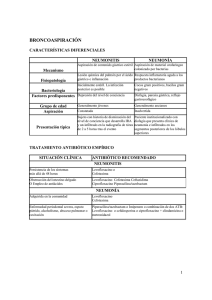
CIPROFLOXACINO Mecanismo de acción Agente antibacteriano perteneciente al grupo de las fluoroquinolonas, la acción bactericida de ciprofloxacino se debe a la inhibición tanto de la topoisomerasa de tipo II (ADN-girasa) como de la topoisomerasa de tipo IV, necesarias para la replicación, la transcripción, la reparación y la recombinación del ADN bacteriano. Forma farmacéutica y formulación: Cada TABLETA contiene: Ciprofloxacino........... 250 y 500 mg (cada 12 horas.) Cada 100 ml de SOLUCIÓN INYECTABLE contienen: Ciprofloxacino..................... 200 mg (cada 12 horas.) Farmacocinética La resistencia al ciprofloxacino se desarrolla lentamente y por etapas. Después de la administración oral es rápidamente absorbido alcanzando una concentración máxima sérica en 1 - 2 horas. Su biodisponibilidad es de aproximadamente 70-80%. Su unión a proteínas es baja (20-30 %). Ciprofloxacino penetra en todos los tejidos resultando en concentraciones que claramente exceden las correspondientes a las concentraciones séricas. Ciprofloxacino se encuentra en altas concentraciones en la bilis y es excretado por vía renal y biliar sin cambios. presentación de la ciprofloxacina es en tabletas normales, para administrarse por vía oral. Farmacodinamia Ciprofloxacino es un agente antibacteriano sintético de amplio espectro, que tiene una acción antibacteriana rápida, no solo en la fase de proliferación, sino también en la fase de descanso. Ciprofloxacino inhibe el DNA girasa, que juega un papel decisivo en la fase de proliferación bacteriana. Hay un arresto del metabolismo bacteriano, ya que información vital no puede ser leída por el cromosoma bacteriano. LA CICLOOXIGENASA (COX) Es la enzima clave en la síntesis de las prostaglandinas, a través de la oxidación del ácido araquidónico. Las prostaglandinas realizan tanto funciones relacionadas con la homeostasis de diversos órganos como con el dolor, la inflamación y el desarrollo de neoplasias. Ciclooxigenasa-1 (COX-1) Tiene como función la regulación de la proliferación de las células normales o neoplásticamente transformadas. La COX-1 es constitutiva en todos los tejidos especialmente en riñón y el tubo gastrointestinal. Participa en la producción de prostaglandinas que intervienen en procesos fisiológicos tales como: protección del epitelio gástrico, mantenimiento del flujo renal, la agregación plaquetaria, la migración de neutrófilos y también se expresan en el endotelio vascular. Se encuentra asociada al retículo endoplásmico de las células. Se han descrito dos pequeñas isoformas (PCOX-1a y PCOX-1b) de la COX-1 Ciclooxigenasa-2 (COX-2) Tiene como función mediar en los procesos de inflamación y en la señalización por prostanoides. La COX-2 se expresa tras inducción inflamatoria, aunque es constitutiva en SNC y riñón. La expresión de la COX-2 es provocada por diversos mediadores inflamatorios (interferón γ, factor de necrosis tumoral α, interleucina 1, factores de crecimiento, etc.) en diversas células (monocitos, macrófagos, células endoteliales, sinoviocitos, condrocitos y osteoblastos) y tejidos (aparato reproductor, sistema nervioso central, estómago, riñón, pulmón y ciertos tejidos afectados por procesos neoplásicos). Se encuentra asociada a la envoltura nuclear de las células. IBUPROFENO Analgésico, antipirético y antiinflamatorio no esteroídico(AINE) derivado del ácido propiónico. Es inhibidor del enzima ciclooxigenasa lo que se traduce en una inhibición de la síntesis de prostaglandinas. Forma Farmacéutica Y Formulación: Cada TABLETA contiene: Ibuprofeno...................................................................... 400 mg Excipiente, c.b.p. 1 tableta. Dosis y Vía de Administración: La administración de IBUPROFENO es por vía oral. En general, se recomiendan dosis de 200 a 400 mg cada 6 horas. Farmacocinética Vía (Oral): Su biodisponibilidad es del 80%. Es absorbido rápidamente (Tmax=1 2 h, 30 min para el arginato). Los alimentos retrasan la absorción oral. El grado de unión a proteínas plasmáticas es del 90 99%. Es ampliamente metabolizado en el hígado, siendo eliminado mayoritariamente con la orina, un 90% en forma de metabolitos inactivos conjugados con ácido glucurónico y un 10% en forma inalterada. Su semivida de eliminación es de 2 h. Farmacología El efecto analgésico es central y periférico y difiere del mecanismo antiinflamatorio. Es un potente inhibidor de la enzima ciclooxigenasa y por lo tanto, un potente reductor de la síntesis de prostaglandinas. Toxicología El único efecto tóxico observado es la aparición de ulceración de la mucosa del tracto gastrointestinal. OMEPRAZOL. Es un medicamento antiulceroso, que inhibe la bomba de protones, mecanismo de producción del ácido del estómago. Se utiliza en el tratamiento de las úlceras benignas del estómago, del duodeno, del reflujo gastroesofágico, profilaxis de la gastropatía por AINES y como coadyuvante en la erradicación de Helicobacter pylori.. También se usa para tratar la enfermedad de Zollinger-Ellison que es una condición en la cual el estómago produce demasiado ácido Forma Farmacéutica Y Formulación: Cada CÁPSULA contiene: Omeprazol......................................................................... 20 mg Cada frasco ámpula contiene: Omeprazol......................................................................... 40 mg DOSIS Y VÍA DE ADMINISTRACIÓN: Inyectable: 40 mg diario o 60 mg. Oral: 1 cápsula de 20 mg al día. 1 cápsula de 20 mg al día, durante 2 ó 3 semanas consecutivas. Propiedades farmacológicas El omeprazol se absorbe rápidamente luego de su administración oral. Su absorción no es afectada por los alimentos y parece ser dosis dependiente; hay un incremento no lineal de la concentración plasmática con dosis por encima de 40 mg, debido al metabolismo de primer paso saturable. La biodisponibilidad del omeprazol aumenta en pacientes de edad avanzada y en aquellos con alteración de la función hepática. RANITIDINA. Es un antagonista H2, uno de los receptores de la histamina, que inhibe la producción de ácido estomacal. Debe utilizarse cuidadosamente en pacientes diabéticos , pacientes de edad avanzada , pacientes con insuficiencia renal y en pacientes con alteraciones de la función hepática. Forma Farmacéutica Y Formulación: Cada ampolleta contiene: Clorhidrato de ranitidina equivalente a............................. 50 mg de ranitidina base Vehículo, c.b.p. 5 ml y 2 ml. Cada TABLETA o GRAGEA contiene: Clorhidrato de ranitidina equivalente a................. 150 Y 300 mg Dosisy Vía de Administración: Dosis recomendada es de 300 mg al acostarse, o bien, 150 mg dos veces al día, durante 4 a 8 semanas; siendo la dosis de mantenimiento de 150 mg por la noche. Síndrome de Zollinger-Ellison la dosis inicial es de 150 mg tres veces al día.En estos pacientes las dosis máximas que se han indicado son de 600 y 900 mg/día, reportándose buena tolerancia. Vía intravenosa: forma lenta en 1 ó 2 minutos, diluyendo los 50 mg en 20 ml de solución salina, glucosada o de Hartman, pudiendo repetir la dosis cada 6 u 8 horas. Infusión continua: 25 mg por hora, durante dos horas, cada 6 u 8 horas. Para prevenir el síndrome de Mendelson, si es cirugía electiva, se deberá administrar 50 mg la noche previa y 50 mg junto con la solución anestésica. En cirugía de urgencia se deberá administrar 50 mg lo antes posible. Indicaciones terapéuticas: • Embarazo: atraviesa la barrera placentaria .Se recomienda usar la Ranitidina durante el embarazo solo si es estrictamente necesario y según el acuerdo con el criterio médico y bajo el control estricto de una especialista , además , que sus beneficios superan el riesgo potencial para el producto de gestación. • Lactancia Materna: se excreta en la leche materna , por lo que debe tenerse sumo cuidado al administrar a madres que lacten , por el potencial daño que pudieran producir en el niño que lacta. Si es inevitable el tratamiento durante la lactancia , esta debe suspenderse. Es excretada por vía renal , por lo que los niveles plasmáticos se incrementan en los pacientes con insuficiencia renal severa . Se han reportado bradicardia asociada a la Ranitidina . Hay escasos reportes en la precipitación de ataques agudos de porfiria en el uso de la Ranitidina , por lo que este medicamento debe evitarse en pacientes con estos antecedentes . Pacientes con intolerancia a la lactosa . EL SÍNDROME COMPARTIMENTA Conjunto de signos y síntomas relacionados con el incremento de la presión dentro de un compartimento definido, que se traduce en una reducción o eliminación de la perfusión vascular y, como consecuencia, en la isquemia de los tejidos en el compartimento afectado. La etiología es secundaria a: Una disminución del compartimento, Un aumento de la presión del contenido del compartimiento o a ambas, El síntoma más importante es la presencia de dolor importante en la extremidad afectada y el aumento de la presión intracompartimental por encima de 30-35 mmHg. Se clasifica habitualmente en: Agudo, cuya causa más habitual suele ser un traumatismo de alta energía que ocasiona una fractura en un hueso largo, y que suele requerir fasciotomía sin cierre para liberar la presión intracompartimental, y Crónico, característico de deportistas que realizan actividad física continuada, causado por hipertrofia muscular, hemorragias o rigidez en la fascia, y susceptible de recibir tratamiento conservador y fisioterápico. Las principales causas son: La acumulación excesiva de líquido, La disminución del volumen provocando una constricción del compartimento y La pérdida de extensibilidad del volumen del compartimento causada por una compresión externa. Es decir, el síndrome compartimental tiene lugar al acumularse líquido a una elevada presión en el interior de un espacio cerrado por fascias. De esta forma disminuye la perfusión capilar por debajo del nivel necesario para la viabilidad de los tejidos. Los síntomas corresponden con las “seis P” en inglés: Presión (pressure): la zona aparece tumefacta y con una gran tensión a la palpación, pero la palpación es un método poco exacto para descubrir el aumento de presión intracompartimental, y además no es fácil de cuantificar. Puede ser enmascarado por un edema subcutáneo. Dolor (pain): es un síntoma difícil de valorar ya que puede variar dependiendo del umbral del dolor del paciente y de su sinceridad. No resulta fácil distinguir el dolor que causa la isquemia del músculo del que causa una fractura. Como ya se ha mencionado, el dolor puede llegar a estar ausente. Paresia (paresis): el músculo puede estar débil debido a la afectación primaria del nervio, a la isquemia muscular o a la defensa al dolor. Parestesia (paresthesia): o déficit sensitivo, que inicialmente se manifiesta como una parestesia, pero que ante la ausencia de tratamiento puede evolucionar una hiperestesia o anestesia. Pulsos presentes y color rosado (pulse alterations – pink): si la lesión arterial no es grave, los pulsos periféricos son palpables, aunque en ocasiones el compartimento puede tener una presión excesiva y puede obstruir una arteria importante CEFOTAXIMA. I Inhibe la síntesis de mucopéptidos en la pared de la célula bacteriana haciéndola defectuosa y osmóticamente inestable Forma farmacéutica y formulación: Cada frasco ámpula contiene: Cefotaxima.............................................................. 500 mg y 1 g Cada ampolleta con diluyente contiene: Vehículo, c.b.p., 2 y 4 ml. CEFOTAXIMA es un antibiótico semisintético de amplio espectro, pertenece al grupo de las cefalosporinas de tercera generación. CEFOTAXIMA está indicada para el tratamiento de infecciones de huesos y articulaciones; genitourinarias, del sistema nervioso central, del tracto respiratorio bajo; de la piel y tejidos blandos; ginecológicas, bacteriemia y septicemia; infecciones intraabdominales y profilaxis en intervenciones quirúrgicas con riesgo de contaminación e infección. Espectro antibacteriano: CEFOTAXIMA es resistente a la mayoría de las betalactamasas, tanto penicilinasas como cefalosporinasas; es activa in vitro, así como en infecciones clínicas contra los siguientes microorganismos: Aerobios grampositivos: Es activa contra Streptococcus pneumoniae, Staphylococcus aureus productores y no productores de penicilinasas, Streptococcus epidermidis y Streptococcus pyogens, Streptococcus agalactiae y Enterococcus sp. Aerobios gramnegativos: Son susceptibles Citrobacter sp, Enterobacter sp, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Proteus vulgaris, Proteus inconstans, Haemophilus influenzae, Haemophilus parainfluenzae, Morganella morganii, Neisseria meningitidis, Neisseria gonorrhoeae, Serratia sp, Providencia rettgeri. Algunas cepas de Pseudomonas aeruginosa, Salmonella sp y Shigella sp. Dosis y vía de administración: Las presentaciones de CEFOTAXIMA son para administración por vía parenteral. La dosis recomendada de CEFOTAXIMA depende del tipo de infección y la susceptibilidad del microorganismo. La dosis máxima diaria recomendada es de 12 g. Como guía general se recomienda que en infecciones leves y no complicadas se administre 1 g de CEFOTAXIMA cada 12 hrs. En infecciones de moderadas a severas, la dosis recomendada es de 1 a 2 g cada 8 horas. En infecciones que requieren dosis mayores se pueden administrar 2 g cada 6 u 8 horas; y en infecciones que ponen en peligro la vida se recomiendan 2 g cada 4 horas. CEFOTAXIMA. I Inhibe la síntesis de mucopéptidos en la pared de la célula bacteriana haciéndola defectuosa y osmóticamente inestable Forma farmacéutica y formulación: Cada frasco ámpula contiene: Cefotaxima.............................................................. 500 mg y 1 g Cada ampolleta con diluyente contiene: Vehículo, c.b.p., 2 y 4 ml. CEFOTAXIMA es un antibiótico semisintético de amplio espectro, pertenece al grupo de las cefalosporinas de tercera generación. Es resistente a la mayoría de las betalactamasas, tanto penicilinasas como cefalosporinasas; es activa in vitro, así como en infecciones clínicas contra los siguientes microorganismos: Aerobios grampositivos: Es activa contra S.pneumoniae, S. aureus , S. epidermidis y S. pyogens, S. agalactiae y Ente-rococcus sp. Aerobios gramnegativos: Son susceptibles Citrobacter sp, Enterobacter sp, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Proteus vulgaris, Proteus inconstans, Haemophilus influenzae, Haemophilus parainfluenzae, Morganella morganii, Neisseria meningitidis, Neisseria gonorrhoeae, Serratia sp, Providencia rettgeri. Algunas cepas de Pseudomonas aeruginosa, Salmonella sp y Shigella sp. Dosis y vía de administración: Las presentaciones de CEFOTAXIMA son para administración por vía parenteral. La dosis recomendada depende del tipo de infección y la susceptibilidad del microorganismo. La dosis máxima diaria recomendada es de 12 g. Como guía general se recomienda que en infecciones leves y no complicadas se administre 1 g de CEFOTAXIMA cada 12 hrs. En infecciones de moderadas a severas, la dosis recomendada es de 1 a 2 g cada 8 horas. En infecciones que requieren dosis mayores se pueden administrar 2 g cada 6 u 8 horas; y en infecciones que ponen en peligro la vida se recomiendan 2 g cada 4 horas.