Anomalías de la córnea
Anuncio

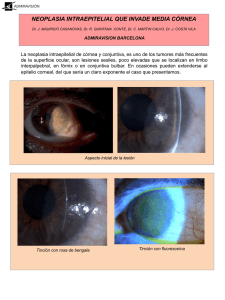
Capítulo 619 Anomalías de la córnea & e619-1 La megalocórnea debe distinguirse del agrandamiento corneal patológico causado por el glaucoma. Cualquier aumento progresivo del tamaño de la córnea, sobre todo si se asocia con fotofobia, lagrimeo o turbidez corneal, requiere un estudio oftalmológico inmediato. MICROCÓRNEA La microcórnea, o microftalmía anterior, es una córnea anormalmente pequeña en un ojo por lo demás normal. Puede ser familiar, con transmisión más a menudo dominante que recesiva, aunque por lo general las córneas pequeñas se observan en ojos microftálmicos o con un desarrollo anormal. Se asocia con colobomas, microfaquia, catarata congénita, glaucoma y aniridia. QUERATOCONO Es una enfermedad de patogenia incierta caracterizada por adelgazamiento y protrusión progresivos de la córnea central, que adopta una forma cónica. Aunque se han descrito casos familiares, la mayoría son esporádicos. Se han propuesto como factores etiológicos el frotarse los ojos y el uso de lentes de contacto, aunque no hay pruebas inequívocas. La incidencia es mayor en personas con atopia, síndrome de Down, síndrome de Marfan y retinitis pigmentosa. La mayoría de los casos son bilaterales, aunque pueden ser asimétricos. El trastorno suele presentarse y progresar con rapidez durante la adolescencia y ralentizarse cuando se completa el crecimiento. En ocasiones se estira la membrana de Desçemet más allá de su punto de elasticidad y esto produce una rotura aguda de la membrana con edema súbito e intenso de la córnea (hidropesía aguda) y disminución de visión. El edema corneal desaparece al cubrir las células endoteliales el área lesionada y, aunque puede quedar cierta cicatrización corneal, la agudeza visual suele ser mejor que antes de la hidropesía. Entre las manifestaciones del queratocono se incluyen también el signo de Munson (abombamiento del párpado inferior al mirar hacia abajo) y el anillo de Fleischer (depósito de hierro en el epitelio que rodea la base del cono). Está indicado el trasplante de córnea si no se consigue una agudeza visual satisfactoria con lentes de contacto. OPACIDADES CORNEALES NEONATALES La pérdida de la transparencia normal de la córnea en los recién nacidos puede ser secundaria a causas hereditarias intrínsecas o a factores ambientales extrínsecos (tabla 619-1). Tabla 619-1 DIAGNÓSTICO DIFERENCIAL DE LAS OPACIDADES CORNEALES EN NEONATOS DIAGNÓSTICO © ELSEVIER. Fotocopiar sin autorización es un delito. Esclerocórnea LATERALIDAD OPACIDAD Unilateral o bilateral PRESIÓN INTRAOCULAR OTRAS ANOMALÍAS OCULARES EVOLUCIÓN NORMAL HERENCIA Vascularizada, se mezcla con la esclerótica, más transparente en el centro DESGARROS ENDOTELIALES Y EN LA MEMBRANA DE DESÇEMET Traumatismo obstétrico Unilateral Edema difuso Normal (o elevada) Córnea plana No progresiva Esporádica Normal Posible hipema, equimosis periorbitaria Esporádica Glaucoma infantil Bilateral Edema difuso Elevada Megalocórnea, fotofobia y lagrimeo, ángulo anormal Mejoría espontánea antes de 1 mes Progresiva si no se trata ÚLCERAS Queratitis por herpes simple Rubéola congénita Unilateral Difusa con defecto epitelial geográfico Edema disciforme o difuso, sin ulceración ostensible Normal Ninguna Progresiva Esporádica Normal o elevada Microftalmos, catarata, moteado del epitelio pigmentario Anomalías palpebrales, neuropatía sensorial congénita Escasas Estable, puede aclararse Esporádica Progresiva Esporádica Progresiva Autosómica dominante Bilateral Autosómica recesiva Neurotróficas exposición Unilateral o bilateral Úlcera central Normal Metabólicas (rara vez presentes al nacer) (mucopolisacaridosis IH, IS; mucolipidosis tipo IV)* Defecto corneal posterior Bilateral Turbidez difusa, más densa periféricamente Normal Unilateral o bilateral Turbidez difusa central o leucoma vascularizado Normal o elevada Síndrome de clivaje de la cámara anterior Estable, a veces aclaramiento precoz o vascularización Esporádica, autosómica recesiva Bilateral Edema corneal difuso, engrosamiento corneal acusado Turbidez difusa, espesor corneal normal Opacidades estromales desflecadas en copos; espesor corneal normal Masa blanca vascularizada, pelo, arco lipídico Normal Ninguna Estable Normal Normal En ocasiones sinequias anteriores periféricas Ninguna Lentamente progresiva Estable Autosómica dominante o recesiva Autosómica dominante Autosómica dominante Normal Ninguna Estable DISTROFIA ENDOTELIAL Distrofia endotelial hereditaria congénita Distrofia polimorfa posterior Distrofia estromal hereditaria congénita Dermoide Bilateral Bilateral Unilateral o bilateral *Mucopolisacaridosis IH (síndrome de Hurler); mucopolisacaridosis IS (síndrome de Scheie). De Nelson LB, Calhoun JH, Harley RD: Pediatric ophthalmology, 3.a ed., Filadelfia, 1991, WB Saunders, pág. 210. Esporádica e619-2 & Parte XXIX Enfermedades oculares ESCLEROCÓRNEA En la esclerocórnea, la córnea transparente normal es reemplazada por un tejido parecido a la esclerótica. En vez de una córnea claramente delimitada, se forma un tejido blanco desflecado, a menudo mal definido y vascularizado en la córnea periférica, de manera que parece mezclarse y proceder de la esclerótica. La córnea central suele ser más transparente, pero puede producirse la sustitución total de la córnea por esclera. La curvatura corneal a menudo es más plana, similar a la de la esclerótica. Entre las posibles anomalías asociadas se encuentran el aplanamiento de la cámara anterior, anomalías del iris y microftalmía. Esta enfermedad suele ser bilateral. En aproximadamente el 50% de los casos se ha descrito un patrón de herencia dominante o recesivo. Se han comunicado casos de esclerocórnea asociados con numerosas alteraciones sistémicas, como deformidad de extremidades, defectos craneofaciales y trastornos genitourinarios. En la esclerocórnea difusa debe valorarse la realización de una queratoplastia precoz para intentar conseguir cierto grado de visión. ANOMALÍA DE PETERS La anomalía de Peters consiste en una opacidad corneal central (leucoma) presente al nacer (fig. 619-1). A menudo se asocia con adherencias iridocorneales que se extienden desde el collarete del iris hasta el borde de la opacidad corneal. Aproximadamente la mitad de los pacientes presenta otras anomalías oculares, como cataratas, glaucoma y microcórnea. Es bilateral hasta en el 80% de los casos y en el 60% se asocia con malformaciones sistémicas que pueden afectar a cualquier sistema orgánico importante. Algunos investigadores han dividido la anomalía de Peters en 2 tipos: una forma mesodérmica o neuroectodérmica (tipo I), que no se asocia con alteraciones del cristalino, y una forma ectodérmica superficial (tipo II), que sí lo hace. Los hallazgos histológicos incluyen ausencia focal de la membrana de Desçemet y el endotelio en la región de la opacidad. La anomalía de Peters puede deberse a migración y diferenciación incompletas de las células precursoras del endotelio y la membrana de Desçemet del centro de la córnea, o a una separación defectuosa entre el cristalino y la córnea primitivos durante la embriogenia. DERMOIDES Los dermoides epibulbares son coristomas. Suelen estar presentes en el nacimiento y aumentar de tamaño con el tiempo. Se localizan por lo general en el cuadrante temporal inferior. Suelen sobrepasar el limbo y extenderse hacia la córnea periférica. Rara vez están totalmente confinados en la conjuntiva o la córnea. Los dermoides epibulbares pueden afectar a la visión si invaden el eje visual o favorecen la aparición de astigmatismo, con el consiguiente riesgo de ambliopía. El dermoide suele observarse como una masa redondeada u oval y circunscrita de color gris o amarillo-rosáceo, con superficie seca, de la que pueden asomar pelos cortos. Aunque a veces sólo afecta a las capas superficiales de la córnea, es frecuente que abarque todo su espesor. Entre las anomalías oculares asociadas pueden encontrarse colobomas palpebrales y del iris, microftalmía y defectos retinianos y coroideos. En el 30% de los casos se asocian con anomalías sistémicas, sobre todo defectos del desarrollo del primer arco branquial (anomalías vertebrales, disostosis de los huesos faciales y anomalías dentales, y síndrome de Goldenhar). Existen dermoides epibulbares en el 75% de los casos de síndrome de Goldenhar. QUERATITIS DENDRÍTICA La infección de la córnea por el virus del herpes simple da lugar a una lesión característica del epitelio corneal, conocida como dendrita, con un patrón ramificado que puede demostrarse mediante tinción con fluoresceína. El episodio agudo se acompaña de dolor, fotofobia y lagrimeo, blefaroespasmo e hiperemia conjuntival. El tratamiento específico suele consistir en el desbridamiento mecánico del epitelio corneal afectado, con el fin de eliminar la fuente de infección y el estímulo antigénico para la inflamación del estroma adyacente. El tratamiento médico se basa en el uso de trifluridina o aciclovir sistémico. Además, pueden emplearse agentes ciclopléjicos para aliviar el dolor por el espasmo del músculo ciliar. El tratamiento antivírico tópico intensivo resulta a veces tóxico para la córnea, por lo que debe evitarse. La infección recidivante y la afectación estromal profunda pueden producir cicatrización corneal y pérdida de visión. El uso de corticoides tópicos exacerba la infección herpética superficial del ojo y desemboca en ocasiones en una perforación corneal; por tanto, deben evitarse los colirios con combinaciones de corticoides y antibióticos para tratar el ojo rojo, salvo que existan indicaciones específicas para su uso y se vigile de manera cercana al paciente. Los recién nacidos de madres infectadas por herpes simple deben ser explorados cuidadosamente para detectar signos de afectación ocular. El herpes ocular en los neonatos debe ser tratado con aciclovir intravenoso. ÚLCERAS CORNEALES [(Figura_1)TD$IG] Figura 619-1 Anomalía de Peters. Opacidad central en un paciente con anomalía de Peters. Los signos y síntomas habituales consisten en turbidez corneal focal o difusa, hiperemia, edema palpebral, dolor, fotofobia, lagrimeo y blefaroespasmo. Es frecuente que exista hipopión (pus en la cámara anterior). Las úlceras corneales requieren tratamiento inmediato. Suelen deberse a lesiones traumáticas que se infectan de manera secundaria. Son muchos los organismos capaces de infectar la córnea. Uno de los más problemáticos es Pseudomonas aeruginosa, que produce a veces una rápida destrucción del tejido estromal con la consiguiente perforación corneal. Neisseria gonorrhoeae también es especialmente peligrosa. Las úlceras tórpidas pueden deberse a hongos, a menudo asociados con el uso de lentes de contacto. En cualquier caso, deben realizarse raspados de la córnea para identificar el agente infeccioso y determinar el mejor tratamiento. Aunque suele requerirse una terapia tópica intensiva para salvar el ojo, en ocasiones también se precisa antibioterapia sistémica. La perforación y la cicatrización secundarias a ulceraciones corneales son una causa importante de ceguera en todo el mundo y representan el 10% de los casos en EE.UU. Las úlceras corneales sin causa aparente en bebés y niños pequeños deben hacer pensar en defectos sensitivos, como los síndromes de Riley-Day o Goldenhar-Gorlin, o en trastornos metabólicos como la tirosinemia. Capítulo 619 Anomalías de la córnea & e619-3 FLICTÉNULAS Son pequeñas lesiones amarillentas y levemente elevadas que suelen localizarse en el limbo corneal; pueden invadir la córnea y extenderse hacia el centro. A menudo se encuentra una pequeña úlcera corneal en el borde de avance de la lesión, con un fascículo de vasos sanguíneos detrás de la cabeza de la lesión. Aunque antes se creían causadas por infección tuberculosa sistémica, en la actualidad sabemos que la queratoconjuntivitis flictenular representa una reacción de hipersensibilidad retardada frente a diversos antígenos. En niños se debe por lo común a reacciones de la córnea o la conjuntiva frente a sustancias bacterianas. El tratamiento suele consistir en eliminar el trastorno subyacente, como blefaritis estafilocócica o meibomitis, y suprimir la respuesta inmune con corticoterapia tópica. A veces persiste un pannus y una cicatrización estromal superficiales después del tratamiento. algunas mucopolisacaridosis (MPS; cap. 82), sobre todo la MPS IH (Hurler), la MPS IS (Scheie), la MPS I H/S (complejo Hurler-Scheie), la MPS IV (Morquio), la MPS VI (Maroteaux-Lamy) y, en ocasiones, la MPS VII (Sly). Pueden aparecer depósitos corneales en pacientes con gangliosidosis GM1 (generalizada) (cap. 80.4). En la enfermedad de Fabry se observan opacidades finas que irradian con un patrón en remolino o abanico y que permiten la identificación de los portadores (cap. 80.4). También se observa a veces un patrón de opacidades en remolino en el síndrome de Bloch-Sulzberger. En la enfermedad de Wilson, el signo corneal característico es el anillo de Kayser-Fleischer, un anillo marrón-dorado en la córnea periférica secundario a alteraciones en la membrana de Desçemet (cap. 349.2). Los recién nacidos con colestasis pueden presentar anillos corneales pigmentados. El hipoparatiroidismo autoinmune se asocia con alteraciones corneales, y en los pacientes con hipercalcemia se forma en ocasiones una queratopatía en banda. Puede haber una queratitis transitoria en el sarampión y, a veces, en la rubéola. QUERATITIS INTERSTICIAL Es una inflamación del estroma corneal, en general causada por sífilis; se trata de una de las manifestaciones tardías típicas de la sífilis congénita. Las alteraciones corneales de la sífilis congénita se presentan en 2 fases. La fase aguda aparece a los 5-10 años de edad, con una queratitis intensa que puede durar varios meses y causar una grave reducción de visión. Se debe principalmente a una reacción inmunitaria del huésped, con infiltración de células mononucleares, proliferación vascular y, en ocasiones, formación de granulomas. La inflamación profunda produce dolor, fotofobia, lagrimeo, hiperemia periquerática y turbidez corneal. Tras este episodio agudo viene un estadio crónico con regresión importante de las alteraciones corneales y mejoría de la agudeza visual. Aunque los signos corneales pueden desaparecer con el tiempo, persisten los «vasos fantasma», que representan los cambios vasculares previos, y parches cicatriciales en la córnea como estigmas permanentes de la enfermedad. El síndrome de Cogan es una queratitis intersticial no luética asociada con pérdida de audición y síntomas vestibulares. Aunque se desconoce su causa, parece deberse a una vasculitis sistémica. Es necesario su tratamiento inmediato para impedir la sordera irreversible. Tanto las alteraciones corneales como la afectación auditiva responden en ocasiones a agentes inmunosupresores. Con menor frecuencia, la queratitis intersticial se debe a otras enfermedades infecciosas, como tuberculosis o lepra. MANIFESTACIONES CORNEALES DE ENFERMEDADES SISTÉMICAS © ELSEVIER. Fotocopiar sin autorización es un delito. Diversas enfermedades sistémicas producen alteraciones típicas en la córnea durante la infancia. En la cistinosis (cap. 79.4) se depositan cristales policromáticos refringentes por toda la córnea. También puede haber depósitos corneales que inducen turbidez variable en BIBLIOGRAFÍA Beauchamp GR, Gillette TE, Friendly DS: Phlyctenular keratoconjunctivitis, J Pediatr Ophthalmol Strabismus 18:22-28, 1981. Chang DC, Grant GB, O’Donnell K, et al: Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution, JAMA 296:953-963, 2006. Comer RM, Daya SM, O’Keefe M: Penetrating keratoplasty in infants, J AAPOS 5:285-290, 2001. Dana MR, Moyes AL, Gomes JA, et al: The indications for and outcome in pediatric keratoplasty. A multicenter study, Ophthalmology 102: 1129-1138, 1995. Garg P, Krishna PV, Stratis AK, et al: The value of corneal transplantation in reducing blindness, Eye (Lond) 19:1106-1114, 2005. Mackey DA, Buttery RG, Wise GM, et al: Description of X-linked megalocornea with identification of the gene locus, Arch Ophthalmol 109:829-833, 1991. Michaeli A, Markovich A, Rootman DS: Corneal transplants for the treatment of congenital corneal opacities, J Pediatr Ophthalmol Strabismus 42:34-44, 2005. Mohandessan MM, Romano PE: Neuroparalytic keratitis in Goldenhar- Gorlin syndrome, Am J Ophthalmol 85:111-113, 1978. Reidy JJ: Congenital corneal opacities, Ophthalmol Clin North Am 2:199-213, 1996. Traboulsi EI, Maumenee IH: Peters’ anomaly and associated congenital malformations, Arch Ophthalmol 110:1739-1742, 1992. Yang LL, Lambert SR, Fernhoff PM, et al: Peters’ anomaly: associated congenital malformations and etiology, Invest Ophthalmol Vis Sci 36:S41, 1995. Yang LL, Lambert SR, Lynn MJ, et al: Long-term results of corneal graft survival in infants and children with Peters anomaly, Ophthalmology 106:833-848, 1999.