Esclerosis
Anuncio

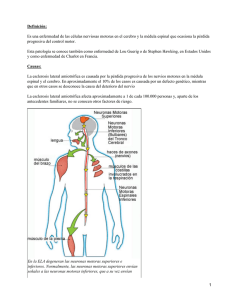
Esclerosis Lateral Amiotrófica Definición Enfermedad degenerativa progresiva de las motoneuronas superior (tractos corticoespinales) e inferior (de las células del asta anterior de la médula espinal) y ciertos núcleos motores de los pares craneales. Etiología La etiología de la enfermedad es desconocida Cerca del 5 % de los casos son familiares y se heredan como un rasgo autosómico dominante. El 20 % de los casos familiares se localiza en el cromosoma 21 donde existen mutaciones del gen de la enzima superóxido dismutasa-1 (SOD -1), que está encargada de los procesos de destoxificación celular Los aminoácidos exitotóxicos, en especial el glutamato, se consideran sospechosos, pueden provocar una reacción tóxica para las motoneuronas y en consecuencia, las destruirían Epidemiología La ELA ataca principalmente a adultos de entre 40 y 70 años. Los varones sufren el trastorno en una proporción dos veces mayor que las mujeres. La prevalencia es de 5 por 100.000 La duración promedio de sobrevida es de 3 a 5 años Anatomía Patológica Se observan fenómenos degenerativos de las células de las astas anteriores, las máximas alteraciones se sitúan en médula cervical y luego, lumbosacra o a nivel del bulbo raquídeo las motoneuronas de los nervios craneales 12, 11, 10 y luego 5 y 7 Degeneración de los Haces piramidales directo y cruzado a nivel de los pedúnculos cerebelosos en brazo posterior de cápsula interna Troncoencefálico 1° en bulbo, 2° en protuberancia y 3° en pedúnculos Manifestaciones clínicas Síndrome de la neurona motora inferior Síndrome bulbar Síndrome piramidal Síndrome de la neurona motora inferior en la médula cervical y bulbo raquídeo que se manifiesta como amiotrofia. Se inicia generalmente en los músculos de la mano con signos de torpeza y debilidad para efectuar movimientos finos Amiotrofia objetiva en el primer espacio ínteróseo y eminencia tenar La mano se deforma como consecuencia de las amiotrofias Las amiotrofias progresan extendiéndose al antebrazo (músculos flexores) y a veces al brazo (bíceps) Las amiotrofias son habitualmente simétricas Cuando la ELA comienza a nivel lumbosacro, la sintomatología aparece en miembros inferiores comúnmente con hipotonía y arreflexia profunda Si la lesión de la neurona motora inferior no se acompaña en los primeros períodos de lesión piramidal, habrá hiporreflexia profunda en los miembros superiores; cuando coexisten los dos síndromes motores, hay una parálisis atrófica en los miembros superiores con hiperreflexia profunda Fasciculaciones o fibrilaciones: se producen en los músculos que aún no están atróficos, más evidente en la eminencia tenar, primer espacio interóseo y músculo deltoides, pectoral mayor y espinales. En conclusión, los signos y síntomas de neurona motora periférica en la ELA comprenden el síndrome completo, amiotrofias distales, fibrilaciones, hipotonía, reacciones de denervación total o parcial y arreflexia profunda, si no existen signos piramidales concomitantes Comienzo Ausencia de trastornos sensitivos objetivos Síndrome Bulbar Respeta la motilidad de la mitad superior de la cara y la motilidad ocular, aunque puede dar oftalmoplejía nuclear progresiva, según cual sea el par craneal afectado. Los núcleos que se afectan son: hipogloso (XII), el espinal (XI), el neumogástrico (X), y también el facial y el trigémino Hay una parálisis atrófica de los músculos de la lengua y de la región peribucal (XII), los músculos ecom y trapecio (XII), de los músculos intrínsecos y extrínsecos de la faringe, laringe y velo del paladar (X y XI), de los músculos faciales (VII) y de los maceteros temporales y pterigoideos (V). Aparecerán así trastornos de la palabra, de deglución, de la masticación y fonación (disartria, disfagia) La primera afectada es la lengua La respiración se suele afectar tardíamente pero, a veces, puede constituir una manifestación temprana o incluso la primera. La respiración se afecta por la paresia de los músculos intercostales y del diafragma: otras veces, la disfagia determina aspiración y neumonitis, que puede constituir la complicación Terminal. Síndrome Piramidal Lo común es que los signos y síntomas piramidales aparezcan con posterioridad a las amiotrofias. Los reflejos osteotendinosos son vivos, exagerados con hipertonía espásticas y a veces policinéticos Cuando hay una degeneración que predomina en el haz geniculado, se ven crisis de risa y llanto espasmódico Cuando la lesión recae sobre los haces corticoespinales se evidencia una parálisis de tipo espástico con predominio de hiperreflexia profunda en los cuatro miembros. En conclusión, en la ELA, en su período de estado, se asistirá a una tetraparesia en donde los miembros superiores, la paresia será atrófica e hipotónica con fibrilaciones, pero con hiperreflexia; en los miembros inferiores habrá una paresia espástica pero con franca hiperreflexia profunda, faltando con cierta frec el signo de babinsky y la arreflexia cutáneo abdominal, y casi siempre el clonus de pié o rótula. ELA – Exámenes complementarios El LCR puede ser normal o tener el contenido de proteínas aumentadas por encima de 50 mg/dl; esto se da en un 30 % de los casos y por encima de 75 mg/dl en un 10 % de los casos El examen eléctrico (electromiografía) confirma la lesión neurológica, amplitud reducida. ELA – Formas Clínicas Forma clásica: comprende la asociación de signos piramidales y de neurona motora inferior Forma miotrófica: ARAN DUCHENNE, se observa la parálisis atrófica de los miembros superiores con predominio distal con atrofias, hipotonía, arreflexia profunda y reacción de degeneración Forma piramidal: solo signos piramidales, con paraparesia espástica Forma bulbar: raramente la enfermedad comienza con signo de parálisis atrófica de los pares craneales: es la parálisis bulbar progresiva Forma pseudopolineurítica: las lesiones comienzan en el ensanchamiento lumbosacro Diagnóstico La presencia de signos generales de la motoneurona inferior es prácticamente diagnóstica de enfermedad de la motoneurona de un adulto, sobre todo si aparecen el signo de babinsky o el clonus aún cuando falten estos signos el diagnóstico parece igualmente seguro si se observa una actividad inapropiada de los reflejos tendinosos o signo de Hoffmann en los miembros superiores y debilidad, atrofia y fasiculaciones musculares. Electromiograma. RNM. Análisis de laboratorio. Biopsia muscular.