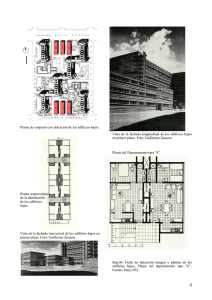
Tesis_Juan Carlos García Gallegos - RUA
Anuncio

SISTEMAS HÍBRIDOS DE POLIANILINA Y NANOESTRUCTURAS DE CARBONO PARA SU APLICACIÓN EN MÚSCULOS ARTIFICIALES Y SUPERCONDENSADORES Juan Carlos García Gallegos Departamento de Ingeniería Química SISTEMAS HÍBRIDOS DE POLIANILINA Y NANOESTRUCTURAS DE CARBONO PARA SU APLICACIÓN EN MÚSCULOS ARTIFICIALES Y SUPERCONDENSADORES Memoria que, para optar al grado de Doctor en Ingeniería Química, presenta: Juan Carlos García Gallegos Alicante, julio de 2012 1 2 D. Ignacio Martín Gullón, Profesor Titular del Departamento de Ingeniería Química y D. Juan A. Conesa, Catedrático del Departamento de Ingeniería Química de la Universidad de Alicante, CERTIFICAN: Que D. Juan Carlos García Gallegos, Ingeniero en Biónica, ha realizado en el Departamento de Ingeniería Química de la Escuela Politécnica Superior de la Universidad de Alicante, bajo nuestra dirección, el trabajo que lleva por título: SISTEMAS HÍBRIDOS DE POLIANILINA Y NANOESTRUCTURAS DE CARBONO PARA SU APLICACIÓN EN MÚSCULOS ARTIFICIALES Y SUPERCONDENSADORES, que constituye su Memoria para aspirar al Grado de Doctor, reuniendo a su parecer, las condiciones necesarias para ser presentada y defendida ante el tribunal correspondiente. Y para que así conste a los efectos oportunos, en cumplimiento de la legislación vigente, firmo el presente certificado en Alicante a 19 de junio de 2012. Fdo.: Ignacio Martín Gullón Profesor Titular de la Universidad de Alicante Fdo.: Juan A. Conesa Catedrático de la Universidad de Alicante 3 4 ÍNDICE 1. Resumen........................................................................................................................... 13 2. Introducción ..................................................................................................................... 17 2.1. Polímeros conductores .............................................................................................. 17 2.1.1. Conductividad eléctrica ...................................................................................... 19 2.1.2. Dopaje y dopantes de los polímeros conductores ............................................... 20 2.1.3. Tipos de dopaje de los polímeros conductores ................................................... 22 2.2. La polianilina ............................................................................................................ 22 2.2.1. Síntesis y procesado de la PAni.......................................................................... 26 2.2.2. Cristalinidad de la PAni...................................................................................... 28 2.2.3. Propiedades eléctricas ....................................................................................... 29 2.2.4. Conductividad eléctrica de la sal de esmeraldina ............................................... 30 2.2.5. Propiedades mecánicas ....................................................................................... 31 2.2.6. Propiedades electroquímicas .............................................................................. 31 2.2.7. Morfología de la PAni ........................................................................................ 32 2.2.7.1. PAni granular ............................................................................................... 32 2.2.7.2. Nanofibras de PAni ...................................................................................... 33 2.2.8. Aplicaciones de la PAni ..................................................................................... 35 2.3. Nanoestructuras de carbono como cargas en materiales compuestos ....................... 35 2.3.1. Nanotubos de carbono ........................................................................................ 36 2.3.1.1. Propiedades eléctricas .................................................................................. 37 2.3.1.2. Propiedades térmicas ................................................................................... 38 2.3.1.3. Propiedades mecánicas ................................................................................ 38 2.3.1.4. Algunas aplicaciones ................................................................................... 39 2.3.1.5. Síntesis de nanotubos de carbono ................................................................ 39 2.3.1.5.1. Descarga en arco eléctrico .................................................................... 39 5 2.3.1.5.2. Ablación láser ........................................................................................ 40 2.3.1.5.3. Producción por CVD ............................................................................. 40 2.3.2. Nanotubos de carbono dopados con nitrógeno (CNx) ........................................ 41 2.3.2.1. Síntesis de CNx............................................................................................. 43 2.3.2.2. Aplicaciones de los CNx .............................................................................. 44 2.3.3. Nanotubos de carbono multicapa funcionalizados con grupos basados en oxígeno (COx-MWCNTs) ............................................................................................. 44 2.3.4. Nanotubos de carbono exfoliados (exMWCNTs) .............................................. 46 2.3.5. Nanofibras de carbono de listones grafíticos helicoidales (HR-CNF) ............... 48 2.3.6. Óxido de grafeno (GO) ....................................................................................... 51 2.3.6.1. Grafeno......................................................................................................... 51 2.3.6.2. Producción de grafeno ................................................................................. 53 2.3.6.3. Óxido de grafeno (GO) ................................................................................ 54 2.4. Compuestos de polianilina ........................................................................................ 55 2.5. Músculos artificiales.................................................................................................. 57 2.5.1. Músculos artificiales de PAni ............................................................................. 60 2.5.2. Músculos artificiales de compuestos de PAni .................................................... 62 2.6. Electrodos para supercondensadores ......................................................................... 63 REFERENCIAS ............................................................................................................... 67 3. Síntesis mecánica de nanocompuestos de polianilina (PAni) y nanotubos de carbono multicapa (MWCNT) y nanotubos de carbono multicapa dopados con nitrógeno (CNxMWCNT) ............................................................................................................................. 89 3.1. ANTECEDENTES .................................................................................................... 89 3.2. EXPERIMENTACIÓN ............................................................................................. 91 3.2.1. Procedimiento general de síntesis de los compuestos ........................................ 91 3.2.2. Experimentos exploratorios ................................................................................ 92 3.2.3. Diseño experimental de superficie de respuesta ................................................. 92 3.2.4. Caracterización ................................................................................................... 92 6 3.3. RESULTADOS Y DISCUSIÓN............................................................................... 93 3.3.1. Experimentos exploratorios ................................................................................... 93 3.3.2. Experimento de optimización ............................................................................. 95 3.4. CONCLUSIONES .................................................................................................. 108 REFERENCIAS ............................................................................................................. 110 4. Síntesis y procesado de nanocompuestos de PAni con distintos tipos de nanotubos de carbono, nanofibras de carbono y óxido de grafeno .......................................................... 115 4.1. ANTECEDENTES .................................................................................................. 115 4.2. EXPERIMENTACIÓN ........................................................................................... 117 4.2.1. Materiales ......................................................................................................... 117 4.2.2. Obtención de los exMWCNTs y los Ani-exMWCNTs .................................... 118 4.2.3. Corte de MWCNTs........................................................................................... 118 4.2.4. Intercalación de Li en los cutMWCNTs ........................................................... 118 4.2.5. Exfoliación de los cutMWCNTs-Li ................................................................. 119 4.2.6. Funcionalización de los exMWCNTs con anilina ............................................ 120 4.3. Síntesis de compuestos de PAni .............................................................................. 120 4.3.1. Proceso 1 .......................................................................................................... 120 4.3.2. Proceso 2 .......................................................................................................... 121 4.3.2.1. Síntesis de PAni con distintos ácidos ........................................................ 122 4.3.2.2. Síntesis de compuestos con nanofilamentos de carbono y GO ................. 122 4.3.2.3. Síntesis de compuestos de PAni con MWCNTs y GO con varios niveles de carga. ....................................................................................................................... 122 4.3.2.4. Síntesis de compuestos de PAni con carga híbrida de GO-MWCNTs. ..... 123 4.4. RESULTADOS ....................................................................................................... 123 4.4.1. Corte de MWCNTs........................................................................................... 123 4.4.2. Exfoliación de cutMWCNTs ............................................................................ 126 4.4.3. Funcionalización de los cutMWCNTs con anilina ........................................... 131 4.4.4. Síntesis de nanocompuestos de PAni ............................................................... 132 7 4.4.4.1. Proceso 1 .................................................................................................... 132 4.4.4.2. Proceso 2 .................................................................................................... 136 4.4.4.2.1. Síntesis de PAni con distintos ácidos .................................................. 136 4.4.4.2.2. Síntesis de compuestos con nanofilamentos de carbono y GO ........... 139 4.4.4.2.3. Síntesis de compuestos de PAni con MWCNTs y GO con varios niveles de carga................................................................................................................ 159 4.4.4.2.4. Síntesis de compuestos de PAni con carga híbrida de GO-MWCNTs. ............................................................................................................................. 161 4.5. CONCLUSIONES................................................................................................... 166 REFERENCIAS ............................................................................................................. 168 ............................................................................................................................................ 174 5. Actuadores lineales y flexionantes de nanocompuestos de PAni y distintas nanoestructuras de carbono ................................................................................................ 175 5.1. ANTECEDENTES .................................................................................................. 175 5.2. EXPERIMENTACIÓN ........................................................................................... 177 5.2.1. Materiales ......................................................................................................... 177 5.2.2. Métodos ............................................................................................................ 177 5.2.3. Actuadores lineales ........................................................................................... 178 5.2.3.1. Elaboración de las probetas cilíndricas ...................................................... 178 5.2.3.2. Medición de la elongación de las probetas. ............................................... 178 5.2.4. Actuadores flexionantes.................................................................................... 179 5.2.4.1. Preparación de los actuadores bicapa (flexionantes) ................................. 179 5.2.4.2. Medición de flexión de los actuadores ....................................................... 179 5.2.4. Optimización..................................................................................................... 180 5.3. RESULTADOS Y DISCUSIÓN ............................................................................. 180 5.3.1. Actuadores lineales. .......................................................................................... 180 5.3.2. Actuadores flexionantes.................................................................................... 184 5.3.3. Optimización..................................................................................................... 187 8 5.4. CONCLUSIONES .................................................................................................. 198 REFERENCIAS ............................................................................................................. 200 ........................................................................................................................................... 204 6. Electrodos de PAni y óxido de grafeno para su aplicación en supercondensadores ..... 205 6.1. ANTECEDENTES .................................................................................................. 205 6.2. EXPERIMENTACIÓN ........................................................................................... 206 6.2.1. Materiales ......................................................................................................... 206 6.2.2. Métodos ............................................................................................................ 206 6.2.2.1. Preparación de electrodos .......................................................................... 206 6.2.2.2. Voltamperometría cíclica de barrido ......................................................... 207 6.2.2.3. Pruebas de Carga/Descarga galvanostática................................................ 208 6.2.2.4. Medición de la conductividad eléctrica de las películas de compuestos ... 208 6.2.2.5. Análisis termogravimétrico de los compuestos de PAni ........................... 208 6.2.2.6. Análisis termomecánico de películas de compuestos de PAni tratadas térmicamente ........................................................................................................... 209 6.3. RESULTADOS Y DISCUSIÓN............................................................................. 209 6.3.1. Voltametría cíclica de películas de compuestos de PAni sin tratamiento térmico .................................................................................................................................... 209 6.3.2. Voltametría cíclica de películas de compuestos de PAni con tratamiento térmico .................................................................................................................................... 214 6.3.3. Capacidades específicas de películas de compuestos de PAni ......................... 221 6.3.4. Carga/descarga de películas de compuestos de PAni/GO (1.1 wt%) con TT . 227 6.3.5. Análisis termogravimétrico de los compuestos de PAni .................................. 230 6.3.6. Análisis termomecánico de compuestos de PAni ............................................. 237 6.4. CONCLUSIONES ............................................................................................... 238 REFERENCIAS ......................................................................................................... 239 ........................................................................................................................................... 242 7. Conclusiones .................................................................................................................. 243 9 7.1. Mecanosíntesis de compuestos de PAni y CNTs .................................................... 243 7.2. Síntesis de compuestos de PAni y nanoestructuras de carbono por el método de polimerización interfacial in situ. ................................................................................... 243 7.3. Actuadores lineales y flexionantes de compuestos de PAni y nanoestructuras de carbono. .......................................................................................................................... 244 7.4. Electrodos de supercondensadores de compuestos de PAni y nanoestructuras de carbono. .......................................................................................................................... 245 10 1. Resumen 11 12 1. Resumen La presente tesis es el resultado del estudio de la síntesis de compuestos de polianilina (PAni) con nanoestructuras de carbono para su aplicación en músculos artificiales y electrodos de supercondensadores. Las nanoestructuras que se emplearon en los compuestos de PAni son nanotubos de carbono multicapa (MWCNT), nanotubos de carbono multicapa dopados con nitrógeno (CNx-MWCNT), nanotubos de carbono multicapa funcionalizados con grupos oxigenados (COx-MWCNT) nanotubos de carbono multicapa exfoliados (exMWCNT), nanofibras de carbono de listones grafíticos helicoidales (HR-CNF) y óxido de grafeno (GO). Antes de abordar el trabajo experimental, se ha llevado a cabo una revisión bibliográfica acerca del estado del arte de la PAni, las nanoestructuras de carbono empleadas, los compuestos de PAni y la utilización de estos materiales como músculos artificiales y electrodos de supercondensadores. Más adelante se muestran y discuten los resultados experimentales de la síntesis de los compuestos de PAni mediante un método mecánico, al principio (utilizando únicamente MWCNTs y CNx como cargas), y después utilizando la polimerización interfacial in situ. Se analiza la dispersión de las cargas en la matriz polimérica así como su efecto en la conductividad eléctrica de los compuestos. En la sección correspondiente al empleo de los compuestos de PAni como músculos artificiales se describe la construcción de actuadores lineales (constituidos de polvos de compuestos aglomerados con acetato de polivinilo) y actuadores flexionantes bicapa (de compuestos procesados con N-metil-2-pirrolidona) así como el efecto de cada nanoestructura de carbono en el accionamiento de estos dispositivos. Además de los experimentos de accionamiento, a las películas de compuestos de PAni (procesada con NMP) se les realizó una caracterización termomecánica. En el capítulo siguiente se discuten los resultados experimentales sobre la utilización de películas de compuestos de PAni (procesadas con NMP) como electrodos de supercondensadores. Además de las pruebas de voltamperometría cíclica y de 13 carga/descarga (típicas en aplicaciones electroquímicas) se realizó una caracterización termogravimétrica a los compuestos junto a una caracterización termomecánica. Se describe con detalle el efecto de las nanoestructuras de carbono en las propiedades electroquímicas y mecánicas de los electrodos de compuestos de PAni. Las conclusiones más importantes se compendian en un capítulo al final de esta memoria. La bibliografía empleada se lista al final de cada capítulo. 14 2. Introducción 15 16 2. Introducción En este capítulo se hará una descripción general del estado del arte referente a la polianilina (PAni), incluyendo la síntesis y sus propiedades. Asimismo, se realiza una descripción bibliográfica referente al procesado de materiales compuestos de PAni con distintas nanoestructuras de carbono (nanotubos, nanofibras de listones grafíticos helicoidales y óxido de grafeno), así como su aplicación en músculos artificiales y electrodos de supercondensadores. 2.1. Polímeros conductores Los polímeros conductores, también llamados polímeros conductores conjugados o conductores orgánicos poliméricos, son materiales intrínsecamente conductores de la electricidad (conformados por C, H y heteroátomos como N y S) debido a su estructura de conjugación π, es decir, una conjugación deslocalizada y extensa originada por la superposición de los electrones π en sus cadenas poliméricas. Esta conjugación es mostrada esquemáticamente en la Figura 1 con el poliacetileno, un polímero conductor típico. Figura 1. Representación esquemática de la conjugación π en el poliacetileno. La conductividad de los polímeros conductores se produce a través de una oxidación química o electroquímica, o en algunos casos, a través de una reducción, mediante especies aniónicas o catiónicas llamadas “dopantes”. Cuando en la literatura se habla de polímeros conductores, generalmente se trata de polímeros conductores en estado oxidado. 17 En la Figura 2 se muestran representaciones típicas de bloques monoméricos de los polímeros conductores. En todos los casos, n es un entero muy grande que se refiere al grado de polimerización. Figura 2. Ejemplos de polímeros conductores. Muchos de los polímeros conductores fueron conocidos en sus formas no conductoras mucho antes de que su conductividad y otras características de interés fueran descubiertas. Algunos sí fueron conocidos en sus formas conductoras, pero mal caracterizados, y no se mostró mucho interés en esta propiedad [1]. La polimerización oxidativa de la anilina fue descrita por Letheby en 1862 [2], y estudiada con mayor detalle por Mohilner y colaboradores en 1962 [3]. El polipirrol fue conocido por formar “pirrol negro” conductor [4] debido a una polimerización química espontánea (en ambiente de aire) en las paredes de los contenedores de pirrol. Su polimerización química fue estudiada con mayor detalle en 1916 [5]. Desde 1957, se ha estudiado la oxidación electroquímica de monómeros aromáticos (utilizados ahora ampliamente como uno de los métodos de síntesis de polímeros conductores) [6-8]. En 1967, se prepararon polímeros eléctricamente conductores a partir del pirrol, tiofeno y furano [9] y se estudió la conductividad eléctrica de la polianilina [10]. En 1968, dall’Ollio [11] describió la electropolimerización del pirrol. En cierto sentido, los polímeros conductores se habían “redescubierto”. 18 Una serie de estudios de gran importancia que enfocó la atención en los polímeros conductores como nuevos materiales con una prometedora alta conductividad (entre otras propiedades) comenzó con un descubrimiento fortuito, en un trabajo de colaboración entre Shirakawa y los grupos de Heeger/McDiarmid [12], donde el poliacetileno, expuesto a vapores de yodo desarrolló una conductividad muy alta (para un material orgánico) la cual fue bien caracterizada. De esta manera se estableció un método químico de síntesis para un nuevo material conductor. Más adelante, el grupo de Diaz en IBM [13], realizó un profundo estudio sobre la electropolimerización del pirrol, originalmente estudiado por dall’Ollio [11]. El poliacetileno fue inicialmente el polímero conductor más estudiado desde los puntos de vista de las comunidades científica y tecnológica. Sin embargo, debido a su inestabilidad química ambiental y otros factores relacionados, el interés en este polímero conductor se ha trasladado totalmente a sus aspectos científicos. Así, los polímeros conductores más estudiados son la polianilina (PAni), el polipirrol (PPy) y el politiofeno (PT) y sus derivados desde los puntos de vista científico y comercial. 2.1.1. Conductividad eléctrica Los materiales en el mundo circundante pueden ser clasificados en tres grandes categorías de acuerdo a sus propiedades de conductividad eléctrica a temperatura ambiente: aislantes, semiconductores y conductores. La superposición de estados electrónicos de moléculas individuales en estos materiales produce bandas electrónicas. Es decir, de manera esquemática, los electrones de valencia se superponen para producir la banda de valencia, mientras que los niveles electrónicos inmediatamente por encima de estos niveles también coalescen para generar la banda de conducción. La energía requerida para que los electrones “salten” de la banda de valencia a la de conducción, se le llama gap o bandgap (generalmente denotado por Eg). Si el gap es grande, por ejemplo 10 eV, los electrones difícilmente se excitarían de la banda de valencia a la de conducción, por lo que resulta un aislante a temperatura ambiente. Si el gap es pequeño, por ejemplo de 1 eV, entonces los electrones de la banda de valencia pueden ser excitados dentro de la banda de conducción, mediante excitación térmica, vibracional, o fotónica; entonces los electrones son móviles en un sentido y el material es llamado semiconductor. Para que la conducción tenga lugar en semiconductores inorgánicos convencionales, los electrones deben ser excitados desde la banda de valencia a la de 19 conducción. Normalmente, la excitación térmica a temperatura ambiente produce cierta conductividad en algunos semiconductores inorgánicos. Los polímeros conductores “dopados” (en un apropiado estado oxidado o reducido) son semiconductores como resultado de su única y extensa conjugación π. Realmente, la extensa superposición de las bandas π produce la banda de valencia y las bandas π* producen la banda de conducción en los polímeros conductores. Finalmente, si el gap disminuye, con la red parcialmente llena, ocurre una superposición de las bandas de valencia y de conducción, produciéndose una conducción metálica. Estas tres clases de conductividad, en términos de sus bandgaps, se muestran esquemáticamente en la Figura 3. Los electrones en la banda de conducción tienen niveles de energía incompletos (en la parte superior de la banda de conducción) que fácilmente se pueden llenar, y en respuesta a un campo eléctrico pueden moverse produciendo conducción. Esta es la situación en los conductores metálicos. El nivel más alto ocupado de la banda de conducción, se conoce como nivel de Fermi, con una correspondiente energía de Fermi. Figura 3. Esquema de los tipos de conductividad eléctrica, en función de sus bandgaps (Eg): (a) aislante, (b) semiconductor, (c) conductor, donde las bandas de conducción y de valencia se superponen. 2.1.2. Dopaje y dopantes de los polímeros conductores La banda electrónica semiconductora de los polímeros conductores permite una excitación electrónica (una substracción/adición de electrones) de la banda de valencia a la de conducción, produciéndose de esta manera las interesantes propiedades de estos materiales. La excitación de electrones desde la banda de valencia a la de conducción, por fotones por ejemplo, podría generar fotoluminiscencia y propiedades ópticas no lineales. 20 Por otro lado, una oxidación química o electroquímica de los polímeros conductores en un medio electrolítico, esencialmente produce una substracción de electrones de la banda de valencia y por lo tanto, se obtiene la presencia de contraiones en las cadenas poliméricas para compensar su carga eléctrica. Estas cargas (contraiones) en general están fuertemente deslocalizadas sobre algunos bloques monoméricos del polímero. Estas cargas también causan una relajación de la geometría del polímero (ahora cargado) a una conformación energética más favorable. La carga podría ser también donada a la banda de conducción del polímero conductor, con la consiguiente reducción del mismo. La oxidación causada por especies químicas genera un polímero conductor positivamente cargado y un anión asociado. La reducción, similarmente genera un polímero conductor cargado negativamente y un catión asociado. Ejemplos de estos procesos, pueden observarse en las siguientes ecuaciones: P(Py) + MClO4 → P(Py)+ClO4-M+ (oxidación) P(Ac) +NaA Na+P(Ac) + A- (reducción) → (1) (2) Ambos procesos dan propiedades conductoras al polímero. Debido a la analogía con las impurezas (causantes de la remoción o inserción de cargas de las bandas de valencia y de conducción) que otorgan mayor conductividad en los semiconductores inorgánicos como el Si o el CdSe, la oxidación química de los polímeros conductores por aniones, o su reducción por cationes, fue originalmente llamado “dopaje”. Los aniones/cationes asociados (los contraiones) fueron llamados “dopantes”. Los dopantes (oxidantes o reductores químicos) son generalmente incorporados dentro del polímero conductor al tiempo de su síntesis. Aunque éstos pueden ser incorporados después química o electroquímicamente. Los dopantes pueden ser pequeños aniones o cationes (ClO4- o Na+) o especies poliméricas grandes (“polielectrolitos” como el ácido poliestireno sulfónico o el ácido polivinil sulfónico). El nivel de oxidación/reducción suele llamarse nivel de dopaje, y es medido generalmente como la proporción de los iones dopantes o moléculas incorporadas por unidad de monómero. Esto se expresa como fracción o porcentaje molar. Por ejemplo, un polímero conductor con un anión dopante por cuatro unidades de monómero tendría un nivel de 21 dopaje de 0.25 o 25%. Generalmente no es posible llegar a un dopaje 1:1 (por las características de las cadenas poliméricas). Un incremento en el nivel de dopaje, produce un incremento en la conductividad eléctrica mediante la creación de cargas móviles. Los niveles máximos de dopaje varían para diferentes polímeros conductores y dopantes. Por ejemplo, el nivel de dopaje para el poliacetileno típicamente varía de 0.5 a 8%. El máximo nivel de dopaje de la polianilina, utilizando Cl- es de 42%. 2.1.3. Tipos de dopaje de los polímeros conductores En caso de que los polímeros conductores no se sinteticen en estado dopado, éstos se podrían dopar químicamente a posteriori, por exposición a una solución o vapor del dopante, o también electroquímicamente, manteniendo al polímero conductor dentro de una solución a un potencial determinado. Cuando se aplica un potencial positivo, los aniones dopantes se mueven de la solución hacia sitios de carga deslocalizada en el polímero conductor, produciéndose un dopaje aniónico. Este dopaje suele denominarse dopaje del tipo-p, otra vez haciendo la analogía con la terminología de la física de estado sólido. Similarmente, si se aplica un potencial negativo al polímero conductor en solución, se introduciría un número de cationes dentro del polímero conductor. Esto suele denominarse dopaje catiónico o dopaje del tipo-n. En el dopaje químico, el dopante (oxidante o reductor), debería tener un potencial redox adecuado para oxidación/reducción (dopaje) del polímero conductor. Durante el proceso de dopaje/desdopaje, el dopante se mueve hacia dentro o fuera de la red polimérica del polímero conductor. Esto conduce a deformaciones volumétricas (contracción/expansión). 2.2. La polianilina Las primeras noticias acerca de la anilina se remontan a 1826, donde algunos investigadores la aislaron del índigo, utilizando ácido sulfúrico o fosfórico (colorante vegetal) [14], del alquitrán de hulla [15, 16], mediante la reducción del nitrobenceno [17] y mediante un tratamiento al índigo con potasa realizada por el alemán C. J. Fritzsche quien denominó al aceite obtenido Anilin [18] (nombre derivado del de la planta Indigofera Anil). Posteriormente, en 1856, William Henry Perkin [19] sintetizó el primer colorante sintético llamado púrpura de anilina mediante la oxidación química de anilina con dicromato potásico en medio ácido, la cual hasta el momento es el método habitual para 22 obtener polianilina. En esos mismos años Henry Letheby obtuvo polianilina (PAni) a la que describió como una sustancia oscura depositada en el ánodo durante la electrólisis de sulfato de anilina [2] y la cual después se identificó como negro de anilina. En la segunda mitad del siglo XIX e inicios del XX, el negro de anilina se produjo en cantidades industriales como colorante en la industria textil. Desde los primeros estudios del negro de anilina, se dieron cuenta de que podía estar en varios estados debido a su cambio de color siguiendo tratamientos de oxidación, reducción y adición de ácidos y bases. Se le dieron nombre a estos estados diferentes como esmeraldina [20] (por su color verde), leucoesmeraldina (del griego leuco: claro o blanco) o negranilina (por su color oscuro, casi negro) de acuerdo a su estado de oxidación. A inicios del siglo XX, después de algunos estudios donde se establecieron diferentes fórmulas moleculares del negro de anilina (se había identificado la relación C:N de 6:1), fue el de Willstäter y Moore [21] con la fórmula C48H36N8 el más cercano a la realidad. De hecho esta fórmula corresponde a lo que conocemos ahora como sal de esmeraldina. A pesar de que aún no hacía su aparición el concepto de macromolécula, ellos propusieron anillos bencenoides o quinoides, enlazados por nitrógenos en posición para. En ese mismo tiempo, el grupo de Arhur G. Green [22, 23] propuso diversas estructuras de octámeros del negro de anilina. Después de estos trabajos, el estudio de la polianilina (PAni) fue escaso hasta llegar a la década de los 80, donde comenzó a estudiarse ampliamente. En la primera mitad de la década de los 80, algunos estudios evidenciaron que la PAni tiene dos estados de oxidación límite, el menos oxidado, de color amarillo claro y el más oxidado de color prácticamente negro [24]. El hecho de que este cambio de estado estuviera muy relacionado con la conductividad eléctrica, hizo que se comenzara a emplear el término “dopado”. Asimismo, se estudiaron las propiedades anticorrosivas de la PAni en el hierro [25, 26]. En 1985 y 1986, Alan G. MacDiarmid y colaboradores [27-30] reportaron sus descubrimientos sobre la estructura de la PAni, en los que se describió el dopado mediante protonación (se utilizó HCl) del polímero, observando un ascenso de la conductividad del orden de 1010 al variar el pH de 4 a 1. La PAni había pasado de un estado aislante a uno metálico, el cual fue el primer ejemplo de dopado no redox en los polímeros conductores. Este dopado, a diferencia de otros estados de la PAni e incluso a diferencia de estados dopados de otros polímeros conductores, es más estable (no se degrada en condiciones medioambientales). El estado dopado de la PAni se podía obtener por dos caminos, el oxidativo y el químico (mediante ácidos próticos). 23 MacDiarmid propuso que la estructura de la PAni podía estar formada por cualquier combinación de las unidades estructurales que aparecen en la Figura 4 a, las cuales podían transformarse en unas y otras mediante la oxidación/reducción y protonación/desprotonación. También estableció que la PAni conformada por estas unidades estructurales se denominaría base si estuviera formada por unidades neutras, o bien por sales con unidades protonadas puesto que deberían estar acompañadas de aniones para compensar su carga positiva. La PAni base se observa en la Figura 4 b. La PAni completamente reducida recibió el nombre de base de leucoesmeraldina. La parcialmente oxidada se denominó base de esmeraldina y a la totalmente oxidada base de pernigranilina. Figura 4. (a) Estructura de las unidades repetitivas de la polianilina, (b) fórmula general de la PAni (MacDiarmid). En la transición de esmeraldina base a la protonada, hay dos tipos de nitrógeno que pueden protonarse: los tipo amina e imina. Los nitrógenos del primer tipo, podrían tener mayor basicidad, por lo que se obtendría la estructura (a) de la Figura 5. La estructura que propuso MacDiarmid fue la mostrada en la Figura 5 b (con los nitrógenos tipo imina protonados). Esta estructura explica la alta conductividad de la esmeraldina protonada (sal de esmeraldina) y tiene algunas formas de resonancia donde la carga positiva puede localizarse en cualquiera de los nitrógenos, lo cual explica su estabilización energética y la deslocalización de las cargas positivas a lo largo de la cadena polimérica. En cambio, la estructura (a), no admite ningún tipo de resonancia que origine desplazamiento de la carga 24 positiva por la presencia de nitrógenos cuaternarios (nitrógenos cuaternarios con hibridación sp3). Figura 5. Productos de la protonación de la base de esmeraldina. La estructura de la sal de esmeraldina (Figura 5 b) puede representarse por otras formas de resonancia denominados radicales catiónicos (polarones) como la que se muestra en la Figura 6. Esto provoca una mayor movilidad de las cargas debido a que las cargas no deben estar en pareja. Se suele llamar radical semiquinona a la estructura polarónica con un número de electrones intermedio entre la estructura oxidada (quinoide) y la reducida (bencenoide), la cual es la que más se representa debido a que se ajusta más a las observaciones experimentales [31, 32]. La estructura bipolarónica supone la existencia de cargas positivas en pareja. Figura 6. (a) Estructuras que representan a la sal de esmeraldina, (b) bloques repetitivos presentes en la sal de esmeraldina. 25 Alan G. MacDiarmid fue galardonado con el premio Nobel de química en el año 2000 junto a Alan J. Heeger y Hideki Shirakawa por “el descubrimiento y desarrollo de los polímeros conductores”. 2.2.1. Síntesis y procesado de la PAni La PAni se sintetiza a partir del monómero de anilina o de alguna sal de anilio mediante una polimerización oxidativa [33], la cual puede ser electroquímica (en el ánodo de una celda electroquímica) o química (mediante un agente oxidante en una solución acuosa). La polimerización química admite una gran variedad de oxidantes. El oxidante más común es el persulfato de amonio (NH4)2S2O8 (APS), utilizado desde los primeros trabajos de MacDiarmid [28]. Algunos de los oxidantes alternativos al APS son el cloruro de hierro (III) (FeCl3), el perclorato de cobre (II) (Cu[ClO4]2), biyodato de potasio (KH[IO3]2) y el peryodato de tetrabutilamonio (NBu4)IO4 (el cual puede utilizarse en disolvente orgánico) [34-37], entre otros. La reacción de polimerización se ve favorecida por disolventes polares (especialmente agua), aunque, como se verá más adelante, se han desarrollado métodos de síntesis de “estado sólido”. Asimismo, la polimerización de la anilina es más eficiente a pHs ácidos, ya que en pHs básicos se producen oligómeros de anilina que a su vez producen heterociclos condensados [38-40]. 26 Figura 7. Estequiometría de la formación de polianilina por oxidación con persulfato de amonio (APS) en un medio ácido. La reacción de polimerización de la anilina en medio ácido consiste de dos fases principales: de inducción y de polimerización rápida. El primero es endotérmico y lento (se piensa que se generan oligómeros de anilina del tipo fenacina), en cambio el segundo es exotérmico, produciéndose la sal de esmeraldina. El pH del medio de reacción desciende al transcurrir la polimerización (debido a la generación de H+ al polimerizarse la anilina) [33, 41, 42]. Se podría acelerar la polimerización en presencia de algunas trazas en el medio de reacción, como oligómeros de anilina, aminas aromáticas, cationes de metales de transición o nanotubos de carbono [43-45]. La sal de esmeraldina producida, suele ser muy regioregular: sólo se han observado acoplamientos monoméricos del tipo “cabeza-cola” (nitrógenos en disposición para). Durante el periodo de inducción en la reacción, se forman oligómeros de anilina que posiblemente acelerarán la polimerización [41, 46], ya que son más fácilmente oxidables que los monómeros de anilina (debido a su potencial de oxidación más bajo en el medio ácido). En esa situación el crecimiento de la cadena comenzaría con la adición de anilina a los oligómeros de anilina oxidados, lo cual produce polianilina con un alto grado de oxidación (pernigranilina) [47, 48] hasta que se consume el oxidante disponible. Después siguen adicionándose monómeros de anilina hasta llegar al estado de oxidación de la esmeraldina, cuyo poder oxidante es ya insuficiente para adicionar más anilina a su cadena. 27 La sal de esmeraldina obtenida de la polimerización (insoluble en medio acuoso), se lava (comúnmente con metanol) y se filtra. En el caso de que el polímero se haya sintetizado electroquímicamente, éste se encontrará depositado en forma de película en el ánodo. Si se llegara a realizar la reacción a pH > 4, la mayor parte de la anilina se consumiría en las primeras fases de oxidación (se formarían oligómeros), por lo que se obtendría mayoritariamente oligómeros oxidados en detrimento de la polianilina. Al parecer, los oligómeros del tipo fenacina, deben estar protonados para comenzar el ciclo de oxidaciónadición de anilina que genera una cadena polimérica regular y de alto peso molecular. La PAni sintetizada en medio ácido presenta una regularidad estructural muy alta, en gran parte, los acoplamientos de la anilina son de “cabeza-cola”, por lo que podría decirse que tiene una estructura homogénea. La cadena polimérica estaría formada por anillos aromáticos alternados con nitrógenos (Figura 4) con distintos grados de protonación y oxidación. En el caso de existir nitrógenos protonados existirán aniones (contraiones) que compensarán en desequilibrio de carga eléctrica, como ya se vio previamente al explicar el dopaje de los polímeros conductores. La síntesis clásica de la PAni (implementada por MacDiarmid) utiliza anilina, un oxidante y un ácido mineral fuerte como dopante y su morfología es granular. Mediante la polimerización interfacial (o bifásica) [49, 50] se ha logrado obtener PAni nanofibrilar. Este método, consta de dos soluciones inmiscibles, una orgánica que contiene al monómero, y otra acuosa en la que se ha disuelto el agente oxidante. La reacción ocurre en la superficie de contacto de ambas soluciones. J. Huang y colaboradores [49], obtuvieron nanofibrillas de PAni de 30-50 nm de diámetro. Utilizaron como fase orgánica varios disolventes como hexano, benceno, tolueno, xileno, entre otros. Como oxidante utilizaron persulfato de amonio (APS) disuelto en la solución ácida dopante. Los ácidos que probaron fueron: HCl, H2SO4, HNO3, entre otros, y algunos ácidos orgánicos sulfónicos. Encontraron que el disolvente no afectó el tamaño y forma de las nanofibras, y el ácido que produjo fibras más delgadas fue el HCl. La morfología se verá con mayor detalle más adelante. 2.2.2. Cristalinidad de la PAni La atracción entre las cadenas poliméricas de la PAni es alta, lo cual explica su baja solubilidad. Esta atracción es debido a interacciones de apilamiento π-π, puentes de hidrógeno, fuerzas de Van der Waals y de dispersión de London, y en el caso de la sal de 28 esmeraldina, interacciones coulómbicas. El estudio de difracción de rayos X (XRD) de la PAni ha revelado que tiene una estructura semicristalina, teniendo más ordenamiento el estado de sal de esmeraldina [51-53]. Los factores más importantes que afectan la cristalinidad de la PAni son: temperatura de reacción (entre más baja es la temperatura, logra obtenerse un mayor ordenamiento en las cadenas poliméricas), tipo de contraión (con el ion canforsulfónico se logra obtener cierta quiralidad en las cadenas) y su concentración, y la fuerza iónica del medio de reacción [54-58]. 2.2.3. Propiedades eléctricas Únicamente el estado de sal de esmeraldina de la PAni presenta una alta conductividad eléctrica. Los demás estados son aislantes. La conductividad es debida a la alta movilidad de los transportadores de carga (huecos en las cadenas poliméricas). La carga positiva de éstas se ha confirmado por evaluaciones de efecto Hall, siendo la sal de esmeraldina un semiconductor de tipo p [59] (transportador de huecos). La sal de esmeraldina puede generarse mediante dos tipos de dopaje: el dopaje por oxidación a partir de leucoesmeraldina, o el dopaje por protonación a partir de base de esmeraldina (ver Figura 8). En el primer caso, la carga positiva sucede por la pérdida de electrones de las cadenas poliméricas y en el segundo, con la polianilina en un estado semioxidado y neutro ocurre la protonación de nitrógenos del tipo imina, lo cual genera la carga positiva. Esto último conlleva a la formación de bipolarones (cargas positivas en pareja) y polarones (radicales catiónicos) (Figura 6). En la Figura 8 se observa un esquema en donde se muestran las seis formas de la PAni: leucoesmeraldina (estado reducido), esmeraldina (estado semioxidado) y la pernigranilina (estado totalmente oxidado), donde cada una puede estar en forma de base (pH alto) o sal (pH bajo, con los contraiones insertados en las cadenas poliméricas). Las flechas indican el paso de un estado a otro en función del pH o de un potencial redox. Cada estado es reversible. 29 Figura 8. Estados de oxidación electroquímica de la PAni. En los estados completamente protonados (derecha) el polímero consiste solamente de unidades x, pero en muchos ácidos, las cadenas poliméricas son una mezcla de unidades x e y, la proporción dependerá del pH. Durante la oxidación electroquímica en un electrolito acuoso, la ganancia/pérdida de aniones y protones, depende del pH. Los polarones y bipolarones se encuentran en un equilibrio termodinámico (en el esquema de la Figura 8, en medio a la derecha, se muestran ambos tipos de configuraciones), aún se estudia que especie es la que está más presente en las cadenas poliméricas y cuál es su función particular en la conducción de la electricidad. 2.2.4. Conductividad eléctrica de la sal de esmeraldina Se ha reportado una conductividad eléctrica de la polianilina que va desde 0.001 hasta 1000 S/cm. Esta variabilidad tan grande es debida a diferencias en la morfología, el grado de cristalinidad y el contraión dopante. En la sal de esmeraldina hay una resistencia al paso de corriente en función del grado de granulación que presente. Cuanto más pequeños sean los gránulos, mayor es la resistencia por contacto entre ellos [60], incrementándose la resistencia total. Debido a esto, en general, las conductividades más bajas se encontrarán con la polianilina nanoestructurada, las conductividades medias con la morfología granular 30 estándar o “clásica” [61] y las altas de polianilina procesada con algún disolvente (por ejemplo sal de esmeraldina dopada con ácido canforsulfónico y disuelta en m-cresol). El contraión o dopante, tiene un efecto en la conductividad eléctrica de la PAni en cuanto logra alterar la conformación y configuración electrónica de las cadenas poliméricas. Esta interacción puede modificar el equilibrio existente entre polarones y bipolarones, por ejemplo, los contraiones con dos electrones de carga tienden a incrementar la presencia de bipolarones [62]. Como se comentará más adelante, el contraión también influye en el tamaño y forma de partícula de la sal de esmeraldina, así como su cristalinidad. El nivel de protonación influye directamente en la conductividad de la sal de esmeraldina, puesto que la protonación es la que produce las cargas en las cadenas poliméricas. El estiramiento de PAni procesada y conformada en películas o fibras también incrementa la conductividad de la sal de esmeraldina puesto que se produce una alta anisotropía en el material (las cadenas poliméricas tienden a alinearse a la dirección de estiramiento) por lo que los valores de la conductividad eléctrica en dirección al estiramiento son varias veces superiores a la medición perpendicular [63-65]. 2.2.5. Propiedades mecánicas Hay pocos estudios sobre las propiedades mecánicas de la PAni pura debido a las dificultades en su procesado (fibras o probetas). El procesado en disolución presenta problemas por la baja solubilidad de la PAni. Los resultados que se han realizado para medir sus propiedades mecánicas han sido relativamente malos [66-68], por lo que se ha estudiado más en compuestos al mezclarse o dispersarse en otras matrices poliméricas [6971]. A pesar de esto, se han realizado pruebas mecánicas (o termomecánicas) a la PAni procesada con algún plastificante como el N-metil-2-pirrolidona (NMP) o el N-N-dimetil propileno urea (DMPU) y conformada en película o fibra. 2.2.6. Propiedades electroquímicas El estado redox de la PAni más estable ambientalmente es el estado semioxidado (esmeraldina) por lo que puede ser reducido a leucoesmeraldina u oxidado a pernigranilina. Sus tres estados reducción/oxidación contrastan con los dos estados típicos del resto de los polímeros conductores (oxidado y reducido) y semiconductores inorgánicos. Los potenciales redox para la PAni son bajos por lo que pueden aplicarse en disoluciones 31 acuosas, de manera reversible y numerosas veces (la PAni suele ser estable al someterse a un número elevado de ciclos redox). La caracterización electroquímica suele realizarse mediante la Voltamperometría Cíclica (CV, por sus siglas en inglés). Mediante esta técnica, el material activo, sumergido en un electrolito se somete a un barrido de potencial controlado (utilizando un potenciostato). Generalmente los barridos son lineales entre dos niveles de potencial (realizándolos de manera alternante de mayor a menor nivel y viceversa). Los resultados se presentan en gráficos de intensidad de corriente contra potencial en donde en el barrido creciente se pueden observar picos de oxidación y en el decreciente de reducción. La PAni, en estos estudios, se encuentra fija en un electrodo (se le denomina de trabajo) sobre el que se aplica el potencial con respecto al electrodo de referencia. La corriente circula con respecto al contraelectrodo, la cual compensará la carga eléctrica en el electrodo de trabajo. Los procesos redox de la polianilina son dependientes de un valor concreto del pH de la solución [72, 73] (a pH básicos no hay procesos redox). En solución acuosa, la transición sal de esmeraldina-leucoesmeraldina suele ocurrir de 0 a 0.2 V. En cambio, la transición esmeraldina-pernigranilina, suele suceder en ~0.7 V. Además de los ciclos redox reversibles, la PAni puede transportar carga (huecos) de modo eficiente si se encuentra cerca del estado sal de esmeraldina (semioxidado), por lo que puede actuar como “puente redox” acumulando cierto nivel de carga y transportándola por su volumen. Esta propiedad es una de las razones del efecto auto-catalítico de la PAni en su síntesis [74, 75]. Las propiedades redox de la PAni son notables en su aplicación de dispositivos de almacenamiento y conversión de energía (baterías, condensadores, celdas de combustible, músculos artificiales) [76-79], como se comentará posteriormente. 2.2.7. Morfología de la PAni La PAni adquiere una morfología particular durante el proceso de síntesis ya que el polímero se genera en el medio de reacción. Por tanto los diferentes tipos de morfología se describen a continuación. 2.2.7.1. PAni granular Como se explicó brevemente, la morfología granular de la PAni obtenida en la década de los 80 (y vigente en los 90) por MacDiarmid suele llamársele morfología clásica y consiste en gránulos de PAni, de varios µm de diámetro, unidos entre sí. Debido al 32 empaquetamiento de los gránulos se logran tener conductividades eléctricas relativamente altas. Esta reacción de polimerización se estandarizó, aunque cualquier reacción en donde se adicione lentamente el oxidante producirá PAni de morfología granular. En el caso de la PAni sintetizada electroquímicamente [80], la sal de esmeraldina depositada en el ánodo suele tener una morfología granular también. La morfología de la PAni en la década pasada comenzó a obtenerse nanoestructuradamente. La polimerización clásica de la PAni lleva inicialmente a tener pocas partículas de nucleación debido a la baja cantidad de oxidante inicial (debido a su adición lenta) en las cuales crecen cadenas poliméricas, conforme se va adicionando más oxidante, se van creando nuevos puntos de nucleación sobre la superficie de las partículas de PAni (la agitación del medio de reacción también contribuye en esto), por lo que al finalizar la reacción se obtendrían partículas de PAni relativamente grandes formadas por la unión de gránulos que crecieron unos sobre otros [81]. 2.2.7.2. Nanofibras de PAni Las nanofibras de PAni se caracterizan por tener forma elongada y un diámetro nanométrico (estructura 1D). Estas características variarán según el método de síntesis empleado. Se han obtenido nanofibras de PAni en presencia de: a) sustratos (ópalos, alúmina, membranas porosas) con poros nanométricos [82-84]; b) con surfactantes [85]; c) mediante síntesis electroquímica [86]; d) mediante síntesis interfacial [49] entre dos soluciones inmiscibles (una acuosa y otra orgánica); e) mediante síntesis mecanoquímica [87] donde los reactivos cloruro de anilinio y cloruro férrico hexahidratado son molidos en estado sólido; y f) mediante síntesis por la acción de ultrasonidos [88]. Se dice que la morfología nanofibrilar es un tipo de morfología intrínseca de la síntesis oxidativa de la PAni. En la interfase de los puntos de nucleación se produce el crecimiento de las cadenas poliméricas, durante gran parte del tiempo de reacción estos puntos de nucleación se encuentran suspendidos en el medio, y si la concentración de reactivos es baja y la acidez es alta, el tamaño de estas partículas tiende a ser nanométrico. Sobre estas partículas comenzará a crecer una cierta cantidad de cadenas poliméricas, las cuales mediante la interacción π-π tenderán a apilarse hasta constituir la fibra. De este mecanismo se infiere que cuanto más pequeña sea la partícula, más delgada será la nanofibra. La formación de nanofibras en la polimerización interfacial se podría explicar por la formación de una capa de cationes anilinio que provienen de la fase orgánica y se han 33 protonado al estar en contacto con la fase acuosa ácida. También se formarían partículas de nucleación por la oxidación de los cationes. En la Figura 9 se observan distintas morfologías nanofibrilares obtenidas mediante la polimerización interfacial en función del ácido utilizado en la fase acuosa (HCl, H2SO4, HNO3, HClO4). La reacción se llevó a cabo en un sistema bifásico de agua/cloruro de metileno. Las fibras producidas con HCl y H2SO4 tuvieron ~30 nm de diámetro; con HNO3 tuvieron ~30-50 nm; con HClO4 las fibras tuvieron un diámetro de ~120 nm [49]. Figura 9. Micrografías SEM de nanofibras de PAni sintetizadas mediante polimerización interfacial en un sistema bifásico de agua/cloruro de metileno. Los ácidos utilizados en la fase acuosa fueron: (a) HCl, (b) H2SO4, (c) HNO3, (d) HClO4 [49]. La PAni nanofibrilar destaca por su alta superficie específica que le permite actuar como sensor químico [89, 90] y condensador electroquímico [91] debido a su elevada área superficial. Los métodos de polimerización interfacial y mecanoquímico de PAni son los que se emplean en esta tesis para la síntesis de PAni. Con respecto al método mecanoquímico, se hace una evaluación en el Capítulo II del efecto del agua en la reacción de “estado sólido” 34 entre los cloruros de anilinio y férrico, ya que el cloruro férrico es altamente higroscópico. La polimerización interfacial es la que se utilizará en el resto de la tesis. 2.2.8. Aplicaciones de la PAni La elevada conductividad eléctrica de la PAni y su estabilidad ambiental han hecho que se aplique como material con propiedades antiestáticas en pinturas, tintas, tejidos y adhesivos [92-95]. También se ha utilizado como conductor en circuitos electrónicos [96] y Las propiedades electrocrómicas de la PAni (aprovechando la reversibilidad de sus procesos redox) [97] podrían aplicarse en ventanas o películas transparentes puesto que en estado de leucoesmeraldina es de color claro y en pernigranilina es de color oscuro. Se han aprovechado las propiedades semiconductoras de la PAni en aplicaciones como transistor de efecto de campo [98, 99] y como diodos emisores de luz orgánicos (OLED, por sus siglas en inglés) [100, 101]. En estos dispositivos las transiciones redox de la PAni implican transferencias de carga muy rápidas por lo que sólo hay cambios muy localizados del estado de oxidación del polímero. Los cambios de estado de oxidación en la PAni, que producen una deformación reversible del polímero, se han aprovechado para construir actuadores o músculos artificiales. El mecanismo de funcionamiento de estos dispositivos se verá con mayor detalle más adelante. 2.3. Nanoestructuras de carbono como cargas en materiales compuestos Durante el desarrollo de esta tesis se trabajó con distintas nanoestructuras de carbono que por sus características estructurales, se pueden dividir en tres: nanotubos de carbono (CNT), nanofibras de carbono (CNF) y óxido de grafeno (GO). Dentro de los CNTs (forma alotrópica del carbono que consiste en uno o varios cilindros concéntricos de estructura hexagonal e hibridación sp2) se utilizaron distintos tipos de nanotubos: nanotubos multicapa (MWCNTs), nanotubos multicapa dopados con nitrógeno (CNx-MWCNTs) en los cuales hay una sustitución parcial de átomos de carbono por átomos de N, nanotubos funcionalizados con grupos oxigenados (COx-MWCNTs) y nanotubos exfoliados (exMWCNTs) que consisten en una mezcla de nanolistones grafíticos, nanotubos cortos parcialmente abiertos y pequeñas hojuelas de grafeno. 35 Con respecto a las CNFs, se utilizaron filamentos del tipo “stacked-cup” (conformados por un listón grafítico enrollado helicoidalmente con un amplio claro en el centro) que comercializa el Grupo Antolín Ingeniería bajo el nombre comercial de GANF. De hecho, estas CNFs en diversas publicaciones están recibiendo el nombre HR-CNF (helical ribbon carbon nanofiber) que resulta más correcto. Finalmente, el GO (láminas de grafeno funcionalizados con grupos oxigenados que le otorgan hidrofilidad) se obtuvo de la exfoliación química y mecánica (por ultrasonidos) de HR-CNFs. El GO utilizado se comercializa actualmente bajo el nombre de GRAnPH, también por el Grupo Antolín Ingeniería. A continuación se describen las características principales de estas estructuras, algunas propiedades importantes y los métodos de síntesis. 2.3.1. Nanotubos de carbono Los nanotubos de carbono (CNTs) son estructuras tubulares de diámetro nanométrico cuya pared son cilindros formados por una red de átomos de carbono en un ordenamiento hexagonal. La denominación y descubrimiento de los nanotubos de carbono de pared simple (SWCNTs) fue debida a S. Iijima en 1991 [102], pocos años después del descubrimiento de los fullerenos, por H. W. Kroto y colaboradores [103]. Los fullerenos son estructuras cerradas formadas por átomos de carbono sp2, de los cuales el más conocido es la estructura de icosaedro truncado denominado C60 que se asemeja a las costuras de un balón de futbol. No obstante, la primera imagen de los CNTs de lo que luego resultaron ser nanotubos de carbono de pared múltiple (MWCNTs) fue obtenida por Morinobu Endo y colaboradores [104] a mediados de la década de los 70 con la denominación de fibras de carbono. Un SWCNT también es una estructura cerrada derivada de la “elongación” de un fullereno en una dirección de tal forma que su longitud es varias veces mayor que su diámetro, o puede ser visto como un grafeno enrollado y cerrado en sus extremos. Teóricamente, es posible construir un tubo de carbono enrollando una lámina de grafeno hexagonal en varias formas. Dos de estas no son quirales, por lo que los hexágonos localizados en los extremos del tubo son siempre paralelos. Estas configuraciones son conocidas como "zigzag" y "armchair". En la estructura zigzag, dos enlaces C-C en los lados opuestos de cada hexágono son paralelos al eje del tubo (Figura 10 a). En la 36 estructura armchair dos de los lados opuestos del hexágono son perpendiculares al eje del tubo (Figura 10 b). Todas las otras conformaciones, donde los enlaces C-C están en inclinación con el eje del tubo, se conocen como quirales o estructuras helicoidales (Figura 10 c). Los cálculos teóricos indican que todos los tubos del tipo armchair son metálicos, el resto son semiconductores. Figura 10. Estructura zigzag (a), armchair (b) y quiral (c) [105]. Los CNTs tienen unas extraordinarias propiedades eléctricas, térmicas y mecánicas con una muy baja densidad (~1.4 g/cm3para SWCNTs), lo cual los hacen materiales con un potencial alto en aplicaciones electrónicas, en el diseño de instrumental físico y en compuestos poliméricos, entre otras. A continuación se describen estas propiedades. 2.3.1.1. Propiedades eléctricas En los SWCNT metálicos (tipo armchair), mediante cálculos teóricos, se ha encontrado una capacidad de conducción eléctrica mayor que la de cualquier material conocido [106] 37 con valores de densidad de corriente de ~4·109 A/cm2 (en los metales es de ~105 A/cm2). Los CNTs metálicos son conductores casi perfectos en su dirección axial (armchair), en los que el transporte de carga se genera de modo balístico [107] (sin pérdidas por difusión térmica). Este transporte de carga es válido sólo para un nanotubo aislado. En la conducción electrónica de un nanotubo a otro se debe de superar una barrera energética que implica una generación de calor conforme se conduce la corriente. Además, hasta el momento, los CNTs producidos y listos para utilizarse, contienen una mezcla de CNTs metálicos y semiconductores. Los CNTs se están empleando ampliamente como carga en materiales compuestos [108] (con un muy bajo nivel de percolación debido a su estructura casi unidimensional); en electrodos en baterías, [109] supercondensadores, [110-112] celdas de combustible, [113], entre otras aplicaciones. La morfología y alta conductividad eléctrica de los CNTs ha hecho que se apliquen en la emisión electrónica por efecto campo [114], donde han tenido aplicación como emisores de electrones para microscopía electrónica o puntas de AFM o STM [115]. Las propiedades semiconductoras de los CNTs también se han aprovechado en la fabricación de transistores de efecto campo (FET) con nanotubos individuales [116]. 2.3.1.2. Propiedades térmicas La propagación de los modos normales de vibración (fonones) a lo largo del eje longitudinal de los CNTs ocurre prácticamente sin dispersión por lo que poseen una alta conductividad térmica. Se han predicho valores (a temperatura ambiente) de hasta de 6000 W/m∙K para SWCNT individuales [117] y medido valores superiores a 3000 W/m∙K para MWCNT y SWCNT [118, 119]. 2.3.1.3. Propiedades mecánicas Las excelentes propiedades mecánicas de los CNTs fueron predichas teóricamente poco después de su descubrimiento. Las mediciones mecánicas experimentales en CNTs individuales son difíciles, se realizan normalmente por técnicas de microscopía (AFM o TEM) que permiten aplicar una tensión o transmitir una vibración a nanotubos individuales. Se han reportado valores cercanos a 1 TPa para el módulo de elasticidad [120-122] que es comparable al módulo de elasticidad del grafito (y del grafeno) en su plano basal (1,06 TPa). La resistencia a la tracción de los CNTs también es muy elevada (del orden de varios GPa al deformarse más de 10%) [123]. En el caso de los MWCNTs, estos suelen ceder a la 38 tracción al deslizarse sus capas concéntricas antes de que se rompa el nanotubo en dos partes, manteniendo la estructura de las capas individuales [124]. 2.3.1.4. Algunas aplicaciones Los CNTs se pueden conformar en fibras (con nanotubos preferentemente alineados en la dirección de hilado) o en forma de hojas, denominados buckypapers [125, 126] (en los que los nanotubos forman una red semejante a la de las fibras de celulosa en las hojas de papel). Estos materiales obviamente presentan propiedades mecánicas bajas con respecto a los CNTs individuales. De momento el área donde se han aplicado las propiedades mecánicas de los CNTs ha sido en la síntesis de compuestos poliméricos [108]. En estos materiales se ha observado una mejora significativa del módulo de elasticidad con un nivel bajo de carga de nanotubos pero sólo si se consigue una dispersión adecuada, que no es tarea fácil. Las propiedades mecánicas de los CNTs son anisotrópicas, siendo mucho mejores en la dirección axial. El control selectivo de la dirección de los CNTs embebidos en la matriz polimérica podría mejorar aún más las propiedades mecánicas de los compuestos al aplicarse a tareas específicas. Asimismo, la elevada área superficial de los CNTs es un factor de importancia en dispositivos como supercondensadores [111], sensores químicos, [127, 128]. Ésta se ha aprovechado en la construcción de baterías de litio [129] y también para el almacenamiento de hidrógeno [130]. 2.3.1.5. Síntesis de nanotubos de carbono 2.3.1.5.1. Descarga en arco eléctrico Mediante este método se aplica una diferencia de potencial elevada a dos electrodos (al menos uno es de grafito) en un ambiente inerte, por lo que se produce un arco eléctrico entre ambos, obteniéndose una temperatura local muy elevada, que provoca la formación de plasma entre los electrodos. En estas condiciones, una fracción del material de grafito del ánodo se sublima y autoensambla formando CNTs. Si el ánodo es de grafito puro, se obtienen MWCNTs, pero si contiene impurezas metálicas (catalizadores) se obtienen SWCNTs (las impurezas formarán parte de este tipo de tubos). La síntesis de estos nanotubos ha sido optimizada para obtener CNTs con una mayor pureza y perfección estructural [131, 132]. El mayor inconveniente de este método, es la baja producción que se obtiene, por lo que difícilmente se podría escalar. 39 2.3.1.5.2. Ablación láser Este método fue desarrollado por Smalley y colaboradores [133] donde se sublima grafito (en ambiente inerte) que tiene incorporado catalizadores metálicos mediante un láser de alta intensidad [134, 135]. El carbono vaporizado se reorganiza para formar SWCNTs. Éstos se agrupan en haces. Por este método también se pueden producir MWCNTs en ausencia de catalizador metálico [136]. La calidad de los nanotubos producidos es comparable a la de los nanotubos producidos por arco eléctrico. 2.3.1.5.3. Producción por CVD En general, para producir CNTs y CNFs mediante CVD es necesaria la presencia de nanopartículas metálicas. Estas partículas son formadas por reducción, durante el proceso CVD, a partir de un precursor en un paso previo (por tratamiento químico y/o químico en el propio horno de reacción). La fuente de carbono es introducida en el horno y calentada a una temperatura determinada. Al estar en contacto con las partículas catalíticas, las moléculas se descomponen produciendo carbono elemental que se deposita en éstas. Las partículas metálicas se saturan de carbono por lo que en la "precipitación" del carbono alrededor de la partícula genera al nanofilamento [137]. Mientras que el proceso de descomposición/deposición se mantenga, el "depósito" de carbono crece con estructura grafítica. Es decir, se forma una nanofibra por cada nanopartícula activa de catalizador [138-140]. El proceso de descomposición de hidrocarburos en fase vapor ha sido utilizado durante más de un siglo para producir filamentos de carbono [141]. Básicamente hay tres técnicas: el método del sustrato, el método del catalizador flotante y el reactor de lecho fluidizado. El primer método es un proceso discontinuo en el que se deposita el catalizador sobre un sustrato y a partir de éste crecen los nanotubos al hacer pasar por el reactor una fuente de carbono a temperaturas entre los 300 y 1200º C (en una atmósfera inerte o con hidrógeno). Por el método de sustrato se pueden producir los llamados "bosques" de nanotubos (SWCNTs y MWCNTs) alineados perpendicularmente a la superficie de los sustratos que pueden ser de níquel, acero, silicio, entre otros. La longitud de los CNTs puede controlarse ajustando el tiempo de reacción. Se han llegado a producir bosques de nanotubos de varios milímetros de altura [142]. La versatilidad del método CVD permite producir nanotubos dopados con N o B, empleando fuentes de carbono que contengan estos átomos (como la benzilamina en el caso del N) [143]. 40 El método de catalizador flotante, ideado por Endo y Koyama en 1983 es un proceso que produce nanotubos de manera continua a escala industrial [144]. Se introduce en el reactor continuamente el catalizador (o su precursor), la fuente de carbono y el gas portador. Primero ocurre la nucleación de las partículas y posteriormente el crecimiento de las nanofibras conforme avanzan por el reactor. Al ser el tiempo de residencia menor en comparación al primer método, se requieren temperaturas más altas (800-1200º C). Esta técnica proporciona MWCNTs de buena calidad utilizando ferroceno y una fuente gaseosa aromática a 725-825º C [145]. Bajo condiciones específicas se forman CNF tipo HR-CNFs (con metano o CO como fuente de carbono, independientemente del metal usado) [146]. Por último, puede obtenerse nanotubos a escala industrial introduciendo el catalizador soportado en un reactor de lecho fluidizado, utilizándose la fuente de carbono con el gas de fluidización. El reactor de lecho fluidizado tiene la ventaja de su elevado coeficiente de transferencia de calor que junto con la agitación, permite mantener una buena isotermicidad. Por último, el método de reactor de lecho fluidizado, se conoce desde principios del siglo XX y es ampliamente utilizada en la industria en aplicaciones como incineradores de residuos, gasificadores de biomasa y craqueo en la industria del refino [147]. Mediante esta técnica se producen CNTs y CNFs [148-150]. La ventaja de este método es que la transferencia del calor entre las partículas del lecho y los gases es excelente, por lo que el control de la temperatura es muy alto. La eficiencia es más alta con respecto al anterior método [147]. 2.3.2. Nanotubos de carbono dopados con nitrógeno (CNx) La modificación de las propiedades cristalinas de los CNTs mediante la generación de defectos o inclusión de heteroátomos (P, Br, N) implica un potencial tecnológico amplio [151-154] ya que se pueden modificar las propiedades, aunque éstas dependerán de la posición de cada heteroátomo (al ser un objeto de escala nanométrica) en la red [155-162]. Teniendo en mente que el nitrógeno (N) posee un electrón adicional comparado con el carbono, podría esperarse que al sustituir un átomo de N por uno de C, se tendría un material del tipo-n [163]. Sin embargo, una sustitución directa de los átomos de C no es la única posibilidad de incorporar N en la estructura del nanotubo. Debido a su tamaño, el N, al formar parte de las paredes, podría también producir defectos en la estructura del tubo de tal forma que los átomos de C cercanos al N tendrían que reordenarse para llegar a un 41 equilibrio energético. En este caso, el comportamiento material de tipo-n, no puede ser deducido con facilidad. El comportamiento electrónico dependerá de la nueva geometría generada (habría una diferente estructura en las paredes de los nanotubos), por lo que no se podría descartar, asimismo, un material con características de tipo-p. La identificación de los diferentes tipos de enlace y evaluación del nivel de dopaje en los CNTs con N, ha sido interpretada en términos de resultados de sistemas dopados muy grandes, por lo que seguramente debe de existir diferencias en la estructura molecular en los CNTs dopados con respecto a los modelos propuestos. Lo que sí se conoce con certeza es que en los sistemas grafíticos grandes dopados con N, los átomos de N tienden a introducir desorden en los planos grafíticos. Estudios teóricos han mostrado que la sustitución de N por átomos de C genera curvatura en las láminas grafíticas [164]. En el caso de los CNTs (en particular los SWCNTs), se ha visto que son extremadamente sensibles a la incorporación local de N. Los primeros estudios donde se evaluó el dopaje en los CNTs ocurrió en 1993 [165], poco después de que Iijima publicara sus observaciones mediante la microscopía electrónica de transmisión [166, 167]. Una nanoestructura de carbono tubular que contiene átomos de carbono, no siempre deberá llamarse un CNT dopado con N. El dopaje por sustitución de átomos se relaciona con la eliminación de átomos de C de la estructura de un CNT por átomo de N. Esto ocasionará un cambio en la organización de sus paredes. Esta segunda posibilidad, donde se remueven varios átomos de C, afectará las propiedades de los CNTs en distintas maneras. Realmente, el N no solamente actuará como una impureza por sustitución. Czerw y colaboradores [168] sugirieron un segundo tipo de enlace de N en el CNT, correspondiendo a una configuración piridínica basada en cálculos teóricos (Figura 11). También llevaron a cabo experimentos con XPS y con la técnica de microscopía de efecto túnel (STM) encontrando en los CNTs dopados con N, una morfología de tipo bambú. La estructura piridínica implica a dos átomos de N coordinados creando una vacancia de un átomo de C, lo cual se asume como causa del comportamiento metálico de los CNTs dopados con N. Aunque la configuración por sustitución lleve a un material del tipo n, los estudios teóricos han sugerido que el dopaje con N podría llevar a una estructura del tipo p o n, dependiendo de la posición atómica de los átomos de N [169]. CNTs dopados con N ya han sido analizados mediante técnicas de espectroscopía donde se han encontrado configuraciones piridínicas [169, 170]. 42 Figura 11. Átomos de nitrógeno sustituyen a átomos de carbono, también se muestran enlaces piridínicos [171]. 2.3.2.1. Síntesis de CNx En la actualidad es posible producir CNx en una forma controlada mediante métodos convencionales. Para aplicaciones prácticas, en general el objetivo no es la máxima incorporación de N en los nanotubos, sino la suficiente para que tengan la porosidad y reactividad deseada. En general estos nanotubos se sintetizan por el método de CVD debido a su potencial de escalarse e incrementar la producción [172-174]. Evidentemente, además de una fuente de carbono hay que añadir una fuente de N. Suelen emplearse hidrocarburos con grupos funcionales nitrogenados, como la piridina o la bencilamina. Se pueden sintetizar CNx relativamente largos y limpios con contenidos debajo del 1% o por encima del 20% [175]. Sin embargo, la morfología difiere notoriamente al compararlos con los MWCNTs (los cuales son cilindros concéntricos con el interior hueco), los CNx en toda su estructura no muestran su interior libre [176], además se ha mostrado una fuerte correlación entre la cantidad de N y la morfología estructural de los nanotubos. Los CNx están divididos en compartimentos huecos a lo largo de su estructura separados por algunas capas de grafeno (se le ha llamado a esta estructura de tipo bambú) (Figura 12 a, b). Los 43 CNx también se han sintetizado mediante el método de descarga de arco alimentando con una mezcla de nitrógeno/helio [177] o N2 [178] durante el crecimiento de los nanotubos. Figura 12. Morfología de CNx obtenida por HRTEM, (a, b) tipo bambú (serie de compartimentos a lo largo del tubo); (c) helicoidal [171]. 2.3.2.2. Aplicaciones de los CNx Los defectos y rugosidad de los nanotubos dopados con N, ayudan a mejorar su funcionalización química para el anclaje de otras especies químicas o partículas [179, 180]. Los métodos de funcionalización en los nanotubos son muy importantes para las aplicaciones químicas o biológicas que podrían requerir un decorado o recubrimiento polimérico [181-183] para alguna función determinada (nanocompuestos). La particular reactividad de los CNx los ha convertido en componentes ideales para sensores rápidos de gas, como lo ha demostrado Villalpando-Paez y colaboradores [184]. Una de las áreas donde los CNx podrían aplicarse ampliamente sería en la de los semiconductores, donde se necesitaría un dopaje bajo y muy controlado. 2.3.3. Nanotubos de carbono multicapa funcionalizados con grupos basados en oxígeno (COx-MWCNTs) La funcionalización química de MWCNTs con grupos basados en oxígeno (carbonilos, hidroxilos, carboxilos), en general se ha realizado mediante tratamiento con ácido concentrado de una relación de H2SO4:HNO3. El ácido ocasiona un daño evidente y no deseado a los nanotubos. Botello-Méndez y colaboradores [185] encontraron una forma de 44 obtener MWCNTs funcionalizados con estos grupos (basados en oxígeno) sin daño, e incluso con características morfológicas particulares. Al adicionar 2.5 wt% de etanol directamente en la solución precursora de tolueno/ferroceno (5 wt% de ferroceno) a utilizar para la síntesis de nanotubos mediante CVD, encontraron nanotubos de casi 1 mm de longitud (e incluso de 5–10 mm en reacciones que duraron más de 1 h). La funcionalización química de la superficie de estos nanotubos la demostraron al formar suspensiones homogéneas de COx en isopropanol, por lo que su solubilidad aumentó con respecto a los MWCNTs sin funcionalización. En un estudio posterior llevado a cabo por Castle y colaboradores [186], se comparó la reactividad de los COx y los CNx mediante análisis termogravimétrico (TGA) (Figura 13). Encontraron que los CNx tuvieron una mayor oxidación que los MWCNTs y los COx, sugiriendo que los CNx y los COx tienen más sitios reactivos que los MWCNTs. Los materiales revelaron una descomposición a temperaturas de 370º para CNx, 420º para COx y 460º para MWCNTs. Figura 13. Análisis TGA de CNx, COx y MWCNTs no dopados en oxígeno [186]. Para confirmar la funcionalización de estos nanotubos, realizaron caracterización mediante la técnica de Rayos X de Energía Dispersiva (EDX, por sus siglas en inglés). Los resultados pueden observarse en la Tabla 1. La reactividad observada puede resumirse como sigue: MWCNTs < COx < CNx. Tabla 1. Análisis EDX y otras características de los nanotubos de carbono [186]. 45 2.3.4. Nanotubos de carbono exfoliados (exMWCNTs) La extensa investigación que actualmente se lleva a cabo para conocer las fascinantes propiedades del grafeno y su potencial aplicación en la física de estado sólido, electrónica, química y la nanociencia en general, ha llevado al desarrollo de muchos métodos para obtenerlo. Particularmente, una lámina de grafeno con uno de sus lados varias veces mayor que el otro, llamado nanolistón grafítico (nanoribbon), podría tener también aplicaciones interesantes en electrónica (como semiconductor), en el desarrollo de sensores (mediante una funcionalización química adecuada), como carga en compuestos poliméricos (aprovechando su reactividad debido a los bordes libres), entre otras más. Los CNTs a menudo son descritos como láminas de grafeno enrolladas, por lo que era natural que se intentara desenrollarlos de alguna manera para obtener grafeno (Figura 14). No fue sino hasta el 2009 que se publicó el primer método para obtener nanolistones grafíticos de nanotubos [187]. Otros dos estudios más, relacionados con la apertura longitudinal de los CNTs aparecieron después [188, 189]. Un cuarto método, primeramente sugerido por Terrones [190] sobre la utilización de partículas metálicas catalíticas como “nanotijeras”, fue demostrado experimentalmente [191]. Asimismo, se demostró que los CNTs pueden ser abiertos mediante una corriente eléctrica elevada dentro del microscopio [192]. Estos métodos diferentes son expuestos en la figura B. 46 Figura 14. Esquema que muestra las diferentes maneras de abrir longitudinalmente a los CNTs para obtener nanolistones grafíticos. (a) intercalación/exfoliación de MWCNTs (tratamiento en amoníaco líquido junto con Li, y subsecuente tratamiento ácido con HCl y choque de temperatura [187]); (b) método químico que tiene que ver con las reacciones ácidas que comienzan a romper los enlaces C-C (H2SO4 y KMnO4 como agente oxidante [188]); método catalítico, en el cual partículas metálicas cortan al tubo como si fueran tijeras [190, 191]; (d) método eléctrico, pasando una corriente eléctrica a través del nanotubo [192]; y (e) método físico-químico donde los tubos se embeben en una matriz polimérica tratándose posteriormente con un plasma de Ar [189]. Uno de los métodos, expuesto en la Figura 14 a, se refiere a la intercalación de Li en amoníaco líquido seguido por un tratamiento ácido y térmico (Figura 15). El material resultante (exMWCNTs) contiene una cantidad de nanolistones grafíticos por encima del 60%, con el resto de nanotubos parcialmente abiertos y muy dañados, junto con hojuelas de grafeno que se desprendieron de la superficie de los CNTs [187]. Esta mezcla de productos se obtiene debido al corte químico (con un tratamiento previo de H2SO4:HNO3 concentrado) de los MWCNTs para facilitar la apertura longitudinal (un MWCNT mayor a 2 μm de longitud raramente es abierto en su totalidad). Este método necesita mejorarse para obtener una mayor eficiencia de nanolistones, así como controlar sus dimensiones en función de los CNTs a utilizar. De momento, la mezcla de los listones con los subproductos obtenidos por la exfoliación, limita a este material para su aplicación en dispositivos electrónicos, sin embargo, podría ser de interés como carga en compuestos 47 poliméricos (debido a que sus abundantes bordes libres actuarían de interfase con la matriz polimérica). Además, su más elevada reactividad, lo hacen un material idóneo para una posterior funcionalización química. Por otra parte, el proceso de intercalación de Li ha sido utilizada para separar cuerdas de SWCNTs y funcionalizarlos por grupos haluros de arilo y alquilo [193-195] y también con monómero de metil metacrilato [196] para la síntesis de materiales compuestos poliméricos. Figura 15. Esquemas e imágenes representativas de la intercalación de Li-NH3 en los nanotubos y su exfoliación para obtener nanolistones grafíticos. El proceso comienza cortando CNTs (para abrir los extremos) y así, permitir la intercalación de iones Li y NH3 en los CNTs cargados negativamente (a). Los iones intercalados estresan las paredes del CNT duplicando la distancia entre capas e iniciando la fractura de las paredes (b). Después del tratamiento ácido y térmico la exfoliación es consumada, obteniéndose los exMWCNTs. Una micrografía HRTEM (d) muestra un MWCNT parcialmente abierto. El detalle de la zona donde comienza a abrirse muestra que después de los tratamientos, las láminas grafíticas pueden reordenarse y cristalizar. Los listones grafíticos pueden comprender varias capas de diferente grosor (f, g). El ordenamiento grafítico es confirmado por el patrón hexagonal de la Transformada Rápida de Fourier (dentro de g). 2.3.5. Nanofibras de carbono de listones grafíticos helicoidales (HR-CNF) Las nanofibras de carbono (CNFs), se diferencian esencialmente de los CNTs por la orientación de las láminas grafénicas respecto al eje longitudinal. La Figura 16 muestra las distintas estructuras, donde vienen comparadas con los SWCNTs y MWCNTs en 16 a y 16 48 b. Las CNFs no se consideran estructuras alotrópicas del C porque las láminas de grafeno que las conforman necesitan de otros elementos (hidrógeno u otros grupos funcionales) para estabilizarse. Sus diámetros pueden ir de los 15 a los 100 nm. Las CNFs tipo “platelet” (Figura 16 c) están formadas por un apilamiento largo de láminas de grafeno pequeñas. La partícula catalítica puede encontrarse en el centro de la fibra, obteniéndose un crecimiento bidireccional [197]. Algunos de estos filamentos pueden formar una serie de bucles [198] (Figura 16 d). Las CNFs del tipo “fishbone” pueden ser huecas [199] (Figura 16 e) o sólidas (Figura 16 f), con los planos grafénicos inclinados respecto a su eje longitudinal, con una sección poligonal, cuadrada o hexagonal [200]. Con respecto a los CNFs del tipo “ribbon” (Figura 16 g) los planos del grafeno suelen ser paralelos al eje longitudinal [197, 201]. Es posible que la partícula catalítica esté en el centro de la fibra. Por último, las fibras “stacked-cup” fueron descritas inicialmente por Endo y colaboradores [202] como fibras de diámetro ancho y un amplio hueco central, formadas por conos truncados, discontinuos y apilados. Sin embargo, otros estudios confirman un tipo de estructura en espiral continua [203-206]. Egksioglu y Nadarajah [206] a través de cálculos teóricos, encontraron que la formación de los conos apilados sólo podía producirse para determinados ángulos de inclinación, aunque experimentalmente (por microscopía electrónica de transmisión, TEM) se había observado una amplia variedad de ángulos. Esta discrepancia sólo podía resolverse considerando una hoja o listón de grafeno enrollada en forma de hélice. Asimismo, la elevada conductividad eléctrica de estos filamentos tiene más sentido considerando una estructura continua en vez de una truncada. 49 Figura 16. Esquema de las diferentes estructuras de nanotubos de carbono y nanofibras de carbono [207]. En 2007, Vera-Agulló y colaboradores [203] publicaron unas micrografías de TEM (Figura 17) definitivas sobre la estructura de las fibras stacked-cup. En las imágenes puede observarse una cinta saliendo del extremo de una fibra, cuyo ancho coincide con el de los planos de grafeno que conforman la pared del filamento. La cinta tiene una serie de repliegues (Figura 17 c) que se repiten constantemente. La longitud de la parte superior e inferior de esos pliegues coincide con el perímetro superior e inferior de los “conos” que conforman las nanofibras. Por lo tanto, resulta evidente que esa cinta es parte de la espiral continua que constituye la nanofibra. 50 Figura 17. Listón de láminas de grafeno desenrollándose de la fibra de carbono [203]. 2.3.6. Óxido de grafeno (GO) 2.3.6.1. Grafeno Como ya se ha comentado, el grafeno es una lamina bidimensional (2D) constituida por átomos de C con hibridación sp2 (1.42 Å de distancia entre átomos). La red hexagonal del grafeno se puede considerar la estructura base de otras formas alotrópicas de C con la misma hibridación, como lo es el grafito (apilamiento de láminas de grafeno con estructura 3D), los CNTs (grafeno enrollado cilíndricamente con estructura 1D), y los fullerenos (grafeno cerrado o envuelto con estructura 0D). Estas estructuras se esquematizan en la Figura 18. 51 Figura 18. El grafeno (2D) genera las demás formas alotrópicas del C, como los fullerenos (0D), los nanotubos de carbono (1D) y el grafito (3D) (abajo, de izquierda a derecha, respectivamente). El sistema conjugado de tipo π del grafeno, le otorga unas propiedades eléctricas, térmicas y mecánicas muy particulares. Fue el primer material bidimensional aislado con un átomo de espesor (posteriormente se prepararon láminas 2D de disulfuro de molibdeno y nitruro de boro). Hasta el año 2004 se concebía su existencia desde un punto de vista teórico y se pudo calcular algunas de sus propiedades [208-210]. En 2004, Andre Geim, Konstantin Novoselov y colaboradores [211] (en la Universidad de Manchester, Reino Unido, y el Instituto de Tecnología en Microelectrónica de Chernogolovka, Rusia) consiguieron aislar cristales de grafeno que fueron estables y pudieron caracterizarse. Lo lograron mediante el desprendimiento mecánico de láminas de grafeno del grafito con cinta adhesiva, que después transfirieron a un sustrato de Si. Por la relevancia de este descubrimiento y por las singulares propiedades de este material, Geim y Novoselov obtuvieron el premio Nobel de física en 2010. 52 Muchos de los estudios sobre el grafeno se han centrado en sus particulares propiedades electrónicas las cuales permiten que los transportadores de carga se comporten como partículas relativistas (cuasipartículas llamadas fermiones de Dirac por ser partículas sin masa, con carga, con espín semientero y que se pueden describir adecuadamente por la ecuación de Dirac). También permite que los transportadores produzcan el efecto de campo ambipolar (campos eléctricos de la misma intensidad pero signo opuesto inducen la formación del mismo número de electrones y huecos en el grafeno) [212]. Asimismo, se ha estudiado el efecto Hall cuántico anómalo a baja y alta temperatura [213] y la movilidad extraordinaria de transportadores de carga [214], entre otras propiedades. En cuanto a las propiedades del grafeno, es de destacar que tiene una densidad muy baja, de 0.77 mg/m2 (cada celda de grafeno contiene 2 átomos de C y un área de 0.052 nm2), al ser casi bidimensional y por consiguiente un área superficial específica muy alta (2600 m2/g). Debido a la alta movilidad de transportadores de carga (~200000 cm2V-1s-1), el grafeno tiene una conductividad eléctrica muy elevada [214]. Asimismo, la conductividad térmica del grafeno es muy alta, comparable e incluso superior a los CNTs (5000 Wm-1K-1) a temperatura ambiente [215]. En cuanto a las propiedades mecánicas del grafeno, estas son extraordinarias también, el módulo de elasticidad y resistencia a la tensión alcanzan valores de 1 TPa y 130 GPa, respectivamente, no superados hasta el momento por ningún otro material [216]. Las láminas de grafeno son flexibles y elásticas (puede estirarse elásticamente hasta en un 20%). 2.3.6.2. Producción de grafeno A pesar de las dificultades que aún existen para la producción de grafeno, existen tres vías principales para obtenerlo. La primera sería la exfoliación mecánica a partir de grafito (técnica empleada por Geim y Novoselov en 2004), aunque a pesar de la calidad de los cristales que suelen obtenerse con este método, no deja de ser artesanal. Únicamente ha sido útil para la caracterización del grafeno. La segunda forma sería mediante el crecimiento epitaxial de grafeno sobre una superficie plana, empleándose CVD para la descomposición de hidrocarburos sobre sustratos metálicos como Ni y Cu [217, 218]. El C producido de la descomposición de hidrocarburos se reorganiza y forma sobre la superficie metálica láminas de grafeno de una o varias capas. También se ha utilizado tratamiento térmico en carburo de silicio, donde los átomos superficiales de Si se volatilizan de tal modo, que los átomos de C que estaban unidos al Si 53 se reorganizan formando grafeno [219, 220]. Estos métodos de crecimiento epitaxial deben mejorarse para escalarse y producir más grafeno. En otra estrategia, se ha exfoliado grafito o sus derivados en fase líquida para obtener dispersiones de grafeno. Se ha utilizado N-metil-2-pirrolidona (NMP) con sonicación para exfoliar el grafito [221]. El derivado de grafito más empleado para la obtención de dispersiones de grafeno es el óxido de grafito, del cual se obtienen dispersiones acuosas de láminas de óxido de grafeno (GO), el cual puede considerarse un derivado del grafeno. 2.3.6.3. Óxido de grafeno (GO) Desde mediados del siglo XIX se conoce que la oxidación en medio ácido del grafito conduce a un material aislante, higroscópico y con un alto contenido en oxígeno, se le llama óxido de grafito [222, 223]. Aún no existe consenso sobre cuál es la estructura del óxido de grafito, ni cuales son sus grupos funcionales oxigenados ni su proporción en el material. En la Figura 19 se muestra uno de los modelos propuestos de la estructura del óxido de grafito [224], en donde aparecen grupos funcionales carboxilo, hidroxilo (en los bordes de la lámina de grafeno) o epóxido. Figura 19. Modelo esquemático de la estructura del óxido de grafito [224]. 54 El óxido de grafito ha recobrado relevancia debido a la facilidad con que puede ser exfoliado en medio acuoso por sonicación, y así obtener dispersiones de láminas de óxido de grafeno (GO). Se ha convertido en uno de los métodos más empleados para obtener derivados de grafeno. El GO puede ser reducido, obteniéndose óxido de grafeno reducido (R-G-O, por sus siglas en inglés), que se caracteriza por contener menos oxígeno y en cierta medida recupera la estructura conjugada π del grafeno. Así, el RGO tiene una conductividad eléctrica más alta que el GO, y características hidrofóbicas en vez de hidrofílicas (como en el caso del GO) [225]. El RGO es una alternativa muy viable al uso del grafeno debido al alto coste del grafeno de mayor calidad estructural obtenido por otros métodos. En el caso del GO utilizado en esta tesis, en lugar de partir de grafito, se utilizaron nanofibras de carbono de listones grafíticos helicoidales (HR-CNF denominadas GANF, con fines de comercialización por Grupo Antolín) como fuente del GO. Estas nanofibras también se someten a un proceso de oxidación, con oxiácidos concentrados, y exfoliación mecánica por ultrasonidos. Después de un proceso de purificación se obtiene GO. A partir del GO, el RGO se obtiene por diversos métodos como la reducción química (empleando hidracina, dimetilhidracina, borohidruro de sodio, entre otros) [226, 227], el tratamiento térmico [228, 229] y la reducción fotocatalítica [230]. Debido a las interacciones π-π que se establecen entre las láminas de RGO que ocasiona que formen agregados, es muy difícil la dispersión (sobre todo en medios acuosos). Por lo que en muchas ocasiones, se procesa al GO en la forma deseada y posteriormente se reduce. La presencia de grupos funcionales activos en el GO permite la funcionalización covalente de las láminas de modo similar a la funcionalización covalente de los CNTs oxidados (incluyendo los COx) lo cual, hasta cierto punto les permite dispersarse en disoluciones orgánicas (o incluso acuosas), para un posterior procesado, que les permita formar parte de matrices poliméricas y así formar compuestos. 2.4. Compuestos de polianilina Los materiales compuestos de polímeros con nanofilamentos de carbono y grafeno es un área en constante desarrollo en donde se ha aprovechado el bajo coste y facilidad de procesado de los polímeros con las extraordinarias propiedades de estas nanoestructuras. Debido a que estos materiales contienen nanoestructuras se les ha denominado nanocompuestos para diferenciarlos de los compuestos convencionales. 55 La dispersión de las nanoestructuras es un factor muy importante en la preparación de nanocompuestos. Éstas, en una situación ideal, deberían encontrarse separados y distribuidas homogéneamente en la matriz polimérica. Una mejor dispersión de nanoestructuras, además de otorgarle homogeneidad al compuesto, maximiza la superficie de contacto efectiva entre las nanoestructuras y el polímero, lo cual mejora la transferencia de propiedades de éstas a la matriz, tales como la conductividad eléctrica, térmica, resistencia mecánica, entre otras. Asimismo, una buena dispersión minimiza la cantidad de carga necesaria para que el compuesto obtenga las propiedades deseadas. En general, hay tres estrategias para sintetizar compuestos con nanofilamentos: mezclado en disolución, mezclado en fundido, polimerización in situ. En el primer método, los filamentos se dispersan en una disolución del polímero, obteniéndose el compuesto tras la eliminación del disolvente. Comúnmente se utilizan ultrasonidos para dispersar las nanoestructuras, aunque también se utilizan agitadores de alto corte. Mediante este método se han obtenido compuestos de CNTs con policarbonato, poliestireno [231, 232], poliuretano [233], PVA [234], entre otros. Este método no es aplicable a polímeros insolubles. El método de mezclado en fundido es uno de los más utilizados a nivel industrial en la preparación de compuestos poliméricos termoplásticos. El polímero y la carga son mezclados por la acción de alta cizalla en extrusoras a una temperatura suficiente para fundir el termoplástico. La dispersión de los filamentos suele ser más baja que en anterior método, dado que los termoplásticos tienen una elevada viscosidad, lo cual limita la introducción de nanoestructuras en el compuesto. Mediante este método se han sintetizado compuestos de CNTs con polietileno [235, 236], polipropileno [237, 238], PMMA [239242], poliamidas [243], entre otros polímeros. En el caso del método de polimerización in situ, las nanofibras se dispersan junto con el monómero para luego realizar la polimerización. Como el polímero se forma en presencia de la carga, se favorece la interacción carga/polímero. Una de las ventajas de este método es que puede ser aplicado a polímeros para los cuales los dos primeros métodos (mezclado en disolución o fundido) son difíciles o inviables, como en el caso de los polímeros insolubles o térmicamente inestables (como la PAni). Mediante la polimerización in situ, se han producido compuestos de CNTs con poliolefinas [244, 245], poliamidas [246, 247], poliésteres [248], poliuretanos [249, 250], entre otros. 56 En algunos casos, para estas polimerizaciones se han empleado filamentos funcionalizados con algún grupo funcional reactivo para formar enlaces covalentes con las moléculas del monómero. Por ejemplo se ha llegado a funcionalizar a SWCNTs con grupos metil metacrilato, obteniéndose una elevada dispersión en el PMMA donde el enlace covalente de los nanotubos con la matriz polimérica es muy alta [196]. La polimerización in situ es práctica y efectiva en la síntesis de compuestos con CNTs y polímeros conductores. La fuerte interacción entre los electrones π, altamente deslocalizados de los CNTs y los electrones π correlacionados con la red del esqueleto polimérico, ha favorecido la transferencia electrón/hueco entre los CNTs y los polímeros conductores. Las interacciones π-π entre las moléculas del monómero y las paredes de los CNTs facilitan su dispersión en el medio de reacción y favorecen la polimerización sobre la superficie de los CNTs. Este método se emplea comúnmente par la síntesis de compuestos con polipirrol [251, 252], politiofeno [253], PAni [254], entre otros. Algunos estudios han mostrado que la anilina interactúa fuertemente con los nanotubos de carbono (CNTs) mediante interacciones tipo donador-aceptor. Los CNTs son relativamente buenos aceptores de electrones, mientras que la PAni podría ser considerada como un buen donador [255]. Estos compuestos de PAni/CNTs han encontrado aplicación potencial en sensores de gas, tintas conductoras, supercondensadores, celdas fotovoltaícas, músculos artificiales, entre otras aplicaciones [256]. El método de polimerización in situ de compuestos de PAni es precisamente el utilizado en la preparación de los compuestos (con las nanoestructuras de carbono) en esta tesis. 2.5. Músculos artificiales Un actuador es un dispositivo que puede responder mecánicamente ante un estímulo físico o químico. Ejemplo de ello son los motores (eléctricos, neumáticos o de combustión interna), pistones o cilindros y piezoeléctricos. Los motores en general transforman la energía eléctrica, neumática o química, en mecánica. Los motores eléctricos poseen un torque bajo comparados con los músculos biológicos. Los motores de combustión interna, generalmente son más eficientes si se operan continuamente, por lo que son inadecuados para aplicaciones en las que el movimiento es interrumpido frecuentemente. Los pistones o cilindros (neumáticos, hidráulicos, magnéticos o de combustión), responden linealmente 57 ante una presión de un fluido o a una fuerza electromagnética. Los piezoeléctricos, que son cristales que ante un campo eléctrico se polarizan y deforman, alcanzan densidades de potencia altas pero su distensión es pequeña (los ferroeléctricos cerámicos se distienden ~0.15%). Además de los actuadores mencionados, en las últimas décadas se han desarrollado otros actuadores, que tratando de emular a los músculos biológicos, han recibido el nombre de “músculos artificiales”. Están basados en materiales poliméricos con propiedades especiales y generalmente consisten de elastómeros o polímeros conductores. Estos materiales se han dividido en dos grupos: polímeros electroactivos (EAP, por sus siglas en inglés) y polímeros electroactivos iónicos (EAP iónicos). Los EAP suelen ser materiales de elastómeros (silicona o elastómero acrílico que son a veces rellenados con partículas, como el TiO2, para incrementar su constante dieléctrica [257]) que se colocan en medio de “placas” conductoras elásticas (funcionando como dieléctrico de un condensador de placas paralelas), que ante un potencial y una consiguiente atracción electrostática entre las placas (producto de un elevado potencial eléctrico del orden de los KV), obligan al elastómero a deformarse (Figura 20). Los electrodos elásticos pueden elaborarse de carbono (como partículas de grafito esparcidas), pasta de Ag, gomas conductoras, o láminas de CNTs elásticas [257-259]. En cambio, los EAP iónicos están conformados por polímeros conductores, como el polipirrol, la polianilina, y los derivados del politiofeno que cuando están inmersos en un electrolito, y se les aplica un potencial bajo (de ~0.6 a 7 V), logran elongarse o contraerse debido a la inserción o desinserción de iones en la fase polimérica. En la Figura 21, se observa el mecanismo de accionamiento de un músculo artificial elaborado de polipirrol, en el cual, mediante un potencial redox determinado, el polímero puede incorporar o expulsar iones de sus cadenas poliméricas en el medio electrolítico. Esto permite expandir o reducir su volumen. Existen también actuadores elaborados de cuerdas de CNTs [260] que se elongan o contraen por inserción de iones en su estructura. 58 Figura 20. Músculos artificiales del tipo de polímero electroactivo (EAP) [261]. Figura 21. Posible mecanismo de accionamiento de un músculo artificial de polímero electroactivo iónico (EAP iónico) basado en polipirrol, donde, ante un potencial redox, ocurre una inserción de iones en las cadenas poliméricas que provienen del medio electrolítico [261]. La ventaja de utilizar EAP, es que suelen obtenerse mayores elongaciones (10-100%), las cuales son del orden, o superiores, a las elongaciones del músculo esquelético (~20%). Aunque debido a los altos voltajes que se utilizan para su accionamiento, se requiere de un cuidado especial en su aplicación para evitar altas descargas. En cambio, las ventajas que se obtienen al utilizar EAP iónicos, es que se requieren voltajes mucho más bajos y los materiales poliméricos son más económicos. Las características que desfavorecen el uso de polímeros conductores es que suelen obtenerse menores elongaciones (2-10%), que deben de estar inmersos en un electrolito (líquido o sólido) y que su velocidad de accionamiento tiende a ser baja (<<1%/s) debido al transporte relativamente lento de iones en el polímero y su nivel de dopaje [262]. Aunque la velocidad de accionamiento puede incrementarse a 59 ~10%/s al utilizar contactos metálicos, polímeros porosos o películas delgadas y fibras [263-266]. El mecanismo de accionamiento de los EAP iónicos es más semejante al de los músculos biológicos debido a que la interacción de los iones en el electrolito con las cadenas poliméricas (ante una señal eléctrica determinada), recuerda la interacción del plasma sanguíneo con las fibrillas musculares (miosina/microtúbulos) ante una señal eléctrica nerviosa. 2.5.1. Músculos artificiales de PAni El primer investigador que mencionó la posible aplicación de los polímeros conductores como músculos artificiales fue R. H. Baughman [267] en 1990, quien junto a su grupo de investigación, construyó el primer dispositivo en 1987. Se trataba de una delgada película rectangular de un copolímero de 3-metil-tiofeno y 3-n-octil-tiofeno con un recubrimiento de oro por un lado. El contraelectrodo utilizado fue de Li y el electrolito fue de LiClO4/carbonato de propileno. Se lograron flexiones reversibles de la película al aplicarle un potencial. A partir de entonces diferentes grupos de investigación comenzaron a evaluar actuadores flexionantes de polipirrol [268-270], destacando un estudio de Smela y colaboradores [271] quienes construyeron microactuadores (10 X 20 μm) utilizando técnicas de microlitografía en una oblea de Si (los actuadores se recubrieron de Au por un lado). Lograron obtener varios miles de ciclos de trabajo antes de que fallaran por delaminación, y tuvieron respuestas de accionamiento de 5 s. Los estudios con polipirrol desde entonces han continuado. Los primeros estudios del cambio de volumen de la PAni en estado oxidado los llevó a cabo Okabayashi y colaboradores [272] en un electrolito de LiClO4/carbonato de propileno. Observaron un incremento significativo del volumen de la PAni al insertarse los iones ClO4- en las cadenas poliméricas al ocurrir la oxidación. Más adelante, al comenzar la década de los 90 se probaron en distintos electrolitos protónicos, las primeras películas de PAni plastificadas y adecuadas (mediante un drop-casting en sustrato de vidrio de PAni disuelta en NMP y luego estiradas con aplicación de temperatura) para los estudios de deformación electroquímica. Se estudió el efecto de la orientación de las cadenas poliméricas (se realizaron mediciones en forma perpendicular y paralela a la dirección de estiramiento al que fueron sometidas las películas) obteniéndose mayores deformaciones 60 en las películas cuyas cadenas poliméricas estaban orientadas más perpendicularmente al eje axial del actuador [273]. En otros estudios, las películas de PAni plastificadas con NMP, fueron adheridas a películas no activas para obtener flexiones (sistemas bicapa, o tricapa con la capa no activa en medio, las dos exteriores siendo de PAni y accionadas alternativamente como ánodo y cátodo con 1.5 V), en general, el electrolito estándar utilizado fue el HCl 1 o 2 M. Se obtuvieron elongaciones de ~1% y observaciones hasta de 200 ciclos de trabajo [274, 275]. En evaluaciones subsiguientes, donde ya no se utilizó una capa inactiva adherida a la película de PAni, se logró obtener hasta un 3% de deformación. Asimismo, se mostró experimentalmente que la elongación de la película fue proporcional a la carga eléctrica almacenada en el polímero [276, 277]. Posteriormente, además de producir películas, se comenzó a conformar fibras mediante la técnica del hilado húmedo (wet spinning), el cual consiste en el alargamiento de una solución altamente concentrada de PAni (sal de esmeraldina) con m-cresol, con una viscosidad apropiada que se va enredando en unas bobinas al mismo tiempo que se sumergen en una o varias soluciones de coagulación (acetona, 4-metil-2-pentanona, entre otras). Las fibras pueden secarse y alargarse hasta un diámetro apropiado mediante aplicación de calor. Se obtuvieron conductividades de hasta 1900 S/cm [278]. Por otra parte, también se han extruído fibras utilizando base de esmeraldina disuelta en dimetil– propileno–urea (DMPU) y se han dopado con ácidos sulfónicos [279-282]. Basándose en el anterior método, pero ahora utilizando ácido 2-acrilamido-2-metilpropano sulfónico (AMPSA, por sus siglas en inglés) como plastificante y dopante (junto con ácido dicloroacético o fórmico) de la PAni, se obtuvieron películas (mediante drop-casting) y fibras (mediante el método de hilado húmedo). Con respecto al accionamiento de las películas, se les colocó una capa delgada de oro por un lado, evitando así, que la resistencia eléctrica intrínseca del polímero pudiera afectar el accionamiento en zonas alejadas del conector [283-287]. Las elongaciones de estos músculos artificiales fue de ~2%, destacándose aquellos en los que se ciclaron en ácido metanosulfónico [286], ya que se encontró que este ácido, estabiliza mecánicamente el estado más oxidado de la PAni (pernigranilina), obteniéndose una vida útil del actuador más larga. Finalmente, Gu y colaboradores [288], elaboraron un músculo artificial de PAni (emulando las miofibrillas de los músculos biológicos) que consistió en una cuerda de fibras de PAni 61 (de 900 nm de diámetro y con un núcleo de poliuretano cada una) que se accionaron en paralelo, logrando distensiones de ~1.65%. 2.5.2. Músculos artificiales de compuestos de PAni Las fibras de PAni producidas por hilado húmedo han mostrado propiedades eléctricas y mecánicas adecuadas para desarrollo de músculos artificiales. En un estudio se produjeron fibras plastificadas con DMPU para probarlas como actuadores lineales. Se comprobó que las fibras con un 2% (w/w) de SWCNTs (la máxima concentración de nanotubos de carbono que pudieron dispersarse adecuadamente en la PAni) se elongaron hasta un 2.2% y tuvieron mejores propiedades eléctricas (tuvieron una conductividad eléctrica 30 veces superior a la de las fibras sin nanotubos) y mecánicas que las fibras sin nanotubos [289]. En las pruebas de accionamiento, hubo una disminución en la elongación del material (0.3%), con respecto al de PAni sin nanotubos. Evaluaron con detalle el efecto del dopaje de las fibras en sus propiedades mecánicas. Por ejemplo, la resistencia a la tracción fue de 260 MPa y de 197 MPa, antes y después del dopaje, respectivamente. Asimismo, el dopaje ocasionó que el módulo de Young disminuyera de 19.7 a 17 GPa, pero a pesar de esta disminución, este nivel estuvo muy por encima de los valores de 2–4 GPa que se tuvieron para las fibras de PAni sin nanotubos. Poco después, Geoffrey M. Spinks y colaboradores [290], bajo las mismas condiciones experimentales, elaboraron fibras a las que se les midió sus propiedades electromecánicas y de accionamiento. Encontraron que, aunque las fibras de compuestos con SWCNTs (0.76 wt%) tuvieron una menor elongación (0.8%) que las de PAni sin nanotubos (1.3%), éstas mejoraron significativamente sus propiedades mecánicas y eléctricas. No se encontraron más estudios de síntesis de compuestos de PAni con otras nanoestructuras de carbono dispersas en su matriz polimérica para aplicaciones de accionamiento, por lo que uno de los principales objetivos de esta tesis, será precisamente evaluar el efecto de distintas nanoestructuras de carbono en actuadores de PAni (lineales y flexionantes). 62 2.6. Electrodos para supercondensadores Los condensadores convencionales, consisten en un par de placas paralelas separadas por un aislante (dieléctrico). La carga eléctrica de signo opuesto se almacena en las placas ante la aplicación de una diferencia de potencial. Las capacidades de estos dispositivos suelen ser de μF (microfaradios) o mF. En tiempos recientes, se han desarrollado condensadores que han aumentado su capacidad de carga hasta llegar a los cientos de F. Se les ha denominado supercondensadores o ultracondensadores. Inicialmente se construyeron de materiales carbonosos de un área superficial elevada (se les conoce como condensador electrolítico de doble capa – EDLC, por sus siglas en inglés–) [291]. La carga eléctrica en estos dispositivos se almacena en la capa eléctrica doble cercana a la interfase del material con el electrolito que se genera para balancear la carga del material. Otro tipo de supercondensador, denominado pseudocondensador, almacena la carga eléctrica en el volumen de su cuerpo, como respuesta a un potencial redox. Esta reacción redox rápida [292-294] actúa como capacidad o capacitancia (de aquí el nombre de pseudocapacidad). Un pseudocondensador almacena mucha más carga eléctrica que un condensador de doble capa debido a que la almacena en su volumen y no en la superficie. Los polímeros conductores son materiales pseudocapacitivos, los cuales fueron por primera vez utilizados como supercondensadores a mediados de la década de los 90 [295]. Los supercondensadores podrían ser el “eslabón” que une a dispositivos tales como condensadores convencionales (los cuales entregan la carga eléctrica almacenada de forma muy rápida pero tienen limitaciones importantes en el almacenamiento de energía) y las baterías (las cuales pueden otorgar energía eléctrica durante largos periodos pero no pueden entregarla en grandes cantidades durante cortos tiempos). De esta forma los supercondensadores pueden almacenar una cantidad relativamente grande de carga y pueden otorgarla en tiempos relativamente cortos. Estos dispositivos serían ideales para vehículos híbridos o puramente eléctricos, donde podrían contribuir a la aceleración/desaceleración de éstos, puesto que se requiere una cantidad de energía elevada en periodos relativamente cortos. El concepto de eslabón de los supercondensadores con respecto a las baterías y condensadores tradicionales se ilustra en la Figura 22. 63 Figura 22. Diferentes tipos de dispositivos que almacenan carga eléctrica; CP se refiere a polímeros conductores [296]. Los polímeros conductores también encuentran aplicación como supercondensadores. Como se ha visto a lo largo de la Introducción, los polímeros conductores ante un potencial redox determinado, causa una inserción iónica del medio electrolítico donde esté el material activo. Este es el mecanismo mediante el cual los polímeros conductores almacenan carga y se deforman (con aplicaciones en músculos artificiales, como se vio anteriormente). Los polímeros más estudiados como supercondensadores han sido la PAni, el polipirrol y el politiofeno [297], de los cuales sobresale la PAni debido a su buena estabilidad ambiental, facilidad de síntesis y procesado y su alta capacidad específica (400500 F/g en un medio protónico) [296, 298-300]. La diferencia de almacenamiento de carga entre supercondensadores de doble capa y supercondensadores basados en polímeros conductores, se muestra esquemáticamente en la Figura 23. En (A) puede observarse la doble capa de cargas opuestas en la interfase del material carbonoso y el electrolito en donde está inmerso. En (B) se aprecia la inserción de contraiones en las cadenas poliméricas del material provenientes también de un electrolito. 64 Figura 23. Comparación del almacenamiento de carga entre (A) condensador de doble capa (carbono) y (B) pseudocondensador (polímero conductor) [296]. Los condensadores de doble capa poseen una vida útil elevada, mayor al medio millón de ciclos [291] mientras que los condensadores de polímeros conductores comienzan a degradarse a partir de algunos cientos de ciclos debido a los cambios que sufre su estructura interna [301]. A pesar de esto, la elevada energía específica de los polímeros conductores sigue siendo de interés para el desarrollo de supercondensadores. La PAni ha sido estudiada extensamente como electrodo para supercondensadores [302307]. Se ha publicado que la PAni tiene una variación amplia en el rango de capacidad (la más alta de todos los PC) que va de 44 a 270 mAh/g [297]. Esta variación se debe a varios factores como el método de síntesis, la morfología del polímero, la cantidad y tipo de aglomerante, el espesor del electrodo (material activo), entre otros. Se ha observado que resulta beneficioso que la PAni sea lo más porosa posible para favorecer el intercambio 65 iónico con el electrolito, por lo que la síntesis de PAni nanoestructurada, mediante el método de polimerización interfacial, es cada vez más frecuente [308-310]. Se ha estudiado también la preparación de electrodos de PAni con distintas cargas de carbono para mejorar sus propiedades eléctricas y mecánicas. Por ejemplo se han fabricado compuestos de PAni como nanotubos de carbono de capa simple (SWCNTs) [311, 312] y múltiple (MWCNTs) [313, 314], óxido de grafeno (GO) [315] y óxido de grafeno reducido (RGO) [316]. Estas cargas dispersas en el polímero han mejorado las propiedades eléctricas y rigidez del material compuesto [317-320]. La mejora conseguida en la rigidez, ha contribuido a disminuir la degradación de la PAni y así evitar un desprendimiento de la película del colector. 66 REFERENCIAS [1] Ito, T.; Shirakawa, H.; Ikeda, S. Simultaneous polymerization and formation of polyacetylene film on the surface of concentrated soluble Ziegler-type catalyst solution. J. Polym. Sci. Polym Chem. Edu. 12, 1974, 11-20. [2] Letheby, H. On the production of a blue substance by the electrolysis of sulphate of aniline. Journal of the Chemical Society 15, 1862, 161-163. [3] Mohilner, D. M.; Adams, R. N.; Argersinger, Jr.; W. J. Investigation of the Kinetics and Mechanism of the Anodic Oxidation of Aniline in Aqueous Sulfuric Acid Solution at a Platinum Electrode. J. Am. Chem. Soc. 84, 1962, 3618-3622. [4] Gardini, G. P. The oxidation of monocyclic pyrroles. Adv. Heterocycl. Chem. 15, 1973, 67-98. [5] Angeli, A. Sopra I neri di pirrolo. Gazz. Chim. Ital. 46, 1916, 279. [6] Lund, H. Electroorganic preparations. IV. Oxidation of aromatic hydrocarbons. Acta Chem. Scand. 11, 1957, 1323-1330. [7] Peover, M. E.; White, B. S. J. The electro-oxidation of polycyclic aromatic hydrocarbons in acetonitrile studied by cyclic voltammetry. Electroanal. Chem. 13, 1967, 93-99. [8] Osa, T.; Yildiz, A.; Kuwana, T. Electrooxidation of benzene. J. Am. Chem. Soc. 91, 1969, 3994-3995. [9] Armour, M.; Davies, A. G.; Upadhyay, J.; Wasserman, A. J. Colored electrically conducting polymers from furan, pyrrole, and thiophene. Polym. Sci. A1. 5, 1967, 15271538. [10] Josefowicz, M.; Yu, L. T.; Belorgey, G.; Buvet, R. J. Polym. Sci. Part C: Polym. Symp. 16, 1969, 2943. [11] Dall’Ollio, A.; Dascola, Y.; Varacca, V.; Bocchi, V. Comptes Rendus, C267, 1968, 433. [12] (a) Chiang, C. K.; Fincher, C. R.; Park, Y. W.; Heeger, A. J.; Shirakawa, H.; Louis, E. J.; Gau, S. C.; McDiarmid, A. G. Phys. Rev. Lett. 39, 1977, 1089. (b) Genies, W. M.; Bidan, G.; Diaz, A. Spectroelectrochemical study of polypyrrole films. J. Electroanal. Chem. 149, 1983, 101-113. [13] (a) Diaz, A. F.; Kanazawa, K. K.; Gardini, G. P. Electrochemical polymerization of pyrrole. J. Chem. Soc. Chem. Commun. 1979, 635-636. (b) Scott, J. C.; Pfluger, P.; Krounbi, M. T.; Street, G. B. Phys. Rev. B, 28, 1983, 2140. (c) Chen, J.; Heeger, A. J. In situ electron spin resonance experiments on polyacetilene during electrochemical doping. Synthetic Metals 24, 1988, 311-327. [14] Unverdorben, O. Ueber das Verhalten der organischen Körper in höheren Temperaturen. Annalen der Physik 84, 1826, 397-410. [15] Runge, F. F. Ueber einige Produkte der Steinkohlendestillation. Annalen der Physik und Chemie 107, 1834, 65-78. 67 [16] Runge, F. F. Ueber einige Producte der Steinkohlendestillation. Annalen der Physik und Chemie 108 (20-25), 1834, 328-333. [17] Zinin, N. Beschreibung einiger neuer organischer Basen, dargestellt durch die Einwirkung des Schwefelwasserstoffes auf Verbindungen der Kohlenwasserstoffe mit Untersalpetersäure. Journal für Praktische Chemie 27, 1842, 140-153. [18] Fritsche, J. Ueber das Anilin, ein neues Zersetzungsproduct des Indigo. Journal für Praktisch Chemie 20, 1840, 453-459. [19] Travis, A. S. Mauve 1856: Perkin's purple and where it led. AATCC Review 6, 2006, 28-31. [20] Travis, A. S. From Manchester to Massachusetts via Mulhouse: The Transatlantic Voyage of Aniline Black. Technology and Culture 35, 1994, 70-99. [21] Willstätter, R.; Moore, C. W. Über Anilinschwarz. I. Berichte der deutschen chemischen Gesellschaft 40, 1907, 2665-2689. [22] Green, A. G.; Woodhead, A. E. CCXLIII. - Aniline-black and allied compounds. Part I. Journal of the Chemical Society, Transactions 97, 1910, 2388-2403. [23] Green, A. G.; Woodhead, A. E., CXVII. - Aniline-black and allied compounds. Part II. Journal of the Chemical Society101, Transactions 1912, 1117-1123. [24] Kobayashi, T.; Yoneyama, H.; Tamura, H. Electrochemical reactions concerned with electrochromism of polyaniline film-coated electrodes. Journal of electroanalytical Chemistry 177(1-2), 1984, 281-291. [25] Mengoli, G.; Munari, M. T.; Bianco, P.; Musiani, M. M. ANODIC SYNTHESIS OF POLYANILINE COATINGS ONTO Fe SHEETS. Journal of Applied Polymer Science 26, 1981, 4247-4257. [26] DeBerry, D. W. Modification of the Electrochemical and Corrosion Behavior of Stainless Steels with an Electroactive Coating. Journal of the Electrochemical Society 132, 1985, 1022-1026. [27] Macdiarmid, A. G.; Chiang, J. C.; Huang, W. S.; Humphrey, B. D.; Somasiri, N. L. D. POLYANILINE - PROTONIC ACID DOPING TO THE METALLIC REGIME. Molecular Crystals and Liquid Crystals 125 (1-4), 1985, 309-318. [28] Chiang, J. C.; MacDiarmid, A. G. Polyaniline: Protonic acid doping of the emeraldine form to the metallic regime. Synthetic Metals 13 (1-3), 1986, 193-205. [29] Huang, W.; Humphrey, B. D.; MacDiarmid, A. G. J. Polyaniline, a novel conducting polymer. Morphology and chemistry of its oxidation and reduction in aqueous electrolytes. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 82 (8), 1986, 2385-2400. [30] Macdiarmid, A. G.; Chiang, J. C.; Halpern, M.; Huang, W. S.; Mu, S. L.; Somasiri, N. L. D.; Wu, W. Q.; Yaniger, S. I. POLYANILINE - INTERCONVERSION OF METALLIC AND INSULATING FORMS. Molecular Crystals and Liquid Crystals 121 (1-4), 1985, 173-180. [31] Genies, E. M.; Lapkowski, M. Electrochemical in situ epr evidence of two polaronbipolaron states in polyaniline. Journal of Electroanalytical Chemistry 236 (1-2), 1987, 199-208. 68 [32] Monkman, A. P.; Bloor, D.; Stevens, G. C.; Stevens, J. C. H.; Wilson, P. Electronic structure and charge transport mechanisms in polyaniline. Synthetic Metals 29 (1), 1989, 277-284. [33] Sapurina, I.; Setjskal, J. The mechanism of the oxidative polymerization of aniline and the formation of supramolecular polyaniline structures. Polym. Int. 57, 2008, 1295-1325. [34] David, S.; Nicolau, Y. F.; Melis, F.; Revillon, A. Molecular weight of polyaniline synthesized by oxidation of aniline with ammonium persulfate and with ferric chloride. Synthetic Metals 69 (1-3), 1995, 125-126. [35] Inoue, M.; Brown, F.; Muñoz, I. C.; Muñoz, F. O. Polymerization of anilines by the use of copper(II) perchlorate as an oxidative coupling agent. Polymer Bulletin (Berlin) 1991, 26 (4), 403-408. [36] Rahy, A.; Yang, D. J. Synthesis of highly conductive polyaniline nanofibers. Materials Letters 62 (28), 2008, 4311-4314. [37] Klavetter, F. L.; Cao, Y. Homogeneous route to polyaniline. Synthetic Metals 55 (2 -3 pt 2), 1993, 989-994. [38] Cao, Y.; Li, S.; Xue, Z.; Guo, D. Spectroscopic and electrical characterization of some aniline oligomers and polyaniline. Synthetic Metals 16(3), 1986, 305-315. [39] Ahmed, S. M., Mechanistic investigation of the oxidative polymerization of aniline hydrochloride in different media. Polymer Degradation and Stability 85(1), 2004, 605-614. [40] Ciric-Marjanovic, G.; Trchova, M.; Stejskal, J. J. Theoretical study of the oxidative polymerization of aniline with peroxydisulfate: Tetramer formation. International Journal of Quantum Chemistry 108(2), 2008, 318-333. [41] Tzou, K.; Gregory, R. V., Kinetic study of the chemical polymerization of aniline in aqueous solutions. Synthetic Metals 47(3), 1992, 267-277. [42] Fu, Y.; Elsenbaumer, R. L. Thermochemistry and kinetics of chemical polymerization of aniline determined by solution calorimetry. Chemistry of Materials 6(5), 1994, 671-677. [43] Stejskal, J.; Kratochvil, P.; Spirkova, M. Accelerating effect of some cation radicals on the polymerization of aniline. Polymer 36 (21), 1995, 4135-4140. [44] Ayad, M. M.; Rehab, A. F.; El-Hallag, I. S.; Amer, W. A. J. Preparation and characterization of polyaniline films in the presence of N-phenyl-1,4-phenylenediamine. European Polymer Journal 43(6), 2007, 2540-2549. [45] Ma, X.; Zhang, X.; Li, Y.; Li, G.; Wang, M.; Chen, H.; Mi, Y., Preparation of nanostructured polyaniline composite film via "carbon nanotubes seeding" approach and its gasresponse studies. Macromolecular Materials and Engineering 2006, 291 (1), 75-82. [46] Wei, Y.; Tang, X.; Sun, Y.; Focke, W. W., Study of mechanism of aniline polymerization. Journal of Polymer Science, Part A: Polymer Chemistry 1989, 27 (7), 2385-2396. [47] Manohar, S. K.; Macdiarmid, A. G.; Epstein, A. J. Polyaniline: Pernigranile, an isolable intermediate in teh conventional chemical synthesis of emeraldine. Synthetic Metals 41 (1-2), 1991, 711-714. [48] MacDiarmid, A. G.; Manohar, S. K.; Masters, J. G.; Sun, Y.; Weiss, H.; Epstein, A. J. Polyaniline: Synthesis and properties of pernigraniline base. Synthetic Metals 41 (1-2), 1991, 621-626. 69 [49] Huang, J.; Kaner, R. B. A General Chemical Route to Polyaniline Nanofibers. J. Am. Chem. Soc. 126 (3), 2004, 851-855. [50] King, R. C. Y.; Roussel, F. Morphological and electrical characteristics of polyaniline nanofibers. Synthetic Metals 153, 2005, 337-340. [51] Jozefowicz, M. E.; Epstein, A. J.; Pouget, J. P.; Masters, J. G.; Ray, A.; Sun, Y.; Tang, X.; MacDiarmid, A. G. X-ray structure of polyanilines. Synthetic Metals 41 (1-2), 1991, 723-726. [52] Pouget, J. P.; Jozefowicz, M. E.; Epstein, A. J.; Tang, X.; MacDiarmid, A. G., X-ray structure of polyaniline. Macromolecules 24 (3), 1991, 779-789. [53] Pouget, J. P.; Laridjani, M.; Jozefowicz, M. E.; Epstein, A. J.; Scherr, E. M.; MacDiarmid, A. G. J. Structural aspects of the polyaniline family of electronic polymers. Synthetic Metals 51 (1-3), 1992, 95-101. [54] Plesu, N.; Kellenberger, A.; Mihali, M.; Vaszilcsin, N. Effect of temperature on the electrochemical synthesis and properties of polyaniline films. Journal of Non-Crystalline Solids 356 (20-22), 2010, 1081-1088. [55] Stejskal, J.; Riede, A.; Hlavata, D.; Prokes, J.; Helmstedt, M.; Holler, P. The effect of polymerization temperature on molecular weight, crystallinity, and electrical conductivity of polyaniline. Synthetic Metals 96(1), 1998, 55-61. [56] Łuzny W k , E. Relations between the structure and electric conductivity of polyaniline protonated with camphorsulfonic acid. Macromolecules 33(2), 2000, 425-429. [57] Raghunathan, A.; Rangarajan, G.; Trivedi, D. C. 13C CPMAS NMR, XRD, d.c. and a.c. electrical conductivity of aromatic acids doped polyaniline. Synthetic Metals 81 (1), 1996, 39-47. [58] Zhang, Z.; Wei, Z.; Wan, M. Nanostructures of polyaniline doped with inorganic acids. Macromolecules 35 (15), 2002, 5937-5942. [59] Ghosh, P.; Sarkar, A.; Ghosh, M.; Meikap, A. K.; Chattopadhyay, S. K.; Chatterjee, S. K.; Chowdhury, P.; Saha, B. A study on Hall voltage and electrical resistivity of doped conducting polyaniline. Czechoslovak Journal of Physics 53 (12), 2003, 1219-1227. [60] Yoo, J. E.; Krekelberg, W. P.; Sun, Y.; Tarver, J. D.; Truskett, T. M.; Loo, Y. L., Polymer conductivity through particle connectivity. Chemistry of Materials 21 (9), 2009, 1948-1954. [61] Zuo, F.; Angelopoulos, M.; MacDiarmid, A. G.; Epstein, A. J., ac conductivity of emeraldine polymer. Physical Review B 39 (6), 1989, 3570-3578. [62] Joo, J.; Chung, Y. C.; Song, H. G.; Baeck, J. S.; Lee, W. P.; Epstein, A. J.; MacDiarmid, A. G.; Jeong, S. K.; Oh, E. J. Charge transport studies of doped polyanilines with various dopants and their mixtures. Synthetic Metals 84 (1-3), 1997, 739-740. [63] Fischer, J. E.; Tang, X.; Scherr, E. M.; Cajipe, V. B.; MacDiarmid, A. G. J. Polyaniline fibers and films: Stretch-induced orientation and crystallization, morphology, and the nature of the amorphous phase. Synthetic Metals 41 (1-2), 1991, 661-664. [64] Annis, B. K.; Lin, J. S.; Scherr, E. M.; MacDiarmid, A. G. J. Evidence for the development of a one-dimensional array of crystallites in stretched polyaniline and the effect of Cl- doping. Macromolecules 25 (1), 1992, 429-433. 70 [65] Wang, Y. Z.; Joo, J.; Hsu, C. H.; Epstein, A. J., Charge transport of camphor sulfonic acid-doped polyaniline and poly(o-toluidine) fibers: role of processing. Synthetic Metals 68 (3), 1995, 207-211. [66] Macdiarmid, A. G.; Min, Y.; Wiesinger, J. M.; Oh, E. J.; Scherr, E. M.; Epstein, A. J. Towards optimization of electrical and mechanical properties of polyaniline: Is crosslinking between chains the key?. Synthetic Metals 55 (2 -3 pt 2), 1993, 753-760. [67] Pomfret, S. J.; Adams, P. N.; Comfort, N. P.; Monkman, A. P., "Electrical and mechanical properties of polyaniline fibres produced by a one-step wet spinning process". Polymer 2000, 41 (6), 2265-2269. [68] Tzou, K. T.; Gregory, R. V. Improved solution stability and spinnability of concentrated poly niline solutions using N N′-dimethyl propylene urea as the spin bath solvent. Synthetic Metals 69 (1-3), 1995, 109-112. [69] Liu, Z.; Luo, Y.; Zhang, K. P(AAm-co-MAA) semi-IPN hybrid hydrogels in the presence of PANI and MWNTs-COOH: Improved swelling behavior and mechanical properties. Journal of Biomaterials Science, Polymer Edition 19 (11), 2008, 1503-1520. [70] Lee, W. P.; Brenneman, K. R.; Hsu, C. H.; Shih, H.; Epstein, A. J. J. Charge transport properties of high-strength, high-modulus sulfonated polyaniline/poly(p-phenylene terephthalamide) fibers. Macromolecules 34 (8), 2001, 2648-2652. [71] Bhadra, S.; Singha, N. K.; Khastgir, D. Mechanical, dynamic mechanical, morphological, thermal behavior and processability of polyaniline and ethylene 1-octene based semi-conducting composites. Journal of Applied Polymer Science 107 (4), 2008, 2486-2493. [72] Focke, W. W.; Wnek, G. E.; Wei, Y. Influence of oxidation state, pH, and counterion on the conductivity of polyaniline. Journal of Physical Chemistry 91 (22), 1987, 58135818. [73] Shaolin, M.; Jincui, L. Electrochemical activity and electrochromism of polyaniline in the buffer solutions. Chemical Journal on Internet 1 (1), 1999, 1-9. [74] Stilwell, D. E.; Park, S.-M. Electrochemistry of conductive polymers. II. Electrochemical studies on growth properties of polyaniline. Journal of the Electrochemical Society 135 (9), 1988, 2254-2262. [75] Shim, Y. B.; Park, S. M. Electrochemistry of conductive polymers VII. Autocatalytic rate constant for polyaniline growth. Synthetic Metals 29 (1), 1989, 169-174. [76] MacDiarmid, A. G.; Yang, L. S.; Huang, W. S.; Humphrey, B. D. Polyaniline: Electrochemistry and application to rechargeable batteries. Synthetic Metals 18 (1-3), 1987, 393-398. [77] Laborde, H.; Léger, J. M.; Lamy, C. Electrocatalytic oxidation of methanol and C1 molecules on highly dispersed electrodes Part 1: Platinum in polyaniline. Journal of Applied Electrochemistry 24 (3), 1994, 219-226. [78] Fusalb ou rec, P.; Villers, D.; Belanger, D. Electrochemical characterization of polyaniline in nonaqueous electrolyte and its evaluation as electrode material for electrochemical supercapacitors. Journal of the Electrochemical Society 148 (1), 2001, A1A6. [79] Herod, T. E.; Schlenoff, J. B. Doping-Induced Strain Stretchoelectrochemistry. Chem. Mater. 5, 1993, 951-955. in Polyaniline: 71 [80] Lu, Y.; Li, J.; Wu, W. Morphological investigation of polyaniline. Synthetic Metals 30 (1), 1989, 87-95. [81] Huang, J.; Kaner, R. B. Nanofiber formation in the chemical polymerization of aniline: A mechanistic study. Angewandte Chemie - International Edition 43 (43), 2004, 5817-5821. [82] Rahman, F.; Khokhar, A. Z. Thermochromic effect in synthetic opal/polyaniline composite structures. Applied Physics A: Materials Science and Processing 2009, 94 (2), 405-410. [83] Choi, J.; Kim, S. J.; Lee, J.; Lim, J. H.; Lee, S. C.; Kim, K. J. Controlled selfassembly of nanoporous alumina for the self-templating synthesis of polyaniline nanowires. Electrochemistry Communications, 9 (5), 2007, 971-975. [84] Chiou, N. R.; Lee, L. J.; Epstein, A. J. J. Porous membrane controlled polymerization of nanofibers of polyaniline and its derivatives. Journal of Materials 18 (18), Chemistry 2008, 2085-2089. [85] Stejskal, J.; Omastova, M.; Fedorova, S.; Prokes, J.; Trchova, M. Polyaniline and polypyrrole prepared in the presence of surfactants: A comparative conductivity study. Polymer 44 (5), 2003, 1353-1358. [86] Xavier, M. G.; Venancio, E. C.; Pereira, E. C.; Leite, F. L.; Leite, E. R.; MacDiarmid, A. G.; Mattoso, L. H. C., Synthesis of nanoparticles and nanofibers of polyaniline by potentiodynamic electrochemical polymerization. Journal of Nanoscience and Nanotechnology 9 (3), 2009, 2169-2172. [87] Huang, J.; Moore, J. A.; Acquaye, J. H.; Kaner, R. B. Mechanochemical route to the conducting polymer polyaniline. Macromolecules 38 (2), 2005, 317-321. [88] Jing, X.; Wang, Y.; Wu, D.; She, L.; Guo, Y. Polyaniline nanofibers prepared with ultrasonic irradiation. Journal of Polymer Science, Part A: Polymer Chemistry 44 (2), 2006, 1014-1019. [89] Huang, J.; Virji, S.; Weiller, B. H.; Kaner, R. B., Polyaniline nanofibers: Facile synthesis and chemical sensors. Journal of the American Chemical Society 125 (2), 2003, 314-315. [90] Virji, S.; Kaner, R. B.; Weiller, B. H. Hydrogen sensors based on conductivity changes in polyaniline nanofibers. Journal of Physical Chemistry B 110 (44), 2006, 2226622270. [91] Subramania, A.; Devi, S. L. Polyaniline nanofibers by surfactant-assisted dilute polymerization for supercapacitor applications. Polymers for Advanced Technologies 19 (7), 2008, 725-727. [92] Hosoda, M.; Hino, T.; Kuramoto, N. Facile preparation of conductive paint made with polyaniline/dodecylbenzenesulfonic acid dispersion and poly(methyl methacrylate). Polymer International 56 (11), 2007, 1448-1455. [93] Shim, G. H.; Han, M. G.; Sharp-Norton, J. C.; Creager, S. E.; Foulger, S. H. Inkjetprinted electrochromic devices utilizing polyaniline-silica and poly(3,4ethylenedioxythiophene)-silica colloidal composite particles. Journal of Materials Chemistry 18 (5), 2008, 594-601. [94] Oh, K. W.; Hong, K. H.; Kim, S. H. Electrically conductive textiles by in situ polymerization of aniline. Journal of Applied Polymer Science 74 (8), 1999, 2094-2101. 72 [95] Hino, T.; Taniguchi, S.; Kuramoto, N., Syntheses of conductive adhesives based on epoxy resin and polyanilines by using N-tert-butyl-5-methylisoxazolium perchlorate as a thermally latent curing reagent. Journal of Polymer Science, Part A: Polymer Chemistry 44 (2), 2006, 718-726. [96] Matveeva, E. S.; Diaz Calleja, R.; Parkhutik, V. Equivalent circuit analysis of the electrical properties of conducting polymers: Electrical relaxation mechanisms in polyaniline under dry and wet conditions. Journal of Non-Crystalline Solids 1998, 235237, 772-780. [97] Shim, G. H.; Han, M. G.; Sharp-Norton, J. C.; Creager, S. E.; Foulger, S. H., Inkjetprinted electrochromic devices utilizing polyaniline-silica and poly(3,4ethylenedioxythiophene)-silica colloidal composite particles. Journal of Materials Chemistry 18 (5), 2008, 594-601. [98] Pinto, N. J.; Johnson Jr, A. T.; MacDiarmid, A. G.; Mueller, C. H.; Theofylaktos, N.; Robinson, D. C.; Miranda, F. A. Electrospun polyaniline/polyethylene oxide nanofiber field-effect transistor. Applied Physics Letters 83 (20), 2003, 4244-4246. [99] Bhadra, J.; Sarkar, D. J., Field effect transistor from dispersion polymerized aniline. Indian Journal of Physics 82 (6), 2008, 795-799. [100] Yang, Y.; Heeger, A. J. Polyaniline as a transparent electrode for polymer lightemitting diodes: Lower operating voltage and higher efficiency. Applied Physics Letters 64 (10), 1994, 1245-1247. [101] Yang, Y.; Westerweele, E.; Zhang, C.; Smith, P.; Heeger, A. J. Enhanced performance of polymer light-emitting diodes using high-surface area polyaniline network electrodes. Journal of Applied Physics 77 (2), 1995, 694-698. [102] Iijima, S. Helical microtubules of graphitic carbon. Nature 354 (6348), 1991, 56-58. [103] Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. C60: Buckminsterfullerene. Nature 318 (6042), 1985, 162-163. [104] Oberlin, A.; Endo, M.; Koyama, T. Filamentous growth of carbon through benzene decomposition. J. Cryst Growth, 1976, 32, 335–349. [105] Terrones, M. Carbon nanotubes: synthesis and properties, electronic devices and other emerging applications. International Material Reviews, 6, 2004, 325-377. [106] Collins, P. G.; Avouris, P. Nanotubes for electronics. Scientific American 283 (6), 2000, 62-69. [107] Bockrath, M.; Cobden, D. H.; Lu, J.; Rinzler, A. G.; Smalley, R. E.; Balents, L.; McEuen, P. L. Luttinger-liquid behaviour in carbon nanotubes. Nature 397 (6720), 1999, 598-607. [108] Coleman, J. N.; Khan, U.; Blau, W. J.; Gun'ko, Y. K. Small but strong: A review of the mechanical properties of carbon nanotube-polymer composites. Carbon 44 (9), 2006, 1624-1652. [109] Gao, B.; Kleinhammes, A.; Tang, X. P.; Bower, C.; Fleming, L.; Wu, Y.; Zhou, O. Electrochemical intercalation of single-walled carbon nanotubes with lithium. Chemical Physics Letters 1999, 307 (3-4), 153-157. [110] Che, G.; Lakshmi, B. B.; Fisher, E. R.; Martin, C. R., Carbon nanotubule membranes for electrochemical energy storage and production. Nature 393 (6683), 1998, 346-349. 73 [111] Frackowiak, E.; Jurewicz, K.; Delpeux, S.; Béguin, F. Nanotubular materials for supercapacitors. Journal of Power Sources 97-98, 2001, 822-825. [112] Niu, C.; Sichel, E. K.; Hoch, R.; Moy, D.; Tennent, H. High power electrochemical capacitors based on carbon nanotube electrodes. Applied Physics Letters 70 (11), 1997, 1480-1482. [113] Thomassin, J. M.; Kollar, J.; Caldarella, G.; Germain, A.; Jérôme, R.; Detrembleur, C. Beneficial effect of carbon nanotubes on the performances of Nafion membranes in fuel cell applications. Journal of Membrane Science 303 (1-2), 2007, 252-257. [114] Rinzler, A. G.; Hafner, J. H.; Nikolaev, P.; Lou, L.; Kim, S. G.; Tománek, D.; Nordlander, P.; Colbert, D. T.; Smalley, R. E. Unraveling nanotubes: Field emission from an atomic wire. Science 269 (5230), 1995, 1550-1553. [115] Hafner, J. H.; Cheung, C. L.; Lieber, C. M. Growth of nanotubes for probe microscopy tips. Nature 398 (6730), 1999, 761-762. [116] Tans, S. J.; Verschueren, A. R. M.; Dekker, C. Room-temperature transistor based on a single carbon nanotube. Nature 393, 1998, 49-52. [117] Berber S.; Kwon Y. K.; Tománek, D. Unusually High Thermal Conductivity of Carbon Nanotubes. Physical Review Letters 84 (20), 2000, 4613-4616. [118] Kim, P.; Shi, L.; Majumdar, A.; McEuen, P. L. Thermal transport measurements of individual multiwalled nanotubes. Physical Review Letters 87 (21), 2001, 21550212155024. [119] Pop, E.; Mann, D.; Wang, Q.; Goodson, K.; Dai, H. Thermal conductance of an individual singlewall carbon nanotube above room temperature. Nano Letters 6 (1), 2006, 96-100. [120] Treacy, M. M. J.; Ebbesen, T. W.; Gibson, J. M. Exceptionally high Young's modulus observed for individual carbon nanotubes. Nature 381 (6584), 1996, 678-680. [121] Wong, E. W.; Sheehan, P. E.; Lieber, C. M. Nanobeam mechanics: Elasticity, strength, and toughness of nanorods and nanotubes. Science 277 (5334), 1997, 1971-1975. [122] Hernández, E.; Goze, C.; Bernier, P.; Rubio, A. Elastic properties of C and BxCyNz composite nanotubes. Physical Review Letters 80 (20), 1998, 4502-4505. [123] Yu, M. F.; Files, B. S.; Arepalli, S.; Ruoff, R. S. Tensile loading of ropes of single wall carbon nanotubes and their mechanical properties. Physical Review Letters 84 (24), 2000, 5552-5555. [124] Yu, M. F.; Lourie, O.; Dyer, M. J.; Moloni, K.; Kelly, T. F.; Ruoff, R. S. Strength and breaking mechanism of multiwalled carbon nanotubes under tensile load. Science 287 (5453), 2000, 637-640. [125] Endo, M.; Muramatsu, H.; Hayashi, T.; Kim, Y. A.; Terrones, M.; Dresselhaus, M. S. Nanotechnology: 'Buckypaper' from coaxial nanotubes. Nature 433 (7025), 2005, 476. [126] Wang, D.; Song, P.; Liu, C.; Wu, W.; Fan, S. Highly oriented carbon nanotube papers made of aligned carbon nanotubes. Nanotechnology 19, 2008, 1-6. [127] Kong, J.; Franklin, N. R.; Zhou, C.; Chapline, M. G.; Peng, S.; Cho, K.; Dai, H. Nanotube molecular wires as chemical sensors. Science 287 (5453), 2000, 622-625. [128] Zhao, Q.; Gan, Z.; Zhuang, Q. Electrochemical sensors based on carbon nanotubes. Electroanalysis 14 (23), 2002, 1609-1613. 74 [129] Wu, Y. P.; Rahm, E.; Holze, R. Carbon anode materials for lithium ion batteries. Journal of Power Sources 114 (2), 2003, 228-236. [130] Liu, C.; Fan, Y. Y.; Liu, M.; Cong, H. T.; Cheng, H. M.; Dresselhaus, M. S. Hydrogen storage in single-walled carbon nanotubes at room temperature. Science 286 (5442), 1999, 1127-1129. [131] Journet, C.; Maser, W. K.; Bernier, P.; Loiseau, A.; Lamy de la Chapelle, M.; Lefrant, S.; Deniard, P.; Lee, R.; Fischer, J. E. Large-scale production of single-walled carbon nanotubes by the electric-arc technique. Nature 388 (6644), 1997, 756-758. [132] Benito, A. M.; Maser, W. K.; Martínez, M. T. Carbon nanotubes: From production to functional composites. International Journal of Nanotechnology 2 (1-2), 2005, 71-89. [133] Thess, A.; Lee, R.; Nikolaev, P.; Dai, H.; Petit, P.; Robert, J.; Xu, C.; Lee, Y. H.; Kim, S. G.; Rinzler, A. G.; Colbert, D. T.; Scuseria, G. E.; Tománek, D.; Fischer, J. E.; Smalley, R. E. Crystalline ropes of metallic carbon nanotubes. Science 273 (5274), 1996, 483-487. [134] Maser, W. K.; Benito, A. M.; Martínez, M. T. Production of carbon nanotubes: The light approach. Carbon 40 (10), 2002, 1685-1695. [135] Maser, W. K.; Muñoz, E.; Benito, A. M.; Martínez, M. T.; De La Fuente, G. F.; Maniette, Y.; Anglaret, E.; Sauvajol, J. L. Production of high-density single-walled nanotube material by a simple laserablation method. Chemical Physics Letters 292 (4-6), 1998, 587-593. [136] Journet, C.; Bernier, P. Production of carbon nanotubes. Applied Physics A: Materials Science and Processing 67 (1), 1998, 1-9. [137] Dai, H. Carbon nanotubes: Opportunities and challenges. Surface Science 500 (1-3), 2002, 218-241. [138] Huang, S.; Woodson, M.; Smalley, R.; Liu, J. Growth mechanism of oriented long single walled carbon nanotubes using "fast-heating" chemical vapor deposition process. Nano Letters 4 (6), 2004, 1025-1028. [139] Du, G.; Feng, S.; Zhao, J.; Song, C.; Bai, S.; Zhu, Z. Particle-wire-tube mechanism for carbon nanotube evolution. Journal of the American Chemical Society 128 (48), 2006, 15405-15414. [140] Little, R. B. Mechanistic Aspects of Carbon Nanotube Nucleation and Growth. Journal of Cluster Science 14 (2), 2003, 135-185. [141] Tibbetts, G. G. Vapor-grown carbon fibers: Status and prospects. Carbon 27 (5), 1989, 745-747. [142] Botello-Méndez, A.; Campos-Delgado, J.; Morelos-Gómez, A.; Romo-Herrera, J. M.; Rodríguez, A. G.; Navarro, H.; Vidal, M. A.; Terrones, H.; Terrones, M. Controlling the dimensions, reactivity and cristallinity of multiwalled carbon nanotubes using low ethanol concentrations. Chem. Phys. Lett. 453, 2008, 55-61. [143] Terrones, M.; Souza Filho, A. G; Rao, A. M. Doped Carbon Nanotubes: Synthesis, Characterization and Applications Vol. 111, 531–566 (Springer Topics in Appl. Phys., Springer, 2008). [144] Endo, M.; Koyama, T. 1983, Japan Patent. 75 [145] Endo, M.; Kim, y. A.; Fukai, Y.; Hayashi, T.; Terrones, M.; Terrones, H.; Dresselhaus, M. S. Comparison study of semi-crystalline and highly crystalline multiwalled carbon nanotubes. Applied Physics Letters 79, 2001, 1531-1533. [146] Lee, J.-O.; Park, C.; Kim, J.-J.; Kim, J.; Park, J. W.; Yoo, K.-H. Formation of low resistance ohmic contacts between carbon nanotube and metal electrodes by a rapid thermal annealing method. Journal of Physics D: Applied Physics 33, 2000, 1953-1956. [147] See, C. H.; Harris, A. T. A Review of Carbon Nanotube Synthesis via Fluidized-Bed Chemical Vapor Deposition. Industrial & Engineering Chemistry Research 46, 2007, 9971012. [148] Wei, F.; Zhang, Q.; Qian, W.-Z.; Yu, H.; Wang, Y.; Luo, G.-H.; Xu, G.-H.; Wang, D.-Z. The mass production of carbon nanotubes using a nano-agglomerate fluidized bed reactor: A multiscale space-time analysis. Powder Technology 183, 2008, 10-20. [149] Serp, P.; Corrias, M.; Kalck, P. Carbon nanotubes and nanofibers in catalysis. Applied Catalysis A: General 253, 2003, 337-358. [150] Perez-Cabero, M.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. Characterization of carbon nanotubes and carbon nanofibers prepared by catalytic decomposition of acetylene in a fluidized bed reactor. Journal of Catalysis 215, 2003, 305-316. [151] Duesberg, G. S.; Graham, A.; Kreupl, A.; Liebau, M.; Seidel, M.; Unger, E. Ways towards the scalable integration of carbon nanotubes into silicon based technology. Diam Rel Mat 13, 2006, 354–61. [152] Dresselhaus, M.; Dresselhaus, G.; Avouris, P. Carbon nanotubes: synthesis, structure, properties, and applications. Germany: Springer-Verlag; 2001. [153] Tsang, J.; Freitag, M.; Perebeinos, V.; Liu, J.; Avouris, P. Doping and phonon renormalization in carbon nanotubes. Nature Nanotech 2, 2007, 725–730. [154] Dresselhaus, M. S.; Dresselhaus, G. Intercalation compounds of graphite. Adv in Phys 30, 1981, 139–326. [155] Choi, H.; Ihm, J.; Louie, S.; Cohen, M. Defects, quasibound states, and quantum conductance in metallic carbon nanotubes. Phys. Rev. Lett. 84, 2000, 2917–2920. [156] Liu, X.; Pichler, T.; Knupfer, M.; Fink, J.; Kataura, H. Electronic properties of FeCl3-intercalated single-wall carbon nanotubes. Phys Rev B 70, 2004, 205405. [157] Ma, J.; Guan, S.; Lai, C. H. Disorder effect on electronic and optical properties of doped carbon nanotubes. Phys Rev B 74, 2006, 205401. [158] Chun, K. Y; Lee, C. J. Potassium doping in the double-walled carbon nanotubes at room temperature. J Phys Chem C 112, 2008, 4492–4497. [159] Smith, B.; Monthioux, M.; Luzzi, D. Encapsulated C60 in carbon nanotubes. Nature 396, 1998, 323–324. [160] Shinohara, H. Endohedral metalofullerenes. Rep Prog Phys 64, 2000, 843–892. [161] Prasad, B. L. V.; Sato, H.; Enoki, T.; Hishiyama, Y.; Kaburagi, Y.; Rao A. M. Intercalated nanographite: structure and electronic properties. Phys Rev B 64, 2001, 235407. [162] Liu, X.; Pichler, T.; Knupfer, M.; Fink, J. Electronic and optical properties of alkalimetal-intercalated single-wall carbon nanotubes. Phys Rev B 67, 2003, 125403. 76 [163] Stephan, O.; Ajayan, P. Doping graphitic and carbon nanotube structures with boron and nitrogen. Science 266, 1994, 1863–1865. [164] Mandumpal, J.; Gemming, S.; Seifert, G. Curvature effects of nitrogen on graphitic sheets: Structures and energetics. Chem Phys Lett 447, 2007, 115–120. [165] Yi, J. Y.; Bernholc, J. Atomic structure and doping of microtubules. Phys Rev B 47, 1993, 1708–1711. [166] Iijima, S. Helical microtubules of graphitic carbon. Nature 354, 1991, 56–58. [167] Iijima, S.; Ichihashi, T. Single-shell carbon nanotubes of 1-nm diameter. Nature 363, 1993, 603–605. [168] Czerw, R.; Terrones, M.; Charlier, J.; Blasé, X.; Foley, B, Kamalakaran, R. Identification of electron donor states in N-doped carbon nanotubes. Nano Lett 1, 2001, 457–460. [169] Kim, S. Y.; Lee J.; Na, C. W.; Park, J.; Seo, K.; Kim, B. N-doped doublewalled carbon nanotubes synthesized by chemical vapor deposition. Chem Phys Lett 413, 2005, 300–305. [170] Ayala P.; Grüneis A.; Gemming T.; Grimm D.; Kramberger C.; Rümmeli M. H. Tailoring N-doped single and double wall carbon nanotubes from a non-diluted carbon/nitrogen feedstock. Jour Phys Chem C 101, 2007, 2879–84. [171] Ayala, P.; Arenal, R.; Rümmeli, M.; Rubio, A.; Pichler, T. The doping of carbon nanotubes with nitrogen and their potential applications. Carbon 48, 2010, 575-586. [172] Ayala P, Gru¨ neis A, Gemming T, Büchner B, Rümmeli MH, Grimm D, et al. Influence of the catalyst hydrogen pre-treatment on the growth of vertically aligned nitrogen-doped carbon nanotubes. Chem Mat 19, 2007, 6131–6137. [173] Koos, A. A.; Dowling, M.; Jurkschat, K.; Crossley, A.; Grobert, N. Effect of the experimental parameters on the structure of nitrogen-doped carbon nanotubes produced by aerosol chemical vapour deposition. Carbon 47, 2009, 30–37. [174] Sen, R.; Satishkumar, B.; Govindaraj, A.; Harikumar, K. R.; Renganathan, M.; Rao, C. Nitrogen-cointainig carbon nanotubes. J Mater Chem 7, 1997, 2335–2337. [175] Glerup, M.; Castignolles, M.; Holzinger, M.; Hug, G.; Loiseau, A.; Bernier, P. Synthesis of highly nitrogen-doped multi-walled carbon nanotubes. Chem Comm 20, 2003, 2542–2543. [176] Sun, C.; Wang, H.; Hayashi, M.; Chen, L.; Chen, K. Atomic-scale deformation in Ndoped carbon nanotubes. J Amer Chem Soc 128, 2006, 8368–8369. [177] Droppa Jr, R.; Hammer, P.; Carvalho, A.; Alvarez, F. Incorporation of nitrogen in carbon nanotubes. J Non-Crys Sol 874, 2002, 299–302. [178] Cui, S.; Scharff, P.; Siegmund, C.; Schneider, D.; Risch, K.; Klötzer, S. Investigation on prepartion of multiwalled carbon nanotubes by dc arc discharge under N2 atmosphere. Carbon 42, 2004, 931–939. [179] Zamudio, A.; Elias, A. L.; Rodriguez-Manzo, J.; Lopez-Urias, F.; RodriguezGattorno G.; Lupo, F. Efficient anchoring of silver nanoparticles on N-doped carbon nanotubes. Small 2, 2006, 346–350. 77 [180] Ayala, P.; Freire Jr, F. L.; Gu, L.; Smith, D.J.; Solorzano, I. G. Decorating carbon nanotubes with nanostructured nickel particles via chemical methods. Chem. Phys. Lett. 431, 2006, 104–109. [181] Carrero-Sanchez, J. C.; Elias A. L.; Mancilla, R.; Arrellin, G.; Terrones, H.; Laclette, J. P. Biocompatibility and toxicological studies of carbon nanotubes doped with nitrogen. Nano Lett. 6, 2006, 1609–1616. [182] Elias, A. L.; Carrero-Sanchez, J. C.; Terrones, H.; Endo, M.; Laclette, J. P.; Terrones, M. Viability studies of pure carbon- and nitrogen-doped nanotubes with entamoeba histolytica: From amoebicidal to biocompatible structures. Small 3, 2007, 1723–1729. [183] Fragneaud, B.; Masenelli-Varlot, K.; Gonzalez-Montiel, A.; Terrones, M.; Cavaille, J. Efficient coating of N-doped carbon nanotubes with polystyrene using atomic transfer radical polymerization. Chem Phys Lett 419, 2006, 567–573. [184] Villalpando-Paez, F.; Romero, A.; Muñoz-Sandoval, E.; Martinez, L.; Terrones, H.; Terrones, M. Fabrication of vapor and gas sensors using films of aligned CNx nanotubes. Chem Phys Lett 386, 2004, 137–143. [185] Botello-Méndez, A.; Campos-Delgado, J.; Morelos-Gómez, A.; Romo-Herrera, J. M.; Rodríguez, A. G.; Navarro, H.; Vidal, M. A.; Terrones, H.; Terrones, M. Controlling the dimensions, reactivity and cristallinity of multiwalled carbon nanotubes using low etanol concentrations. Chem. Phys. Let. 453, 2008, 55-61. [186] Castle, A. B.; Gracia-Espino, E.; Nieto-Delgado, C.; Terrones, H.; Terrones, M.; Hussain, S. Hydroxil-Functionalized and N-Doped Multiwalled Carbon Nanotubes Decorated with Silver Nanoparticles Preserve Cellular Function. ACS Nano, 4, 2011, 2458-2466. [187] Cano-Marquez, A.G.; Rodriguez-Macias, F.J.; Campos-Delgado, J.; EspinosaGonzalez, C.G.; Tristan-López, F.; Ramirez-Gonzalez, D.; Cullen, D.A.; Smith, D.J.; Terrones, M.; Vega-Cantu, Y.I. ExMWNTs: Graphene sheets and ribbons produced by lithium intercalation and exfoliation of carbon nanotubes. Nano Lett. 9, 2009, 1527-1533. [188] Kosynkin, D. V.; Higginbotham, A. L.; Sinitskii, A.; Lomeda, J. R.; Dimiev, A.; Price, B. K.; Tour, J. M. Longitudinal unzipping of carbon nanotubes to form graphene nanoribbons. Nature 458, 2009, 872-876. [189] Jiao, L.; Zhang, L.; Wang, X.; Diankov, G.; Dai. H. Narrow graphene nanoribbons from carbon nanotubes. Nature 458, 2009, 877-880. [190] Terrones, M. Materials Science: Nanotubes unzipped. Nature 458 (2009) 845-846. [191] Elias, A.L.; Botello-Mendez, A.R.; Meneses-Rodriguez, D.; Gonzalez, V.J.; Ramirez-Gonzalez, D.; Ci, L.; Muñoz-Sandoval, E.; Ajayan, P.M.; Terrones, H.; Terrones, M. Nano Lett. 9, 2009, 31371. [192] Kim, K.; Sussman, A.; Zettl, A. ACS Nano 4 (2010) 13626. [193] Liang, F.; Sadana, A. K.; Peera, A.; Chattopadhyay, J.; Gu, Z.; Hauge, R. H.; Billups, W. E. A Convenient Route to Functionalized Carbon Nanotubes. Nano Letters, 7, 2004, 1257-1260. [194] Liang, F.; Alemany, L. B.; Beach, J. M.; Billups, W. E. Structure Analyses of Dodecylated Single-Walled Carbon Nanotubes. J. Am. Chem. Soc. 127, 2005, 1394113948. 78 [195] Chattopadhyay, J.; Sadana, A. K.; Liang, F. Beach, J. M.; Xiao, Y.; Hauge, R. H.; Billups, W. E. Carbon Nanotube Saltas. Arylation of Single-Wall Carbon Nanotubes. Organic Letters, 19, 2005, 4067-4069. [196] Liang, F.; Beach, J. M.; Kobashi, K.; Sadana, A. K.; Vega-Cantu, Y. I.; Tour, J. M.; Billups, W. E. In Situ Polimerization Initiated by Single-Walled Carbon Nanotube Salts. Chem. Mater. 18, 2006, 4764-4767. [197] Rodríguez, N. M.; Chambers, A.; Baker, R. T. K. Catalytic Engineering of Carbon Nanostructures. Langmuir, 11, 1995, 3862-3866. [198] Zheng, G.-B.; Kouda, K.; Sano, H.; Uchiyama, Y.; Shi, Y.-F. Quan, H.-J. A model for the structure and growth of carbon nanofibers synthesized by the CVD method using nickel as a catalyst. Carbon 42, 2004, 635-640. [199] Helveg, S.; López-Cartes, C.; Sehested, J.; Hansen, P. L.; Clausen, B. S.; RostrupNielsen, J. R.; Abild-Pedersen, F.; Norskov, J. K. Atomic-scale imaging of carbon nanofibre growth. Nature 427, 2004, 426-429. [200] Tanaka, A.; Yoon, S.-H.; Mochida, I. Preparation of highly crystalline nanofibers on Fe and Fe-Ni catalysts with a variety of graphene plane alignments. Carbon 42, 2004, 591597. [201] Yoon, S.-H.; Lim, S.; Hong, S.-h.; Mochida, I.; An, B.; Yokogawa, K. Carbon nanorod as a structural unit of carbon nanofibers. Carbon 42, 2004, 3087-3095. [202] Endo, M.; Oshida, K.; Terrones, M.; Yanagisawa, T.; Higaki, S.; Dresselhaus, M. S. Structural characterization of cup-stacked-type nanofibers with an entirely hollow core. Applied Physics Letters 80, 2002, 1267-1269. [203] Vera-Agulló, J.; Varela-Rizo, H.; Conesa, J. A.; Almansa, C.; Merino, C.; MartínGullón, I. Evidence of growth mechanism and helix-spiral cone structure of stacked-cup carbon nanofibers. Carbon 45, 2007, 2751-2758. [204] Eksioglu, B.; Nadarajah, A. Structural analysis of conical carbon nanofibers. Carbon 44, 2006, 360-373. [205] Weisenberger, M.; Martín-Gullón, I.; Vera-Agulló, J.; Varela-Rizo, H.; Merino, C. Andrews, R.; Qian, D.; Rantell, T. The effect of graphitization temperature on the structure of helical-ribbon carbon nanofibers. Carbon 47, 2009, 2211-2218. [206] Endo, M.; Kim, Y. A.; Hayashi, T.; Yanagisawa, T.; Muramatsu, H.; Ezaka, M.; Terrones, H.; Terrones, M.; Dresselhaus, M. S. Microstructural changes induced in “stacked-cup” carbon nanofibers by heat treatment. Carbon 41, 2003, 1941-1947. [207] Martín-Gullón, I.; Vera, J.; Conesa, J. A.; González, J. L.; Merino, C. Differences between carbon nanofibers produced using Fe and Ni catalysts in a floating catalyst reactor. Carbon 44, 2006, 1572-1580. [208] Wallace, P. R. The band theory of graphite. Physical Review 71 (9), 1947, 622-634. [209] McClure, J. W. Diamagnetism of graphite. Physical Review 104 (3), 1956, 666-671. [210] Semenoff, G. W. Condensed-Matter simulation of a three-Dimensional anomaly. Physical Review Letters 53 (26), 1984, 2449-2452. [211] Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Electric field in atomically thin carbon films. Science 306 (5696), 2004, 666-669. 79 [212] Castro Neto, A. H.; Guinea, F.; Peres, N. M. R.; Novoselov, K. S.; Geim, A. K. The electronic properties of graphene. Reviews of Modern Physics 81 (1), 2009, 109-162. [213] Bolotin, K. I.; Ghahari, F.; Shulman, M. D.; Stormer, H. L.; Kim, P. Observation of the fractional quantum Hall effect in graphene. Nature 462 (7270), 2009, 196-199. [214] Bolotin, K. I.; Sikes, K. J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H. L. Ultrahigh electron mobility in suspended graphene. Solid State Communications 146 (9-10), 2008, 351-355. [215] Balandin, A. A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C. N. Superior thermal conductivity of single-layer graphene. Nano Letters 8 (3), 2008, 902907. [216] Lee, C.; Wei, X.; Kysar, J. W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321 (5887), 2008, 385-388. [217] Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; Banerjee, S. K.; Colombo, L.; Ruoff, R. S. Large-area synthesis of highquality and uniform graphene films on copper foils. Science 324 (5932), 2009, 1312-1314. [218] Reina, A.; Jia, X.; Ho, J.; Nezich, D.; Son, H.; Bulovic, V.; Dresselhaus, M. S.; Jing, K. Large area, few-layer graphene films on arbitrary substrates by chemical vapor deposition. Nano Letters 9(1), 2009, 30-35. [219] Gilje, S.; Han, S.; Wang, M.; Wang, K. L; Kaner, R. B. A Chemical Route to Graphene for Device Applications. Nano Letters 7 (11), 2007, 3394-3398. [220] de Heer, W. A.; Berger, C.; Wu, X.; First, P. N.; Conrad, E. H.; Li, X.; Li, T.; Sprinkle, M.; Hass, J.; Sadowski, M. L.; Potemski, M.; Martinez, G. Epitaxial graphene. Solid State Communications 143 (1-2), 2007, 92-100. [221] Hernandez, Y.; Nicolosi, V.; Lotya, M.; Blighe, F. M.; Sun, Z.; De, S.; McGovern, I. T.; Holland, B.; Byrne, M.; Gun'ko, Y. K.; Boland, J. J.; Niraj, P.; Duesberg, G.; Krishnamurthy, S.; Goodhue, R.; Hutchison, J.; Scardaci, V.; Ferrari, A. C.; Coleman, J. N. High-yield production of graphene by liquid phase exfoliation of graphite. Nature Nanotechnology 3 (9), 2008, 563-568. [222] Staudenmaier, L. Verfahren zur darstellung der graphitsaure. Ber. Dtsch. Chem. Ges 31, 1898, 1481-1487. [223] Brodie, B. C. On the Atomic Weight of Graphite. Philosophical Transactions of the Royal Society of London 149, 1859, 249-259. [224] Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited. J. Phys. Chem. B 102, 1998, 4477-4482. [225] Stankovich, S.; Dikin, D. A.; Piner, R. D.; Kohlhaas, K. A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S. T.; Ruoff, R. S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45 (7), 2007, 1558-1565. [226] Stankovich, S.; Dikin, D. A.; Dommett, G. H. B.; Kohlhaas, K. M.; Zimney, E. J.; Stach, E. A.; Piner, R. D.; Nguyen, S. T.; Ruoff, R. S. Graphene-based composite materials. Nature 442 (7100), 2006, 282-286. [227] Shin, H. J.; Kim, K. K.; Benayad, A.; Yoon, S. M.; Park, H. K.; Jung, I. S.; Jin, M. H.; Jeong, H. K.; Kim, J. M.; Choi, J. Y.; Lee, Y. H. Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance. Advanced Functional Materials 19 (12), 2009, 1987-1992. 80 [228] Jung, I.; Vaupel, M.; Pelton, M.; Pinery, R.; Dikin, D. A.; Stankovich, S.; An, J.; Ruoff, R. S. Characterization of thermally reduced graphene oxide by imaging ellipsometry. Journal of Physical Chemistry C 112 (23), 2008, 8499-8506. [229] Zhu, Y.; Stoller, M. D.; Cai, W.; Velamakanni, A.; Piner, R. D.; Chen, D.; Ruoff, R. S. Exfoliation of graphite oxide in propylene carbonate and thermal reduction of the resulting graphene oxide platelets. ACS Nano 4 (2), 2010, 1227-1233. [230] Akhavan, O. Photocatalytic reduction of graphene oxides hybridized by ZnO nanoparticles in ethanol. Carbon 49 (1), 2011, 11-18. [231] Qian, D.; Dickey, E. C.; Andrews, R.; Rantell, T. Load transfer and deformation mechanisms in carbon nanotube-polystyrene composites. Applied Physics Letters 76 (20), 2000, 2868-2870. [232] Kota, A. K.; Cipriano, B. H.; Duesterberg, M. K.; Gershon, A. L.; Powell, D.; Raghavan, S. R.; Bruck, H. A. Electrical and rheological percolation in polystyrene/MWCNT nanocomposites. Macromolecules 40 (20), 2007, 7400-7406. [233] Chen, W.; Tao, X. Self-organizing alignment of carbon nanotubes in thermoplastic polyurethane. Macromolecular Rapid Communications 26 (22), 2005, 1763-1767. [234] Shaffer, M. S. P.; Windle, A. H. Fabrication and characterization of carbon nanotube/poly(vinyl alcohol) composites. Advanced Materials 11 (11), 1999, 937-941. [235] McNally, T.; Pötschke, P.; Halley, P.; Murphy, M.; Martin, D.; Bell, S. E. J.; Brennan, G. P.; Bein, D.; Lemoine, P.; Quinn, J. P. Polyethylene multiwalled carbon nanotube composites. Polymer 46 (19 SPEC. ISS.), 2005, 8222-8232. [236] Tang, W.; Santare, M. H.; Advani, S. G. Melt processing and mechanical property characterization of multi-walled carbon nanotube/high density polyethylene (MWNT/HDPE) composite films. Carbon 41(14), 2003, 2779-2785. [237] Manchado, M. A. L.; Valentini, L.; Biagiotti, J.; Kenny, J. M. Thermal and mechanical properties of single-walled carbon nanotubes-polypropylene composites prepared by melt processing. Carbon 43(7), 2005, 1499-1505. [238] Zhang, H.; Zhang, Z. Impact behaviour of polypropylene filled with multi-walled carbon nanotubes. European Polymer Journal 43 (8), 2007, 3197-3207. [239] Jin, Z.; Pramoda, K. P.; Xu, G.; Goh, S. H. Dynamic mechanical behavior of meltprocessed multiwalled carbon nanotube/poly(methyl methacrylate) composites. Chemical Physics Letters 337 (1-3), 2001, 43-47. [240] Gorga, R. E.; Cohen, R. E. Toughness enhancements in poly(methyl methacrylate) by addition of oriented multiwall carbon nanotubes. Journal of Polymer Science, Part B: Polymer Physics 42 (14), 2004, 2690-2702. [241] Zeng, J.; Saltysiak, B.; Johnson, W. S.; Schiraldi, D. A.; Kumar, S. Processing and properties of poly(methyl methacrylate)/carbon nanofiber composites. Composites Part B: Engineering 35 (3), 2004, 245-249. [242] Haggenmueller, R.; Gommans, H. H.; Rinzler, A. G.; Fischer, J. E.; Winey, K. I. Aligned singlewall carbon nanotubes in composites by melt processing methods. Chemical Physics Letters 330 (3-4), 2000, 219-225. [243] Meincke, O.; Kaempfer, D.; Weickmann, H.; Friedrich, C.; Vathauer, M.; Warth, H. Mechanical properties and electrical conductivity of carbon-nanotube filled polyamide-6 and its blends with acrylonitrile/butadiene/styrene. Polymer 45 (3), 2004, 739-748. 81 [244] Jia, Z.; Wang, Z.; Xu, C.; Liang, J.; Wei, B.; Wu, D.; Zhu, S. Study on poly(methylmethacrylate)/carbon nanotube composites. Materials Science and Engineering A 271 (1-2), 1999, 395-400. [245] Putz, K. W.; Mitchell, C. A.; Krishnamoorti, R.; Green, P. F. Elastic modulus of single-walled carbon nanotube/poly(methyl methacrylate) nanocomposites. Journal of Polymer Science, Part B: Polymer Physics 42 (12), 2004, 2286-2293. [246] Gao, J.; Itkis, M. E.; Yu, A.; Bekyarova, E.; Zhao, B.; Haddon, R. C. Continuous spinning of a single-walled carbon nanotube-nylon composite fiber". Journal of the American Chemical Society 127(11), 2005, 3847-3854. [247] Qu, L.; Veca, L. M.; Lin, Y.; Kitaygorodskiy, A.; Chen, B.; McCall, A. M.; Connell, J. W.; Sun, Y. P. Soluble nylon-functionalized carbon nanotubes from anionic ringopening polymerization from nanotube surface. Macromolecules 38 (24), 2005, 1032810331. [248] González-Vidal, N.; de Ilarduya, A. M.; Muñoz-Guerra, S.; Castell, P.; Martínez, M. T. Synthesis and properties of poly(hexamethylene terephthalate)/multiwall carbon nanotubes nanocomposites. Composites Science and Technology 70 (5), 2010, 789-796. [249] Xia, H.; Song, M. Preparation and characterization of polyurethane-carbon nanotube composites. Soft Matter 1 (5), 2005, 386-394. [250] Yoo, H. J.; Jung, Y. C.; Sahoo, N. G.; Cho, J. W. Polyurethane-carbon nanotube nanocomposites prepared by in-situ polymerization with electroactive shape memory. Journal of Macromolecular Science, Part B: Physics 45 B (4), 2006, 441-451. [251] Fan, J.; Wan, M.; Zhu, D.; Chang, B.; Pan, Z.; Xie, S. Synthesis, characterizations, and physical properties of carbon nanotubes coated by conducting polypyrrole. Journal of Applied Polymer Science 74 (11), 1999, 2605-2610. [252] Sahoo, N. G.; Jung, Y. C.; So, H. H.; Cho, J. W. Polypyrrole coated carbon nanotubes: Synthesis, characterization, and enhanced electrical properties. Synthetic Metals 157 (8-9), 2007, 374-379. [253] Guo, H.; Zhu, H.; Lin, H.; Zhang, J.; Yu, L. Synthesis and characterization of multiwalled carbon nanotube/polythiophene composites. Journal of Dispersion Science and Technology 29 (5), 2008, 706-710. [254] Li, X.-h.; Wu, B.; Huang, J.-e.; Zhang, J.; Liu, Z.-f.; Li, H.-l. Fabrication and characterization of well-dispersed single-walled carbon nanotube/polyaniline composites. Carbon 411, 2002, 1645-1687. [255] Smith, D. W.; Huseyin-Zengin; Wensheng-Zhou; Jianyong-Jin. Carbon nanotube Doped Polyaniline. Advanced Materials, 2002, 14, No. 20, 1480-1483, Sun Y., Wilson, R., Schuster, D. I., J. Am. Chem. Soc. 2001, 123, 5348. [256] Zhang, X. Comparison of chiral polyaniline carbon nanotube nanocompuestos synthesized by aniline dimer-assisted and electrochemistry methods, Synthetic Metals 158, 2008, 336-344. [257] Carpi, F.; Rossi, D. D. Improvement of Electromechanical Actuating Performances of a Silicone Dielectric Elastomer by Dispersion of Titanium Dioxide Powder. IEEE Trans. Dielec. Elec. Insul. 11, 2004, 835-842. 82 [258] Kornbluh, R., et al., EAP Actuators, Devices and Mechanisms – Application of Dielectric Elastomers. In Electroactive polymer (EAP) actuators as artificial muscle, 2nd edition, Bar-Cohen, Y., (ed.), SPIE Press, Bellingham, WA, (2004). [259] Zhang, M.; Fang, S.; Zakhidov, A. A.; Lee, S. B.; Aliev, A. E.; Williams, C. D.; Atkinson, K. R.; Baughman, R. H. Strong, Transparent, Multifunctional, Carbon Nanotube Sheets. Science 309, 2005, 1215-1219. [260] Mirfakhrai, T.; Oh, J., Kozlov, M.; Fok, E. C. W.; Zhang, M.; Fang, S.; Baughman, R. H.; Madden, J. D. W. Electrochemical actuation of carbon nanotube yarns. Smart. Mater. Struct. 16, 2006, S243. [261]Mirfakhari, T.; Madden, J. D. W.; Baughman, R. H. Polymer artificial muscles. Materials Today 4, 2007, 30-38. [262] Warren, M. R.; Madden, J. D. Electrochemical switching of conducting polymers: A variable resistance transmission line model. J. Electroanal. Chem. 590, 2006, 76-81. [263] Madden, J. D.; Cush, R. A.; Kanigan, T. S.; Hunter, I. W. Fast contracting polypyrrole actuators. Synthetic Metals 113, 2000, 185-192. [264] Hara, S.; Zama, T.; Takashima, W.; Kaneto, K. Free-standing polypirrole actuators with response rate of 10.8%s-1. Synth. Met. 149, 2005, 199-201. [265] Ding, J.; Liu, L.; Spinks, G. M.; Zhou, D.; Wallace, G. G.; Gillespie, J. High performance conducting polymer actuator utilising a tubular geometry and helical wire interconnects. Synthetic Metals 138, 2003, 391-398. [266] Smela, E.; Inganäs, O.; Lundström, I. Controlled Folding of Micrometer-Size Structures. Science 268, 1995, 1735-1738. [267] Baughman, R. H.; Shacklette, L. W.; Elsenbaumer, R. L.; Plitcha, E. J.; Becht, C.; in Brédas, J. L.; Chance, R. R. (eds.) Conjugated Polymeric Materials: Opportunities in Electronics, Optoelectronics, and Molecular Electronics. Kluwer, Dordrecht, 1990, 559582. [268] Pei, Q.; Inganäs, O. Electrochemical muscles: Bending strips built from conjugated polymers. Synthetic Metals 57, 1993, 3718-3723. [269] Otero, T. F.; Rodríguez, J.; Angulo, E.; Santamaría, C. Artificial muscles from bilayer structures. Synthetic Metals 57, 1993, 3713-3717. [270] Smela, E.; Inganäs, O.; Pei, Q.; Lundström, I. Electrochemical muscles: Micromachining fingers and corkscrews. Adv. Mater. 5, 1993, 630-632. [271] Smela, E.; Inganäs, O.; Lundström, I. J. Conducting polymers as artificial muscles: challenges and possibilities. Micromech. Microeng. 3, 1993, 203-205. [272] Okabayashi, K.; Goto, F.; Abe, K.; Yoshida, T. Electrochemical Studies of polyaniline and its application. Synthetic Metals 18, 1987, 365-370. [273] Herod, T. E.; Schlenoff, J. B. Doping-Induced Strain in Polyaniline: Stretchoelectrochemistry. Chem. Mater. 5, 1993, 951-955. [274] Takashima, W.; Kaneko, M.; Kaneto, K.; MacDiarmid, A. G. The electrochemical actuator using electrochemically-deposited poly-aniline film. Synthetic Metals 71, 1995, 2265-2266. 83 [275] Kaneto, K.; Kaneko, M.; Min, Y.; MacDiarmid, A. G. “Artificial muscle”: Electromechanical actuators using polyaniline films. Synthetic Metals 71, 1995, 22112212. [276] Takashima, W.; Uesugi, T.; Fukui, M.; Kaneko, M.; Kaneto, K. Mechanochemoelectrical Effect of Polyaniline Film. Synthetic Metals 85, 1997, 13951396. [277] Kaneko, M.; Kaneto, K. Electrochemomechanical deformation of polyaniline films doped with self-existent and giant anions. Reactive & Functional Polym. 37, 1998, 155161. [278] Pomfret, S. J.; Adams, P. N.; Comfort, N. P.; Monkman, A. P. Electrical and mechanical properties of polyaniline fibres produced by a one-step wet spinning process. Polymer 41, 2000, 2265-2269. [279] Salvia, M.; Emirkhanian, R.; Ferreira, J.; Jaffrezic, N.; Chazeau, L.; Mazzoldi, A. Characterization of thermal relaxations of polyaniline fibers by dynamic mechanical thermal analysis. Materials Science & Engineering, C 26, 2006, 227-231. [280] Guang, L.; Jianming, J.; Wei, P.; Shenglin, Y.; Electrically conductive PANIDBSA/Co-PAN composite fibers prepared by wet spinning. Synthetic Metals 149, 2005, 181-186. [281] Roussel, F.; Chan Y. K. Morphological and electrical characteristic of polyaniline nanofibers. Synthetic Metals, 153, 2005, 337-340. [282] Mattes, R. B.; Lu, W. Factors Influencing Electrochemical Actuation of Polyaniline Fibers in Ionic Liquids. Synthetic Metals 152, 2005, 53-56. [283] Qi, B.; Lu, W.; Mattes, B. R. Strain and energy efficiency of polyaniline fiber electrochemical actuators in aqueous electrolytes. J. Phys. Chem. B 108, 2004, 6222-6227. [284] Lu, W.; Smela, E.; Adams, P. N.; Zuccarello, G.; Mattes, B. R. Development of Solid-in-Hollow Electrochemical Linear Actuators Using Highly Conductive Polyaniline. Chem. Mater. 16, 2004, 1615-1621. [285] Smela E.; Lu, W.; Mattes, B. R. Polyaniline actuators Part. 1. PANI (AMPS) in HCl. Synthetic Metals 151, 2005, 25-42. [286] Smela, E.; Mattes, R. B. Polyaniline actuators Part 2. PANI (AMPS) in methanesulfonic acid. Synthetic Metals 151, 2005, 43-48. [287] Lu, W.; Smela, E.; Mattes, B. R.; Adams, P. N.; Zuccarello, G. Electrochemical devices incorporating high-conductivity conjugated polymers. U. S. Patent 6,982,514, B1, Jan. 3, 2006. [288] Gu, B. K.; Ismail, Y. A.; Spinks, G. M.; Kim, S. I.; So, I.; Kim, S. J. A Linear Actuation of Polymeric Nanofibrous Bundle for Artificial Muscles. Chem. Mater. 21, 2009, 511-515. [289] Mottaghitalab, V.; Xi, B.; Spinks, G. M. Wallace, G. G. Polyaniline fibres containing single-walled carbon nanotubes: Enhanced performance artificial muscles. Synth. Met. 2006, 156, 796–803. [290] Spinks, G. M.; Mottaghitalab, V.; Bahrami-Samani, M.; Whitten, P. G.; Wallace, G. G. Carbon-Nanotube-Reinforced Polyaniline for High-Strength Artificial Muscles. Adv. Mater. 18, 2006, 18, 637-640. 84 [291] Pandolfo, A. G.; Hollenkamp, A. F. Carbon properties and their role in supercapacitors. J. Power Sources 157, 2006, 11-27. [292] Snook, G.A.; Chen, G.Z.; Fray, D.J.; Hughes, M.; Shaffer, M. Studies of deposition of and charge storage in polypyrrole-chloride and polypyrrole-carbon nanotube composites with an electrochemical quartz crystal microbalance. J. Electroanal. Chem. 568, 2004, 135142. [293] Arbizzani, C.; Catellani, M.; Mastragostino, M.; Mingazzini, C. N- and P-doped Polydithieno[3,4-B:3’,4’-D] thiophene: A narrow band gap polymer for redox supercapacitors. Electrochim. Acta 40, 1995, 1871-1876. [294] Lota, K.; Khomenko, V.; Frackowiak, E. Capacitance properties of poly(3,4ethylenedioxytiophene)/carbon nanotubes composites. J. Phys. Chem. Solids 65, 2004, 295-301. [295] Carpi, F.; Rossi, D. D. Improvement of electromechanical actuating performances of a silicone dielectric elastomer by dispersion of titanium dioxide powder. IEEE Trans. Dielec. Elec. Insul. 12, 2005, 835-843. [296] Snook, G. A.; Kao, P.; Best, A. S. Conducting-polymer-based supercapacitor devices and electrodes. J. of Power Sources 196, 2011, 1-12. [297] Sivakkumar, S. R.; Saraswathi, R. Performance evaluation of poly(N-methylaniline) and polyisothianaphthene in charge-storage devices. J. Power Sources 137, 2004, 322-328. [298] Rudge, A.; Davey, J.; Raistrick, I.; Gottesfeld, S.; Ferraris, J. P. Conducting polymers as active materials in electrochemical capacitors. J. Power Sources 47, 1994, 89. [299] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P. Synthesis and characterization of new polyaniline/nanotube composites. Materials Science and Engineering C 23, 2003, 87-91. [300] Wu, T. M.; Lin, Y. W.; Liao, C. S. Preparation and characterization of polyaniline/multi-walled carbon nanotube composites. Carbon 43, 2005, 734–740. [301] Talbi, H.; Just, P.-E.; Dao, L.H. Electropolymerization of aniline on carbonized polyacrylonitrile aerogel electrodes: applications of supercapacitors. J. Appl. Electrochem. 33, 2003, 465-473. [302] Ryu, K.S.; Kim, K.M.; Park, Y.J.; Park, N.G.; Kang, M.G.; Chang, S.H. Redox supercapacitor using polyaniline doped with Li salts as electrode. Solid State Ionics 152, 2002, 861-866. [303] Gomez-Romero, P.; Chojak, M.; Cuentas-Gallegos, K.; Asensio, J.A.; Kulesza, P.J.; Casañ-Pastor, N.; Lira-Cantú, M. Hybrid organic-inorganic nanocomposite materials for application in solid state electrochemical supercapacitors. Electrochem. Commun. 5, 2003, 149-153. [304] Hussain, A. M. P.; Kumar, A.; Singh, F.; Avasthi, D. K. Effects of 160 MeV Ni12+ ion irradiation on HCl doped polyaniline electrode. J. Phys. D: Appl. Phys. 39, 2006, 750755. [305] Khomenko, V.; Frackowiak, E.; Beguin, F. Determination of the specific capacitance of conducting polymer/nanotubes composite electrodes using different cell configurations. Electrochim. Acta 50, 2005, 2499-2506. 85 [306] Jang, J.; Bae, J.; Choi, M.; Yoon, S.H. Fabrication and characterization of polyaniline carbon nanofiber for supercapacitor. Carbon 43, 2005, 2730-2736. [307] Zhang, J.; Shan, D.; Mu, S. L. A rechargeable Zn-poly(aniline-co-m-aminophenol) battery. J. Power Sources 161, 2006, 685-691. [308] Wang, Y.; Jing, X. Formation of Polyaniline Nanofibers: A morphological Study. J. Phys. Chem. B 112, 2008, 1157-1162. [309] King, R. C. Y.; Roussel, F. Morphological and electrical characteristics of polyaniline nanofibers. Synthetic Metals 153, 2005, 337-340. [310]doNascimento, G. M.; Kobata, P. Y. G.; Temperini, M. L. A. Structural and vibrational characterization of polyaniline nanofibers prepared from interfacial polymerization. J. Phys. Chem. B 112, 2008, 11551-11557. [311] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P. Synthesis and characterization of new polyaniline/nanotube compuestos. Materials Science and Engineering C 23, 2003, 87-91. [312] Wallace, G. G.; Mottaghitalab, V.; Spinks, G. F. The influence of carbon nanotubes on mechanical and electrical properties of polyaniline fibers. Synthetic Metals 152, 2005, 77-80. [313] Wu, T. M.; Lin, Y. W.; Liao, C. S. Preparation and characterization of polyaniline/multi-walled carbon nanotube compuestos. Carbon 43, 2005, 734–740. [314] Wu, T. M.; Lin, Y. W. Doped polyaniline/multi-walled carbon nanotube compuestos: Preparation, characterization and properties. Polymer 47, 2006, 3576-3582. [315] Wang, H.; Hao, Q.; Wang, H.; Yang, X.; Lu, L. Graphene oxide doped polyaniline for supercapacitors. Electrochemistry communications 11, 2009, 1158-1161. [316] Manthiram, A.; Murugan, A. V.; Muralingath, T. Rapid, facile microwavesolvothermal synthesis of graphene nanosheets and their polyaniline nanocompuestos for energy storage. Chem. Mater. 21, 2009, 5004-5006. [317] Chow, T. W.; Thostenson, E. T.; Li, C. Sensors and actuators based on carbon nanotubes and theircompuestos: A review. Composite Science and Technology 68, 2008, 1212-1249. [318] Dong-Won K.; Sivakkumar, S. R.; Kima W. J.; Ji-Ae C.; MacFarlane D. R.; Forsyth, M. Electrochemical performance of polyaniline nanofibres and polyaniline/multi-walled carbon nanotube composite as an electrode material for aqueous redox supercapacitors. J. Power Sources 171, 2007, 1062–1068. [319] Martinez, M. T.; Ansón, A.; Cendoya, I.; Ganborena, L.; Murillo, N.; Miguel, O.; Ochoteco, E.; Pomposo, J. A.; Grande, H.; Sainz, R.; Maser, W. K.; Benito, A. M. Compuestos of carbon nanotubes and conducting polymer as electrodes of supercapacitors. Instituto de Carboquímica (CSIC), Miguel LuesmaCastán 4, 50018, Zaragoza, Spain. [320] Wu, J.; Zhao, X. S.; Zhang, K.; Zhang, L. L. Graphene/polyaniline nanofiber composites supercapacitor electrodes. Chem. Mater. 22, 2010, 1392-1401. 86 3. Síntesis mecánica de nanocompuestos de polianilina (PAni) y nanotubos de carbono multicapa (MWCNT) y nanotubos de carbono multicapa dopados con nitrógeno (CNx-MWCNT) 87 88 3. Síntesis mecánica de nanocompuestos de polianilina (PAni) y nanotubos de carbono multicapa (MWCNT) y nanotubos de carbono multicapa dopados con nitrógeno (CNx-MWCNT) 3.1. ANTECEDENTES Se tiene un interés particular en compuestos de polianilina (PAni) debido a su buena procesabilidad, estabilidad ambiental y control en su conductividad eléctrica basado en reacciones ácido-base, así como por su variedad de aplicaciones de este polímero conductor [1, 2]. Las propiedades extraordinarias de los nanotubos de carbono (CNTs) como su elevado módulo de Young, buena flexibilidad y elevada conductividad eléctrica y térmica, han despertado un gran interés en su utilización como refuerzo de compuestos poliméricos. Actualmente se puede encontrar una amplia literatura sobre los avances, desafíos y aplicaciones de CNTs en este tipo de materiales [3-7]. Los CNTs pueden mejorar tanto las propiedades mecánicas como la conductividad eléctrica de este polímero [8-11]. Por ejemplo, los CNTs se han utilizado para mejorar las propiedades eléctricas de electrodos de batería [12]; el rendimiento electroquímico de supercondensadores [13, 14]; y las propiedades mecánicas y eléctricas de actuadores [15]. La PAni puede ser sintetizada mediante una polimerización oxidativa electroquímica de la anilina, obteniéndose una película depositada en el ánodo [7, 16]. En la polimerización química, donde el monómero puede oxidarse en solución, se pueden obtener mayores cantidades de polímero, y bajo ciertas condiciones, ésta puede estar nanoestructurada [1719]. En este inicio de la tesis se decidió estudiar uno de los métodos de síntesis de PAni menos comprendidos: el mecanoquímico. Huang y colaboradores, fueron los primeros en reportar un método mecanoquímico para obtener nanofibras ramificadas de PAni conductora en donde la reacción fue realizada en un molino de bolas, moliendo una sal de anilinio y un oxidante bajo condiciones ambientales normales y sin ningún solvente [20]. Otros grupos de investigación han estudiado la síntesis mecanoquímica de la PAni, o sus derivados, mediante una molienda manual de sales de anilinio y oxidantes en un mortero [21-24]. Particularmente, Du y colaboradores [25], sintetizaron nanofibras ramificadas de PAni moliendo manualmente 89 cloruro férrico e hidrocloruro de anilina, obteniendo un rendimiento máximo de síntesis de ~8%, comparable al del método interfacial (6-10%) [17]. Por otro lado, se han preparado compuestos de CNTs-PAni mediante una polimerización in situ al dispersar los CNTs en el medio ácido [26]. En otras síntesis se han mezclado la PAni y los CNTs aprovechando las interacciones electrostáticas entre la PAni, cargada positivamente, y los nanotubos oxidados, cargados negativamente, para lograr una buena dispersión [27]; en otros trabajos, se han dispersado CNTs en disoluciones de PAni utilizando N-metil 2 pirrolidona (NMP) o N,N’-dimetil propileno urea (DMPU) [15]. Estos procedimientos suelen consistir de reacciones lentas e implican el uso de disolventes orgánicos, así como de ácidos fuertes y oxidantes. De acuerdo a la revisión bibliográfica que se realizó, hasta el momento ha habido un solo reporte de síntesis de compuestos utilizando un método mecanoquímico. Ubul y colaboradores [28] describieron la síntesis de un composite de PAni-MWCNTs moliendo manualmente MWCNTs impregnados con monómero de anilina, ácido p-tolueno sulfónico y persulfato de amonio (APS) en un mortero (~50 min). Obtuvieron rendimientos de PAni de 68-82%. En esta tesis se reporta por primera vez un método en el cual mediante una molienda de una mezcla de CNTs, hidrocloruro de anilina y cloruro férrico en un molino de bolas, se produce un composite con una buena dispersión en un solo paso. Ni disolventes ni ácidos fueron utilizados. El dopaje de la PAni se obtuvo por el hidrocloruro de anilina. Un diseño experimental de superficie de respuesta (RS) [29] fue utilizado para seleccionar las condiciones óptimas de rendimiento polimérico y dispersión de los CNTs en función de algunos factores que en breve se describirán. Se encontró que este método podría ser útil para producir compuestos en un solo paso con MWCNTs convencionales y CNTs dopados con nitrógeno (CNx-MWCNTs). Se decidió utilizar CNx debido a que son más reactivos debido al dopaje y además, ha habido reportes en los que ha tenido mejores interacciones con las matrices en compuestos poliméricos [30]. 90 3.2. EXPERIMENTACIÓN Como reactivos de síntesis se emplearon hidrocloruro de anilina ([C6H7N HCl] o Ani·HCl, al 97%, de la casa Aldrich) y cloruro de hierro (III) hexahidratado (FeCl3·6H20 grado ACS de la casa Fermont) como oxidante, el cual se referirá como FeCl3 por conveniencia a lo largo del texto. Se utilizó agua destilada en todos los experimentos. Se utilizó etanol para eliminar la anilina sin reaccionar y los subproductos. Los MWCNTs fueron producidos alimentando una corriente de ferroceno/tolueno (2.5% wt/vol), en un inerte, a un reactor de CVD para el crecimiento de los nanotubos en sustrato a 800ºC durante 30 min, según el método descrito por Kamalakaran y colaboradores [31]. Los CNx fueron crecidos en un sistema idéntico, excepto que se alimentó bencilamina en tolueno (2.5%) a 800ºC durante 30 min, según lo descrito por Terrones y colaboradores [32]. Posteriormente, los CNTs se purificaron dispersándolos en agua, bajo reflujo, con una sonda de irradiación pulsada de ultrasonidos, seguido de un reflujo en HCl (6M) y filtración. Este proceso está basado en el método hidrotérmico de Alvizo-Páez y colaboradores [33] para eliminar el carbono amorfo y facilitar la interfase del filamento y la matriz polimérica. 3.2.1. Procedimiento general de síntesis de los compuestos La molienda se llevó a cabo en un molino de bolas orbital 8000D (SPEX Certiprep) utilizando viales de acero de 62 mL (se llevaron a cabo dos experimentos al mismo tiempo), y tres bolas de acero de 12.4 mm de diámetro y tres de 5 mm de diámetro. El Ani·HCl, el FeCl3, los CNTs, y el agua destilada se añadieron al vial en cantidades que se verán a continuación. Después de la molienda, la pasta húmeda fue removida del vial, y junto con las bolas, se enjuagó con agua destilada para recuperar los residuos. Los compuestos fueron lavados y filtrados con membranas de nylon de tamaño de poro de 0.45 µm bajo vacío. El lavado se realizó con ~120 mL de etanol, seguido por ~100 mL de agua destilada y finalmente con~40 mL de etanol. Los compuestos se secaron en un horno a 100° C antes de pesarse. 91 3.2.2. Experimentos exploratorios Para probar si el ultrasonido ayudaba a la dispersión de CNTs, se sonicaron en un baño ultrasónico 40.4 mg de CNx en 7 mL de agua (ya que los nanotubos dopados con nitrógeno son más dispersables que los MWCNTs convencionales) durante 60, 30 o 0 min, siguiendo con una molienda durante 20 min, con 8.34 g de FeCl3 y 2.02 g de anilina (razón molar FeCl3:Ani·HCl de 2:1). Se utilizó un diseño factorial ½2k para detectar los efectos en la eficiencia de polimerización (%η) y la dispersión de nanotubos utilizando dos niveles de k = 3 variables: volumen de agua (2 y 12 mL), razón molar de oxidante con respecto a anilina (1:1 con 4.17 g de FeCl3; 2:1 utilizando 8.34 g) y tiempo de molienda (20 y 60 min). 3.2.3. Diseño experimental de superficie de respuesta Se eligió un diseño experimental de superficie de respuesta de Box-Behnken para evaluar el efecto de tres factores en la %η de la PAni y la conductividad eléctrica de los compuestos. Se eligieron 2, 7 y 12 mL de volumen de agua, y tres niveles (0, 20.2 y 40.4 mg) de dos tipos de nanotubos (MWCNTs y CNx). En este diseño se mantuvo constante la relación molar FeCl3:Anilina 2:1 (8.34 g FeCl3·6H20, 2.02 g Ani·HCl) y también el tiempo de molienda de 20 min. 3.2.4. Caracterización La morfología y dispersión de los CNTs en los compuestos se observó mediante microscopía electrónica de barrido (SEM) en un FEI-XL-30-SFEG con un accesorio de microscopía electrónica de barrido y de transmisión (STEM), y también en el modo SEM regular. Al utilizar el modo STEM, se prepararon rejillas de carbono con gotas de los compuestos diluidos en una suspensión. También se caracterizaron las muestras por difracción de rayos X utilizando un BruckerD8 ADVANCE (ánodo de Cu, λ=1.5406 Å, operando a 35 kV) y por espectroscopía Raman con un Renishaw InVia microRaman utilizando un láser HeNe (633 nm). La conductividad eléctrica se midió, por quintuplicado, con una sonda de cuatro puntas (Jandel CYL-1.0-100-TC-100-1M) utilizando una fuente Keithley 2400. Se prepararon películas cuadradas de los compuestos (5 mm de lado, ~ 0,5 mm de espesor) cortando la película depositada en la membrana de nylon en la microfiltración. 92 3.3. RESULTADOS Y DISCUSIÓN 3.3.1. Experimentos exploratorios Se encontró que el tratamiento previo con ultrasonido no afectó la dispersión final en el compuesto. Teniendo en cuenta que, durante la molienda, se tuvo un mezclado altamente energético, esto no fue sorpresa. Los rendimientos de polimerización en estos experimentos fueron muy similares (para ts=0, 1.86 %± 0.19; ts=30,1.75%± 0.25; ts =60,1.63% ±0.05). Es pertinente señalar que después de la sonicación durante 30 y 60 min en agua desionizada, el CNx aún formaba flóculos. Así que se decidió eliminar el paso de la sonicación. Du y colaboradores [26] llamaron a su síntesis método "seco", aunque ellos utilizaron también FeCl3 como oxidante, una sal que, como se conoce, es delicuescente [34]; como la reacción fue realizada en condiciones ambientales normales, el FeCl3 debió haber absorbido fácilmente la humedad del aire, y de hecho ellos informan que al final de la molienda manual en el mortero, obtuvieron una pasta húmeda. Ante esto, se consideró que el agua podría tener un efecto en la polimerización, por lo que en el diseño experimental exploratorio, se eligió como uno de los factores, junto con la relación molar oxidante:anilina y el tiempo de molienda. Para el experimento exploratorio, se decidió utilizar un diseño factorial ½2k-1, apelando al Principio de Efectos Esparcidos: la mayoría de los sistemas están dominados por algunos de los efectos principales y las interacciones de orden inferior, la mayor parte de las interacciones de orden superior son insignificantes. Asimismo, este diseño permitió reducir a la mitad el número de corridas experimentales. Los niveles de cada variable y los rendimientos poliméricos de cada corrida se muestran en la Tabla 1, en donde se muestra que el agua tuvo un claro efecto en el aumento de la %η. Es importante señalar que, en un experimento preliminar donde el agua no se añadió, el rendimiento fue de 0.3%, confirmando que bajo estas condiciones es importante añadir agua en la reacción. El efecto de las otras variables no es tan evidente a partir de la tabla, pero por el análisis estadístico de los resultados, se obtuvo un modelo de regresión lineal múltiple (Ecuación 1). El modelo predijo una %η promedio de 2.21% (de anilina polimerizada a PAni). Tabla 1. Diseño experimental ½2k donde se observan los niveles de las variables bajo estudio y el rendimiento de conversión de anilina en polianilina. 93 Corrida tm (Tiempo de molienda, min) Vw (Volumen de H2O, mL) Ox (Razón molar oxidante*: anilina) %η (Eficiencia de polimerización de la PAni) 1 2 20 60 2 2 2:1 1:1 1.7 1.54 3 4 20 60 12 12 1:1 2:1 2.72 2.87 *FeCl3·6H20, 4.17 or 8.34 g to 2.02 g Ani·HCl %η = 2.21 ‒ 0.0025(tm) + 0.585(Vw) + 0.0775(Ox) (1) donde %η es la eficiencia de polimerización, tm el tiempo de molienda, Vw el volumen de agua y Ox la razón molar oxidante:anilina. Los niveles factoriales se codificaron como +1 y -1 (niveles alto y bajo) para el análisis [29]. Los resultados en la Tabla 1 muestran que no hay ninguna ventaja al utilizar tiempos de molienda mayores a 20 min. El modelo de regresión refleja esto con un muy pequeño coeficiente en su respectiva variable, el cual podría considerarse muy cerca del cero, dentro del posible error experimental. La ecuación también muestra un signo negativo para el tiempo de molienda, lo cual podría indicar que un tm alto podría disminuir la %η, pero se conoce, que bajo las condiciones experimentales utilizadas no debería ocurrir a una despolimerización. El signo negativo del tiempo de molienda podría ser debido al hecho de que la combinación en el nivel bajo, tanto para el volumen de H2O y la relación de oxidante (corrida 2) resultó en el menor rendimiento. El modelo de regresión muestra un coeficiente pequeño para la razón oxidante:anilina. Se decidió mantener la proporción molar mayor, de 2:1, para garantizar un exceso estequiométrico de FeCl3. El mayor coeficiente en el modelo de regresión fue para V w, confirmando la hipótesis de que éste es un factor importante cuando se utiliza FeCl3 como oxidante. Por lo tanto, este factor se utilizó en los experimentos subsecuentes para estudiar con mayor profundidad su efecto. En la síntesis de estado sólido la difusión de precursores sólidos podría limitar el rendimiento [20]. La adición de agua en estos experimentos podría reducir esta limitación 94 al disolver el hidrocloruro de anilina y el cloruro férrico. Pero se sospechó que el agua podría contribuir a la polimerización oxidativa de la anilina al facilitar la reducción del hierro como iones Fe+3 solvatados. 3.3.2. Experimento de optimización En este experimento se decidió utilizar los dos tipos de nanotubos para evaluar si el dopaje de nitrógeno de los CNx pudiera favorecer alguna interacción con la PAni debido a su mayor reactividad. Asimismo, el dopaje con nitrógeno provoca estados electrónicos en el grafeno que los vuelve metálicos a pesar de su quiralidad [35, 36], por lo que los CNx podrían ser eléctricamente más conductores que una mezcla de MWCNTs metálicos y semiconductores, lo cual podría ocasionar diferencias en la conductividad eléctrica de los compuestos. Se ha seleccionado un diseño experimental de superficie de respuesta para tres variables: peso de MWCNTs, peso de CNx y volumen de agua. Las cantidades de los dos tipos de CNTs utilizados y los valores del volumen de agua se muestran en la Tabla 2, junto con los resultados de la eficiencia de polimerización (%η) y conductividad eléctrica (σ). El número de corrida experimental mostrado en la Tabla 2 indica el orden nominal del diseño experimental sólo como referencia; los experimentos fueron realizados en orden aleatorio, y los datos se ordenaron de tal forma que se muestran los niveles de agua en forma ascendente. En la Tabla 2 se observa otra vez que es evidente que el volumen de agua (V w) es una variable importante para la %η la PAni, calculada mediante gravimetría. %η varió de 1.45 a 3.15% (media de 2.05 ± 0.48%) como puede observarse en la tabla. El mejor modelo de regresión cuadrático para %η, mediante el análisis de varianza (ANOVA), se muestra en la Ecuación 2, el cual tiene un coeficiente de determinación R2=0.776. Este modelo ajustado, refleja adecuadamente las características principales del proceso, incluyendo solamente los efectos principales (no se incluyeron las interacciones entre las variables ni los términos cuadráticos de los factores, los cuales no fueron significativos). Otra vez, el mayor coeficiente es para Vw. En cuanto a las cantidades de los tipos de CNTs se esperaba que no interfirieran en la eficiencia de polimerización puesto que no participarían en la oxidación de la anilina. En concordancia con esto, los coeficientes de sus respectivas variables fueron similares y pequeños. 95 %η = 2.05 + 0.064(MWCNT) + 0.065(CNx) + 0.59(Vw) (2) Tabla2. Resultados de los experimentos de superficie de respuesta para la eficiencia polimérica (%η) y la conductividad (σ) de los compuestos de PAni-CNTs. Corrida MWCNTs CNx Volumen %η % σ (S/cm) (mg) (mg) de agua CNTs (mL) 5 9 7 11 16 2 15 13 4 1 17 3 14 10 8 6 12 0 20.2 40.6 20.3 20.1 0 20.3 20.3 40.9 0 20.4 40.4 20.4 20.3 40.4 0 20.8 20.1 0 20.5 40.4 20.1 40.4 20.1 20.3 40.4 0 20.2 0 20.1 0 20.3 20.4 40.6 2 2 2 2 7 7 7 7 7 7 7 7 7 12 12 12 12 1.45 1.49 1.50 1.46 2.12 2.02 2.07 1.69 2.22 1.95 2.12 2.03 2.11 2.11 3.15 2.97 2.40 40.8 40.16 66.74 67.33 48.56 49.75 49.08 54.16 64.33 0 48.5 49.57 48.67 32.11 48.75 25.22 55.54 1.337 ±0.118 4.439 ±1.03 4.512 ±0.43 4.153 ±0.517 1.727 ±0.161 2.415 ±0.334 3.008 ±0.429 3.869 ±0.226 2.14 ±0.253 10-4 2.025 ±0.251 3.407 ±0.919 2.545 ±0.206 4.426 ±0.507 2.863 ±0.28 2.096 ±0.141 2.006 ±0.131 La conductividad eléctrica (σ) varió de ~10-4 a 4.51 S·cm-1, con una media de 2.76 ± 1.25% S·cm-1. En la Figura 1 se observan los resultados de la conductividad en función de la cantidad total de los CNTs. Los dos tipos de nanotubos incrementaron significativamente la conductividad eléctrica en órdenes de magnitud similares. Los contenidos de CNTs por arriba del 25% incrementaron la conductividad ligeramente, indicando que a estos niveles de concentración de nanotubos se está ya por arriba del nivel de percolación. La dispersión en los resultados de la conductividad eléctrica posiblemente se debió a que ambos tipos de MWCNTs, dopados con nitrógeno y sin dopaje, contribuyeron de manera distinta a la conductividad eléctrica: teóricamente, los CNx tienen una mayor conductividad eléctrica que los MWCNTs no dopados, pero en la práctica, los CNx tienen defectos estructurales que dispersan a los electrones, haciendo que su conductividad sea más baja. Además, es probable que la longitud de los CNTs disminuya 96 debido al proceso de dispersión, lo cual decrecería también la conductividad eléctrica [37, 38]. Adicionalmente a los defectos de los CNx, y al error experimental intrínseco de las mediciones de conductividad, otra fuente de dispersión en estos resultados pudo haber sido el error experimental en la medición del contenido de CNTs (e. g. debido a la manipulación de la muestra). Es evidente que ante la elevada cantidad de CNTs en los compuestos, éstos tuvieron una conductividad eléctrica significativamente alta en comparación al de la PAni sin nanotubos (~4 órdenes de magnitud). El modelo de resultados para σ (Ecuación 3) mostró que (MWCNT) fue un factor más significativo en el incremento de la conductividad que (CNx). El coeficiente negativo para Vw es consistente con su marcado efecto en la %η: una mayor cantidad de polímero es producido con niveles de agua altos y un menor % de CNTs disminuye la conductividad del composite. La R2=0.748 del modelo proveniente del ANOVA, indica que representa ampliamente la variabilidad factorial del experimento. Figura1. Gráfico de la conductividad eléctrica en función del porcentaje de CNTs obtenida a partir del diseño experimental de la tabla 2. Los niveles bajo, medio y alto (L, M, H, respectivamente) de las variables se muestran como referencia, de acuerdo a MWCNT/CNx/ Vw. 97 σ = 2.72 + 0.88(MWCNT) - 0.20(CNx) - 0.38(Vw) - 0.92(MWCNT)(CNx) 0.60(MWCNT)(Vw) – 0.84(MWCNT)2 + 0.92(Vw)2 (3) donde σ es la conductividad eléctrica, MWCNT es la cantidad de MWCNTs, CNx es la cantidad de CNx y Vw es la cantidad de agua. El efecto de las tres variables en la eficiencia polimérica (%η) y la conductividad eléctrica (σ) puede ser visualizado en los gráficos de superficie de respuesta de la Figura 2. En el gráfico del rendimiento, Figura 2 A, se observa que la cantidad de CNTs prácticamente no tuvo efecto alguno. En cambio, la %η se incrementa al hacerlo el volumen de agua. Como era de esperarse, la σ se incrementa debido a las propiedades eléctricas de los CNTs. Las superficies de respuesta de la σ, Figuras 2 B, 2 C y 2 D, confirman que la mayor conductividad ocurrió al tenerse una mayor cantidad de MWCNTs. También muestran que la σ ligeramente aumenta de 1.6 a 2.9 S·cm-1 al incrementarse la cantidad de CNx. A primera vista, en el modelo de la Ecuación 3, podría parecer contradictorio que el coeficiente del término (CNx) tenga un signo negativo, pero considerando el coeficiente y signo de (MWCNT)(CNx), si se tiene, por ejemplo un nivel bajo de MWCNTs, entonces, mediante las consiguientes operaciones algebraicas, el signo cambia y (CN x) tendrá contribuciones positivas a la respuesta σ, como puede observarse en la Figura 2 B. El efecto negativo del agua en la conductividad puede apreciarse más fácilmente en el extremo del más alto nivel de MWCNT en la Figura 2 C. 98 Figura2. Superficies de respuesta del rendimiento y la conductividad eléctrica de los nanocompuestos de PAni/CNTs. (A) Rendimiento de los compuestos en función de los CNTs y el volumen de agua, Vw. (B) Conductividad eléctrica de los compuestos en función de las cantidades de CNx y MWCNT (Vw=7mL). (C) Conductividad eléctrica en función de MWCNT y Vw. (D) Conductividad eléctrica en función de CNx y Vw. Los resultados obtenidos muestran que la reacción fue mucho más efectiva en una mezcla con agua que en una mezcla seca. Estudios previos no han considerado esta variable, la cual podría incluso incrementar rendimientos al utilizar otros oxidantes. Es conocido que al utilizar cloruro férrico se obtienen menores rendimientos que al utilizar persulfato de amonio [39]. Sin embargo, se ha reportado que el persulfato de amonio (APS) oxida a los CNTs [40], lo cual podría resultar en una menor conductividad eléctrica, aunque en este trabajo no se probó si al añadir APS en la molienda el rendimiento se incrementa. Se apreció que en ausencia de CNTs, este método produce PAni en la forma de nano y micropartículas, como puede observarse en la Figura 3. 99 Figura 3. Micrografía SEM de PAni sin CNTs obtenida de la molienda de hidrocloruro de anilina y cloruro férrico con agua. La Figura 4 muestra algunas imágenes representativas de SEM y un par SEM/STEM de los compuestos. En general se observó que de manera irregular se formaron nanopartículas de PAni entre los CNTs; algunos CNTs mostraron nanopartículas de PAni adheridas en su superficie, esto fue más evidente en el par de imágenes SEM/STEM (Figuras 4 B y C, E y F, y H e I). Aunque se esperaba observar más diferencias, debido a la mayor interacción de PAni con los CNx, las micrografías SEM no mostraron alguna diferencia notable. Los puntos oscuros que aparecen en las micrografías STEM (Figuras C, F, I) podrían ser nanopartículas catalíticas de Fe que provocaron el crecimiento de los CNTs, como las que se observan dentro de los círculos en F, pero otras nanopartículas metálica parecen estar fuera de los nanotubos, las cuales podrían ser resultado del impacto entre las bolas de acero y el vial. En todas las muestras, las nanopartículas de PAni parecen estar bien mezcladas y dispersas. Algunas nanopartículas de PAni se observaron adheridas a las paredes de los CNTs, sin una diferencia notable entre uno u otro tipo de CNTs. 100 Figura 4. Imágenes SEM/STEM de compuestos de PAni/CNTs. A, B, C corresponden al composite con 49.75% de CNx. D, E, F corresponden al composite con 49.57% de MWCNTs. G, H, I son pertenecen al composite con 54.16% de una mezcla de CNx/MWCNTs. Para confirmar los resultados de los diseños experimentales, se sintetizaron compuestos de PAni/CNx con un mayor volumen de agua (17 mL). Al utilizarse una razón molar [FeCl3·6H20]:[Ani] de 2:1 la eficiencia de polimerización se incrementó a 3.58%, confirmándose nuevamente que el agua facilitó la polimerización. Esta muestra tuvo una conductividad de 2.23 S/cm. Asimismo, al incrementarse la razón de oxidante a 3:1 con 17 mL de agua, la %η aumentó a 4.37%. Este composite tuvo una conductividad de 2.14 S/cm, y se muestra en la Figura 5. Una mayor cantidad de PAni tiende a formar partículas más grandes, aunque la morfología es similar a las otras muestras. Los CNTs estuvieron bien dispersos y mezclados entre los aglomerados de polímero. Las conductividades de estas muestras son ligeramente más bajas debido a las menores cantidades relativas de CNTs, aunque están en el mismo rango que las mostradas en la Tabla 2. 101 Figura 5. Micrografía SEM del composite con 31.4% de CNx obtenido por mecanosíntesis con una razón molar [FeCl3·6H20]:[Ani] de 3:1 y un volumen de agua de 17 mL. La espectroscopía Raman (Figura 6a) muestra un pico en 1336 cm-1 perteneciente al alargamiento de C-N·+ de la estructura bipolarónica de la sal de esmeraldina, forma conductora de la PAni [8, 9, 13, 41], lo cual podría indicar que la PAni sintetizada fue parcialmente conductora por lo que su conductividad eléctrica fue muy baja. La PAni también mostró un pico en 1583 cm-1 (tensión C-C del anillo benzenoide) y en ~1163 cm-1 (deformación C-H en el plano del anillo quinoide). Estos picos se superpusieron con los picos D (de “desorden” de hibridación sp3 en ~1337 cm-1) y G (“grafítico” de hibridación sp2 en ~1590 cm-1) de los CNTs en los compuestos (perfiles b, c y e de la Figura 6). En general, los perfiles de los compuestos muestran picos bien definidos correspondientes a la PAni y a sus CNTs respectivos. Los perfiles de los CNx y MWCNTs son típicos, con una razón ID/IG más grande en los CNx (1.018) que en los MWCNTs (0.595). En ambos tipos de CNTs, la banda 2D (en 2665 cm-1) aparece junto con las bandas D y G (curvas d y f de la Figura 6, respectivamente). La banda de 1481 cm-1 (alargamiento del anillo quinoide C=N) decreció en su intensidad en los perfiles de los compuestos. Esto ha sido reportado como una evidencia de la fuerte interacción del anillo quinoide de la PAni con la superficie de los CNTs [15, 26, 42]. Sean I1163 e I1481 las intensidades de las bandas de 1163 (~constante) y 1481 cm-1 (variable), respectivamente, la disminución de la razón I1481/I1163 es baja para los compuestos de PAni/MWCNTs (I1481/I1163=1.547), con respecto al de la PAni (1.884), pero es más baja en 102 los compuestos de PAni/CNx (1.436) y PAni/MWCNT/CNx (1.383), lo cual sugiere una mejor interacción de la PAni con los CNx que con los MWCNTs, aunque no se encontró alguna diferencia evidente en las micrografías de sus respectivos compuestos. Figura 6. Espectrogramas Raman de (a) PAni, (b) PAni/MWCNT/CNx, (c) PAni/CNx, (d) CNx purificados, (e) PAni/MWCNTs y (f) MWCNTs purificados. La PAni mecánicamente sintetizada por este método no es cristalina como puede observarse en el amplio pico de 12 a 28º de la Figura 7 A [19, 41]. Es notable que el pico (002) de los CNTs no muestra un apreciable ensanchamiento en los difractogramas de los compuestos (Figura 7 B, C, D) comparado con el de CNx (Figura 7 E) y el de MWCNTs (Figura 7 F). Esto indica que no hay un daño significativo en los CNTs debido a la molienda. El pico ancho de la PAni es menos visible en los compuestos, donde el pico (002) de los CNTs tiene una intensidad más alta que la PAni amorfa; los compuestos también muestran picos intensos provenientes de las partículas de acero. En el caso de los perfiles de los CNx y MWCNTs los pequeños picos que se observan, son de las partículas catalíticas de Fe. 103 Figura 7. Difractogramas de rayos X de (A) PAni pura, sintetizada sin CNTs, muestra un pico típico y ancho de PAni no cristalina; los picos observados en 43.6 y 44.4º, sonde las partículas de acero provenientes del vial y mezcladas con los productos durante la mecanosíntesis. (B) composite con 49.8% de CNx, (C) composite de 54.2% de CNTs (CNx + MWCNTs), (D) composite con 49.6% de MWCNT, y CNx purificados (E) y MWCNTs (F). La caracterización por SEM y rayos X no mostraron CNTs dañados. Sin embargo, se ha reportado que la molienda mediante molino de bolas produce daño significativo a los CNTs [43]. Incluso Hilding y colaboradores [44] reportaron que la molienda manual de CNTs en un mortero durante aproximadamente una hora resultó en una reducción significativa de sus longitudes y con daño en sus capas, ocasionándoles defectos estructurales. Esto tiene que ver directamente con los resultados obtenidos por Ubul y colaboradores [28] quienes, mediante una molienda manual durante 50 min, obtuvieron compuestos con MWCNTs altamente fragmentados, como pudo observarse en sus micrografías TEM, y además confirmado en las imágenes SEM donde no se apreciaron nanotubos largos. En las muestras producidas en este estudio no fue posible explorar si hubo daño en los CNTs mediante espectroscopía Raman: los cambios en la razón ID/IG, la cual podría indicar daño mecánico por la molienda (o funcionalización con anilina u oxidación) no puede ser medida directamente debido a la superposición de bandas de la PAni y los CNTs. Para 104 reunir evidencia de que los CNTs no fueron dañados durante la molienda, se realizaron dos experimentos sin hidrocloruro de anilina. Se utilizaron tiempos de molienda de 20 y 60 min, 8.34 g de FeCl3, 12 mL de H2O, y 40.4 mg de CNx. En la Figura 8 se muestra claramente que en ausencia de monómero, los CNTs son severamente dañados. Después de 20 minutos (Figura 8 A) muchos nanotubos se fragmentaron, otros se mantuvieron largos pero con sus superficies muy irregulares, algunos nanotubos quedaron aparentemente sin daño. Los fragmentos de los nanotubos muestran diferencias en su tamaño y aspecto superficial al compararlos con las partículas de PAni de las Figuras 4 y 5, hasta algunos mantuvieron la morfología cilíndrica (flechas de la Figura 8 A). Incluso una partícula de acero puede observarse en la misma figura. Estas impurezas son comunes en la molienda con molino de bolas (como se mencionó arriba, en las imágenes STEM de la Figura 4, los puntos oscuros podrían deberse a nanopartículas pequeñas de acero). Asimismo, después de la molienda durante 60 min de los CNx, se obtuvo un pequeño incremento (~4%) en el peso de los nanotubos recuperados. Esto ocurrió debido a que las nanopartículas metálicas provenientes del vial o de las bolas de acero, se mezclaron con las muestras. Después de 60 minutos, solamente fragmentos cortos e irregulares de CNx fueron observados (Figura 8 B), e incluso, pareció que la morfología cilíndrica hubiera desaparecido al aparecer fragmentos aplanados e irregulares (Figura 8 C). 105 Figura 8. Micrografías SEM de CNx molidos sin C6H7N·HCl: (A) Molienda de 20 min (B, C) Molienda de 60 minutos. La espectroscopía Raman de estas muestras blanco (producidas sin anilina) muestra principalmente los picos de las bandas D, G y 2D. La razón ID/IG de los CNx se incrementa de 1.018 (la ID/IG de CNx purificados de la Figura 6) a 1.080 después de 20 min de molienda (Figuras 9 a), pero, no se incrementa más después de 60 min de molienda (Figura 9 b). Esto indica que la presencia de agua protege a los CNTs de daño, y podría indicar que su oxidación es muy baja en estas condiciones, incluso después de una hora de molienda. En contraste, cuando se realiza la molienda de CNx sin ningún reactivo durante 60 min (Figura 9 c) la razón ID/IG se incrementa a 1.349, revelando que hubo un daño severo debido a la molienda en seco, asimismo, se confirma que el agua no solamente incrementa el rendimiento sino que disminuye significativamente el daño a los CNTs. La explicación más probable de esto, es que el agua absorbe parte de la energía mecánica y la disipa, evitando daño a los CNTs, la cual, adicionalmente, podría actuar como lubricante de tal forma que los CNTs son empujados a los lados en lugar de ser aplastados entre las bolas y el vial. 106 Figura 9. Espectrogramas Raman de las muestras blanco de CNx molidos sin anilina, pero con FeCl3 y agua durante (A) 20 y (B) 60 min, y (C) CNx molidos en seco durante 60 min. Para la descripción de estos resultados experimentales, se propone el siguiente mecanismo de reacción ilustrado en la Figura 10, en donde en el inicio de la reacción se produce una disociación entre iones de Fe3+ y Cl– (provenientes de las sales empleadas en la molienda: cloruro férrico e hidrocloruro de anilina) debido a la energía mecánica en presencia de agua. Los monómeros de anilina y el HCl también comienzan a aparecer en el medio. La polimerización de la anilina inicia cuando los iones Fe3+ comienzan a oxidar sus aminas, las cuales se van a ir enlazando [39]. Algunos de los protones, H+, liberados en el medio, comienzan a dopar algunas secciones de las cadenas poliméricas de la PAni. Finalmente, se obtiene el polímero de PAni con una fracción de ella en forma de sal de esmeraldina (forma conductora de la PAni). En el caso de la síntesis de los nanocompuestos con CNTs, las partículas de PAni se adherirán en algunas zonas de la superficie de los CNTs, tal como se observó en las micrografías. 107 Figura 10. Mecanismo de reacción de la mecanosíntesis de la PAni. 3.4. CONCLUSIONES La mecanosíntesis de nanocompuestos de PAni/CNTs en un solo paso ha sido desarrollada sin el uso de surfactantes, disolventes orgánicos ni ácidos. Este método de mezclado de alta energía en el molino de bolas provee una simple y relativamente rápida producción de nanocompuestos de PAni/CNTs con una buena interacción carga/matriz polimérica. Los CNTs no sufrieron daño significativo. Además de que los tiempos relativamente cortos de síntesis ayudan a evitar un daño mecánico a los nanotubos, la adición de agua en la molienda (un disolvente ambientalmente amigable) es el principal factor que evita el daño a los CNTs. 108 La adición de agua en la reacción promueve la eficiencia en la polimerización, además, podría facilitar la reducción de Fe+3 por la disociación de la sal cloruro, facilitando así su difusión y por lo tanto la oxidación de la anilina. Los compuestos resultantes, tienen PAni en su forma conductora y la presencia de CNTs incrementa la conductividad por 4 órdenes de magnitud, siendo los MWCNTs mejores que los CNx en este sentido. La espectroscopía Raman sugiere que los nanotubos dopados con nitrógeno tienen una mejor interacción con la PAni al compararlos con los MWCNT sin dopaje. 109 REFERENCIAS [1] Salaneck, W. R.; Lundstrom, I.; Huang, W.-S.; MacDiarmid, A. G. A two-dimensionalsurface ‘state diagram’ for polyaniline. Synthetic Metals 13, 1986, 291–297. [2] Zarras, P.; Irvin, J. Electrically Active Polymers. In Encyclopedia of Polymer Science and Technology, Concise; Mark, H.F. Ed.; John Wiley & Sons Inc.: Hoboken, New Jersey, 2007, 351–358. [3] Harris, P. J. F. Carbon nanotubes composites. Int. Mater. Rev. 49, 2004, 31–43. [4] Du, J.-H.; Bai, J.; Cheng, H.-M. The present status and key problems of carbon nanotube based polymer composites. Express Polym. Lett 1, 2007, 253–273. [5] Schaefer, D. W.; Justice, R. S. How Nano Are Nanocomposites? Macromolecules 40, 2007, 8501–8517. [6] Spitalsky, Z.; Tasis, D.; Papagelis, K.; Galiotis, C. Carbon nanotube polymer compuestos: Chemistry, processing, mechanical and electrical properties. Prog. Polym. Sci. 35, 2010, 357–401. [7] Li, C.; Thostenson, E. T.; Chow, T.-W. Sensors and actuators based on carbon nanotubes and their composites: A review. Comp. Sci. Tech. 68, 2008, 1227–1249. [8] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P.Synthesis and characterization of new polyaniline/nanotube composites. Mater. Sci. Eng. C 23, 2003, 87– 91. [9] Wu, T.-M.; Lin, Y.-W.; Liao, C.-S. Preparation and characterization of polyaniline/multi-walled carbon nanotube composites. Carbon 43, 2005, 734–740. [10] Mottaghitalab, V.; Spinks, G. F.; Wallace, G. G. The influence of carbon nanotubes on mechanical and electrical properties of polyaniline fibers. Synthetic Metals 152, 2005, 77–80. [11] Wu, T.-M.; Lin, Y.-W. Doped polyaniline/multi-walled carbon nanotube composites: Preparation, characterization and properties. Polymer 47, 2006, 3576–3582. [12] Wang, C. Y.; Mottaghitalab, V.; Too, C. O.; Spinks, G. M.; Wallace, G. G. J. Polyaniline and polyaniline-carbon nanotube composite fibres as battery materials in ionic liquid electrolyte. Power Sources 163, 2007, 1105–1109. [13] Sivakkumar, S. R.; Kim W. J.; Choi, J.-A.; MacFarlane, D. R.; Forsyth, M.; Kim, D.W. J. Electrochemical performance of poly-aniline nanofibres and polyaniline/multiwalled carbon nanotube composite as an electrode material for aqueous redox supercapacitors. Power Sources 171, 2007, 1062–1068. [14] Zhang, J.; Kong, L.-B.; Wang, B.; Luo, Y.-C.; Kang, L. In-situ electrochemical polymerization of multi-walled carbon nanotube/polyaniline composite films for electrochemical supercapacitors. Synthetic Metals 159, 2009, 260–266. [15] Mottaghitalab, V.; Xi, B.; Spinks, G. M. Wallace, G. G. Polyaniline fibres containing single-walled carbon nanotubes: Enhanced performance artificial muscles. Synthetic Metals 156, 2006, 796–803. [16] Zotti, G.; Cattarin, S.; Comisso, N. J. Cyclic potential sweep electropolymerization of aniline: The role of anions in the polymerization mechanism. Electroanal. Chem. 239, 1988, 387-396. 110 [17] Huang, J.; Virji, S.; Weiller, B. H.; Kaner, R. B. Polyaniline nanofibers: Facile synthesis and chemical sensors. J. Am. Chem. Soc. 125, 2003, 314–315. [18] Huang, J.; Kaner, R. B. A general chemical route to polyaniline nanofibers. J. Am. Chem. Soc. 126, 2004, 851–855. [19] Stejskal, J.; Sapurina, I.; Trchová, M.; Konyushenko, E. N. Oxidation of Aniline: Polyaniline Granules, Nanotubes, and Oligoaniline Microspheres. Macromolecules 41, 2008, 3530–3536. [20] Huang, J.; Moore, J. A.; Acquaye, J. H.; Kaner, R. B. Mechanochemical Route to the Conducting Polymer Polyaniline. Macromolecules 38, 2005, 317–321. [21] Abdiryim, T.; Xiao-Gang, Z.; Jamal, R. Comparative studies of solid-state synthesized polyaniline doped with inorganic acids. Mater. Chem. Phys. 90, 2005, 367–372. [22] Abdiryim, T.; Xiao-Gang, Z.; Jamal, R. Synthesis and Characterization of Poly(oToluidine) Doped with Organic Sulfonic Acid by Solid-State Polymerization. J. Appl. Polym. Sci. 96, 2005, 1630–1634. [23] Jamal, R.; Abdiryim, T.; Ding, Y. Nurulla, I. J. Comparative studies of solid-state synthesized poly(o-methoxyaniline) doped with organic sulfonic acids. Polym. Res. 15, 2008, 75–82. [24] Ding, Y.; Abdiryim, T.; An, S. Nurulla, I. Effect of β-Cyclodextrin on the Solid-State Synthesized Polyaniline Doped with Hydrochloric Acid. J. Appl. Polym. Sci. 107, 2008, 3864–3870. [25] Du, X.-S.; Zhou, C.-F.; Wang, G.-T.; Mai, Y.-M. Novel Solid-State and TemplateFree Synthesis of Branched Polyaniline Nanofibers. Chem. Mater. 20, 2008, 3806–3808. [26] Cochet, M.; Maser, W. K.; Benito, A. M.; Callejas, M. A.; Martinez, M. T.; Benoit J.M.; Schreiber, J.; Chauvet, O. Synthesis of a new polyaniline/nanotube composite: “insitu” polymerization and charge transfer through site-selective interaction. Chem. Commun. 16, 2001, 1450–1451. [27] Yan, X.-b.; Y.; Han, Z.-J.; Yang, Y.; Tay, B.-K. Fabrication of Carbon NanotubePolyaniline Composites via Electrostatic Adsorption in Aqueous Colloids. J. Phys. Chem. C 111, 2007, 4125–4131. [28] Ubul, A.; Jamal, R.; Rahman, A.; Awut, T.; Nurulla, I.; Abdiryim, T. Solid-state synthesis and characterization of polyaniline/multi-walled carbon nanotubes composite. Synth. Met. 161, 2011, 2097–2102. [29] Montgomery, D. C. Design and Analysis of Experiments. 5th Edition, John Wiley & Sons, 2004. [30] Fragneaud, B.; Masenelli-Varlot, K.; Gonzalez-Montiel, A.; Terrones, M.; Cavaillé, J.-Y. Efficient coating of N-doped carbon nanotubes with polystyrene using atomic transfer radical polymerization. Chem. Phys. Lett. 419, 2006, 567–573. [31) Kamalakaran, R.; Terrones, M.; Seeger, T.; Kohler-Redlich, Ph.; Rühle, M.; Kim, Y. A.; Hayashi, T.; Endo, M. Synthesis of Thick and Crystalline Nanotube Arrays by Spray Pyrolysis. Appl. Phys. Lett. 77, 2000, 3385–3387. [32] Terrones, M.; Kamalakaran, R.; Seeger, T.; Rühle, M. Novel nanoscale gas containers: encapsulation of N2 in CNx nanotubes. Chem. Commun. 2000, 2335–2336. 111 [33] Alvizo-Paez, E. R.; Romo-Herrera, J. M.; Terrones, H.; Terrones, M.; Ruiz-García, J.; Hernández-López, J. L. Soft purification of N-doped and undoped multiwall carbon nanotubes. Nanotechnology 19, 2008, 155701 (9 pp). [34] O’Neil, M. J. (Ed.) The Merck Index. 13th edition, Merck &Co., Inc., Whitehouse Station, NJ, 2001, p. 712. [35] Ewels, C.P.; Glerup, M. Nitrogen doped in carbon nanotubes. J. Nanosci. Nanotechnol. 5, 2005, 1345–1363. [36] Ayala, P.; Arenal, R.; Rümmeli, M.; Rubio, A.; Pichler, T. The doping of carbon nanotubes with nitrogen and their potential applications. Carbon 48, 2010, 575–586. [37] Czerw, R.; Terrones, M.; Charlier, J.-C.; Blase, X.; Foley, B.; Kamalakaran, R.; Grobert, N.; Terrones, H.; Tekleab, D.; Ajayan, P. M.; Blau, W.; Rühle, M.; Carroll, D. L. Identification of electron donor states in n-doped carbon nanotubes. Nano Lett. 1, 2001, 457–460. [38] Wiggings-Camacho, J. D.; Stevenson, K. J. Effect of nitrogen concentration on capacitance, density of states, electronic conductivity, and morphology of N-doped carbon nanotube electrodes. J. Phys. Chem. C 113, 2009, 19082–19090. [39] Nestorovic, G. D.; Jeremic, K. B.; Jovanovic, S. M. Kinetics of anline polymerization initiated with iron (III) chloride. J. Serb.Chem. Soc. 71, 2006, 895–904. [40] Niu, C.; Moy, D.; Ma, J.; Chishti, A. Modification of Nanotubes Oxidation with Peroxygen Compounds. U.S. Patent7, 070,753, July 4, 2006. [41] Dallas, P.; Stamopoulos, D.; Boukos, N.; Tzitzios, V.; Niarchos, D. Petridis, D. Characterization, magnetic and transport properties of polyaniline synthesized through interfacial polymerization. Polymer 48, 2007, 3162–3169. [42] Baibarac, M.; Baltog, I.; Lefrant, S.; Mevellec, J. Y.; Chauvet, O. Polyaniline and carbon nanotubes based compuestos containing whole units and fragments of nanotubes. Chem. Mater. 15, 2003, 4149–4156. [43] Li, Y. B.; Wei, B. Q.; Liang, J.; Yu, Q.; Wu, D. H. Transformation of carbon nanotubes to nanoparticles by ball milling process. Carbon 37, 1999, 493–497. [44] Hilding, J.; Grulke, E.A.; Zhang, Z.G. and Lockwood, F. Dispersion of Carbon Nanotubes in Liquids. J. Dispersion Sci. Techno. 24, 2003, 1-41. 112 4. Síntesis y procesado de nanocompuestos de PAni con distintos tipos de nanotubos de carbono, nanofibras de carbono y óxido de grafeno 113 114 4. Síntesis y procesado de nanocompuestos de PAni con distintos tipos de nanotubos de carbono, nanofibras de carbono y óxido de grafeno 4.1. ANTECEDENTES Como se comentó en la introducción, la polianilina (PAni) puede sintetizarse de forma nanoestructurada bajo ciertas condiciones. La polimerización interfacial de la anilina, permite una morfología nanofibrilar en la PAni [1-5], la cual, para aplicaciones electroquímicas, tales como supercondensadores [6, 7], ha resultado ideal por el incremento del área superficial del polímero que le ha permitido un mayor intercambio iónico con el medio [1]. A pesar de esta ventaja, aún no se han reportado la utilización de PAni nanoestructurada en otros dispositivos electroquímicos como actuadores o músculos artificiales [8-10]. Se decidió utilizar este método de síntesis porque se conjeturó que si se desarrollaban actuadores con este material nanofibrilar, podrían tener una mayor velocidad de accionamiento. Además, la nanoestructuración de la PAni no es obstáculo a su posterior disolución para obtener películas o fibras. Asimismo, a pesar de que el persulfato de amonio (APS) es el oxidante más comúnmente utilizado, se quiso conocer cómo afectaría en la morfología otro oxidante como el cloruro férrico (FeCl3), ya probado en la mecanosíntesis (Capítulo II). A partir del descubrimiento de los nanotubos de carbono (CNTs) [11] se ha reportado la obtención de compuestos de PAni utilizando nanotubos de capa simple (SWCNTs) y multicapa (MWCNTs) [12-17], lográndose mejorar las propiedades eléctricas de la PAni, y también sus propiedades mecánicas (después de ser procesada y conformada en fibras o películas [14, 18]). Una de las principales dificultades, como ha ocurrido al utilizar una amplia variedad de polímeros, ha sido la dispersión de los CNTs en la matriz polimérica [14-16], debido a su poca reactividad con los medios dispersantes. En parte se ha solucionado este problema al funcionalizar químicamente sus superficies [21, 22] o al modificar su estructura dopándolos con B o N [23-25], o exfoliando sus capas [26, 27]. Desde hace unos años, el abanico de elección de cargas carbonosas en compuestos poliméricos ha aumentado, desde el negro de carbono [28], pasando por las nanofibras [29] y nanotubos de carbono, hasta el óxido de grafeno [30] y el grafeno [31]. Cada una de ellas, de acuerdo a sus características, podría tener una aplicación concreta para algún polímero determinado. Por lo que uno de los objetivos principales de esta tesis, es 115 comparar el efecto de algunas de estas cargas en la PAni en la modalidad de actuador electro-químico-mecánico (Capítulo IV) o como electrodo para supercondensadores (Capítulo V). En la presente sección, se describe la síntesis y caracterización de compuestos de PAni, mediante la polimerización interfacial in situ, con diferentes nanoestructuras de carbono: nanotubos (CNTs), nanofibras de listones grafíticos helicoidales (HR-CNF) [32] y óxido de grafeno (GO). En cuanto a los CNTs se utilizaron diferentes tipos: nanotubos de carbono multicapa (MWCNTs), nanotubos de carbono dopados con nitrógeno (CNx) [33], nanotubos de carbono multicapa oxidados (COx) [34], nanotubos de carbono exfoliados (exMWCNTs) [26] y exMWCNTs funcionalizados con anilina (Ani-exMWCNTs). Asimismo, se prepararon muestras con MWCNTs y CNx con anclaje de nanopartículas de Ag para averiguar si éstas contribuían de alguna manera en las propiedades electromecánicas de los compuestos con PAni. Las nanoestructuras de carbono se dispersaron en la solución acuosa antes de llevar a cabo la polimerización. Pablo Jiménez y colaboradores [35] reportan que mediante la polimerización interfacial in situ obtuvieron malas dispersiones de sus cargas en la PAni, pero esto se debió a que sus reacciones las realizaron sin agitación. En el presente estudio, todas las síntesis de compuestos, se realizaron bajo agitación magnética. Cano-Márquez y colaboradores [26] obtuvieron exMWCNTs de la exfoliación de MWCNTs debido a la intercalación de Li-NH3 entre sus capas grafíticas, y posterior tratamiento térmico. Esta intercalación de Li ha sido utilizada para deshacer cuerdas de nanotubos de carbono de capa simple (SWCNTs) puesto que los nanotubos, dispersos en NH3 líquido forman “sales de nanotubos” con el Li, el cual transfiere electrones a los SWCNTs [36], por lo que pueden reaccionar con haluros de alquilo o de arilo [37]. Basados en los estudios de Cano-Márquez, se incluye en el presente trabajo una evaluación de algunas de las variables utilizadas en el proceso de exfoliación a partir de MWCNTs cortados (mediante sonicación en una mezcla de ácido concentrado de HNO3/H2SO4, reportado por Liu y colaboradores [38]), con el objetivo de optimizar el proceso y si fuera posible, incrementar la cantidad de nanolistones grafíticos (la cual fue ~50%). Estas estructuras, debido a sus bordes libres, son más reactivas que los MWCNTs por lo que podrían dispersarse mejor en la matriz polimérica. Asimismo, estos exMWCNTs se funcionalizaron con anilina (Ani-exMWCNTs), mediante una reacción de arilación 116 reductiva basada en el mismo método empleado (sales de nanotubos), con el objeto de mejorar aún más su interacción con la matriz polimérica. Los compuestos se caracterizaron principalmente con espectrografía Raman y microscopía electrónica de barrido (SEM) y transmisión (TEM). Mediante la difracción de rayos X, se corroboró la estructura semicristalina típica de la PAni en su estado conductor: sal de esmeraldina (ES). La conductividad eléctrica de los compuestos se midió mediante el método de las cuatro puntas, en pastillas preparadas con aglomerante (PAni dopada desde la síntesis), y en películas (PAni procesada). 4.2. EXPERIMENTACIÓN 4.2.1. Materiales Como reactivos de síntesis de polimerización y procesado de la PAni se utilizaron anilina (al 99.5% calidad ACS, de la casa Sigma-Aldrich), Cloruro Férrico Hexahidratado (calidad ACS de la casa Fermont, SA de CV), Amoniaco, litio (al 99%, de la casa Aldrich), Tetrahidrofurano (de la casa Tedia Company, EEUU), 4-yodoanilina (al 98%, de la casa Aldrich), acetato de polivinilo (de la empresa Resistol SA de CV), ácido clorhídrico, ácido nítrico, ácido sulfúrico, ácido metanosulfónico (MSA), ácido 2-acrilamido-2-metilpropanosulfónico (AMPSA) y ácido dodecil-benceno sulfónico (DBSA), nanofibras de listones grafíticos helicoidales (HR-CNF o GANF, Grupo Antolín) y óxido de grafeno (GO o GRAnPH, Grupo Antolín). Los nanotubos de carbono multicapa (MWCNTs) fueron producidos alimentando una corriente de ferroceno en tolueno (2.5% wt/vol), en un inerte, a un reactor de CVD para el crecimiento de los nanotubos en sustrato a 800ºC durante 30 min, según el método descrito por Kamalakaran y colaboradores [39]. Los nanotubos de carbono multicapa dopados con nitrógeno (CNx) fueron crecidos en un sistema idéntico, excepto que se alimentó un flujo de ferroceno en bencilamina (2.5% wt/vol) a 800ºC durante 30 min, según lo descrito por Terrones y colaboradores [33]. Igualmente, los nanotubos de carbono multicapa oxidados (COx) fueron sintetizados en un sistema similar, excepto que se preparó una solución de etanol en tolueno (5 % v/v), y se utilizó para alimentar una corriente de ferroceno en tolueno/etanol (2.5% wt/vol) a 800ºC durante 30 min, según lo descrito por BotelloMéndez y colaboradores [34]. Posteriormente, los CNTs se purificaron dispersándolos en agua, bajo reflujo, con una sonda de irradiación pulsada de ultrasonidos, seguido de un reflujo en HCl (6M) y filtración. Este proceso está basado en el método hidrotérmico de 117 Alvizo-Páez y colaboradores [40] para eliminar el carbono amorfo y facilitar la interfase del filamento y la matriz polimérica. Las nanofibras de listones grafíticos helicoidales (HR-CNF) y el óxido de grafeno (GO), fueron suministrados por Grupo Antolín Ingeniería. Se utilizó un homogeneizador, baño ultrasónico y horno cilíndrico Barnsted Thermolyne 21100, moldes de caucho de silicón (Poliformas, SA de CV), cabeza de cuatro puntas (Jeol, 1 mm entre puntas) y fuente de poder Keithley 2400, espectrógrafo Micro Raman Renishaw, microscopio electrónico de barrido SEM XL30 SFEG, microscopio electrónico de transmisión HRTEM 300KV FEI TECNAI F30 STWIN G2. 4.2.2. Obtención de los exMWCNTs y los Ani-exMWCNTs En el proceso de exfoliación de MWCNTs, Cano-Márquez y colaboradores [26], observaron que la apertura longitudinal de los nanotubos era de ~700 nm por lo que se conjeturó que cortándolos a esta longitud, se podría incrementar la eficiencia de nanolistones grafíticos a valores mayores que ~50%. Para obtener los MWCNTs cortados (cutMWCNTs) de ~700 nm de longitud, se sonicaron MWCNTs durante distintos tiempos, en una mezcla de ácido concentrado de H2SO4/HNO3. Posteriormente, aunado al corte específico de MWCNTs, se estudió el efecto de 4 variables en el proceso de exfoliación de cutMWCNTs mediante un diseño experimental ½2k (de “tamizado”) [41] y, finalmente, los MWCNTs exfoliados (exMWCNTs) se funcionalizaron con anilina mediante la arilación reductiva basada en el método de “sales de nanotubos” [36]. 4.2.3. Corte de MWCNTs 500 mg de MWCNTs se sonicaron en un baño ultrasónico durante 12, 18, 24, 48 y 72 horas en 500 mL de ácido concentrado de H2SO4:HNO3 (2:1, 2.5:1 y 3:1). Mediante una inspección visual de micrografías SEM y STEM, se obtuvo una estadística de longitudes y diámetros de los MWCNTs cortados (cutMWCNTs). 4.2.4. Intercalación de Li en los cutMWCNTs Este proceso se realizó en un matraz de bola de tres bocas (500 mL), en el cual, mediante un refrigerante de vidrio y acetona solidificada con nitrógeno líquido, se condensó amoniaco a –76°C. Se acumularon 350 mL de amoníaco (en una atmósfera de argón) y después se adicionó Li (100 o 1000 mg), adquiriendo el amoníaco un color azul oscuro 118 debido a la disolución del Li y a la solvatación de los electrones libres por los iones amonio. Después se adicionaron 100 mg de cutMWCNTs sonicados previamente en tetrahidrofurano (THF) seco (1 h). Se agitó la mezcla cinco veces durante dos minutos mediante el homogeneizador (DremelMultipro395). Al terminar el proceso y después de que el amoniaco se hubo evaporado, los cutMWCNTs-Li se lavaron con 100 mL de HCl al 15% (v/v) para remover el exceso de Li. Finalmente los cutMWCNTs-Li se lavaron y filtraron con metanol en una membrana de teflón con tamaño de poro de 0.45 μm bajo vacío. 4.2.5. Exfoliación de los cutMWCNTs-Li Los cutMWCNTs-Li se colocaron en un recipiente de porcelana dentro de un tubo de cuarzo con un flujo de argón para aplicarles choques térmicos de ~1000ºC (Figura 1), tal y como lo reportó Cano-Márquez y colaboradores [26]. Para evaluar el efecto de algunas variables que se consideraron importantes en el proceso, se utilizó un diseño experimental de tamizado ½2K para cuatro factores: relación de Li:cutMWCNTs (10:1 y 1:1), temperatura del tratamiento térmico (800 y 1000°C), duración del pulso de temperatura (1 y 5 min), número de tratamientos térmicos (1 y 5). En cada una de las ocho corridas experimentales se utilizaron 25 mg de cutMWCNTs-Li. Los cutMWCNTs exfoliados (exMWCNTs) se caracterizaroncon el microscopio electrónico de transmisión (HRTEM 300KV FEI TECNAI F30 STWIN G2). Figura 1. Esquema del sistema de tratamiento térmico para la exfoliación de cutMWCNTs con intercalación de Li. 119 4.2.6. Funcionalización de los exMWCNTs con anilina Los exMWCNTs se funcionalizaron con anilina, utilizando la reacción de sales de nanotubos [36]. Se utilizó prácticamente el mismo procedimiento de intercalación de Li. La única diferencia es que después de adicionar 25 mg de exMWCNTs al reactor, con la disolución de Li-amoniaco, se adicionaron 125 mg de yodoanilina. Al finalizar la reacción y después de que el amoniaco se hubo evaporado, los exMWCNTs funcionalizados con anilina (Ani-exMWCNTs) se lavaron en 100 mL de HCl al 15% (v/v) para quitar el exceso de Li y posteriormente con cloroformo para remover el exceso de yodoanilina. Los AniexMWCNTs se lavaron y filtraron con metanol en una membrana de teflón (microporo de 0.45 μm). 4.3. Síntesis de compuestos de PAni Debido a que la selección de las mejores nanoestructuras de carbono en compuestos de PAni (con un enfoque a actuadores) fue realizándose continuamente a través de distintas síntesis, y no en una sola síntesis total, se describirán los dos procesos principales que se realizaron. Ambos, basados en la polimerización interfacial in situ, la cual, como se vio en el Capítulo I, consiste en una fase orgánica (donde se disuelve la anilina) y otra acuosa (pH<2.0 y conteniendo al oxidante). La reacción ocurre en la interfase. 4.3.1. Proceso 1 Los nanocompuestos de PAni se sintetizaron con las siguientes nanoestructuras: MWCNTs, CNx, MWCNTs-Ag, CNx-Ag, exMWCNTs y Ani-exMWCNTs. Inicialmente, se dispersaron 20 mg de cargas de carbono en 180 mL de agua en baño de ultrasonidos durante 3 h. Se añadió 20 mL de HCl para obtener la concentración de 10% (v/v). Esta mezcla constituyó la fase acuosa. La fase orgánica consistió de 100 mL de tolueno con 7.86 mL de anilina. Poco antes de iniciar la reacción, se adicionaron 8 g de oxidante (FeCl3·6H2O) a la solución acuosa. La relación molar oxidante:anilina fue de 1:1. La duración de la reacción fue de 3 h bajo agitación magnética. Posteriormente, se produjeron compuestos de PAni con MWCNTs, MWCNTs-Ag, CNx, CNx-Ag, COx y Ani-exMWCNTs. En esta síntesis se dispersaron 10 y 30 mg de estas cargas en 270 mL de agua en un baño de ultrasonidos durante 3 h. Se añadió 30 mL de HCl para obtener una concentración de 10% (v/v). La fase orgánica fue de 100 mL de tolueno con 3.9 mL de anilina. Antes de iniciar la reacción, se adicionaron 4.9 g de persulfato de 120 amonio [(NH4)2S2O8] a la solución acuosa. La relación molar oxidante:anilina en este caso de fue de 2:1. Las muestras se lavaron y filtraron con metanol y agua bidestilada utilizando una membrana de teflón (poro de 0.45 µm). Los compuestos se guardaron húmedos para preparar pastillas a las cuales se les pudiera medir la conductividad eléctrica mediante el método de las cuatro puntas. Las probetas se prepararon tomando 70 mg de compuesto de PAni, se les adicionó 0.05 mL de una suspensión de aglutinante de acetato de polivinilo en agua (PVA) al 5% (v/v). Con la pasta obtenida se rellenaron discos en bajorrelieve de un molde de caucho (Figura 2 A) y se secaron en el horno (50ºC, 4 h). Al deshidratarse el material (Figura 2 B), se obtuvieron pastillas de 48 mm de diámetro por 1 mm de espesor. En la Figura 2 C se observa una muestra de compuesto de PAni a punto de medírsele su conductividad eléctrica mediante el método de las cuatro puntas (0.5 mA, 1 mm de distancia entre puntas). Las mediciones se realizaron por septuplicado. Figura 2. (A) y (B): Preparación de pastillas de compuestos de PAni en un molde de caucho; C) muestra de compuesto de PAni a punto de medírsele la conductividad eléctrica mediante el método de las cuatro puntas. 4.3.2. Proceso 2 En este apartado se describe la síntesis de PAni con distintos ácidos dopantes; la síntesis de compuestos de PAni con nanofilamentos de carbono como: MWCNTs, CNx, HR-CNF y también con GO, utilizando dos niveles de carga; la síntesis de compuestos de PAni con MWCNTs o GO con 5 niveles de carga; y la síntesis de compuestos de PAni con una carga híbrida de GO-MWCNTs. 121 4.3.2.1. Síntesis de PAni con distintos ácidos Se sintetizó PAni utilizando como solución acuosa HCl, ácido metanosulfónico (MSA), ácido 2-acrilamido-2-metilpropano-sulfónico (AMPSA) y ácido dodecil-benceno sulfónico (DBSA), todos a un pH=1.0 y también utilizando agua a un pH=7.0. 4.3.2.2. Síntesis de compuestos con nanofilamentos de carbono y GO 10 y 30 mg de MWCNTs, CNx, HR-CNF y GO se dispersaron en 135 mL de agua. En las tres primeras nanoestructuras se utilizó un homogeneizador (Silverson) a 8000 rpm (8 min), el GO se dispersó con una punta ultrasónica (pulsos de 40% de potencia, 3 h). Para la acidificación se adicionó 15 mL de HCl concentrado. La fase orgánica consistió en 50 mL de tolueno junto con 1.95 mL de anilina. Se adicionó 2.45 g de persulfato de amonio como oxidante a la solución acuosa con las cargas dispersas, y, junto a la orgánica reaccionaron durante 3 h bajo agitación magnética. La relación molar oxidante:anilina fue de 2:1. El lavado y filtración fue idéntico a los anteriores procesos aunque antes de recuperar las muestras de la membrana, se desdoparon adicionando 40 mL de NaOH 0.2 M, posteriormente, con lavados de agua, se disminuyó el pH hasta ~7.0. Las muestras se secaron en el horno (70º, 4h). Se disolvieron las muestras en N-metil-2-pirrolidona (NMP) (10% w/v) y mediante un casting sobre un sustrato de vidrio se obtuvieron películas de compuestos de ~50 μm de espesor. Por último, se redoparon con HCl o MSA (1 M) y se les midió la conductividad eléctrica mediante el método de las cuatro puntas (0.5 mA, 0.7 mm de distancia entre puntas). Se realizaron pruebas termomecánicas a tiras de películas de PAni y sus compuestos (50 μm de espesor, 1.6 mm de ancho) a tensión constante de 0.1 N (esfuerzo inicial de 1.25 MPa) en un rango de temperatura de 30 a 380º C (5º C/min) en un ambiente de argón. Se utilizó un equipo TMA (Thermo-Mecanical Analysis) de la marca TA Instruments Q400. 4.3.2.3. Síntesis de compuestos de PAni con MWCNTs y GO con varios niveles de carga. Se prepararon compuestos de PAni con MWCNTs o con GO. Se dispersaron 4.7, 8, 16, 24 y 33.9 mg de MWCNTs o GO en 135 mL de agua, utilizando el homogeneizador para la primera carga (8000 rpm, 8 min) y la punta ultrasónica (pulsos de 40% de potencia, 3 h) para la segunda. A las dispersiones se adicionó 15 mL de HCl concentrado para obtener HCl (10% v/v). La fase orgánica consistió en 50 mL de tolueno junto con 1.95 mL de anilina. Se adicionó 2.45 g de persulfato de amonio como oxidante a la solución acuosa (la 122 relación molar oxidante:anilina fue de 2:1). La reacción tuvo una duración de 3 h bajo agitación magnética. El lavado y filtración y desdopaje de la PAni fue idéntico a los anteriores procesos. Las muestras se secaron en el horno (70º, 4h). Se disolvieron las muestras en N-metil-2-pirrolidona (NMP) (10% w/v) y mediante un casting sobre un sustrato de vidrio se obtuvieron películas de compuestos de ~50 μm de espesor. 4.3.2.4. Síntesis de compuestos de PAni con carga híbrida de GOMWCNTs. Por último, se dispersaron 4.1, 6.2. 8.3 y 12.4 mg de GO en 135 mL de agua mediante una punta ultrasónica (pulsos de 40% de potencia, 3 h). En las dispersiones de 4.1, 6.2 y 8.3 mg de GO, se adicionó 8.3, 6.2 y 4.1 mg de MWCNTs, respectivamente, además, en 135 mL de agua sin GO se adicionaron 12.4 mg de MWCNTs. Estas mezclas se dispersaron utilizando un homogeneizador (Silverson) a 8000 rpm (8 min). Las dispersiones de GOMWCNTs quedaron como sigue: 1:2, 1:1 y 2:1. Para la acidificación se adicionó 15 mL de HCl concentrado. La fase orgánica consistió en 50 mL de tolueno junto con 1.95 mL de anilina. Se adicionó 2.45 g de persulfato de amonio como oxidante a la solución acuosa con las cargas dispersas, y, junto a la orgánica reaccionaron durante 3 h bajo agitación magnética. La relación molar oxidante:anilina fue de 2:1. El lavado, filtración y desdopaje de la PAni fue idéntico a lo anteriormente descrito. Las muestras se secaron en el horno (70º C, 4h). Se obtuvieron compuestos de PAni/MWCNTs (~1.3 wt%), PAni/GO (~1.3 wt%), e híbridos de PAni/GO/MWCNTs (~1.3 wt%) con una relación GO:MWCNTs de 1:2, 1:1 y 2:1. 4.4. RESULTADOS 4.4.1. Corte de MWCNTs Después de sonicar 500 mg de MWCNTs en 500 mL de ácido concentrado de H2SO4:HNO3 (2:1, 2.5:1 y 3:1) durante periodos de 12, 18, 24, 48 y 72 horas, se obtuvieron rendimientos (Tabla 1) desde ~0.8 (72 h de sonicación, 3:1) hasta 69% (18 h, 2:1). Se obtuvo una estadística de longitudes y diámetros de los MWCNTs cortados (cutMWCNTs) mediante una inspección visual de micrografías SEM y STEM de los materiales. En la Figura 3 se muestran micrografías SEM de cutMWCNTs con diferentes tiempos de sonicación. Las longitudes obtenidas variaron de ~530 (12 h, 3:1) hasta ~2560 nm (18 h, 2:1) (Tabla 1). 123 Una mayor cantidad de H2SO4 favoreció una menor longitud de los nanotubos. No se apreció diferencia significativa entre los cutMWCNTs obtenidos de los procesos de 12 h (a) y 24 h, en donde la diferencia en tiempo fue compensada por la relación de ácidos (mayor cantidad de H2SO4 en el primer proceso). El rendimiento de 36.2% para el proceso de 24 h fue ligeramente mayor que el de 12 h (a) (36.2%). Tabla 1. Corte de MWNT en H2SO4:HNO3 concentrado Tiempo de Relación de Producto Rendimiento Longitud sonicación (h) H2SO4:HNO3 obtenido (%) promedio (μm) (mg) 124 72 3:1 4 0.8 - 48 2:1 42 8.4 0.49±0.33 24 2:1 181 36.2 0.72±0.31 18 2:1 345 69 2.56±2.5 12 (a) 3:1 157 31.4 0.53±0.4 12 (b) 2.5:1 166 33.2 0.71±0.35 Figura 3. Micrografías SEM de MWCNTs cortados (cutMWCNTs) en sonicación ácida de H2SO4:HNO3 durante (A) 48 h (2:1), (B) 24 h (2:1), (C) 18 h (2:1), (D) 12 (a) h (3:1) y (E) 12 (b) h (2.5:1). Las bandas Raman D (de “desorden” de hibridación sp3), G (“grafítico” de hibridación sp2) y 2D (segundo armónico de la banda D) (Figura 4-I) para cutMWCNTs con tiempos de sonicación de 24 (A) y 48 h (B), y MWCNTs (C), se ubicaron en 1336, 1583 y 2661 cm-1, respectivamente. Las razones ID/IG para A, B y C fueron, 1.424, 1.499, y 0.595, respectivamente. El incremento tan evidente de la razón ID/IG de los cutMWCNTs indica que los tratamientos dañaron significativamente a los MWCNTs, sobre todo en el de 48 h de sonicación. Los respectivos difractogramas (Figura 4-II) de cutMWCNTs sonicados en ácido concentrado durante 24 (A) y 48 h (B), y de MWCNTs sin tratamiento (C) muestran el pico principal entre 25.5 y 26° (002) que corresponde a la estructura grafítica de los 125 CNTs. Los picos en ~43°, corresponden al contenido de Fe catalítico que originaron el crecimiento de los MWCNTs. Como puede observarse en los difractogramas, el pico principal de las muestras tratadas (Figura 4-II A, B) se ensancha ligeramente con respecto al pico de los MWCNTs sin tratamiento (C). Esto fue debido a que el tamaño de los MWCNTs disminuyó conforme se aumentó el tiempo de sonicación en ácido concentrado. Figura 4. (I) Espectrogramas Raman de cutMWCNTs obtenidos de la sonicación ácida (H2SO4:HNO3 de 2:1) de (A) 24 horas, (B) 48 horas y (C) MWCNTs sin tratamiento. (II) Difractogramas de MWCNTs obtenidos de la sonicación de (A) 24 h, (B) 48 h y MWCNTs sin tratamiento. 4.4.2. Exfoliación de cutMWCNTs De acuerdo a los resultados estadísticos de la longitud de los cutMWCNTs, se decidió exfoliar los cutMWCNTs provenientes de la sonicación en H2SO4:HNO3 de 2:1, (24 h), y los provenientes de la sonicación en H2SO4:HNO3 de 2.5:1, (12 h) ya que ambos tuvieron una longitud promedio de ~700 nm. En una prueba preliminar, antes de comenzar el experimento ½2K (de “tamizado”), se les intercaló Li a 100 mg de cutMWCNTs (mediante el método descrito en la sección experimental). El peso de la muestra se incrementó a 141 mg. Al someter a los exMWCNTs-Li al tratamiento térmico de 1000º C durante 5 min (Figura 5 A), su peso disminuyó a 60 mg. Al parecer, independientemente de las pérdidas debido a la manipulación, la mayor parte del peso de la muestra se perdió debido al tratamiento 126 térmico. Residuos del material expulsado durante el choque térmico puede observarse en la Figura 5 B. Figura 5. (A) Colocación de los cutMWCNTs-Li en una bandeja de porcelana dentro de un tubo de cuarzo, poco antes de ser exfoliados mediante un choque térmico de 1000°C en un flujo de argón. (B) Residuos que se desprendieron durante la exfoliación. Basándose en el diseño experimental ½2K, se evaluaron los 4 factores (2 niveles) siguientes: Razón en peso de Li/cutMWCNTs (1, 10), temperatura del tratamiento térmico (800, 1000ºC), duración de tratamiento térmico (1, 5 minutos) y el número de tratamientos térmicos (1, 5). El objetivo fue identificar a los factores con mayor efecto en la exfoliación de MWCNTs. Por cada corrida experimental (Tabla 2) se utilizaron 25 mg de cutMWCNTs-Li. Las respuestas experimentales de este diseño: peso de exMWCNTs y eficiencia de exfoliación (%exMWCNTs), se estimaron por gravimetría y mediante el análisis morfológico (microscopía SEM y STEM) (Figura 6 A, B). Un análisis más minucioso de la morfología de las hojas de grafeno y nanotubos abiertos se realizó por HRTEM (Figura 6 C, D). Debido a pérdidas por manipulación de las muestras, no se tomó en cuenta su peso ni su posible correlación con el %exMWCNTs. 127 Tabla 2. Diseño experimental ½2K para cuatro factores y sus respuestas experimentales. Corrida Factores a evaluar Respuestas TT t Li/C Temp Peso %exMWCNTs (tratamientos (tiempo) (Li/cutMWCNTs) (temperatura) (mg) térmicos) (min) 1 1 1 1 800 16.25 27.7 2 5 1 1 1000 18.2 55.3 3 1 5 1 1000 8.74 25.5 4 5 5 1 800 11.76 27.1 5 1 1 10 1000 15.77 22 6 5 1 10 800 12.66 23.4 7 1 5 10 800 12.88 7.22 8 5 5 10 1000 13.06 39.33 (°C) El modelo de regresión de la eficiencia de exfoliación (%exMWCNTs), fue %exMWCNTs = 28.44 + 7.84(TT) – 3.66(t) – 5.46(Li/C) + 7.09(Temp) + 0.59(TT)(t) + 0.54(TT)(Li/C) + 3.95(TT)(Temp) (1) donde TT es el número de tratamientos térmicos, t es la duración del tratamiento térmico, Li/C es la razón entre la cantidad de Li y los cutMWCNTs (utilizada en la reacción de intercalación de Li), y Temp es la temperatura del tratamiento térmico. El primer término después del signo igual de la Ecuación 1, 28.44, es la media del %exMWCNTs. El coeficiente de cada término cuantifica el efecto que tuvo dicho factor (la codificación de los factores establece que éstos tendrán valor de -1 para el nivel bajo y +1 para el alto) o interacción en %exMWCNTs. Como se observa en la ecuación, el factor que más efecto tuvo fue el número de tratamientos térmicos (TT), la exfoliación aumenta cuando se utilizan 5. El otro factor que más efecto tuvo, fue la temperatura del tratamiento térmico (Temp): la exfoliación aumenta con 1000°C. La razón Li/cutMWCNTs (Li/C), contribuyó de manera inversa (por el signo negativo), es decir, se requirió de una relación Li/cutMWCNTs de 1, en la reacción de intercalación de Li, para obtener una mayor exfoliación de nanotubos. En cuanto a la duración del tratamiento térmico (t), es preferible utilizar el nivel bajo: 1 min. 128 Por otra parte, el coeficiente de la interacción (TT)(Temp) indica que para que exista una mayor exfoliación, se requiere que estos dos factores estén a un nivel alto o a un nivel bajo, siendo correcto el primer caso por tener un sentido físico. Por lo tanto, bajo estas condiciones experimentales, para obtener una mayor exfoliación de nanotubos, se necesita una temperatura de 1000°C, una relación Li/cutMWCNTs de 1, cinco tratamientos térmicos con duración de 1 min cada uno. Como conclusión parcial, se llegó prácticamente al mismo resultado de eficiencia de más de 50% en la exfoliación de MWCNTs, a la que Abraham-Cano y colaboradores llegaron. Ellos utilizaron 5 pulsos de temperatura (1 min) de 1000ºC cada uno, aunque con una cantidad 10 veces mayor de Li en la reacción de intercalación. Un par de micrografías SEM/STEM (Figura 6 A, B) muestra MWCNTs parcialmente abiertos (exMWCNTs) junto con algunas hojas de grafeno en el fondo. En las micrografías HRTEM (Figura 6 C, D) se observan algunas hojas de grafeno apiladas y algunas otras desprendidas de los exMWCNTs. En general, los exMWCNTs se observan con una significativa cantidad de bordes reactivos. Figura 6. Micrografías SEM (A), STEM (B) y HRTEM (C, D) de los exMWCNTs. En las micrografías A y B además de los MWCNTs, con aberturas en algunos puntos a lo largo de su longitud (exMWCNTs), se aprecian algunas hojas de grafeno. En las micrografías C y D también se observan hojas de grafeno apiladas y desprendidas de los nanotubos. 129 La razón ID/IG del espectrograma Raman es un estimador del nivel de cristalinidad de las estructuras de carbono, que se obtiene de las intensidades de los picos en las bandas D (hibridación sp3) y G (hibridación sp2). Estas bandas se localizaron en 1334 cm-1 y 1582 cm-1, respectivamente (Figura 7-I). Las muestras que mostraron una menor cristalinidad fueron la 5/5/1/800 y la 5/5/10/1000 (ID/IG=1.93 y 1.82, respectivamente) que fueron sometidas al tratamiento térmico más largo, de cinco pulsos (5 min) cada uno (como puede verse en su nomenclatura). En cambio, las muestras 5/1/10/800 y 1/5/10/800 (ID/IG=1.50 y 1.46, respectivamente) fueron las que casi mantuvieron el nivel de cristalinidad de los cutMWCNTs (ID/IG ~1.5) (ver Figura 4). A estas muestras se les trató con un choque térmico de 800°C, una sola vez (5 min) para la primera, y cinco veces (1 min) para la segunda. El resto de las razones ID/IG fluctuaron entre estos valores. Prácticamente no hubo corrimiento en las bandas D, G y 2D (2663 cm-1). Por otra parte, el pico principal de los difractogramas de las muestras (Figura 7-II) se ubicó en 2θ=25.4° (002) que corresponde a la estructura grafítica de los CNTs. Al comparar el ancho de estos picos, se observa que el de las muestras 1/1/1/800 y 1/1/10/1000 son los más estrechos, indicando que están conformadas por cristales de exMWCNTs más grandes. Éstas se prepararon con un solo choque térmico (800º C para la primera y 1000º C para la segunda, 1 min). En cambio, los difractogramas de las muestras 1/5/1/1000 y 5/5/10/1000, con sus picos más anchos, mostraron que fueron los que tuvieron una menor cristalinidad. Estas muestras se sometieron a 1 pulso de 1000°C (5 min), la primera, y 5 pulsos de 1000ºC (5 min) la segunda. Estos resultados en la estructura de los exMWCNTs y su cristalinidad apoyan hasta cierto punto lo encontrado en la eficiencia de exfoliación en cuanto a que una mayor temperatura y un mayor número de pulsos, tienden a incrementarla. Por ser estas evaluaciones un experimento de “tamizado”, no se obtiene la suficiente información que permita correlacionar la duración de los pulsos y la cantidad de Li en el medio de reacción de intercalación. 130 Figura 7. Perfiles Raman (I) y de XRD (II) de las 8 muestras obtenidas en el experimento ½2K (Tabla 2). Nomenclatura: TT/t/Li:C/Temp donde TT es el número de tratamientos térmicos, t la duración del tratamiento térmico (min), Li/C la razón entre las cantidades de Li y exMWCNTs, y Temp es la temperatura del tratamiento térmico (º C). 4.4.3. Funcionalización de los cutMWCNTs con anilina Los exMWCNTs se funcionalizaron con anilina mediante la reacción de “sales de nanotubos” [37] en la que los iones Li, intercalados en los exMWCNTs, fueron sustituidos por iones de anilina provenientes de la yodoanilina disuelta en amoníaco líquido (Figura 8). De 25 mg de exMWCNTs utilizados en la reacción se recuperaron 23 mg al finalizarla. El efecto de la funcionalización se observó directamente de su compuesto con PAni, como se verá más adelante. 131 Figura 8. Esquema de la sustitución de los iones de anilina por los de Li en la reacción de sales de nanotubos en amoníaco líquido (reacción de arilación reductiva). 4.4.4. Síntesis de nanocompuestos de PAni 4.4.4.1. Proceso 1 Compuestos de PAni (~2.3 wt%) con: MWCNTs, CNx, MWCNTs-Ag, CNx-Ag, exMWCNTs y Ani-exMWCNTs fueron obtenidos de la polimerización interfacial in situ (20 mg de cada carga). La eficiencia de polimerización fue de ~11% utilizando al FeCl3 como oxidante. En cambio, el rendimiento polimérico al utilizar persulfato de amonio (APS) fue mayor, ~31%, debido a su mayor poder de oxidación y a la mayor razón molar oxidante:anilina utilizada (2:1). En este último caso se obtuvieron compuestos de PAni (~0.16 o ~0.48 wt%) con 10 o 30 mg de: MWCNTs, MWCNTs-Ag, CNx, CNx-Ag, COx, y Ani-exMWCNTs. Las micrografías SEM más representativas de estos compuestos (Figuras 9-11) se colocaron en pares (cuando los hubo) que consistieron en imágenes de los compuestos polimerizados utilizando cloruro férrico (FeCl3) y persulfato de amonio (APS), para poder apreciar mejor la diferencia morfológica en el compuesto debido al uso de uno u otro oxidante. La imagen SEM de la PAni sin carga polimerizada con FeCl3 (Figura 9 A1) muestra una morfología irregular que posiblemente comenzó siendo nanofibrilar pero después las fibras engruesaron hasta casi unirse entre ellas. Este fenómeno podría explicarse debido a la agitación magnética, ya que permite el crecimiento secundario de la PAni en las fibras previamente formadas [42]. En cambio, la morfología de la PAni sintetizada con APS, fue más nanofibrilar (Figura 9 A2). 132 En cuanto a la morfología de compuestos con MWCNTs, preparados con FeCl 3 (Figura 9 B1) se observa un recubrimiento polimérico parcial en los CNTs y también regiones donde predomina más la PAni o los CNTs (prácticamente sin daño), indicando que hubo una mala dispersión. En el compuesto sintetizado con APS (Figura 9 B2), la morfología fue más similar a la de la PAni sin carga, posiblemente debido a la menor cantidad de nanotubos en la matriz (~0.48 wt%), aunque pueden apreciarse algunas puntas sobresaliendo de los extremos de las fibras, las cuales podrían ser extremos de MWCNTs. Figura 9. Micrografías SEM de PAni, y sus compuestos sintetizados, con FeCl 3 o persulfato de amonio (APS) como oxidantes. A1) PAni sintetizada con APS, A2) PAni sintetizada con FeCl3, B1) PAni/MWCNTs producida con FeCl3, y B2) PAni/MWCNTs sintetizada con APS. Los compuestos con CNx preparados con FeCl3 (Figura 10 A1), mostraron una morfología similar a la de los compuestos con MWCNTs, se observan nanotubos recubiertos de PAni y también regiones donde predominan los CNx y otras donde lo hace el polímero. En el caso de los compuestos con CNx sintetizados con APS (Figura 10 A2), su morfología fue más homogénea puesto que fue difícil encontrar un nanotubo, o una parte de éste, no 133 recubierto o aglomerado con otros, y a pesar de su parecido con el compuesto equivalente con MWCNTs, fue difícil encontrar puntas o extremos de CNx sobresaliendo de las fibras de PAni. Esto posiblemente sucedió debido a la mayor reactividad que tienen estos nanotubos [43, 44]. Los compuestos con MWCNTs y CNx con anclaje de Ag mostraron una morfología idéntica a los CNTs sin Ag (no se incluye aquí). Debido a que la Ag en los CNTs no mostró efecto alguno en la morfología, estructura y conductividad eléctrica (como se verá más adelante) del compuesto, se decidió descartar su utilización en subsecuentes experimentos. Una morfología más peculiar se observa en el compuesto de PAni/COx (Figura 10 B), preparado con APS como oxidante, en donde el polímero rodeó homogéneamente a los nanotubos hasta llegar a formar fibras de más de 200 nm de diámetro. Algunos extremos de COx aparecen sin polímero. La interacción superior entre los COx y la PAni, obedeció a la funcionalización de su superficie con grupos basados en oxígeno, que posiblemente permitió un anclaje radial de las cadenas poliméricas. A pesar de esto, en algunas regiones se vieron solamente agregados irregulares de PAni, indicando una mala dispersión. La micrografía del compuesto PAni/exMWCNTs, sintetizado con FeCl3 (Figura 10 C) mostró una mejor interacción de la carga con la PAni que entre los MWCNTs y la PAni (ver Figura 9 B1), e independientemente de la menor longitud de los exMWCNTs, no se apreciaron agregados. 134 Figura 10. Micrografías SEM de compuestos de PAni. (A1) PAni/CNx sintetizado con FeCl3, (A2) PAni/CNx sintetizado con APS, (B) PAni/COx sintetizado con APS, (C) PAni/exMWCNTs sintetizado con FeCl3. Una mejor interacción aún, entre carga/matriz polimérica, se observó en los compuestos PAni/Ani-exMWCNT, sintetizada con FeCl3 y con APS (Figura 11 A, B) en donde una homogeneidad morfológica alta se apreció en todas sus micrografías, indicando que la PAni rodeó totalmente a estas nanoestructuras (más visible aún en la A, donde el contenido de carga, ~2.3 %, fue mayor que la B, ~0.48%). Las imágenes HRTEM del compuesto preparado con FeCl3 confirman lo observado (Figura 11 C, D), las cuales muestran a los exMWCNT totalmente rodeados de polímero. 135 Figura 11. Micrografías SEM y HERTEM de compuestos de PAni/Ani-exMWCNTs. (A) SEM de compuesto preparado con FeCl3, (B) SEM de compuesto preparado con APS. (C, D) HRTEM del compuesto preparado con FeCl3. 4.4.4.2. Proceso 2 4.4.4.2.1. Síntesis de PAni con distintos ácidos El rendimiento de la PAni sintetizada con HCl, ácido metanosulfónico (MSA), ácido 2acrilamido-2-metilpropano-sulfónico (AMPSA), ácido dodecil-benceno sulfónico (DBSA) y sólo con agua, se muestra en la Tabla 3. El mayor rendimiento se obtuvo con HCl (48.6%), con MSA fue ligeramente inferior (46%). En cambio con el AMPSA (22.6%), y sobre todo con el DBSA (3%), el rendimiento fue bajo. Con un pH neutro se obtuvo un rendimiento de 5.1%, y por el tono pardo claro de este producto (Figura 12 D), al parecer se conformó sólo de oligoanilinas, ya que, como se ha reportado [45], con un pH arriba de 4 se obtienen cadenas poliméricas muy cortas. En el caso de la PAni obtenida con DBSA y AMPSA (Figura 12 A, B), el color negro indicaría que se obtuvo pernigranilina: una sal de PAni altamente oxidada [46]. En cambio, el color de la PAni obtenida con HCl y MSA fue verde oscuro (Figura 12 C), el tono típico de la sal de esmeraldina. Por lo tanto, se decidió 136 utilizar HCl para síntesis posteriores ya que fue con el que se obtuvo la mayor eficiencia polimérica. La PAni (obtenida de la síntesis con HCl) se desdopó con NaOH 0.2 M. El color de la PAni pasó de un verde oscuro a un color azul oscuro, típico de la PAni en estado base de esmeraldina. El polímero se disolvió en N-metil-2-pirrolidona (NMP) (10% w/v) y se obtuvieron películas de PAni (y también sus compuestos) mediante “drop-casting” en un sustrato de vidrio (retirándose el disolvente bajo vacío a 70ºC). Se ha reportado que, a pesar de la evaporación del NMP de la película, de un 15 a un 25% de NMP residual, permanece en la PAni como plastificante [47, 48]. Los espectrogramas Raman de PAni sintetizada con distintos ácidos (Figura 13 d, e, f, g) no muestran diferencias significativas. El único pico bien definido es el de la banda de 1366 cm-1, aunque es tan amplio que sin duda se superpone al pico que indica la presencia de la estructura bipolarónica de la PAni (~1336 cm-1), típica en la sal de esmeraldina. Este pico es un tanto más ancho en el perfil de la PAni sintetizada con HCl (Figura 13 d). El espectrograma correspondiente a la PAni sintetizada a un pH=7.0 muestra el mismo pico (1366 cm-1) más intenso y estrecho (Figura 13 c). También resalta el pico en 1590 cm 1 (tensión C-C) del anillo benzenoide y se muestra un poco oculto el pico en 1481 cm-1 (tensión C-N) del anillo quinoide. Los perfiles de PAni desdopada (Figura 13 b) y plastificada con NMP (13 a) no muestran el pico en 1366 cm-1, pero sí el de 1165 (deformación C-H en el plano) y 1481 cm-1 (tensión C-N) del anillo quinoide, y también en 1590 cm-1 (tensión C-C) del anillo benzenoide. Como cabría de esperarse, estos picos son menos intensos en la PAni plastificada con NMP (Figura 13 a). Tabla 3. Producto y rendimiento de la polimerización interfacial de anilina (2 g en 50 mL de tolueno), 100 mL de solución acuosa, razón molar oxidante:anilina de 2:1, tiempo de reacción: 3 h. Ácido dopante Producto (mg) Rendimiento (%) HCl 973 mg 48.6 MSA 920 mg 46.0 AMPSA 451 22.6 (pH=1.0) 137 DBSA 59 3.0 Agua (pH=7.0) 102 5.1 Figura 12. PAni obtenida mediante la polimerización interfacial utilizando distintos ácidos como DBSA (A), AMPSA (B), HCl y MSA (C), y sin ácido (D). 138 Figura 13. Espectrogramas Raman de PAni: (a) desdopada y plastificada con NMP, (b) PAni desdopada con NaOH 0.2 M, (c) PAni sintetizada a un pH neutro, y PAni sintetizada utilizando: (d) HCl, (e) ácido metasulfónico (MSA), (f) ácido 2-acrilamido-2metilpropano-sulfónico (AMPSA) y (g) ácido dodecil-benceno sulfónico (DBSA). 4.4.4.2.2. Síntesis de compuestos con nanofilamentos de carbono y GO Los compuestos tuvieron un rendimiento polimérico de ~45.5% en estado de sal de esmeraldina. Al desdoparse la PAni con NaOH 0.2 M, se perdió casi ¼ de su peso (debido a la expulsión de los iones Cl- de sus cadenas poliméricas). Se sintetizaron compuestos (~1.1 y ~3.3 wt%, con respecto a sal de esmeraldina) de PAni con 10 y 30 mg de MWCNTs, CNx, HR-CNF y GO. El homogeneizador permitió una mejor dispersión de los CNTs en la PAni, al compararla con la obtenida en el Proceso 1, aunque en detrimento de su longitud (Figuras 14 C, D, y 15 C, D). La mejor dispersión (sobre todo con los MWCNTs) incrementó la interacción entre los CNTs y la PAni, como se verá más adelante. 139 Las micrografías SEM de los compuestos sin procesar (micrografías A de las Figuras 1417) muestra una morfología similar observada en los compuestos del Proceso 1, sintetizados con APS (como en este caso). También algunos extremos de MWCNTs (Figura 14 A) sobresalieron del polímero, y la interacción de los CN x (Figura 15 A) con la PAni pareció superior a la que tuvieron con los MWCNTs. En cuanto al compuesto PAni/HR-CNF (Figura 16 A), su morfología es fibrilar aunque heterogénea en longitudes y diámetros de las fibras (de hecho los HR-CNF son de por sí muy heterogéneos en sus diámetros). Es posible que esta heterogeneidad haya sido debido al aglomeramiento de HR-CNF (recubiertos de PAni) como puede verificarse en las micrografías TEM de este composite (Figura 16 C y D) donde se puede apreciar una mala dispersión. La morfología del compuesto de PAni/GO (Figura 17 A) muestra una morfología fibrilar aunque también pueden observarse fragmentos aplanados en los que, posiblemente en su interior, haya láminas relativamente grandes de GO. Los grupos funcionales del GO sin duda permitieron una elevada interacción con las cadenas poliméricas de la PAni, tal y como ocurrió con los COx. En las micrografías B de la Figuras 14-17 se observa la morfología de las películas (~50 μm de espesor) de compuestos procesados mediante el casting de una disolución con NMP (10% w/v). El compuesto con MWCNTs (Figura 14 B) mostró algunas de sus puntas ligeramente sobresaliendo de la película, y como puede corroborarse con las micrografías TEM (Figura 14 C y D), su dispersión fue relativamente buena. En el caso del compuesto con CNx (Figura 15 B), éstos interactuaron tan fuertemente con la PAni que posiblemente al disolver la PAni del composite en el NMP, éste no alcanzó a penetrar en algunas de sus intrincadas regiones (sobre todo en los aglomerados) como la que se observa en la micrografía. Las imágenes TEM (Figura 15 C y D) mostraron una mayor cantidad de aglomerados (de mayor tamaño) que en el caso de los MWCNTs. La morfología de la película del compuesto con HR-CNF (Figura 16 B) fue lisa y altamente homogénea a pesar de la mala dispersión de las nanofibras, como ya se mencionó. Sus diámetros son tan heterogéneos y en su mayoría, relativamente pequeños (Figura 16 C y D) al compararlos con los CNTs, que sus aglomerados fueron de menor tamaño y posiblemente no alcanzaron a notarse en la morfología superficial de la película. La morfología superficial de la película de PAni/GO fue relativamente lisa (Figura 17 B) y la dispersión excelente (debido a la hidrofilidad del GO) como puede verse en la Figura 17 140 C y D. Incluso algunos fragmentos de HR-CNF (que es de donde proviene el GO) pudieron observarse (izquierda, ligeramente arriba, de la micrografía de la Figura 17 C). Figura 14. Micrografías SEM (A, B) y TEM (C, D) de compuesto de PAni/MWCNTs. (A) Compuesto antes del procesado, (B) después del “drop-casting”, (C) dispersión de los nanotubos en la película, (D) detalle de los nanotubos en la matriz polimérica. 141 Figura 15. Micrografías SEM (A, B) y TEM (C, D) de compuestos de PAni/CNx. (A) Compuesto antes del procesado, (B) después del “drop-casting”, (C) dispersión de los nanotubos en la película, (D) detalle de los nanotubos en la matriz polimérica. 142 Figura 16. Micrografías SEM (A, B) y TEM (C, D) de compuestos de PAni/HR-CNF. (A) Compuesto antes del procesado, (B) después del “drop-casting”, (C) dispersión de los filamentos en la película, (D) detalle de los HR-CNF en la matriz polimérica. 143 Figura 17. Micrografías SEM (A, B) y TEM (C, D) de compuestos de PAni/GO. (A) Compuesto antes del procesado, (B) después del “drop-casting”, (C) dispersión del óxido de grafeno en la película, (D) detalle de una hojuela de GO en la matriz polimérica. El difractograma de la PAni (Figura 18 a), del Proceso 1, muestra la semicristalinidad típica de la PAni (picos en 2θ = 14.7, 21.2 y 25.5°) en su forma de sal de esmeraldina. Como el pico en 25.5° es más alto que el de 21°, el dopaje de la PAni por el HCl fue alto [49], el cual ocasiona un ordenamiento de las cadenas poliméricas, aumentando la cristalinidad. Este nivel de cristalinidad se observó, en mayor o menor medida, en todos los difractogramas de los compuestos (Figura 18 b, d, e), excepto en el de PAni/AniexMWCNTs (Figura 18 c), donde el pico en 25.5° está muy por debajo del de 21°, indicando una pérdida de cristalinidad notable en el compuesto. Sin duda, la funcionalización con la anilina incrementó notablemente la interacción de la carga con la matriz polimérica logrando “desordenar” a las cadenas poliméricas de la PAni. Esta 144 característica concuerda con lo observado en su morfología, donde se observa una interacción sobresaliente entre carga y polímero. Los difractogramas de CNx (Figura 18 f), exMWCNTs (18 g) y MWCNTs (18 h) muestran picos del carbono grafítico (26.3°, 002) y de Fe (44.5°) catalítico. El pico en 26.3° es el que se superpone principalmente al perfil de la PAni en sus respectivos compuestos (Figura 18 b, c, e). Figura 18. Difractogramas de (a) PAni, (b) PAni/CNx, (c) PAni/Ani-exMWCNTs, (d) PAni/exMWCNTs, (e) PAni/MWCNTs, (f) CNx, (g) exMWCNTs y (h) MWCNTs. 145 Los espectrogramas Raman de los compuestos del Proceso 1 (Figuras 19, 21, 22) muestran claros picos en la banda de ~1336 cm-1 indicando la presencia de bipolarones en la estructura de sal de esmeraldina [12, 13, 50, 51]. Los picos en las bandas de 1596 (tensión C-C) y 1217 cm-1 (deformación C-N-C), muestran la presencia de anillos benzoides, y los de 1162 (deformación C-H en el plano) y ~1475 cm-1 (tensión C=N), la presencia de anillos quinoides. Los perfiles de los compuestos producidos utilizando FeCl3 como oxidante (Figura 19 b-e) en general no muestran diferencias significativas entre sí. Incluso, la diferencia de estos perfiles no es tan notable con respecto al de PAni sin carga (19 a). Figura 19. Espectrogramas Raman de PAni y compuestos de PAni sintetizados con FeCl3. (a) PAni, (b) PAni/CNx, (c) PAni/Ani-exMWCNTS, (d) PAni/exMWCNTs, y PAni/MWCNTs (e). Los perfiles de las cargas, como era de esperarse, sí muestran diferencias importantes en las intensidades de las bandas G (hibridación sp2) y D (hibridación sp3). El pico G es mucho mayor que el D para los MWCNTs y los COx (Figura 20 b, e), no sucede así en los CNx (Figura 20 a) que debido al dopaje con N, ambos picos están casi al mismo nivel. En contraste, en el perfil de los exMWCNTs (Figura 20 d), el pico de la banda D es mayor al G, debido a la exfoliación de los nanotubos. En el caso de los Ani-exMWCNTs (Figura 20 146 c) el pico D es aún mayor que el G debido al “desorden” adicional que se creó en la estructura grafítica con la funcionalización con anilina. Figura 20. Espectrogramas Raman de las cargas de los compuestos. (a) CN x (ID/IG=1.041), (b) COx (0.603), (c) Ani-exMWCNTs (2.097), (d) exMWCNTs (1.855), y (e) MWCNTs (0.596). Los espectrogramas Raman de los compuestos producidos utilizando APS como oxidante (Figuras 21 y 22), mostraron un poco más de diferencia entre sí, a pesar de haber tenido un contenido de carga más bajo (~0.17 y ~0.48 wt%) que en el caso de los anteriores compuestos. Los perfiles de los compuestos con MWCNTs y Ani-exMWCNTs (Figura 21) muestran un decrecimiento del pico de la banda de 1470 cm-1 (tensión C=N del anillo quinoide) en el nivel bajo de carga (0.17 wt%), con respecto al pico correspondiente del perfil de la PAni sin carga. La intensidad de esta banda ha sido correlacionada con la interacción entre la PAni y los CNTs [52, 53, 54]. Solamente que en este caso, el decrecimiento debió haber sido para los compuestos con el nivel alto de carga (~0.48%). Es posible que los bajos niveles de carga en los compuestos y una deficiente dispersión hayan contribuido a incrementar el error experimental. Con respecto a los compuestos con CNx y COx, sus espectrogramas (Figura 22) fueron muy similares con respecto a los dos niveles de carga, no así, en el tipo de carga. Tomando nuevamente como indicador de la interacción PAni/CNTs a la intensidad de la banda de 147 1470 cm-1, puede observarse que en los compuestos con COx, la intensidad fue menor que en los compuestos con CNx, lo cual podría indicar que la interacción de los primeros con la PAni, fue superior, tal y como se apreció en la morfología de este compuesto. En cambio, los demás picos, prácticamente permanecieron invariables en ambos compuestos. Figura 21. Perfiles Raman de compuestos de PAni preparados con persulfato de amonio (APS) como oxidante. (a) PAni, (b) PAni/MWCNTs (alto), (c) PAni/MWCNTs (bajo), (d) PAni/Ani-exMWCNTs (alto), y (e) PAni/Ani-exMWCNTs (bajo), de donde alto=0.48 wt% y bajo=0.18 wt% son los contenidos de carga en los compuestos. 148 Figura 22. Espectrogramas Raman de compuestos de PAni preparados con APS como oxidante. (a) PAni, (b) PAni/CNx (alto), (c) PAni/CNx (bajo), (d) PAni/COx (alto), y (e) PAni/COx (bajo), de donde bajo=0.18 wt% y alto=0.48% son los contenidos de carga en los compuestos. Los perfiles Raman de compuestos de PAni dopada de la síntesis del Proceso 2 (Figuras 23-II, 24-II), fueron similares entre sí, tal y como ocurrió en el anterior proceso. En cambio, los espectrogramas de los compuestos de PAni desdopada (Figuras 23-I, 24-II), mostraron más variación en la intensidad relativa de sus picos, lo cual permitió conocer un poco más la interacción de cada carga particular con la matriz polimérica. El perfil de PAni desdopada (Figura 23-I a), muestra principalmente los picos de las bandas de 1165 (deformación C-H en el plano) y 1481 cm-1 (tensión C-N) del anillo quinoide, y también de 1590 cm-1 (tensión C-C) del anillo benzenoide. Particularmente en el compuesto con GO, puede observarse que la carga en la PAni atenúa en gran medida a estos picos (Figura 23-I b, c), y aparece un pico, no muy definido, que podría estar centrado en la banda de 1351 cm-1. Es interesante observar que cuando la PAni de este compuesto está dopada (Figura 23-II b, c), es precisamente el pico en ~1350 cm-1 el que más destaca. Además, el pico en 1572 cm-1 (tensión C-C del anillo benzenoide) se incrementa notablemente y prácticamente los demás picos desaparecen. En el perfil de la PAni dopada (Figura 23-II-a), es el pico de 1366 cm-1 el que más destaca en comparación a los pequeños picos de la bandas de 1481 y 1590 cm-1. Particularmente un pico estrecho en 149 1366 cm-1 ya se había observado en el espectrograma de la PAni sintetizada con agua (pH=7.0) (Figura 13 c), al igual este pico aparece, aunque menos intenso, en los perfiles de los compuestos preparados con distintos ácidos (Figura 13 d, e, f, g). En contraste, los perfiles de los compuestos de PAni desdopada con HR-CNF (Figura 23-I d, e) no muestran el pico o un “hombro” centrado en la banda de 1351 cm-1, aunque sí se aprecia una atenuación de los picos de la PAni sin carga. También es notable que un nivel mayor de HR-CNF en el compuesto (Figura 23-I d) atenúa menos los picos típicos de la PAni (siendo el más notorio aún el de 1481 cm-1), como puede verse al observar el perfil del compuesto con un nivel bajo de carga (Figura 23-I d). Esto pudo haber ocurrido porque posiblemente el haz de luz, utilizada en la técnica Raman, impactó en una región con menor cantidad de filamentos en el compuesto con un nivel alto de carga, ya que, como se vio en sus micrografías correspondientes, la dispersión de los HR-CNF no fue buena (Figura 16 c, d). Pero es llamativo que en el perfil del compuesto de PAni dopada con un alto nivel de carga (Figura 23 II d), también hay una menor atenuación de los picos de la PAni, con respecto al perfil del compuesto con un nivel bajo de carga (Figura 23 II e). Lo cual podría sugerir que un nivel bajo de carga podría favorecer su dispersión en la matriz polimérica. Asimismo, en el perfil del compuesto con menor cantidad de HR-CNF, destacan los picos de 1350 y 1572 cm-1 como sucedió con los perfiles de los compuestos con GO. 150 Figura 23. Espectrogramas Raman de compuestos de PAni desdopada (I) y dopada (II). (a) PAni, (b) PAni/GO (3.3 wt%), (c) PAni/GO (1.1 wt%), (d) PAni/HR-CNF (3.3 wt%), (e) PAni/HR-CNF (1.1 wt%). Los espectrogramas de los compuestos de PAni desdopada con CNx (Figura 24-I b, c) muestran también una atenuación de los picos de la PAni (Figura 24-I a), sobre todo ante una mayor cantidad de nanotubos en la matriz polimérica (Figura 24-I b). Es más notoria la disminución del pico de la banda de 1481 cm-1 (tensión C=N del anillo quinoide) por lo que, como ya se observó en el Proceso 1, podría indicar una mayor interacción entre la superficie de los nanotubos y la PAni. En cambio, los perfiles de estos compuestos con la PAni dopada (Figura 24-II b, c) prácticamente son idénticos, con los picos de 1350 y 1572 cm-1 sobresaliendo, como en los anteriores compuestos de la Figura 23-II. En los perfiles de los compuestos de PAni desdopada con MWCNTs (Figura 24-I d, e), los picos sobresalientes son los de las bandas de 1351 (más intenso que en los perfiles de PAni desdopada con GO) y 1570 cm-1, sobre todo en los correspondientes al compuesto con mayor nivel de carga (Figura 24-I d). Incluso en este perfil (d), puede apreciarse más nítidamente el pico en 2641 cm-1 correspondiente a la banda 2D de los MWCNTs. Sus correspondientes compuestos con PAni dopada (Figura 24-II d, e) muestran los picos más intensos en las bandas de 1350 y 1572 cm-1. Es imposible dejar de notar el parecido entre los perfiles de estos compuestos en estado dopado y desdopado. Más aún es notable cuando se observa la evidente diferencia entre los espectrogramas de PAni dopada y desdopada (Figura 24 I, II, a). 151 Es posible que la relativamente buena dispersión de los MWCNTs en la matriz polimérica en este Proceso 2, haya permitido una mayor interacción de los electrones deslocalizados de su superficie con las cadenas poliméricas, produciéndose polarones en éstas (a pesar de la no protonación de la PAni). La principal diferencia entre los perfiles de los compuestos con PAni dopada es que el pico de la banda de 1572 cm-1 disminuye cuando el nivel de carga es menor (el cual, en parte, podría ser ocasionado por el pico de la banda G, en 1578 cm-1 de los MWCNTs, ver Figura 22 d). En cambio, el pico en 1350 cm-1 es similar en ambos niveles de carga. Por otro lado, los perfiles de compuestos de PAni desdopada muestran que, además de disminuir el pico de 1570 cm-1 conforme el nivel de carga lo hace, también lo hace el de 1351 cm-1, lo cual podría sugerir que el nivel de “dopaje” de la PAni disminuye también. El dopaje de la PAni, no protonada, por CNTs ha sido reportado ya por Baibarc y colaboradores [52], y por Zengin y colaboradores [55]. Sobre todo los primeros sintetizaron compuestos de PAni con fragmentos de CNTs, logrando obtener una mejor dispersión y una mejor interacción entre la carga y la PAni. Algo similar pudo haber ocurrido en este trabajo al dispersarse los CNTs mediante el homogeneizador, puesto que, como se observa en las micrografías TEM (Figuras 14, 15, C, D), se favoreció la dispersión en la matriz polimérica en detrimento de su longitud, es decir, los CNTs hasta cierto punto se fragmentaron. Asimismo, como ya se observó, los perfiles de los compuestos de PAni/GO tienen un pico (aunque no intenso) en la banda de 1350 cm-1, lo que podría sugerir que, al igual que los MWCNTs, los grupos funcionales del GO (con una mejor dispersión aún en el polímero), hayan dopado a la PAni. Este dopaje de la PAni debido al GO, ha sido estudiado en aplicaciones para supercondensadores por Wang y colaboradores [56]. 152 Figura 24. Espectrogramas Raman de compuestos de PAni desdopada (I) y dopada (II). (a) PAni, (b) PAni/CNx (3.3 wt%), (c) PAni/CNx (1.1 wt%), (d) PAni/MWCNTs (3.3 wt%), (e) PAni/MWCNTs (1.1 wt%). En la Figura 25 se muestran los espectrogramas Raman de las nanoestructuras de carbono utilizadas. El pico de la banda G del espectrograma del GO (Figura 25 a) se observa en 1587 cm-1. En el resto de las cargas (25 b, c, d) este pico se observa en 1578 cm-1. El pico de la banda D si aparece en la misma posición en todos los perfiles de las cargas (1330 cm-1). Como era de esperarse, sólo en el perfil de los MWCNTs, el pico G fue mayor que el D (ID/IG = 0.684), al igual, el pico 2D (2630 cm-1) fue mucho más intenso que en los demás espectrogramas. Las razones ID/IG para GO, HR-CNF y CNx fueron 1.378, 1.439 y 1.283, respectivamente, las cuales indican que las HR-CNF mostraron un mayor “desorden” estructural debido a los bordes libres de los listones que conforman su estructura. 153 Figura 25. Espectrogramas Raman de (a) GO (ID/IG=1.378), (b) HR-CNF (1.439), (c) CNx (1.283) y (d) MWCNTs (0.684). La medición de la conductividad eléctrica de los compuestos sintetizados en el Proceso 1 (pastillas preparadas como se describe en la sección de Métodos) y en el Proceso 2 (directamente sobre las películas conformadas redopadas con HCl 1 M) se realizó mediante el método de las cuatro puntas. La conductividad eléctrica de los compuestos del Proceso 1, preparados utilizando FeCl 3 como oxidante (Tabla 4), tuvo un valor desde ~0.5 (PAni) hasta 3.97 S/cm (PAni/MWCNTs). Indudablemente los CNTs (con y sin anclaje de nanopartículas de Ag) mejoraron la conductividad en los compuestos, incluyendo al de PAni/exMWCNTs, el cual tuvo una conductividad de 2.215 S/cm. El compuesto de PAni/Ani-exMWCNTs, tuvo una conductividad similar al de la PAni sin carga, de 0.573 S/cm. Este resultado se podría explicar debido a que la funcionalización con anilina, privó a los exMWCNTs de carga deslocalizada que les permitiera conducir la electricidad y quedando además embebidos en la matriz polimérica (como ya se vio en el análisis morfológico, Figura 11). Esto no ocurrió con el compuesto de PAni/exMWCNTs, en donde los exMWCNTs mantuvieron sus propiedades eléctricas. A pesar de que se encontró una excelente interacción entre los Ani-exMWCNTs y la PAni, observada en su respectivo difractograma (Figura 18 c) y en el análisis morfológico 154 (Figura 11), la conductividad eléctrica del compuesto fue casi la misma que la de la PAni sin carga. Los Ani-exMWCNTs no ayudaron a incrementar la conductividad eléctrica del compuesto. En general, la conductividad eléctrica de la PAni y sus compuestos fue relativamente baja en comparación con la encontrada en la literatura (~5 S/cm [49, 56] para PAni, ~30 S/cm para sus compuestos [14] y ~170, 400 o 1000 S/cm para PAni procesada y conformada en fibras [9, 18]), lo cual pudo haber ocurrido por la elevada porosidad en estos materiales. Estos compuestos se conformaron como actuadores lineales (utilizando PVA como aglomerante) y se probaron en un electrolito de HCl 1 M. Lo cual se verá en el siguiente capítulo. Tabla 4. Peso, densidad y conductividad eléctrica de compuestos de PAni (Proceso 1) Muestra (~2.3 Peso (mg) Densidad 3 wt%) Conductividad (mg/cm ) eléctrica (S/cm) PAni 8.9±0.05 4.92 0.497±0.123 PAni/MWNT 6.4±0.15 3.54 3.973±0.743 PAni/MWNT/Ag 6.7±0.1 3.7 3.930±1.607 PAni/CNx 8.6±0.2 4.75 2.097±0.543 PAni/CNx/Ag 7.3±0.1 4.03 3.103±0.57 PAni/exMWNT 7.9±0.1 4.36 2.215±0.640 PAni/exMWNT-Ani 9.35±0.05 5.17 0.573±0.730 En los compuestos sintetizados con APS como oxidante, la conductividad eléctrica más baja (Tabla 5) fue de la PAni (~0.3 S/cm), la cual fue ligeramente menor a la de la PAni preparada con FeCl3 (Tabla 4). Esto se debió a que la PAni tuvo una estructura más nanofibrilar en este caso (Figura 9 A2) por lo que la cantidad de vías de conducción eléctrica disminuyó. En los compuestos, a niveles altos de carga, se tuvo una mayor conductividad, destacándose la de PAni/MWCNTs y PAni/CNx (la Ag en los CNTs, como en el caso 1, no tuvo efecto en la conductividad eléctrica), con 1.823 y 1.363 S/cm, respectivamente. Por otra parte, la conductividad del compuesto de PAni/Ani-exMWCNTs fue de 0.437, para el 155 nivel bajo, y 0.787 S/cm, para el alto, concordando con lo encontrado en el compuesto equivalente preparado con FeCl3. Los COx tuvieron un efecto no significativo en la conductividad de los compuestos de PAni, lo cual es atribuible a la funcionalización (con grupos basados en el oxígeno) de la superficie de estos nanotubos, por lo que la transferencia de carga eléctrica disminuye considerablemente. Por lo tanto, las cargas con algún tipo de funcionalización (Ani-exMWCNTs y COx), aunque hayan mejorado la interacción con la PAni, tienen muy poco efecto en el incremento de la conductividad eléctrica del compuesto (en el caso de los CNx, no están funcionalizados, sino dopados con nitrógeno, por lo que teóricamente, no deberían tener una conductividad más baja que los MWCNTs [24, 25, 44]). Ante estos resultados, se decidió prescindir de los exMWCNTs y los COx, puesto que teniendo como meta el desarrollo de actuadores, es particularmente importante el incremento en la conductividad eléctrica total del material. Tabla 5. Conductividad eléctrica de compuestos de PAni (proceso 2) Muestra % de carga Conductividad eléctrica (% wt) (S/cm) PAni - 0.293 ± 0.153 PAni/MWCNTs 0.15 1.823 ± 0.63 0.5 3.110 ± 0.707 0.15 1.047 ± 0.357 0.5 2.581 ± 0.507 0.15 1.363 ± 0.337 0.5 11.92 ± 1.65 0.15 1.040 ± 0.341 0.5 2.12 ± 0.418 0.15 0.437 ± 0.203 0.5 0.787 ± 0.213 0.15 0.223 ± 0.110 0.5 0.397 ± 0.287 PAni/MWCNTs-Ag PAni/CNx PAni/CNx-Ag PAni/Ani-exMWCNTs PAni/COx 156 En el Proceso 2, en lugar de pastillas, se tuvieron películas de compuestos que fueron redopadas con HCl 1 M a las que se les midió la conductividad eléctrica (Tabla 6). El nivel general de conductividad en los compuestos fue similar al observado en las mediciones del Proceso 1, aunque la conductividad de la PAni, de 1.6 S/cm, fue superior a la de los anteriores casos debido a la menor porosidad de la película. La conductividad de la película de PAni/MWCNTs (1 wt%) fue de ~2.05 S/cm, aunque una mayor cantidad de CNTs no incrementó este valor sino que lo disminuyó a casi la mitad (1.05 S/cm). Algo similar ocurrió con el compuesto con CNx. Con la carga de HRCNF, el valor de conductividad fue similar en ambos niveles de carga (1.41 S/cm para el nivel bajo, 1.66 S/cm para el alto). En contraste, el GO fue la carga que más incrementó la conductividad eléctrica (2.73 y 2.84 S/cm) con respecto a la de la PAni. Esto puede explicarse por el hecho de que los grupos funcionales del, prácticamente aislante, GO contribuyeron a un mayor dopaje de la PAni, ocasionando más vías para la transferencia de carga [56]. Esto refuerza lo encontrado en la espectrografía Raman, donde sus respectivos perfiles muestran un posible dopaje por el GO (Figura 20 d, e). Tabla 6. Conductividad eléctrica de compuestos de PAni (proceso 3) Muestra % de carga (% Conductividad eléctrica wt) (S/cm) PAni - 1.602 ± 0.183 PAni/MWCNTs 1.1 2.047 ± 0.380 3.3 1.052 ± 0.243 1.1 1.507 ± 0.857 3.3 0.980 ± 0.553 1.1 1.411 ± 0.173 3.3 1.657 ± 0.223 1.1 2.732 ± 1.451 3.3 2.84 ± 0.964 PAni/CNx PAni/HR-CNF GO 157 Las tiras de películas de PAni y sus compuestos (espesor ~50 μm, ancho = 1.6 mm, área transversal = 8 X 10-8 m2), fueron sometidas a pruebas termomecánicas (TMA), por duplicado, para evaluar su deformación en función a la temperatura (Figura 26). La máxima deformación de las películas (entre 50 y 60%) ocurrió en el intervalo de ~140 a ~230ºC, dentro de la cual podría ubicarse la temperatura de transición vítrea (T G) de la PAni plastificada con NMP (~100-220º) [47, 48]. La película que más repetibilidad mostró en los ensayos, fue la de PAni/HR-CNF y la que menos, la de PAni/CNx. Esto se vio reflejado en el corte y preparación de las tiras, ya que fue más sencillo cortar películas de PAni/HR-CNF y mucho más complicado cortar películas con CNx. La dificultad fue extrema con las películas de PAni/GO (3 wt%) ya que fue imposible llevar a cabo las pruebas en el equipo. Como se observa en la Figura 26 B, un nivel alto de carga produjo una mayor resistencia a la ruptura, sobre todo en las tiras de películas con MWCNTs, las cuales soportaron sin romperse hasta arriba de los 350ºC (e incluso hubo una contracción ligera). Asimismo, este nivel de carga ocasionó un incremento en la resistencia a la deformación de las películas, por debajo de los 140º C, como puede observarse en la Figura 26 B, donde los perfiles de los compuestos ya se encuentran por debajo de los de la PAni. En el intervalo de máxima deformación de las películas (~140-230º C), los perfiles de los compuestos con un nivel alto de carga son más parecidos entre sí que en el caso del nivel bajo de carga (Figura 26 A). Estos resultados indican que las películas más resistentes fueron las que tuvieron un nivel alto de carga, sobre todo aquellas con MWCNTs. Las que mayor repetibilidad mostraron en los ensayos fueron las de HR-CNF (dos niveles de carga) y GO (nivel bajo de carga). 158 Figura 26. Análisis termomecánico (TMA, Tensión = 0.1 N, Esfuerzo inicial = 1.25 MPa) por duplicado, de películas de PAni (línea continua) y sus compuestos. A) Nivel de carga de 1.1 wt%. B) Nivel de carga de 3.3 wt%. Las líneas horizontales indican la ruptura de las películas. 4.4.4.2.3. Síntesis de compuestos de PAni con MWCNTs y GO con varios niveles de carga. Los compuestos de PAni con MWCNTs y GO que se prepararon con distintos niveles de carga, tuvieron un contenido aproximado de 0.55, 0.91, 1.84 2.6 y 3.94 wt%. Únicamente se les realizó caracterización por espectroscopía Raman a los compuestos con 0.55, 1.84 y 3.94 wt% para poder evaluar el efecto del nivel de carga en su interacción con la PAni. Estos compuestos se sintetizaron con el objetivo de utilizarlos en el desarrollo de actuadores flexionantes, como se podrá ver en el siguiente capítulo. En los espectrogramas de los compuestos (Figuras 27 para PAni/MWCNTs y 28 para PAni/GO), además de observar el perfil de la PAni (a), es posible observar el efecto de las cargas en el polímero conforme su contenido aumenta: 0.55 (b), 1.84 (c) y 3.94 wt% (d). Se colocó al final el perfil de las cargas (e) de los compuestos para facilitar el análisis general. En los perfiles de los compuestos con MWCNTs, a diferencia de los perfiles de la Figura 24 (d, e), el pico de la banda de 1351 cm-1 no aparece, sino que conforme aumenta el nivel de carga, los picos de la PAni se atenúan (Figura 24-I c, d) mientras se van traslapando con los de la carga (Figura 27-I e) en 1330, 1587 y 2641 cm-1, los cuales son las bandas D, G y 2D, respectivamente. Al parecer, en estos perfiles no se observa el “dopaje” de los CNTs en la PAni como si se apreció con los de la Figura 24. En los espectrogramas con la PAni dopada (Figura 24-II) se observa que el pico principal de la PAni en 1366 cm-1 pareció 159 desplazarse a la banda de 1346 cm-1 ante un contenido de MWCNTs de 0.55 wt% en el compuesto (Figura 24-II b). Conforme el nivel de carga en los compuestos aumenta (Figura 24-II c, d), los picos de las bandas D, G y 2D comienzan a traslaparse más con los atenuados picos de la PAni en estado dopado. Figura 27. Espectrogramas Raman de compuestos de PAni, desdopada (I) y dopada (II), con MWCNTs. (a) 0 wt%, (b) 0.55 wt%, (c) 1.84 wt%, (d) 3.94 wt%, (e) MWCNTs El pico de la banda de 1346 cm-1 de los espectrogramas Raman de los compuestos de PAni, desdopada, con GO (Figura 28 I), comienza a destacar en el compuesto con 0.55 wt% de contenido de carga (Figura 28-I b). Este pico se incrementó en los compuestos con mayor nivel de carga (c, d), aunque sin variar en uno u otro perfil. Lo mismo ocurrió con el pico de la banda de 1565 cm-1, el cual es muy parecido en ambos espectrogramas. Es notable que a diferencia de los perfiles de los compuestos con MWCNTs de la figura anterior, los picos de la PAni se atenúan marcadamente a partir del perfil del compuesto con 0.55 % de contenido de GO. Esto indica una alta interacción del GO con el polímero, como ya fue apreciado en los perfiles de la Figura 23 (b, c). Como en los compuestos con MWCNTs, los picos de las bandas D y G (Figura 28-I e) parecen traslaparse con el perfil de la PAni. En los perfiles de los compuestos con la PAni dopada (Figura 25 II b, c, d), sólo predominan los picos de las bandas de 1346 y 1569 cm-1, 160 sobre todo el primero es más intenso. Como puede observarse, no hay mucha variación de los perfiles en función del contenido de GO. Son muy similares. Figura 28. Espectrogramas Raman de compuestos de PAni desdopada (I) y dopada (II) con GO. (a) 0 wt%, (b) 0.55 wt%, (c) 1.84 wt%, (d) 3.94 wt%, (e) GO. 4.4.4.2.4. Síntesis de compuestos de PAni con carga híbrida de GO-MWCNTs. En algunos estudios se han realizado dispersiones en medio acuoso de GO y CNTs en proporción GO:CNTs de 1:1 [57]; en proporción 3:1 [58] (para aplicaciones de supercondensadores de carbono de doble capa); y en proporción 2:1 [59] (con una decoración de PAni en la carga de un ~9 wt% para aplicación de supercondensadores). Se encontró que el GO funciona como agente dispersante de los CNTs en medio acuoso debido a la hidrofilidad del GO. Al parecer, las láminas de GO se intercalan entre los CNTs. Debido a estos resultados se decidió realizar una síntesis de compuestos de PAni con una carga híbrida de GO y MWCNTs. Los compuestos de PAni con MWCNTs, GO y carga híbrida de GO-MWCNTs (2:1, 1:1 y 1:2) con contenido de carga de 1.3 wt% fueron caracterizados morfológicamente mediante microscopía TEM para conocer la dispersión de la carga en la matriz polimérica. 161 En la Figura 29 se muestran las micrografías de los compuestos de PAni (1.3 wt%) únicamente con MWCNTs (29 A, B) y GO (29 C, D). No se observaron aglomerados de MWCNTs, la dispersión y longitud de los nanotubos fue similar a la obtenida con anterioridad. Asimismo, la dispersión del GO en la matriz polimérica fue alta. En la Figura 29 C se observan algunos restos de HR-CNF (fuente del GO) dispersos; en la 29 D puede observarse una lámina de GO. Figura 29. Difractogramas de compuestos de PAni con MWCNTs (A, B) y GO (C, D) con un contenido de carga de 1.3 wt%. En la Figura 30 se observan las micrografías de compuestos de PAni con GO-MWCNTs. Las micrografías de compuestos con una carga GO:MWCNTs de 1:2 (30 A, B) muestran la presencia de nanotubos y láminas de GO (a la derecha de la micrografía 30 A). La dispersión de los MWCNTs fue similar a la de los compuestos únicamente con nanotubos. 162 Las imágenes de compuestos con una relación de GO:MWCNTs de 1:1 (Figura 30 C, D) muestran una menor cantidad de MWCNTs y aunque una mayor presencia de GO no es muy visible, se observa una mayor cantidad de residuos de HR-CNF (sobre todo en 30 C). Las micrografías de compuestos con un contenido de GO:MWCNTs de 2:1 (Figura 30 E, F) muestran una disminución de CNTs dispersos en la matriz polimérica, así como un incremento de residuos de HR-CNFs. Esto último indica una mayor presencia de GO en el compuesto. Las sombras que acompañan al fragmento de nanotubo de la imagen 30 F podrían ser apilamientos de GO. En la parte baja del nanotubo, parece ser un trozo de HRCNF con láminas de GO unidas a ella. En general, el efecto dispersante del GO en los MWCNTs no se apreció en los compuestos de PAni. Esto posiblemente ocurrió debido al nivel tan bajo de carga en el compuesto. En los estudios donde se han trabajado con sistemas híbridos de GO-MWCNTs no se han dispersado en matrices poliméricas, como se vio al inicio de este apartado. 163 Figura 30. Micrografías de compuestos de PAni con carga híbrida de GO-MWCNTs (1.3 wt%) con una relación GO-MWCNTs de 1:2 (A, B), de 1:1 (C, D) y de 2:1 (E, F). En la caracterización por espectrografía Raman de los compuestos de PAni con carga híbrida de GO-MWCNTs (Figura 31), se observa que el pico de la banda de 1481 cm-1 (tensión C=N del anillo quinoide) disminuye en intensidad conforme la proporción de GO 164 en la carga aumenta. Como ya se mencionó previamente, la disminución de este pico indica que la interacción carga/matriz polimérica es alta. De manera similar que en los perfiles Raman de compuestos anteriores, hay una atenuación de los picos de la PAni en los compuestos (31-I b-f), y conforme la proporción de GO aumenta, el pico de la banda en 1346 cm-1 se incrementa, llegando al máximo nivel cuando en el compuesto únicamente hay GO (31-I f). Este perfil del compuesto de PAni/GO desdopado, guarda cierto parecido con los perfiles de compuestos dopados de la derecha (31-II b-f). Esto se debe a que los grupos funcionales oxigenados del GO están “dopando” a la PAni desdopada. En cambio, parece que los MWCNTs tampoco están dopando a la PAni como se había visto con anterioridad (Figura 24-I d, e). Los perfiles de compuestos de PAni dopados (Figura 31-II b-f) son similares entre sí y a los otros perfiles de compuestos dopados vistos con anterioridad. Sobresalen los picos de las bandas de 1346 y 1570 cm-1. En cambio, el pico más intenso de la PAni sin carga (31-II a) se ubica en la banda de 1366 cm-1. Los perfiles Raman de las cargas de MWCNTs y GO son los que se observaron con anterioridad. El pico de la banda D en ambas cargas se ubica en 1330 cm-1, y la G en 1578 y 1587 cm-1 para GO y MWCNTs, respectivamente. La banda 2D está en la banda de 2641 cm-1 para los MWCNTs. 165 Figura 31. Difractogramas Raman de compuestos de PAni con carga híbrida de GOMWCNTs (1.3 wt%) dopados (I) y desdopados (II). a) PAni, b) PAni/MWCNTs, c) PAni/MWCNTs-GO (2:1), d) PAni/MWCNTs-GO (1:1), e) PAni-GO (1:2), f) PAni/GO, g) MWCNTs prístinos, h) GO prístinos. 4.5. CONCLUSIONES En el proceso de exfoliación de MWCNTs se confirmó la importancia de la temperatura de 1000ºC, así como el número de tratamientos térmicos (5), previamente encontrados por Cano-Márquez y colaboradores, aunque se encontró que con una relación Li:MWCNTs de 1:1 (en la reacción de intercalación) se obtienen resultados similares a la de 10:1 previamente utilizada. Se funcionalizó de manera apropiada a los exMWCNTs con anilina mediante la reacción de “sales de nanotubos”, lo cual fue constatado en la morfología, difractograma y conductividad eléctrica del composite PAni/Ani-exMWCNTs. A pesar de haber obtenido una buena interacción entre Ani-exMWCNTs o COx con la PAni, se descartaron estas cargas debido a que no incrementaron 166 significativamente la conductividad eléctrica de los compuestos, la cual, es una propiedad importante para el desarrollo de actuadores. Se obtuvieron eficiencias de polimerización cercanas al 50% utilizando HCl 1 M en la polimerización interfacial de la anilina. Se obtuvieron películas con un buen control de su espesor, mediante la disolución de la PAni desdopada en N-metil-2-pirrolidona (NMP) y un casting en sustrato de vidrio. Se logró mejorar la dispersión de los nanotubos de carbono en los compuestos utilizando un homogeneizador, aunque en detrimento de su longitud. La dispersión del GO fue muy elevada debido a su hidrofilicidad, asimismo, sus grupos funcionales, contribuyeron a un dopaje extra en las cadenas poliméricas de la PAni, ocasionando un incremento de su conductividad eléctrica. Las cargas en los compuestos, incrementaron la resistencia a la deformación de sus respectivas películas, sobre todo a un nivel de carga del 3.3 wt%. Una mayor resistencia a la deformación se obtuvo con las películas de PAni/MWCNTs (3.3 wt%). 167 REFERENCIAS [1] Huang, J.; Virji, S.; Weiller, B. H.; Kaner, R. B. Polyaniline nanofibers: Facile synthesis and chemical sensors. J. Am. Chem. Soc. 125, 2003, 314–315. [2] Huang, J.; Kaner, R. B. A general chemical route to polyaniline nanofibers. J. Am. Chem. Soc. 126, 2004, 851–855. [3] Stejskal, J.; Sapurina, I.; Trchová, M.; Konyushenko, E. N. Oxidation of Aniline: Polyaniline Granules, Nanotubes, and Oli-goaniline Microspheres. Macromolecules 41, 2008, 3530–3536. [4] Du, X.-S.; Zhou, C.-F.; Wang, G.-T.; Mai, Y.-M. Novel Solid-State and Template-Free Synthesis of Branched Polyaniline Nanofibers. Chem. Mater. 20, 2008, 3806–3808. [5] Dallas, P.; Stamopoulos, D.; Boukos, N.; Tzitzios, V.; Niarchos, D. Petridis, D. Characterization, magnetic and transport properties of polyaniline synthesized through interfacial polymerization. Polymer 48, 2007, 3162–3169. [6] Zhou, H.; Chen, H.; Luo, S.; Lu, Gewu, Wei, W.; Kuang, Y. The effect of the polyaniline morphology on the performance of polyaniline supercapacitors. J. Solid State Electrochem. 9, 2005, 574–580. [7] Gupta, V.; Miura, N. High performance electrochemical supercapacitor from electrochemically synthesized nanostructured polyaniline. Materials Letters 60, 2006, 1466-1469. [8] Lu, W.; Smela, E.; Adams, P. N.; Zuccarello, G.; Mattes, B. R. Development of Solidin-Hollow Electrochemical Linear Actuators Using Highly Conductive Polyaniline. Chem. Mater. 16, 2004, 1615-1621. [9] Smela E.; Lu, W.; Mattes, B. R. Polyaniline actuators Part. 1. PANI (AMPS) in HCl. Synthetic Metals, 151, 2005, 25-42. [10] Smela E., Mattes R. B. Polyaniline actuators Part 2. PANI (AMPS) in methanesulfonic acid. Synthetic Metals, 151, 2005, 43-48. [11] Iijima, S.; Ichihashi, T. Single shell carbon nanotubes of one nanometer diameter. Nature 1993, 363, 603-605. [12] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P. Synthesis and characterization of new polyaniline/nanotube composites. Mater. Sci. Eng. C 23, 2003, 87– 91. [13] Wu, T.-M.; Lin, Y.-W.; Liao, C.-S. Preparation and characterization of polyaniline/multi-walled carbon nanotube composites. Carbon 43, 2005, 734–740. [14] Mottaghitalab, V.; Spinks, G. F.; Wallace, G. G. The influence of carbon nanotubes on mechanical and electrical properties of polyaniline fibers. Synth. Met. 152, 2005, 77– 80. [15] Wu, T.-M.; Lin, Y.-W. Doped polyaniline/multi-walled carbon nanotube composites: Preparation, characterization and properties. Polymer 47, 2006, 3576–3582. [16] Lafuente, E.; Callejas, M. A.; Sainz, R.; Benito, A. M.; Maser, W. K.; Sanjuán, M. L.; Saurel, D.; de Teresa, J. M.; Martínez, M. T. The influence of single-walled carbon nanotube functionalization on the electronic properties of their polyaniline composites. Carbon 46, 2008, 1909-1917. 168 [17] Zelikman, E.; Narkis, M.; Siegmann, A.; Valentini, L.; Kenny, J. M. Polyaniline/Multiwalled Carbon Nanotube Systems: Dispersion of CNT and CNT/PANI Interaction. Polym. Eng. and Sci. 2008, 1872-1877. [18] Mottaghitalab, V.; Spinks, G. M.; Wallace, G. G. The development and characterization of polyaniline – single walled carbon nanotube composite fibres using 2acrylamido-2-methyl-1-propane sulfonic acid (AMPSA) through one step wet spinning process. Polymer 47, 2006, 4996-5002. [19] Harris, P.J.F. Carbon nanotubes composites. Int. Mater. Rev. 49, 2004, 31–43. [20] Du, J.-H.; Bai, J.; Cheng, H.-M. The present status and key problems of carbon nanotube based polymer composites. Express Polym. Let. Vol 1, 5, 2007, 253-273. [21] Spitalsky, Z.; Tasis, D.; Papagelis, K.; Galiotis, C. Carbon nanotube polymer composites: Chemistry, processing, mechanical and electrical properties. Prog. Polym. Sci. 35, 2010, 357–401. [22] Gopalan, A. I.; Lee, K.-P.; Santosh, P.; Kim, K. S.; Nho, Y. C. Different types of molecular interactions in carbon nanotube/conducting polymer composites – A close analysis. Comp. Sci. and Tech. 67, 2007, 900-905. [23] Terrones, M. Carbon nanotubes: synthesis and properties, electronic devices and other emerging applications. Int. Mat. Review 6, 49, 2004, 325-377. [24] Ewels, C.P.; Glerup, M. Nitrogen doped in carbon nanotubes. J. Nanosci. Nanotechnol. 5, 2005, 1345–1363. [25] Ayala, P.; Arenal, R.; Rümmeli, M.; Rubio, A.; Pichler, T. The doping of carbon nanotubes with nitrogen and their potential applications. Carbon 48, 2010 575–586. [26] Cano-Márquez, A. G.; Rodríguez-Macías, F. J.; Campos-Delgado, J.; EspinosaGonzález, C. G.; Tristán-López, F.; Ramírez-González, D.; Cullen, D. A.; Smith, D. J.; Terrones, M.; Vega-Cantú, Y. I. ExMWNTs: Graphene Sheets and Ribbons Produced by Lithium Intercalation and Exfoliation of Carbon Nanotubes. Nano Lett. 9 (4), 2009, 1527– 1533. [27] Terrones, M.; Botello-Méndez, A. R.; Campos-Delgado, J.; López-Urías, F.; VegaCantú, I.; Rodríguez-Macías, F. J.; Elías, A. L.; Muñoz-Sandoval, E.; Cano-Márquez, A. G.; Charlier, J.-C.; Terrones, H. Graphene and graphite nanoribbons: Morphology, properties, synthesis, defects and applications. Nano Today 5, 2010, 351-372. [28] Huang, J.-C. Carbon Black Filled Conducting Polymers and Polymer Blends. Adv. in Polym. Tech. 21(4), 2002, 299-313. [29] Eksiouglu, B.; Nadarajah, A. Structural analysis of conical carbon nanofibers. Carbon 44(2), 2006, 360-373. [30] Varela-Rizo, H.; Rodríguez-Pastor, I.; Merino, C.; Terrones, M.; Martin-Gullon, I. Graphene oxide nanoplatelets of different crystallinity synthesized from helical-ribbon carbon nanofibers and multiwall carbon nanotubes. J. of Mat. Research. 26, 2011, 26322641. [31] Ju, H.-M.; Huh, S. H.; Choi, S.-H.; Lee, H.-L. Structures of thermally and chemically reduced graphene. Mat. Let. 64, 2010, 357-360. [32] Vera-Agullo, J.; Varela-Rizo, H.; Conesa, J. A.; Almansa, C.; Merino, C.; MartinGullon, I. Evidence for growth mechanism and helix-spiral con structure of stacked-cup carbon nanofibers. Carbon 45, 2007, 2751-2758. 169 [33] Terrones, M.; Kamalakaran, R.; Seeger, T.; Rühle, M. Novel nanoscale gas containers: encapsulation of N2 in CNx nanotubes. Chemical Communications, 2000, 2335–2336. [34] Botello-Méndez, A.; Campos-Delgado, J.; Morelos-Gómez, A.; Romo-Herrera, J. M.; Rodríguez, A. G.; Navarro, H.; Vidal, M. A.; Terrones, H.; Terrones, M. Controlling the dimensions, reactivity and cristallinity of multiwalled carbon nanotubes using low ethanol concentrations. Chem. Phys. Lett. 453, 2008, 55-61. [35] Jiménez Manero, Pablo. (2011). Materiales nanoestructurados basados en polianilina, nanotubos de carbono y grafeno, Tesis de Doctorado, España: Universidad de Zaragoza. [36] Liang, F.; Sadana, A. K.; Peera, A.; Chattopadhyay, J.; Gu, Z.; Hauge, R. H.; Billups, W. E. A Convenient Route to Functionalized Carbon Nanotubes. Nano Lett. 4, 2004, 12571260. [37] Chattopadhyay, J.; Sadana, A. K.; Liang, F.; Beach, J. M.; Xiao, Y.; Hauge, R. H.; Billups, W. E. Carbon Nanotube Salts. Arylation of Single-Wall Carbon Nanotubes. Org. Let. 7, 19, 2005, 4067-4069. [38] Liu, J.; Rinzler, A. G.; Dai, H.; Hafner, J. H.; Bradley, R. K.; Boul, P. J.; Lu, A.; Iverson, T.; Shelimov, K.; Huffman, C. B.; Rodriguez-Macias, F.; Shon, Y.-S.; Lee, T. R.; Colbert, D. T.; Smalley, R. E. Fullerene Pipes. Science 280, 1998, 1253-1256. [39] Kamalakaran, R.; Terrones, M.; Seeger, T.; Kohler-Redlich, Ph.; Rühle, M.; Kim, Y. A.; Hayashi, T.; Endo, M. Synthesis of Thick and Crystalline Nanotube Arrays by Spray Pyrolysis. Appl. Phys. Lett. 77, 2000, 3385–3387. [40] Alvizo-Páez, E. R.; Romo-Herrera, J. M.; Terrones, H.; Terrones, M.; Ruiz-García, J.; Hernández-López, J. L. Soft purification of N-doped and undoped multiwall carbon nanotubes. Nanotechnology 19, 2008, 155701. [41] Montgomery, D. C. Design and Analysis of Experiments. 5th Edition, John Wiley & Sons, 2004. [42] Huang, J.; Kaner, R. B. The intrinsic nanofibrilar morphology of polyaniline. Chem. Commun. 2006, 367-376. [43] Czerw, R.; Terrones, M.; Charlier, J.-C.; Blase, X.; Foley, B.; Kamalakaran, R.; Grobert, N.; Terrones, H.; Tekleab, D.; Ajayan, P. M.; Blau, W.; Rühle, M.; Carroll, D. L. Identification of electron donor states in n-doped carbon nanotubes. Nano Lett. 1, 2001, 457–460. [44] Wiggings-Camacho, J. D.; Stevenson, K. J. Effect of nitrogen concentration on capacitance, density of states, electronic conductivity, and morphology of N-doped carbon nanotube electrodes. J. Phys. Chem. C 113, 2009, 19082–19090. [45] Sapurina, I.; Setjskal, J. The mechanism of the oxidative polymerization of aniline and the formation of supramolecular polyaniline structures. Polym. Int. 2008, 57, 1295-1325. [46] Manohar, S. K.; Macdiarmid, A. G.; Epstein, A. J. Polyaniline: Pernigranile, an isolable intermediate in teh conventional chemical synthesis of emeraldine. Synthetic Metals 1991, 41(1-2), 711-714. [47] Wei, Y.; Jang, G.-W.; Hsueh, K.; Scherr, E. M.; MacDiarmid, A.G.; Epstein, A. J. Thermal transitions and mechanical properties of films of chemically prepared polyaniline. Polymer 33, 1992, 314-322. 170 [48] Milton, A. J.; Monkman, A. P. A comparative study of polyaniline films using thermal analyses and IR spectroscopy. J. Phys. D: Appl. Phys. 26, 1993, 1468-1474. [49] Abdiryim, T.; Xiao-Gang, Z.; Jamal, R. Comparative studies of solid-state synthesized polyaniline doped with inorganic acids. Mater. Chem. Phys. 90, 2005, 367–372. [50] Sivakkumar, S. R.; Kim W. J.; Choi, J.-A.; MacFarlane, D. R.; Forsyth, M.; Kim, D.W. J. Electrochemical performance of poly-aniline nanofibres and polyaniline/multiwalled carbon nanotube composite as an electrode material for aqueous redox supercapacitors. Power Sources, 171, 2007, 1062–1068. [51] Dallas, P.; Stamopoulos, D.; Boukos, N.; Tzitzios, V.; Niarchos, D. Petridis, D. Characterization, magnetic and transport properties of polyaniline synthesized through interfacial polymerization. Polymer 48, 2007, 3162–3169. [52] Baibarac, M.; Baltog, I.; Lefrant, S.; Mevellec, J. Y.; Chauvet, O. Polyaniline and Carbon Nanotubes Based Composites Containing Whole Units and Fragments of Nanotubes. Chem. Mater. 15, 2003, 4149-4156. [53] Mottaghitalab, V.; Xi, B.; Spinks, G. M. Wallace, G. G. Polyaniline fibres containing single-walled carbon nanotubes: Enhanced performance artificial muscles. Synth. Met. 156, 2006, 796–803. [54] Cochet, M.; Maser, W. K.; Benito, A. M.; Callejas, M. A.; Martinez, M. T.; Benoit J.M.; Schreiber, J.; Chauvet, O. Synthesis of a new polyaniline/nanotube composite: “insitu” polymerization and charge transfer through site-selective interaction. Chem. Commun. 16, 2001, 1450–1451. [55] Zengin, H.; Zhou, W.; Jin, J.; Czerw, R.; Smith, D. W.; Echegoyen, L.; Caroll, D. L.; Foulger, S. H.; Ballato, J. Carbon Nanotube Doped Polyaniline. Adv. Mater. 14, 2002, 1480-1483. [56] Wang, H.; Hao, Q.; Yang, X.; Lu, L.; Wang, X. Graphene oxide doped polyaniline for supercapacitors. Electrochem. Comm. 11, 2009, 1158-1161. [57] Qiu, L.; Yang, X.; Gou, X.; Yang, W.; Ma, Z.-F.; Wallace, G. G.; Li, D. Dispersing Carbon Nanotubes with Graphene Oxide in Water and Synergistic Effects between Graphene Derivatives. Chem. Eur. J. 16, 2010, 10653-10658. [58] Aboutalebi, S. H.; Chidembo, A. T.; Salari, M.; Konstantinov, K.; Wxler, D.; Liu, H. K.; Dou, S. X. Comparison of GO, Go/MWCNTs composite and MWCNTs as potential electrode materials for supercapacitors. Energy Environ. Sci. 4, 2011, 1855-1865. [59] Lu, X.; Dou, H.; Yang, S.; Hao, L.; Zhang, L.; Shen, L.; Zhang, F.; Zhang, X. Fabrication and electrochemical capacitance of hierarchical graphene/polyaniline/carbon nanotube ternary composite film. Electrochimia Acta 56, 2011, 9224-9232. 171 172 5. Actuadores lineales y flexionantes de nanocompuestos de PAni y distintas nanoestructuras de carbono 173 174 5. Actuadores lineales y flexionantes de nanocompuestos de PAni y distintas nanoestructuras de carbono 5.1. ANTECEDENTES Los actuadores de polianilina (PAni) comenzaron a estudiarse en forma de película plastificada con N-metil-2-pirrolidona (NMP) [1-3]. Frecuentemente, la película de PAni se adhiere a otra no activa (en general de celofán), de tal manera que al expandirse o contraerse en un electrolito protónico, resultaba en una flexión del sistema bicapa en un sentido o en otro [4]. Asimismo se fabricaron dispositivos con una capa delgada de oro en contacto con la película de PAni, con objeto de que hubiera la misma electroactividad a lo largo del actuador [5]. También llegaron a formarse sistemas tricapa con celofán en medio y capas de PAni en ambos exteriores, siendo ánodo y cátodo de manera alternativa [6]. Recientemente, se ha estudiado el accionamiento y la recuperación a la deformación, aplicando distintos esfuerzos, a este tipo de películas plastificadas con NMP [7]. Asimismo, utilizando ácido 2-acrilamido-2-metilpropano sulfónico (AMPSA, por sus siglas en inglés) como plastificante y dopante (junto con ácido dicloroacético o fórmico) de la PAni, se han llegado a elaborar películas y fibras (mediante el método de hilado húmedo), las cuales se han estirado ~100–500% su longitud (sobre todo las fibras) a temperaturas entre 90–130º C con el objetivo de orientar las cadenas poliméricas longitudinalmente, incrementando así su conductividad eléctrica (de ~50 a ~600 S/cm a lo largo de los actuadores). Para el accionamiento de estas películas, se les ha colocado una capa delgada de oro por un lado, evitando así, que la resistencia eléctrica intrínseca del polímero, pudiera afectar el accionamiento en zonas alejadas del conector [8-14]. Bon K. Gu y colaboradores [15], elaboraron un músculo artificial de PAni (emulando las miofibrillas de los músculos biológicos) que consistió en una cuerda de fibras de PAni (de 900 nm de diámetro y con un núcleo de poliuretano cada una) que se accionaron en paralelo, logrando distensiones de ~1.65%. Hasta el momento hay pocos estudios sobre el efecto de distintas nanoestructuras de carbono en la PAni para el desarrollo de actuadores o músculos artificiales. Mottaghitalab y colaboradores [16, 17], evaluaron las propiedades mecánicas y eléctricas de fibras, de compuestos de PAni (polimerizados ex situ) con nanotubos de carbono de pared simple (SWCNTs), conformadas por hilado húmedo, donde los plastificantes fueron dimetil 175 propileno urea (DMPU) o AMPSA con ácido dicloroacético. Poco después, Spinks y colaboradores [18], bajo las mismas condiciones experimentales, elaboraron fibras a las que se les midió sus propiedades electromecánicas y de accionamiento. Encontraron que, aunque las fibras de compuestos con SWCNTs (0.76 wt%) tuvieron una menor elongación (0.8%) que las de PAni sin nanotubos (1.3%), éstas mejoraron significativamente sus propiedades mecánicas y eléctricas. En un estudio posterior, se determinó el aporte de este tipo de fibras, plastificadas con DMPU, también como músculos artificiales. Se comprobó que las fibras con un 2% (w/w) de SWCNTs se elongaron hasta un 2.2% y tuvieron mejores propiedades eléctricas y mecánicas que las fibras sin nanotubos [19]. No se encontraron más reportes de síntesis de compuestos de PAni con otras nanoestructuras de carbono dispersas en su matriz polimérica para aplicaciones de accionamiento. Por lo que uno de los principales objetivos de esta tesis, es evaluar el efecto de distintas nanoestructuras de carbono (además de los SWCNTs) en actuadores de PAni (lineales y flexionantes), donde la síntesis de los compuestos se realiza por polimerización in situ. A lo largo de este trabajo, se probaron distintas nanoestructuras en los actuadores de compuestos de PAni, descartándose las menos adecuadas y manteniendo al final las que tuvieron un mayor efecto en el accionamiento electromecánico. Estas nanoestructuras fueron: nanotubos de carbono multicapa (MWCNTs), nanotubos de carbono multicapa oxidados (COx-MWCNTs), nanotubos de carbono multicapa dopados con nitrógeno (CNxMWCNTs), nanotubos de carbono multicapa exfoliados (exMWCNTs), nanotubos de carbono multicapa exfoliados y funcionalizados con anilina (Ani-exMWCNTs), nanofibras de listones grafíticos helicoidales (HR-CNF) y óxido de grafeno (GO). A lo largo del presente capítulo se mantuvo prácticamente invariable el método de polimerización in situ de los compuestos comentado anteriormente en el capítulo 3. Asimismo, las condiciones experimentales del accionamiento de los actuadores se mantuvieron constantes, exceptuando las variables de interés. En un inicio se desarrollaron probetas cilíndricas de compuestos de PAni obtenidos directamente desde la síntesis (dopados y nanoestructurados hasta cierto punto por la polimerización interfacial in situ) llevándolos a un molde de caucho y utilizando un aglomerante para el material (acetato de polivinilo, PVA, por sus siglas en inglés). Únicamente se variaron los porcentajes de contenido de la carga. Posteriormente, debido a la fragilidad de las fibras (ocasionada por su porosidad) se abandonó este método y se 176 decidió disolver los compuestos (desdopados) en NMP, y, mediante la técnica de “dropcasting” en un sustrato de vidrio, obteniendo películas. Éstas se utilizaron para formar actuadores bicapa con una película de celofán y se evaluaron en un electrolito protónico (HCl o ácido metil sulfónico, MSA, por sus siglas en inglés). En las pruebas con estos actuadores también se trató de mantener en lo posible las mismas condiciones experimentales. Solamente se varió el porcentaje de carga de los compuestos. En la última parte del estudio, se eligieron las dos cargas que mejor ayudaron a la respuesta de los actuadores que como se verá posteriormente son: los MWCNTs y el GO, y se realizó un experimento de optimización para obtener el nivel de carga y voltaje más adecuados para el funcionamiento de estos dispositivos bicapa. En este trabajo, las pruebas a los actuadores lineales y flexionantes, se realizan en una celda de dos electrodos. En el caso de los actuadores flexionantes un actuador se conectó como ánodo y otro como cátodo. Para los lineales se utilizó Cu como contraelectrodo. En ambos casos se invirtió sucesivamente esta configuración, y así poder observar sus elongaciones/flexiones (elongación/contracción) en función de los potenciales redox aplicados con una fuente de voltaje convencional. 5.2. EXPERIMENTACIÓN 5.2.1. Materiales Compuestos de PAni con distintas nanoestructuras de carbono, acetato de polivinilo (PVA), caucho para elaboración de los moldes, HCl, ácido metil sulfónico (MSA), ácido dodecil benceno sulfónico (DBSA), ácido 2-acrilamido-2-metilpropano sulfónico (AMPSA) y fuente variable de voltaje. 5.2.2. Métodos A lo largo de esta investigación, el accionamiento de actuadores de compuestos de PAni se evaluó en tres partes. En la primera, se midió el accionamiento lineal de probetas cilíndricas, formadas con polvos de los compuestos y un aglomerante; en la siguiente, se midió la flexión y la velocidad de flexión de películas de compuestos unidas a películas de celofán; y en la última parte, se realizó un experimento de optimización, también con actuadores bicapa de películas (compuestos con MWCNTs o con GO) y celofán. Los electrolitos utilizados fueron HCl, en las dos primeras partes, y MSA en la última. 177 5.2.3. Actuadores lineales 5.2.3.1. Elaboración de las probetas cilíndricas Se aglutinaron polvos hidratados de nanocompuestos mezclándolos con una suspensión de acetato de polivinilo (PVA) diluida (5% v/v) en una relación de 1.4 g/mL. La mezcla se vació en moldes de caucho (figura 1) para conformar probetas cilíndricas de dos diámetros distintos (2 y 4 mm). Se obtuvieron probetas cilíndricas de 4 mm de diámetro de los compuestos sintetizados con FeCl3 (Cap. III, proceso 1, con cargas de MWCNTs, MWCNTs-Ag, CNx, CNx-Ag, exMWCNTs, Ani-exMWCNTs, a 2.3 wt%) y probetas de 2 y 4 mm cuando se sintetizaron con persulfato de amonio (APS) (Cap. III, proceso 1, con cargas de MWCNTs, MWCNTs-Ag, CNx, CNx-Ag, COx, Ani-exMWCNTs a 0.16 y 0.48 wt%). En los extremos de los surcos del molde, se colocó un alambre de Cu para que quedara embebido al vaciarse el compuesto. El material se secó en el horno (50° C, 4 h) y se desmoldaron las probetas (longitud ~15 mm) para su caracterización electromecánica. Figura 1. Molde de caucho utilizado para obtener probetas cilíndricas de nanocompuestos de PAni. 5.2.3.2. Medición de la elongación de las probetas. La probeta (electrodo de trabajo) se sumergió verticalmente, casi en su totalidad (~90%), en una solución de HCl (10% v/v). Se utilizó alambre de Cu como contra-electrodo. Se dejó a la probeta permearse en la solución unos 25 min y después se aplicaron voltajes de ±1.25 V cada dos minutos. Se tomaron fotografías de la probeta bajo accionamiento para determinar su % de elongación. 178 5.2.4. Actuadores flexionantes. 5.2.4.1. Preparación de los actuadores bicapa (flexionantes) Tal y como se describe en el capítulo anterior, los compuestos de PAni (con cargas de MWCNTs, CNx, HR-CNF, GO, a 1.1 y 3.3 wt%) sintetizados con persulfato de amonio y desdopados con NaOH 0.2 M, se disolvieron en NMP (10% w/v), y mediante un “dropcasting” sobre vidrio, se prepararon películas (5 X 20 mm) de espesor ~30 μm. Las tiras obtenidas, después de redoparlas (con HCl o MSA), se fijaron en cinta adhesiva de celofán, de tal forma que entre las películas, en un extremo, quedó adherido un trocito de papel de aluminio, directamente en contacto con la película de compuesto, y también un alambre de cobre muy delgado, por el lado de contacto con el celofán (figura 2). De esta forma el conector quedó entre las películas de PAni y de celofán. Se prepararon actuadores flexionantes con dos niveles de carga (1.1 y 3.3 wt%). Figura 2. Esquema de la preparación de los actuadores bicapa (flexionantes). 5.2.4.2. Medición de flexión de los actuadores Estos actuadores bicapa se sumergieron en el electrolito (~90%) y se alternaron como ánodo o cátodo en las mediciones. Los niveles de voltaje utilizados fueron de 3 y 5 V. Se probaron 4 electrodos al mismo tiempo, utilizándose HCl 1M como electrolito. Se midió el potencial óptimo de accionamiento (voltaje al cual se obtuvieron flexiones máximas), la flexión de los actuadores (mediante análisis de fotografías), el tiempo de respuesta y el número de ciclos. 179 5.2.4. Optimización. Los actuadores bicapa flexionantes de MWCNTS y GO fueron preparados exactamente igual que en la sección anterior. Se utilizó MSA 1.5 M como electrolito. Las mediciones se realizaron por cuadruplicado y simultáneamente. Finalmente se realizó un experimento de corroboración del punto óptimo de rendimiento encontrado con estos experimentos. Se sintetizaron compuestos de PAni/MWCNTs y PAni/GO con un nivel de carga de ~1.3 wt%, y se elaboraron actuadores que se probaron a un potencial de 2.6 V. 5.3. RESULTADOS Y DISCUSIÓN 5.3.1. Actuadores lineales. La figura 2 muestra algunas fotografías de los pasos necesarios para la preparación de los actuadores. La fragilidad de las probetas de compuestos fue alta puesto que la mayoría de ellas se rompió al retirárseles el agua dentro del horno (figura 2B). A pesar de esto, se obtuvieron fibras suficientes (figura 2C) de casi todos los compuestos para realizar las pruebas de accionamiento. Figura 2. A) Vaciado de compuestos de PAni en un molde de caucho; B) fibras de compuestos obtenidas al sacarse el molde del horno donde se observan los alambres de Cu en un extremo; C) fibras listas para las pruebas. La elongación y contracción (oxidación, reducción, respectivamente) de las probetas al someterse a ±1.25 V, fue baja. La mayor deformación fue para la probeta de PAni (~2%) (figura 3 A, B). En el caso de los compuestos, la elongación fue casi imperceptible (Figura 3 C, D), aunque mediante el análisis de las fotografías digitales, pudo determinarse en algunos una cierta deformación (detalles en Tabla 1). 180 Figura 3. Actuador lineal de PAni de 4 mm de diámetro, el cual muestra una elongación de 2% (A, B) y de compuesto de PAni/MWCNTs de 2 mm de diámetro, con ~0.5% de elongación (C, D). Las probetas obtenidas de los compuestos sintetizados con cloruro férrico (FeCl3) tuvieron elongaciones de ~0.5 a ~2% (Tabla 1), ésta última muy similar al resultado de Mottaghitalab y colaboradores de 2.2% para la fibra de compuesto de PAni/SWCNTs [19]. Particularmente, las probetas con exMWCNTs y Ani-exMWCNTs tuvieron las elongaciones más bajas (~0.5%), aunque la conductividad eléctrica fue casi 4 veces más alta en las probetas con exMWCNTs con respecto a las de Ani-exMWCNTs (las conductividades eléctricas vistas en el capítulo III se muestran en la Tabla 1 a fin de relacionarlas con la elongación). Posiblemente la longitud relativamente corta de los exMWCNTs y Ani-exMWCNTs, y su mejor dispersión, produjo más resistencia a la deformación del compuesto. En contraste, la probeta de PAni sin carga, se elongó ~2% a pesar de tener una conductividad eléctrica similar al del compuesto con Ani-exMWCNTs. Por otra parte, las probetas con MWCNTs y CNx, tuvieron elongaciones similares entre sí (~1.2%), e igualmente, sus conductividades eléctricas fueron parecidas. Es posible que la mayor longitud de los CNTs y su peor dispersión en la matriz polimérica, hayan influido en una mayor elongación al compararlas con las probetas con nanotubos exfoliados. Las probetas provenientes de compuestos sintetizados con persulfato de amonio (APS por sus siglas en inglés), fueron mucho más frágiles que las preparadas en presencia de FeCl 3, de modo que casi se obtuvo una probeta entera por cada cuatro o cinco segmentadas. Esto 181 pudo haberse debido a la mayor porosidad del material, puesto que su nanoestructuración fue mayor (como se vio en el capítulo previo). Asimismo, en general mostraron menores elongaciones que las probetas anteriores, que fueron de ~0.2 a ~1.8% (Tabla 1). Además, algunas probetas no mostraron elongación alguna (en su mayoría de 4 mm) y otras no fueron lo suficientemente largas para probarlas (con CNx y Ani-exMWCNTs, nivel de carga de 0.15%, y diámetro de 4 mm). Como en el caso anterior, la elongación mayor se obtuvo con la probeta de PAni de 2 mm sin carga, aunque la probeta de 4 mm (equivalente a la obtenida utilizando FeCl3), se elongó ~1%. En general, un diámetro menor de las probetas favoreció una mayor elongación. La mayor deformación de la PAni sin carga confirma que las cargas dispersas en la matriz polimérica ejercen resistencia a la deformación, en concordancia a lo encontrado por Spinks y colaboradores [18], donde las fibras con SWCNTs (0.76 wt%) se elongaron 0.8% y las de PAni sin nanotubos 1.3%. Sin embargo, en el estudio de Mottaghitalab y colaboradores concluyeron que las fibras de compuestos con SWCNTs (2 wt%) tuvieron una mayor elongación que las fibras de PAni sin carga [19]. Los SWCNTs tienden a formar cuerdas [20], lo cual puede disminuir su interacción con la matriz polimérica, a pesar de aumentar la conductividad eléctrica de la fibra. También se observa, a diferencia de las probetas de compuestos sintetizados con FeCl3, que no hubo una relación directa entre la razón de aspecto de la carga y la elongación de las probetas. No hay una distinción clara del efecto de cada carga en la elongación de las probetas, pero sí es notable el efecto en la conductividad. Puede concluirse que una baja porosidad en las probetas de compuestos de PAni las vuelve más resistentes e incrementa la interacción carga/polímero para aplicaciones mecánicas; una mayor relación de aspecto de las cargas (como los CNTs en comparación a los exMWCNTs) ofrece menos resistencia a la elongación; así como un menor diámetro de las probetas permite la elongación con mayor facilidad. En los experimentos realizados no se ha podido medir la velocidad de elongación debido las limitaciones en el análisis por imágenes digitales, puesto que las elongaciones de las probetas fueron muy bajas. El siguiente paso en este estudio, sería el desarrollo de actuadores de mucho menor espesor, y que permitan detectar con más facilidad sus elongaciones mediante imágenes 182 digitales. Por ello se decidió construir actuadores flexionantes con películas de los compuestos, cuyos resultados se describen en la siguiente sección. Tabla 1. Conductividades eléctricas y elongaciones de probetas de compuestos de PAni (ante un potencial de ±1.25 V) sintetizadas con cloruro férrico y persulfato de amonio. Agente Carga Nivel de σ (S/cm) Diámetro Elongación oxidante en la carga (%) de la síntesis de la probeta PAni (mm) (%) Cloruro férrico - 0.0 0.497±0.123 4 2 (FeCl3) MWCNTs 2.3 3.973±0.743 4 1.2 CNx 2.3 2.097±0.543 4 1.2 exMWCNTs 2.3 2.215±0.640 4 0.5 Ani- 2.3 0.573±0.730 4 0.5 0.0 0.293 ± 2 1.8 0.153 4 1.0 1.823 ± 0.63 2 0.5 4 0.2 3.110 ± 2 0.4 0.707 4 0.0 1.363 ± 2 0.0 0.337 4 X* 3.795 ± 2 0.4 0.525 4 0.0 0.223 ± 2 0.4 0.110 4 0.0 0.397 ± 2 0.2 0.287 4 0.0 0.437 ± 2 0.2 0.203 4 X 0.787 ± 2 0.5 0.213 4 0.0 exMWCNTs Persulfato de - amonio (APS) MWCNTs 0.15 0.50 CNx 0.15 0.50 COx 0.15 0.50 Ani- 0.15 exMWCNTs 0.50 *No se logró obtener fibras completas. 183 Nota: MWCNTs y CNx con anclaje de nanopartículas de Ag fueron utilizados también como cargas en estos experimentos, pero los resultados no se incluyeron debido a que la Ag no tuvo efecto significativo en la conductividad eléctrica ni en la elongación de las fibras. 5.3.2. Actuadores flexionantes. Como se vio en el capítulo anterior, la disolución de la PAni (en la forma de base de esmeraldina) en NMP, ocasionó la pérdida de la morfología nanofibrilar obtenida desde la síntesis, aunque se ganó plasticidad (debido al NMP que permaneció en la PAni) y firmeza en las películas conformadas por drop-casting. Esto hizo factible la elaboración de actuadores bicapa flexionantes. Los actuadores, accionados en HCl 1M (sumergidos en ~90%), se probaron (como ánodo o cátodo) con 3 o 5 V (Tabla 2). Al funcionar como ánodo (potencial positivo), la película se flexionó ~47–154º (o elongó 0.4 – 1.8%, donde la elongación de las películas se obtuvo mediante el análisis geométrico descrito en [4]), ocurriendo lo contrario al accionarse como cátodo (figura 4). Atendiendo a los datos de la Tabla 2, los mejores actuadores fueron los de PAni/MWCNTs y PAni/GO, con un voltaje de accionamiento de 3 V, una flexión media de ~95–150º (excepto el actuador de 1.1 wt% de GO), un tiempo de respuesta menor a 2 min, y casi 20 ciclos de trabajo. Es notable que a pesar de la diferencia estructural de los MWCNTs en comparación al GO, se hayan obtenido resultados tan similares en el accionamiento. Es posible que la excelente dispersión del GO en la matriz polimérica y el dopaje adicional que provocó en la PAni, hayan llevado a su compuesto al nivel del de PAni/MWCNTs. En contraste, los peores actuadores fueron los de PAni/CNx, los cuales se mostraron rígidos (flexiones de ~50º), solamente soportando ~6 ciclos de trabajo. Los actuadores de PAni/HR-CNF estuvieron casi en el nivel de los de PAni sin carga, con 5 V de accionamiento, ~100º de flexión media, y ciclos de trabajo casi de 10. Ante estos resultados, se eligió a los actuadores de PAni/MWCNTs y PAni/GO para la experimentación subsecuente. 184 Figura 4. Actuadores de PAni/MWCNTs accionados en HCl 1 M con un voltaje de 3 V. A) El actuador de la izquierda es el cátodo (reducción de la PAni, contracción) y el de la derecha el ánodo (oxidación de la PAni, elongación); B) el voltaje se invirtió, siendo ahora el ánodo el actuador de la izquierda y el cátodo el de la derecha. 185 Tabla 2. Flexión media, tiempo de respuesta y ciclos de trabajo de actuadores en función del tipo de carga su nivel de contenido en el compuesto de PAni. Carga % de σ (S/cm) Voltaje Flexión Tiempo Ciclos carga óptimo* media (°)** de de respuesta trabaj (min) o - - 4.80 ± 0.55 5 109.2 ± 22.7 2 10 ± 3 MWCNTs 1.1 6.14 ± 1.14 3 154.2 ± 33.4 1 40 ± 11 3.3 3.12 ± 0.73 3 95 ± 25.6 1.5 18 ± 7 1.1 4.52 ± 2.57 5 61.7 ± 29.1 2 7±4 3.3 2.94 ± 1.65 5 46.7 ± 19.5 2 5±3 1.1 4.23 ± 0.52 5 110 ± 22.9 2 12 ± 4 3.3 4.97 ± 0.67 5 73.3 ± 21.4 2 10 ± 3 1.1 8.20 ± 4.37 3 47.15 ± 15 1.5 7±3 3.3 8.52 ± 2.90 3 130.8 ± 31.7 1.5 28 ± 11 CNx HR-CNF GO *Voltaje al cual la máxima flexión ocurrió en dos minutos a lo sumo. **La flexion fue el ángulo entre el extremo móvil del actuador cuando se elonga y se contrae. Se indica en la bibliografía [13, 22, 23] que los ácidos sulfónicos como el canforsulfónico (CSA), el naftalensulfónico (NSA) y el metanosulfónico (MSA) han impedido la degradación de la PAni en su estado de pernigranilina (arriba del segundo pico de oxidación de la PAni). Debido a esto, particularmente el MSA ha beneficiado el rendimiento de los actuadores de PAni [13]. Se realizó una prueba con actuadores de PAni/MWCNTs (1.1 wt%) y PAni/GO (3.3 wt%) en un electrolito de MSA 1.5 M. Ambos actuadores se activaron con 3 V. Para el actuador con MWCNTs, se obtuvo una flexión media, un tiempo de respuesta y ciclos de trabajo de 124 ± 32º, 1.5 min y 43 ± 8, respectivamente. Para el actuador con GO se obtuvo para las mismas respuestas, 119 ± 24.5º, 1 min y 37 ± 12, respectivamente. Aunque no se notó una mejoría en la flexión media ni en los tiempos de respuesta (con respecto a los actuadores equivalentes de la Tabla 2), sí hubo una mejoría en el número de ciclos de trabajo. Por lo tanto, para llevar a cabo la optimización con estos actuadores, se optó por trabajar con MSA en lugar de HCl. 186 5.3.3. Optimización. Se accionaron actuadores flexionantes de PAni/MWCNTs y PAni/GO (por cuadruplicado, figura 5 A) utilizando un diseño central compuesto (DCC) de dos factores: nivel de carga en el compuesto (Carga) y nivel de voltaje (Potencial), y tres respuestas: flexión media (FM), velocidad de flexión (VF), y número de ciclos de trabajo (CT) (Tabla 3), utilizándose MSA 1.5 M como electrolito. Los actuadores se sumergieron ~90% en el electrolito y fueron alternadamente ánodo (oxidación de la PAni) y cátodo (reducción de la PAni) (figura 5 B). Al accionarlos, con el potencial de prueba conectado entre ellos, durante más de 2 min, el ánodo se elongó y el cátodo se contrajo. Al invertir el voltaje, el que había sido cátodo y ahora ánodo, se elongó, y el nuevo cátodo se contrajo, y así se invirtió el voltaje sucesivamente (figura 6 A, B). Figura 5. A) Fuente de voltaje y celdas de dos electrodos para las pruebas de accionamiento de los actuadores flexionantes; B) Detalle de una celda donde se observan a los actuadores funcionando como ánodo o cátodo. 187 Figura 6. A) Actuadores flexionantes de PAni/MWCNTs accionados a un potencial de 1.84 V; B) mismos actuadores con el potencial invertido, la flexión ahora ocurrió en el sentido opuesto. Cuando la PAni se oxida en un electrolito protónico (pasando de leucoesmeraldina a esmeraldina y luego a pernigranilina [1-14]), en este caso en MSA 1.5 M, se expande debido a la inserción de iones CH3SO3- que tratarán de neutralizar la carga positiva dada por la fuente de voltaje (figura 7). Asimismo, las cadenas poliméricas, cargadas positivamente, se repelerán electrostáticamente, favoreciendo esta elongación (o flexión en el sentido de la película de celofán). En el caso opuesto, cuando el voltaje se invierte, ocurre la reducción de la PAni (pasa de pernigranilina a esmeraldina y luego a leucoesmeraldina), en la cual los aniones son expulsados de la PAni, propiciando una contracción del material (o una flexión hacia el lado opuesto de la película de celofán), y así sucesivamente. La carga (MWCNTs o GO) dispersa en la matriz polimérica, de acuerdo a sus características y propiedades, afectará la elongación (o flexión), la velocidad de elongación y la resistencia del actuador al desgaste. 188 Figura 7. Esquema del accionamiento de dos actuadores flexionantes configurados como ánodo (el de la izquierda, que contiene MWCNTs) y cátodo (el de la derecha, que contiene GO), en un electrolito de ácido metano sulfónico (MSA) 1.5 M. Para la optimización se utilizó un Diseño Central Compuesto (DCC por sus siglas en inglés) [21] de dos factores: nivel de carga en el compuesto (0.55, 0.91, 1.84, 2.6, 3.94%) y nivel de voltaje (0.88, 1.5, 2.5, 3.5, 4.98 V). Con la siguiente codificación: -1.414, -1, 0, +1, +1.414, para los niveles de los factores. Se midió la flexión, la velocidad de flexión y el número de ciclos de trabajo de los actuadores. Los resultados del DCC de los actuadores flexionantes de compuestos de PAni con MWCNTs (Tabla 3), mostraron, de manera general, que una mayor carga en el compuesto (2.6 y 3.9 wt%, de las pruebas 3, 4, 10), volvió más rígidas a las películas, aunque una carga muy baja (0.55 wt% de prueba 11) tampoco ocasionó una flexión alta. En cambio, una carga media (1.84%) junto con un voltaje medio (2.5 V) provocaron las máximas flexiones. Las mayores flexiones se lograron con un voltaje alto (3.5 V y 4.98 V de las pruebas 2 y 12, respectivamente). Las velocidades de flexión más altas se obtuvieron también con potenciales altos (3.5 y 4.98 V) junto con una carga baja o media (0.91 o 1.84 wt%); las menores, se tuvieron con potenciales bajos (0.88 y 1.5 V), siendo más baja aún, con un nivel alto de carga (2.6 wt%). 189 Tabla 3. Diseño central compuesto (DCC) de dos factores: Carga en el compuesto y potencial, y tres respuestas: flexión media (FM), velocidad de flexión (VF), y ciclos de trabajo (CT), para actuadores flexionantes de PAni/MWCNTs. Prueba Carga (wt%) Potencial (V) FM (°)* VF (°/min) CT (valores (valores codificados) codificados) 1 0.91 (-1)** 1.5 (-1) 19.6 ± 13.7 14.3 ± 10.6 116 2 0.91 (-1) 3.5 (1) 63.7 ± 30.4 62.4 ± 37.5 18 3 2.6 (1) 1.5 (-1) 13.9 ± 3.5 9.4 ± 2.4 42 4 2.6 (1) 3.5 (1) 33.3 ± 11.4 24.4 ± 9.5 12 5 1.84 (0) 2.5 (0) 74.6 ± 18.0 49.0 ± 17.1 91 6 1.84 (0) 2.5 (0) 65.8 ± 17.2 47.6 ± 19.0 70 7 1.84 (0) 2.5 (0) 50.6 ± 15.1 33.8 ± 8.6 66 8 1.84 (0) 2.5 (0) 56.9 ± 11.3 36 ± 13.4 77 9 1.84 (0) 2.5 (0) 69.5 ± 15.2 44.6 ± 13.3 81 10 3.94 (1.414) 2.5 (0) 12.3 ± 2.3 9.3 ± 2.3 5 11 0.55 (-1.414) 2.5 (0) 32.7 ± 7.8 23.7 ± 7.3 40 12 1.84 (0) 4.98 (1.414) 71.4 ± 25.2 69.1 ± 38.7 13 13 1.84 (0) 0.88 (-1.414) 19.4 ± 11.8 15.4 ± 11.0 36 *La flexión se midió con la ubicación del extremo móvil del actuador, desde la elongación máxima hasta la contracción máxima. **Entre paréntesis se muestra el valor codificado del diseño experimental. Mediante el análisis de varianza (ANOVA) del experimento de optimización, se obtuvieron modelos cuadráticos empíricos de cada respuesta experimental (ecuaciones 1, 2, 3): FM= 63.48 - 8.11(Carga) + 17.13(Potencial) - 6.18(Carga)(Potencial) - 20(Carga)29.4(Potencial)2, R2=0.941 (1) VF= 42.2 - 7.91(Carga) + 17.4(Potencial) - 8.28(Carga)(Potencial) - 13.29(Carga)20.41(Potencial)2, R2=0.940 (2) CT= 77 - 16.19(Carga) - 20.07(Potencial) + 17(Carga)(Potencial) - 21.38(Carga)220.38(Potencial)2, R2=0.812 (3) 190 donde FM, VF y CT son flexión media, velocidad de flexión y ciclos de trabajo, respectivamente; los factores nivel de carga en el compuesto y voltaje, se denotan como Carga y Potencial, respectivamente. El primer término de las tres ecuaciones, es la media de sus respectivas respuestas experimentales, la cual fue 63.48º para FM, 42.2º/min para VF, y 77 ciclos para CT. Los coeficientes de los factores, interacciones y términos cuadráticos, cuantifican su respectivo efecto adicionándosele a la media mediante su valor numérico y su signo, ya que los valores codificados de los tres niveles (bajo, medio y alto) de cada factor tienen valores -1, 0 y +1 (Tabla 3). Las R2 para las ecuaciones 1 y 2 es muy alta, 0.94, y para la 3 es buena, 0.812, lo cual indica que los modelos representan muy bien la variabilidad factorial de los experimentos. En la ecuación 1, la Carga y su término cuadrático, con coeficientes de -8.11 y -20, es el factor que más efecto negativo tiene en la media, de 63.48, cuando está en su nivel alto (Carga = +1). Es decir, un nivel alto de carga decrecerá la flexión de los actuadores. Por otra parte, el Potencial, con un coeficiente de 17.13, y su término cuadrático con -9.4, indican una mayor flexión a niveles altos de este factor, aunque a un nivel bajo (Potencial = -1), el decrecimiento de la flexión será más significativo aún. La interacción (Carga)(Potencial), con un coeficiente de -6.18, indica que si ambos factores, tienen niveles altos o bajos a la vez, la flexión de los actuadores se vería afectada negativamente. Aunque si el Potencial tiene nivel alto, y la Carga nivel bajo, entonces sólo así se contribuirá a incrementar la flexión. El caso opuesto, Potencial bajo y Carga alta, no tiene sentido físico. Por lo tanto, para una mayor flexión de los actuadores, se necesitará un Potencial alto (3.5 V) y una Carga de 1.46%. La representación gráfica de este modelo aparece en la figura 8 I a. 191 Figura 8. Superficies de respuesta de actuadores flexionantes de I) PAni/MWCNTs y II) PAni/GO. a) Flexión, b) Velocidad de Flexión, c) Ciclos de Trabajo (en función del Potencial y el % de Carga del compuesto). La ecuación de la velocidad de flexión (VF) (ecuación 2) sigue una tendencia muy parecida al modelo anterior, donde el factor con más efecto es la Carga, con coeficientes de -7.91, y -13.29 para su término cuadrático. Un nivel alto de Carga, disminuirá significativamente la velocidad de flexión del actuador. En cambio, el coeficiente del término lineal del Potencial (+17.4), incrementaría o disminuiría notablemente a la media de 42.2, según tenga un nivel alto o bajo, puesto que el coeficiente de su término cuadrático es bajo, -0.41 (habiendo una mayor “linealidad” en este factor). La interacción (Carga)(Potencial), con un coeficiente de -8.28, puede explicarse de manera idéntica que 192 en el caso anterior, sólo que en este caso, tiene una mayor significancia debido al menor valor de la media (42.2). Los parámetros con los que se obtiene una VF mayor es con un Potencial alto (3.5 V) y una Carga de 1.23%. La superficie de respuesta de este modelo, puede observarse en la figura 8 I b. El modelo de los ciclos de trabajo (CT) (ecuación 3) es distinto a los anteriores puesto que el Potencial, en este caso, afecta negativamente a la durabilidad de los actuadores. La media, como puede verse en la ecuación, es de 77. Los coeficientes de la Carga, y de (Carga)2, de -16.19 y -21.38, respectivamente, indican que un nivel alto de carga tenderá a disminuir el número de ciclos de trabajo. Asimismo un Potencial alto, con un coeficiente de -20.07, y -20.38, para su término cuadrático, será aún más significativo que el factor anterior en cuanto a la disminución de los ciclos. En el caso del coeficiente de la interacción (Carga)(Potencial), ahora con un signo positivo (+17), contribuye positivamente a la media solamente si ambos factores son altos o bajos, teniendo sólo un sentido físico el segundo caso. Su representación gráfica, puede observarse en la figura 8 I c. Las condiciones experimentales para una cantidad de ciclos de trabajo mayor son: Un potencial de 1.72 V y un nivel de carga de 1.18%. Las respuestas experimentales del Diseño Central Compuesto de los actuadores de PAni/GO (Tabla 4), muestran una tendencia parecida a los resultados de los actuadores de PAni/MWCNTs. Los niveles altos de potencial (3.5 V y 4.98 V, de las pruebas 2, 4 y 12) incrementaron la flexión (FM) y la velocidad de flexión (VF) de los actuadores, igualmente, la Carga, en un nivel alto, tuvo un efecto negativo en estas respuestas experimentales. La cantidad de ciclos de trabajo (CT) fue menor que la de los actuadores con MWCNTs. Esta respuesta no pareció tan sensible al variar el nivel de Carga (pruebas 10 y 11) aunque sí un poco más al hacerlo el Potencial, donde a niveles altos, el número de CT fue menor (pruebas 2, 4, y 12). Puede observarse con más claridad en este caso, que FM y VF, son variables antagónicas a CT, por lo que los modelos matemáticos empíricos fueron de mucha utilidad para encontrar el punto óptimo de máximo rendimiento de estos actuadores. 193 Tabla 4. Diseño central compuesto (DCC) de dos factores: Carga en el compuesto y potencial, y tres respuestas: flexión media (FM), velocidad de flexión (VF), y ciclos de trabajo (CT), para actuadores flexionantes de PAni/GO. Prueba Carga (wt%) Potencial (V) FM (°)* VF (°/min) CT (valores (valores codificados) codificados) 1 0.91 (-1)** 1.5 (-1) 23.4 ± 9.8 11.8 ± 3.9 9 2 0.91 (-1) 3.5 (1) 58.5 ± 20.0 80.1 ± 37.2 20 3 2.6 (1) 1.5 (-1) 17.0 ± 8.4 11.5 ± 5.9 22 4 2.6 (1) 3.5 (1) 75.0 ± 22.0 57.8 ± 30.5 23 5 1.84 (0) 2.5 (0) 31.9 ± 10.0 20.4 ± 9.2 37 6 1.84 (0) 2.5 (0) 68.4 ± 21.7 49.1 ± 35.0 35 7 1.84 (0) 2.5 (0) 106.6 ± 24.2 77.6 ± 30.2 44 8 1.84 (0) 2.5 (0) 44.1 ± 19.7 20.3 ± 17.3 51 9 1.84 (0) 2.5 (0) 40.8 ± 10.3 29.1 ± 12.3 49 10 3.94 (1.414) 2.5 (0) 23.9 ± 7.4 18.9 ± 11.8 42 11 0.55 (-1.414) 2.5 (0) 62.1 ± 19.7 23.5 ± 8.0 40 12 1.84 (0) 4.98 (1.414) 57.1 ± 18.3 79.7 ± 33.2 20 13 1.84 (0) 0.88 (-1.414) 15.4 ± 10.2 11.2 ± 7.4 14 *La flexión se midió con la ubicación del extremo móvil del actuador, desde la elongación máxima hasta la contracción máxima. **Entre paréntesis se muestra el valor codificado del diseño experimental. Los modelos obtenidos a través del ANOVA fueron ajustados retirándose términos (exceptuando los efectos principales) con nula significancia. Estos se muestran a continuación: FM= 49.5 - 5.49(Carga) + 19(Potencial) - 6.49(Potencial)2, R2=0.601 (4) VF= 33.42 - 3.64(Carga) + 26.43(Potencial) - 5.5(Carga)(Potencial) - 4.37(Carga)27.76(Potencial)2, R2=0.866 (5) CT= 43.2 + 2.35(Carga) + 2.56(Potencial) - 3.73(Carga)2- 15.73(Potencial)2, (6) 194 R2=0.807 El primer término de las ecuaciones después del signo igual (49.5º, 33.42º/min, 43.2 ciclos de trabajo) es la media de sus respectivas respuestas experimentales. La R2 = 0.601 de la ecuación 4 fue relativamente baja, por lo que el modelo describe en un 60% la variabilidad de los factores en el experimento, lo cual se debe a un error experimental más significativo que en el caso de los actuadores con MWCNTs, donde la certidumbre alcanzó un 94% (ecuación 1). En cambio, las R2 de los modelos 5 y 6, tienen una mayor predictibilidad con valores de 0.866 y 0.807, respectivamente. En el modelo de la flexión media (ecuación 4), el coeficiente -5.49 de la Carga indica que en un nivel alto, se contribuirá negativamente en la flexión, como ha sido la característica general de los actuadores flexionantes. Más significativo aún es el Potencial con un coeficiente lineal de +19, y cuadrático de -6.49 los cuales establecen que un voltaje a un nivel alto, favorecerá la flexión de los actuadores. Al no haber término cuadrático de la Carga, prácticamente su efecto en FM será lineal (figura 8 II a). La máxima flexión media se obtendría en un actuador con un Potencial alto (3.5 V) y un nivel de Carga bajo (0.91%). En la velocidad de flexión (VF), con una media de 33.42 (ecuación 5), el factor que más efecto tiene es el Potencial, con un coeficiente de 26.43, por lo que a un nivel alto, la velocidad se incrementa, aunque limitado por el coeficiente de -7.76 de su término cuadrático. En cambio, la Carga, con un coeficiente de -3.64 y -4.37 para su término cuadrático, indican que a un nivel alto disminuye la velocidad de flexión. La menor magnitud relativa de los coeficientes de Carga con respecto al Potencial, comparándolo con el modelo de actuadores con MWCNTs (ecuación 2) indica que el GO tiene menor efecto en la velocidad de flexión que los nanotubos. La interacción (Carga)(Potencial) con un coeficiente de -5.5, establece que cuando el nivel de ambos factores es alto o bajo, la VF decrecerá, no así cuando los dos tengan niveles opuestos, en donde solamente un nivel alto de Potencial (y bajo para Carga) tendría sentido físico. El punto óptimo donde el actuador tendría la máxima velocidad de flexión, sería con un Potencial alto (3.5 V) y un nivel de Carga bajo (0.91%). La superficie de respuesta de este modelo, puede apreciarse en la figura 8 II b. El modelo empírico de los ciclos de trabajo (CT), muestra una media de 43.2, donde un nivel alto de Carga, apenas influiría en ésta, puesto que sus coeficientes lineal y cuadrático son 2.35 y -3.73, respectivamente. En contraste, el Potencial tendría más efecto ya que sus coeficientes lineal y cuadrático, de 2.56 y -15.73, indican que un nivel alto de este factor, 195 disminuiría significativamente la vida útil del actuador, pero también lo haría un nivel bajo. Las condiciones experimentales para un actuador que pudiera soportar la mayor cantidad de ciclos de trabajo, sería con un potencial de 2.58 V y un nivel de Carga de 2.03%. La representación gráfica de este modelo puede observarse en la figura 8 II c. Las superficies de respuesta de los modelos cuadráticos empíricos obtenidos del ANOVA (figura 8), en general muestran una similaridad en ambos actuadores, sobre todo para las respuestas flexión media (FM) y velocidad de Flexión (VF) (figura 8 I, II, a y b), donde un Potencial alto y niveles medios para la Carga (sólo para los actuadores de PAni/MWCNTs) incrementó a estas respuestas. Las superficies de respuesta para los ciclos de trabajo (CT) de ambos tipos de actuadores divergieron un poco más entre sí (figura 8 I, II c), aunque es evidente, que el punto óptimo, se encuentra dentro de la región experimental, los cuales, como ya se vió previamente son 1.72 V y 1.18 wt%, para los actuadores con MWCNTs, y 2.58 V y 2.03 wt% para los actuadores con GO, teniendo en cuenta, que para estos últimos, el nivel de carga no fue tan significativo como en el caso de los actuadores con MWCNTs. Al probarse los actuadores de PAni con MWCNTs y GO, se observó que con esta última carga, una parte importante de las películas se seccionaron (figura 9 D, E, F), inutilizándolos sobre todo cuando esta rotura se localiza cerca del conector (figura 9 F). En cambio, los actuadores con MWCNTs, fueron más resistentes en este sentido, la mayoría se conservó sin seccionarse (figura 9 G, H, I). Esto concuerda con el mayor número de ciclos de trabajo logrados por estos últimos (Tablas 3 y 4). 196 Figura 9. A) Actuadores flexionantes de compuestos de PAni; B, C) Actuadores poco antes de sumergirlos en el electrolito; D, E, F) Actuadores de PAni/GO después de ser probados; G, H, I) Actuadores de PAni/MWCNTs después de ser ciclados. En el experimento final (Tabla 5), en el cual se buscó corroborar lo encontrado en la optimización, se decidió utilizar un nivel de carga de ~1.3 wt% para ambos compuestos con MWCNTs y GO. Esta carga, más cercana al nivel de carga del modelo de los CT de los actuadores con MWCNTs, se eligió así debido a que el nivel de GO pareció tener menos efecto en el accionamiento de los actuadores, por lo que pareció más importante ajustarse al nivel de carga de los primeros. El nivel de potencial, se eligió de 2.6 V, más cercano al potencial óptimo del modelo de los CT de los actuadores con GO. Este nivel estuvo por encima de 1.72 V (nivel de potencial óptimo de los CT de los actuadores con MWCNTs), porque se consideró importante aumentarlo debido al resultado de la flexión y la velocidad de flexión, las cuales fueron superiores en un nivel medio (2.5 V) y alto (3.5 V) de voltaje. El accionamiento de los actuadores se llevó a cabo por cuadruplicado. Los valores de flexión (FM), velocidad de flexión (VF) y ciclos de trabajo (CT), de ambos 197 actuadores (Tabla 5), estuvieron muy cercanos a los resultados obtenidos cuando se utilizaron niveles medios de Carga y Potencial (1.84 wt%, 2.6 V) (Tablas 3 y 4). En general, puede concluirse que los actuadores de PAni/MWCNTs tuvieron un mejor rendimiento, puesto que el número de ciclos fue significativamente superior. Como ya se había mencionado, el problema de los actuadores con GO fue que se seccionaron más rápidamente (figura 9 D, E, F), disminuyendo así su rendimiento. Tabla 5. Experimento de corroboración de los resultados de optimización de actuadores de compuestos de PAni/MWCNTs y PAni/GO con un nivel de carga de 1.3 wt% y accionados con un potencial de 2.6 V. FM (°) VF (°/min) CT Actuador PAni/MWCNTs 66.8 ± 15.4 37.5 ± 11.3 79 PAni/GO 59.3 ± 19.6 45.6 ± 15.7 43 5.4. CONCLUSIONES Una menor porosidad o nanoestructuración, obtenida utilizando FeCl3 como oxidante en la polimerización de la PAni, benefició a los actuadores lineales en su facilidad de conformado y en una elongación que permitió apreciar el efecto de las cargas dispersas en su matriz polimérica. Las cargas que mostraron tener una excelente interacción con la PAni, como los exMWCNTs, Ani-exMWCNTs y los COx, no contribuyeron a mejorar las características de los actuadores, como su conductividad eléctrica y la elongación de los mismos. El procesado (disolución en NMP) de los compuestos de PAni para obtener películas permitió observar mucho mejor la elongación (flexión) de los actuadores en función de las cargas dispersas en el polímero. A pesar de que los CNx mostraron una buena interacción con la PAni, sus aglomerados perjudicaron a la flexibilidad de las películas, por lo que, bajo las condiciones experimentales utilizadas, fueron rechazados como carga en los actuadores. 198 Los HR-CNF no mostraron efecto alguno en los actuadores donde estuvieron dispersos con respecto a los de la PAni sin carga, a pesar de que la flexibilidad de las películas fue buena. El GO y los MWCNTs resultaron ser las mejores cargas para su aplicación como actuadores. El GO fue la carga que mejor se dispersó en la matriz polimérica, y a pesar de que es prácticamente aislante, sus grupos funcionales incrementaron el dopaje de la PAni. En el caso de los MWCNTs, la dispersión no fue tan alta pero sí la suficiente para contribuir al dopaje en la PAni y además servir como puentes de transporte electrónico entre algunas regiones del polímero. Las condiciones experimentales óptimas para los actuadores flexionantes de PAni/MWCNTs y PAni/GO fueron: nivel de carga de 1.3 wt% y nivel de potencial de 2.6 V. En general, los actuadores de PAni/MWCNTs tuvieron un mejor rendimiento que los actuadores de PAni/GO, debido a una mayor resistencia de la película al ciclado redox. 199 REFERENCIAS [1] Herod, T. E.; Schlenoff, J. B. Doping-Induced Stretchoelectrochemistry. Chem. Mater. 5, 1993, 951–955. Strain in Polyaniline: [2] Kaneko, M.; Fukui, M.; Takashima, W.; Kaneto, K. Electrolyte and strain dependences of chemomecanical deformation of polyaniline film. Synthetic Metals 84, 1997, 795-796. [3] Takashima, W.; Uesugi, T.; Fukui, M.; Kaneko, M.; Kaneto, K. Mechanochemoelectrical Effect of Polyaniline Film. Synthetic Metals 85, 1997, 13951396. [4] Takashima, W.; Kaneko, M.; Kaneto, K.; MacDiarmid, A. G. The electrochemical actuator using electrochemically-deposited poly-aniline film. Synthetic Metals 71, 1995, 2265-2266. [5] Kaneko, M.; Kaneto, K. Electrochemomechanical deformation of polyaniline films doped with self-existent and giant anions. Reactive & Functional Polym. 37, 1998, 155161. [6] Kaneto, K.; Kaneko, M.; Min, Y.; MacDiarmid, A. G. “Artificial muscle”: Electromechanical actuators using polyaniline films. Synthetic Metals 71, 1995, 22112212. [7] Tominaga, K.; Hashimoto, H.; Takashima, W.; Kaneto, K. Training and shape retention in conducting polymer artificial muscles. Smart Mater. Struct. 20, 2011, 124005 (6pp). [8] Kaneko, M.; Kaneto, K. Electrochemomechanical deformation in polyaniline and poly(o-methoxyaniline). Synthetic Metals 102, 1999, 1350-1353. [9] Pomfret, S. J.; Adams, P. N.; Comfort, N. P.; Monkman, A. P. Electrical and mechanical properties of polyaniline fibres produced by a one-step wet spinning process. Polymer 41, 2000, 2265-2269. [10] Qi, B.; Lu, W.; Mattes, B. R. Strain and energy efficiency of polyaniline fiber electrochemical actuators in aqueous electrolytes. J. Phys. Chem. B 108, 2004, 6222-6227. [11] Lu, W.; Smela, E.; Adams, P. N.; Zuccarello, G.; Mattes, B. R. Development of Solidin-Hollow Electrochemical Linear Actuators Using Highly Conductive Polyaniline. Chem. Mater. 16, 2004, 1615-1621. [12] Smela E.; Lu, W.; Mattes, B. R. Polyaniline actuators Part. 1. PANI (AMPS) in HCl. Synthetic Metals 151, 2005, 25-42. [13] Smela E., Mattes R. B. Polyaniline actuators Part 2. PANI (AMPS) in methanesulfonic acid. Synthetic Metals 151, 2005, 43-48. [14] Lu, W.; Smela, E.; Mattes, B. R.; Adams, P. N.; Zuccarello, G. Electrochemical devices incorporating high-conductivity conjugated polymers. U. S. Patent 6,982,514, B1, Jan. 3, 2006. [15] Gu, B. K.; Ismail, Y. A.; Spinks, G. M.; Kim, S. I.; So, I.; Kim, S. J. A Linear Actuation of Polymeric Nanofibrous Bundle for Artificial Muscles. Chem. Mater. 21, 2009, 511-515. [16] Mottaghitalab, V.; Spinks, G. M.; Wallace, G. G. The influence of carbon nanotubes on mechanical and electrical properties of polyaniline fibers. Synthetic Metals 152, 2005, 77-80. 200 [17] Mottaghitalab, V.; Spinks, G. M.; Wallace, G. G. The development and characterization of polyaniline–single walled carbon nanotube compuesto fibres using 2acrylamido-2-methyl-1-propane sulfonic acid (AMPSA) through one step wet spinning process. Polymer 47, 2006, 4996-5002. [18] Spinks, G. M.; Mottaghitalab, V.; Bahrami-Samani, M.; Whitten, P. G.; Wallace, G. G. Carbon-Nanotube-Reinforced Polyaniline for High-Strength Artificial Muscles. Adv. Mater. 18, 2006, 18, 637-640. [19] Mottaghitalab, V.; Xi, B.; Spinks, G. M. Wallace, G. G. Polyaniline fibres containing single-walled carbon nanotubes: Enhanced performance artificial muscles. Synthetic Metals 156, 2006, 796–803. [20] Liang, F.; Sadana, A. K.; Peera, A.; Chattopadhyay, J.; Gu, Z.; Hauge, R. H.; Billups, W. E. A Convenient Route to Functionalized Carbon Nanotubes. Nano Let. 4(7), 2004, 1257-1260. [21] Montgomery, D. C. Design and Analysis of Experiments. 5th Edition, John Wiley & Sons, 2004. [22] Bernard, M.-C.; Goff, A. H.-L.; Bich, V. T.; Zeng, W. Study by optical multichannel analysis of the electrochromic phenomena in polyaniline doped with camphorsulfonic acid. Synthetic Metals 81, 1996, 215-219. [23] Tawde, S.; Mukesh, D.; Yakhmi, J. V. Redox behavior of polyaniline as influenced by aromatic sulphonate anions: cyclic voltammetry and molecular modeling. Synthetic Metals 125, 2002, 401-413. 201 202 6. Electrodos de PAni y óxido de grafeno para su aplicación en supercondensadores 203 204 6. Electrodos de PAni y óxido de grafeno para su aplicación en supercondensadores 6.1. ANTECEDENTES Desde hace más de una década, la pseudocapacitancia de los polímeros conductores (CP) ha despertado interés para el desarrollo de supercondensadores [1]. Se han llevado a cabo muchos estudios en los que sobresale la utilización de la polianilina (PAni) debido a su buena estabilidad ambiental, facilidad de síntesis y procesado y su alta capacidad específica [2-4]. Se ha visto que conviene que la PAni sea lo más porosa posible para favorecer el intercambio iónico con el electrolito, por lo que la síntesis de PAni nanoestructurada, mediante el método de polimerización interfacial, es cada vez más frecuente [5-7]. Asimismo, la utilización de distintas cargas de carbono en compuestos de PAni como nanotubos de carbono de capa simple (SWCNTs) [8, 9] y múltiple (MWCNTs) [10, 11], óxido de grafeno (GO) [12] y óxido de grafeno reducido (RGO) [13] ha mejorado las propiedades eléctricas y rigidez del material [14-17]. Esta última propiedad ha contribuido a evitar que la contracción/expansión del compuesto, debido al proceso de óxido/reducción de la PAni, provoque un desprendimiento de la película. En general, los electrodos de polvos de PAni y sus compuestos se elaboran mezclándolos con negro de carbono o grafito en un 10 wt% y posteriormente, se adiciona un ~5 wt% de teflón [12, 18-21] o nafión [22] o caucho [15, 23] (como aglomerantes) para forma una pasta que se unta sobre un sustrato conductor que suele ser de acero. La película recién formada, mediante presión y temperatura se adhiere en el acero. Una morfología nanoestructurada o porosa en la PAni, beneficiaría la velocidad del flujo iónico hacia el material. La desventaja de esta morfología, es que, como se vio en el Capítulo 1, la conductividad tiende a ser menor, existiendo una caída de potencial en las regiones del electrodo más alejadas del acero. Por lo que la electroactividad del material se vería afectada. Los tres métodos principales para determinar la capacidad de los supercondensadores se denominan: Voltamperometría Cíclica (CV, por sus siglas en inglés), Carga/Descarga Galvanostática (corriente constante), y espectroscopía de impedancia electroquímica (EIS, por sus siglas en inglés). 205 En el presente estudio se formaron y adhirieron películas de compuestos de PAni (mediante un drop-casting utilizando N-metil-2-pirrolidona, NMP, como disolvente) sobre placas de acero (~2 cm2). El espesor y la masa de las películas depositadas fue de ~10 μm y ~0.1 mg, respectivamente. Estas películas adheridas en el acero se utilizaron como material activo en los electrodos. A pesar de la evidente disminución de la porosidad de las películas debido al procesado, se obtuvo un buen control del espesor de la película por lo que se pudo estudiar mejor el efecto de la carga dispersa en el compuesto en el almacenamiento de carga eléctrica en un electrolito protónico (H2SO4 1 M). Asimismo, se aplicó un tratamiento térmico (210º C en vacío, 20 min) a las películas para mejorar la adherencia. Las cargas que se probaron en los compuestos de PAni fueron: nanotubos de carbono multicapa (MWCNTs), nanotubos de carbono multicapa dopados con nitrógeno (CNx), nanofibras de carbono de listones grafíticos helicoidales (HR-CNF) y óxido de grafeno (GO). A fin de evitar que una fracción de carga muy alta pueda enmascarar el efecto de la carga dispersa, se optó por trabajar a niveles bajos (1-3.3 wt%). Se utilizaron las técnicas de Voltamperometría Cíclica y Carga/Descarga Galvanostática para determinar la capacidad de carga de los electrodos de los compuestos de PAni. 6.2. EXPERIMENTACIÓN 6.2.1. Materiales Compuestos de PAni con distintas nanoestructuras de carbono, N-metil-2-pirrolidona (NMP), ácido sulfúrico (H2SO4) y un potenciostato. 6.2.2. Métodos 6.2.2.1. Preparación de electrodos Los electrodos se prepararon depositando (por la técnica de drop-casting) ~0.01 mL de disolución de compuestos de PAni en NMP (6% v/w) sobre placas de acero de 1.9 cm 2. El disolvente se retiró, en lo posible, de las películas de compuestos en una estufa de vacío (70°C, 6 h). Una parte de este material fue sometida a un tratamiento térmico (TT) de 210º C durante 20 min también en vacío (Tabla 1). Posteriormente, se conectó un alambre de Cu a un extremo de las placas de acero mediante resina epóxica conductora y se aisló con resina epóxica todo lo que no fuera material activo. El alambre de Cu se introdujo a una varilla hueca de vidrio (Figura 1). La superficie efectiva de los electrodos fue de ~1.7 cm2. 206 Tabla 1. Películas de compuestos de PAni utilizados en la preparación de los electrodos Tipo de carga Contenido de carga (wt %) MWCNTs 1.1 y 3.3 CNx 1.1 y 3.3 HR-CNFs 1.1 y 3.3 GO 1.1 y 3.3 GO/MWCNTs (2:1) 1.3 GO/MWCNTs (1:2) 1.3 La mitad de estas películas (~0.1 mg) se sometieron a un tratamiento térmico de 210º durante 20 min en vacío. Figura 1. Electrodo de compuesto de polianilina. 6.2.2.2. Voltamperometría cíclica de barrido Con todos los materiales preparados se llevaron a cabo mediciones de voltametría cíclica (series de 10 ciclos) con una celda de tres electrodos, donde la referencia, el 207 contraelectrodo y el electrodo de trabajo fueron AgCl/Ag, Pt y compuestos de PAni, respectivamente. Se utilizó H2SO4 1 M como electrolito. Las velocidades de barrido de potencial fueron 20, 50, y 80 mV/s, con una ventana de potencial máxima de -0.5 a 1.1 V. El peso de la película depositada en el colector fue de ~0.1 mg. El cálculo de la capacitancia específica se llevó a cabo mediante la siguiente ecuación (utilizando el menor barrido de potencial): Cesp = ∫(IdV) / 2mυV (1) donde Cesp es la capacidad específica (F/g), I es la corriente (A), V es el potencial (V), υ es la velocidad de barrido de potencial (V/s) y m es la masa del material activo (g). 6.2.2.3. Pruebas de Carga/Descarga galvanostática Electrodos de PAni/GO fueron evaluados en las mismas condiciones experimentales utilizadas en los experimentos de voltametría cíclica. Las corrientes de prueba fueron 0.1, 0.2 y 0.4 mA. El cálculo de la capacitancia específica se realizó con la ecuación siguiente: (2) donde Cesp es la capacidad específica (F/g), I es la corriente (A), ΔV/Δt la razón la pendiente de la variación del potencial con respecto el tiempo (V/s) y m la masa del material activo (g). 6.2.2.4. Medición de la conductividad eléctrica de las películas de compuestos La conductividad eléctrica de las películas dopadas con ácido metanosulfónico (MSA) con y sin TT, se midió mediante el método de las cuatro puntas (0.7 mm entre ellas) utilizando una corriente constante de 0.3 mA. 6.2.2.5. Análisis termogravimétrico de los compuestos de PAni Compuestos de PAni desdopadas con NaOH 0.2 M y plastificadas con N-metil-2pirrolidona fueron descompuestos térmicamente mediante un análisis termogravimétrico (TGA) de 35 a 1000º C los primeros y de 35 a 700º C los segundos, en un ambiente de nitrógeno. Se utilizó un equipo simultáneo TG-DTA METTLER TOLEDO modelo TGA/SDTA851e/SF/1100. 208 6.2.2.6. Análisis termomecánico de películas de compuestos de PAni tratadas térmicamente Tiras de películas de PAni y sus compuestos (espesor ~30 μm, ancho = 1.6 mm, área transversal = 4.8 X 10-8 m2), con tratamiento térmico (TT, 210ºC en vacío, 20 min), fueron probadas por duplicado mediante un análisis termo-mecánico (TMA), para evaluar su deformación en función a la temperatura (35-350º C) en un ambiente de nitrógeno. Se utilizó un equipo TMA de la marca TA Instruments modelo Q400. 6.3. RESULTADOS Y DISCUSIÓN Tras una serie de pruebas, para conocer la cantidad de material óptima depositada en el sustrato de acero mediante drop-casting, se llegó a películas de 1.7 cm2, con un espesor de ~30 μm y una masa de ~0.1 mg, con una disolución de PAni en NMP de 6% (w/v). La adherencia en el colector de las películas de PAni y sus compuestos es uno de los principales retos en el desarrollo de electrodos para supercondensadores, por lo que se evaluó en esta tesis el efecto de la temperatura en la adherencia de las películas de compuestos de PAni plastificadas con NMP. Antes de mostrar estos resultados, se verá el comportamiento electroquímico de estas películas sin tratamiento térmico. 6.3.1. Voltametría cíclica de películas de compuestos de PAni sin tratamiento térmico La voltametría cíclica de electrodos de PAni a 20 mV/s (figura 2 a) se observan los picos iniciales (líneas azules) de reducción (~0.05 V) y oxidación (~0.45 V), los cuales se desplazan a la derecha (0.7 y 5.6 V) y pierden intensidad (líneas rojas) conforme transcurre la serie de 10 ciclos. Esto indica que el sistema tiende a la inestabilidad (sus picos redox se van alejando entre sí) y la capacidad de almacenamiento de carga de la película decae rápidamente. Al aplicar un barrido de potencial de 50 mV/s (Figura 2 b), los picos redox se observan más alejados entre sí (~0.6 V) y sigue observándose su decaimiento. Este comportamiento electroquímico de la película podría deberse a que la PAni, conforme pasa del estado de leucoesmeraldina (reducido) al de esmeraldina (semioxidado), y viceversa, sufre una deformación (contracción/expansión) [24-26], ante lo cual la película va desprendiéndose del acero conforme transcurren los ciclos redox. En la Figura 2 se muestra también el resultado del cálculo de la capacidad específica del material. La capacidad específica a un barrido de 20 mV/s, de 326.4 F/g (en el ciclo 10), 209 disminuyó a 187.3 F/g (en el ciclo 20) a 50 mV/s. Esta disminución, además de la pérdida de eficiencia del sistema, se debe a que cuanto mayor sea el barrido de potencial, menos oportunidad hay de que los aniones del medio, se introduzcan a las cadenas poliméricas. Figura 2. Voltamogramas de PAni con barridos de potencial de (a) 20 mV/s, (b) 50 mV/s. En la Figura 3 se muestra la voltametría cíclica de los compuestos de PAni con MWCNTs a dos niveles distintos de carga. Se muestra también un decaimiento de los picos redox conforme transcurren los ciclos, pero éstos se muestran más cercanos entre sí, sobre todo los del compuesto con un nivel de carga de 1.1 wt% a 20 mV/s (figura 3 a) en 0.8 y 0.27 V, respectivamente. Sin duda, los nanotubos de carbono, al incrementar la conductividad eléctrica del compuesto, mejoraron su actividad electroquímica, como ya ha sido mostrado con anterioridad [8-11, 15, 18, 19, 23]. El voltamograma del compuesto con un nivel de MWCNTs de 3.3% (Figura 3 c) muestra una disminución/alejamiento de los picos redox de una manera más marcada. Como era de esperar, los voltamogramas de los compuestos a 50 mV/s (Figura 3 b, d) muestran un módulo de densidad de corriente mayor, y aunque se aprecia un decaimiento de la intensidad de los picos, éste es menor que el de los ciclos iniciales (hay una cierta estabilización). En este caso, el voltamograma correspondiente a un nivel de carga de 3.3% (Figura 3 d) muestra un mayor decaimiento conforme transcurren los ciclos al compararlo con el del compuesto con 1.1 % de carga (Figura 3 b), e incluso puede apreciarse cierto ruido (mayor en la región de oxidación, 0.5-0.75 V). 210 A pesar de la inestabilidad electroquímica observada, estas películas mostraron un mejor comportamiento que las de PAni. Figura 3. Voltamogramas de compuestos de PAni/MWCNTs. (a) 1.1 wt%, 20 mV/s, (b) 1.1 wt%, 50 mV/s, (c) 3.3 wt%, 20 mV/s, (d) 3.3 wt%, 50 mV/s. El efecto de los CNx en los compuestos de PAni fue muy similar al de los MWCNTs como puede observarse en la Figura 4. La diferencia más notable es que en este caso los voltamogramas de los compuestos con mayor cantidad de nanotubos (Figura 4 c, d) muestran unos picos redox más intensos. Esta discrepancia entre nivel de carga de nanotubos en los compuestos y sus voltamogramas podría ser debido al error experimental. Otro aspecto que resalta, es la mejor definición de los picos redox en el voltamograma del compuesto con 1.1 % de CNx a 20 mV/s (Figura 4 a), aunque el decaimiento de los picos es mayor que su equivalente de la Figura 4 a con MWCNTs. 211 Figura 4. Voltamogramas de compuestos de PAni/CNx. (a) 1.1 wt%, 20 mV/s, (b) 1.1 wt%, 50 mV/s, (c) 3.3 wt%, 20 mV/s, (d) 3.3 wt%, 50 mV/s. La voltametría cíclica de los compuestos de PAni con HR-CNF (Figura 5) muestra que el comportamiento electroquímico de las películas fue similar al de los CNTs (Figuras 3 y 4): se observa la disminución entre los picos redox, su decaimiento al transcurrir los ciclos y un decaimiento menor a barridos de potencial de 50 mV/s (Figura 5 b, d). Una característica notable en los voltamogramas b y d, es que en el d (correspondiente al compuesto con 3.3% de filamentos), el pico de oxidación se observa más definido y cercano a 0.4 V. En contraste, este pico se ubica en el b en ~0.46 V, a nivel de carga inferior, lo cual podría deberse también al error experimental. 212 Figura 5. Voltamogramas de compuestos de PAni/HR-CNF. (a) 1.1 wt%, 20 mV/s, (b) 1.1 wt%, (c) 3.3 wt%, 20 mV/s, (d) 3.3 wt%, 50 mV/s. Los voltamogramas de compuestos de PAni y GO (Figura 6) en general muestran un mayor decaimiento de los picos redox a un barrido de 20 mV/s (Figura 6 a, c) y un mayor distanciamiento entre ellos (sobre todo el del compuesto con 1.1% de GO, el cual tuvo que probarse con una ventana de potencial de -0.55 a 0.75 V) al compararlos con los anteriores voltamogramas. Incluso, los voltamogramas en rojo, recuerdan a los obtenidos de la PAni sin carga (Figura 2). En base a estos resultados, el GO es la carga que menos efecto electroquímico tiene en las películas de compuestos de PAni, sobre todo si se observan los voltamogramas a 50 mV/s (Figura 6 b, d), donde los picos redox están ya muy alejados entre sí (mucho más en el b) y la intensidad de éstos no es tan grande. Sin embargo, es destacable la mayor estabilidad de los voltamogramas a un nivel de carga mayor (Figura 6 d) al transcurrir los ciclos, donde el decaimiento es bajo. 213 Figura 6. Voltamogramas de compuestos de PAni/GO. (a) 1.1 wt%, 20 mV/s, (b) 1.1 wt%, 50 mV/s, (c) 3.3 wt%, 20 mV/s, (d) 3.3 wt%, 50 mV/s. 6.3.2. Voltametría cíclica de películas de compuestos de PAni con tratamiento térmico El tratamiento térmico (TT) de las películas de compuestos de PAni depositadas sobre acero se llevó a cabo con todos los compuestos de PAni hasta ahora vistos incluyendo además dos compuestos con carga “híbrida” de MWCNTs y GO (1.3 wt%) en proporción de 1:2 y 2:1 (ver Capítulo 2). No todas las películas soportaron el TT (210ºC en vacío durante 20 min) porque sufrieron daño inmediato al aplicarse la voltametría cíclica. Esto sucedió con las películas de PAni, las de compuestos de PAni/MWCNTs (en ambos niveles de carga, 1.1 y 3.3 wt%) y parcialmente con las de PAni/CNx (3.3 wt%). Solamente las películas de compuestos de PAni con HR-CNF o GO soportaron el TT en ambos niveles de carga (1.1 y 3.3 wt%) 214 ganando adherencia en el sustrato de acero y estabilidad electroquímica, como se verá a continuación. En la Figura 7 se muestran los voltamogramas del compuesto de PAni con CNx (1.1 wt%) con TT. En el detalle amplificado dentro del círculo, del voltamograma con un barrido de potencial de 20 mV/s (Figura 7 a) puede observarse que el pico de oxidación, en ~0.5 V, se va incrementando ligeramente conforme transcurren los ciclos, lo cual nunca ocurrió con las películas de compuestos sin TT. La repetibilidad y estabilidad electroquímica durante los ciclos se observan superiores a su equivalente sin TT (Figura 4 a) aunque la intensidad de los picos redox se ve significativamente disminuida (por lo tanto, su capacidad específica también). Lo mismo puede decirse del voltamograma a 50 mV/s (Figura 7 b), pues se muestra estable al transcurrir los ciclos y sus picos redox se observan menos intensos, aunque, a diferencia de su equivalente sin TT (figura 4 b) la separación entre estos es mucho mayor (-1.7 y 0.750.85 V para el de reducción y oxidación, respectivamente). Es posible que el pico de oxidación de leucoesmeraldina a esmeraldina (~0.68 V) y el otro de esmeraldina a pernigranilina (~0.85 V), se estén solapando debido a la ampliación de la ventana de potencial (-0.55 a 1 V). Figura 7. Voltamogramas de compuestos de PAni/CNx (1.1 wt%) con tratamiento térmico (TT). (a) 20 mV/s, (b) 50 mV/s. 215 La voltametría cíclica de la película del compuesto de PAni/HR-CNF (1.1 wt%) a 20 mV/s (Figura 8 a) muestra un decaimiento de los picos redox no tan evidente como el de su equivalente sin TT (Figura 5 a), por lo que muestra una mayor estabilidad y reversibilidad electroquímica. En el caso de los voltamogramas a 50 mV/s (Figura 8 b), los picos redox se alejan entre sí y su decaimiento es aún menor que el de los voltamogramas anteriores. De una capacidad específica de 120.4 F/g (ciclo 10) a 20 mV/s, pasó a 70.8 F/g (ciclo 20) a 50 mV/s. En el caso de los compuestos con 3.3% de contenido de HR-CNF, los voltamogramas a un barrido de 20 mV/s (Figura 8 c) muestran que los picos redox disminuyen conforme los ciclos transcurren, aunque no de manera tan evidente como su equivalente sin TT (Figura 5 c). Asimismo, la intensidad de estos picos es significativamente menor. A un barrido de 50 mV/s (Figura 8 d) la distancia entre los picos redox fue mayor (1.2 y 0.7 V, respectivamente) e igualmente se aprecia un menor decaimiento entre estos al transcurrir los ciclos con respecto a los voltamogramas a 20 mV/s. La capacidad específica de ambas películas fue inferior a los 100 F/g. 216 Figura 8. Voltamogramas de compuestos de PAni/HR-CNF con tratamiento térmico (TT). (a) 1.1 wt%, 20 mV/s, (b) 1.1 wt%, 50 mV/s, (c) 3.3 wt%, 20 mV/s, (d) 3.3 wt%, 50 mV/s. Los voltamogramas de la película de compuesto de PAni/GO (1.1 wt%) a 20 mV/s (Figura 9 a) muestran un incremento y una mejor definición de los picos redox conforme transcurren los ciclos, llegando a niveles de intensidad similares a los de las películas de compuestos sin TT. Incluso en el detalle de los voltamogramas a 20 mV/s de los ciclos 1120 (b) se observa un muy ligero incremento. Puede observarse que la estabilidad y reversibilidad electroquímica es alta, comparada con los anteriores voltamogramas. También existe un incremento de la capacidad específica, por encima de los 300 F/g. En los voltamogramas a 50 mV/s (c) se observa el alejamiento entre los picos redox que ya se había mostrado a este barrido de potencial y logran definirse los dos picos de oxidación de la PAni en ~0.7 y ~0.85 V debido al incremento de la ventana de potencial. También comienza a notarse una ligera disminución de la intensidad de los picos. En la figura 8 d se observan los voltamogramas de los ciclos 91-100 a 20 mV/s donde es significativo el 217 decrecimiento y alejamiento de los picos redox, aunque aún se observa una relativa buena estabilidad electroquímica en el sistema debido a que el decaimiento de los picos no es tan notorio como en las películas de compuestos sin TT. Figura 9. Voltamogramas de compuestos de PAni/GO (1.1 wt%). (a) 20 mV/s, (b) 20 mV/s, (c) 50 mV/s, (d) 20 mV/s. En la figura 10 se muestran los voltamogramas de películas de PAni/GO (3.3 wt%) donde se observa un comportamiento electroquímico similar al de las películas con un contenido de GO de 1.1 %. En el caso de 3.3% de carga, la intensidad de los picos redox fue menor, y en el caso de los voltamogramas de 50 mV/s (Figura 10 c) el decaimiento de los picos es ligeramente más pronunciado. Los voltamogramas de los ciclos 71-80 a 20 mV/s (Figura 10 d) muestran los picos redox más alejados entre sí y menos intensos al compararlos con los voltamogramas de los ciclos iniciales (Figura 10 a). 218 Figura 10. Voltamogramas de compuestos de PAni/GO (3.3 wt%). (a) 20 mV/s, (b) 20 mV/s, (c) 50 mV/s, (d) 20 mV/s. En la Figura 11 se muestran los voltamogramas de películas de compuestos híbridos de GO-MWCNTs (1.3 wt%) en proporción 1:2 (a, b) y 2:1 (c, d) con TT. Los voltamogramas de compuestos GO-MWCNTs de proporción 1:2 muestran que, a pesar de que las películas no se dañaron más allá de los ciclos iniciales, el comportamiento electroquímico de la película tiende a la inestabilidad. En el detalle de los voltamogramas a un barrido de 20 mV/s (a) se aprecia ruido en el estado oxidado de la PAni, y más adelante, en el barrido a 50 mV/s (b), el pico de oxidación se pierde comenzando a sobresalir solamente el de la oxidación del agua (1 V). En cambio, los picos de reducción se muestran más nítidos aunque su paulatina disminución de intensidad es significativa. Las capacidades específicas de 110.7 F/g (20 mV/s, ciclo 10) y 62.9 F/g (50 mV/s, ciclo 20) en esta película fueron relativamente bajas al compararlas con las de compuestos con GO. 219 En el caso de la película con una proporción GO-MWCNTs de 2:1, sus voltamogramas (Figura 12 c, d) muestran una mayor estabilidad electroquímica que en el caso anterior, e incluso, como se observa en los voltamogramas con un barrido de potencial de 20 mV/s (Figura 12 c), los picos redox van aumentando conforme transcurren los ciclos. Al parecer, al haber una mayor cantidad de GO en este compuesto, se logra estabilizar la película, aunque como puede observarse, la intensidad de la densidad de corriente de los picos redox es significativamente menor a la de los compuestos solamente con GO (figuras 9, 10). Los voltamogramas a 50 mV/s (Figura 12 d) muestran una repetibilidad y estabilidad electroquímica superiores a la de la película de los voltamogramas b. Asimismo, las capacidades específicas de estas películas de 162.1 F/g (20 mV/s, ciclo 10) y 91.3 F/g (50 mV/s, ciclo 20) son mayores que las de las películas anteriores (con una proporción GOMWCNTs de 1:2). Se concluye entonces que el GO podría ser un estabilizante electroquímico de las películas de PAni sometidas a un TT de 210ºC durante 20 min. Es posible que la buena dispersión de esta carga en la matriz polimérica refuerce mecánicamente a la película y también impida hasta cierto punto la degradación de la PAni a esta temperatura como se verá más adelante. 220 Figura 12. Voltamogramas de compuestos de PAni/GO-MWCNTs con tratamiento térmico. (a) 1.3 wt% (1:2), 20 mV/s, (b) 1.3 wt % (1:2), 50 mV/s, (c) 1.3 wt% (2:1), 20 mV/s, (d) 1.3 wt% (2:1), 50 mV/s. 6.3.3. Capacidades específicas de películas de compuestos de PAni La conductividad eléctrica y la capacitancia específica de las películas de compuestos de PAni sin y con TT se observan en las Tablas 1-4. Los cálculos de la capacitancia específica se llevaron a cabo utilizando el barrido de potencial a 20 mV/s. Como se observa, el TT ocasionó una disminución de la conductividad eléctrica de las películas. Es posible que, además de una cierta degradación del polímero con el TT, una parte importante del plastificante (NMP) se haya retirado a esta temperatura, ocasionando una mayor rigidez y agrietamiento y por consiguiente una disminución del contacto eléctrico en la película. 221 La conductividad eléctrica de la PAni (Tabla 1) sin TT (1.528 S/cm) es prácticamente 15 veces mayor que la película con TT (0.104 S/cm), lo cual podría explicarse por la pérdida de plasticidad de la película (al retirarse parte del NMP residual) y consecuente pérdida del continuo volumétrico de la película al someterla al TT. Reza Ansari y colaboradores [27] estudiaron el efecto del tratamiento térmico en la conductividad de la PAni. Encontraron que a temperaturas superiores a los 200º C, el polímero sufre un cambio estructural irreversible tal como la apertura de anillos en sus cadenas poliméricas. Esto ocasiona que la conductividad de la PAni decrezca rápidamente. Como se mencionó previamente, fue imposible aplicar voltametría cíclica a las películas de PAni con TT debido al daño inmediato que sufrieron, por lo que en la tabla sólo se muestran las capacitancias específicas calculadas para las películas sin TT. Después de 10 ciclos, la película mostró una capacitancia específica de 229.3 F/g, al cabo de 50, la capacitancia cayó a 118.5 F/g. La caída fue de casi 50 %. Sin duda, el desprendimiento de la película conforme transcurrieron los ciclos fue el principal factor que ocasionó que la capacitancia disminuyera. Tabla 1. Conductividades eléctricas y capacitancias específicas de las películas de PAni después de 10 y 50 ciclos voltamométricos. Material Nivel de Conductividad C10 C50 Caída de carga (%) eléctrica (S/cm) (F/g) (F/g) C (%) PAni - 1.528 ± 0.175 229.3 118.5 48.3 PAni (TT) - 0.104 ± 0.102 X X X Barrido de 20 mV/s, TT: tratamiento térmico, X: La película sufrió daño al llevarse a cabo la voltametría cíclica. En general, las películas con nanotubos de carbono (MWCNTs, CNx) con TT también sufrieron una disminución de su conductividad eléctrica (Tabla 2), no siendo tan evidente en el caso del compuesto PAni/MWCNTs al 1.1 wt% con 1.34 S/cm, aunque su desviación estándar de 0.862 es de las mayores. La capacitancia específica (al cabo de 10 ciclos) del compuesto con MWCNTs (3.3 wt%) de 280.5 F/g fue superior a la del compuesto con 1.1 % de nanotubos que fue de 208.7 F/g. 222 Aunque la caída de capacitancia de 44.4 % en el primero fue mayor al finalizar los 50 ciclos que el segundo con una caída de 35.7 %. Con respecto a los CNx, en el ciclo 10, se obtuvo una mayor capacidad específica para un nivel menor de carga (1.1 wt%) en el compuesto de 292.7 F/g que en el caso del compuesto con 3.3 % de nanotubos, que mostró una capacidad de 222 F/g. Al cabo de 50 ciclos, la capacidad para el compuesto con 1.1 % de CNx cayó en un 8.6% y en el segundo caso, 17%, mostrando así estas películas una mejor estabilidad que las de PAni/MWCNTs. En cuanto a la aplicación del TT a las películas, solamente la de PAni/CNx (1.1 wt%) resultó adecuada para las mediciones. En el ciclo 10 mostró una capacidad de 186.7 F/g, cayendo a 163.75 F/g en el ciclo 50, teniendo una pérdida de eficiencia de 12.3 %. Tabla 2. Conductividad eléctrica y capacidad específica de películas de compuestos de PAni con MWCNTs y CNx, después de 10 y 50 ciclos. Material PAniMWCNT PAniMWCNT (TT) PAni-CNx PAni-CNx (TT) Nivel de Conductividad C10 C50 Caída de carga (%) eléctrica (S/cm) (F/g) (F/g) C (%) 1.1 1.954 ± 0.595 208.7 134.2 35.7 3.3 0.993 ± 0.232 280.5 156 44.4 1.1 1.342 ± 0.862 X X X 3.3 0.153 ± 0.144 X X X 1.1 1.439 ± 0.818 292.7 267.4 8.6 3.3 0.936 ± 0.525 222 184.3 17.0 1.1 0.087 ± 0.024 186.7 163.8 12.3 3.3 0.485 ± 0.166 X X X Barrido de 20 mV/s, TT: tratamiento térmico, X: La película sufrió daño al llevarse a cabo la voltametría cíclica. La disminución de la conductividad eléctrica en las películas de compuestos de PAni/HRCNF con TT (Tabla 3) fue más evidente al no superar 0.1 S/cm en ambos niveles de carga 223 (1.1 y 3.3 wt%). En el caso de compuestos con GO, la conductividad de las películas disminuyó ~10 veces con respecto a las películas sin TT. La película de compuesto de PAni con 1.1% de HR-CNF mostró una capacidad específica de 265.4 F/g a los 10 ciclos, y de 223.7 F/g a los 50, por lo que la pérdida de eficiencia fue de 15.7%. La película con un nivel de carga de 3.3%, mostró una menor capacidad a los 10 ciclos, de 170.1 F/g, con una caída de capacidad de 13.1% a los 50 ciclos. El TT en las películas de compuestos con HR-CNF, ocasionó una disminución de la capacidad específica, puesto que en el ciclo 10, fueron de 97.2 y 88.7 F/g para los compuestos con 1.1 y 3.3% de contenido de carga, respectivamente. Asimismo, la caída de capacidad en estas películas, de 28.6 y 35.7% fue aún más notoria que la de las películas sin TT. Con respecto a las películas de compuestos de PAni/GO sin TT, la capacidad específica en el ciclo 10, para ambos niveles de carga, fue parecida a la de las películas anteriores aunque la pérdida de eficiencia en el ciclo 50 fue de más de 30% para las dos. En contraste, en la película de PAni/GO (1.1 wt%) con TT, la caída de capacidad (en el ciclo 50 con respecto al 10) fue de tan solo 0.5%, la cual inicialmente fue de 356.4 F/g. Puesto que esta película fue la que más estabilidad mostró (Figura 9), se realizaron más ciclos de voltametría. En el ciclo 100, la película tuvo 141.15 F/g, por lo que la caída de capacitancia con respecto al ciclo 10 fue de 60.4%. La película con 3.3% de GO mostró también cierta estabilidad, aunque la caída de capacidad en el ciclo 10 (283.7 F/g) con respecto al 50 (215.1 F/g) fue de 24.2%. Ante estos resultados, se eligió a la película de compuesto de PAni/GO (1.1 wt%) con TT para la experimentación subsecuente. 224 Tabla 3. Conductividad eléctrica y capacidad eléctrica de las películas de compuestos de PAni con GO y HR-CNF, después de 10, 50 y 100 ciclos. Material PAni-HRCNF PAni-HRCNF (TT) PAni-GO PAni-GO Nivel de Conductividad C10 C50 Caída C100 (F/g) carga eléctrica (F/g) (F/g) de C (caída de (%) (S/cm) (%) C) 1.1 1.346 ± 0.166 265.4 223.7 15.7 - 3.3 1.582 ± 0.213 170.1 147.2 13.1 - 1.1 0.079 ± 0.008 97.2 88.8 28.6 - 3.3 0.058 ± 0.005 88.6 63.3 35.7 - 1.1 2.610 ± 1.391 218.8 148.2 32.3 - 3.3 2.706 ± 0.923 242.3 145.7 39.9 - 1.1 0.326 ± 0.023 356.4 354.6 0.5 141.2 (60.4%) (TT) 3.3 0.124 ± 0.031 283.7 215.1 24.2 - Barrido de 20 mV/s, TT: tratamiento térmico, X: La película sufrió daño al llevarse a cabo la voltametría cíclica En la Tabla 4 se presentan las conductividades eléctricas de las películas de compuestos de PAni y una mezcla de MWCNTs y GO (1.3 wt%) en proporción de 2:1 y 1:2. Las conductividades obtenidas en ambos casos sin TT fueron similares (1.73 S/cm y 1.63 S/cm, respectivamente), estuvieron prácticamente en el mismo nivel que las conductividades de los compuestos de PAni/MWCNTs y PAni/GO (ver Tabla 5 del Capítulo II). El TT a estas películas ocasionó una disminución de la conductividad de prácticamente un orden de magnitud. La capacidad específica del ciclo 10 de las películas sin TT fueron de 157.8 y 133.5 F/g, con un decaimiento en el ciclo 50 de 27.6 y 37.5%, para las proporciones MWCNTs/GO de 2:1 y 1:2, respectivamente. Estos valores son relativamente bajos si se comparan con las capacidades específicas previas. En el caso del compuesto con una proporción MWCNTs/GO de 2:1, el TT disminuyó su capacidad específica (81.9 F/g en el ciclo 10) 225 con una pérdida de eficiencia en el ciclo 50 de 13.1%. En cambio, la película del compuesto con relación 1:2, mostro un incremento en su capacidad, con respecto a su equivalente sin TT, el cual fue de 162.1% (ciclo 10), aunque el decaimiento de 43.7% en el ciclo 50 fue mayor. Estas películas de compuestos de PAni con carga híbrida, además de mostrar una menor capacidad específica, resultaron hasta cierto punto inestables electroquímicamente (sobre todo la película con relación MWCNTs/PAni de 2:1, Figura 12 a, b), por lo que fueron descartados para la experimentación subsecuente. Tabla 4. Conductividad eléctrica y capacidad específica de compuestos de PAni y una mezcla de GO y MWCNTs (2:1, 1:2) después de 10 y 50 ciclos. Material PAni- Nivel de Conductividad C10 C50 Caída de carga (%) eléctrica (S/cm) (F/g) (F/g) C (%) 1.3 1.725 ± 0.321 157.8 114.3 27.6 1.3 0.117 ± 0.051 81.9 71.2 13.1 1.3 1.634 ± 0.29 133.5 83.5 37.5 1.3 0.209 ± 0.037 162.1 91.3 43.7 MWCNTsGO (2:1) PAniMWCNTsGO (2:1) (TT) PAniMWCNTsGO (1:2) PAniMWCNTsGO (1:2) (TT) Barrido de 20 mV/s, TT: tratamiento térmico, X: La película sufrió daño al llevarse a cabo la voltametría cíclica. 226 6.3.4. Carga/descarga de películas de compuestos de PAni/GO (1.1 wt%) con TT En la Figura 13 se muestran las pruebas de carga/descarga galvanostática de películas de compuestos de PAni con GO (1.1 wt%) con TT. Las pruebas se realizaron a 0.1 mA con límites de potencial desde -0.2 a 0.75 y 0.8 V. Se observa que las pendientes son simétricas (en general, la de carga comenzando en ~0.26 V y la de descarga en ~0.5 V), sobre todo después del ciclo inicial en el caso de llegar a 0.8 V (Figura 13 A). Los cálculos de la capacidad específica mediante estas pendientes (utilizando la ecuación 2) arrojaron los siguientes resultados: 351.8 ± 34.6 F/g para la primera película (A) y 534.7 ± 40.8 F/g para la segunda (B). Al promediar la capacidad de las dos películas, se obtiene una capacidad específica de 456.3 ± 100.9 F/g, para una corriente constante de 0.1 mA. El tiempo total de las pruebas fue de 3.89 h en A y 5 h en B. Las películas, al sacarse del electrolito de H2SO4, y lavarse con agua, apenas mostraron daño. 227 Figura 13. Gráficos de la carga/descarga galvanostática de películas de compuesto de PAni/GO (1.1 wt%) a una densidad de corriente de 1 A/g. (A) Rango de potencial de -0.2 a 0.8 V, (B) rango de -0.2 a 0.75 V. Los gráficos de las pruebas de carga/descarga aplicadas a las películas de PAni/GO (1.1 wt%) a corriente constante de 0.2 mA (Figura 14 A) muestran también unas pendientes simétricas, la carga de la película comenzó en ~0.22 V, y la descarga en ~0.47 V. La capacidad específica promedio calculada utilizando estas pendientes fue de 237.4 ± 10.8 F/g. El gráfico de la prueba a corriente constante de 0.4 mA (Figura 14 B), también muestra pendientes de carga y descarga simétricas. Las primeras iniciando en ~0.22 V y las segundas en ~0.5 V. El promedio de la capacidad específica de las pendientes fue de ~96.3 ± 6.3 F/g. Al igual que en las pruebas con una corriente constante de 0.1 mA (Figura 13), las películas sufrieron poco daño al terminar los ciclos y lavarse con agua destilada. 228 Las capacidades específicas calculadas con las pruebas de carga/descarga galvanostática, variaron de ~530 a ~100 F/g, en función de la corriente eléctrica, que varió de 0.1 a 0.4 mA (densidades de corriente de 1, 2 y 4 A/g). Estas cantidades concuerdan con las calculadas en la voltametría cíclica (a 20 mV/s), que variaron de ~350 a ~140 F/g conforme transcurrieron los ciclos de 10 a 100. Este es aproximadamente el orden de magnitud de la capacidad específica de los electrodos de PAni, o sus compuestos, que puede encontrarse en la literatura. Por ejemplo, H. Wang y colaboradores [12], encontraron capacidades de 216, 284 y 531 F/g (a una corriente de 0.2 A/g) para PAni, PAni/GO (1.6 wt%) y PAni/GO (1 wt%), respectivamente. Es destacable que las capacidades específicas de las películas obtenidas en este trabajo hayan sido casi del mismo nivel que las de H. Wang pero con una densidad de corriente ~10 veces mayor. Figura 14. Gráficos de la carga/descarga galvanostática de películas de compuesto de PAni/GO (1.1 wt%) a una densidad de corriente y unos límites de potencial de (A) 2 A/g, 0.2 a 0.75 V, (B) 4 A/g, -0.2 a 0.8 V. 229 Como se observó en las pruebas de voltametría cíclica y de carga/descarga, cuanto más alta es la corriente en las pruebas de carga/descarga o mayor el barrido de potencial, respectivamente, menor es la capacidad específica del material activo. Esto se debe a que al utilizar barridos de potencial bajos en la voltametría o al utilizar corrientes pequeñas en la carga/descarga, se necesita más tiempo para terminar las pruebas, por lo que hay más almacenamiento de carga eléctrica (hay una mayor cantidad de aniones que se incorporan o se expulsan del polímero). 6.3.5. Análisis termogravimétrico de los compuestos de PAni Se realizó un análisis termogravimétrico (TGA, por sus siglas en inglés) a los compuestos de PAni con procesamiento (drop-casting utilizando al NMP como disolvente y posteriormente, plastificante de las películas) y sin él (solamente desdopados con NaOH 0.2 M) a una velocidad de calefacción de 5º C/min en atmosfera inerte. El objetivo de estas pruebas fue averiguar si el GO como carga dispersa en la matriz polimérica, de alguna manera pudiera haber “protegido” a la PAni de una posible degradación debido al tratamiento térmico (TT) en las películas. A fin de obtener una mayor información, también se realizaron análisis al resto de los compuestos probados como electrodos. La Figura 15 muestra un TGA de PAni plastificada con NMP. Se puede observar una pérdida inicial de peso hasta ~120ºC debido a la pérdida de moléculas de agua. A partir de ~170º C comienza otro declive pronunciado en el peso de la muestra que no se detiene hasta poco antes de ~270º C. Es en este intervalo de temperatura donde el TT se localiza (210ºC), por lo que, el TT de las películas de los compuestos de PAni, provoca que una fracción del NMP residual se volatilice (la temperatura de ebullición del NMP es de 202204ºC) y así, disminuye su plasticidad. A temperaturas superiores de ~400ºC comenzaría la degradación total de la PAni, la cual podría producir gases como acetileno o amonio [28, 29]. A los 1000ºC, el material disminuye su peso en casi 58%. 230 Figura 15. Análisis termogravimétrico de PAni plastificada con NMP. En las Figuras 16 y 17 se muestran los perfiles gravimétricos de los compuestos de PAni (a 1.1 y 3.3 wt% de carga) con y sin NMP como plastificante, respectivamente. Se muestra el intervalo de temperatura (380 – 600º C) donde ocurre la descomposición del polímero. El perfil de descomposición de la PAni, plastificado con NMP, sin carga (s/c) (Figura 16 a, b) es el mismo que se observó en la figura anterior (se colocó a manera de comparación con los perfiles de los compuestos plastificados con NMP). Como se muestra en el perfil, a 600º C la muestra pierde 50.1% de peso. La descomposición del polímero en los compuestos ocurre en el intervalo de ~450–600º C. Los compuestos con 1.1 wt% de carga (Figura 16 a) pierden de ~17 a 25% de peso a 450º C. Siendo el compuesto con MWCNTs el que menos peso pierde, y el que contiene CNx el que más lo hace. La pérdida de peso de los compuestos con HR-CNF y GO se mantiene entre estos valores, estando ligeramente arriba el del compuesto con GO. Sin embargo, a 600º C, el compuesto que menos peso pierde es el de GO (50.5%), casi en el nivel de la PAni s/c con 50.1%. A este compuesto le siguen los de CNx y MWCNTs (~51.4%) y finalmente el de HR-CNF con 54.4%. Los compuestos que tuvieron una menor pérdida de peso en este intervalo de descomposición del polímero, son los que contienen CNx y GO. Además, el perfil de descomposición de este último, es el que más está desplazado a la derecha. Al comparar 231 este perfil con el de la PAni sin carga nos podría indicar que el GO, principalmente, podría estar protegiendo hasta cierto punto al polímero de la degradación térmica. En la Figura 16 b, inmediatamente resalta que una mayor cantidad de carga en el compuesto (3.3%) tiene un efecto “protector” en el polímero puesto que después de la descomposición de la PAni (~600º C), los compuestos pierden menos peso. En general los perfiles de los compuestos son similares a los anteriores, un poco más parecidos entre sí antes de comenzar la descomposición térmica (450º C), sobre todo los de compuestos con CNx y GO. Al cabo de la degradación (600º C), los compuestos con GO y CNx son los que menos peso pierden (~44.5%), por debajo de los compuestos con MWCNTs y HR-CNF (con una pérdida de peso de ~50.5%). Al igual que en el caso anterior, los compuestos con GO y CNx fueron los que menos peso perdieron en este proceso térmico. Es posible que la mayor reactividad de estas cargas dispersas en la matriz polimérica haya contribuido a aumentar la resistencia del polímero a la degradación térmica. Figura 16. Análisis termogravimétrico de compuestos de PAni (sin NMP) con un nivel de carga de (a) 1.1 wt%, (b) 3.3 wt%. En la Figura 17 se muestran los perfiles gravimétricos de los compuestos de PAni sin plastificante (NMP). La temperatura de descomposición del polímero parece iniciar en 450 232 o 460º C. Los perfiles en general muestran más parecido entre ellos a esta temperatura, que en el caso de los perfiles de compuestos plastificados de la figura anterior. En el caso de los compuestos con 1.1 wt% de carga (Figura 17 a), los perfiles muestran que, a 600º C, perdieron menos peso (46.6–49.2%) que la muestra sin carga (50.05%). Siendo el compuesto con CNx el que menos peso perdió (46.6%). Además, su perfil de descomposición térmica es el que se encuentra más a la derecha, indicando que es necesaria más energía térmica para descomponer al polímero. Este resultado del compuesto con CNx, hasta cierto punto coincide con el análisis anterior de compuestos de PAni con plastificante. Los perfiles de compuestos con el nivel de carga mayor (3.3%) (Figura 17 b) muestran a una temperatura de 600º C, una pérdida de peso aún menor (37.3–46.5 %) que el de la PAni sin carga (50.5%), destacando significativamente el compuesto con GO que fue el que perdió menos peso (37.3%). Los otros tres compuestos con HR-CNF, CNx y MWCNTs tuvieron una pérdida de peso similar de ~54%. Asimismo, puede observarse que los compuestos con GO requieren de una energía térmica de descomposición mayor que los demás (por la ubicación del perfil más a la derecha). El resto de compuestos, necesita una energía térmica de descomposición similar. Por último, la PAni sin carga es la que menos energía necesita (su perfil es el que se ubica más a la izquierda). Puede concluirse que las cargas de CNx y GO protegieron hasta cierto punto a la PAni de la degradación térmica (destacando aún más el GO en este último análisis). Además, se observó que a un mayor nivel de carga en los compuestos, hay una tendencia de éstos a resistir la descomposición del polímero. 233 Figura 17. Análisis termogravimétrico de compuestos de PAni (sin NMP) con un nivel de carga de (a) 1.1 wt%, (b) 3.3 wt%. En la Figura 18, se muestran los perfiles de TGA (mostrados ya en la Figura 17) completos, de 30 a 1000º C, de los compuestos de PAni sin plastificante (NMP) con nanotubos de carbono (MWCNTs y CNx), y en la Figura 19 se muestran los perfiles de los compuestos de PAni con HR-CNF y GO. En los gráficos de compuestos con MWCNTs (Figura 18 a, b), la derivada de los perfiles muestra cuatro inflexiones en ~63, ~177, ~285 y ~486º C. La primera corresponde a la evaporación de agua, la segunda a la volatilización de agua residual y a restos de polímero degradado. Las dos restantes (sobre todo la última) se deben a la descomposición total del polímero. En el compuesto con 1.1% de MWCNTs (Figura 18 a), la pérdida de peso a 1000º C fue de 56.4%. En el compuesto con 3.3 wt% (Figura 18 b), la pérdida fue de 58.6%. Aunque por lo visto anteriormente (en la Figura 16), hubo una mayor pérdida de peso para el compuesto con menor nivel de carga (a 600º C). Por otra parte, los perfiles de compuestos con CNx (Figura 18 c, d) son similares a los de los compuestos con MWCNTs. Aunque por lo visto en la Figura 16 y por la menor intensidad del pico de la derivada de la pérdida de peso (en el intervalo de descomposición principal del polímero, ~492º C), se aprecia que hay una menor pérdida de peso en este caso que en el anterior (con MWCNTs), debido posiblemente a la mayor reactividad de los 234 CNx. La pérdida de peso del compuesto a 1000º C, con 1.1 y 3.3% de CNx, fue de 56.65 y 54.88 %, respectivamente. Figura 18. Análisis termogravimétrico de compuestos de PAni con MWCNTs, (a) 1.1 wt%, (b) 3.3 wt%, y con CNx, (c) 1.1 wt%, (d) 3.3 wt%. Los perfiles termogravimétricos de los compuestos de PAni/HR-CNF (Figura 19 a, b) son similares a los anteriores compuestos con nanotubos de carbono. Las cuatro inflexiones aproximadamente siguen apareciendo en la misma ubicación. La única variación notable, es que la última inflexión (perteneciente a la descomposición total de la PAni) se desplazó de 488 (Figura 19 a) a 494º C (Figura 19 b) conforme aumentó la cantidad de nanofilamentos en la matriz polimérica. El peso a los 1000º C se perdió en 53.8 y 55.2% para el nivel de carga de 1.1 y 3.3 wt%, respectivamente. Los perfiles de los compuestos de PAni/GO (Figura 19 c, d), similares a los de los anteriores compuestos, sólo variaron significativamente en la ubicación de la cuarta inflexión de pérdida de peso, la cual es de 487º C para 1.1% de contenido de GO y 506º C para el de 3.3%. La diferencia, de casi 20º C, fue la mayor de todos los compuestos, 235 además, se puede observar en el perfil que la pérdida de peso en el compuesto con 3.3% de GO (d), fue menor (pico bajo de línea de trazos en 506º C). El porcentaje de pérdida de peso a 1000º C de las muestras con 1.1 y 3.3 wt% de GO fueron 58 y 52%, respectivamente, resultando en una diferencia de 6%. Aunque como se observó en la Figura 17, la pérdida de peso a 600º C para el compuesto con 1.1 y 3.3 wt% de GO fue de 48.6% y 37.3%, respectivamente. La diferencia entre ambas fue de ~11%, por lo que un contenido alto de GO, hasta cierto punto impidió que el polímero se degradara. Con lo cual podría explicarse que las películas de compuestos de GO depositadas en acero y sometidas al TT, hayan tenido una respuesta electroquímica superior a la del resto de los compuestos también con TT. Figura 19. Análisis termogravimétrico de compuestos de PAni con HR-CNF, (a) 1.1 wt%, (b) 3.3 wt%, y con GO, (c) 1.1 wt%, (d) 3.3 wt%. 236 6.3.6. Análisis termomecánico de compuestos de PAni Se han probado tiras de películas de PAni y sus compuestos (espesor ~30 μm, ancho = 1.6 mm, área transversal = 4.8 X 10-8 m2), con TT, mediante un análisis termo-mecánico (TMA). Las pruebas se realizaron por duplicado y tuvieron como objetivo evaluar su deformación en función de la temperatura. La Figura 20 a muestra los resultados más relevante para las películas con 1.1% de contenido de carga. La máxima deformación de ~17% (380º C) se obtuvo con el compuesto de PAni/HR-CNF, aunque su duplicado no alcanzó 13 % (225º C) cuando se rompió. En general, la repetibilidad de las pruebas por cada compuesto fue baja, sucediendo lo mismo con las películas de PAni sin carga (una se deformó 8% a 380º C y la otra poco más de 12% a 340º C). Las películas que mayor repetibilidad mostraron fueron las de PAni/MWCNTs, sobrepasando un poco ambas la deformación de 8% (a 370º C). En contraste, la repetibilidad fue aún más baja con las películas de compuestos con un 3.3% de carga (b) donde la mayoría se rompieron antes de llegar a los 270º C y con deformaciones menores al 8%. Únicamente una película de PAni/GO logró deformarse poco más del doble llegando a los 380º C. Es notable, que en las pruebas de TMA realizadas a tiras de películas sin TT (Figura 26 A, B, del capítulo III) las deformaciones ocurrieron entre un 50 y 60%. Es decir, el TT volvió menos plásticas a las películas, lo cual podría explicarse por la volatilización de una fracción del NMP residual a 210º C en vacío. Pero a pesar de esta pérdida de NMP, quedó el suficiente para seguir plastificando las películas, ya que, como se ve en los perfiles de la Figura 20, las máximas deformaciones se alcanzaron de 200 a 260º C, coincidiendo en parte con la temperatura de transición vítrea de la PAni plastificada (~100-220º) [30, 31]. Fue difícil preparar probetas de películas de compuestos con CNx, las cuales se rompieron antes de llevar a cabo las pruebas (por ello no se muestran sus perfiles en la figura). En el caso de las películas sin TT (del Capítulo III) sí se pudieron probar películas de compuestos con CNx, aunque en esa ocasión no se pudieron preparar películas de prueba con un 3.3% de GO. 237 Figura 20. Análisis termo-mecánico de las películas de PAni y sus compuestos con tratamiento térmico (210ºC, 20 min). (a) Nivel de carga de 1.1 wt%, (b) nivel de carga de 3.3 wt%. Las líneas verticales indican el rompimiento de las películas. 6.4. CONCLUSIONES El tratamiento térmico (TT, 210º C en vacío, 10 min) disminuyó la conductividad eléctrica de las películas de compuestos de polianilina (PAni) hasta en un orden de magnitud (después de haber sido redopadas con MSA 1.5 M). El TT perjudicó significativamente a las películas de PAni, PAni-MWCNT y parcialmente a las de PAni-CNx como electrodos en las pruebas de voltametría cíclica en H2SO4 1 M. En el caso de los electrodos de PAni-HR-CNF, a pesar de no perjudicarlas, su capacitancia se vio disminuida. El TT mejoró la adherencia de las películas de PAni-HR-CNF y de PAni-GO al sustrato de acero. En las segundas, se necesitaron ~10 ciclos voltamométricos para que la película, en casi su totalidad, participara en los ciclos redox. El TT mejoró la estabilidad electroquímica y vida útil de los electrodos de PAniGO, principalmente con un contenido de carga de 1.1 wt%. El TT volatilizó parte del NMP residual de las películas, perdiendo así una parte de su plasticidad. El GO en cierta forma protegió de la degradación a la PAni debido a la temperatura, ya que sus compuestos a un nivel alto de carga (3.3 wt%) tuvieron la menor pérdida de peso en las pruebas termogravimétricas. 238 REFERENCIAS [1] Rudge, A.; Davey, J.; Raistrick, I.; Gottesfeld, S.; Ferraris, J. P. “Conducting polymers as active materials in electrochemical capacitors”, J. Power Sources 47, 1994, 89. [2] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P. “Synthesis and characterization of new polyaniline/nanotube composites”, Materials Science and Engineering C 23, 2003, 87-91. [3] Wu, T. M.; Lin, Y. W.; Liao, C. S. Preparation and characterization of polyaniline/multi-walled carbon nanotube composites. Carbon 43, 2005, 734–740. [4] Snook, G. A.; Kao, P.; Best, A. S. Conducting-polymer-based-supercapacitor devices and electrodes. J. Power Sources 196, 2011, 1-12. [5] Huang, J. Kaner, R. B.; A general chemical route to polyaniline nanofibers. J. Am. Chem. Soc. 126, 2004, 851-855. [6] King, R. C.Y.; Roussel, F. Morphological and electrical characteristics of polyaniline nanofibers. Synthetic Metals 153, 2005, 337-340. [7] doNascimento, G. M.; Kobata, P. Y. G.; Temperini, M. L. A. Structural and vibrational characterization of polyaniline nanofibers prepared from interfacial polymerization. J. Phys. Chem. B 112, 2008, 11551-11557. [8] Maser, W. K.; Benito, A. M.; Callejas, M. A.; Seeger, T.; Martínez, M. T.; Schreiber, J.; Muszinsky, J.; Chauvet, O.; Osváth, Z.; Kóos, A. A.; Biró, L. P. Synthesis and characterization of new polyaniline/nanotube compuestos. Materials Science and Engineering C 23, 2003, 87-91. [9] Wallace, G. G.; Mottaghitalab, V.; Spinks, G. F. The influence of carbon nanotubes on mechanical and electrical properties of polyaniline fibers. Synthetic Metals 152, 2005, 7780. [10] Wu, T. M.; Lin, Y. W.; Liao, C. S. Preparation and characterization of polyaniline/multi-walled carbon nanotube composites. Carbon 43, 2005, 734–740. [11] Wu, T. M.; Lin, Y. W. Doped polyaniline/multi-walled carbon nanotube composites: Preparation, characterization and properties. Polymer 47, 2006, 3576-3582. [12] Wang, H.; Hao, Q.; Wang, H.; Yang, X.; Lu, L. Graphene oxide doped polyaniline for supercapacitors. Electrochemistry communications 11, 2009, 1158-1161. [13] Manthiram, A.; Murugan, A. V.; Muralingath, T. Rapid, facile microwavesolvothermal synthesis of graphene nanosheets and their polyaniline nanocomposites for energy storage. Chem. Mater. 21, 2009, 5004-5006. [14] Chow, T. W.; Thostenson, E. T.; Li, C. Sensors and actuators based on carbon nanotubes and their composites: A review. Composite Science and Technology 68, 2008, 1212-1249. [15] Dong-Won K.; Sivakkumar, S. R.; Kima W. J.; Ji-Ae C.; MacFarlane D. R.; Forsyth, M. Electrochemical performance of polyaniline nanofibres and polyaniline/multi-walled carbon nanotube composite as an electrode material for aqueous redox supercapacitors. J. Power Sources 171, 2007, 1062–1068. [16] Martinez, M. T.; Ansón, A.; Cendoya, I.; Ganborena, L.; Murillo, N.; Miguel, O.; Ochoteco, E.; Pomposo, J. A.; Grande, H.; Sainz, R.; Maser, W. K.; Benito, A. M. 239 Composites of carbon nanotubes and conducting polymer as electrodes of supercapacitors. Instituto de Carboquímica (CSIC), Miguel Luesma Castán 4, 50018, Zaragoza, Spain. [17] Wu, J.; Zhao, X. S.; Zhang, K.; Zhang, L. L. Graphene/polyaniline nanofiber composites supercapacitor electrodes. Chem. Mater. 22, 2010, 1392-1401. [18] Dong, B.; He, B.-L.; Xu, C.-L.; Li, H.-L. Preparation and electrochemical characterization of polyaniline/multi-walled carbon nanotubes composites for supercapacitors. Materials Science and Engineering B 143, 2007, 7-13. [19] Zhou, Y.-k.; He, B.-l.; Zhou, W.-j.; Huang, J.; Li, X.-h.; Wu, B.; Li, H.-l. Electrochemical capacitance of well-coated single-walled carbon nanotube with polyaniline composites. Electrochimia Acta 49, 2004, 257-262. [20] Park, J. H.; Park, O. O. Hybrid electrochemical capacitors based on polyaniline and activated carbon electrodes. J. Power Sources, 111, 2002, 185-190. [21] Kong, L.-B.; Zhang, J.; An, J.-J.; Luo, Y.-C.; Kang, L. MWNTs/PANI composite materials prepared by in-situ chemical oxidative polymerization for supercapacitor electrode. J. Mater. Sci. 43, 2008, 3664-3669. [22] Zhang, K.; Zhang, L.L.; Zhao, X. S.; Wu, J. Graphene/Polyaniline Nanofiber Composites as Supercapacitor Electrodes. Chem. Mater. 22, 2010, 1392-1401. [23] Sivakkumar, S. R.; Kim, W. J.; Choi, J.-A.; MacFarlane, D. R.; Forsyth, M.; Kim, D.W. Electrochemical performance of polyaniline nanofibres and polyaniline/multi-walled carbon nanotube composite as an electrode material for aqueous redox supercapacitors. J. Power Sources 171, 2007, 1062-1068. [24] Herod, T. E.; Schlenoff, J. B. Doping-Induced Strain Stretchoelectrochemistry. Chem. Mater. 5, 1993, 951–955. in Polyaniline: [25] Kaneko, M.; Fukui, M.; Takashima, W.; Kaneto, K. Electrolyte and strain dependences of chemomecanical deformation of polyaniline film. Synthetic Metals 84, 1997, 795-796. [26] Takashima, W.; Uesugi, T.; Fukui, M.; Kaneko, M.; Kaneto, K. Mechanochemoelectrical Effect of Polyaniline Film. Synthetic Metals 85, 1997, 13951396. [27] Ansari, R.; Keivani, M. B. Polyaniline Conducting Electroactive Polymers: Thermal and Enviromental Stability Studies. E-Journal of Chemistry Vol. 3, No 4, 2006, 202-217. [28] Wei, Y.; Hsueh, K. F. Thermal analysis of chemically synthesized polyaniline and effects of thermal aging on conductivity. J. Polym. Sci.: Part A: Polym. Chem. 27, 1989, 4351-4363. [29] Ansari, R.; Price, W. E.; Wallace, G. G. Effect of thermal treatment on the electroactivity of polyaniline. Polymer 37, 1996, 917-923. [30] Wei, Y.; Jang, G.-W.; Hsueh, K.; Scherr, E. M.; MacDiarmid, A.G.; Epstein, A. J. Thermal transitions and mechanical properties of films of chemically prepared polyaniline. Polymer 33, 1992, 314. [31] Milton, A. J.; Monkman, A. P. A comparative study of polyaniline films using thermal analyses and IR spectroscopy. J. Phys. D: Appl. Phys. 26, 1993, 1468-1474. 240 7. Conclusiones 241 242 7. Conclusiones 7.1. Mecanosíntesis de compuestos de PAni y CNTs La mecanosíntesis de nanocompuestos de PAni/CNTs en un solo paso ha sido desarrollada sin el uso de surfactantes, disolventes orgánicos ni ácidos. Este método de mezclado de alta energía en el molino de bolas provee una simple y relativamente rápida producción de nanocompuestos de PAni/CNTs con una buena interacción carga/matriz polimérica. Los CNTs no sufrieron daño significativo. Además de que los tiempos relativamente cortos de síntesis ayudan a evitar un daño mecánico a los nanotubos, la adición de agua en la molienda (un disolvente ambientalmente amigable) es el principal factor que evita el daño a los CNTs. La adición de agua en la reacción promueve la eficiencia en la polimerización, además, podría facilitar la reducción de Fe+3 por la disociación de la sal cloruro, facilitando así su difusión y por lo tanto la oxidación de la anilina. Los compuestos resultantes, tienen PAni en su forma conductora y la presencia de CNTs incrementa la conductividad por 4 órdenes de magnitud, siendo los MWCNTs mejores que los CNx en este sentido. La espectroscopía Raman sugiere que los nanotubos dopados con nitrógeno tienen una mejor interacción con la PAni al compararlos con los MWCNT sin dopaje. 7.2. Síntesis de compuestos de PAni y nanoestructuras de carbono por el método de polimerización interfacial in situ. En el proceso de exfoliación de MWCNTs se confirmó la importancia de la temperatura de 1000ºC, así como el número de tratamientos térmicos (5), previamente encontrados por Cano-Márquez y colaboradores, aunque se encontró que con una relación Li:MWCNTs de 1:1 (en el medio de reacción de intercalación) se obtienen resultados similares a la de 10:1 previamente utilizada. Se funcionalizó de manera apropiada a los exMWCNTs con anilina mediante la reacción de “sales de nanotubos”, lo cual fue constatado en la morfología, difractograma y conductividad eléctrica del composite PAni/Ani-exMWCNTs. 243 A pesar de haber obtenido una buena interacción entre Ani-exMWCNTs o COx con la PAni, se descartaron estas cargas debido a que no incrementaron significativamente la conductividad eléctrica de los compuestos, la cual, es una propiedad importante para el desarrollo de actuadores y electrodos de supercondensadores. Se obtuvieron eficiencias de polimerización cercanas al 50% utilizando HCl 1 M en la polimerización interfacial de la anilina. Se obtuvieron películas con un buen control de su espesor, mediante la disolución de la PAni desdopada en N-metil-2-pirrolidona (NMP) y un drop-casting en sustrato de vidrio. Se logró mejorar la dispersión de los nanotubos de carbono en los compuestos utilizando un homogeneizador, aunque en detrimento de su longitud. La dispersión del GO fue muy elevada debido a su hidrofilidad, asimismo, sus grupos funcionales, contribuyeron a un “dopaje extra” en las cadenas poliméricas de la PAni, ocasionando un incremento de su conductividad eléctrica. Las cargas en los compuestos, incrementaron la resistencia a la deformación de sus respectivas películas, sobre todo a un nivel de carga de 3.3 wt%. Una mayor resistencia a la deformación se obtuvo con las películas de PAni/MWCNTs (3.3 wt%). 7.3. Actuadores lineales y flexionantes de compuestos de PAni y nanoestructuras de carbono. Una menor porosidad o nanoestructuración, obtenida utilizando FeCl3 como oxidante en la polimerización de la PAni, benefició a los actuadores lineales en su facilidad de conformado y en una elongación que permitió apreciar el efecto de las cargas dispersas en su matriz polimérica. Las cargas que mostraron tener una excelente interacción con la PAni, como los exMWCNTs, Ani-exMWCNTs y los COx, no contribuyeron a mejorar las características de los actuadores, como su conductividad eléctrica y la elongación de los mismos. El procesado (disolución en NMP) de los compuestos de PAni para obtener películas permitió observar mucho mejor la elongación (flexión) de los actuadores en función de las cargas dispersas en el polímero. 244 A pesar de que los CNx mostraron una buena interacción con la PAni, sus aglomerados perjudicaron a la flexibilidad de las películas, por lo que, bajo las condiciones experimentales utilizadas, fueron rechazados como carga en los actuadores. Los HR-CNF no mostraron efecto alguno en los actuadores donde estuvieron dispersos con respecto a los de la PAni sin carga, a pesar de que la flexibilidad de las películas fue buena. El GO y los MWCNTs resultaron ser las mejores cargas para su aplicación como actuadores. El GO fue la carga que mejor se dispersó en la matriz polimérica, y a pesar de que es prácticamente aislante, sus grupos funcionales incrementaron el dopaje de la PAni. En el caso de los MWCNTs, la dispersión no fue tan alta pero sí la suficiente para contribuir al dopaje en la PAni y además servir como puentes de transporte electrónico entre algunas regiones del polímero. Las condiciones experimentales óptimas para los actuadores flexionantes de PAni/MWCNTs y PAni/GO fueron: nivel de carga de 1.3 wt% y nivel de potencial de 2.6 V. En general, los actuadores de PAni/MWCNTs tuvieron un mejor rendimiento que los actuadores de PAni/GO, debido a una mayor resistencia de la película al ciclado redox. 7.4. Electrodos de supercondensadores de compuestos de PAni y nanoestructuras de carbono. El tratamiento térmico (TT, 210º C en vacío, 10 min) disminuyó la conductividad eléctrica de las películas de compuestos de polianilina (PAni) hasta en un orden de magnitud (después de haber sido redopadas con MSA 1.5 M). El TT perjudicó significativamente a las películas de PAni, PAni-MWCNT y parcialmente a las de PAni-CNx como electrodos en las pruebas de voltametría cíclica en H2SO4 1 M. En el caso de los electrodos de PAni-HR-CNF, a pesar de no perjudicarlas, su capacitancia se vio disminuida. El TT mejoró la adherencia de las películas de PAni-HR-CNF y de PAni-GO al sustrato de acero. En las segundas, se necesitaron ~10 ciclos voltamométricos para que la película, en casi su totalidad, participara en los ciclos redox. El TT mejoró la estabilidad electroquímica y vida útil de los electrodos de PAniGO, principalmente con un contenido de carga de 1.1 wt%. 245 El TT volatilizó parte del NMP residual de las películas, perdiendo así una parte de su plasticidad. El GO en cierta forma protegió de la degradación a la PAni debido a la temperatura, ya que sus compuestos a un nivel alto de carga (3.3 wt%) tuvieron la menor pérdida de peso en las pruebas termogravimétricas. 246 Reunido el Tribunal que suscribe en el día de la fecha acordó otorgar, por de Don/Dña. Juan Carlos García Gallegos Alicante de la calificación de a la Tesis Doctoral . de El secretario, El presidente, UNIVERSIDAD DE ALICANTE CEDIP La presente Tesis de D.__ Juan Carlos García Gallegos_________ ha sido registrada con el nº ____________ del registro de entrada correspondiente. Alicante______de____________de_______ El Encargado del Registro, 247