(48,/,%5,265('2;
,1752'8&&,Ï1
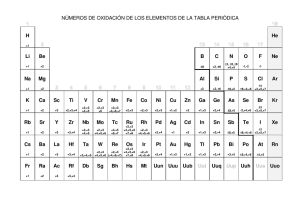
Ë1',&(2(67$'2'(2;,'$&,Ï1
$-867('((&8$&,21(65('2;
3,/$'$1,(//2&(/'$*$/9È1,&$
7,326'((/(&752'26
327(1&,$/'((/(&752'2
&/$6,),&$&,21'(/26$*(17(62;,'$17(6<
5('8&725(6
(&8$&,Ï1'(1(567
62%5(327(1&,$/
&$032'((67$%,/,'$''(/$*8$
'(6352325&,21&20352325&,21
(/(&752/,6,6
&25526,Ï1
3,/$6
9$/25$&,215('2;
6(5,((/(&75248,0,&$
1
,1752'8&&,Ï1
Antiguamente se entendía por oxidación a aquellos procesos en que una
sustancia tomaba oxígeno y por reducción a aquellos otros en que éste se liberaba.
Posteriormente, se aceptó la reducción como aquellas reacciones en las que se fijaba
hidrógeno y oxidación en las que éste se liberaba. Actualmente se considera como
reacciones redox, o de oxidación reducción a aquellas en las que cambia el estado o
grado de oxidación de las especies reaccionantes porque se produce un intercambio de
electrones entre los reactivos, aunque no intervengan en ellas ni el oxígeno ni el
hidrógeno.
Para que se produzca una reacción redox es necesario la presencia de una
especie que ceda electrones (reductor) y otra especie que acepte electrones (oxidante).
Tras la reacción redox, el reductor se transforma en su forma oxidada y el oxidante en
su forma reducida:
2[LGDQWHQH )RUPDUHGXFLGDGHOR[LGDQWH 5HGXFFLyQ 5HGXFWRUQH )RUPDR[LGDGDGHOUHGXFWRU 2[LGDFLyQ Reacción global:
2[LGDQWH5HGXFWRU5HGXFWRU2[LGDQWH
El concepto de reacciones redox recuerda al de las reacciones ácido base de
Bronsted-Lowry. Ambas implican la transferencia de una o más partículas cargadas
desde un dador a un aceptor, siendo éstas los electrones en las redox y los protones en
las de neutralización.
Ejemplos de reacciones redox:
El Ce4+ es un oxidante fuerte que oxida el Fe2+ a Fe3+, reduciendose a Ce3+:
Ce4+ + Fe2+ Ce3+ + Fe3+
Si se introduce hierro metálico en una disolución que contenga Sb3+ se forman copos de
Sb metálico y se puede detectar la presencia de Fe2+ en la disolución:
3Fe + 2Sb3+ 3Fe2+ + 2Sb
La disolución de Aluminio metálico en ácido clorhídrico es también una reacción redox:
2Al + 6H+ 2Al3+ + 3H2
Puede observarse que:
2
En las reacciones redox pueden intervenir, bien como reactivos o como
productos de reacción, átomos, iones o moléculas que pueden encontrarse en estado
sólido, en disolución y en forma gaseosa.
/DR[LGDFLyQHVXQSURFHVRHQHOTXHXQDHVSHFLHSLHUGHXQRRPiVHOHFWURQHV
GHIRUPDTXHFXDQGRXQHOHPHQWRVHR[LGDVXHVWDGRGHR[LGDFLyQWRPDYDORUHVPiV
SRVLWLYRV8QDHVSHFLHR[LGDQWHHVDTXHOODTXHJDQDHOHFWURQHVUHGXFLpQGRVHGXUDQWH
HOSURFHVR.
(QODUHGXFFLyQKD\JDQDQFLDGHHOHFWURQHVHOHOHPHQWRTXHVHUHGXFHWRPD
YDORUHV PiV QHJDWLYRV GH VX HVWDGR GH R[LGDFLyQ 8Q DJHQWH UHGXFWRU HV DTXHO TXH
SLHUGH HOHFWURQHV HQ XQD UHDFFLyQ R[LGiQGRVH HQ HO SURFHVR $PERV SURFHVRV
R[LGDFLyQ\UHGXFFLyQWLHQHQTXHYHULILFDUVHVLPXOWiQHDPHQWH
,1',&(2(67$'2'(2;,'$&,Ï1
Cuando en un proceso redox intervienen sustancias covalentes es dificultoso
detectar las especies involucradas en la transferencia electrónica. Para salvar esta
adversidad se introdujo el concepto de número de oxidación, también llamando índice o
estado de oxidación. Este representa la carga que tendría un átomo en cuestión
considerando que los únicos enlaces que forman la especie química en la interviene
dicho elemento son iónicos. En los compuestos iónicos, el número de oxidación
coincide con la carga eléctrica de los iones. En compuestos covalentes el estado de
oxidación representa una carga eléctrica fictícea, ya que en este enlace la transferencia
de electrones entre los átomos involucrados no es total, cuya determinación puede
llevarse a cabo aplicando las siguientes reglas:
a) La carga eléctrica total de una molécula es nula.
b) El estado de oxidación de los elementos en estado fundamental o sin combinar es
nulo.
c) El H tiene un estado de oxidación ±1.
d) El O tiene un estado de oxidación -2, excepto en los peróxidos O22- en los que es -1 y
en los superóxidos O2- que es -1/2.
e) El estado de oxidación de los alcalinos es +1.
f) El estado de oxidación de los alcalinotérreos es +2.
g) El estado de oxidación del flúor es siempre -1.
3
h) El estado de oxidación de una especie iónica es igual a la suma algebraica de los
estados de oxidación de todos los átomos que la forman.
i) El estado de oxidación de un átomo central en una molécula se calcula por diferencia,
de forma que, la carga total de la molécula sea nula.
j) En compuestos orgánicos se ha de tener en cuenta que:
j1) Al N se le asigna estado de oxidación -1, -2 ó -3 según esté unido a un
carbono por un enlace sencillo, doble o triple.
j2) A los halógenos se les asigna un estado de oxidación -1.
j3) A los grupos alquilo o arilo se les asigna un estado de oxidación +1.
j4) Al oxígeno se le asigna un estado de oxidación -2 si está unido al carbono
por un doble enlace y -1 si se une mediante un enlace sencillo.
$-867('((&8$&,21(65('2;
Existen dos métodos para ajustar ecuaciones redox:
1) Método del número de oxidación
2) Método del ión electrón
En la reacción ajustada se ha de cumplir la ley de conservación de la masa,
conservación de las cargas eléctricas (la suma algebraica de las cargas eléctricas en los
dos miembros de la ecuación ha de ser la misma) y que el número de electrones cedidos
por el agente reductor sea igual al número de electrones captados por el agente oxidante.
(QDPERVPpWRGRVFRPSUREDUDOILQDOTXHVHKDDMXVWDGRODFDUJD\ODPDVD
D 0pWRGRGHOFDPELRHQHOQ~PHURGHR[LGDFLyQ
a1) Asignar números de oxidación a los átomos que cambian su número de oxidación.
a2) Escribir las semirreacciones con los átomos que cambian su estado de oxidación,
ajustando con electrones la carga en ambos miembros.
a3) Multiplicar cada semirreacción por un número que haga que los electrones cedidos
por los átomos que se oxidan sean iguales a los electrones ganados por los átomos que
se reducen.
a4) Sumar las semirreacciones eliminando los electrones.
a5) Disponer en la ecuación molecular los exponentes obtenidos en el paso anterior.
a6) Ajustar adecuadamente las demás sustancias que intervienen en la reacción.
Ajustar las siguientes reacciones:
4
Cu + HNO3
:Cu(NO3)2 + NO + H2O
3(Cu0 – 2e- Cu2+) oxid
2(N5+ + 3e- N2+) red
3Cu0 + 2N5+ 3Cu2+ + 2N2+
3Cu + 8HNO3 3Cu(NO3)2 + 2NO + 4H2O
Sb2S5 + HNO3
HSbO3 + S + NO + H2O
CH3CH2OH + KMnO4 + H2SO4
CH3COOH + K2SO4 + MnSO4 + H2O
P4 + KOH + H2O
KH2PO2 + PH3
E 0pWRGRGHOLyQHOHFWUyQ
b1) Asignar números de oxidación a los átomos que cambian su estado de oxidación.
b2) Escribir las semirreacciones para las especies oxidantes y reductoras. Si la reacción
tienen lugar en medio ácido, añadir H2O al miembro de la ecuación deficiente en H+. Si
la reacción tiene lugar en medio básico, por cada O en exceso se añade 1 H2O a ese
miembro y 2HO- en el otro. Entonces, ajustar con electrones la carga eléctrica en ambos
miembros.
b3) Multiplicar cada semirreacción por un no que haga que los electrones cedidos por
los átomos que se oxidan sean iguales a los electrones ganados por los átomos que se
reducen.
b4) Sumar las semirreacciones eliminando los electrones y las especies comunes en
ambos miembros.
b5) Disponer en la ecuación molecular los exponentes obtenidos en el paso anterior.
Ajustar las siguientes reacciones:
K2Cr2O7 + HI + HClO4 Cr(ClO4)3 + I2 + H2O + KClO4
Cr2O72- + 14H+ + 6e- 2Cr3+ + 7H2O (Reducción)
3 (2I- I2 + 2e-) (Oxidación)
K2Cr2O7 + 6HI + 8HClO4 2Cr(ClO4)3 + 3I2 + 7H2O + 2KClO4
5
La reacción entre el permanganato potásico y el ioduro potásico en presencia de
potasa acuosa conduce a la formación de manganato de potasio, iodato potásico y agua.
Ajustar la reacción:
MnO4- + I-+ HO- MnO42- + IO3- + 3H2O
6(MnO4- + 1e- MnO42-) (red)
I-+ 6HO- IO3- + 3H2O +6e- (Ox)
6MnO4- + I-+ 6HO- 6MnO42- + IO3- + 3H2O
Cu + HNO3
Cu(NO3)2 + NO + H2O
Cr3+ + I- + HO- + Cl2
CH3CH2OH + KMnO4 + H2SO4
CH3CH2OH + Ag(NH3)2OH
CrO42- + IO4- + Cl- + H2O
CH3COOH + K2SO4 + MnSO4 + H2O
CH3COONH4 + Ag0 + NH3 + H2O
En la preparación de disoluciones de agentes oxidantes y reductores debemos
tener en cuenta el número de electrones que cada especie intercambia a la hora de
calcular la normalidad. Por ejemplo, el MnO4- actúa como oxidante en medio ácido
reduciéndose a sal manganosa mientras que en medio neutro lo hace a bióxido de
manganeso:
Mn7+ + 5e- Mn2+ (medio ácido)
Mn7+ + 3e- Mn4+ (medio neutro o básico)
3,/$'$1,(//2&(/'$*$/9$1,&$
Celda galvánica o voltaica es aquella en la que una reacción química espontánea
genera una tensión eléctrica. Para lograrlo, uno de los reactivos debe oxidarse y otro
reducirse simultáneamente. No debe haber contacto entre ambos, pues de lo contrario,
los electrones fluirían directamente del agente reductor al agente oxidante. Por tanto, el
agente reductor y oxidante deben estar físicamente separados y unidos solamente por
una conexión eléctrica, de forma que los electrones estén forzados a circular por un
circuito externo para ir de un reactivo al otro (del reductor al oxidante).
6
Consideremos una pila que implique la reacción:
Zn(s) + Cu2+ Cu(s) + Zn2+
Cuyas semireacciones son:
Zn(s) Zn2+ + 2e- (ox)
Cu2+ +2e- Cu0 (red)
La celda galvánica consiste en dos vasos de reacción, uno de los cuales contiene
una solución de Cu2+ y una varilla de Cu y, el otro, una disolución de Zn2+ y una varilla
de Zn. Ambos recipientes se encuentran unidos entre si mediante un puente salino o
tabique poroso que contiene KCl o NO3NH4. La finalidad de este puente salino es
asegurar el contacto eléctrico entre las disoluciones de electrolitos evitando su contacto
físico. Cada uno de los recipienes que forman una celda galvánica se denomina
semicelda o semipila.
Si cerramos el circuito a través de un amperímetro, se observa el paso de una
corriente eléctrica. La varilla de zinc empieza a disolverse y los iones cobre(II) se
deposita sobre la varilla de cobre como cobre metálico. La solución de Zn2+ se hace más
concentrada y la solución de Cu2+ más diluida. La corriente eléctrica fluye de la varilla
de Zn a la varilla de Cu. En el vaso de Zn es donde ocurre la oxidación por lo que, su
disolución quedaría cargada positivamente perdiendo la electroneutralidad. Para evitar
esto, se establece un flujo aniónico en el puente salino que descarga aniones en el vaso
de Zn, motivo por el cual se denomina ánodo (donde ocurre la oxidación). En el vaso de
Cu ocurre la reducción por lo que, su disolución iría perdiendo progresivamente cargas
positivas por lo que el puente salino descarga cationes en el para no perder la
7
electroneutralidad. Por este motivo se denomina cátodo. Luego la semicelda de Zn es el
ánodo y la de Cu es el cátodo.
Los dispositivos que unen disoluciones con el circuito eléctrico externo (las
varillas de Cu y Zn) y en cuya superficie se produce el intercambio de electrones se
llaman electrodos.
El puente salino consiste en un tubo de vidrio curvado en forma de U,
conteniendo una disolución concentrada de electrolito inerte que no experimenta
modificación alguna en el proceso redox (electroactivamente inertes). No se mezcla con
las disoluciones de Zn2+ y Cu2+, permitiendo en cambio el tránsito de los iones SO42- de
una a otra disolución, lo que hace posible el mantenimiento de la neutralidad eléctrica
en ambos compartimentos.
Las pilas galvánicas suelen utilizar como notación la siguiente:
$QRGR'LVROXFLyQDQyGLFDSXHQWHVDOLQR'LVROXFLyQFDWyGLFD&iWRGR
Ejemplo:
Zn / Zn2+ (1.0 M) // Cu2+ (1.0 M) / Cu
Las disoluciones anódicas y catódicas quedan simbolizadas por las especies
electroactivas y sus respectivas concentraciones.
Muchas veces, los electrodos son metales activos como en el caso anterior. Es
decir, los electrodos metálicos se forman o se disuelven a medida que se produce la
reacción de la celda. Sin embargo, son comunes los electrodos inertes, que no
experimentan cambio por la reacción neta. Por ejemplo el electrodo de platino.
8
La reacción neta de la celda es:
Ag+ + Fe2+ Fe3+ + Ag0
Cuyas semireacciones son:
(catódica) Ag+ + 1e- Ag0
(anódica) Fe2+ Fe3+ + 1eLa notación sería:
Cu / Cu2+ (1.0 M) // Fe3+, Fe2+ (1.0 M) / Pt
Un tercer tipo de electrodo es el llamado electrodo de gas. El electrodo de gas
hidrógeno consiste en una lámina de platino cubierta por negro de platino introducida en
un tubo de vidrio, por cuyo interior se hace burbujear hidrógeno gaseoso a la presión de
1 atmósfera y a 25ºC de temperatura, y sumergida en el seno de una disolución en la que
la concentración de iones H+ es 1 M. El negro de platino absorbe el hidrógeno, que de
esta forma está en contacto con los protones de la disolución, estableciéndose el
equilibrio:
H2(g) 2H+ + 2eLa notación simbólica del electrodo normal de hidrógeno es:
(Pt) H2 (1atm) / H+ (1M)
327(1&,$/'((/(&752'2)8(5=$(/(&7520275,='(81$3,/$
Al introducir un metal en una disolución de sus iones aparece inmediatamente
una diferencia de potencial entre el metal y la disolución, llamada potencial de contacto
o potencial de electrodo. Dicho potencial depende de la naturaleza del metal y de la
concentración de la disolución, además de la temperatura, por tratarse de un equilibrio
químico. El potencial de un electrodo aislado no se puede medir directamente, pues al
introducir uno de los bornes del voltímetro en la disolución ya habría dos metales en
ella; para subsanar el problema hay que recurrir a medir la diferencia de potencial entre
el electrodo en cuestión y otro elegido arbitrariamente como referencia y al que se le
asigna el potencial cero. Este electrodo de referencia es el de hidrógeno.
La fuerza electromotriz de una pila es la diferencia de potencial entre sus
electrodos, medida mediante un potenciómetro.
∆Epila = Ecátodo – Eánodo
9
Como el movimiento de los electrones tiene lugar en el sentido de potenciales
crecientes, en el funcionamiento real de toda pila se ha de cumplir que: Ecátodo > E
ánodo.
327(1&,$/(61250$/(6'((/(&752'2
El potencial de un electrodo depende de la concentración de la disolución en la
que se encuentra sumergido y de la temperatura y, además, su medida requiere la
elección de un electrodo de referencia al cual se le asigna valor cero. Este electrodo de
referencia es el electrodo normal de hidrógeno.
Una vez fijado este criterio se puede determinar el potencial normal o estándar
(Eo) de cualquier electrodo sin más que hallar la fuerza electromotriz de la pila formada
por el electrodo en cuestión sumergido en una disolución 1 M de sus iones y el
electrodo normal de hidrógeno, efectuando la medida a la temperatura de 25ºC. El
potencial normal de dicho electrodo es la diferencia de potencial entre dicho electrodo y
el electrodo normal de hidrógeno.
Al construir una pila con un electrodo cualquiera y el estándar de hidrógeno
puede suceder que el flujo de electrones tenga lugar desde dicho electrodo al de
referencia, en este caso la fuerza electromotriz es negativa y también lo será el potencial
de reducción del electrodo, teniendo lugar en él su oxidación. Por el contrario si el flujo
de electrones tiene lugar en sentido contrario, la fuerza electromotriz resulta positiva, al
igual que el potencial de reducción del electrodo, verificándose en él la reducción.
Procediendo de esta manera, se han obtenido los potenciales normales o estándar
de los distintos electrodos o semipilas, constituyendo lo que se denomina serie
electromotriz o serie electroquímica de los elementos cuyos potenciales corresponden al
proceso de reducción:
Xn+ + me- X(n-m)+ + E
Cuando el potencial de reducción del electrodo anterior es positivo, la reacción
que en él ocurre tiene más tendencia a producirse que la de reducción del hidrógeno,
mientras que si el potencial es negativo, la reacción tiende a verificarse en sentido
contrario.
Cuanto más elevado sea el potencial de reducción de un elemento mayor será su
tendencia a reducirse, es decir, su carácter oxidante, y viceversa. Del análisis de esta
10
serie, se deduce que el mejor oxidante es el fluor (F2 + 2e- 2F- ; Eo = +2.85 v) y el
mejor reductor el lítio (Li+ + 1e- Li Eo = -3.05 v).
Las tablas de potenciales sirven para calcular la fuerza electromotriz estándar de
una pila cualquiera, a partir de los potenciales conocidos de sus electrodos, teniendo en
cuenta que el electrodo de menor potencial de reducción será el polo negativo (ánodo),
en el que tiene lugar la oxidación, y el de mayor potencial el positivo (cátodo), donde se
verifica la reducción. La reacción global que ocurre en la pila será la suma de ambas
semirreacciones, y su fuerza electromotriz se obtendrá sumando los correspondientes
potenciales de electrodo.
Si el potencial de una reacción global es positivo, tal reacción se verifica
espontáneamente en le sentido en que está escrita. Si por el contrario, su potencial es
negativo, la reacción transcurrirá en sentido contrario.
&/$6,),&$&,21'(/26$*(17(62;,'$17(6<5('8&725(6
D $JHQWHVR[LGDQWHV
1.- El iFLGR SHUFOyULFR, HCLO4, concentrado y caliente es uno de los oxidantes más
fuertes. Se utiliza frecuentemente en la disolución de aceros aleados; oxida al cromo y
al vanadio y sus estados de oxidación más elevados.
Su acción oxidante puede
interrumpirse por dilución y /o enfriamiento.
2.- El SHUR[LGLVXOIDWR, [K2S2O8 o (NH4)2S2O8], es uno de los oxidantes más fuertes, su
acción lenta puede ser catalizada con ion plata.
Se utiliza para la oxidación de
manganeso (II) a permanganato y para la oxidación del carbono en muestras de aceros.
3.- El SHU\RGDWR SRWiVLFR, KIO4, utilizado en la oxidación de manganeso (II) a
permanganato.
4.- El ELVPXWR VyGLFR, NaBiO3, utilizado en oxidación de manganeso (II) a
permanganato.
5.- El FORUDWR SRWiVLFR, KClO3, en disolución ácida o en fusiones alcalinas en un
oxidante poderoso.
6.- El SHUPDQJDQDWR SRWiVLFR, KMnO4, además de utilizarse como reactivo
oxidimétrico, puede utilizarse en oxidaciones previas. Es un poderoso oxidante en
disoluciones ácidas o neutras.
7.- El SHUy[LGRGHKLGUyJHQR, H2O2 , es un poderoso oxidante y un reductor de fuerza
media.
11
8.- El R[LGR GH SODWD ,, , Ag2O2, en disolución ácida es adecuado para oxidar el
manganeso (II) a permanganato, el cromo (III) a dicromato y el cerio (III) a cerio (IV) a
la temperatura ambiente.
9.- El R]RQR, O3, puede prepararse haciendo circular, oxígeno gaseoso por un tubo de
vidrio entre cuyas paredes se mantiene una descarga eléctrica.
10.- El iFLGRQtWULFR, HNO3, puede originar diversos productos de reducción (NO2, NO,
N2O, etc...); el producto formado depende de la fuerza del reductor, de la concentración
del ácido y de la temperatura.
11.- +DOyJHQRV. El cloro y el bromo encuentran cierta utilización en oxidaciones
previas. El yodo es un oxidante más bien débil; sin embargo, se utiliza extensamente
como reactivo oxidimétrico de reductores fuertes.
E $JHQWHVUHGXFWRUHV
1.- 0HWDOHV. Debido a la facilidad con que pierden electrones los metales activos, son
buenos reductores.
Los metales pueden presentarse en forma de alambre, lámina,
gránulos, polvo y amalgamas líquidas; pueden llegar a ser muy selectivos.
El UHGXFWRU-RQHV consiste en gránulos de cinc amalgamado (aleado con mercurio) para
minimizar la reducción del ion hidrógeno de la disolución sulfúrica de la muestra.
El UHGXFWRU:DOGHQ es plata metálica y ácido clorhídrico 1 M.
Otros metales usados como reductores son aluminio, cadmio, plomo, bismuto y sodio;
los tres últimos se emplean en forma de amalgamas líquidas.
2.- El FORUXURHVWDQQRVR. Para reducir Fierro (III) a Fierro (II) en la determinación del
hierro.
3.- Las VDOHVIHUURVDV que son reductores de fuerza media, se añaden usualmente como
cantidad media en exceso de su disolución patrón en forma de FeSO4 * [NH4]2SO4 *
6H2O pesado en exceso.
El exceso se determina después por valoración con un
oxidante.
4.- El GLy[LGRGHD]XIUH o un sulfato en medio ácido es un reductor de fuerza media,
utilizándose en reducciones similares a las que se efectúan con plata.
5.- El VXOIXURGHKLGUyJHQR es comparable al dióxido de azufre en poder reductor y en
la forma de eliminar su exceso.
6.- El KLSRVXOILWRVyGLFR Na2S2O4, es un reductor poderoso especialmente en disolución
alcalina.
12
7.- El iFLGRFORUKtGULFR (concentrado) se utiliza como reductor principalmente para la
disolución de óxidos de plomo y manganeso.
,1)/8(1&,$ '( /$ &21&(175$&,Ï1 (1 /26 327(1&,$/(6 '(
(/(&752'2(&8$&,Ï1'(1(567
Los potenciales normales son indicadores de cambios en condiciones estándar.
Pero si se quiere estudiar la tendencia de una reacción es preciso conocer el signo y el
YDORU GH * HQ GLFKDV FRQGLFLRQHV 3DUD REWHQHU GLFKD LQIRUPDFLón se utiliza la
siguiente expresión termodinámica para una reacción genérica:
Ox1 + n1e- Red1 (cátodo)
Red2 + n2e- Ox2 (ánodo)
Ajustando los electrones, multiplicamos la primera por n2 y la segunda por n1, de
forma que la reacción global es:
n2Ox1 + n1Red2 n2Red1 + n1Ox2
G = Gº + RTlnQ
donde Q es el cociente de reacción:
[ Red 1 ] 2 [Ox 2 ] 1
4=
[Ox 1 ] 2 [ Red 2 ] 1
La reacción será espontánea si *.
Llamando Q al número total de electrones intercambiados: Q Q Q, y teniendo en
cuenta que * Q)∆( la expresión anterior queda de la siguiente forma:
Q)∆( Q)∆(57OQ4
∆( ∆( 57Q) OQ4
Esta expresión es conocida como laHFXDFLyQGH1HUVW.
Sustituyendo los valores de la temperatura (25 ºC) y las constantes y expresando el
logaritmo neperiano como decimal se tiene:
13
R: constante de los gases ideales (8´314 JK-1mol-1)
T: temperatura absoluta, 298 K
F: la constante de Faraday (96485 coulombios).
∆( ∆( Q ORJ4
La ecuación de Nerst se expresa normalmente en términos positivos: (en el
numerador se encuentran los reactivos ya que al aumentar su concentración hacen que el
equilibrio se desplace hacia la derecha aumentando el valor de ∆E):
∆( ∆( Q ORJ 4 Es decir:
[Ox 1 ] 2 [ Red 2 ] 1
0.059
log
∆( = ∆( +
Q
[ Red 1 ] 2 [Ox 2 ] 1
La diferencia de potencial de una reacción redox espontánea es positivo, ∆(!,
y su ¨* . Para una reacción en equilibrio ∆( (¨* ) y su cociente de
reacción, 4, es igual a la constante de equilibrio, .:
E = 0 = Eº - (RT/nF)lnK Æ ( 57Q) OQ.(¨*º = -RTlnK)
La ecuación de Nerst da la relación entre la fuerza electromotriz de una pila y la
constante de equilibrio de la reacción que tiene lugar. Sea, por ejemplo, la siguiente
reacción:
Zn(s) + Cu2+ Zn2+ + Cu(s) . =
∆( = ( − ( = ( +
0.059
/RJ[&X 2 + ] − ( 2
∆( = ∆( +
−
[ =Q 2 + ]
[&X 2 + ]
0.059
/RJ[ =Q 2 + ]
2
0.059
[&X 2+ ]
log
2
[ =Q 2 + ]
14
En el equilibrio la fuerza electromotriz de la pila es nula (∆E =0), por lo que:
2∆
0.059
[ =Q 2 + ] 0.059
∆( = 0 ∴ ∆( =
/RJ
=
/RJ . ∴ . = 10 0.059
2
2
[&X 2 + ]
Para la reacción general anteriormente considerada:
n2Ox1 + n1Red2 n2Red1 + n1Ox2
[Ox 1 ] 2 [ Red 2 ] 1
0.059
∆( = ∆ ( +
log
Q
[ Red 1 ] 2 [Ox 2 ] 1
Es decir:
En el equilibrio:
∆( = ∆ ( −
0.059
log .
Q
∆
0.059
∆( = 0 ∴ ∆( =
log . ∴ . = 10 0.059
Q
Conocida cualquiera de las tres magnitudes ∆* , ∆( R . las otras dos quedan
determinadas.
Para una semirreacción, el potencial de electrodo es una medida del grado en
que las concentraciones reales existentes en una semicelda difieren de los valores de
equilibrio. Así, por ejemplo, hay una mayor tendencia a que el proceso:
Ag+ + 1e- Ag(s)
ocurra en una disolución concentrada del ión plata que en una disolución diluida. En
consecuencia, el valor numérico del potencial de electrodo de este proceso debe hacerse
también más grande a medida que aumenta la concentración del ión Ag+ en la
disolución.
Para la semirreacción reversible genérica:
aOx + ne- bRed
la ecuación de Nerst toma la forma:
0.059
[Ox ]
(=( +
log
Q
[Red ]
15
Se observa que para que un sistema redox actúe como oxidante o como reductor
no solo hay que tener en cuenta el valor de Eo, sino que también influye el pH, la
temperatura y cualquier factor que actúe sobre las actividades o concentraciones de las
formas oxidadas y reducidas.
62%5(327(1&,$/
La Termodinámica sirve para predecir la espontaneidad de una reacción en unas
determinadas condiciones, pero puede predecir si la reacción transcurrirá a velocidades
apreciables o no. Una regla empírica útil (que encuentra numerosas excepciones) dice
que “los pares con potenciales inferiores a –0.6 V pueden reducir al ion H+ a H2 a
velocidad apreciable”. Esta regla se puede ampliar diciendo que una reacción redox se
producirá a una velocidad apreciable siempre que su potencial sea superior a 0.6 V. Por
ejemplo para que se pueda producir la reacción de oxidación del agua a tal velocidad:
O2 (g) + 4H+(ac) + 4e- 2 H2O (l), Eº = 1.23 V
se necesita un par redox Mn+/M cuyo potencial sea de Eº = 1.23 + 0.6 = 1.83 V.
Este valor de potencial de 0.6 V en exceso se denomina VREUHSRWHQFLDO. La
existencia de este sobrepotencial explica por qué ciertos metales reducen al H+ en medio
ácido y no en agua a pH neutro. Tales metales (entre los que se encuentra el Fe y el Zn)
poseen potenciales de reducción negativos, pero no poseen un valor suficientemente
grande como para conseguir un sobrepotencial de –0.6 V para la reducción del H+ a pH
neutro.
Ejemplo: reducción del H+ por Fe(s).
La ecuación de Nerst para el hidrógeno, a P = 1atm, es:
H+ (ac) + e- ½ H2 (g)
E = Eº + 0.059log [H+]
Como Eº = 0 y log[H+] = pH, se stiene:
E = -0.059pH
16
A pH neutro, pH = 7
EH+/H2, pH = 7 = -0.41
Suponiendo que el par redox Fe2+/Fe se encuentra en condiciones estándar:
EFe2+/Fe = EºFe2+/Fe = -0.44 v.
Luego:
∆E = EH+/H2, pH = 7 - EFe2+/Fe = -0.41 - (-0.44) = 0.03 v
Por lo que a pH neutro, el sistema Fe2+/Fe puede reducir los protones del agua, pero la
reacción será lenta, por no sobrepasar la diferencia de potencial el valor del
sobrepotencial (0.03 << 0.6).
A pH ácido, por ejemplo pH =2 el sistema Fe2+/Fe puede reducir también los protones
del agua pero la reacción es aún lenta:
EH+/H2, pH = 2 = -0.118
∆E = EH+/H2, pH =2 - EFe2+/Fe = -0.118 - (-0.44) = 0.32 v
&$032'((67$%,/,'$''(/$*8$
Un ion o molécula en disolución puede ser inestable al sufrir reacciones de
oxido-reducción, debido a la presencia de cualquiera de las otras especies presentes en
disolución, o incluso al mismo disolvente. Cuando el disolvente empleado es H2O, éste
puede actuar como agente reductor, liberando O2 (se oxida el ion O2-), o como oxidante
produciendo H2 (el H+ se reduce). Las especies que pueden existir en H2O poseen
potenciales de reducción que se encuentran entre los límites definidos por estos dos
procesos:
17
La oxidación de los metales por el agua o por los ácidos en medio acuoso,
generalmente, responde al siguiente tipo de proceso:
M(s) + H2O (l) M+ (ac) + ½ H2 (g) + OH- (ac)
M(s) + H+ (ac) M+ (ac) + ½ H2 (g)
Estos procesos son termodinámicamente favorables cuando el metal, M, es un
elemento del bloque s, distinto del berilio, o un metal de la primera parte (Grupos 4-7:
Ti, V, Cr, Mn) de la primera serie de transición. Algunos metales pueden sufrir
reacciones semejantes, pero transfieren un número de electrones superior
2Sc(s) + 6H+ (ac) 2Sc3+ (ac) + 3H2 (g)
De manera general, cuando el potencial normal de reducción de un ion metálico
a metal es negativo, el metal se oxida en un ácido 1M, con desprendimiento de H2. Sin
embargo, como ya se ha comentado anteriormente, la reacción puede ser lenta. Por
ejemplo, las reacciones del Mg y del Al con el aire húmedo son espontáneas, pero estos
metales se pueden usar durante años en presencia de H2O y oxígeno. No se atacan por
oxígeno porque se SDVLYDQ, es decir, se recubren de una capa impermeable de óxido que
los aísla del exterior e impide que progrese la reacción de oxidación. Este fenómeno de
pasivado también ocurre en los metales como el Fe, Cu y Zn.
El agua puede actuar como reductora mediante la semirreacción:
O2 (g) + 4H+(ac) + 4e- 2 H2O (l) Eº = 1.23 V
Este potencial de reducción muy positivo muestra que el H2O se comporta como
un reductor muy débil, excepto frente a agentes oxidantes enérgicos como por ejemplo
el ion Co3+ (ac), para el que Eº(Co3+/Co2+) = +1.82 V. Este ion se reduce en H2O con
liberación de O2.
4Co3+ (ac) + 2H2O (l) 4Co2+ (ac) + O2 (g) + 4H+ (ac)
∆Eº = Eº(Co3+/Co2+) – Eº(H+,O2/H2O) =1.82-1.23 = 0.59 v
18
Este valor es muy próximo al valor del sobrepotencial necesario para que la velocidad
de reacción sea significativa. Como se producen H+ en la reacción, un cambio de pH
desde neutro a pH altos favorece la reacción.
Existen otros sistemas como el Ce4+/Ce3+ (Eº = 1.61 v), Cr2O72-/Cr3+ (Eº = 1.33
v, medio ácido) y MnO4-/Mn2+ (Eº = 1.51 v, medio ácido), para los que no se alcanza el
sobrepotencial, y son estables en disolución acuosa, aunque para todos ellos ∆Gº < 0.
El origen de esta dificultad del agua para actuar como reductor reside en la necesidad de
transferir cuatro electrones a fin de un doble enlace oxígeno-oxígeno.
El campo de estabilidad del agua se define como el intervalo de valores del
potencial de reducción y de pH para el que el H2O es termodinámicamente estable a la
reducción y a la oxidación. Los límites superior e inferior del campo de estabilidad se
identifican hallando la dependencia del potencial con el pH para las semirreacciones
correspondientes:
a) H2O actuando como reductor
O2 (g) + 4H+(ac) + 4e-
+2O (l), Eº = 1.23 v
E = 1.23 – 0.059pH
Cualquier especie con un potencial de reducción más alto que este valor puede ser
reducida por el agua, liberándose O2. Éste es el límite superior del campo de estabilidad
del H2O como reductor.
b) H2O como oxidante:
2H+(ac) + 2e-
+2 (g), Eº = 0
E = -0.059pH
Cualquier especie con un potencial de reducción inferior a este valor puede reducir al
H+(ac) a H2, por lo que éste es el límite inferior del campo de estabilidad.
La siguiente figura muestra el campo de estabilidad del agua. El eje vertical representa
el Eº de reducción de los pares redox en agua: Los que caen por encima de la línea
19
superior pueden oxidar al agua; aquellos que caen por debajo del línea inferior pueden
reducir al agua. La zona sombreada representa la zona de estabilidad del agua pura.
&DPSRGHHVWDELOLGDGGHODJXD
'(6352325&,21&20352325&,21
Como Eº(Cu+/Cu) = 0.52 v y Eº(Cu2+/Cu+) = 0.16 v ambos potenciales se
encuentran dentro del campo de estabilidad del H2O. Sin embargo, el Cu(I) no es estable
en disolución acuosa porque sufre una GHVSURSRUFLyQ o GLVPXWDFLyQ, que consiste en
una reacción en la que el número de oxidación del elemento aumenta y disminuye. Es
decir, el elemento que se desproporciona actúa simultáneamente como oxidante y como
reductor:
2 Cu+ (ac) Cu2+ (ac) + Cu (s)
Esta reacción es espontánea ya que el ∆Eº = Eº( Eº(Cu+/Cu) - Eº(Cu2+/Cu+) = 0.36 v con
una constante de equilibrio, K = 1.3x106, que indica que la reacción está muy
desplazada a la derecha.
20
El ácido hipocloroso también está sometido a la desproporción:
5 HClO (ac)
&O2 (g) + ClO3- (ac) + 2H2O (l) + H+ (ac)
Esta reacción es la suma de dos semirreacciones:
4 HClO (ac) + 4H+ (ac) + 4eClO3- (ac) + 5H+ (ac) + 4e-
&O2 (g) + 4H2O (l), Eº = 1.63 v
+ClO (ac) + 2H2O (l), Eº = 1.43 v
∆Eº = +1.63 –1.43 V = 0.20 v, K = 3x1013
La reacción inversa a la desproporción es la FRPSURSRUFLyQ. En esta reacción,
dos especies del mismo elemento en estado de oxidación diferentes forman un producto
en el que el elemento se encuentra en un estado de oxidación intermedio:
Ag2+ (ac) + Ag (s)
$J+ (ac), ∆Eº = 1.18 v, K = 9x1019
(/(&752/,6,6
La electrólisis o electrolisis es un método de separación de los elementos que
forman un compuesto aplicando electricidad. Consiste en llevar a cabo una reacción
química no espontánea en la que la fuerza impulsora externa es la corriente eléctrica.
Cuando la reacción redox no es espontánea en un sentido, podrá suceder si desde el
exterior se suministra el potencial necesario.
Electrólisis procede de dos radicales, electro que hace referencia a electricidad y lisis
que quiere decir rotura.
En el proceso electrolítico se disuelve una sustancia en un determinado disolvente, con
el fin de que los iones que constituyen dicha sustancia estén presentes en la disolución.
Posteriormente se aplica una corriente eléctrica a un par de electrodos conductores
colocados en la disolución. Cada electrodo atrae a los iones de carga opuesta. Así, los
iones positivos, o cationes, son atraídos al cátodo, mientras que los iones negativos, o
aniones, se desplazan hacia el ánodo. La energía necesaria para separar a los iones e
incrementar su concentración en los electrodos, proviene de una fuente eléctrica externa
que mantiene la diferencia de potencial en los electrodos.
En los electrodos, los electrones son absorbidos o emitidos por los iones,
formando concentraciones de los elementos o compuestos deseados. Por ejemplo, en la
electrólisis del agua, se forma hidrógeno en el cátodo, y oxígeno en el ánodo. Esto fue
descubierto en 1820 por el físico y químico inglés 0LFKDHO)DUDGD\.
21
Las UHDFFLRQHVGHHOHFWUyOLVLV,se llevan a cabo enFHOGDVHOHFWUROtWLFDV como la
representada en la siguiente figura. Obsérvese que no se requiere la separación de las
semirreacciones por un puente salino y que la polaridad se invierte respecto a una pila
electro química. El ánodo constituye el polo positivo de la celda por encontrarse unido
al polo positivo de la pila externa. El cátodo constituye el polo negativo y se encuentra
unido al polo negativo de la pila externa. No obstante en el ánodo se produce la
oxidación y en el cátodo la reducción, al igual que en las pilas electroquímicas.
Las leyes que describen la electrólisis son las Leyes de Faraday:
1) El cambio químico producido en la electrólisis es proporcional a la carga de
electricidad que pasa por la celda.
2) La carga requerida para depositar o liberar una masa, m, es (para depositarse un
equivalente de cualquier sustancia se precisan 96500 coulombios):
donde:
P es la masa de la sustancia producida en el electrodo (en gramos),
4 es la carga eléctrica total que pasó por la solución (en Culombios),
T es la carga del electrón = 1.602 x 10-19 culombios por electrón,
Q es el numero de valencia de la sustancia como ion en la solución (electrones por ion),
es la Constante de Faraday,
0 es la masa molar de la sustancia (en gramos por mol), y
1 es el Numero de Avogadro = 6.022 x 1023 iones por mol.
Teniendo en cuenta que 4 ,W resulta:
m(g) =
M× I× t
n × 96500
Siendo , la intensidad de corriente y W el tiempo que ésta ha circulado.
22
En la electrolisis de disoluciones acuosas se ha de tener en cuenta las reacciones
de reducción y oxidación del agua, así, en la electrolisis del NaCl se obtendrá Cl2 en el
ánodo e hidrógeno en el cátodo:
&iWRGR
2H2O + 2e- H2 + 2OH-Eo 0,83 v
Na+ + e- Na
Eo 2,71 v
“El sodio no se deposita en el cátodo en la electrolisis de sus disoluciones
acuosas ya que antes habría que reducir todo el agua”.
ÈQRGR
2Cl- Cl2 + 2e-
2 H2O 4H+ + O2 + 4e-
Eo 1,36 v
Eo 1,23 v (1.83 con sobrepotencial)
23
Las aplicaciones de la electrolisis son variadas, entre ellas destacan:
a) Obtención industrial de metales a por reducción de las sales fundidas de dichos
metales. El mejor ejemplo es el caso de la Célula Down para la producción
comercial de sodio metálico. Como subproducto se obtiene cloro en el ánodo.
(ver figura):
La fuente de potencial externa permite la reacción no espontánea de
descomposición de NaCl en sus elementos Na y Cl2, que ocurre en el proceso
electrolítico. El potencial mínimo que se precisa es de 4.07 v (–2’71 – 1’36).
5HGXFFLyQ FiWRGR 2Na+(aq) + 2e– → 2Na (s)
2[LGDFLyQ iQRGR 2Cl–(aq) → Cl2(g) + 2e–
24
b) Obtención industrial de otras sustancias a partir de disoluciones de sus sales. El
ejemplo más conocido es la obtención de sosa y cloro.
La salmuera consiste en una disolución concentrada de NaCl. Al producirse la
electrolisis se produce la oxidación del cloruro a cloro en el ánodo y la reducción de los
protones del agua, generándose una disolución concentrada de NaOH.
c) Galvanoplastia o electrodeposición, proceso de recubrir un objeto metálico con
una capa fina de otro metal. Tal es el caso del cincado Zn2+ + 2e– → Zn o niquelado
Ni2+ + 2e– → Ni y los baños de plata Ag+ + 1e- → Ag u oro Au3+ + 3e– → Au .
25
d) Electrorrefinado de metales:
La pieza metálica impura actúa como ánodo produciéndose la oxidación del metal que
pasa a la disolución como ion. Éste de reduce en el cátodo, electrodepositándose puro.
&25526,21
Es la tendencia que tienen los metales a volver al estado combinado, es decir, al
mismo estado en que se encontraban en la naturaleza, que es, en términos
termodinámicos, el estado más estable.
En el caso del acero o del hierro la corrosión se pone de manifiesto con la conocida
"herrumbre".
Exceptuando la corrosión a temperaturas elevadas, que es un proceso puramente
químico, los restantes procesos de corrosión son siempre de naturaleza electroquímica,
tratándose de la formación de una pila, con una corriente eléctrica que circula entre
determinadas zonas de la superficie del metal, conocidas con el nombre de ánodos y
cátodos, y a través de una solución llamada electrolito capaz de conducir dicha
corriente. El funcionamiento de estas pilas da lugar a la corrosión de las zonas anódicas
(oxidación del metal).
En la formación de la herrumbre, el hierro metálico y la gota de agua en su
superficie constituyen una celda galvánica, en la cual, el hierro se oxida a ión ferroso, en
la superficie de hierro (región anódica) y el oxígeno atmosférico se reduce cerca del
26
menisco de la gota (región catódica). El ión ferroso se oxida a férrico por el oxígeno
disuelto antes de que se deposite como herrumbre (Fe2O3.H2O).
Las semirreacciones que tienen lugar son:
ÈQRGRFe(s) Fe2+ + 2e– posteriormente se oxida a Fe3+.
&iWRGRO2(g) + 4H+ + 4e– 2H2O
5HDFFLyQJOREDO:
2Fe(s) + O2(g) + 4H+ 2Fe2+ + 2H2O
Oxidación posterior del ion ferroso por el oxígeno disuelto:
2Fe(s) + 3H2O Fe2O3 + 6H+ + 4eO2(g) + 4H+(aq) + 4e– 2H2O
5HDFFLyQJOREDO:
4Fe2+ + O2 + 4H2O 2Fe2O3 + 8H+
La corrosión puede evitarse tanto por SURWHFFLyQ FDWyGLFD como DQyGLFD. La
SURWHFFLyQ FDWyGLFD ocurre cuando un metal es forzado a ser el cátodo de la celda
corrosiva adhiriéndole (acoplándolo o recubriéndolo) de un metal que se corroa más
fácilmente que él, de forma tal que esa capa recubridora de metal se corroa antes que el
metal que está siendo protegido y así se evite la reacción corrosiva. Una forma conocida
de protección catódica es la JDOYDQL]DFLyQ (cubrir un metal con Zinc para que éste se
corroa primero) en la que el zinc pasa a ser el ánodo de sacrificio, porque él ha de
corroerse antes que la pieza metálica protegida. Otra forma muy extendida de
protección catódica consiste en soldar a la tubería de hierro un ánodo de sacrificio de
27
magnesio, que evita que el hierro se oxide al oxidarse el magnesio. Este procedimiento
tiene como fundamento la polarización, a potenciales más negativos, de la superficie
metálica hasta alcanzar un grado de polarización, en el cual se acepta que dicha
superficie metálica es inmune a la corrosión.
Por otro lado, la SURWHFFLyQ DQyGLFD, es un método similar que consiste en
recubrir el metal con una fina capa de óxido para que no se corroa. Existen metales
como el Aluminio que al contacto con el aire son capaces de generar espontáneamente
esta capa de óxido y por lo tanto, se hacen resistentes a la corrosión. Aún así, la capa de
óxido que recubre al metal no puede ser cualquiera. Tiene que ser adherente y muy
firme, ya que de lo contrario no serviría para nada. Por ejemplo, el óxido de hierro no es
capaz de proteger al hierro, porque no se adquiere a él en la forma requerida.
&(/8/$692/7$,&$635,0$5,$6
Las pilas son dispositivos generadores de electrones, con una vida limitada por
el agotamiento de las sustancias redox reaccionantes ya que a medida que una célula
voltaica produce corriente (se descarga), se consumen productos químicos. Las células
voltaicas primarias no se pueden recargar. Una vez que los productos químicos se han
consumido, no es posible reacción química posterior. Los electrolitos y/o los electrodos
no se pueden recargar invirtiendo el flujo de corriente a través de la célula usando una
fuente externa de corriente continua. Los ejemplos más familiares de células voltaicas
primarias son las “pilas secas”, que son las pilas ordinarias que se usan como fuente de
energía en linternas y en otros aparatos pequeños. Aunque la celda Zn-Cu fue empleada
como generador de corriente durante mucho tiempo, se demostró que eran mucho más
eficaces y prácticas la pilas sin disolución líquida en su interior. La más conocida es la
pila seca de Leclanché. El recipiente de esta pila, hecho de cinc, actúa como uno de los
electrodos. El otro electrodo es una barra de carbono inerte en el centro de la célula. El
recipiente de cinc está recubierto de papel poroso para separarlo de los demás materiales
de la célula. El espacio entre los electrodos se llena con una mezcla húmeda de coluro
amónico (NH4Cl), óxido de manganeso (IV), (MnO2), cloruro de cinc (ZnCl2), y un
filtro poroso e inerte. Las pilas secas se sellan para evitar pérdidas de humedad por
evaporación. Una vez conectados los electrodos externamente, el Zn metálico se oxida
a Zn2+, y los electrones fluyen a lo largo del recipiente hasta el circuito exterior. Así, el
electrodo de cinc es el ánodo (electrodo negativo).
28
Zn Zn2+ + 2e- (oxidación anódica)
2NH4+ + 2e- 2NH3 + H2 (reducción catódica, barra grafito).
Reacción global de la célula:
Zn + 2NH4+ Zn2+ + 2NH3(g) + H2(g) (Ecélula = 1.6 v)
Se observa que se producen dos gases en esta reacción. A medida que se forma
H2, se oxida por el MnO2 de la célula, evitando recoger gas en el cátodo, que pararía la
reacción:
H2 + 2MnO2 2MnO(OH) Mn2O3 (s) + H2O (l)
El amoniaco producido en el cátodo se combina con los iones cinc y forma un
compuesto soluble que contiene el ión complejo tetraaminzinc(II):
Zn2+ + 4NH3 + 2Cl- [Zn(NH3)4]Cl2
Esta reacción evita la polarización (acumulación de productos de reacción sobre
un electrodo) debida a la acumulación de amoníaco, y evita que la concentración de
Zn2+ aumente sustancialmente, lo que haría disminuir el potencial de la pila.
El potencial que genera esta pila tiene un valor inicial de 1.5 voltios y su
duración depende principalmente de la calidad y cantidad de MnO2.
Las pilas alcalinas secas son parecidas a la pila seca de Leclanchè, excepto que
el electrolito es básico porque contiene KOH, la superficie interior del recipiente de Zn
es rugosa, lo que proporciona mayor area superficial, y tienen una vida más larga que
las pilas secas ordinarias, soportando mejor el uso continuado. El ánodo es de Zn (o a
veces de acero) y el cátodo también de acero.
El voltaje de una pila alcalina es de unos 1,5 voltios y durante la descarga, las
reacciones que tienen lugar son:
Anodo:
Zn(s) + 2(OH-) (aq) Zn(OH)2(s) + 2e- ZnO(s) + H2O(l) + 2eCátodo:
29
2MnO2(s) + 2H2O(l) + 2e- 2MnO(OH)(s) + 2(OH-) (aq) ' Mn2O3 + H2O + 2OHGlobal:
Zn (s) + 2MnO2(s) + 2H2O(l) Zn(OH)2(s) + 2MnO(OH)(s)
Otro tipo de pilas primarias utilizadas son las pilas de botón, de muy pequeño
tamaño y con capacidad de liberar una gran cantidad de corriente. Son de 1 cm de
diámetro y 4 mm de espesor. Están formadas por capas separadas por fibras plásticas y
los agentes oxidantes más utilizados son HgO y Ag2O.
Anodo: Zn en polvo amalgamado, impregnado en KOH al igual que en la pila alcalina.
La reacción es la misma:
Zn(s) + 2(OH-) (aq) Zn(OH)2(s) + 2e- ' ZnO(s) + H2O(l) + 2e-
Cátodo: El compartimento es de acero niquelado y está separado del ánodo por la parte
central mediante fibra de nylón. Se pone HgO o Ag2O mezclado con polvo de grafito e
impregnado en KOH conc.
HgO + 2e- + H2O Hg0 + 2OH- (1.35 v)
Ag2O + 2e- + H2O 2Ag0 + 2OH- (1.40 v)
Hay otras pilas de botón que se conocen como pilas de litio debido a que utilizan
como reductor el Li en vez de Zn. El principal problema de estas pilas de Li es que este
metal no puede entrar en contacto con el aire o con el agua, por lo que llevan electrolitos
orgánicos que son mucho menos conductores de la corriente que los electrolitos
acuosos. Son pilas de dimensiones menores que las de HgO o Ag2O y la reacción
anódica se basa en la oxidación del litio: Li → Li+ + 1e- . El cátodo es un Pb3O4, PbO,
CuO, MnO2 meclado con grafito en polvo. El electrolito es de tipo orgánico,
30
generalmente mezclas de esteres y éteres (carbonato de propileno con 1,2-dimetoxietano
embebidos de perclorato de litio, que es esponjoso).
&e/8/$692/7$,&$66(&81'$5,$6$&808/$'25(6
En las células voltaicas secundarias, o células reversibles, los reactivos
originales se pueden regenerar. Esto se hace pasando una corriente continua a través de
la célula en el sentido opuesto a la corriente de descarga, proceso que se conoce como
cargado o recargado de una célula o batería. El ejemplo más común es la batería
acumuladora de plomo usada en la mayoría de los automóviles.
Los acumuladores son dispositivos que pueden utilizarse indistintamente como
generadores de corriente eléctrica y también como receptores. De ahí que puedan
cargarse de nuevo empleando la energía eléctrica de una fuente exterior. Son
especialmente útiles en aplicaciones que precisan energía o en un momento determinado
pero que producen energía en otras ocasiones como es el caso del automóvil. La batería
proporciona energía para la puesta en marcha del vehículo y posteriormente se carga
gracias al alternador cuando el coche está en movimiento.
La batería acumuladora de plomo consiste en un grupo de placas de plomo
conteniendo plomo esponjoso que alternan con un grupo de placas de plomo que
contienen oxido de plomo(IV), PbO2. Los electrodos están sumergidos en una
disolución del 40% en ácido sulfúrico. Cuando la célula se descarga, el plomo
esponjoso se oxida a iones plomo, y las placas de plomo acumulan una carga negativa.
Durante la descarga, en el ánodo, la oxidación del plomo metálico genera iones
plumbosos, que se combinan con los aniones sulfato, procedentes del ácido sulfúrico,
produciendo sulfato de plomo(II) insoluble:
Proceso anódico en descarga:
Pb Pb2+ + 2e- (oxidación)
Pb2+ + SO42- PbSO4 ↓ (precipitación)
Pb + SO42- PbSO4↓ (s) + 2e- (-Eo = 0.356 v) (global)
Los electrones se mueven por el circuito exterior y re-entran al electrodo de
PbO2, que actúa de cátodo durante la descarga. En presencia de iones hidrógeno, el
óxido de plomo(IV) se reduce iones plomo(II), que se combinan con los sulfatos del
31
ácido sulfúrico para formar una capa de PbSO4 insoluble sobre el electrodo de
plomo(IV).
Proceso catódico en descarga:
PbO2 + 4H+ + 2e- Pb2+ + 2H2O (reducción)
Pb2+ + SO42- PbSO4↓ (s) (precipitación)
PbO2 + 4H+ + SO42- + 2e- PbSO4(s) + 2H2O (Eo = 1.685 v) (proceso global)
Reacción neta de la celda en descarga:
Pb(s) + PbO2(s) + 2H2SO4 ' 2PbSO4(s) + 2H2O Eo = 2.041 v.
Cada célula crea un potencial de unos 2 voltios. Las baterías de 12 voltios de los
automóviles tienes seis células conectadas en serie. El potencial disminuye sólo
ligeramente durante su uso, porque se están consumiendo reactivos sólidos. A medida
que la célula se usa, se consume algo de H2SO4 disminuyendo su concentración.
Cuando a través de los electrodos se le impone a la batería un potencial algo
mayor que la batería puede generar, el flujo de corriente se invierte. Entonces la batería
se puede recargar mediante la inversión de todas las reacciones. El alternador o
generador aplica este potencial cuando el motor está funcionando. Las reacciones que
tienen lugar en una batería acumuladora de plomo se asumen de la manera siguiente:
Pb(s) + PbO 2 (s) + 2H 2 SO 4
Descarga
→
←
Carga
+ 2PbSO 4 + 2H 2 O
Durante muchos ciclos de carga-descarga, parte del PbSO4 se deposita en el
fondo del recipiente y la concentración de H2SO4 permanece en consecuencia baja. Con
el paso del tiempo la batería no se puede recargar totalmente y se debe cambiar la
batería. Si bien, el plomo se puede reciclar.
32
&(/8/$'(1,48(/&$'0,2 1,&$' La célula de niquel-cadmio tiene una vida útil mucho más larga que las pilas
secas ordinarias ya que se pueden recargar. Las baterías de níquel se usan en relojes
electrónicos, calculadoras y equipo fotográfico.
El ánodo es cadmio, y el cátodo es óxido de níquel(IV), siendo la disolución
electrolítica básica.
Reacciones de descarga:
Anodo:
Cd(s) + 2OH-(aq) Cd(OH)2(s) + 2eCátodo:
NiO2(s) + 2H2O(l) + 2e- Ni(OH)2(s) + 2OHGlobal:
Cd(s) + NiO2(s) + 2H2O(l) Cd(OH)2 (s) + Ni(OH)2(s)
El producto de reacción sólido en cada electrodo se adhiere a la superficie del
electrodo. Por tanto, una batería de nicad se puede recargar mediante una fuente externa
de electricidad que invierta las reacciones de los electrodos. Debido a que no se
producen gases en las reacciones de una batería de nicad, la unidad se puede sellar. El
voltaje de esta celda es de 1,4 voltios, ligeramente inferior a la de la pila de Leclanchè.
33
9$/25$&,215('2;
La valoración redox es muy similar a la valoración ácido base. Se ha de ajustar
la reacción de valoración para determinar el número de moles de especie oxidante y
reductora que reaccionan entre sí. El peso equivalente de una sustancia oxidante o
reductora se determina dividiendo su masa molecular por el nº de e– ganados o perdidos
En el punto de equivalencia de la valoración se cumple:
HTXLYDOHQWHVGHR[LGDQWH HTXLYDOHQWHVGHUHGXFWRU
por tanto:
9R[u1R[ 9UHGu1UHG
La selección adecuada del indicador redox requiere la construcción de la curva
de valoración: potencial frente a volumen de agente valorante. A continuación se
muestra un ejemplo en el que la curva se construye de manera sencilla trabajando con
porcentajes de valoración (este método no siempre es posible).
34
'LEXMDUODFXUYDGHYDORUDFLyQGHP/GHXQDGLVROXFLyQGH)H 0FRQ&H GH
"!
# !
ODPLVPDFRQFHQWUDFLyQPRODU
Primero se exponen las semirreacciones y se estudia la espontaneidad de la reacción
global
Según las tablas de potenciales redox estándar:
Fe3+ + 1e- Æ Fe2+ Eo1 = 0.77 v
Ce4+ + 1e- Æ Ce3+ Eo2 = 1.44 v
La reacción de valoración es:
Fe2+ + Ce4+ ' Fe3+ + Ce3+ Eo = 1.44 - 0.77 = 0.67v
Su potencial indica espontaneidad hacia la derecha, con una constante, . = 2.27*1011.
La estequiometría de la reacción es 1:1, es decir, cada vez que añadimos un mol
de Ce4+ se consume un mol de Fe2+ y se produce un mol de Ce3+ y un mol de Fe3+,
respectivamente.
La forma de trabajar es con porcentaje de la valoración de tal forma que
independientemente al final, con un simple cálculo se establece el volumen. No es
necesario calcular concentraciones. Los valores de potenciales así calculados valen para
cualquier valoración en las que estén implicadas las mismas semirreacciones, lo que
cambiará, si acaso, serán los volúmenes.
Antes de llegar al punto de equivalencia, el potencial se calcula teniendo en
cuenta el par redox donde existe exceso, en este caso, el par redox del valorato, es decir,
el del sistema Fe3+/Fe2+. En el punto de equivalencia es necesario trabajar con los dos
pares redox simultáneamente evitando así el cálculo de concentraciones. Pasado el
punto de equivalencia se calcula el potencial del par redox del valorante, ya que se
encuentra en exceso. Realizando así la construcción de la curva de valoración, los
cálculos resultan bastante sencillos.
-Antes del punto de equivalencia: teniendo en cuenta que la curva es sigmoidal se
seleccionan los siguientes porcentajes: 5, 50, 90, 95 y 99%
El cálculo se basa en que [Fe3+] / [Fe2+] es el %valoración / (100-%valoración) que se
lleva a la ecuación de Nerst.
Por ejemplo, al 5% de la valoración [Fe3+] / [Fe2+] = 5/95. El potencial:
E = Eo1 + 0.059*log [Fe3+]/[Fe2+] = 0.77 + 0.059*log(5/95) = 0.695v
35
El potencial al 50% de la valoración es igual al normal, ya que [Fe3+] = [Fe2+], y el
logaritmo de 1 es 0. Esto no significa que estemos en condiciones normales,
simplemente que coinciden los valores.
Procediendo igualmente se obtienen todos los valores hasta llegar al punto de
equivalencia. En este punto, al ser la valoración completa y la reacción estequiométrica
se cumple:
[Fe3+] = [Ce3+]
[Fe2+] = [Ce4+]
Calculando el potencial por un sistema u otro, este debe tomar el mismo valor:
E1 = Eo1 + 0.059*log[Fe3+]/[Fe2+]
E2 = Eo2 + 0.059*log[Ce4+]/[Ce3+]
2Epe = Eo1 + Eo2 + 0.059*log ([Fe3+]*[Ce4+]) / ([Fe2+]*[Ce3+])
Epe = (Eo1 + Eo2 ) / 2 = (0.77 + 1.44) / 2 = 1.105v.
El volumen del punto de equivalencia, al ser de estequiometría 1:1 es:
40 * 0.1 = x* 0.1; x = 40 mL de Ce4+ 0.1 M.
-Después del punto de equivalencia el potencial se calcula con el sistema Ce4+/Ce3+. Se
seleccionan los siguientes porcentajes: 101, 105, 110, 150, y 200%.
El cálculo se basa en que [Ce4+] / [Ce3+] es (%valoración-100) / 100.
Por ejemplo, al 101 % de la valoración [Ce4+] / [Ce3+] = 1/100. El potencial:
E = Eo2 + 0.059*log [Ce4+]/[Ce3+] = 1.44 + 0.059*log(1/100) = 1.322v
De la misma forma se procede para los demás porcentajes.
El potencial al 200% de la valoración es igual al normal, ya que [Ce4+] = [Ce3+], y el
logaritmo de 1 es 0. Al igual que ocurría al 50%, esto no significa que estemos en
condiciones normales, simplemente que coinciden los valores.
36
&H
$&%
9DORUDFLyQ P/ 1.50
( Y 1.40
5
2
0.695
50
20
0.77
90
36
0.826
95
38
0.845
99
39.6
0.888
100
40
1.105
101
40.4
1.322
105
42
1.363
110
44
1.381
0.70
150
60
1.422
0.60
200
80
1.44
1.30
Potencial (v)
1.20
1.10
1.00
0.90
0.80
0
10
20
30
40
50
4+
Volumen Ce
60
70
80
(mL)
37
'LEXMDUODFXUYDGHYDORUDFLyQGHP/GHXQDGLVROXFLyQGH)H 0FRQ.0Q2# "!
PRODU5HDOL]DUORDS+ \
Semirreacciones y espontaneidad de la reacción global
Según las tablas de potenciales redox estándar:
Fe3+ + 1e- Æ Fe2+ Eo1 = 0.77 v
MnO4- + 8H+ + 5e- Æ Mn2+ + 4H2O Eo2 = 1.52 v
La reacción de valoración es:
5Fe2+ + MnO4- + 8H+ ' 5Fe3+ + Mn2+ + 4H2O Eo = 1.52 - 0.77 = 0.75v
Su potencial indica espontaneidad hacia la derecha, con una constante, . = 3.631*1063.
La estequiometría de la reacción es 1:5, es decir, cada vez que añadimos un mol
de MnO4-se consumen cinco moles de Fe2+ y se produce un mol de Mn2+ y cinco moles
de Fe3+, respectivamente.
-Antes de llegar al punto de equivalencia, el potencial se calcula teniendo en cuenta el
par redox Fe3+/Fe2+ donde existe exceso del valorato. Se seleccionan los siguientes
porcentajes: 5, 50, 90, 95 y 99%
El cálculo se basa en que [Fe3+] / [Fe2+] es el %valoración / (100-%valoración).
Por ejemplo, al 5% de la valoración [Fe3+] / [Fe2+] = 5/95. El potencial:
E = Eo1 + 0.059*log [Fe3+]/[Fe2+] = 0.77 + 0.059*log(5/95) = 0.695v
Es decir, son los mismos valores que en el caso 3. Independientemente del pH, estos
valores son los mismos para todas las curvas.
Al igual que en el caso 3, el potencial al 50% de la valoración es igual al normal.
-En el punto de equivalencia, al ser la valoración completa y la reacción estequiométrica
se cumple:
[Fe3+] = 5 [Mn2+]
[Fe2+] = 5[MnO4-]
Calculando el potencial por un sistema u otro, este debe tomar el mismo valor:
E1 = Eo1 + 0.059*log[Fe3+]/[Fe2+]
E2 = Eo2 + 0.059/5*log[MnO4-][H+]8/[Mn2+]
6Epe = Eo1 + 5Eo2 + 0.059*log ([Fe3+]*[MnO4-][H+]8/ ([Fe2+]*[Mn2+])
Reagrupando:
Epe = (Eo1 + 5Eo2) / 6 + 0.059/6 *log ([Fe3+]*[MnO4-] / ([Fe2+]*[Mn2+]) – 8/6 *0.059*pH
38
Que teniendo en cuenta las relaciones estequiométricas de la reacción:
Epe = (Eo1 + 5Eo2) / 6 – 7.87*10-2*pH
Esta expresión nos indica que el potencial en el punto de equivalencia depende
linealmente del pH, de forma que al aumentar éste, el potencial en el punto de
equivalencia se hace menor y por tanto, menos cuantitativa es la reacción y menor es el
salto en las proximidades del punto de equivalencia. Esta expresión nos indica también
que el potencial en el punto de equivalencia se aproxima más al del agente valorante ya
que éste intercambia 5 electrones por cada electrón que intercambia el valorato. Esto
hace que el MnO4- sea mejor agente valorante que el Ce4+, por ejemplo, pues, aunque
dependiente del pH del medio, provoca mayor salto de potencial en las proximidades
del punto de equivalencia.
Calculamos el potencial en el punto de equivalencia para los pHs propuestos:
-A pH = 0, Epe = (0.77+5*1.52)/6 = 1.395v
-A pH = 2, Epe = 1.395 – 0.157 = 1.238 v
-A pH = 4, Epe = 1.395 – 0.314 = 1.081 v (0.314 = 2*0.157)!!!
Para calcular el volumen en el punto de equivalencia: ya que la reacción es 1:5,
necesitamos 5 veces menos de lo que hay de Fe2+ para valorarlo. Por tanto:
(50*0.1)/5 = 0.025*x de donde x = 40 mL.
Igualmente se puede obtener por equivalentes, pero es mejor trabajar con los
coeficientes estequiométricos para que se refuerce su comprensión.
En este caso, que también se les explica:
V1N1 = V2N2
50 * 0.1 = V2 * 0.025*5 (son 5 los electrones intercambiados, lo que otorga la valencia
al permanganto). V2 = 40 mL.
Después del punto de equivalencia distinguimos entre los tres casos de pH. Sin
embargo, si se presta atención, vasta calcular la curva a pH = 0 y para pH = 2 y pH = 4
simplemente se restan los valores obtenidos antes de 0.157 y 0.314v, respectivamente.
Incluso para ahorrar tiempo en su trazado, vasta con dibujar la de pH = 0 y la de pH = 4
y la de pH = 2 es justamente la intermedia entre las ya dibujadas.
Veamos los cálculos a pH = 0 para los siguientes porcentajes: 101, 105, 110, 150, y
200%:
La relación [MnO4-] / [Mn2+] es igual a (%valoración-100) / 100.
Por ejemplo, al 101 % de la valoración, el potencial toma el valor:
E = Eo2 + 0.059/5 *log [MnO4-] / [Mn2+] = 1.52 + 0.059/5*log(1/100) = 1.496v
39
De la misma forma se procede para los demás porcentajes, hasta llegar al 200% de la
valoración, donde [MnO4-] = [Mn2+] y el potencial es 1.52v.
-A pH = 2 los cálculos son los mismos, restando a cada valor la cantidad de 0.157v
-A pH = 4 los cálculos son los mismos, restando a cada valor la cantidad de 0.314 v
El resto de los volúmenes se obtienen proporcionalmente al obtenido para el punto de
equivalencia.
Se construye la tabla de datos y se superponen en una gráfica.
0Q2$('
( Y ( Y ( Y 5
2
0.695
0.695
0.695
50
20
0.77
0.77
0.77
90
36
0.826
0.826
0.826
95
38
0.845
0.845
0.845
99
39.6
0.888
0.888
0.888
100
40
1.395
1.238
1.081
101
40.4
1.496
1.339
1.182
105
42
1.505
1.348
1.191
110
44
1.508
1.351
1.194
150
60
1.516
1.359
1.202
200
80
1.52
1.363
1.206
9DORUDFLyQ
P/ S+ S+ S+ 40
1.60
pH = 0
1.50
1.40
Punto equivalencia a pH = 0
pH = 2
Potencial (v)
1.30
Punto de equivalencia a pH = 2
1.20
1.10
pH = 4
Punto de equivalencia a pH = 4
1.00
0.90
0.80
0.70
0.60
0
10
20
30
Volumen
40
50
MnO4
(mL)
60
70
80
Cuando el agente valorante es el dicromato potásico no se puede utilizar el
método de los porcentajes y la construcción de la curva de valoración requiere el cálculo
de las concentraciones a cada una de las adiciones propuestas de agente valorante.
41