Hemocromatosis: etiopatogenia, diagnóstico y estrategia terapéutica
Anuncio

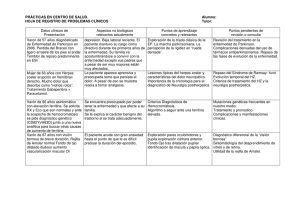
ACTUALIZACIÓN Hemocromatosis: etiopatogenia, diagnóstico y estrategia terapéutica I. Ortiz Polo, J.M. Paredes Arquiola, A. López Serrano y E. Moreno-Osset Servicio de Medicina Digestiva. Hospital Universitario Dr. Peset. Valencia. España. Universidad de Valencia. Valencia. España. Palabras Clave: Resumen - Hemocromatosis - Flebotomía La hemocromatosis hereditaria (HH) engloba varios trastornos hereditarios caracterizados por un aumento de la absorción intestinal de hierro, y una acumulación posterior en los tejidos. La mayoría de los pacientes (aproximadamente el 90%) con HH tienen mutaciones en el gen HFE. Aproximadamente el 95% de las personas con HH con mutación HFE son homocigotos para la mutación C282Y. Estudios poblacionales indican que la mutación C282Y tiene una penetrancia incompleta. Las mutaciones en los genes relacionados con el hierro que codifican la hemojuvelina, hepcidina, ferroportina, receptor de transferrina 2 y la ferritina constituyen las HH no relacionadas con el gen HFE. En HH-HFE el exceso de hierro se deposita preferentemente en el citoplasma de las células de los múltiples órganos, incluyendo el hígado, páncreas, corazón, glándulas endocrinas, la piel y las articulaciones. Los síntomas están relacionados con el daño de estos órganos. El diagnóstico incluye estudios del metabolismo del hierro, pruebas genéticas y biopsia hepática para evaluar la concentración de hierro hepático y el grado de lesión hepática. Las flebotomías previenen y revierten la acumulación del exceso de hierro y son el tratamiento de elección. Keywords: Abstract - Hemochromatosis Hemochromatosis: pathogenesis, diagnosis and therapeutic strategy - Hierro - Hepcidina - Gen HFE - Mutación - Iron - Hepcidin - HFE gene - Mutation - Phlebotomy Hereditary hemochromatosis (HH) constitutes several inherited disorders characterized by an increased intestinal absorption of iron with its subsequent accumulation in tissues. Most (approximately 90%) patients with HH have mutations in HFE. Aproximately 95% of persons with HFE-related HH are homozygous for the C282Y mutation. Population studies indicate that the penetrane of the C282Y mutation is incomplete. Mutations in the iron-related genes encoding for hemojuvelin, hepcidin, ferroportin, trasnferrin receptor 2 and ferritin result in non-HFE related HH. In HFE-related HH the excess iron is preferentially deposited in the cytoplasm of parenhymal cells of varios organs, including the liver, pancreas, heart, endocrine glands, skin and joints. Symptoms are related to damage of this organs. The diagnosis includes iron studies, genetic testing and liver biopsy to assess the hepatic iron concentration and degree of liver injury. Because phlebotomy prevents and reverses the accumulation of excess iron, is the treatment of choice. Medicine. 2012;11(19):1153-61 1153 05 ACT 5 (1153-1161).indd 1153 04/10/12 08:40 ENFERMEDADES ENDOCRINOLÓGICAS Y METABÓLICAS Introducción En sentido estricto, el término hemocromatosis denota el trastorno producido por la acumulación patológica de hierro (Fe) en el organismo. En la práctica clínica, este término se reserva para denotar aquellas situaciones en las que la acumulación de Fe es debida a un trastorno congénito de su metabolismo (hemocromatosis hereditaria [HH]). La acumulación de Fe, por otra parte, puede ser secundaria a otras causas, en cuyo caso se denomina sobrecarga férrica secundaria. TABLA 1 Clasificación de los síndromes de hemocromatosis hereditaria (HH) HH relacionada con el gen HFE o HH tipo I Mutación homocigota C282Y Mutación heterocigota H63D/C282Y HH no relacionada con el gen HFE HH tipo II o HH juvenil Mutación hemojuvelina (HJV). HH tipo IIa Mutación hepcidina. HH tipo IIb HH tipo III, asociada al receptor 2 de la transferrina (TfR2) HH tipo IV, mutación de la ferroportina (SLC40A1) HH tipo V, mutación de la H-ferritina Hemocromatosis hereditaria Es un síndrome caracterizado por una absorción intestinal excesiva de Fe que establece un estado de sobrecarga férrica en distintos órganos, especialmente hígado, corazón y páncreas, de carácter progresivo, causando un deterioro anatómico y funcional de dichos órganos. Es el defecto genético más frecuente en la población de raza blanca y se transmite en la mayor parte de los casos por herencia autosómica recesiva. Clasificación La HH incluye diferentes formas clínicas que responden a distintos tipos de alteraciones genéticas; todas ellas comparten una absorción intestinal de Fe anormalmente elevada y, adicionalmente, los cuatro tipos con herencia recesiva comparten unos niveles bajos de hepcidina urinaria y plasmática. En la tabla 1 se detallan las diferentes formas clínicas de la HH. Hemocromatosis hereditaria relacionada con el gen HFE o tipo I Está causada por una mutación del gen HFE, localizado en el brazo corto del cromosoma 6. El significado del acrónimo HFE proviene de la contracción del término en inglés relacionado con HLA-H que es la región del sistema HLA cercano al gen que codifica la proteína, y FE como símbolo del Fe. Se transmite de forma autosómica recesiva. Con una prevalencia de 1/200-300 habitantes caucásicos, este es el tipo más frecuente de HH, constituyendo más del 90% de los casos1. Las mutaciones del gen HFE que originan la HH tipo I pueden adoptar las siguientes formas: Mutación homocigota C282Y. El 90-95% de los pacientes con HH tipo I tienen una mutación homocigota del gen HFE, por la que la proteína resultante tiene sustituida una tirosina por una cisteína en la posición 282 (C282Y). Esta mutación tiene una gran variabilidad geográfica, siendo muy frecuente en Estados Unidos, Gran Bretaña, Francia, Canadá y Australia. Mutación heterocigota H63D/C282Y. El 5% de los pacientes con HH tipo I son homocigotos compuestos (C282Y en un alelo y en el otro alelo la mutación H63D). En esta última, el aminoácido aspartato ha sido sustituido por la his- Sobrecarga de hierro en África Subsahariana Miscelánea: otras formas de HH HH asociada a la mutación del gen transportador metal divalente 1(DMT1) Atransferrinemia congénita Aceruloplasminemia hereditaria tidina en la posición 63. Esta variante es más frecuente en la Europa mediterránea y en la India, y es responsable de sobrecarga moderada de Fe y de baja penetrancia. Otras mutaciones HFE. En casos aislados, se han detectado otras mutaciones como la S65C, donde la cisteína es sustituida por la serina en la posición 65, y se ha relacionado con sobrecarga férrica cuando se asocia con la mutación C282Y en el otro alelo2. Hemocromatosis hereditaria no relacionada con el gen HFE En los casos restantes (10%) la excesiva absorción intestinal de Fe es causada por mutaciones de otros genes, como el de ferroportina, hemojuvelina (HJV), hepcidina (HAMP) o del receptor de la transferrina 2(TfR2)3. Como en la mutación del gen HFE, la mutación de cualquiera de estos genes ocasionará una disminución en la producción de HAMP y, como consecuencia, se producirá una liberación inadecuada de Fe desde los macrófagos y enterocitos al plasma. En este grupo de HH se incluyen los siguientes tipos: Hemocromatosis hereditaria tipo II o juvenil. La HH juvenil puede ser debida a una mutación del gen HJV, dando lugar a la HH tipo IIa, o a una mutación de la HAMP (gen HAMP [hepcidin antimicrobial peptide]) que origina la HH tipo IIb. En ambas, la herencia es autosómica recesiva. En la HH tipo IIa la mutación se localiza en el cromosoma 1q21 y en la HH tipo IIb en el cromosoma 19q13. Las mutaciones de los genes HJV y HAMP originan las formas clínicas más agresivas de HH. Afectan a niños o adultos jóvenes de menos de 30 años, de ambos sexos. Se presentan en sujetos de raza blanca y procedencia europea, y se caracterizan clínicamente por originar hipogonadismo hipogonadotropo, cardiomiopatía, hepatomegalia, cirrosis hepática y pigmentación melánica de la piel. Hemocromatosis hereditaria tipo III. Se produce por mutación del receptor 2 de la transferrina (TfR2), localizado en el cromosoma 7q22. Clínicamente es similar a la HH tipo I, 1154 Medicine. 2012;11(19):1153-61 05 ACT 5 (1153-1161).indd 1154 04/10/12 08:40 HEMOCROMATOSIS: ETIOPATOGENIA, DIAGNÓSTICO Y ESTRATEGIA TERAPÉUTICA con transmisión autosómica recesiva. Esta mutación se ha detectado sobre todo en familias con consanguinidad del sur de Italia, aunque también se ha identificado en Japón, Portugal y Francia. Hemocromatosis hereditaria tipo IV por mutación del gen de la ferroportina (SLC40A1). Esta variante se transmite de forma autosómica dominante. El defecto genético se halla en el gen de la ferroportina (SLC40A1 [solute carrier family 40-iron regulated transporter, member 1]), localizado en el cromosoma 2q32. Existe una alteración del mecanismo de liberación del Fe desde el macrófago, ocasionando retención y depósito de Fe en los tejidos pero con un déficit de Fe plasmático. En estos enfermos es muy común que la tasa de ferritina en sangre esté muy elevada, sin que se acompañe de un aumento de la saturación de la transferrina. La hemoglobina puede estar disminuida en sangre. Es muy característico el aumento de Fe en las células del sistema retículo endotelial (SRE) localizadas en el hígado y sobre todo en el bazo. En la analítica se observa anemia ferropénica leve. Hemocromatosis hereditaria tipo V por mutación de la H-ferritina. La ferritina está compuesta por las subunidades L y H y tiene un papel importante en el almacenamiento y distribución intracelular del Fe. Esta mutación, de transmisión autosómica dominante, se describió en el año 2001 en una familia japonesa que presentaba hiperferritinemia, aumento del Fe sérico y de la saturación de la transferrina, así como siderosis a nivel de los hepatocitos de las áreas 1 y 2 de lobulillo hepático4. Sobrecarga de hierro en el África Subsahariana Sobrecarga férrica que ocurre en los africanos subsaharianos, atribuida al consumo de dietas muy ricas en Fe, sobre todo al beber cervezas fermentadas en recipientes de Fe no galvanizado. Sin embargo, en los últimos años se ha demostrado que además de este factor dietético existe un factor genético, no relacionado con el HFE, que está implicado en la patogenia de esta sobrecarga de Fe5. Miscelánea Otras variantes poco frecuentes de sobrecarga hereditaria de Fe incluyen: Mutación del transportador de metales divalente 1. El transportador de metales divalente 1 (DMT1) (divalent metal transporter 1) se localiza en el borde en cepillo intestinal y además presenta el Fe a las células eritrocitarias precursoras. La mutación homocigota de su gen regulador E399D, en la que el aminoácido aspartato ha sido sustituido por ácido glutámico, limita la disponibilidad de Fe para los precursores eritocitarios y la anemia microcítica grave resultante estimula la absorción intestinal del Fe dietético mediada por el DMT16. Atransferrinemia congénita. Es una enfermedad rara consecuencia de la mutación en el gen de la transferrina. Dispo- TABLA 2 Características de los principales tipos de hemocromatosis hereditaria (HH) de acuerdo al registro OMIM (Online Mendenlian Inheritance in Man) OMIM Tipo 1 Tipo 2 A Tipo 2 B Tipo 3 Tipo 4 235200 602390 613313 604250 606069 Mutación gen HFE HJV HAMP TfR2 SLC40A1 Herencia Recesiva Recesiva Recesiva Recesiva Dominante Década de inicio 4.a o 5.a 2.a o 3.a 2.a o 3.a 4.a o 5.a 4.a o 5.a Clínica principal Hepatopatía Endocrina Endocrina Hepatopatía Hepatopatía Curso clínico Leve Grave Grave Leve Leve HFE: gen de la hemocromatosis; HJV: gen de la hemojuvelina; HAMP: gen de la hepcidina; TfR2: gen del receptor 2 de la transferrina; SLC40A1: gen de la ferroportina. nemos de pocos estudios genéticos de esta enfermedad en el hombre. La enfermedad se transmite de forma autosómica recesiva, originando intensa ferropenia y aumento muy marcado de la ferritina sérica, junto a niveles de transferrina muy bajos o indetectables7. Aceruloplasminemia hereditaria. Esta enfermedad rara se transmite de forma autosómica recesiva, y se debe a una mutación en el gen de la ceruloplasmina, localizado en el cromosoma 3, que ocasiona la pérdida de función o la desaparición de la ceruloplasmina plasmática. La ceruloplasmina es esencial para la oxidación del Fe y para que la transferrina pueda transportar el Fe hasta los órganos donde es necesario. En ausencia de ceruloplasmina, el Fe no puede ser transportado por la transferrina, y queda retenido en las células del SRE, depositándose en los tejidos (hígado, páncreas, sistema nervioso central, etc.)8. En la tabla 2 se muestran las características de los principales tipos de HH de acuerdo al registro OMIM (Online Mendelian Inheritance in Man) que constituye una base de datos que cataloga todas las enfermedades conocidas con un componente genético. En el presente artículo se expondrán preferentemente los aspectos más relevantes de la HH tipo I dada su mayor frecuencia. Etiopatogenia La característica principal de la HH es un incremento en la absorción intestinal de Fe9. La homeostasis del Fe depende estrechamente de las necesidades fisiológicas del organismo, que son aproximadamente 1 mg al día. La absorción de la forma iónica del Fe a través del enterocito ocurre mediante dos pasos: en primer lugar, se produce la captación del Fe dietético a través de la membrana apical y a continuación es transferido al plasma a través de la membrana basolateral. Antes de la captación, el Fe iónico requiere reducirse de la forma férrica a la ferrosa, mediante las reductasas férricas que se expresan en la superficie luminal de los enterocitos duodenales. El Fe ferroso cruza la membrana apical usando el transportador DMT1. El Fe captado por el enterocito se almacena en su interior como ferritina y se excreta con las heces o bien se transfiere a través de la membrana basolateral al plasma mediante el transportador denominado ferroportina, una vez que el Fe ha sido oxidado a su forMedicine. 2012;11(19):1153-61 1155 05 ACT 5 (1153-1161).indd 1155 04/10/12 08:40 ENFERMEDADES ENDOCRINOLÓGICAS Y METABÓLICAS ma férrica por la ferroxidasa hefaestina. A continuación el Fe pasa a la circulación, uniéndose a la Condiciones normales transferrina. Esta última interacciona con su receptor en los hepaEnterocito Hígado tocitos, el receptor de la transferriHFE HJV na (TfR), para depositar el Fe que transporta en sangre. HAMP Ferritina-Fe TfR2 En los pacientes con HH se ha Fe detectado un incremento de la tasa de Hepcidina Fe que se extrae de la dieta, un incremento de la ferroportina y un aumento en la expresión del DMT1. Se han propuesto dos mecanisFerroportina Macrófago Ferroportina mos por los que la proteína de la hemocromatosis regula la absorción intestinal de Fe. Uno de ellos se rePlasma laciona con el papel que se asigna a las células de las criptas duodenales Ferritina-Fe como sensoras del contenido corporal de Fe. La proteína HFE junto con β2-microglobulina forma un complejo en las criptas de los enteHemocromatosis hereditaria tipo I rocitos duodenales con los receptores de transferrina-1 (TfR1); este Enterocito complejo se consideraba el responHígado sable principal de la regulación de HJV la absorción del Fe procedente de la HFE dieta, pero en la actualidad esta teoHAMP ría ha perdido vigencia a partir del TfR2 descubrimiento del papel de la HAMP en esta regulación. La HAMP es la principal hormona reguladora de la absorción del Fe10. Es un péptido de 25 aminoácidos producido por el hígado que posee propiedades antimicrobianas, de ahí Plasma su nombre (hepatic bactericidal proMacrófago tein). Está codificada por el gen HAMP y los genes HFE, HJV y TfR2 regulan su formación y su respuesta. La HAMP se une a la ferroportina, localizada tanto en el Fig. 1. Patogenia de la hemocromatosis hereditaria. En sujetos sanos, la hepcidina secretada por el hígado enterocito como en el macrófago, modula la tasa de hierro (Fe) liberada desde los macrófagos y enterocitos. Los genes HFE, TfR2, HJV regulan la respuesta de la hepcidina. La mutación del gen HFE ocasiona un déficit de hepcidina y una liberación se une a ella y causa su degradainapropiada de hierro desde los macrófagos y desde los enterocitos al plasma. ción11. De esta forma, la HAMP a través de la disminución de la actividad de la ferroportina produce el secuestro celular de Fe en las células del SRE y en los enterocitos duodenales, disminuyendo la retenido en las células del SRE y a continuación depósito de absorción del Fe de la dieta. Fe en los tejidos y daño tisular. En condiciones fisiológicas, la HAMP es producida por En la figura 1 se esquematizan estos aspectos de la patolos hepatocitos en respuesta a la inflamación, a la sobrecarga genia de la HH tipo 1. de Fe y a la interleuquina 6, actuando, por lo tanto, como reactante de fase aguda. Disminuye en la hipoxia o cuando aumentan las necesidades de eritropoyesis12. Penetrancia clínica del defecto genético Cuando se produce la mutación genética (HFE, HJV y TfR2), disminuye la producción de HAMP, lo que conllevará La penetrancia es la proporción de individuos que presenta el aumento de la absorción intestinal de Fe, liberación del Fe un cambio genético determinado y padecen la enfermedad13. 1156 Medicine. 2012;11(19):1153-61 05 ACT 5 (1153-1161).indd 1156 04/10/12 08:40 HEMOCROMATOSIS: ETIOPATOGENIA, DIAGNÓSTICO Y ESTRATEGIA TERAPÉUTICA La susceptibilidad genética de la forma más frecuente de HH (HH por homocigosis C282Y del gen HFE) se observa en aproximadamente una de cada 250 personas caucásicas. La expresión fenotípica se produce en aproximadamente el 70% de los homocigotos C282Y, y menos del 10% de los homocigotos C282Y desarrollan una sobrecarga de Fe lo suficientemente intensa como para originar daño orgánico. Es imposible predecir si un homocigoto C282Y expresará la enfermedad fenotípica. Clínica de la hemocromatosis hereditaria Paciente sintomático Paciente asintomático Adultos familiares de primer grado de pacientes con HH IST Ferritina IST < 45% Ferritina normal IST > 45% ± Ferritina elevada Fin de evaluación Genotipo HFE C282Y/H63D C282Y/salvaje salvaje/salvaje H63D/H63D Excluir enfermedades hematológicas Excluir enfermedades hepáticas C282Y/C282Y Ferritina < 1.000 µg/l y transaminasas normales Ferritina > 1.000 µg/l y transaminasas elevadas Flebotomías Biopsia hepática y/o Las manifestaciones clínicas típicas Flebotomías de la HH, descritas en los estudios Biopsia hepática más antiguos, son cirrosis hepática, y/o Flebotomías diabetes mellitus y la pigmentación de la piel (la llamada “diabetes bronceada”). En estas series, la enfermedad Fig. 2. Algoritmo diagnóstico-terapéutico de la hemocromatosis hereditaria. El diagnóstico inicial se realiza era más frecuente en los hombres y mediante los marcadores séricos de sobrecarga férrica: el índice de saturación de la transferrina (IST) y la ferritina. Un IST menor del 45% con una ferritina normal excluye el diagnóstico de sobrecarga férrica. La se manifestaba 10 años antes que en detección de IST ≥ 45% y una ferritina elevadas nos obligan a realizar a continuación un análisis del genolas mujeres, debido al efecto “protipo HFE. Si el paciente presenta el genotipo C282Y se confirma el diagnóstico de hemocromatosis hereditatector” de las pérdidas menstruales ria (HH) tipo I y entrará en un programa de flebotomías. Si el paciente tiene unos niveles de ferritina sérica > 1.000 μg/l o elevación de transaminasas, deberá realizarse una biopsia hepática para descartar la existende Fe. En la actualidad, la mayoría de cia de fibrosis avanzada o cirrosis hepática. De otra manera, si en el análisis del genotipo el paciente tiene los pacientes están asintomáticos, y cualquier otro genotipo distinto del homocigoto C282Y, deben investigarse otras causas de sobrecarga férrise llega al diagnóstico debido al haca. Los adultos familiares de primer grado de pacientes con HH deberán realizarse un estudio del genotipo llazgo de resultados anormales del HFE. metabolismo férrico en análisis rutinarios o por haberse realizado un cribado familiar; lógicamente, cuando los pacientes se identifican en estudios de cribado, la edad En la figura 2 se muestra el algoritmo diagnóstico de la de diagnóstico para hombres y mujeres es similar. HH. La enfermedad sintomática se caracteriza por una o varias de las siguientes manifestaciones: astenia, dolor en el Determinaciones de laboratorio cuadrante superior derecho abdominal, artralgias, condroEl diagnóstico de la enfermedad se basa en detectar la exiscalcinosis, impotencia, disminución de la libido y síntomas tencia de un aumento de las reservas de Fe, también llamado de insuficiencia cardiaca o diabetes mellitus. manifestación fenotípica, mediante la determinación de la Entre los hallazgos de la exploración física destacan hecapacidad de fijación de Fe de la transferrina (estimada por patomegalia, hiperpigmentación cutánea, artritis (especialel índice de saturación de la transferrina [IST]) y la determimente de la segunda y tercera articulación metacarpofalánginación de la ferritina sérica. Un IST igual o mayor al 45% es ca), hipogonadismo y alteraciones cardiacas14. el marcador fenotípico más precoz de HH, precediendo a la eleLos síntomas y signos de la enfermedad se producen como vación de la ferritina y a la alteración de las transaminasas15,16. consecuencia del depósito progresivo de Fe en distintos órgaLa ferritina sérica tiene una tasa elevada de resultados nos: hígado, páncreas, corazón, articulaciones, hipófisis. falsos positivos debido a que se eleva en pacientes con otras enfermedades crónicas inflamatorias o neoplásicas y en hepatopatías con actividad necroinflamatoria, como la enferDiagnóstico medad hepática alcohólica (EHA), la hepatopatía crónica por virus C (HVC), la hepatopatía crónica por virus B (HVB), El diagnóstico de la HH se basa en demostrar la existencia hígado graso no alcohólico (HGNA) y la porfiria cutánea de sobrecarga férrica tanto en la sangre como en los órganos, tarda (PCT). De hecho, en general, la sobrecarga de Fe no es especialmente el hígado. la causa más común de un nivel de ferritina elevada. AsimisMedicine. 2012;11(19):1153-61 1157 05 ACT 5 (1153-1161).indd 1157 04/10/12 08:40 ENFERMEDADES ENDOCRINOLÓGICAS Y METABÓLICAS TABLA 3 Valores del metabolismo del hierro en sujetos sanos y en pacientes con hemocromatosis hereditaria (HH) dependiendo de la presencia/ausencia de enfermedad y de manifestaciones clínicas Sujetos sanos Pacientes con HH Asintomáticos Sintomáticos Hallazgos en sangre Fe sérico (μg/dl) 60-80 150-280 180-300 IST (%) 20-50 45-100 80-100 Hombres 20-200 150-1.000 500-6.000 Mujeres 15-150 120-1.000 500-6.000 Ferritina sérica (μg/l) Hallazgos en el hígado Concentración hepática de hierro g/g de peso seco 300-1.500 2.000-10.000 8.000-30.000 mol/g de peso seco 7-27 36-179 140-550 < 1,0 > 1,9 > 1,9 0-1+ 2+ a 4+ 3+,4+ Índice de Fe hepático* Histología hepática Tinción de Perls Fe: hierro sérico; HH: hemocromatosis hereditaria; IST: índice de saturación de la transferrina. *El índice de hierro hepático representa el cociente de la concentración hepática de hierro (en μ/g de peso seco) y la edad del paciente (en años). mo, la ferritina es también un factor predictor de fibrosis hepática avanzada y cirrosis en pacientes con HH, de forma que niveles de ferritina sérica superiores a 1.000 μg/l apoyan la existencia de cirrosis17. Unos niveles de ferritina en suero superiores a 1.000 μg/l y/o una elevación de transaminasas predicen la presencia de cirrosis en el 80% de los pacientes homocigotos C282Y18. La valoración fenotípica está indicada en aquellos sujetos pertenecientes a grupos de alto riesgo de padecer HH19: 1. Familiares de pacientes diagnosticados de HH. 2. Pacientes con sospecha de afectación de órganos por el depósito de Fe. 3. Pacientes con detección bioquímica y/o alteraciones radiológicas sugestivas de sobrecarga de Fe. Cuando hay alteración de los marcadores fenotípicos se debe realizar un estudio de los marcadores genotípicos, definidos por la ocurrencia familiar de homocigosis C282Y o heterocigosis compuesta C282Y/H63D20. En la tabla 3 se muestran los valores del metabolismo del Fe en sangre e hígado, útiles para el diagnóstico de HH, en sujetos sanos y pacientes con HH asintomáticos y sintomáticos. Biopsia hepática Desde que se dispone del análisis genético de la mutación HFE, la biopsia hepática tiene un papel secundario en el diagnóstico de la enfermedad, y su principal aplicación es con fines pronósticos. La identificación de cirrosis va a modificar el tratamiento clínico de los pacientes, porque a partir de ese momento se debe realizar un cribado de varices esofágicas y de carcinoma hepatocelular (CHC). A En los pacientes homocigotos C282Y se debe realizar una biopsia cuando haya elevación de transaminasas hepáticas o un nivel de ferritina sérica superior a 1.000 μg/ml en ausencia de consumo de alcohol. Los pacientes con elevación del IST y ferritina sérica que carecen de homocigosis C282Y deben ser considerados para el diagnóstico mediante biopsia hepática si tienen niveles elevados de enzimas hepáticas u otra evidencia clínica de enfermedad hepática con un fin sobre todo diagnóstico. El estudio histológico de la muestra hepática obtenida requiere que esta sea sometida a las tinciones de hematoxilina-eosina, tricrómico de Masson y azul de Perls para evaluar el grado y la distribución celular de los depósitos de Fe. Para cuantificar la concentración hepática de Fe (CHH) se necesita una muestra de 4 mg de tejido hepático seco. Existen distintos métodos para evaluarlo, uno de ellos (sistema BattsLudwig) utiliza una estimación de la proporción de los hepatocitos que se tiñen por contener Fe. La clasificación de los rangos de tinción varía del grado 1 al 4, siendo el grado 4 el que representa un depósito panlobulillar. El índice de Fe hepático (IHH) cuantifica la tasa de acumulación de Fe hepático dividiendo la CHH (en μmol/g) por la edad del paciente en años. La mayoría de los pacientes homocigotos con HH tienen un IHH > 1,9 μmol/g al año, mientras que los pacientes con otras enfermedades crónicas tienen un IHH < 1,9 μmol/g/año21,22. La disponibilidad de las pruebas genéticas ha demostrado que la expresión fenotípica de la homocigosis puede ocurrir con un IHH mucho menor y, por lo tanto, este parámetro ya no se utiliza rutinariamente. En el examen histológico de la biopsia hepática se observa depósito de Fe en los hepatocitos, preferentemente de la zona periportal (zona acinar 1) con disminución del gradiente hacia la región central (zona acinar 3). A medida que aumenta la sobrecarga férrica, aparecen nódulos sideróticos que son agregados de células de Kupffer, depósito de Fe en células epiteliales biliares y fibrosis en los tractos portales. En la figura 3 se muestra el parénquima hepático tras tinción de Perls de un paciente con HH tipo I. En pacientes con la enfermedad de la ferroportina hay datos distintivos en la biopsia hepática, ya que el depósito de Fe se produce principalmente en el SRE, o bien puede presentar un patrón mixto con depósito en los hepatocitos y en las células reticuloendoteliales sin predominio periportal. B Fig. 3. A. Muestra de biopsia hepática (100 HPF) tras tinción de Perls de un paciente con hemocromatosis hereditaria tipo I. El círculo abarca el área ampliada. B. Ampliación (400HPF) de la zona circunscrita en A, donde se observa el lobulillo hepático con depósito férrico intracitoplasmático en los hepatocitos. 1158 Medicine. 2012;11(19):1153-61 05 ACT 5 (1153-1161).indd 1158 04/10/12 08:40 HEMOCROMATOSIS: ETIOPATOGENIA, DIAGNÓSTICO Y ESTRATEGIA TERAPÉUTICA Fig. 4. Imagen de resonancia magnética de un paciente con hemocromatosis hereditaria tipo I. Se observa parénquima hepático (H) muy hipointenso por depósito de hierro respecto al músculo y la grasa en secuencia STIR. Esta pérdida de señal mejora la detección de carcinoma hepatocelular multifocal (flechas). En otras patologías hepáticas que asocian un depósito incrementado de Fe, tales como EHA, HGNA, HVC, HVB la distribución del Fe es panlobular y generalmente leve (1 + a 2 +), con depósito de Fe en las células de Kupffer y en los hepatocitos. Métodos de diagnóstico no invasivo Resonancia magnética. Debido a las propiedades paramagnéticas del Fe, esta técnica permite cuantificar el CHH. Es el mejor método diagnóstico no invasivo, y tiene una excelente correlación inversa con el CHH, con una sensibilidad del 84-91% y una especificidad del 80-100% según el punto de corte del CHH. Puede confirmar el diagnóstico, es capaz de distinguir entre la HH primaria y las formas secundarias de sobrecarga férrica, puede determinar la gravedad de la lesión y monitorizar el efecto del tratamiento, con la ventaja añadida de que puede detectar lesiones neoplásicas pequeñas hepáticas23. Además aporta información veraz sobre el grado de fibrosis hepática, en este aspecto ofrece una sensibilidad del 100% con una especificidad del 67%. No obstante, sólo tiene utilidad en la estimación de la fibrosis cuando la biopsia hepática está contraindicada24. En la figura 4 se muestra una imagen de resonancia magnética de un paciente con HH y hepatocarcinoma multinodular. Elastografía. La elastrografía se ha introducido en los últimos años como un método no invasivo eficaz en la valoración de la fibrosis de diferentes hepatopatías. También ha demostrado su utilidad en la HH. Tratamiento El objetivo del tratamiento de la HH es reducir la sobrecarga de Fe mediante medidas dietéticas y flebotomías25. Como medidas dietéticas, a los pacientes se les recomienda realizar una dieta sana y variada, evitar polivitamínicos con Fe y cereales, así como suprimir o disminuir el consumo de vitamina C y de alcohol. Se han descrito infecciones por Vibrio vulnificus en pacientes con HH que ingieren mariscos crudos, por lo que se recomienda evitar estos alimentos. El tratamiento de elección son las flebotomías, que se deben instaurar de forma precoz, ya que reducen significativamente la morbilidad y la mortalidad cuando se inician antes del desarrollo de la cirrosis hepática y/o de la diabetes mellitus. Las flebotomías están indicadas en pacientes asintomáticos con HH homocigotos y marcadores fenotípicos de sobrecarga de Fe, en pacientes sintomáticos (el tratamiento va a reducir la progresión del daño orgánico), y en pacientes con criterios clínico-histológicos de HH no relacionados con el gen HFE. Cada unidad de sangre (500 ml) contiene aproximadamente 200-250 μg de Fe y se deben realizar flebotomías una o dos veces por semana si son bien toleradas. La ferritina empieza a disminuir progresivamente a medida que se reducen las reservas férricas pero, por el contrario, el IST permanece elevado hasta que los depósitos de Fe se agotan. El objetivo principal es conseguir unos niveles de ferritina de 50 μg/ml y un IST inferior al 50%, tras lo cual se pueden detener las flebotomías y se debe evaluar si el paciente va a necesitar tratamiento depletivo de mantenimiento. Se desconoce la causa, pero no todos los pacientes con HH reacumulan Fe, por lo que la frecuencia de las flebotomías de mantenimiento varía en cada paciente. La realización de flebotomías es seguida de una mejoría del dolor abdominal, de la astenia, de los requerimientos de insulina, de la hiperpigmentación de la piel, de la cardiopatía, de la superviviencia y de la fibrosis hepática que en algunos casos puede llegar a desaparecer. Otras manifestaciones no mejoran, como la atrofia testicular, la artropatía y la cirrosis avanzada. La realización de flebotomías antes del desarrollo de fibrosis avanzada es una estrategia importante para prevenir el CHC, ya que si el paciente ha desarrollado cirrosis hepática tiene un riesgo aumentado de presentar un CHC, y debe someterse a programas de vigilancia26. El CHC representa aproximadamente el 30% de las muertes relacionadas con HH, mientras que las complicaciones de la cirrosis hepática determinan el 20%, por lo que en ambas situaciones existe indicación de trasplante hepático. El tratamiento con quelantes se recomienda cuando la flebotomía no se tolera o está contraindicada, por ejemplo, en pacientes con insuficiencia cardíaca o anemia. Cribado de la enfermedad Una vez que un paciente con HH se ha identificado (caso índice), se recomienda realizar a todos los familiares de primer grado el estudio tanto del genotipo (mutación HFE) como del fenotipo (ferritina e IST)27. Para los hijos de un caso índice se recomienda realizar el análisis genotípico al cónyuge, de forma que si el resultado es negativo, el descendiente(s) es un heterocigoto obligado y no tiene que someterse a más pruebas. Si el cónyuge presenta homocigosis C282Y o heterocigosidad compuesta (C282Y/ H63D) y en el hijo(s) la ferritina es normal se recomienda determinar el IST y la ferritina sérica anualmente. El hallazgo de heterocigosis C282Y y heterocigosis H63D no implica riesgo de desarrollar una sobrecarga de Fe progresiva. De forma ocasional, los homocigotos H63D pueden tener alteraciones menores en las mediciones del IST o de la ferritina. Cualquiera de los genotipos mencionados puede ser un cofactor para el desarrollo de la enfermedad de hígado cuando se producen en conjunción con otras enfermedades hepáticas28. Medicine. 2012;11(19):1153-61 1159 05 ACT 5 (1153-1161).indd 1159 04/10/12 08:40 ENFERMEDADES ENDOCRINOLÓGICAS Y METABÓLICAS TABLA 4 Principales mecanismos y causas de sobrecarga férrica secundaria Eritropoyesis ineficaz Talasemia mayor mente, se administra por vía oral y ha demostrado ser eficaz en el tratamiento de la HH en ensayos clínicos fase I/II, pero la preocupación sobre sus efectos adversos cuestiona su indicación para pacientes con HH30. Anemia sideroblástica Anemia hemolítica crónica Sobrecarga de hierro parenteral Transfusiones de glóbulos rojos Inyecciones de hierro-dextrano Conflicto de intereses Los autores declaran no tener ningún conflicto de intereses. Hemodiálisis a largo plazo Hepatopatías crónicas Porfiria cutánea tarda Hepatitis crónicas virales C y B Enfermedad hepática alcohólica Enfermedad de hígado graso no alcohólico El cribado de la población general no está indicado. Asimismo, tampoco se recomienda la detección de las mutaciones en las HH no asociadas al gen HFE, ya que representan menos del 5% de los casos encontrados, y las pruebas genéticas son muy difíciles de practicar, salvo en los laboratorios de investigación. Sobrecarga férrica secundaria En la tabla 4 se muestran las principales causas de sobrecarga férrica secundaria. En estas patologías, el incremento de la absorción de Fe es consecuencia de otra enfermedad. En las anemias asociadas a sobrecarga férrica un rasgo común es la refractariedad. Esta situación se caracteriza por hipercelularidad de la médula ósea, y por la existencia de eritropoyesis ineficaz. En estos pacientes, las transfusiones hemáticas repetidas a las que son sometidos pueden agravar el estado de sobrecarga férrica. Enfermedades hepáticas crónicas como la HVC, HVB, EHA y el HGNA cursan con necroinflamación del hígado y disminución de la HAMP. En la PCT, los mecanismos implicados en el exceso de Fe son el déficit de uroporfirinógeno decarboxilasa y su frecuente asociación a la infección por virus C y al consumo de alcohol. La sobrecarga de Fe parenteral es iatrogénica, y es el resultado de transfusión de glóbulos rojos, inyecciones de Fe y diálisis a largo plazo. La flebotomía está indicada en pacientes con PCT, en los que mejoran las manifestaciones cutáneas. En la HVC, la flebotomía reduce los niveles de transaminasas y consigue una mejoría marginal en la histopatología, pero no tiene ningún efecto sobre el aclaramiento virológico. En estos pacientes, la flebotomía no se recomienda cuando la sobrecarga de Fe es leve. En el HGNA mejoran los parámetros de resistencia a la insulina, y hay una reducción de las transaminasas29. No hay evidencia de que la flebotomía sea beneficiosa en la EHA. Los quelantes constituyen el tratamiento de elección en los pacientes con sobrecarga férrica adquirida debida a eritropoyesis ineficaz. Desferoxiamina (Desferin®) es un fármaco de administración subcutánea en infusión continua. Otro quelante, deferasirox (Exjade®), comercializado reciente- Bibliografía • Importante •• Muy importante ✔ Metaanálisis ✔ Artículo de revisión Ensayo clínico controlado ✔ ✔ Guía de práctica clínica Epidemiología ✔ 1. •• Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki ✔ MB, Nicoll AJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-30. 2. • Wallace DF, Walter AP, Pietrangel A, Clare M, Bomford AB, Dixon ✔ JL, et al. Frequency of the S65C mutation of HFE and iron overload in 309 subjects hetererozygous for C282Y. J Hepatol. 2002;36:474-9. 3. •• Pietrangelo A. Non-HFE hemochromatosis. Hepatology. 2004; ✔ 39:21-29. 4. •• Solís Herruzo JA, Solís Muñoz P. Hemocromatosis no ligada al ✔ HFE. Rev Esp Enferm Dig. 2005;97:266-86. 5. •• Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diag✔ nosis and management or hemochromatosis:2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-43. Mims MP, Prchal JT. Divalent metal transporter 1. Hematology. 2005;10:339-45. Knisely AS, Gelbart T, Beutler E. Molecular characterization of a third case of human atransferrinemia. Blood. 2004;104:2607. Loréal O, Turlin B, Pigeon C, Moisan A, Ropert M, Morice P, et al. Aceruloplasminemia: new clinical, pathophysiological and therapeutic insights. J Hepatol. 2002;36:851-6. Pietrangelo A. Hereditary hemochromatosis: Pathogenesis, diagnosis and treatment. Gastroenterology. 2010;139:393-408. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783-8. Nemeth E, Tuttle MS, Powelson J, Vaugh MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004; 306:2090-3. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461-3. Pietrangelo A. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatology. 2010;53:3-22. Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-30. Adams PC, Reboussin DM, Press RD, Barton JC, Acton RT, Moses GC, et al. Biological variability of transferrin saturation and unsaturated iron-binding capacity. Am J Med. 2007;120:999.e1-e7. Tavill AS. Diagnosis and management of hemochromatosis. Hepatology. 2001;33:1321-8. Morrison ED, Brandhagen DJ, Phatak PD, Barton JC, Krawitt EL, El-Serag HB, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627-33. Beaton M, Guyader D, Deugnier Y, Moirand R, Chakrabarti S, Adams P. Noninvasive prediction of cirrhosis in C282Y-linked hemochromatosis. Hepatology. 2002;36:673-8. Powell LW, Dixon JL, Ramm GA, Purdie DM, Lincoln DJ, Anderson GJ, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294-301. Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology. 2007;46:1291-301. Summers KM, Halliday JW, Powell LW. Identification of homozygous hemochromatosis subjects by measurement of hepatic iron index. Hepatology. 1990;12:20-5. Sallie RW, Reed WD, Shilkin KB. Confirmation of the efficacy of hepatic tissue iron index in differentiating genetic haemochromato- 6. • ✔ 7. • ✔ 8. • ✔ 9. •• ✔ 10. •• ✔ 11. •• ✔ 12. •• ✔ 13. •• ✔ 14. •• ✔ 15. • ✔ 16. •• ✔ 17. • ✔ 18. • ✔ 19. • ✔ 20. •• ✔ 21. • ✔ 22. • ✔ 1160 Medicine. 2012;11(19):1153-61 05 ACT 5 (1153-1161).indd 1160 04/10/12 08:40 HEMOCROMATOSIS: ETIOPATOGENIA, DIAGNÓSTICO Y ESTRATEGIA TERAPÉUTICA sis from alcoholic liver disease complicated by alcoholic haemosiderosis. Gut. 1991;32:207-10. Gandon Y, Olivie D, Guyader D, Aube C, Oberti F, Sebille V, et al. Non-invasive assessment of hepatic iron stores by MRI. Lancet. 2004;363:357-62. Olynyk JK, St Pierre TG, Britton RS, Brunt EM, Bacon BR. Duration of hepatic iron exposure increases the risk of significant fibrosis in HCC: a new role for magnetic resonance imaging. Am J Gastroenterol. 2005;100:837-41. Bokhoven MA, Deursen BM, Swinkels. Diagnosis and management of hereditary haemochromatosis. BMJ. 2011;342:218-23. Farrell GC, Chan HL, Yuen MF, Amarapurkar DN, Chutaputti A, Fan JG, et al. Prevention of hepatocellular carcinoma in the Asia-Pacific region: consensus statements. J Gastroenterol Hepatol. 2010;4:657-63. 23. • ✔ 24. • ✔ 25. •• ✔ 26. • ✔ 27. • Bulaj ZJ, Ajioka RS, Phillips JD, LaSalle BA, Jorde LB, Griffen ✔ LM, et al. Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med. 2000;343:1529-35. 28. •• Adams P, Brissot P, Powell LW. EASL International Consensus ✔ Conference on Haemochromatosis. J H Hepatol. 2000;33:485-504. 29. • Valenti L, Fracanzani AL, Dongiovanni P, Bugianesi E, Marchesi✔ ni G, Manzini P, et al. Iron depletion by phlebotomy improves insu- lin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case-control study. Am J Gastroenterol. 2007;102:1251-8. 30. Phatak P, Brissot P, Wurster M, Adams PC, Bonkovsky HL, Gross J, et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52:1671-779. ✔• Medicine. 2012;11(19):1153-61 1161 05 ACT 5 (1153-1161).indd 1161 04/10/12 08:40