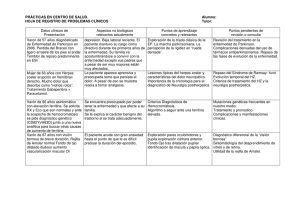
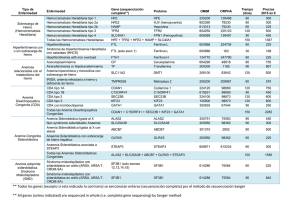
Manual para pacientes con hemocromatosis
Anuncio

Asociación Española de Hemocromatosis Manual para pacientes con hemocromatosis Dr. Albert Altés Hernández Dra. Susana Vives Polo Manual para pacientes con hemocromatosis Dr. Albert Altés Hernández Dra. Susana Vives Polo Asociación Española de Hemocromatosis Con la colaboración de: Dra. SUSANA VIVES POLO Licenciada en Medicina y Cirugía. Especialista en Hematología Dr. ALBERT ALTÉS HERNÁNDEZ Licenciado en Medicina y Cirugía. Especialista en Hematología Presidente de la Asociación Española de Hemocromatosis Primera edición: Septiembre 2008 © Asociación Española de Hemocromatosis Producción e impresión: aureagràfic, s.l. Dep. legal.: B-38065-2008 Ninguna parte de esta publicación, incluido el diseño de la cubierta, puede ser reproducida, almacenada o transmitida de ningún modo ni por ningún medio, ya sea eléctrico, químico, mecánico, óptico, de grabación o xerocopia, sin permiso previo de la Asociación Española de Hemocromatosis. Agradecimientos Este libro no hubiera podido ser confeccionado sin la participación de todos los socios de la Asociación Española de Hemocromatosis. Asimismo, Novartis ha aportado fondos para su financiación, y la Asociación Española de Hematología y Hemoterapia nos ha avalado el interés científico del texto. Por último, hemos tenido el apoyo de las becas del Fondo de Investigaciones Sanitarias PI (04/1120) y de la Agència d'Avaluació de Tecnologia i Recerca Mèdica (005/29/2004) para confeccionarlo. 3 Indice Prólogo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 1 Introducción: hemocromatosis hereditaria y secundaria . . . . . . . . . . . . . . . . . . . . . . . 9 2 Metabolismo del hierro. Absorción, transporte, reciclado . . . . . . . . . . . . . . . . . . . . 13 3 Hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 3.1 Definición de hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 3.2 Causas de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18 3.3 Origen de la mutación C282Y del gen HFE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 3.4 Incidencia de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22 3.5 Otros tipos raros de hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . 23 3.6 Escrutinio poblacional de la hemocromatosis hereditaria . . . . . . . . . . . . . . . 25 3.7 Recomendaciones generales del escrutinio y estudio familiar . . . . . . . . . . 25 3.8 Diagnóstico de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . 27 3.8.1 Signos y síntomas de la hemocromatosis hereditaria . . . . . . . . . . . 27 3.8.1.1 Enfermedad hepática . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 3.8.1.2 Alteraciones endocrinológicas . . . . . . . . . . . . . . . . . . . . . . . . . . 29 3.8.1.3 Enfermedad cardíaca . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 3.8.1.4 Enfermedad en las articulaciones . . . . . . . . . . . . . . . . . . . . . . . 30 3.8.1.5 Afectación de la piel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 3.8.2 Estudios complementarios en la hemocromatosis hereditaria . . 31 3.8.2.1 Determinación de hierro sérico . . . . . . . . . . . . . . . . . . . . . . . . . 32 3.8.2.2 Determinación de transferrina y del índice de saturación de transferrina en suero . . . . . . . . . . . . . . . . . . . . . 32 3.8.2.3 Determinación de ferritina sérica . . . . . . . . . . . . . . . . . . . . . . . 33 3.8.2.4 Determinación del genotipo . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 3.8.2.5 Biopsia hepática . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 3.8.2.6 Resonancia magnética nuclear . . . . . . . . . . . . . . . . . . . . . . . . . . 36 3.8.2.7 Flebotomía cuantitativa (sangrías) . . . . . . . . . . . . . . . . . . . . . . 38 3.8.3 Diagnóstico diferencial de la hemocromatosis hereditaria . . . . . . . . . . . 38 3.8.3.1 Enfermedades hematológicas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 3.8.3.2 Enfermedades hepáticas crónicas . . . . . . . . . . . . . . . . . . . . . . . . 40 3.8.3.3 Otras causas de sobrecarga de hierro . . . . . . . . . . . . . . . . . . . 40 5 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 4 5 6 7 6 3.9 Tratamiento de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40 3.9.1 Flebotomías terapéuticas en la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 3.9.1.1 Fase de reducción del hierro . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42 3.9.1.2 Fase de mantenimiento . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43 3.9.1.3 Monitorización clínica del paciente durante las flebotomías terapéuticas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43 3.9.2 Quelación del hierro como tratamiento de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 3.9.3 Trasplante hepático en la hemocromatosis hereditaria . . . . . . . . . . . 45 3.9.4 Modificaciones de la dieta en la hemocromatosis hereditaria . . 45 3.9.5 Pronóstico de la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . 47 3.10 Otros aspectos a considerar en el paciente con hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 3.10.1 Impacto del diagnóstico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 3.10.2 Avances en la hemocromatosis hereditaria . . . . . . . . . . . . . . . . . . . . . . 49 Hemocromatosis secundaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51 4.1 ¿Qué es la hemocromatosis secundaria? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51 4.2 ¿Qué causa la hemocromatosis secundaria? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51 4.3 Fisiopatología de la hemocromatosis secundaria . . . . . . . . . . . . . . . . . . . . . . . . . 53 4.4 Diagnóstico de la hemocromatosis secundaria . . . . . . . . . . . . . . . . . . . . . . . . . . . 54 4.4.1 Signos y síntomas de la hemocromatosis secundaria . . . . . . . . . . . 54 4.4.2 Evaluación analítica de la hemocromatosis secudaria . . . . . . . . . . . 54 4.4.3 Diagnóstico diferencial de la hemocromatosis secundaria . . . . . . 55 4.5 Tratamiento de la hemocromatosis secundaria . . . . . . . . . . . . . . . . . . . . . . . . . . 56 Historia y objetivos de la Asociación Española de Hemocromatosis . . . . . . . . . 59 Bibliografía . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 Anexo 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 Prólogo Este es el primer libro que se ha escrito sobre hemocromatosis dirigido al público hispanohablante. Se ha intentado que tenga un nivel científico elevado pero a la vez, que sea comprensible para los pacientes. De hecho, el libro nace en la Asociación Española de Hemocromatosis que tiene por principal objetivo la información a los pacientes con estas enfermedades. Así pues, se ha intentado que el libro responda a las necesidades de éstos. Hace 10 años, con la colaboración activa y desinteresada de compañeros y pacientes, contribuí a la creación de la Asociación Española de Hemocromatosis. Hoy me llena de orgullo y satisfacción que haya sido esa Asociación la que ha patrocinado este libro. Por otra parte, es evidente que los costes de publicación del mismo no habrían podido ser cubiertos si no fuera por la ayuda que Novartis Oncology nos ha ofrecido. Por último, disponer del aval para este libro de una organización de prestigio contrastado como la Asociación Española de Hematología y Hemoterapia (AEHH) constituye una demostración de calidad para este libro y un aporte de seguridad para los pacientes que lo lean. Por todo ello, los autores no podemos más que dar las gracias a todos por haber contribuido en la edición de este libro que esperamos sirva a muchos pacientes y a sus familiares. Nada nace sin errores, y este libro no va a ser una excepción. Esperamos que los lectores nos ayuden a subsanar los mismos con notas que pueden enviar al correo electrónico de nuestra Asociación, aeh@hes.scs.es Muchas gracias a todos Dr. A LBERT A LTÉS H ERNÁNDEZ Presidente de la AEH 7 1 Introducción: hemocromatosis hereditaria y secundaria Las hemocromatosis son enfermedades por sobrecarga de hierro en el organismo. El hierro a pesar de ser un elemento imprescindible para la vida, en exceso resulta tóxico y produce enfermedad. La sobrecarga de hierro en el organismo puede ser debida al incremento inapropiado de la absorción intestinal del hierro (el hierro procedente de la comida que adquirimos por el intestino) o al aumento de los aportes de hierro por vías no fisiológicas, como el uso de hierro medicinal por vía parenteral (endovenosa) o el procedente de las transfusiones de sangre. El hierro se almacena en las células en forma de un pigmento que se llama hemosiderina, precisamente la sustancia que podemos observar cuando teñimos una muestra de tejido. Resulta lógico que nos refiramos al término hemosiderosis cuando queremos describir la presencia de hierro en abundancia en los tejidos que se puede teñir El Atomium, edificio emblemático de Bruselas, en Bélgica, representa un cristal de hierro aumentado millones de veces. 9 MANUAL PARA PACIENTES CON HEMOCROMATOSIS con tinciones específicas. Además de observar el acumulo de hierro mediante métodos puramente morfológicos, también podemos cuantificarlo con precisión mediante métodos bioquímicos para obtener una estimación precisa del hierro corporal. Frente al término más bien histológico de hemosiderosis, usualmente utilizamos un término más clínico, hemocromatosis, para describir la sobrecarga progresiva y potencialmente grave de hierro que tiene como consecuencia el daño de los órganos. Las complicaciones más frecuentes de este cuadro clínico son la cirrosis hepática (fase terminal de las enfermedades hepáticas), diabetes mellitus (falta de insulina por el mal funcionamiento del páncreas), artritis (inflamación de las articulaciones), miocardiopatía (enfermedad del corazón) e hipogonadismo (disminución de las funciones sexuales). Se distinguen dos tipos de sobrecarga de hierro: • Hemocromatosis hereditaria, genética o primaria: habitualmente, este trastorno está motivado por la herencia de un gen (denominado HFE) alterado (mutante), que está situado en una zona del material genético denominada brazo corto del cromosoma 6. Como ya explicaremos en este manual, también se han descrito formas raras de hemocromatosis hereditaria no relacionadas con el gen HFE causadas por mutaciones en otros genes relacionados con el metabolismo del hierro. Mediante estudios de laboratorio (bioquímicos) la enfermedad puede reconocerse y diagnosticarse en sus etapas iniciales, cuando son mínimas la sobrecarga de hierro y la lesión de órganos. En una segunda fase, tecnologías genéticas pueden proporcionarnos el diagnóstico de certeza. • Hemocromatosis secundaria: Como veremos, la causa más frecuente de éste tipo de patología es la acumulación de hierro procedente de transfusiones sanguíneas reiteradas en pacientes con anemia crónica grave (por ejemplo aquejados de talasemia mayor o síndrome mielodisplásico). Menos frecuentemente, algunos pacientes que no se trasfunden pueden sufrir hemocromatosis secundaria debida a un incremento de la absorción del hierro de la dieta. Esta hiperabsorción de hierro se produce 10 INTRODUCCIÓN: HEMOCROMATOSIS HEREDITARIA Y SECUNDARIA como consecuencia del estímulo a la absorción intestinal que se genera en situaciones de eritropoyesis ineficaz (formación anómala y rotura de precursores de glóbulos rojos por la médula ósea) que se da en algunas anemias crónicas como la talasemia intermedia o la anemia sideroblástica. Sea cual sea la causa y tipo de hemocromatosis, las consecuencias del acúmulo orgánico de hierro pueden ser parecidas, y por tanto, las manifestaciones clínicas también lo serán (afección del hígado, páncreas, corazón y glándulas endocrinas). 11 2 Metabolismo del hierro. Absorción, transporte, reciclado El hierro (Fe) es un elemento natural de nuestro entorno que es esencial para la formación de hematíes y muchos compuestos en el organismo. Una dieta equilibrada contiene de 10 a 20 milígramos (mg) de hierro, de los cuales el cuerpo absorbe (introduce en el organismo a través del intestino) aproximadamente de 1 a 2 mg al día (aproximadamente el 10%), mientras que el resto pasa a través del tubo digestivo y se elimina con las heces. La misma cantidad de hierro diariamente absorbida en el intestino se elimina cada día a través de la descamación de la piel muerta, pelo y uñas, por lo que normalmente existe un equilibrado balance (homeostasis) entre el hierro absorbido y el eliminado. La mayor parte del hierro corporal es exquisitamente reciclado en un proceso en el que prácticamente nada se gana ni se pierde. La regulación de esta homeostasis viene dada en su totalidad por la regulación de la absorción intestinal del hierro. El cuerpo humano no dispone de mecanismos fisiológicos para excretar (eliminar) el exceso de hierro (por ejemplo el hierro adquirido a través de múltiples transfusiones). El organismo puede únicamente regular, y sólo hasta cierto punto, la cantidad de hierro que se absorbe, pero de ninguna forma puede eliminar un exceso de este metal. Cualquier proceso que altere la absorción normal de hierro puede conducir a una patología. La falta de suficiente hierro en el cuerpo (déficit de hierro) puede producir anemia, que hace que las personas se sientan débiles y cansadas. Por el contrario, si el cuerpo absorbe y almacena demasiado hierro, se producirá una enfermedad por sobrecarga de hierro en la cual el hierro acumulado puede producir daño en órganos vitales como el hígado, corazón y páncreas que son esenciales para la salud y el bienestar. Puede parecer extraño que el hierro, un elemento usualmente asociado a la salud, pueda producir daño en los tejidos y órganos vitales. La razón de este hecho es que cuando existe 13 MANUAL PARA PACIENTES CON HEMOCROMATOSIS un importante acumulación de hierro se sobrepasan los mecanismos fisiológicos de control. En esa situación no existen suficientes proteínas para almacenar el exceso de hierro y éste inicia un proceso de reacciones químicas descontroladas. Estas reacciones tienden a generar radicales libres, sustancias químicas de alto poder reactivo que con gran facilidad lesionan los tejidos. Entender el metabolismo del hierro es importante para poder entender la enfermedad generada por la acumulación del metal y las situaciones atribuidas a la sobrecarga férrica. El hierro de la dieta se absorbe a través del tubo digestivo y entra en el sistema circulatorio que lo repartirá a los tejidos que lo necesiten. Uno de los tejidos que precisa más hierro es la médula ósea, lugar donde se forma la sangre. Allí, el hierro se combina con una proteína de los glóbulos rojos para formar hemoglobina (el componente de los glóbulos rojos que transporta el oxígeno). Aproximadamente el 75% del hierro corporal se une a la hemoglobina de los glóbulos rojos. Será este pigmento el que se una al oxígeno en los pulmones y después de viajar por los vasos sanguíneos hará posible que las células de los diversos tejidos sean capaces de realizar sus funciones metabólicas. Sin el hierro nuestros glóbulos rojos no serían capaces de unirse ni de transportar oxígeno y, por lo tanto, no seríamos capaces de sobrevivir. El otro 10-20% del hierro corporal forma parte de otras substancias importantes para la vida (proteínas, enzimas) o se almacena y se utiliza sólo si es preciso. Hay dos proteínas importantes, llamadas ferritina y transferrina, que juegan un papel importante en la capacidad del cuerpo para utilizar el hierro. La transferrina es la proteína transportadora del hierro por excelencia. Se sintetiza (se “fabrica”) en el hígado, se une muy fuertemente al hierro que hay en la sangre (no el que está unido a la hemoglobina que a ése ya no hay quien le separe) y lo transporta por el torrente sanguíneo a todos los tejidos que lo precisan, y tal como se ha dicho anteriormente muy especialmente a la médula ósea donde se producen los glóbulos rojos. La cantidad de hierro que se une a la transferrrina se puede medir y expresar como el índice de saturación de transferrina en suero. 14 METABOLISMO DEL HIERRO. ABSORCIÓN, TRANSPORTE, RECICLADO Duodeno 1-2 mg/día Mioglobina 300 mg Transferrina del plasma 3 mg Médula ósea 300 mg Eritrocitos 1800 mg Hígado 1000 mg Descamación duodenal Menstruación Otras pérdidas 1-2 mg/día Macrófagos 600 mg Ciclo corporal del hierro. Cada día se absorben a nivel duodenal 1-2 mg de hierro, que serán los que aproximadamente se perderán diariamente. El resto del hierro corporal se halla en compartimentos funcionales (hemoglobina, mioglobina, médula ósea), formando parte de enzimas, en proceso de reciclaje en macrófagos o en depósito fundamentalmente hepático Adaptado de la Biblioteca John Hopkins, USA. La ferritina es una proteína que se une al hierro y que sirve para almacenar de forma segura éste metal en el interior de la célula. Grandes cantidades de ferritina acumulada formarán la hemosiderina. La ferritina se encuentra sobre todo concentrada en el hígado, músculo esquelético, bazo, médula ósea y en una pequeñísima parte en la sangre. A parte de la ferritina que se acumula en los tejidos, tenemos una pequeña cantidad de ferritina libre en el suero (un componente de la sangre), que podemos medir mediante el parámetro ferritina sérica. El nivel de ferritina sérica es directamente proporcional a la cantidad de hierro almacenada en el hígado (como veremos, en realidad es muy proporcional cuando la cantidad de hierro es baja y mucho menos pro- 15 MANUAL PARA PACIENTES CON HEMOCROMATOSIS porcional cuando la cantidad de hierro es excesiva). El cuerpo incrementa la producción de ferritina cuando hay exceso de hierro absorbido para facilitar su almacenamiento. Así pues, medimos el nivel de transferrina para saber la capacidad existente de transporte de hierro, mientras que nos fijamos en la ferritina para conocer el tamaño de los depósitos de hierro del organismo. Curiosamente, un tercer parámetro denominado sideremia (o hierro sérico) muy solicitado por los médicos tan sólo nos informa de la cantidad de hierro que hay en ese momento en la sangre, pero de ninguna forma nos informa de las reservas corporales de hierro. 16 3 Hemocromatosis hereditaria 3.1 Definición de hemocromatosis hereditaria La hemocromatosis hereditaria es una alteración genética que comporta sobrecarga corporal de hierro debida a incremento en la absorción intestinal de este metal y daño en los tejidos. El término hemocromatosis fue acuñado en el siglo XIX por el anatomopatólogo alemán von Recklinghausen. En pacientes con hemocromatosis here- Fotografía de von Recklinghausen, creador del término Hemocromatosis. ditaria, el intestino absorbe demasiado hierro procedente de la comida. El exceso de hierro se distribuye a través del cuerpo y se acumula a lo largo de la vida en diversos órganos produciendo, si no se trata, alteración en la función de los mismos. Parámetros bioquímicos clásicos de hemocromatosis son la elevación de los niveles de saturación de transferrina y de los niveles de ferritina sérica. El diagnóstico y tratamiento precoces de la enfermedad pueden eliminar la sobrecarga de hierro, prevenir el daño de los órganos y permi- 17 MANUAL PARA PACIENTES CON HEMOCROMATOSIS tir que los pacientes tengan una vida normal y productiva. Incluso si los daños orgánicos ya se han producido, el inicio del tratamiento suele ralentizar mucho la progresión de sus complicaciones. Sin embargo, si el paciente continúa sin tratamiento, se produce un progresivo acumulo de hierro en el hígado, páncreas, corazón, articulaciones y glándula pituitaria que potencialmente causan graves enfermedades. 3.2 Causas de la hemocromatosis hereditaria Casi siempre, la hemocromatosis hereditaria es una enfermedad hereditaria autosómica recesiva. El término autosómica recesiva hace El cromosoma 6, el mismo que aparece en el título de un libro de Robin Cook, alberga el gen HFE en su brazo corto. referencia a un modo de transmisión de los caracteres heredados en el cual hay dos copias de un gen alterado (mutado), residentes en dos cromosomas homólogos (cromosomas que portan los mismos genes, uno del padre y el otro de la madre). Por tanto, para sufrir una enfermedad que es autosómica recesiva es necesario heredar un gen mutado (enfermo) procedente del padre y otro también mutado de la madre. Un porcentaje elevado de las personas que portan esta muta- 18 HEMOCROMATOSIS HEREDITARIA ción recesiva por partida doble (en homozigosis) sufrirán la enfermedad. Durante mucho tiempo los médicos sabían que la alteración genética causal de la hemocromatosis hereditaria se hallaba en una zona concreta del genoma, exactamente en el brazo corto del cromosoma humano número 6, en las cercanías de una zona llamada complejo mayor de histocompatibilidad. En 1996, se avanzó en la comprensión del patrón genético de la hemocromatosis hereditaria, cuando por fin y tras un gran esfuerzo internacional se descubrió el gen HFE (efectivamente localizado en el brazo corto del cromosoma 6) que se asocia con la enfermedad. Dos mutaciones (alteraciones genéticas o variantes) del gen HFE, conocidas como C282Y y H63D, se asociaron al aumento de la absorción y del depósito del hierro que es tan característico en la hemocromatosis hereditaria. En el capítulo dedicado al diagnóstico de la enfermedad explicaremos cómo deben interpretarse estas mutaciones. Artículo original de Feder i col. en el que se identifican las mutaciones genéticas usualmente asociadas a la hemocromatosis hereditaria. En los países anglosajones, el presentar la mutación C282Y en forma homozigota (es decir, que la alteración genética se recibe por partida doble, por parte de padre y de madre) explica la presencia de hemocromatosis hereditaria en el 80% de los casos. Dicho de otra forma, el 80% de los pacientes con hemocromatosis hereditaria son homocigotos C282Y. En Europa este porcentaje es en general menor y oscila mucho más. Entre el 50 y el 90 % de las personas con esta enfer- 19 MANUAL PARA PACIENTES CON HEMOCROMATOSIS medad son homozigotas C282Y dependiendo de la localización geográfica exacta. Concretamente en España se ha calculado que este porcentaje es del 70%, pero también varía entre Comunidades Autónomas. Es comprensible que las personas que son homozigotas para el genotipo C282Y (C282Y/C282Y) tienen un alto riesgo de desarrollar hemocromatosis hereditaria. No está claro qué porcentaje de individuos homozigotos desarrollará realmente hemocromatosis hereditaria (en un principio se estimó que el 80%, pero sin embargo, investigaciones recientes muestran que el porcentaje es quizás mucho menor, sobre el 20%). Parece ser que la homozigosidad para la mutación C282Y es necesaria para el desarrollo de la hemocromatosis hereditaria pero no suficiente en muchos casos. No se conoce, en este momento, qué otros factores determinan qué pacientes homocigotos C282Y desarrollarán hemocromatosis hereditaria clínica (es decir, con afectación de órganos). Es posible que algunos factores sean de carácter genético, es decir, otras alteraciones genéticas sumadas a las alteraciones del gen HFE. No obstante existen algunos factores clínicos que sí influyen en la presentación de la enfermedad que son: • Edad: la hemocromatosis hereditaria con afectación clínica suele darse a partir de los 50-60 años de edad. • Género: los varones son diagnosticados típicamente de hemocromatosis hereditaria a una edad más temprana que las mujeres, puesto que la menstruación provoca una pérdida natural de sangre que reduce los depósitos de hierro. De hecho un estudio reciente establece que casi la totalidad de los afectados con complicaciones clínicas son hombres. • Hábitos alimentarios: como el consumo de alcohol, el uso de vitamina C o los suplementos de hierro. • Ciertas mutaciones genéticas añadidas a las alteraciones del gen HFE (causa muy infrecuente, ver más adelante). • Presencia de otras enfermedades (situaciones de comorbididad). • Hábitos de vida, por ejemplo ser donante de sangre (previene la aparición de la enfermedad). Aproximadamente el 2% de los pacientes afectos de hemocromatosis hereditaria son homozigotos para la mutación H63D 20 HEMOCROMATOSIS HEREDITARIA (H63D/H63D) y un 5% son heterozigotos dobles o compuestos (presencia de dos mutaciones diferentes o variantes de un gen en el mismo cromosoma) y expresan el genotipo C282Y/H63D. Actualmente se considera que el genotipo homozigoto C282Y es el que más predispone a sufrir hemocromatosis. Los heterozigotos dobles sólo tienen un ligero incremento de riesgo de sufrir la enfermedad frente a la población general. Por otra parte se ha descartado que el resto de genotipos (incluido los homozigotos H63D) presenten ningún tipo de predisposición a sufrir hemocromatosis, algo que se ha puesto en duda durante años. 3.3 Origen de la mutación C282Y del gen HFE La mutación C282Y del gen HFE, mutación clave en la hemocromatosis hereditaria, parece que se originó en épocas no demasiado lejanas (aproximadamente hace unos 2.000-3.000 años). Posiblemente la O GR EN LA ND IA F IN LAN NO RU EG A SU EC IA ISLAS FEROE D IA INSLANDIA ESTÓNIA DINAMARCA DINAMARCA FRANCIA EUROPA POLONIA A N IA REINO UNIDO PAISES BAJOS ALEM REPÚBLICA DE IRLANDA SUDOESTE RUSSIA REPÚBLICA CHECA A RI T R IA NG AUS HU BULGARIA POR TUG AL IT AL IA TURQUIA ESPAÑA GRECIA mas del 10% 1-5% 5-10% 0-1% CHIPRE La mutación C282Y del gen HFE muestra una distribución desigual en distintos países, con un gradiente de Norte a Sur y mayor frecuencia en poblaciones originalmente célticas. El mapa reproduce las frecuencias de la mutación C282Y en diversos países europeos. mutación apareció espontáneamente en un individuo de origen céltico del norte de Europa, y se extendió con relativa celeridad de norte a sur. Algunos investigadores han sugerido que quizá la mutación apareció en poblaciones vikingas del norte de Europa y que la rápida expansión 21 MANUAL PARA PACIENTES CON HEMOCROMATOSIS de esta mutación puede explicarse por las invasiones que este pueblo realizó en zonas muy alejadas de su país. Otros autores niegan este punto y basan la expansión de la enfermedad en los movimientos migratorios realizados por las poblaciones célticas del norte y centro de Europa. El hecho que la mutación se haya extendido por una zona geográfica tan grande y en un tiempo relativamente corto sugiere que puede conferir algún tipo de ventaja evolutiva a sus portadores. Estamos todavía lejos de esclarecer cual pudo ser esa ventaja. 3.4 Incidencia de la hemocromatosis hereditaria A pesar de que la mayoría de las personas no ha oído hablar nunca de la hemocromatosis hereditaria, sin duda muchas de ellas están a riesgo de sufrir esta enfermedad dado que se trata de una de la condiciones genéticas patológicas más frecuentes. Por otra parte se ha demostrado que se trata de una patología de diagnóstico difícil. Los Centros de Control de Enfermedades (CDC) de EE.UU. han estimado que el término medio para el diagnóstico de la enfermedad es de 7 años y los pacientes han requerido en general consultar con tres doctores antes de ser diagnosticados correctamente de hemocromatosis hereditaria. A pesar de que tanto los varones como las mujeres pueden adquirir la mutación genética que causa la hemocromatosis hereditaria, los síntomas de la enfermedad suelen ser mucho más frecuentes en varones que en mujeres. La edad de diagnóstico en los varones suele ser entre los 50 y los 60 años. Las mujeres son diagnosticadas con más frecuencia en la postmenopausia, ya que con la menstruación los niveles corporales de hierro disminuyen. Se estima que aproximadamente el 0,5 % de la población caucasiana de EE.UU. (1 cada 200 habitantes), especialmente aquellos descendientes del norte y oeste de Europa, presenta dos copias mutadas del gen HFE y tiene riesgo de desarrollar la enfermedad. En España la proporción de personas homozigotas para la mutación C282Y del gen HFE es de una por cada 1.000 habitantes, y la proporción de heterocigotos dobles C282Y/H63D es de 1%. Se trata de una patología muy poco frecuente en otras razas a parte de la 22 HEMOCROMATOSIS HEREDITARIA caucásica, y raramente se encuentra en poblaciones indígenas de África, Asia o Islas del Pacífico. 3.5 Otros tipos raros de hemocromatosis hereditaria Se han identificado cuatro tipos bien establecidos de hemocromatosis hereditaria: • Tipo I: Éste es el tipo “clásico” de hemocromatosis hereditaria y es el tema fundamental de esta guía. • Tipo II: También llamada hemocromatosis juvenil. La hemocromatosis juvenil es una enfermedad tan grave como rara. No se dispone de estadísticas fiables de la prevalencia de esta enfermedad, dada la extrema rareza de la misma. Afecta a personas a partir de la adolescencia y siempre jóvenes (menos de 30 años de edad). Es una forma de hemocromatosis caracterizada por un depósito de hierro mucho más importante y sobre todo por una acumulación de hierro mucho mas rápida que en la hemocromatosis “tradicional”. A diferencia de la hemocromatosis “tradicional” hombres y mujeres resultan igualmente afectados en esta enfermedad. La incidencia de afectación cardíaca y hipogonadismo hipogonadotrófico es mayor en estos pacientes, mientras que presentan menor frecuencia de afectación hepática y diabetes. Usualmente, la atrofia testicular y la desaparición de la menstruación son los síntomas iniciales y la principal causa de muerte es el fallo cardíaco secundario a cardiomiopatia. Desgraciadamente, cuando la miocardiopatia se manifiesta el deterioro del paciente suele ser muy rápido y suelen existir alteraciones irreversibles. Es frecuente la aparición de arritmias graves. Hace pocos años se descubrió la causa de esta enfermedad. Alteraciones genéticas de dos genes (HAMP y hemojuvelina) explican la patogenia de esta enfermedad. El déficit en la síntesis de estos genes implica una casi nula o nula síntesis del péptido hepcidina, y ello conlleva un incremento muy notable en la absorción del hierro. El tratamiento de estos pacientes es similar al de los pacientes con hemocromatosis hereditaria, pero más difícil dado que la presencia 23 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Árbol familiar de una familia española afecta de hemocromatosis tipo IV debida a una mutación situada en el gen de la ferroportina. Los individuos en rojo subren sobrecarga intensa de hierro, mientras que los familiares en blanco no la sufren. La presencia en enfermedad en generaciones contiguas demuestra que la enfermedad se transmite de forma autonómica dominante de miocardiopatia y arritmias complica mucho los procedimientos de flebotomía. En ocasiones debe recurrirse a las eritroaféresis. En ocasiones debe realizarse un trasplante de corazón para mantener al paciente con vida. • Tipo III: Ésta es una alteración autosómica recesiva causada por la mutación en el gen del receptor 2 de la transferrina que se localiza en el cromosoma 7. Se han descrito muy pocos casos, fundamentalmente en Italia y sur de Francia. • Tipo IV: Se trata del único tipo de hemocromatosis con transmisión genética autosómica dominante que se evidencia fácilmente con el estudio del árbol familiar. Se debe a la presencia de mutaciones en un gen situado en el cromosoma 2 que codifica para una proteína denominada ferroportina (una proteína que transporta el hierro desde la luz del intestino al torrente circulatorio y desde el interior de unas células de depósito denominadas macrófagos a la médula ósea). Parece tratarse de una enfermedad más benigna que el tipo clásico, con un acúmulo de hierro muy importante pero que suele localizarse más en los macrófagos que en las células nobles del tejido hepático. Muchos de estos pacientes presentan un nivel de ferritina sérica muy elevado, pero sin sufrir no obstante elevaciones importantes del nivel de saturación de la transferrina. 24 HEMOCROMATOSIS HEREDITARIA 3.6 Escrutinio poblacional de la hemocromatosis hereditaria Las guías para la detección en la población de hemocromatosis hereditaria han sido publicadas por varias organizaciones médicas incluyendo: • American College of Physicians. • American College of Gastroenterology. • American Association for the Study of Liver Diseases. • U.S. Preventive Services Task Force. • U.S. Centers for Disease Control an Prevention. A pesar de la disponibilidad de numerosas guías, hay todavía una ausencia general de consenso sobre la conveniencia de realizar un cribado de la enfermedad en la población general. Esto es así por las siguientes razones: • No es posible conocer con precisión cuáles de las personas con mutaciones asociadas a hemocromatosis desarrollarán realmente la enfermedad • Los estudios sugieren que el número de personas que efectivamente desarrollarán hemocromatosis hereditaria con alteración orgánica podría ser menor de lo que se piensa, lo cual reduciría el valor del cribado a gran escala. • Los individuos que han sido identificados con la mutación en el gen HFE mediante una prueba de detección y que están asintomáticos podrían ser estigmatizados en sus puestos de trabajo, o en aspectos relativos a sus seguros de salud con primas altas o bien podrían sufrir estrés psicológico por percibir sobre su persona el hecho de estar enfermos o de tener un elevado riesgo de enfermedad. 3.7 Recomendaciones generales del escrutinio y estudio familiar Los médicos que proponen realizar las pruebas de detección de la hemocromatosis hereditaria en la población general lo hacen porque el diagnóstico precoz de la enfermedad implica un gran beneficio en la salud de las personas diagnosticadas, con una prevención efectiva 25 MANUAL PARA PACIENTES CON HEMOCROMATOSIS de las complicaciones de la enfermedad. No obstante, la mayoría de expertos opinan que dichas pruebas deben realizarse en pacientes seleccionados, incluyendo: • Familiares de primer grado de los pacientes que se han diagnosticado de hemocromatosis hereditaria incluyendo hijos mayores de 18 años, hermanos y padres. Varios expertos sugieren retrasar las pruebas diagnósticas en los hijos de los individuos que son homozigotos para C282Y hasta la adolescencia porque las manifestaciones de la sobrecarga de hierro en los niños son extremadamente raras excepto en el caso de la hemocromatosis juvenil. En general, el estudio a realizar en los parientes de primer grado es directamente el estudio genético. No obstante si el estudio genético es negativo puede ser necesario analizar en los familiares los parámetros bioquímicos de sobrecarga de hierro (saturación de transferrina y ferritina sérica). Una forma de evitar el análisis genético en los hijos cuando éstos son pequeños es realizar el análisis en el cónyuge de la persona afecta y valorar con esta información si es realmente imprescindible realizar el estudio genético en los hijos. Si el cónyuge no presenta ninguna de las mutaciones del gen HFE posiblemente no será necesario estudiar a los hijos porque éstos sólo podrán ser portadores pero no enfermos de esta patología. • Pacientes con presencia de síntomas asociados con hemocromatosis hereditaria, como hepatomegalia (aumento de tamaño de hígado), elevación de las enzimas hepáticas, miocardiopatía (cambios estructurales o funcionales del corazón) y artritis. • Presencia de síntomas no específicos como astenia (cansancio), dolor abdominal, pérdida de peso o pigmentación cutánea que no se atribuyen a ninguna otra patología médica. • Individuos con marcadores séricos con valores anormales (como elevación significativa y mantenida del índice de saturación de la transferrina o de la ferritina sérica) descubiertos en análisis de rutina o por pruebas diagnósticas referentes a otras situaciones médicas. El índice de saturación de transferrina sérica se considera la prueba clínica más útil en el diagnóstico de la sobrecarga del hierro. No obstante, para que este test tenga valor debe demostrarse francamente elevado en 26 HEMOCROMATOSIS HEREDITARIA dos determinaciones individuales separadas por tres meses. Si los niveles de saturación de transferrina y los niveles de ferritina sérica están alterados, debe considerarse la realización de test genético y, si fuera necesario, debe realizarse una biopsia hepática como diagnóstico confirmatorio de hemocromatosis hereditaria. Los pacientes que poseen mutaciones del gen HFE pero que tienen niveles normales de ferritina y de saturación de transferrina, deberían seguir un control evolutivo para determinar la posible progresión de la enfermedad. No hay guías estrictas que nos digan cuan frecuentes deben ser las determinaciones séricas en la sobrecarga del hierro. 3.8 Diagnóstico de la hemocromatosis hereditaria En la actualidad, la mayoría de los pacientes con hemocromatosis hereditaria permanecen asintomáticos en el momento del diagnóstico. Normalmente son los niveles altos de saturación de transferrina o ferritina sérica los que disparan la alarma y llevan al diagnóstico. En muchas ocasiones dicho hallazgo proviene de análisis solicitados por otra razón. Sin embargo, en pocos pero significativos casos, el diagnóstico se produce en fases avanzadas, en las que el tratamiento sólo obtiene respuestas parciales. 3.8.1 Signos y síntomas de la hemocromatosis hereditaria La primera manifestación de la hemocromatosis hereditaria suele aparecer como una elevación de la saturación de transferrina en suero o incremento notable de ferritina, mucho antes de la aparición de signos y síntomas. Los varones presentan típicamente síntomas iniciales de hemocromatosis hereditaria (p.e. fatiga o pigmentación cutánea) en las edades comprendidas entre los 50 y los 60 años. Sin embargo, un análisis pertinente de parámetros del hierro habría detectado la enfermedad a los 30 años. Las mujeres no suelen presentar síntomas de hemocromatosis hereditaria hasta pasada la menopausia, si es que los presentan (es infrecuente la aparición de enfermedad clínica en mujeres). Las diferencias en 27 MANUAL PARA PACIENTES CON HEMOCROMATOSIS la presentación de signos y síntomas de la enfermedad entre hombres y mujeres están relacionadas con la pérdida de sangre menstrual, que produce una disminución natural de los depósitos de hierro. Otros factores que pueden influir en el período de tiempo de aparición de la clínica podrían ser el consumo de alcohol en los pacientes con hemocromatosis y la presencia de otras situaciones médicas como las infecciones. Los síntomas iniciales de aparición de enfermedad son muy sutiles y poco específicos. Estos signos y síntomas incluyen: • Fatiga generalizada. • Dolor articular. • Dolor abdominal. • Pérdida de peso. • Disminución del apetito sexual. Mientras la hemocromatosis hereditaria progresa, otros síntomas adicionales pueden incluir fatiga grave, dolor abdominal crónico y depresión. Si no se diagnostica ni se trata de forma precoz, la hemocromatosis hereditaria puede provocar complicaciones con relación a la afección de órganos vitales (enfermedad avanzada): • Enfermedad hepática. • Enfermedad cardíaca. • Alteraciones endocrinológicas. • Enfermedades articulares. • Alteraciones cutáneas. 3.8.1.1 Enfermedad hepática El hígado suele ser el primer órgano vital afectado en la hemocromatosis hereditaria. En algunas series recientes parece que se trata quizá de la única alteración primariamente ligada a la sobrecarga férrica, y posiblemente la primera en aparecer. Pueden darse diferentes alteraciones: • Hepatomegalia (aumento del tamaño del hígado). • Alteración bioquímica de las pruebas hepáticas. • Cirrosis hepática: Ésta es la complicación más temida de las comúnmente observada en los pacientes con hemocromatosis hereditaria. Se trata de una desestructuración progresiva de la 28 HEMOCROMATOSIS HEREDITARIA arquitectura del tejido hepático que conduce progresivamente a una disfunción del órgano, con múltiples consecuencias clínicas (déficit de coagulación, alteraciones cerebrales, acumulo de agua en el abdomen, coloración amarilla de la piel e incluso evolución potencialmente mortal). • Hepatocarcinoma (cáncer primario de hígado): Se ha calculado que los pacientes con hemocromatosis hereditaria que desarrollan cirrosis hepática tienen un riesgo anual del 5% de desarrollar un hepatocarcinoma. 3.8.1.2 Alteraciones endocrinológicas • Diabetes mellitus: Esta es una de las alteraciónes más comúnes en los pacientes con hemocromatosis hereditaria. La diabetes puede ser causada tanto por la disminución en la producción de insulina debida a la acumulación de hierro en las células pancreáticas que regulan la producción de insulina (células beta) como por la alteración en la sensibilidad de la insulina por exceso de hierro. Esta complicación aparece aproximadamente en el 25% de los pacientes con la enfermedad y se desarrolla más si el paciente presenta historia familiar de diabetes. Algunos estudios recientes indican que quizá la diabetes en los pacientes con hemocromatosis esté más ligada a la presentación de hepatopatía que a la propia sobrecarga de hierro. • Hipogonadismo: El hipogonadismo es una situación en la que se ve reducida o ausente la secreción de hormonas sexuales por las gónadas (glándulas sexuales) y es la segunda alteración endocrinológica más frecuente en los pacientes con hemocromatosis hereditaria. En varones, el hipogonadismo afecta a los testículos produciendo síntomas como caída del pelo corporal, disminución de la masa muscular, disfunción sexual, y aumento del tamaño de las mamas. En las mujeres, el hipogonadismo afecta a los ovarios causando disminución anormal de los niveles de la hormona sexual llamada estrógeno que produce síntomas como alteraciones en la menstruación, menopausia precoz, disminución del apetito sexual, sofocos y osteoporosis. Diversas alteraciones pueden causar hipogonadismo en los pa- 29 MANUAL PARA PACIENTES CON HEMOCROMATOSIS cientes con hemocromatosis hereditaria, pero lo más frecuente es que ésta esté causada por el acumulo de hierro en la glándula pituitaria. El tratamiento del hipogonadismo consiste en terapia hormonal sustitutiva con testoterona (en los varones) o estrógeno con o sin progesterona (en las mujeres). 3.8.1.3 Enfermedad cardíaca El corazón puede afectarse en los pacientes con hemocromatosis hereditaria, aunque este tipo de alteración es más frecuente y tiene mayor gravedad en los pacientes con hemocromatosis secundaria. Las complicaciones de la hemocromatosis hereditaria pueden incluir: • Arritmias: latidos irregulares del corazón • Hipertrofia cardíaca: aumento del tamaño del músculo del corazón • Insuficiencia cardíaca congestiva: Fallo del corazón para bombear suficiente sangre al resto del cuerpo. Las alteraciones cardíacas ocurren aproximadamente en el 15% de los pacientes con hemocromatosis hereditaria siendo las arritmias la manifestación más común. Las miocardiopatías (condiciones que afectan a la estructura y a la función del corazón) y las arritmias, puede causar muerte súbita en individuos con hemocromatosis hereditaria. Las alteraciones cardíacas no necesariamente son permanentes y se suelen tratar de forma independiente del resto de síntomas de la hemocromatosis hereditaria. 3.8.1.4 Enfermedad en las articulaciones La artropatía (enfermedad de las artriculaciones) ocurre en más del 50% de los pacientes con hemocromatosis hereditaria y aparece frecuentemente en los hombros, muñecas, caderas y rodillas, pero sobre todo es típica la afectación de las articulaciones de los dedos de las manos. Con frecuencia no se conoce el mecanismo por el cual se desarrolla una artropatía en los pacientes con hemocromatosis, puesto que algunos pacientes desarrollan esta complicación sin tener una 30 HEMOCROMATOSIS HEREDITARIA sobrecarga férrica importante. Menos del 10% de los pacientes con hemocromatosis y artropatía mejoran sus síntomas de artritis al realizarse tratamiento de disminución férrica. La afectación más frecuente se da en las articulaciones metacarpofalángicas de los dedos índice y corazón. Radiológicamente es frecuente observar la presencia de condrocalcinosis o calcificación de los tejidos blandos próximos a la articulación afectada. 3.8.1.5 Afectación de la piel Principalmente puede observarse un oscurecimiento anormal de la piel (melanoderma), causado por el depósito de un pigmento llamado melanina, que suele aparecer en forma de “bronceado” de la piel y puede estar presente al diagnóstico de la hemocromatosis hereditaria. Con más frecuencia la pigmentación cutánea es generalizada, sin embargo, en algunos pacientes puede localizarse en áreas específicas, fundamentalmente áreas que reciben irradiación solar como la cara, el cuello o a lo largo de las piernas. En países anglosajones se da mucho énfasis en esta alteración, pero en países como España donde la piel bronceada es habitual no resulta un rasgo tan distintivo. En su forma más severa, los cambios de color de la piel incluyen áreas grises o marrones en la boca. En pacientes con hemocromatosis hereditaria los cambios en la pigmentación de la piel están en relación con el incremento de los depósitos de hierro en las glándulas sudoríparas. Además, los pacientes pueden presentar signos de debilidad en las uñas, reducción o pérdida del pelo corporal y descamación de la piel. 3.8.2 Estudios complementarios en la hemocromatosis hereditaria El diagnóstico de la hemocromatosis es fundamentalmente analítico y no clínico, y se basa en una batería de pruebas relativas al metabolismo férrico: • Hierro sérico (sideremia) • Índice de saturación de transferrina en suero • Ferritina sérica 31 MANUAL PARA PACIENTES CON HEMOCROMATOSIS • Genotipo HFE • Biopsia hepática • Flebotomía cuantitativa 3.8.2.1 Determinación de hierro sérico El hierro sérico mide la cantidad de hierro circulante en el torrente circulatorio que está ligado a la transferrina (proteína de transporte del hierro). La cantidad de hierro circulante es muy variable según muchas circunstancias como la dieta, el momento del día en que se realiza el análisis, los suplementos de hierro y, por lo tanto, no es una medida precisa del estado del hierro corporal. La determinación de hierro sérico se utiliza en combinación con otras determinaciones para medir detalladamente la presencia de sobrecarga de hierro. 3.8.2.2 Determinación de transferrina y del índice de saturación de transferrina en suero Como ya se ha comentado en otra sección, la transferrina es la proteína que transporta al hierro en la sangre. Por tanto, la medición de la transferrina equivale a medir la capacidad de la sangre de transportar hierro. De hecho, la transferrina se puede medir directamente por pruebas inmunológicas (determinación directa de la concentración de la proteína) o bien se puede medir de forma indirecta saturando el suero de hierro para determinar “cuanto hierro puede transportar”, es decir, su capacidad de transporte de hierro. En cuanto al diagnóstico de hemocromatosis hereditaria, más interesante que medir la transferrina es medir el denominado índice de saturación de transferrina, que no es otra cosa que determinar qué porcentaje de la transferrina sérica está cargada de hierro para transportar. Sería algo así como investigar qué porcentaje de taxis en una ciudad transportan un viajero (en este caso el taxi es la transferrina y el viajero el hierro). Analíticamente se obtiene dividiendo la sideremia por la capacidad de transporte del hierro y multiplicando por 100. En el año 2002, los centros de control de enfermedades de los EE.UU. propusieron unas recomendaciones en la interpretación del 32 HEMOCROMATOSIS HEREDITARIA seguimiento de los resultados de la saturación de transferrina en suero: • Menos del 16% de saturación indica déficit de hierro. • Entre el 16 y el 45% se considera como normal sin sospecha de sobrecarga de hierro. • Más del 45% indica valores elevados y puede ser indicativo de sobrecarga de hierro. Sin embargo, este dato debe confirmarse en dos ocasiones separadas tres meses. Un valor de saturación de transferrina del 50% puede considerarse suficientemente sensible para detectar la mayor parte de casos de hemocromatosis hereditaria (además es una cifra fácil de recordar). Si la determinación de la saturación de la transferrina se realiza con el paciente completamente en ayunas deben considerarse valores algo superiores de saturación de la transferrina para sospechar una sobrecarga de hierro. La determinación del índice de saturación de la transferrina en suero es considerada por la mayoría de especialistas como la prueba principal de escrutinio en el diagnóstico de la hemocromatosis hereditaria. 3.8.2.3 Determinación de ferritina sérica Esta prueba mide la cantidad de hierro depositado en el hígado. Un valor de ferritina sérica igual o superior a 200 µg/l se considera como elevado en mujeres premenopausicas, y un valor superior a 300 µg/l o más se considera elevado en varones y en mujeres postmenopáusicas. Un valor elevado de ferritina sérica alerta al médico de la posibilidad de presentar una hemocromatosis y requiere seguimiento posterior con pruebas genéticas que descarten una hemocromatosis hereditaria. Niveles de ferritina sérica de 1.000 µg/l o superiores sugieren de forma contundente la presencia de riesgo de presentar daño hepático, por lo que en esta situación debe recomendarse al paciente la realización de biopsia hepática para descartar una cirrosis – fibrosis hepática. La elevación de la ferritina sérica no es específica de la hemocromatosis hereditaria y puede estar alterada en otras situaciones y, por lo tanto, esta prueba se utiliza conjuntamente con la determinación del índice de saturación de transferrina en suero para diagnosticar hemocromatosis hereditaria. 33 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 3.8.2.4 Determinación del genotipo El genotipo (prueba genética) hace referencia a una prueba especial de carácter genético para determinar si una persona es portadora de ciertas mutaciones genéticas que incrementen el riesgo de presentar una enfermedad específica, alteración o síndrome. En el caso de los pacientes con sospecha de hemocromatosis hereditaria el médico debe solicitar una prueba especial en sangre para determinar si el paciente es portador de mutaciones significativas de un gen denominado HFE. La determinación genotípica de la mutación en HFE se considera como la prueba de diagnóstico definitiva de hemocromatosis hereditaria en un paciente con sobrecarga férrica. La prueba genética para la determinación de la mutación de HFE es fácil de realizar en España y se costea por el sistema público de salud. Dicha prueba se realiza en una muestra de sangre usando una técnica llamada reacción en cadena de la polimerasa (PCR). El objetivo del estudio genético es detectar la presencia de las mutaciones en HFE (C282Y y H63D) que se asocian con hemocromatosis hereditaria. Las investigaciones han demostrado que el genotipo más comúnmente asociado con la hemocromatosis hereditaria es el genotipo homozigoto para el gen C282Y (C282Y/C282Y). La presencia de un genotipo homozigoto para C282Y indica que el individuo está genéticamente predispuesto o bien que es susceptible de desarrollar una hemocromatosis hereditaria, pero no es indicativo de que vaya a pre- El estudio genético ha mejorado el diagnóstico de la hemocromatosis hereditaria. 34 HEMOCROMATOSIS HEREDITARIA sentar la enfermedad clínica. Los individuos que son homozigotos para el gen H63D (H63D/H63D) no están predispuestos a desarrollar una hemocromatosis hereditaria. Los heterozigotos dobles (C282Y/H63D) presentan una ligera predisposición a sufrir la enfermedad. Puede calcularse con facilidad que el riesgo hereditario que tiene una persona de tener dos copias mutadas C282Y del gen HFE dependiendo del status genético de sus familiares respecto a dicha mutación es el siguiente: • Riesgo del 100%: si ambos progenitores han sido diagnosticados de hemocromatosis hereditaria y son homozigotos C282Y. • Riesgo del 50%: si uno de los progenitores y algún hermano ha sido diagnosticado de hemocromatosis hereditaria homozigota C282Y. • Riesgo del 25%: si alguno de sus hermanos (pero ninguno de sus progenitores) ha sido diagnosticado de hemocromatosis hereditaria homozigota C282Y. 3.8.2.5 Biopsia hepática Biopsia hepática de un paciente con hemocromatosis y gran sobrecarga de hierro. En la actualidad, la biopsia hepática no se considera esencial para el diagnóstico de hemocromatosis hereditaria. Habitualmente es suficiente un estudio bioquímico y genético para diagnosticar la enfermedad. 35 MANUAL PARA PACIENTES CON HEMOCROMATOSIS La biopsia hepática se suele restringir a aquellos pacientes en los que se sospecha una enfermedad hepática grave (cirrosis) o en aquellos en que la genética del gen HFE no es diagnóstica de hemocromatosis a pesar de presentar claros signos y síntomas de la enfermedad. El médico debe aconsejar la realización de una biopsia hepática si el paciente presenta una de las siguientes circunstancias: • Un valor de ferritina sérica igual o superior a 1.000 µg/l. • Unos niveles elevados de una enzima llamada alanino aminotransferasa (AST o GOT). • Un aumento del tamaño de hígado (hepatomegalia). • Otras razones que hagan sospechar que pudiera existir una cirrosis hepática. Además de realizar un estudio anatomopatológico del hígado, la práctica de la biopsia hepática permite la cuantificación precisa de la concentración del hierro en hígado por método bioquímico. Este era el procedimiento habitual para diagnosticar la hemocromatosis antes del desarrollo de técnicas genéticas de diagnóstico. El resultado de la cuantificación suele dividirse por la edad en años del paciente, para obtener el índice de hierro hepático. Se considera un índice de hierro hepático superior a 1.9 como diagnóstico de hemocromatosis. 3.8.2.6 Resonancia magnética nuclear En los últimos diez años se ha perfeccionado mucho la cuantificación del hierro almacenado en el hígado mediante la técnica denominada resonancia magnética nuclear. Esta técnica se basa en la aplicación de un campo magnético muy potente a la zona del cuerpo que se quiere investigar (en este caso, el hígado). Dicho campo magnético orienta en una dirección precisa los pequeños dipolos magnéticos que constituyen los propios átomos de hidrógeno del organismo. Al variar bruscamente el campo magnético generado por la máquina se produce una alteración en el magnetismo de los átomos de hidrógeno que desprenden energía. Dicha energía es captada por detectores del instrumento que generan una imagen del organismo a partir de los átomos de hidrógeno presentes. En tejidos con mucho hierro (que 36 HEMOCROMATOSIS HEREDITARIA Imagen de resonancia magnética nuclear de un paciente con hemocromatosis hereditaria e intensa sobrecarga de hierro. Obsérvese como el hígado (H) aparece mucho más oscuro que el bazo (B). tiene capacidad paramagnética, es decir, capaz de conservar un campo magnético) puede demostrarse una demora en la ejecución de estos cambios y la liberación de energía. Este fenómeno es mayor cuanto mayor es la cantidad de hierro en el órgano. Ello se traduce en una falta de información sobre los átomos de hidrógeno en esa zona, que se aprecia en la imagen como una zona poco informativa (de tono negro). En definitiva, es típico en la hemocromatosis que el hígado aparezca más oscuro de lo habitual. El cálculo de estos datos mediante complejos algoritmos da como resultado una evaluación del hierro hepático que puede ser tan precisa como la cuantificación bioquímica de la propia biopsia. No hay duda de que en unos años, la resonancia magnética nuclear constituirá una prueba de referencia en la cuantificación férrica de los pacientes con hemocromatosis hereditaria. 37 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 3.8.2.7 Flebotomía cuantitativa (sangrías) Las flebotomías cuantitativas, extraen un volumen fijo de sangre (aproximadamente 450 ml), que suponen una pérdida de 0,2 gr de hierro por extracción. De este modo, cuantificando el número de sangrías realizadas en el paciente hasta presentar anemia ferropénica o ferritina inferior a 50 µg/l es posible cuantificar la cantidad de hierro corporal presente. Un paciente con hemocromatosis hereditaria necesitará 20 o más sesiones, que se realizarán de forma semanal, antes de que aparezca evidencia de déficit de hierro (anemia). La población sin hemocromatosis hereditaria desarrollará anemia (déficit de hierro) únicamente con la realización de 3 o 4 flebotomías. Así pues, la flebotomía cuantitativa es a veces un método indirecto para diagnosticar la sobrecarga grave de hierro y la hemocromatosis. El siguiente esquema resume el proceso diagnóstico en esta enfermedad (Ver página 39). 3.8.3 Diagnóstico diferencial de la hemocromatosis hereditaria Varias situaciones pueden causar sobrecarga férrica y deberían ser consideradas en el diagnóstico diferencial de la hemocromatosis hereditaria. Estas situaciones incluyen: • Enfermedades hematológicas. • Enfermedades hepáticas crónicas. • Otras causas de sobrecarga férrica. 3.8.3.1 Enfermedades hematológicas • Talasemia mayor: enfermedad hereditaria de la sangre que afecta la capacidad de las personas para producir hemoglobina. • Anemia sideroblástica: alteración en una enzima que previene al organismo de incorporar hierro en la hemoglobina llevando a una sobrecarga de hierro. • Anemia hemolítica crónica: alteración hemolítica crónica en la que los glóbulos rojos se destruyen a más velocidad de la que la médula ósea es capaz de producirlos. 38 HEMOCROMATOSIS HEREDITARIA 1: La biopsia hepática, en esta situación, tiene como objetivo conocer el grado de afectación histológica hepática y, con ello, el pronóstico del paciente. 2: Cuando no se dispone del utillaje necesario para valorar el grado de sobrecarga mediante técnicas no invasivas o bien el paciente no consiente realizar la biopsia hepática, puede procederse a las flebotomías y valorar el grado de sobrecarga mediante el recuento de las mismas hasta la depleción férrica (ver texto). Figura 1: Diagnóstico diferencial de la hemocromatosis. 39 MANUAL PARA PACIENTES CON HEMOCROMATOSIS • Anemia aplásica: situación en la que la médula ósea produce cantidades insuficientes de glóbulos rojos, glóbulos blancos y plaquetas. Estas alteraciones pueden conllevar hemocromatosis, pero de tipo secundario, incluso en el caso de no recibir transfusiones. 3.8.3.2 Enfermedades hepáticas crónicas • Hepatitis. • Enfermedades hepáticas de causa alcohólica. • Porfiria cutánea tarda: enfermedad resultante de la alteración de una enzima que lleva a la acumulación de porfirinas (pigmentos) en el hígado y causa epidermolisis (aparición de ampollas) de la piel cuando se expone a la luz del sol. 3.8.3.3 Otras causas de sobrecarga de hierro • Transfusiones de glóbulos rojos (ver hemocromatosis secundaria). • Hemodiálisis prolongada. • Aceruloplasminemia: enfermedad muy rara del metabolismo del hierro, con genética autosómica recesiva, caracterizada por diabetes, degeneración de la retina y síntomas neurológicos. • Exceso de ingestión oral de suplementos de hierro. • Trasplante de progenitores hematopoyéticos. 3.9 Tratamiento de la hemocromatosis hereditaria La primera medida en el tratamiento de la hemocromatosis hereditaria es prevenir el daño secundario a la sobrecarga férrica de órganos vitales. Los pacientes que ya presenten signos de daño orgánico secundario a la exposición prolongada a los niveles elevados de hierro son tratados con la intención de prevenir más daño o ralentizar la presentación de nuevas complicaciones. En algunos casos, puede convenir complementar el tratamiento con modificaciones dietéticas para redu- 40 HEMOCROMATOSIS HEREDITARIA cir la cantidad de hierro ingerido en la dieta diaria. La hemocromatosis hereditaria suele ser tratada por hematólogos experimentados en el procedimiento de las flebotomías. A pesar que las flebotomías constituyen la base del tratamiento en la hemocromatosis primaria, existen otras opciones de tratamiento como: • Terapia con quelantes del hierro. • Trasplante hepático. • Modificación de la dieta. 3.9.1 Flebotomías terapéuticas en la hemocromatosis hereditaria Una vez el diagnóstico de hemocromatosis hereditaria se ha establecido, el tratamiento de elección aceptado son las flebotomías terapéuticas (extraer sangre del cuerpo) con el objetivo de reducir los niveles de exceso de hierro. Ésta es la manera más fácil, efectiva y exenta de toxicidad de extraer el exceso de hierro. Si las flebotomías terapéuticas se inician de forma precoz en el curso de la hemocromatosis hereditaria, el paciente puede tener una esperanza de vida normal y evitar gran parte de las alteraciones asociadas a la enfermedad. Aún en el caso de que exista evidencia de daño en algún tejido u órgano, la flebotomía puede detener la progresión de daños orgánicos subsiguientes y sus complicaciones asociadas. El inicio de flebotomías terapéuticas para el tratamiento de la hemocromatosis hereditaria puede evitar la alteración hepática, cardíaca, hormonal y cutánea si éstas todavía no están instauradas. Sin embargo, es incierto el impacto en la afectación articular y en los síntomas de cansancio y fatiga. En general, toda afectación grave instaurada sólo puede ralentizar su progresión con la práctica de flebotomias, pero difícilmente regresará. El paciente hemocromatósico con cirrosis hepática, diabetes o miocardiopatia seguirá con estas complicaciones a pesar del tratamiento. El tratamiento de los pacientes con hemocromatosis hereditaria con flebotomías terapéuticas se divide en dos fases: • Fase de reducción del hierro. • Fase de mantenimiento. 41 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 3.9.1.1 Fase de reducción del hierro El objetivo de esta fase inicial del tratamiento es extraer el exceso de hierro del organismo y volver los niveles de hierro a valores normales. La intención de esta fase intensiva de tratamiento es inducir un déficit de hierro temporal que elimine depósitos de hierro. Típicamente, las flebotomías terapéuticas, también llamadas sangrías, consisten en extracciones de sangre idénticas a las realizadas en los bancos de sangre (aproximadamente de 450 a 500 mL por unidad, que corresponde sobre unos 200 mg de hierro) una o dos veces por semana, hasta que los niveles de ferritina sérica sean bajos (menos de 50 µg/l). En caso de pacientes jóvenes con grandes depósitos de hierro, la cadencia de flebotomías se intenta que sea de 2 por semana. La duración del tratamiento suele ser de 3 meses a un año, o incluso más, hasta alcanzar una suficiente disminución del hierro. La cantidad de sangre que se extrae y la frecuencia con la que se realizan las flebotomías varía de acuerdo con la constitución corporal, la edad o la presencia de enfermedades concomitantes. Los niveles de ferritina sérica son la forma más segura y barata de monitorizar la evolución de las flebotomías terapéuticas. Es recomendable monitorizar los niveles de ferritina sérica durante las flebotomías cada 8 semanas durante las fases iniciales del tratamiento y pasar al final, cuando los niveles de hierro ya son bajos, a monitorizar cada 4 semanas. Es imprescindible también medir el nivel de hemoglobina Imagen flebotomia: Las flebotomías pueden proporcionar sangre apta para transfusión. 42 HEMOCROMATOSIS HEREDITARIA para descartar que se esté causando anemia al paciente. No existe un acuerdo generalizado entre los investigadores sobre si debe medirse o no la saturación de la transferrina durante la monitorización. 3.9.1.2 Fase de mantenimiento Una vez los niveles de hierro restablecidos son aceptables, los pacientes requieren flebotomías de mantenimiento, unas 3 a 4 veces al año según la tolerancia del paciente. El objetivo es mantener unos niveles de ferritina de menos de 50 µg/l. La monitorización debe continuar durante toda la vida del paciente. Dado que la Sociedad Española de Transfusión sanguínea ha reconocido que la sangre extraída en estas flebotomías de mantenimiento es apta para transfusión heteróloga (para donar a otras personas que la necesitan), debería recomendarse que estas flebotomías de mantenimiento se realicen en un banco de sangre siempre que no existan las contraindicaciones habituales a la donación. Los hombres suelen requerir flebotomías de forma más frecuente que las mujeres para mantener sus niveles de ferritina sérica. Ellos suelen necesitar por lo menos unas tres flebotomías al año y se les extrae, aproximadamente, de 1,5 a 2,5 litros de sangre al año. Las personas muy mayores pueden prescindir de realizar más sangrías, aunque es lógico seguir una monitorización más suave. Quien suele realizar la flebotomía terapéutica es un/a enfermero/a con experiencia o un profesional sanitario bajo supervisión de un médico. El procedimiento suele realizarse en 45 minutos o una hora. 3.9.1.3 Monitorización clínica del paciente durante las flebotomías terapéuticas Los pacientes deben ser vigilados para que no presenten signos de hipovolemia (síntomas asociados con la disminución del volumen de sangre en el organismo, como hipotensión, taquicardia, aumento de la frecuencia respiratoria, mareo o alteración de la consciencia) durante y después de la flebotomía. Después del procedimiento, es conveniente que el paciente beba agua y permanezca unos 15 minutos más en observación. 43 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Muchos pacientes no presentan problemas mayores con las flebotomías, sin embargo, pueden presentarse algunas reacciones adversas: • Cansancio (éste es el efecto más comúnmente descrito), con presencia de debilidad o mareo debido a la pérdida de volumen de sangre. • Hematomas (marcas negras y azules) en la zona de punción de la aguja. • Molestias gástricas. • Lesión nerviosa (muy raro). • Espasmos musculares. • Infección en la zona de punción de la aguja. • Anemia (si se extrae mucha sangre, el organismo no puede producir suficientes glóbulos rojos para mantener unos niveles adecuados de hemoglobina y aparece la anemia). Es muy conveniente instruir a los pacientes para que beban mucho liquido antes de la flebotomía, aproximadamente 1 o 2 litros. Esto disminuirá el riesgo de hipovolemia y debilidad. Es importante también que los pacientes después de las flebotomías no beban alcohol, no fumen y es mejor que no conduzcan. 3.9.2 Quelación del hierro como tratamiento de la hemocromatosis hereditaria La quelación de hierro es otro método utilizado en la disminución del exceso de hierro en el cuerpo y es una opción en aquellos pacientes que no toleran o bien no son candidatos a la realización de flebotomías terapéuticas repetidas. Este tratamiento consiste en la administración intravenosa, subcutánea u oral de un medicamento llamado quelante del hierro, que se une preferentemente al hierro formando un complejo soluble que se excreta en orina o en heces. Hasta hace pocos años, el único quelante utilizable en seres humanos era la desferoxamina o desferrioxamina. La desferoxamina se administra con una inyección, intravenosa o bien mediante una infusión continua con una bomba subcutánea. Este tipo de tratamiento es terriblemente engorroso y duro para el paciente. Además el tratamiento quelante es menos eficaz que las flebotomías. Además es mucho más caro que las fleboto- 44 HEMOCROMATOSIS HEREDITARIA mías. Los efectos adversos potenciales del tratamiento quelante con desferoxamina son: • Dolor, inflamación y picor en el lugar de la punción. • Reacciones alérgicas. • Alteración en la coloración de la orina (color rojizo). • Alteraciones oculares o auditivas. 3.9.3 Trasplante hepático en la hemocromatosis hereditaria La asociación americana para el estudio del hígado recomienda el trasplante hepático en pacientes cuidadosamente seleccionados con hemocromatosis hereditaria. Esta recomendación se basa en la observación de que los pacientes con hemocromatosis hereditaria que desarrollan cirrosis hepática tienen un alto riesgo de desarrollar un carcinoma hepatocelular (cáncer primario de hígado). El trasplante hepático es el único tratamiento efectivo para los pacientes con estado avanzado de hepatopatía debido a cirrosis hepática o con carcinoma hepatocelular resecable quirúrgicamente. Algunos estudios han demostrado que pacientes trasplantados por afección hepática por hemocromatosis hereditaria tienen una supervivencia menor que aquellos pacientes trasplantados por otras razones, especialmente si la sobrecarga de hierro no se había controlado antes del trasplante. Las causas más frecuentes de mortalidad en pacientes con hemocromatosis hereditaria que han recibido un trasplante hepático son las infecciones fúngicas y las complicaciones cardíacas. 3.9.4 Modificaciones de la dieta en la hemocromatosis hereditaria Como ya se ha remarcado, el tratamiento básico de la hemocromatosis hereditaria son las flebotomías. Cualquier modificación en la dieta únicamente optimizará el tratamiento, pero de ninguna forma disminuirá los depósitos de hierro y no puede de ninguna forma substituir a las flebotomías. Es importante para los pacientes que sean bien educados sobre los factores dietéticos que pueden ayudarles a tener un mejor control de 45 MANUAL PARA PACIENTES CON HEMOCROMATOSIS su situación. En general los pacientes con hemocromatosis hereditaria pueden seguir dietas regulares, pero también deben aprender a evitar ciertos tipos de comidas que pueden influir en sus niveles de hierro. Algunas recomendaciones dietéticas generales incluyen: • Limitar o reducir la ingesta de alimentos que son ricos en hierro, por ejemplo: – Carnes rojas: la carne roja contiene un tipo de hierro conocido como hierro hemo que se absorbe de forma muy fácil en el cuerpo. – Órganos y vísceras: algunos órganos como el hígado, contienen abundantes cantidades de hierro y deberían ser evitados. – Comidas enriquecidas con hierro: algunas comidas que están enriquecidas con hierro, como ciertos cereales y panes, deberían ser evitadas. • Evitar tomar suplementos de hierro o complejos multivitamínicos que contengan hierro. • Dado que el alcohol aumenta la cantidad de hierro absorbido por el cuerpo, su consumo debe ser limitado. Los pacientes con enfermedad hepática deberían evitar completamente su consumo. • Como los suplementos de vitamina C aumentan la absorción de hierro, algunos profesionales de la salud recomiendan evitar completamente la vitamina C mientras que otros recomiendan limitar su consumo a menos de 500 mg por día. La vitamina C natural que se encuentra en muchas frutas y verduras, no aumenta la absorción de hierro y deberían consumirse. • Algunos investigadores han apoyado que el consumo de té podría ser una manera de reducir hierro. El té es rico en sustancias llamadas tanatos que han sido descritas, en un número limitado de estudios, como inhibidoras de la absorción de hierro en el intestino. Otras comidas que podrían inhibir la absorción de hierro incluyen huevos, fibra y suplementos de calcio. • Debe incrementarse el consumo de frutas y verduras que contengan vitaminas y minerales. Se puede aumentar la ingesta de frutos secos, cereales, arroz y legumbres. Estos alimentos contienen hierro no hemo por lo que es más difícil de absorber que el hierro hemo. 46 HEMOCROMATOSIS HEREDITARIA • Evitar comer o manipular marisco crudo (cocinado sí que se puede comer) ya que pueden contener un microorganismo que puede ser fulminante en individuos que tienen niveles altos de hierro. • Evitar tomar paracetamol si las enzimas hepáticas están elevadas. • Algunos médicos recomiendan tomar ácido fólico a pacientes a los que se les realizan flebotomías para ayudar en la producción de la nueva hemoglobina. • Los pacientes con hemocromatosis hereditaria deberían discutir con su médico o con su dietista las modificaciones y restricciones específicas que deben seguir. 3.9.5 Pronóstico de la hemocromatosis hereditaria Como se ha explicado previamente, el diagnóstico y el tratamiento precoz con flebotomías terapéuticas son cruciales para prevenir el daño de órganos vitales como el hígado, el páncreas, el corazón y la glándula pituitaria en pacientes con hemocromatosis hereditaria. Con la extracción del exceso de hierro con flebotomías repetidas, los pacientes presentan una mejoría sintomática de su fatiga, cansancio y aletargamiento, la hepatomegalia puede disminuir y las pruebas que determinan la función hepática pueden normalizarse. Las investigaciones han demostrado que la presencia o ausencia de cirrosis hepática es el factor pronóstico (predice el curso o la evolución de la enfermedad) más importante en los pacientes con hemocromatosis hereditaria. En general, los estudios han demostrado que si las flebotomías terapéuticas se han iniciado antes de que se desarrolle una cirrosis hepática la supervivencia de los enfermos con hemocromatosis hereditaria es similar a la de la población de su mismo sexo y edad. En presencia de cirrosis hepática, el pronóstico para los pacientes con hemocromatosis hereditaria es menos bueno. Por el contrario, las investigaciones han demostrado que la tasa de supervivencia a los 10 años en los pacientes con hemocromatosis hereditaria que desarrollan cirrosis hepática es alrededor del 60% incluso con flebotomías terapéuticas de mantenimiento. La menor supervivencia en los pacientes cirróticos se atribuye al riesgo de desarrollar un car- 47 MANUAL PARA PACIENTES CON HEMOCROMATOSIS cinoma hepatocelular, hecho que sucede en aproximadamente un 25-30% de los pacientes con cirrosis hepática. En los pacientes con hemocromatosis y cirrosis hepática es importantísimo iniciar cuanto antes las flebotomías y disminuir los depósitos de hierro, porque el tiempo en que éstos se mantienen es un riesgo importante para la aparición de hepatocarcinoma. 3.10 Otros aspectos a considerar en el paciente con hemocromatosis hereditaria 3.10.1 Impacto del diagnóstico Después de ser diagnosticados de hemocromatosis hereditaria, es importante para los pacientes que sean educados por especialistas en la enfermedad para realizar un consejo genético y pruebas de cribado a los familiares de primer grado, como hermanos e hijos. Hay ventajas e inconvenientes de la determinación de la presencia de la mutación en el gen HFE. Si la prueba es positiva, pero el paciente está asintomático, como ya se ha dicho, puede repercutir en su vida en forma de discriminación laboral, aumento de primas en su seguro de vida o bien cancelación de pólizas. Además el individuo puede precisar consejo psicológico para enfrentarse a los efectos emocionales y psicológicos por el hecho de tener unas pruebas genéticas positivas a pesar de estar asintomático. En estos aspectos es muy relevante el papel que pueden jugar las asociaciones de pacientes. En ellas, las personas afectadas por la enfermedad pueden contrastar sus experiencias con otras personas en situación similar. Es también frecuente que los pacientes sufran problemas en el ámbito del trabajo y las experiencias de otros enfermos en este aspecto les pueden ayudar. Suele ser un alivio para los pacientes disponer de información adicional sobre la enfermedad relativa a ellos mismos o a sus familiares y esta información puede ser aportada por instituciones de ayuda como las asociaciones. Por último, el paciente con hemocromatosis suele sentirse mejor cuando puede contribuir a la realización de proyectos que facilitarán el diagnóstico, el tratamiento, 48 HEMOCROMATOSIS HEREDITARIA la información, la integración social, etc de otras personas afectadas por la enfermedad. 3.10.2 Avances en la hemocromatosis hereditaria En los últimos años se han realizado grandes estudios que investigan, mediante un cribado extenso, la prevalencia exacta de la hemocromatosis hereditaria, el impacto del estudio genético, la importancia de los determinantes medioambientales para desarrollar la hemocromatosis, así como la clínica y el impacto personal en pacientes con hemocromatosis hereditaria. En estos estudios se ha observado: • Es frecuente la sobrecarga férrica en las personas homozigotas para la mutación C282Y del gen HFE. Sin embargo, la mayoría de estas personas no sufren síntomas de la enfermedad. • El 93% de los participantes que accedieron a ser cribados consintieron que la información del riesgo genético pudiera ser compartida con los miembros de la familia. • La hepatitis vírica y otras enfermedades hepáticas pudieron ser reveladas en el transcurso del cribado de la sobrecarga férrica con las determinaciones de la ferritina sérica y el índice de saturación de transferrina. Otra área de interés es la de desarrollar mejores pruebas para el escrutinio de la enfermedad. Se ha comparado el clásico test de saturación de transferrina con la “capacidad de hierro-ligado insaturado” (UIBC) como prueba de escrutinio para detectar la hemocromatosis en los homozigotos C282Y. La determinación de UIBC es un proceso de un solo paso y es más barato que la determinación tradicional de la saturación de transferrina. La conclusión es que la medida de la UIBC parece superar a la saturación de la transferrina como prueba de escrutinio. Se está avanzando mucho en la comprensión del complejo sistema de absorción del hierro y de la interacción de los péptidos hepcidina y ferroportina. En este sentido, los investigadores están aprendiendo más sobre el papel de la hepcidina, un péptido sintetizado en el hígado que 49 MANUAL PARA PACIENTES CON HEMOCROMATOSIS controla la absorción intestinal del hierro y la salida del hierro de sus depósitos en unas células especializadas llamadas macrófagos. La hepcidina puede jugar un papel clave en el desarrollo de la sobrecarga férrica y entender su funcionamiento podría llevar a comprender mejor la hemocromatosis hereditaria. Además, su medida puede contribuir al diagnóstico de este tipo de enfermedades. 50 4 Hemocromatosis secundaria 4.1 ¿Qué es la hemocromatosis secundaria? La hemocromatosis secundaria es una alteración que se manifiesta con sobrecarga de hierro y que se asocia a un grupo de enfermedades crónicas (hereditarias o adquiridas) y no motivada por un defecto de los sistemas de regulación del hierro (a diferencia de lo que sucede en la hemocromatosis hereditaria o primaria). Suele darse en pacientes que sufren anemias que precisan transfusión repetida. La sangre aportada en dichas transfusiones conlleva una sobrecarga férrica que causa la enfermedad como veremos a continuación. 4.2 ¿Qué causa la hemocromatosis secundaria? A continuación se mencionan algunas enfermedades que con más frecuencia se asocian a la hemocromatosis secundaria: • Enfermedades hereditarias: – Talasemia mayor (alteración en una de las proteínas de la hemoglobina que como consecuencia produce anemia). – Anemias diseritropoyéticas congénitas (grupo heterogéneo de anemias donde los glóbulos rojos no funcionan correctamente). – Anemias hemolíticas severas (alteración que se puede dar por diferentes causas y que produce destrucción de los glóbulos rojos y como consecuencia anemia). – Anemia sideroblástica hereditaria (grupo heterogéneo de anemias donde hay un aumento de producción de glóbulos rojos y de sus precursores pero que son ineficaces). – Déficit de glucosa-6-fosfato deshidrogenasa y déficit de piruvato kinasa (déficit de unas proteínas necesarias para que la membrana que recubre los glóbulos rojos sea estable y no se destruya). 51 MANUAL PARA PACIENTES CON HEMOCROMATOSIS – Porfirias (grupo heterogéneo de enfermedades hereditarias producidas por un déficit en alguna de las proteínas que intervienen en la correcta síntesis del grupo hemo que es indispensable para la formación de hemoglobina–proteína transportadora del oxígeno). • Enfermedades adquiridas: – Aplasia medular (enfermedad normalmente secundaria a algún medicamento o bien de causa desconocida que consiste en la alteración en la producción de glóbulos rojos, glóbulos blancos y plaquetas). – Síndromes mielodisplásicos (enfermedad de causa desconocida que consiste en la alteración en la calidad y la cantidad de glóbulos rojos, glóbulos blancos y plaquetas). – Hepatopatía (enfermedad del hígado). – Yatrogénico (secundaria a actuaciones médicas). – Tras tratamiento intensivo con transfusiones (por ejemplo en las leucemias agudas o durante la realización de un trasplante de progenitores hematopoyéticos (trasplante de células madre que se realiza en enfermedades malignas de la sangre). El origen de la sobrecarga férrica en estas enfermedades suele ser por acumulo de hierro secundario a la administración intravenosa (ya sea como tratamiento con hierro medicinal o mediante transfusión), aunque con menos frecuencia puede deberse a un aumento en la absorción a nivel del tubo digestivo secundario a una formación ineficaz de glóbulos rojos en la médula ósea, o bien una combinación de ambos mecanismos. Se aprecia hemocromatosis severa en aquellos pacientes con anemia crónica que precisan transfusión. Esta situación se observa con más frecuencia en aquellos pacientes que presentan anemias hereditarias graves. El incremento en la supervivencia que actualmente tienen estos pacientes gracias a las transfusiones terapéuticas ha motivado que la hemocromatosis secundaria sea hoy su principal causa de morbi-mortalidad y el principal enemigo a batir. Los pacientes con síndromes mielodisplásicos (alteración en la cantidad y en la calidad de las células de la médula ósea) y alteraciones adquiridas de la eritropoyesis (alteraciones en la formación de precursores de los glóbulos rojos), cuando pre- 52 HEMOCROMATOSIS SECUNDARIA cisan tratamiento transfusional crónico, presentan problemas graves en función de las transfusiones que precisen y de la duración de su enfermedad. Otro tipo de pacientes con hemocromatosis secundaria que actualmente está apareciendo, son aquellos pacientes que han sido diagnosticados de enfermedades malignas de la sangre y que con tratamiento quimioterápico intensivo o bien con un trasplante de progenitores hematopoyéticos han logrado curarse, estos tratamientos precisan muchas transfusiones que comportan por su parte un aumento del aporte de hierro. 4.3 Fisiopatología de la hemocromatosis secundaria El hierro es un elemento esencial que interviene en procesos necesarios para la formación de las células de nuestro cuerpo. El hierro también interviene en el transporte del oxígeno desde los pulmones al resto de los tejidos. Por ello, el hierro del organismo está sujeto a un proceso de reutilización donde casi nada se pierde. Aproximadamente 20 mg de hierro son reciclados diariamente procedentes de la destrucción de los glóbulos rojos viejos, y la misma cantidad se utiliza para la formación de nueva hemoglobina (proteína transportadora del oxígeno que precisa hierro para su formación). Durante el proceso de evolución de las especies no se han desarrollado mecanismos para la excreción de hierro sobrante, y solamente se regula el mecanismo de la absorción de hierro de los alimentos. Este hecho conlleva un enorme problema en el tratamiento con trasfusiones de sangre, dado que cada bolsa de sangre supone un aporte neto de hierro de 0,2-0,25 gr. El metal se acumulará progresivamente en el organismo, alcanzando niveles tóxicos a partir de haber transfundido unas 20-30 bolsas de sangre (aproximadamente 5 g de hierro). El hierro acumulado interfiere con el resto de las células y se acumula en los tejidos y en los órganos provocando daños vitales. No está claro el mecanismo por el que se produce la hemocromatosis asociada a hepatopatía (enfermedad del hígado) y a síndrome 53 MANUAL PARA PACIENTES CON HEMOCROMATOSIS metabólico, aunque parece claro que ambas se relacionan con un incremento de la absorción de hierro a nivel intestinal. 4.4 Diagnóstico de la hemocromatosis secundaria 4.4.1 Signos y síntomas de la hemocromatosis secundaria Los pacientes con hemocromatosis secundaria, además de presentar los signos y síntomas correspondientes a sus enfermedades de base, tienen, además, signos y síntomas específicos por presentar además una sobrecarga férrica. En general, la sintomatología clínica es semejante a la presente en la hemocromatosis hereditaria, pero suele existir una afectación cardíaca más severa en forma de insuficiencia cardíaca y/o arritmias (alteración en el ritmo de los latidos del corazón) que puede llegar a causar la muerte. Asimismo pueden presentarse síntomas asociados a insuficiencia hepática por mal funcionamiento del hígado y es frecuente la aparición de diabetes debida a daño pancreático (por acumulo de hierro en el páncreas). Puede darse amenorrea en mujeres (retirada de la menstruación) e impotencia en varones debidas a daño hipotalámico (órgano endocrino situado en el cerebro que regula algunas funciones sexuales). En los niños son frecuentes las alteraciones del crecimiento (en parte debidas al uso de quelantes). 4.4.2 Evaluación analítica de la hemocromatosis secudaria La biopsia hepática revela la presencia de precipitados de hemosiderina (depósitos de hierro) mediante tinción de Perls, que se distribuyen en las células del hígado. También se podría determinar la presencia de acumulo de hierro anormal, con o sin destrucción del tejido, en el corazón y en órganos endocrinos. En estas biopsias también se podrán observar las alteraciones propias de cada enfermedad que cause la sobrecarga de hierro. Cuando el origen de la hemocromatosis secundaria es transfusional, el diagnóstico debe sospecharse al sobrepasar la cifra de 20-30 bolsas de 54 HEMOCROMATOSIS SECUNDARIA sangre transfundidas. Un valor de ferritina sérica de 1.000-2.000 µg/l suele considerarse indicativo de inicio de tratamiento quelante (fármacos que se unen al hierro y favorecen su excreción por la orina o por las heces). También pueden usarse métodos más sofisticados como la cuantificación de la concentración del hierro hepático mediante resonancia magnética nuclear (prueba de imagen) o mediante métodos bioquímicos en biopsia hepática: • Ferritina sérica: Constituye un buen índice de los depósitos orgánicos de hierro. Pero hay que recordar que estos niveles aumentan desproporcionadamente con relación al depósito orgánico de hierro en individuos con inflamación. • Índice de saturación de la transferrina: Consiste en medir la saturación en hierro que presenta su proteína transportadora en suero. Valores superiores a un 50% en dos ocasiones son sugestivos de la enfermedad. Se trata de un test barato aunque no específico, apto para el cribado de la población. • La biopsia hepática es la prueba de referencia para la determinación del hierro corporal y consiste en la cuantificación bioquímica del hierro en la muestra del hígado. Aunque preciso y útil, este método es invasivo y no exento de morbilidad. Permite la revisión del tejido de la arquitectura hepática, lo cual mejora la información sobre la situación clínica del paciente. No obstante, los riesgos que conlleva hacen que esta prueba se use cada vez menos. • La resonancia magnética nuclear es un método sensible, certero y no invasivo para la determinación de hierro (cuando está tres veces por encima de los valores normales) especialmente indicada en hígados que no se pueden biopsiar y también indicada en el seguimiento del tratamiento con flebotomías o quelantes. 4.4.3 Diagnóstico diferencial de la hemocromatosis secundaria En aquellos casos en los que la hemocromatosis secundaria no se deba a las transfusiones terapéuticas, el diagnóstico diferencial deberá establecerse con la hemocromatosis hereditaria o primaria. En general, los análisis genéticos específicos (determinación de la presencia de 55 MANUAL PARA PACIENTES CON HEMOCROMATOSIS mutaciones en el gen HFE) y la presentación de otros familiares con la enfermedad diferenciará ambos procesos. 4.5 Tratamiento de la hemocromatosis secundaria • Flebotomías terapéuticas: En los pocos pacientes sin anemia, el tratamiento más eficaz, barato y con menos efectos secundarios son las flebotomías terapéuticas. El procedimiento consiste en la extracción una o dos veces por semana de 4-7 ml/kg de sangre del paciente (habitualmente 450 ml en un adulto), realizando controles periódicos de hemoglobina y ferritina. Es recomendable continuar con el procedimiento hasta obtener valores de ferritina sérica inferiores a 50 µg/l. • Tratamiento quelante: El tratamiento de quelación del hierro consiste en la utilización de un fármaco capaz de unirse al hierro en el organismo para formar una molécula con hierro que pueda ser eliminado del cuerpo fácilmente. Existe un fármaco de referencia que ha demostrado de forma inequívoca, tras más de 40 años de aplicación clínica, beneficios en morbilidad y mortalidad. Se ha demostrado que mientras que la práctica totalidad de los pacientes afectos de talasemia mayor bien quelados con desferoxamina cumplen los 25 años de edad, sólo un tercio de los mal quelados lo consiguen. Hasta muy recientemente, la desferoxamina ha sido el único agente quelante del hierro aprobado en todo el mundo para el tratamiento de la hemocromatosis transfusional. Sin embargo, la desferoxamina es una molécula grande que tiene una mala absorción oral. Este fármaco debe administrarse mediante infusión intravenosa lenta a lo largo de un periodo de 8-12 horas al día, de cinco a siete días por semana. Aunque el fármaco es eficaz, muchos pacientes no soportan esta forma de administración y abandonan el tratamiento. Posteriormente, se introdujo un segundo fármaco quelante (deferiprona) con una mejor absorción por vía oral. Desgraciadamente, el programa de desarrollo clínico de este fármaco resultó insuficiente, por lo que su uso no fue aprobado ni en EE.UU. ni en Canadá. En Europa la 56 HEMOCROMATOSIS SECUNDARIA deferiprona obtuvo aprobación para tratamiento de pacientes con talasemia mayor en los que el tratamiento con desferoxamina esté contraindicado o resulte inadecuado. La aparición de efectos secundarios como artropatías (enfermedades de las articulaciones) y agranulocitosis (interacción en la formación de glóbulos blancos en la médula ósea que conlleva a una disminución en la cantidad de glóbulos blancos fabricados), así como la escasa información disponible para el uso de este fármaco en pacientes menores de 6 años, ha limitado mucho su uso en todo el mundo. Recientemente, ha sido aprobado en la mayoría de países el uso de un nuevo quelante oral de una sola toma diaria (deferasirox). Deferasirox es una molécula pequeña con buena absorción vía oral. Además presenta una vida media en el organismo larga, por lo que su administración oral en una sola toma al día permite mantener niveles terapéuticos del fármaco en sangre. A dosis de 20-30 mg/Kg/día, deferasirox parece ser tan eficaz y seguro como desferoxamina, pero su facilidad en la administración asegura un mejor cumplimiento del tratamiento. En este fármaco, la excreción de hierro es fundamentalmente fecal. El nuevo quelante puede afectar la función renal, por lo que no debe administrarse en pacientes con insuficiencia renal avanzada y debe realizarse seguimiento estrecho de la función renal. En los pacientes tratados debe realizarse una evaluación otorrinolaringológica y oftalmológica anual, dado que se han comunicado (al igual que con el tratamiento con desferoxamina) casos de cataratas y disminución de la audición inducidos por el fármaco. En general, deferasirox debe utilizarse en pacientes que han recibido entre 20 y 40 unidades de sangre y con niveles de ferritina superiores a 1000-2000 µg/l. En nuestro entorno, la causa más frecuente de hemocromatosis secundaria es la asociada a pacientes que sufren síndromes mielodisplasicos (un tipo de enfermedad degenerativa de la médula ósea) que deben ser politrasfundidos. Atendiendo a ello, la Asociación Española de Hematologia y Hemoterapia elaboró en el año 2008 unas guías de tratamiento de este tipo de pacientes con fármacos quelantes. Con el fin de mejorar la información disponible sobre este tema en este manual hemos añadido dicha guía como anexo 1 al presente libro. 57 5 Historia y objetivos de la Asociación Española de Hemocromatosis La Asociación Española de Hemocromatosis, con anagrama AEH, es una asociación sin ánimo de lucro que se funda en el Hospital de l'Esperit Sant, en Santa Coloma de Gramanet (Barcelona), el 11 de mayo de 1999, con el objetivo primordial de luchar contra la Hemocromatosis. Bajo el patrocinio de la Fundación del Hospital de l'Esperit Sant se propone utilizar todos los medios legales a su alcance para conseguir sus fines fundacionales. La Asociación obtuvo el reconocimiento del Ministerio del Interior del Estado Español el 23 de octubre de 2000. El ámbito de actuación de la AEH es todo el territorio español. Además, desde el año 2005 La AEH es socio fundador de la Federación Europea de Asociaciones contra la Hemocromatosis, y en este sentido el ámbito de actuación de la Federación es Europa. Por último y dadas las características de la AEH, también mantenemos comunicación e intentamos ayudar a todas aquellas personas o entidades de habla hispana afectadas por este grupo de enfermedades. Hospital de l'Esperit Sant, sede de la Asociación Española de Hemocromatosis. 59 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Los fines fundacionales de la AEH son: • Sensibilizar a la opinión pública y a las instituciones públicas y privadas respecto a las Hemocromatosis. • Promover la investigación clínica y farmacéutica de la enfermedad • Promover el diagnóstico precoz • Facilitar la interrelación entre las personas afectadas, sus familiares y los profesionales de esta enfermedad • Facilitar a sus socios toda la información disponible sobre la enfermedad, velar por un tratamiento adecuado y por la no discriminación social, laboral o de cualquier otro tipo de los afectados. • Relacionarse con otras Asociaciones afines. • Promover que la sangre obtenida mediante flebotomía de los pacientes con hemocromatosis hereditaria se utilice en transfusión heteróloga (es decir, se utilice como sangre válida para transfundir a pacientes). Los inicios de la Asociación se remontan al año 1997, cuando un grupo de facultativos del Hospital de l'Esperit Sant reflexionaron sobre la escasa información existente sobre este tipo de patología. Comprobaron además que este problema se daba en muchos países, y que ello había propiciado la aparición de Asociaciones de Hemocromatosis en Gran Bretaña, Francia, Canadá, EE.UU. y otros países. Tras ponerse en contacto con algunas de estas asociaciones se tomó conciencia del interés de crear una Asociación similar en España. La naciente Asociación obtuvo la ayuda técnica del Dr. Enric Gimferrer, hematólogo del Hospital de Sant Pau de Barcelona, docente de la Universidad Autónoma de Barcelona y fundador de la primera Unidad de Ferropatología y Radicalosis del mundo y de la Dra. Montserrat Baiget, directora del Servicio de Genética del Hospital de Sant Pau para presentar fundamentos y convencer a la Fundación del Hospital de l'Esperit Sant sobre la conveniencia de crear una Asociación Española de Hemocromatosis. Los argumentos fueron escuchados y se iniciaron los trámites que culminaron en la creación de la AEH. Inicialmente y de forma artesanal se creó una página web “sin coste. A pesar que los medios con los que se creó esa página eran cier- 60 HISTORIA Y OBJETIVOS DE LA ASOCIACIÓN ESPAÑOLA DE HEMACROMATOSIS El Dr. Gimferrer fue un incansable luchador por el estudio de las enfermedades por sobrecarga de hierro. Fundó la primera Unidad de Ferropatología y Radicalosis que existió en el mundo. tamente rudimentarios, la web sirvió como mecanismo inicial de comunicación con los primeros socios y con la sociedad en general. A partir de este momento relacionamos a continuación las principales acciones realizadas por la AEH: 1997: Se inició un servicio de consultorio médico mediante e-mail y desde entonces se han atendido miles peticiones. 1998: Se consigue el apoyo para la realización del proyecto de organizaciones extranjeras, como las Asociaciones Americana, Canadiense, Francesa e Inglesa para las enfermedades por sobrecarga de hierro y del Dr. Arturo Rolla, Profesor clínico asociado de la escuela médica de Harvard. Se consigue difundir noticias sobre la enfermedad mediante algunos medios de comunicación. El patronato del Hospital del Esperit Sant aprueba la creación de una Asociación de lucha contra la hemocromatosis. 1999: Se inicia un trabajo en el Servicio de Hematología del Hospital de Sant Pau para valorar las causas de sobrecarga férrica en una serie de pacientes con esta patología. Una vez finalizado, el estudio se publica internacionalmente. Se crean los estatutos de la Asociación Española de Hemocromatosis y se constituye la junta directiva. Se elabora un primer material informativo para los asociados, entre ellos un manual (el anterior al presente) sobre la enfermedad. 61 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 2000: El 11/1/2000 se presenta la Asociación en la Sociedad Catalana de Hematologia y Hemoterapia. En Octubre de 2000 se registra oficialmente la AEH. El mismo año se obtiene la primera aportación de una empresa privada al proyecto. También el mismo año se inicia en el Servicio de Hematología del Hospital de Sant Pau una línea de trabajo que intenta investigar la posible relación entre la sobrecarga de hierro y la muerte tóxica (fundamentalmente infecciosa) en el trasplante de progenitores hematopoyéticos. Este trabajo se publicó al final en una revista internacional y ha resultado pionero en este campo. 2001: Se encarga a un diseñador la creación del logo de la Asociación y del diseño del material informativo. Se crea un boletin de información para los socios de aparición periódica. Se inicia el soporte administrativo de la Asociación. El 23/5/2001 se realiza rueda de prensa para anunciar la creación de la Asociación. Esta información se difunde en prensa especializada (Diario Médico) y diversos medios de comunicación escritos y televisión local (TV5, TV3, Catalunya avui, Canal Sur, TVE2, TV Badalona, El Punt, MEDifusión; y algunos medios de la red). Se envía por correo un tríptico informativo sobre la enfermedad y la Asociación a todos los médicos especialistas en Digestivo y Hematología de Cataluña (en total más de 450 especialistas). Se reparten más de 500 trípticos informativos a pacientes y familiares 2002: Se vincula la Asociación con el Departamento de Ingenieria Electrónica de la Universidad Politécnica de Catalunya para la creación de un sistema no invasivo de medida del hierro hepático. El proyecto culmina en una publicación científica internacional y la tesis doctoral del Dr. Casañas. El presidente de la AEH participó como moderador de una mesa en el Simposio Internacional sobre hemocromatosis hereditaria celebrado en el Hospital Gregorio Marañon y soportado por la Universidad Complutense y la Comunidad de Madrid. Se realiza un programa de televisión (TV2) de la serie Línea 900, de 30 minutos de duración, divulgativo sobre la enfermedad. El programa se llamó “Los hombres de hierro”, y fue emitido dos veces para covertura nacional y una vez en América Latina mediante conexión por el satélite Hispasat. Este año la Asociación interviene por primera vez como perito en un 62 HISTORIA Y OBJETIVOS DE LA ASOCIACIÓN ESPAÑOLA DE HEMACROMATOSIS Asistentes a la reunión del European Iron Club en Rennes, en el año 2004. Allí se gestó la creación de la Federación Europea de Asociaciones de Hemocromatosis. caso de invalidez total por hemocromatosis (el primero en España), caso que el paciente gana. En el año 2002 el presidente de la AEH participó en el programa educacional de la XLIV Reunión Nacional de la Asociación Española de Hematologia y Hemoterapia con la ponencia “Diagnóstico y tratamiento de la hemocromatosis”. 2003: Se inician contactos con el Servei d'Educació Sanitaria i Programes de Salut del Departament de Sanitat i Seguretat Social. Dichos contactos culminan en la realización de un estudio epidemiológico para la determinar la situación del metabolismo férrico en Cataluña. Dicho estudio se publica en la revista “Medicina Clínica”. Asimismo, se inician contactos con la Agencia de Evaluación de Tecnología Médica del Instituto de Salud Carlos III para la realización de un estudio de coste-efectividad del cribado genético de la enfermedad. Estos contactos culminan en una beca FIS concedida para este proyecto, mediante la que puede estudiarse el genotipo de más de 1.000 neonatos catalanes. Los resultados de coste-efectividad serán finalizados en breve por el Instituto de Salud Carlos III. Este proyecto ha sido aceptado para publicación en la revista norteamericana Genetic Testing. 2004: Se interviene como péritos en el segundo caso en España de incapacidad laboral por hemocromatosis. El paciente gana el caso. Se 63 MANUAL PARA PACIENTES CON HEMOCROMATOSIS publica un estudio en el que se analizó si la dieta rica en magnesio contribuye a disminuir la absorción de hierro en los pacientes con hemocromatosis. Además se participa en Septiembre de 2004 en la reunión del European Iron Club (Rennes, Francia). La reunión sirvió además para establecer las bases de la futura Federación Europea de Asociaciones de Hemocromatosis. En 2004 se inicia un servicio de atención personalizada a pacientes, sin ánimo de lucro, en el propio Hospital de l'Esperit Sant. Desde ese momento se han atendido pacientes procedentes de toda la geografía nacional, Francia y Chile. 2005: Rueda de prensa celebrada en el Hospital de l'Esperit Sant el 12 de mayo de 2005. “Un nou impuls per combatre l'Hemocromatosi: beques de recerca i congrés de l'European Iron Club (Club Europeo del Hierro) a casa nostra”. Explicando las becas recibidas para investigación en Hemocromatosis y la futura celebración en Barcelona del Congreso del Europea Iron Club. Se inicia además una colaboración con el Departamento de Economía y Empresa de la Universidad de la Rioja en la asesoría científica para la elaboración de: “Evaluación económica de la detección precoz de la hemocromatosis hereditaria en España”. La asociación asiste el mismo año al Bioiron 2005, los días 22-27 de mayo 2005 y se interviene en diversos congresos nacionales. Se constituye la federación Europea de Asociaciones de Hemocromatosis, con España como uno de los tres socios fundadores. 2006: Organización del simposio sobre patología del metabolisimo del hierro en el XLVIII Congreso de la Asociación Española de Hematología y Hemoterapia. Granada. Se organiza el congreso del “European Iron Club” en Barcelona con gran éxito de participación. Además se organiza la II Reunión de la Federación Europea de Asociaciones de Hemocromatosis. El mismo año se obtiene la aprobación por parte de la Sociedad Española de Transfusión Sanguínea para que la sangre procedentes de las personas con hemocromatosis hereditaria pueda usarse en transfusión heteróloga. 2007: Es el año de la inauguración del nuevo hospital de l'Esperit Sant. Se inicia la confección de este manual, y se crea una nueva y moderna web www.hemocromatosis.es con nuevos contenidos y gestor de noticias. 64 HISTORIA Y OBJETIVOS DE LA ASOCIACIÓN ESPAÑOLA DE HEMACROMATOSIS No sabemos lo que nos deparará el futuro, pero intuimos que será aún mejor que el pasado. Para seguir las noticias que se dan día a día lo mejor es consultar nuestra página web. 65 Bibliografía 1. Adams PC, Reboussin DM, Barton JC, et al. (2005) Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 352,176978 2. Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, Bahlo M, Nisselle AE, Vulpe CD, Anderson GJ, Southey MC, Giles GG, English DR, Hopper JL, Olynyk JK, Powell LW, Gertig DM. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008 ;358:221-30 3. Altes A, Remacha A, Sureda A, Martino R, Briones J, Brunet S, Sierra J, Gimferrer E. Iron overload might increase TRM in haematopoietic stem cell transplantation. Bone Marrow Transplantation. In press. 4. Altes A, Ruiz A, Barceló MJ, et al (2004) Prevalence of C282Y, H63D and S65C mutations of HFE gene in 1146 newborns from a region of Northern Spain. Genetic Testing 8:407-10 5. Altes A et al. (2007). The relationship between iron overload and clinical characteristics in a Spanish cohort of 100 C282Y homozygous hemochromatosis patients. Ann Hematol 86, 831-835 6. Andrews NC. Disorders of iron metabolism. N Engl J Med 1999;341:1986-95 7. Angelucci E, Muretto P, Lucarelli G, Ripalti M, Baronciani D, Erer B. Treatment of iron overload in the “exthalassemic”. Report from the phlebotomy program. Ann New York Acad Sci 1998;850:288-293 8. Arija V, Viteri F. Deficiencias de nutrientes conducentes a anemia, su prevención y tratamiento. En: Serra LL, Aranceta J. Nutrición y salud pública 2ª ed. Barcelona: Masson; 2006. p. 393-705 9. Asberg A, Hveem K, Thorstensen K, et al. (2001) Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol. 36,1108-15 10. Bradley S, Gosriwitana I, Srichairatanakool S, Hider R, Porter J. Non-transferrinbound iron induced by myeloablative chemotherapy. Br J Haematol 1997;99:337343 11. Barry M, Sherlock SA (1971) Measurements of liver-iron concentration in needlebiopsy specimens. Lancet 1,100-103 12. Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845GíA (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002;359:211-218 13. Beutler E, Beutler L, Lee PL, Barton JC. (2004) The mitochondrial nt 16189 polymorphism and hereditary hemochromatosis. Blood Cells Mol Dis 33, 344-345 67 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 14. Bonkovsky HL, Ponka P, Bacon BR, Drysdale J, Grace ND, Tavill AS. An Update on iron metabolism: Summary of the fifth international conference on disorders of iron metabolism. Hepatology 1996; 24:718-729 15. Brittenham GM, Sheth S, Allen CJ, Farrell DE. Noninvasive methods for quantitative assessment of transfusional iron overload in sickle cell disease. Semin Hematol 2001;38:37-56 16. Bulaj ZJ, Ajioka RS, Phillips JD, LaSalle BA, Jorde LB, Griffen LM et al. Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med 2000;343:1529-35 17. Burt RB, Wilson WH. Conditioning (preparative) regimens. En: Burt RK, Deeg HJ, Santos GW(eds) On call in Bone marrow Transplantation. New York: Chapman and Hall; 1996; p. 94-108 18. Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat genet 2000;25:14-15 19. Casañas R, Scharfetter H, Altes A, Rosell J. Magnetic induction system for the noninvasive measurement of susceptibility and conductivity of biological tissues June 2001, XI Conference in Electrical Bio-Impedance, Oslo, Norway. 20. Conrad ME, Umbreit JN, Moore EG, Uzel C, Berry MR. Alternate iron transport pathway. J Biol Chem 1994;269:7169-7173 21. Donovan A, Lima CA, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1:191-2000, 2005 22. Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J et al. Positional cloning of zebrafish ferroportin 1 identifies a conserved vertebrate iron exporter. Nature 2000;403:776-81 23. EASL International Consensus Conference on Haemochromatosis. J Hepatol 2000; 33:485-504 24. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat gen 1996; 13:399-408 25. Fleming DJ, Jacques PF, Tucker KL, et al. Iron status of the free-living, elderly Framingham Heart Study cohort: an iron-replete population with a high prevalence of elevated iron stores. Am J Clin Nutr 2001; 73:638-646 26. Fletcher LM, Dixon JL, Purdie DM, Powell LW, Crawford DH. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 2002;122:281-289 27. Finch CA, Huebers H, Eng M, Miller L. Effect of transfused reticulocytes on iron exchange. Blood 1982;59:364-369 28. Finch CA, Huebers HA, Cazzila M, Bergamaschi G, Belloti V. Storage iron. In: Albertini A, Arosio P, Chiancone E, Crysdale J, eds. Ferritins and isoferritins as biochemical markers. Amsterdam: Elsevier;1984. p. 3-21 68 BIBLIOGRAFÍA 29. Finch CA, Belloti V Stray S. Plasma ferritin determination as a diagnostic tool. West J Med 1986;145:657-663 30. Gimferrer E, Ubeda J, Royo MT. El receptor de la transferrina. Bioferrum 1996;1:49-50 31. Goldman J, Schmitz N, Niethammer D, Gratwohl A. Allogeneic and autologous transplantation for haematological diseases, solid tumors and immune disorders: current practice in Europe in 1998. Bone Marrow Transplant 1998;21:1-7 32. Halliwelll B, Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease: an overview. Meth Enzymol 1990;186:1-85 33. Hernández C, Genesca J, Ignasi JE, García L, Simó R. Relación entre reserva de hierro y diabetes mellitus en pacientes con infección por virus de la Hepatitis C: un estudio de casos y controles. Med Clin (Barc) 2000; 115:21-22 34. Huebers HA, Finch CA. The physiology of transferrin and transferrin receptors. Physiol Rev 1987;67:520-82 35. Jacobs A. Ferritin: An interim review. Curr top hematol 1985;15:25-62 36. Jacolot S, Le Gac G, Scotet V, Quere I, Mura C, Ferec C. (2004) HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood. 103,2835-40 37. Le Gac G, Férec C. (2005) The molecular genetics of haemochromatosis. European Journal of Human Genetics 13,1172-1185 38. Le Gac G, Scotet V, Ka C, Gourlaouen I, Bryckaert L, Jacolot S, Mura C, Férec C. (2004). The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Human Molecular Genetics 13,1913-1918 39. Lee PL, Gelbart T, West C, Halloran C, Felitti V, Beutler E. (2001). A study of genes that may modulate the expression of hereditary hemochromatosis: transferrin receptor-1, ferroportin, ceruloplasmin, ferritin light and heavy chains, iron regulatory proteins (IRP)-1 and -2, and hepcidin. Blood Cells Mol and Dis 27, 783-802 40. Leggett BA, Halliday JW, Brown NN, Bryant S, Powell LW. Prevalence of haemochromatosis amongst asymptomatic australians. Br J Haematol 1990; 74:525-530 41. Livesey KJ, et al. (2004). The 16189 variant of mitochondrial DNA occurs more frequently in C282Y homozygotes with haemochromatosis than those without iron loading. J Med Genet 41, 6-10 42. McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000;5:299-309 43. Merryweather-Clarke A, Pointon JJ, Shearman JD, Robson KJH (1997) Global prevalence of putative haemochromatosis mutations. J Med Genet. 34, 275-8. 44. Merryweather-Clarke AT et al. (2003) Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet 12,2241-7 69 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 45. Milet J et al. (2007). Common Variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. The American Journal of Human Genetics. 81,799-807 46. Moirand R, Mortaji AM, Loreal O, Paillard F, Brissot P, Deugnier Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997;349:95-97 47. Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A (2001). Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 108, 619-623. 48. Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 2001;28:213-214 49. Olivieri NF, Brittenham GM. Iron-Chelating therapy and the treatment of thalassemia. Blood 1997;89:739-761 50. Papanikolaou G, Sauels ME, Ludwig EH, MacDonald MLE, Franchini PL, Dubé MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP. (2004). Mutation in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 36, 77-82 51. Papanikolau G, Pantopoulos K. Iron metabolism and toxicity. Toxicology and Applied Pharmacology 2005;202;199-211 52. Pérez G, Vittori D, Pregi N, Garbossa G, Nesse A. Homeostasis del hierro, mecanismos de absorción, captación celular y regulación. Acta Bioquím Clín Latinoam 2005;39:301-314 53. Petersen KM, Parkinson AJ, Nobmann ED, Bulkow L, Yip R, Mokdad A. Iron deficiency anemia among Alaska Natives may be due to fecal loss rather than inadequate intake. J Nutr 1996;126:2774-2784 54. Pietrangelo A. (2004) Hereditary hemochromatosis--a new look at an old disease. N Engl J Med. 350,2383-97 55. Pietrangelo A. (2004) The ferroportin disease. Blood Cells, Molecules and Diseases 32, 131-138 56. Pré J. La lipoperoxydation. Pathol Biol 1991;39:716-736 57. Remacha AF, Carrasco M, Sarda MP, Barceló MJ, Baiget M. HFE mutation analysis in patients with hepatitis C virus with positive screening for iron overload. Haematologica 1999; 84:284-285. 58. Remacha AF, Barcelo MJ, Sarda MP, Blesa I, Altes A, Baiget M. The S65C mutation in Spain. Implications for iron overload screening. Haematologica. 2000;85:1324-1325 59. Remacha AF, Altes A, Baiget M. Iron overload diagnosis. Role of HFE mutations and dysmetabolic syndrome. 7th Congress of the European Haematology Association. Florence 2002. Abstr 495 70 BIBLIOGRAFÍA 60. Roetto A, Papanikolau G, Politou M, et al. (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 33, 21-22 61. Sanchez M, Bruguera M, Bosch J, Rodes J, Ballesta F, Oliva R. (1998). Prevalence of the Cys282Tyr and His63Asp HFE gene mutations in Spanish patients with hereditary hemochromatosis and in controls. J Hepatol 29, 725-8 62. Sanchez AM, Schreiber GB, Bethel J, McCurdy PR, Glynn SA, Williams AE et al. Prevalence, donation practices, and risk assessment of blood donors with hemochromatosis. JAMA 2001;286:1475-1481 63. Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N et al. Identification of an intestinal heme transporter. Cell 2005;122:789-801 64. Sierra J. Fundamento, tipos y principales indicaciones del TPH. En Carreras E, Brunet S, Ortega JJ, Rovira M, Sierra J, Urbano-Ispizua A (eds) Manual de Trasplante Hemopoyético, 2ª edición. Barcelona: Antares; 2000. p. 9-11 65. Sturrock A, Alexander J, Lamb J, Craven CM, Kaplan J. Characterization of a transferrin-independent uptake system for iron in HeLa cells. J Biol Chem 1990;265: 3139-3145 66. Thorburn D, Curry G, Spooner R, Spence E, Oien K, Halls D, Fjox R, McCruden EA, McSween RN, Mills PR. The role of iron and haemochromatosis gene mutations in the progression of liver disease in chronic hepatitis C. Gut 2002;50:248-252 67. Urbano-Ispizua A. Fuentes de Progenitores Hemopoyéticos. En Carreras E, Brunet S, Ortega JJ, Rovira M, Sierra J, Urbano-Ispizua A (eds) Manual de Trasplante Hemopoyético, 2ª edición. Barcelona: Antares; 2000. p. 9-11 68. Vulpe CD, Kuo YM, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999;21:195-9 69. Waalen J, Nordestgaard BG, Beutler E. (2005) The penetrance of hereditary hemochromatosis. Best Pract Res Clin Haematol. 18, 203-20 70. Waheed A, Grubb JH, Zhou XY, Tomatsu S, Fleming RE, Costaldi ME, et al. Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci USA 2002;99:3117-3122 71. Wallace DF, Pedersen P, Dixon JL, Stephenson P, Searle JW, Powell LW (2002). Subramaniam VN. Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood 100, 692-694 72. Wardman P, Candelas LP. Fenton chemistry: an introduction. Radiat Res 1996;145: 523-531 73. Wick M, Pinggera W, Lehmann P. Iron metabolism, diagnosis and therapy of anemias. 3th ed. New York: Springer; 1996 71 Anexo Guía clínica de quelación del paciente con síndrome mielodisplásico* B. Arrizabalaga1, C. del Cañizo2, A. Remacha3, G. Sanz4, A. Villegas5 Introducción Los síndromes mielodisplásicos (SMD) son una serie de enfermedades clonales de la célula progenitora hematopoyética con manifestaciones clínicas muy variadas que dependen del subtipo de SMD y del estadio evolutivo de la enfermedad. Pese a esta variabilidad, una característica clínica muy frecuente en estos pacientes es la anemia. De hecho, el 80 % de los casos presentan anemia en el momento del diagnóstico, y en la mayoría de dichos casos el síndrome anémico es la causa de la consulta. Estos hechos condicionan que el objetivo fundamental del tratamiento en estos pacientes sea controlar la sintomatología provocada por el descenso de hemoglobina (Hb). Para lograr este objetivo, evitar el daño tisular debido a la anemia y mejorar la calidad de vida de los pacientes, en el momento actual disponemos de distintos tipos de tratamiento (agentes inmunomoduladores, agentes eritropoyéticos, etc.), pero no cabe duda de que la hemoterapia continúa siendo el más ampliamente utilizado. Las necesidades transfusionales, generalmente, se incrementan con el paso del tiempo, originando, antes o después –dependiendo del número de transfusiones que se administren–, una sobrecarga férrica. Servicio de Hematología. 1Hospital de Cruces. Barakaldo (Bizkaia). 2Hospital Clínico Universitario de Salamanca. 3Hospital de la Santa Creu i Sant Pau. Barcelona. 4Hospital Universitario La Fe. Valencia. 5Hospital Clínico Universitario San Carlos. Madrid. * Publicado por Grupo Acción médica en “Hematología” (Volumen 93, suplemento 1, enero 2008) con el patrocinio científico de la Asociación Española de Hematología y Hemoterapia (AEHH) y la Fundación Española de Hematología y Hemoterapia (FEHH). 73 MANUAL PARA PACIENTES CON HEMOCROMATOSIS En la actualidad no se dispone de información con un nivel de evidencia suficiente para establecer el beneficio potencial en términos de mejora de supervivencia de un programa de quelación en los pacientes con SMD. En niños con talasemia son necesarios varios años para que la quelación inadecuada tenga un impacto negativo en la supervivencia, con una caída dramática de su curva de supervivencia a partir de los 15 años1-3. Es altamente probable que la edad avanzada, la presencia frecuente de enfermedades asociadas y una peor tolerancia cardíaca a la anemia crónica por parte de los pacientes con SMD aumenten su susceptibilidad a los efectos tóxicos de la sobrecarga de hierro, por lo que el efecto deletéreo sobre la supervivencia podría ser evidente en un tiempo sustancialmente inferior en estos pacientes. Esto parecen indicar diversos estudios que han mostrado que las consecuencias clínicas de la sobrecarga de hierro en adultos con dependencia transfusional regular no debida a talasemia pueden aparecer antes de los 4 años y que los pacientes que han recibido más de 75 transfusiones presentan depósitos de hierro en el miocardio. Es bien conocido por todos que, cuando el contenido hepático de hierro (LIC, liver iron content) excede los 7 mg/g de peso seco (aproximadamente después de haber recibido 40 transfusiones), se comienza a observar toxicidad por el mismo en varios órganos, fundamentalmente hígado, corazón y órganos endocrinos1,3,4. Hasta la fecha, no disponemos de estudios que permitan definir con claridad la morbimortalidad debida a sobrecarga de hierro en SMD. Ello se ha debido tanto a la existencia de otras múltiples causas de morbimortalidad en este tipo de pacientes (infección, hemorragia, anemia crónica, edad avanzada y enfermedades asociadas) como a la inexistencia hasta la fecha de un tratamiento quelante apropiado. En un estudio publicado recientemente5, el 75 % de los pacientes con SMD habían recibido a lo largo de su vida más de 40 transfusiones, y el 30 % más de 160, lo que sugiere que la mayoría de estos pacientes pueden acabar padeciendo una sobrecarga férrica. Otro estudio un poco más antiguo4 referido previamente evidenció que el 40 % de los pacientes con SMD que habían recibido más de 74 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO 50 transfusiones mostraban signos de sobrecarga férrica, fundamentalmente disfunción cardíaca. El grupo de Pavía, en varias publicaciones muy recientes y con una amplia serie de casos, ha mostrado que el número de transfusiones y la ferritina sérica tienen impacto pronóstico sobre la supervivencia de pacientes con SMD6-8. Pese a la ausencia de estudios prospectivos sobre el posible impacto de la utilización de quelantes en la supervivencia de estos pacientes, el conocimiento que se posee de otras enfermedades con sobrecarga férrica como la -talasemia hace considerar la conveniencia de utilizar tratamiento quelante en pacientes con SMD y necesidades transfusionales. Además, los resultados de dos estudios muy recientes han mostrado mayor supervivencia en los pacientes con SMD que reciben quelación9,10. En el más extenso de los dos estudios, 76 pacientes recibieron tratamiento quelante de hierro y 89 no lo recibieron. La mediana de supervivencia del grupo sometido a quelación, que tenía menor edad, mayor intensidad transfusional y peor pronóstico según el Índice Pronóstico Internacional (IPSS), fue claramente superior (115 meses vs. 51 meses). Esta ventaja en supervivencia también fue apreciable en los pacientes con una puntuación IPSS de 0-1 puntos. En el análisis multivariante el tratamiento quelante mantuvo su impacto pronóstico favorable de forma independiente. Los pacientes que recibieron una quelación adecuada presentaron una supervivencia más larga que los que fueron quelados con pautas de intensidad inadecuada10. El reto actual es definir qué pacientes podrían beneficiarse realmente de dicho tratamiento. Objetivos El objetivo de la presente guía es proveer a los facultativos de una herramienta de fácil manejo y apropiada para averiguar qué pacientes con SMD deberían recibir tratamiento quelante, cuándo habría que iniciarlo, qué tipo de quelante se debería emplear, cómo se tendría que utilizar, y controlar su uso y sus resultados. 75 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Estudio del paciente candidato a quelación Monitorización de la sobrecarga férrica Se debe monitorizar la sobrecarga de hierro desde el inicio del diagnóstico de SMD. Diagnóstico inicial de SMD a) Sideremia, transferrina, índice de saturación de la transferrina (IST). b) Ferritina. Con qué frecuencia debe monitorizarse a) Enfermo no transfundido: cada 6 meses (sideremia, transferrina, IST, ferritina). b) Enfermo transfundido: cada 3 meses (sideremia, transferrina, IST, ferritina). Estudio del paciente, previo a la quelación Valoración del estado férrico a) Sideremia, transferrina, IST. b) Ferritina: dos determinaciones en un intervalo comprendido entre 15 días y 1 mes . c) LIC (estudio de hierro hepático, resonancia magnética): recomendable. d) T2* cardíaco: recomendable. Otros estudios recomendables para valorar una posible toxicidad del hierro o de la quelación. a) Hemograma. b) Audiometría. c) Estudio oftalmológico. d) Bioquímica (incluyendo glucemia, pruebas de función hepática, serología hepática, creatinina, aclaramiento de creatinina y proteinuria). e) Fracción de eyección por ecocardiograma u otras técnicas. * Riesgo bajo: riesgo bajo o intermedio 1 del IPSS; riesgo muy bajo, bajo o intermedio del WPSS, o riesgo bajo del IPE. 76 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO Criterios de inclusión y exclusión de quelación El grupo de Pavía ha demostrado que la dependencia transfusional es un factor pronóstico independiente de la morfología y citogenética para la supervivencia en SMD6,8. Asimismo, en el primer estudio las cifras séricas de ferritina se asociaron a la supervivencia en el análisis multivariante independientemente de la dependencia transfusional. Aunque es posible que estos hallazgos reflejen algún aspecto desfavo- Riesgo bajo* Candidato a TPH Anemia dependiente de transfusión Sí Edad muy avanzada con enfermedades asociadas y/ o comorbilidades que limiten de forma notoria la calidad o esperanza de vida No Ferritina sérica > 1.000 ng/mL Sí Quelación de hierro Algoritmo 1. Tratamiento quelante del hierro en el SMD. 77 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Tabla 1. Índice pronóstico IPSS Variable Blastos en médula ósea (%) 0 0 Puntuación Intermedio 1 0,5 1,0 5-10 – Intermedio Desfavorable 2/3 – Bajo 0 <5 Bueno 0/1 Intermedio 2 1,5 2,0 11-20 21-30 – – – – Grupos de riesgo: puntuación 0: bajo; 0,5-1,0: intermedio 1; 1,5-2,0: intermedio 2; 2,5: alto. a Bueno = normal, -Y, del(5q), del(20q). Desfavorable = cariotipo complejo (3 anomalías o más) o anomalías del cromosoma 7. Intermedio = otras alteraciones cariotípicas. b Hb < 10 g/dL; neutrófilos < 1.800/µL; plaquetas < 100.000/µL. Tabla 2. Índice pronóstico WPSS Variable Puntuación 0 1 2 3 Categoría OMS AR, ARSA, 5q- CRDM, CRDM-SA AREB-1 AREB-2 Cariotipoa Bueno Intermedio Desfavorable – No Regular – Requerimiento transfusionalb Grupos de riesgo: muy bajo (puntuación: 0), bajo (1), intermedio (2), alto (3-4) y muy alto (5). AR: anemia refractaria; ARSA: anemia refractaria con sideroblastos en anillo; 5q-: síndrome mielodisplásico con del(5q) aislada y menos del 5 % de blastos en médula ósea; CRDM: citopenia refractaria con displasia multilínea; CRDM-SA: citopenia refractaria con displasia multilínea y sideroblastos en anillo; AREB-1: anemia refractaria con exceso de blastos 1; AREB -2: anemia refractaria con exceso de blastos 2. a Bueno = normal, -Y, del(5q), del(20q). Desfavorable = cariotipo complejo (3 anomalías o más) o anomalías del cromosoma 7. Intermedio = otras alteraciones cariotípicas. b Dependencia transfusional: una transfusión o más cada 8 semanas durante un período de 4 meses. Tabla 3. Índice Pronóstico Español (IPE) Variable Blastos en médula ósea (%) Plaquetas (°Edad (años) 78 109/L) Puntuación Grupo de riesgo Puntuación 0 1 2 <5 5-10 > 10 Bajo 0-1 50 Intermedio 2-3 Alto 4-5 100 51-100 < 60 60 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO rable de la enfermedad de base que desconocemos, es también verosímil que ello pueda deberse, al menos en parte, a las consecuencias de la sobrecarga de hierro. Por otro lado, varias series indican que la cifra de ferritina sérica antes del trasplante alogénico de progenitores hematopoyéticos se asocia a la mortalidad del procedimiento, claramente superior en los pacientes con niveles de ferritina muy elevados11. En una serie reciente, este impacto en la supervivencia fue especialmente notable en pacientes con leucemia mieloblástica aguda y SMD12. Este hecho sugiere que un programa de quelación de hierro podría estar indicado particularmente en este tipo de pacientes. Los criterios de selección para tratamiento quelante del hierro (Algoritmo 1) que se muestran a continuación tratan de identificar a los pacientes con SMD en los que cabe esperar mayor beneficio potencial de un programa de este tipo. Estos criterios, basados en las recomendaciones actuales de los expertos13, tienen en cuenta la heterogeneidad pronóstica de los SMD, la intensidad de las transfusiones, la edad, el estado general, las enfermedades asociadas de los pacientes y la modalidad seleccionada para tratar la enfermedad hematológica de base. Criterios de inclusión a) Anemia dependiente de transfusión. b) Nivel de ferritina sérica superior a 1.000 µg/L. c) Subtipo de SMD (uno o varios de los siguientes criterios): • Índice pronóstico IPSS14 (Tabla 1) de riesgo bajo o intermedio (puntuación: 0-1). • Índice pronóstico WHO classification-based Prognostic Scoring System (WPSS)8 (Tabla 2) de riesgo muy bajo, bajo o intermedio (puntuación: 0-2). • Índice Pronóstico Español15 (Tabla 3) de riesgo bajo (puntuación: 0-1) para pacientes sin estudio citogenético disponible. • Candidatos a trasplante alogénico de progenitores hematopoyéticos, independientemente del grupo de riesgo pronóstico al que pertenezcan. 79 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Tabla 4. Propiedades de los fármacos Principio activo (nombre comercial) Indicación aprobada Dosisb Vía de administración NTBI (24 h) Adhesión Deferoxamina Deferiprona Deferasirox Sobrecarga Fe Sobrecarga Fe en TM > 10 añosa TM > 6 años Sobrecarga transfusionala 25-50 mg/kg/día 75 mg/kg/día 10-30 mg/kg/día Perfusión subc. 8-10 h/díac Oral/8 h Oral/24 h Persiste Indicios Negativo Mala Buena Buena Agranulocitosis Alteración renal 10 años 3 años Principales efectos adversos Problemas locales Experiencia clínica en TM 30-40 años a Cuando la deferoxamina está contraindicada o es inadecuada. En relación con el ritmo transfusional (± 2 C.H./3-4 s). c En el SMD se ha utilizado como pauta alternativa 1 g/12 h diario en inyección s.c. lenta. La vía i.v. en perfusión continua se reserva para sobrecargas severas con afectación cardíaca. NTBI: non-transferrin-bound iron; TM: talasemia major. b Criterios de exclusión Pacientes de edad muy avanzada con enfermedades asociadas y/o comorbilidades que limiten de forma notoria su calidad o esperanza de vida. Fármacos quelantes del hierro Los fármacos quelantes atrapan el hierro tóxico y lo eliminan por la orina y/o las heces. El diseño de un quelante idóneo requiere unas características químicas a veces contrapuestas, lo que ha restringido considerablemente el mercado2. 80 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO Actualmente, los fármacos quelantes aprobados en Europa son: a) Deferoxamina. b) Deferiprona. c) Deferasirox. Consideraciones básicas en el tratamiento quelante: a) La toxicidad de todo fármaco quelante es inversa al nivel de sobrecarga. b) El objetivo no es "eliminar" la sobrecarga de hierro, sino mantener unos niveles que no sean dañinos a nivel visceral. En la talasemia major los niveles recomendados se han establecido en ferritina 1.500 µg/L y/o hierro hepático 7 mg/g. c) La dosis del fármaco debe guardar relación con la gravedad de la sobrecarga y de los requisitos transfusionales del paciente. d) El objetivo teórico de un buen quelante es la eliminación continua del LPI (labile plasma iron), que es la fracción tóxica a nivel celular. El LPI forma parte de una molécula compleja denominada NTBI (non-transferrin-bound iron), que sólo aparece en el plasma cuando existe sobrecarga. e) El tratamiento quelante en las anemias transfusionales habitualmente va a durar toda la vida del paciente, por lo que un fármaco quelante ha de confirmar su eficacia y falta de toxicidad a muy largo plazo. En la Tabla 4 se recogen las características más importantes de los tres fármacos. Experiencia clínica en SMD Deferoxamina Todos los grupos de expertos en SMD (MDS Consensus Statement, Nagasaki 2005; NCCN Clinical Practice Guidelines; UK MDS Guidelines Group; Sociedad Italiana de Hematología) aconsejan quelación con deferoxamina en pacientes con SMD de buen pronóstico que reciben terapia transfusional periódica. Sin embargo, faltan todavía estudios validados que demuestren un aumento de supervivencia en los pacientes que reciben deferoxamina. 81 MANUAL PARA PACIENTES CON HEMOCROMATOSIS La falta de datos al respecto se puede atribuir a que la pauta de deferoxamina (25-50 mg/kg/día, 5 días/semana) que se requiere para un tratamiento eficaz (medida por disminución de ferritina sérica) debe ser administrada en perfusión subcutánea durante 8-10 horas o bien en inyección subcutánea prolongada cada 12 horas, lo cual es difícil de mantener a largo plazo. Deferiprona No se han publicado estudios controlados en SMD. Tabla 5. Propiedades de los fármacos quelantes Principio activo (nombre comercial) Deferoxamina Deferiprona Deferasirox Tratamiento oral — +++ +++ Afinidad por el Fe +++ + ++ Especificidad por el Fe ++ ++ ++ Penetración celular + ++ ++ Quelación de Fe hepático ++ + ++ Quelación de Fe cardíaco + ++ ++ Balance negativo de Fe ++ + ++ Toxicidad hematológica* — + — Cobertura 24 h quelación — — + Deferasirox El deferasirox tiene una experiencia clínica todavía limitada en SMD en relación con su reciente incorporación al mercado. Los ensa- 82 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO yos clínicos que han permitido su aprobación recogen su eficacia (medida por ferritina y hierro hepático) en 47 pacientes con SMD tratados con deferasirox al menos durante 1 año16. Su administración es muy cómoda, pero su seguridad exige control renal. Selección del tratamiento A la hora de seleccionar un quelante hay que tener en cuenta: a) Las indicaciones establecidas: • La deferoxamina está indicada en el tratamiento de la sobrecarga férrica de cualquier etiología. • La deferiprona está indicada sólo en los pacientes con talasemia major mayores de 10 años, cuando la deferoxamina está contraindicada o su tratamiento es inadecuado. No está aprobada su indicación como tratamiento quelante en la sobrecarga férrica postransfusional de los SMD. • El deferasirox está indicado en la sobrecarga férrica postransfusional en pacientes con talasemia major mayores de 6 años. También está indicado cuando la deferoxamina está contraindicada o su tratamiento es inadecuado en: – Talasemia major con sobrecarga férrica debida a transfusiones infrecuentes. – Niños entre los 2 y los 6 años de edad con talasemia major que requieren quelación. – Sobrecarga férrica postransfusional asociada a otros tipos de anemia, incluyendo SMD. b) Características del fármaco: • En la Tabla 5 aparecen las características de los diferentes quelantes. A modo de resumen, comentamos: Deferoxamina • Su inconveniente principal es la forma de administración endovenosa o subcutánea, en bombas de infusión continua. Muchos 83 MANUAL PARA PACIENTES CON HEMOCROMATOSIS pacientes son incapaces de tolerar este tipo de tratamiento, por lo cual incumplen el tratamiento. • Farmacológicamente la deferoxamina tiene dos inconvenientes: – Penetra poco intracelularmente. – No ofrece una cobertura quelante diaria. Deferiprona • En muchos casos no consigue un balance férrico negativo. • No mantiene una cobertura quelante diaria en muchos pacientes. • Puede inducir agranulocitosis/neutropenia, lo que hace que la deferiprona sea poco adecuada en los SMD. Deferasirox • • • • Ofrece cobertura quelante durante las 24 horas del día. Penetra intracelularmente. Consigue un balance negativo férrico. Su tolerancia es buena, pero puede provocar alteraciones leves de la función renal. Seguridad y toxicidad La toxicidad depende del grado de sobrecarga férrica; por este motivo se recomienda suspender la quelación cuando se llega a niveles de ferritina sérica de 500 µg/L. La deferoxamina presenta los siguientes efectos adversos17: • Problemas locales de administración subcutánea (40 %). • Pérdida de capacidad auditiva (18 %). • Trastornos visuales (ceguera nocturna, cataratas, etc.) (6 %). • Trastornos del crecimiento (3 %). • Reacciones alérgicas (2 %). • Síntomas gastrointestinales (2 %). • Infecciones por Yersinia enterocolitica (1 %). Con la deferoxamina hemos de esperar un alto índice de abandonos del tratamiento quelante, debidos a su forma de administración. 84 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO Si aumento por encima del 33% de la creatinina sérica con respecto a la media basal en dos determinaciones consecutivas (7 días) Si descenso del aclaramiento de creatinina por debajo de 90 mL/min en dos determinaciones consecutivas (7 días) Mantener una buena hidratación oral (2 L/día). Vigilar interacciones con fármacos nefrotóxicos Reducir dosis en 10 mg/kg Si no mejora, interrumpir tratamiento Valorar reintroducción a dosis más baja si normalización. Contactar con nefrólogo si no mejoría Algoritmo 2. Ajuste de dosis de deferasirox (I): tolerancia renal. Interrumpir deferasirox Desaparición Persiste más de 8 días Reiniciar deferasirox en dosis del 60% de la inicial ± corticoides No reaparece el rash Reaparece el rash Aumentar dosis 5 mg/kg cada 2 semanas hasta dosis inicial Detención definitiva No reaparece el rash Reaparece el rash Detención definitiva Continuar con deferasirox Algoritmo 3. Ajuste de dosis de deferasirox (II): rash cutáneo agudo. 85 MANUAL PARA PACIENTES CON HEMOCROMATOSIS La deferiprona presenta los siguientes efectos adversos18,19: • Alteraciones gastrointestinales (náuseas, vómitos, dolor abdominal) (35 %). • Artralgias/Artropatías (13 %). • Neutropenia/Agranulocitosis (5 % y 0,5 %, respectivamente). • Alteraciones de las transaminasas. • Déficit de zinc. Cuando se administra deferiprona hemos de vigilar la cifra de neutrófilos de forma secuencial. El deferasirox presenta los siguientes datos de seguridad y toxicidad que provienen de los más de 1.000 pacientes incluidos en estudios clínicos20,21: • Un aumento de la creatinina (36 %). • Trastornos gastrointestinales (26 %). • Rash cutáneo (8 %). • Elevación de las transaminasas (2 %). • Sordera/Hipoacusia o cataratas (raras). El deferasirox es bien tolerado y de fácil administración. Requiere un seguimiento de la función renal. Teniendo en cuenta la reciente comercialización del deferasirox, es necesario vigilar los registros postcomercialización, sobre todo en lo referente a una posible toxicidad renal, interacciones con fármacos y citopenias. Ajuste de dosis Dependerá de los cambios en la sobrecarga férrica o de los efectos adversos del fármaco. Ajuste de dosis según sobrecarga férrica Deferoxamina: Dosis inicial de 40 mg/kg/día de deferoxamina en bomba de perfusión subcutánea durante 5 noches/semana. 86 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO • La dosis se ajustará según las variaciones de las cifras de ferritina sérica en un período de 6 meses o por posible toxicidad. • Por debajo de 500 µg/L de ferritina sérica hay que suspender el tratamiento para evitar toxicidad. Deferiprona: No tiene indicación establecida ni hay datos disponibles de eficacia y seguridad en el manejo de la sobrecarga férrica postransfusional de los SMD. Deferasirox: La dosis inicial es de 20 mg/kg/día en 1 sola toma diaria. • El objetivo terapéutico es mantener una ferritina sérica de entre 1.000 y 1.500 µg/L, usando dosis de entre 10 y 30 mg/kg/día dependiendo de las necesidades transfusionales del paciente y de la eficacia del tratamiento. ALT (GPT) > 3 veces valor basal o si ALT > 500 Ul/L Interrumpir deferasirox Control de ALT 1 semana después de interrupción. Después, control 2 semanas ALT < 2 valor basal Reiniciar deferasirox al 50% de la dosis inicial Continua el aumento Detención definitiva Control 1 vez/semana durante un més. Si no cambio ALT, aumento progresivo de dosis hasta dosis inicial Algoritmo 4. Ajuste de dosis de deferasirox (III): alteraciones en pruebas hepáticas. ALT: alanina aminotransferasa; GPT: transaminasa glutámico-pirúvica. 87 MANUAL PARA PACIENTES CON HEMOCROMATOSIS Tabla 5. Propiedades de los fármacos quelantes • Los trastornos digestivos son frecuentes. Informar al paciente para evitar que deje la medicación, ya que suelen ser leves y se autolimitan • Frecuencia y duración de los efectos secundarios: – Náuseas: 10 % – Vómitos: 9 % – Dolores abdominales: 14 % – Diarrea: 12 % • Prescribir tratamiento sintomático • Retrasar la toma de deferasirox por la tarde-noche (30 min antes de la comida o, preferiblemente, 2 h después) • Hacer coincidir con la ingestión de alimentos* • Tomar con agua en vez de con zumo • Para el manejo de la diarrea, véase Algoritmo 5 * Aumenta la absorción. En caso de alimentos ricos en lípidos puede doblarse la absorción. Ajuste de dosis según efectos adversos Deferasirox: • En caso de toxicidad renal, seguir el Algoritmo 2. • En caso de rash cutáneo, seguir el Algoritmo 3. • En caso de alteración en las pruebas hepáticas, seguir el Algoritmo 4. • En caso de alteraciones digestivas, seguir las recomendaciones de la Tabla 6. • En caso de diarrea, seguir el Algoritmo 5 y usar medidas de soporte. Como medidas de soporte en la diarrea, además de una adecuada hidratación, evitar laxantes, una dieta astringente y loperamida, se puede administrar el deferasirox por la noche y disolverlo en agua en vez de en zumo. Con estas medidas suele controlarse la diarrea sin necesidad de ajuste de dosis. • Los comprimidos de deferasirox contienen lactosa: valorar intolerancia a la lactosa. • Si hay que reescalar el deferasirox se hará con incrementos de 5 mg/kg/día semanales. 88 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO En el caso del deferasirox se han de vigilar posibles interacciones farmacológicas con: • Fármacos nefrotóxicos. • Fármacos que afecten la glucuronización. Monitorización del paciente durante el tratamiento quelante Medida del hierro • Ferritina sérica: cada 3 meses. • Resonancia magnética hepática: anualmente (recomendable). • Resonancia magnética del corazón con técnica T2*: anualmente (recomendable). Medida de otras pruebas para monitorizar las complicaciones (sobrecarga férrica y complicaciones del quelante empleado) Deferoxamina • Enzimas hepáticas: cada 6 meses. • Fracción de eyección por ecocardiografía/otra técnica: anualmente. • Audiometría: anualmente. • Estudio oftalmológico: anualmente. • Si fiebre y sospecha clínica de infección (Yersinia enterocolitica): cultivo de heces/hemocultivo/serología. Deferasirox • Hemograma: mensualmente. • Creatinina: el primer mes, como mínimo cada 15 días; después mensualmente. • Aclaramiento de creatinina: el primer mes, como mínimo cada 15 días; después mensualmente. • Proteinuria: mensualmente. • Estudio de enzimas hepáticas: mensualmente. • Audiometría: anualmente. 89 MANUAL PARA PACIENTES CON HEMOCROMATOSIS • Estudio oftalmológico: anualmente. • Fracción de eyección por ecocardiograma/otra técnica: anualmente. Diarrea Leve Persistencia Diarrea Moderada Persistencia Diarrea Grave Persistencia Tratamiento de soporte: deferasirox 20 mg/kg/día Valorar intolerancia a lactosa Discontinuar loperamida tras 12h sin diarrea Resolución Tratamiento de soporte: deferasirox 10 mg/kg/día Discontinuar loperamida Reescalar deferasirox Resolución Tratamiento de soporte: suspensión del deferasirox Resolución Suspensión de la loperamida. Deferasirox 10 mg/kg/día y reescalar Detención definitiva. Valorar otras etiologías Algoritmo 5. Ajuste de dosis de deferasirox (IV): diarrea. 90 GUÍA CLÍNICA DE QUELACIÓN DEL PACIENTE CON SÍNDROME MIELODISPLÁSICO Bibliografía 1. Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood 1997; 89: 739-61. 2. Hershko Ch. Oral iron chelators: new opportunities and new dilemmas. Haematologica 2006; 91 (10): 1307-12. 3. Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, et al. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med 1994; 331: 567-73. 4. Schafer AI, Cheron RG, Dluhy R, Cooper B, Gleason RE, Soeldner JS, et al. Clinical consequences of acquired transfusional iron overload in adults. N Engl J Med 1981; 304: 319-24. 5. Takatoku M, Uchiyama T, Okamoto S, Kanakura Y, Sawada K, Tomonaga M, et al.; Japanese National Research Group on Idiopathic Bone Marrow Failure Syndromes. Retrospective nationwide survey of Japanese patients with transfusion-dependent MDS and aplastic anemia highlights the negative impact of iron overload on morbidity/mortality. Eur J Haematol 2007; 78: 487-94. 6. Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol 2005; 23: 7594-603. 7. Malcovati L, Della Porta MG, Cazzola M. Predicting survival and leukemic evolution in patients with myelodysplastic syndromes. Haematologica 2006; 91: 1588-90. 8. Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25: 3503-10. 9. Leitch HA, Wong DHC, Leger CS, et al. Improved leukemia-free and overall survival in patients with myelodysplastic syndrome receiving iron chelation therapy: a subgroup analysis. Blood 2007; 110 (Suppl 1): 1469 (abstract). 10. Rose C, Brechignac S, Vassilief D, et al. Positive impact of iron chelation therapy (CT) on survival in regularly transfused MDS patients. A prospective analysis by the GFM. Blood 2007; 110 (Suppl 1): 249 (abstract). 11. Altes A, Remacha AF, Sarda P, Baiget M, Sureda A, Martino R, et al. Early clinical impact of iron overload in stem cell transplantation. A prospective study. Ann Hematol 2007; 86: 443-7. 12. Armand P, Kim HT, Cutler CS, Ho VT, Koreth J, Alyea EP, et al. Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood 2007; 109: 4586-8. 13. Gattermann N, Porter JB, Lopes LF, Seymour J. Consensus statement on iron overload in myelodysplastic syndromes. Hematol Oncol Clin North Am 2005; 19 (Suppl 1): 18-25. 91 MANUAL PARA PACIENTES CON HEMOCROMATOSIS 14. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079-88. 15. Sanz GF, Sanz MA, Vallespí T, Cañizo MC, Torrabadella M, García S, et al. Two regression models and a scoring system for predicting survival and planning treatment in myelodysplastic syndromes: a multivariate analysis of prognostic factors in 370 patients. Blood 1989; 74: 395-408. 16. Gattermann N, et al. P-113. MDS Symposium 2005, Nagasaki. Leukemia Res; 29 (Suppl1)S67; AbsP-113; 2005. 17. Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR; Thalassemia Clinical Research Network. Complications of beta-thalassemia major in North America. Blood 2004; 104: 34-9. 18. Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood 2003; 102: 17-24. 19. Pennell DJ, Berdoukas V, Karagiorga M, Ladis V, Piga A, Aessopos A, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006; 107: 3738-44. 20. Neuferld EJ. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: new data, new questions. Blood 2006; 107: 3436-41. 21. Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with betathalassemia. Blood 2006; 107: 3455-62. 92 Con el aval de: Asociación Española de Hematología y Hemoterapia Con el patrocinio de: