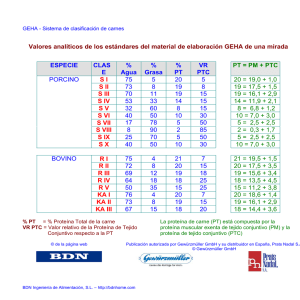
expresión y purificación de las proteínas recombinantes
Anuncio

EXPRESIÓN Y PURIFICACIÓN DE LAS PROTEÍNAS RECOMBINANTES ESTRUCTURALES VP5, VP6 Y VP8 DEL ROTAVIRUS LUZ YURANY MORENO RUEDA UNIVERSIDAD NACIONAL DE COLOMBIA FACULTAD DE MEDICINA DEPARTAMENTO DE CIENCIAS BIOLÓGICAS MAESTRIA EN BIOQUÍMICA BOGOTÁ D.C 2010 EXPRESIÓN Y PURIFICACIÓN DE LAS PROTEÍNAS RECOMBINANTES ESTRUCTURALES VP5, VP6 Y VP8 DEL ROTAVIRUS LUZ YURANY MORENO RUEDA Código: 597660 Trabajo de grado presentado para optar el título de Magister en Bioquímica Director CARLOS ARTURO GUERRERO FONSECA, MD., MSc., PhD. UNIVERSIDAD NACIONAL DE COLOMBIA FACULTAD DE MEDICINA DEPARTAMENTO DE CIENCIAS BIOLÓGICAS MAESTRIA EN BIOQUÍMICA BOGOTÁ D.C 2010 AGRADECIMIENTOS Después de culminar una de las etapas más importantes de mi vida, quiero agradecer a Dios por darme la fortaleza y sabiduría que se requieren para continuar día tras día y afrontar cada uno de los desafíos que se ponen en nuestro camino; a mi madre querida, quien con su amor, dedicación y apoyo incondicional ha logrado que cada una de las metas que me he propuesto se lleven a cabo; a mi hermanita, a quien amo con todo el corazón por ser quien es hoy en día y a quien espero poder brindarle lo mejor de mí. Así mismo, quiero agradecer a mis amigos y compañeros participes de este proceso, con quienes compartí toda clase de momentos y a quienes quiero y admiro profundamente, especialmente a Luz Marina Castillo, por acompañarme y apoyarme en los momentos difíciles. Por último, quiero manifestar mis más sinceros agradecimientos a la Universidad Nacional de Colombia y especialmente a las personas que conforman el laboratorio de Biología Molecular de Virus. Particularmente, al Doctor Carlos Arturo Guerrero Fonseca por permitirme realizar mi trabajo de grado y por compartir su valiosa experiencia durante el desarrollo de este trabajo; al Doctor Orlando Acosta, por ser partícipe en mi proceso de formación durante la maestría y a Miguel Ospino Marquez, a quien aprecio por su valiosa colaboración y preocupación. RESUMEN La infección con rotavirus se ha convertido en una de las principales causas de gastroenteritis, mal nutrición y diarrea en niños y animales jóvenes. La mortalidad promedio en países desarrollados es baja; sin embargo, cada año mueren más de 600.000 niños en el mundo y aún no existen estrategias terapéuticas lo suficientemente eficientes para disminuir los casos de diarrea aguda ocasionada por la infección en infantes y en animales de importancia económica como bovinos, cerdos y aves. Por esta razón, el laboratorio de Biología Molecular de Virus de la Universidad Nacional ha intentado dilucidar las moléculas virales y celulares involucradas en los pasos de unión y penetración viral y la secuencialidad de estas interacciones para que en un futuro, se logren proponer diferentes estrategias que permitan reducir la infección con rotavirus en los eventos iniciales del proceso infeccioso. De esta manera, el objetivo general de este trabajo consistió en la expresión y purificación de tres proteínas estructurales del rotavirus, VP5, VP6 y VP8, para determinar la interacción de estas con proteínas celulares candidatas a receptores del rotavirus en los eventos de unión y penetración viral, como Hsc70 y PDI. La metodología que se utilizó para el desarrollo de este trabajo consistió en expresar las proteínas recombinantes VP5 y VP8 del rotavirus y la proteína celular Hsc70 mediante un sistema de expresión bacteriano, para lo cual se realizó la transfección de los plásmidos pGEX-4T o pET 28a, que presentan las secuencias génicas que codifican para cada una de las proteínas, en la cepa bacteriana de E. coli BL21(DE3) y se optimizaron las condiciones de expresión para cada una de ellas, los factores que se tuvieron en cuenta fueron: medio de crecimiento, número de bacterias antes de inducir la expresión, concentración del inductor y tiempo de inducción. La expresión de la proteína VP6 se llevó a cabo mediante la infección de células MA104 con el virus vaccinia, el cual fue modificado genéticamente y presenta la secuencia que codifica la proteína viral. Posteriormente, se procedió a la purificación de las proteínas expresadas teniendo en cuenta las características bioquímicas de las mismas mediante técnicas cromatográficas, electroelución y tratamiento con detergentes. La identificación de la interacción de las proteínas recombinantes estructurales del rotavirus con PDI y Hsc70 en un sistema acelular se determinó mediante ensayos de ELISA indirectos y co-inmunoprecipitación; mientras que los ensayos de interacción en un sistema celular fueron llevados a cabo con células MA104, para lo cual se identificó la presencia de las proteínas virales por coinmunoprecipitación de la proteína Hsc70. Por otra parte, se realizaron ensayos de inhibición de la infección con las proteínas virales en células MA104 y se identificó mediante inmunocitoquímica. Finalmente, la producción de anticuerpos policlonales se realizó con las proteínas recombinantes obtenidas en conejos Nueva Zelanda. La mayor concentración de la proteína viral recombinante VP8 se obtuvo cuando las bacterias transformadas se cultivaron en medio LB y la inducción se llevó a cabo cuando el cultivo alcanzó una DO 600 nm de 0,5 con 1 mM de IPTG durante 6 h. En el caso de la de la proteína recombinante VP5, se alcanzó la mayor concentración de esta proteína cuando el cultivo alcanzó una DO 600 nm de 0,2 y la expresión se indujo con 0,5 mM de IPTG durante 4 h en medio 2XYT en presencia de glucosa al 2 %. Mientras que la proteína celular recombinante Hsc70 fue expresada cuando el bacteriano alcanzó una DO 600 nm de 0,6 con 1 mM de IPTG durante 4 horas medio LB. Las proteínas recombinantes virales y celulares obtenidas en este trabajo fueron obtenidas de forma insoluble, para lo cual fue necesario el uso de detergentes iónicos y no iónicos para su posterior proceso de purificación mediante cromatografía de afinidad (VP5, VP8 y Hsc70) o solubilización diferencial con detergentes y agentes caotrópicos (VP6). Así mismo, se obtuvieron anticuerpos policlonales dirigidos contra las proteínas recombinantes virales, demostrando la actividad antigénica de las proteínas purificadas. Estos anticuerpos fueron empleados para el desarrollo de este trabajo y serán utilizados en el laboratorio de Biología Molecular del Virus en estudios posteriores puesto que fueron caracterizados y se demostró el reconocimiento de las proteínas nativas en la partícula viral tanto en condiciones denaturantes como no denaturantes. Finalmente, se logró identificar la interacción de las proteínas virales recombinantes VP5 y VP6 con las proteínas celulares recombinantes Hsc70 y PDI en dos sistemas libres de células. La interacción de las proteínas virales VP5 y VP6 con Hsc70 fue determinada en células MA104 y localizadas en microdominios lípidicos de la membrana. Por otra parte, se demostró que la presencia de la proteína VP6 durante la unión y penetración del virus a las células, inhibió la infección viral casi en un 100%. Estos resultados sugieren la participación de las proteínas celulares Hsc70 y PDI durante los eventos iniciales del proceso infeccioso mediado por interacciones con las proteínas estructurales de la partícula viral. Cabe resaltar, que la proteína estructural VP6 puede estar implicada en las etapas iniciales de la infección y al parecer es un factor determinante en la entrada del virus a la célula. CONTENIDO Pág. 1. INTRODUCCIÓN 16 ROTAVIRUS 19 1.1 CARACTERÍSTICAS GENERALES DEL ROTAVIRUS 19 1.2 PROTEÍNAS ESTRUCTURALES QUE FORMAN LA PARTÍCULA VIRAL 20 1.3 CARACTERÍSTICAS GENERALES DE LA INFECCIÓN CON ROTAVIRUS 23 1.4 MECANISMOS DE UNIÓN Y ENTRADA DEL ROTAVIRUS 24 2 27 SISTEMAS DE EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS 2.1 EXPRESIÓN DE PROTEÍNAS RECOMBINANTES EN Escherichia coli 27 2.2 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN LEVADURAS 32 2.3 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN CÉLULAS DE 33 INSECTO, LARVAS Y CÉLULAS DE MAMÍFERO MEDIANTE BACULOVIRUS 2.4 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN CÉLULAS DE 34 MAMÍFERO MEDIANTE EL VIRUS VACCINIA 3 37 SISTEMAS DE PURIFICACIÓN DE PROTEÍNAS HETERÓLOGAS 3.1 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LAS 41 CARACTERÍSTICAS DE SUPERFICIE DE LAS PROTEÍNAS 3.2 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LA 41 ESTRUCTURA TOTAL DE LA PROTEÍNA: FORMA Y TAMAÑO 3.3 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LA CARGA 42 NETA DE LA PROTEÍNA 3.4 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN 42 BIOPROPIEDADES (AFINIDAD) 4 OBJETIVOS 4.1 OBJETIVO GENERAL 44 44 4.2 OBJETIVOS ESPECÍFICOS 44 5 METODOLOGÍA 45 5.1 AMPLIFICACIÓN Y PURIFICACIÓN DE LO S VECTORES QUE 45 PRESENTAN LAS SECUENCIAS GÉNICAS QUE CODIFICAN LAS PROTEÍNAS VIRALES VP5 Y VP8 Y LA PROTEÍNA CELULAR Hsc 70 5.1.1 Características de los plásmidos que presentan las secuencias génicas 45 de las proteínas recombinantes 5.1.2 Cepas bacterianas de Escherichia Coli. 46 5.1.3 Preparación de medios de cultivo para el crecimiento de las cepas bacterianas de E. coli. 5.1.4 Transfección de los plásmidos que presentan las secuencias de las proteínas VP5 y VP8. 5.1.5 Purificación de los plásmidos que presentan las secuencias de las proteínas estructurales VP5 y VP8. 5.1.6 Corte de los plásmidos con la enzima de restricción Pst I 47 47 48 49 5.2 EXPRESIÓN DE LAS PROTEÍNAS RECOMBINANTES VP5, VP8 Y VP6 50 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 5.3 EXPRESIÓN DE LA PROTEÍNA RECOMBINANTE VP6 EN UN SISTEMA 51 DE INFECCIÓN DE CÉLULAS EUCARIOTAS 5.3.1 Cultivo celular. 51 5.3.2 Infección de la línea celular MA104 con el virus vaccinia recombinante 51 que presenta la secuencia que codifica la proteína estructural del rotavirus VP6. 5.3.3 Análisis de la expresión de la proteína viral estructural VP6 mediante el 52 conteo de Unidades Formadoras de Foco (UFF). 5.4 SOLUBLIZACIÓN Y PURIFICACIÓN DE LA S PROTEÍNAS 53 ESTRUCTURALES VP5, VP6, VP8 Y Hsc 70 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc70 5.4.1 Solubilización de las proteínas recombinantes VP5, VP8 y Hsc70 a 53 partir del lisado de bacterias BL21(DE3) transformadas con los vectores de expresión. 5.4.2 Purificación de las proteínas estructurales VP5 y VP8 del rotavirus 54 mediante cromatografía de afinidad. 5.4.3 Purificación de la proteína celular Hsc 70 mediante cromatografía de 55 afinidad. 5.4.4 Solubilización e identificación de la localización de la proteína 55 estructural VP6. 5.4.5 Purificación de la proteína estructural VP6 del rotavirus mediante el 56 tratamiento con detergentes. 5.5 OBTENCIÓN DE ANTICUERPOS POLICLONALES DIRIGIDOS CONTRA 57 LAS PROTEÍNAS ESTRCUTURALES VP5, VP6 Y VP8 DEL ROTAVIRUS 5.5.1 Preparación de las proteínas estructurales VP5 y VP8 del rotavirus 57 mediante electroelución para la inmunización de conejos Nueva Zelanda. 5.5.2 Corte del polipéptido GST de las proteínas virales. 58 5.5.3 Preparación de la proteína estructural VP6 del rotavirus para la 58 inmunización de conejos Nueva Zelanda. 5.5.4 Inoculación en conejos para la obtención de anticuerpos policlonales 59 contra las proteínas estructurales del rotavirus VP5, VP6 y VP8 5.5.5 Caracterización de los anticuerpos policlonales contra las proteínas 59 virales VP5, VP6 y VP8 y la proteína celular Hsc 70. 5.6 ENSAYOS DE INTERACCIÓN ENTRE LAS PROTEÍNAS ROTAVIRALES Y 62 LAS PROTEÍNAS CELULARES Hsc70 Y PDI 5.6.1 Ensayo de ELISA indirecto. 62 5.6.2 Co - inmunoprecipitación 63 5.6.3 Interacción de las proteínas virales con la proteína Hsc70 en los 63 dominios rafts lipídicos 5.6.4 Ensayos de inhibición de la infección en células MA104 con el 64 rotavirus de la cepa RRV 6 66 RESULTADOS 6.1 AMPLIFICACIÓN Y PURIFICACIÓN DE LO S VECTORES QUE 66 PRESENTAN LAS SECUENCIAS GÉNICAS QUE CODIFICAN LAS PROTEÍNAS VIRALES VP5 Y VP8 6.2 EXPRESIÓN DE LAS PROTEÍNAS RECOMBINANTES VP5, VP8 Y VP6 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 67 6.3 6.2.1 Expresión de la proteína recombinante estructural del rotavirus VP8 68 6.2.2 Expresión de la proteína recombinante estructural del rotavirus VP5 73 6.2.3 Expresión de la proteína recombinante Hsc 70. 79 6.2.4 Expresión de la proteína estructural del rotavirus VP6. 84 PURIFICACIÓN DE LA S PROTEÍNAS RECOMBINANTES 85 ESTRUCTURALES VP5, VP8 Y VP6 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 6.3.1 Purificación de la proteína VP8-GST mediante cromatografía de 85 afinidad. 6.3.2 Purificación de la proteína VP5-GST mediante cromatografía de 87 afinidad. 6.3.3 Purificación de la proteína recombinante Hs c 70 mediante 89 Purificación de la proteína VP6 mediante tratamiento con detergentes. 91 cromatografía de afinidad. 6.3.3 6.4 OBTENCIÓN DE ANTICUERPOS POLICLONALES CONTRA LA S 93 PROTEÍNAS ESTRUCTURALES VP5, VP8 Y VP6 DEL ROTAVIRUS EN CONEJOS 6.5 INTERACCIÓN DE LAS PROTEÍNAS RECOMBINANTES PURIFICADAS 100 VP5, VP8 Y VP6 CON LAS PROTEÍNAS CELULARES Hsc 70 Y PDI 6.5.1 Interacción de las proteínas celulares PDI y Hsc70 con las proteínas 100 recombinantes VP5, VP6 y VP8 en condiciones acelulares. 6.5.2. Interacción de la proteína celular Hsc70 con las proteínas virales VP5, 105 VP6 y VP8 en condiciones celulares. 6.5.3 Inhibición de la infección con RRV de células MA104 con las proteínas 107 recombinantes estructurales del rotavirus. 7 ANÁLISIS DE RESULTADOS 110 8 CONCLUSIONES 125 BIBLIOGRAFÍA 128 LISTA DE FIGURAS P ág . Figura 1 Representación gráfica de una partícula viral de rotavirus. 20 Figura 2 Composición proteíca de la partícula del rotavirus. 22 Figura 3 Representación gráfica de las proteínas estructurales del rotavirus 23 VP7 y VP4, sitio de corte de la proteína VP4, dominios de unión con integrinas y ácido siálico. Figura 4 Modelo propuesto entre las interacciones de las proteínas virales y las 25 moléculas celulares en el proceso de adhesión y penetración viral. Figura 5 Activación e inactivación del la transcripción del operón Lac tras el 29 estímulo con Lactosa o Glucosa, respectivamente. Figura 6 a. Estructura química de la ampicilina, b. Reacción catalizada por las 30 enzimas que confieren resistencia a la kanamicina. Figura 7 Versatilidad del vector de expresión baculovirus. 34 Figura 8 Representación gráfica del vector de expresión utilizado para la 46 inserción de las secuencias de ADN que codifican las proteínas estructurales del rotavirus, VP5 y VP8. Figura 9 Montaje para la separación y purificación de ADN plasmídico. 49 Figura 10 Montaje para la electroelución de las proteínas virales recombinantes 57 a partir de geles de poliacrilamida. Figura 11 a) Purificación de los plásmidos pGEX-4T que presentan las 67 secuencias que codifican las proteínas virales VP5 y VP8 purificados y digeridos con Pst I. b) Secuencia génica que codifica la proteína viral VP5. Figura 12 a) Curva de crecimiento de las bacterias BL21(DE3) – VP8 en medio LB medido por turbidimetría a 600 nm. b) Expresión de la proteína viral recombinante VP8 – GST variando el número de bacterias 69 transformadas antes de inducir la expresión. Figura 13 a) Expresión de la proteína viral VP8 – GST variando el tiempo de 72 inducción con 1 mM de IPTG. b) Expresión de la proteína viral VP8 – GST variando la concentración de IPTG. Figura 14 Expresión de la proteína viral VP8 – GST variando la temperatura a la 73 cual se realizó el crecimiento bacteriano y la inducción de la expresión de la proteína recombinante. Figura 15 Expresión de la proteína viral VP5 - GST en medio LB y 2XYT en 74 presencia y ausencia de glucosa al 2 %. Figura 16 a) Curva de crecimiento de las bacterias BL21(DE3) – VP5 en medio 2XYT y 2 % de Glucosa 75 medido por turbidimetría a 600 nm. b) Expresión de la proteína viral recombinante VP5 – GST variando el número de bacterias transformadas antes de inducir la expresión. Figura 17 a) Expresión de la proteína viral VP5 – GST variando el tiempo de 78 inducción con 1 mM de IPTG. b) Expresión de la proteína viral VP5 – GST variando la concentración de IPTG. Figura 18 Expresión de la proteína viral VP8 - GST (a) y VP5 – GST (b) en 79 cuerpos de inclusión. Figura 19 a) Curva de crecimiento de las bacterias BL21(DE3) – Hsc 70 en 80 medio 2XYT medido por turbidimetría a 600 nm. b) Expresión de la proteína recombinante Hsc70 variando el número de bacterias transformadas antes de inducir la expresión. Figura 20 a) Expresión de la proteína recombinante Hsc70 variando el tiempo 83 de inducción con 1 mM de IPTG. b) Expresión de la proteína recombinante Hsc70 variando la concentración del inductor IPTG. Figura 21 Localización de la proteína viral VP6. 85 Figura 22 Purificación de la proteína recombinante VP8 – GST mediante 87 cromatografía de afinidad. El lisado bacteriano fue tratado con N – Sarcosina y DTT para solubilizar los cuerpos de inclusión y tratado con: (a) tritón X-100 al 3 % o (b) diluido 10 veces con PBS, las fracciones obtenidas de la cromatografía fueron analizadas por SDSPAGE. Figura 23 Purificación de la proteína recombinante VP5 – GST mediante 88 cromatografía de afinidad a partir de lisados bacterianos tratados con N – sarcosina y DTT para solubilizar los cuerpos de inclusión y con diferentes detergentes renaturantes. Figura 24 Purificación de la proteína recombinante VP5 – GST mediante 89 cromatografía de afinidad a partir de un lisado bacteriano tratado con N – sarcosina y DTT para solubilizar los cuerpos de inclusión. Figura 25 Purificación de la proteína Hsc 70 mediante cromatografía de afinidad 90 para lo cual se empleó una resina de sefarosa unida a Ni2+. Figura 26 Solubilización de la proteína viral VP6 posiblemente precipitada 91 intracelularmente. Figura 27 Purificación parcial de la proteína viral VP6 mediante el tratamiento 92 del pellet obtenido después de la lisis de células MA104 infectadas con el virus vaccinia recombinante con diferentes detergentes de manera secuencial. Figura 28 Preparación de los antígenos de las proteínas virales para la 93 generación de anticuerpos policlonales en conejos. Figura 29 Digestión con trombina del polipéptido GST de las proteínas virales 94 VP8 y VP5 obtenidas a partir de las bandas identificadas en los geles preparativos, electroeluidas y precipitadas con acetona. Figura 30 Caracterización de los anticuerpos policlonales contra las proteínas 97 recombinantes VP5, VP6 y VP8 obtenidos en conejos Nueva Zelanda utilizando diferentes cantidades de las proteínas recombinantes. Figura 31 Identificación de las proteínas virales recombinantes y en TLPs con 99 diferentes diluciones de los anticuerpos policlonales dirigidos contra estas proteínas. Figura 32 Cuantificación por SDS – PAGE de las proteínas virales (VP5, VP6 y 100 VP8) y las proteína celulares Hsc70 y PDI frente a una curva de BSA teñido con azul de coomassie. Figura 33 Determinación de la interacción de las proteínas estructurales del 103 rotavirus con las proteínas celulares Hsc70 y PDI mediante el ensayo de ELISA indirecto en condiciones acelulares. Figura 34 Identificación de la interacción de las proteínas estructurales del rotavirus con las proteínas celulares Hsc70 y PDI mediante co – 105 inmunoprecipitación. Figura 35 a) Identificación de la presencia de las proteínas celulares Hsc70, 107 Integrina 3 y PDI en rafts lipídicos. b) Identificación de la interacción de las proteínas estructurales del rotavirus en condiciones celulares. Figura 36 Efecto de la presencia de diferentes concentraciones de las proteínas 108 virales VP5 – GST, VP8 – GST, VP6 y DLPs sobre la infección de RRV en células MA104. Figura 37 Caracterización de las TLPs y DLPs de RRV utilizadas en los ensayos de inhibición de la infección en presencia de las proteínas virales recombinantes. 109 LISTA DE TABLAS P ág . Tabla 1 Determinación aproximada de la concentración de la proteína viral 70 VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes densidades ópticas al momento de inducir la expresión de la proteína VP8 con IPTG. Tabla 2 Determinación aproximada de la concentración de la proteína viral 70 VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de que el cultivo alcanzó una DO 600 nm de 0,5. Tabla 3 Determinación aproximada de la concentración de la proteína viral 71 VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana con diferentes concentraciones de IPTG después de que el cultivo alcanzó una DO 600 nm de 0,5. Tabla 4 Concentración aproximada de la proteína viral VP5 - GST en las 76 fracciones de proteínas insolubles después de la lisis bacteriana a diferentes densidades ópticas antes de la inducción con 1 mM de IPTG. Tabla 5 Concentración aproximada de la proteína viral VP5 unida a GST en 77 las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de alcanzar una DO 600 nm de 0,2. Tabla 6 Concentración aproximada de la proteína viral VP5 unida a GST en 77 las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes concentraciones de IPTG después de alcanzar una DO nm Tabla 7 600 de 0,2. Concentración aproximada de la proteína recombinante Hsc70 en las fracciones de proteínas solubles e insolubles después de la lisis 81 bacteriana a diferentes densidades ópticas antes de la inducción con 1 mM de IPTG. Tabla 8 Concentración aproximada de la proteína recombinante Hsc 70 en las 82 fracciones de proteínas solubles e insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de alcanzar una DO 600 nm de 0,6. Tabla 9 Concentración aproximada de la proteína recombinante Hsc 70 en las fracciones de proteínas solubles e insolubles después de la lisis bacteriana con diferentes concentraciones de IPTG después de alcanzar una DO 600 nm de 0.6. 83 INTRODUCCIÓN El rotavirus es una partícula viral no envuelta en membrana lipídica formada por tres capas proteicas, entre las cuales se encuentran: la capa más externa compuesta por las proteínas estructurales VP4 y VP7, la capa media formada por la proteína VP6 y la capa interna formada por VP1, VP2 y VP3. Esta última capa encierra el genoma viral compuesto por 11 segmentos de ARN de cadena doble (en inglés dsRNA), los cuales codifican las seis proteínas estructurales y seis proteínas no estructurales (NSP1 – NPS6) involucradas en el ciclo viral del rotavirus, incluyendo replicación, transcripción, traducción y formación de nuevas partículas infecciosas. La infección con rotavirus en niños y animales es principalmente restringido a los enterocitos localizados en las microvellosidades intestinales y aunque el rotavirus puede infectar niños de mayor edad y adultos, la diarrea severa es observada principalmente en niños menores a dos años de edad. El marcado tropismo del rotavirus por los enterocitos maduros del intestino delgado in vivo sugiere que estas células expresan una serie de moléculas que pueden actuar como receptores del rotavirus que facilitan su penetración al interior de la célula. Adicionalmente, el rotavirus interacciona e infecta una serie de líneas celulares in vitro, que incluyen células de riñón o de origen intestinal, estómago, hueso y pulmón, entre otras. Actualmente, se han identificado mediante estudios in vitro en las líneas celulares MA104 (línea celular epitelial de riñón de mico) y Caco-2 (línea celular de un adenocarcinoma humano) diferentes moléculas celulares que están involucradas en la unión y penetración del virus y que actúan como receptores de adhesión e internalización, entre las cuales se encuentran: ácido siálico (AS), integrinas 21, 41, v3 y x2, la proteína de choque térmico (en inglés hsc) 70 y algunos gangliósidos. Sin embargo, el mecanismo por el cual el rotavirus penetra a la célula, incluyendo complejidad y temporalidad de las interacciones con las moléculas celulares, no ha sido definido aunque se ha propuesto un mecanismo de penetración directa y otro mediado por endocitosis 1. La infección con rotavirus es la causa más común de diarrea severa en niños menores de 5 años y tiene un gran impacto sobre la morbilidad y mortalidad de la población infantil, siendo responsable de aproximadamente 600.000 muertes anuales, predominantemente en países en vía de desarrollo. En Colombia, se han desarrollado diferentes estudios sobre rotavirus desde el año 1979 – 2003, indicando que el 70% de las pruebas que resultaron positivas para este virus correspondían a niños menores de 24 meses 2 - 3. Actualmente, la vacunación se ha convertido en el único control que tiene un impacto notable sobre la incidencia de la diarrea deshidratante causada por la infección con rotavirus. En 1999, una vacuna contra rotavirus altamente eficiente fue patentada en Estados Unidos (RotaShield) y retirada del mercado después de 14 meses por su asociación con invaginación del intestino delgado. Dos nuevas vacunas orales fueron patentadas en el año 2.006, la vacuna de rotavirus rearreglado de origen humano – bovino (Rotateq) y la vacuna monovalente de rotavirus humano (Rotatrix), las cuales han mostrado una alta eficiencia en la disminución de la infección en diferentes estudios clínicos y en América Latina, además no han revelado incrementos de riesgo de invaginación. Sin embargo, el porcentaje de eficiencia en la disminución de la infección después de la vacunación es de cerca del 65% 4 y no se encuentra accesible a toda la población, por lo tanto es necesario la creación de nuevas estrategias terapéuticas que permitan prevenir o disminuir la infección casi en su totalidad, que sea de mayor accesibilidad y que no sea necesariamente preventiva. Por esta razón, en el laboratorio de Biología Molecular de Virus de la Universidad Nacional se están llevando a cabo diferentes proyectos de investigación que pretenden dilucidar los mecanismos básicos bioquímicos y moleculares, las interacciones entre el rotavirus y las células huésped y las vías de señalización que se activan o inhiben y que favorecen la replicación e infección viral a fin de encontrar posibles blancos terapéuticos involucrados en el proceso infeccioso. Adicionalmente, se ha demostrado la participación de diferentes moléculas en este proceso, como son Hsc70 y la integrina αv3. Recientemente, se logró identificar a la proteína Disulfuro Isomerasa (PDI) como un posible receptor del rotavirus mediante ensayos de infección en presencia o ausencia de agentes que afectan la actividad isomerasa de esta proteína y ensayos de interacción con la partícula viral en un ambiente acelular y celular; sin embargo, no se ha identificado con cual de las proteínas estructurales del virus interacciona. Por lo tanto, resulta necesario diseñar estrategias experimentales viables que permitan el desarrollo y culminación de estos proyectos de investigación. A partir del presente trabajo, se estandarizaron protocolos para la expresión de proteínas estructurales del rotavirus con actividad antigénica en un sistema de transfección bacteriana (VP5 y VP8) y mediante un virus recombinante (VP6) y se obtuvieron anticuerpos policlonales contra estas proteínas para uso en el Laboratorio de Biología Molecular de Virus para el apoyo y desarrollo de otros proyectos de investigación en pruebas de identificación mediante ensayos de ELISA, Western Blot, inmunofluoresecencia e inmunocitoquímica de ensayos de inhibición de la infección. Así mismo, se determinó la interacción de las proteínas celulares, PDI y Hsc70, con las proteínas virales recombinantes en un sistema in vitro y en condiciones celulares mediante ensayos de unión en dominios microdominos lipídicos y de competencia en células MA104. Estos resultados permiten dirigir la atención a estas chaperonas como posibles moléculas determinantes en el proceso infeccioso, particularmente durante la unión y penetración del virus a la célula, convirtiéndolas en moléculas blanco para el diseño de nuevas estrategias terapeúticas que permitan reducir los casos de diarrea deshidratante ocasionados por la infección con este virus. 1. ROTAVIRUS 1.1 CARACTERÍSTICAS GENERALES DEL ROTAVIRUS El rotavirus, el cual hace parte de la familia de los Reovirus, es ahora reconocido como la principal causa de gastroenteritis viral severa en humanos y animales jóvenes. Las principales características bioquímicas y morfológicas del rotavirus incluyen las siguientes: a) las partículas virales maduras no presentan envoltura lipídica y posee una tricapa proteica icosahedral de aproximadamente 75 nm de diámetro, b) el genoma del virus consta de 11 segmentos de ARN de cadena doble (ARN ds), los cuales codifican seis proteínas estructurales (VP1, VP2,VP3, VP4, VP6 Y VP7) y seis proteínas no estructurales (NSP1 – NSP6), c) las partículas contienen una ARN polimerasa dependiente de ARN y otras enzimas capaces de replicar y transcribir el ARN, d) la replicación del virus ocurre en el citoplasma de células infectadas, e) el cultivo de estos virus es facilitado por el tratamiento de enzimas proteolíticas que aumenta la infección por corte de la proteína VP4 de la capa más externa, f) las partículas virales son ensambladas en el retículo endoplasmático (RE) y g) las partículas infecciosas son liberadas de la células huésped mediante lisis. 5 Los rotavirus se clasifican serológicamente en grupos o serogrupos que contienen los virus que muestran reactividad cruzada antigénica detectable por ensayos serológicos tales como, inmunofluorescencia, ELISA y microscopia inmunoelectrónica. De acuerdo con esto, seis grupos distintos de rotavirus (A - F) han sido identificados. Los grupos A, B y C han sido encontrados tanto en animales como en humanos; mientras que los grupos D, E y F solo han sido caracterizados en animales. Así mismo, los rotavirus de un grupo son divididos en serotipos, los cuales son definidos por ensayos de reducción de placa y neutralización reducción de focos fluorescentes usando antisueros de animales hiperinmunizados con partículas virales purificadas. Estos ensayos miden la reactividad de anticuerpos con las dos proteínas de la capa más externa del rotavirus (VP4 y VP7), las cuales inducen la producción de anticuerpos con actividad neutralizante. De lo anterior, los rotavirus de un grupo se clasifican en serotipos G (denota a la proteína VP4, sensible a proteasas), P (denota a VP7, glucoproteína) y SI o SII (denotan la proteína VP6). 6 1.2 PROTEÍNAS ESTRUCTURALES QUE FORMAN LA PARTÍCULA VIRAL Las partículas virales constan de tres capas concéntricas de proteínas. La capa más interna está formada por la proteína estructural VP2, la cual rodea el genoma viral compuesto por 11 segmentos de ARN de cadena doble, y las proteínas VP1 y VP3; en conjunto estas proteínas constituyen el centro viral. La adición de la proteína VP6 a la capa de VP2 produce partículas de doble capa. La capa más externa, característica de partículas infecciosas de triple capa, está compuesta por dos proteínas, VP4 y VP7. La superficie del virus está compuesta por 260 trímeros de VP7 y 60 dímeros de VP4 que se extiende en estructuras con forma de espigas en la superficie del virus (figura 1). 7 Conjuntamente, estas proteínas presentan actividad replicasa en sistemas de centros derivados de viriones o baculovirus que expresan estas proteínas y permiten la formación de CLPs (Partículas similares al centro viral). Por otra parte, una señal esencial para la replicación fue encontrada en los extremos 3´ del gen 8 y 9 del rotavirus que son altamente conservados, sugiriendo que puede ser una característica común de los 11 segmentos de ARN viral para su eficiente replicación y transcripción. 8 - 16 Figura 1. Representación gráfica de una partícula viral de rotavirus. Los círculos de color amarillo representan la proteína estructural VP7, las estructuras de color rojo representan la proteína VP4 (capa externa), los círculos de color azul representan la proteína VP6 (capa intermedia) y la capa interna está reprensada de color verde. VP1: Esta proteína es codificada por el segmento 1 del genoma del rotavirus y hace parte del centro de la cápside viral. Un pequeño número de monómeros de la proteína hacen parte del virión, sugiriendo que esta proteína no tiene una importante función estructural y que hace parte de un complejo enzimático. Además, evidencias experimentales han permitido demostrar que esta proteína es una ARN polimerasa dependiente de ARN, teniendo en cuenta los siguientes aspectos: a) presenta dominios homólogos a otras ARN polimerasas de otros virus, 8 - 10 b) se une a nucleótidos y el uso de nucleótidos análogos azido-ATP inhibe la actividad polimerasa, 11 y c) partículas virales semejantes al centro viral (CLPs) que constan de VP1 y VP2 pueden catalizar la síntesis de ARN ds a partir de ARNm (actividad replicasa), mientras que partículas que solo presentan la proteína VP2 carecen de esta actividad polimerasa 12. VP2: Esta proteína es codificada por el segmento 2 del genoma y es la más abundante en el centro viral. VP2 es la única proteína que tiene sitios de unión de ácidos nucleícos (DNA, ssRNA y dsRNA), preferencialmente por dsRNA, pero la unión no es secuencia específica 5. Esta proteína tiene la capacidad de ensamblarse y formar CLPs 13 - 14. VP3: Esta proteína es codificada por el segmento 3 del genoma del rotavirus que hace parte del centro viral de la cápside en cantidades mínimas y se une covalentemente a GTP, lo que ha permitido proponerla como la guanililtransferasa viral 15. VP6: Es una proteína codificada por el segmento 6 del genoma viral y forma la capa media del virión. La caracterización bioquímica de VP6 ha permitido identificar la formación de trímeros y túbulos como una capacidad intrínseca de la proteína extraída y purificada de viriones y mediante ensayos estructurales tridimensionales de la superficie de estas partículas. Estudios in vitro e in vivo indican que VP6 es requerida para la actividad polimerasa pero no se encuentra directamente involucrada en la trascripción; lo cual ha permitido sugerir que es un componente estructural de las partículas para mantener la organización del complejo transcripcional, compuesto por las proteínas del centro viral. Adicionalmente, VP6 es altamente inmunogénica y antigénica siendo ampliamente usada para ensayos de diagnóstico para la identificación de partículas virales 5. VP4: Es una proteína codificada por el segmento 4 del genoma del rotavirus, hace parte de la capa externa del rotavirus, no es glucosilada y constituye la hemaglutinina de algunas cepas virales 5. VP4 está implicada tanto en el proceso de unión y penetración a la célula huésped como en la hemaglutinación, neutralización, virulencia y aumento de la infectividad del rotavirus por proteasas 17 - 19. Este último fenómeno es particularmente importante teniendo en cuenta que el rotavirus se replica preferencialmente en enterocitos maduros del intestino delgado, un ambiente rico de proteasas. El corte proteolítico de VP4 aumenta la eficiencia de entrada viral a las células y por tanto la capacidad infectiva 20 - 21. Durante la proteólisis, VP4 es clivada en dos fragmentos, VP5 (60 kDa) y VP8 (28 kDa) (figura 2), los cuales permanecen asociados al virión 22. Datos experimentales han mostrado que VP4 sufre cambios conformacionales transitorios de un estado desorganizado a uno organizado después de la tripsinización que parecen ser los responsable del aumento en la capacidad infectiva del rotavirus (figura 3) 23. En el proceso de entrada del rotavirus a la célula se han evidenciado una serie de pasos que involucra los dos fragmentos de la proteína VP4, en donde el domino de VP8 está involucrado en procesos de unión a ácido siálico (AS), mientras que VP5 está implicada en las interacciones con la integrina 21 y Hsc70 (figura 2) 24. VP7: Es la segunda proteína más abundante de la cápside viral y se localiza en la capa más externa del rotavirus. Esta proteína es codificada por el segmento 9 de la cepa SA11 de simios y segmento 8 de la cepa UK de bovinos. Además, es una proteína glucosilada, altamente inmunogénica e induce anticuerpos neutralizantes. Al igual que VP4, la proteína VP7 se encuentra involucrada en el proceso de penetración y entrada del rotavirus a la célula huésped (figura 3). Figura 2. Composición proteíca de la partícula del rotavirus a. Representación gráfica de una partícula rotaviral TLP, las estructuras de color amarillo representan la proteína estructural VP7 y las de color rojo, a la proteína VP4, b. DLP, las estructuras de color azul representan la proteína estructural VP6 y c. Corte de la proteína VP4 que genera dos fragmentos VP8 y VP5. 7 Figura 3. Representación gráfica de las proteínas estructurales del rotavirus VP7 y VP4, sitio de corte de la proteína VP4, dominios de unión con integrinas y ácido siálico. 7 1.3 CARACTERÍSTICAS GENERALES DE LA INFECCIÓN CON ROTAVIRUS El rotavirus presenta un específico tropismo celular in vivo, infectando inicialmente enterocitos maduros y sin capacidad proliferativa de la vellosidad del intestino delgado, sugiriendo que estas células tiene receptores específicos para el virus. Por otra parte, en estudios in vitro, el rotavirus se une a una gran variedad de líneas celulares aunque solo un grupo de estas (incluyendo células de origen renal o intestinal y líneas celulares transformadas derivadas de seno, estómago, hueso y pulmón) son eficientemente infectadas, sugiriendo que la unión del rotavirus a la célula es promiscua y que la interacción con receptores celulares que son responsables de la entrada viral probablemente ocurre en un paso post-unión 26. Por tanto, la infección con rotavirus en niños se localiza en el epitelio de la vellosidad del intestino delgado, resultando en la atrofia total de la misma. Aunque el rotavirus puede infectar niños y adultos, la diarrea es generalmente observada en niños menores a dos años de edad, la cual es ocasionada por una serie de factores, incluyendo: reducción del área de superficie epitelial, reemplazo de enterocitos maduros por inmaduros (células de la cripta), un efecto osmótico resultante de la incompleta absorción de carbohidratos del lumen intestinal en combinación con fermentación bacteriana de los compuestos no absorbidos, secreción del fluido intestinal y electrolitos a través de la activación del sistema nerviosos entérico y el efecto de la proteína no estructural NSP4, la cual ha sido considerada como la enterotoxina viral 27. 1.4 MECANISMOS DE UNIÓN Y ENTRADA DEL ROTAVIRUS La entrada del rotavirus a la célula es un proceso de múltiples pasos, en los cuales diferentes dominios de las proteínas de la superficie del rotavirus interactúan con diferentes dominios de las proteínas de la superficie de la célula, los cuales actúan como receptores de unión y entrada viral 7. Diversas estrategias experimentales han sugerido que el rotavirus interactúa secuencialmente con diversas moléculas de la superficie de la célula para entrar a la célula, usando diferentes dominios de las proteínas que forman la capa más externa del rotavirus, entre los cuales se encuentran los siguientes: AS – VP8: Rotavirus dependientes de SA inicialmente interaccionan con AS sobre la superficie celular mediante una región localizada entre los aminoácidos 93 – 208 del dominio VP8 de VP4 45 - 46. Integrina α2β1 – VP5: La caracterización de un mutante de rotavirus. Conocido como nar3, indica que este se une a la superficie celular por interacción con la integrina α2β1, mientras que el rotavirus parental RRV de simio interactúa con esta integrina en un paso post-unión, después de su unión a AS. Así mismo, se evidenció en cepas de rotavirus independientes de AS o resistentes a neuraminidasa, los cuales interactúan con esta integrina en un paso post-unión 42. La interacción de RRV y nar3 con la integrina α2β1es mediada por un motivo de reconocimiento DGE, localizado en los aminoácidos 308 – 310 de VP4, en el dominio VP5 43. Hsc70 – VP5: Después del contacto inicial del rotavirus con AS y la integrina α2β, el virus interactúa con tres proteínas adicionales de la célula huésped: hsc 70, αvβ3 y αxβ2. La interacción viral con hsc 70 es mediada por un dominio en VP5 que está localizado entre los aminoácidos 642 – 659. Un péptido sintético que asemeja esta región y anticuerpos contra hsc 70 bloquean la infección con rotavirus pero no su unión a la célula 47. Por otra parte, una región similar (aminoácidos 650 - 657) del VP5 de la cepa CRW8 fue seleccionada por su habilidad para unirse a células MA104 y especialmente a la proteína Hsc 70, es representado por el péptido KID y no conservado en diferentes cepas virales, sugiriendo que la interacción hsc 70 – VP5 no es estrictamente secuencia específica 48. Integrina αvβ3 – VP7: La interacción del rotavirus con esta proteína aparentemente es independiente del dominio RGD que está presente en los ligandos naturales de esta integrina 49. Un estudio permitió comparar las secuencias de las proteínas de superficie del rotavirus y hantavirus (el cual usa la integrina αvβ3 de una manera independiente de del dominio RGD) y se identificó una región que consta de 9 aminoácidos (entre 161 - 169) con alta homología entre la proteína VP7 de RRV y la proteína G1G2 del hantavirus L99. Esta secuencia en VP7 es altamente conservada en diferentes cepas de rotavirus y un péptido sintético de esta región se une a la integrina αvβ3 en estudios in vitro, un sitio diferentes de unión al dominio RGD y eficientemente bloqueó la infección pero no la unión del rotavirus a la célula 50. Integrina αxβ2 – VP7: Esta interacción se basa en que VP7 contiene el dominio de unión GPR a la proteína αxβ2, entre los aminoácidos 253 – 255. La caracterización de esta interacción fue demostrada mediante el uso de péptidos sintéticos GPRP que bloquearon la infección de las cepas Wa y RRV pero no su unión a la célula. Figura 4. Modelo propuesto entre las interacciones de las proteínas virales y las moléculas celulares en el proceso de adhesión y penetración viral. Este modelo se basa en estudios realizados con una cepa sensible a neuramidasa. 7 Adicionalmente, ensayos experimentales de inhibición con anticuerpos dirigidos contra la integrina 41 y sus ligandos naturales ha permitido sugerir que esta proteína también participa en los eventos iniciales de la infección con rotavirus 1. Por otra parte, la infección con rotavirus es parcialmente bloqueada por el uso de inhibidores metabólicos de N-glucosilación y síntesis de glucolípidos y eliminación de colesterol de la membrana celular 51. Estos resultados sugieren que regiones o dominios ricos de esfingolípidos y colesterol en la membrana, conocidos como microdominios lipídicos, pueden estar involucrados en la entrada viral 52. La participación de los de los microdominios lipídicos se basa en que el gangliósido GM1, las subunidades de las integrinas α2 y β3, y hsc70 están asociadas a estas regiones y que las partículas infecciosas también se asocian con estos dominio en interacciones tempranas del virus con la células. Por lo tanto, los microdominios lipídicos pueden servir como plataformas para facilitar la eficiente interacción de los receptores celulares con la partícula viral mencionados anteriormente se resumen en la figura 4. 53. Los mecanismos 2. SISTEMAS DE EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS 2.1 EXPRESIÓN DE PROTEÍNAS RECOMBINANTES EN Escherichia coli Escherichia coli es una bacteria en forma de varilla con un cromosoma circular de cerca 3 millones de pares de bases (pb) de longitud, que puede crecer rápidamente en medio mínimo que contenga una fuente de carbono y sales que proporcionen nitrógeno, fósforo y algunas trazas de metales. Esta bacteria ha sido el huésped procarionte más utilizado para la expresión de proteínas recombinantes debido a que ha su genética molecular, fisiología y sistema de expresión han sido ampliamente caracterizados 54 - 55, de costos bajos respecto a otros métodos de expresión y facilidad de obtener cultivos con una alta densidad celular que han facilitado la obtención de proteínas recombinantes y productos biomoleculares no proteicos, tales como: aminoácidos, metabolitos primarios y secundarios con alto rendimiento 56 - 59. La expresión de proteínas recombinantes mediante este sistema requiere de vectores específicos, conocidos como plásmidos, de los cuales existe una gran variedad con una gran diversidad genética que facilitan la localización y purificación de la proteína expresada. Estos vectores son pequeñas moléculas de ADN circular que se encuentran en bacterias de forma independiente al ADN cromosomal que no son esenciales para la sobrevivencia de las mismas; sin embargo, contienen genes que codifican proteínas que les confieren resistencia a antibióticos. El gen de interés puede ser insertado a la secuencia del ADN plasmídico para formar un híbrido o plásmido recombinante que es capaz de replicarse y ser expresado en las bacterias puesto que presentan secuencias de iniciación de la replicación, promotores de la transcripción, regiones de iniciación de la traducción así como secuencias de terminación de la transcripción y traducción reconocidos por la maquinaria bacteriana. La unión del gen de interés en el vector requiere el uso de enzimas de restricción que reconocen una secuencia específica presente tanto el gen como en el plásmido, los fragmentos generados son realineados por complementariedad de las bases nitrogenadas y unidos mediante una DNA ligasa. Las plásmidos recombinantes formados son introducidos en las bacterias, por un procedimiento de transformación como: el método de sales (generalmente, CaCl2), polietilenglicol y electroporación, que se fundamentan en la formación de poros en la membrana y pared celular para permitir la entrada del material genético hacia el interior de la célula 60 - 61. Por otra parte, la expresión de proteínas heterólogas en estas bacterias requiere que el vector presente un promotor transcripcional fuerte e inducible para controlar la expresión del gen a un alto nivel. El promotor transcripcional más comúnmente utilizado es el operón Lac que consta de tres genes LacZ, LacY y LacA. Cuando las células crecen en un medio mínimo en presencia de glucosa, la transcripción es bloqueada por el represor Lac (producto del gen LacI) el cual se une a un único sitio corriente arriba del gen LacZ y evita que la RNA polimerasa transcriba los genes LacZ, LacY y LacA, de los cuales dos de estos productos son necesarios para el crecimiento en presencia de lactosa. Para la expresión de proteínas recombinantes generalmente se emplean inductores químicos o térmicos, de los cuales el más común es el Isopropil D tioglactopiranósido (IPTG) el cual interacciona con el represor Lac evitando que se inhiba la transcripción de genes que se encuentren bajo el control de este promotor 62. La transcripción y expresión basal en la ausencia de un inductor es minimizado en presencia de un inhibidor o represor, lo cual es de vital importancia cuando la proteína expresada produce una situación de estrés celular y permite la pérdida del plásmido o la muerte celular. El inhibidor de la transcripción basal más empleado durante la expresión de proteínas recombinantes, es la glucosa, puesto que disminuye la concentración de AMPc, el cual se une a la proteína de unión a AMPc (CAP) activando la transcripción de este operón (figura 5) 63 - 64. El operón LacUV es una versión mutada del operón Lac, cuya actividad basal es menos sensible al incremento de niveles intracelulares de AMPc y al igual que el operón Lac, se activa tras el uso de análogos de lactosa. Los marcadores de resistencia más comúnmente utilizados en los vectores de expresión confieren resistencia a diversos antibióticos, como: ampicilina, puesto que expresa una enzima -lactamasa, la cual es secretada al periplasma en donde cataliza la hidrólisis del anillo -lactam del antibiótico; Kanamicina, la cual es inactivada en el periplasma por la actividad de aminoglucósido fosfotransferasa (figura 6); Cloranfenicol, el cual es inactivado por el producto del gen CAT (cloranfenilcol acetiltransferasa) y tetraciclina, el cual es inactivado por el producto de varios genes 62. Figura 5. Activación e inactivación del la transcripción del operón Lac tras el estímulo con Lactosa o Glucosa, respectivamente 62. Otro factor fundamental que se debe tener en cuenta para la expresión de proteínas recombinantes es la cepa de E. coli huésped, puesto que se debe caracterizar por tener una baja expresión de proteasas a fin de evitar la degradación de la proteína de expresión. La cepa de E. coli que es generalmente utilizada en estos protocolo es la cepa BL21(DE3), la cual se caracteriza por alcanzar una alta densidad óptica, no es patogénica y es deficiente en la expresión de ompT y Lon, dos proteasas extracelular e intracelular respectivamente, que pueden degradar la proteína de interés. Adicionalmente, esta cepa presenta la secuencia que codifica la RNA polimerasa T7 que depende del promotor lacUV5 y la expresión del represor LacI, por esta razón los vectores de expresión utilizados con esta cepa deben presentar el promotor con la secuencia de reconocimiento para esta RNA polimerasa. Algunos vectores generan proteínas de fusión, proteínas ligadas a péptidos o proteínas (His6 y GST) de características conocidas, que proporcionan un marcador de reconocimiento, simplifica el proceso de purificación y el replegamiento de proteínas denaturadas mediante cromatografía de afinidad; sin embargo, esta molécula puede afectar la estructura tridimensional y generalmente el corte del sitio específico de esta molécula en el proceso de purificación, no es del 100% 62. a. b. Acetiltransferasa Fosfotrasnferasa Adeniltrasnferasa Figura 6. a. Estructura química de la ampicilina, b. Reacción catalizada por las enzimas que confieren resistencia a la kanamicina. Adicionalmente, esta bacteria consta de una membrana interna que divide la célula en tres compartimentos: el citoplasma, el periplasma y el espacio extracelular. Se han desarrollado estrategias para dirigir la síntesis de las proteínas recombinantes a un compartimento definido. Cuando el destino final es el citoplasma, un alto nivel en la producción de la proteína frecuentemente permite la formación de cuerpos de inclusión, los cuales contienen la proteína recombinante con plegamientos intermedios y no nativos o desnaturalizada 67, 75 Además, E. coli no es conveniente para la expresión de proteínas de gran tamaño o complejos proteícos que contengan enlaces disulfuros o modificaciones pos–transduccionales y la estabilidad de la proteína foránea puede ser baja debido a degradación proteolítica. Sin embargo, diversas estrategias han sido desarrolladas para permitir la formación de la estructura nativa de la proteína, esto incluye el uso de diferentes promotores y cepas bacterianas 76 - 77, 66, 68 - 69, la co-expresión de chaperonas 70 – 71, el uso de cepas deficientes en tioredoxina para mantener un potencial redox favorable o proteasa, para disminuir la degradación de la proteína la velocidad de síntesis de la proteína 67, 80, 78 - 79, la reducción de disminución de la temperatura durante el cultivo 72 – 74, 81 y uso de polipéptidos altamente solubles para producir proteínas de fusión 82 - 83 Cabe mencionar que la expresión de proteínas en cuerpos de inclusión tiene una serie de ventajas, entre ellas: estas agregaciones proteicas son acumuladas en el citoplasma con una alta concentración (aproximadamente el 25% de la proteína total); pueden ser purificados y concentrados por simple centrifugación, lo que reduce los procesos para eliminar proteínas contaminantes; no tienen actividad biológica, facilitando la producción de proteínas que pueden ser tóxicas para las bacterias y por último, los cuerpos de inclusión son resistentes a proteólisis; sin embargo, para obtener laproteína activa se requieren de procesos adicionales de solubilización y replegamiento que no garantizan el 100 % de eficiencia. Otro problema que debe tenerse en cuenta es que la proteína final puede presentar una metionina formilada en el N-terminal 65, 84. Por otra parte, la producción de la proteína recombinante en el periplasma presenta varias ventajas, entre ellas: la identidad del aminoácido del N- terminal se mantiene, la actividad proteasa es menor respecto al citoplasma, la purificación de la proteína recombinante es más fácil puesto que no hay un gran contenido de proteínas contaminantes, la formación de puentes disulfuro puede ser facilitada porque el espacio periplásmico proporciona el ambiente oxidante requerido y las proteínas pueden ser usadas en ensayos de actividad in vivo 84 - 86. Sin embargo, para que una proteína recombinante se dirija al espacio periplásmico debe presentar una secuencia señal, la cual puede no ser completamente procesada o puede ser removida; además, la proteína de interés puede ser expresada en bajas concentraciones. En algunos casos, la células se lisan porque la membrana externa se debilita y la eficiente secreción varía dependiendo de la proteína recombinante. Adiconalmente, se ha evidenciado la formación de cuerpos de inclusión y los puentes disulfuro pueden formarse inapropiadamente 84. Finalmente, la expresión de proteínas recombinantes en el medio extracelular se deben emplear cepas de E. coli que carezcan de pared bacteriana o bacterias Gram – positivas 87. Este mecanismo de expresión presenta una serie de ventajas respecto a los dos anteriores puesto que la proteína de interés se mantiene soluble y por tanto no se evidencia la formación de cuerpos de inclusión, el replegamiento se favorece, la identidad del N-terminal se mantiene, se disminuye significativamente la degradación por proteasas y se facilita el proceso de purificación; sin embargo, la concentración de la proteína excretada es muy poca y en algunos casos no se excreta, se aumenta la lisis celular y la proteína recombinante que se obtiene se encuentra muy diluida 84. 2.2 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN LEVADURAS Las levaduras como sistema de expresión de proteínas heterólogas han sido ampliamente usadas por cerca de dos décadas en investigación básica y en la industria biotecnológica para la producción y secreción de proteínas humanas, de animales, de plantas o virales. En muchos casos, este sistema ha sido empleado para la producción de proteínas foráneas que no se pueden extraer de su fuente natural debido a que no pueden ser purificadas en grandes cantidades o porque son proteínas que presentan mutaciones puntuales y no se encuentran naturalmente, lo que ha permitido el desarrollo de estudios de proteínas de importancia médica y farmacológica 88. Las levaduras se han convertido en un buen sistema de expresión de proteínas heterólogas por ciertas razones, entre ellas: el crecimiento microbiano es rápido y se obtiene una alta densidad óptica a bajos costos, establece una mezcla entre la manipulación genética encontrada en bacterias en un ambiente eucariota, como procesamiento proteolítico, plegamiento, formación de enlaces disulfuro y glucosilación 89; mientras que las bacterias carecen de la propiedad de realizar estas modificaciones y por tanto producen proteínas eucariotas con plegamientos no nativos, insoluble e inactivas y otros sistemas eucariontes, como células de ovario de hámster (CHO) y líneas celulares de insectos infectadas con baculovirus resultan ser más costosos, el rendimiento en la expresión es menor y demandan más tiempo y esfuerzo 90, 96.Sin embargo, no puede realizar modificaciones pos-transduccionales más complejas, como: prolil hidroxilación, amidación y algún tipo de glucosilación y fosforilación 91. La expresión de proteínas foráneas en levaduras consta de cuatro pasos: clonación de la secuencia génica que codifica la proteína de interés en un casete de expresión que contiene un promotor y sitio de terminación transcripcional de levaduras, transformación estable del DNA recombinante en la célula huésped, síntesis de la proteína foránea en condiciones específicas del cultivo y finalmente, la purificación de la proteína heteróloga y comparación con la proteína nativa. Generalmente, un promotor regulable es usado para controlar la síntesis de la expresión de la proteína foránea puesto que se debe evitar la expresión basal de la misma, lo que minimiza la selección de células que no la expresen durante la fase de crecimiento celular 90, 96. Diversas cepas de levaduras han sido empleadas en la expresión de proteínas heterólogas 92 - 95; sin embargo, las cepas más empleadas para este propósito son Pichia pastoris y Saccharomyces cerevisiae 2.3 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN CÉLULAS DE INSECTO, LARVAS Y CÉLULAS DE MAMÍFERO MEDIANTE BACULOVIRUS Los vectores de expresión Baculovirus han sido ampliamente utilizados para la obtención de una gran variedad de proteínas recombinantes en células de insecto, incluyendo proteínas citoplasmáticas, nucleares, mitocondriales o unidas a membrana. La expresión de proteínas heterólogas mediante este sistema permite el plegamiento, modificaciones pos-transduccionales, transporte y ensamblaje de polipéptidos sintetizados para producir proteínas solubles e idénticas a las nativas 97 - 99; sin embargo, estas células tienen un perfil de glucosilación diferente en comparación con células de eucariontes superiores, debido que permite la formación de N – glicanos con un alto contenido de manosa o paucimanosa 100. Por otra parte, se puede obtener un alto nivel de expresión de la proteína de interés cuando es co-transfecta con chaperonas, las cuales intervienen en el plegamiento y modificaciones de estas proteínas 101. Así mismo, es un proceso seguro y manipulable a pequeña y gran escala. Por otra parte, se han diseñado diversas estrategias metodológicas para la obtención de proteínas heterólogas utilizando este vector, para lo cual el virus ha sido modificado genéticamente y permite la expresión de proteínas en células de mamífero, entre ellas: condrocitos primarios de rata, hepatocitos de ratón y humano y ciertas líneas celulares procedentes de osteocarcinomas y hepatomas 102 - 105. Adicionalmente, péptidos o proteínas foráneos pueden ser obtenidos sobre la superficie de las partículas virales por fusión del péptido o la proteína de interés a la glicoproteína gp64 del baculovirus 106 - 107. Por último, este virus recombinante ha sido empleado para la infección de larvas y aunque se obtiene una gran concentración de la proteína heteróloga a gran escala, el mantenimiento de los cultivos resulta dispendioso y costoso 108 - 109. Figura 7. Versatilidad del vector de expresión baculovirus. Los vectores baculovirus tienen una variedad de aplicaciones, entre ellas: infectan larvas, células de insecto y células de mamífero. Las células de insecto y de mamífero fueron tratadas con un baculovirus que expresa GFP. Los virus también pueden ser modificados genéticamente y expresar la proteína heteróloga unida a la superficie de la partícula viral 110. 2.4 EXPRESIÓN DE PROTEÍNAS HETERÓLOGAS EN CÉLULAS DE MAMÍFERO MEDIANTE EL VIRUS VACCINIA La expresión de altas concentraciones de proteínas de mamífero puede ser obtenida por la amplificación de un gen en líneas celulares transformadas establemente. El uso de virus como vectores de expresión proporciona ciertas ventajas como la diversidad en la infección de diferentes tipos de células inclusive de modelos animales. Los virus de ADN y retrovirus han sido preferencialmente utilizados como vectores, incluyendo virus de tamaño intermedio o pequeños, entre ellos: SV40, virus de papiloma bovino, retrovirus y adenovirus, los cuales son fácilmente manipulables para la construcción del vector y proporcionan relativamente alto nivel de expresión de la proteína de interés; sin embargo, estos virus tienen un limitado número de células huésped y pueden reacomodar un fragmento de información genética que puede afectar la replicación viral. Los virus de ADN de mayor tamaño, tales como vaccinia y herpes simple, son más difíciles de manipular genéticamente, pero tienen un amplio rango de células huésped y pueden retener ADN foráneo sin perder su capacidad infectiva. Los Poxvirus comprenden una gran familia con miembros que infectan tanto células huésped de vertebrados e invertebrados 111. A diferencia de otros virus de ADN, ellos replican en el citoplasma de las células infectadas. Los miembros más conocidos de esta familia son el virus vaccinia y variola. El virus vaccinia consta de un genoma de ADN de doble cadena de 185000 pb y presenta estructura horquilla (en inglés “hairpin”) en los dos extremos del genoma uniéndolo. El ADN se encuentra en el centro del virus, el cual contiene un sistema completo de transcripción, incluyendo una RNA polimerasa específica del virus y enzimas que modifican el ARN m (cap, metilación y poliadenilación). La fracción externa del virus está compuesta por membrana formada por lipoproteinas. Cuando el virus penetra la célula huésped, el sistema de transcripción viral es activado y aproximadamente 100 genes son expresados durante la fase temprana de la infección. Las proteínas virales sintetizadas incluyen la DNA polimerasa y otros factores necesarios para la replicación del genoma del virus. Las secuencias que regulan la expresión temprana y tardía de determinados genes, están localizadas corriente arriba de la región codificante y difiere significativamente del promotor eucariota 112. Los productos de los genes de expresión tardía incluyen la mayoría de las proteínas estructurales ensambladas en la partícula viral en el citoplasma, algunos viriones permanecen intracelularmente mientras que otros, se localizan en el aparato de golgi y son llevados fuera de la membrana plasmática con una envoltura adicional. La inserción de ADN foráneo se realiza en regiones no esenciales del genoma viral por recombinación homóloga 113 - 114. Los plásmidos que facilitan la inserción y expresión de genes foráneos presentan un promotor transcripcional del virus y uno o más sitios de restricción con endonucleasas para la inserción de la secuencia que codifica la proteína foránea, flanqueada por ADN de una región no esencial del genoma del virus vaccinia 115 116. La elección del promotor determina tanto el tiempo (temprano o tardío del proceso infeccioso) como el nivel de expresión, mientras que la secuencia de ADN que flanquea determina el sitio de recombinación homóloga. Sin embargo, solo se obtiene una partícula viral recombinante de mil viriones por este método y la identificación de esta partícula se realiza mediante un ensayo de placa o hibridación con una sonda de DNA. La cinética de la síntesis de la proteína de interés depende del promotor vaccinia seleccionado. Cuando la señal transcripcional temprana es usada, la expresión ocurre durante las seis primeras horas del proceso infeccioso; si la señal es tardía, la expresión de la proteína se da después de las 6 horas de infección 117. El promotor más usado, P7.5, contiene señales de expresión temprana y tardía y permite continuar la expresión durante 1 a 2 días dependiendo de las condiciones 118; sin embargo, periodos más largos de incubación no se llevan a cabo, puesto que la infección con este virus induce la muerte celular. Adicionalmente, cuando la proteína expresada por el vector virus vaccinia recombinante es de mamífero, las modificaciones pos-transduccionales parecen ser muy similares a su estructura nativa 119 - 121. 3. SISTEMAS DE PURIFICACIÓN DE PROTEÍNAS HETERÓLOGAS Antes de iniciar cualquier proceso de purificación de la proteína de interés resulta necesario identificar su localización a nivel celular y de esta manera seleccionar la técnica apropiada para su posterior aislamiento. Las proteínas heterólogas pueden ser expresadas en forma soluble o insoluble y se pueden localizar en el espacio extra o intracelular o en forma de agregaciones insolubles o cuerpos de inclusión. A continuación se describen algunas ventajas y desventajas de la localización y solubilidad de la proteína de interés durante su purificación. a. Proteínas localizadas en el espacio extracelular: Las proteínas expresadas en el espacio extracelular procedentes del medio de cultivo de bacterias, hongos, células de animales o plantas, generalmente no contienen un gran número de proteínas contaminantes y la proteína de interés es la más abundante, especialmente si es producida como una proteína recombinante. No obstante, la proteína puede encontrarse muy diluida en la muestra inicial y se requieran de grandes volúmenes para ser procesados y obtener una mayor concentración de la proteína. b. Proteínas Intracelulares (citoplasmáticas): Para obtener una proteína intracelular soluble (principalmente enzimas), las células deben ser lisadas para liberar su contenido. Las células procedentes de animales y bacterias son fácilmente lisadas a diferencia de células de plantas y hongos debido a la presencia de la pared celular. El contenido macromolecular soluble de las células está principalmente constituido por proteínas con ácidos nucleicos en menor proporción. Los extractos bacterianos pueden ser viscosos debido a la presencia de ADN cromosomal para lo cual debe ser tratado con una DNasa la cual rompe el material genético. Aunque procesos cormatográficos pueden ser aplicados a los extractos crudos, en ocasiones se requiere que algunos compuestos, incluyendo proteínas inestables, sean retirados de la muestra puesto que se pueden unir a la proteína de interés o a la resina y no son difícilmente retirados 122 - 125. c. Proteínas asociadas a membrana: Existes dos estrategias metodológicas para el aislamiento de proteínas asociadas a membrana. En el primer método, la fracción que corresponde a membranas puede ser preparada y usada para la purificación de la proteína de interés. Alternativamente, la muestra completa puede ser usada para la extracción, en donde se solubilizan las membranas y el contenido citoplasmático es liberado en el mismo procedimiento. La purificación de la proteína asociada a membrana es mejor cuando se realiza el aislamiento de las membranas, puesto que la actividad específica de la membrana solubilizada es mucho mayor respecto al segundo método. Sin embargo, este proceso de purificación puede permitir la perdida sustancial de la proteína. De esta manera, si es más importante obtener la mayor cantidad de proteína que su pureza, utilizar el extracto del tejido o lisado celular total es más apropiado. Las proteínas periféricas se encuentran débilmente unidas a la membrana y pueden ser liberadas mediante cambios de pH, adición de EDTA o bajas concentraciones de detergentes no iónicos y una vez se encuentre en solución, se deben emplear detergentes para mantener su solubilidad. Las proteínas integrales de membrana, a diferencia de las proteínas periféricas, requieren de tratamientos más fuertes, entre ellos concentraciones más altas de detergentes para solubilizar por completo la membrana. Estas proteínas generalmente son insolubles e inestables en ausencia de detergentes. En algunos casos, es necesario mantener los fosfolípidos naturales en asociación con la proteína con el objetivo de mantener su actividad biológica. Algunos procedimiento para la purificación de la proteína de interés son afectados por la presencia de detergentes y de esta manera se dificulta el aislamiento de proteínas integrales de membrana 122 - 125. d. Proteínas insolubles: Las proteínas que son insolubles en solventes comunes son generalmente proteínas estructurales, las cuales son entrecuzadas para formar un polímero por modificaciones pos-transduccionales. El primer paso de purificación consiste en retirar todas las proteínas solubles y de esta manera obtener el pellet remanente que contiene la proteína deseada. Sin embargo, la obtención de la proteína en su estado nativo puede ser difícil puesto que su solubilización requiere de agentes denaturantes 122 - 125. e. Proteínas recombinantes solubles: Las proteínas recombinantes que no son expresadas en formas de agregaciones insolubles, se encuentran de forma soluble en el espacio citoplasmático, pero si un vector de excreción es usado, se localizará en el espacio extracelular o en el espacio periplásmico bacteriano. En algunos sistemas, la expresión es tan buena que la proteína recombinante es la más abundante y el proceso de purificación es relativamente simple. En sistemas en donde la expresión de la proteína recombinante es baja, los procesos de purificación pueden ser más complejos. Actualmente, muchas proteínas recombinantes pueden ser obtenidas como proteínas de fusión con el objetivo de facilitar los procedimientos de purificación mediante cromatografía de afinidad a partir del fragmento unido a la proteína. Una gran variedad de polipéptidos o tags han sido empleados para la obtención de proteínas recombinantes, tales como: proteína A, glutatión S transferasa (GST), proteína de unión a maltosa, así como péptidos que pueden ser reconocidos por anticuerpos específicos, biotinilados en la células huésped o unirse específicamente a metales inmovilizados. Adicionalmente, la purificación casi completa de la proteína recombinante puede obtenerse al pasar el extracto crudo por la resina con el ligando específico y un eluyente adecuado. Otra ventaja de la expresión de proteínas recombinantes es que la expresión de la proteína puede ser determinada por las señales de transcripción y traducción inducidas por el péptido de fusión (el cual ha sido optimizado en el vector). La principal desventaja del uso de estos polipétidos para la expresión de una proteína en particular, consiste en retirar este tag de la proteína de interés. Para esto se requiere de un paso porteolítico, en el cual se emplean proteasas altamente específicas, por ejemplo: factores de la cascada de coagulación, para lo cual su sitio de reconocimiento ha sido insertado entre la secuencia del péptido y la de la proteína de interés 122 - 125. f. Proteínas recombinantes insolubles (Cuerpos de inclusión): La expresión de proteínas recombinantes en un sistema bacteriano, por ejemplo: E. coli, generalmente se da en forma de cuerpos de inclusión o agregados densos al interior de la célula constituidos principalmente por la proteína de interés, pero en un estado no nativo. Los cuerpos de inclusión se pueden formar por una diversidad de razones, entre ellas: la insolubilidad de la proteína a las concentraciones que está siendo producida, inhabilidad del correcto plegamiento en el ambiente bacteriano o inhabilidad de formar puentes disulfuro. La purificación es simple puesto que los cuerpos de inclusión pueden ser separados por centrifugación diferencial de otros constituyentes celulares, dando un producto casi puro; el problema radica en que su contenido proteíco es inactivo e insoluble. Los cuerpos de inclusión formados y separados pueden ser solubilizados en presencia de agentes denturantes como hidrocloruro de guanidina o úrea y tioles, como mercaptoetanol o glutatión con el objeto de romper los puentes disulfuro formados y prevenir la formación de otros. El plegamiento correcto de la proteína puede requerir una cantidad de aditivos, así como la remoción lenta del agente denaturante, lo cual puede ser realizado mediante una simple dilución o diálisis. El replegamiento correcto es facilitado cuando se emplean bajas concentraciones de la proteína y si la proteína nativa no presenta enlaces disulfuro, entonces es importante crear condiciones redox (pero no en exceso), de tal manera que la oxidación de grupos tiol se lleve a cabo 125, 131. Una vez la proteína recombinante ha sido correctamente replegada, el proceso de purificación consiste en remover la pequeñas cantidades de proteína que no fue plegada adecuadamente y otras proteínas contaminantes atrapadas en los cuerpos de inclusión. Los métodos desarrollados para la purificación de proteínas se basan desde simples procedimientos de precipitación hasta métodos cromatográficos y de afinidad mas avanzados. Las técnicas empleadas para este propósito se pueden clasificar en cuatros grupos dependiendo de las propiedades bioquímicas de la proteína que se desea purificar, entre ellas: características de superficie, tamaño y forma, carga neta y biopropiedades. 3.1 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LA S CARACTERÍSTICAS DE SUPERFICIE DE LAS PROTEÍNAS Las características de superficie de una proteína incluyen la distribución de la carga, localización de cadenas laterales de aminoácidos hidrofóbicos e hidrofílicos y exteriorización de dominios proteícos. Estas propiedades dependen del solvente y el pH usado para solubilizar la proteína de interés. Los métodos que se basan en estas propiedades generalmente dependen de la solubilidad de la proteína. Diferencias en la solubilidad permiten la precipitación por manipulación de diversos solventes en el cual las proteínas son solubilizadas. Estos solventes, los cuales generalmente son un buffer que contiene una baja concentración de sal, pueden alterar propiedades como fuerza iónica y constante dieléctrica, con el objetivo de precipitar selectivamente algunas de las proteínas presentes en la muestra. Adicionalmente, algunas de ellas pueden ser solubilizadas a partir de un estado insoluble, cuando al solvente se le agrega un detergente o un agente caotrópico 126 - 131. La distribución de los aminoácidos hidrofóbicos es determinante en la solubilidad de estas proteínas y puede ser utilizado para la purificación de una proteína en particular por cromatografía hidrofóbica. Así mismo, la cromatografía de inmunoafinidad hace parte de este grupo, en la cual un anticuerpo dirigido contra un epítope de la superficie de la proteína es usado para purificar la proteína de una mezcla. 3.2 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LA ESTRUCTURA TOTAL DE LA PROTEÍNA: FORMA Y TAMAÑO Las técnicas que se basan en estas propiedades para el aislamiento y purificación de proteínas específicas son: la cromatografía de exclusión por tamaño y la electroforesis preparativa. Muchas proteínas en su estado bioactivo son oligómeros de más de un polipéptido y estos pueden ser disociados con pérdida de su estructura. De esta manera, muchas proteínas pueden presentar dos pesos moleculares correspondientes al oligómero en su estado nativo y a los polipéptidos disociados y denaturados. Los métodos de cromatografía de exclusión por tamaño generalmente permiten la purificación de la proteína en su estado nativo; mientras que la electroforesis preparativa involucra la separación de los polipéptidos disociados 126 - 131. 3.3 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN LA CARGA NETA DE LA PROTEÍNA Las dos técnicas que se fundamentan en la carga neta de una proteína para su purificación son: cromatografía de intercambio iónico y electroforesis. La cromatografía de intercambio iónico consta de un grupo cargado inmovilizado positiva (aniónico) o negativamente (catiónico) en una resina, de tal manera que pueda retener proteínas con carga contraria. La carga neta de una proteína depende del pH de la solución o solvente en el cual se encuentre solubilizada. De esta manera, una proteína se carga positivamente cuando se encuentra en pH muy ácido o negativamente cuando el pH de la solución es muy alcalino. La carga neta cero de una proteína se adquiere a un determinado pH, denominado pI, en el cual las cargas negativas son exactamente iguales a las cargas positivas. El estado más cargado de una proteína (sin discernir en el signo de la carga) se encuentra en el rango de pH de 6.0 a 9.0, siendo el rango de pH en el que la mayoría de las proteínas son más estables. La purificación de una proteína a partir de cromatografía de intercambio iónico principalmente se realiza con grupos cargados positivamente debido a que la mayoría de las proteínas a pH neutro se cargan negativamente y por tanto tienen un pI más bajo 126 - 131. 3.4 MÉTODOS DE PURIFICACIÓN DE PROTEÍNAS BASADOS EN BIOPROPIEDADES (AFINIDAD) Un método eficaz en la purificación de una proteína en particular es el bioespecífico, en el cual la propiedad biológica particular de la proteína es utilizada. La afinidad es limitada a proteínas que tienen propiedades de unión específicas, excepto las proteínas que son teóricamente capaces de ser purificadas por técnicas de inmunoafinidad. Muchas proteínas de interés tienen un ligando específico, por ejemplo: las enzimas reconocen un sustrato y un cofactor, proteínas de unión a hormonas y receptores se unen a hormonas y otros factores específicos. Por tanto, la inmovilización de un ligando que sea reconocido por la proteína de interés (o un anticuerpo dirigido contra la proteína de interés), selectivamente retiene la proteína. Esta técnica es conocida como cromatografía de afinidad; sin embargo, existen técnicas no cromatográficas que se fundamentan en el mismo principio 126 - 131. 4. OBJETIVOS 4.1 OBJETIVO GENERAL Expresar y purificar las proteínas recombinantes estructurales del rotavirus VP5, VP6 y VP8 para realizar ensayos biológicos y obtener anticuerpos policlonales contra estas proteínas. 4.2 OBJETIVOS ESPECÍFICOS 4.2.1 Amplificar y purificar los vectores que presentan las secuencias que codifican las proteínas estructurales del rotavirus VP5 y VP8 en una cepa de E. coli deficiente en endonucleasas. 4.2.2 Expresar y purificar las proteínas estructurales del rotavirus VP5 y VP8 a partir de vectores de expresión en una cepa bacteriana de E. coli deficiente en proteasas. 4.2.3 Expresar y purificar la proteína recombinante VP6 del rotavirus en el sistema de vaccinia mediante la infección de células MA104. 4.2.4 Obtener anticuerpos policlonales contra cada una de las proteínas recombinantes en conejos. 4.2.5 Identificar la interacción entre las proteínas recombinantes y las proteínas Hsc70 y PDI en un sistema acelular y durante el proceso infeccioso en células MA104 mediante ensayos de unión y competencia de la infección. 5. METODOLOGÍA La estrategia experimental que se utilizó para desarrollar este trabajo de investigación se dividió en 5 etapas: 1) Amplificación y purificación de los vectores que presentan la secuencia génica que codifica las proteínas VP5 y VP8; 2) Expresión de las proteínas recombinantes en un sistema de transfección bacteriana (VP5 y VP8) e infección de células de mamífero con el virus vaccinia recombinante (VP6); 3) Purificación de las proteínas recombinantes mediante cromatografía de afinidad, electroelución y tratamiento con detergentes; 4) Interacción de las proteínas recombinantes estructurales VP5, VP6 y VP8 con las proteínas celulares PDI y Hsc70 en un sistema acelular y celular y 5) Obtención de anticuerpos policlonales en conejos Nueva Zelanda. 5.1 AMPLIFICACIÓN Y PURIFICACIÓN DE LOS VECTORES QUE PRESENTAN LAS SECUENCIAS GÉNICAS QUE CODIFICAN LAS PROTEÍNAS VIRALES VP5 Y VP8 Y LA PROTEÍNA CELULAR Hsc 70 5.1.1 Características de los plásmidos que presentan las secuencias génicas de las proteínas recombinantes. Para la expresión de las proteínas estructurales del rotavirus VP5 y VP8 se utilizó el plásmido pGEX-4T (figura 8a), en los cuales se clonó las secuencias de ADN copia que codifican estas proteínas procedentes de la cepa de rotavirus RRV de simio, los cuales fueron donados por el Doctor Carlos Arias del Departamento de Genética del Desarrollo y Fisiología Molecular del Instituto de Biotecnología de la Universidad Autónoma de México. El plásmido pGEX presenta una secuencia que codifica el polipéptido de Glutation S Transferasa (GST) unida a la secuencia génica que codifica cada una de las proteínas estructurales, VP5 o VP8, en la región amino terminal y un gen que codifica una βlactamasa que le confiere resistencia a ampicilina a la cepa bacteriana después de la transfección, la cual rompe el anillo - lactámico de la ampicilina desactivando su actividad antimicrobiana. La inserción de las secuencias génicas se realizó corriente abajo del promotor tac. Para la expresión de la proteína Hsc70 se utilizó el plásmido pET28a (figura 8b), en el cual se clonó la secuencia que codifica esta proteína. Este vector presenta una secuencia que codifica una segmento de seis histidinas que quedan unidas mediante un enlace peptídico a la región amino terminal de la proteína celular después de la traducción; adicionalmente codifica una proteína que confiere resistencia a Kanamicina. La inserción de la secuencia génica de Hsc70 se realizó corriente abajo del promotor para la RNA polimerasa del Bacteriófago T7. a. b. Figura 8. Representación gráfica del vector de expresión utilizado para la inserción de las secuencias de ADN que codifican las proteínas estructurales del rotavirus, VP5 y VP8. 5.1.2 Cepas bacterianas de Escherichia Coli. Las cepas bacterianas de E. coli que se utilizaron para el desarrollo de este trabajo de investigación fueron: XL-1BLUE y BL21(DE3). La cepa bacteriana XL-1 BLUE ha sido modificada genéticamente y es deficiente en la expresión de enzimas que catalizan el rearreglo y deleción de estructuras secundarias y terciarias que ocurren frecuentemente en el ADN, es deficiente en endonucleasas (endA) y enzimas de recombinación (recA) y presenta la mutación en el gen hsdR, la cual previene el corte del ADN clonado por el sistema de endonucleasa EcoK, lo que asegura la correcta y eficiente amplificación de los vectores. Esta cepa fue utilizada para la amplificación de los plásmidos y obtención de una mayor concentración de los mismos. La cepa bacteriana BL21(DE3) fue utilizada para la expresión de los plásmidos transfectados puesto que es una cepa deficiente en la expresión de proteasas OmpT (proteasa extracelular) y Lon (proteasa intracelular). Adicionalmente, presenta la secuencia de que codifica la RNA polimerasa T7 del fago DE3 y su expresión es dependiente del promotor lacUV5 y del represor LacIq. Las cepas bacterianas fueron donadas por el Doctor Carlos Arias del Departamento de Genética del Desarrollo y Fisiología Molecular del Instituto de Biotecnología de la Universidad Autónoma de México. 5.1.3 Preparación de medios de cultivo para el crecimiento de las cepas bacterianas de E. coli. Uno de los medio de cultivo de uso general más utilizado es el Luria – Bertani (LB) constituido por triptona (hidrolizado de proteínas) 1 %, extracto de levaduras 0.5 % y cloruro de sodio (NaCl) 1 %. Para la preparación de un caldo de cultivo se pesó cada uno de los nutrientes y se disolvieron en H2O, la solución fue ajustada a pH 7.0 – 7.5 y se aforó a volumen total. Para la preparación de un medio sólido se adicionó a la mezcla anterior agar- agar a una concentración final de 1.2 %, se calentó para disolver y se autoclavó a 20 psi durante 20 min. Por último, se dejó enfriar (en este paso se adicionaron los antibióticos necesarios) y se sirvió en cajas de petri estériles hasta que solidificó. Como control de esterilidad se dejó una caja de petri con medio sólido a 37ºC durante varios días en ausencia del antibiótico. Para favorecer la expresión de la proteína recombinante estructural VP5 del rotavirus se empleó otro medio líquido, 2XYT, el cual contenía triptona al 1.6 %, extracto de levadura al 1% y NaCl al 0.5%. 5.1.4 Transfección de los plásmidos que presentan las secuencias de las proteínas VP5 y VP8. Preparación de células competentes. La metodología empleada para preparar las células competentes fue una modificación del protocolo publicado por Chung C. T., et al, en 1989 132. Una colonia de bacterias XL1BLUE o BL21(DE3) fue inoculada en 100 ml de medio LB y cultivada a 37 ºC con agitación constante a 150 rpm durante toda la noche. Al siguiente día se inoculó 1 ml del cultivo anterior en 100 ml de medio LB (fd: 100) hasta alcanzar una densidad óptica (DO) de 0.3 – 0.4 a 600 nm. Posteriormente, se obtuvo el pellet de células mediante centrifugación a 5000 rpm durante 10 min y fue resuspendido en 5 ml (1/20 respecto al volumen inicial de medio LB) de medio de congelación que contenía Polietilenglicol (PEG) 3350 al 10%, Dimetilsulfóxido (DMSO) al 5%, NaCl 100 mM, MgCl2 10 mM, MgSO4 10 mM y Glicerol al 10% disuelto en medio LB al 60% v/v en H2O estéril pH 6,7 – 7,0. La mezcla se incubó en hielo durante 10 min, se hicieron alícuotas de a 200 l en crioviales de 2 ml y se congelaron en nitrógeno líquido. Antes de iniciar el proceso de transformación, fue importante comprobar la viabilidad de las bacterias competentes para lo cual se sembró una alícuota de estas bacterias en una placa de agar LB y se incubó a 37 ºC durante toda la noche. Transformación de las células competentes. Una de las alícuotas anteriores de 200 l de bacterias XL1BLUE o BL21(DE3) competentes se mezclaron con aproximadamente 10 ng de ADN plasmídico (pGEX-4TVP5 o pGEX-4T-VP8) disuelto en 100 µl de una solución de KCl 100 mM, CaCl2 30 mM y MgCl2 50 mM y cuantificado a 260 nm en un GeneSpec I. La mezcla de bacterias y ADN se agitaron suavemente y posteriormente se incubaron en hielo durante 20 min. Con el objeto de proceder a la transformación, las células fueron expuestas a choque térmico a 42 ºC durante 2 minutos para garantizar la entrada del vector a la célula. Las células transformadas se sembraron en placas LB – agar con ampicilina o kanamicina a una concentración final de 100 g/ml y se incubaron durante 14 horas a 37 ºC. Así mismo, se empleó como control negativo de transformación un cultivo de células XL1BLUE o BL21(DE3), a las cuales se les realizó los procedimientos descritos anteriormente pero sin plásmido que indujera la transformación bacteriana y se crecieron en placas de LB – agar en presencia de ampicilina o kanamicina. 5.1.5 Purificación de los plásmidos que presentan las secuencias de las proteínas estructurales VP5 y VP8. La extracción del ADN plasmídico se realizó con el kit PurelinkTM Quick Plasmid Miniprep de Invitrogen. Una vez transformadas las bacterias XL1BLUE con cada uno de los plásmidos, se realizó el crecimiento de una cepa transformada hasta alcanzar una concentración de 1 – 2 x 109 células por 5 ml de medio LB con 100 g/ml de ampicilina, se centrifugó durante 10 minutos a 5000 rpm y se descartó el sobrenadante. El pellet de células fue resuspendido en 250 l de buffer Tris HCl 50 mM pH 8.0, EDTA 10 mM y RNAasa A 0.2 mg/ml (R3, numeración del catálogo del kit). A la mezcla anterior se adicionó 250 l de buffer de lisis que contiene NaOH 200 mM y SDS 1% (L7, numeración del kit), se mezcló 5 veces por inversión y se dejó en reposo durante 5 min. Posteriormente, se adicionó 350 l de buffer de precipitación (N4, numeración del catálogo del kit), se mezcló 5 veces por inversión y se centrifugó a 12000 x g durante 10 min a temperatura ambiente. El aislamiento del ADN plasmídico se realizó con una mini columna de membrana de silica (figura 9), a la cual se le adicionó la mezcla obtenida en el paso anterior, se centrifugó a 12000 x g durante 1 min y se descartó la solución que quedó en el tubo de lavado. Se realizaron dos lavados al ADN plasmídico adherido a la columna con un buffer de lavado que tiene etanol (W9, numeración del catálogo del kit) y se centrifugó a 12000 x g por 1 min. Finalmente, el ADN fue eluido de la columna mediante la adición de 75 l de buffer TE (Tris – HCl 10 mM pH8.0 y EDTA 0,1 mM), el cual había sido previamente calentado a 65ºC, y centrifugación a 12000 x g por 1 min. Una alícuota de 5 l de los plásmidos purificados fueron mezclados con 1 l de buffer muestra tipo I (Xilencianol 0,25%, Azul de bromofenol 0,25% y sacarosa 40 %), separados en geles de agarosa al 1.2 % a 100 voltios durante 45 min y teñidos con 0,5 g/ml de bromuro de etidio (BrEt). Por último, la cuantificación de las muestras obtenidas se realizó espectrofotométricamente a 260 nm en un GeneSpec I y se tuvo en cuenta la relación 260/280 a fin de determinar su pureza. Figura 9. Montaje para la separación y purificación de ADN plasmídico. 5.1.6 Corte de los plásmidos con la enzima de restricción Pst I. En tubos de 0,2 ml estériles se mezclaron 2 l de buffer de restricción H 10X, 0.5 l de PstI 10 U/l, 1 g de ADN plasmídico y H2O desionizada estéril para completar un volumen final de 20 l. La mezcla fue incubada a 37 ºC durante 2 h en un termociclador PTC – 100 TM MJ RESEARCH, Inc. Posteriormente, la mezcla fue incubada a 65 ºC por 15 min para inactivar la enzima y las muestras fueron analizadas por electroforesis en geles de agarosa al 1.2 % y teñidos con BrEt 0.5 g/ml, como se describió en el numeral anterior. 5.2 EXPRESIÓN DE LAS PROTEÍNAS RECOMBINANTES VP5, VP8 Y VP6 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 Para la expresión de las proteínas recombinantes se utilizó la cepa de E. coli, BL21(DE3) puesto que es deficiente en la expresión de proteasas y disminuye el riesgo de proteólisis de la proteína de interés; además, presenta la secuencia que codifica la RNA polimerasa T7 indispensable para la expresión de los vectores pGEX-4T y pET-28a. Una vez transformadas las bacterias BL21(DE3) con cada uno de los plásmidos, se procedió a inducir la expresión de cada una de las proteínas virales. Las alícuotas congeladas de las cepas transformadas fueron sembradas en una caja de petri en presencia del antibiótico. Una colonia de las cepas transformadas fue sembrada en 25 ml de medio líquido LB o 2XYT en presencia de 100 g/ml de ampicilina, para las cepas transformadas con el vector pGEX-4T o 50 g/ml de Kanamicina, para las cepas transformadas con el vector pTE-28a, y se incubaron a 37ºC con agitación constante a 150 rpm durante 12 h. Adicionalmente, se crecieron dos controles negativos de expresión de células BL21(DE3) sin transformar y las bacterias transformadas que no fueron inducidas. Posteriormente, se adicionaron 5 ml del cultivo anterior a 500 ml de medio LB o 2XYT (Fd: 100) y se incubaron a 37 ºC con agitación constante a 150 rpm durante 1 – 8 h hasta alcanzar una densidad óptica correspondiente a la fase exponencial del crecimiento bacteriano. Posteriormente, se adicionó el inductor Isopropil β-D-Tioglactopiranósido (IPTG) a una concentración entre 0.5 – 2 mM durante 2, 4, 6 y 8 h. Para la expresión de la proteína viral VP5 fue necesario la adición de 0.2% de glucosa durante todo el procedimiento de crecimiento celular. Al finalizar el tiempo de inducción, el cultivo líquido se centrifugó a 5000 rpm durante 10 min. El pellet de células fue lavado dos veces con buffer PBS y tratado con 100 g/ml de lisozima y se incubó durante 15 min en hielo con agitación constante. A fin de completar la lisis celular, la mezcla fue sonicada con una amplitud de 30% durante 5 ciclos de 30 segundos, se centrifugó a 10000 rpm durante 10 min a 4ºC y se separó el sobrenadante (proteínas solubles) del pellet (proteínas insolubles). El pellet fue tratado con 5 ml de úrea 1, 2, 4 y 8 M en PBS de manera consecutiva, a 4ºC con agitación constante durante 2 h cada uno, con el objeto de evidenciar si la proteína viral se encontraba en forma de cuerpos de inclusión. Las proteínas del pellet solubilizadas en úrea fueron centrifugadas a 12000 rpm durante 10 min después de cada tratamiento con las diferentes concentraciones del agente caotrópico. A los controles negativos de células no transformadas y transformadas pero sin inducción se les realizó el mismo procedimiento. La expresión de las proteínas recombinantes fue evaluada y analizada mediante SDSPAGE y Western Blot frente a una marcador de peso molecular, un control de bacterias BL21(DE3) sin transformar y un control de células BL21(DE3) transformadas pero en ausencia del inductor, para lo cual se cuantificó la concentración de proteínas totales de cada una de las fracciones a 280 nm respecto a una curva de calibración de BSA de 2 mg/ml, con el fin de sembrar la misma cantidad de proteínas totales en el gel. 5.3 EXPRESIÓN DE LA PROTEÍNA RECOMBINANTE VP6 EN UN SISTEMA DE INFECCIÓN DE CÉLULAS EUCARIOTAS 5.3.1 Cultivo celular. La línea celular MA104 fue cultivada en Medio Eagle Dulbecco's Modified (DMEM), suplementado con 2% (v/v) de suero fetal bovino (SFB) y 2 mM de L-Glutamina en condiciones de atmósfera de aire enriquecido con 5% de CO2 a 37°C en frascos de cultivo de 25, 75 y 150 cm2 . Todos los pasos anteriores se realizaron en una cabina de flujo laminar bajo estrictas condiciones de esterilidad. La línea celular fue donada por los Doctores Manuel Franco y Juanita Ángel Uribe del Instituto de Genética Humana de la Pontificia Universidad Javeriana. 5.3.2 Infección de la línea celular MA104 con el virus Vaccinia recombinante que presenta la secuencia que codifica la proteína estructural del rotavirus VP6. Las cajas de células MA104 con una confluencia entre el 80 – 100 % fueron lavadas dos veces con PBS y posteriormente infectadas con una alícuota de virus Vaccinia – VP6 durante 1 h. Transcurrido este tiempo, se retiró el remanente de lisado viral y se incubaron con 2, 4 o 8 ml de medio MEM para las cajas de 25, 75 y 150 cm2, respectivamente. Se incubaron durante 24 h o hasta que se evidenciara un efecto citopático, como desprendimiento de la monocapa. La alícuota del virus Vaccinia – VP6 fue donada por los Doctores Manuel Franco y Juanita Ángel Uribe del Instituto de Genética Humana de la Pontificia Universidad Javeriana. Después de terminar el tiempo de infección, el cultivo fue congelado a – 70ºC y descongelado a temperatura ambiente, tres veces a fin de liberar las proteínas citoplasmáticas de las membranas celulares y se centrifugó a 12000 rpm durante 15 minutos a 4°C para separar la fracción de proteínas solubles de las proteínas insolubles. La fracción de proteínas solubles fue tratada con acetona fría y se incubó a -70ºC durante toda la noche, se centrifugó a 12000 rpm por 20 min a 4ºC. El pellet de proteínas obtenido fue resuspendido en PBS después de que se evaporó las trazas del solvente. La fracción de proteínas insolubles fue tratada con úrea 8M y centrifugada a 10000 rpm por 10 min a 4ºC. Las fracciones fueron analizadas por electroforesis y western blot frente a un marcador de peso molecular y un control de células sin infectar a las cuales se les realizó el procedimiento descrito. 5.3.3 Análisis de la expresión de la proteína viral estructural VP6 mediante el conteo de Unidades Formadoras de Foco (UFF). Para determinar la concentración viral de un lisado de células infectadas con vaccinia recombinante y a la vez, verificar la expresión de la proteína rotaviral VP6, se realizaron diluciones sucesivas del lisado viral y se adicionaron a células MA104 confluentes al 100% en una caja de 96 pozos. Transcurrida una hora de infección, el virus se retiró y se adicionó 100 l de medio a la monocapa de células. El proceso infeccioso se dejo incubar durante 14 h a 37ºC. Una vez finalizado el tiempo de infección, las células fueron fijadas y permeabilizadas con 50 l de metanol preenfríado durante 40 min a 4ºC. La monocapa fue lavada dos veces con PBS y se incubó con un anti-rotavirus en una dilución 1:1000 en PBS a 37ºC durante 1h. Posteriormente, se lavó tres veces con PBS, se adicionó un anticuerpo secundario conjugado con peroxidasa y se incubó durante 45 minutos a 37 ºC. La monocapa de células fue lavada tres veces con PBS y se reveló utilizando como sustrato aminoetilcarbazol (AEC) disuelto en N-dimetilfomamida. El AEC se diluyó a una concentración final de 0.268 % (a partir de una solución patrón de 0,4 % de AEC disuelto en dimetilformamida) en buffer de acetato de sodio 0,1 M pH 5.2, se adicionó H2O2 a una concentración final de 0.03% y se filtró tres veces hasta retirar el precipitado que se forma. El conteo se realizó en un microscopio invertido y se eligió el pozo en donde se encontraron 100 focos contados una sola vez, desde un extremo del pozo, en línea recta, hasta el otro extremo. Luego se aplicó la siguiente fórmula: 20 x número de focos contados x la dilución donde había 100 focos de infección x 5.5 En donde, 20 corresponde al fd en el cual se diluye el virus y 5.5, es el número de campos que tiene el pozo al recorrer el lente 20X en el microscopio invertido (Euromex, Holanda). Adicionalmente, se utilizaron como controles negativos células sin infectar y se les realizó el procedimiento descrito en el párrafo anterior. 5.4 SOLUBLIZACIÓN Y PURIFICACIÓN DE LAS PROTEÍNAS ESTRUCTURALES VP5, VP6 Y VP8 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc70 5.4.1 Solubilización de las proteínas recombinantes VP5, VP8 y Hsc70 a partir del lisado de bacterias BL21(DE3) transformadas con los vectores de expresión. La solubilización de las proteínas recombinantes VP5, VP8 y Hsc 70 fue desarrollada mediante el protocolo publicado por Frangioni et al., en 1993 176, el cual fue modificado como se muestra a continuación. Cada cepa bacteriana transformada con los plásmido que presentaban las secuencias de las proteínas virales (VP5 y VP8) o la proteína celular Hsc 70, de manera independiente, fueron cultivadas y las proteínas fueron expresadas teniendo en cuenta las condiciones estandarizadas en el numeral 5.2.1. Finalizado el tiempo de expresión, las bacterias fueron centrifugadas a 5000 rpm por 10 min y lavadas dos veces con buffer STE (Tris – HCl pH 8.0 10 mM, NaCl 150 mM y EDTA 1 mM) para la bacterias que expresaron las proteínas virales y buffer PBS para las bacterias que expresaron la proteína Hsc 70. Posteriormente, el pellet de células fue resuspendido en 50 ml de buffer STE o PBS y tratado con 100 g/ml de lizosima durante 15 min con agitación constante a 4 ºC. A la mezcla anterior, se adicionó 5 mM de DTT, 1% de N-Laurilsarcosina y para completar la lisis celular, la mezcla fue sonicada durante 4 ciclos de 15 segundos a 20% de amplitud y centrifugada a 10000 rpm por 15 min a 4 ºC. Para el lisado de las bacterias que expresaron la proteína viral VP5, el sobrenadante obtenido fue tratado con Tritón X-100, Tween 20, NP40 y Octilglucósido (detergentes no iónicos) y CHAPS (zwiteriónico) en concentraciones del 1 y 3 %, como agentes renaturantes y de manera independiente. En el caso del lisado obtenido de bacterias que expresaron la proteína viral VP8, el sobrenadante fue diluido 10 veces en PBS, aunque también se realizaron tratamientos con los diferentes detergentes como se mencionó para el lisado procedente de bacterias que expresaron la proteína viral VP5. Los sobrenadantes obtenidos fueron analizados por SDS-PAGE y western blot para verificar la presencia de las proteínas virales. En el caso de las bacterias que expresaron la proteína recombinante Hsc 70, fueron lavadas y resuspendidas en buffer PBS, puesto que el buffer STE contiene EDTA, el cual quela iones metálicos como Ni2+, indispensables para la purificación de la proteína. Una vez se obtuvo el lisado bacteriano de las células transformadas de la misma manera que el procedimiento anterior, se trató con tritón X-100 al 1 % como agente renaturante y la presencia de la proteína fue identificada mediante SDS-PAGE y western blot. 5.4.2 Purificación de las proteínas estructurales VP5 y VP8 del rotavirus mediante cromatografía de afinidad. Las proteínas virales expresadas VP5 y VP8 tienen un polipéptido de la proteína GST unido covalentemente al dominio amino terminal de cada una de las proteínas virales, el cual fue empleado para la purificación de las proteínas recombinantes. Una vez se obtuvo el sobrenadante de los lisados bacterianos que presentaron la proteína viral VP5 o VP8, se procedió a purificar las proteínas mediante una resina de agarosa conjugada a glutatión (Molecular Probes). En una columna cromatográfica se cargaron 5 ml de la resina, la cual fue lavada con 10 volúmenes de columna de PBS, con el objeto de retirar la azida presente en la resina; posteriormente se equilibró con 5 volúmenes de buffer STE en presencia de DTT 5 mM y N-Laurilsarcosina al 1 % y se incubó con el lisado bacteriano. Para la unión de las proteínas recombinantes virales con la resina se estandarizó el tiempo de interacción para cada una de ellas. De esta manera, para la purificación de la proteína viral VP5, el lisado fue adicionado a la resina en la columna cromatográfica y se pasó dos veces el mismo lisado con el fin de retener la mayor cantidad de proteína. En el caso de la proteína viral VP8, el lisado diluido 10 veces con PBS se incubó toda la noche a 4 ºC con la resina puesto que presentó menor afinidad por el glutatión de la resina. Posteriormente, se realizaron varios lavados con PBS (aproximadamente 10 volúmenes de columna o hasta no evidenciar color con el reactivo de Bradford) y se procedió a la elución de cada proteína, para lo cual se emplearon concentraciones crecientes de glutatión entre 5 – 50 mM. El glutatión fue preparado en Tris – HCl pH 8.0 50 mM, 100 mM y 150 mM, para concentraciones de glutatión de 5 -15 mM, 20 mM y 50 mM, respectivamente. Los eluídos fueron recolectados a un flujo de 1 ml cada 3 min para VP5 ó 1 ml cada min para VP8. Finalmente, la resina fue tratada con 5 volúmenes de PBS que contenía NaCl 3M y se lavó con 10 volúmenes de PBS. Paralelamente, cada una de las fracciones obtenidas en el corrido cromatográfico, entre ellas: no retenido, lavados con PBS, eluídos y lavados con NaCl fueron tratadas con el reactivo de Bradford con el objetivo de evidenciar la presencia de proteínas, mediante el cambió de color rojo – café a azul. Por otra parte, las fracciones fueron analizadas por SDS-PSGE y western blot. 5.4.3 Purificación de la proteína celular Hsc 70 mediante cromatografía de afinidad. Para la purificación de esta proteína se empleó una resina HisTrap puesto que la proteína recombinante presenta en su amino terminal, una cola de seis histidinas que interaccionan con el níquel (Ni2+) cargado en la resina. Se cargaron 10 ml de resina en una columna de cromatografía y se lavó con 5 volúmenes de buffer fosfatos (NaH2PO4 50 mM y NaCl 300 mM) y se equilibró con 5 volúmenes de buffer fosfatos que contenía 10 mM de Imidazol. Paralelamente, al lisado celular se le adicionó imidazol a una concentración final de 10 mM a partir de una solución patrón 1 M. La muestra que contenía la proteína recombinante fue adicionada a la resina, y se realizaron lavados con buffer fosfatos con 10 mM de imidazol para retirar lo que no se unió a la resina hasta que no se detectara la presencia de proteínas por el método de Bradford. Finalmente, la proteína recombinante fue eluída con concentraciones crecientes de imidazol en buffer fosfatos, entre 0,1 - 1 M. La resina fue lavada con 10 volúmenes de H2Od y 5 volúmenes de etanol al 30%. 5.4.4 Solubilización e identificación de la localización de la proteína estructural VP6. Las células MA104 infectadas fueron congeladas a – 70 ºC y descongeladas a 37 ºC tres veces. El lisado obtenido fue transferido a un tubo de 15 ml, sonicado durante 2 ciclos de 15 seg a 20% de amplitud y centrifugado para separar la fracción soluble del pellet, correspondiente a la fracción de membranas. Las proteínas presentes en el sobrenadante fueron precipitadas a fin de concentrarlas con cinco volúmenes de acetona fría y almacenadas a -70 ºC durante toda la noche. La mezcla anterior fue centrifugada a 12000 rpm durante 10 min a 4 ºC, el sobrenadante fue descartado y el pellet fue resuspendido en PBS después de dejar evaporar las trazas de acetona; mientras que las proteínas presentes en el pellet obtenido después de la sonicación, fueron disueltas en úrea 8M. Finalmente, cada una de las fracciones fueron analizadas por SDS-PAGE y western blot para identificar la presencia de la proteína rotaviral VP6. 5.4.5 Purificación de la proteína estructural VP6 del rotavirus mediante el tratamiento con detergentes. La fracción de proteínas adheridas a membranas de células MA104 proveniente de 10 cajas de cultivo 75 cm2, fue lavada dos veces con 2 ml buffer tris –HCl 20 mM pH 8.0 que contenía KCl 150 mM. Luego se adicionó 1 ml de buffer RIPA frío (Tris HCl pH 7.4 10 mM, SDS 0.1%, Tritón X-100 1%, Deoxicolato de sodio 1% y NaCl 150 mM), incubado a 4 ºC durante 1 h y centrifugado a 10000 rpm durante 10 min a 4 ºC. El pellet remanente fue tratado con 500 l de buffer Tris HCl 20 mM pH 8.0, KCl 150 mM y diferentes detergentes al 1 %, entre ellos: CHAPS, Octilglucósido, NP40, Tween 20 y Tritón X-100, de manera independiente con el objetivo de evaluar la solubilidad de esta proteína. Los pellet solubilizados fueron incubados toda la noche a 4 ºC y centrifugados a 10000 rpm durante 10 min a 4 ºC. El pellet remanente fue tratado con una solución de 500 l úrea 8 M disuelta en Tris HCl 20 mM y KCl 150 mM pH 8.0 y N-laurilsarcosina al 1%. Las muestras solubilizadas fueron dializadas frente a un gradiente decreciente de úrea en PBs, diluidas 10 veces en buffer PBS y concentradas mediante un amicón Ultra – 4 (Milipore). 5.5 OBTENCIÓN DE ANTICUERPOS POLICLONALES DIRIGIDOS CONTRA LAS PROTEÍNAS ESTRUCTURALES VP5, VP6 Y VP8 DEL ROTAVIRUS 5.5.1 Preparación de las proteínas estructurales VP5 y VP8 del rotavirus mediante electroelución para la inmunización de conejos Nueva Zelanda. Los lisados bacterianos transfectados e inducidos para las proteínas VP5 y los cuerpos de inclusión de VP8 fueron corridos en geles preparativos de poliacrilamida al 12% en condiciones denaturantes a 40 mAmp por lámina de gel (16.5 cm x 17.5 cm x 3 mm) toda la noche. Un extremo del gel fue cortado y transferido a una membrana de fluoruro de polivinilideno (PVDF) para la localización de la proteína viral con un anticuerpo policlonal para cada proteína. El resto del gel se tiñó con Azul de Coomassie y después de identificada la banda correspondiente a cada proteína mediante Western blot, se cortó el segmento donde se ubicaba la proteína, se colocó en trozos pequeños en un tubo de vidrio y se realizó el montaje de la figura 10. La electroelución de las proteínas se llevó a cabo en una cámara de electroelución (modelo 422 de Bio Rad) durante 5h a 10 mA constantes por tubo en buffer de corrido (192 mM de Glicina, 25 mM de Tris y SDS 0.1% pH 8.3). La proteínas electroeluídas se precipitaron con cinco volúmenes de acetona fría durante toda la noche a -70 ºC y se centrifugaron a 10000 rpm por 10 min. Las proteínas precipitadas fueron resuspendidas en un buffer que contenía Tris HCl 50 mM pH 8.0 y CaCl2 10 mM para el posterior corte de la región GST de cada proteína. Figura 10. Montaje para la electroelución de las proteínas virales recombinantes a partir de geles de poliacrilamida. 5.5.2 Corte del polipéptido GST de las proteínas virales. Después de electroeluídas las proteínas de fusión VP5 y VP8, se procedió a digerirlas con trombina para retirar el péptido de GST, puesto que las proteínas recombinantes presentan la secuencia que codifica el sitio de reconocimiento para esta enzima. El corte de GST se realizó con el kit THROMBIN CleanCleave (Sigma), el cual consta de una resina de agarosa unidad a la trombina y un buffer de digestión 10X que contiene Tris – HCl 500 mM pH 8.0 y CaCl2 100 mM . Se tomaron 50 l de la resina y se centrifugaron a 20 x g durante 2 min a fin de retirar el sobrenadante que contiene Tris – HCl 20 mM y Glicerol al 50% y se realizaron dos lavados con buffer de digestión 1X. Posteriormente, se adicionó la proteína viral electroeluida (500 ng), la cual estaba resuspendida en el buffer de digestión que contenía tris – HCl 50 mM pH 8.0 y CaCl2, se incubó durante 4 h a temperatura ambiente con agitación constante, se centrifugó a 500 rpm por 5 minutos y se separó el sobrenadante que contenía la proteína rotaviral y la fracción GST; por último, se realizó un lavado de la resina con el buffer de digestión 1X para retirar el remanente de la proteína. Posteriormente, las fracciones que contenían tanto la proteína rotaviral como la fracción GST fueron corridas en un gel de electroforesis SDS-PAGE al 12 % en un único pozo de siembra y al igual que en los casos anteriores, se cortó la banda que correspondía a la proteína viral identificada por western blot, la cual fue utilizada para la inmunización de los conejos. 5.5.3 Preparación de la proteína estructural VP6 del rotavirus para la inmunización de conejos Nueva Zelanda. Para el caso de VP6, la proteína viral fue parcialmente purificada, como se mencionó en el numeral 5.4.5 y sembrada en geles de poliacrilamida al 12 % en condiciones denaturantes en un único pozo de siembra a 20 mAmp por lámina de gel con dimensiones de 8 cm ancho por 7 cm de largo y 1.5 mm de grosor durante 1.5 horas. Un extremo del gel fue cortado y transferido a una membrana de PVDF para identificar la proteína mediante western blot. El resto del gel fue teñido con una solución de azul de Coomassie y la banda identificada por western blot fue cortada para inocularla en el conejo. 5.5.4 Inoculación en conejos para la obtención de anticuerpos policlonales contra las proteínas estructurales del rotavirus VP5, VP6 y VP8. Las bandas del gel que contenían aproximadamente 50 g / banda de las proteínas recombinantes VP5, VP6, VP8 y Hsc70 fueron cortadas en trozos pequeños y transferidas a una jeringa de 5 ml, luego se pasaron varias veces a través de agujas de diferentes calibres (No.18, 21 y 25) a fin de formar una suspensión en buffer PBS. Posteriormente, se inocularon 15 bandas cortadas y homogenizadas de cada una de las proteínas a conejos machos raza Nueva Zelanda de 2 - 3 meses de edad, administradas en tres dosis con periodos de tiempo de 20 días entre cada inmunización. Las dosis fueron aplicadas vía subcutánea, distribuidas paralelamente a la columna vertebral. Después de terminado el periodo de inoculación, los conejos fueron sangrados a blanco 20 días después de la tercera inmunización. Las muestras de sangre fueron incubadas a 37°C por una hora y se incubaron a 4 ºC toda la noche con el objetivo de retraer el coágulo. Después la muestra fue centrifugada a 5000 rpm durante 10 min y se recuperó el suero, el cual contenía los anticuerpos contra cada una de las proteínas rotavirales. El suero fue diluido con glicerol en una proporción 1:1, dispensado en tubos de 1.5 ml estériles y almacenados a – 20 ºC. 5.5.5 Caracterización de los anticuerpos policlonales contra las proteínas virales VP5, VP6 y VP8 y la proteína celular Hsc 70. Los anticuerpos obtenidos contra las proteínas estructurales VP5, VP6 y VP8 del rotavirus y la proteína celular Hsc 70 fueron caracterizados mediante diferentes técnicas de inmunodetección, entre ellas: western blot, ELISA e inmunocitoquímica. Western Blot. Diferentes cantidades de las proteínas purificadas (500 – 62,5 ng) y lisados bacterianos (100 – 25 g), para VP5 y VP8, así como, lisados celular infectados con vaccinia recombinante, para VP6, fueron separados en geles SDS-PAGE y transferidos a membranas de PVDF a 50 mAmp durante 2h en una cámara Mini Trans Blot Cell (BioRad Labs, USA) con buffer de transferencia que contenía Glicina 129 mM, Tris 25 mM y Metanol al 10 %. Finalizada la transferencia, las membranas fueron teñidas con rojo Ponceau al 0,1 % en ácido acético al 5 %, con el fin de verificar la transferencia. Posteriormente, las membranas fueron lavadas con PBS para retirar la tinción y bloqueadas con leche descremada al 5 % en PBS durante 2 h a 37 ºC, se lavaron con PBS dos veces y se incubaron con una dilución de 1:1000 de los anticuerpos correspondientes en PBS, Tween 20 al 0,05 % y leche descremada al 0.1 %. El exceso de anticuerpo no unido fue retirado mediante cinco lavados de 5 min cada uno con PBS Tween 20 al 0.1 % y se incubaron con un anticuerpo secundario anti – conejo conjugado a peroxidasa en una dilución 1:1000 en PBS que contenía leche descremada al 0.1 % y Tween 20 al 0,05 % y al igual que el paso anterior, se realizaron cinco lavados con PBS Tween 20 al 0,1 % de 5 minutos cada uno para retirar el anticuerpo no unido. La detección se llevó a cabo utilizando como sustrato aminoetilcarbazol (AEC) disuelto en N-dimetilformamida. El AEC se diluyó a una concentración final de 0.268 % en buffer de acetato de sodio 0,1 M pH 5.2, se adicionó H2O2 a una concentración final de 0.03% y se filtró tres veces hasta retirar el precipitado que se forma. Finalmente, las membranas fueron lavadas con PBS. Por otra parte, se analizó el reconocimiento de las proteínas en las partículas virales, para lo cual se emplearon 500 ng de TLPs, pero a diferencia del ensayo anterior, se variaron las diluciones de los anticuerpos policlonales, entre 1:250 a 1:1000, con el objetivo de identificar el límite de detección. Inmunocitoquímica. Se cultivaron células MA104 en cajas de 96 pozos con una confluencia del 100 %, la monocapa fue lavada dos veces con medio de cultivo, infectadas con 50 l de TLPs purificadas de RRV en una dilución 1:3200 que tenían una concentración de 1.8 x 108 uff/ml, incubadas a 37 ºC durante 1 h, lavadas dos veces e incubadas con 100 l de medio MEM a 37 ºC durante 12 h. Finalizado el tiempo de infección, la monocapa fue lavada dos veces con PBS y fijadas con 50 l de metanol frío y refrigeradas a 4 ºC durante 40 min. El metanol fue retirado y las células lavadas con PBS. Posteriormente, las células fijadas fueron incubadas de manera independiente con diferentes diluciones de los anticuerpos policlonales, entre 1:250 a 1:2000 en PBS e incubadas a 37 ºC durante 1 h. La monocapa celular fue lavada 5 veces con PBS tween 20 0,05 %, incubadas con un anti-conejo conjugado a peroxidasa en una dilución 1:3000 en PBS durante 45 min a 37ºC y lavadas cinco veces con PBS tween al 0,05%. El proceso de detención se realizó con AEC, como se describió en el procedimiento para western blot. Por último, las células fueron lavadas con H2O y se realizó el conteo de la uff en cada pozo. ELISA. En una placa de 96 pozos para ELISA se incubaron concentraciones crecientes de las proteínas virales recombinantes (50 ng – 2000 ng) en PBS en un volumen final de 50 l durante toda la noche a 4 ºC. Posteriormente, la proteína no unida fue retirada lavando dos veces con 200 l de PBS cada uno, la caja fue bloqueada con 200 l de albúmina al 1 % por pozo durante 2 h a 37 ºC y lavada con 200 l de PBS dos veces. Las proteínas adheridas fueron incubadas con los anticuerpos policlonales respectivos en una dilución 1:1000 en PBS, Tween 20 0,05 % y albúmina al 0,1 % e incubadas durante 2 h a 37 ºC. El anticuerpo no unido fue retirado mediante el lavado de los pozos con 200 l de PBS Tween 20 al 0,1 % tres veces. A continuación, se adicionó a cada pozo un anticuerpo anti – conejo conjugado a peroxidasa en un dilución 1:3000 en PBS Tween 20 0,05 % y albúmina 0,1 % e incubado durante 1 h a 37 ºC. El exceso de anticuerpo secundario fue retirado con tres lavados de 200 l de PBS Tween 20 al 0,1 %. Finalmente, la detección se llevó a cabo mediante la adición de 50 l por pozo de una solución de 1mg /ml de orto – fenilendiamina (OPD) disuelto en H2O2 al 0,03 % y buffer fosfato/citrato pH 5.0, que contiene 25 mM de ácido cítrico y 50 mM de Na2PO4. Cuando se evidenció la formación de un color amarillo intenso, la reacción se detuvo mediante la adición de 50 l de H2SO4 2,5 M. Como controles negativos de la técnica, se realizaron las incubaciones mencionadas en ausencia de alguno de los componentes y en lugar de ellos se reemplazó por 50 l de albúmina al 1 %. Los ensayos fueron realizados por duplicado y el procedimiento se realizó dos veces de manera independiente. Para las gráficas se tabuló el DO, el cual resulta de la diferencia de la absorbancia obtenida para los experimentos menos la absorbancia de los controles negativos. 5.6 ENSAYOS DE INTERACCIÓN ENTRE LAS PROTEÍNAS ROTAVIRALES Y LAS PROTEÍNAS CELULARES Hsc70 Y PDI Para identificar la interacción de las proteínas recombinantes virales VP5, VP6 y VP8 con las proteínas celulares recombinantes Hsc 70 y PDI en condiciones acelulares se empleó la técnica de ELISA indirecto y ensayos de competencia de la interacción. 5.6.1 Ensayo de ELISA indirecto. Las placas de ELISA fueron incubadas con 40 ng/l por pozo de la proteína recombinante Hsc 70 o PDI en un volumen final de 50 diluida en PBS e incubadas a 4 ºC toda la noche. Posteriormente, las placas con la proteína adherida fueron bloqueadas con 200 l de BSA al 1 % en PBS e incubadas a 37 ºC durante 2 h o a 4 ºC toda la noche. Se realizaron dos lavados con PBS y se incubaron con concentraciones crecientes de las proteínas virales VP5, VP6 y VP8, entre 1 – 20 ng/l en un volumen final de 50 l diluidas en PBS e incubadas a 37 ºC durante 2 h. Finalizado el tiempo de interacción, 50 l de los anticuerpos policlonales contra las proteínas virales o contra la fracción GST fueron adicionados a la placa de ELISA en una dilución 1:1000 en PBS que contenía BSA al 0.1 % y Tween 20 al 0,05 % y se incubó durante 2 h a 37 ºC para llevar a cabo el reconocimiento de la proteína viral. Se realizaron tres lavados con 200 l de PBS Tween 20 al 0,1 %, se adicionaron 50 l de los anticuerpos anti-conejo conjugado a peroxidasa en PBS que contenía BSA al 0.1 % y Tween 20 al 0,05 %, se incubaron a 37 ºC durante 1h y se realizaron los cincos lavados correspondientes. Finalmente, el revelado se realizó con 50 l de 1 mg/ml de orto – parafenilendiamina (OPD) disuelto en un buffer de Na2HPO4 50 mM y ácido cítrico 25 mM pH 5,0 y H2O2 al 0,03 %. Después de evidenciar la formación de un color amarillo, producto de la actividad de la HRP, la reacción se detuvo con 50 l de H2SO4 2,5 M. La lectura de las absorbancias se realizó a 492 nm. Como controles negativos de la técnica, se realizaron las incubaciones mencionadas en ausencia de alguno de los componentes y en lugar de ellos se reemplazó por 50 l de albúmina al 1 %. Adicionalmente, los ensayos fueron realizados por duplicado y el procedimiento se realizó dos veces de manera independiente. Para las gráficas se tabuló el DO, el cual resulta de la diferencia de la absorbancia obtenida para los experimentos menos la absorbancia de los controles negativos. 5.6.2 Co – inmunoprecipitación. Aproximadamente 2,5 ng/l de la proteína viral VP6 o 10 ng/l de VP5 o VP8 fueron incubadas con 10 ng/l de las proteínas celulares, PDI y Hsc70, de manera independiente en presencia de albúmina al 0,1 % en un volumen final de 200 l. Las mezclas proteícas fueron incubadas durante toda la noche a 4 ºC, se les adicionó del anticuerpo policlonal de conejo dirigido contra la proteína viral respectiva en una dilución 1:200 y se incubó a 37 ºC durante 1 h. La mezcla fue tratada con 10 l de proteína A unida a agarosa (Bio Rad) previamente tratada con albúmina al 4 % e incubada a temperatura ambiente durante 1 h. Finalmente, la resina fue lavada tres veces con PBS – tween al 0,05 % y centrifugada a 1500 rpm durante 10 min después de cada lavado. El complejo proteíco unido a la resina fue analizado por SDS – PAGE y western blot, para lo cual se empleó un anticuerpo policlonal contra las proteínas celulares. Como controles negativos, las proteínas celulares fueron incubadas con los anticuerpos dirigidos contra las proteínas virales e inmunoprecipitadas a fin de evidenciar si la proteína celular se unía inespecíficamente a la resina o era reconocida por el anticuerpo. En tubos independientes, la mezcla de proteínas celulares y virales fue incubada, pero a diferencia del ensayo anterior, se inmunoprecipitó la proteína celular con 1 ng/l de un anticuerpo policlonal dirigido contra Hsc70 o PDI y en el western blot se identificaron las proteínas virales. Como controles negativos, las proteínas virales fueron incubadas con los anticuerpos dirigidos contra las proteínas celulares con el objetivo de evidenciar si estas proteínas se unían a la resina en ausencia de la proteína celular. 5.6.3 Interacción de las proteínas virales con la proteína Hsc70 en los dominios rafts lipídicos. Para identificar la interacción de las proteínas virales con la proteína Hsc 70 en condiciones celulares, se crecieron células MA104 en 2 cajas T - 160 hasta alcanzar un confluencia del 100 %, las cuales fueron tratadas con 1 ml de tripsina al 0.1 mg /ml e incubadas 10 min a 37 ºC a fin de desprenderlas de la caja. Las células fueron mezcladas, transferidas a un tubo de 15 ml, centrifugadas a 1400 rpm durante 10 min y lavadas con medio MEM para retirar el remanente de la enzima. Posteriormente, las células fueron resuspendidas en 1400 l de medio, divididas en 4 tubos aproximadamente 9,2 x 10 6 de 1.5 ml con células y mezcladas de manera independiente con 15 ng/l de cada una de las proteínas virales VP5 o VP8 y 1.5 ng/l de VP6, durante 1 hora a temperatura ambiente en un volumen final de 700 l. El tubo de células remanente fue empleado como control del ensayo, el cual no fue incubado con las proteínas virales. Finalizado el tiempo de incubación, las células fueron lavadas dos veces con PBS y lisadas en 120 l buffer lisis tritón frio, el cual contenía 25 mM de Tris HCl pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1 mM DTT, 1 % Tritón X 100 y 1 mM de PMSF, durante 30 min a 4 ºC. El lisado se mezcló 1:1 con sacarosa al 80% disuelta en buffer TNC (25 mM de Tris HC l pH 7.5, 150 mM NaCl y 1 mM CaCl2) y puesto al final de un tubo de ultracentrífuga. Sobre el lisado se formó un gradiente discontinuo de sacarosa del 30 al 5 % en buffer TNC y se ultracentrifugó a 170000 x g por 8 h a 4 ºC. Posteriormente, fueron recolectadas 8 fracciones de 500 l de cada ensayo, las cuales fueron mezcladas con 10 mM de ciclodextrina y 0,8 % de Octilglucósido e incubadas a 37 ºC durante 1 h. Cada una de las fracciones fue incubada con 2 ng/l de un anticuerpo policlonal de conejo dirigido contra la proteína celular Hsc70 (Santa cruz) durante 1 h a 37 ºC, luego se adicionó 30 l de proteína A unida a agarosa (Bio Rad) y las fracciones fueron incubadas durante 1 hora a temperatura ambiente. Las muestras fueron centrifugadas a 1500 rpm por 10 min y lavadas con PBS tres veces. Las fracciones inmunoprecipitadas fueron analizadas por western blot con anticuerpos policlonales dirigidos contra las proteínas virales. 5.6.4 Ensayos de inhibición de la infección en células MA104 con el rotavirus de la cep a R R V Para determinar si las proteínas recombinantes virales inhibían el proceso infección con TLPs de RRV, diferentes concentraciones, entre 1,25 - 40 ng/l, de las proteínas virales fueron adicionadas a un cultivo de células MA104 adheridas a placas de 96 pozos durante 1 h a 37ºC. Posteriormente, se adicionaron 50 l de TLPs purificadas en una dilución 1:3600 (1.8 x 108 uff/ml) a la monocapa y se incubaron durante 1 h a 37 ºC, luego se lavó con medio para retirar el exceso de virus no adherido y se incubó durante 12 h en 100 l de medio MEM cada pozo a 37ºC. La identificación de la infección se realizó como se describió en el numeral 5.5.5 (Inmunocitoquímica). El porcentaje de infección se comparó respecto a células que no fueron incubadas con ninguna proteína viral. 6. RESULTADOS 6.1 AMPLIFICACIÓN Y PURIFICACIÓN DE LOS VECTORES QUE PRESENTAN LAS SECUENCIAS GÉNICAS QUE CODIFICAN LAS PROTEÍNAS VIRALES VP5 Y VP8 Los vectores que presentan las secuencias génicas que codifican las proteínas virales VP5 y VP8 fueron amplificados mediante la transfección de los mismos en bacterias E. coli de la cepa XL1BLUE deficiente en la expresión de enzimas que catalizan el rearreglo, deleción, recombinación o corte del ADN. Los plásmidos pGEX-4T-VP5 y pGEX-4T-VP8 fueron purificados con el kit PurelinkTM Quick Plasmid Miniprep (Invitrogen) como se describió en metodología numeral 5.1.5. Como se observa en la figura 11 a, carriles 2 y 3, las fracciones purificadas de los plásmidos presentaron tres bandas de ADN de alto peso molecular, que pueden corresponder a diferentes enrrollamientos de los vectores o a una contaminación con material genético bacteriano. Para descartar esta última posibilidad, los plásmidos fueron tratados con la enzima de restricción Pst I, puesto que estos vectores presentan un único sitio de corte para esta enzima (…CTGCA/G…) (figura 10, carriles 4 y 5). En esta misma figura, se observa un corrido diferencial entre las muestras que no habían sido tratadas (carriles 2 y 3) y las que fueron digeridas (carriles 4 y 5). El plásmido pGEX4T-VP5 al ser digerido con la enzima de restricción Pst I generó tres fragmentos con pesos moleculares de 5997.9, 1733.8 y 501.19 pb (figura 11 a, carril 4), puesto que la secuencia que codifica la proteína viral presenta dos sitios de corte para esta enzima, como se muestra en la figura 11 b. En el caso del plásmido pGEX-4T-VP8 digerido con la enzima de restricción Pst I, se observó la presencia de una sola banda de 6839.1 pb, correspondiente al vector en su estructura lineal (figura 11 a, carril 5). Figura 11. a) Purificación de los plásmidos pGEX-4T que presentan las secuencias que codifican las proteínas virales VP5 y VP8 purificados y digeridos con Pst I. En el gel de agarosa al 1,2 % teñido con BrEt se observa en los carriles de izquierda a derecha: 1. Marcador de peso molecular; 2, Plásmido pGEX-4TVP5 sin digerir con Pst I; 3, Plásmido pGEX-4T-VP8 sin digerir con Pst I; 4, pGEX-4T-VP5 digerido con Pst I y 5, pGEX-4T-VP8 digerido con Pst I. b) Secuencia génica que codifica la proteína viral VP5. En color rojo se identifican los sitios de corte para la enzima Pst I presentes en la secuencia que codifica para esta proteína viral. 6.2 EXPRESIÓN DE LAS PROTEÍNAS RECOMBINANTES VP5, VP8 Y VP6 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 La expresión de las proteínas estructurales del rotavirus VP8 y VP5 y la proteína celular Hsc 70 se realizó en un sistema de transfección de células procariontes, Escherichia coli, de la cepa BL21(DE3), la cual se caracteriza por ser una cepa modificada genéticamente que es deficiente en la expresión de proteasas. Por otra parte, la proteína VP6 fue expresada mediante la infección de células eucariotas de riñón de mono verde MA104 con el virus recombinante vaccinia. Para la expresión de las proteínas del rotavirus VP5 y VP8 y la proteína Hsc 70 fue necesario establecer las condiciones óptimas de expresión para obtener el mayor rendimiento de la proteína de interés. Las variables analizadas fueron: número de bacterias antes de inducir la expresión, tiempo de inducción, concentración del inductor, medio de crecimiento y presencia de cuerpos de inclusión. La expresión de las proteínas recombinantes fue determinada mediante SDS – PAGE y western blot a partir de los lisados bacterianos. 6.2.1 Expresión de la proteína recombinante estructural del rotavirus VP8. La primera variable que se tuvo en cuenta para optimizar la expresión de la proteína viral VP8, fue el número de bacterias presentes antes de inducir la expresión de esta proteína, para lo cual se empleó un método indirecto que consistió en determinar la densidad óptica a 600 nm (DO600 nm) por unidad de tiempo a fin de identificar las diferentes fases del crecimiento microbiano (latencia, exponencial y estacionaria). De esta manera, se realizó una curva de crecimiento de la bacteria transformada, para lo cual se determinó la DO600 nm cada hora, teniendo en cuenta la relación directa entre el incremento del número de células y el valor de absorbancia registrado sin discriminar la viabilidad de las mismas. A partir de esta curva de crecimiento se pudo definir la fase de latencia, comprendida entre 0 – 1 h de crecimiento, la fase exponencial, entre 1 – 4 h y por último, la fase estacionaria, a partir de las 4 horas de crecimiento. De esta manera, se decidió evaluar la expresión de la proteína viral VP8 en rangos de absorbancia entre 0,1 – 0,6 (en la curva se graficó el log (DO x 100) vs tiempo) que corresponden a la fase exponencial y el inicio de la fase estacionaria del crecimiento bacteriano (figura 12 a). Varias alícuotas de 25 ml de las bacterias transformadas fueron cultivadas en medio LB hasta alcanzar una DO 600 nm entre 0.1 a 0.6 y se indujo la expresión de la proteína viral con 1 mM de IPTG durante 4 h. El pellet de células de cada ensayo fue sonicado y centrifugado, de lo cual se obtuvo una fracción de proteínas solubles y una de proteínas insolubles que fueron tratadas con úrea 8M. Estas fracciones fueron analizadas por SDSPAGE y la presencia de la proteína viral recombinante VP8 - GST fue detectada mediante un anticuerpo policlonal contra GST y un anticuerpo policlonal dirigido contra un péptido de la proteína viral (figura 12 b). Los carriles 2 y 3 de la figura 12 b, corresponden a la fracción soluble e insoluble del lisado bacteriano que no fue tratado con IPTG, respectivamente, en los cuales se observó una baja expresión basal de la proteína viral en la fracción de proteínas insolubles con un peso molecular de 49,6 KDa. En los carriles 4, 6, 8, 10 y 12, que corresponden a las fracciones solubles de los lisados bacterianos que fueron tratados con IPTG una vez habían alcanzado las diferentes DO 600 nm, no se observó la presencia de la proteína recombinante. En los carriles 5, 7, 9, 11 y 13 se sembraron las fracciones insolubles de los lisados bacterianos correspondientes y se observó que a medida que se incrementaba el número de bacterias, la cantidad de la proteína VP8 era mayor y se mantuvo constante a partir del carril 7, el cual corresponde a un cultivo que alcanzó una DO 600 nm de 0.3 en el momento de inducir la expresión con IPTG. De esta manera, la expresión de la proteína viral VP8 se localizó en la fracción insoluble, independientemente de la densidad óptica a la cual se realizó la inducción de la expresión de la proteína viral. Figura 12. a) Curva de crecimiento de las bacterias BL21(DE3) – VP8 en medio LB medido por turbidimetría a 600 nm. b) Expresión de la proteína viral recombinante VP8 – GST variando el número de bacterias transformadas antes de inducir la expresión. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fueron analizadas por SDS-PAGE al 12% y western blot con un anticuerpo primario contra GST. En los carriles de los geles y membranas correspondientes se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS DO 600 nm 0,1; 5, FI DO 600 nm 600 nm 0,1; 6, FS DO 600 nm 0,2; 7, FI DO 600 nm 0,2; 8, FS DO 600 nm 0,4; 9, FI DO 600 nm 0,4; 10, FS DO 0,5; 11, FI DO 600 nm 0,5; 12, FS DO 600 nm 0,6 y 13, FI DO 600 nm 0,6. La concentración aproximada de la proteína viral se determinó mediante la cuantificación de proteínas totales a 280 nm y se sembró en el gel 100 g de proteínas totales de cada fracción. La banda correspondiente a la proteína VP8 fue comparada visualmente con una curva de BSA corrida en un gel de electroforesis teñido con azul de coomasie y se estableció la concentración aproximada de la misma respecto al volumen sembrado en el gel (tabla 1). Teniendo en cuenta los datos mostrados en la tabla 1, se observa que la mayor concentración de la proteína viral se obtuvo cuando el cultivo alcanzó una DO 600 nm de 0,5 al momento de inducir la expresión de la proteína, con una concentración aproximada de la proteína viral de 0,141 g/l. Tabla 1. Determinación aproximada de la concentración de la proteína viral VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes densidades ópticas al momento de inducir la expresión de la proteína VP8 con IPTG. Carril de siembra DO 600 nm en el gel de la Cantidad VP8 – GST Volumen (g) sembrado (l) figura 12 b Concentración aproximada VP8 – GST (g/l) 0,1 5 0.2 8,63 0,023 0,2 7 0,8 11,3 0,071 0,4 9 0,8 7,87 0,102 0,5 11 0,8 5,66 0,141 0,6 13 0,8 7,67 0,104 Otros factores que afectan la cantidad y solubilidad de las proteínas recombinantes durante su expresión, es el tiempo de inducción y la concentración del inductor. De esta manera, se evaluaron diferentes tiempos de inducción, entre 2 a 8 h, con 1 mM de IPTG cuando el cultivo alcanzó una DO 600 nm de 0,5 (figura 13 a) y diferentes concentraciones de IPTG, entre 0,5 - 2 mM (figura 13 b). Como se muestra en la figura 13 a, en las fracciones insolubles correspondientes a los lisados bacterianos que fueron tratados durante 2, 4, 6 y 8 horas con 1mM de IPTG (carriles 5, 7, 9 y 11), se observó la presencia de la proteína viral en el gel, correspondiente a la banda más ancha, la cual fue identificada mediante western blot; mientras que en las fracciones solubles correspondientes (carriles 4, 6, 8 y 10) no se evidenció la presencia de la proteína viral. Al igual que en el ensayo anterior, la concentración de la proteína viral fue determinada visualmente frente a una curva de BSA con relación al volumen de siembra que contenía 100 g de proteína total en geles de poliacrilamida (tabla 2). Tabla 2. Determinación aproximada de la concentración de la proteína viral VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de que el cultivo alcanzó una DO 600 nm de 0,5. Concentración Tiempo de Carril de siembra en Cantidad VP8 – Volumen Inducción (h) el gel de la figura 13 a GST (g) sembrado (l) 2 5 2 11,8 0,169 4 7 2 9,48 0,211 aproximada VP8 – GST (g/l) Concentración Tiempo de Carril de siembra en Cantidad VP8 – Volumen Inducción (h) el gel de la figura 13 a GST (g) sembrado (l) 6 9 2 8,00 0,250 8 11 2 8,20 0,244 aproximada VP8 – GST (g/l) De acuerdo con los resultados calculados en la tabla 2, se determinó que la concentración de la proteína viral VP8 - GST fue mayor cuando el cultivo bacteriano transformado fue inducido durante 6 a 8 h, con una concentración aproximada de 0,250 g/l de VP8 – GST, respecto a 2 y 4 h con concentraciones de 0,169 – 0,211 g/l de VP8 – GST, respectivamente. Así mismo, se determinó el efecto de la concentración de IPTG sobre la expresión de la proteína viral. Teniendo en cuenta los resultados obtenidos en la figura 13 b, se observa que la mayor cantidad de la proteína viral se obtuvo cuando el cultivo bacteriano fue tratado con 1 mM de IPTG y se localizó en la fracción insoluble del lisado bacteriano (carril 7); sin embargo, a una menor o mayor concentración del inductor, la cantidad de proteína viral expresada fue menor (carril 5 y 9). Por otra parte, las fracciones de proteínas solubles correspondientes no presentaron la proteína recombinante (carriles 4, 6 y 8). La tabla 3 indica las concentraciones aproximadas calculadas para la proteína viral VP8 – GST en cada uno de los ensayos, de lo cual se obtuvo 0,252 g/l cuando el cultivo fue inducido con 1 mM de IPTG. Tabla 3. Determinación aproximada de la concentración de la proteína viral VP8 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana con diferentes concentraciones de IPTG después de que el cultivo alcanzó una DO 600 nm de 0,5. Concentración de IPTG (mM) Carril de siembra en el gel de la figura 13 b Cantidad VP8 – Volumen GST (g) sembrado (l) Concentración aproximada VP8 – GST (g/l) 0,5 5 1 5,79 0,172 1,0 7 2 7,93 0,252 2,0 9 1 7,71 0,129 En resumen, las bacterias transformadas con el vector pGEX-4T-VP8 fueron cultivadas hasta alcanzar una DO 600 nm de 0.5 al momento de inducir la expresión de la proteína con 1 mM de IPTG durante 6 h. Figura 13. a) Expresión de la proteína viral VP8 – GST variando el tiempo de inducción con 1 mM de IPTG. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y Western Blot con un anticuerpo primario contra GST. En los carriles del gel y membrana correspondiente se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido 2h; 5, FI inducido 2h; 6, FS inducido 4h; 7, FI inducido 4h; 8, FS inducido 6h; 9, FI inducido 6h; 10, FS inducido 8h y 11, FS inducido 8h. b) Expresión de la proteína viral VP8 – GST variando la concentración de IPTG. En los carriles del gel y membrana correspondiente se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido con 0,5 mM IPTG; 5, FI inducido 0,5 mM IPTG; 6, FS inducido 1 mM IPTG; 7, FI inducido 1 mM IPTG; 8, FS inducido 2 mM IPTG y 9, FI inducido 2 mM IPTG. Teniendo en cuenta que la expresión de la proteína viral recombinante VP8 se localizó en la fracción de proteínas insolubles, se analizó la expresión de la proteína pero a una temperatura menor (25 ºC) durante el crecimiento bacteriano y la inducción de la proteína recombinante. Teniendo en cuenta los resultados obtenidos en la figura 14, se puede evidenciar que a pesar de disminuir la temperatura durante la inducción de la proteína recombinante, esta se sigue localizando en la fracción de proteínas insolubles y su rendimiento fue menor (carril 4), en comparación a 37ºC (carril 5). Figura 14. Expresión de la proteína viral VP8 – GST variando la temperatura a la cual se realizó el crecimiento bacteriano y la inducción de la expresión de la proteína recombinante. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y Western Blot con un anticuerpo primario contra GST y contra un péptido de VP8. En los carriles del gel y membranas correspondientes se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS inducido a 25 ºC; 3, FS inducido a 37 ºC; 4, FI inducido a 25 ºC y 5, FI inducido a 37 ºC. 6.2.2 Expresión de la proteína recombinante estructural del rotavirus VP5. Para la expresión de la proteína viral VP5 – GST se emplearon las condiciones previamente estandarizadas para la expresión de la proteína viral VP8 – GST; sin embargo, el rendimiento no fue el adecuado teniendo en cuenta los resultados obtenidos para VP8 – GST. Por tanto, se evaluaron las variables descritas anteriormente y dos variables adicionales que permitieron aumentar la expresión de la proteína recombinante, entre ellas: el medio de cultivo y la adición de glucosa durante el crecimiento bacteriano. Teniendo en cuenta lo anterior, se analizó la expresión de la proteína viral VP5 – GST cuando las bacterias transformadas fueron cultivadas en dos medios diferentes: LB y 2XYT. El primero tiene los nutrientes esenciales para el crecimiento bacteriano y el segundo, es un medio enriquecido, el cual presenta una mayor concentración de triptona y extracto de levadura respecto a LB. La presencia de la proteína viral fue identificada en la fracción de proteínas insoluble mediante western blot con un anticuerpo policlonal dirigido contra la proteína viral y otro anticuerpo policlonal dirigido contra GST y presentó un peso molecular de 75 KDa (figura 15). Teniendo en cuenta los resultados obtenidos en la figura 15, se pudo observar que cuando las bacterias fueron cultivadas en medio LB pero no se empleó inductor, no se evidenció la expresión basal de la proteína viral (carril 2); sin embargo, cuando el cultivo se realizó en medio 2XYT, se incrementó la expresión basal de la misma (carril 3), pero en presencia de glucosa al 2% se inhibió (carril 4). Así mismo, la mayor concentración de la proteína viral se obtuvo cuando el cultivo se realizó en medio 2XYT en presencia de glucosa al 2 % con 1 mM de IPTG (carril 8), respecto a las bacterias que fueron cultivadas en este mismo medio pero en ausencia de glucosa (carril 7) y en medio LB con y sin glucosa en presencia del inductor (carril 5 y 6). Figura 15. Expresión de la proteína viral VP5 - GST en medio LB y 2XYT en presencia y ausencia de glucosa al 2 %. En los carriles del gel se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FI No inducido en medio LB; 3, FI No inducido en medio 2XYT; 4, FI No inducido en medio 2XYT y glucosa; 5, FI inducido en medio LB; 6, FI inducido en medio LB y glucosa; 7; FI inducido en medio 2XYT; 8, FI inducido en medio 2XYT y glucosa. En la parte inferior se encuentra la membrana correspondiente, para lo cual se empleó un anticuerpo dirigido contra el polipéptido GST. Con el fin de incrementar la concentración de proteína viral VP5 – GST y de ser posible en forma soluble, se analizó la expresión de la proteína recombinante cuando la inducción se realizó a diferentes DO 600nm al momento de adicionar el IPTG. Para esto se realizó la curva de crecimiento para las bacterias BL21(DE3) transfectadas con el plásmido pGEX- 4T-VP5 con el fin de identificar la fase de crecimiento exponencial y estacionaria de las bacterias transformadas en medio 2XYT y en presencia de glucosa al 2 % (figura 16 a). A partir de la figura 16 a fue posible definir la fase de latencia, entre 0 – 1 h de crecimiento, la fase exponencial, entre 1 – 4h y la fase estacionaria, después de 4h en adelante, del cultivo bacteriano. Después de identificado el tiempo en el cual se llevó a cabo la fase de crecimiento exponencial y estacionaria de las bacterias transformadas, fue posible evaluar la expresión de la proteína viral cuando los cultivos alcanzaron las diferentes DO 600 nm (0,1 – 0,7) al momento de adicionar el inductor. En los carriles 2 y 3 de la figura 16 b, se sembraron las fracciones soluble e insoluble, respectivamente, del lisado bacteriano que no fue inducido con IPTG, en los cuales no se evidenció expresión basal de la proteína recombinante. Los carriles 4, 6, 8, 10 y 12 corresponden a las fracciones de proteínas solubles de cada uno de los lisados bacterianos que fueron tratados con el inductor una vez habían alcanzado una DO 600 nm de 0.1, 0.2, 0.4, 0.6 y 0.7, respectivamente, en los cuales tampoco se detectó la presencia de la proteína viral. Mientras que en los carriles 5, 7, 9, 11 y 13, en los que se sembró las fracciones insolubles de los lisados anteriores, se identificó la presencia de esta proteína. Por lo tanto, la proteína viral VP5 – GST fue expresada en la fracción de proteínas insolubles a pesar de variar el número de bacterias antes de estimular su expresión. Figura 16. a) Curva de crecimiento de las bacterias BL21(DE3) – VP5 en medio 2XYT y 2 % de Glucosa medido por turbidimetría a 600 nm. b) Expresión de la proteína viral recombinante VP5 – GST variando el número de bacterias transformadas antes de inducir la expresión. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y western blot con un anticuerpo primario contra GST. En los carriles de los geles y membranas correspondientes se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS DO 600 nm 0,1; 5, FI DO 600 nm 600 nm 0,1; 6, FS DO 600 nm 0,2; 7, FI DO 600 nm 0,2; 8, FS DO 600 nm 0,4; 9, FI DO 600 nm 0,4; 10, FS DO 0,6; 11, FI DO 600 nm 0,6; 12, FS DO 600 nm 0,7 y 13, FI DO 600 nm 0,7. Así mismo, la concentración aproximada de la proteína recombinante fue determinada teniendo en cuenta la intensidad de la banda obtenida para esta proteína y comparándola con una curva de BSA con relación al volumen sembrado en el gel de electroforesis (tabla 4). Tabla 4. Concentración aproximada de la proteína viral VP5 - GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes densidades ópticas antes de la inducción con 1 mM de IPTG. Carril de siembra DO 600 nm en el gel de la figura 16 b Cantidad VP5 – Volumen GST (g) sembrado (l) Concentración aproximada VP5 – GST (g/l) 0,1 5 0,2 10,0 0,020 0,2 7 1,0 10,0 0,100 0,4 9 0,5 4,69 0,107 0,6 11 0,1 4,35 0,023 0,7 13 0,1 3,44 0,029 En la tabla 5 se calculó que la mayor concentración de la proteína VP5 – GST se obtuvo cuando el cultivo alcanzó una DO 600 nm entre 0.2 – 0.4 y se indujo con 1 mM de IPTG durante 4 h, con una concentración de la proteína viral de 0.100 – 0.106 g/l, respectivamente. Por otra parte, la expresión de la proteína viral VP5 fue evaluada a diferentes tiempos de inducción, para lo cual las bacterias transformadas fueron cultivadas hasta alcanzar una DO 600 nm de 0,4 en medio 2XYT en presencia de glucosa al 2 % y tratadas con 1 mM de IPTG durante 2 – 8 h. Como se observa en la figura 17 a, la banda correspondiente a la proteína VP5 – GST fue detectada solo en las fracciones insolubles de los lisados bacterianos inducidos durante 2, 4, 6 y 8 h (carriles 5, 7, 9 y 11, respectivamente). Mientras que en los carriles 4, 6, 8 y 10, correspondientes a las fracciones solubles de los lisados anteriores, no se observó la proteína viral. La concentración de la proteína viral VP5 – GST fue calculada en cada una de las fracciones insolubles de los lisados bacterianos como se mencionó anteriormente. En la tabla 5, se puede observar que la mayor concentración de la proteína viral fue obtenida cuando el cultivo fue inducido durante 4 h, con un valor aproximado de 0,297 g/l; sin embargo, no se observó cambios drásticos en la cantidad de proteína expresada en los diferentes tiempos analizados. Tabla 5. Concentración aproximada de la proteína viral VP5 unida a GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de alcanzar una DO 600 nm de 0,2. Carril de siembra Tiempo de en el gel de la Inducción (h) figura 17 a Concentración Cantidad VP5 – Volumen GST (g) sembrado (l) 2 7,00 0,286 aproximada VP5 – GST (g/l) 2 5 4 7 1 3,37 0,297 6 9 0,8 3,03 0,264 8 11 1 4,65 0,215 Otra variable que afecta la expresión de una proteína recombinante es la concentración del inductor. Para esto, se crecieron las bacterias transformadas en medio 2XYT en presencia de 2% de glucosa y se indujo la expresión de la proteína con diferentes concentraciones de IPTG, entre 0,5 – 2 mM, durante 4 h. Como se observa en la figura 17 b, la proteína viral VP5 – GST se mantuvo en la fracción de proteínas insolubles (carriles 5, 7 y 9), aun cuando se varió la concentración del inductor. Al igual que en los ensayos anteriores, no se observó la presencia de la proteína viral en las fracciones solubles de los lisados anteriores (carriles 4, 6 y 8). Tabla 6. Concentración aproximada de la proteína viral VP5 unida a GST en las fracciones de proteínas insolubles después de la lisis bacteriana a diferentes concentraciones de IPTG después de alcanzar una DO 600 nm de 0,2. Concentración de IPTG (mM) 0,5 Carril de siembra en el gel de la figura 17 b 5 Cantidad VP5 – Volumen GST (g) sembrado (l) 2 5,15 Concentración aproximada VP5 – GST (g/l) 0,388 Concentración de IPTG (mM) Carril de siembra en el gel de la figura 17 b Cantidad VP5 – Volumen GST (g) sembrado (l) Concentración aproximada VP5 – GST (g/l) 1,0 7 2 5,23 0,382 2,0 9 1 5,02 0,199 La concentración de la proteína viral VP5 – GST fue determinada en los ensayos anteriores y se tabuló en la tabla 6. A partir de los datos obtenidos, se determinó que la mayor concentración de la proteína fue obtenida cuando al cultivo se le adicionó 0.5 y 1 mM de IPTG, con un valor aproximado de 0,388 g/l. Sin embargo, concentraciones de 2 mM del inductor disminuyó la concentración de la proteína viral, con un valor aproximado de 0,199 g/l. Figura 17. a) Expresión de la proteína viral VP5 – GST variando el tiempo de inducción con 1 mM de IPTG. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y western blot con un anticuerpo primario contra GST. En los carriles del gel y membrana correspondiente se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido 2h; 5, FI inducido 2h; 6, FS inducido 4h; 7, FI inducido 4h; 8, FS inducido 6h; 9, FI inducido 6h; 10, FS inducido 8h y 11, FI inducido 8h. b) Expresión de la proteína viral VP5 – GST variando la concentración de IPTG. En los carriles del gel y membrana correspondiente se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido con 0,5 mM IPTG; 5, FI inducido 0,5 mM IPTG; 6, FS inducido 1 mM IPTG; 7, FI inducido 1 mM IPTG; 8, FS inducido 2 mM IPTG y 9, FI inducido 2 mM IPTG. En resumen, las bacterias transformadas con el vector pGEX-4T-VP5 fueron cultivadas hasta alcanzar una DO 600 nm de 0.2 al momento de inducir la expresión de la proteína con 0,5 mM de IPTG durante 4 h. La expresión de la proteína viral VP8 y VP5 fue localizada en la fracción de proteínas insolubles en las diferentes condiciones analizadas, lo cual sugiere la formación de agregados insolubles de la proteína recombinante, denominados cuerpos de inclusión. Para confirmar este evento, el pellet obtenido después de la lisis celular fue tratado con concentraciones crecientes de úrea (1 – 8M), teniendo en cuenta que solo a altas concentraciones del agente caotrópico se solubilizan este tipo de agregaciones. Como se puede evidenciar en la figura 18, las proteínas recombinantes VP8 – GST y VP5 - GST fueron solubles solo cuando se empleó 8 M de úrea (carriles 8), sugiriendo que la expresión de estas proteínas se dió en forma de cuerpos de inclusión. Figura 18. Expresión de la proteína viral VP8 - GST (a) y VP5 – GST (b) en cuerpos de inclusión. En los carriles de los geles se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido; 5, FI tratada con úrea 1 M; 6, FI tratada con úrea 2 M; 7; FI tratada con úrea 4 M; 8, FI tratada con úrea 8 M. La inducción de la expresión de la proteína viral VP8 – GST se llevó a cabo cuando el cultivo bacteriano alcanzó una DO 600 nm: 0,5, con 1 mM de IPTG durante 6 horas; mientras que la expresión de la proteína viral VP5 – GST se realizó cuando el cultivo alcanzó una DO 600 nm de 0,4 con 0,5 mM de IPTG durante 4 h. 6.2.3 Expresión de la proteína recombinante Hsc 70. Para optimizar las condiciones de expresión de la proteína recombinante Hsc 70 se tuvieron en cuenta las variables anteriormente mencionadas para las proteínas virales VP8 y VP5. Inicialmente, se realizó la curva de crecimiento de las bacterias BL21(DE3) trasnformadas con el vector pET-28a-Hsc70 en medio LB, la cual permitió definir las fases del crecimiento con relación al tiempo que corresponde a la fase de latencia, entre 0 – 1h, la la fase exponencial, entre 1 – 4h, y la fase estacionaria, de 5 h en adelante (figura 19 a). Por tanto, los ensayos de inducción de la expresión de la proteína recombinante Hsc 70 se realizaron con cultivos que tuvieran absorbancia entre 0,1 – 0,7 que corresponden a la fase exponencial y el inicio de la fase estacionaria del crecimiento celular. La presencia de la proteína fue identificada con un anticuerpo policlonal dirgido contra la proteína celular y se calculó un peso molecular de 65 KDa. Teniendo en cuenta los resultados obtenidos en la figura 19 b, no se observó expresión basal de la proteína Hsc70 en la fracción soluble ni en la insoluble procedente de un cultivo bacteriano al cual no se le adicionó IPTG (carriles 2 y 3, respectivamente). Por otra parte, se evidenció una banda correspondiente a la proteína recombinante tanto en las fracciones solubles como en las insolubles de los cultivos bacterianos que alcanzaron una DO 600 nm de 0.1 a 0.4 (carriles 4 - 11) y que fueron tratados con 1 mM de IPTG durante 4 h. Sin embargo, la presencia de Hsc70 fue identificada únicamente en las fracciones insolubles procedentes de cultivos que alcanzaron una DO 600 nm de 0.5 a 0.7 al momento de inducir la expresión con IPTG (carriles 13, 15 y 17). Figura 19. a) Curva de crecimiento de las bacterias BL21(DE3) – Hsc 70 en medio 2XYT medido por turbidimetría a 600 nm. b) Expresión de la proteína recombinante Hsc70 variando el número de bacterias transformadas antes de inducir la expresión. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y western blot con un anticuerpo primario contra la proteína. En los carriles de los geles y membranas correspondientes se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS DO 600 nm 0,1; 5, FI DO 600 nm 600 nm 0,1; 6, FS DO 0,4; 11, FI DO 600 nm 600 nm 0,2; 7, FI DO 0,4; 12, FS DO 600 nm 600 nm 0,2; 8, FS DO 0,5; 13, FI DO 600 nm 600 nm 0,3; 9, FI DO 0,5; 14, FS DO 600 nm 600 nm 0,3; 10, FS DO 0,6; 15, FI DO 600 nm 0,6; 16, FS DO 600 nm 0,7 y 17, FI DO 600 nm 0,7. En la tabla 7 se muestran los datos de concentración aproximada de la proteína Hsc70 obtenida en los diferentes cultivos analizados, tanto en la fracción soluble como en la insoluble. Los resultados obtenidos muestran que la mayor concentración de la proteína celular se obtuvo cuando el cultivo bacteriano alcanzó una DO 600 nm de 0.6 al momento de inducir la expresión, con un valor de 0,118 g/l, en comparación con los datos obtenidos en los otros cultivos. Tabla 7. Concentración aproximada de la proteína recombinante Hsc70 en las fracciones de proteínas solubles e insolubles después de la lisis bacteriana a diferentes densidades ópticas antes de la inducción con 1 mM de IPTG. Carril de siembra DO 600 nm en el gel de la Fracción figura 19 b 0,1 0,2 0,3 Concentración Cantidad Volumen Hsc 70 (g) sembrado (l) aproximada Hsc 70 (g/l) 4 Soluble 0,2 9,63 0,021 5 Insoluble 0,2 11,7 0,017 6 Soluble 0,2 11,3 0,018 7 Insoluble 0,2 10,1 0,020 8 Soluble 0,1 10,2 0,010 9 Insoluble 0,2 8,69 0,023 10 Soluble 0,1 9,57 0,010 11 Insoluble 0,2 8,29 0,024 0,5 13 Insoluble 0,5 4,91 0,102 0,6 15 Insoluble 0,4 3,40 0,118 0,7 17 Insoluble 0,4 6,93 0,058 0,4 Con el objeto de incrementar la concentración de la proteína recombinante y aumentar la proporción de la misma en la fracción soluble, se evaluó el tiempo de inducción, entre 2 a 8 h, con 1 mM de IPTG después de que el cultivo alcanzó una DO 600 nm de 0,6. Teniendo en cuenta los resultados en la figura 20 a, la presencia de la proteína recombinante fue identificada en todas las fracciones procedentes de los lisados bacterianos obtenidos en cada ensayo (carriles 4 - 11); sin embargo, se evidenció una mayor cantidad de la proteína en las fracciones insolubles en comparación con las fracciones solubles. La concentración de Hsc70 en las fracciones obtenidas de cada uno de los cultivos fue determinada y tabulada en la tabla 8. De acuerdo con los resultados obtenidos en la tabla, la mayor concentración de Hsc70 (0,134 g/l) fue obtenida cuando el cultivo bacteriano alcanzó una DO 600 nm de 0,6 y la inducción se realizó con 1 mM de IPTG durante 4 h, teniendo en cuenta las dos fracciones. Tabla 8. Concentración aproximada de la proteína recombinante Hsc 70 en las fracciones de proteínas solubles e insolubles después de la lisis bacteriana a diferentes tiempos de inducción con 1 mM de IPTG después de alcanzar una DO 600 nm de 0,6. Tiempo de inducción (h) 2 4 6 8 Carril de siembra en el gel de la Fracción figura 20 a Cantidad Hsc 70 (g) Volumen sembrado (l) Concentración aproximada Hsc 70 4 Soluble 0,1 9,45 0,011 5 Insoluble 0,5 9,73 0,051 6 Soluble 0,1 7,26 0,014 7 Insoluble 1,0 8,33 0,120 8 Soluble 0,1 9,97 0,010 9 Insoluble 0,5 6,75 0,074 10 Soluble 0,1 8,15 0,012 11 Insoluble 0,5 5,48 0,091 Así mismo, se evaluó el efecto sobre la concentración y localización de la proteína recombinante Hsc 70 al utilizar diferentes concentraciones de IPTG, entre 0,5 – 2 mM. Como se muestra en la figura 20 b, la proteína recombinante fue identificada tanto en la fracción insoluble y soluble de cada uno de los cultivos inducidos con las diferentes concentraciones de IPTG (carriles 4 - 9) y al igual que el ensayo anterior, la mayor cantidad de Hsc70 fue observada en la fracción insoluble de los lisados bacterianos (carriles 5, 7 y 9). De la misma manera, la concentración de la proteína Hsc70 fue determinada en cada una de las fracciones obtenidas a partir de los diferentes cultivos. Como se muestra en la tabla 10, la mayor concentración de la proteína se obtuvo cuando el cultivo se trató con 1 mM de IPTG, con un valor de 0,089 g/l; sin embargo, no se observaron diferencias radicales en cuanto a cantidad ni localización de la proteína aun al variar la concentración del inductor. Tabla 9. Concentración aproximada de la proteína recombinante Hsc 70 en las fracciones de proteínas solubles e insolubles después de la lisis bacteriana con diferentes concentraciones de IPTG después de alcanzar una DO 600 nm de 0.6. Concentración Carril de siembra del inductor en el gel de la (mM) figura 20 b 0,5 1,0 2,0 Fracción Cantidad Hsc 70 (g) Volumen Concentración sembrado aproximada (l) Hsc 70 (g/l) 4 Soluble 0,1 8,05 0,012 5 Insoluble 0,5 7,21 0,069 6 Soluble 0,1 5,83 0,017 7 Insoluble 0,5 6,93 0,072 8 Soluble 0,1 7,31 0,014 9 Insoluble 0,5 7,62 0,066 Figura 20. a) Expresión de la proteína recombinante Hsc70 variando el tiempo de inducción con 1 mM de IPTG. La fracción de proteínas solubles (FS) e insolubles (FI) de cada uno de los cultivos fue analizado por SDS-PAGE al 12% y Western Blot con un anticuerpo primario contra GST. En los carriles del gel y membrana se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido 2h; 5, FI inducido 2h; 6, FS inducido 4h; 7, FS inducido 4h; 8, FS inducido 6h; 9, FI inducido 6h; 10, FS inducido 8h y 11, FI inducido 8h. b) Expresión de la proteína recombinante Hsc70 variando la concentración del inductor IPTG. En los carriles del gel y membrana se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FS No inducido; 3, FI No inducido; 4, FS inducido con 0,5 mM IPTG; 5, FI inducido 0,5 mM IPTG; 6, FS inducido 1 mM IPTG; 7, FI inducido 1 mM IPTG; 8, FS inducido 2 mM IPTG y 9, FI inducido 2 mM IPTG. Teniendo en cuenta los resultados obtenidos para optimizar la expresión de la proteína Hsc70, se puede evidenciar que la mayor concentración de la proteína recombinante se obtuvo cuando el cultivo alcanzó una DO 600 nm de 0,6 y la inducción se llevo a cabo durante 4 h con 1 mM de IPTG. 6.2.4 Expresión de la proteína estructural del rotavirus VP6 La expresión de la proteína estructural VP6 del rotavirus se realizó mediante la infección de células MA104, procedentes de células epiteliales de riñón de mono verde, con un virus recombinante vaccinia que tiene la secuencia que codifica esta proteína. Las células infectadas fueron lisadas a partir del tratamiento con diferentes detergentes y sonicación. Una vez separado el pellet (proteínas adheridas a membrana) del sobrenadante (proteínas solubles) se procedió a identificar la presencia de la proteína rotaviral VP6 en cada una de las fracciones mediante SDS – PAGE y western blot con un anticuerpo policlonal contra rotavirus. Como se puede observar en la figura 21, se identificó la presencia de la proteína viral VP6 en células infectadas con el virus vaccinia recombinante con un peso molecular de 39 KDa, comparado con un control positivo de TLPs (carril 2) y un control negativo de células sin infectar que incluye una fracción soluble (carril 7), fracción solubilizada con tritón X 100 (carril 8) y fracción solubilizada con úrea 8 M (carril 9). La proteína viral VP6 se localizó en la fracción de proteínas insolubles (carril 6) de las células infectadas, posiblemente adheridas a membranas o agregaciones proteícas de VP6 producidas tras el proceso infección y la sobreexpresión de la misma, mientras que la fracción soluble (carril 5) y la fracción solubilizada con el detergente (carril 5) no presentaron la proteína rotaviral. Figura 21. Localización de la proteína viral VP6. a. En los carriles del gel de electroforesis se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, TLPs purificadas; 3, lisado de células MA104 infectadas con rotavirus RRV; 4, FS obtenida a partir de un lisado de células MA104 infectadas con el virus vaccinia recombinante; 5, FI tratada con tritón al 1% obtenida a partir de un lisado de células MA104 infectadas con el virus vaccinia recombinante; 6, Pellet remanente solubilizado con úrea 8M obtenido a partir de un lisado de células MA104 infectadas con el virus vaccinia recombinante; 7, FS obtenida a partir de un lisado de células MA104 sin infectar; 8, FI tratada con tritón al 1% obtenida a partir de un lisado de células MA104 sin infectar y 9, Pellet remanente solubilizado con úrea 8M obtenido a partir de un lisado de células MA104 sin infectar. En la parte inferior se observa la membrana correspondiente para lo cual se empleó un anticuerpo policlonal dirigido contra la partícula viral del rotavirus. 6.3 PURIFICACIÓN DE LAS PROTEÍNAS RECOMBINANTES ESTRUCTURALES VP5, VP8 Y VP6 DEL ROTAVIRUS Y LA PROTEÍNA CELULAR Hsc 70 Después de optimizar la expresión de cada una de las proteínas virales (VP5, VP6 y VP8) y la proteína celular Hsc70, se procedió a estandarizar el proceso de purificación de las proteínas. 6.3.1 Purificación de la proteína VP8-GST mediante cromatografía de afinidad. La expresión de esta proteína viral se dio en forma de cuerpos de inclusión, los cuales fueron solubilizados con úrea 8M. El uso de este agente caotrópico dificultó la separación de la proteína recombinante, puesto que su purificación por cromatografía de afinidad dependía de la estructura tridimensional del polipéptido GST unido a la proteína viral y por tanto del reconocimiento e interacción con el glutatión unido a una resina de agarosa. Teniendo en cuenta lo anterior, fue necesario emplear diferentes estrategias experimentales que permitieran la interacción de la proteína con la resina de agarosa. En primera instancia, las fracciones insolubles de los lisados bacterianos solubilizadas con úrea 8M fueron dializados frente a PBS a 4 ºC durante 24 horas con recambios del buffer cada 4 horas. Posteriormente, los lisados dializados fueron incubados con la resina, la proteína recombinante fue eluída con 20 mM de glutatión y las fracciones obtenidas fueron analizados mediante SDS – PAGE. Con este método, la proteína no fue separada en los eluídos y se mantuvo en el lisado bacteriano; es decir, no se unió a la resina. Otra metodología empleada para el replegamiento de la proteína recombínate consistió en dializar las muestras en concentraciones decrecientes del agente caotrópico en PBS, el cual fue reemplazado cada 4 horas hasta dializar con PBS en ausencia de úrea. Posteriormente, las muestras dializadas fueron tratadas con la resina y la proteína eluída con glutatión; sin embargo, no fue separada la proteína viral de la fracción insoluble del lisado bacteriano en los eluídos obtenidos. Por este motivo, se empleó una metodología publicada por Frangioni et al., en 1993, para lo cual se utilizó N-Laurilsarcosina y DTT para lisar bacterias y solubilizar los cuerpos de inclusión, como se mencionó en metodología, numeral 5.4.1. Adicionalmente, se empleó un detergente no iónico tritón, X-100 al 1 % como agente renaturante; sin embargo, la cantidad de proteína recombinante purificada fue muy poca respecto a la cantidad de proteína total identificada en el lisado bacteriano. Con el objeto de aumentar la cantidad de proteína de fusión unida a la resina, se incrementó la concentración del detergente renaturante al 3 % y los resultados fueron analizados por SDS-PAGE; sin embargo, no se incrementó la cantidad de proteína retenida significativamente (figura 22 a, carril 4). Paralelamente, se realizó el mismo tratamiento con N-laurilsarcosina y DTT a un pellet bacteriano, pero no se empleó un detergente renaturante, sino que la muestra fue diluida 10 veces en buffer PBS y después sometida a la cromatografía de afinidad. Como se observa en la figura 22 b, la cantidad de proteína recombinante recuperada y el número de fracciones eluidas (carriles 4 - 6) fue mayor respecto al ensayo anterior, teniendo en cuenta que se partió de un lisado bacteriano con la misma concentración de proteínas y el volumen de siembra en el gel fue igual en los dos tratamientos. Figura 22. Purificación de la proteína recombinante VP8 – GST mediante cromatografía de afinidad. El lisado bacteriano fue tratado con N – Sarcosina y DTT para solubilizar los cuerpos de inclusión y tratado con: (a) tritón X-100 al 3 % o (b) diluido 10 veces con PBS, las fracciones obtenidas de la cromatografía fueron analizadas por SDS-PAGE. En los carriles de los geles se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Lisado bacteriano total; 3, Lisado que no fue retenido por la resina; 4, Eluido 1; 5, Eluido 2; 6, Eluido 3; 7, Eluido 4 y 8, resina lavada después de obtener los eluidos. Los eluidos fueron obtenidos con 5 – 20 mM glutatión. 6.3.2 Purificación de la proteína VP5-GST mediante cromatografía de afinidad. Al igual que la proteína viral VP8, la proteína VP5 - GST fue localizada en la fracción de proteínas insolubles y por tanto se empleó úrea 8 M para su solubilización. A pesar de repetidos intentos por purificar la proteína recombinante mediante cromatografía de afinidad después de dializarla en PBS con el objetivo de renaturarla, no se evidenció interacción entre la proteína y la resina. Para la purificación de la proteína viral VP5 - GST, se empleó la última metodología utilizada para VP8 – GST; sin embargo, se evaluaron diferentes posibles agentes renaturantes, incluyendo: Trintón X100, Tween 20, NP40, CHAPS y octilglucósido. Las fracciones obtenidas después del corrido cromatográfico se analizaron mediante SDSPAGE y western blot. En la figura 23, se observa que de los detergentes que se utilizaron como agentes renaturantes y facilitaron la interacción de la proteína recombinante con el glutatión, fueron: CHAPS y NP40 al 1 % (carriles 6 y 15); sin embargo, la cantidad total de la proteína de fusión no fue retenida por la resina, como se evidenció en los carriles 5 y 14. Los detergentes restantes (octilglucósido, tween 20 y tritón X100) no permitieron la interacción de la proteína viral unida a GST con el glutatión presente en la resina (carriles 3, 9 y 12, respectivamente). Figura 23. Purificación de la proteína recombinante VP5 – GST mediante cromatografía de afinidad a partir de lisados bacterianos tratados con N – sarcosina y DTT para solubilizar los cuerpos de inclusión y con diferentes detergentes renaturantes. Las fracciones obtenidas de la cromatografía de afinidad fueron analizadas por SDS – PAGE y western blot. En los carriles de los geles se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Lisado bacteriano tratado con - octilglucósido al 1 % no retenido; 3, Eluido obtenido a partir del lisado tratado con - octilglucósido; 4, resina lavada después de obtener el eluido; 5, Lisado bacteriano tratado con CHAPS al 1 % no retenido; 6, Eluido obtenido a partir del lisado tratado con CHAPS; 7, resina lavada después de obtener el eluido; 8, Lisado bacteriano tratado con tween 20 al 1 % no retenido; 9, Eluido obtenido a partir del lisado tratado con tween 20; 10, resina lavada después de obtener el eluido; 11, Lisado bacteriano tratado con tritón X 100 al 1 % no retenido; 12, Eluido obtenido a partir del lisado tratado con tritón X 100; 13, resina lavada después de obtener el eluido; 14, Lisado bacteriano tratado con NP40 al 1 % no retenido; 15, Eluido obtenido a partir del lisado tratado con NP40; 16, resina lavada después de obtener el eluido. Los eluidos fueron obtenidos con 5 mM glutatión. En la parte inferior se encuentra las membranas de western blot correspondientes. Por esta razón, se evaluó una concentración mayor de cada uno de los detergentes; sin embargo, al utilizar 3 % de CHAPS en el lisado total, se observó la formación de una suspensión, que después de centrifugar y analizar por SDS-PAGE y western blot, se identificó la presencia de la proteína viral junto con otras proteínas bacterianas, el restante de proteína que quedó soluble en el lisado fue incubado con la resina, pero el proceso de purificación permitió la recuperación de una baja cantidad de proteína viral respecto a la encontrada en el lisado total, como se observó en los carriles 4 a 8 de la figura 24 a. Al utilizar NP40 al 3 % como agente renaturante, no se observó la formación de la suspensión anterior y se logró purificar una mayor cantidad de la proteína recombinante VP5 – GST en comparación con CHAPS al 3 % teniendo en cuenta que se partió del mismo lisado bacteriano (figura 24 b, carriles 4 - 8). Figura 24. Purificación de la proteína recombinante VP5 – GST mediante cromatografía de afinidad a partir de un lisado bacteriano tratado con N – sarcosina y DTT para solubilizar los cuerpos de inclusión. El lisado bacteriano fue tratado con CHAPS al 3 % (a) o con NP40 al 3 % (b), las fracciones obtenidas después de la cromatografía de afinidad fueron analizadas por SDS – PAGE. En los carriles de los geles se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Lisado bacteriano total tratado con el detergente correspondiente; 3, Lisado bacteriano no retenido por la resina; 4, Eluido 1; 5, Eluido 2; 6, Eluido 3; 7, Eluido 4; 8, Eluido 5; 9, resina lavada después de obtener los eluidos. Los eluidos fueron obtenidos con 5 – 20 mM glutatión. 6.3.3 Purificación de la proteína recombinante Hsc 70 mediante cromatografía de afinidad. La proteína recombinante Hsc 70 tiene una cola de histidinas unida en la región amino terminal de la proteína, lo cual fue útil en el proceso de purificación puesto que al emplear una resina de sefarosa cargada con Ni2+, interacciona con los nitrógenos presentes en los anillos imidazólicos de los residuos de histidina permitiendo su purificación. En el proceso de estandarización de la expresión, la proteína recombinante Hsc70 fue identificada tanto en la fracción soluble como en la insoluble. Por tanto, el pellet de células fue tratado de la misma manera que las bacterias que expresaron las proteínas recombinantes VP5 y VP8. Es importante resaltar, que las bacterias que expresaron las proteínas rotavirales fueron tratadas con el buffer STE, N-sarcosina y DTT con el objeto de solubilizar los cuerpos de inclusión para el proceso de purificación, el cual contiene EDTA en su composición; mientras que las células que expresaron la proteína recombinante Hsc70 fueron resuspendidas en buffer PBS, N-sarcosina y DTT en ausencia de EDTA, puesto que esta molécula puede quelar el Ni2+ de la resina y evitar la purificación de la proteína recombinante. Después de la lisis celular y la solubilización de los posibles cuerpos de inclusión formados durante la expresión de la proteína, el lisado celular fue tratado con Tritón X100 al 1 % como agente renaturante y para eliminar interacciones inespecíficas de otras proteínas bacterianas con la resina. Por último, la elución de la proteína se realizó con concentraciones crecientes de imidazol y las fracciones obtenidas fueron analizadas por SDS-PAGE. Teniendo en cuenta los resultados obtenidos en la figura 25, la mayor cantidad de la proteína Hsc70 presente en el lisado bacteriano total (carril 2) fue retenida por la resina puesto que la banda correspondiente a esta proteína fue visiblemente reducida en el lisado bacteriano no retenido (carril 3). Por otra parte, se logró eluir la proteína celular con concentraciones crecientes de imidazol (carriles 7 - 12); sin embargo, los eluidos correspondiente a los carriles 8, 9 y 10 presentaron bandas inespecíficas de alto y bajo peso molecular. Figura 25. Purificación de la proteína Hsc 70 mediante cromatografía de afinidad para lo cual se empleó una resina de sefarosa unida a Ni2+. Las fracciones obtenidas de la cromatografía de afinidad fueron analizadas por SDS – PAGE. En los carriles del gel se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Lisado bacteriano total; 3, Lisado bacteriano no retenido por la resina; 4, Lavado con 10 mM de Imidazol en PBS antes de realizar la elución; 5, Eluido 1; 6, Eluido 2; 7; Eluido 3; 8, Eluido 4; 9, Eluido 5; 10, Eluido 6; 11, Eluido 7; 12, Eluido 8. Los eluidos 1 a 8 fueron obtenidos con 50 mM de Imidazol en PBS. 6.3.4 Purificación de la proteína recombinante VP6 mediante tratamiento con detergentes. Como se describió en metodología numeral 5.4.4, el pellet obtenido después de la lisis celular fue tratado con N-Laurilsarcosina, Deoxicolato de Sodio, Octilglucósido, Tween 20, Tritón X100, CHAPS al 1 %, úrea 5 M o con buffer RIPA (el cual contiene 0.1 % de SDS y 1 % de Tritón X 100) a 4 ºC con el objeto de identificar cuál de ellos permitía la solubilización total de la proteína viral. Sin embargo, solo los detergentes denaturantes (NLaurilsarcosina y Deoxicolato de Sodio) y úrea 5 M lograron solubilizar parte de la proteína viral (figura 26 a, carril 2, 4 y 10); mientras que los detergentes no iónicos, zwiteriónicos y el buffer RIPA (el cual contiene 0.1 % de SDS y 1 % de Tritón X 100) no la solubilizaron (figura 26 a, carril 3, 5, 6, 7, 8 y 9); sin embargo, se evidenció la solubilización de proteínas celulares, lo cual fue útil para la purificación de VP6 puesto que eliminó contaminantes celular, lo que facilita la recuperación de la proteína recombinante. En todos los ensayos realizados se obtuvo un pellet que se mantenía insoluble después de los tratamientos realizados con los detergentes, la úrea o el buffer RIPA, el cual fue completamente solubilizado con úrea 8 M y fueron analizados por SDS – PAGE y western blot. Como se observa en la figura 26 b, estas fracciones proteícas presentaron la proteína VP6 parcialmente purificada que corresponde a la banda de 39 KDa identificada con un anticuerpo dirigido contra rotavirus. Cabe resaltar, que el tratamiento del pellet con octilglucósido al 1 % permitió la solubilización de proteínas de alto peso molecular a diferencia de otros detergentes, mientras que VP6 permaneció en el pellet remanente solubilizado con úrea (figura 26 a y b, carril 6). Adicionalmente, se observó que los carriles 2, 4 y 10 presentaron una cantidad menor de la proteína viral respecto a los otros carriles, puesto que parte de la proteína fue solubilizada en el tratamiento inicial con N – laurilsarcosina, DOC y úrea, debido a que son altamente denaturantes a las concentraciones empleadas. Figura 26. Solubilización de la proteína viral VP6 posiblemente precipitada intracelularmente. (a) Tratamiento de los pellets obtenidos después de la lisis de células MA104 infectadas con el virus vaccinia recombinante con diferentes detergentes al 1 %. Las proteínas solubilizadas por cada uno de los detergentes fueron sometidas a SDS – PAGE y la presencia de la proteína viral VP6 fue identificada por western blot con un anticuerpo policlonal dirigido contra rotavirus. En los carriles del gel y membrana se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FI tratada con úrea 5 M; 3, FI tratada con buffer RIPA; 4, FI tratada con N - laurilsarcosina; 5, FI tratada con CHAPS; 6, FI tratada con - octilglucósido; 7, FI tratada con tritón X 100; 8, FI tratada con tween 20; 9, FI tratada con NP40 y 10, FI tratada con DOC. (b) Solubilización con úrea 8 M de los pellet obtenidos a partir del tratamiento anterior. De acuerdo con los resultados obtenidos en los ensayos anteriores, se estableció un protocolo para la purificación parcial de la proteína VP6, como se observa en la figura 27, que consistió en solubilizar la mayor cantidad de proteínas celulares en el tratamiento inicial con buffer RIPA (carril 3 y 4) y posteriormente con - octilglucósido al 1 % (carril 6). Una vez retiradas las proteínas que no eran de interés, VP6 fue solubilizada con úrea 8M y N – sarcosina al 1 % (carriles 7 y 8, respectivamente). Figura 27. Purificación parcial de la proteína viral VP6 mediante el tratamiento del pellet obtenido después de la lisis de células MA104 infectadas con el virus vaccinia recombinante con diferentes detergentes de manera secuencial. La fracción de proteínas insolubles fue tratada con buffer RIPA, octilglucósido y úrea 8 M, secuencialmente. De cada uno de los tratamientos se obtuvieron diferentes fracciones con proteínas solubilizadas, las cuales fueron analizadas por SDS – PAGE. En los carriles del gel se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, FI solubilizada con úrea 8 M; 3, FS precipitada con acetona fría; 4, FI tratada con RIPA a 4 ºC durante 4 horas; 5, Pellet remanente del procedimiento anterior nuevamente tratado con RIPA a 4 º C durante 4 h; 6, pellet remanente del procedimiento anterior tratado con - octilglucósido al 1 %; 7, pellet remanente del tratamiento anterior solubilizado con úrea 8 M y 8, pellet remanente del tratamiento anterior solubilizado con úrea 8 M en presencia de N – sarcosina al 1 %. Con el objeto de disminuir la concentración de los agentes denaturantes y emplear las proteínas en los ensayos de interacción en condiciones acelulares y celulares, las fracciones que contenían la proteína viral parcialmente purificada fueron diluidas en PBS y concentradas mediante un amicon ultra – 4 (Millipore). 6.4 OBTENCIÓN DE ANTICUERPOS POLICLONALES CONTRA LAS PROTEÍNAS RECOMBINANTES ESTRUCTURALES VP5, VP8 Y VP6 DEL ROTAVIRUS EN CONEJOS En el caso de la proteína de fusión VP8 – GST, los pellets obtenidos después de la lisis celular fueron tratados con concentraciones crecientes de úrea (1 M – 8 M) con el objeto de separar y solubilizar los cuerpos de inclusión. Las muestras solubles en úrea 8 M fueron corridas en geles preparativos y la banda correspondiente a la proteína recombinante, identificada por western blot con un anticuerpo policlonal contra GST (figura 28 a), fue cortada y electroeluída como se mencionó en el numeral 5.5.1 de metodología. Para la proteína VP5 – GST, se partió de la fracción insoluble tratada con úrea 8M (figura 28 b). Figura 28. Preparación de los antígenos de las proteínas virales para la generación de anticuerpos policlonales en conejos. (a) Gel preparativo en el cual se corrieron las fracciones correspondientes a cuerpos de inclusión formados durante la expresión de la proteína viral VP8 – GST, extraídos y solubilizados con concentraciones crecientes de úrea. (b) Gel preparativo en el cual se corrió la fracción insoluble de los lisados bacterianos que expresaron la proteína viral VP5 – GST. En el extremo derecho de los geles se encuentran las membranas correspondientes en las cuales se localizó la proteína viral para cortar la banda de interés con un anticuerpo policlonal contra GST. Las fracciones electroeluídas fueron concentradas con acetona fría y el pellet obtenido fue disuelto en buffer de digestión que contenía calcio para ser cortadas con el kit THROMBIN CleanCleave (Sigma) (figura 29) para retirar el polipéptido GST que se encontraba unido en sus dominios amino terminal. Por último, la fracción que contenía los polipéptidos cortados fue separada en geles preparativos y la banda correspondiente a la proteína viral fue cortada e inoculada en los conejos. Figura 29. Digestión con trombina del polipéptido GST de las proteínas virales VP8 y VP5 obtenidas a partir de las bandas identificadas en los geles preparativos, electroeluidas y precipitadas con acetona. (a) 2 g de la proteína viral VP8 – GST fueron digeridos con trombina y las muestras obtenidas fueron separadas mediante SDS – PAGE, la proteína viral fue identificada con un anticuerpo policlonal dirigido contra un péptido de VP8. En los carriles del gel y la membrana correspondiente se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Proteína VP8 – GST sin digerir; 3, Proteína VP8 – GST digerida durante 2 h; 4, Proteína VP8 – GST digerida durante 4 h y 5, Proteína VP8 – GST digerida durante 8 h. Las incubaciones fueron realizadas a temperatura ambiente. (b) 500 ng de la proteína viral VP5 – GST fueron digeridos con trombina y las muestras fueron separadas por SDS – PAGE. En los carriles del gel se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Proteína VP5 – GST sin digerir y 3, Proteína VP5 – GST digerida durante 4 h a temperatura ambiente. La proteína viral VP6 fue purificada parcialmente como se mencionó anteriormente y separada en geles preparativos. La banda correspondiente a esta proteína fue identificada por western blot y utilizada como antígeno para la inoculación en conejos. Los conejos fueron inoculados con aproximadamente 750 g de cada una de las proteínas virales administradas en tres dosis. Finalizado el tiempo de inmunización, los conejos fueron desangrados y los sueros hiperinmunes fueron caracterizados por western blot, inmunocitoquímica y ELISA, para lo cual se empleó como antígeno TLPs purificadas, las proteínas recombinantes purificadas, lisados bacterianos y celulares que expresaron las proteínas recombinantes. En la figura 30 se puede observar la caracterización de los anticuerpos polilconales obtenidos contra las proteínas virales mediante western blot y ELISA indirecto utilizando concentraciones crecientes de las proteínas recombinantes. Para el caso del anticuerpo policlonal dirigido contra la proteína viral VP5, se puede evidenciar que el anticuerpo permitió el reconocimiento mediante western blot de una banda de aproximadamente 75 KDa en los lisados bacterianos en todas las concentraciones proteicas utilizadas en este ensayo (figura 30 a); sin embargo, se observó el reconocimiento inespecífico de varias bandas adicionales correspondientes a proteínas del lisado bacteriano que no expresaba la proteína viral (figura 30 a, carril 5). Así mismo, se determinó que concentraciones mayores a 62,5 ng de la proteína VP5 purificada por cromatografía pueden ser identificadas con este anticuerpo en una dilución 1:1000 (figura 30 d); sin embargo, se obtuvo un mayor reconocimiento a 500 y 250 ng de la proteína (figura 30 d, carriles 3 y 4). También se observó una banda de menor peso molecular, posiblemente un producto de degradación de la proteína recombinante o a un reconocimiento inespecífico del mismo. Adicionalmente, no se evidenció reconocimiento inespecífico por BSA (figura 30 d, carril 2). En el caso del anticuerpo policlonal obtenido contra la proteína viral VP6 se puede evidenciar el reconocimiento de la proteína mediante western blot en todas las concentraciones proteícas de los lisados celulares infectados (figura 30 b). Adicionalmente, se observa una banda de menor peso molecular, la cual puede corresponder a un producto de degradación o del proceso de síntesis de la proteína viral; teniendo en cuenta que el control del lisado celular sin infectar no mostró la aparición de ninguna banda apreciable empleando como método de revelado carbazol (figura 30 b, carril 5). Así mismo, el anticuerpo dirigido contra VP6 reconoció todas las concentraciones evaluadas de la proteína viral y no presentó reconocimiento por BSA (figura 30 e). Por otra parte, el anticuerpo policlonal obtenido contra la proteína viral VP8 reconoció la presencia de la proteína recombinante VP8 – GST tanto en los lisados bacterianos como la proteína purificada mediante western blot. En los lisados bacterianos no se observó reconocimiento inespecífico de proteínas bacterianas puesto que no se identificó ninguna banda en el lisado bacteriano control (figura 30 c). Así mismo, el anticuerpo reconoció todas las concentraciones analizadas de la proteína purificada por cromatografía de afinidad; sin embargo, se observó un mayor reconocimiento cuando se utilizaron 500 250 ng de la proteína viral (figura 30 f, carril 3 y 4). Adicionalmente, el anticuerpo no identificó inespecíficamente la proteína BSA (figura 30 f, carril 2). Otra de las técnicas utilizadas para la caracterización de los anticuerpos policlonales obtenidos contra las proteínas recombinantes estructurales del rotavirus, fue el ensayo de ELISA. Para esto, diferentes cantidades de las proteínas virales fueron incubadas en la placa en un volumen final de 50 l, la detección de la proteína se llevó a cabo con una dilución 1:1000 de los anticuerpos policlonales contra cada una de las proteínas respectivas, un anticuerpo secundario conjugado a peroxidasa y revelado con OPD. Teniendo en cuenta los resultados obtenidos en la figura 30 g, h e i, se puede observar el incremento en la DO al aumentar la cantidad de proteína recombinante en cada uno de los pozos. El reconocimiento de la proteína recombinante VP5 tuvo un valor de DO superior a 0,2, cuando se emplearon 800 ng de la proteína viral (figura 30 g). En el caso de los anticuerpos policlonales dirigidos contra las proteínas recombinantes VP6 (figura 30 h) y VP8 (figura 30 i), se obtuvo una DO superior a 0.2 cuando se emplearon cantidades mayores a 500 ng de cada una de las proteínas virales. Con el objeto de identificar la dilución límite a la cual se podían utilizar cada uno de los anticuerpos policlonales dirigidos contra las proteínas virales mediante western blot, fueron corridas 250 ng de las proteínas recombinantes purificadas mediante cromatografía de afinidad o tratamiento con detergentes y transferidas a membranas de PVDF. La identificación de las proteínas recombinantes se realizó con diluciones de los anticuerpos entre 1:500 a 1:3000, obteniéndose un reconocimiento por cada una de las proteínas virales con el anticuerpo correspondiente, aún con las diluciones mayores (1:3000), como se observa en la figura 31, carril 2 de a, b y c. De la misma manera, se evaluó el reconocimiento de las proteínas virales en las TLPs tratadas con tripsina, por los anticuerpos policlonales obtenidos en este trabajo, anti-VP5, anti-VP6 y anti-VP8 de conejo. Para esto, se emplearon 500 ng de las partículas virales y se analizó el reconocimiento de cada una de las proteínas con diferentes diluciones de los anticuerpos, entre 1:250 a 1:1000 comparado con un anti – rotavirus policlonal que reconoce las proteínas estructurales del virus generado en el laboratorio. Figura 30. Caracterización de los anticuerpos policlonales contra las proteínas recombinantes VP5, VP6 y VP8 obtenidos en conejos Nueva Zelanda utilizando diferentes cantidades de las proteínas recombinantes. Diferentes cantidades proteícas de lisados bacterianos que expresaron la proteína viral recombinante o lisados celulares infectados con el virus vaccinia recombinante fueron separados en geles SDS – PAGE y transferidos a una membrana de PVDF. En los carriles de las membranas se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, 100 g; 3, 50 g; 4, 25 g de proteína total del lisado que expresa la proteína viral y 5, 100 g de proteína total de un lisado control que no expresaba la proteína viral correspondiente. La presencia de las proteínas virales fue identificada con los anticuerpos policlonales correspondientes. (a) anti – VP5, (b) anti – VP6 y (c) anti – VP8. Diferentes cantidades de las proteínas virales purificadas fueron corridas en geles SDS – PAGE y transferidas a una membrana de PVDF. En los carriles de las membranas se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, 500 ng de BSA; 3, 500 ng; 4, 250 ng; 5, 125 ng y 6, 62,5 ng de las proteínas virales. La presencia de las proteínas virales fue identificada con los anticuerpos policlonales correspondientes. (c) anti – VP5, (d) anti – VP6 y (f) anti – VP8. Por último, concentraciones crecientes de cada una de las proteínas recombinantes fueron adheridas a una placa de ELISA y fueron identificadas con el anticuerpo policlonal dirigido contra la proteína viral correspondiente. (g) anti – VP5, (h) anti – VP6 y (i) anti – VP8 y un anticuerpo secundario conjugado a peroxidasa. Los anticuerpos policlonales dirigidos contra las proteínas virales recombinantes fueron utilizados en una dilución 1:1000 en todos los ensayos y la concentración de proteína total fue determina mediante la cuantificación a 280 nm en relación a una curva de BSA. Como se puede observar en la figura 31 d, e y f, los anticuerpos obtenidos contras las proteínas recombinantes del rotavirus permitieron el reconocimiento de todas las proteínas virales analizadas (VP5, VP6 y VP8) procedentes de las TLPs en una dilución 1:250 y 1:500 mediante western blot, revelado con un anticuerpo secundario conjugado con peroxidasa y carbazol (carriles 4 y 5); sin embargo, la dilución de 1:1000 de los anticuerpos no permitió el reconocimiento de estas proteínas (carril 6). En comparación con el anti – rotavirus, los anticuerpos dirigidos contra las proteínas virales recombinantes presentaron un mejor reconocimiento por las proteínas virales estructurales de la partícula viral, como se puede observar en la figura 31 d, e y f, carriles 1, 2 y 3. Finalmente, se llevó a cabo el reconocimiento de las proteínas virales estructurales del rotavirus mediante inmunocitoquímica. Para esto, células MA104 fueron cultivadas en cajas de 96 pozos hasta alcanzar un confluencia del 100 % y fueron infectadas con TLPS de RRV e incubadas a 37 ºC durante 12 h. Una vez fijadas las células, se incubaron concentraciones crecientes de los anticuerpos policlonales dirigidos contra las proteínas recombinantes VP5, VP6 y VP8 obtenidos en este trabajo (figura 31 g, h e i). En el caso del anticuerpo policlonal dirigido contra la proteína recombinante VP5 se evidenció el reconocimiento de esta proteína en células infectadas con RRV aún con la mayor dilución empleada (1:4000) con un promedio de 6,34 x 107 uff/ml (figura 31 g). De la misma manera, el anticuerpo policlonal dirigido contra la proteína viral VP6 reconoció esta proteína en células MA104 infectadas con rotavirus en todas las concentraciones evaluadas, obteniéndose un promedio de 4,49 x 107 uff/ml con una dilución de 1:2000 (figura 29 h). Por último, el anticuerpo policlonal dirigido contra VP8 permitió la identificación de esta proteína viral en todas las diluciones empleadas del anticuerpo, identificándose 5,56 x 107 uff/ml con una dilución de 1:1000 (figura 31 i). Figura 31. Identificación de las proteínas virales recombinantes y en TLPs con diferentes diluciones de los anticuerpos policlonales dirigidos contra estas proteínas. 250 ng de las proteínas virales recombinantes fueron corridas mediante SDS – PAGE y transferidas a membranas de PVDF. En los carriles de las membranas se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, anticuerpo policlonal contra la proteína viral (a) VP5 – GST, (b) VP6 o (c) VP8 – GST en una dilución 1:3000; 3, 1:2000; 4, 1:1000; 5, 1:500. 500 ng de TLPs fueron separadas mediante SDS – PAGE y transferidos a membranas de PDVF. En los carriles de las membranas se observa de izquierda a derecha: 1, anti - rotavirus en una dilución 1:250; 2, 1:500; 3, 1:1000; 4, anticuerpo contra la proteína viral (d) VP5, (e) VP6 o (f) VP8 en una dilución 1:250; 5, 1:500; 6, 1:1000. Células MA104 fueron infectadas con 1.8 x 108 uff/ml de TLPs de RRV purificadas en una dilución 1:3200 e incubadas durante 12 h a 37 ºC. El reconocimiento de las proteínas estructurales en las partículas virales se realizó con diluciones entre 1:250 a 1:4000 de los anticuerpos policlonales contras (g) VP5, (h) VP6 y (i) VP8. 6.5 INTERACCIÓN DE LAS PROTEÍNAS RECOMBINANTES PURIFICADAS VP5, VP8 Y VP6 CON LAS PROTEÍNAS CELULARES Hsc 70 Y PDI 6.5.1 Interacción de las proteínas celulares PDI y Hsc70 con las proteínas recombinantes VP5, VP6 y VP8 en condiciones acelulares. Para determinar la posible interacción de las proteínas celulares Hsc 70 y PDI con cada una de las proteínas recombinantes VP5, VP6 o VP8 en condiciones acelulares se realizaron diferentes ensayos, entre ellos: ELISA indirecto y co-inmunoprecipitación. Las fracciones de las proteínas purificadas utilizadas para estos ensayos fueron separadas en geles SDS – PAGE con el objetivo de determinar la concentración aproximada de las mismas frente a una curva de BSA. Como se muestra en la figura 32, la banda correspondiente a la proteína viral VP8 (carril 6) tuvo una intensidad similar a la banda de 500 ng de albúmina (carril 2), por tanto la concentración calculada de la proteína viral purificada con relación al volumen sembrado (10 l) fue de 50 g/ml. La concentración calculada de la proteína viral VP5 (carril 7) y VP6 (carril 9) fue de 25 g/ml y para las proteínas celulares Hsc70 y PDI fue de 200 g/ml (carril 10 y 11, respectivamente). La proteína PDI utilizada en estos ensayos fue expresada en bacterias E. coli de la cepa BL21(DE3) con el vector pET28a y purificada mediante cromatografía de interacción hidrofóbica con la resina t – butil (BioRad). Figura 32. Cuantificación por SDS – PAGE de las proteínas virales (VP5, VP6 y VP8) y las proteína celulares Hsc70 y PDI frente a una curva de BSA teñido con azul de coomassie. En los carriles del gel se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, 500 ng de BSA; 3, 250 ng de BSA; 4, 125 ng de BSA; 5, 62,5 ng de BSA; 6, 500 ng / 10 l de VP8 - GST; 7, 250 ng / 10 l de VP5 - GST; 8, 62,5 ng / 10 l de VP6; 9, 250 ng / 10 l de VP6; 10, 2 g / 10 l de Hsc70 y 11, 2 g / 10 l de PDI. Para el ensayo de ELISA, se adhirió a cada pozo de la placa 40 ng/l de la proteína celular (Hsc70 o PDI) que se incubaron con concentraciones crecientes de las proteínas virales purificadas. En el caso de la proteína viral VP5, se evidenció que cuando se utilizaron 1 ng/l (50 ng) de la proteína recombinante se registró un DO de 0,159; con 2 ng/l (100 ng), un DO de 0,180; con 4 ng/l (200 ng), un DO de 0,294 y con 8 ng/l ng (400 ng), un DO de 0,451(figura 33 a). Para la proteína viral VP6, se observó que cuando se emplearon concentraciones de 0,078 ng/l (3,90 ng) de la proteína viral se registró un DO de 0,0265; con 0,156 ng/l (7,80 ng), un DO de 0,1595; con 0,312 ng/l (15,62 ng), un DO de 0,4035; con 0,625 ng/l (31,25 ng), un DO de 0,5955 y con 1,25 ng/l (62,50 ng) se obtuvo un DO de 1,0415 (figura 33 c). Los DO graficados fueron obtenidos luego de restar las DO de los ensayos de interacción con los controles de cada placa. Estos resultados sugieren que las proteínas virales VP5 y VP6 interaccionan con la proteína celular Hsc70 en condiciones acelulares. Respecto a la proteína VP8, el pozo que contenía la mayor concentración de la proteína viral y la proteína recombinante Hsc70 se registró un valor de DO similar al obtenido en uno de los pozos control, en el cual se incubó la proteína viral, el anticuerpo primario y el anticuerpo secundario conjugado a peroxidasa en ausencia de la proteína celular que fue reemplazada por BSA al 1 %. Este resultado sugiere que VP8 no interacciona con Hsc70 en las condiciones evaluadas y que el incremento en la DO se debe probablemente a la interacción de la proteína viral con BSA (figura 33 e). De la misma manera, se evaluó la interacción de las proteínas virales con la proteína celular PDI mediante un ensayo de ELISA indirecto. Para este ensayo, 40 ng/l de PDI fueron adheridos a la placa e incubada con diferentes concentraciones de las proteínas virales. La detección de la interacción se llevó a cabo con un anticuerpo policlonal que reconocía las proteínas virales, un anticuerpo secundario conjugado a peroxidasa y revelado con OPD. Como se observó en la figura 33 b y d, hay un aumento en el DO cuando se incrementan las cantidades de las proteínas virales VP5 y VP6, cuando la placa había sido cubierta con PDI. En el caso de la proteína viral VP5, cuando se utilizó 1,25 ng/l (62,5 ng) se registró un valor del DO de 0,084; con 2,5 ng/l (125 ng), un DO de 0,100; con 5 ng/l (250 ng), un DO de 0,122 y con 10 ng/l (500 ng) se obtuvo un DO de 0,184 (figura 33 b). Para la proteína viral VP6, cuando se empleó 0,156 ng/l (7,80 ng) de la proteína se registró un DO de 0,029; con 0,312 ng/l (15,62 ng), un DO de 0,058; con 0,625 ng/l (31,25 ng), un DO de 0,100; con 1,25 ng/l (62,50 ng), un DO de 0,215 y con 2,5 ng/l (125 ng), un DO de 0,210 (figura 33 d). Estos resultados sugieren que existe una interacción directa entre las proteínas virales recombinantes y PDI; sin embargo, teniendo en cuenta que los valores de DO son inferiores a 0,2 se realizó otro ensayo que permitió verificar esta interacción, como se mencionará más adelante en este mismo ítem. En el caso de la proteína viral VP8, se obtuvo el mismo resultado que el registrado con la proteína celular Hsc70. Como se puede observa en la figura 33 f, el control que presentaba la proteína viral, el anticuerpo primario contra VP8 y el anticuerpo secundario conjungado a peroxidasa en ausencia de PDI y reemplazada por 1 % de BSA, registró un valor de DO similar al pozo en donde se pretendía evaluar la posible interacción de la proteína viral con la proteína celular, mostrando que el incremento de la DO era dependiente del reconocimiento de la proteína viral unida a BSA y no era dependiente de una posible interacción con PDI (figura 33 f). Este resultado sugiere que VP8 no interacciona con la proteína celular PDI. Con el objetivo de confirmar los resultados obtenidos en los ensayos de ELISA indirecto, se procedió a realizar un ensayo de co-inmunoprecipitación que permitió corroborar la interacción entre las proteínas virales recombinantes VP5, VP6 y VP8 con las proteínas recombinantes, Hsc70 y PDI purificadas. Para este ensayo se incubaron 10 ng/l de las proteínas celulares con 2,5 ng/l de la proteína viral VP6 o 10 ng/l de las proteínas VP5 y VP8 en ensayos independientes. A la mezcla proteíca se le adicionó un anticuerpo dirigido contra la proteína celular y se inmunoprecipitó, las fracciones obtenidas fueron separadas mediante SDS – PAGE, transferidas a membranas de PVDF y la presencia de la proteína viral se identificó con el anticuerpo policlonal correspondiente. Como se evidencia en la figura 34 a y b, las proteínas virales VP5 y VP6 fueron co – inmunoprecipitadas con las proteínas celulares Hsc70 y PDI (carriles 3 y 4) en el western blot; sin embargo, VP8 no se pudo identificar puesto que tiene un peso molecular similar a la de la cadena pesada del anticuerpo empleado para la inmunoprecipitación de las proteínas celulares y el anticuerpo secundario reconoce esta cadena, por tanto no se puede asegurar que VP8 no interacciona con Hsc70 o PDI mediante este ensayo (figura 34 c, carriles 3 y 4). Así mismo, se evaluó la interacción de cada una de las proteínas virales y del anticuerpo dirigido contra las proteínas celulares con la resina en ausencia de Hsc70 o PDI, como controles del ensayo, a fin de evidenciar si la proteína viral se adhería a la resina sin necesidad de la proteína celular. Se observó que ninguna de las proteínas virales se unió por sí sola a la resina que estaba bloqueada con BSA al 4 % antes de realizar el ensayo de inmunoprecipitación (figura 34 a, b y c, carriles 5 y 6). Figura 33. Determinación de la interacción de las proteínas estructurales del rotavirus con las proteínas celulares Hsc70 y PDI mediante el ensayo de ELISA indirecto en condiciones acelulares. 40 ng/l de la proteínas celulares fueron adheridos a la placa de ELISA de manera independiente y se incubaron con diferentes concentraciones de las proteínas virales (a) VP5 – GST y Hsc70, (b) VP5 – GST y PDI, (c) VP6 y Hsc70, (d) VP6 y PDI, (e) VP8 – GST y Hsc 70 y (f) VP8 – GST y PDI. Se emplearon como controles, pozos de la placa en los cuales se realizaron las incubaciones correspondientes en ausencia de alguno de los componentes (proteína viral, proteína celular o anticuerpo primario) y se reemplazó por 1 % de albúmina. En las figuras a - d se tabuló el DO, el cual resulta de la diferencia de la absorbancia obtenida para los experimentos y la absorbancia de los controles negativos; mientras que en la figura e y f se tabularon los datos de absorbancia de los controles negativos y de los experimentos (* en ausencia de anti – GST, ** en ausencia de VP8 – GST, *** en ausencia de Hsc70 o PDI). Adicionalmente, se analizó al interacción de las proteínas virales con las proteínas celulares, invirtiendo el procedimiento anterior, para lo cual la inmunoprecipitó se relizó con un anticuerpo dirigido contra la proteína viral y en el western blot se identificó la proteína celular. Como se observa en la figura 34 d, la presencia de Hsc70 fue identificada cuando se inmunoprecipitó la proteína viral VP5 y VP6 (carriles 4 y 6), pero no con la proteína VP8 (carriles 8). Así mismo, se evaluó la posible interacción de Hsc70 con la resina en ausencia de las proteínas virales, como control del ensayo, y no se identificó la proteína celular en la membrana (carriles 3, 5 y 7). Estos resultados confirman la interacción de la proteína celular, Hsc70, con las proteínas virales VP5 y VP6, pero no con VP8. De la misma manera, se evaluó la interacción de las proteínas virales con PDI, para lo cual la mezcla se inmunoprecipitó utilizando un anticuerpo dirigido contra la proteína celular y en el western blot se identificó la proteína viral. Sin embargo, no fue posible identificar la proteína celular PDI puesto que tiene un peso muy cercano a la de la cadena pesada del anticuerpo usado en la inmunoprecipitación. Figura 34. Identificación de la interacción de las proteínas estructurales del rotavirus con las proteínas celulares Hsc70 y PDI mediante co – inmunoprecipitación. 2,5 ng/l de la proteína viral VP6 o 10 ng/l de VP5 o VP8 fueron incubadas con 10 ng/l de las proteínas celulares de manera independiente y fueron coinmunoprecipitadas con un anticuerpo policlonal dirigido contra las proteínas celulares o contra las proteínas virales. Co-inmunoprecipitación de Hsc70 o PDI e identificación en la membrana de (a) VP5 – GST, (b) VP6 y (c) VP8 - GST. En los carriles de las membranas se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Proteína viral; 3, Co – inmunoprecipitación de Hsc70 e identificación de la proteína viral; 4, Co – inmunoprecipitación de PDI e identificación de la proteína viral; 5, control negativo en ausencia de Hsc70 y carril 6, control negativo en ausencia de PDI. (d) Co-inmunoprecipitación de las proteínas virales e identificación en la membrana de Hsc70. En los carriles de las membranas se observa de izquierda a derecha: 1, Marcador de peso molecular; 2, Hsc70; 3, control negativo en ausencia de VP5 – GST; 4, Co – inmunoprecipitación de VP5 – GST; 5, control negativo en ausencia de VP6; 6, Co – inmunoprecipitación de VP6; 7, control negativo en ausencia de VP8 – GST y 8, Co – inmunoprecipitación de VP8 – GST. Los westerns blot fueron revelados con un anticuerpo secundario conjugado a fosfatasa alcalina. 6.5.2 Interacción de la proteína celular Hsc70 con las proteínas virales VP5, VP6 y VP8 en condiciones celulares. Aproximadamente 9.2 x 106 células MA104 fueron incubadas con 15 ng/l de VP5, VP8 o 1,5 ng/l de VP6, de manera independiente. Las células fueron lisadas a 4 ºC en un buffer que contenía tritón X 100 al 1% con el objetivo de no disgregar fracciones de membrana resistentes a detergentes o rafts lipídicos. Los lisados celulares fueron ultracentrifugados en un gradiente de sacarosa y las fracciones obtenidas fueron tratadas con ciclodextrina y octilglucósido, para desintegrar los microdominios lipídos y de esta manera poder determinar interacciones proteína – proteína sin interferencia de los lípidos presentes en los rafts 193. Posteriormente, las fracciones fueron inmunoprecipitadas con un anticuerpo policlonal dirigido contra Hsc70, separadas mediante SDS – PAGE y transferidas a membranas de PVDF. La presencia de las proteínas virales fue identificada en el western blot con el anticuerpo dirigido contra cada una de las proteínas virales. Como control se emplearon células MA104 tratadas similarmente sin incubar con las proteínas virales recombinantes. Esto se hizo para identificar la presencia de las proteínas Hsc70, PDI e integrina 3 en los rafts, las cuales fueron detectadas mediante western blot (figura 35 a). En la figura 35 b y c se observó que las proteínas virales VP5 y VP6 fueron co – inmunoprecipitadas con la proteína celular Hsc70. Para el caso de la proteína viral VP5, fue identificada en las siete primeras fracciones del gradiente de sacarosa; mientras que la proteína viral VP6 se localizó en la fracciones 2 a 6. Estos resultados muestran que una fracción de las proteína virales VP5 y VP6 se localizaron en los rafts unidas directa o indirectamente con la proteína celular Hsc70, teniendo en cuenta resultados previos del laboratorio que han determinado que Hsc70 forma un complejo proteico con la integrina 3 y PDI en estos microdominios lipídicos y se encuentra involucrado en los eventos iniciales del proceso infeccioso del rotavirus 197. En el caso de la proteína viral VP8 resultó difícil su identificación teniendo en cuenta que su peso molecular coincide con el peso de la cadena pesada del anticuerpo empleado para la inmunoprecipitación (figura 35 d). Las fracciones recuperadas de células MA104, utilizadas como control, fueron inmunoprecipitadas con anti-Hsc70, separadas mediante SDS – PAGE, transferidas a una membrana de PVDF y tratadas con una mezcla de los anticuerpos policlonales dirigidos contra las proteínas virales con el objetivo de identificar si existía un reconocimiento inespecífico por las proteínas celulares. Teniendo en cuenta los resultados obtenidos en este ensayo se descartó la posible inespecificidad (figura 35 e). Figura 35. a) Identificación de la presencia de las proteínas celulares Hsc70, Integrina 3 y PDI en rafts lipídicos. Células MA104 fueron lisadas con buffer tritón a 4 ºC y ultracentrifugadas a 38000 xg durante 8 h a 4 ºC a fin de separar fracciones de membranas resistentes a detergentes. Cada una de las fracciones fueron separadas en geles SDS – PAGE y transferidas a membranas de PVDF y se identificó la presencia de las proteínas celulares con anticuerpos policlonales específicos. b) Identificación de la interacción de las proteínas estructurales del rotavirus en condiciones celulares. Células MA104 fueron incubadas con 15 ng/l de las proteínas virales VP5 – GST, VP8 – GST o 1,5 ng/l de VP6 y los rafts lipídicos fueron separados. Las fracciones fueron inmunoprecipitadas con 2 ng/l de un anticuerpo policlonal dirigido contra Hsc70 y la presencia de las proteínas virales fue identificada con el anticuerpo policlonal correspondiente (b) VP5 – GST, (c) VP6, (d) VP8 – GST y (e) Control de células en ausencia de las proteínas virales e inmunoprecipitadas con anti – Hsc70. 6.5.3 Inhibición de la infección con RRV de células MA104 con las proteínas recombinantes estructurales del rotavirus. Resultados previos publicados en el laboratorio permitieron demostrar que al incubar las células MA104 con DLPs, el proceso infeccioso de las TLPs es inhibido, sugiriendo que la proteína estructural más abundante de esta partícula (VP6) puede estar involucrada en el paso de penetración del virus a la células 84. Con el objetivo de confirmar este evento, células MA104 fueron incubadas con las proteínas virales VP5, VP6 o VP8 durante 1 h a 37 ºC, infectadas con TLPs de RRV y cosechadas por 12 h. El rotavirus fue detectado por inmunocitoquímica utilizando un anticuerpo policlonal contra la partícula viral. Se observó una inhibición de la infección con rotavirus de más del 90 % cuando las células fueron incubadas con 0,5 ng/l (25 ng) de la proteína viral VP6 (figura 36 c). No se observó inhibición de la infección con las proteínas recombinantes VP5 – GST y VP8 – GST, a pesar de emplear mayor concentración de estas proteínas (figura 36 a y b). Como control positivo de la inhibición se realizó el mismo ensayo, incubando las células MA104 con concentraciones crecientes de DLPs y posteriormente incubando con TLPs. Las DLPs empleadas para este ensayo fueron previamente analizadas por SDS – PAGE y por inmunocitoquímica a fin de evaluar si estaban completamente purificadas o presentaban TLPs que pudieran interferir con los resultados obtenidos (figura 37). Se realizaron ensayos de integridad de la membrana con azul de trypan cuando las células fueron incubadas con las proteínas virales o las DLPs con el objeto de verificar que la disminución de la infección era ocasionada por la presencia de la proteína viral y no por posible daño celular. Figura 36. Efecto de la presencia de diferentes concentraciones de las proteínas virales VP5 – GST, VP8 – GST, VP6 y DLPs sobre la infección de RRV en células MA104. Células MA104 fueron incubadas con diferentes concentraciones de las proteínas estructurales del rotavirus (a) VP5 – GST, (b) VP8 – GST, (c) VP6 y (d) DLPs durante 1 hora a 37 ºC y posteriormente se llevó a cabo la infección con 50 l de1.8 x 10 8 uff /ml de RRV (en una dilución 1:3200) durante 1 h a 37 ºC, se retiró el virus no unido y se incubó durante 12 h a 37 ºC. La identificación de la infección viral se llevó a cabo mediante inmunocitoquímica con un anticuerpo policlonal dirigido contra rotavirus. La viabilidad celular fue determinada mediante azul de tripán concentración de las proteínas fue determinada a 280 nm en relación a una curva de BSA. y la Figura 37. Caracterización de las TLPs y DLPs de RRV utilizadas en los ensayos de inhibición de la infección en presencia de las proteínas virales recombinantes. (a) Electroforesis SDS – PAGE teñido con Nitrato de plata y (b) Western blot de las partículas virales reveladas con un anticuerpo dirigido contra rotavirus. En los carriles del gel y la membrana se observa de izquierda a derecha: 1. TLPs y 2. DLPs. 7. ANÁLISIS DE RESULTADOS La infección con rotavirus se ha convertido en una de las causas más comunes de enfermedades gastrointestinales en niños menores de 2 años de edad y en animales jóvenes de importancia económica en países subdesarrollados. Sin embargo, no se han desarrollados estrategias terapéuticas lo suficientemente eficientes para combatir la infección. Por esta razón, se han intentado dilucidar los mecanismos moleculares involucrados en los procesos de unión y penetración del virus en células susceptibles a la infección rotaviral. Para poder identificar la participación de las proteínas virales estructurales y celulares en estos procesos se han empleado diferentes técnicas que han permitido dilucidar algunos componentes fundamentales para que los eventos iniciales del ciclo infeccioso se lleven a cabo, entre ellos: ensayos de inhibición de la unión del virus a la célula en presencia de anticuerpos dirigidos contra las proteínas celulares candidatas a receptores del rotavirus o moléculas químicas que afecten la actividad de estas proteínas. Igualmente, la expresión y purificación de proteínas heterólogas ha sido una estrategia útil para la identificación de este tipo de interacciones, en este caso entre las proteínas virales recombinantes VP5, VP6 y VP8 con los posibles receptores del rotavirus, Hsc70 y PDI, tanto en condiciones celulares como acelulares. Inicialmente, los vectores que presentan las secuencias génicas que codifican las proteínas virales VP5 y VP8 fueron amplificados con el objetivo de obtener mayor cantidad de los plásmidos. Para esto, bacterias E. coli de la cepa XL1BLUE (deficiente en la expresión de endonucleasas) fueron transformadas con los vectores, para lo cual se empleó un método convencional para hacerlas competentes con CaCl2. Sin embargo, no se logró la transformación bacteriana con esta metodología. Se utilizó el método publicado por Chung C. T., et al, en 1989, utilizando Polietilenglicol (PEG) e iones metálicos mono y divalentes, los cuales facilitan la entrada de moléculas de ADN circular al modificar la membrana bacteriana haciéndola más permeable a estas moléculas. Así mismo, el uso de DMSO facilita el almacenaje de las bacterias competentes en único procedimiento 132. De esta manera se lograron transformar bacterias E. coli de la cepa XL1BLUE para la amplificación de los vectores y bacterias E. coli de la cepa BL21(DE3) utilizadas para la expresión de las proteínas VP5, VP8 y Hsc70. Al analizar los vectores pGEX-4T-VP5 y pGEX-4T-VP8 en geles de agarosa inicialmente se observó 3 bandas de alto peso molecular (figura 11 a, carriles 1 y 2), pero al digerir el vector pGEX-4T-VP8 con la enzima de restricción Pst I, se obtuvo una única banda correspondiente al vector linealizado (figura 11 a, carril 5). Para el plásmido pGEX-4T-VP5 las tres bandas corresponden a productos de digestión de la secuencia génica que codifica la proteína viral que presenta dos sitios de corte para esta enzima, como se muestra en la figura 11 b. Estos resultados demuestran que los vectores fueron purificados y no presentaron contaminación con ADN cromosomal bacteriano. Durante la expresión de proteínas recombinantes se deben tener en cuenta la concentración y la solubilidad de la proteína de interés, debido a que estos factores afectan el rendimiento de la expresión y la estructura tridimensional dificultando el proceso de purificación por cromatografía de afinidad y por consiguiente los ensayos de interacción proteína – proteína o de actividad enzimática. Para obtener la mayor concentración de las proteínas recombinantes, las variables que se tuvieron en cuenta fueron: número de bacterias antes de inducir la expresión de la proteína medida por el incremento de la DO 600 nm, tiempo d-e inducción y concentración del inductor IPTG. En el caso de la proteína viral VP8, la mayor concentración de proteína, 0,141 g/l, se obtuvo al realizar la inducción cuando el cultivo alcanzó una DO 600 nm de 0.5 en medio LB (aproximadamente 5 horas de crecimiento), que corresponde al inicio de la fase estacionaria del crecimiento bacteriano (figura 12 a y tabla 1). Este resultado muestra que existe una relación directa entre la concentración de proteína expresada y el número de células presentes al momento de inducir la expresión durante la fase exponencial del crecimiento bacteriano. Así mismo, sugiere que el mayor rendimiento en la expresión no dependió de células que se encuentren en duplicación constante y no fue afectada por la concentración de nutrientes o productos de desecho presentes en el medio. Respecto al tiempo de inducción, la concentración de la proteína recombinante se incrementó a medida que aumentaba el tiempo de inducción de 2 a 6 h; mientras que a 8 h de inducción, la concentración de la proteína se mantuvo constante respecto a 6 h (figura 13 a y tabla 2). Otro factor analizado fue la concentración del inductor, de lo cual se obtuvo que a 1 mM de IPTG se obtuvo el mayor rendimiento en la expresión de la proteína recombinante VP8, respecto a 0,5 y 2 mM de IPTG (figura 13 b y tabla 3). Este comportamiento ya ha sido reportado previamente, al cuantificar la expresión de la enzima -Galactosidasa en presencia de diferentes concentraciones del inductor (0,0 – 7,5 mM) y se encontró que aunque la cantidad del transcrito de interés se incrementa ligeramente a partir de 1 mM de IPTG, la estabilidad del mismo se ve afectada al igual que la cantidad de RNA r y proteína total, sugiriendo que altas concentraciones de este compuesto se asocian con una disminución en la maquinaria encargada de la traducción celular y por tanto, una reducción en la concentración de la proteína recombinante 133 - 137. Para expresar la proteína viral VP5, se empleó inicialmente medio LB y posteriormente el medio 2XYT, el cual contiene una mayor concentración de triptona y extracto de levadura; sin embargo, se observó un incrementó en la expresión basal de la proteína recombinante (en ausencia del inductor) (figura 15, carril 3). Esto se controló empleando glucosa al 2%, la cual disminuye los niveles de AMPc intracelular que son requeridos para activar la expresión de la RNA polimerasa T7 después de su interacción con la proteína de unión a AMPc (CAP) (figura 15, carril 4) 60, 138. En presencia del inductor se observó una mayor concentración de la proteína viral en medio 2XYT con glucosa en comparación con el medio LB (figura 15, carril 8 y 5, respectivamente). La mayor concentración de VP5 se obtuvo al realizar la inducción cuando el cultivo se encontraba en la fase exponencial y alcanzó una DO 600 nm entre 0,2 a 0,4 (figura 16 a y tabla 4). Estos resultados sugieren que la expresión de esta proteína recombinante requiere que las bacterias se encuentren metabólicamente activas cuando los nutrientes no se hayan agotado y los productos generados no inhiban la duplicación o incrementen la muerte celular. En cuanto al tiempo de inducción, no se evidenció un cambio significativo en la concentración de VP5 a los diferentes tiempos evaluados, lo que muestra que a partir de 2 h de inducción se logra obtener la mayor concentración de VP5. La poca disminución en la concentración de la proteína recombinante a las 6 y 8 h de inducción, puede deberse a la degradación de la misma; sin embargo, no se evidenciaron bandas de menor peso molecular en el western blot (figura 17 a y tabla 5). Con concentraciones del IPTG de 0,5 y 1 mM no variaron significativamente el rendimiento en la expresión de la proteína recombinante VP5, 0,388 y 0382 g/l, respectivamente; mientras que concentraciones de 2 mM redujeron la producción de la misma (0,199 g/l) (figura 17 b y tabla 6). Por otra parte, las proteínas recombinantes VP8 y VP5 no fueron identificadas en el sobrenadante y se localizaron en la fracción que se denominó “fracción insoluble” haciendo referencia al pellet obtenido después de centrifugar el lisado celular, que solo fue soluble cuando se utilizaron concentraciones de úrea de 8M (figura 18). Estas agregaciones proteícas se denominan cuerpos de inclusión y se forman en el espacio citoplasmático y periplásmico de la célula bacteriana cuando se obtienen altos niveles de expresión de proteínas heterólogas (más del 2 % del total de proteínas bacterianas) o cuando la proteína expresada es hidrofóbica. Estas agregaciones son principalmente formadas por interacciones hidrofóbicas no – nativas entre estructuras con plegamiento intermedio, en las cuales los dominios hidrofóbicos se exponen. Para la solubilización de estas agregaciones proteícas se requiere de altas concentraciones de agentes caotrópicos, que generan estructuras en forma de rollos aleatorios (en inglés, random coil) en donde los aminoácidos hidrofóbicos se exteriorizan y se pierde la estructura nativa de la proteína dificultando su proceso de purificación por técnicas de afinidad que involucren su estructura tridimensional 139 - 141. Estudios de cristalografía de rayos X revelaron que la proteína viral VP8 presenta en su estructura 9 % de hélices y 55 % de hojas plegadas 142 y aunque estas características bioquímicas no explican la formación de cuerpos de inclusión durante la expresión de la proteína recombinante, puesto que no es determinante en su posible carácter hidrofóbico, la producción de aproximadamente 0,25 mg/ml de VP8 en el cultivo puede estar relacionada con este evento. Por otra parte, en diferentes estudios se ha expresado esta proteína mediante varios sistemas, entre ellos: infección de células de insecto Sf9 con un baculovirus recombinante 19, 22, 143 - 144, inducción de la expresión en bacterias E. coli JM109 transfectadas con el vector pGEMEX-1-VP8 o pGEX-4T-2-VP8 145, 146 y bacterias BL21(DE3) transfectadas con el vector pGEX-4T-2-VP8 o pET28a(+) 147 - 149. Todos los sistemas de expresión permitieron obtener la proteína viral de forma soluble; excepto, cuando la expresión se realizó en bacterias BL21(DE3) con el vector pGEX-4T-2-VP8 y la inducción se llevó a cabo cuando el cultivo bacteriano alcanzo una DO 600 nm entre 0.2 – 0.4 con 1 mM de IPTG durante 3 h a 37 ºC, datos que confirman los resultados reportados en este trabajo. Sin embargo, en un estudio posterior se demostró que la disminución en la temperatura a 25 ºC durante la inducción de la proteína, incrementa su concentración en el sobrenadante o fracción de proteínas solubles 150. Esta última variable se debe tener en cuenta puesto que durante el desarrollo de este trabajo, la temperatura más baja utilizada durante el crecimiento bacteriano fue de 28 ºC (figura 14), factor que no afectó la solubilización de la proteína pero se observó una disminución en su expresión, por tanto, es necesario realizar ensayos de expresión de esta proteína pero a temperaturas más bajas durante la inducción. Estos resultados muestran que la solubilidad de la proteína recombinante no solo depende de las características bioquímicas o de la concentración de la proteína en el cultivo; sino del sistema de expresión empleado 145, 149. En el caso de la proteína viral VP5, diversos estudios han demostrado que esta proteína presenta dos dominios requeridos para inducir la permeabilidad celular: un dominio que permite la asociación periférica de la proteína a la membrana y un segundo dominio hidrofóbico, que comprende los residuos 248 a 474, requerido para la formación del poro 151. Así mismo, este dominio presenta 1 % de hélices y 56 % de hojas plegadas que se asocian en forma de dímeros y trímeros durante la penetración viral determinado por cristalografía de rayos X 152. Estas características bioquímicas y la producción de aproximadamente 0,3 mg/ml de la proteína viral puede sustentar la formación de cuerpos de inclusión aun cuando se variaron las condiciones de expresión de la proteína. La insolubilidad de esta proteína recombinante en forma de agregaciones insolubles ha sido previamente reportada en bacterias E. coli BL21(DE3) y JM109 transformadas con los vectores pET28a(+) y pGEMEX-1 recombinantes, respectivamente 146, 153; sin embargo, al expresar este dominio en células de insecto Sf9 a partir de un baculovirus recombinante, la proteína fue obtenida de forma soluble y purificada mediante cromatografía de intercambio iónico 152, 154, sugiriendo que la insolubilidad de VP5 no fue solo dependiente de las propiedades de su secuencia proteíca sino también del sistema de expresión empleado. Por otra parte, existen reportes en los cuales se demuestra que la expresión de esta proteína viral en bacterias E. coli BL21(DE3) con el vector pGEX-4T2 puede darse en forma soluble si la inducción de la expresión se realiza a 20 ºC, con bajas concentraciones de IPTG (0,01 mM) y es co-purificada con la chaperona GroEL 155; sin embargo, este procedimiento requiere de un paso adicional que consiste en retirar la chaperona con ATP 156. Cabe mencionar que esta variable no fue tenida en cuenta durante el proceso de optimización de la expresión de esta proteína recombinante. Para optimizar la expresión de Hsc70 se analizaron las variables mencionadas anteriormente, encontrándose que las condiciones que permitieron una mayor concentración de la proteína recombinante se obtuvieron cuando el cultivo alcanzó una DO 600 nm de 0,6 y la inducción se llevo a cabo durante 4 h con 1 mM de IPTG. Cabe mencionar que eta proteína fue obtenida tanto en la fracción soluble como en la insoluble. Su solubilidad puede depender en gran medida en la homología que existe con chaperonas bacterianas, como Dna K 157; sin embargo, se observó que a medida que el número de bacterias era mayor antes de inducir la expresión, la proteína se localizó únicamente en la fracción insoluble posiblemente debido a la sobreexpresión de la misma (figura 19, carriles 13, 15 y 17). Así mismo, un estudio previo permitió confirmar los resultados obtenidos, puesto que el lisado bacteriano fue disuelto con úrea 8M para solubilizar los cuerpos de inclusión formados durante la expresión de esta proteína 158. Sin embargo, otros reportes han mostrado que esta proteína fue expresada en forma soluble mediante el mismo sistema empleado en este trabajo, en el cual la inducción se realizó cuando el cultivo alcanzó una DO 600 nm de 0,3 con 0,4 mM de IPTG durante 2 horas. Estos resultados corroboran los datos obtenidos, puesto que a DO inferiores a 0,4 al momento de inducir la expresión de la proteína recombinante, una fracción de esta es obtenida en el sobrenadante 159 - 162. Por otra parte, para la obtención de la proteína VP6 del rotavirus se han empleado diferentes sistemas de expresión en estudios previos, entre ellos: infección con baculovirus recombinante de células Sf9 163 - 164 y Spodoptera frugiperda transfección de bacterias BL21(DE3) con el plásmido pMAL-C2X/EDIM6 167 - 169, 165 - 166, infección de células CV-1 170 - 172 o MA104 173 con el virus vaccinia recombinante y transfección de Solanum tuberosum con el vector pPCV70 174. En casi todos los reportes muestran que para la posterior purificación de esta proteína fue necesario solubilizar los lisados en úrea 5 M o detergentes iónicos, como deoxicolato de sodio al 1 %, o se observó la formación de estructuras tubulares retiradas por ultracentrifugación a 100000 x g, sugiriendo que la expresión de esta proteína se dio en forma insoluble, lo cual está de acuerdo con los resultados obtenidos en este trabajo (figura 21). La insolubilidad de esta proteína se debe a su carácter altamente hidrofóbico y puede estar relacionada con la formación de trímeros, como ocurre de forma nativa en la estructura de las partículas virales 175. La purificación de las proteínas recombinantes VP5 - GST y VP8 – GST se realizó mediante cromatografía de afinidad. Para lo cual, se emplearon dos técnicas de solubilización de las agregaciones proteícas: el primero, se realizó con úrea 8 M y el segundo, con el detergetne iónico, N- laurilsarcosina. La úrea es un agente caotrópico que se une a la proteína dependiendo de la concentración empleada generando la formación de estructuras proteicas más flexibles y desorganizadas perdiendo su estructura nativa. El replegamiento de las proteínas recombinantes se realizó mediante diálisis, la cual fue desarrollada a baja temperatura y con intercambios de buffer con concentraciones decrecientes de úrea en lapsos de 4 horas a fin de evitar la precipitación de la proteína por plegamientos incorrectos que suelen ocurrir durante la disminución de la concentración del agente caotrópico 72; sin embargo, no se observó la retención de la proteína recombinante durante el corrido cromatográfico. Estos resultados sugieren que el replegamiento del polipéptido GST no fue el apropiado y por tanto no reconoció el glutatión presente en la resina de agarosa (datos no mostrados). Por otra parte, los pellet celulares fueron tratados con N- laurilsarcosina, detergente iónico alcalino que ha sido ampliamente utilizado para solubilizar casi todas las proteínas unidas a GST y en general agregaciones proteícas 176 - 177, debido a que dispersa los cuerpos de inclusión en estructuras monomoleculares por la fuerte repulsión electrostática del complejo proteína – detergente. La proteína en este complejo puede adquirir estructuras secundarias no nativas permitiendo la formación de interacciones intra – moleculares nativas y no nativas. La unión de un detergente a una proteína es determinada por la concentración micelar crítica (CMC), cuando la concentración del detergente es superior a la CMC, se da la formación de una micela con la proteína en donde la proteína denaturada es altamente soluble, mientras que concentraciones menores a la CMC, reducen la solubilidad de la misma 178. La concentración empleada de N – laurilsarcosina para solubilizar las proteínas bacterianas y los cuerpos de inclusión fue de 1 %, superior al valor de CMC determinado para este detergente (0,427 %), el cual permitió la solubilización de las agregaciones proteícas. Sin embargo, el uso de esta concentración del detergente aniónico afecta considerablemente la estructura de la proteína y por tanto su interacción con la resina de agarosa unida a glutatión, puesto que depende básicamente de la estructura tridimensional del polipéptido GST. Estudios previos han demostrado que detergentes no iónicos pueden formar micelas mezcladas con el detergente iónico y previenen su precipitación en presencia de iones divalentes, como calcio y magnesio 177, 179, 200. Adicionalmente, se ha demostrado que este tipo de detergentes no afectan la interacción de las proteínas unidas a GST a la resina de agarosa 180. Por esta razón, fue necesario utilizar un detergente no iónico a los lisados bacterianos que contenían 1 % de N – laurilsarcosina que permitiera la renaturación o replegamiento de las proteínas presentes en el lisado bacteriano, incluyendo la proteína de fusión solubilizada. El detergente utilizado en el proceso de purificación de la proteína viral VP8 fue tritón X-100 en concentraciones del 1 - 3 % 176 y se observó la unión de la proteína recombinante a la resina (figura 22 a); sin embargo, la cantidad de proteína retenida fue muy baja en comparación con el total de la proteína recombinante presente en el lisado bacteriano total y la encontrada en la fracción no retenida, obtenida después de la interacción del lisado con la resina (figura 22 a, carriles 2 y 3). Por lo tanto, se empleó una estrategia experimental diferente para inducir el replegamiento correcto del polipéptido, con el objetivo de aumentar la cantidad de proteína unida a la resina, que consistió en diluir 10 veces el lisado celular solubilizado con N – laurilsarcosina al 1 % en buffer PBS y posteriormente realizar el paso de purificación 77. De acuerdo con los resultados obtenidos en la figura 22 b, es posible evidenciar que este tratamiento resultó más eficiente, puesto que se logró obtener un mayor número de eluidos de la proteína VP8 - GST a partir de un lisado bacteriano con las mismas condiciones al utilizado en el procedimiento anterior. La diferencia en la cantidad de proteína recombinante VP8 purificada a partir de las metodologías empleadas para el replegamiento proteico, posiblemente se debe a que las concentraciones empleadas de Tritón X 100 (1 y 3 %) superan su CMC (0,021 %) y por tanto forman micelas que tienen un peso molecular (PM) de 90 KDa, superior al peso de la proteína recombinante (54 KDa). Sin embargo, es importante mencionar que las micelas formadas son una mezcla del detergente iónico y el no iónico, por tanto el PM de una micela, suponiendo que la relación fuera 1:1, sería de aproximadamente 50 KDa (PM de una micela de N-laurilsarcosina es de 0,6 KDa) ocasionando que la proteína quede atrapada dentro de la micela y no se expongan completamente sus dominios de interacción con glutatión para su purificación 181 - 182. Para la purificación de la proteína recombinante VP5 – GST se empleó la misma estrategia utilizada para VP8 – GST; sin embargo, la proteína viral no se unió al glutatión de la resina cuando el lisado fue tratado con Tritón X-100. De esta manera, se emplearon otros detergentes no iónicos (tween 20, NP40 y -octilglucósido) y un detergente zwiteriónico (CHAPS), como agentes renaturantes 183. Como se puede evidenciar en la figura 23, carriles 6 y 15, los detergentes que indujeron la interacción del polipéptido GST a la resina, fueron CHAPS y NP40, respectivamente, obteniéndose mayor rendimiento con el primero de ellos. Estos detergentes tienen características químicas diferentes, entre ellas: estructura química, carácter iónico o no – iónico, CMC, peso molecular del monómero y en forma micelar, lo que no logra explicar los resultados obtenidos respecto a los otros detergentes analizados, teniendo en cuenta que muestran características similares. Por otra parte, cuando el tratamiento de purificación se realizó con cultivos a mayor escala en presencia de CHAPS, la cantidad de proteína retenida fue poca respecto a la cantidad de proteína de fusión expresada en el lisado bacteriano total. Por tanto, se incrementó la concentración del detergente al 3 %; sin embargo, se observó la formación de un precipitado en el lisado bacteriano correspondiente al CHAPS, puesto que a la temperatura a la cual se estaba trabajando (4 ºC) y la concentración del mismo, se favorece su insolubilidad en solución acuosa. Este efecto puede ser ocasionado porque el detergente se disolvió en forma de agregados a temperatura ambiente, pero cuando se incubó a 4 ºC, estos agregados se precipitaron debido a que nunca fueron realmente disueltos (efecto cinético) o por una sobresaturación, en la cual el detergente pudo mantenerse soluble a temperatura ambiente, pero al disminuir la temperatura, el detergente se precipitó 181 - 185. El lisado fue centrifugado con el objeto de retirar estas partículas insolubles y el sobrenadante fue tratado con la resina. Los resultados obtenidos en la figura 24 a, muestran que la cantidad de proteína unida a la resina fue baja en comparación con la proteína total encontrada en el lisado total antes de la adición del CHAPS; sin embargo, al analizar el precipitado formado se observó que gran cantidad de la proteína recombinante junto con otras proteínas bacterianas fueron precipitadas de la misma manera. Por otra parte, el uso de NP40 al 3 % permitió mayor retención de la proteína viral por la resina y a diferencia del CHAPS, no se precipitó a pesar de la baja temperatura y la concentración empleada (figura 24 b). En comparación con la literatura, las proteínas recombinantes VP5 y VP8 obtenidas con el mismo sistema de expresión utilizado en este trabajo, fueron purificadas mediante cromatografía de afinidad sin dificultad puesto que se obtuvieron de forma soluble; mientras que en los ensayos en donde se encontraron insolubles, se aplicaron técnica de diálisis que permitieron el replegamiento del polipéptido GST y su interacción con la resina, aunque esta metodología no funcionó en nuestros ensayos 145, 147, 148. Respecto a la purificación de la proteína celular Hsc70, se empleó cromatografía de afinidad, para lo cual se empleó una resina que contiene iones metálicos de Ni2+ que tiene gran afinidad por los anillos imidazólicos de las colas de histidinas que presentó la proteína de fusión. Por esta razón, el pellet bacteriano fue resuspendido y lisado en un buffer que no contenía EDTA, a diferencia de las bacterias que expresaron las proteínas virales recombinantes VP5 y VP8, debido a que por su propiedad de quelar iones metálicos, no permitiera la purificación de Hsc70 186. Así mismo, se empleó N- laurisarcosina y tritón x-100 para la solublización de los cuerpos de inclusión formados durante su expresión 187. La purificación de esta proteína fue realizada de acuerdo con reportes realizados previamente 158, 162. Teniendo en cuenta los resultados obtenidos en la figura 23, se puede observar que la mayor parte de la proteína expresada fue retenida por la resina al comparar la cantidad de proteína presente en el lisado bacteriano total (figura 25, carril 2) y el lisado bacteriano no retenido obtenido después de pasar el lisado por la resina (figura 25, carril 3). El procedimiento de elución se llevó a cabo con concentraciones crecientes de imidazol entre 50 mM a 1 M; las fracciones obtenidas con 50 mM presentaron un gran contenido de bandas inespecíficas (figura 25, carriles 7 a 12) y al incrementar la concentración de imidazol, menor era el número de bandas inespecíficas. Cabe mencionar que aunque no se realizaron ensayos de actividad enzimática para las proteínas recombinantes purificadas (VP5, VP8 y Hsc70), el protocolo utilizado para la solubilización de los cuerpos de inclusión formados durante su expresión permitió la interacción del polipéptido GST, en el caso de VP5 y VP8, y la cola de His, para Hsc70, con la resina empleada para la purificación de cada una. Estos resultados sugieren que esta estrategia experimental facilita el correcto plegamiento de las proteínas recombinantes. Adicionalmente, estudios recientes han permitido demostrar que la solubilización de estas agregaciones proteicas, mediante el uso de N-laurilsarcosina, encapsulan las proteínas y las disgregan; mientras que los detergente no iónicos o zwiteriónicos forman grandes micelas o bicelas mezcladas que incorporan las moléculas del detergente iónico (N-laurilsarcosina), disminuyendo su concentración alrededor de la proteína recombinante, dejando sitios activos libres o facilitando el replegamiento de la proteína, en los cuales demuestran que las proteínas purificadas mediante esta metodología presentaron una actividad mayor al 80 % tras el uso de diferentes detergentes y mezcla de ellos 134, 154 - 156, 188 - 190. En el caso de la proteína recombinante VP6, se han empleado diferentes técnicas que han permitido la purificación de esta proteína, incluyendo cromatografía de afinidad - 169, cromatografía de intercambio iónico 163, inmunoprecipitación ultracentrifugación de estructuras tubulares formadas por la proteína 166. 170, 165, 167 73 y Sin embargo, en este trabajo se empleó una técnica de purificación basada en detergentes y en el carácter apolar de la proteína recombinante viral. El pellet que contenía la proteína viral obtenido después de la lisis celular fue tratado con diferentes detergentes, incluyendo iónicos, no – iónicos y zwiteriónicos. Como se observa en la figura 21 y 26 a, los detergentes que solubilizaron esta proteína fueron los iónicos (N-laurisarcosina y DOC); mientras que los detergentes no – iónicos o zwiteriónicos, no la solubilizaron aun cuando se utilizaron concentraciones superiores al CMC correspondiente de cada detergente; sin embargo, solubilizaron una gran cantidad de proteínas celulares posiblemente adheridas a membrana. Estos resultados indican que la proteína recombinante VP6 permitió la formación de agregaciones proteicas que no se encontraban adheridas a membrana, teniendo en cuenta que los detergentes iónicos son agentes denaturantes que rompen interacciones proteína – proteína, los detergentes no iónicos permiten la disociación de interacciones proteínas – lípidos y lípido – lípido, mientras que los zwiteriónicos, aunque no son altamente denaturantes, pueden disociar interacciones proteína – proteína; sin embargo, el detergente CHAPS, a la concentración y temperatura analizada, no permitió la solubilización de esta proteína 191 - 192. Teniendo en cuenta lo anterior y como se muestra en la figura 29, el buffer RIPA, el cual contiene SDS al 0,1%, DOC al 1% y Tritón X 100 al 1%, permitió la solubilización de un conjunto de proteínas celulares (carriles 4 y 5), las cuales fueron retiradas casi que completamente con - Octilglucósido al 1% (carriles 6). El pellet remanente que contenía la proteína viral recombinante VP6 solo fue soluble cundo se trató con úrea 8M y Nlaurilsarcosina al 1%, compuestos altamente denaturantes (carriles 7 y 8). Los resultados obtenidos sugieren que esta proteína formó agregaciones proteicas altamente insolubles debido a su capacidad de polimerización y de formar estructuras tubulares cuando es sobreexpresada 166 o trímeros cuando se encuentra en las partículas infecciosas del rotavirus 175. Para la caracterización de los anticuerpos policlonales dirigidos contra las proteínas recombinantes del rotavirus VP5, VP6 y VP8 se realizaron diferentes técnicas de inmunodetección, entre ellas: western blot, inmunocitoquímica y ELISA. En el caso del anticuerpo policlonal obtenido contra VP5, se logró identificar hasta 250 ng de la proteína recombinante purificada (figura 30 d, carril 3 y 4), cantidades menores fueron débilmente reconocidas (figura 30 d, carril 5 y 6); sin embargo, en los lisados bacterianos que expresaron esta proteína, se observa la identificación de una serie de bandas inespecíficas correspondientes a proteínas bacterianas puesto que este mismo perfil fue evidente en el lisado de bacterias que no expresaron la proteína viral (figura 30 a, carril 5). El reconocimiento de proteínas bacterianas por este anticuerpo puede ser consecuencia del método de extracción de la proteína viral utilizada como antígeno para la inoculación de los conejos, puesto que se partió de un lisado total y aunque se identificó la banda correspondiente a la proteína mediante western blot, resulta importante mencionar que muchas proteínas bacterianas pueden tener el mismo peso molecular y de esta manera contaminar el antígeno que se intentó purificar. Por otra parte, se evidenció el reconocimiento de la proteína recombinante por el método de ELISA, utilizando una dilución de 1:1000 del anticuerpo; sin embargo, valores de DO 492 nm mayores a 0,2, solo fueron obtenidos cuando se emplearon cantidades mayores a 200 ng de la proteína (figura 30 g). Así mismo, se analizó el reconocimiento del anti – VP5 por las proteínas nativas procedentes de las partículas virales TLPs y durante el proceso infeccioso mediante western blot e inmunocitoquímica, respectivamente. Los resultados muestran que este anticuerpo permite el reconocimiento de la proteína viral VP5 mediante western blot en condiciones denaturantes y en la partícula viral infecciosa en condiciones nativas en el proceso infeccioso (figura 31 d y g). En el caso del anticuerpo dirigido contra la proteína viral VP6, se observó la presencia de una banda inespecífica de menor peso molecular, probablemente producto de la degradación de la proteína viral teniendo en cuenta que el lisado celular que no fue infectado con el virus recombinante no presentó estas bandas. Así mismo, se logró el reconocimiento de las proteínas procedentes de las partículas virales tanto en condiciones denaturantes como en su estado nativo (figura 31 e y h). La dilución empleada del anti – VP6 en cada uno de los ensayos fue de 1:1000. Finalmente, el anticuerpo dirigido contra la proteína VP8 permitió el reconocimiento de la proteína viral en cada una de las muestras analizadas en diluciones 1:1000 y en diferentes concentraciones los diferentes antígenos utilizados, como: proteína recombinante purificada (figura 30 f e i) y lisados bacterianos (figura 30 c). Así mismo, se logró la identificación de la proteína en las partículas virales denaturadas mediante western blot, siendo mayor el reconocimiento respecto a un anticuerpo policlonal contra rotavirus del laboratorio (figura 31 f) y en condiciones no denaturantes mediante inmunocitoquímica (figura 31 i). Estos resultados muestran que las proteínas recombinantes del rotavirus VP5, VP8 y VP6 indujeron la respuesta inmune en los conejos que fueron inoculados. Finalmente, la identificación de la interacción entre las proteínas recombinantes virales VP5 - GST, VP6 y VP8 - GST con las proteínas candidatas a receptores del rotavirus, Hsc70 y PDI, se realizó mediante ensayos de ELISA indirecto y co-inmunoprecipitación. De acuerdo con los resultados obtenidos en las figuras 33 a y 34 a, se pudo evidenciar que la proteína recombinante VP5 – GST interacciona directamente y dependiente de concentración con la proteína recombinante Hsc70 en las condiciones analizadas. Estos resultados confirman datos previamente reportados, en los cuales se demuestra que el dominio carboxi terminal de la proteína viral interacciona con el dominio KID de esta chaperona, lo cual fue determinado mediante ensayos de ELISA y de competencia con péptidos 46, 162, 201. Adicionalmente, se pudo establecer que esta interacción sucede en condiciones celulares, al co-inmunoprecipitar Hsc70 procedente de células MA104 con VP5, la cual fue determinada en regiones específicas de la membrana celular, conocidos como rafts lipídicos 193 - 194 (figura 35 b). Resulta importante mencionar que la interacción de esta proteína recombinante con la proteína celular no fue dependiente del polipéptido GST, puesto que la proteína recombinante viral VP8 – GST no interaccionó con Hsc70 en los ensayos realizados, aun en presencia de este polipéptido (figura 33 e, 34 c, 35 d). Paralelamente, la interacción entre la proteína viral VP6 y la proteína recombinante Hsc70 fue identificada mediante los ensayos mencionados en condiciones acelulares y al igual que con la proteína VP5, esta interacción es dependiente de concentración. Adicionalmente, se logró establecer que esta interacción sucede en condiciones celulares en raft lipídicos. Estos resultados complementan datos obtenidos previamente en el laboratorio, en los cuales se demostró que las DLPs y péptidos sintéticos de VP6 se unen a esta proteína de choque térmico e inhiben el proceso infeccioso con diferentes cepas rotavirales 43, 195, así como la co-inmunoprecipitación de la proteína viral con Hsc70 en células MA104 196. Por otra parte, se determinó el efecto en la infección con rotavirus cuando las células fueron expuestas a las proteínas virales recombinantes. Teniendo en cuenta los resultados obtenidos en la figura 36, se evidenció una inhibición de la infección de aproximadamente del 90 % cuando se utilizaron 25 ng de la proteína recombinante VP6, lo que fue comparable con ensayos previos en los que se demostró que las partículas DLPs pueden inhibir el proceso infeccioso del rotavirus 43; sin embargo, este efecto no fue observado con las otras proteínas virales expresadas. Estos resultados sugieren que la proteína viral VP6 es indispensable en los primeros eventos de la infección con rotavirus RRV en células MA104, puesto que el tratamiento previo de las células con esta proteína puede ocupar receptores específicos del virus que inhiban la entrada o penetración del virus a la célula, entre ellos Hsc70, el cual ha sido previamente reconocido que participa durante un evento pos-unión del rotavirus a la célula 43, 162. Mientras que las otras proteínas virales pueden interaccionar con diferentes moléculas de la superficie celular, lo que hace que su presencia no inhiba el proceso infeccioso 40. Adicionalmente, se identificó la interacción de las proteínas recombinantes virales VP5 – GST y VP6 con la proteína recombinante PDI (figura 33b, d y 34). La PDI cataliza la reducción, oxidación e intercambio de enlaces disulfuro, además presenta actividad de chaperona 202 - 203 y ha sido implicada en la reducción de los enlaces disulfuro del heterodímero de la toxina diftérica 204 y en la entrada del virus de la inmunodeficiencia humana (HIV) a las células linfoides 205. Respecto al rotavirus, se ha reportado que la PDI interacciona con la VP7 glicosilada para asegurar la formación y el plegamiento correcto de la VP7 en el ER, y por tanto del ensamblaje viral 206. El silenciamiento de la expresión de chaperonas del ER, entre ellas la PDI, interfiere la formación o el rearreglo de los enlaces disulfuro de la VP7 y la formación de TLPs 207. Se ha demostrado que la interferencia en la formación de los puentes de disulfuro en VP7 afecta la formación de partículas virales infecciosas debido al bloqueo en el ensamblajede la capa más externa del virion 208. Recientemente, estudios realizados en el Laboratorio de Biología Molecular del Virus reportaron la inhibición de la infección rotaviral mediante el tratamiento previo de células MA104 con inhibidores del intercambio tiol-disulfuro tales como bacitracina y DTNB [5,5dithio-bis-(2-nitrobenzoic acid)]. Dado que estos inhibidores no son transportados a través de la membrana celular, se sugirió la participación de la PDI de la superficie celular en el proceso de entrada de los rotavirus. Ensayos de biotinilación con reactivos que no permeabilizan la membrana celular y análisis de citometría de flujo (FACS) confirmaron la presencia de PDI en la superficie de la célula. La especificidad del bloqueo de la infección con bacitracina y DTNB se confirmó con la adición de anticuerpos dirigidos contra PDI o de péptidos sintéticos correspondientes a VP4 y VP7 que contenían cisteínas en su secuencia, los cuales inhibieron significativamente la infección con rotavirus RRV 197, 199. Estos resultados permiten sugerir que la interacción de estas proteínas celulares, Hsc70 y PDI, con las proteínas recombinantes virales analizadas puede ser un paso determinante en el proceso infeccioso del rotavirus, que sucede durante los eventos iniciales del ciclo viral puesto que se lograron identificar en los microdominios lipídicos de células MA104, en donde se ha establecido que existe una co-localización de PDI, Hsc70, integrina 3 197 y las partículas rotavirales 198, siendo fundamentales para la unión y penetración del virus a las células hospederas. Estas chaperonas pueden estar involucradas en cambios conformacionales que sufren las proteínas de la partícula viral durante estos eventos 209, lo cual puede ser sustentado a partir de estudios cristalográficos de un fragmento de VP5* realizados previamente, en los cuales indican que esta proteína modifica su estructura al retraerse sobre sí misma, pasando de una estructura dimérica a una trimérica, para permitir su interacción con la membrana celular a través de la exposición de su dominio hidrofóbico 152 - 210. Adicionalmente, experimentos en solución realizados para definir la interacción de Hsc70 con las proteínas de la cápside externa del rotavirus, permitieron evidenciar cambios conformacionales en las proteínas virales al estar en contacto con esta chaperona 162 - 209. Aunque no es posible afirmar que las interacciones establecidas entre las proteínas virales y las proteínas celulares ocurran particularmente durante el paso de unión o postunión del virus a la célula con los datos mostrados, previamente se ha demostrado que Hsc 70 y PDI participan en el evento de pos-unión del virus, puesto que al utilizar anticuerpos dirigidos contra estas proteínas o ligandos naturales se observa una disminución de la infección pero no de la unión del virus a la célula 43, 162, 199. Así mismo, no se puede establecer si estas interacciones suceden in vivo puesto que es necesario tener en cuenta factores como la estructura tridimensional de las proteínas purificadas teniendo en cuenta la insolubilidad que presentaron durante el proceso de expresión y purificación, por tanto resulta necesario realizar estos ensayos de interacción pero en un modelo animal, como ratón. Por último, para definir la especificidad de las interacciones identificadas se requieren de ensayos de competencia a partir del uso de otras moléculas que naturalmente se asocien o actúen como sustratos de las proteínas celulares candidatas a receptores del rotavirus. 8. CONCLUSIONES El mayor rendimiento en la expresión de la proteína viral recombinante VP8 – GST se obtuvo cuando las bacterias BL21(DE3) transformadas con el vector pGEX-4T-VP8 fueron cultivadas en medio LB a 37 ºC con agitación hasta alcanzar una DO 600 nm de 0,6 e inducidas con 1 mM de IPTG durante 6 horas. En el caso de la proteína viral recombinante VP5 - GST, la mayor concentración se obtuvo cuando las bacterias BL21(DE3) transformadas con el vector pGEX-4T-VP5 fueron cultivadas en medio 2XYT en presencia de glucosa al 2% a 37 ºC con agitación constante hasta alcanzar una DO 600 nm de 0,2 e inducidas con 0,5 mM de IPTG durante 4 horas. Para obtener la mayor concentración de la proteína recombinante Hsc70, bacterias BL21(DE3) transformadas con el vector pET-28a-Hsc70 fueron cultivadas en medio LB a 37 ºC con agitación constante hasta alcanzar una DO 600 nm de 0,6 e inducidas con 1 mM de IPTG durante 4 horas. Las proteínas recombinantes obtenidas fueron expresadas como cuerpos de inclusión insolubles, los cuales fueron solubilizados con un detergente iónico, altamente denaturante. Las proteínas fueron replegadas con un detergente no iónico, como NP40 en el caso de VP5 o tritón X 100, en el caso de Hsc70; mientras que para VP8, la renaturación se llevó a cabo por dilución. El replegamiento del polipéptido GST, para las proteínas recombinantes VP5 y VP8, fue demostrado al purificar estas proteínas mediante cromatografía de afinidad usando glutatión como ligando. La purificación parcial de la proteína viral VP6 se basó en su carácter apolar, puesto que no fue soluble en ningún detergente no iónico, sugiriendo la formación de polímeros insolubles teniendo en cuenta la formación de trímeros en la partícula viral. Para su solubilización se emplearon altas concentraciones de úrea que fue retirada por diálisis y dilución. Las proteínas virales recombinantes VP5, VP8 y VP6 mostraron ser inmunogénicas al inducir la producción de anticuerpos policlonales dirigidos contra estas proteínas en conejos Nueva Zelanda. Los anticuerpos policlonales dirigidos contra las proteínas virales recombinantes permitieron el reconocimiento de las proteínas en su estado nativo en las partículas virales mediante diferentes técnicas de inmunodetección. Las proteínas virales recombinantes VP5 – GST y VP6 interaccionan con las proteínas recombinantes Hsc70 y PDI en sistemas libres de células y en condiciones celulares, específicamente en microdominios lipídicos, sugiriendo la participación de estas chaperonas durante los eventos de iniciales del proceso infeccioso del rotavirus, posiblemente asociadas a cambios conformacionales de las proteínas virales requeridas para la unión y penetración de la partícula viral a la célula huésped. Por otra parte, la proteína recombinante VP8 – GST no interaccionó con estas proteínas celulares en las condiciones acelulares evaluadas. Finalmente, la proteína viral VP6 inhibió eficientemente la infección con rotavirus, sugiriendo que esta proteína participa durante los primeros eventos del ciclo infeccioso siendo una molécula determinante en el proceso. BIBLIOGRAFÍA 1. ISA P, GUTIERREZ M, ARIAS C and LOPEZ S. Rotavirus Cell Entry. Future Medicine (2008), vol. 3, p. 135 – 146. 2. Ministerio de Protección Social, Instituto Nacional de Salud. 2003. Enfermedad diarreica aguda e infección por rotavirus, Colombia, Inf. Quina. Epidemiol. Nac., 8(20):321-36Boletín Epidemiológico Semanal. 2002. Ministerio de Salud, Colombia Julio p. 5 3. GUERRERO C, MORENO L, PEÑARANDA J, ACOSTA O. Caracterización electrofóretica de rotavirus aislados de infantes con gastroenteritis en la ciudad de Bogotá. Revista Facultad de Medicina (1994), vol. 42, p. 203-208. 4. DENNEHY P. Rotavirus Vaccines: an Overview. Clinical Microbiology Reviews (2008), vol. 21, p. 198 – 208. 5. ESTES M, COHEN J. Rotavirus Gene Structure and Function. Microbiological Reviews (1989), vol. 53, p. 410 – 449. 6. LUNDGREN O., SVENSSON L. Pathogenesis of Rotavirus Diarrhea. Microbes and Infection (2001), vol. 3, p. 1145 – 1156. 7. LÓPEZ S., ARIAS C. Multistep Entry of Rotavirus into Cells: a Versaillesque Dance. Trends in Microbiology (2004), vol. 12, p. 271 – 277. 8. COHEN J., CHARPILIENNE A., CHILMONCYZK S., ESTES M. K. Nucleotide sequence of bovine rotavirus gene 1 and expression of the gene product in baculovirus. Virology (1989), vol. 171, p.131–140. 9. FUKUHARA N., NISHIKAWA K., GORZIGLIA M. Nucleotide sequence of the gene segment 1 of a porcine rotavirus strain. Virology (1989), vol. 173, p. 743–749. 10. MITCHELL D. B., BOTH G. W. Completion of the genomic sequence of the simian rotavirus SA11: nucleotide sequences of segments 1, 2, and 3. Virology (1990), vol. 177, p. 324–331. 11. VALENZUELA S., PIZARRRO J., SANDINO A. M., VAZQUES M. Photoaffinity labeling of rotavirus VP1 with 8-azido-ATP: identification of the viral RNA polymerase. Journal of Virology (1991), vol. 65, p. 3964–3967. 12. ZENG C. Q., WENTZ M. J., COHEN J., ESTES M. K. Characterization and replicase activity of double-layered and single-layered rotavirus-like particles expressed from baculovirus recombinants. Journal of Virology (1996), vol. 70, p. 2736 – 2742. 13. LABBE M., BAUDOUX A., CHARPILIENNE D., PONCET D., COHEN J. Identification of the nucleic acid binding domain of the rotavirus VP2 protein. Journal General of Virology (1994), vol. 75, p. 3423 – 3430. 14. LABBE M., CHARPILIENNE D., CRAWFORD S. E. ESTES M. K., COHEN J. Expression of rotavirus VP2 produces empty corelike particles. Journal of Virology (1991), vol. 65, p. 2946 – 2952. 15. LUI M., MATTION N. M., ESTES M. K. Rotavirus VP3 expressed in insect cells possesses guanylyltransferase activity. Virology (1992), vol. 188, p. 77 – 84. 16. PATTON J. Rotavirus VP1 Alone Specifically Binds to the 39 End of Viral mRNA, but the Interaction Is Not Sufficient To Initiate Minus-Strand Synthesis. Journal of Virology (1996), vol. 70, p. 7940 – 7947. 17. KALICA A. R., FLORES J., GREENBERG B. H. Identification of the Rotaviral Gene that Codes for Hemagglutination and Protease-enhanced Plaque Formation. Virology (1983), vol. 125, p. 194 – 205. 18. LIZANO M., LÓPEZ S., ARIAS C. F. The Amino-terminal Half of Rotavirus SA114fM VP4 Protein Contains a Hemagglutination Domain and Primes for Neutralizing Antibodies to the Virus. Journal of Virology (1991), vol. 65, p. 1383 – 1391. 19. MACKOW E. R., BARNETT J. W., CHAN H., GREENBERG H. B. The Rhesus Rotavirus Outer capsid Protein VP4 Functions as a Hemagglutinin and is Antigenically Conserved when Expressed by a Baculovirus Recombinant. Journal of Virology (1989), vol. 63, p. 1661 – 1668. 20. ARIAS C. F., ROMERO P., ALVAREZ V., LÓPEZ S. Trypsin Activation Pathway of Rotavirus Infectivity. Journal of Virology (1996), vol. 70, p. 5832 – 5839. 21. ESTES M. K., GRAHAM D. Y., MASON B. Proteolityc Enhancement of Rotavirus Infectivity: Molecular mechanisms. Journal of Virology (1981), vol. 39, p. 879 – 888. 22. FLORE L., GREEBERG H. B., MACKOW E. R. The VP8 Fragment of VP4 is the Rhesus Rotavirus Hemagglutinin. Virology (1991), vol. 181, p. 553 – 563. 23. CRAWFORD S. E., MUKHERJEE S. K., ESTES M. K., PRASAD B. V. Trypsin Cleavage Stabilizes the Rotavirus VP4 Spike. Journal of Virology (2001), vol. 75, p. 6052 – 6061. 24. PESAVENTO J. B., CRAWFORD S. E., ROBERTS E., ESTES M. K. pH-Induced Conformational Change of the Rotavirus VP4 Spike: Implications for Cell Entry and Antibody Neutralization. Journal of Virology (2005), vol. 79, p. 8572 – 8580. 25. LOO D. D. F., WRIGHT E. M. Water Pumps. Journal Physiological (2002), vol. 542, p. 53 – 60. 26. RAMING R. Pathogenesis of Intestinal and Systemic Rotavirus infection. Journal of Virology (2004), vol. 78, p. 10213 – 10220. 27. BOSHUIZEN J., REIMERINK J., EINERHAND W. Changes in Small Intestinal Homeostasis, Morphology and Gene Expression during Rotavirus Infection of Infant Mice. Journal of Virology (2003), vol. 77, p. 13005 – 13016. 28. MIRKOVITCH V., WINISTORFER B., SAEGESSER F. Response of the Intestinal Mucosa to Ischemia. Gut (1981), vol. 22, p. 512 – 527. 29. HALAIHEL N., LIEVIN V., ALVARADO F. Rotavirus Infection Impairs Intestinal Brush Border Membrane Na+ Solute Cotransport Activities in Young Rabbits. Am. J. Physiol. Gastrointest. Liver Physiol. (2000), vol. 279, p. G587 – G596. 30. HALAIHEL N., LIEVIN V., ALVARADO F. Direct Inhibitory Effect of Rotavirus NSP4 (114 - 135) Peptide on the Na+ - D- Glucose Symporter of Rabbit Intestinal Brush Border Membrane. Journal of Virology (2000), vol. 74, p. 9464 – 9470. 31. COLLINS J., WALLIS T., STEPHEN J. Intestinal Enzymes Profiles in Normal and Rotavirus – Infected Mice. J. Pediatric Gastroenterology Nutr. (1988), vol. 7, p. 264 – 272. 32. KATYAL R., RANA S., SINGH V. Effect of Rotavirus Infection on Small Gut Pathophysiology in a Mouse Mode. Journal Gastroenterology Hepatology (1999), vol. 14, p. 779 – 784. 33. CIARLET M., ESTES M. K. Human and Most Animal Rotavirus strains do not Require the Presence of Sialic Acid on the Cell Surface for Efficient Infectivity. Journal of General Virology (1999), vol. 80, p. 943 – 948. 34. DELORME C., et al. Glycosphingolipid Binding Specificities of Rotavirus: Identification of a Sialic Acid – Binding Epitope. Journal of Virology (2001), vol. 75, p. 2276 – 2287. 35. CIARLET M. et al. Initial Interaction of Rotavirus Strains with N-acetylneuraminic (sialic) Acid Residues on the Cell Surface Correlates with VP4 Genotype, not Species of Origin. Journal of Virology (2002), vol. 76, p.4087–4095. 36. ROLSMA M.D. et al. Structure and Function of a Ganglioside Receptor for Porcine Rotavirus. Journal of Virology (1998), vol. 72, p. 9079–9091. 37. GUO, C. et al. Ganglioside GM1a on the Cell Surface is Involved in the Infection by Human Rotavirus KUN and MO Strains. Journal of Biochemistry. (Tokyo) (1999), vol. 126, p. 683–688. 38. COULSON, B.S. et al. Rotavirus Contains Integrin Ligand Sequences and a Disintegrin-like Domain that are Implicated in Virus Entry into Cells. Proc. Natl. Acad. Sci. U. S. A (1997), vol. 94, p. 5389–5394. 39. HEWISH M.J. et al. Integrins α2β1 and α4β1 can mediate SA11 Rotavirus Attachment and Entry into Cells. Journal of Virology (2000), vol. 74, p. 228–236. 40. GRAHAM K. L. et al. Integrin-using Rotaviruses Rind α2β1 Integrin alpha2 I Domain via VP4 DGE Sequence and Recognize αXβ2 and αVβ3 by using VP7 During Cell Entry. Journal of Virology (2003), vol. 77, p. 9969–9978. 41. CIARLET M. et al. VLA-2 (α2β1) Integrin Promotes Rotavirus Entry into Cells but is not Necessary for Rotavirus Attachment. Journal of Virology (2002), vol. 76, p. 1109– 1123. 42. ZÁRATE, S. et al. Integrin α2β1 Mediates the Cell Attachment of the Rotavirus Neuraminidase-Resistant Variant nar3. Virology (2000), vol. 278, p. 50–54. 43. GUERRERO, C.A. et al. Heat Shock Cognate Protein 70 is Involved in Rotavirus Cell Entry. Journal of Virology (2002), vol. 76, p. 4096–4102. 44. FIORE L. et al. The VP8 Fragment of VP4 is the Rhesus Rotavirus Hemagglutinin. Virology (1991), vol. 81, p. 553–563. 45. FUENTES PANANA E.M. et al. Mapping the Hemagglutination Domain of Rotaviruses. Journal of Virology (1995), vol. 69, p. 2629–2632. 46. ZÁRATE S. et al. Interaction of Rotaviruses with Hsc70 During Cell Entry is Mediated by VP5. Journal of Virology (2003), vol. 77, p. 7254–7260 47. JOLLY C.L. et al. Selection of Rotavirus VP4 Cell Receptor Binding Domains for MA104 Cells using a Phage Display Library. Journal of Virology Methods (2001), vol. 98, p. 41–51. 48. GUERRERO C.A. et al. Integrin alpha(v)beta(3) Mediates Rotavirus Cell Entry. Proc. Natl. Acad. Sci. U. S. A (2000), vol. 97, p. 14644–14649. 49. GAVRILOVSKAYA I.N. et al. β3 Integrins Mediate the Cellular Entry of Hantaviruses that cause Respiratory Failure. Proc. Natl. Acad. Sci. U. S. A (1998), vol. 95, p. 7074– 7079. 50. GUERRERO C.A. et al. Biochemical Characterization of Rotavirus Receptors in MA104 Cells. Journal of Virology (2000), vol. 74, p. 9362–9371. 51. SIMONS K. and IKONEN E. Functional Rafts in Cell Membranes. Nature (1997), vol. 387, p. 569–572. 52. ISA P. et al. Rotaviruses Associate to Lipid Membrane Microdomains During Cell Entry. Virology (in press). 53. MANES S. et al. Pathogens: raft hijackers. Nat. Rev. Immunol (2003), vol. 3, p. 557– 568. 54. CHOI J and LEE S. Secretory and Extracellular Production of Recombinant Proteins Using Escherichia coli. Applied Microbiology and Biotechnology (2004), vol. 64, p. 625–635. 55. MAKRIDES C. Strategies for Achieving High-level Expression of Genes in Escherichia coli. Microbiological Review (1996), vol. 60, p. 512–538. 56. GERIGK M, BUJNICKI R, GANPO-NKWENKWA E, BONGAERTS J, SPRENGER G and TAKORS R. Process Control for Enhanced L-phenylalanine Production Using Different Recombinant Escherichia coli. Biotechnology and Bioengineering (2002), vol. 80, p. 746–754. 57. JEONG J and LEE Y. High-level Production of Human Leptin by Fedbatch Cultivation of Recombinant Escherichia coli and its Purification. Applied and Environmental Microbiology (1999), vol. 65, p. 3027–3032. 58. JEONG J, CHOI H, YOO M, KEUM C, YOO C, LEE Y and SUNG H. Constitutive Production of Human Leptin by Fed-batch Culture of Recombinant rpoS− Escherichia coli. Protein Expression and Purification (2004), vol. 36, p. 150–156. 59. MIJTS N and SCHMIDT-DANNERT C. Engineering of secondary metabolite pathway. Current Opinion in Biotechnology (2003), vol. 14, p. 597–602. 60. BANEYX F. Recombinant Protein Expression in Escherichia coli. Current Opinion in Biotechnology (1999), vol. 10, p. 411 – 421. 61. PEREDELCHUK M and BENNETT G. A Method for Construction of E. coli Strain with Multiple DNA Insertions in the Chromosome. Gene (1997), vol. 187, p. 231 – 238. 62. SORENSE H and MORTENSE K. Advanced Genetic Strategies for Recombinant Protein Expression in Escherichia coli. Journal of Biotechnology (2005), vol. 115, p. 113 – 128. 63. GROSSMAN T, KAWASAKI S, PUNREDDY R and OSBURNE S. Spontaneous cAMP-dependet Derepression of Gene Expression in Stationary Phase Plays a Role in recombinant Expression Instability. Gene (1998), vol. 209, p. 95 – 103. 64. KUO J, CHANG Y, TSENG C. Growth Rate Regulation of lac Operon Expression In Escherichia coli Is Cyclic AMP Dependent. FEBS Letters (2003), vol. 553, p. 397 – 401. 65. HANNING G and MAKRIDES C. Strategies for Optimizing Heterologous Protein Expression in Escherichia coli. Trends Biotechnology (1998), vol. 16, p. 54 – 60. 66. JONASSON P, LILIJEQVIST S, NYGREN A and STAHL S. Genetic Design for Facilitated Production and Recovery of Recombinant Proteins in Escherichia coli. Biotechnology and Applied Biochemistry (2002), vol. 35, p. 91–105. 67. VILLAVERDE A and CARRIO M. Protein Aggregation in Recombinant Bacteria: Biological Role of Inclusion Bodies. Biotechnology Letters (2003), vol. 25, p. 1385– 1395. 68. MORGAN – KISS R, WADLER C and CRONAN J. Long-term and Homogeneous Regulation of the Escherichia coli araBAD Promoter by Use of a Lactose Transporter of Relaxed Specificity. Proc. Natl. Acad. Sci. U. S. A. (2002), vol. 99, p. 7373–7377. 69. KHLEBNIKOV A and KEASLING D. Effect of lacY Expression on Homogeneity of Induction from the P(tac) and P(trc) Promoters by Natural and Synthetic Inducers. Biotechnology Progress (2002), vol.18, p. 672–674. 70. EHRNSPERGER M, GRABER S, GAESTEL M and BUCHNER J. Binding of Nonnative Protein to Hsp25 During Heat Shock Creates a Reservoir of Folding Intermediates for Reactivation. EMBO J. (1997), vol. 16, p. 221–229. 71. VEINGER L, DIAMANT S, BUCHNER J and GOLOUBINOFF P. The Small Heatshock Protein IbpB From Escherichia coli Stabilizes Stress-denatured Proteins for Subsequent Refolding by a Multichaperone Network. Journal of Biological Chemistry (1998), vol. 273, p. 11032–11037. 72. TERPE K. Overview of Tag Protein Fusions: From Molecular and Biochemical Fundamentals to Commercial Systems. Applied Microbiology and Biotechnology (2003), vol. 60, p. 523–533. 73. SORENSE H, SPERLING-PETERSEN H and MORTENSEN K. A Favorable Solubility Partner for the Recombinant Expression of Streptavidin. Protein Expression and Purification (2003), vol. 32, p. 252–259. 74. CARBONELL X and VILLAVERDE A. Protein Aggregated into Bacterial Inclusion Bodies does not Result in Protection From Proteolytic Digestion. Biotechnology Letters (2002), vol. 24, p. 1939–1944. 75. KANE F and HARTLEY L. Formation of Recombinant Protein Inclusion Bodies in Escherichia coli. Trends in Biotechnology (1988), vol. 6, p. 95–101. 76. BESSETTE H, ASLUND F, BECKWITH J and GEORGIOU G. Efficient Folding of Proteins with Multiple Disulfide Bonds in the Escherichia coli Cytoplasm. Proceedings of the National Academy of Sciences of the United States of America (1999), vol. 96, p. 13703–13708. 77. WALL G and PLUCKTHUN A. Effects of Overexpressing Folding Modulators on the in vivo Folding of Heterologous Proteins in Escherichia coli. Current Opinion in Biotechnology (1995), vol. 6, p. 507–516. 78. DERMAN I, PRINZ A, BELIN D and BECKWITH J. Mutations that allow Disulfide Bond Formation in the Cytoplasm of Escherichia coli. Science (1993), vol. 262, p. 1744–1747. 79. PROBA K, GE M and PLUCKTHUN A. Functional Antibody Singlechain Fragments from the Cytoplasm of Escherichia coli: Influence of Thioredoxin Reductase (TrxB). Gene (1995), vol. 159, p. 203–207. 80. BOWDEN A and GEORGIOU G. Folding and Aggregation of Beta lactamasa in the Periplasmic Space of Escherichia coli. Journal of Biological Chemistry (1990), vol. 265, p. 16760–16766. 81. SCHEIN H. Production of Soluble Recombinant Proteins in Bacteria. Biotechnology (1989), vol. 7, p. 1141–1149. 82. MURBY M, UHLEN M and STAHL S. Upstream Strategies to Minimize Proteolytic Degradation Upon Recombinant Production in Escherichia coli. Protein Expression and Purification (1996), vol. 7, p. 129–136. 83. UHLEN M and MOKS T. Gene Fusions for Purpose of Expression Introduction. Methods in Enzymology (1990), vol. 185, p. 129–143. 84. CHOI J, CHANG K and YUP S. Production of recombinant proteins by high cell density culture of Escherichia coli. Chemical Engineering Science (2006), vol. 61, p. 876 – 885. 85. CHEN G, HAYHURST A, THOMAS G, GARVEY R, IVERSON L and GEORGIOU G. Isolation of high-affinity Ligand Binding Proteins by Periplasmic Expression with Cytometric Screening (PECS). Nature Biotechnology (2001), vol. 19, p. 537–542. 86. SROGA E and DORDICK J. A Strategy for in vivo Screening of Subtilisin E Reaction Specificity in E. coli Periplasm. Biotechnology and Bioengineering (2002), vol. 78, p. 761–769. 87. KUJAU J, HOISCHEN C, RIESENBERG D and GUMPERT J. Expression and Secretion of Functional Miniantibodies McPC603scFvDhlx in Cellwall- Less L-form Strains of Proteus mirabilis and Escherichia coli: a Comparison of the Synthesis Capacities of L-form Strains with an E. coli Producer Strain. Applied Microbiology and Biotechnology (1998), vol. 49, p. 51–58. 88. CEREGHINO G and CREGG J. Applications of Yeast in Biotechnology: Protein Production and Genetic Analysis. Current Opinion in Biotechnology (1999), vol, 10, p. 422–427. 89. ECKART R and BUSSINEAU M. Quality and Authenticity of Heterologous Proteins Synthesized in Yeast. Current Opinion Biotechnology (1996), vol. 7, p. 525-530. 90. CREGG M. Expression in the methylotrophic yeast Pichia pastoris. Gene Expression Systems: Using Nature for the Art of Expression (1999). Edited by Fernandez JM, Hoeffler JP. San Diego, CA: Academic Press, p. 157-191. 91. CREGG M and HIGGINS R. Production of foreign proteins in the yeast Pichia pastoris. Can J Bot (1995), vol. 73 (suppl 1) p .891-897. 92. GELLISEN G, HOLLENBERG P and JANOWICZ A. Gene Expression in Methylotrophic Yeasts. Bioprocess Technology (1995), vol. 22, p.195-239. 93. MULLER S, SANDAL S, KAMP-HANSEN P and DALBOGE H. Comparison of Expression Systems in the Yeasts Saccharomyces cerevisiae, Hansenula polymorpha, Klyveromyces lactis, Schizosaccharomyces pombe and Yarrowia lipolytica. Cloning of two novel promoters from Y. lipolytica. Yeast (1998), vol. 14, p. 1267-1283. 94. PIONTEK M, HAGEDORN J, HOLLENBERG CP, GELLISSEN G and STRASSER M. Two Novel Gene Expression Systems Based on the Yeasts Schwanniomyces occidentalis and Pichia stipitis. Applied Microbiology and Biotechnology (1998), vol. 50, p. 331-338. 95. GELLISEN G and HOLLENBERG C. Application of Yeasts in Gene Expression Studies: A Comparison of Saccharomyces cerevisiae, Hansenula polymorpha, and Kluyveromyces lactis — a review. Gene (1997), vol. 190, p. 87-97. 96. SUDBERY P. The Expression of Recombinant Proteins in Yeasts. Current Opinion in Biotechnology (1996), vol. 7, p. 517-524. 97. LUCKOW V and SUMMER M. Trends in the Development of Baculovirus Expression Vector. Biotechnology (1988), vol. 6, p. 47 – 55. 98. O´REILLY D, MILLER L and LUCKOW V. Baculovirus Expression Vectors: A Laboratory Manual. W. H. Freeman, New York (1992). 99. JAVIS D. Baculovirus Expression Vectors. The Baculoviruses, ed. Miller L.K, Plenum Press, New York (1997), p. 389 – 431. 100. ALTMANN F, SCHWIHLA H, STAUDACHER E, GLOSSL J and MARZ L. Insect Cells Contain an Unusual, Membrane – Bound - N – Acetylglucosaminidase Probably Involved in the Processing of Protein N – Glycans. The Journal of Biological Chemistry (1995), vol. 270, p. 17344 – 17349. 101. HIGGINS M, DERMIR M and TATE C. Calnexin Co – expression and the Use of Weaker Promoters Increase Expression of Correctly Assambled Shaker Potassium Channel in Insect Cells. Biochimia et Biophysica Acta (2003), vol. 1610, p. 124 – 132. 102. HOFMANN C et al. Efficient Gene Transfer Into Human Hepatocytes by Baculovirus Vectors. Proc. Natl. Acad. Sci. USA (1995), vol. 92, p. 10099 – 10103. 103. BOYCE F and BUCHER N. Baculovirus – mediated Gene Transfer Into Mammalian Cells. Proc. Natl. Acad. Sci. USA (1996), vol. 93, p. 2348 – 2352. 104. KOST T and CONDREAY J. Recombinant Baculovirus as Mammalian Cell Gene – delivery Vectors. Trends Biotechnology (2002), vol. 20, p. 173 – 180. 105. CONDREAY J, WITHERSPOON S, CLAY W and KOST T. Transient and Stable Gene Expression in Mammalian Cells Transduced with a Recombinant Baculovirus Vector. Proc. Natl. Acad. Sci. USA (1999), vol. 96, p. 127 – 132. 106. BOUBLIK Y, BONITO P and JONES I. Eucaryotic Virus Display: Engineering the Major Surface Glycoprotein of the Autographa californica Nuclear Polyhedrosis Virus (AcNPV) for the Presentation of Foreign Proteins on the Virus Surface. Biotechnology (1995), vol. 13, p. 1079 – 1084. 107. OKER – BLOM C, AIRENNE K and GRABHERR R. Baculovirus Display Strategies: Emerging Tools for Eucaryotic Libraries and Gene Delivery. Brief. Funct. Genomic. Proteomic (2003), vol. 2, p. 244 – 253. 108. MAEDA S, et al. Production of Human Alpha – Interferon in Silkworn Using a Baculovirus Vector. Nature (1985), vol. 315, p. 592 – 594. 109. WU D, et al. Expression of Biologically Active Recombinant Equine Interferon - by Two Different Ecpression Systems Using Insect Cells and Silkworm Larvae. Cytokine (2002), vol. 20, p. 63 – 69. 110. KOST T, PATRICK J and JARVIS D. Baculovirus as Versatile Vectors for Protein Expression in Insect and Mammalian Cells. Nature Biotechnology (2005), vol. 23, p. 567 – 575. 111. FENNER F. Poxviruses. In Virology, ed. B. N. Fields, D. M. Knipe, R. M. Chanock, J. L. Melnick, B. Roizman, R. E. Shope, New York (1985) p. 661 – 84. 112. COCHRAN A, PUCKETT C and MOSS B. In Vitro Mutagenesis of the Promoter Region for a Vaccinia Virus Gene: Evidence for Tandem Early and Late Regulatory Signals. Journal of Virology (1985), vol. 54, p 30 – 37. 113. PANICALI D and PAOLETTI E. Construction of Poxviruses as Cloning Vectors: Insertion of the Thymidine Kinase Gene from Herpes Simplex Virus into the DNA of Infectious Vaccinia Virus. Proc. Natl. Acad. Sci. USA (1982), vol. 79, p. 4927-4931. 114. MACKETT M, SMITH L and MOSS, B. Vaccinia Virus: A Selectable Eukaryotic Cloning and Expression Vector. Proc. Natl. Acad. Sci. USA (1982), vol. 79, p. 74157419. 115. MACKETT M, SMITH L and MOSS, B. A General Method for the Production and Selection of Infectious Vaccinia Virus Recombinants Expressing Foreign Genes. Journal of Virology (1982), vol. 49, p. 857 – 864. 116. MACKETT M, SMITH L and MOSS, B. The Construction and Characterization of Vaccinia Virus Recombinants Expressing Foreign Genes. DNA Cloning, vol. II (1985), ed. D. M. Glover, Oxford: IRL Press. p. 191 - 212. 117. WEIR P and MOSS B. Regulation of Expression and Nucleotide Sequence of a Late Vaccinia Virus Gene. Journal of Virology (1984), vol. 51, p. 662 – 669. 118. SMITH L, MACKETT M and MOSS B. Infectious Vaccinia Virus Recombinants that Express Hepatitis B Virus Surface Antigen. Nature (1983), vol. 302, p. 490 -495. 119. STEPHENS B, COMPANS W, EARL P and MOSS B. Surface Expression of Viral Glycoproteins is Polarized in Epithelial Cells Infected with Recombinant Vaccinia Viral Vectors. EMBO J. (1986), vol. 5, p. 237 – 245. 120. CHAKRABARTI S, ROBERT - GUROFF M, WONG - STALL F, GALLO C and MOSS, B. Expression of the HTLV-III Envelope Gene by a Recombinant Vaccinia Virus. Nature (1986), vol. 320, p. 535 – 537. 121. THOMAS G, HERBERT E and HRUBY E. Expression and Cell-Type-Specific Processing of Human Preproenkephalin With a Vaccinia Recombinant. Science (1986), vol. 232, p. 1641 – 1646. 122. COLIGAN J, DUNN M, PLOEGH L, SPEICHER W and WINGFIELD T. Current Protocols in Protein Science (1998), ed. John Wiley & Sons, New York. 123. OKA M, YANG S, NAGATA S, ESAKI N, TANAKA M and SODA K. Overproduction of Thermostable Leucine Dehydrogenase of Bacillus stearothermophilus and its Onestep Purification from Recombinant Cells of Escherichia coli. Biotechnology Applied Biochemistry (1989), vol. 11, p. 307-316. 124. PAIN H. Overview of protein folding. Current Protocols in Protein Science (1995), ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield, John Wiley & Sons, New York, p. 6.4.1 – 6.4.7. 125. PALMER I and WINGFIELD T. Preparation and Extraction of Insoluble (InclusionBody) Proteins from Escherichia coli. Current Protocols in Protein Science (1995), ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield, John Wiley & Sons, New York, p. 6.3.1 – 6.3.15 126. SASSENFELD M. Engineering Proteins for Purification. Trends Biotechnology (1990), vol. 8, p. 88-93. 127. WALTER H and JOHANSSON G. Aqueous Two-phase Systems. Methods Enzymology (1994), vol. 228, p. 1 - 725. 128. WINGFIELD T. Use of Protein Folding Reagents. Current Protocols in Protein Science (1995), ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield. John Wiley & Sons, New York, p. A.3A.1- A.3A.4. 129. WINGFIELD T. Preparation of Soluble Proteins from Escherichia coli. Current Protocols in Protein Science (1995), ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield. John Wiley & Sons, New York, p. 6.2.1-6.2.15. 130. WINGFIELD T. Overview of the Purification of Recombinant Proteins Produced in Escherichia coli. Current Protocols in Protein Science, ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield. John Wiley & Sons, New York, p. 6.1.1- 6.1.22. 131. WINGFIELD T, PALMER I and LIANG S. Folding and Purification of Insoluble (Inclusion Body) Proteins from Escherichia coli. Current Protocols in Protein Science (1995), ed. J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield. John Wiley & Sons, New York, p. 6.5.1-6.5.27. 132. CHUNG C.T, SUZANNE L, NIEMELA, MILLER R. One – Step Preparation Of Competent Escherichia coli: Transformation And Storage Of Bacterial Cells In The Same Solution. Proc. Natl. Acad. Sci. USA (1989), vol. 86, p. 2172 – 2175. 133. WOOD T AND PERETTI S. Effect of Chemically-Induced, Cloned-Gene Expression on Protein Synthesis in E. coli. Biotechnology and Bioengineering (1991), vol. 38, p. 397-412. 134. JINKS-ROBERTSON S, GOURSE R and NOMURA M. Expression of rRNA and tRNA Genes in Escherichia coli: Evidence for Feedback Regulation by Products of rRNA Operons. Cell (1983), vol. 33, p. 865. 135. CASADABAN M, MARTINEZ-ARIAS A, SHAPIRA S and CHOU J. -Galactosidase Gene Fusions for Analyzing Gene Expression in Escherichia coli and Yeast. Methods Enzymol (1983), vol. 100, p. 293 - 315. 136. GAUSING K. Regulation of Ribosome Production in Escherichia coli: Synthesis and Stability of Ribosomal RNA and of Ribosomal Protein Messenger RNA at Different Growth Rates. Journal Molecular Biology (1977), vol. 115, p. 335 – 342. 137. NILSSON G, BELASCO J, COHEN S and VON GABAIN A. Effect of Premature Termination of Translation on mRNA Stability Depends on the Site of Ribosome Release. Proc. Nat. Acad. Sci (1987), vol. 84, p. 4890 – 4898. 138. KUO J, CHANG Y, TSENG C. Growth Rate Regulation of lac Operon Expression In Escherichia coli Is Cyclic AMP Dependent. FEBS Letters (2003), vol. 553, p. 397 – 401. 139. MOHAN S, KUMAR A. Solubilization And Refolding Of Bacterial Inclusion Body Proteins. Journal of Bioscience and Bioengineering (2005), vol. 99, p. 303 – 310. 140. MAEDA Y, KOGA H, YAMADA H, UEDA T and IMOTO T. Effective Renaturation of Reduced Lysozyme by Gentle Removal of Urea. Protein Eng. (1995), vol. 8, p. 201– 205. 141. EJIMA D, WATANABE M, SATO T, DATE M, YAMADA N and TAKAHARA Y. High Yield Refolding and Purification Process for Recombinant Human Interleukin-6 Expressed in Escherichia coli. Biotechnology and Bioengineering (1999), vol. 62, p. 301–310. 142. BLANCHARD H, YU X, COULSON B and ITZSTEIN M. Insight Into Host Cell Carbohydrate – Recognition By Human And Porcine Rotavirus From Crystal Structures Of The Virion Spike Associated Carbohydrate – Binding Domain (VP8*). Journal Molecular Biology (2001), vol. 367, p. 1215 - 1226. 143. GORZIGILIA M, LARRALDE G, KAPIKIAN A and CHANOCK R. Antigenic Relationships Among Human Rotaviruses as Determined by Outer Capsid Protein VP4. Proc. Nati. Acad. Sci. USA (1990), vol. 87, p. 7155 – 7159. 144. KHODABANDEHLOO M, SHAHRABADI M, KEYVANI H and BAMBAI B. Cloning and Expression of Simian Rotavirus Spike Protein (VP4) in Insect Cells by Baculovirus Expression System. Iran. Biomed. J. (2009), vol. 13, p. 9-18. 145. KOVACS – NOLAN J, SASAKI E, YOO D and MINE Y. Cloning And Expression Of Human Rotavirus Spike Protein, VP8*, In Escherichia coli. Biochemical and Biophysical Research Communications (2001), vol. 282, p. 1183 – 1188. 146. LARRALDE G, LI B, KAPIKIAN A and GORZIGLIA M. Serotype – Specific Epitope(s) Present on the VP8 Subunit of Rotavirus VP4 Protein. Journal of Virology (1991), vol. 65, p. 3213 – 3218. 147. MONNIER N, HIGO – MORIGUCHI K, SUN Z, VENKATARAM B, TANIGUCHI K and DORMITZER P. High – Resolution Molecular and Antigen Structure of the VP8* Core of a Sialic Acid Independent Human Rotavirus Strain. Journal of Virology (2006), vol. 80, p. 1513 – 1523. 148. NAVA P, LOPEZ S, ARIAS C, ISLAS S and GONZALEZ L. The Rotavirus Surface Protein Modulates the Gate and Fence Function of Tight Junctions in Epithelial Cells. Journal of Cell Science (2004), vol. 117, p. 5509 – 5519. 149. FAVACHO A, KURTENBACH E, SARDI S and GOUVEA V. Cloning, Expression and Purification Of Recombinant Bovine Rotavirus Hemagglutinin, VP8*, In Escherichia coli. Protein Expression and Purification (2006), vol. 46, p. 196 – 203. 150. DORMITZER P, SUN Z, WAGNER G and HARRISON S. The Rhesus Rotavirus VP4 Sialic Acid Binding Domain Has a Galectin Fold With a Novel Carbohydrate Binding Site. The EMBO Journal (2002), vol. 21, p. 885 – 897. 151. GOLANTSOVA N, GORBUNOVA E and MACKOW R. Discrete Domains within the Rotavirus VP5* Direct Peripheral Membrane Association and Membrane Permeability. Journal of Virology (2004), vol. 78, p. 2037 – 2044. 152. YODER J and DORMITZER P. Alternative Intermolecular Contacts Underlie The Rotavirus VP5* Two – To – Three Fold Rearrangement. The EMBO Journal (2006), vol. 25, p. 1559 – 1568. 153. DOWLING W, DENISOVA E, LAMONICA R and MACKOW E. Selective Membrane Permeabilization by the Rotavirus VP5* Protein Is Abrogated by Mutations in an Internal Hydrophobic Domain. Journal of Virology (2000), vol. 74, p. 6368 – 6376. 154. DORMITZER P, GREENBERG H and HARRISON S. Proteolysis of Monomeric Recombinant Rotavirus VP4 Yields an Oligomeric VP5* Core. Journal of Virology (2001), vol. 75, p. 7339 – 7350. 155. KAMATA T and TAKAD Y. Direct Binding of Collagen to the I Domain of Integrin 21 (VLA-2, CD49b/CD29) in a Divalent Cation-independent Manner. The Journal of Biological Chemistry (1994), vol. 269, p. 26006 – 26010. 156. ROHMAN M and HARRISON – LAVOIE K. Separation of Copurifying GroEL from Glutathione-S Transferase Fusion Proteins. Protein Expression and Purification (2000), vol. 20, p. 45 – 47. 157. KIANG J and TSOKOS G. Heat Shock Protein 70 kDa: Molecular Biology, Biochemistry, and Physiology. Pharmacol. Ther. (1998), Vol. 80, p. 183–201. 158. ESPEJO J, SOSA A, CARRASCAL A, CIFUENTES C and BARRETO A. Producción de la Proteína Recombinante Humana Hsc70 y su Aplicación en el Desarrollo de un Sistema de Detección por ELISA. Revista Facultad de Ciencias Pontificia Universidad Javeriana (2004), vol. 9, p. 69 – 80. 159. WANG C and LEE M. High-level Expression of Soluble Rat Hsc7O in Escherichia coli: Purification and Characterization of the Cloned Enzyme. Biochem. J. (1993), vol. 294, p. 69-77. 160. IMAMOTO N, MATSUOKA T, KURIHARA K, KOHNO M, MIYAGI F, SAKIYAMA Y, OKADA S and YONEDA Y. Antibodies against 70-kD heat shock cognate protein inhibit mediated nuclear import of karyophilic proteins. The Journal of Cell Biology (1992), vol. 119, p. 1047–1061. 161. KOSE S, IMAMOTO N, TACHIBANA T, SHIMAMOTO T and YONEDA Y. Ranunassisted Nuclear Migration of a 97-kD Component of Nuclear Poretargeting Complex. The Journal of Cell Biology (1997), vol. 139, p. 841–849. 162. PEREZ J, ROMERO P, LÓPEZ S and ARIAS C. The Peptide-Binding and ATPase Domains of Recombinant hsc70 Are Required To Interact with Rotavirus and Reduce Its Infectivity. Journal of Virology (2006), vol. 80, p. 3322 – 3331. 163. TOSSER G, LABBE M, BREMONT M and COHEN J. Expression of the Major Capsid Protein VP6 of Group C Rotavirus and Synthesis of Chimeric Single-Shelled Particles by Using Recombinant Baculoviruses. Journal of Virology (1992), vol. 66, p. 5825 – 5831. 164. CRAWFORD S, LABBE M, COHEN J, BURROUGHS M, ZHOU Y and ESTES M. Characterization of Virus-Like Particles Produced by the Expression of Rotavirus Capsid Proteins in Insect Cells. Journal of Virology (1994), vol. 68, p. 5945 – 5952. 165. ESTES M, CRAWFORD S, PENARANDA M, PETRIE B, BURNS J, CHAN W, ERICSON B, SMITH G and SUMMERS M. Synthesis and Immunogenicity of the Rotavirus Major Capsid Antigen Using a Baculovirus Expression System. Journal of Virology (1987), vol. 61, p. 1488 – 1494. 166. PETITPAS I, LEPAULT J, VACHETTE P, CHARPILIENNE A, MATHIEU M, KOHLI E, POTHIER P, COHEN J and REY F. Crystallization and Preliminary X-Ray Analysis of Rotavirus Protein VP6. Journal of Virology (1998), vol. 72, p. 7615 – 7619. 167. CHOI A, BASU M, MCNEAL M, FLINT J, VANCOTT J, CLEMENTS J and WARD R. Functional Mapping of Protective Domains and Epitopes in the Rotavirus VP6 Protein. Journal of Virology (2000), vol. 74, p. 11574 – 11580. 168. STROUP E. Synthesis of the Major Inner Capsid Protein VP6 of the Human Rotavirus Wa in Escherchia coli. Gene (1988), vol. 68, p. 345 – 356. 169. SMITH R, KISTER S and CAROZZI N. Cloning and Expression of the Major Inner Capsid Protein of SA-11 Simian Rotavirus in Escherichia coli. Gene (1989), vol. 79, p. 239 – 248. 170. AFFRANCHINO J and GONZÁLEZ S. Deletion Mapping of Functional Domains in the Rotavirus Capsid Protein VP6. The Journal of General Virology (1997), vol. 78, p. 1949 – 1955. 171. GONZÁLEZ S and AFFRANCHINO J. Assembly of Double-layered Virus-like Particles in Mammalian Cells by Coexpression of Human Rotavirus VP2 and VP6. Journal of General Virology (1995), vol.76, p. 2357 – 2360. 172. GONZÁLEZ S, AFFRANCHINO J, GELDERBLOM H and BURNY A. Assembly of the matrix protein of simian immunodeficiency virus into virus-like particles. Virology (1993), vol. 194, p. 548 - 556. 173. PONCET D, CORTHIER G, CHARPILIENNE A and COHEN J. A Recombinant Vaccinia Virus Expressing the Major Capsid Protein of Simian Rotavirus – Induced anti–rotavirus Antibodies. Virus Research (1990), vol. 15, p. 267 – 274. 174. YU J and LANGRIDGE W. Expression of Rotavirus Capsid Protein VP6 in Transgenic Potato and its Oral Immunogenicity in Mice. Transgenic Research (2003), vol. 12, p. 163 – 169. 175. MATHIEU M, PETITPAS I, NAVAZA J, LEPAULT J, KOHLI E, POTHIER P, COHEN J and REY F. Atomic Structure Of The Major Capsid Protein Of Rotavirus: Implications For The Architecture Of The Virion. The EMBO Journal (2001), vol. 20, p. 1485 – 1497. 176. FRANGIONI J, NEEL B. Solubilization And Purification Of Enzymatically Active Glutathione S – Transferase (pGEX) Fusion Proteins. Analytical Biochemistry (1993), vol. 210, p. 179 – 187. 177. McNALLY E, SOHN R, FRANKEL S and LEINWAND L. Expression of myosin and actin in Escherichia coli. Methods Enzymology (1991), vol. 196, p. 368–389. 178. TSUMOTO K, EJIMA D, KUMAGAI I and ARAKAWA T. Practical Considerations In Refolding Proteins From Inclusion Bodies. Protein Expression and Purification (2003), vol. 28, p. 1 – 8. 179. TSUMOTO K, ABE R, EJIMA D and ARAKAWA T. Non – Denaturing Solubilization of Inclusion Bodies. Current Pharmaceutical Biotechnology (2010), vol. 4, p. 309 – 312. 180. SMITH D and JOHNSON K. Single-step Purification Of Polypeptides Expressed In Escherichia coli As Fusions With Glutathione S-Transferase. Gene (1988), vol. 67, p. 31 – 40. 181. SEDDON A, CURNOW P and BOOTH P. Membrane Proteins, Lipids and Detergents: Not Just a Soap Opera. Biochim Biophys Acta (2004), vol. 1666, p. 105-17. 182. TANFORD C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. 1st ed. 1973, New York: John Wiley & Sons, Inc. 183. LUNDBÄCK A. K, HANESKOG L, ANDERSSON L, HEIJBEL A and BIRSE D. Optimal Solubilization Screening Strategies For GST-Fusion Membrane Proteins. Life Science News (2003), vol. 15, p. 10 – 12. 184. HELENIUS A, et al. Properties of detergents. Methods Enzymol (1979), vol. 56, p. 734-49. 185. LINKE D. Chapter 34 Detergents: An Overview. Methods in Enzymology (2009), vol. 463, p. 603 – 617. 186. AMERSHAM BIOSCIENCES. Affinity Chromatography: Principles and Methods. p. 47 – 51. 187. PRYOR K and LEITING B. High-level Expression of Soluble Protein in Escherichia coli Using a His6-tag and Maltose-Binding-Protein Double-Affinity Fusion System. Protein Expression and Purification (1997), vol. 10, p. 309-319. 188. LILIE H, SCHWARZ E and RUDOLPH M. Advances in Refolding of Proteins Produced in E. coli. Curr. Opin. Biotechnol. (1998), vol. 9, p. 497-501. 189. BURGESS R. Purification of Overproduced Escherichia coli RNA Polymerase Sigma Factors by Solubilizing Inclusion Bodies and Refolding from Sarkosyl. Methods Enzymol. (1996), vol. 273, p. 145-149. 190. TAO H, WENJUN L, BRANDI N, SIMMONS H, HARRIS T and MASSIAH M. Purifying Natively Folded Proteins from Inclusion Bodies Using Sarkosyl, Triton X-100, and CHAPS. Benchmarks (2010), vol. 48, p. 61 – 64. 191. BANERJEE P, JOO J, BUSE J and BET D. Differential Solubilization of Lipids Along with Membrane Proteins by Different Classes of Detergents. Chemistry and Physics of Lipids, 1995. 77(1): p. 65-78. 192. GARAVITO R and FERGUSON – MILLER S. Detergents as Tools in Membrane Biochemistry. The Journal of Biological Chemistry (2001), vol. 276, p. 32403 - 32406. 193. CUADRAS M, GREENBERG H. Rotavirus Infectious Particles Use Lipid Rafts During Replication For Transport To The Cell Surface in vitro and in vivo. Virology (2003), vol. 313, p. 308 – 321. 194. PIKE L. Lipids Rafts: Heterogeneity On The High Seas. Biochemical Journal (2004), vol. 378, p. 281 – 292. 195. GUERRERO C, GUALTEROS D, GUZMÁN F, RODRIGUEZ L and ACOSTA O. Determinación de la Función de la Proteína VP6 y los Péptidos (280-297) de VP6 (530-559) de VP4 en la Entrada del Rotavirus. Area de Ciencias Básicas y Experimentales. 196. RODRIGUEZ L and GUERRERO C. La Proteína Viral VP6 de Rotavirus Coinmunoprecipita con HSC70 en Células MA104. Revista Facultad de Medicina Universidad Nacional de Colombia (2006), vol. 54, p. 24-31. 197. ACOSTA O, CALDERÓN M, MORENO L and GUERRERO C. UN Modelo del Mecanismo de Entrada de los Rotavirus a la Célula Hospedera. Revista Facultad de Medicina Universidad Nacional de Colombia (2009), vol. 57, p. 124 - 147. 198. ISA P, REALPE M, ROMERO P, LÓPEZ S and ARIAS C. Rotavirus RRV Associates with Lipid Membrane Microdomains During Cell Entry. Virology (2004), vol. 322, p. 370-381. 199. Calderon MN, Acosta O, Guerrero CA, Guzman F. Protein Disulfide Isomerase Activity is Involved in Rotavirus Entry to MA104 Cells. Proceedings of the XIV International Congress of Virology, Istanbul. (2008), p. 163-164. 200. STEWART F, REGINA S and LEINWAND L. The Use Of Sarkosyl In Generating Soluble Protein After Bacterial Expression. Proc. Natl. Acad. Sci. U. S. A (1991), vol. 88, p. 1192 – 1196. 201. GUALTERO D, GUZMÁN F, ACOSTA O and GUERRERO F. Amino Acid Domains 280–297 of VP6 and 531–554 of VP4 are Implicated in Heat Shock Cognate Protein Hsc70-mediated Rotavirus Infection. Archives of Virology (2007), vol. 152, p. 2183 – 2196. 202. TURANO C, COPPARI S, ALTIERI F and FERRARO A. Proteins of the PDI family: Unpredicted non-ER Locations and Functions. J Cell Physiol. (2002), vol. 193, p. 154 - 163. 203. APPENZELLER – HERZOG C and ELLGAARD L. The Human PDI Family: Versatile Packed into a Single Fold. Bichim Biophys Acta (2008), vol. 1783, p. 535 - 548. 204. MANDEL R, RYSERJ, GHANI F, WU M and PEAK D. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide isomerase. Proc Natl Acad Sci USA (1993), vol. 90, p. 4112 - 4116. 205. MARKOVIC I, STANCHEY S, FIELDS H, TIFFANY J, TOMIC M, WEISS D, BRODER C, et al. Thiol/disulfide Exchange is a Prerequisite for CXCR4-tropic HIV-1 Envelopemediated T-cell Fusion During Viral Entry. Blood (2004), vol. 103, p. 1586 - 1594. 206. MIRAZIMI A and SVENSSON L. Carbohydrates Facilitate Correct Disulfide Bond Formation and Folding of Rotavirus VP7. Journal of Virology (1998), vol, 75, p. 3887 3892. 207. MARURI - AVIDAL L, LÓPEZ S and ARIAS C. Endoplasmic Reticulum Chaperones are Involved in the Morphogenesis of Rotavirus Infectious Particles. Journal of Virology (2008), vol. 82, p. 5368 - 5380. 208. SVENSSON L, DORMITZER P, VON BONSDORFF C, MAUNULA L and GREENBERG H. Intracellular Manipulation of Disulfide Bond Formation in Rotavirus Proteins During Assembly. Journal of Virology (1994), vol. 68, p. 5204 - 5215. 209. ISA P, GUTIÉRREZ M, ARIAS C and LÓPEZ S. Rotavirus cell entry. Future Virology (2008), vol. 3, p. 135 - 146. 210. DORMITZER R, NASON B, PRASAD V and HARRISONC. Structural Rearrangements in the Membrane Penetration Protein of a Non-enveloped Virus. Nature (2004), vol. 430, p. 1053 - 1058.