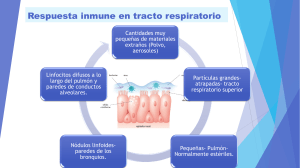
FÚNGICA INVASORA PATOGÉNESIS DE LA ENFERMEDAD
Anuncio

PATOGÉNESIS DE LA ENFERMEDAD FÚNGICA INVASORA Carlos Humberto Saavedra*, Pilar Rivas**, Javier Pemán*** Colaboración: Guillermo Quindós*** *Departamento de Medicina Interna, Facultad de Medicina, Universidad Nacional de Colombia; Servicio de Infectología, Hospital Universitario Clínica San Rafael (Bogotá, Colombia) ** Grupo de Micología Médica y Diagnóstica, Facultad de Medicina, Universidad Nacional de Colombia; Micología Médica, Microbiología, Hospital Central de la Policía (Bogotá, Colombia) ***Unidad de Micología, Servicio de Microbiología Clínica, Hospital Universitario y Politécnico La Fe (Valencia, España) ****Unidad de Formación e Investigación 11/25 " Microbios y Salud", Departamento de Inmunología, Microbiología y Parasitología, Facultad de Medicina y Odontología, Universidad del País Vasco-Euskal Herriko Unibertsitatea (UPV/EHU) (Bilbao, España) Los pacientes inmunodeprimidos o críticamente enfermos son más susceptibles a las enfermedades fúngicas invasoras. El conocimiento de la capacidad patógena de los hongos implicados, así como el tipo de respuesta inmune del paciente frente al proceso infeccioso son herramientas imprescindibles para el conocimiento y manejo de estas enfermedades infecciosas. Aunque solo una mínima proporción de hongos son capaces de producir enfermedad infecciosa, estos pueden presentar diferentes factores de virulencia que les permiten desarrollar su capacidad invasora sobre todo cuando los mecanismos de defensa inmune del paciente están comprometidos. Micelio fúngico atacando una célula eucariota humana (Shutterstock). El reino Fungi se compone de un gran número de especies que se asocian a un amplio espectro de enfermedades humanas, que van desde alergia y autoinmunidad a infecciones invasoras, potencialmente mortales. Hongos como Aspergillus fumigatus, Cryptococcus neoformans, Pneumocystis jirovecii e Histoplasma capsulatum son ubicuos en el medio ambiente, mientras que otros como Candida albicans, establecen para toda la vida comensalismo en las superficies del cuerpo humano. Los seres humanos están constantemente expuestos a los hongos, principalmente por inhalación o implantación traumática donde el oportunismo fúngico ocurre en aquellos pacientes con defectos de la inmunidad. La enfermedad fúngica se produce cuando estos microorganismos invaden el nicho natural, o cuando se presenta una alteración en el precario equilibrio que evita que un agente comúnmente colonizante se torne patógeno. 1 La enfermedad subsecuente puede presentarse en dos escenarios ampliamente reconocidos: el primero, como infección primaria que afecta a individuos aparentemente inmunocompetentes y de manera habitual solo a una pequeña proporción de la población, está particularmente relacionado con agentes endémicos; en el segundo, la enfermedad solo se presenta en aquellos individuos con alteración de la inmunidad innata o adquirida, manifestándose como infección oportunista secundaria a su estado inmune. En los últimos años, debido a los avances en el cuidado sanitario, ha aumentado la supervivencia en condiciones de inmunosupresión relativa o absoluta y, secundariamente, se ha ampliado el riesgo para el desarrollo de una enfermedad fúngica invasora (EFI) primaria u oportunista (Figura 1).1,2 El sistema inmune no ignora a los hongos " comensales o sapró tos, los acepta y mantiene una relación estable de acogida al hongo 2.1. RESPUESTA INMUNE FRENTE A LA INFECCIÓN FÚNGICA La mayoría de los hongos patógenos necesitan un estado de interacción hospedador-hongo, que se caracteriza por una respuesta inmune tolerante y de acogida, para permitir la supervivencia del hospedador sin necesariamente eliminar al patógeno, lo que establece una relación de comensalismo o latencia. Por lo tanto, el equilibrio entre señales pro-inflamatorias y anti-inflamatorias es un requisito previo para el éxito de este tipo de interacción, y donde la responsabilidad para la virulencia del hongo es compartida por el hospedador. La mayoría de los hongos patógenos humanos no pueden sobrepasar la capacidad inmune del hospedador como para producir enfermedad Cuando los hongos sobrepasan estos mecanismos de defensa generan colonización e infección Los mecanismos de defensa del hospedador frente a las infecciones por hongos son numerosos, desde mecanismos de protección o “inmunidad innata” hasta sofisticados mecanismos de adaptación o “inmunidad adaptativa” inducidos específicamente durante la infección y enfermedad (Figuras 2 y 3). Los mecanismos de respuesta innata están diseñados para detectar patrones generales y conservados, que difieren entre los diferentes organismos patógenos, inducibles tras la infección, y su activación requiere la identificación de los patrones moleculares asociados a patógenos (PAMPS) por los Separata 2 31 Patogénesis de la enfermedad fúngica invasora AMBIENTE: Nuevos nichos ecológicos, construcciones y catástrofes Antecedentes de exposición antimicrobiana PACIENTE: Antecedentres y factores de riesgo, ocupación, exposición HONGO: Capacidad de adaptación, patogenicidad, virulencia DESARROLLO DE LA ENFERMEDAD FÚNGICA INVASORA Figura 1. Factores a tener en cuenta para el desarrollo de una EFI. receptores de reconocimiento de PAMPS (PRR) a través de los receptores tipo Toll (TLR) (Tabla 1).2-5 Si el patógeno infeccioso traspasa estas primeras líneas de defensa, se produce una respuesta inmune adaptativa con generación de anticuerpos (Acs) específicos, linfocitos T ayudadores (Helper) (Th) y células tipo B, que atacan al patógeno y establecen la memoria para prevenir la infección subsiguiente por el mismo microorganismo. Los dos sistemas están íntimamente vinculados y controlados por un conjunto de moléculas y receptores que actúan para generar un procedimiento coordinado para la protección contra patógenos fúngicos (Tabla 2).3,4 las células dendríticas (DC). Entre las funciones efectoras de los fagocitos se encuentra el efecto fungicida y la inhibición de crecimiento, así como los procesos para resistir la inefectividad de los hongos que incluyen los efectos inhibitorios sobre el dimorfismo y la promoción del cambio fenotípico. La restricción óptima del crecimiento de hongos se produce a través de una combinación de mecanismos oxidativos y no oxidativos que son complementarios. Entre los mecanismos no oxidativos se encuentra la liberación intracelular o extracelular de moléculas efectoras, defensinas, péptidos catiónicos y secuestro de hierro (Tabla 1). Las principales barreras contra lasinfecciones fúngicas son la piel y las super cies mucosas del tracto digestivo y respiratorio Enzimas como la nicotinamida adenina dinucleótido fosfato oxidasa (NADPH) y la óxido nítrico sintetasa inducen el inicio de las vías oxidativas, conocidas como el estallido respiratorio. La mieloperoxidasa (una hemoproteína lisosomal presente en los gránulos azurófilos de los neutrófilos, los monocitos y los macrófagos) es una mediadora en la muerte oxígeno-dependiente de los hongos.6 Los macrófagos tienen como atributos la presentación de antígenos; sin embargo, su principal contribución defensiva la realizan mediante la fagocitosis y muerte de los patógenos fúngicos. Pero también pueden ser utilizados por los hongos dimórficos para protegerse del entorno, permitirles su multiplicación y facilitar su difusión desde el pulmón a otros órganos. El sistema inmune innato distingue lo propio de lo ajeno y activa los mecanismos inmunes adaptativos, mediante la provisión de señales específicas.5 Los mecanismos constitutivos de defensa se encuentran presentes en aquellos sitios de interacción continua con los hongos e incluyen la función de barrera de las superficies de cuerpo y la mucosa epitelial del tracto respiratorio, gastrointestinal y genitourinario. Los factores humorales contribuyen a mejorar los mecanismos de defensa innatos. Las proteínas de unión a manosa o lectinas (MBL), las colectinas y el sistema de complemento junto con los Acs, promueven la unión (opsonización) del hongo, y constituyen un mecanismo de reconocimiento llevado a cabo por una serie de PRR. Las actividades biológicas especificas del sistema de complemento y los Acs, que contribuyen a la resistencia de las infecciones fúngicas, son múltiples e interdependientes. Los Acs contribuyen en gran medida a la activación del sistema del complemento y este es esencial para la protección mediada por Acs. La fracción de complemento CR3 (también conocida como CD11b/CD18) es uno de los medios más eficientes para fagocitar el hongo opsonizado.3,4 El reconocimiento, independiente del antígeno de los hongos, por el sistema inmune innato conduce a la movilización inmediata de los mecanismos inmunes efectores y reguladores que proporcionan tres ventajas de supervivencia importante: Durante la mayoría de las infecciones fúngicas se produce un buen nivel de Acs, muy útiles para pruebas diagnósticas, pero que tienen muy poca participación en la defensa contra ellos, salvo por su acción opsonizante y reguladora de la respuesta inmune.5 a) Inicio rápido de la respuesta inmune y generación de un entorno co-estimulador para el reconocimiento antigénico. b) Establecimiento de una primera línea de defensa que controla al hongo patógeno durante la maduración de la respuesta inmune adaptativa. c) Direccionamiento de la respuesta inmune adaptativa, ya sea celular o humoral. El complemento, los Acs y las colectinas no sólo ayudan con una primera línea de defensa contra los hongos, también participan en la respuesta inflamatoria y la respuesta inmune adaptativa a través de la regulación en la secreción de citoquinas y la expresión de moléculas co-estimuladoras sobre los fagocitos. La liberación local de estas moléculas efectoras inicia una respuesta inflamatoria, activando las células fagocíticas a un estado microbicida y dirigiendo el desarrollo de las células Th / Treg (linfocitos T reguladores).6-8 1) Inmunidad Innata Por lo tanto, con el fin de lograr la activación óptima antígenoespecífica de la inmunidad adaptativa, es necesario activar los mecanismos de detección de los patógenos por parte de la inmunidad innata.5 En esto juegan un papel esencial, los fagocitos profesionales, que consiste en leucocitos polimorfonucleares (neutrófilos), leucocitos mononucleares (monocitos y macrófagos) y 32 Separata 2 2) Detección de los hongos- Sistemas de reconocimiento TLR y No-TLR El sistema TLR es, sin duda, el mejor de los sensores inmunes ante posibles patógenos, participando en las vías de señalización que se activan la inmunidad innata y que ayudan a reforzar la inmunidad Tabla 1. Componentes de la respuesta innata a las infecciones fúngicas. Función principal Componentes Barreras Epitelio Defensinas Linfocitos intraepiteliales Previene la entrada del patógeno Destrucción del patógeno Destrucción del patógeno Células efectoras circulantes Neutró los Macrófagos NK Fagocitosis temprana, muerte del patógeno Fagocitosis temprana, muerte del patógeno, activación de la respuesta in amatoria Muerte de las células infectadas, activación de macrófagos DC Fagocitosis, conexión de la respuesta innata y adaptativa Proteínas efectoras circulantes Complemento Colectinas (lectinas de unión a manosa) Proteína C reactiva (Pentraxina) Muerte del patógeno, opsonización, activación de leucocitos Opsonización, activación del complemento (vía de las lectinas) Fagocitosis temprana, muerte del patógeno Factores de coagulación Localización del tejido infectado Citoquinas FNT In amación local, activación del endotelio/linfocitos IFN-γ Formación de granulomas, activación de macrófagos TGF-β Inhibición de la activación de linfocitos T y macrófagos Inducción de síntesis de moléculas de adhesión In amación, activación de linfocitos T, activación de macrófagos IL-1 IL-4 IL-5 Activación y diferenciación de células B, potente inhibidor de la apoptosis Promueve la diferenciación de linfocitos Th2 Activación, reconocimiento y/o diferenciación de linfocitos B, eosinó los y precursores hematopoyéticos IL-10 Inhibición de macrófagos y de la producción de citoquinas de células T Activación y diferenciación de linfocitos T Promueve la diferenciación de linfocitos Treg IL-12 Diferenciación de linfocitos T Inducción de síntesis y secreción de citoquinas Proliferación de células T y NK Promueve la diferenciación de linfocitos Th1 IL-17 Inducción de IL6, IL8 y adhesión (ICAM-1) Proliferación de linfocitos T IL-18 Producción de IFN-γ por las NK y células T IL-23 Promueve la in amación de los tejidos y el reclutamiento de neutró los al sitio de la in amación Promueve la diferenciación de linfocitos Th17 NK: Células asesinas naturales; DC: Células dendríticas; FNT: Factor de necrosis tumoral; IFN-γ: Interferón gamma; IL: Interleuquina; TGF- : Factor de crecimiento tumoral beta; Th: Linfocito T ayudador; ICAM: Moléculas de adhesión celular; Treg: Linfocitos T reguladores. Adaptado de: Garry C1, Kavanagh K5 y Romani L3,4 adaptativa. Los TLR pertenecen a la super-familia TIR (Toll/interleuquina-1 (IL-1)) que se divide en dos subgrupos: los receptores IL-1y la TLR. Las vías de señalización comunes utilizados por IL-1R y TLR implican el reclutamiento de varias proteínas adaptadoras, incluida la MyD88 (proteína de diferenciación mieloide de respuesta primaria), que activan a su vez una serie de quinasas que son cruciales para la inmunidad innata. Los miembros individuales de la familia TLR y los PRR interactúan entre sí y los efectos acumulativos de sus interacciones mejoran la respuesta inmune contra el patógeno (Tabla 1).9 Sin embargo, la activación de los TLR es un arma de doble filo ya que, son esenciales para provocar la respuesta innata y mejorar la inmunidad adaptativa pero, a su vez, también están involucrados en la patogénesis de enfermedades autoinmunes y trastornos inflamatorios (asma, artritis reumatoide, enfermedades infecciosas). Los receptores lectina, tipo C (es decir, dectina-1 y 2, DC-SIGN y la familia de galectina) son importantes PRR para varios componentes fúngicos y no tienen como vía de señalización a los TLR. Separata 2 33 Patogénesis de la enfermedad fúngica invasora Tabla 2. Subpoblaciones de linfocitos T en la respuesta inmune a las infecciones fúngicas. Factor de diferenciación Th1 Th2 Th17 Treg IL-12 IFN-γ IL-4 IL-4, IL-5, IL-13 IL-17 IL10, TGF-β IL-23 IL-10 Función Citoquina secretada Tipo celular Promoción de la eliminación fúngica - In amación Inhibición de la eliminación fúngica - Alergia Inhibición de la eliminación fúngica - In amación Limita la patología infecciosa - Persistencia fúngica Inmunidad a largo plazo IFN-γ: Interferón gamma; IL: Interleuquina; TGF-β: Factor de crecimiento tumoral beta; Th: Linfocito T: Linfocito T ayudador. Adaptado de: Garry C1, Kavanagh K5 y Romani L3,4 COMPONENTES DE LA RESPUESTA INMUNE CONTRA LAS INFECCIONES FÚNGICAS Inmunidad Innata • Barreras físicas: epitelio de piel y mucosas • Barreras químicas: pH de uidos corporales, péptidos antimicrobianos (defensinas), proteínas (lisozimas, péptidos catiónicos) • Células fagocíticas: neutró los, monocitos/macrófagos, células asesinas naturales, células dendríticas Inmunidad Adaptativa • Respuesta humoral: anticuerpos, citoquinas, etc. • Respuesta celular: linfocitos T y B Figura 2. Componentes de la respuesta inmune. 3) A nación de la respuesta inmune adaptativa: el papel instructivo de las células dendríticas Las DC están equipadas con varios TLR, y son los conectores principales entre la respuesta innata y adaptativa. La función dual de activar/tolerar está mediada por su capacidad de cambiar del contexto de célula presentadora de antígeno y el poder comunicar a las células T la naturaleza de los antígenos que están presentando. El sistema DC tiene la capacidad de responder de una manera flexible a los diferentes estímulos. Las DC son las únicas expertas en decodificar la información asociada a los hongos y de traducirlo a diferentes respuestas asociadas a las células T. Los diferentes PRR determinan plasticidad funcional de las DC en respuesta a los hongos y contribuyen al reconocimiento de la discriminación de los diferentes morfotipos fúngicos (Tabla 2).1,5 34 Separata 2 Las DC reconocen e internalizan un número variado de hongos (A. fumigatus, C. albicans, C. neoformans, H. capsulatum, Malassezia furfur y Saccharomyces cerevisiae) y captan antígenos fúngicos exógenos a través de los macrófagos apoptóticos, siendo las únicas capaces de decodificar la información asociada al hongo en la interfaz del hospedador-hongo y capturar los diferentes elementos fúngicos producido a través de diferentes receptores y formas de fagocitosis. El reconocimiento y la internalización de las levaduras y conidias no opsonizados ocurren a través de los receptores de manosa (MR), DC-SIGN (proteína integral de membrana tipo II de las DC), dectina-1 y CR3. En contraste, la entrada de hifas se produce por una fagocitosis tipo cremallera que cuenta la acción cooperativa de FcγR (fracción cristalizable de las inmunoglobulinas por las cuales se adhiere a células) II y III y CR3 (sin intervención de los TLR/ MyD88). La participación de los distintos receptores asociados a los diferentes morfotipos de hongos se traduce en la regulación de la producción de la citoquinas y la co-estimulación, un evento muy influenciado por opsoninas fúngicas tales como la MBL, C3, y/o Acs. Por lo tanto, los TLR colaboran con otros receptores de inmunidad innata para la activación de las DC contra los hongos a través de vías dependientes e independientes del MyD88 (Figura 4). La activación a través de MR y dectina-1 desencadena la producción de citoquinas pro-inflamatorias (incluyendo la IL-12), la regulación positiva de moléculas co-estimuladoras y la de antígenos del complejo mayor de histocompatibilidad (CMH) clase II. La activación de IL-12 por DC requiere de la vía de MyD88 con la implicación de distintos TLR. En contraste, la coligación de CR3 con FcγR, que sucede en la fagocitosis de las hifas, activa la producción de IL-4/IL-10 y la regulación positiva de moléculas co-estimuladoras y antígenos del CMH clase II. La producción de IL-10 es independiente de MyD88. Una característica notable e importante de las DC es su capacidad de producir IL-10 en respuesta a los hongos. Las IL-10 activan las Treg Cd4+ y CD25+ que son componentes esenciales de resistencia antifúngica. Así, al subvertir el programa morfotipo-específico de la activación de las DC, opsoninas, Acs, y otros factores ambientales, pueden afectar el funcionamiento cualitativo de las DC y las Th / Treg y alterar la virulencia fúngica. En este escenario, el desarrollo cualitativo de la respuesta de las células Th frente a un hongo no depende de la naturaleza y la forma del los hongos, sino del tipo de señalización celular iniciada por las DC y de la interacción ligando-receptor en las DC. La capacidad de las DC para activar los Th1 y Th2 se correlaciona con la resistencia y susceptibilidad a las infecciones fúngicas (Figura 4). Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Candida / Aspergillus Barreras físicas, mecánicas y químicas Colonización RESPUESTA INMUNE INNATA Primera barrera de defensa: mucosa epitelial Activación de las defensas químicas y celulares Invasión Reconocimiento del hongo a través de diferentes receptores Receptores transmembrana (TLR y dectina-1) que activan macrófagos, células dendríticas, neutró los y monocitos Liberación de citoquinas Maduración de células dendríticas y presentación del antígeno RESPUESTA INMUNE ADAPTATIVA Receptores solubles (lectinas, pentraxina) Complemento/Lisozimas Péptidos catiónicos Maduración de los macrófagos, neutró los y monocitos Activación de procesos de degradación, oxidación y acidi cación Respuesta humoral y celular Producción de la respuesta Th1, Th2, Th17 y Treg Producción de anticuerpos Destrucción del hongo TLR: Receptores tipo Toll; Th: Linfocitos T ayudadores; Treg: Linfocitos T reguladores. Adaptado de : Garcia-Vidal C y Carratalá J.2 Figura 3: Respuesta inmune del hospedador frente a la infección fúngica. 4) Inmunidad adaptativa: Th1, Th2 y Th17 Los linfocitos de sujetos sanos muestran una fuerte respuesta proliferativa después de la estimulación con antígenos fúngicos y la producción de diferentes citoquinas. Para muchas infecciones fúngicas, la respuesta eficaz de los tejidos al proceso invasivo es una inflamación granulomatosa, un sello de la inmunidad mediada por células (IMC). La heterogeneidad del repertorio de las células T CD4+ y CD8+ demuestran la multiplicidad y la redundancia de los mecanismos efectores a través de los cuales los linfocitos T participan en el control de infecciones fúngicas. El programa flexible de los linfocitos T también implica la producción de un número de mediadores, incluyendo citoquinas (Figura 5). Debido a su acción sobre los leucocitos circulantes, las citoquinas producidas por hongos específicos de las células T son decisivas en la movilización y activación de efectores antifúngicos, proporcionando así control rápido y efectivo cuando el hongo se ha establecido en tejidos o se ha propagado a órganos internos.10-13 La generación de una respuesta Th1 dominante generada por la IL-12, es escencial para la protección contra las infecciones fúngicas (Figura 4). La resistencia a las infecciones fúngicas es dependiente de la inducción de la inmunidad celular, mediada por linfocitos T, citoquinas y los fagocitos efectores A través de la producción de interferón gamma (IFN-γ) y con la ayuda de Acs opsonizados, la activación de las células Th1 permite la activación óptima de los fagocitos en los sitios de infección. Por lo tanto, la ausencia de las señales activadoras de los fagocitos efectores puede predisponer a las EFI, limitar la eficacia terapéutica de los antifúngicos y Acs y favorecer la persistencia y/o comensalismo. La generación de una respuesta Th2 generada por la IL-4 se relaciona con la gravedad de la enfermedad y es un marcador de mal pronóstico ya que permite la susceptibilidad a las infecciones fúngicas (Tabla 2). Separata 2 35 Patogénesis de la enfermedad fúngica invasora Hongos Levaduras Conidias Inmunidad Innata Micelios Lectinas de unión a manosa Inmunidad Adaptativa Th1 Carga fúngica Th2 Procesos alérgicos Th17 Expresión de mediadores de reclutamiento de neutró los Tregs Inmunidad celular y alergia Pentraxina-3 Reconocimiento de los PAMPS TLR-4 TLR-9 Super cie Celular TLR-2 Dectina-1 Producción de citoquinas pro-in amatorias Producción de IL-12 por las células dendríticas Inducción de la explosión respiratoria y degranulación Diferenciación de las células T ayudadoras 1 (Th1) Citoplasma Activación de NAPH oxidasa Generación de reactivos oxidativos PAMPS: Patrones moleculares asociados a patógenos; TLR: Receptores tipo Toll; Th: Linfocitos T ayudadores; Tregs: Lintocitos T reguladores; IL: Interleuquina. Adaptado de: Garry C1, Kavanagh K5 y Romani L3,4 Figura 4. Respuesta inmune frente a las infecciones fúngicas. Reconocimiento del agente patógeno. Las células Th17, un linaje de células Th efectoras, contribuyen a la patogénesis inmune previamente atribuida al linaje Th1, donde la IL-23 es una citoquina crítica para la generación y mantenimiento de este linaje y desempeña un papel fundamental en la antifúngica, desarrollando un efecto pro-inflamatorio que les permite hacer de puente entre la inmunidad innata y la adaptativa y donde los niveles altos de IL-23/IL-17 se correlacionan con la gravedad de la enfermedad y la inmunopatología (Tabla 2).14 2.1.1. Respuesta inmunológica a la infección por Candida Candida puede ser un comensal en las mucosas oral, respiratoria, gastrointestinal y genitourinaria, así como de la piel sin que esto genere ningún tipo de enfermedad, dependiendo de la capacidad de hospedador de mantener todas sus líneas defensivas activas. Este agente etiológico puede permanecer como comensal o progresar y producir una infección local, con formas mucocutáneas, o producir candidiasis invasora (CI) potencialmente fatal. Los mecanismos de defensa contra este tipo de infecciones son tan o más complejos que los mecanismos patogénicos y la interacción hospedador-patógeno mantiene un equilibrio a veces precario en múltiples niveles y no necesariamente independientes entre sí.9,13,15 La defensa contra la invasión por Candida puede ser innata o adaptativa. La innata puede ser mediada física y químicamente o inmunológicamente, la inmunidad adaptativa puede ser celular o humoral. 36 Separata 2 TENGA PRESENTE: C. albicans y otras especies de Candida, además de ser comensales inocuos, son patógenos oportunistas y responsables de diferentes tipos de infecciones, desde super ciales hasta procesos invasivos y diseminados. 2.1.1.1. Respuesta inmune innata Normalmente, en un hospedador inmunocompetente la respuesta innata anti-Candida es eficaz y lo protege contra el microorganismo. Esta respuesta natural comprende barreras físicas como la piel y las mucosas intactas. Tras superar el epitelio empieza la infección invasora por Candida. Como una primera respuesta del hospedador, el endotelio vascular secreta mediadores proinflamatorios y péptidos antimicrobianos, como las defensinas, que estimulan el reclutamiento y la activación de los leucocitos. Una vez que atraviesan las barreras mucocutáneas, los neutrófilos y monocitos son las células claves en los estadios iníciales de la respuesta frente a la Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Hongos Mucosa epitelial PRR Respuesta innata PRR PRR Células dendríticas inmaduras Maduración Macrófagos Drenaje de ganglios linfáticos Células dendríticas maduras RCT CMH LT L-10 L-12 L-18 Presentación antigénica LB LT Producción de anticuerpos Opsoninas Neutró los Destrucción del patógeno Respuesta in amatoria LB Inicio de las funciones de la respuesta innata : Fagocitos y degranulación de los neutró los Anticuerpos IL-12 Th1 IFN-γ FNT IL-4 Th2 IL-4 IL-5 IL-10 Treg TGF- IL-10 Activación de linfocitos y liberación de citoquinas Respuesta adaptativa Las líneas continuas representan señales positivas Las líneas discontinuas representan señales negativas PRR: Receptores de reconocimiento de PAMPS; IFN-γ: Interferón gamma; IL: Interleuquina; TCR: Receptor de células T; CMH: Complejo mayor de histocompatibilidad; LT: Linfocitos T; LB: Linfocitos B; TGF-: Factor de crecimiento tumoral beta; FNT: Factor de necrosis tumoral. Adaptado de: Romani L.3 Figura 5. Respuesta inmune ante las infecciones fúngicas. Respuesta innata y adaptativa. infección. El tamaño de la levadura puede dificultar la fagocitosis del hongo, por lo que son necesarios otros componentes extracelulares para marcar al patógeno y favorecer su ingestión y destrucción.2,15-22 Para que los fagocitos profesionales (macrófagos, neutrófilos y células dendríticas) puedan unirse al hongo se necesita el reconocimiento previo del patógeno mediante la acción de las PRR. Los PRR más importantes en el reconocimiento de Candida spp. por los neutrófilos y monocitos son los TLR, los RM y la dectina-1. Los TLR interaccionan con diferentes proteínas, entre las que destaca por su papel regulador la MyD88, y activan una serie de factores de trascripción que lideran la producción de citoquinas pro o anti-inflamatorias y consecuentemente, la activación de una u otra respuesta inmune adaptativa. Los TLR más importantes en la respuesta frente a Candida spp. son el TLR2y el TLR4 que reconocen dos PAMPS diferentes. El PAMP que reconoce el TLR2 es el fosfolipomanano de la pared del hongo y se asocia con la activación de la respuesta Th2. Por su parte, el TLR4 se une al manano y facilita respuesta mediada por las células Th1.3,4 En la respuesta a la infección por Candida spp. mediada por la activación de los TLR, la proteína MyD88 juega un papel imprescindible para la activación de los macrófagos. Por otro lado, los RM permiten el reconocimiento y fagocitosis de Candida spp. que no ha sido opsonizada previamente. La dectina-1 se une a los β-glucanos y actúa favoreciendo la maduración de las células dendríticas, monocitos y macrófagos así como en la activación de diferentes citoquinas, en especial la IL-2 y la IL-10. La unión de Candida spp. a estas proteínas induce una respuesta mediada por linfocitos Th17. La activación simultánea de múltiples PRR por Candida spp. dibuja un amplio espectro de posibilidades en la respuesta del hospedador. Para una óptima respuesta es necesario un equilibrio entre los diferentes componentes, la dectina-1 según su activación puede estimular la respuesta del TLR2 o la mediada por el TLR4, los RM pueden activar el TLR2 y el propio TLR2 inhibe el TLR4. Como consecuencia final de todos estos procesos se activará una u otra respuesta inmunitaria adaptativa y una serie de procesos dirigidos a producir la muerte de Candida spp., entre los que destacan los procesos oxidativos que incluyen la generación de radicales libres de oxígeno y nitrógeno, así como los no oxidativos.6 Los factores humorales también participan en la defensa frente a la infección por Candida spp. Este hongo activa el complemento por su vía clásica y alternativa, facilitando el reclutamiento y activación de células fagocíticas e incrementando su efecto fungicida. 2.1.1.2. Respuesta inmune adaptativa Las DC juegan un papel muy importante para unir la inmunidad innata y la adaptativa, el tipo de respuesta de estas células depende Separata 2 37 Patogénesis de la enfermedad fúngica invasora mucho de la morfología de Candida spp. Ante la presencia de estructuras levaduriformes o pseudomiceliales, las DC utilizan diferentes receptores para interaccionar con Candida spp. y establecen diferentes respuestas. Si fagocitan estructuras levadurifornes inducen una diferenciación de las células CD4+ a células Th1. Si fagocitan formas pseudomiceliales inducen una respuesta Th2. Una respuesta mediada por las células Th1 se asocia a protección frente a la infección fúngica. Por el contrario, la respuesta Th2 se relaciona con la capacidad del microorganismo de evadir o inhibir la respuesta inmune del hospedador. El resultado final de una u otra respuesta Th influirá en activación de los linfocitos B y en la maduración del resto de células fagocíticas.14 Los Treg, que disminuyen la inmunidad celular y favorecen los procesos de alergia, junto con los Th17 son también importantes en la respuesta del hospedador frente a la infección candidiásica. El papel de la formación de Acs en la respuesta a la infección por Candida spp. es poco conocido. Clínicamente, un déficit en la IMC de las células B no se asocia a un aumento en la susceptibilidad a la infección. Sin embargo, se ha observado que existen algunos Acs que son capaces de potenciar de manera considerable la respuesta de las células fagocíticas frente a la infección fúngica e incluso de activar por sí mismos acciones beneficiosas del complemento. 2.1.2. Respuesta inmunológica a la invasión por Aspergillus La concentración media de conidias de Aspergillus en el aire está entre 0,2 a 15 conidias/m3 y puede llegar hasta 106 conidias/m3, por lo que, normalmente, los seres humanos se exponen de forma habitual a las mismas sin desarrollar enfermedad. En los pocos casos de enfermedad, la forma de la manifestación clínica dependerá del daño continuo de bajo nivel y de la progresión de la alteración de la respuesta inmune.19 2.1.2.1. Respuesta inmune innata La respuesta inmune innata es esencial en el control de la invasión. El mecanismo de protección inicial y fundamental en prevención de la penetración de las conidias es la barrera de la vía aérea, constituida por los cornetes nasales y la película de moco de la pequeña vía aérea, (encargada de la captación y barrido de las conidias.) Las conidias de pequeño tamaño (2-6 µm) pueden sobrepasar el epitelio ciliado y quedar en contacto con el epitelio desnudo, activando el segundo sistema de defensa basado en el reconocimiento del agente como extraño.1,23,24 Tras la barrera anatómica del epitelio respiratorio y las defensas mucociliares, los macrófagos alveolares son la siguiente línea de defensa fagocítica frente a las esporas inhaladas. Posteriormente, las diferentes células sanguíneas del sistema inmune (células dendríticas, monocitos y neutrófilos), llegan al sitio de la infección jugando un papel fundamental en la destrucción inicial del hongo y la activación de las posteriores etapas de la respuesta inmunitaria. Los macrófagos y monocitos tienen una acción esencial en la fagocitosis y la muerte de las esporas, impidiendo así su transición a las formas invasivas de las hifas. Los neutrófilos son imprescindibles en la respuesta del hospedador frente a las conidias en proceso de germinación y también frente a las formas hifales (Tabla 1).25-29 Los PRR más importantes en la respuesta inmune innata son los TLR y la dectina-1. Entre los TLR, el TLR2 y el TLR4 juegan un papel fundamental, aunque la activación de cada receptor mediará respuestas 38 Separata 2 totalmente diferentes y no se conoce que PAMPS de Aspergillus son los reconocidas por cada TLR. Cuando Aspergillus se une al TLR4, se genera una respuesta de citoquinas pro-inflamatorias (FNT-α, IL-1, IL12, IL-15 y IFN-γ) que se asocia a un efecto de protección frente la infección y se genera una respuesta adaptativa mediada por los linfocitos Th1. Por el contrario, la activación del TLR2 favorece una respuesta anti-inflamatoria, mediada por la IL-10 y la IL-4, y como consecuencia se promueve una respuesta del sistema inmune adaptativo mediado por los Th2, relacionado con una mayor susceptibilidad a padecer una EFI.26 La dectina-1, un tipo de lectina, juega un papel imprescindible en el reconocimiento y comunicación celular. Es específica para los βglucanos presentes en la pared celular de los hongos; este receptor aumenta su expresión cuando el hospedador está continuamente expuesto al patógeno. Su activación favorece la fagocitosis del hongo, inicia la cascada inflamatoria de citoquinas y activa los procesos oxidativos. Una vez que Aspergillus es reconocido por los diferentes receptores se producen dos fenómenos importantes: La producción de citoquinas para activar la respuesta inmune adaptativa y el inicio de diferentes procesos que tienen como objetivo la muerte del hongo. Las células de defensa fagocitan las diferentes formas fúngicas y, según se trate de una espora o de una hifa, se activan diferentes procesos. Los macrófagos eliminan las esporas mayoritariamente mediante procesos de oxidación o de acidificación. Dentro de los procesos oxidativos destacan los mediados por la NADPH-oxidasa con la formación de radicales libres con capacidad antimicrobiana, sobre todo en los neutrófilos y frente a las hifas. Además de la acción de la NADPH-oxidasa, los neutrófilos utilizan otros mecanismos oxidativos para eliminar las partículas fúngicas. Estos procesos están mediados por substancias que se generan en sus gránulos (proteasas, lisoenzimas, lactoferrina, pentraxina-3). Algunos factores humorales también participan en la respuesta inmune innata frente a la infección por Aspergillus. El sistema del complemento actúa frente a las diferentes formas del hongo, ya sea por su vía alternativa (que reconoce y elimina esporas) o por la clásica (que reconoce las conidias en germinación y las hifas). En el fluido alveolar, existen algunas proteínas (lectinas tipo C) que favorecen la fagocitosis y aglutinan las esporas de Aspergillus, inmovilizando al patógeno y 27 favoreciendo la acción del sistema inmune. En ocasiones, el tamaño de las hifas es demasiado grande para ser fagocitado y estas pueden ser dañadas a través de mecanismos extracelulares tales como lisozimas (que favorecen la fagocitosis) o péptidos catiónicos (que forman canales en la pared del hongo favoreciendo su lisis). 2.1.2.2. Respuesta inmune adaptativa En la infección por Aspergillus, la respuesta adaptativa tiene relevancia tanto en la defensa contra nuevas infecciones como en el aumento de las manifestaciones clínicas en la aspergilosis broncopulmonar alérgica. El sistema inmune adaptativo permite una respuesta inmunitaria más contundente y el establecimiento de la denominada memoria inmunológica. Las células del sistema inmune adaptativo son los linfocitos T, que generan la respuesta IMC, y las células B, encargadas de la respuesta inmune humoral mediada por Acs. En la aspergilosis invasora (AI), la inmunidad celular es clave mientras que el papel de la inmunidad humoral es menor. Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Atributos de virulencia seleccionados Estrés ambiental Competencia Metabolitos secundarios (Af) Privación de nutrientes Sideróforos (Af) Temperatura Termotolerancia (Af, Cn, Ca) Radiación UV Melanina (Af, Cn) Oxidación Cápsula (Cn) Predación Adherencia (Ca, Cn, Af) Incremento signi cativo en inmunocompromiso Patogenésis de micosis oportunista Incremento en su capacidad de adaptación Estrés In vivo Atributos de virulencia presentes en la población Micro-evolución In vivo Propagación clonal Regreso al medio ambiente Incremento de la torencia Transposones Recombinación sexual Af: Aspergillus fumigatus; Cn: Cryptococcus neoformans; Ca: Candida albicans. Adaptado de: Anaissie, McGinnis & Pfaller. Clinical Mycology (2nd ed.) Elsevier, inc. 2009. Figura 6. Evolución de los hongos patógenos humanos de acuerdo a sus atributos de virulencia. La respuesta inmune adaptativa requiere el reconocimiento de antígenos del microorganismo invasor durante la presentación de antígenos, en este proceso las DC son importantes para entrelazar la respuesta inmune innata y la adaptativa. Las DC procesan el antígeno y lo presentan a los linfocitos a través del CMH. Los Th se encargan de regular y amplificar la respuesta del hospedador frente Aspergillus, y las sub-poblaciones Th1 y Th2, a través de la secreción de citoquinas, son responsables de coordinar la respuesta inmune celular. La respuesta mediada por las células Th1 se asocia con la producción de citoquinas pro-inflamatorias que activan los macrófagos; a su vez, genera más linfocitos T CD4+, favorece la producción de Acs, retrasa las reacciones de hipersensibilidad y se asocia con una respuesta protectora frente a la infección fúngica. La respuesta Th2 se asocia a la secreción de citoquinas anti-inflamatorias, entre las que destaca la IL10, que regulan la inflamación causada por las citoquinas dependientes de la respuesta Th1 y se asocia a diferentes mecanismos de las reacciones alérgicas. Solo de un óptimo balance entre la respuesta Th1 y Th2 se obtendrá una respuesta inmunitaria que permita hacer frente a la infección por Aspergillus. Esta respuesta Th influirá en activación de linfocitos B y en la maduración del resto de células fagocíticas. Los Treg disminuyen la inmunidad celular y favorecen los procesos alérgicos.2 2.2. CONTEXTO DE LA ENFERMEDAD INFECCIOSA 2.2.1. El patógeno La colonización suele ser el primer paso para una enfermedad invasora. El microorganismo debe alcanzar un microambiente en el individuo con suficientes sustratos, donde pueda reproducir y de alguna forma evitar la respuesta inmune normal. El periodo de colonización puede ser de pocas semanas a muchos años, y en muchos casos es imposible determinar si la enfermedad invasora se presenta secundaria a la diseminación de un nicho antiguo que se activa, o corresponde a la progresión de una exposición y colonización reciente.1,2 El proceso de colonización y posterior infección está relacionado con la capacidad del hongo para adherirse eficazmente al tejido expuesto, incluso en condiciones en las cuales no es posible que otros microorganismos puedan sobrevivir en el hospedador, este fenómeno se conoce como Factor de virulencia (Figura 6). Muchas especies de hongos patógenos tienen la capacidad de cambiar su morfología Separata 2 39 Patogénesis de la enfermedad fúngica invasora COLONIZACIÓN -defensinas INVASIÓN Blastoconidias Blastoconidias y pseudomicelios -defensinas Mucosa epitelial Pseudomicelios Fagocitosis Formación de micelios Neutró los Células dendríticas Quimiocinas, citoquinas, alarminas Exposición de -glucano Respuesta innata IL-17 Diseminación fúngica Respuesta adaptativa Th 17 Monocitos/Macrófagos Activación de in amasoma Respuesta innata Diferenciación de células Th Presentación de antígeno Las líneas negras indican los mecanismos de defensa del hospedador Las líneas rojas indican invasión por Candida albicans y sus mecanismos de evasión de la respuesta inmune IL: Interleuquina; Th: Linfocitos T ayudadores. Adaptado de: Cheng SC, Joosten LA, Kullberg BJ, Netea MG.10 Figura 7. Interacción entre Candida albicans y la respuesta inmune innata en la mucosa epitelial. Los factores de virulencia de los hongos son aparentemente, accidentes de la naturaleza y el resultado del proceso de evolución que permitieron la supervivencia del microorganismo en condiciones saprófitas pero que, en forma incidental, permiten la supervivencia en el tejido de los mamíferos. Además de los factores de virulencia, la enfermedad invasora se puede favorecer por el tamaño del inoculo fúngico ya que grandes inóculos pueden afectar al individuo a pesar de encontrarse expuesto a agentes aparentemente poco virulentos (Tabla 3).1,2,7,23 TENGA PRESENTE: La capacidad de un hongo patógeno para producir enfermedad depende de: · La virulencia de la partícula infectante. · El tamaño del inóculo fúngico. · La vía de infección. · La inmunidad del hospedador. · El órgano afectado. · La coexistencia con otras infecciones o enfermedades. Una vez el microorganismo alcanza el hospedador potencialmente susceptible es necesario que el hongo burle los múltiples sistemas de defensa del organismo que van desde las barreras de la piel y 40 Separata 2 mucosas hasta la inmunidad específica, pasando por los mecanismos pro-inflamatorios y la inmunidad no específica (Figura 6).3,4 Las estructuras fúngicas son extremadamente complejas y su interacción con el hospedador es diferente dentro de una misma especie de acuerdo al estado en que se encuentra, siendo aun más heterogénea entre diferentes especies. Los hongos desarrollan diferentes mecanismos para tolerar y superar distintas condiciones adversas (temperatura, pH, fuentes de carbono y nitrógeno, adquisición de hierro, niveles de oxígeno y dióxido de carbono) y así producir invasión y enfermedad.1,7 La pared celular fúngica desempeña un papel crucial en la interacción hospedador-hongo y participa activamente en los procesos de adhesión fúngica y fagocitosis celular. Es una estructura muy dinámica que se adapta a los diferentes cambios en su entorno y depende del ciclo de vida del hongo. Véase también: Introducción al estudio de los hongos como patógenos humanos La gran ventaja de los hongos reside en su capacidad de adaptación a diferentes ambientes Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós TENGA PRESENTE: Etapas necesarias para que un hongo sea patógeno: 1. Entrada y adhesión al tejido del hospedador (estrato córneo o super cies mucosas). 2. Invasión del tejido an trión: Por su capacidad de penetración. 3. Multiplicación, colonización y diseminación a tejidos (termotolerancia y adaptación a las condiciones físico-químicas del hospedador). 4. Evasión del sistema inmune del hospedador y daño a los tejidos. 2.2.1.1. Candida albicans La capacidad de las especies de Candida para permanecer como flora normal de la piel y mucosas en diferentes nichos anatómicos, su adaptación a ambientes extremos (fluctuaciones de pH, supresión de nutrientes) y la manifestación de de factores de virulencia (transición morfológica, cambio fenotípico, expresión de adhesinas e invasinas, tigmotropismo, formación de bio-películas y/o secreción de enzimas hidrolíticas) las hacen el modelo de mecanismo patogénico de los hongos levaduriformes. Este puede ser dividido en cuatro componentes: 1) Adhesión o anclaje; 2) Invasión; 3) Destrucción celular y alteración del sistema inmune y 4) Diseminación hematógena y formación del bio-película (Figura 7).7,15 1) Mecanismos patogénicos de Candida spp. a) Adhesión: Las proteínas de adhesión son fundamentales en la interacción de las células fúngicas; se localizan en la pared celular y permiten la comunicación con el exterior. Su acción ha sido descrita como “social”, que es el componente agregativo entre colonias, y “antisocial” que es el componente patógeno. Estas proteínas permiten la realización de los cambios morfológicos de las colonias, la formación de bio-películas, la gemación y la interacción con el hospedador.19,29 La capacidad de C. albicans para adherirse a las células epiteliales tiene componentes específicos (ej. unión proteína–proteína) y no específicos (actividad hidrofóbica). C. albicans tiene comportamiento transicional entre levadura e hifa y hay una multiplicidad de expresión de adhesinas de acuerdo a su pared estructurada. A pesar de que la colonización suele ser iniciada por las levaduras, la adhesión de las formas miceliales es mayor. Por lo tanto, las formas salvajes de Candida, incapaces de producir hifas, tienen poca capacidad de producir infección. 15,29 La interacción de las levaduras con las células epiteliales es el mayor estímulo para la formación de hifas. Una vez se inicia la conformación de las hifas, la adhesión es más fuerte y se puede continuar el proceso de invasión, que se podrá realizar dependiendo de la expresión de las moléculas en la pared celular (al estimular la destrucción celular y la invasión). 13 b) Invasión: Después de la adhesión, las hifas optimizan esta unión y desencadenan la invasión en el hospedador susceptible. Esta puede realizarse por dos mecanismos: i) Endocitosis, mediada por la célula epitelial y ii) Penetración activa, mediada por el propio hongo. 16 El mecanismo predominante depende del epitelio afectado: en las mucosas oral y vaginal se presentan los dos mecanismos, mientras que en la intestinal solo se ha descrito la penetración activa. i) Endocitosis: Es el proceso mediante el cual una invasina de la pared del hongo estimula a la célula epitelial para formar pseudópodos que engloban las hifas fuertemente adheridas. 16,17 ii) Penetración activa: Este mecanismo se presenta tardíamente con relación a la endocitosis y es independiente de la misma; en la penetración activa las hifas pueden invadir los epitelios a través de los espacios epiteliales o penetrando las células mismas. A la fecha no se ha podido determinar con precisión cuales son las moléculas de Candida involucradas en la penetración activa; pero este mecanismo es fundamental en la penetración de los epitelios estratificados, donde la funcionalidad de las células de la superficie es baja y tienen poca capacidad de realizar una endocitosis inducida. In vivo, la invasión favorece la penetración activa hasta la región submucosa, donde las células epiteliales, jóvenes y activas, pueden desarrollar la endocitosis inducida. 9,11,16 c) Destrucción celular: Una vez se ha establecido la invasión, el siguiente paso es la destrucción de las células, la cual puede presentarse mediante la necrosis o la apoptosis. Durante la invasión por penetración activa y también por endocitosis, Candida puede producir destrucción de la célula epitelial mediante proteínasas aspárticas secretadas (PAS). Estas proteínas destruyen las células epiteliales y favorecen la adhesión y penetración de nuevas hifas activas. 18 d) Diseminación hematógena y formación de bio-película: Después de completar la destrucción celular, Candida puede acceder a la circulación sanguínea a través de la filtración intersticial por penetración activa. En la sangre, la supervivencia de Candida depende de múltiples factores como la evasión de los mecanismos de defensa del hospedador (blindaje de sus proteínas de membrana, inhibición de la fagocitosis, del sistema del complemento, de la producción de citoquinas inflamatorias y de la formación de radicales libres) y la formación de bio-películas donde prácticamente es inalcanzable por los antimicrobianos (Figura 8).19,22 TENGA PRESENTE: Factores de virulencia de Candida albicans: · Termotolerancia: Tolera de 37°C a 39°C. · Presencia de adhesinas. · Expresión de proteasas, fosfolipasas y otras enzimas. · Cambios morfológicos (switching). · Hidrofobicidad. · Producción de melanina. · Formación de bio-películas. · Tigmotropismo. · Mecanismos de resistencia antifúngica. 2) Evasión de los mecanismos de defensa por Candida spp: Para evitar ser reconocida por el sistema PRR, Candida tiene recubierto el β-glucano por las proteínas de pared externa impidiendo el reconocimiento de la dectina-1, parte fundamental de PAMPS.18 Separata 2 41 Patogénesis de la enfermedad fúngica invasora Tabla 3. Papel de los factores de virulencia en la patogenicidad fúngica. Factor de virulencia Patógeno Adhesinas (familia Als, HWP1) Dimor smo (phr1, hyr1, chs2, chs3, rbf1) Cambio (switching) fenotípico (gen EFG1) Candida albicans Aspergillus spp. Cryptococcus neoformans Histoplasma capsulatum Papel en la patogenicidad Adherencia a las células epiteliales, bronectina, establecimiento de bio-películas Fase hifal, necesaria para la invasión y la adhesión Fase de levadura para diseminación Conversión a formas más virulentas que muestran un aumento de las PAS, adhesión y evasión de la respuesta del hospedador Proteinasas aspárticas secretadas (PAS 1-10) Fosfolipasas A, B, C, D (plb1, 2,3) Absorción de nutrientes, invasión de tejidos, adherencia y diseminación Invasión tisular y adherencia Farnesol Detección de quórum, formación de bio-películas Catalasa, superóxido dismutasa SUN41, GCN4, MKc1p Componente de la pared celular β-1,3-glucano Tamaño de conidias (2-3 µm) cAMP, rasA, rasB Prevención de daños oxidativos Formación de bio-películas Adhesión celular Escape de la extrusión mucociliar Absorción nutricional y crecimiento del patógeno, germinación de los conidias, rami cación hifal Degradación de la elastina en el tejido pulmonar Prevención del daño oxidativo de los macrófagos Elastasa- Proteinasa serina alcalina Catalasas (catA, catB y cat2), superóxido dismutasa Fosfolipasa C (plb1, 2,3) Gliotoxina, ácido helvolico Ribotoxina Sideróforo (gen sidA) Crecimiento a 37 ° C, hsp1, cgrA Cápsula Daño tisular y penetración Propiedades inmunosupresoras, prevención del estallido oxidativo de los macrófagos Escisión del enlace fosfodiéster en 28s rRNA eucariota Absorción del hierro del grupo heme de la sangre Capacidad de invasión tisular y supervivencia a una temperatura elevada Inhibición de la fagocitosis Melanina Manitol Fosfolipasas A, B, C, D (plb1, 2,3) Proteínasas acidas Prevención de los daños oxidativos Manipulación de los radicales hidroxilo durante estallido respiratorio Invasión tisular y adhesión Invasión tisular y diseminación Dimor smo Alteración de la super cie celular para adhesión, invasión tisular por la fase hifal, diseminación por la fase levaduriforme α-1,3-glucano de la pared celular Crecimiento en el interior de los macrófagos Catalasa Necesaria para adhesión Evasión de las células inmunes, diseminación a otros tejidos Protección contra la muerte oxidativa Adaptado de: Ahmad I, Owais M, Shahid M, Aqil F. (Eds). Combating fungal Infections: Problems and Remedy. New York, Springer-Verlag, 2010. Se ha descrito inhibición de la fagocitosis mediada por varias moléculas secretadas por Candida, entre ellas las PAS (que degradan el factor C3b), el factor H1 y la proteína Pra1(que inhiben la activación del complemento y la transformación de C3 y C4b). Dentro del macrófago la hifa se ubica en el aparato lisosomal donde inhibe de forma activa el sistema de comunicación intracelular y estructuración del fagolisosoma.10,15 Otra forma de evitar el sistema inmunológico es la interacción con el sistema oxidativo de radicales libre (ROS) del macrófago; Este bloqueo se relaciona con el metabolismo activo y la conformación vacuolar de la hifa mediante la catalasa-1 y las superóxidos dismutasas, particularmente SOD1, SOD2 y SOD4 de la superficie de Candida. En tanto que hay un proceso de activación del ROS y 42 Separata 2 disminución de la supervivencia del macrófago a través del farnesol, una molécula recientemente reconocida como miembro del quórum de detección (QMS). El farnesol favorece la destrucción del macrófago por ROS, protegiendo a la hifa mediante la activación de la catalasa1 y el sistema de superóxidos dismutasas (Tabla 3).7 Finalmente, C. albicans también puede inhibir el sistema de las citoquinas por dos mecanismos diferentes. Una glicoproteína secretada, reconocida simplemente como factor soluble, inhibe la IL–12 y el IFN-γ. Esta inhibición es dependiente de las formas viables de C. albicans (su estructura y receptor extracelular no han sido identificados hasta la fecha). Otro mecanismo involucra la inhibición del metabolismo del triptófano con aumento de los metabolitos de 5hidroxi-triptofano, que a su vez inhibe la producción de la IL-17.4,15 Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós a. Transición de levadura a hifa d. Inhibición de la degradación o sistema del complemento Candida Proteínas de super cie Hifas de Candida PRA-1 Reclutamiento de reguladores de complemento (C4-BP, factor H, FHL-1, etc.) Bloqueo de la activación/conversión de C3 Daño Evasión β-defensinas β-defensinas Escisión de los componentes del complemento Proteinasas aspárticas secretadas (PAS) Mucosa epitelial e. Inhibición de la formación del fagolisosoma b. Baja regulación de la expresión epitelial de TLR4 Fagosomas Mucosa epitelial Lisosomas Hifas de Candida c. Blindaje de PAMPS - No reconocimiento por los PRR f. Modulación de la función de las células T Dectina-1 Blindaje del β-glucano Monocitos/ Macrófagos Exposición de β-glucano Dectina-1 Células dendríticas Th 1 IFNy Th 2 IL-10 Th 17 IL-17 Las líneas negras indican los mecanismos de defensa del hospedador Las líneas rojas indican invasión por Candida albicans y sus mecanismos de evasión de la respuesta inmune TLR: Receptores tipo Toll; PAMPS: Patrones moleculares asociados a patógenos; Th: Linfocitos T ayudadores; C: Complemento; IL: Interleuquina; IFN-γ: Interferon gama PRR: Receptores de reconocimiento de PAMPS. Adaptado de: Cheng SC y col.10 Figura 8: Estrategias de evasión de Candida albicans a la respuesta inmune innata del hospedador. a) Formación de bio-película: Un paso fundamental en la patogenia de Candida, particularmente asociada a las infecciones intrahospitalarias, es la formación de bio-películas sobre los dispositivos protésicos como catéteres endovasculares y prótesis articulares, o sobre estructuras orgánicas como las válvulas cardiacas. Las bio-películas fúngicas son comunidades de levaduras o mohos embebidas en una matriz extracelular fundamentalmente polisacárida, altamente hidratada, comunicadas al exterior mediante canales de agua, y desarrolladas sobre superficies biológicas e inertes. La mayor resistencia de las bio-películas de Candida al tratamiento antifúngico convencional hacen necesaria, casi siempre, la retirada del dispositivo. En la formación de la bio-película ha sido involucrada principalmente la proteína de choque térmico de 90 kD, perteneciente a la familia de las adhesinas (Als); 19 esta proteína es esencial para la diseminación del bio-película. La producción de la matriz extracelular es controlada por factores adicionales como el factor de transcripción dependiente de zinc (Zap1). Este factor regula negativamente el β 1,3glucano, componente esencial de la matriz; en tanto que las glucamilasas (Gca 1, Gca 2), las glucan-transferasas (Bgl2, Phr 1) y la exo-glucasa Xog1 son reguladores positivos de la producción de β 1,3glucano. La bio-película confiere protección a Candida contra la acción de los neutrófilos bloqueando la actividad de los ROS. El contacto directo de las hifas con la superficie epitelialo endotelial y, en particular, la presencia de “crestas” genera el crecimiento direccional de las hifas (tigmotropismo) y la conformación de las bio-películas (Figura 9).22 2.2.1.2. Aspergillus spp. Los hongos del género Aspergillus son saprofitos y juegan un papel en el sistema global de intercambio de carbono y nitrógeno; su nicho natural es el suelo y la vegetación en putrefacción, sus conidias se dispersan fácilmente y resisten las condiciones medioambientales. Se reconocen más de 200 especies diferentes, incluso con actividades esenciales para la industria alimentaria, farmacéutica y agrícola. Entre las especies patógenas para el hombre destacan A. fumigatus, Aspergillus flavus, Aspergillus terreus, Aspergillus niger y Aspergillus nidulans. El espectro de enfermedad en humanos depende de múltiples factores, desde la hipersensibilidad a sus componentes hasta la inmunosupresión extrema. En individuos con asma o fibrosis quística la manifestación más frecuente es la aspergilosis broncopulmonar alérgica. Cuando hay lesiones cavitadas preexistentes, la exposición recurrente a conidias puede asociarse a bolas de hongos no invasivas. Finalmente, la forma más grave de aspergilosis se presenta en pacientes muy inmunodeprimidos; En especial los que presentan enfermedad maligna hematológica, los sometidos a trasplantes de órganos, los usuarios crónicos de esteroides y los pacientes con sida (Figura 10).23 Separata 2 43 Patogénesis de la enfermedad fúngica invasora Adhesión Dimor smo Células del hospedador Invasión Cambio fenotípico Tigmotropismo Blanca Opaca Daño Formación de bio-película Capacidad de adaptación Respuesta al estrés Captación de aminoácidos Mediada por HsPs Regulación del pH (alcalinización) Excreción de NH3 Catéter Absorción de Fe, Zn, Cu, Mn Captura de C, N HsPs: Proteínas de choque térmico; NH3: Amoníaco; C: Carbono; N: Nitrógeno; Fe: Hierro; Zn: Zinc; Cu: Cobre; Mn: Manganeso. Adaptado de: Mayer FL y col.19 Figura 9. Mecanismos de patogenicidad de Candida albicans. En pacientes inmunocompetentes, la inhalación de esporas de Aspergillus es rápidamente neutralizada por los neutró los y macrófagos, frenando la invasión o el desarrollo de la infección El ciclo infeccioso se inicia con la inhalación de las conidias que se depositan en los bronquiolos y alvéolos. En las personas sanas estas conidias son removidas por el sistema ciliar, las no removidas son fagocitadas por los macrófagos alveolares, desencadenando una respuesta inflamatoria de defensa y estimulando el reclutamiento de polimorfonucleares que infiltran el lugar de la infección y terminan destruyendo las hifas que habían escapado a la actividad alveolar. La pérdida de la capacidad de responder en cada una de estas líneas se asocia a diferentes manifestaciones de la enfermedad (Figuras 11 y 12).23 1) Mecanismos patogénicos de Aspergillus spp. a) Tamaño de las conidias: Aunque potencialmente todas las especies de Aspergillus pueden producir enfermedad, A. fumigatus tiene conidias de 2-3 µm que penetran más profundamente en el árbol 44 Separata 2 TENGA PRESENTE: Factores de virulencia que facilitan la infección por mohos: · Termotolerancia. · Conidias infectantes de pequeño tamaño. · Crecimiento acelerado in vivo. · Crecimiento apical de las hifas. · Tendencia a la angioinvasión. · Presencia de adhesinas. · Producción de enzimas líticas. · Expresión de proteasas. · Secreción de micotoxinas. · Producción de melanina. bronquial, mientras que las conidias de mayor tamaño de A. flavus y A. niger son fácilmente capturadas en el moco y barridas por el sistema ciliar. En condiciones normales una persona puede inhalar diariamente cerca de 200 conidias.23,27,30,31 b) Termotolerancia y estrés metabólico: A. fumigatus es más Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Cornetes nasales Conidias inhaladas por el aire Epitelio de las vías respiratorias Membrana basal Lumen de la tráquea Fluido en la interfase aérea Lumen del espacio aéreo Receptores unidos a las células Receptores solubles Macrófago alveolar Epitelio de las vías respiratorias Epitelio alveolar Células dendríticas Espacio intersticial Células dendríticas Nódulo linfático Lumen capilar PMN Diferenciación de células Th NK Monocitos Th: Linfocitos T ayudadores; PMN: Polimorfonucleares; NK: Células asesinas naturales. Adaptado de: Park SJ y Mehrad B.25 Figura 10. Respuesta inmune del hospedador a la inhalación de conidias de Aspergillus spp. termotolerante que otras especies, crece bien a 37oC y soporta temperaturas por encima de 50 o C, propias de la materia en descomposición. El estrés generado a altas temperaturas podría inducir en A. fumigatus, la expresión de genes que aumentan su capacidad de virulencia. De otra parte, se ha demostrado que la capacidad de crecer en forma radiada a 37oC se relaciona con una mayor patogénesis. Además, la albúmina humana parece favorecer el crecimiento de A. fumigatus a temperaturas mayores de 37oC, pero no a 25oC.30 condiciones de pH alcalino, muy lejano de las condiciones de los mamíferos. Se ha descrito un factor regulador del pH inducido por el gen pac-C que activa un sistema de expresión alcalina y reduce la acción acidificante. No se ha identificado claramente los genes involucrados en el proceso de adaptación; sin embargo, se reconocen algunas familias de factores de transcripción como los elementos reguladores de esterol unidos a proteínas, que son fundamentales para la adaptación del metabolismo en condiciones bajas de oxígeno (propias de los tejidos infectados).23,27 c) Crecimiento y transformación de conidias: Se realiza en d) Adhesión al epitelio: Durante la injuria pulmonar aguda y el Separata 2 45 Patogénesis de la enfermedad fúngica invasora Inhalación de conidias y fragmentos de micelio Limpieza mucociliar Hifas Serina proteasa y otras proteasas Gliotoxina, ácido hevolico, fumagilina Verruculogen Conidias Colonización Invasión Epitelio alveolar Residuos de acido siálico Daño de la membrana basal Epitelio respiratorio Revestimiento de la tráquea, bronquios y bronquiolos Los productos fúngicos (en rojo) pueden aumentar la colonización por lesión tisular y la adhesión a las células epiteliales o a la membrana basal dañada Adaptado de: Dagenais TR y Keller NP.23 Figura 11. Interacción de Aspergillus fumigatus con el epitelio respiratorio. distress respiratorio, el ácido síalico de la conidia de A. fumigatus metabólicas que involucran a estos elementos en un factor de facilita la unión y captación por las células epiteliales y la posterior invasión del epitelio lesionado (por adhesión mediada por la laminina y fibronectina). Las especies más patogénicas presentan mayores cantidades de ácido siálico. Además, en el epitelio sano A. fumigatus puede inducir daño a través de la producción de una proteasa dependiente de serina y cisteína.28 patogénesis (debido al escaso contenido de nitrógeno en los mamíferos). Por su parte, el carbono es tomado directamente del hospedador por múltiples vías, siendo llamativa la capacidad de crecimiento de A. fumigatus en presencia de lípidos. e) Evasión de la fagocitosis: i) Melanina: Aspergillus puede enmascarar el β 1, 3-glucano y retardar la activación de la fagocitosis y destrucción de las conidias gracias al barrido de radicales libres de oxígeno producido por la melanina. ii) Enzimas proteolíticas: Las cepas patógenas de Aspergillus son ricas en la producción de elastasas. Todos los aislamientos clínicos producen esta proteasa y solo dos terceras partes de los aislamientos saprófitos tienen esa capacidad. La producción de proteinasa ácida y de fosfolipasa también pueden ser expresadas por las diferentes especies de Aspergillus en proporción directa con su actividad patogénica; A. fumigatus puede expresar todas las enzimas proteolíticas en tanto que A. flavus solo elastasas y A. niger solo fosfolipasa.23,27 iii) Componentes nutricionales: Las necesidades metabólicas naturales de Aspergillus de carbono y nitrógeno convierten las vías 46 Separata 2 iv) Captación del hierro: La habilidad de captación de hierro por los hongos patógenos es un factor de virulencia reconocido en múltiples especies. A. fumigatus usa dos sistemas de captación de hierro, uno mediado por cuatro pequeños sideróforos: triacetilfuricina C y fusaricina C (que actúan extracelularmente) y ferricrocina e hidroxiferricocina (que almacenan el hierro dentro de las hifas y conidias). Otros mecanismos de captación se basan en la reducción y asimilación. v) Metabolitos secundarios: La síntesis de metabolitos secundarios puede contribuir a la patogénesis de A. fumigatus durante la formación de las hifas, el más representativo de este grupo es una glicotoxina (epipolitiodioxopiperazina) aislada en la AI. Su actividad biológica se relaciona con la unión y desactivación de proteínas a través de un puente disulfuro en su interior, el cual también afecta el sistema de radicales libres de oxígeno. La inactivación de estos sistemas por la gliotoxina se relaciona con un proceso de inmunosupresión de múltiples pasos que involucra el sistema de fagocitosis, la actividad mitogénica y la citotoxicidad de las células T, la capacidad de reconstitución inmunológica secundaria a la irradiación y la actividad del aclaramiento Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Tabla 4. Factores que incrementan el riesgo de EFI en el paciente hematológico. Factores de riesgo de EFI Factor favorecedor de la siopatogenia de la infección Candidiasis invasora Colonización Aumento del inóculo Uso previo de antibióticos Alteración en la ora digestiva con aumento del inóculo fúngico Aspergilosis invasora Edad avanzada Sustitución progresiva con la edad de la respuesta Th1 por Th2 Enfermedad de base Diferente cinética de reconstitución de la respuesta inmunitaria ante un mismo procedimiento de trasplante según la enfermedad de base del paciente Mayor compatibilidad entre donante y receptor se asociará a menor alteración inmunológica Alteración de la mucosa respiratoria favoreciendo la invasión Inmunomodulación: Producción de IL-10 por el virus Aumento de la respuesta Th2 Características del trasplante Infección por virus respiratorios Número de transfusiones Candidiasis y aspergilosis invasora Citopenia Enfermedad de injerto contra el hospedador Disminución de las células de defensa Alteración de la mucosa digestiva favoreciendo la invasión Alteración en el balance Th1 y Th2 Alteración funcional de linfocitos y monocitos Inmunomodulación: Producción de IL-10 por el virus Múltiples efectos a diferentes niveles del sistema inmunitario Infección por citomegalovirus Corticoterapia Th: Linfocitos T ayudadores; IL: Interleuquina Tomado de: Garcia-Vidal C. y Carratalà J.2 Neutró los β (1,3)- Glucano Conidias Dectina-1 PRRs Melanina ROS Gliotoxina (Otros metabolitos secundarios) Catalasas Superóxido dismutasa Acidi cación lisosomal ROS Superóxido dismutasa Proteasas, elastasas, fosfolipasas Hifas Macrófagos alveolares Invasión tisular Adquisición de nutrientes Los nombres y líneas rojas indican los productos fúngicos que pueden contribuir a la patogenicidad fúngica PRR: Receptores de reconocimiento de PAMPS; ROS: Sistema oxidativo de radicales libres. Adaptado de: Dagenais TR y Keller NP.23 Figura 12. Interacción de Aspergillus fumigatus con los fagocitos. Separata 2 47 Patogénesis de la enfermedad fúngica invasora ciliar. Adicionalmente, induce daño epitelial e inducción de apoptosis en linfocitos, fagocitos, células dendríticas, hepáticas y fibroblastos a través de la inducción de FNT, el sistema de caspasas y los radicales libres de oxígeno. La gliotoxina también puede inhibir la presentación de antígenos por monocitos y células dendríticas así como la activación del sistema oxidativo NADPH de los neutrófilos. Se han descrito otros 13 metabolitos secundarios, pero su capacidad de virulencia no ha sido completamente elucidada; dentro de los más relevantes se incluyen la restritoxina (una ribonucletoxina que puede afectar la acción de los neutrófilos y parece ser relevante en individuos no neutropénicos), la festuclavina (un alcaloide de ergot que podría interferir el sistema de regulación de los mamíferos mediante la captación de serotonina, dopamina y adrenalina), y la fumagilina (un sesquiterpeno inhibidor potente de la angiogénesis asociada con la invasión de las hifas) (Tabla 3).30,31 TENGA PRESENTE: Atributos de virulencia de Aspergillus fumigatus: · Termotolerancia. · Morfología de conidias. · Producción de metabolitos secundarios. · Producción de enzimas de degradación. · Resistencia al estrés oxidativo. No es muy virulento, pero tiene atributos para sobrevivir en pacientes inmunodeprimidos 2.2.2. El paciente La manifestación de una enfermedad invasora dependerá en gran medida del tipo de hospedador implicado. Los pacientes de mayor riesgo para una EFI son los pacientes inmunodeprimidos (con enfermedades hematológicas malignas, receptores de trasplante de progenitores hematopoyéticos o de órganos sólidos y aquellos con inmunodeficiencias congénitas o adquiridas) y los pacientes críticos. La prevalencia, epidemiología y evolución de una EFI dependerá de factores relacionados con el paciente como: los defectos inmunitarios inherentes, la presencia de factores biológicos o condiciones subyacentes o los diferentes regímenes de acondicionamiento para tratar la enfermedad de base, siendo importante el antecedente de una EFI previa (Tabla 4).2 Aunque la infección por hongos es común la enfermedad fúngica invasora es rara 2.2.2.1. Candidiasis invasora La principal fuente de infección por Candida spp. es endógena (previa colonización de la piel o mucosas), aunque también puede trasmitirse a través de material infectado, personal sanitario o desde otros pacientes. La supresión de la flora bacteriana habitual del tracto intestinal, por la acción de antibacterianos de amplio espectro, facilita la proliferación de levaduras en el tubo digestivo y aumenta el riesgo del paso al torrente sanguíneo (a través del epitelio intestinal) por fenómenos de translocación. Una estancia hospitalaria prolongada, la presencia de accesos venosos o sondas vesicales, la 48 Separata 2 TENGA PRESENTE: Mecanismos de resistencia del Homo sapiens frente a las infecciones fúngicas: · Temperatura central: 37-39°C: los hongos patógenos se diseminan entre 25-35°C. · pH alcalino en los uidos corporales: los hongos patógenos pre eren pH levemente ácido a neutro. · Inmunidad innata y adaptativa que elimina rápidamente las partículas fúngicas. Véase también: Evaluación del riesgo de la Enfermedad Fúngica Invasora administración de nutrición parenteral, los antecedentes de una cirugía gastrointestinal y el uso previo de antibióticos de amplio espectro o antiácidos gástricos son situaciones habituales en pacientes hospitalizados y se han descrito como los factores de riesgo más frecuentes que favorecen una EFI por levaduras (Tabla 4).1 Existen dos poblaciones con un alto riesgo de padecer una CI, los pacientes con una neoplasia hematológica y los pacientes que se encuentran en una unidad de cuidados críticos (UCI), aunque no siempre es comparable su manejo y pronóstico. Al ingreso hospitalario entre un 5-15% de pacientes están colonizados por Candida, porcentaje que aumenta a un 50-85% si el paciente ingresa en una UCI o sufre una estancia prolongada. Ambas situaciones se asocian con un mayor número de complicaciones, mayor número de intervenciones invasivas y uso de antibióticos de amplio espectro. Las intervenciones invasivas (uso de catéter vasculares, sondas vesicales, nutrición parenteral o cirugía gastrointestinal) provocan una disrupción de la piel y mucosas favoreciendo la invasión fúngica. Además, el uso de antibióticos y antiácidos se relaciona con un cambio en la flora intestinal que favorece el crecimiento de Candida spp. La ausencia o disfunción de los neutrófilos está relacionada con una disminución en la capacidad de hospedador para oponer resistencia a las infecciones. Los corticoides alteran la función de linfocitos, neutrófilos, monocitos, macrófagos y células dendríticas y disminuyen la capacidad para crear una respuesta inmunológica mediada por Th1, aumentando la producción de citoquinas relacionadas con los Th2. Además, actúan sobre el propio hongo favoreciendo la capacidad de adherencia las mucosas y los procesos de translocación de Candida del tubo digestivo a la sangre. 2.2.2.2. Aspergilosis invasora La AI es la principal causa de EFI causada por mohos debido al incremento continuo de las poblaciones en riesgo (Figura 13). La principal fuente de infección por Aspergillus spp. es exógena (vía inhalación). La evolución final de la AI depende de diferentes factores concurrentes como la enfermedad subyacente, los procedimientos a los que es sometido el paciente, el estado neto de inmunosupresión, localización geográfica, virulencia y sensibilidad antifúngica del propio patógeno y, obviamente, de la estrategia terapéutica seleccionada y del agente antifúngico elegido en el marco de una terapia integral.2,23 Carlos Humberto Saavedra, Pilar Rivas, Javier Pemán, Guillermo Quindós Sin olvidar el papel que juega la profilaxis antimicrobiana prolongada y el antecedente de tratamiento antifúngico que pueden seleccionar cepas con resistencia a los antifúngicos disponibles. También debe considerarse el ambiente externo e interno del hospedador como resultado de las enfermedades de base, los posibles hábitos del hospedador que aumenten el riesgo de exposición a partículas fúngicas, así como posibles cambios en explotaciones agrícolas que faciliten el contacto accidental con hongos emergentes.1,19 Figura 13: Micelios septados en ángulo agudo compatibles con una aspergilosis pulmonar en tejido pulmonar (Tinción de H&E, 1000X). (Cortesía: Dra. Pilar Rivas) Los factores específicos promotores de un mayor riesgo en estas poblaciones más susceptibles son muy variados e incluyen: neutropenia (recuento de polimorfonucleares <500/µl), antibioterapia prolongada de amplio espectro, corticoterapia y uso de otros agentes inmunosupresores, quimioterapia, colonización por Aspergillus, infección por citomegalovirus o por P. jirovecii, fuente de progenitores hematopoyéticos, enfermedad del injerto frente al hospedador, grado de concordancia en el HLA (anticuerpos de los leucocitos humanos) del trasplante de progenitores hematopoyéticos, así como el uso de terapias biológicas y de análogos de nucleósidos. El colectivo con mayor riesgo de AI lo constituyen los pacientes receptores progenitores hematopoyéticos, ya que este tipo de trasplante supone un proceso complejo asociado a una alteración del sistema inmunitario. La manifestación de la EFI dependerá de las características del hospedador, el tipo de trasplante, las complicaciones o y las terapias inmunosupresoras pos-trasplante (Tabla 4).23 Aquellas enfermedades que comporten una alteración más prolongada del sistema inmune se relacionan con un mayor riesgo de EFI Las características del trasplante tiene una relación directa con el riesgo de una AI, el grado de compatibilidad entre el donante y el receptor, está relacionado de manera directa con la alteración inmunitaria del receptor: a mayor disparidad del HLA, mayor grado de rechazo del injerto y mayor necesidad de intensificar la terapia inmunosupresora. 2.2.3. El medio ambiente El entorno circundante del paciente y con los esquemas de profilaxis antimicrobiana también influyen en el riesgo de EFI. Los diferentes patógenos (Candida spp., Aspergillus spp., Paecilomyces spp., Fusarium spp., Scedosporium spp. y otros hongos filamentosos) pueden ser adquiridos en la comunidad o en el medio hospitalario, ya sea por condiciones micro-ambientales, fómites o personal sanitario. La virulencia de los hongos patógenos está relacionada con su capacidad de adaptación y supervivencia a condiciones ambientales extremas. Estos determinantes de virulencia se expresan, con gran diversidad, en plantas y animales susceptibles. La interacción microbiana favorece la selección de especies con fuerte capacidad de adaptación, donde las etapas de colonización, comensalismo e infección también son un proceso continuo de adaptabilidad. La evolución de este proceso tiene muchos determinantes, pero los factores de virulencia del agente son de gran importancia (Figura 6). TENGA PRESENTE: Condiciones ambientales que facilitan las infecciones fúngicas: · Sistemas eco-biológicos nuevos. · Destrucción de bosques. · Construcciones, remociones de tierras. · Desvío de corrientes uviales. · Exposición a aerosoles con conidias. · Traumatismos. · Exposición ocupacional o habitacional. · Calamidades naturales (tornados y terremotos). La mayoría de aislamientos de hongos emergentes causantes de EFI tienen una fuente exógena y sus esporas pueden persistir en el medio ambiente durante largo tiempo TENGA PRESENTE: La comprensión de los mecanismos de patogenicidad durante una infección invasora, es fundamental para un diagnóstico y manejo adecuado de la patología infecciosa. Separata 2 49 Patogénesis de la enfermedad fúngica invasora AI Acs C CI DC CMH EFI FNT HLA IFN-γ IMC IL MBL MR MyD88 PAMPS PAS PRR ROS TGF-β Th TLR Treg Aspergilosis invasora Anticuerpos Complemento Candidiasis invasora Células dendríticas Complejo mayor de histocompatibilidad Enfermedad fúngica invasora Factor de necrosis tumoral Anticuerpos de los leucocitos humanos Interferón gamma Inmunidad mediada por células Interleuquina Lectina ligadora de manosa Receptor de manosa Proteína de diferenciación mieloide de respuesta primaria Patrones moleculares asociados a patógenos Proteinasas aspárticas secretadas Receptores de reconocimiento de PAMPS Sistema oxidativo de radicales libres Factor de crecimiento tumoral beta Linfocitos T ayudadores (Helper) Receptores tipo Toll Linfocitos T reguladores LECTURAS SUGERIDAS 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 50 Garry C. Fungal Pathogenesis. En Anaissie Elias, McGinnis Ml, Pfaller M. (Eds.) Clinical Mycology. New York, Churchill Livistone, 2003. García-Vidal C, Carratalà J. Patogenia de la infección fúngica invasora. Enferm Infecc Microbiol Clin. 2012; 30(3): 151-8. Romani L. Immunity to fungal infections. Nat Rev Immunol. 2004; 4: 1-13. Romani L. Immunity to fungal infections. Nat Rev Immunol. 2011; 11: 275-288. Kavanagh K. (Ed). New Insights in Medical Mycology. Dordrechet, The Nehtherlands, Springer Science, 2007. Netea MG, Marodi L. Innate immune mechanisms for recognition and uptake of Candida species. Trends Immunol. 2010; 31: 346 353. Calderone RA, Fonzi WA. Virulence factors of Candida albicans. Trends Microbiol. 2001; 9: 327 335. Weindl G, Wagener J, Schaller M. Epithelial Cells and Innate Antifungal Defense. J Dent Res. 2010; 89(7): 666-675. Jouault T, Ibata-Ombetta S, Takeuchi O, Trinel PA, Sacchetti P, L e f e b v r e P, A k i r a S , P o u l a i n D. C a n d i d a a l b i c a n s phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003; 188: 165 172. Cheng SC, Joosten LA, Kullberg BJ, Netea MG. Interplay between Candida albicans and the mammalian innate host defense. Infect Immun. 2012 Apr; 80(4): 1304-13. Brown G. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011; 29: 1-21. Gow NA, et al. Immune recognition of Candida albicans betaglucan by dectin-1. J Infect Dis. 2007; 196: 1565 1571. Ashman RB, Papadimitriou JM. Production and function of cytokines in natural and acquired immunity to Candida albicans infection. Microbiol Rev. 1995; 59(4): 646-72. Separata 2 14. Ashman RB, Papadimitriou JM, Ott AK, Warmington JR. Antigens and immune responses in Candida albicans infection. Immunol Cell Biol. 1990; 68: 1 13. 15. Whiteway M, Bachewich C. Morphogenesis in Candida albicans. Annu Rev Microbiol. 2007; 61: 529 553. 16. Zhu W, Filler SG. Interactions of Candida albicans with epithelial cells. Cell Microbiol. 2010; 12: 273 282. 17. Naglik JR, Moyes DL, Wächtler B, Hube B. Candida albicans interactions with epithelial cells and mucosal immunity. Microbes Infect. 2011; 13(12-13): 963-76. 18. Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev. 2003; 67(3): 400-28. 19. Mayer FL, Wilson D, Hube B. Candida albicans pathogenicity mechanisms. Virulence. 2013; 15; 4(2): 119-28. 20. Garlanda, C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, Maccagno A, Riva F, Bottazzi B, Peri G, Doni A, Vago L, Botto M, De Santis R, Carminati P, Siracusa G, Altruda F, Vecchi A, Romani L, Mantovani A. Non-redundant role of the long pentraxin PTX3 in antifungal innate immune response. Nature; 2002; 420: 182 186. 21. Lawson PR, Reid KB. The roles of surfactant proteins A and D in innate immunity. Immunol Rev. 2000; 173: 66 78. 22. Brand A, Shanks S, Duncan VM, Yang M, Mackenzie K, Gow NA. Hyphal orientation of Candida albicans is regulated by a calciumdependent mechanism. Curr Biol. 2007; 17: 347-52. 23. Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis. Clin Microbiol Rev. 2009; 22(3): 447-65. 24. Falvey DG, Streifel AJ. Ten-year air sample analysis of Aspergillus prevalence in a university hospital. J Hosp Infect. 2007; 67: 35 41. 25. Park SJ, Mehrad B. Innate immunity to Aspergillus species. Clin Microbiol Rev. 2009; 22(4): 535-51. 26. Gersuk GM, Underhill DM, Zhu L, Marr KA. Dectin-1and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J. Immunol. 2006; 176: 3717 3724. 27. Behnsen J., Hartmann A, Schmaler J, Gehrke A, Brakhage A, Zipfel PF. The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect Immun. 2008; 76: 820 827. 28. Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells ght against respiratory pathogens. Eur Respir J. 2004; 23: 327 333. 29. Dranginis AM, Rauceo JM, Coronado JE, Lipke PN. A biochemical guide to yeast adhesins: glycoproteins for social and antisocial occasions. Microbiol Mol Biol Rev. 2007; 71(2): 282-94. 30. Bhabhra R, Miley MD, Mylonakis E, Boettner D, Fortwendel J, Panepinto JC, Postow M, Rhodes JC,Askew DS. Disruption of the Aspergillus fumigatus gene encoding nucleolar protein CgrA impairs thermotolerant growth and reduces virulence. Infect Immun. 2004; 72: 4731 4740. 31. Bok JW, Chung D, Balajee SA, Marr KA, Andes D, Nielsen KF, Frisvad JC, Kirby KA, Keller NP. Gli Z, a transcription al regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect Immun. 2006; 74: 6761 6768. Material de uso exclusivo para el cuerpo médico COECA1413033 Glosario de Abreviaturas