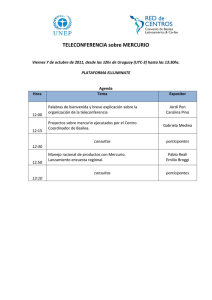
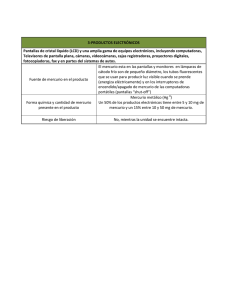
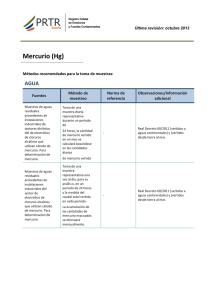
Determinación de mercurio por generación de vapor frío y
Anuncio

UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE QUÍMICA DETERMINACIÓN DE MERCURIO POR GENERACIÓN DE VAPOR FRÍO Y DETECCIÓN VÍA ABSORCIÓN Y FLUORESCENCIA ATÓMICA Por: Br. Grecia Carolina García Niño TRABAJO DE GRADO Presentado ante la Ilustre Universidad Simón Bolívar como requisito parcial para optar al título de Licenciada en Química Sartenejas, Octubre de 2010 UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE QUÍMICA DETERMINACIÓN DE MERCURIO POR GENERACIÓN DE VAPOR FRÍO Y DETECCIÓN VÍA ABSORCIÓN Y FLUORESCENCIA ATÓMICA Por: Br. Grecia Carolina García Niño Realizado con la asesoría de: Dr. José I. Alvarado TRABAJO DE GRADO Presentado ante la Ilustre Universidad Simón Bolívar como requisito parcial para optar al título de Licenciada en Química Sartenejas, Octubre de 2010 DETERMINACIÓN DE MERCURIO POR GENERACIÓN DE VAPOR FRÍO Y DETECCIÓN VÍA ABSORCIÓN Y FLUORESCENCIA ATÓMICA RESUMEN Se diseño y evaluó una celda de reacción hecha en vidrio para la generación de vapor frío de mercurio para ser acoplada a un espectrómetro de fluorescencia atómica y a un espectrómetro de absorción atómica. La evaluación se centró en la optimización de las condiciones instrumentales de análisis para la obtención de las mejores señales en lo que concierne a exactitud, precisión y sensibilidad. Se evaluó el tiempo de medición, la cantidad de reactivos necesarios para la generación del vapor frío, los flujos del gas de arrastre así como el tipo de gas más apropiado para llevar el vapor de Hg0 hacia la zona de detección. Con estos ensayos se obtuvieron las distintas figuras de mérito. Para el estudio se seleccionaron muestras líquidas de agua y muestras sólidas de sedimentos marinos. El contenido de mercurio en muestras líquidas se determinó utilizando espectrometría de fluorescencia y absorción atómica con vapor frío; mientras que el contenido de mercurio en muestras sólidas de sedimentos marinos se determinó mediante espectrometría de absorción atómica con vapor frío. Los resultados indicaron que el nuevo sistema de generación de vapor frío permite obtener límites de detección a nivel de traza y se puede aplicar fácilmente a muestras tanto sólidas como líquidas. Se recomienda la utilización de este sistema de generación de vapor frío para la determinación de mercurio, en forma rutinaria, en los tipos de muestra aquí estudiados, especialmente si se quiere determinar el contenido de mercurio que es extraíble mediante lixiviación de muestras sólidas. iii DEDICATORIA Dedicada a un hombre que consideraba que la educación es parte fundamental para todo ser humano, quien creyó que para las mujeres es indispensable tener una profesión universitaria. Dedicada al nono Emiliano Niño. Grecia Carolina García Niño iv AGRADECIMIENTOS A Dios, a la Virgen y a José Gregorio Hernández, mis guías espirituales. A mi madre, Esperanza Niño, una digna representante del género femenino, luchadora, aguerrida, inteligente, fuerte y sobretodo una excelente mamá que me dio los conocimientos básicos de la vida para salir a desafiar al mundo. A mi nona, otra madre ejemplar, que con su cariño, paciencia y amor supo alimentarme el alma y con sus manos de mujer trabajadora siempre me ofrece su deliciosa comida. Por supuesto, a mis tías, Aude, Judith y Olga. Mujeres tenían que ser. Gracias por enseñarme a vivir, gracias porque ustedes con amor me dieron grandes lecciones. Al parecen tengo muchas mamás! A una niña muy especial, María Valentina, una prima, casi una hermanita, que con su gracia siempre me hace reír y con su amor siempre me llena el corazón. Espero servir como ejemplo para que en los venideros años puedas obtener un título universitario. Sé que lo lograrás. A mi novio Jorge Luis, ¡te amo muchísimo! has sido un apoyo fundamental en mi carrera. Siempre estás en el sitio y en el momento indicado, gracias por tu ayuda. Pronto seremos colegas. Gracias por todo el cariño y el amor que me brindas todos los días. Gracias también por la paciencia en aquellos días de desespero. Tu conviertes los malos momentos en los mejores, la tristeza en alegría. Definitivamente mi vida no sería lo mismo sin ti. Un especial agradecimiento a mi tutor, el profesor José Alvarado, quien me dio los conocimientos básicos de la química y quien confió en mí para la realización de este proyecto. Gracias por apoyarme y enseñarme el maravilloso mundo de la química analítica. Al profesor José Ramón Domínguez, un cuasi tutor, gracias por animarme a lo largo de este tiempo y por ayudarme a formarme como Químico. Profe, usted me enseño millones de cosas y siempre se lo agradeceré. Gracias por apoyarme, ayudarme y darme esas palabras de aliento siempre que lo necesite. Un especial agradecimiento a los Profesores Fernando Morales y Julio Herrera por darme la orientación adecuada para realizar este trabajo. Al gran amigo, Roberto Villagrán, excelente con el arte de moldear el vidrio. Gracias por el gran apoyo que me diste, siempre con tanta amabilidad. Eficiente, responsable, trabajador, honesto. Es incalculable el agradecimiento que te tengo. A unos grandes amigos, María Luisa Altuve y Estefan Baran, quienes me llevaron hace años a conocer esta maravillosa casa de estudios donde pronto saldré egresada. v A mis tíos-padrinos, Emiliano y Nelly. A mis tíos, Marcelo, Edgar, Omar, Gerardo, Carlos, Isolde, Cuña, América, Diana. A mis primos, Johan, Gianco, Carlos, Rafael, Beatriz, Omar Leonel, Omar Rances, Yusmelis, Yusleidi, Junior, Nelson, Jairo, María Angélica, Luis, José Manuel, Marianita, Carolina y al más chiquitico, Carlucho, ¡la nueva adquisición de la casa! Fue por culpa de este proyecto que no pude estar en tu nacimiento. A mi nuevo hermano Jahzeel y a mi sobrina Sarita, gracias por llegar a mi vida, los recibo con los brazos abiertos, al igual que a mi cuñada Mildred. A mi mejor amiga, Jormary, gracias por acompañarme en los sufrimientos y alegrías de estos cinco años, amiga te quiero muchísimo, eres y siempre serás la mejor amiga del mundo, cómo olvidar nuestros sufrimientos con física, nuestras rumbas en semana cuatro, ocho y doce, nuestros desayunos en casa del estudiante, memorables esos momentos. A un excelente Ingeniero Químico, mi gran amigo Francisco Blanco, gracias porque siempre estuviste allí para ayudarme, jamás olvidaré tus explicaciones de Mate cinco y el famoso philly chesse de Miga´s. Gracias también a la Sasha Barhh, cómo olvidarla, compartimos momentos maravillosos. A mis futuros colegas Xin Yan, Norbis, Ivan, Naymar, Mariale, Osmin, Ren, Inexis, Leo, Rosa, Marcel, Pedro y Luis Ospino, se que seremos excelentes profesionales, nos queda un camino enorme por recorrer, éxito para todos. Jamás olvidaré aquellas clases interminables, aquellos sufrimientos por las notas, y por supuesto las alegrías al finalizar los laboratorios. Fue excelente compartir mi carrera con ustedes. Los quiero muchísimo. A una gran amiga de la casa, Judith León que siempre estuvo presente en los momentos importantes con sus oraciones y sus valiosas palabras. A Nairobi, mi compañera de laboratorio, gracias por ayudarme y acompañarme en el lab. También gracias a mi otra compañera María por los momentos vividos en el lab. No puedo dejar de mencionar a mi gran amiga Maritza Sotillo, quisiera agradecerte por todo el apoyo que me diste. Por los consejos, por las palabras, por todo. Gracias, gracias. Gracias también a mis amigos del colegio, aquellos con los que de niña soñé estar en este momento, Juan Carlos, Milagros, Aleyna, Cindy y Andreina. Gracias también a mis amigos Silvana, Sofía, Ismael, y Yari, me recibieron con muchísimo cariño en los primeros años de la carrera, jamás los olvidaré. No podía olvidarme de mis amigos, Victor, Mineyomar, Claudia y Jorge Zegarra. Finalmente quisiera agradecer a toda la comunidad de la Universidad Simón Bolívar, personal administrativo, obrero y profesores, se que sin ellos nada de esto hubiese sido posible, gracias por su colaboración. Así mismo quisiera agradecer a todos los venezolanos que han puesto su confianza en mí y me dieron la oportunidad de estudiar gratuitamente en esta excelente casa de estudios, daré todo mi esfuerzo y mi trabajo para contribuir en hacer de Venezuela un gran país. vi INDICE GENERAL RESUMEN…………………………………………………………………………....………........iii DEDICATORIA...……………………………………………………………….....…………........iv AGRADECIMIENTOS…………………………………………………………………………....v INDICE DE TABLAS……………………………………………………………….……….........ix INDICE DE FIGURAS……………………………………………………………….………........x INDICE DE GRÁFICOS…………………………………………………………….……….......xii TABLA DE ABREVIATURAS……………………………………………………………...........xiv INTRODUCCIÓN .........................................................................................................................................1 CAPÍTULO 1 ...................................................................................................................................................3 MARCO TEÓRICO .......................................................................................................................................3 1.1. El mercurio como agente agresor del medio ambiente .......................................................................3 1.2. Toxicidad del mercurio y valores aceptados ......................................................................................... 5 1.3. Metodología de análisis: almacenamiento, digestión y determinación cuantitativa de mercurio por técnicas de espectrometría atómica ........................................................................................................6 1.3.1. Almacenamiento ....................................................................................................................................6 1.3.2. Métodos de digestión ............................................................................................................................ 8 1.3.3. Determinación cuantitativa de mercurio por técnicas de espectrometría atómica con vapor frío. .....................................................................................................................................................................9 1.4. Conceptos teóricos fundamentales para optimización y validación de los métodos de análisis 16 1.5. Análisis de muestras sólidas.................................................................................................................. 24 1.5.1. Digestión .............................................................................................................................................. 25 1.5.2. Combustión Directa ........................................................................................................................... 26 1.5.3. Lixiviación ............................................................................................................................................ 27 CAPÍTULO 2 ................................................................................................................................................ 28 SECCIÓN EXPERIMENTAL .................................................................................................................. 28 2.1. Contaminación de Hg durante los ensayos. ....................................................................................... 29 2.2. Tratamiento de los desechos de mercurio. ......................................................................................... 29 2.3. Análisis previos....................................................................................................................................... 29 2.3.1. Material de laboratorio ....................................................................................................................... 29 2.3.2. Preparación de las soluciones a utilizar en los análisis .................................................................. 29 2.4. Metodología de análisis ......................................................................................................................... 31 2.4.1. Diseño de la celda de reacción utilizada para la generación de vapor frío ................................ 31 vii 2.4.1.1. Primer diseño .................................................................................................................................. 31 2.4.1.2. Segundo diseño ............................................................................................................................... 32 2.4.1.3. Tercer diseño ................................................................................................................................... 34 2.4.2. Instalación de la celda de reacción utilizada para la generación de vapor frío al detector de fluorescencia atómica .................................................................................................................................... 35 2.4.3. Condiciones experimentales para las mediciones por fluorescencia atómica ............................ 37 2.4.4. Instalación de la celda de reacción utilizada para la generación de vapor frío al detector de absorción atómica ......................................................................................................................................... 39 2.4.5. Condiciones experimentales para las mediciones por absorción atómica .................................. 41 CAPÍTULO 3 ................................................................................................................................................ 43 RESULTADOS Y DISCUSIONES .......................................................................................................... 43 3.1. Evaluación de la celda de reacción para determinación de mercurio por generación de vapor frío y detección mediante fluorescencia atómica en muestras líquidas. ................................................ 43 3.1.1. Optimización de las condiciones de medida................................................................................... 43 3.1.2. Determinación de las figuras de mérito ........................................................................................... 47 3.2. Evaluación de la celda de reacción NCR para determinación de mercurio por generación de vapor frío y detección mediante absorción atómica en muestras líquidas y sólidas. ........................... 53 3.2.1. Determinación de las figuras de mérito para muestras líquidas ................................................... 57 3.2.2. Determinación de las figuras de mérito para muestras sólidas. ................................................... 62 3.2.3. Comparación de la sensibilidad del método en muestras líquidas y sólidas. .............................. 65 CONCLUSIONES ....................................................................................................................................... 68 RECOMEDACIONES ............................................................................................................................... 69 BIBLIOGRAFÍA ...................................................................................... Error! Bookmark not defined. viii INDICE DE TABLAS Tabla 2.1 Tabla 2.2 Tabla 2.3 Tabla 2.4 Tabla 2.5 Tabla 3.1 Tabla 3.2 Tabla 3.3 Tabla 3.4 Tabla 3.5 Tabla 3.6 Tabla 3.7 Tabla 3.8. Tabla 3.9 Reactivos empleados durante la realización del Trabajo de Grado Equipos empleados durante la realización del Trabajo de Grado Tiempos usados por el generador de vapor para determinar mercurio por fluorescencia atómica con el Espectrómetro Merlin P.S. Analytical LTD. PSA 10.023 Flujos de gas utilizados para la determinación de mercurio por fluorescencia atómica con el Espectrómetro Merlin P.S. Analytical LTD. PSA 10.023 Condiciones experimentales mediante las cuales se hicieron las mediciones del vapor de mercurio por absorción atómica con el Espectrómetro Perkin Elmer 460. Condiciones instrumentales para los flujos gas utilizados en las mediciones. Resultados obtenidos para el cálculo de la precisión del nuevo método analítico para la generación de vapor frío con la celda NCR y su detección vía fluorescencia atómica. ANOVA del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. Condiciones instrumentales de análisis para determinación de mercurio mediante vapor frío usando la celda de reacción. Resultados obtenidos para el cálculo de la precisión del nuevo método para la generación de vapor frío en muestras líquidas con la celda NCR y su detección vía absorción atómica. ANOVA del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Resultados obtenidos para el cálculo de la precisión del nuevo método analítico para la generación de vapor frío en muestras sólidas con la celda NCR y su detección vía absorción atómica. ANOVA del intervalo lineal para la determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica Resumen de los logros obtenidos en las determinaciones de mercurio en muestra sólida y líquida utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia y absorción atómica. ix 28 28 37 37 41 45 47 51 53 57 61 63 65 67 INDICE DE FIGURAS Figura 1.1 Ciclo del mercurio en el medioambiente 4 Figura 1.2 Esquema de transiciones electrónicas en absorción atómica 10 Figura 1.3 Esquema de la disposición de los componentes de un sistema de medición por espectrometría de absorción atómica con vapor frío 10 Figura 1.4 Sistema de generación de vapor frío que puede ser acoplado al sistema de medición por absorción atómica 12 Figura 1.5 Esquema de transiciones electrónicas en fluorescencia atómica 13 Figura 1.6 Esquema de la disposición de los componentes de un sistema de medición por espectrometría de fluorescencia atómica con vapor frío 14 Figura 1.7 Sistema de generación de vapor frío que puede ser acoplado al sistema de medición por fluorescencia atómica 15 Figura 1.8 Distribución de valores alrededor de cero y nivel crítico. 18 Figura 1.9 Nivel crítico y límite de detección para = (pequeños) 19 Figura 2.1 Primer montaje para la generación de vapor frío de mercurio. 32 Figura 2.2 Segundo montaje para la generación de vapor frío de mercurio. 33 Figura 2.3 Celda de reacción utilizada para la generación de vapor frío (NCR). 34 Figura 2.4 Esquema de un sistema de medición por fluorescencia atómica 36 Figura 2.5 Esquema del acoplamiento de la celda de reacción utilizada para la generación de vapor frío al detector de fluorescencia atómica. 36 Figura 2.6 Esquema de la membrana higroscópica de Nafión para secar la humedad del vapor de mercurio antes de llegar al detector 38 Figura 2.7 Chimenea donde llega el vapor de mercurio seco cubierto por el gas de escudo. 38 Figura 2.8 Esquema de un espectrómetro de absorción atómica. 39 Figura 2.9 Esquema del reservorio de átomos de Hg0. 40 Figura 2.10 Adaptación de la base de un quemador de premezcla para colocar y alinear el tubo de plástico en la zona de detección del espectrómetro. 40 Figura 2.11 Esquema del acoplamiento de la celda de reacción utilizada para la 41 x generación de vapor frío al espectrómetro de absorción atómica. Figura 2.12 Esquema del acoplamiento de la bomba de vacio con válvula antirretorno al del reservorio de átomos de Hg0. 42 Figura 3.1 Señal típica obtenida con el generador de vapor frío convencional. 44 Figura 3.2 Señal típica obtenida con la celda de reacción para la generación de vapor frío. 44 xi INDICE DE GRÁFICOS Gráfico 3.1 Optimización de la cantidad de agente reductor utilizado para la reducción de una solución de mercurio de 1,00 µg/L. 46 Gráfico 3.2 Intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. 50 Gráfico 3.3 Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. 50 Gráfico 3.4 Representación gráfica para el estudio de la exactitud del nuevo método para determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. 52 Gráfico 3.5 Representación gráfica para la escogencia del flujo de gas de arrastre utilizado en el nuevo método para determinación de mercurio utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 54 Gráfico 3.6 Representación gráfica para la escogencia del gas de arrastre para determinación de Hg utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 55 Gráfico 3.7 Representación gráfica para la escogencia del agente reductor utilizado en el nuevo método para determinación de mercurio utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 56 Gráfico 3.8 Intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 60 Gráfico 3.9 Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 60 Representación gráfica para el estudio de la exactitud del nuevo Gráfico 3.10 método para determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección xii 62 vía absorción atómica Intervalo lineal para la determinación de mercurio en muestras sólidas Gráfico 3.11 utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 64 Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras sólidas utilizando la celda Gráfico 3.12 NCR para la generación del vapor frío y detección vía absorción atómica. 64 Representación gráfica para el estudio de la exactitud del nuevo Gráfico 3.13 método para determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Representación gráfica que permite comparar la sensibilidad del nuevo método para determinación de mercurio en la misma sólida y disuelta Gráfico 3.14 muestra utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica xiii 65 66 TABLA DE ABREVIATURAS ANOVA b CV CV-AFS CV-AAS NCR NCR-EFA NCR-EAA G.V. Merlin PSA 10.023 H0 Hl ISO LC LD LC m LDM LDC MHS10 UV WHO x y %R R2 r S s SST SSP SSR Nivel de significación Análisis de Varianza “ Analysis of Variance” Intercepto de la recta de calibrado lineal Coeficiente de Variación Espectrometría de Fluorescencia Atómica con Vapor Frío “Cold Vapor Atomic Fluorescense Spectrophotometry” Espectroscopia de Absorción Atómica con Vapor Frío “Cold Vapor Atomic Absorption Spectrophotometry” Nueva Celda de Reacción para la Generación de Vapor Frío Nueva Celda de Reacción acoplada a un espectrómetro de fluorescencia atómica Nueva Celda de Reacción acoplada a un espectrómetro de absorción atómica Generador de Vapor Merlin PSA 10.023 Hipótesis Nula Hipótesis Alternativa Organización Internacional para la Estandarización “International Organization for Standardization” Valor crítico Límite de detección “Limit of Detection” Límite de cuantificación “Limit of Quantification” Pendiente de la recta de calibrado lineal Límite de detección del Método. “Method Detection Limit” Límite de cuantificación del Método. “Method Quantification Limit” Generador de Hidruros Perkin Elmer modelo MHS10 Ultravioleta Organización Mundial de la Salud “World Health Organization” Variable independiente de la recta de calibrado lineal Variable dependiente de la recta de calibrado lineal Porcentaje de Recuperación Coeficiente de determinación Coeficiente de correlación Sesgo Desviación estándar Suma de cuadrados total Suma de cuadrados explicada por el modelo lineal Suma de cuadrados de los residuales xiv INTRODUCCIÓN Desde hace varios años se conoce que los metales pesados, como mercurio, a niveles de trazas son elementos tóxicos para el ser humano y pueden causar desordenes en la salud de los mismos. Desde que se descubrió que el consumo de mercurio por seres humanos es de alta peligrosidad, se ha llevado un seguimiento de las distintas fuentes de procedencia. Distintos estudios han demostrado que este metal puede ser determinado en seres humanos con el análisis de cabello, uñas u orina de la persona contaminada. Para la determinación de mercurio a niveles de trazas son requeridas técnicas de alta sensibilidad que permitan determinar y cuantificar su presencia. Entre las técnicas más utilizadas se encuentran la Espectrometría de absorción atómica con vapor frio y la Espectrometría de fluorescencia atómica con vapor frio. Estas han demostrado tener alta sensibilidad en determinaciones exactas de mercurio. Sin embargo, la selectividad y sensibilidad de estas técnicas dependen de los componentes de la matriz de la muestra. Es por ello que se acostumbra realizar digestiones de las muestras para tener un mejor acceso al analito en una muestra líquida de la cual el mismo se pueda separar y evitar o minimizar así el efecto de la matriz. Este proceso de disolución trae consigo un aumento en el riesgo de contaminación de la muestra, una mayor laboriosidad en el análisis y por ende un mayor tiempo de trabajo y un mayor costo. Debido a los efectos deletéreos que puede causar una matriz compleja en la exactitud y precisión de las determinaciones de un dado analito, en los últimos años, se han introducido al mercado nuevas técnicas mediante las cuales el elemento de interés puede ser separado de la matriz de la muestra mediante formación de un compuesto volátil del mismo. Después de la digestión de la muestra se llevan a cabo las reacciones necesarias para formar el compuesto volátil y esta especie conteniendo al analito se lleva mediante un flujo de argón hacia la zona de detección. Estas alternativas analíticas han encontrado especial aplicación en la detección de mercurio, con alta exactitud y alta sensibilidad, lo cual ha dado origen a la opción conocida como “generación de vapor frío”. Sin embargo, esta opción ha sido aplicada casi exclusivamente a muestras líquidas o a muestras sólidas que han sido previamente disueltas. En este último caso, aunque se ha eliminado sustancialmente el efecto de la matriz sobre el resultado de los análisis, la laboriosidad en la preparación de la muestra, la posibilidad de contaminación con los reactivos utilizados para la disolución, el tiempo de análisis y su mayor costo no han sido mejorados. Se desprende de lo anterior que la situación ideal sería el análisis directo de la muestra sólida sin que la matriz de la misma tuviese ningún efecto apreciable en los resultados. Este objetivo ha sido logrado casi en un cien por ciento en algunos equipos modernos especialmente diseñados con este propósito en mente. Ejemplo de 1 estos equipos es el Analizador Directo de Mercurio DMA-80 (Milestone Inc., Monroe, CT). Pero, el costo de los equipos adecuados para este tipo de análisis es elevado y por lo tanto el análisis de muestras sólidas se hace poco accesible en laboratorios con presupuestos limitados. En estos casos queda la posibilidad de recurrir a alternativas más económicas, aunque no tan sofisticadas, desarrolladas en los propios laboratorios interesados en mejorar la calidad de sus mediciones. Dado el interés en la determinación de mercurio a nivel de traza que se ha tenido en nuestro laboratorio, se planteó el desarrollo y evaluación de una vía sencilla y económica que fuese útil para ese tipo de determinación. Con los conocimientos básicos de la generación de vapor frío y el funcionamiento de los equipos de Absorción y fluorescencia atómica se quiso proponer un nuevo sistema de generación de vapor frío de mercurio que obviara el proceso de digestión de la muestra y todos los inconvenientes ya mencionados. Es decir, una celda de reacción que permitiera formar el vapor frío de mercurio cuantitativamente a partir de la muestra sólida. Dicha celda se acoplaría a un espectrofotómetro de absorción o a uno de fluorescencia para la determinación del mercurio. Para ello en el presente trabajo se plantearon los objetivos detallados a continuación. Objetivo General Determinación de mercurio en muestras sólidas mediante generación de vapor frío y detección vía espectrometría de absorción y fluorescencia atómicas. Objetivos Específicos: Diseño, evaluación y acople de una celda de reacción por cargas para generar vapor frío de mercurio a partir de muestras sólidas a un sistema detector de mercurio mediante absorción atómica y mediante fluorescencia atómica Optimización de las distintas condiciones de medida para las cuales se encuentra la mejor sensibilidad del detector usando la celda y soluciones acuosas del analito. Determinación de la figuras de mérito usando soluciones acuosas de mercurio. Adaptar las condiciones de análisis óptimas para las soluciones acuosas a las determinaciones de mercurio en muestras sólidas. Para esto se hará el chequeo de la exactitud de los resultados y determinación de figuras de mérito comparando con un material certificado y analizando la muestra sólida. Aplicación de estas condiciones de análisis en la determinación de Hg en muestras sólidas reales. 2 CAPÍTULO 1 MARCO TEÓRICO 1.1. El mercurio como agente agresor del medio ambiente El mercurio es un metal de transición que se encuentra presente en estado líquido a temperatura ambiente y es de color gris brillante. En la corteza terrestre se puede encontrar como sulfuro de mercurio (HgS), también llamado cinabrio. Además, se pueden encontrar más de 20 minerales que contienen dicho metal. En Venezuela existe un sólo yacimiento de sulfuro de mercurio y se encuentra ubicado en San Jacinto, Estado Lara. (López, V. M. y J. H. Brineman Jr., 1943.) A lo largo de los años el mercurio ha sido ampliamente utilizado en el ámbito científico e industrial ya que este elemento posee propiedades físicas y químicas muy atractivas. El uso más común es en aparatos de medida como termómetros. Sin embargo, se usa también en electrodos de calomelanos, en tubos de rayos X y en barómetros. También se puede utilizar como catalizador y como cátodo en la electrólisis de cloruros alcalinos. En la medicina, específicamente en la odontología, es usado para realizar empastes dentales aunque en la actualidad no se emplea por su gran toxicidad. (Halbach, 2008) El mercurio constituye un contaminante sumamente agresivo del medio ambiente. Se puede encontrar en sus estados de oxidación, Hg (0), Hg (I), Hg (II), y en todos los estados de agregación, sólido, líquido o gaseoso. Es más común la presencia de estas especies en medios acuáticos que en suelos exceptuando los suelos aledaños a zonas acuáticas contaminadas. Las emisiones estimadas de Hg a la atmósfera están entre 2700 y 6000 toneladas por año, de las cuales de 2000 a 4500 toneladas se deben a las actividades del hombre. (Nielsen J.B., 2000) En las zonas donde se han encontrado altas concentraciones de mercurio se debe principalmente a derrames hechos por industrias cercanas. El peor desastre de contaminación por mercurio se reportó en la ciudad japonesa Minamata, la compañía Chisso. (Shigeo Ekino, 2007). En Venezuela, la explotación minera de oro ha contaminado la cuenca del rio Caroní ya que el mercurio tiene la capacidad de formar amalgamas con este metal precioso. Además, desde el año 1974 se conoce que 3 la industria petroquímica ha contaminado de mercurio un área del noroccidente del país conocida como Golfo Triste donde se encuentra una de las sedes de la Petroquímica de Venezuela S.A. (PEQUIVEN). Investigadores han reportado que sedimentos recogidos del Caño Alpargatón en el Rio Yaracuy están contaminados con altos niveles de mercurio (mayores a 3 ppm). (Pardi, 1974). Esta corriente de agua sirvió como colector de los desechos de la planta de cloro-soda que se ubicaba en Morón y perteneciente a la industria mencionada anteriormente. Esta planta producía para los años sesenta hidróxido de sodio y ácido clorhídrico, para ello se utilizaba un proceso electrolítico con electrodos de mercurio metálico. Las descargas de mercurio a este río se produjeron durante el mantenimiento y limpieza de las celdas electrolíticas. Debido a los graves problemas ambientales ocasionados, actualmente no se usa este tipo de proceso industrial pero todavía persiste la contaminación ambiental generada. Debido a la naturaleza volátil del mercurio, este puede ser transportado grandes distancias por las lluvias y por el aire al mover los sedimentos en donde se ha depositado. La capacidad de retener mercurio por parte de los sedimentos puede retrasar la eliminación de la contaminación por muchos años. Se sabe, asimismo, que el mercurio inorgánico puede ser metilado por parte de los microorganismos que están en los sedimentos, y que las formas orgánicas del mercurio son aún más tóxicas que las inorgánicas. La Fig. 1.1 es una representación esquemática de estas transformaciones. Figura 1.1. Ciclo del mercurio en el medioambiente. (Winfrey, 1990) La forma orgánica más común de encontrar este metal pesado es como metilmercurio (MeHg), el cual tiene una alta solubilidad en lípidos, atraviesa fácilmente las membranas celulares y entra rápidamente en las cadenas alimentarias acuáticas. El mercurio, en ninguna de sus formas, tiene función benéfica alguna en los humanos, y cualquier exposición de largo o aún de corto plazo puede conducir a una alteración progresiva del funcionamiento normal de los órganos donde se acumula (Gochfeld, 2003) 4 1.2. Toxicidad del mercurio y valores aceptados Los mecanismos biológicos para excretar el mercurio son muy ineficientes lo que lo hace extremadamente tóxico tanto para los animales como para las plantas, aún en bajas concentraciones. Es así que el aumento de los niveles de mercurio puede provocar daños en cualquier organismo expuesto. Los órganos que suelen acumular mercurio son los riñones, el hígado y el sistema nervioso central y la exposición excesiva a mercurio orgánico o inorgánico puede dañar estos órganos de manera permanente (Shigeo Ekino, 2007) Los seres humanos están expuestos a contaminación por mercurio de varias formas. Por consumo de peces y moluscos provenientes de aguas contaminadas, por inhalación de vapores de mercurio provenientes de la incineración de las amalgamas en la minería, por inhalación de las emisiones de industrias que utilicen catalizadores de mercurio u otros compuestos que contengan dicho metal, por contacto directo con el mercurio proveniente de los termómetros utilizados en casa y en algunos países existe un grave peligro pues se siguen utilizando amalgamas de mercurio para los empastes dentales. Exposiciones continuas pueden provocar trastornos en niños y adultos. Se conoce que una madre contaminada por mercurio le transmite al feto gran cantidad del mercurio presente en su cuerpo ya que los tejidos fetales nuevos tienen mayor afinidad para unirse al metilmercurio que los tejidos viejos de la madre. Por lo tanto, al entrar en contacto el feto con este compuesto, se ve afectada la formación de su sistema nervioso central lo que trae graves consecuencias cerebrales. (Shigeo Ekino, 2007), (Harada, 1968) En adultos expuestos continuamente puede provocar una disminución en la capacidad visual y auditiva así como deficiencias en los sentidos del olfato, gusto y tacto. También se pueden presentar atrofias musculares, temblores involuntarios, alteraciones del aparato digestivo, pérdida del apetito y de peso, daños en los riñones, malformaciones y hasta la muerte. (Li, 2008) Existen diversas opiniones que establecen los niveles de mercurio permitidos. La Organización Mundial de la Salud (WHO/IPCS), concluyó, a través de distintos estudios, que valores menores a 50 µg de mercurio/g de cabello no representan un riesgo significativo a la salud de la población adulta (hombres o mujeres no embarazadas). A través de estudios clínicos hechos en Irak, la misma organización concluye que una mujer embarazada es más sensible a los efectos del metilmercurio porque es posible que se cause un daño neurológico al feto. En 1990 se hicieron estudios estadísticos donde se concluye que 30% de los infantes con signos neurológicos anormales, son hijos de madres con valores mayores a 70 µg de mercurio/gramo de cabello. A partir de un análisis estadístico 5 adicional, la WHO/IPCS estima que los infantes con madres que poseen valores entre 10 y 20 µg de mercurio/ gramo de cabello, poseen un 5% de riesgo de sufrir desordenes neurológicos. ( United States Environmental Protection Agency, 1997) Tal como se expuso anteriormente, los suelos adyacentes a sistemas acuáticos contaminados con mercurio se ven afectados de la misma manera y la acumulación progresiva de las distintas formas contaminantes del mercurio producen niveles inadecuados para dichos suelos, por lo tanto cualquier especie que habite en estos espacios se verá afectada, ya sean las plantas que crecen en los alrededores, o los animales y los humanos que habiten la zona. Los niveles de mercurio asociados a suelos y sedimentos no contaminados están entre 0,03 mg/kg a 0,5 mg/kg (Bryan, 1992). Este intervalo de concentraciones implica que la determinación de Hg debe realizarse con técnicas que puedan detectar cantidades realmente bajas del metal. Para lograr la determinación de los niveles mencionados han pasado gran cantidad de años de desarrollo científico y técnico desde el punto de vista metodológico si consideramos que las mediciones deben realizarse rápidamente y con alta calidad analítica, es decir con exactitud y precisión. (Skoog D, 2005) 1.3. Metodología de análisis: almacenamiento, digestión y determinación cuantitativa de mercurio por técnicas de espectrometría atómica Los resultados de un análisis cuantitativo se obtienen a partir de dos medidas. La primera es la masa (o volumen) de la muestra que se analiza. La segunda es la medida de alguna cantidad proporcional a la del analito en la muestra, como la intensidad luminosa o carga eléctrica. Existen métodos gravimétricos, volumétricos, electroanalíticos así como espectroscópicos que permiten cuantificar especies atómicas o moleculares, según sea el caso. Por lo general este tipo de análisis se realiza a alícuotas de las muestras, es decir, se seleccionan cantidades minúsculas que representan el total de muestra analizada. Es por ello que se debe considerar la forma en la cual se almacena la muestra así como el método de digestión pues lo más común es que las técnicas requieran tener a la muestra en disolución. 1.3.1. Almacenamiento En las determinaciones analíticas, el tiempo es un factor muy importante para la obtención de medidas de calidad. Generalmente, las muestras no se pueden analizar inmediatamente después de ser tomadas. Tanto las muestras reales como los patrones preparados y/o certificados se encuentran 6 condicionados por el tiempo desde el momento en que se recogen o se preparan hasta el momento en que se hace la medición. En patrones certificados es común que el fabricante sugiera condiciones para conservarlas, por ejemplo, la temperatura y el material del cual debe ser el recipiente donde se guardará. Además el fabricante garantiza el valor del analito por un tiempo específico. El mercurio metálico Hg (0), tal como se dijo anteriormente, tiene una presión de vapor de 0,002 mm Hg/m3 a temperatura ambiente, lo cual indica que debe almacenarse a bajas temperaturas y en recipientes cerrados para evitar la evaporación. Otras formas de mercurio, orgánico e inorgánico, han sido estudiadas por distintos investigadores, explicando las propiedades que deben tener los envases en los cuales se almacenen dichos compuestos. Por ejemplo, el cloruro de metilmercurio fue encontrado como el más volátil de un grupo de compuestos de mercurio. (Juanes J., 1996) En soluciones que contienen distintas especies de mercurio, al estar expuestas al aire, se pierde mercurio por la reducción de Hg (II) a Hg (I) y Hg (0), siendo esta última especie sumamente volátil. Por lo tanto, se deben agregar compuestos que mantengan el mercurio en su más alto estado de oxidación. Para ello se puede agregar un agente oxidante con el fin de reducir la pérdida. Por otra parte, la acidez es un factor importante pues investigaciones demostraron que al incrementar el pH de la solución de Hg aumenta la perdida de mercurio. (Juanes J., 1996). Investigaciones señalan que se observa la perdida de mercurio al marcar con 203 Hg. Se observa una disminución de la radioactividad de este isótopo a medida que se alcaliniza la solución con hidróxido de sodio o cloruro estannoso. El efecto contrario se obtiene al usar agentes oxidantes como hipoclorito de sodio o permanganato de potasio. (Morita, Tanaka, & Shimomura, 1995) Por lo tanto, agentes oxidantes como permanganato de potasio o una mezcla bromuro de potasio/ bromato de potasio, entre otros deben ser utilizados para determinaciones de mercurio, También, debe mantenerse un bajo pH acidificando con ácidos orgánicos o inorgánicos, por ejemplo ácido clorhídrico. Estas condiciones permitirán mantener al mercurio en su forma bivalente. El material de los envases utilizados para almacenar las muestras que contienen mercurio es de gran importancia, pues es necesario que no sea permeable, que no sea adsorbente y que soporte las condiciones fuertemente acidas en las cuales se debe mantener la muestra. Los materiales más utilizados para la fabricación de envases son el vidrio de borosilicato, el policarbonato, el politetrafluoroetileno o teflón así como el polietileno. La pérdida de mercurio por adsorción en las paredes de los recipientes puede evitarse al acidificar la solución, y si además se coloca un agente oxidante se puede prevenir la adsorción del mercurio en las paredes. Los materiales más adecuados 7 son el vidrio de borosilicato, el policarbonato y el politetrafluoroetileno. (Chilov, 1975) (Bothner, 1978) Para la determinación de mercurio, el vidrio de borosilicato presenta la ventaja que se puede limpiar fácilmente, además es inerte excepto a la presencia de fuertes álcalis. Los plásticos mencionados presentan como ventajas que son materiales duraderos y ligeros además de ser muy económicos (excepto el teflón). 1.3.2. Métodos de digestión La mayor parte de las medidas analíticas se realizan con disoluciones acuosas del analito. Algunas muestras se disuelven fácilmente en agua o en soluciones acidificadas o alcalinizadas. Sin embargo, existen otras muestras que requieren de reactivos más eficientes y tratamientos más rigurosos. Esta etapa del análisis es muy delicada cuando se trabaja a nivel de traza pues se puede contaminar la muestra o en algunos casos se puede perder el analito. Errores como estos limitan la exactitud del análisis. En muestras con mercurio es delicado su tratamiento pues se debe considerar la volatilidad de las especies. Además la cantidad de mercurio determinada es tan minúscula que la muestra puede contaminarse con los reactivos utilizados para su digestión. Los métodos más utilizados para la digestión son: por calentamiento con soluciones acuosas ácidas, por calentamiento con microondas con ácidos, por ignición a altas temperaturas en aire u oxígeno y por fusión en medios de sales fundidas. Para la determinación de mercurio, si hace falta disolver la muestra, las técnicas de digestión ácida mediante calentamiento convencional y con microondas son las más utilizadas. (Landi & Fagioli, 1994) (Joaquim A. Nóbrega, 2002) Dado que es preciso evitar las pérdidas del analito durante estos procesos, lo cual no siempre es fácil de lograr y dado que son procesos largos, laboriosos, que utilizan considerable cantidad de ácidos y otros reactivos, las posibilidades de pérdidas y de contaminación son altas. En caso de ser posible evitar estos procesos de digestión, esta opción sería lo más conveniente. Sin embargo, las técnicas instrumentales de análisis, las más apropiadas para determinaciones a bajos niveles de analito con suficiente exactitud y precisión, con muy pocas excepciones, requieren muestras disueltas. Ese es el caso de la determinación de mercurio mediante generación de vapor frío y detección mediante absorción y fluorescencia atómica, que han sido las técnicas más usadas en la determinación de mercurio, siendo también las escogidas para las determinaciones que se describen en este trabajo. Para ceñirnos a lo expresado arriba como una alternativa, es decir evitar los procesos de digestión, el trabajo desarrollado tuvo como reto la generación del vapor frío de mercurio a partir de la muestra 8 sólida. Se explicarán a continuación la espectrometría de absorción y fluorescencia atómica teniendo como objetivo la determinación de mercurio 1.3.3. Determinación cuantitativa de mercurio por técnicas de espectrometría atómica con vapor frío. En los últimos años se han aplicado una gran cantidad de técnicas para la determinación de mercurio, incluyendo espectrometría de absorción atómica con atomización a la llama (Garrido I., 2001), espectrometría de absorción atómica con atomización electrotérmica (Hafez M. A. H., 2001) (Jorge Moreda-Piñeiro, 2001), espectrometría de masas con fuente de plasma de acoplamiento inductivo (Nixon D. E., 1999), espectrometría de emisión atómica con fuente de plasma de acoplamiento inductivo (Valde´s-Hevia M. C., 1993), espectrometría de absorción atómica con vapor frío (María Liva Garrido, 1998), y espectrometría de fluorescencia atómica con vapor frío (Rahman L., 2000) Tradicionalmente se ha empleado como técnica para la detección de mercurio, la espectrometría atómica con vapor frío, debido a que este analito existe como elemento neutro en fase de vapor a temperatura ambiente. Esto implica la existencia de considerable número de átomos en fase vapor sobre la superficie del metal líquido sin necesidad de suministrar energía térmica por una llama o por otro sistema de atomización, lo cual dio origen al término “vapor frío”. La generación del vapor frío de mercurio persigue reducir el mercurio presente en la muestra líquida o disuelta, en cualquier forma que éste se encuentre, a mercurio elemental por la acción de un agente reductor, por ejemplo, cloruro de estaño (II). El vapor de mercurio elemental generado se transfiere, con la ayuda de una corriente de gas inerte (argón), hasta el camino óptico del espectrómetro. La cuantificación se puede efectuar mediante absorción, emisión o fluorescencia atómica comparando con una curva de calibración obtenida de forma similar con estándares acuosos de concentración conocida (PSAnalytical, 1997) Espectrometría de absorción atómica con Vapor Frío (CV-AAS) La absorción atómica es el proceso mediante el cual el átomo en estado fundamental puede absorber energía de una radiación a una longitud de onda específica y pasar al estado excitado. La espectrometría de absorción atómica usa este proceso para cuantificar la cantidad de energía, en 9 forma de fotones de radiación, absorbidos por una muestra En la Figura 1.2 se muestra las transiciones electrónicas en absorción atómica. (Beaty, 1979) Figura 1.2. Esquema de transiciones electrónicas en absorción atómica Instrumentación de absorción atómica con vapor frío Descripción del equipo de absorción atómica En la Figura 1.3, se muestra la disposición de los componentes de un sistema de medición por espectrometría de absorción atómica con vapor frío. La característica de interés en las medidas por absorción atómica, es el monto de radiación que es absorbida, a la longitud de onda resonante, cuando esta radiación pasa a través de una nube atómica. Conforme el número de átomos se incrementa en el paso de la radiación, la cantidad que de ésta será absorbida se incrementará en una forma proporcional. Se puede efectuar una determinación cuantitativa del analito presente, midiendo la cantidad de radiación absorbida. El uso de fuentes especiales de radiación y la selección cuidadosa de la longitud de onda, permite la determinación cuantitativa específica de elementos individuales en la presencia de otros. En el caso del mercurio, dicha longitud de onda corresponde a 253,7 nm. (Beaty, 1979) Figura 1.3. Esquema de la disposición de los componentes de un sistema de medición por espectrometría de absorción atómica con vapor frío La fuente de energía debe poder emitir esta longitud de onda específica, esto hace de la absorción atómica una técnica analítica con pocas interferencias espectrales. La lámpara de cátodo hueco es una 10 excelente fuente de energía discreta para la mayoría de los elementos determinables por absorción atómica. El cátodo de la lámpara es un cilindro hueco. El ánodo y el cátodo se encuentran en un cilindro de vidrio cerrado y lleno ya sea de neón o argón. Al extremo del cilindro se ha fundido una ventana transparente a la radiación emitida. Al aplicar un potencial eléctrico entre el ánodo y el cátodo, algunos de los átomos del gas de relleno se ionizan. Los iones cargados positivamente se aceleran a través del campo eléctrico y colisionan con el cátodo cargado negativamente, desalojando átomos metálicos individuales del mismo proceso. Los átomos de metal desalojados son entonces excitados para la emisión, por los impactos subsecuentes que tienen con más iones del gas de relleno. (Beaty, 1979) Para la medición de la radiación específica se necesitan dos componentes, un monocromador y un detector. El primero puede dispersar las distintas longitudes de onda de la radiación que es emitida de la fuente y separa la línea particular que se emplea para medir cierto elemento en presencia de otros. Una vez aislada la longitud de onda por el monocromador el detector, recibe esta radiación. Este segundo componente es un tubo fotomultiplicador, que produce una corriente eléctrica que depende de la intensidad de la radiación incidente. Finalmente esta corriente será amplificada y procesada por componentes electrónicos. Descripción y funcionamiento del generador de vapor frio comúnmente acoplado al equipo de absorción atómica Uno de los espectrómetros de absorción atómica más conocidos es el Perkin Elmer modelo 460. A este sistema se le puede acoplar un sistema de generación de hidruros llamado MHS-10 Perkin Elmer. Se utiliza como agente reductor borohidruro de sodio en una solución al 3% en NaOH al 1%. El vapor de mercurio generado se traslada al detector mediante el uso de argón como gas de arrastre. En la Figura 1.4 se muestra un esquema de este sistema de generación de vapor frío. El agente reductor, Borohidruro de Sodio, se encuentra en el recipiente de la izquierda y el patrón o muestra que contiene mercurio se encuentra en el recipiente de la derecha. Para hacer pasar el agente reductor a la solución con Hg al pulsar el botón que se encuentra en la parte superior del aparato. El vapor de mercurio Hg0 generado, según la ecuación (1.1), es arrastrado con el gas de arrastre hasta el sistema de detección. (Cabrera-Vique, 2007) BH 4 3H 2O H Hg 2 H 3 BO3 Hg 0 8 H (1.1) 11 Limitaciones del generador de vapor frio comúnmente acoplado al equipo de absorción atómica. Requiere gran cantidad de reactivos y patrones acuosos. Aproximadamente 15 mL de agente reductor y 10 mL de patrón acuoso por cada medición. Requiere la utilización de borohidruro de sodio que es un reactivo altamente corrosivo, tóxico e inflamable. Utiliza argón como gas de arrastre lo cual incrementa los costos de las mediciones. Es de difícil acceso la adquisición del generador de hidruros MHS10. Figura 1.4. Sistema de generación de vapor frío que puede ser acoplado al sistema de medición por absorción atómica Espectrometría de fluorescencia atómica con vapor frío (CV-AFS) La espectrometría de fluorescencia atómica se basa en el uso de fotones de energía más elevada para excitar una muestra que emitirá fotones de inferior energía. Esta técnica incorpora aspectos de absorción y emisión atómica. Igual que en absorción atómica, los átomos en estado fundamental, son excitados al hacerse incidir la radiación sobre el vapor atómico. Se mide la emisión resultante del decaimiento de los átomos excitados por la fuente, a este proceso se le denomina fluorescencia. La intensidad de esta “fluorescencia” se incrementa conforme se incrementa la concentración de los átomos, lo que sirve de base para una determinación cuantitativa. En la Figura 1.5 se muestran las transiciones electrónicas en fluorescencia atómica. (Beaty, 1979) 12 Figura 1.5. Esquema de transiciones electrónicas en fluorescencia atómica La aplicación de la fluorescencia se basa también en los mismos aspectos que la absorción, con la ventaja que produce señales más intensas para la misma cantidad de analito. La eficiencia de la generación de vapor atómico de mercurio es mayor desde soluciones de Hg2+, por lo que sea cual fuere la forma de detección, antes de adicionar el agente reductor se aconseja agregar un exceso de agente oxidante a la muestra. (Tsalev, 1995) La espectrometría de fluorescencia atómica con vapor frío se ha utilizado más en la determinación de mercurio en muestras ambientales con contenidos bajos del elemento (Stockwell P. B., 1994) ya que, como se mencionó antes, permite tener niveles de sensibilidad mayores comparada con la espectrometría de absorción atómica con vapor frío. Esta técnica es de gran utilidad ya que se ha demostrado que es posible obtener límites de detección de partes por trillón. (Cizdziel James V, 2010) Instrumentación de fluorescencia atómica con vapor frío Descripción del equipo de fluorescencia atómica Tal como se muestra en la Figura 1.6, la fuente de energía para fluorescencia atómica, está fuera del plano del resto del sistema óptico, de manera tal que el detector lumínico, cuantifica sólo la fluorescencia que sucede en el vapor de mercurio y no la radiación de la lámpara en sí. Normalmente son más brillantes las lámparas de fluorescencia atómica que las utilizadas en absorción atómica, con el objeto de incrementar el grado de excitación del átomo y suministrar una más alta sensibilidad con fluorescencia. (Beaty, 1979) En el caso de las determinaciones de mercurio por fluorescencia atómica, generalmente son utilizadas lámparas de mercurio de baja presión. Esta tiene una fuente de radiación UV eficiente a 13 bajas longitudes de onda. La línea principal de emisión es a 253,7 nm. El sistema de contiene un monocromador que, tal como se dijo anteriormente, dispersa las longitudes de onda emitidas por la fuente y separa la línea particular que se emplea para medir el mercurio. Y el detector utilizado es igualmente un tubo fotomultiplicador, que produce una corriente eléctrica que depende de la intensidad de la radiación incidente. (Beaty, 1979) Figura 1.6. Esquema de la disposición de los componentes de un sistema de medición por espectrometría de fluorescencia atómica con vapor frío Descripción y funcionamiento del generador de vapor frio comúnmente acoplado al equipo de fluorescencia atómica Uno de los equipos más conocidos para determinación por fluorescencia atómica es el Merlin PSA. Este consta de un sistema de generación de vapor frío que sigue el esquema mostrado en la Figura 1.7. Con el fin de hacer mediciones de manera automatizada, este sistema fue diseñado de forma tal que una bomba peristáltica pueda bombear distintas soluciones, en este caso son tres, agente reductor, blanco y patrón (o muestra). Además consta de una válvula de mezcla que permite hacer pasar agente reductor y patrón para generar la reacción (1.2) o agente reductor y blanco para generar una línea base y limpiar las vías con el fin de evitar el efecto memoria. (PSAnalytical, PSA Introduces the 10.004 Hydride/Vapour Generator, 1997) Sn 2 Hg 2 Sn 4 Hg 0 (1.2) 14 Figura 1.7. Sistema de generación de vapor frío que puede ser acoplado al sistema de medición por fluorescencia atómica Limitaciones del generador de vapor frio comúnmente acoplado al equipo de fluorescencia atómica Es inevitable el desgaste de algunas de las piezas internas como la bomba peristáltica y la válvula de mezcla. Las mangueras usadas para hacer pasar las soluciones se deterioran con facilidad ya que son presionadas constantemente por la bomba peristáltica. Los costos de reparación son muy altos y las piezas son difíciles de conseguir en nuestro país. Además no existe una empresa representante de la marca comercial del equipo que ofrezca servicio técnico confiable. Este sistema requiere el uso de soluciones acuosas para poder ser transportadas por las mangueras, lo cual representa una desventaja para muestras sólidas ya que es necesario la realización de digestiones donde se requiere calentar para acelerar el proceso, aumentando así la posibilidad de pérdida por volatilización ya que el mercurio metálico así como otras especies de dicho metal, son muy volátiles. Actualmente se ha implementado el uso de un horno microondas especializado para la realización de digestiones de muestras sólidas, incrementando así los costos de las mediciones. 15 1.4. Conceptos teóricos fundamentales para optimización y validación de los métodos de análisis Las mediciones analíticas se hacen con métodos y equipos que deben ser probados para asegurar la calidad de los análisis. Para esto es necesario validar el método utilizado con el fin de confirmar, mediante evidencias experimentales objetivas, el cumplimiento de los requisitos para considerar que el método propuesto es apto para su utilización o aplicación. Todas las mediciones analíticas son importantes y por lo tanto determinar el resultado correcto y ser capaz de demostrarlo es de gran relevancia. Los análisis están condicionados al equipo, el cual debe funcionar y estar calibrado correctamente. Pero además el operador debe ser competente y tener suficiente conocimiento para tomar decisiones apropiadas a partir de las observaciones que él realice. Los parámetros que determinan el desempeño de un método son: La identidad y selectividad o especificidad. Exactitud y precisión. Incertidumbre. Límite de detección y límite de cuantificación. Intervalo de trabajo y ámbito lineal. Sensibilidad y recuperación. Dada la importancia de estos parámetros, se ahondará un poco en los detalles más relevantes de cada uno a fin de destacar su contribución al logro de un buen análisis. Identidad. Confirmar la identidad implica establecer que la señal o respuesta obtenida se debe al analito y no a la presencia de otro ente químico o físico. (Eurachem, 1998) Selectividad y especificidad. La selectividad se refiere a la aptitud del método para determinar exacta y específicamente el analito de interés en presencia de otros componentes en la matriz de la muestra. Mientras que la especificidad es la aptitud del método para medir solamente lo que se busca medir. (Eurachem, 1998) 16 Exactitud y precisión La exactitud corresponde por su parte a la proximidad entre el resultado de una medición y el valor verdadero del analito. La certeza de un método es una expresión de cuan cerca está la media de un conjunto de resultados del valor verdadero. La precisión es una medida de cuán cerca están los resultados unos de otros por mediciones replicadas en condiciones especificas y generalmente se expresa por medidas como la desviación estándar que describe la dispersión de los resultados. Las dos medidas de precisión más comunes son repetibilidad y reproducibilidad, la primera indica la variabilidad de los resultados obtenidos por un método con el mismo operador en un mismo equipo a lo largo de un periodo de tiempo corto. Mientras que la segunda corresponde a la variabilidad de resultados obtenidos para una misma muestra en las mismas condiciones descritas para repetibilidad pero sin esperar variaciones respecto al tiempo. (ISO, 1994) Para evaluar la exactitud y precisión de un método analítico es necesario el uso de un material de referencia certificado. Según la Organización Internacional para la Estandarización (ISO) “es un material, al que acompaña un certificado, con una o más de sus propiedades certificadas por un procedimiento que establezca su trazabilidad a una realización precisa de la unidad en la que los valores de la propiedad se expresan, y para las cuales cada valor certificado se acompaña por una incertidumbre a un determinado nivel de confianza”. (Eurachem, 1998) Para llevar a cabo una medición de la exactitud del método se debe usar material de referencia certificado. Se compara el valor obtenido (x), a partir de la recta de calibración, con el valor de referencia certificado (µ). El sesgo del método será la diferencia entre dichos valores: s x (1.1) Por otro lado, tal como se dijo anteriormente, la repetibilidad es una medida de la precisión. Esta puede ser calculada como un promedio de las mediciones y , con una desviación estándar “s”, asociada a dicho promedio: N y y i 1 N N i (1.2) _ ( y i y )2 s i 1 N 1 17 (1.3) Una forma alternativa de expresar el error asociado al promedio de las mediciones es como un coeficiente de variación o desviación estándar relativa: %CV s _ 100% (1.4) y Incertidumbre La incertidumbre es un parámetro asociado al resultado de una medición, que caracteriza la dispersión de los valores que podrían ser razonablemente atribuidos al mesurando. (Eurachem, 1998) Límite de detección y Límite de cuantificación. El límite de detección es la menor cantidad de analito que puede ser distinguida de la señal de fondo con cierto nivel de confianza especificado. (OAA, 2003) Figura 1.8. Distribución de valores alrededor de cero y nivel crítico. Los valores de concentración se distribuirían alrededor de cero con una desviación estándar 0. Quiere decir que como resultado de la medida de un blanco, y debido a los errores experimentales del método (reflejados en 0) podríamos obtener una concentración que no fuese cero. La distribución se acota en un punto llamado valor crítico, L c, que permite, una vez medida la muestra, tomar la decisión de si el analito se halla presente o no. (Boqué, 2004) LC z 1 0 (1.5) Donde z1 es el valor de la distribución normal de una cola para un nivel de significación y 0 es la desviación estándar de la concentración neta cuando el analito no está presente en la muestra L C se define para marcar un valor mínimo a partir del cual una concentración predicha se considera 18 causada por el analito. De esta forma, existe un riesgo de cometer un error de tipo I (o falso positivo). Sin embargo, si además se quiere mantener un riesgo (llamado ) pequeño de cometer un falso negativo, el límite de detección del método, LD, debe ser mayor. (Boqué, 2004) Figura 1.9. Nivel crítico y límite de detección para = (pequeños) Así pues, tomando en consideración ambas probabilidades de error: [45] L D z 1 0 z 1 D (1.6) Donde z1 es el valor de la distribución normal de una cola para un nivel de significación y D es la desviación estándar de la concentración cuando el analito está presente en la muestra al nivel del límite de detección. En las ecuaciones anteriores se ha asumido que las concentraciones siguen una distribución normal con varianza conocida. Tomando como valores por defecto para ==0.05, y asumiendo que la varianza es constante entre c=0 y c=LD, la ecuación anterior se transforma en: [(Boqué, 2004) L D 3.3 0 (1.7) Si no se conocen las varianzas y deben estimarse a partir de análisis replicados, entonces los valores de 0 y D deben ser reemplazados por sus correspondientes estimaciones, s0 y sD. Así mismo, los valores de z deben ser reemplazados por los correspondientes valores de t de una distribución tStudent con grados de libertad. (Boqué, 2004) Existen otros autores que recomiendan definir el límite de detección (LD) como la concentración de analito que proporciona una señal igual a la señal del blanco, yB, más tres veces la desviación estándar del blanco, sB. (Miller, 2002) 19 LD y B 3s B (1.8) Es necesario aclarar que en la ecuación (1.5) la probabilidad de que ocurra cada uno de los tipos de error (tipo I y tipo II)es sólo del 5%. Mientras que para la ecuación (1.6) la probabilidad es de un 7% aproximadamente. (Miller, 2002) Existe otro llamado límite de cuantificación (LC) que representa la menor cantidad que puede ser determinada cuantitativamente con una incertidumbre asociada, para un dado nivel de confianza. Este valor se emplea por lo general en determinaciones de analitos a nivel de trazas. (Miller, 2002) LC y B 10 s B (1.9) En la práctica se obtienen los términos yB y sB. Con el uso de la recta de calibración se conduce a un límite de detección reportado en términos de la variable independiente “x” ya sea como concentración o cantidad de analito. (Miller, 2002) Por otra parte, existe un concepto llamado “Límite de Detección del Método (LDM)” que se define como la mínima concentración de una sustancia que puede ser medida y reportada con un 99% de confianza que la concentración del analito es mayor que cero y es determinado del análisis de una muestra en una matriz dada que contiene el analito. (Ripp, 1996) El procedimiento utilizado para calcular el LDM es: 1. Estimar el límite de detección usando una de las siguientes opciones: a) La concentración correspondiente a una relación Señal/Ruido en el rango de 2.5 a 5 b) La concentración correspondiente a tres veces la desviación estándar del promedio de una serie de mediciones del blanco. c) La concentración correspondiente a la región de la curva donde hay un cambio significativo de sensibilidad. d) La concentración correspondiente a la limitación instrumental. 2. Preparar un blanco que esté lo más libre de analito posible. 3. Para determinar el LDM se puede hacer a partir de un reactivo preparado con un estándar que contenga el analito. La concentración a la cual se preparará será igual a la obtenida al estimar el límite de detección. (Se recomienda que sea entre 1 y 5 veces el límite de detección estimado.) 4. Se toman siete alícuotas de la muestra preparada y es analizada con el método que se está evaluado, se hacen los cálculos para reportar los resultados en unidades de concentración. 20 5. Se calcula la desviación estándar del promedio “S” de las siete mediciones realizadas según la ecuación 1.3. 6. Se calcula el LDM según: LDM S t ( n 1,1 0 , 99 ) (1.10) Siendo t ( n 1,1 0 , 99 ) la t de Student apropiada para un nivel de confianza del 99% y una desviación estándar estimada para n-1 grados de libertad. Finalmente se puede calcular el límite de cuantificación como: LCM 10 S (1.11) Intervalo de trabajo y ámbito lineal. La linealidad del un método define su aptitud para obtener resultados proporcionales a la concentración del analito y el rango lineal es el intervalo de concentraciones de analito para la cuales el método brinda resultados proporcionales a la concentración. El extremo inferior del rango por lo general son los límites de detección y/o cuantificación. El extremo superior dependerá de la respuesta del instrumento. (Miller, 2002) En química analítica, calibración es una operación que determina la relación funcional entre valores medibles (intensidad de la señal) y cantidades de analito (concentración). Este proceso tiene dos pasos. En el primero se mide grafican las intensidades de las señales obtenidas para cada solución de concentración conocida (estándares). En el segundo paso, se deduce la concentración de una muestra desconocida usando la medida de la intensidad de la señal de dicha muestra en el gráfico de calibración obtenido en el primer paso. Si se usan más de dos estándares es necesario llevar a cabo una Regresión que puede ser lineal o curva, dependiendo del comportamiento de los datos. (Danzer, 1998) Una regresión se basa comúnmente en el uso del Método de los Mínimos Cuadrados (LSM) el cual consiste en minimizar la suma de los cuadrados de la diferencia entre el valor calculado y el valor experimental. El mejor gráfico de calibración para un analista es aquel que provee la menor diferencia entre el valor calculado y el valor experimental. (Boqué, 2004) El modelo lineal es el modelo más usado en calibración analítica. Consiste en encontrar la recta de calibrado donde cada punto se encuentre definido por una variable “x” (variable independiente) y 21 una variable “y” (variable dependiente). La recta de calibrado se encuentra definida por una ordenada en el origen (b) y una pendiente (m) a través de la ecuación: y mx b (1.12) El Método de los Mínimos Cuadrados, mencionado anteriormente, permite obtener las estimaciones de la ordenada en el origen y la pendiente: n (x i x )( y i y ) i 1 m (1.13) n ( x i x) 2 i 1 b y m x (1.14) Donde x e y corresponden, respectivamente, al valor medio de las coordenadas “x” e “y” de los n puntos experimentales. Para validar el modelo lineal tradicionalmente se utiliza mediante la comprobación del coeficiente de determinación (r2). El coeficiente de determinación es el cuadrado del coeficiente de correlación (r) (Boqué, 2004): n ( x r i x )( y i y ) i 1 n (x i 1 (1.15) n i x) 2 ( y i y) 2 i 1 El coeficiente de determinación proporciona la correlación entre las variables dependiente e independiente. Este valor se encuentra comprendido entre -1 y 1, este valor permite al analista verificar si el modelo es aplicable para describir los datos. Sin embargo, la IUPAC afirma que este no es el único parámetro usado en calibración para medir la relación entre dos variables, es tan solo una herramienta para determinar de forma preliminar la linealidad de la recta de calibrado. (Boqué, 2004) Otro de las herramientas que pueden ser utilizadas para la determinación del intervalo lineal es el análisis visual del gráfico de los residuales. El residual de cada punto experimental es la distancia vertical de cada punto a la recta de calibrado obtenida. Un grafico de residuales es por lo tanto una representación de la coordenada “x” de cada punto experimental respecto a su valor residual. Es por ello que para considerar que un modelo lineal es apropiado, se debe observar en el gráfico de residuales que existe aproximadamente el mismo número de residuales positivos y negativos. 22 También dichos residuales deberían estar distribuidos aleatoriamente y tener aproximadamente el mismo valor absoluto y además no deberían mostrar tendencia alguna. (Boqué, 2004) Otra herramienta estadística más rigurosa para asegurar la validez del modelo lineal es el análisis de varianza (ANOVA), este requiere de replicados por cada punto experimental. Para esto se debe descomponer la variabilidad de la variable respuesta (y) en variabilidad explicada por el modelo más la variabilidad no explicada o residual. Bajo la hipótesis que existe una relación lineal entre la variable respuesta (y) y la independiente (x), se realiza el siguiente contraste de hipótesis: H o : y b (1.16) H1 : y b mx (1.17) Por tanto, si se acepta Ho, la variable independiente no influye y no hay relación lineal entre ambas variables. En caso contrario, si existe una dependencia lineal de la variable respuesta con la independiente. (Navarro, 2009) Matemáticamente, la suma de los cuadrados total es: SST SSR SS P (1.18) SST es la suma de cuadrados total. SSP es la suma de cuadrados explicada por el modelo lineal. SSR es la suma de cuadrados de los residuales. MSR y MSP son los cuadrados medios que se obtienen dividiendo SSP y SSR entre sus respectivos grados de libertad. (Boqué R. A., 2004) Para comparar MSR y MSP se realiza una prueba de hipótesis F. Si no existe diferencia estadísticamente significativa entre ellas, la presencia de “x” no influye en el modelo y se acepta Ho. Si, por el contrario existe alguna relación entre las variables dependiente e independiente es decir, la x si influye y se rechaza Ho, MSP será mucho mayor que MSR, con lo cual el valor calculado de F será mayor que el tabulado para el nivel de significación escogido y los grados de libertad mencionados. (Boqué R. A., 2004) Sensibilidad Según la IUPAC la sensibilidad es el cambio en la respuesta de un instrumento de medición dividido por el cambio de la concentración del analito, en el modelo lineal se puede observar con la pendiente de la recta, a mayor pendiente habrá una mayor sensibilidad. Por otra parte la robustez es la medida de la capacidad de un procedimiento analítico para no ser afectado por variaciones 23 pequeñas pero deliberadas en los parámetros del método y que provee una indicación de su fiabilidad durante su uso habitual. (Danzer, 1998) Recuperación La recuperación permite evaluar la eficiencia del método para detectar todo el analito presente en una muestra y por ende su exactitud. [38]. Es una alternativa viable cuando no se dispone de un material certificado de referencia apropiado para simular la matriz de la muestra que se analiza. Esencialmente lo que se hace es dopar alícuotas de la muestra con cantidades conocidas del analito y analizar las alícuotas para chequear cuánto del analito se determina con el método en uso. Los resultados generalmente se expresa como porcentaje. (OAA, 2003) %R x 100 (1.19) 1.5. Análisis de muestras sólidas Una de las principales preocupaciones de la química analítica en la actualidad es el desarrollo de métodos sostenibles desde el punto de vista medioambiental. Sin embargo, es necesario mantener las características analíticas como precisión, exactitud, especificidad, etc., pero también que puedan reducirse la cantidad de residuos generados en los laboratorios, minimizando de esta manera el consumo de reactivos y aumentando la rapidez de los análisis. El avance hacia este tipo de técnicas permite el ahorro en el costo de reactivos y mejora las condiciones de seguridad e higiene en el trabajo. A lo largo de los años, han sido propuesto distintos métodos para la determinación de mercurio mediante espectroscopia atómica pueden clasificarse en dos grupos, atendiendo a la manipulación que se requiere realizar sobre la muestra: Los que contemplan la extracción del analito con un disolvente adecuado y posterior medida del extracto. Los basados en medidas directas sobre muestras sólidas empleando combustión. Los métodos donde es necesario extraer el analito con un disolvente han sido ampliamente desarrollados, usándose por lo general mezclas de ácidos. La selección del disolvente depende de la matriz de la muestra. Es decir, se debe considerar si la muestra es liquida o solida, y que cantidad de 24 materia orgánica tiene. También es necesario considerar el tiempo requerido que dependerá del tipo de muestra. 1.5.1. Digestión Por lo general las técnicas analíticas requieren la muestra en solución acuosa antes de los análisis. El proceso de digestión se basa en convertir el analito en formas químicas que permanezcan estables en disolución. La aplicabilidad de los métodos de digestión conocidos depende de la matriz de la muestra. Se sabe que dichos métodos requieren una gran cantidad de tiempo y reactivos para poder eliminar toda la materia orgánica presente y/o disolver en su totalidad la matriz de la muestra. Además existe un gran riesgo de contaminación de la muestra en cualquier paso que se debe llevar a cabo en este proceso. En el caso del mercurio metálico así como otras especies de dicho metal, existe un problema adicional y es la pérdida por volatilización, lo cual representa una desventaja para la realización de digestiones por vía húmeda, ya que en estas se requiere calentar para acelerar el proceso. Es por ello que la utilización de métodos de digestión para la determinación de mercurio a nivel de trazas exige una cuidadosa escogencia. Desventajas de la digestión con ácido perclórico. Uno de los ácidos más utilizados para la digestión de muestras solidas es el ácido perclórico. Es muy eficiente debido a que es un oxidante muy poderoso. Sin embargo, es inflamable y corrosivo. Lo cual representa grandes riesgos para la persona que los manipule. No se pueden respirar los vapores, por lo tanto, este debe ser calentado solo en una campana química de construcción incombustible. Además, es necesario que los materiales orgánicos presentes en la muestra, sean digeridos previamente con ácido nítrico antes de la acción del ácido perclórico pues puede explotar violentamente con trazas de material orgánico, por lo tanto se gasta una gran cantidad de tiempo en este tipo de digestión lo cual representa un inconveniente adicional pues para realizar trabajos de gran exactitud y precisión se requieren gran número de mediciones. Las campanas químicas en las que se caliente ácido perclórico deben ser inspeccionadas frecuentemente para detectar la acumulación de percloratos. Los depósitos deben ser lavados con agua y retirados. Para operaciones de rutina se debe utilizar una bomba de agua conectada con un equipo de vidrio de eliminación de humos. Las campanas especiales para trabajar con ácido son campanas extractoras de acero inoxidable con instalaciones para lavado. 25 Es necesario tener en cuenta que debe ser almacenado en botellas de vidrio sobre bandejas no combustibles, como cerámica o plástico, que sean lo suficientemente grandes como para contener completamente el contenido de la botella. Por último se recuerda que no se debe calentar ácido perclórico con ácido sulfúrico. Desventajas de la digestión por microondas Debido a todos los inconvenientes que trae la digestión húmeda, se han diseñado hornos microondas que evitan el uso de grandes cantidades de ácido pero incrementan el costo de las mediciones pues los equipos tienen un elevado valor monetario. Una de las ventajas que tiene este tipo de digestión es que seguro al utilizarlo a presión atmosférica. Pueden manejarse muestras con grandes materia orgánica que generan enormes cantidades de gas. En hornos de alto costo se puede programar para la adición de reactivos en cualquier instante durante la digestión lo que permite un ataque secuencial con ácidos. Sin embargo, la limitación principal está en que la cantidad de muestra que se puede digerir es limitada. No se pueden digerir grandes cantidades pues la potencia máxima no es suficiente. Otro de los problemas es que la peligrosidad en la manipulación de los reactivos sigue estando presente. Pues aunque las microondas pueden ayudar a romper la matriz, no son eficientes de manera aislada, es necesario utilizar un ácido o mezclas de ácidos para ayudar a la digestión. Sumado a esto, existe una gran dificultad para la adición de reactivos si es un horno convencional de bajo costo. Por último, aunque es menor el tiempo utilizado, sigue considerándose muy largo el tiempo para la preparación previa de la muestra lo cual limita los análisis. 1.5.2. Combustión Directa El análisis directo de muestra solida empleando un analizador de mercurio surge como una alternativa que evita el uso de los métodos convencionales de digestión (vía húmeda y microondas). Esta nueva herramienta tiene la capacidad de hacer mediciones rápidas de muestras líquidas y sólidas sin necesidad de hacerle un tratamiento previo a las mismas. Las medidas directas sobre muestras sólidas por combustión se han aplicado a mercurio debido a su volatilidad en la forma Hg 0. Sin embargo, la destrucción total de la muestra es una desventaja pues no se puede conservar para determinaciones posteriores de otros metales como por ejemplo, plomo o cadmio. El elevado costo de estos equipos hace poco viable su uso en análisis de tipo general ya que son muy selectivos, es 26 decir, solo se puede determinar mercurio y no se puede modificar para medir otros metales. (Cizdziel James V, 2010) 1.5.3. Lixiviación El diseño y puesta a prueba de procedimientos sencillos basados en medidas directas y no destructivas de muestras solidas busca eliminar parcial o completamente el uso de disolventes y reactivos con lo cual se reduce, el tiempo de preparación previo de la muestra. Además, la muestra puede ser almacenada sin las mismas consideraciones (ej: refrigeración) que deben tenerse para las muestras disueltas para análisis posteriores. La reducción en el tiempo de análisis permite la realización de mayor cantidad de mediciones con lo cual se mejora la productividad al incrementar la frecuencia de análisis. (Akira Iwashita, 2004) Asi mismo el costo de cada determinación se reduce. Los tipos de muestras que se suelen analizar son sólidas, líquidas o gaseosas. Las muestras más habituales son del tipo sólido, entre ellas están los suelos, sedimentos, sedimentos marinos como en el caso que nos ocupa, cabello, uñas, muestras biológicas como órganos de animales desecados, etc. Mientras que las muestras liquidas son principalmente aguas, lixiviados y residuos líquidos incluyéndose también fluidos biológicos tales como orina y sangre. El aire del ambiente es la muestra gaseosa por excelencia. La mayoría de las muestras liquidas contaminadas con mercurio corresponden a aguas de ríos, afluentes, mares, etc. Los sólidos en el medio ambiente tienen la capacidad de retener, a lo largo del tiempo, contaminantes como el mercurio. En el caso de sedimentos marinos, que es de nuestro interés, esta contaminación es notoria y preocupante en el caso de zonas afectadas por la industria petroquímica. Después de la adsorción de estos contaminantes, se deben utilizar procesos físicos y químicos para purificar estos suelos o sedimentos y poder utilizarlos. Una forma de purificar sólidos es mediante lixiviación. Una lixiviación es la extracción de algún compuesto soluble presente en un sólido con la ayuda de un solvente líquido. Este sólido debe ser pulverizado previamente. El solvente líquido dependerá de las propiedades del compuesto contaminante. En un gran número de casos pueden utilizarse solventes orgánicos como alcoholes o cetonas. En el caso particular del mercurio pueden utilizarse soluciones acidificadas con ácido clorhídrico, nítrico, etc. (Akira Iwashita, 2004) 27 CAPÍTULO 2 SECCIÓN EXPERIMENTAL En este trabajo se diseñó una celda de reacción para formar vapor atómico de Hg a partir de muestras líquidas y sólidas, para que éste fuese detectado vía espectrometría de absorción o fluorescencia atómica. En las Tablas 2.1 y 2.2 se presenta una lista de los reactivos y una lista de aparatos usados para la realización de este Trabajo de Grado. Tabla 2.1. Reactivos empleados durante la realización del Trabajo de Grado Reactivo Suministrado por Acido Clorhídrico Pro-Análisis Riedel-deHaёn Ácido Nítrico Pro-Análisis Riedel-deHaёn Ácido Sulfúrico Pro-Análisis Riedel-deHaёn 3 Argón de alta pureza (cilindros de 6 m ) AGA Bromato de Potasio Pro-análisis Merck Bromuro de Potasio Pro-análisis Riedel-deHaёn Cloruro de Hidroxilamonio Pro-análisis Riedel-deHaёn Cloruro de Estaño (II) 99,6% Riedel-deHaёn Estándar de Hg 100 ppm en Fixanal Riedel-deHaёn Material Certificado de Referencia PACS-2 de Hg 3,04 ±0,20 ppm NRC∙CNRC Arena lavada de mar. 0,03% de metales pesados Fisher Scientific Tabla 2.2. Equipos empleados durante la realización del Trabajo de Grado Equipo Marca / Modelo Balanza analítica digital. 0,0001 g de apreciación Mettler. / AJ180 Balanza analítica digital. 0,01 g de apreciación Mettler. / PJ400 Detector Fluorescente de Mercurio Merlín PS Analytical / PSA 10.023 Espectrofotómetro de absorción atómica Perkin Elmer / 460 28 Celda de reacción utilizada para la generación de vapor frío. NCR Bomba de vacio con válvula antirretorno. 2.1. Contaminación de Hg durante los ensayos. Las trazas de mercurio, en estado gaseoso, generadas durante los análisis son atrapadas por una trampa de carbón activado que se encuentra dispuesta justo al salir del camino óptico tanto del equipo de absorción atómica como del equipo de fluorescencia atómica, inmediatamente antes de que el vapor de mercurio se difunda en el ambiente de laboratorio. 2.2. Tratamiento de los desechos de mercurio. En caso de que haya necesidad de eliminar los desechos provenientes de las soluciones sobrenadantes conteniendo mercurio después de hacer cada medición deber ser neutralizadas con hidróxido de calcio hasta un pH de 14, para seguidamente adicionarles sulfuro de sodio al 20% para precipitar sulfuro de mercurio. 2.3. Análisis previos 2.3.1. Material de laboratorio El material de vidrio utilizado fue lavado según el siguiente procedimiento: a) Enjuagar el material con agua y jabón. b) Llenar el material con HNO3 al 10% y dejarlo tapado durante toda la noche. c) Al día siguiente, enjuagar el material 3 veces con agua desionizada. d) Dejar secar el material a temperatura ambiente y taparlo con papel parafilm hasta el momento de su uso. 2.3.2. Preparación de las soluciones a utilizar en los análisis • HCl 33% v/v Se diluyen 167 mL de ácido clorhídrico (HCl) 37% v/v a 500 mL con agua desionizada. 29 • Agente Oxidante. Mezcla KBr/ KBrO3 0,1N Se disuelven 1,19 g de bromuro de potasio (KBr) en 25 mL de agua desionizada y luego se afora a 50 mL. Se disuelven 0,278 g de bromato de potasio (KBrO3) en 25 mL de agua desionizada y luego se afora a 50 mL. Se mezclan ambas soluciones en una proporción 1:1. Esta solución se prepara diariamente. • Agente reductor. SnCl2∙2H2O 2% m/v-HCl 10% v/v Se añaden 5 g de cloruro estannoso dihidratado (SnCl2∙2H2O) en 25 mL de ácido clorhídrico (HCl) al 37% v/v. Se calienta esta disolución hasta disolver completamente el sólido y luego se afora a 250 mL con agua desionizada. Esta solución se prepara diariamente. • Agente reductor. NaBH4 3% m/v-NaOH 1,5% m/v Se disuelven 3,75 g de hidróxido de sodio (NaOH) en 100 mL de agua desionizada. Se añaden a esta disolución 7,5 g de borohidruro de sodio (NaBH4). Se afora a 250 mL con agua desionizada. Esta solución se prepara diariamente. • OHNH3Cl 12% m/v Se disuelven 3 g de clorhidrato de hidroxilamina (OHNH3Cl) en 10 mL de agua desionizada. Se afora a 25 mL con agua desionizada. Esta solución se prepara diariamente. • Muestra Líquida La solución estándar de mercurio para los análisis de muestra líquida es preparada a partir de una ampolla Fixanal, que es una disolución espectrométrica de mercurio, 0.100 g de Hg+2 en HCl 1%, marca Riedel de Haen. A partir de esta solución se realizan disoluciones sucesivas en agua desionizada para obtener patrones a distintas concentraciones. Se añaden 2 mL de mezcla bromuro de potasio/bromato de potasio (KBr/ KBrO3) y 15 mL de ácido clorhídrico (HCl) al 33% v/v a una alícuota de solución madre y se deja tapado por una hora. Luego, se decolora la solución colocando 60 µL de clorhidrato de hidroxilamina (OHNH3Cl) al 12% m/v. Se afora a 100 mL con agua desionizada. Estas soluciones deben ser analizadas en las siguientes 3 horas, por lo tanto se debe preparar diariamente. 30 • Blanco para Muestra Líquida Se añaden 5 mL de la mezcla bromuro de potasio/bromato de potasio (KBr/ KBrO3) a 37,5 mL de ácido clorhídrico (HCl) al 33% v/v. Se decolora la solución colocando 150 µL de clorhidrato de hidroxilamina (OHNH3Cl) al 12% m/v. Se afora a 250 mL con agua desionizada. Esta solución se prepara diariamente. • Muestra Sólida Se pesa la cantidad de Material Estándar Certificado de Referencia PACS-2. Se agrega 1 mL de HNO3. Se tapa con papel parafilm y se deja reposar por 10 minutos. Una vez transcurrido el tiempo se procede a medir. • Blanco para Muestra Sólida Se pesa una cantidad aproximada a 0,0300 g de arena lavada de mar. Se agrega 1 mL de HNO3. Se tapa con papel parafilm y se deja reposar por 10 minutos. Una vez transcurrido el tiempo se procede a medir. 2.4. Metodología de análisis 2.4.1. Diseño de la celda de reacción utilizada para la generación de vapor frío 2.4.1.1. Primer diseño Inicialmente se evaluó la forma de trabajo del generador de vapor frío utilizado por el espectrómetro de fluorescencia atómica marca Merlin P.S. Analytical LTD. Modelo PSA 10.023. Una vez evaluado el funcionamiento del equipo se diseñó un sistema de generación de vapor alternativo con el que se pudiesen llevar a cabo las etapas que requiere la medición con el equipo convencional. Con el material existente en el laboratorio se realizó el montaje mostrado en la Figura 2.1. Se utilizaron dos kitazatos conectados entre sí con mangueras y dos tubos de vidrio en forma de “T” y “Y” que permitían separar las corrientes de gas y vapor. El funcionamiento consistía en tener el blanco en el kitazato 1, y tener el patrón en el kitazato 2. El gas de arrastre (argón) entraba al sistema a través de una manguera y dependiendo del instante de la medición, este gas era llevado al 31 kitazato 1 ó 2 usando un tubo en forma de “Y”. Así mismo el gas, con o sin vapor, que llegaba al detector era separado antes por otro tubo en forma de T con mangueras donde unas pinzas de metal dejaban o no pasar el gas correspondiente al instante de la medición. Inicialmente se hacía pasar el gas de arrastre por el kitazato 1 para generar la línea base, luego se cambiaba y se hacía pasar el gas al kitazato 2 donde estaba el patrón de mercurio y se hacía reaccionar con SnCl2 que era inyectado por la parte superior del kitazato. Durante todo este proceso se mantenían ambas soluciones en agitación magnética con el fin de que rápidamente ocurriera la reacción. Figura 2.1. Primer montaje para la generación de vapor frío de mercurio. El montaje anterior sirvió para demostrar la viabilidad de la generación de vapor de Hg0 y su transporte hacia un detector mediante un flujo controlado de Ar. Las primeras mediciones así realizadas adolecían de alta irreproducibilidad. 2.4.1.2. Segundo diseño Se diseñó un segundo sistema de medición para que fuese realizado en el taller de vidrio de la Universidad Simón Bolívar (Figura 2.2). Se mantuvo el principio general utilizado en el diseño del primer prototipo. En este nuevo diseño, se sustituyeron las mangueras y las pinzas por llaves de teflón con el fin de hacer más fácil su manipulación y también garantizar la llegada al detector de todo el vapor generado. 32 Se usaron dos recipientes de vidrio conectados por tubos de vidrio con llaves de teflón. En el recipiente 1 se encontraba el blanco al cual se le inyectaba el cloruro estannoso SnCl2 por la parte superior del mismo, el gas de arrastre entra por un costado de dicho recipiente y es llevado al detector al estar la llave A abierta en las vías 1 y 2, y cerrada en la vía 3. Luego de generar la línea base, se abre la llave B y se deja pasar el SnCl2 al recipiente 2 que contiene el patrón de mercurio o la muestra. Se abre la llave A en los sentidos 2 y 3, y permanece cerrada en el sentido 1. El vapor generado es arrastrado por la corriente de argón hasta el detector. Es necesario recordar que estos vapores antes de llegar al detector son pasados por el separador gas líquido con el fin de que no pase humedad al detector, además son pasados por una membrana higroscópica de nafión donde la humedad remanente en el vapor es eliminada antes de llegar al detector. Figura 2.2. Segundo montaje para la generación de vapor frío de mercurio. En este segundo prototipo se observó que no se podía garantizar una agitación reproducible para cada una de las mediciones. Además, una vez se genera el vapor de mercurio, las distancias que éste recorre son muy largas (50 cm aproximadamente), por lo tanto, existe una alta probabilidad que el vapor se condense en este camino y no alcance la llegada al detector. 33 2.4.1.3. Tercer diseño Es por lo antes expuesto que se probó un tercer y último montaje donde se tomaban en cuenta todas las observaciones hechas anteriormente, este sistema se optimizó y fue usado para las mediciones finales con espectrometría de fluorescencia atómica con vapor frío y espectrometría de absorción atómica con vapor frío. A diferencia del segundo diseño (Figura 2.2) las longitudes de las conexiones se mantuvieron lo más cortas posible y se conectaron con la celda de reacción manteniendo cierto ángulo entre ellas a fin de facilitar el desplazamiento por gravedad del agente reductor. Figura 2.3. Celda de reacción utilizada para la generación de vapor frío (NCR). Para obtener la señal de absorción o fluorescencia se deben cumplir tres etapas: 1. En la primera se añade el reductor a una solución que no debe contener el analito (Hg2+) pero que contenga todos los reactivos con los que se preparan los patrones. Se insufla el gas de arrastre y se realiza así la medición de la línea base o medición del blanco. 34 2. En la segunda se cambia el envase removible por otro que contiene un volumen exactamente conocido del estándar o la muestra disuelta. Se añade el reductor y los vapores formados se transfieren hacia el detector para la medición. 3. Finalmente, en la tercera etapa se repite la medición de la línea base tal como se hizo en la primera etapa con el fin de limpiar las vías que fueron recorridas por el mercurio y evitar un posible efecto memoria. Así, se tiene ya la celda lista para cambiar el envase removible por otro con otro estándar o con otra muestra. La celda de reacción mostrada en la Figura 2.3, permite llevar a cabo estas etapas. Esta consta de dos llaves de teflón, denominadas en la figura “A” y “B”. La llave A es una llave de tres vías, permite separar las tres etapas. Si se comunican las vías 1 y 3, manteniendo cerrada la llave B, se permite generar la línea base. Se inyecta blanco a través de un septum y el cloruro estannoso es suministrado a través de la bureta. Luego se cambia la llave permitiendo comunicar las vías 2 y 3. Se abre la llave B y se deja caer el cloruro estannoso sobre la solución de Hg que previamente fue colocada en el envase removible. Para cumplir la tercera etapa se cierra la llave B y se comunican nuevamente las vías 1 y 3. Observando detalladamente la Figura 2.3, se aprecia que el conducto que permite el paso del agente reductor y del gas de arrastre, queda sumergido dentro de la solución del estándar, de la muestra o del blanco. Se diseñó de esta forma para que el reductor entrara directamente en contacto con el seno de cualquiera de las tres soluciones, lo cual incrementa el contacto entre el analito, en los casos del estándar y la muestra, y el reductor. Adicionalmente, el flujo del gas de arrastre además de cumplir esta función, sirve como medio de agitación de la solución para contribuir a la máxima eficiencia de la reacción de reducción y expulsar del seno del líquido el Hg0 formado. 2.4.2. Instalación de la celda de reacción utilizada para la generación de vapor frío al detector de fluorescencia atómica Para las determinaciones por espectrometría de fluorescencia atómica se utilizó un detector de fluorescencia atómica marca Merlin P.S. Analytical LTD. Modelo PSA 10.023. En la Figura 2.4 se muestra que los componentes principales de este instrumento son el automuestreador, el generador de vapor frio que contiene un sistema de mangueras flexibles, una bomba peristáltica y una válvula 35 de mezcla, el separador gas líquido, la membrana higroscópica y el detector de fluorescencia. (PSAnalytical, PSA Introduces the 10.004 Hydride/Vapour Generator, 1997) Figura 2.4. Esquema de un sistema de medición por fluorescencia atómica Con la instalación, mostrada en la Figura 2.5, de la celda de reacción utilizada para la generación de vapor frío al detector de fluorescencia atómica se evita el uso del automuestreador, del generador de vapor frio y del separador gas líquido que requieren convencionalmente. Por lo tanto el sistema se transforma en una alternativa más sencilla para la determinación de mercurio. Es necesario dejar la membrana higroscópica ya que esta permite el secado del vapor de mercurio húmedo antes de llegar al detector y así evitar el “quenching”. Figura 2.5. Esquema del acoplamiento de la celda de reacción utilizada para la generación de vapor frío al detector de fluorescencia atómica. 36 2.4.3. Condiciones experimentales para las mediciones por fluorescencia atómica Tiempos El generador de vapor del sistema convencional utiliza cuatro tiempos en los cuales se cumplen las etapas descritas a continuación. Tabla 2.3. Tiempos usados por el generador de vapor para determinar mercurio por fluorescencia atómica con el Espectrómetro Merlin P.S. Analytical LTD. PSA 10.023 Tiempo Definición Tiempo de retraso Tiempo en el cual se hace pasar blanco para generar la línea base Tiempo de subida Tiempo que tarda el detector en alcanzar la máxima señal Tiempo de análisis Tiempo total en el que la señal es máxima Tiempo de memoria Tiempo en el que se limpian las vías con el uso de un blanco Gases El gas utilizado para las determinaciones de mercurio con este equipo es Argón. Existen tres tipos de flujos que son descritos a continuación: Tabla 2.4. Flujos de gas utilizados para la determinación de mercurio por fluorescencia atómica con el Espectrómetro Merlin P.S. Analytical LTD. PSA 10.023 Tipo de flujo Definición Valor Gas de secado Permite secar el vapor húmedo de mercurio 2,5 L min Gas de arrastre Arrastra al vapor de mercurio 0,5 L min Gas de escudo Rodea la zona donde ocurre la fluorescencia para evitar el “quenching” (Ver Figura 2.7) 0,2 L min -1 -1 -1 El detector de mercurio en su parte posterior tiene dos entradas y una salida de gases. El analito llega a través de una manguera arrastrado mediante una corriente de argón, tal como se dijo anteriormente, este vapor de mercurio previamente debe pasar por la membrana higroscópica (Figura 2.6). La otra entrada de gas corresponde al Gas de escudo que cubre en vapor de mercurio que llega al detector evitando el “quenching” (Figura 2.7). 37 Figura 2.6. Esquema de la membrana higroscópica de Nafión para secar la humedad del vapor de mercurio antes de llegar al detector Esta membrana higroscópica, mostrada en la Figura 2.6, es de intercambio iónico y continuamente realiza el proceso de secado para remover selectivamente el vapor de agua de una mezcla de gases que pase por ella. Está hecha de un polímero fluorado denominado “Nafión” y se encuentra diseñada en forma de tubo. Además, está cubierta por otro tubo impermeable de mayor diámetro, el cual tiene dos aberturas que corresponden a la entrada del gas de secado (Ar) y la salida del vapor de agua extraído de la muestra. Si una muestra con vapor de mercurio entra húmeda a la membrana, el polímero fluorado absorberá selectivamente el vapor de agua debido a sus propiedades iónicas y lo transferirá a través de las paredes del tubo de nafión al exterior del mismo, para que finalmente sea arrastrado por el gas de secado y lo lleve al ambiente externo. Figura 2.7. Chimenea donde llega el vapor de mercurio seco cubierto por el gas de escudo. 38 Funciones de ganancia: Estos controles permiten aumentar la capacidad de detección del sistema. Es una combinación de dos factores: el rango y la ganancia fina. Permite amplificar la señal y de esta forma detectar concentraciones más bajas. Sin embargo, es necesario considerar que al aumentar la ganancia así como se amplifica la señal proveniente de la fluorescencia, también se amplifican las señales provenientes de ruido, por lo tanto si se amplifica demasiado la señal de entrada es muy probable que la señal de salida salga distorsionada, con mucho ruido, lo que disminuye la relación señal/ruido haciendo las mediciones imprecisas. Esto implica que existe un límite en el uso de esta función que no debe ser sobrepasado para poder realizar mediciones confiables. (PSAnalytical, Explanation of the gain funtions of the Merlin System, 1994) Cantidades de solución estándar de mercurio y agente reductor: Se quiso establecer la cantidad de cloruro estannoso necesario para completar la reducción del mercurio presente en una solución estándar. Para ello se realizaron distintas mediciones variando el volumen de agente reductor añadido a 1 mL de solución estándar a una concentración de 1,00 µg/L. 2.4.4. Instalación de la celda de reacción utilizada para la generación de vapor frío al detector de absorción atómica Tal como se muestra en el esquema de la Figura 2.8, la mayoría de los espectrómetros de absorción atómica contienen como fuente de radiación una lámpara de cátodo hueco para mercurio que fue alineada para obtener la mayor cantidad de radiación proveniente de la misma. El detector que posee este instrumento es un tubo fotomultiplicador. Para aislar la línea de mercurio de 253,7 nm el sistema consta de un monocromador donde se seleccionó un ancho de banda espectral de 0,7 nm. Figura 2.8. Esquema de un espectrómetro de absorción atómica. 39 Para las determinaciones por espectrometría de absorción atómica realizadas en el presente trabajo se utilizó un espectrómetro de absorción atómica Perkin Elmer Modelo P.E. 460 que mantiene el esquema mostrado anteriormente en la Figura 2.8 La adaptación de la celda de reacción en estudio al espectrofotómetro se esquematiza en la 2.11. La Figura 2.9 representa el reservorio de átomos de mercurio generados en la celda después de la reacción del analito con el agente reductor. El reservorio es un tubo plástico de 15 cm de largo con un diámetro de 3 cm, cerrado en ambos extremos mediante ventanas de cuarzo que permiten el paso de la radiación de la lámpara de cátodo hueco. El tubo permite la entrada y salida del flujo de gas que acarrea al Hg0 generado en la celda de reacción. Figura 2.9. Esquema del reservorio de átomos de Hg0. En la Figura 2.10 se muestra la adaptación de la base de un quemador de premezcla para colocar el tubo de plástico. El arreglo principal consistió en colocar una adaptación de metal que podía ser atornillada a la base del quemador de premezcla que se usa comúnmente para determinaciones por la técnica de atomización por llama. A esta adaptación se le instalaron unas pinzas de acrílico que permitían colocar fijamente y alinear el tubo de plástico en el camino óptico. Estas pinzas permiten también remover el tubo para ser limpiado cuando se desee. En la Figura 2.11 se muestra de forma global como están acoplados la celda de reacción y el tubo de plástico al espectrómetro de absorción atómica. Figura 2.10. Adaptación de la base de un quemador de premezcla para colocar y alinear el tubo de plástico en la zona de detección del espectrómetro. 40 Figura 2.11. Esquema del acoplamiento de la celda de reacción utilizada para la generación de vapor frío al espectrómetro de absorción atómica. 2.4.5. Condiciones experimentales para las mediciones por absorción atómica Utilizando el arreglo esquematizado en la Figura 2.11, se procedió, adoptando la opción y error, a definir las mejores condiciones instrumentales para las mediciones de Hg en estándares y muestras disueltas. En la tabla 2.5 se agrupan estas condiciones junto con el flujo óptimo de gas de arrastre, los tiempos de medición, el volumen de agente reductor y su concentración, y los volúmenes de estándar o muestra. Tabla 2.5. Condiciones experimentales mediante las cuales se hicieron las mediciones del vapor de mercurio por absorción atómica con el Espectrómetro Perkin Elmer 460. Variable Corriente de la lámpara Ganancia Longitud de onda Ancho de banda espectral Modo de lectura Tiempo de medición Flujo de gas de arrastre Volumen de estándar/muestra Volumen de solución reductora Concentración de solución reductora 41 Designación 12 mA 75% de la escala arbitraria 253,7 nm 0,7 nm (Normal) Continuo 50 s 1,96 L/min 2 mL 2 mL 2%m/v Para seleccionar las condiciones óptimas del gas de arrastre se hicieron ensayos con argón y con aire proveniente de un compresor. Se evaluó también la posibilidad de usar vacio para succionar el vapor de mercurio desde la celda de reacción hasta la zona de observación. Para esto se utilizó una válvula para creación de vacio mediante un flujo continuo de agua. La bomba de vacio con válvula antirretorno permite extraer las moléculas de aire que ingresan a la celda a través de la “Entrada de aire” mostrada en la Figura 2.12. Una vez ocurre la reacción mostrada en la ecuación (1) ó (2) los átomos de Hg0 generados también son arrastrados junto con el aire. Se realizó la determinación de la cantidad de flujo de gas de arrastre necesario para transportar el vapor de mercurio generado y realizar la determinación de este elemento por absorción atómica. En ella se demuestra que el valor ideal es 1,96 L/min del flujometro utilizado. Figura 2.12. Esquema del acoplamiento de la bomba de vacio con válvula antirretorno al del reservorio de átomos de Hg0. 42 CAPÍTULO 3 RESULTADOS Y DISCUSIONES 3.1. Evaluación de la celda de reacción para determinación de mercurio por generación de vapor frío y detección mediante fluorescencia atómica en muestras líquidas. 3.1.1. Optimización de las condiciones de medida Las optimizaciones mencionadas en forma general en el capítulo anterior se describen en detalle y se discuten en este capítulo. Se empezará por comparar la determinación de Hg en soluciones acuosas usando el sistema Merlin convencional y el sistema constituido por la celda de reacción que se propone en este estudio, acoplada al detector de fluorescencia del sistema Merlin. (Ver Figuras 2.4 y 2.5) Posteriormente se optimizaron las condiciones de análisis para la determinación de mercurio vía la generación de su vapor a partir de muestras sólidas. Dado que para esta fecha el equipo de fluorescencia dejó de funcionar correctamente y no hubo posibilidad de una rápida reparación, esta optimización y evaluación se hará acoplando la celda de reacción al detector de fluorescencia del equipo Merlin y a un espectrofotómetro de absorción atómica. Tiempos de análisis La imagen que es registrada en la pantalla de la computadora del sistema Merlin convencional corresponde a un gráfico de señal de fluorescencia, representada como “altura de pico” en el eje de las ordenadas, frente al tiempo en segundos en el eje de las abscisas. Tal como se aprecia en la Figura 3.1, se diferencian tres tipos de tiempo para la realización de una medición. Estos lapsos ya fueron definidos anteriormente (Ver Tabla 2.3) 43 Figura 3.1. Señal típica obtenida con el generador de vapor frío convencional. Es necesario cumplir las cuatro etapas en su totalidad para obtener resultados reproducibles. En la Figura 3.2 se presenta la señal típica que se obtiene usando el acoplamiento entre la celda de reacción bajo estudio NCR y el detector del sistema Merlin. Es evidente que esta última señal es mucho más sencilla que la anterior y se completa en la mitad del tiempo. En términos de promedios para mediciones de soluciones acuosas con concentración de mercurio alrededor de la zona central de las curvas de calibración para cada caso, la lectura convencional requiere 100 segundos mientras que la otra requiere sólo 50 segundos. Por lo tanto, usando el acoplamiento entre la celda de reacción bajo estudio y el detector del sistema Merlin se pueden realizar mediciones en la mitad del tiempo en comparación con el generador de vapor Merlin. Esto representa una gran ventaja ya que se duplica el número de análisis por unidad de tiempo. Figura 3.2. Señal típica obtenida con la celda de reacción NCR para la generación de vapor frío. 44 Flujos de Argón Tal como se explicó en el capítulo dos, el equipo convencional utiliza tres flujos de argón. Uno para secado, otro para arrastre y el tercero como protección “antiquenching”. Los ensayos realizados acoplando la celda de reacción al detector de fluorescencia, indicaron que los valores óptimos para dichos flujos son similares a los recomendados para la operación convencional. Por lo tanto, dichos valores no se modificaron, siendo los siguientes: Tabla 3.1. Condiciones instrumentales para los flujos gas utilizados en las mediciones. Propósito Φ (L min-1) Gas de secado 2,5 Gas de arrastre 0,5 Gas de escudo 0,2 Se pudo comprobar que para el caso del gas de arrastre a valores más pequeños, el vapor de mercurio generado no lograba llegar en su totalidad al detector, pues posiblemente se condensaba en el camino. Mientras que para valores mayores, el vapor de mercurio llegaba tan rápido que no era posible la detección ya que duraba muy poco tiempo en el camino óptico. Funciones de ganancia Las condiciones de amplificación de señal recomendadas por el fabricante para la operación convencional del sistema Merlin son: Rango (ganancia gruesa) 100 unidades y ganancia fina 5 unidades. Esta última tuvo que duplicarse cuando se acopló a la celda de reacción que estamos proponiendo ya que la sensibilidad para esta opción es menor que la correspondiente a la opción convencional. Una de las razones para esta diferencia en su sensibilidad es el hecho que en la opción convencional se trabaja a flujo continuo mientras que usando la celda el proceso es por cargas. A pesar que la ganancia gruesa puede ser aumentada al igual que la ganancia fina, no resultó conveniente una mayor ampliación en ninguno de los dos casos para evitar distorsión de la señal analítica. Con los valores mencionados se logran determinar cantidades de Hg en el orden de los microgramos con el sistema que proponemos. 45 Cantidades de patrón de Hg y cloruro estannoso utilizado En el Gráfico 3.1 se muestran los resultados obtenidos al reducir el mercurio presente en 1,0 mL de solución de Hg+2 conteniendo 1,00 µg/L del analito, utilizando como agente reductor una solución al 2% de cloruro estannoso. Se hicieron aumentos continuos del volumen de SnCl2 entre 0,1 y 4,0 mL. Gráfico 3.1. Optimización de la cantidad de agente reductor utilizado para la reducción de una solución de mercurio de 1,00 µg/L. En el Gráfico 3.1 se puede observar que para valores mayores a 1 mL de solución de SnCl2, la señal de fluorescencia representada como altura de pico disminuye considerablemente, es posible que esto se deba a que al aumentar el volumen de líquido presente en el envase que contiene al Hg2+ se hace más difícil extraer el vapor de mercurio generado en el seno de la solución. El líquido mismo actúa como una barrera que impide el arrastre el Hg0 formado. Por otro lado, a volúmenes menores entre 0,1 y 1 mL se mantienen valores parecidos de la señal de fluorescencia ya que el agente reductor dada su concentración siempre se encontrará en exceso con respecto al patrón de mayor concentración del analito correspondiente al valor máximo del intervalo lineal de las curvas de calibración. La medición de volúmenes de SnCl2 por debajo de 1 mL requiere mayor cuidado y del uso de micropipetas. Es por ello que se estableció que 1,0 mL de cloruro estannoso al 2% es suficiente para reducir la cantidad de mercurio presente en cualquiera de las soluciones estándar ya que el agente reductor siempre se encontrará en exceso y además este valor se puede medir fácilmente con una apreciación de hasta 0,1 mL. 46 3.1.2. Determinación de las figuras de mérito Las figuras de mérito y sus definiciones fueron tratadas en el primer capítulo de este libro. En esta sección se ahondará en los detalles experimentales para su determinación y se presentarán los resultados obtenidos. • Precisión Para su evaluación se utilizó 1 mL de solución de Hg+2 a una concentración de 1,00 µg/L, se realizaron 8 medidas consecutivas con las mismas condiciones experimentales, los resultados obtenidos se muestran en la Tabla 3.2. Tabla 3.2. Resultados obtenidos para el cálculo de la precisión del nuevo método analítico para la generación de vapor frío con la celda NCR y su detección vía fluorescencia atómica. Medición Altura de pico 1 19,90 2 20,50 3 20,90 4 19,90 5 20,30 6 20,10 7 19,90 8 20,00 Utilizando la ecuación 1.2 se obtuvo para la altura de pico un valor promedio de: N y y i i 1 20 ,19 N La desviación estándar del promedio se obtuvo utilizando la ecuación 1.3: N ( y s _ i y )2 i 1 N 1 47 0,91 Coeficiente de variación: El coeficiente de variación o desviación estándar relativa correspondiente a los ocho datos de altura de pico de la Tabla 3.2 y usando la desviación estándar obtenida antes se calculó utilizando la ecuación 1.4: CV s _ 100% 4 ,50% y Con este CV se refleja que el método es preciso bajo las condiciones de análisis establecidas. Otros autores que han empleando la técnica CV-AFS utilizando el generador de vapor P.S. Analytical LTD han encontrado resultados para la desviación estándar relativa entre 2,74 y 7,46 %, vemos que estos son comparables con los obtenidos en el presente trabajo. (Juanes J., 1996) Por lo tanto los errores cometidos son del tipo aleatorio o indeterminado. • Límite de detección del método (LDM) y Límite de cuantificación del método (LCM) Para los análisis realizados generando el vapor frío con la celda NCR y detectando dicho vapor por espectrometría de fluorescencia atómica se realizó el cálculo del límite de detección del método (LDM) mediante la ecuación 1.10. Se consideró como límite de detección a la concentración correspondiente a la limitación instrumental que corresponde a 10 partes por trillón, es decir 0,010 µg/L (PSAnalytical, PSA Introduces the 10.004 Hydride/Vapour Generator, 1997). Se preparó un solución que tuviese 8 veces esta concentración, es decir, 0,08 µg/L. Se tomaron 8 alícuotas de esta solución de mercurio, y para el cálculo se utilizó una t de Student de 2,998 que corresponde a siete grados de libertad: LDM S t ( 6 ,1 0 ,99 ) 0 ,016 g / L Mientras que el límite de cuantificación del método (LCM) que sería la menor cantidad de analito en la muestra que puede ser cuantificada con precisión y exactitud se calculó mediante la ecuación 1.11: LCM 10 S 0,055 g / L Estos valores se determinaron utilizando la ecuación de la curva lineal representada en el Gráfico 3.2 por el método de extrapolación de la respuesta a concentración cero. Se tomó como “y” a la 48 respuesta promedio reportada como fluorescencia mientras que “x” corresponde a la concentración en microgramos sobre litros. Según el límite de detección calculado con la celda, 0,016 µg/L, esta alternativa es menos sensible que la opcion convencional que permite límites de detección inclusive menores a 0,010 µg/L. Este resultado es consecuencia de una serie de factores que limitan la sensibilidad de las mediciones con la nueva celda. Entre los factores más importantes está el hecho que la nueva alternativa es un proceso por cargas mientras que en la opción convencional se trabaja con flujos continuos. Esta modalidad permite una mayor estabilización de la señal analítica en función del tiempo, lo cual redunda en mayor precisión de las mediciones. Como se ve en la Figura 3.2, la señal obtenida con la nueva celda es una señal transiente mucho más difícil de reproducir en el tiempo. De estas consideraciones se desprende que debe diseñarse una nueva celda que permita trabajar en la modalidad de flujo continuo. Adicionalmente, se pueden realizar otras mejoras tendientes a minimizar la posibilidad de condensación del analito en los volúmenes muertos de la celda evaluada. • Intervalo lineal Para la determinación del intervalo lineal se realizó el Gráfico 3.2 donde se representa la señal de fluorescencia representada como altura de pico frente a la concentración de mercurio. Se utilizaron disoluciones de mercurio a concentraciones entre 0,25 y 20,00 µg/L, que fueron preparadas a partir de diluciones de la solución madre de Hg (FIXANAL). Para esta etapa de calibración se utilizó el modelo lineal, que según la ecuación 1.12 se define como y mx b , donde “y” es la variable dependiente es decir, la respuesta del equipo, que en este caso será la señal de fluorescencia representada como altura de pico; “x” es la variable independiente que para este caso corresponde a la concentración de mercurio reportada en µg/L o partes por billón (ppb). Mientras que “m” es la pendiente de la recta calculada según el método de los mínimos cuadrados (ecuación 1.13) que mostrará la sensibilidad del método. “b” es el intercepto. El resultado de los primeros ensayos donde se cubrió el intervalo entre 0,25 y 20,00 µg/L de Hg, no es muy ilustrativo se puede apreciar que la linealidad, estrictamente hablando, se comienza a perder a partir de los 5,00 µg/L. Se obtuvo entonces un gráfico considerando el intervalo entre 0,25 y 5,00 µg/L. Tal como se ve en la página 50 este nuevo gráfico si demuestra que el intervalo hasta 4,00 µg/L es estrictamente lineal. 49 Gráfico 3.2. Intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. Para validar el modelo lineal propuesto se utilizó inicialmente el valor arrojado por el coeficiente de determinación (r2) calculado a partir del cuadrado de la ecuación 1.15. El intervalo lineal entre (0,25 a 4,00) µg/L, está definido por la ecuación y = 2,05E+01x-7,09E-01 con un coeficiente de determinación R2 = 9,98E-01. Conjuntamente con la determinación del intervalo lineal se realizó un análisis visual del gráfico de los residuales. Vemos que el modelo lineal propuesto es apropiado ya que en el Gráfico 3.3 se observa que existe aproximadamente el mismo número de residuales positivos y negativos, Estos están distribuidos aleatoriamente y tiene aproximadamente el mismo valor absoluto y además no muestran tendencia alguna. Gráfico 3.3. Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. 50 La última herramienta estadística utilizada para asegurar la validez del modelo lineal es el análisis de varianza (ANOVA). Para ello fue necesario hacer tres replicados por cada punto experimental. Se planteó que bajo la hipótesis de que existe una relación lineal entre la variable respuesta (y) y la independiente (x), se puede realizar el siguiente contraste: H o : y b , H 1 : y b mx Tabla 3.3. ANOVA del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. Fuente de Variación SS df MS F P-valor F crit Entre grupos 9,46x1004 8 1,18 x1004 2,21 x1004 1,87 x10-49 2,30 Dentro de grupos 14,4 27 0,540 Total 9,46 x1004 35 Según la Tabla 3.3, la prueba de hipótesis F nos dice que existe alguna relación entre las variables dependiente e independiente es decir, la x si influye y se rechaza Ho, ya que el valor calculado de F es mayor que el Fcrit para un de 0,05. Esto avala el uso de la expresión propuesta antes. • Exactitud La exactitud, que mide el error sistemático o determinado del método analítico, se evaluó preparando una solución de mercurio Hg, a partir de la solución madre FIXANAL, para tener una concentración final de 1,50 µg/L. Esta solución se “analizó”, es decir que se tomó como muestra para determinar su contenido de Hg. Esta determinación se llevó a cabo en paralelo con la determinación del intervalo lineal cuyos resultados se representaron en el Gráfico 3.2. El mismo gráfico ahora con el valor obtenido para la solución de 1,50 µg/L. de Hg, se presenta como Gráfico 3.4. Para tener una estimación de la exactitud del método se comparó el valor obtenido (x), a partir de la recta de calibración, con el valor de referencia (µ). Se encontró para “y” un valor de 29,80 en altura de pico. De la curva de calibración se sabe que la pendiente “m” es 2,05E+01, mientras que el intercepto es -7,09E-01. Por lo tanto, “x” que es la variable independiente será: x y b 1,49 g / L m 51 Se obtuvo un intervalo de confianza, al 95%, del valor encontrado de ±0,18 µg/L. Lo cual significa que 5 de cada 100 veces, la media experimental puede que se desvíe ±0,18 µg/L. El sesgo del método que es la diferencia entre el valor verdadero (aceptado) y el valor encontrado, se determinó a partir de la ecuación 1.1, se sabe que el valor aceptado es 1,50 µg/L mientras que el valor encontrado es 1,49 µg/L . Entonces, el sesgo será S x 0,01g / L Otra alternativa para evaluar la exactitud del método es calculando el porcentaje de recuperación. Este fue calculado según la ecuación 1.19, usando los valores obtenidos para x y el valor de concentración de la solución preparada de mercurio. %R x 100 99,3% Los valores obtenidos para el sesgo y el porcentaje de recuperación evidencian que el método propuesto para generar vapor frio de mercurio utilizando la celda NCR es capaz de medir un valor muy cercano al valor real, es por ello que se demuestra la exactitud de dicho método. Gráfico 3.4. Representación gráfica para el estudio de la exactitud del nuevo método para determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia atómica. 52 3.2. Evaluación de la celda de reacción NCR para determinación de mercurio por generación de vapor frío y detección mediante absorción atómica en muestras líquidas y sólidas. La celda de reacción se acopló a un espectrómetro de absorción atómica y se llevaron a cabo varias mediciones bajo las condiciones especificadas en la tabla 3.4. Tabla 3.4. Condiciones instrumentales de análisis para determinación de mercurio mediante vapor frío usando la celda de reacción. Variable Condición Longitud de onda 253,7 nm. Ganancia 75% de la escala en unidades arbitrarias. Corriente de la lámpara de cátodo hueco marca Perkin Elmer para análisis de mercurio. 15 mA. Apertura de la rendija (slit) 0,7 nm. Modo de lectura Contínuo. Como el modo de lectura utilizado fue el modo contínuo, se observó la señal de absorción hasta que se alcanzara su máximo valor, una vez que esto ocurre se considera completa la etapa de reducción del mercurio y el proceso de transferencia de todo el mercurio cero Valente hacia la zona de detección. El tiempo necesario para que la señal alcanzara un máximo osciló entre 20 y 50 segundos dependiendo de la concentración de la solución del analito. Gas de arrastre Se quiso comparar tres formas de llevar el vapor de mercurio hacia la zona de detección. Inicialmente se usó Argón que es el gas recomendado en la manera convencional. Luego se ensayó con aire proveniente de un compresor y finalmente se aplicó vacío con una bomba de flujo contínuo de agua basada en el efecto Venturi con la intención de succionar el vapor de mercurio formado en la celda de reacción. Para la optimización en los dos primeros casis se observó la señal de absorción atómica a medida que se varió el flujo de Argón o de aire. Los flujos en ambos casos fueron regulados mediante un 53 flujometro ubicado a la salida de la bombona, en el caso del Argón, y en la salida del compresor en el caso del aire a presión. Para la succión con vacío se reguló el flujo de agua que pasa a través de la bomba, lo que a su vez regula la extensión del vacío que se aplica a la salida del tubo de plástico que finge como reservorio de átomos (Ver Figura 2.12). Los resultados se sumarizan en el Gráfico 3.5. Es evidente que la mayor sensibilidad, mayor lectura de absorbancia, se alcanza a lecturas del flujómetro de 1,96 L/min. Es posible que a flujos menores no llegue todo el vapor frío de mercurio a la zona de detección ya que probablemente parte se condensa. Mientras que a flujos mayores, el vapor frío de mercurio atraviesa muy rápido la zona de detección, lo que implica un menor tiempo de exposición de los átomos a la radiación y por ende una menor eficiencia en la absorción. El flujo de gas de 1,96 L/min transporta eficientemente los átomos y les permite suficiente tiempo de excitación, maximizando así la señal de absorbancia. Gráfico 3.5. Representación gráfica para la escogencia del flujo de gas de arrastre utilizado en el nuevo método para determinación de mercurio utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Con este valor de flujo de gas de arrastre se procedió a evaluar el comportamiento de la señal con cada gas. En el Gráfico 3.6 se muestran los resultados obtenidos. Estos resultados avalan las conclusiones que pueden derivarse del ensayo anterior, representado en le gráfico 3.6. 54 Gráfico 3.6. Representación gráfica para la escogencia del gas de arrastre para determinación de Hg utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. El Gráfico 3.6 indica que la señal de absorbancia es mayor cuando se arrastra el mercurio con aire a presión que cuando se hace con argón a presión. Adicionalmente, estos experimentos permitieron definir un intervalo estrictamente lineal comprendido entre 10,00 y 120,00 µg/L de Hg, Otras investigaciones (Qiuju Yang, 2005) han reportado que es mejor el uso de aire comprimido como gas de arrastre que el uso de argón debido a que el aire tiene un peso molecular más pequeño mayor cantidad de partículas de menor peso, así hay un arrastre más eficiente de mayor cantidad de átomos de mercurio en fase gaseosa. Por lo tanto, el uso de aire comprimido permite mejoras en la sensibilidad y además abarata los costos de las mediciones. Agente reductor Para generar vapor frío de mercurio se utiliza comúnmente como agente reductor borohidruro de sodio o cloruro estannoso. Se quiso estudiar la diferencia que pudiese existir en los resultados al utilizar cualquiera de estos reductores, ya que el borohidruro de sodio se encuentra en medio básico mientras que el cloruro estannoso se encuentra en medio ácido. En el Gráfico 3.7 se muestran dichos resultados. 55 Gráfico 3.7. Representación gráfica para la escogencia del agente reductor utilizado en el nuevo método para determinación de mercurio utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Como puede observarse en el Gráfico 3.7 no existe una diferencia significativa, en términos de sensibilidad, al utilizar como agente reductor borohidruro de sodio o cloruro estannoso. Investigaciones afirman que si la solución de Hg2+se encuentra en medio ácido, la velocidad de la reacción entre Hg2+ y NaBH4 aumenta. Esto pareciera ser favorable ya que se forman inmediatamente átomos de mercurio. Sin embargo, como ocurre la formación de clusters atómicos y partículas coloidales, estas no permiten que se liberen fácilmente los átomos de mercurio reduciendose la sensibilidad de las mediciones (Henglein A., 1991). Los autores recomiendan realizar determinaciones con la solución de Hg2+ en medio básico. Preparación de la muestra sólida Dilución Sólido-Sólido Se diluyó la muestra con SiO2 en una proporción 1:2. No se encontraron resultados precisos debido a que la granulometría del sedimento no es la misma que la del SiO2. Por lo tanto se procedió a medir sin diluir. 56 Sin Dilución Se midió directamente sobre la muestra sólida sin diluir. Se encontró respuesta pero muy baja sensibilidad. En suspensión ácida Se probó hacer una lixiviación con una solución ácida: Se realizaron mediciones con ácido clorhídrico, para 4 mL de ácido no se lograba estabilizar la señal debido a que los vapores de ácido empañaban las ventanas de cuarzo. Se redujo a 1 mL de ácido se evitó que las ventanas de cuarzo de la celda se empañaran. Sin embargo, se encontraban señales dispersas e inexactas. Se cambió por ácido nítrico, observándose que con 4 mL de ácido también se presentó el mismo problema con los vapores de ácido que empañaban las ventanas de cuarzo. Con 1 mL de ácido, se encontró reproducibilidad y exactitud en las mediciones por lo tanto con este procedimiento se realizaron las optimizaciones del método. 3.2.1. Determinación de las figuras de mérito para muestras líquidas • Precisión Con el objeto de estudiar la precisión del nuevo método de generación de vapor frío de mercurio, en muestra líquida y detección vía absorción atómica, se colocaron en la celda de reacción 2 mL de solución de 10,00 µg/L de Hg para cada medición y se realizaron 15 medidas consecutivas bajo las mismas condiciones experimentales que se establecieron según lo explicado anteriormente. Los resultados obtenidos se muestran en la Tabla 3.5. Tabla 3.5. Resultados obtenidos para el cálculo de la precisión del nuevo método para la generación de vapor frío en muestras líquidas con la celda NCR y su detección vía absorción atómica. Medición 1 2 3 4 5 Absorbancia 0,031 0,049 0,042 0,028 0,027 57 6 7 8 9 10 11 12 13 14 15 0,021 0,038 0,042 0,037 0,048 0,028 0,020 0,022 0,046 0,029 Utilizando la ecuación 1.2 se obtuvo para la absorbancia un valor promedio de: N y i i 1 y 0,034 N La desviación estándar del promedio se obtuvo utilizando la ecuación 1.3: N ( y s _ i y )2 i 1 N 1 0,0014 El coeficiente de variación o desviación estándar relativa correspondiente a los quince datos de absorbancia se calculó utilizando la ecuación 1.4: CV s _ 100% 4 ,08% y Con este CV se refleja que el método es preciso bajo las condiciones de análisis establecidas. Por lo tanto los errores cometidos son del tipo aleatorio o indeterminado. Existen investigaciones recientes donde reportan coeficientes de variación mayores para métodos modificados en la determinación de mercurio por CV-AAS. Silva y colaboradores reportan un coeficiente de variación de 4% para la determinación de mercurio (II) a una concentración de 0,20 µg/L . Esta concentración es muy pequeña por lo que existe mayor probabilidad de que existan errores del tipo aleatorio. Mientras que en este trabajo, si se observa el coeficiente de variación, se puede decir que se obtuvo mayor imprecisión pues esta concentración cincuenta veces mayor y debería obtenerse un coeficiente de variación menor. (Silva F. A., 2005) 58 • Límite de detección Para determinar la menor cantidad de analito que puede ser detectado o diferenciado de la señal de fondo, en determinaciones realizadas generando el vapor frío con la celda NCR y detectando el Hg en dicho vapor por espectrometría de absorción atómica, se realizó el cálculo del límite de detección (LD) calculado como tres veces la desviación estándar de medidas del blanco más el promedio de dichas mediciones (ecuación 1.8): LD y B 3s B 4 ,91g / L Mientras que el límite de cuantificación (LC) que sería la menor cantidad de analito en la muestra que puede ser cuantificada con precisión y exactitud se calculó mediante la ecuación 1.9: LC y B 10 s B 6, 26 g / L Estos valores se determinaron utilizando la ecuación de la curva lineal del Gráfico 3.8 por el método de extrapolación de la respuesta a concentración cero. Se tomó como “y” a la respuesta promedio reportada como absorbancia mientras que “x” corresponde a la concentración en microgramos sobre litros. Se puede notar que los límites de detección obtenidos son considerablemente mayores que los que se obtuvieron para la determinación de mercurio vía espectrometría de fluorescencia atómica. Esto se debe a que la fluorescencia siempre ha tenido la ventaja de tener límites de detección menores comparada con la absorción, ya que en fluorescencia se utilizan fuentes (lámparas) con una radiación más intensa que consecuentemente producirá señales más intensas para la misma cantidad de analito, mientras que en absorción se utilizan lámparas de cátodo hueco que tienen intensidad relativamente baja. (Olsen, 1990) Intrinsecamente la fluorescencia puede hacerse más sensible que la absorción ya que ella depende directamente de la intensidad de la radiación que excita a los átomos. Las lámparas de excitación utilizadas en fluorescencia producen radiación de mayor intensidad que las utilizadas en absorcióna tómica debido a que en este proceso no existe una dependencia lineal entre la intensida de la radiación de la lámpara de cátodo hueco y la absorción producida. Sin embargo, se observa que con la celda de reacción NCR se obtienen límites de detección más altos que los obtenidos con otros sistemas de generación de vapor frío como el MHS10, con el que 59 se han obtenido límites de detección para mercurio en agua de 0,10 µg/L. (Cabrera-Vique R.-L. J., 2007) • Intervalo lineal En forma similar a lo ya descrito, se determinó el intervalo lineal, los residuales correspondientes y se realizó un estudio ANOVA. Los gráficos 3.8, 3.9, y la Tabla 3.6 recopilan los resultados Gráfico 3.8. Intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Gráfico 3.9. Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 60 Tabla 3.6. ANOVA del intervalo lineal para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Fuente de Variación SS df MS F P-valor F crit Entre grupos 7,94x1004 1 7,94 x1004 13,0 4,74 x10-03 4,96 Dentro de grupos 6,08 x1004 10 6,08 x1003 Total 14,0 x1004 11 Según la Tabla 3.6, la prueba de hipótesis F nos dice que existe alguna relación entre las variables dependiente e independiente es decir, la x si influye y se rechaza Ho, ya que el valor calculado de F es mayor que el Fcrit para un de 0,05. Al analizar los resultados obtenidos, vemos que el intervalo lineal es más amplio para la determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía CV-AAS en comparación con la detección vía CV-AFS, esto representa una ventaja, pues facilita la determinación de este elemento a distintas concentraciones. Sin embargo, existe una clara desventaja, y es que al realizar la detección vía CV-AAS se tiene un límite de cuantificación considerablemente mayor que detectando vía CV-AFS, lo cual restringe al método pues no permite cuantificar a concentraciones por debajo de 10,00 µg/L, y desde el punto de vista analítico, los nuevos métodos siempre buscan llegar a límites de cuantificación cada vez más pequeños. Las razones para la diferencia entre estos límites de detección fue ya discutido anteriormente. En forma similar a lo ya expuesto, se determinó la exactitud, el sesgo, el porcentaje de recuperación para el método. Los valores se sumarizan en la Tabla 3.9 al finalizar este capítulo y en el Gráfico 3.10. 61 Gráfico 3.10. Representación gráfica para el estudio de la exactitud del nuevo método para determinación de mercurio en muestras líquidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica 3.2.2. Determinación de las figuras de mérito para muestras sólidas. Precisión Con el objeto de estudiar la precisión del nuevo método de generación de vapor frío de mercurio en muestra sólida y detección vía absorción atómica, se empleó un Material Estándar Certificado de Referencia de nombre PACS-2. Este material es un sedimento marino pulverizado y es comercializado por la National Research Council Canada. Se utilizaron aproximadamente 0,0109 g de este sedimento marino que equivale a 33,2 ng de mercurio. Se realizaron 15 medidas consecutivas bajo las mismas condiciones experimentales que se establecieron según lo explicado anteriormente. Los resultados obtenidos se muestran en la Tabla 3.7. Antes de analizar la muestra sólida, a esta se le agrega 1 mL de HNO3, se tapa con papel parafilm y se deja reposar por 10 minutos. Una vez transcurrido el tiempo se procede a medir. 62 Tabla 3.7. Resultados obtenidos para el cálculo de la precisión del nuevo método analítico para la generación de vapor frío en muestras sólidas con la celda NCR y su detección vía absorción atómica. Masa de mercurio (ng) Absorbancia 31,6160 0,024 34,3520 0,014 32,5280 0,028 33,7440 0,029 31,0080 0,017 34,3520 0,010 33,1360 0,007 35,5680 0,013 30,7040 0,027 34,6560 0,011 29,7920 0,028 33,4400 0,026 34,9600 0,008 31,9200 0,005 36,1760 0,010 Utilizando estos valores se determinó el promedio, su desviación estándar, el coeficiente de variación. El límite de detección y el límite de cuantificación se determinó utilizando arena lavada de mar como blanco. Además se determinó el intervalo lineal y los correspondientes residuales y se realizó también un estudio ANOVA. Estos resultados se agrupan en la Tabla 3.8 y en los gráficos 3.11, 3.12, y en la Tabla 3.9. En el Capítulo I se mencionó que para el análisis de muestras sólidas existen métodos para determinar mercurio directamente sobre la muestra sin digerirla previamente. Estos están basados en la combustión total de la muestra para volatilizar el mercurio como oxido, pasarlo a través de una solución reductora y amalgamar el mercurio libre en una malla de oro. Luego, al calentar la amalgama el Hg0 se desprende y se detecta mediante absorción atómica. Aunque es posible alcanzar límites de 63 detección más bajos que los obtenibles con el método que aquí se propone, la aplicación de esos métodos requiere el uso de equipos sumamente costosos en obtención, obtención y mantenimiento, tal como también se mencionó con anterioridad. (Cizdziel James V, 2010) Gráfico 3.11. Intervalo lineal para la determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Gráfico 3.12. Residuales para el establecimiento del intervalo lineal para la determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 64 Tabla 3.8. ANOVA del intervalo lineal para la determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. Fuente de Variación SS df MS F P-valor F crit Entre grupos 2,45 x1005 1 2,45 x1005 27,7 7,59 x10-04 5,32 Dentro de grupos 7,08 x1004 8 8,84 x1003 Total 3,16 x1005 9 Gráfico 3.13. Representación gráfica para el estudio de la exactitud del nuevo método para determinación de mercurio en muestras sólidas utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica. 3.2.3. Comparación de la sensibilidad del método en muestras líquidas y sólidas. Con el fin de saber en qué tipo de muestra el método tiene mayor capacidad para discernir pequeñas variaciones en la cantidad de analito, se quiso comparar la sensibilidad obtenida para determinación de mercurio en muestras líquidas y sólidas. 65 Se obtuvo para muestras líquidas una mayor sensibilidad que para muestras sólidas, entendida esta como el cambio en la respuesta de un instrumento de medición dividido por el cambio de la concentración de analito. Como el modelo propuesto al que se ajustan los datos es un modelo lineal, esta aseveración se hizo teniendo como criterio las pendientes de las rectas de acuerdo al Gráfico 3.14. Era de esperarse que la sensibilidad fuese menor en muestras sólidas, pues con el proceso de lixiviación no se puede garantizar que se logra extraer por completo el mercurio presente en la muestra, mientras que con una digestión exhaustiva para llevar la muestra a solución, la extracción total del analito hacia la solución está garantizada. Gráfico 3.14. Representación gráfica que permite comparar la sensibilidad del nuevo método para determinación de mercurio en muestra sólida y líquida utilizando la celda NCR para la generación del vapor frío y detección vía absorción atómica 66 Tabla 3.9. Resumen de los logros obtenidos en las determinaciones de mercurio en muestra sólida y líquida utilizando la celda NCR para la generación del vapor frío y detección vía fluorescencia y absorción atómica. LD (ng Hg0) LC (ng Hg0) CV (%) Sesgo (ng Hg0) %R Intervalo lineal (ng Hg0) NCR-EFA Muestra Líquida 0,016 0,055 4,50 0,01 99,3 0,25 - 4,00 NCR-EAA Muestra Líquida 9,82 12,52 4,08 7,20 92,7 20,00-100,00 NCR-EAA Muestra Sólida 9,52 10,97 6,42 5,15 104,77 33,60-156,7 67 CONCLUSIONES El estudio realizado permite concluir que: La celda evaluada es apropiada para generar vapor atómico de mercurio en muestras líquidas y sólidas. Su acople a un detector de fluorescencia atómica permite realizar la detección de Hg a concentraciones por encima de 0,05 µg/L, para muestras líquidas. Mientras que su acople a un detector de absorción atómica permite realizar la detección de Hg a concentraciones por encima de 6 µg/L y para muestras sólidas por encima de 9,52 ng de mercurio. Tanto para fluorescencia como para absorción atómica estos valores se obtienen con suficiente exactitud y precisión. Las determinaciones de Hg usando esta celda se pueden realizar en menor tiempo, con menor gasto de reactivos y muestra, que con generadores convencionales. La laboriosidad de la detección y la posibilidad de contaminación se reducen si se opta por el análisis de muestras sólidas. Con el uso de esta celda se elimina la necesidad de recurrir a equipos costosos, sofisticados y complejos (tales como el MHS-10, el Merlin, el AVANTA, etc) que dependen de partes movibles (bombas peristálticas), partes consumibles (mangueras especiales) que han de reemplazarse con relativa frecuencia y que son difíciles de conseguir en nuestro país. Aunque este estudio se limitó al acoplamiento de esta celda a detectores por fluorescencia o por absorción atómica, la misma podría ser acoplada a otros sistemas de detección tal como un espectrómetro de emisión mediante plasma inductivamente acoplado ICP. 68 RECOMEDACIONES Diseñar una nueva celda que permita realizar determinaciones en forma continua y no por cargas. Esto permitiría incrementar la sensibilidad de las mediciones. Se recomienda el uso de esta celda para el análisis de muestras sólidas en aquellos casos donde se quiera conocer la cantidad de Hg extraíble mediante lixiviación y no el contenido total del mismo en la muestra. En este caso se recomienda el uso de la celda para análisis de muestras líquidas o sólidas disueltas. Se recomienda el acople de la celda a un espectrofotómetro de absorción atómica su las concentraciones de mercurio en las muestras está por encima de 10 µg/L. en caso contrario se recomienda el acople de la celda a un detector de fluorescencia. 69 BIBLIOGRAFÍA Akira Iwashita, S. T. (2004). Removal of mercury from coal by mild pyrolysis and leaching behavior of mercury. Fuel , 631-638. Beaty, R. (1979). Conceptos, Instrumentación y Técnicas de Espectrofotometría por Absorción Atómica. Estado Unidos de America: Perkin Elmer Corporation. Boqué, R. A. (2004). El Análisis de Varianza (ANOVA). Recuperado el 12 de 08 de 2010, de Grupo de Quimiometría y Cualimetría. Universitat Rovira i Virgili.: http://www.quimica.urv.es/quimio/general/anovacast.pdf Boqué, R. (2004). El límite de detección de un método analítico. Técnicas de Laboratorio296. Recuperado el 24 de 06 de 2010, de Grupo de Quimiometría y Cualimetría Universitat Rovira i Virgili: http://www.quimica.urv.es/quimio/cast/maincat.html Bothner, M. R. (1978). Mercury contamination of sea water samples stored in polyethylene containers. Analytical Chemistry , 47, 592-595. Bryan, G. L. (1992). Environmental Pollution , 76, 89-131. Cabrera-Vique, R.-L. J. (2007). Mercury in Waters from South-eastern Spain: Possible Sources of Pollution. Ars Pharm , 48, 37-53. Cabrera-Vique, R.-L. M. (2007). Mercury in Waters from South-eastern Spain: Possible Sources of Pollution. Ars Pharmaceutica , 48, 37-53. Chilov, S. (1975). Determination of small amounts of mercury. Talanta , 22, 205. Cizdziel James V, T. C. (2010). Direct analysis of environmental and biological samples for total mercury with comparison of sequential atomic absorption and fluorescence measurements from a single combustion event. Spectrochimica Acta Part B , 65, 176-180. Danzer, K. (1998). Guidelines for Calibration in Analytical Chemistry. En Part 1. Fundamentals and Single Component Calibration (Vol. 70, págs. 993-1014). Gran Bretaña: International Union of Pure and Applied Chemistry. 70 Eurachem. (1998). The Fitness for Purpose of Analytical Methods. A laboratory guide to method validation and related topics. Recuperado el 04 de 09 de 2010, de Eurachem: http://www.eurachem.org/guides/html/mval.htm Garrido I., R. M.-M. (2001). FLAME ATOMIC ABSORPTION SPECTROMETRY WITH FLOW-INJECTION ON-LINE ADSORPTION PRECONCENTRATION USING A KNOTTED REACTOR FOR CADMIUM DETERMINATION IN AQUEOUS SAMPLES. Analytical Letters , 34, 1763. Gochfeld, M. (2003). Cases of mercury exposure, bioavailability, and absorption. Ecotoxicology and Environmental Safety , 56, 174–179. Hafez M. A. H., K. I. (2001). Preconcentration and separation of total mercury in environmental samples using chemically modified chloromethylated polystyrene-PAN (ionexchanger) and its determination by cold vapour atomic. Talanta , 53, 749. Halbach, S. (2008). Blood and urine mercury levels in adult amalgam patients of a randomized controlled trial: Interaction of Hg species in erythrocytes. Environmental Research , 107, 69– 78. Harada, Y. (1968). Congenital (or Fetal) Minamata Disease. In: Study Group of Minamata Disease, editor. Minamata Disease. Kumamoto: Kumamoto University. Henglein A., M. P. (1991). Chemistry of Agn aggregates in aqueous solution: non-metallic oligomeric clusters and metallic particles. Faraday Discuss , 92, 31-44. ISO, 5. (1994). Precision of Test Methods. Geneva. Joaquim A. Nóbrega, L. C. (2002). Focused-microwave-assisted strategies for sample preparation. Spectrochimica Acta Part B: Atomic Spectroscopy , 57 (12), 1855-1876. Jong., J. (. (11 de Octubre de 2010). SPE. Recuperado el 02 de Noviembre de 2010, de SPE: http://netherlands.spe.org/images/netherlands/articles/94/Okt%2011%202010%20Jan%20de%20 Jong%20-20Gulf%20of%20Mexico%20Follow%20up%20status%20in%20the%20Netherlands.pdf 71 Jorge Moreda-Piñeiro, C. M.-P.-M. (2001). Multivariate optimisation of hydride generation procedures for single element determinations of As, Cd, Sb and Se in natural waters by electrothermal atomic absorption spectrometry. Talanta , 53, 871-883. Juanes J. (1996). Evaluación de técnicas de análisis para la determinación de mercurio en muestras naturales. Sartenejas: Universidad Simón Bolívar. Landi, S., & Fagioli, F. (1994). The adaptation of the dichromate digestion method for total mercury by cold-vapour atomic absortion spectrometry to the analysis of soils, sediments and sludges. Analytica Chimica Acta , 298, 363-374. Li, P. (2008). Mercury exposures and symptoms in smelting workers of artisanal mercury mines in Wuchuan, Guixhou, China. Environmental Research , 107, 108–114. López, V. M. y J. H. Brineman Jr. (1943.). Estudio geológico y minero del yacimiento de mercurio de San Jacinto, estado Lara. 5(50), 29-61. María Liva Garrido, R. M.-O. (1998). Determination of cadmium in aqueous media by flow injection cold vapour atomic absorption spectrometry. Application to natural water samples. Journal of Analytical Atomic Spectrometry , 13, 295. Miller, J. M. (2002). Estadística y Quimiometría para Química Analítica. Madrid: Pearson Educación. Morita, H., Tanaka, H., & Shimomura, S. (1995). Atomic fluorescence spectrometry of mercury: principles and developments. Review. Spectrochimica acta 50B , 1, 69. Navarro, A. (28 de 07 de 2009). Análisis de regresión. Recuperado el 15 de 07 de 2010, de Universidad Tecnológica de Izúcar de Matamoros: http://www.utim.edu.mx/~navarrof/Docencia/MatematicasIII/M3UT7/regresion_lineal.htm Nielsen J.B., G. P. (2000). Mercury. Environmental toxicants: Human exposures and their health effects. Chichester, UK: Wiley Interscience. Nixon D. E., B. M. (1999). The determination of mercury in whole blood and urine by inductively coupled plasma mass spectrometry. Spectrochimica Acta, Part B , 54, 1141. 72 OAA, O. A. (2003). Guía para la validación de métodos de ensayo. OAA.DC-LE-05. Olsen, E. D. (1990). Métodos Ópticos de Análisis. España: Editorial Reverté. P, N. J. (2000). Mercury. In: Lippmann M. .Environmental toxicants: Human exposures and their health effects (Segunda Edición ed.). Chichester, UK: Wiley Interscience. Pardi, G. P. (1974). Estudio integral de la cuenca del Rio Yaracuy. Caracas: Dirección de Malariologia y Saneamiento Ambiental M .S.A.S. Petroleum, B. (08 de Septiembre de 2010). British Petroleum . Recuperado el 01 de Noviembre de 2010, de www.bp.com: http://www.bp.com/liveassets/bp_internet/globalbp/globalbp_uk_english/incident_response/ST AGING/local_assets/downloads_pdfs/Deepwater_Horizon_Accident_Investigation_Report.pdf PSAnalytical. (1994). Method for total mercury in drinking, surface, ground, industrial & domestic waste waters and saline waters. PSAnalytical. (1994). Explanation of the gain funtions of the Merlin System. PSAnalytical. (1997). Manual Merlin PSA 10.023. Method for mercury in sludge, soils and sediments. PSAnalytical. (1997). PSA Introduces the 10.004 Hydride/Vapour Generator. Qiuju Yang, Q. T. (2005). Direct detection of mercury in vapor and aerosol from chemical atomization and nebulization at ambient temperature: exploiting the flame atomic absorption spectrometer. Journal of Analytical Atomic Spectrometry , 20, 760-762. Rahman L., C. W. (2000). Determination of mercury, selenium, bismuth, arsenic and antimony in human hair by microwave digestion atomic fluorescence spectrometry. Talanta , 52, 833. Ripp, J. (04 de 1996). Guidance. Recuperado el 05 de 09 de 2010, de Wisconsin Departament of Natural Resources: http://www.dnr.state.wi.us/org/es/science/lc/outreach/- publications/lod%20guidance%20document.pdf Shigeo Ekino, M. S. (2007). Minamata disease revisited: An update on the acute and chronic manifestations of methyl mercury poisoning. Journal of the Neurological Sciences , 262, 131-144. 73 Silva F. A., I. L. (2005). Determination of Hg in water by CVAAS using 2-aminothiazole modified silica. Ecletica Quimica , 30, 47-55. Skoog D, W. D. (2005). Fundamentos de Química Analitica. (Octava ed.). Madrid: THOMSON PARANINFO. Stockwell P. B., C. W. (1994). Environmental sensors based on atomic fluorescence. Analyst , 119, 1641. Tsalev, D. L. (1995). Atomic Absorption Spectrometry in Occupational and Enviromental Health Practice (Vol. III). Estados Unidos de América: CRC Press. United States Environmental Protection Agency. (1997). Mercury Study Report to Congress. Volume I: Executive Summary. EPA-452/R-97-003. Valde´s-Hevia M. C., T. M.-M. (1993). Sensitive method for determination of lead by potassium dichromate–lactic acid hydride generation inductively coupled plasma atomic emission spectrometry. Journal of Analytical Atomic Spectrometry , 8, 847. Winfrey, M. a. (1990). Review -- Environmental Factors Affecting the Formation of Methylmercury in Low pH Lakes. Environmental Toxicology & Chemistry , 9, 853-869. 74