la serie 5β empleados hasta el momento. Además, 13 Revisión Bibliográfica ambos residuos terminales están directamente unidos al esqueleto peptídico con una disposición axial, lo que produce una rigidez conformacional muy superior a los casos antes reportados. Otra de las aplicaciones más importantes de los conjugados péptido-esteroidales es la búsqueda de nuevas propiedades farmacológicas derivadas de la unión de ambas moléculas. 28 En este sentido, la alta especificidad y capacidad de circulación de los ácidos biliares en el sistema enterohepático ha permitido producir fármacos del tipo conjugados peptídicos a ácidos biliares con mejor absorción intestinal que las drogas peptídicas por sí solas.27, 29 Kramer y Wess estudiaron el efecto de la conjugación de diferentes fármacos a las posiciones 3, 7, y 12 del núcleo esteroidal.27 Así demostraron, por ejemplo, que la conjugación del fármaco peptídico oxaprolil – muy empleado para el tratamiento de la fibrosis quística – al ácido cólico produce un fármaco híbrido con mejores propiedades farmacológicas que su homólogo peptídico.(Figura 8)29 Figura 8. Conjugado peptídico oxaprolil- ácido biliar 1.4 Métodos de síntesis 1.4.1 Reacciones multicomponentes Las reacciones multicomponentes (RMC) son un tipo de procesos químicos en los cuales tres o más sustancias reaccionan para formar un producto que contenga átomos de todos y cada uno de los sustratos de partida. 30, 31 En muchos casos, los materiales de partida no reaccionan simultáneamente sino que lo hacen mediante una secuencia de pasos elementales que dan lugar al producto final. De esta forma, a partir de sustratos sencillos y accesibles es posible obtener sustancias con gran complejidad estructural en un solo paso de reacción, por lo que constituyen un procedimiento que se acerca mucho al concepto de la ‘síntesis ideal’. Una ‘síntesis ideal’ es aquella que conduce al producto deseado en pocos pasos de reacción, con un buen rendimiento global y usando reactivos químicos compatibles con el medio ambiente. Otra característica de las RMC es el alto grado de diversidad estructural que se puede lograr cambiando las estructuras de cada uno de los componentes que participan en las mismas. Este hecho ha influido en que las RMC sean de los procesos más empleados en la Química 14 Revisión Bibliográfica Combinatoria y que hayan tenido un impacto significativo en la síntesis química de nuevos fármacos y otros agentes terapéuticos. Hasta hace algunos años las RMC eran consideradas como un exótico tipo de reacciones orgánicas. A partir de la década del 90 muchos grupos de investigación se percataron de que este campo de investigación está lleno de grandes oportunidades, pues permite la producción rápida y eficiente de bibliotecas de un alto número de compuestos por variación de los sustratos de partida, y empleando síntesis automatizada. Dadas las incuestionables ventajas de estos procesos, los mismos son ampliamente utilizados en la síntesis de productos naturales biológicamente activos. 32 Hasta el momento se conocen de 300 a 400 tipos de RMC, y cada año se realizan grandes esfuerzos en el desarrollo de procesos de esta naturaleza. 33 Un tipo muy especial de RMC son las basadas en isonitrilos (RMCI), las que constituyen el 20-25% de todas las RMC empleadas en síntesis química cada año. 34 Este dato demuestra el impacto de las mismas en síntesis orgánica moderna, por lo que para comprender su gran potencial es necesario estudiar en detalle el grupo funcional isonitrilo. 1.4.2 La química de los isonitrilos Los isonitrilos, también conocidos como isocianuros, representan un grupo funcional de naturaleza única y muy interesante. Su inusual valencia y reactividad ha sido muy discutida desde su descubrimiento en 1859, pues además de los carbenos, es el único grupo funcional en química orgánica que presenta un carbono divalente (CII). A diferencia de los carbenos es una especie química estable, pues se comercializan un gran número de ellos y se han aislado cientos de productos naturales que contienen el grupo isonitrilo en su estructura. 35 La propiedad química más importante de estos compuestos, desde el punto de vista sintético, es la reactividad tanto con electrófilos como con nucleófilos en el átomo de carbono del grupo isonitrilo 36, 37 , 38 (Figura 9). Esto se debe al carácter dual (electrofílico y nucleofílico) de su carbono divalente. Con excepción de los carbenos y el monóxido de carbono, ningún otro grupo funcional presenta carácter nucleofílico y electrofílico en el mismo centro. Figura 9. Reactividad química de los isonitrilos. 15 Revisión Bibliográfica La química de los isonitrilos comenzó en 1859 cuando Lieke obtuvo el alil-isonitrilo por reacción entre el yoduro de alilo y el cianuro de plata. 39 Sin embargo, durante todo un siglo solo se lograron preparar 12 isonitrilos, cuyas propiedades químicas no se investigaron a profundidad debido a su olor desagradable. En 1958, cuando estos compuestos se pudieron obtener fácilmente por deshidratación de formamidas comenzó una nueva era en la química de los isonitrilos. Los isonitrilos se caracterizan por tres tipos fundamentales de propiedades químicas a) la acidez de su posición α (carácter electroaceptor), b) la fácil formación de radicales y c) la adición doble de electrófilos y nucleófilos.37b Los isonitrilos polimerizan en presencia de ácidos de Lewis para formar poliiminometilenos y forman compuestos α/metalados con metales de transición. Las aplicaciones más importantes de los isonitrilos se pusieron de, manifiesto cuando en 1921 Mario Passerini reportó la adición de aldehídos y ácidos carboxílicos para dar α/aciloxicarboxamidas (reacción de Passerini 3/componentes), un motivo químico encontrado en diversos productos naturales bioactivos, como los depsipéptidos. Sin embargo, la enorme popularidad de estos compuestos se alcanzó en 1959, cuando Ivar Ugi y col. desarrollaron la primera reacción de cuatro componentes basada en isonitrilos. 40 1.4.3 Reacción de Ugi - 4 componentes A partir de 1961 a esta familia de procesos se les llamó reacción de Ugi 4-componentes (Ugi-4C) y desde entonces pasó a ser una de las más estudiadas y utilizadas en síntesis orgánica. 41 Figura 10. Reacción de Ugi-4C. La versión más simple de la reacción de Ugi-4C es la condensación de un aldehído o cetona, una amina primaria, un ácido carboxílico y un isonitrilo para formar un dipéptido N-sustituido. Aunque el mecanismo detallado de esta reacción aún no se ha determinado con exactitud, se sabe que transcurre a través de una secuencia de pasos elementales reversibles que culmina con la oxidación irreversible del carbono divalente a tetravalente. La secuencia transcurre a través de la formación de la correspondiente base de Schiff, seguida de activación mediante protonación por el ácido carboxílico y adición doble de los iones iminio y carboxilato al isonitrilo dando lugar al αaducto. El paso final es el reordenamiento de Mumm, que se puede considerar como una 16 Revisión Bibliográfica acilación intramolecular del nitrógeno de la amina, seguido de la conversión tautomérica de la hidroxilamina en amida. A pesar de que el orden de adición no se ha determinado, se conoce que la protonación de la imina favorece la adición al isonitrilo debido al aumento de la electrofilicidad del enlace C=N, además de propiciar la desprotonación del carboxilato para generar la especie nucleofílica (Figura 11). Figura 11. Mecanismo propuesto para la reacción de Ugi-4C El carácter iónico de esta reacción permite el empleo de disolventes polares próticos como el metanol, el etanol, etc. Se ha comprobado además que se obtienen mejores rendimientos de la reacción si se procede a preformar la imina por agitación de la amina y el aldehído en MeOH durante 2h. 42 Debido a que la reacción produce un nuevo centro estereogénico y la misma no es estereoselectiva, el empleo de un compuesto carbonílico proquiral conlleva a la formación de mezclas de estereosimómeros, por lo que en muchos casos se emplea paraformaldehído como compuesto carbonílico. 43 1.4.4 Síntesis de péptidos en disolución Las diversas aplicaciones biológicas y médicas de los péptidos ha propiciado un creciente interés en el perfeccionamiento de los métodos de síntesis de oligopéptidos de mediano y grande tamaño. Al igual que la biosíntesis de péptidos en los ribosomas, la síntesis química de estos utiliza una estrategia iterativa en la que los sucesivos aminoácidos se van incorporando a la cadena peptídica creciente en un orden programado. Existen dos metodologías alternativas para la producción de oligopéptidos: las síntesis en disolución y la síntesis en fase sólida. En ambos casos, para permitir la formación del enlace amida entre el grupo carboxilo de un aminoácido y el 17 Revisión Bibliográfica grupo amino del otro es necesario la activación del grupo carboxilo, factor que propicia el ataque nucleofílico de la amina. Tanto la cadena peptídica creciente como el aminoácido que deseamos incorporar a esta son compuestos bifuncionales que poseen un grupo ácido y un grupo amino. Por tanto es necesario la protección de la amina y el carboxilato que no deseamos que reaccionen, para asegurar que el enlace amida se forme entre los grupos deseados y evitar una polimerización incontrolada. En la construcción de la cadena peptídica se deben ir alternando pasos de protección, formación de enlaces amida, y desprotección para liberar los grupos reactivos que van a participar en la formación del siguiente enlace peptídico. Los grupos protectores de aminas más utilizados son los carbamatos, como el fluorenilmetoxicarbonilo (Fmoc) y el t-butoxicarbonilo (Boc), los cuales pueden ser eliminados con facilidad de forma ortogonal cuando no son necesarios (Figura 12). Los ácidos carboxílicos se protegen comúnmente en forma de ésteres, por lo que no abundaremos en estos procesos tan ampliamente estudiados. Figura12. Grupos protectores de amina En cuanto a los métodos de activación del grupo carboxilo, uno de los más efectivos está basado en el uso de carbodiimidas, como la diciclohexilcarbodiimida (DCC) según se muestra en la figura 13. Un inconveniente de estos agentes acoplantes es su gran reactividad, lo que puede provocar reacciones secundarias no deseadas, así como racemización de los aminoácidos y péptidos. Este fenómeno propició la introducción de los agentes supresores de racemización, que no son más que aditivos que se emplean junto con las carbodiimidas para formar ésteres reactivos con la suficiente capacidad para activar el carboxilato frente al ataque nucleofílico de la amina, pero evitando reacciones colaterales y la racemización del carbono α del carboxilato. Los agentes supresores de racemización más empleados en la actualidad son la hidroxisuccinimida y el 1hidroxibenzotriazol (HOBt). Más recientemente se han introducido agentes acoplantes que ya traen incorporados el agente supresor de racemización en su estructura, lo que simplifica el procedimiento experimental de forma significativa, aunque son extremadamente costosos. 44 18 Revisión Bibliográfica Figura 13. Estructuras de agentes acoplantes y supresores de racemización. La síntesis de péptidos en disolución es hoy en día menos empleada que la síntesis en fase sólida, debido a que son necesarios pasos de purificación cada cierto número de acoplamientos peptídicos. No obstante es mucho más barata, fácil de implementar y permite la obtención de oligopéptidos de pequeño y mediano tamaño incluso a nivel de kilogramos en plantas de producción de medicamentos. La metodología más utilizada es el acoplamiento de un aminoácido protegido con Boc en su grupo amino a otro aminoácido con éster metílico en el carboxilato. La sucesiva desprotección del grupo Boc en el dipéptido resultante, seguido de posteriores acoplamientos peptídicos permite el ensamblaje de la cadena peptídica deseada. Para la incorporación de aminoácidos trifuncionales como lisina, arginina, histidina, y los ácidos aspárticos y glutámicos, es necesario el uso de grupos protectores ortogonales tanto al grupo Boc como al éster metílico. Esto es de fácil implementación pues dichos grupos protectores son ampliamente conocidos y utilizados en la síntesis de péptidos. Estos son, por mencionar algunos: el benciloxicarbonilo (Cbz o Z) y el alilcarbamato (Alloc), ortogonales tanto al Boc como al Fmoc; y el éster tert-butílico, ortogonal al éster metílico. La figura 14 muestra un procedimiento típico de síntesis en disolución, en el que se introducen los distintos aminoácidos utilizando la combinación carbodiimida/HOBt como agente acoplante y empleando desprotección del grupo Boc en medio ácido. Es importante señalar que el empleo de diciclohexilcarbodiimida produce diciclohexilurea como producto colateral, por lo que resulta obligatoria la purificación final del tetrapéptido por CC. Figura 14. Síntesis en disolución de un tetrapéptido empleando carbodiimida/HOBt como agente acoplante 19 Revisión Bibliográfica 1.4.5 Funcionalización de esteroides con grupos amino La estructura de los esteroides permite la obtención de sustancias con las más diversas funciones químicas. La introducción de grupos aminos en diversas posiciones del esqueleto esteroidal se puede realizar mediante dos metodologías diferentes: a) la activación de un grupo hidroxilo secundario, por formación de un éster sulfónico (TsO o MsO), seguido de desplazamiento nucleofílico por el grupo azido y posterior reducción a amina y b) oxidación a cetona, seguido de formación de la oxima y reducción a amina. De ambas metodologías, se selecciona la que produzca la amina esteroidal con mejores rendimientos de reacción y en forma estereoquímicamente pura. Se ha reportado que la reducción de oximas en las posiciones 7 y 12 del esqueleto esteroidal produce las aminas con estereoquímica α en más de un 95% de 6 rendimiento. Como se ilustra en la figura 15, la oxidación se realiza fácilmente utilizando el reactivo de Jones (CrO3/H2SO4) como agente oxidante y la oxima se obtiene por tratamiento de la cetona con hidrocloruro de hidroxilamina. La oxima obtenida se somete a reducción por hidrogenación catalítica empleando PtO2 (catalizador de Adams), lo que da lugar a la hidroxilamina correspondiente, que luego se transforma en la amina deseada por tratamiento con Zn(s) en AcOH. Un procedimiento alternativo de reducción de oximas a aminas es el tratamiento con alcohol anhidro a reflujo y sodio metálico, un proceso que genera hidrógeno molecular in situ y permite también la reducción estereoselectiva de la oxima en la posición 12, aunque no en posición 7. 45 Figura 15. Síntesis estereoselectiva de una diamina esteroidal por reducción de las oximas en 7 y 12. Sin embargo, existen posiciones del esqueleto esteroidal en las que la reducción de la función oxima a amina no transcurre de forma estereoselectiva con ninguno de los procedimientos conocidos. Este es el caso de la oxima en el carbono 3, cuya reducción produce una mezcla de las aminas epiméricas 3α y 3β en cantidad equimolares. En este caso, es necesario recurrir al procedimiento estereoespecífico que involucra la introducción de la azida con inversión de la configuración y su posterior reducción a amina. Las azidas son excelentes precursores de grupos amino, son resistentes frente a variadas condiciones de reacción y se introducen con facilidad por 20 Revisión Bibliográfica sustitución nucleofílica (SN) de un grupo hidroxilo activado como éster sulfónico (tosilato o mesilato). Si las condiciones de reacción se ajustan para transcurrir esencialmente por una SN2, la misma procede con inversión total de la configuración del alcohol de partida. También es posible lograr la sustitución de un OH por N3 con retención de la configuración, mediante la sulfonación con inversión por reacción de Mitsunobu y posterior desplazamiento de azida. 46 Figura 16. Esquemas de obtención de 3-aminoesteroides, por reducción de oximas (no estereoselectiva) y por reducción de azidas (estereoselectiva). El paso final de esta ruta alternativa es la reducción del grupo azido a amina. Esta transformación puede realizarse por hidrogenación catalítica o mediante la reacción de Staudinger. El primero de estos métodos es el más generalizado en la síntesis de aminas esteroidales y se lleva a cabo utilizando atmósfera de H2 (g) y Pd sobre carbono al 10% como catalizador. El tratamiento de una hidrogenación a temperatura y presión ambiental es muy sencillo, limpio y produce elevados rendimientos de reacción, lo que se considera como una de sus ventajas fundamentales. Sin embargo, la hidrogenación catalítica no es un proceso quimioselectivo, por lo que pueden afectarse otros grupos funcionales como un doble enlace o un grupo carbonilo. La reacción de Staudinger es también un proceso muy empleado en la transformación N3→NH2. 47 Brosa y col. han obtenido muy buenos resultados en la reducción de azidas esteroidales en el carbono 3 mediante esta reacción, incluso si estas tienen disposición axial. 48 El mecanismo propuesto para esta reacción 49 se representa en la figura 17, lo que explica la alta quimioselectividad de esta reacción que solo afecta a la función azida. 21 Revisión Bibliográfica Figura 17. Mecanismo propuesto para la reacción de Staudinger. No obstante, una desventaja evidente de esta reacción es que se genera óxido de trialquil (o trifenil) fosfina, lo cual obliga a realizar tediosas purificaciones por cromatografía de columna (CC). En caso de que la amina sea el sustrato de partida para otra reacción y no el producto final, la mejor variante es proceder al paso posterior con la amina impurificada con el R3PO, el que es totalmente inerte. 22 Parte Experimental 2. Parte Experimental 2.1 Equipos Materiales y Métodos 2.1.1 Procedimientos generales Los espectros de RMN-1H y RMN-13C fueron registrados en un espectrómetro Varian Mercury, con una frecuencia de resonancia de 300.0 MHz, utilizando TMS como referencia interna. Los corrimientos químicos (δ) están reportados en ppm y las constantes de acoplamiento (J) en Hz. Los desplazamientos de 13 C están referidos a la señal del disolvente (77.0 ppm). Las temperaturas de fusión fueron determinadas en un equipo Electrothermal 9100 y no están corregidas. Los espectros de masa de alta resolución se registraron en un espectrómetro Bruker Apex 70e de resonancia ion ciclotrón a transformada de Fourier (FT-ICR), dotado de una celda InfinityTM con un magneto superconductor de 7.0 T. Todos los espectros fueron registrados en el Instituto de Bioquímica de las Plantas de Halle, Alemania, como parte de un proyecto de colaboración. 2.1.2 Cromatografía, reactivos y disolventes Los productos fueron purificados por cromatografía de columna con gel de sílice 60 (Merck, <230 mesh). El curso de las reacciones fue controlado por cromatografía de capa delgada (CCD) en cromatoplacas de gel de sílice (Merck, 5 x 2.5cm) empleando como sistema revelador una disolución de ácido fosfomolíbdico en EtOH al 5%. Los disolventes utilizados para fines cromatográficos fueron destilados y/o desecados según procedimientos reportados. Los reactivos utilizados para síntesis fueron procedentes de las firmas Merck, Fluka, BDH y Panreac. Las materias primas de naturaleza esteroidal utilizadas fueron suministradas por el Centro de Estudios de Productos Naturales. 23 Parte Experimental 2.2 Síntesis de aminas esteroidales 2.2.1 Síntesis del (25R)-3α-azido-5α-espirostan-5,6β-diol (20) A una disolución de 2.0 g (4.5 mmol) del triol 18 en 10 ml de piridina seca se le adicionan 1.3 g (6.8 mmol) de TsCl. La mezcla se agita a temperatura ambiente, hasta desaparición total del material de partida, indicada por CCD (n-hexano/AcOEt 3:1). La reacción se vierte sobre una solución saturada de KHCO3 y el sólido que precipita se separa por filtración a presión reducida, se lava con una solución de HCl 5 % (2 × 50 ml), con abundante agua y se seca en desecadora a vacío. Una vez seco, el crudo de reacción se disuelve en 35 ml de DMF y se le adicionan 783 mg (12 mmol) NaN3. La mezcla de reacción se agita a 50 ºC durante 48 h, luego se vierte sobre 100 ml de agua fría y el precipitado se colecta por filtración a presión reducida. El producto se purifica por cromatografía de columna (n-hexano/AcOEt 3:1) para obtener la azida 20 pura (1.5 g, 70 %). Rf= 0,28 (n-Hexano/AcOEt – 5:1) Tf (EtOH): 218-219 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.78 (d, 3H, J = 6.4 Hz, H-27); 0.78 (s, 3H, H-18); 0.96 (d, 3H, J = 7.2 Hz, H-21); 1.27 (s, 3H, H-19); 2.40 (dd, 1H, J = 15.2/4.4 Hz); 3.36 (t, 1H, J = 10.8 Hz, H-26ax); 3.47 (dd, 1H, J = 10.9/4.8 Hz, H-26eq); 3.50 (s, 1H, OH); 3.57 (m, 1H, H-3β); 4.09 (m, 1H, H-6α); 4.38 (m, 1H, H-16α). RMN-13C (75 MHz, CDCl3): δ = 14.4, 16.5, 16.6, 17.1 (CH3); 20.4, 25.0 (CH2); 28.3, 28.7 (CH2); 29.7, 30.2 (CH); 31.2, 31.6, 33.8, 34.5 (CH2); 39.0 (C); 39.9 (CH2); 40.6 (C); 41.55, 45.1, 55.6, 58.3; 62.0 (CH); 66.8 (CH2); 74.1 (C); 75.0, 80.8 (CH); 109.2 (C). HRMS (ESI-FT-ICR) m/z: 496.3145 [M+Na]+; calculada para C27H43N3NaO4: 496.3151. 24 Parte Experimental 2.2.2 Síntesis del (25R)-3α-amino-5α-espirostan-5,6β-diol (21) A una disolución de 600 mg (1.3 mmol) de la azida 20 en 10ml de THF, se le añaden 498 mg (1.9 mmol) de PPh3, la mezcla de reacción se agita a temperatura ambiente durante 24h, luego se le adicionan 0.3 ml de H2O, manteniendo la agitación durante 24 h adicionales. El disolvente se evapora a presión reducida y el sirope residuo se disuelve en 50 ml CHCl3 y se lava con disolución de NaOH 1M. La fase orgánica se seca sobre Na2SO4 anhidro y el disolvente se evapora para obtener la amina 21. Esta amina se empleó en el procedimiento de conjugación multicomponente sin previa purificación. Rf= 0,2 (CHCl3/MeOH - 5:1). 2.2.3 Sintesis de la 3α-azido-5α-androstan-17-ona (24) Una disolución que contiene 1.5 g (5.2 mmol) de la cetona 22 en 15 mL de piridina seca, se hace reaccionar con 1.47 g (7.8 mmol) de TsCl según el procedimiento descrito en el epígrafe 2.2.1 para producir el tosilato 23 correspondiente. El producto de reacción se seca en desecadora a vacío, se disuelve en 30ml de DMF y se hace reaccionar con 927 mg (14.3 mmol) NaN3 según el procedimiento descrito en el epígrafe 2.2.1. El producto se purifica por cromatografía de columna (n-hexano/AcOEt 5:1) para obtener la azida 24 (1.38g, 85 %). Tf (AcOEt):155-156 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.72 (s, 3H, H-19); 0.79 (s, 3H, H-18); 3.62 (m, 1H, H-3β). RMN-13C (75 MHz, CDCl3): δ = 11.1 (CH3); 12.2 (CH3); 20.3 (CH2); 22.1, 23.3, 26.8, 28.9, 29.0 (CH2); 30.5 (C); 31.7 (CH2); 35.5 (CH); 36.3, 36.8, 38.7 (CH2); 43.0 (C); 47.1 (CH); 51.1 (CH); 54.8 (CH), 205.3 (CO). HRMS (ESI-FT-ICR) m/z: 316.2395 [M+H]+; calculada para C19H30N3O: 316.2389. 25 Parte Experimental 2.2.4 Síntesis de la 3α-amino-5α-androstan-17-ona (25) 600 mg (1.9 mmol) de la azida 24 y 1.5 g (5.7 mmol) de PPh3 reaccionan en la mezcla THF (10 ml) / H2O (0.3 ml) según el procedimiento descrito en el epígrafe 2.2.2 para obtener la amina 25. Esta amina se empleó en el procedimiento de conjugación multicomponente sin previa purificación. 2.2.5 Síntesis del 3β-azido-5β-colano-24-ato de metilo (28) 800 mg (2.0 mmol) del alcohol 26 disueltos en 15 ml de piridina seca se ponen a reaccionar con 584 mg de TsCl (3.1 mmol) según el procedimiento descrito en el epígrafe 2.2.1 para producir el tosilato 27 correspondiente. El producto de reacción se seca en desecadora a vacío y se hace reaccionar con 328 mg (5.0 mmol) de NaN3 en 30 ml de DMF según el procedimiento descrito en el epígrafe 2.2.1. El compuesto se purifica por cromatografía de columna (n-hexano/AcOEt 3:1) para obtener la azida 28 (689 mg, 81 %). Rf= 0,88(n Hexano/AcOEt - 4:1) T.f. (AcOEt): 171-172 ºC. RMN-1H (300 MHz, CDCl3) δ = 0.64 (s, 3H, H-18); 0.91 (d, 3H, J=6.4 Hz, H-21); 0.93 (s, 3H, H-19); 3.31 (br. m, 1H, H-3β); 3.66 (s, 3H, CH3O). RMN-13C (75 MHz, CDCl3) δ = 12.0, 18.2 (CH3); 20.8 (CH2); 23.4 (CH3); 24.1, 26.3, 26.7, 27.0, 28.1, 30.9, 31.0, 32.4 (CH2); 34.6 (C); 35.3 (CH); 35.5 (CH2); 35.7 (CH); 40.0 (CH2); 40.4 (CH); 42.3 (C); 42.6 (CH); 51.5 (CH3); 55.9, 56.3, 61.2 (CH); 174.7 (C). HRMS (ESI-FT-ICR) m/z: 438.3100 [M+Na]+; calculada para C25H41N3NaO: 438.3096. 26 Parte Experimental 2.2.6 Síntesis del 3β-amino-5β-colano-24-ato de metilo (29) 350 mg (0.84 mmol) de la azida colánica 28 y 331 mg (1.26 mmol) de PPh3 reaccionan en la mezcla THF (10 mL) / H2O (0.3 ml) según el procedimiento descrito en el epígrafe 2.2.2 para obtener la amina 29. Esta amina se empleó en el procedimiento de conjugación multicomponente sin previa purificación. 2.2.7 Síntesis del (25R)-12E-oximino-5α-espirostan-3β-ol (30) A una disolución de 4.5 g (10.5 mmol) de la cetona 4 en 40 ml de piridina seca se le adicionan 1.1g (15.7 mmol) de hidrocloruro de hidroxilamina. La mezcla de reacción se agita a temperatura ambiente durante 8 h y una vez finalizada la reacción se vierte sobre 200 ml de agua fría. El precipitado se colecta a presión reducida, se lava varias veces con agua y se seca en estufa a 60 ºC, para producir la oxima 30 (4.3g, 92%). Una porción de este compuesto fue recristalizado de EtOH para su posterior caracterización. Tf (EtOH): 238–239 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.78 (d, 3H, J=6.2 Hz, H-27); 0.88 (s, 3H, H-18); 0.95 (s, 3H, H19); 1.05 (d, 3H, J=6.8 Hz, H-21); 3.36 (t, 1H, J=11.0 Hz, H-26ax); 3.46 (dd, 1H, J=10.8/4.9 Hz, H26eq); 3.56 (m, 1H, H-3α); 4.39 (m, 1H, H-16α). RMN-13C (75 MHz, CDCl3): δ = 11.8, 13.0, 16.9, 17.0 (CH3); 19.8, 28.2, 28.5 (CH2); 30.0 (CH); 30.7, 30.8, 31.3, 31.6 (CH2); 34.6 (CH); 35.9 (C); 36.5 (CH2); 37.4 (C); 42.4, 44.6 (CH); 47.0 (CH2); 53.4, 55.1, 56.2 (CH); 66.7 (CH2); 70.5 (CH); 80.0 (CH); 109.3 (C); 164.1 (C=N). HRMS (ESI-FT-ICR) m/z: 468.3091 [M+Na] +; calculada para C27H43NaNO4: 468.3089. 27 Parte Experimental 2.2.8 Síntesis del (25R)-12α-amino-5α-espirostan-3β-ol (31) Una disolución de 1.5 g (3.4 mmol) de la oxima 30 en 30 ml de n-propanol seco se calienta a reflujo y se trata con 775 mg (33.7 mmol) de sodio en pequeñas porciones. La mezcla de reacción se mantiene a reflujo durante 4h adicionales y se concentra a presión reducida hasta sequedad. El residuo se disuelve en 200 ml de CH2Cl2, la fase orgánica se lava con agua (2 × 100 ml), se seca sobre Na2SO4 anhidro y se concentra a presión reducida para dar lugar a la amina 31 (1.1 g, 76 %). Una porción de este compuesto fue recristalizado de EtOH para su posterior caracterización. Tf (EtOH): 232-233 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.76 (s, 3 H, H-18); 1.17 (s, 3 H, H-19); 0.78 (d, 3 H, J=6.5 Hz, H27); 0.94 (d, 3 H, J=6.5 Hz, H-21); 3.36 (t, 1H, J=10.9 Hz, H-26ax); 3.49 (dd, 1H, J=10.8/3.9 Hz, H26eq); 3.56 (br. m, 1H, H-3α); 3.19 (br. s, 1H, H-12β); 4.40 (m, 1H, H-16α). RMN-13C (75 MHz, CDCl3): δ = 11.9, 13.2, 16.1, 17.1 (CH3); 27.2, 28.1, 28.8, (CH2); 30.0 (CH); 30.2, 31.2, 31.4, 31.6 (CH2); 33.8, 34.3 (CH); 36.1 (C); 36.2 (CH2); 37.7 (C); 42.2, 44.4 (CH); 53.5, 55.0, 55.6, 59.6 (CH); 66.8 (CH2); 71.1, 79.8 (CH); 109.3 (C). HRMS (ESI-FT-ICR) m/z: 432.3479 [M+H]+; calculada para C27H46NO3: 432.3476. 2.2.9 Síntesis del 17α-amino-5α-androstan-3β-ol (33) 1.2 g (4.1 mmol) de androsterona (22) y 0.428 g (6.2 mmol) de hidrocloruro de hidroxilamina reaccionan siguiendo la técnica descrita en el epígrafe 2.2.7, dando lugar a la oxima 32 (1.2 g, 94 %). Posteriormente la oxima se hacer reaccionar con 0.895 g (38.9 mmol) de sodio según la técnica descrita en el epígrafe 2.2.8, para dar lugar a la amina 33 (0.916 g, 80 %). Una porción de este compuesto fue recristalizado de EtOH para su posterior caracterización. Tf (EtOH): 201-203 ºC. 28 Parte Experimental RMN-1H (300 MHz, CDCl3): δ = 0.74 (s, 3H, H-18); 0.82 (s, 3H, H-19); 2.82 (m, 1H); 3.37 (m, 2H); 3.55 (br. m, 1H, H-3α). RMN-13C (75 MHz, CDCl3): δ = 10.9, 12.0 (CH3); 20.4, 23.4, 27.6, 28.2, 30.7, 31.4 (CH2); 35.2 (C); 35.3 (CH); 36.0, 36.7, 37.3 (CH2); 42.0 (C); 44.6, 52.4, 53.9, 60.6, 70.5 (CH). HRMS (ESI-FT-ICR) m/z: 292.2647 [M+H]+; calculada para C19H34NO: 292.2640. 2.2.10 Síntesis del (25R)-6E-oximino-5α-espirostan-3β-ol (35) 5.0 g (11.6 mmol) de laxogenina (34) y 1.2 g (17.4 mmol) de hidrocloruro de hidroxilamina reaccionan siguiendo la técnica descrita en el epígrafe 2.2.7 dando lugar a la oxima 35 (4.7 g, 93 %). Una porción de este compuesto fue recristalizado de EtOH para su posterior caracterización. Tf (EtOH): 227-229 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.78 (d, 3H, J = 6.2 Hz, H-27); 0.77 (s, 3H, H-18); 1.03 (s, 3H, H19); 0.98 (d, 3H, J = 6.6 Hz, H-21); 3.35 (t, 1H, J = 10.9 Hz, H-26ax); 3.47 (m, 1H, H-26eq); 3.56 (m, 1H, H-3α); 4.41 (m, 1H, H-16α). RMN-13C (75 MHz, CDCl3): δ = 11.8, 14.4, 16.3, 17.0 (CH3); 21.4, 28.7 (CH2); 30.2 (CH); 30.0, 30.7, 31.2, 31.5, 36.6 (CH2); 37.2 (CH); 39.3 (CH2); 41.5 (CH); 40.6, 40.9 (C); 46.6 (CH2); 53.7, 56.3, 56.6, 61.9 (CH); 66.8 (CH2); 70.6, 80.1 (CH); 109.2 (C); 164.5 (C=N). HRMS (ESI-FT-ICR) m/z: 468.3094 [M+Na] +; calculada para C27H43NaNO4: 468.3089. 2.2.11 Síntesis del (25R)-6α-amino (Boc)-5α-espirostan-3β-ol (37) 4.5 g (10.0 mmol) de la oxima 35 y 2.3 g (100.0 mmol) de sodio reaccionan siguiendo la técnica descrita en la sección 2.2.8 obteniéndose la amina 36 (4.05 g, 74%). El compuesto 36, se disuelve en una mezcla de 100 ml de THF y 50 ml de una disolución saturada de NaHCO3. La mezcla se trata con 3.3 g (15 mmol) de di-ter-butildicarbonato (Boc2O), se agita por 3 días a temperatura 29 Parte Experimental ambiente y se concentra a presión reducida. El residuo se disuelve en 200 ml de CH2Cl2 y se lava consecutivamente con disolución acuosa de HCl 2N (2 × 50 mL) y 50 ml de salmuera, se seca sobre Na2SO4 anhidro y se concentra a presión reducida. El producto fue purificado por cromatografía de columna (n-hexano/AcOEt 3:1) para obtener el carbamato 37 (3.9g, 71%) como un sólido blanco. Tf (AcOEt): 213-215 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.76 (s, 3H, H-18); 0.79 (d, 3H, J=6.4 Hz, H-27); 0.96 (d, 3H, J=6.8 Hz, H-21); 0.96 (s, 3H, H-19); 3.08 (br. s, 1H, NH); 3.37 (t, 1H, J=11.0 Hz, H- 26ax); 3.47 (dd, 1H, J= 10.4/2.8 Hz, H-26eq); 3.56 (m, 1H, H-3α); 3.91 (br. m, 1H, H-6β); 4.37 (m, 1H, H-16α); 4.43 (s, 1H, NH). RMN-13C (75 MHz, CDCl3): δ = 14.4, 16.3, 17.0, 17.1 (CH3); 20.1, 20.9, 26.1 (CH2); 28.3 (CH3); 28.7 (CH2); 30.2 (CH); 31.3, 31.4, 31.7 (CH2); 34.1 (CH); 35.0, 39.9 (CH2); 40.7, 41.2 (C); 41.6, 42.7, 53.9, 56.0, 62.0 (CH); 66.8 (CH2); 71.2 (CH); 80.1 (C); 80.6 (CH); 109.3, 157.4 (C). HRMS (ESI-FT-ICR) m/z: 554.3814 [M+Na]+; calculada para C32H53NO5Na: 554.3816. 30 Parte Experimental 2.3 Síntesis de los conjugados péptido-esteroide 2.3.1 Procedimiento General Una disolución que contiene la amina (1mmol) y paraformaldehído (1mmol) en MeOH (volumen necesario para disolver los reaccionantes) se mantiene agitando a temperatura ambiente durante 2 h para formar la imina correspondiente. Se adiciona el ácido (1mmol) y transcurridos unos 15 min se adiciona el isonitrilo (1mmol). La mezcla de reacción se agita por período entre 24 h y 48 h. Posteriormente se diluye con cloroformo y se lava sucesivamente con disoluciones de HCl al 5%, NaHCO3 al 10% y NaCl (sat.). La fase orgánica se seca con Na2SO4 anhidro y se concentra a presión reducida. El crudo de reacción se purifica por cromatografía de columna. Los péptidos Nprotegidos que se utilizan en este trabajo como componente acido (Figura 18) para la conjugación multicomponente, fueron facilitados por un colaborador del Centro de Estudios de Productos Naturales. Figura 18. Tripéptidos (38, 39) y tetrapéptidos (40,41) facilitados utilizados en la conjugación. 31 Parte Experimental 2.3.2 Síntesis del conjugado péptido-esteroide 42 500 mg (1.7 mmol) de la amina 25, 841mg (1.7 mmol) de Boc-Ile-Ala-Leu-Ala-OH (40), 52 mg (1.7 mmol) de paraformaldehído y 0.17ml (1.7 mmol) de isocianoacetato de metilo reaccionan disueltos en 20ml de MeOH según el procedimiento descrito en el epígrafe 2.3.1. La purificación cromatográfica (n-hexano/AcOEt 5:1) permitió obtener el conjugado 42 (966 mg, 63 %). Tf (AcOEt):231-233 ºC. RMN-1H (300 MHz, CDCl3): δ = 0.64 (s, 3H, CH3); 0.85-0.92 (m, 12H, 4×CH3); 096 (s, 3H, CH3); 1.33 (d, 3H, J = 6.0 Hz, CH3); 1.36 (d, 3H, J = 7.0 Hz, CH3); 1.41 (s, 9H, t.Bu-Boc); 2.04 (dd, J=19.0/8.9 Hz, 1H); 2.22 (m, 2H); 2.41 (dd, J=19.2/8.8 Hz, 1H); 3.67 (s, 3H); 3.71 (m, 1H); 3.80 (dd, 1H, J = 17.7/5.7 Hz); 3.98 (m, 1H); 4.06 (m, 2H); 4.08 (m, 1H); 4.12 (m, 1H); 4.31-4.26 (m, 2H); 4.54 (m, 1H); 4.75 (m, 1H); 4.91 (t, 1H, J = 6.5 Hz); 5.47 (sancho, 1H, NH); 7.08 (sancho, 1H, NH); 7.38 (sancho, 1H, NH); 7.44 (m, 1H, NH); 7.92 (sancho, 1H, NH). RMN-13C (75 MHz, CDCl3): δ = 11.9, 11.4, 15.6, 18.2, 18.4, 18.9 (CH3); 21.4 (CH2); 21.9, 22.1 (CH3); 22.6 (CH2); 23.1, 23.4 (CH); 24.1 (CH2); 24.6 (CH); 24.7, 26.0, 26.9, 28.0 (CH2); 28.2 (CH3); 29.3, 29.6, 30.5, 30.9, 31.0, 31.9 (CH2);33.7 (C); 34.5 (CH); 35.1 (CH2); 35.3, 37.6, 38.0 (CH); 39.9, 41.0, 41.5, 42.1 (CH2); 42.5 (C); 45.5 (CH); 46.6 (CH2); 46.8, 48.9, 51.4, 52.1 (CH); 53.0 (CH3); 55.7, 56.6, 59,3 (CH); 65.5 (CH2); 80.0 (C); 156.2, 169.5, 170.1, 171.7, 172.1, 172.8, 174.7, 205.4 (CO). HRMS (ESI-FT-ICR) m/z: 909.5697 [M+Na]+; calculada para C47H78N6NaO10: 909.5672. 32 Parte Experimental 2.3.3 Síntesis del conjugado péptido-esteroide 43 500 mg (1.1 mmol) de la amina 21, 702 mg (1.1 mmol) de Boc-Lys(Cbz)-Val-Phe-OH (38), 33.6 mg (1.1 mmol) de paraformaldehído y 0.12 ml (1.1 mmol) de isocianoacetato de metilo reaccionan disueltos en 20 ml MeOH según el procedimiento descrito en el epígrafe 3.3.1. La purificación cromatográfica (n-hexano/AcOEt 3:1) permitió obtener el conjugado 43 (556 mg, 40 %). Tf (AcOEt): 245-247 °C. RMN-1H (300 MHz, CDCl3): δ = 0.79 (d, 3H, J = 6.4 Hz); 0.78 (s, 3H); 0.87 (d, 3H, J = 7.0 Hz); 0.89 (d, 3H, J = 6.8 Hz); 0.96 (d, 3H, J = 6.8 Hz); 1.26 (s, 3H); 1.43 (s, 9H); 2.90- 2.98 (m, 1H); 3.103.21 (m, 2H); 3.37 (t, 1H, J = 10.9 Hz); 3.47 (dd, 1H, J = 10.10/4.1 Hz); 3.56 (m, 1H); 3.72 (s, 3H); 4.01 (m, 2H); 4.38 (m, 1H); 4.47 (m, 1H); 5.10 (s, 2H); 5.23 (m, 1H); 7.19- 7.35 (mancho, 10H); 7.47 (td, 1H, J = 7.4/2.8 Hz); 7.55 (t, 1H, J = 7.2 Hz); 7.65 (d, 1H, J = 6.8 Hz); 7.68 (d, 1H, J = 7.0 Hz). RMN-13C (75 MHz, CDCl3): δ = 14.1, 14.5, 16.5, 17.1, 17.4, 19.2 (CH3); 20.5, 22.7 (CH2); 28.3, 28.7, 29.0 (CH2); 29.4, 29.7, 30.2, 30.6 (CH); 31.3, 31.6, 31.8 (CH2); 32.0, 34.4, 34.6, 35.6, 36.4, 37.4, 37.7 (CH); 39.1 (CH2); 39.9, 40,0 (C); 40.5 (CH2); 40.8, 41.1, 41.2, 41.5, 45.1, 45.9, 48.4, 49.3, 51.1, 52.1 (CH); 52.3 (CH3); 54.7, 55.6, 55.8 (CH); 55.9, 58.9 (CH2); 59.1, 62.0 (CH); 66.8, 72.7 (CH2); 75.3, 80.3, 109.2 (C); 127.0, 128.0, 128.4, 128.5, 128.7, 129.4, 129.7, 131.9, 132.1, 133.6, 133.8 (CH); 136.4, 137.1 (C); 156.7, 157.0, 170.3, 170.6, 172.0, 172.5, 172.9 (CO). HRMS (ESI-FT-ICR) m/z: 1207.6864 [M+Na]+; calculada para C65H96N6NaO14: 1207.6877. 33 Parte Experimental 2.3.4 Síntesis del conjugado péptido-esteroide 44 300 mg (0.8 mmol) de la amina 29, 375 mg (0.8 mmol) de Boc-Ile-Ala-Leu-Ala-OH (40), 23 mg (0.8 mmol) de paraformaldehído y 0.10 ml (0.8 mmol) de isocianoacetato de metilo reaccionan disueltos en 20 ml MeOH según el procedimiento descrito en el epígrafe 2.3.1. La purificación cromatográfica (n-hexano/AcOEt 4:1) permitió obtener el conjugado 44 (487 mg, 64 %). Tf (AcOEt):227-229 °C. RMN-1H (300 MHz, CDCl3): δ = 0.63 (s, 3H, CH3); 0.83-0.94 (m, 12H, 4×CH3); 1.12 (d, 3H, J = 6.6 Hz, CH3); 1.23 (s, 3H, CH3); 1.31 (d, 3H, J = 6.6 Hz, CH3); 1.35 (d, 3H, J = 6.9 Hz, CH3); 1.41 (s, 9H, t.Bu-Boc); 2.17-2.23 (m, 1H); 2.31-2.36 (m, 2H); 2.41 (dd, 1H, J=19.2/8.8 Hz); 3. 65 (s, 3H); 3.69 (s, 3H); 3.85 (dd, 1H, J=17.8/5.3 Hz); 4.00-4.05 (m, 6H); 4.13 (m, 1H); 4.51 (m, 1H); 4.73 (m, 1H); 4.93 (t, 1H, J = 7.0 Hz); 5.37 (sancho, 1H, NH); 6.89 (sancho, 1H, NH); 7.33 (m, 2H, NH); 7.79 (sancho, 1H, NH). RMN-13C (75 MHz, CDCl3): δ = 11.6, 12.2, 15.6, 16.2, 17.2, 19.4, 20.0 (CH3); 21.0, 21.8 (CH); 23.0, 23.4 (CH3); 23.6 (CH2); 24.6 (CH); 25.3, 26.0, 27.1 (CH2); 28.3, 28.5 (CH3); 30.4, 30.8, 31.0 (CH2); 33.7 (CH); 34.4 (CH2); 34.5 (C); 34.9, 36.1, 37.8 (CH); 39.7, 40.4, 40.8, 41.2 (CH2); 41.5 (C); 43.3, 46.0, 46.4, 47.0, 47.4, 48.0, 51.4 (CH); 51.8, 52.6 (CH3); 58.1 (CH); 78.3 (C); 156.5, 169.8, 171.6, 171.9, 172.6, 173.1, 173.5, 174.9 (CO). HRMS (ESI-FT-ICR) m/z: 1009.6567 [M+Na]+; calculada para C53H90N6NaO11: 1009.6559. 34 Parte Experimental 2.3.5 Síntesis del conjugado péptido-esteroide 45 450 mg (1.0 mmol) del (Boc)-amina 37 se tratan con TFA/CH2Cl2 al 20% a 0ºC y se agita durante 3h. Al finalizar la reacción se concentra a presión reducida y el producto se disuelve en 30 ml CH2Cl2 y se lava con disolución de NaOH 1M (2 x 40ml). La fase orgánica se seca sobre Na2SO4 anhidro y se concentra a presión reducida. El crudo obtenido se disuelve en 20 ml de MeOH y se trata con 578 mg (1.0 mmol) de Boc-Tyr(OBn)-Ala-Leu-OH (39), 31 mg (1.0 mmol) de paraformaldehído y 0.13 ml (1.0 mmol) de isocianoacetato de metilo según el procedimiento descrito en el epígrafe 2.3.1. La purificación cromatográfica (n-hexano/AcOEt 4:1) permitió obtener el conjugado 45 (836 mg, 73 %). Tf (AcOEt): 249-251 °C. RMN-1H(300 MHz, CDCl3): d = 0.79 (d, 3H, J=5.8 Hz); 0.88 (s, 3H); 0.91 (d, 3H, J=6.4 Hz); 0.92 (d, 3H, J=6.4 Hz); 0.97 (d, 3H, J=6.4 Hz); 1.05 (s, 3H); 1.29 (d, 3H, J=7.2 Hz); 1.39 (s, 9H); 3.00 (m, 2H); 3.37 (t, 1H, J= 10.9 Hz); 3.49-3.46 (m, 1H); 3.58 (m, 1H); 3.68 (s, 3H, CH3O); 3.35-4.04 (m, 4H); 4.08-4.20 (m, 4H); 4.29-4.40 (m, 3H); 4.45-4.50 (m, 2H); 4.62-4.65 (m, 1H); 4.68-4.80 (m, 2H); 4.98 (t, 1H, J= 6.9 Hz); 5.03 (s, 2H); 6.69 (d, 1H, J= 7.0 Hz, NH); 6.88 (d, 1H, J= 7.1 Hz, NH); 6.90 (dd, 2H, J= 8.8 /3.0 Hz, Ar); 7.10 (dd, 2H, J= 8.7 /3.2 Hz, Ar); 7.32-7.43 (mancho, 5H, Ar); 7.49 (t, 1H, J= 5.7 Hz); 8.24 (sancho, 1H, NH). RMN-13C (75 MHz, CDCl3): d = 14.1, 14.5, 16.3, 17.1, 17.4 (CH3); 19.9, 20.9 (CH2); 20.9, 21.6, 22.0 (CH3); 22.7 (CH2); 23.2, 23.4, 24.4, 24.6 (CH); 28.2 (CH3); 28.7, 29.0, 29.7 (CH2); 30.2 (CH); 31.3, 31.4, 31.5, 30.0 (CH2); 35.3 (CH); 37.0, 37.1, 37.3, 39.7, 39.8, 40.6, 40.8, 41.1 (CH2); 41.5 (CH); 41.8, 42.3 (C); 43.0, 47.1, 47.8 (CH); 48.3 (CH2); 48.5, 48.7 (CH); 49.8, 50.4 (CH2); 51.0 (CH); 52.3 (CH3); 52.5, 52.7, 55.5, 55.8, (CH); 56.0 (CH2); 60.0, 62.0 (CH); 66.8, 69.9 (CH2); 76.0 (CH); 80.5, 109.1 (C); 114.9, 115.0, 127.4, 127.9 (CH); 128.4, 128.3 (C); 128.5, 130.2, 130.3, 130.4 (CH); 136.8 (C); 157.9, 170.1, 171.1, 171.5, 172.4, 173.6, 175.0 (CO). 35 Parte Experimental HRMS (ESI-FT-ICR) m/z: 1120.6554 [M+Na]+; calculada para C62H91N5NaO12: 1120.6556 2.3.6 Síntesis del conjugado péptido-esteroide 46 400 mg (1.0 mmol) de la amina 31, 471mg (1.0 mmol) de Boc-Phe-Leu-Leu-Ala-OH (41), 27.9mg (1.0 mmol) de paraformaldehído y 0.11 ml (1.0 mmol) de isocianoacetato de metilo reaccionan disueltos en 20 ml de MeOH según el procedimiento descrito en el epígrafe 2.3.1. La purificación cromatográfica (n-hexano/AcOEt 4:1) permitió obtener el conjugado 46 (656 mg, 64 %). Tf (AcOEt): 244-247 °C. RMN-1H (300 MHz, CDCl3): δ = 0.79 (d, 3H, J=5.8 Hz); 0.89 (s, 3H); 0.90 (d, 3H, J=5.6 Hz, CH3); 0.91 (d, 3H, J=5.6 Hz, CH3); 0.97 (d, 3H, J=6.4 Hz); 1.12 (s, 3H); 1.33 (d, 3H, J=7.2 Hz, CH3); 1.40 (s, 9H, (CH3)3C); 2.92 (m, 1H); 3.23-3.11 (m, 2H); 3.42 (m, 1H); 3.52 (m, 1H); 3.71 (s, 3H, CH3O); 3.82 (m, 2H); 4.07-3.94 (m, 4H); 4.18-4.11 (m, 4H); 4.35 (m, 1H); 4.50-4.43 (m, 2H); 4.56 (m, 1H); 4.66 (m, 1H); 4.94 (m, 1H); 5.51 (d, 1H, J=4.6 Hz, NH); 7.09 (m, 1H, NH); 7.12 (m, 1H, NH); 7.287.19 (m, 5H, Phe); 7.53 (d, 1H, J=7.2 Hz, NH); 7.59 (m, 2H, NH). RMN-13C (75 MHz, CDCl3): δ = 12.3, 14.5, 16.4, 17.1, 18.6, 21.6, 22.9 (CH3); 21.1 (CH2); 24.7 (CH); 28.2 (CH3); 28.5, 28.7 (CH2); 30.2 (CH); 31.2, 31.3, 31.4, 31.7, 32.2 (CH2); 35.0 (CH); 35.5 (C); 36.9, 38.1, 40.0 (CH2); 40.5 (C); 40.9, 41.2, 43.4 (CH2); 44.8, 45.3 (CH); 45.8 (CH2); 52.0 (CH); 52.2 (CH3); 54.3, 55.9, 56.3, 57.3, 62.1 (CH); 66.8 (CH2); 71.2 (CH); 80.2 (C); 80.8, 126.8, 128.4, 128.6, 128.8, 129.1 (CH); 136.6 (C); 155.8, 169.5, 169.8, 171.9, 172.5, 172.7, 173.3 (CO). HRMS (ESI-FT-ICR) m/z: 1127.6990 [M+Na]+; calculada para C61H96N6NaO12: 1127.6984 36 Parte Experimental 2.3.7 Síntesis del conjugado péptido-esteroide 47 14 6 mg (0.5 mmol) de la amina 33, 243 mg (0.5 mmol) de Boc-Ile-Ala-Leu-Ala-OH (40), 15 mg (0.5 mmol) de paraformaldehído y 0.16 ml (0.5 mmol) de isocianoacetato de metilo reaccionan disueltos en 20 ml de MeOH según el procedimiento descrito en el epígrafe 2.3.1. La purificación cromatográfica (n-hexano/AcOEt 2:1) permitió obtener el conjugado 47 (303 mg, 68 %). Tf: 229-230 °C. RMN-1H (300 MHz, CDCl3): δ = 8.76 (sancho, 1H); 8.57 (sancho, 1H); 8.43 (sancho, 1H); 7.58 (sancho, 1H); 5.72 (sancho, 1H); 5.15 (m, 1H); 5.02 (m, 1H); 4.86 (m, 1H); 4.77 (m, 1H); 4.54 (m, 1H); 4.224.06 (m, 4H); 3.66 (s, 3H, CH3O); 1.45 (s, 9H, (CH3)3C); 1.36 (d, 1H, J=6.9 Hz, CH3); 1.34 (d, 1H, J=7.0 Hz, CH3); 0.91-0.89 (m, 6H, 2×CH3), 0.88 (d, 3H, J=6.5 Hz, CH3); 0.84 (s, 3H, CH3); 0.72 (s, 3H, CH3). RMN-13C (75 MHz, CDCl3): δ = 11.4, 12.0, 17.2, 17.4 (CH3); 20.5 (CH2); 15.6, 18.2 (CH3); 20.8 (CH2); 23.4, 23.5 (CH3); 26.2, 26.9, 28.0, 28.1 (CH2); 28.3 (CH3); 31.4, 33.8 (CH2); 34.9 (CH); 30.9, 33.9 (CH2); 34.7 (CH); 35.0 (CH2); 35.3 (C); 35.7, 37.0 (CH); 39.9, 40.3, 40.5 (CH2); 40.8 (C); 41.0, 42.0, 42.7, 48.8 (CH); 51.5 (CH3); 55.8, 56.1, 59.4, 61.0 (CH); 80.1 (C); 155.9, 169.0, 170.6, 170.6, 171.4, 172.1, 174.7 (CO). HRMS (ESI-FT-ICR) m/z: 911.5839 [M+Na]+; calculada para C47H80N6NaO10: 911.5834. 37 Discusión de los Resultados 3. Discusión de los Resultados 3.1 Metodología de trabajo El presente trabajo está encaminado al desarrollo de un nuevo y eficiente procedimiento de conjugación de péptidos a esteroides, que permitirá poseer una metodología alternativa al tradicional método de conjugación basado en acoplamiento peptídico. Los conjugados péptidoesteroidales se definen como quimeras moleculares con importantes aplicaciones químicobiológicas derivadas de la unión de ambas familias de moléculas, cada una aportando características estructurales que definen las propiedades del híbrido resultante. Dichas aplicaciones incluyen su empleo como receptores supramoleculares de oligopéptidos, como enzimas artificiales (conjugados con propiedades biocatalíticas), y como miméticos de péptidos catiónicos antibacterianos. Muchas de las propiedades de estos híbridos se deben a la mayor preorganización conformacional que presentan las cadenas peptídicas al estar conjugadas a un esqueleto de gran rigidez como es el núcleo esteroidal. En los trabajos anteriores basados en acoplamiento peptídico, se reportaron compuestos híbridos en los que la cadena peptídica se encuentra conjugada al esteroide por su residuo C-terminal, lo que se logra mediante el empleo de una amina esteroidal como sustrato de partida para la incorporación secuencial de los αaminoácidos N-protegidos, mediante acoplamientos peptídicos consecutivos. La metodología de conjugación desarrollada consiste en el empleo de la reacción de Ugi-4C entre aminas esteroidales y oligopéptidos N-protegidos, para obtener compuestos híbridos con estructuras nunca antes reportadas en la literatura científica. Como se muestra en la figura 19, durante el proceso de conjugación se produce un dipéptido ramificado, por lo que la cadena peptídica original se alarga en dos residuos de aminoácidos, quedando a la vez N-sustituida con el esqueleto esteroidal. En todos los casos se emplea paraformaldehído como componente carbonílico de la reacción de Ugi-4C, para evitar la formación de mezclas de estereoisómeros. Figura 19. Metodología de conjugación de péptidos a esteroides empleando la reacción de Ugi-4C. 38 Discusión de los Resultados La naturaleza multicomponente de la reacción de Ugi-4C permite implementar el concepto de síntesis orientada a la diversidad, en el que varios de los componentes que participan en el proceso se pueden variar para generar estructuras diversas. Por ejemplo, los sustratos esteroidales pueden ser funcionalizados con cada uno de los grupos funcionales que participan en el proceso multicomponente–amino, ácido carboxílico, isonitrilo o aldehído–logrando así una gran diversidad en el esqueleto dipeptídico que uniría el esteroide con el oligopéptido. En este trabajo nos enfocamos exclusivamente en el empleo de aminas esteroidales, por lo que la generación de diversidad estructural se logra mediante la variación de la posición del grupo amino en el esqueleto esteroidal y de los α-amino-ácidos que componen el oligopéptido, manteniendo siempre constantes la estructura del aldehído y del isonitrilo. La figura 20 muestra algunas de las posibles combinaciones utilizadas para obtener híbridos péptidos-esteroidales en los que se ha modificado tanto el sitio de conjugación como la estructura de los péptidos. Figura 20. Ejemplo de algunos conjugados péptido-esteroidales sintetizados en este trabajo Para el desarrollo de este nuevo procedimiento de conjugación de péptidos a esteroides fue necesario dividir el trabajo en dos partes fundamentales: a) funcionalización de los esteroides mediante la introducción de grupos amino en diversas posiciones del esqueleto, b) evaluación de la eficiencia química y versatilidad de la reacción de Ugi-4C como método de conjugación. A continuación describimos el desarrollo de cada una de estas partes del trabajo experimental y la caracterización espectroscópica de los compuestos obtenidos. Todos los compuestos nuevos fueron caracterizados mediante RMN 1H, RMN 13 C, DEPT 135, y espectrometría de masa. No obstante, algunos compuestos han sido sintetizados previamente en el Centro de Estudios de 39 Discusión de los Resultados Productos Natrurales, por lo que en tales casos la identificación se realizó por comparación de las temperaturas de fusión y de los Rf con los de patrones debidamente caracterizados. 3.2 Síntesis de aminas esteroidales Uno de los objetivos de este trabajo es la obtención de aminas esteroidales para su posterior conjugación a oligopéptidos N-protegidos mediante la reacción de Ugi-4C. Como se explicó en la revisión bibliográfica (epígrafe 1.4.5), la incorporación de grupos amino en diferentes posiciones del núcleo esteroidal puede realizarse mediante la reducción de oximas o de azidas esteroidales. Es conocido que la reducción de un grupo oxima en posición 3 del esqueleto esteroidal no es un proceso estereoselectivo con ninguno de los métodos conocidos en la literatura, por lo que la obtención de 3-amino-esteroides requiere de la introducción estereoselectiva de una azida y su posterior reducción a amina. Afortunadamente, este proceso es de fácil implementación mediante la tosilación de un grupo alcohol en dicha posición y posterior sustitución (SN2) por azida con inversión total de la configuración original del alcohol de partida. Figura 21. Secuencia de síntesis de los 3-amino-esteroides 21, 25 y 29. En la figura anterior se muestra un procedimiento que asegura la introducción de grupos azida y amina de forma estereoespecífica en el esqueleto esteroidal, tiene como inconveniente que se obtienen aminas con disposición axial cuando reacciona un alcohol secundario ecuatorial. En el último paso de esta ruta sintética se reduce la azida a amina, para lo cual se utilizó la reacción de de Staudinger con trifenil fosfina y agua como sistema reductor. Esta reducción procede con total 40 Discusión de los Resultados quimioselectividad, pues no afecta otros grupos funcionales (grupos carbonilo y dobles enlaces) que sí son afectados mediante hidrogenación catalítica. Para la síntesis de 3-amino-esteroides se emplearon materias primas suministradas por el Centro de Estudios de Productos Naturales, las cuales contienen un grupo OH en la posición 3 (Tabla 1). Tabla 1. Sustratos de partida para la síntesis de 3-amino-esteroides. Sustrato de partida Amina sintetizada En todos los casos, la introducción del grupo tosilo se llevó a cabo empleando TsCl como reactivo y piridina como disolvente. Los tosilatos se obtuvieron con excelentes rendimientos y pureza, por lo que fueron empleados en la reacción de SN2 sin purificación previa. Para la síntesis de la amina 21 se empleó el triol 18, compuesto en el que se aprovechó la diferencia de reactividad entre los tres grupos hidroxilos presentes, para realizar la tosilación selectiva del grupo 3β-OH sin afectar los grupos 5α-OH y 6β-OH. Es conocido que la tosilación se puede realizar de forma selectiva en alcoholes secundarios ecuatoriales, por lo que el hidroxilo terciario 5α-OH no se afecta debido fundamentalmente al impedimento estérico provocado por las interacciones gauche de un sustituyente en disposición axial. Como se observa en la figura 22, el hidroxilo 6β-OH además de ser axial presenta interacción 1,3-diaxial con el CH3-19, por lo que se encuentra muy impedido para la introducción de un grupo voluminoso como el tosilo. 41 Discusión de los Resultados Figura 22. Diferencia de reactividad entre los grupos hidroxilos de las posiciones 3, 5 y 6. La síntesis de las azidas 20, 24 y 28 se llevó a cabo mediante la reacción de sustitución nucleofílica bimolecular (SN2), empleando azida de sodio como fuente del anión N3¯ y con agitación vigorosa en DMF a 50 ˚C. En estas condiciones de reacción se favorece la SN2, por lo que las azidas resultantes presentan la configuración inversa del alcohol de partida. En todos los casos aparece una pequeña mancha adicional a la mayoritaria de la azida en el análisis del transcurso de la reacción por CCD, ésta se corresponde con el alqueno obtenido como producto del proceso de eliminación que ocurre colateralmente a la SN2 se obtiene solo en 1-5% de rendimiento y son de fácil eliminación mediante la purificación de las azidas por CC. La figura 23 muestra el espectro de RMN 1H de la azida espirostánica 23, en el mismo espectro se observan las señales clásicas de todos los esteroides que presentan una cadena espirocetálica, estos son los protones H-16α, H-26ecuatorial y H-26 axial, y las señales de los metilos 18, 19, 21 y 27. El análisis de dicho espectro también permite la confirmación de la estereoquímica α del grupo azida axial en el carbono 3, pues se corrobora la disposición ecuatorial de la señal de H-3. La forma fina que presenta el multiplete del H-3 es la evidencia que confirma la disposición ecuatorial de dicho protón, debido a que tienen lugar acoplamientos axial-ecuatorial con H-2β y H4α y ecuatorial-ecuatorial H-2α y H-4β, todos con constantes de acoplamiento pequeñas (J~4-5 Hz). Además, se observa la señal del protón del OH-5α, lo que evidencia la formación de puentes de hidrógeno con el grupo azida del C-3, demostrando que el mismo presenta estereoquímica α y una disposición axial. De igual manera, se confirmó la disposición ecuatorial del protón H-3 de la azida 24 (H-3β) y la disposición axial del mismo para el caso de la azida 28 (H-3α). Esta última se reconoce en el espectro de RMN 1H por la presencia de un multiplete ancho debido a los acoplamientos axial-axial con H-2α y H-4α (J~10-11) y axial ecuatorial con H-2β y H-4β; lo que demuestra la inversión total de la configuración con respecto a los alcoholes esteroidales de partida. La reducción del grupo azido de los compuestos 20, 24 y 28 se realizó mediante la reacción de Staudinger, utilizando PPh3 y H2O como sistema reductor. Estas reacciones se siguieron por 42 Discusión de los Resultados CCD, en las que se observó la desaparición de las manchas de las azidas de partida y la aparición de manchas en la zona de aplicación correspondientes a las aminas esteroidales. Como se explicó en el epígrafe 1.4, la reducción con PPh3 presenta el inconveniente de generar óxido de trifenilfosfina (Ph3PO), producto colateral que requiere purificación por CC para su eliminación. Este proceso conlleva a la pérdida de parte de la amina esteroidal, por adsorcion al gel de sílice debido a su elevada polaridad y carácter básico. Consecuentemente, se decidió emplear las aminas 21, 25 y 29 en las reacciones de Ugi-4C sin previa purificación, teniendo en cuenta que el Ph3PO no interfiere en el curso de la reacción y es de fácil eliminación en el proceso final de purificación de los conjugados péptido-esteroidales. Figura 23. Espctro de RMN 1H en CDCl3 del (25 R) -3α –azido-5α espirostano-5,6β- diol (20). Otra variante sintética de gran utilidad, es la reducción de oximas a aminas, este procedimiento no se emplea para la obtención de 3-amino-esteroides porque no transcurre con estereoselectividad y produce los 3α y 3β-amino derivados. Sin embargo, es conocido que en las posiciones 12 y 17 el proceso sí es estereoselectivo y produce el 12α y 17α-amino-esteroide como productos fundamentales en más de un 95% de rendimiento. La estereoselectividad de este proceso de reducción de oximas en las posiciones 12 y 17 – que transcurre con control termodinámico – se debe a la presencia del CH3-18 en la posición vecina. Este grupo voluminoso produciría una desestabilización por impedimento estérico, si el grupo funcional resultante presentara estereoquímica β, por lo que en las condiciones de reducción reportadas se obtienen de forma 43 Discusión de los Resultados casi cuantitativa las aminas con estereoquímica α. Como se observa en la figura 24, se utilizó este método para la introducción de grupos amino en los carbonos 12 y 17, mediante la formación y posterior reducción de las oximas a partir del 12-ceto y el 17-ceto esteroides 4 y 22, respectivamente. Las aminas 31 y 33 se obtuvieron con buenos rendimientos de reacción y fueron purificadas por recristalización de EtOH para su caracterización espectroscópica. Un aspecto a destacar es la obtención de un único isómero configuracional en todas las oximas producidas (presumiblemente el isómero E), un resultado que también coincide con lo reportado en la literatura. La reacción de condensación con el hidrocloruro de hidroxilamina se llevó a cabo empleando piridina como disolvente. En todos los casos la transformación se completó en 24h a temperatura ambiente, al observar en la CCD que el sustrato de partida se consumió totalmente y se formó un producto de mayor polaridad. Figura 24. Secuencia de síntesis de las aminas 31, 33 y 36. Debido a nuestro interés en variar el sitio de conjugación, se procedió a la obtención del 6-aminoesteroide 36, un resultado no reportado en la literatura con anterioridad. Debido a que no se conocía la estereoselectividad de este proceso en el carbono 6, se implementó la misma metodología de formación y reducción de la oxima pero se procedió además a proteger el grupo amino resultante en forma de tert-butilcarbamato (Boc) para facilitar la purificación por CC y la asignación de la estereoquímica por RMN 1H. Así, partiendo del 6-ceto-esteroide, laxogenina (34) se produjo el 6-amino derivado 37 con un 71% de rendimiento total. El análisis por RMN 1H del único producto obtenido demostró que corresponde al 6α-amino-esteroide, lo que es una nueva evidencia de la estereoselectividad de este proceso de reducción en la posición 6 del núcleo 44 Discusión de los Resultados esteroidal. Este resultado es previsible atendiendo al hecho de que la hipotética amina con estereoquímica 6β tendría una disposición axial, por lo que presentaría interacciones 1,3-diaxial con el CH3-19 del esteroide. El impedimento estérico provocado por dichas interacciones de naturaleza desestabilizante, conlleva a que el producto termodinámico de la reacción sea la amina 6α, la cual presenta disposición ecuatorial, por lo que es el único producto de dicha reacción. Figura 25. Espectro de RMN 1H en CDCl3 del Boc-amino-esteroide 37. El análisis del espectro de RMN 1H del compuesto 37 confirma la estereoquímica α del grupo amino en el carbono 6, en este caso protegido en forma de tert-butilcarbamato. Como se muestra anteriormente en la figura 25, la disposición ecuatorial (estereoquímica α) de dicho grupo se demuestra por la forma ancha del multiplete correspondiente a H-6, lo que evidencia la disposición axial del protón H-6β que presenta acoplamientos axial-axial (J~10-12 Hz) con H-5α y H-7α. Otra evidencia de la estereoquímica 6α del grupo tert-butilcarbamato es el bajo desplazamiento químico del CH3-19, cuyo carácter blindado por debajo de 1.00 ppm demuestra la ausencia de impedimento estérico con dicho grupo voluminoso. Como se mencionó anteriormente, de poseer estereoquímica β, tendría lugar un gran impedimento estérico debido a la interacción 1,3-diaxial con el CH3-19, por lo que el singlete de dicho metilo aparecería más desblindado, alrededor de 1.2 ppm, a consecuencia de la polarización electrónica de los enlaces C-H del grupo metilo. Para entender dicho efecto se puede comparar con el espectro RMN 1H del 45 Discusión de los Resultados compuesto 20 (Figura 23), el que presenta interacción 1,3-diaxial entre el CH3-19 y el OH-6β. Todas estas evidencias espectroscópicas, permiten asegurar que en el compuesto 36 (y en su derivado protegido 37) el grupo amino en elcarbono 6 presentan una disposición ecuatorial (estereoquímica 6α), por lo que se demuestra el carácter estereoselectivo de la reducción de oxima en posición 6. 3.3 Síntesis de los híbridos péptido- esteroidales La metodología de obtención de los híbridos péptido-esteroidales consiste en emplear el esteroide funcionalizado como amina para la conjugación a péptidos N-protegidos, mediante la reacción de Ugi-4C (Figura 26). Figura 26. Metodología de obtención de los conjugados péptido-esteroides. Los conjugados obtenidos por este procedimiento se diferencian del resto de los conjugados reportados en la literatura en que el esqueleto peptídico no queda unido de forma lineal al esteroide por su residuo C-terminal, sino que resulta en un péptido ramificado en el que el esteroide se sitúa como N-sustituyente de un enlace amida. Este método tiene la particularidad de que en un sólo paso de reacción la cadena peptídica crece en dos amino-ácidos, lo que se debe al empleo de la reacción de Ugi-4C que genera un esqueleto dipeptídico. El procedimiento general empleado para la obtención de los híbridos fue el mismo en todos los casos. El seguimiento de las reacciones se realizó por CCD, y en todos los casos se observó la formación de un producto de mayor Rf que el ácido y la amina de partida. En algunos casos, después de 24h no se observaron cambios significativos en el transcurso de la reacción, por lo que se decidió detenerla a pesar de quedar sin reaccionar parte de los sustratos de partida. Esto fue más evidente en los casos de aminas con disposición axial, en los que un estudio de diferentes tiempos de reacción permitió concluir que no hubo mejoras significativas en los rendimientos de reacción, a pesar de prolongar la reacción por varios días. Todos los productos finales se purificaron por cromatografía de columna, empleando la mezcla de CH2Cl2/AcOEt de 46 Discusión de los Resultados polaridad creciente, lo que permitió separar el conjugado péptido-esteroidal del poco remanente de material de partida sin reaccionar. La tabla 2 muestra las combinaciones del procedimiento de conjugación por reacción de Ugi-4C de 3-amino-esteroides a péptidos N-protegidos. En todos los casos se empleó paraformaldehído e isocianoacetato de metilo como los otros dos componentes, lo que permitió conservar la naturaleza peptídica del producto. Tabla 2. Conjugación de péptidos a 3 amino-esteroides Amina Ácido Conjugado El rendimiento de dichos procesos de conjugación (42-64%) puede considerarse bueno, si tenemos en cuenta la naturaleza axial del grupo amino en el C-3 y el impedimento estérico que produce la introducción de una cadena peptídica voluminosa en dicha posición. Para evaluar el efecto estructural sobre la eficiencia química de la reacción de conjugación, se emplearon las aminas 21, 25 y 29, todas con el grupo amino en el C-3 con disposición axial, de esqueletos androstánico, espirostánico y colánico, respectivamente. Se puede concluir que la presencia de un grupo hidroxilo en la posición 5α (21) tiene un efecto desfavorable en el rendimiento de la conjugación en la posición 3, pues este rendimiento no mejoró al prolongar el tiempo de reacción. 47 Discusión de los Resultados Esto se puede explicar por la menor reactividad de la amina 3α debido a la interacción 1,3-diaxial que presenta con el grupo OH-5α. Por otra parte, la diferencia estructural entre esteroides de la serie 5α (fusión trans de los anillos A/B) y 5β (fusión cis de los anillos A/B) no tiene un efecto marcado en el rendimiento de la reacción, pues no se observan diferencias significativas en la eficiencia de la conjugación del tetrapéptido 40 a las aminas 25 y 29. La tabla 3 muestra los híbridos péptido-esteroidales obtenidos mediante la conjugación multicomponente en las posiciones 6, 12 y 17 de diferentes esqueletos esteroidales. Estas conjugaciones permitieron estudiar el efecto de la posición y la estereoquímica de los grupos amino en el rendimiento de la reacción. Así, la conjugación en la posición 6 fue la que produjo mayor rendimiento de reacción debido al carácter ecuatorial de la amina de partida, lo que favorece tanto la formación de la imina correspondiente como la incorporación de la cadena peptídica voluminosa. Por otra parte, la obtención del conjugado en posición 12 transcurrió con un rendimiento ligeramente inferior al de la conjugación en posición 17. De forma similar a los casos anteriores, esto se debe a la disposición axial de la amina 12α, la que presenta mayor impedimento estérico que la amina pseudo-ecuatorial 17α. En ambos casos, pudiera considerarse el impedimento estérico que brinda el metilo 18 axial, el cual se encuentra en posición vecina, debido al carácter voluminoso de los péptidos introducidos. No obstante, a pesar de que este efecto de vecindad de un grupo voluminoso no se encuentra en los casos anteriores, el mismo no parece ser tan acentuado al encontrase el metilo 18 en la cara opuesta a los grupos amino 12α y 17α. Así, se puede concluir que el efecto estructural de mayor influencia en la eficiencia química del proceso de conjugación es la diferencia de reactividad de los amino-esteroides debido al carácter axial o ecuatorial de las aminas empleadas, y no a la presencia de los grupos metilos 18 y 19 que presentan una estereoquímica contraria a la de los grupos amino. Tabla 3. Conjugación de péptidos a 6,12 y 17-amino-esteroides. Amina Ácido Conjugado 48 Discusión de los Resultados Para la caracterización estructural de los híbridos obtenidos se utilizaron técnicas de RMN y espectrometría de masa (ESI-MS). El empleo de la primera técnica no resultó de gran utilidad debido a la complejidad estructural de los conjugados sintetizados que conlleva a la superposición de señales y por tanto, se hace difícil la asignación. No obstante, esta técnica permite corroborar la estructura del conjugado pues tanto en RMN 1H como en 13 C se observan las señales características del esqueleto esteroidal, de la cadena peptídica original y del dipéptido Nsubstituido; formado durante la conjugación. Por otra parte la espectrometría de masa permitió la confirmación de las estructuras propuestas.El empleo de la técnica de ionización por electro spray (técnica blanda que provoca pocas fragmentaciones) confirmó la obtención de los conjugados con la masa molecular esperada a través de los picos [M+H]+ y [M+Na]+. En los anexos se presentan los espectros de RMN y de ESI-MS de todos los conjugados obtenidos, como evidencia de la asignación estructural realizada. A continuación se explican algunos ejemplos de espectros de RMN que demuestran el análisis espectroscópico realizado para la asignación estructural. La figura 27 muestra el espectro de RMN 1H del híbrido hexapéptido-androstano 42, en el mismo se observan señales anchas que evidencian la presencia de más de un isómero conformacional para dicha molécula. Esta característica se pone de manifiesto en los espectros de RMN 1H de todos los conjugados obtenidos, debido a que la cadena peptídica se encuentra N-sustituida por un grupo muy voluminoso como es el esqueleto esteroidal. Este hecho propicia la fácil interconversión entre los confórmeros cis y trans de dicho enlace amida, y por tanto la presencia de ambos isómeros en disolución. Lo anterior, unido a la gran complejidad estructural de los conjugados, hace del análisis detallado de estos espectros una tarea muy compleja a pesar de la 49 Discusión de los Resultados alta frecuencia de resonancia del equipo empleado (300 MHz). En este sentido, la asignación espectroscópica de los conjugados se llevó a cabo mediante la identificación de las señales características de cada subunidad estructural presente en la molécula. Por ejemplo, en la zona más desblindada del espectro del conjugado 42 se observan las señales de las amidas, mientras en la zona entre 3 y 5 ppm aparecen las señales de los protones α del péptido, así como el singlete del éster metílico. En la zona blindada del espectro aparecen los complejos multipletes característicos de los protones alifáticos del esteroide y de las cadenas laterales de los aminoácidos, incluyendo las típicas señales de los metilos del grupo Boc, la isoleucina, la leucina, la alanina y los clásicos metilos 18 y 19 del esteroide androstánico. Figura 27. Espectro de RMN 1H en CDCl3 del híbrido péptido-esteroidal 42. La figura 28 muestra el espectro de RMN 1H del híbrido 44 formado por la conjugación del péptido 40 a la amina colánica 29. En este espectro también se observan las señales clásicas de los protones de las amidas en la zona más desblindada, las señales de los protones α de cada uno de los aminoácidos, y los dos singletes de los ésteres metílicos, uno de la cadena peptídica y otro del esqueleto colánico. En la zona más blindada aparecen el singlete del tert-butilo del grupo protector Boc, los dobletes característicos de los metilos de la isoleucina, leucina y alanina, así como las señales de los metilos del esteroide colánico. 50 Discusión de los Resultados Figura 28. Espectro de RMN 1H en CDCl3 del híbrido péptido-esteroidal 44. Con el objetivo de hacer una comparación de los espectros de los diferentes tipos de conjugados, en la figura 29 se muestra el espectro de RMN 1H del híbrido 45. Debido a la elevada complejidad estructural que se logra por conjugación del péptido 39 con la amina espirostánica 37, resulta difícil asignar algunas de las señales tanto de la cadena peptídica como del esqueleto espirostánico. No obstante, el análisis de dicho espectro permite la confirmación inequívoca de la estructura esperada. Así, en la zona más desblindada del espectro se observa el multiplete que integra cinco protones correspondientes al grupo bencilo protector del fenol, y un par de dobletes característicos del fenilo para-disustituido de la tirosina (Tyr). 51 Discusión de los Resultados Figura 29. Espectros de RMN 1H y 13C en CDCl3 del híbrido péptido-esteroidal 45. La zona intermedia del espectro de RMN 1H del híbrido 45 es de difícil asignación debido a la superposición de las señales de la cadena peptídica y del esqueleto esteroidal, algo que no ocurría en esta medida en los conjugados anteriores debido a la menor complejidad estructural de 52 Discusión de los Resultados los esteroides androstánicos y colánicos. Es importante recordar que los esteroides espirostánicos poseen varias funciones oxigenadas en su estructura, por lo que presentan protones que aparecen en la zona entre 3 y 5 ppm al estar unidos a los carbonos funcionalizados con dichas funciones oxigenadas. De esta forma, las señales de los protones H-3α, H-6β, H-16α, H-26 ecuatorial y H-26 axial aparecen superpuestas con las señales de los protones α de los aminoácidos y con la señal de la cadena lateral de la tirosina. Como en los casos anteriores se observa el singlete del éster metílico, el singlete del tert-butilo del Boc, y las señales de los metilos de la leucina y la alanina, así como las señales de los cuatro metilos del esteroide spirostánico. La figura 29 también muestra el espectro de RMN 13 C del híbrido 45, en cuya zona más desblindada aparecen las señales de los carbonilos de las amidas, del éster metílico y del carbamato (Boc). Además se observan las señales de los carbonos aromáticos de la tirosina y del grupo protector bencilo, así como la clásica señal alrededor de 109 ppm del carbono espirocetálico (C-22) del esteroide espirostánico. Por otra parte, en la zona entre 0 y 100 ppm aparecen las numerosas señales de los carbonos alifáticos de la cadena peptídica y del esteroide, incluyendo las señales de los metilos que son las más blindadas del espectro. De forma similar al análisis de los espectros de RMN 1H, la aparición en los espectros de RMN 13 C tanto de las señales típicas de la parte peptídica como esteroidal confirman las estructuras híbridas resultantes de los procesos de conjugación. Indudablemente, la determinación de la masa molecular y su comparación con el valor calculado para el compuesto esperado, constituye un factor importante en la caracterización de los conjugados sintetizados Esto se realizó por espectrometría de masa empleando técnicas de ionización blandas como la ionización por electrospray (ESI-MS), la que produce pocas fragmentaciones del ión molecular y es ideal para la asignación estructural de compuestos peptídicos. La figura 30 muestra un ejemplo de la caracterización por espectrometría de masa (ESI-MS) del híbrido 43, en la que se observa el pico [M+Na]+ como el de mayor intensidad. En todos los casos, la confirmación del peso molecular del compuesto esperado se logra a través de la coincidencia entre los valores de masa de los picos obtenidos con valores teóricos calculados para [M+H]+ y [M+Na]+, por lo que se puede concluir que el compuesto obtenido es el deseado. En algunos casos se realizó caracterización por espectrometría de masa de alta resolución, que es la técnica, de este tipo, que permite afirmar con total seguridad si estamos en presencia del compuesto esperado, al ofrecer un valor de masa molecular muy exacto y con error sólo en la 53 Discusión de los Resultados cuarta cifra decimal. En los anexos se muestra la caracterización por espectrometría de masa (ESI-MS) del los conjugados obtenidos. Figura 30. Espectro de masa (EM-ESI) del hibrido péptido-esteroidal 43. 54 Conclusiones Conclusiones 1. Se desarrolló una nueva metodología de síntesis de híbridos péptidos-esteroidales a través de la reacción de Ugi 4 componentes. 2. La formación de oximas fue un método eficiente y esteroselectivo para la obtención de aminas esteroidales en las posiciones 6, 12 y 17. 3. Se implementó un nuevo método estereoselectivo en la obtención de aminas en el carbono 3 que consistió en la introducción del grupo azida y posterior reducción de Staudinger. 4. El procedimiento de conjugación fue sensible a factores estéricos, pues con aminas ecuatoriales resultó ser mas eficiente que con aminas axiales. 5. Se obtuvieron 6 nuevos conjugados péptidos-esteroidales los cuales fueron caracterizados por técnicas espectroscópicas de RMN (1H y 13C) y ESI-MS. 55 Recomendaciones Recomendaciones 1. Aplicar el método de conjugación desarrollado a la obtención de híbridos con aplicaciones biomedicinales. 2. Emplear otros tipos de reacciones multicomponentes basadas en isonitrilos para el desarrollo de nuevos procedimientos de obtención de híbridos péptidosesteroidales. 56 Bibliografía Bibliografía 1 Sewald, N.; Jakubke, H.-D.; Peptides: Chemistry and Biology, Wiley-VCH, 2002, Cap.1. 2 P. M. Hwang, H. J. Vogel, Biochem. Cell Biol. 1998, 76, 235. 3 Fieser, L. F.; Fieser, M.; Steroids, Reinhold Publishing Co., 1959. 4 Ding, B.; Taotofa, U.; Orsak, T.; Chadwell, M.; Savage, P. B.; Org. Lett., 2004, 6, 3433-3436. 5 Davis, A. P.; Pérez-Payán, M. N.; Synlett 9, 1999, 991–993. 6 Fried, J.; Edwards, J. A.; Organic Reactions in Steroid Chemistry, Van Nostrand Reinhold Co., 1972. 7 Sasse, J. M.; Brassinosteroids: Chemistry, Bioactivity and Applications, Eds. Cluter, H.J.; Yokota, T.; Adam, G.; ACS Symposium Series 474 American Chemical Society, Washington DC, 1991. pp. 158-166. 8 . Sewald, N.; Jakubke, H.-D.; Peptides: Chemistry and Biology, 2002, Wiley-VCH, Cap. 9 9 Yin, H.; Hamilton, A. D.; Angew. Chem., 2005, 4430-4163 10 Castani, A.; Sansone, F.; Ungaro, R.; Acc. Chem. Res., 2003, 36, 246–254 11 Lazzarotto, M.; Sansone, F.; Baldini, L.; Casnati,A.; Cozzini, P.;Ungaro, R.; Eur. J. Org. Chem., 2001, 595–602. 12 Sansone, F.; Barboso, S.; Casnati, A.; Fabbi, M.; Pochini, A.; Ugozzoli, F.; Ungaro, R.; Eur. J. Org. Chem., 1998, 897–905. 13 Park, H. S.; Lin, Q.; Hamilton, A. D.; J. Am. Chem. Soc., 1999, 121, 8–13 14 Hamuro, Y.; Calama, M. C.; Park, H. S.; Hamilton, A. D.; Angew. Chem. Int. Ed., 1997, 36, 2680–2683. 15 Chamorro, C.; Liskamp, R. J. M.; Tetrahedron, 2004, 60, 11145–11157. 16 Chamorro, C.; Liskamp, R. J. M.; J. Comb. Chem., 2003, 5, 794–801. 17 Rump, E. T.; Rijkers, D. T. S.; Hilbers, H. W.; de Groot, P. G.; Liskamp, R. J. M.; Chem. Eur. J., 2002, 8, 4613–4621. 18 Causton, A. S.; Sherman, J. C.; Bioorg. Med. Chem., 1999, 7, 23–27. 57 Bibliografía 19 Wright, A. T.; Anslyn, E. V.; Chem. Soc. Rev., 2006, 35, 14–28. 20 Wright, A. T.; Zhong, Z.; Anslyn, E. V.; Angew. Chem. Int. Ed., 2005, 44, 5679–5682. 21 Wright, A. T.; Griffin, M. J.; Zhong, Z.; MacCleskey, S. C.; Anslyn, E. V.; McDevitt, J. T.; Angew. Chem. Int. Ed., 2005, 44, 6375–6378. 22 (a) Rivera, D. G.; Wessjohann, L. A.; J. Am. Chem. Soc. 2009, 131, 3721-3722. (b) Rivera, D. G.; Wessjohann, L. A.; J. Am. Chem. Soc. 2006, 128, 7122–7123. (c) Feigel, M.; Ladberg, R.; Engels, S.; Herbst-Irmer, R.; Fröhlich, R. Angew. Chem. Int. Ed. 2006, 45, 5698–5702. (d) Wessjohann, L. A.; Voigt, B.; Rivera, D. G.; Angew. Chem. Int. Ed., 2005, 44, 4785–4790. (e) Albert, D.; Feigel, M.; Benet-Buchholz, J.; Boese, R. Angew. Chem. Int. Ed., 1998, 37, 2727–2729. (f) Albert, D.; Feigel, M.; Helv. Chim. Acta., 1997, 80, 2168-2181. (g) Wess, G.; Bock, K.; Kleine, H.; Kurz, M.; Guba, W.; Hemmeric, H.; Lopez-Calle, E.; Baringhaus, K.-H.; Glombic, H.; Enhsen, A.; Kramer, W.; Angew. Chem. Int. Ed., 1996, 35, 2222-2224. (h) Rivera, D. G.; Concepción, O.; Pérez-Labrada, K.; Coll, F.; Tetrahedron, 2008, 64, 5298-5305. 23 (a) del Amo, V.; Siracusa, L.; Markidis, T.; Baragaña, B.; Bhattarai, K. M.; Galobardes, M.; Naredo, G.; Pérez-Payán, M. N.; Davis, A. P.; Org. Biol. Chem., 2004, 2, 3320-3328. (b) Ding, B.; Taotofa, U.; Orsak, T.; Chadwell, M.; Savage, P. B.; Org. Lett., 2004, 6, 3433-3436. (c) Madder, A.; Li L.; De Muynck, H.; Farcy, N.; Van Haver, D.; Fant, F.; Vanhoenacker, G.; Sandra, P.; Davis, A. P.; De Clercq, P. J.; J. Comb. Chem., 2002, 4, 552-562. (d) De Muynck, H.; Madder, A.; Farcy, N.; De Clercq, P. J.; Pérez-Payán, M. N.; Öhberg, L. M.; Davis, A. P.; Angew. Chem. Int. Ed., 2000, 39, 145-148. (e) Li, H.; Wang, L.-X.; Org. Biol. Chem., 2003, 1, 3507-3513. (f) Davis, A. P.; Molecules, 2007, 12, 2106–2122. (g) Cheng, Y.; Suenaga, T.; Still, W. C.; J. Am. Chem. Soc., 1996, 118, 1813-1814. (h) Boyce. R.; Li, G.; Nestler, H. P.; Suenaga, T.; Still, W. C.; J. Am. Chem. Soc., 1994, 116, 7955-7956. 24 Banerjee, A.; Sergienko, E.; Vasile, S.; Gupta, V.; Vouri, K.; Wipf, P.; Org. Lett., 2009, 11 (1), 65–68. 25 Ruoslahti, E.; Pierschbacher, M.; Science, 1987, 238, 491–497 26 Gentilucci, L.; Cardillo, G.; Squassabia, F.; Tolomelli, A.; Spam-pinato, S.; Sparta, A.; Baiula, M.; Bioorg. Med. Chem. Lett., 2007, 17, 2329–2333. 27 Enhsen, A.; Kramer, W.; Wess, G.; Drug Discov. Today, 1998, 3, 409-418. 28 Tamminen, J.; Kolehmainen, E.; Molecules, 2001, 6, 21-46. 58 Bibliografía 29 (a) Kramer, W.; Wess, G.; Neckermann, G.; Schubert, G.; Fink, J.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.-H.; Boger, G.; Enhsen, A.; Falk, E.; Friedirch, M.; Glombik, H.; Hoffmann, A.; Pittius, C.; Urmann, M.; J. Biol. Chem., 1994, 269, 10621-10627. (b) Kramer, W.; Wess, G.; Schubert, G.; Bickel, M.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.-H.; Enhsen, E.; Glombik, H.; Mullner, S.; Neckermann, G.; Schulz, S.; Petzinger, E.; J. Biol. Chem., 1992, 267, 18598-18604. 30 Multicomponent Reactions; Zhu, J.; Bienaymé H., Eds.; Wiley-VCH: Weinheim, Germany, 2005. 31 Ugi, I.; Werner, B.; Dömling, A.; Molecules, 2003, 8, 53-66. 32 Touré, B. B.; Hall, D. G.; Chemical Rev. 2009, 109, 4439-4486. 33 Weber, L.; Illgen, K.; Almstetter, M.; Synlett., 1999, 3, 366-374. 34 Weber, L.; Curr. Med. Chem., 2002, 9(13), 1241-1253. 35 Edenborough, M. S.; Herbert, R. B.; Nat. Prod. Rep., 1988, 5, 229-245. 36 Ugi, I.; Isonitrile Chemistry, Academic Press, New York, 1971. 37 (a) Dömling, A.; Chem. Rev., 17-89, 106, 2006. (b) Dömling, A.; Ugi, I.; Angew. Chem., 2000, 39, 3168-3210. 38 Campo, J.; García, M.; Marcaccini, S.; Rojo, M. J.; Torroba, T.; Org. Biomol. Chem., 2006, 4, 757-765. 39 Ugi, I.; Pure Appl. Chem., 2001, 73(1), 187-191. 40 Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C.; Angew. Chem., 1959, 71, 386. 41 Dömling, A.; Curr. Op. Chem. Biol., 2000, 4, 318-323. 42 Rivera, D. G.; Tesis de doctorado, Universidad Martin-Luther, Alemania, 2007. 43 (a) Rivera, D. G.; Pando, O.; Coll, F.; Tetrahedron, 2006, 62, 8327-8334. (b) Wessjohann, L. A.; Rivera, D. G.; Coll, F.; J. Org. Chem., 2006, 71, 7521-7526. (c) Wessjohann, L. A.; Rivera, D. G.; J. Am. Chem. Soc., 2006, 128, 7122-7123. 44 Han, S. Y.; Kim, Y. A.; Tetraherdon, 2004, 60, 2447-2467. 45 Barry, J. F.; Davis, A. P.; Pérez-Payán, M. N.; Elsegood, M. R. J.; Jackson, R. F. W.; Gennari, C.; Piarulli, U.; Gude, M.; Tetrahedron Lett., 1999, 40, 2849. 46 Rivera, D. G.; León, F.; Coll, F.; Davison, G. P.; Steroids, 2006, 71, 1-11. 59 Bibliografía 47 (a) Scriven, E. F. V.; Turnbull, K.; Chem. Rev., 1988, 88, 351-368. (b) Gololobov, Y. G.; Zhmurova, I. N.; Kasukhin, L. F.; Tetrahedron, 1981, 37, 437-472. (c) Gololobov, Y. G.; Kasukhin, L. F.; Tetrahedron, 1992, 48, 1353-1406. 48 Brosa, C.; Amorós, M.; Molist, M.; Hernández, X.; Tetrahedron, 2004, 60, 8529–8534. 49 Nyffeler, P. T.; Liang, C. H.; Koeller, K. M.; Wong, C. H.; J. Am. Chem. Soc., 2002, 124, 10773- 10778. 60