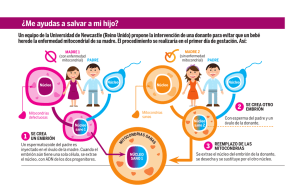
Evaluación de la disminución de la expresión de Pink1 en eventos
Anuncio

Evaluación de la disminución de la expresión de Pink1 en eventos de fusión y fisión mitocondrial en un modelo de neuronas dopaminérgicas Liliana Alexandra Rojas Charry Universidad Nacional de Colombia Facultad de Medicina Departamento de Ciencias Fisiológicas Bogotá D.C., Colombia 2012 Evaluación de la disminución de la expresión de Pink1 en eventos de fusión y fisión mitocondrial en un modelo de neuronas dopaminérgicas Liliana Alexandra Rojas Charry Tesis presentada como requisito parcial para optar al título de: Magíster en Bioquímica Director: Gonzalo Humberto Arboleda Bustos, MD MSc PhD Grupo de Investigación en Muerte Celular Universidad Nacional de Colombia Facultad de Medicina Departamento de Ciencias Fisiológicas Bogotá D.C., Colombia 2012 "It is not knowledge, but the act of learning, not possession, but the act of getting there, which grants the greatest enjoyment" Carl Friedrich Gauss Agradecimientos Expreso mis más sinceros agradecimientos a las personas que hicieron una gran contribución al desarrollo de este trabajo y que me acompañaron en esta etapa de formación tanto académica como personal: A mi familia y a Duván Al profesor Gonzalo Humberto Arboleda A Ruth Sánchez, a Rita Baldrich y a Andrea Niño A Mark Cookson y a Melissa McCoy A los compañeros del grupo de investigación en neurociencias y muerte celular A los profesores de la maestría en bioquímica Resumennn nnnnnnnnnnnnnnnnnnnnnn Las mitocondrias están muy lejos de ser organelos estáticos. Actualmente está bien establecido que las mitocondrias están sometidas a un proceso constante de cambio, al cual se le ha denominado dinámica mitocondrial que incluye procesos de fusión y fisión. Hay muchas evidencias acerca de que la pérdida de función de Pink1 y de la dinámica mitocondrial son responsables de diferentes enfermedades neurodegenerativas, incluyendo el Parkinson tipo 2. En este trabajo se estudió el efecto del silenciamiento de Pink1 eventos de fusión y fisión mitocondrial en neuronas dopaminérgicas. La disminución de la expresión de Pink1 se realizó usando plásmidos lentivirales con dos secuencias de shRNA y mediante qPCR se comprobó que los niveles de Pink1 disminuyeron en más del 80%. Se hicieron ensayos de MTT para evaluar actividad mitocondrial, mientras que las proteínas de fusión fisión fueron analizadas mediante western blot y la morfología mitocondrial con microscopía confocal. Las células transducidas con el constructo shPink1 muestran una disminución en actividad mitocondrial y menores niveles de expresión de Drp1 y de Mfn2, en un 31 y 55%, respectivamente. Sus mitocondrias tienen una morfología principalmente fragmentada y también se encontró que estas células no responden de la misma manera que las células control ante el estímulo generado por la forskolina, un potenciador de la vía del cAMP que fosforila a Drp1, inactivándolo. De esta manera se propone que Pink1 puede ejercer un papel neuroprotector promoviendo fusión mitocondrial. Palabras clave: Pink1, fusión, fisión, mitocondria, neuronas dopaminérgicas, enfermedad de Parkinson VI Abstract Mitochondria are organelles far from being static. It is now well established that mitochondria are subject to a constant process of change, which has been called mitochondrial dynamics, including fusion and fission processes. There are a lot of evidences that show that the loss of function of Pink1 and mitochondrial dynamics are responsible for various neurodegenerative diseases, including Parkinson type 2. In this work, we studied the effect of silencing Pink1 in events of mitochondrial fusion and fission, in dopaminergic neurons. Two lentiviral shRNA sequences were used to knockdown Pink1 and by qPCR it was found that Pink1 levels decreased more than 80%. MTT assays were conducted to assess mitochondrial activity, while fission-fusion proteins were analyzed by western blot and mitochondrial morphology by confocal microscopy. The cells transduced with the construct against Pink1 show a decrease in mitochondrial activity and reduced levels of expression of Drp1 and Mfn2, by 31 and 55%, respectively. Their mitochondrial morphology was mainly fragmented and we also found that these cells do not respond the same way that the control ones to the stimulus generated by forskolin, an enhancer of cAMP pathway that phosphorylates Drp1, inactivating it. Thus it is proposed that Pink1 can exert a neuroprotective role by promoting mitochondrial fusion. Keywords: Pink1, fission, fusion, mitocondria, dopaminergic neurons, Parkinson disease VI Contenido Resumen .......................................................................................................VI Abstract .........................................................................................................VI Lista de Figuras .............................................................................................. X Lista de Tablas ..............................................................................................XI Lista de Abreviaturas .................................................................................. XII 1 2 Capítulo 1 ................................................................................................. 1 1.1 Introducción ........................................................................................ 1 1.2 Problema Científico ............................................................................ 2 1.3 Justificación ........................................................................................ 3 Capítulo 2 ................................................................................................. 5 2.1 Enfermedad de Parkinson .................................................................... 5 2.2 Pink1 ................................................................................................... 6 2.3 Posibles mecanismos neuroprotectores de Pink1 ................................. 8 2.4 Pink1 y función mitocondrial .............................................................. 9 2.5 Pink1 y su interacción con parkin ...................................................... 12 2.6 Pink1 y citoesqueleto ........................................................................ 14 2.7 Función mitocondrial ........................................................................ 16 2.8 Fusión y fisión mitocondrial.............................................................. 18 2.9 C2-Ceramida y Rotenona .................................................................. 26 2.10 Células CAD y M17 .......................................................................... 27 VII 3 4 Capítulo 3 ............................................................................................... 29 3.1 Objetivo General ............................................................................... 29 3.2 Objetivos Específicos ........................................................................ 29 Capítulo 4 ............................................................................................... 30 4.1 Cultivos Celulares ............................................................................. 30 4.2 Conteo y viabilidad celular................................................................ 32 4.3 Establecimiento de las poblaciones de CAD y M17 .......................... 32 4.4 Transformación de E.coli JM109 con los plásmidos lentivirales para shRNA de Pink1......................................................................................... 33 4.5 Purificación de los plásmidos lentivirales .......................................... 33 4.6 Transducción y establecimiento de las líneas celulares CAD y M17 con silenciamiento permanente shRNA de Pink1 ....................................... 33 4.7 Extracción de RNA ........................................................................... 35 4.8 Transcripción reversa ........................................................................ 36 4.9 PCR en tiempo real ........................................................................... 36 4.10 Determinación de la actividad mitocondrial ...................................... 38 4.11 Extracción y cuantificación de proteínas ........................................... 39 4.12 Electroforesis en SDS-PAGE ............................................................ 40 4.13 Detección de proteínas por Western blotting ..................................... 40 4.14 Análisis de la distribución mitocondrial por tinción con Mitotracker 41 4.15 Inmunofluorescencia ......................................................................... 42 4.16 Análisis de los eventos de fusión y fisión mitocondrial por transfección con mito-YFP ......................................................................... 43 VIII 4.17 Análisis estadístico ............................................................................ 44 5 Capítulo 5 ............................................................................................... 45 5.1 La disminución de la expresión de Pink1 disminuye la actividad mitocondrial y aumenta la vulnerabilidad al daño inducido por C2-ceramida 45 5.2 La disminución de la expresión de Pink1 induce cambios en la morfología y distribución mitocondrial, éstos se incrementan en presencia de agentes neurotóxicos .................................................................................. 49 5.3 El silenciamiento de Pink1 afecta la expresión de algunas proteínas involucradas en fusión/fisión ...................................................................... 53 5.4 6 7 Discusión de Resultados.................................................................... 61 Capítulo 6 ............................................................................................... 66 6.1 Conclusiones ..................................................................................... 66 6.2 Perspectivas ...................................................................................... 66 Anexos .................................................................................................... 68 7.1 RT-PCR para determinación de la temperatura óptima de anillamiento de los primers de Pink1 .............................................................................. 68 7.2 Picos de fusión de Pink1 (azul) y de β–actina (verde) tomando como temperatura de anillamiento 51°C .............................................................. 68 7.3 Curvas de amplificación (A) y picos de fusión (B) de Pink1 y de β– actina .......................................................................................................... 69 7.4 Comprobación de la eficiencia de los primers para Pink1 mediante diluciones seriadas ..................................................................................... 70 8 Bibliografía ............................................................................................. 71 IX Lista de Figuras Figura No. 1 Dominio quinasa y mutaciones de Pink1 asociadas a EP. ........... 8 Figura No. 2 Disfunción mitocondrial en la EP. ............................................ 10 Figura No. 3 Esquema de las principales proteínas involucradas en fusión y fisión mitocondrial .................................................................................. 21 Figura No. 4 Esquema de la dinámica mitocondrial ...................................... 22 Figura No. 5 Microfotografías de las células CAD y M17. ........................... 31 Figura No. 6 Programa usado en la amplificación de Pink1 por qPCR .......... 38 Figura No. 7 Determinación de la dosis tóxica mínima de blasticidina para las células CAD. ........................................................................................... 46 Figura No. 8 Niveles de expresión de Pink1. ................................................. 47 Figura No. 9 Disminución de la actividad mitocondrial en células shPink1. .. 48 Figura No. 10 Distribución mitocondrial en células CAD. ............................ 50 Figura No. 11 Procesamiento de las imágenes mitocondriales obtenidas por microscopía confocal. ............................................................................. 51 Figura No. 12 Microfotografías in vivo de las mitocondrias de células M17 Control shPink1 y shPink1. ..................................................................... 53 Figura No. 13 Morfología mitocondrial de las células M17. .......................... 54 Figura No. 14 Cuantificación de la morfología mitocondrial de células M17 control shPink1 y shPink1. ...................................................................... 55 Figura No. 15 Western blot de algunas de las proteínas involucradas en fusión y fisión mitocondrial. .............................................................................. 56 Figura No. 16 Densitometría de las proteínas de fusión/fisión detectadas por western blot. ............................................................................................ 57 Figura No. 17 Inmunofluorescencia de Fis1. ................................................. 58 Figura No. 18 Inmunofluorescencia de p-Drp1. ............................................. 59 Figura No. 19 Inmunofluorescencia de Mfn1. ............................................... 60 X Lista de Tablas Tabla No. 1 Secuencias de los constructos de shRNA para Pink1 y de los controles de silenciamiento. .................................................................... 34 Tabla No. 2 Lista de reactivos y su concentración final en la reacción de transcripción reversa. .............................................................................. 36 Tabla No. 3 Primers específicos usados en la amplificación de Pink1 y de βactina por qPCR. ..................................................................................... 37 Tabla No. 4 Anticuerpos empleados para los ensayos de western blot ........... 41 XI Lista de Abreviaturas m Diferencia de potencial de membrana mitocondrial µg Microgramo µM Micromolar µm Micrómetro AR Relación de aspecto ATCC American Type Culture Collection Aup1 Fosfatasa 2 C BCA Ácido bicinconínico BSA Albúmina sérica bovina cAMP AMP cíclico CCCP Carbonil 3-clrofenil hidrazona cDNA DNA complementario Cit C Citocromo C CL Cardiolipina CMT2A Charcot Marie Tooth tipo 2 A Ct Ciclos necesarios para alcanzar el umbral DIC Contraste de interferencia diferencial DMSO Dimetil sulfóxido DNA Ácido desoxirribonucleico DOA Atrofia óptica dominante dPink1 Pink1 de Drosophila Drp1 Proteína relacionada a la dinamina 1 ECL Quimioluminiscencia mejorada EP Enfermedad de Párkinson ER Retículo endoplasmático XII FF Factor de forma Fis1 Proteína de fisión FK Forskolina FRAP Recuperación de la fluorescencia después de photobleaching HR-1 Repeticiones de héptadas Hsp75 Proteína de choque térmico 75 HtrA2 Proteína de alto requerimiento de temperatura A2 IMM Membrana interna mitocondrial IMS Espacio intermembranal LRRK2 Quinasa con repeticiones ricas en leucina Mff Factor de fisión mitocondrial Mfn1 Mitofusina 1 Mfn2 Mitofusina 2 MiD51 Proteína de dinámica mitocondrial de peso molecular 51 MIEF1 Factor de elongación mitocondrial mL Mililitro mRNA RNA mensajero mtDNA DNA mitocondrial NO Óxido nítrico OMM Membrana externa mitocondrial Opa1 Proteína relacionada a atrofia óptica tipo 1 PAGE Electroforesis PBS Tampón fosfato salino PCR Reacción en cadena de la polimerasa PI3K Fosfatidil inositol 3 quinasa Pink1 Quinasa inducia por PTEN XIII PTEN Fosfatasa y homóloga de tensina p-Drp1 Drp1 fosforilado qPCR PCR cuantitativa RING Nuevo gen realmente interesante RNA Ácido ribonucleico RNAi RNA de interferencia ROS Especies reactivas de oxígeno RT Transcripción reversa RT-PCR Transcripción reversa y PCR SDS Dodecil sulfato de sodio SFB Suero fetal bovino shRNA RNA de horquilla corta TBS Tris buffer salino TBST TBS Tween TH Tirosina hidroxilasa TIM Transportador de membrana interna mitocondrial TNF-1 Factor de necrosos tumoral 1 TOM Transportador de membrana externa mitocondrial TRAP-1 Proteína asociada al receptor de TNF 1 UCHL-1 Ubiquitil carboxil terminal esterasa L1 WB Western blot YFP Proteína amarilla fluorescente α-syn Alfa-sinucleína XIV 1 Capítulo 1 1.1 Introducción La Enfermedad de Parkinson (EP) es un desorden neurodegenerativo caracterizado por la pérdida selectiva de neuronas dopaminérgicas en la substancia nigra del mesencéfalo. Es la segunda enfermedad neurodegenerativa más común después de la Enfermedad de Alzheimer. Afecta tanto a hombres como a mujeres con edad promedio de inicio 60 años y su incidencia aumenta significativamente con la edad. Aproximadamente 5 10 % de los pacientes tienen menos de 40 años de edad y se estima que una de cada 100 personas mayores de 60 años puede tener EP. La EP se caracteriza por generar una gran discapacidad entre los afectados, ya que sus síntomas son temblor en estado de reposo, rigidez y lentitud para realizar los movimientos; también se puede encontrar inestabilidad postural, disminución del balanceo de los brazos, de la expresión de la cara o del tono de voz; dificultad para la deglución, alteraciones del sueño, ansiedad, depresión y otras alteraciones mentales. Todo esto conlleva grandes pérdidas económicas y graves consecuencias sociales y familiares. En Colombia afecta a unos 230.000 individuos aproximadamente y la tendencia demográfica mundial es al aumento de la población anciana por lo que se espera que en los próximos 20 años la EP aumente cuatro veces su prevalencia actual, convirtiéndose en un serio problema de salud pública. La EP es una enfermedad compleja y presenta una amplia heterogeneidad clínico-genética, lo que ha dificultado profundizar en el conocimiento de sus mecanismos fisiopatológicos básicos, a pesar de los avances en el tratamiento sintomático y en su diagnóstico. Algunas formas genéticas mendelianas se han 1 descrito recientemente gracias a los progresos de la genómica; así se han identificado varios genes (α-Sinucleína, Parkin, UCHL-1, DJ-1, Pink1, LRRK2 y HtrA2) que constituyen las únicas causas definidas de la EP hasta la fecha. Estos avances han revolucionado y esclarecido algunos de los procesos subyacentes de su etiología, sin embargo, el papel de sus proteínas en la fisiología neuronal, aún está por aclararse. En este trabajo se analizó en células CAD y M17 el impacto del gen Pink1 sobre las proteínas de fusión y fisión mitocondrial. Se simuló el efecto de pérdida de función de la proteína Pink1 y se evaluó su impacto sobre la homeóstasis mitocondrial a nivel de los mecanismos de fusión-fisión mitocondrial. Como hipótesis de trabajo se propone que la disminución de la expresión de Pink1 con short hairpin RNA (shRNA) de la proteína Pink1 produce una alteración en la fisiología neuronal alterando la homeóstasis mitocondrial (fusión-fisión), lo que conlleva a muerte neuronal. Metodológicamente, las células fueron transducidas con vectores plasmídicos lentivirales shRNA para Pink1 con el fin de disminuir su expresión y posteriormente se caracterizaron sus efectos sobre el mecanismo de fusiónfisión mitocondrial. 1.2 Problema Científico La Enfermedad de Parkinson (EP) es un desorden neurodegenerativo progresivo que se caracteriza por causar una gran discapacidad, generando consecuencias negativas tanto a nivel económico como social. En Colombia la padecen cerca de 230.000 personas y en 20 años incrementará hasta cuatro veces, convirtiéndose en un problema de salud pública. Se ha determinado que la principal causa de esta enfermedad es la muerte de las neuronas 2 dopaminérgicas de la substancia nigra, aunque los mecanismos moleculares por los cuales estas neuronas mueren selectivamente aún son desconocidos. Recientes estudios demuestran que esta susceptibilidad puede deberse a la existencia de factores genéticos que junto con factores de riesgo ambientales podrían contribuir a la forma esporádica de esta enfermedad. La identificación de pacientes de EP con variantes en genes que codifican para proteínas mitocondriales constituye la primera evidencia de una serie de estudios bioquímicos que implican una disfunción mitocondrial en la patogénesis de la EP. Sin embargo, el mecanismo molecular de esta disfunción no se conoce con exactitud, por lo tanto el análisis molecular in vitro de la función mitocondrial y de procesos fisiológicos como fusión-fisión podrían esclarecer aspectos relevantes de esta anomalía. Un gen muy importante asociado a EP es Pink1 el cual parece ejercer un efecto neuroprotector. Sin embargo, su estudio no ha sido fácil debido a que se encuentran niveles endógenos muy bajos, por lo que ha sido necesario emplear una gran variedad de modelos. En este trabajo se estudió el efecto del silenciamiento de Pink1 en las principales proteínas que regulan los procesos de fusión y fisión mitocondrial en neuronas dopaminérgicas. 1.3 Justificación Aunque en los últimos años se han empleado diferentes estrategias terapéuticas para el tratamiento de la EP, ésta sigue siendo considerada una enfermedad incurable. Esto posiblemente se debe al desconocimiento de los mecanismos moleculares involucrados en la muerte de las neuronas dopaminérgicas. En los últimos años se ha avanzado en el conocimiento de 3 estos procesos y se sabe que Pink1 es determinante en la correcta función mitocondrial. Adicionalmente, el grupo de investigación en muerte celular de la Universidad Nacional de Colombia ha obtenido resultados importantes en cuanto a la relación de la proteína Pink1 y la vía de supervivencia de fosfatidil inositol 3 quinasa (PI3K/AKT) indicando la necesidad de continuar con el desarrollo de estudios que se enfoquen en evidenciar los mecanismos de fisión-fusión mitocondrial con el objetivo de que a futuro este conocimiento permita el diseño racional de fármacos contra blancos terapéuticos específicos o la identificación de proteínas y genes que sirvan como marcadores bioquímicos tempranos útiles para la detección de la enfermedad. 4 2 Capítulo 2 Estado del Arte 2.1 Enfermedad de Parkinson Este desorden neurodegenerativo asociado a la edad se caracteriza por la pérdida preferencial de las neuronas que secretan dopamina y por la acumulación de inclusiones intraneuronales (conocidas como cuerpos de Lewy) (Forno, 1996). La enfermedad es lentamente progresiva y causa movimientos lentos, rigidez de las extremidades y temblor en las manos. La EP se presenta de forma esporádica, es decir, sin antecedentes familiares conocidos en el 90 % de los casos y en el 10 % restante la enfermedad puede ser hereditaria, afectando tanto a hombres como a mujeres. La forma esporádica de la enfermedad es más frecuente después de los 60 años mientras que en las formas hereditarias la edad de inicio es más temprana (Forno, 1987). El tratamiento de la enfermedad depende del estado de severidad en que encuentre el paciente. En la actualidad se conoce un grupo numeroso de medicamentos para tratarla entre los cuales la Levodopa con Carbidopa (Sinemet) o con benserazida (Madopar) son los de mayor beneficio. El tratamiento incluye además medidas de rehabilitación física y de los trastornos asociados que puedan afectar al paciente como depresión, insomnio, ansiedad, etc. 5 En las formas familiares de la EP se han encontrado mutaciones en genes que han sido asociadas a formas autosómicas dominantes; como la α-sinucleína (PARK1), Parkin (PARK2), DJ1 (PARK3) y Pink1 (PARK6) (responsables de formas autosómicas recesivas) (Gasser, 2007). A pesar de los múltiples esfuerzos por conocer la causa de la muerte neuronal en el modelo de esta enfermedad, esto todavía es considerado un enigma. Probablemente, la EP es resultado de la interacción de múltiples factores, que incluyen el envejecimiento normal, la susceptibilidad genética y la exposición a factores ambientales. 2.2 Pink1 Pink1 (PTEN-induced kinase) es una proteína de 581 aminoácidos que contiene un dominio serina/treonina quinasa y un dominio de localización mitocondrial. El gen Pink1 se localiza en el cromosoma 1, en la región 1p3536, tiene 8 exones y abarca 1.8 kb. Pink1 fue descubierto en el año 2001 a través de un estudio de perfiles de genes que son activados por la fosfatasa supresora de tumores y homóloga de tensina PTEN (Unoki y Nakamura, 2001). En el año 2004 esta proteína fue identificada como el tercer gen autosómico recesivo asociado a parkinson juvenil. Los autores de este estudio mostraron que Pink1 silvestre, más no el mutante, estabilizaba el potencial de membrana mitocondrial ( m) y protegía a las células de apoptosis cuando eran expuestas a inhibición de proteosoma. acentúan las condiciones de la Las mutantes del gen Pink1 enfermedad, induciendo trastornos mitocondriales y apoptóticos (Valente et al., 2004). 6 Lo anterior sugiere que Pink1 puede funcionar en vías de señalización intracelulares implicadas en la patogénesis de la EP. Tres propiedades de Pink1 son especialmente relevantes; en primer lugar, análisis de secuencias primarias y estudios bioquímicos han demostrado que Pink1 es miembro de una familia de proteínas quinasas eucarióticas involucradas en la transducción y amplificación de señales intracelulares (Silvestri et al., 2005). En segundo lugar, el dominio N-terminal de Pink1 contiene un motivo de localización mitocondrial que sugiere que Pink1 es candidata potencial en la regulación de procesos mitocondriales asociados con apoptosis (Muqit et al., 2006) y en tercer lugar, Pink1 ha mostrado una interacción funcional con otros genes asociados a EP como Parkin y HtrA2 (Clark et al., 2006; Park et al. 2006; Wang et al. 2006; Plun-Favreau et al., 2007). La proteína Pink1 tiene una actividad de autofosforilación in vitro; Beilina y colaboradores explicaron los efectos de las mutaciones del gen Pink1 sobre su actividad quinasa y los niveles de expresión y localización en células mamíferas. Ellos confirmaron la actividad quinasa y demostraron que las proteínas mutantes son estables pero carecen de esta actividad. La mutación L347P desestabiliza y reduce la actividad quinasa, mientras que la mutación G309D tiene un menor efecto en la actividad quinasa in vitro. En este trabajo también demostraron la localización, procesamiento y los niveles de la proteína en células mamíferas, encontrando que Pink1 es procesada en su secuencia N-terminal de una manera consistente con su importación mitocondrial. Además demostraron que la proteína madura también existe en el citosol. Estos resultados implican que una porción de Pink1 podría ser exportada después de procesarse en mitocondria (Beilina et al., 2005). 7 Figura No. 1 Dominio quinasa y mutaciones de Pink1 asociadas a EP. En a) Se muestra un esquema lineal de Pink1, con mutaciones asociadas a EP, subdominios quinasa. Las flechas indican la posible ubicación de hélices α, los cilindros hojas β. En b) se muestra un esquema de un dominio serina-treonina quinasa (Mills et al., 2008). 2.3 Posibles mecanismos neuroprotectores de Pink1 La expresión de la superóxido dismutasa 1 humana suprime la neurodegeneración inducida por la inactivación de Pink1 en Drosophila. 8 Adicionalmente el tratamiento con superóxido dismutasa y vitamina E en moscas con RNAi de dPink1 inhibe la degeneración ommatidial (Wang et al., 2006). De esta manera dPink1 jugaría un rol esencial en la supervivencia neuronal previniendo que las neuronas sigan en estrés oxidativo. ¿A través de qué moléculas estaría ejerciendo este efecto? Hasta ahora se conocen pocos substratos de Pink1 y el mecanismo por el cual las mutaciones de esta proteína conducen a la neurodegeneración también es desconocido. El gen Pink1 es transcripcionalmente activado por fosfatasas y homólogos del gen de la tensina (Pten), un gen supresor tumoral involucrado en la regulación de vías señalizadoras de transducción. Pridgeon et. al, 2007, reportó la identificación de una proteína asociada al receptor TNF-1 (TRAP1), una chaperona molecular mitocondrial conocida como proteína 75 de choque térmico (Hsp75), como substrato celular para la quinasa Pink1. Esta proteína se une y colocaliza con TRAP1 en la mitocondria y la fosforila in vitro e in vivo. Además se desmostró que Pink1 protege contra el estrés oxidativo que induce muerte celular, suprimiendo la liberación del CitC desde la mitocondria. Esta acción protectora del gen Pink1 depende de su actividad quinasa para fosforilar a TRAP1 (Pridgeon et al., 2007). 2.4 Pink1 y función mitocondrial La deleción del gen Pink1 en ratones altera de forma significativa las funciones mitocondriales. Estudios de microscopía electrónica cuantitativa del striatum de estos ratones evidenció que no hay cambios apreciables en la ultraestructura del número total de mitocondrias, pero sí un aumento importante en el número de mitocondrias alargadas. En este mismo estudio también se pudo detectar una reducción en la actividad de la aconitasa, la cual 9 está asociada con el ciclo de Krebs y las actividades respiratorias en la corteza cerebral de estos ratones se ve disminuida en ratones de 2 años de edad con Pink-/- , pero no en los ratones más jóvenes de 2 a 4 meses, lo que indica que la edad exacerba la disfunción mitocondrial en estos ratones (Gautier et al., 2008). Figura No. 2 Disfunción mitocondrial en la EP. Se han identificado muchas alteraciones a nivel de la biología mitocondrial en la EP; a) actividad reducida del complejo I; b) mayor producción de ROS; c) daño en el mtDNA; d) daño bioenergético; e) liberación de citocromo c mediada por Bax, apoptosis mediada por mitocondria; f) mitofagia defectuosa; g) aumento en la capacidad buffer de Ca2+. Se muestran también los genes mutados en EP (Perier et al. 2012). El rol de las proteínas de control de calidad de la mitocondria en la etiología de la EP se sugiere después de hallazgos de mutaciones en las proteínas HtrA2/OMI. HtrA2 se localiza en el espacio intermembranal de la mitocondria 10 y protege del estrés mitocondrial ya que puede degradar polipéptidos mal plegados y hace parte de la cascada de señalización adaptativa al estrés oxidativo. HtrA2 se asocia con Pink1 y tanto Pink1 como HtrA2 parecen interactuar en la misma vía protectora ya que Pink1 es necesario para la fosforilación de HtrA2, la cual incrementa su actividad proteolítica in vitro (Plun-Favreau et al., 2007). Sin embargo, aún no se tiene claro si HtrA2 es un sustrato de Pink1. Llama la atención que aunque los roles fisiológicos de Pink1, HtrA2 y TRAP1 no han sido determinados, la relación entre estrés oxidativo, control de calidad mitocondrial y señalización de estrés está emergiendo como posible patogénesis en EP. En un estudio realizado por Wang y colaboradores en 2007, se demostró que Pink1 silvestre inhibe la muerte celular apoptótica causada por inhibidores del proteosoma, los cuales inducen la liberación de CitC por disminución de la expresión del gen Bax. Este estudio demostró que la sobreexpresión de Pink1 silvestre bloquea la liberación de CitC inducida por un inhibidor de proteasoma (Atractosida) y mostró como las mutantes fallan para inhibir la citotoxicidad producida por éste (Wang et al., 2007). Los resultados han demostrado que Pink1 silvestre expresado en mitocondria ejerce su papel antiapoptótico por inhibición de la apertura del poro mitocondrial y mutaciones en la proteína interfieren en la habilidad para prevenir la apertura del poro (Gautier et al., 2008), este mismo grupo encontró que la pérdida de Pink1 causa un incremento selectivo en la apertura del poro y del calcio mitocondrial y que una apertura excesiva de este poro puede ser la responsable de los defectos mitocondriales encontrados en las células que carecen Pink (Gautier et al., 2012). 11 Fisiológicamente se demostró que Pink1 puede regular el flujo de calcio mitocondrial a través del intercambiador Na +/Ca2+ la deficiencia de Pink1 causa una acumulación mitocondrial de calcio lo que conduce a una “sobrecarga”. Esta sobrecarga estimula la producción de ROS a través de la NADPH oxidasa. La producción de ROS inhibe el transportador de glucosa, reduciendo el envío de este sustrato lo que conlleva a una disfunción de la cadena respiratoria. Cuando se suministran sustratos de los complejos I y II, el daño en la cadena respiratoria puede ser superado. Posiblemente las ROS disminuyen el umbral de apertura del poro de permeabilidad mitocondrial, inducido por calcio. Estos hallazgos permiten proponer un mecanismo por el cual la disfunción de Pink1 hace que las neuronas sean más vulnerables a la muerte celular (Gandhi et al., 2009). 2.5 Pink1 y su interacción con parkin La pérdida del homólogo de Pink1 en Drosophila es indistinguible de moscas deficientes en parkin (PARK2), una proteína ubiquitina ligasa E3 localizada principalmente a nivel citosólico, que media la marcación de porteínas para su posterior degradación. Las moscas exhiben defectos motores debido a la pérdida de neuronas dopaminérgicas por déficits mitocondriales. Este fenotipo debido a la deficiencia de Pink1 puede ser rescatado mediante la sobreexpresión de parkin, pero lo contrario no ocurre, lo que lleva a la conclusión de que Pink1 posiblemente actúa corriente arriba de parkin en la misma vía molecular (Clark et al., 2006;Park et al., 2006;Yang et al., 2006). Otros estudios en Drosophila sugieren que la pérdida de Pink1 y parkin tiene como consecuencia cambios en la morfología mitocondrial, encontrándose que éstas se alargan. Este fenotipo puede ser rescatado mediante la 12 sobreexpresión de la proteína mitocondrial del fisión Drp1, por lo cual se concluye que el homólogo de parkin en Drosophila está promoviendo fisión mitocondrial (Deng et al., 2008;Poole et al., 2008) Sin embargo, en células humanas parkin y Pink1 parecen promover fusión mitocondrial. El silenciamiento de Drp1 puede rescatar el fenotipo mitocondrial inducido por la deficiencia en Pink1 y en parkin (Lutz et al., 2009;Sandebring et al., 2009). Lutz adicionalmente demostró que en células S2 de Drosophila, la fisión mitocondrial es un efecto temprano de parkin y Pink1, que progresa a fusión con el tiempo, lo cual puede explicar la discrepancia entre los modelos. También existen reportes que relacionan el papel que parkin y Pink1 desempeñan en el modelamiento de la mitocondria. Parkin media la degradación de mitocondrias dañadas (Narendra et al., 2010) mientras que Pink1 (silvestre, no los mutantes) es necesario para que parkin se pueda translocar a la mitocondria (Vives-Bauza et al., 2010). Adicionalmente, Gegg demostró que el silenciamiento de Pink1 da como resultado una disminución de la síntesis mitocondrial (Gegg et al., 2009), mientras que Kuroda mostró que parkin se une al DNA mitocondrial lo cual promueve su síntesis en células de neuroblastoma humano (Kuroda et al., 2006;Rothfuss et al., 2009). Pink1 regula el dominio RING1 de parkin (dominio importante para la translocación a la mitocondria) fosforilando directamente la tirosina 175, la cual se localiza en la región de unión de parkin (Kim et al., 2008). VivesBauza confirmó que la relocalización de parkin en mitocondria es dependiente de Pink1, pero esto no involucra la fosforilación de parkin. Sin embargo, un estudio más reciente mostró que Pink1 es activado cuando hay depolarización mitocondrial y directamente fosforila y activa a parkin. El monitoreo de la 13 fosforilación de parkin en la serina 65 y/o en Pink1 en la treonina 257 representa los primeros biomarcadores para examinar la actividad de la vía de señalización Pink-parkin in vivo. En este estudio también se sugiere que pequeñas moléculas activadoras de parkin pueden conferir una nueva aproximación terapéutica para la EP (Kondapalli et al., 2012). En cuanto al reclutamiento de parkin a nivel mitoconcondrial, Lazarou y colaboradores mostraron que Pink1 endógeno forma un complejo de 700 kDa con la translocasa de la membrana externa TOM selectivamente en mitocondrias depolarizadas mientras que Pink1 ectópicamente dirigido a la membrana externa mitocondrial mantiene la asociación con TOM en mitocondrias polarizadas. Cuando Pink1 es dirigido hacia los lisosomas o hacia los peroxisomas, que carecen del complejo TOM, recluta a parkin e induce la actividad ubiquitina ligasa en estos organelos. Una vez allí, parkin induce autofagia organelo-selectiva de peroxisomas pero no de lisosomas. En este estudio se propone que la asociación de Pink1 con el complejo TOM permite el rápido reimporte de Pink1 para rescatar mitocondrias repolarizadas de mitofagia y reducir factores específicos mitocondriales para la translocación y activación de parkin. La unión de Pink1 a TOM puede funcionar para rápidamente reimportar Pink1 y regular la vía Pink1/parkin (Lazarou et al., 2012). 2.6 Pink1 y citoesqueleto El transporte mitocondrial en neuronas y su distribución espacial están directamente relacionadas con la actividad sináptica. Las neuronas requieren de mecanismos especializados para regular el transporte y la retención de las mitocondrias en la proximidad de conos de crecimiento activos, nodos de 14 Ranvier y terminales sinápticas, en donde la producción de energía y la capacidad homeostática del calcio tienen una alta demanda (Hollenbeck y Saxton, 2005). Este transporte de larga distancia depende de proteínas motoras basadas en microtúbulos. Mientras que motores de dineína citoplasmáticas conducen el movimiento retrógrado, la quinesina-1 fue la primera proteína motora identificada para el transporte anterógrado de mitocondrias. Varios adaptadores, incluyendo a Miro-Milton y sintabulina han sido identificados por unir la mitocondria al motor de quinesina en neuronas (Cai y Sheng, 2009). En el año 2009, Weihofen y colegas, reportaron la identificación de un complejo multiproteínico mitocondrial que contiene a Pink1, la GTPasa Miro y la proteína adaptadora Milton. Se sabe que el complejo Miro-Milton tiene un papel importante en el tráfico mitocondrial a lo largo de los microtúbulos. Adicionalmente una variante de Pink1 que no expresa el dominio de localización mitocondrial pudo encontrarse en las fracciones subcelulares mitocondriales al inducir la expresión de Miro y Milton, lo que podría sugerir un papel de Pink1 en este importante proceso (Weihofen et al., 2009). La expresión de Pink1 con el dominio quinasa inactivo da como resultado el aumento en la fosforilación de cofilina, una proteína importante para el remodelamiento del filamento de actina, lo que sugiere que una dinámica alterada de actina y el incremento de la asociación de parkin con el filamento de actina pueden conllevar a condiciones patológicas resultantes de la deficiencia de Pink1 (Kim y Son, 2010). Las células mantienen su balance energético y evitan el estrés oxidativo regulando el movimiento mitocondrial, su distribución y remoción. Pink y parkin participan en esta regulación arrestando el movimiento mitocondrial, ya 15 que Pink1 fosforila a Miro un componente del componente primario del complejo motor/adaptador que ancla a la quinesina a la superficie mitocondrial. La fosforilación de Miro activa su degradación proteosomal dependiente de parkin. Al remover Miro la proteína quinesina se separa de la superficie mitocondrial. Previniendo el movimiento de las mitocondrias la vía Pink1/parkin puede “poner en cuarentena” las mitocondrias dañadas antes de removerlas. En este estudio se propone que la fosforilación de sustratos por parte de Pink1 desencadena la subsecuente acción de Parkin y del proteosoma. (Wang et al., 2011). 2.7 Función mitocondrial Las mitocondrias son organelos celulares muy especializados. Más allá de su clásica función como fuente de energía celular, gracias a contener la cadena respiratoria, desempeñan papeles cruciales en diversos procesos que incluyen apoptosis, almacenamiento de calcio y rutas biosintéticas, que inciden directamente en la homeostasis celular. Por eso no sorprende que su disfunción tenga consecuencias muy serias para la célula y que esté relacionada con envejecimiento y desórdenes neurológicos (Lin y Beal, 2006). La mayoría de sus proteínas residen en la matriz y son importadas a este subcompartimento por la cooperación de dos principales translocasas, el complejo TOM en la membrana externa y el complejo TIM 23 en la membrana interna. Estos complejos interactúan entre sí para trasferir preproteínas al interior de las dos membranas de una manera concertada. El complejo TOM es la translocasa en la membrana externa que media el importe de virtualmente todas las proteínas de la mitocondria. Reconoce proteínas precursoras en el citosol, facilita la liberación de factores de unión citosólicos, 16 contribuye al desplegamiento de proteínas, transfiere polipéptidos a través de los poros de la membrana externa y media la inserción de algunas proteínas que residen en ésta (Neupert y Herrmann, 2007). El complejo TIM23 es la principal translocasa en la membrana interna de la mitocondria. Transloca todos los precursores de proteínas de matriz, la mayoría de las proteínas de la membrana interna y muchas proteínas del espacio intermembranal. La translocación por el complejo TIM 23 es dirigida energéticamente por el potencial eléctrico de la membrana interna y por la hidrólisis de ATP. Los componentes de este complejo pueden ser subdivididos en dos grupos, que operan de una manera secuencial y cooperativa: el sector membranal, que forma el canal conductor de proteínas y el motor de importe, que conduce la translocación en el espacio de la matriz mitocondrial (Donzeau et al., 2000). Tres importantes descubrimientos han corroborado el concepto de que las mitocondrias participan en señalización celular. En primer lugar, las ROS y especies de nitrógeno pueden participar en lo que se conoce como la “señalización celular redox”. En segundo lugar, la interacción entre la oxidasa citocromo c y el óxido nítrico (NO•) constituye una vía de señalización para la producción controlada de peróxido de hidrógeno (H2O2) para transducción de señales. En tercer lugar, proteínas mitocondriales como el citocromo c pueden ser liberadas hacia el citosol e iniciar vías de señalización que conducen a apoptosis. Las ROS representan un reto adicional para la integridad mitocondrial ya que inducen modificaciones en las proteínas, peroxidación de lípidos y daño al DNA. Éstas son un inevitable subproducto de la fosforilación oxidativa. Las 17 mitocondrias disfuncionales deben lidiar con el riesgo de una hidrólisis insignificante de ATP y un aumento del estrés oxidativo. Aun más, un daño extensivo a nivel mitocondrial, puede conducir a la disipación del potencial de membrana a través de la membrana interna e inducir muerte celular por la liberación de proteínas proapoptóticas. Así que es crucial el control de la fosforilación oxidativa y el monitoreo de la funcionalidad de la cadena respiratoria para mantener la integridad del DNA mitocondrial y limitar el daño en la mitocondria (Tatsuta y Langer, 2008). 2.8 Fusión y fisión mitocondrial Las mitocondrias están muy lejos de ser organelos estáticos. Actualmente está bien establecido que las mitocondrias están sometidas a un proceso constante de cambio, al cual se le ha denominado dinámica mitocondrial que incluye procesos de fusión y fisión. Varios estudios han demostrado que las mitocondrias están en movimiento constante e interaccionan con otros orgánulos como el retículo endoplasmático o con otras mitocondrias (Bakeeva et al., 1978). Mediante análisis de grabación en video de imágenes adquiridas por microscopía de fluorescencia, se han visualizado las mitocondrias moviéndose desde las regiones perinucleares de la célula a las regiones más periféricas unidas tanto a microtúbulos como a filamentos de actina (Krendel et al., 1998). Durante su movimiento a través de los elementos del citoesqueleto, las mitocondrias se fusionan con otras formando filamentos, o éstas se disgregan produciendo mitocondrias aisladas, las cuales, a su vez, se dirigen en distintas direcciones para volverse a fusionar con otras mitocondrias. Pero, ¿cuál es el papel fisiológico de este mecanismo? Los estudios realizados por Skulachev y 18 colaboradores mostraron que filamentos extensos y otros filamentos conectados, se despolarizaban cuando se generaba una lesión en la membrana mitocondrial (en un único punto de 0,5 mm de diámetro) demostrando que las redes mitocondriales funcionaban como una unidad eléctrica compartiendo el mismo potencial de membrana. Estas extensas redes son constitutivas en tejidos compuestos de células con gasto energético elevado (como células musculares y neuronas), lo que llevó a proponer la teoría de “los cables transmisores de energía” (Skulachev, 2001). Esta teoría propone que una red mitocondrial extensa es eficiente en condiciones en las que la disponibilidad de oxígeno en la región central de la fibra muscular se convierte en el paso limitante con el objeto de mantener la actividad de la cadena respiratoria. Se han obtenido conclusiones similares por otros grupos, mediante medidas de recuperación de fluorescencia después del photobleaching (FRAP), o siguiendo sucesos sincrónicos de despolarización. Estudios genéticos han revelado la importancia de estos procesos en el funcionamiento normal de las células y también se han descrito varios de los componentes moleculares que los caracterizan. Dentro de la maquinaria de fusión de los mamíferos se encontraron dos proteínas denominadas Mitofusina-1 (Mfn1) y Mitofusina-2 (Mfn2) que son GTPasas altamente conservadas localizadas en la membrana externa mitocondrial (Chen et al., 2003). En ausencia de estas dos proteínas, no hay fusión mitocondrial y se cree que su acción se desarrolla a través de unos dominios hidrofóbicos (HR1) que son 19 esenciales para la asociación con otras mitofusinas en las mitocondrias cercanas. Otra proteína involucrada en este proceso es Opa1, que fue inicialmente identificada como un gen mutado en atrofia óptica dominante autosomal (DOA, por sus siglas en inglés), una causa común de pérdida visual (Delettre et al., 2000). El silenciamiento de Opa1 conduce a aberraciones en la morfología de las crestas mitocondriales y genera fragmentación mitocondrial (Chen y Chan, 2005). Misko y colaboradores presentaron evidencias acerca de que mitofusina 2 es necesaria para el transporte axonal de mitocondrias y además interactúa con el complejo Miro/Milton. En este trabajo se identificó un rol para las mitofusinas hasta el momento desconocido, en regular directamente el transporte mitocondrial, lo cual ofrece una importante contribución al entendimiento del mecanismo molecular de la degeneración axonal en la enfermedad de CMT2A y en la atrofia óptica dominante (Misko et al., 2010). En cuanto a la maquinaria de la fisión mitocondrial las proteínas más importantes que se han caracterizado son Drp1 y Fis 1. Drp1 se encuentra en el citosol, pero una subpoblación se localiza en sitios puntuales de los túbulos mitocondriales, estos sitios marcan lugares en donde se llevará a cabo la fisión (Chan, 2006). Drp1 contiene dominios característicos de la familia de GTPasas de dineína y aparentemente puede constreñir las membranas mitocondriales. Análisis in vitro demostraron que la hidrólisis de GTP es suficiente para que Drp1 constriña y tubule membranas liposomales, lo que indica que Drp1 tiene la 20 capacidad de ejecutar tanto la unión, constricción y liberación de membranas sin la adición de otros factores (Elgass et al., 2012). Figura No. 3 Esquema de las principales proteínas involucradas en fusión y fisión mitocondrial (Chan, 2012) En cuanto a Fis 1 se sabe que es una pequeña proteína de 17 kDa localizada uniformemente en la membrana mitocondrial externa y su sobreexpresión conduce a fragmentación mitocondrial dependiente de Drp1. La importancia fisiológica de la fisión mitocondrial ha sido demostrada por la identificación de un paciente con una mutación dominante negativa en Drp1. Este paciente 21 exhibió varias anormalidades incluyendo un pobre desarrollo cerebral, atrofia óptica, acidosis láctica y mortalidad 37 días después de la gestación. Figura No. 4 Esquema de la dinámica mitocondrial (Perier y Vila, 2012) Recientemente se han identificado nuevas proteínas dentro de la maquinaria de fusión y fisión mitocondrial. En el año 2011 Zhao y colaboradores reportaron que el factor de fisión mitocondrial MIEF1, también conocido como MiD51, al ser sobreexpresado induce una extensiva fusión mitocondrial, mientras que su depleción conduce a fragmentación de las mitocondrias. MIEF1 interactúa y recluta a Drp1 en la mitocondria de una manera independiente de Fis1, de Mff (una proteína que también puede reclutar a Drp1 en la mitocondria y generar fisión mitocondrial) (Gandre-Babbe y van 22 der Bliek, 2008;Otera et al., 2010) y de Mfn2, pero inhibe la actividad de Drp1, ejecutando así un efecto negativo en la fisión mitocondrial. MIEF1 también interactúa con Fis1 y cuando los niveles de expresión de esta última aumentan parcialmente se revierte el fenotipo de fusión inducido por MIEF1. Además de inhibir a Drp1, MIEF1 promueve fusión pero de una manera independiente de las mitofusinas. Estos hallazgos develan un nuevo mecanismo que controla la maquinaria de fusión y fisión mitocondrial en vertebrados. Al ser MIEF1 una proteína específica de vertebrados, también se muestra que hay importantes diferencias en la regulación de la dinámica mitocondrial entre levaduras y vertebrados (Zhao et al., 2011). Hay muchas evidencias acerca de que la pérdida de función y de la dinámica mitocondrial son responsables de diferentes enfermedades neurodegenerativas, incluyendo el parkinson tipo 2. Mutaciones en Mfn2 se han descrito como causa de la enfermedad CMT2A (Charcot-Marie-Tooth subtipo 2A), una neuropatía periferal caracterizada por debilidad muscular y pérdida sensorial en el limbo distal (Zuchner et al., 2004). La disminución de la expresión de Opa1 hace a las células más susceptibles a la inducción de apoptosis, mientras que la ausencia de Fis1 inhibe la muerte celular. El silenciamiento de Drp1, también aumenta la viabilidad celular aunque en una menor proporción que el silenciamiento de Fis1, lo que sugiere un rol proapoptótico para Drp1 y Fis1, y uno antiapoptótico para Opa1 (Lee et al., 2004). No sólo los genes recesivos causantes de EP tipo dos se han relacionado con efectos en la morfología mitocondrial, también se ha encontrado α-syn puede participar de este proceso. Una proteína muy importante ya que no sólo es el componente predominante de los cuerpos de Lewy (deposiciones 23 intracelulares proteínicas, muy características de la patología) sino que como se mencionó anteriormente también presenta mutaciones causantes de la enfermedad. Esta proteína de 140 aminoácidos está ampliamente distribuida en el cerebro y se expresa en altos niveles en las neuronas, en donde puede alcanzar concentraciones entre 0.5 y 1% de proteína total. Se sabe que interactúa con membranas lipídicas y que adopta conformaciones de tipo amiloide. Su toxicidad puede ser consecuencia de interacciones membranales anormales y alteraciones en el tráfico vesicular. Una de las propiedades bioquímicas bien descritas de α-sinucleína es su unión a membranas, lo que se asocia con cambios estructurales. En un estudio publicado en el año 2010 se demostró por primera vez que α-sinucleína inhibe la fusión de membranas y que su expresión induce fragmentación mitocondrial, mientras que la disminución de la expresión de esta proteína conduce a la elongación de la mitocondria. Sorpresivamente el fenotipo mitocondrial causado por la expresión de α-sinucleína es rescatado por la coexpresión de tres genes asociados a la EP recesiva: Pink1, Parkin y DJ1(Kamp et al., 2010). A nivel fisiológico, la dinámica parece participar en muchas funciones. Como las mitocondrias son organelos altamente móviles y dinámicos que continuamente se funden y se dividen, pueden intercambiar contenidos como el DNA mitocondrial. En un estudio realizado en músculo esquelético de ratón se mostró que la pérdida de función de las mitofusinas causa una severa disfunción mitocondrial y atrofia muscular. Los ratones mutantes también mostraron depleción en el mtDNA y sus genomas rápidamente acumularon mutaciones puntuales y deleciones. Posiblemente la fusión mitocondrial es un factor proactivo en enfermedades humanas asociadas a mutaciones en el DNA mitocondrial (Chen et al., 2010). 24 La yuxtaposición entre el retículo endoplasmático y la mitocondria es una característica estructural común, que provee la base física para la intercomunicación durante la señalización por Ca 2+; los mecanismos moleculares que controlan esta interacción aún son desconocidos. Un estudio mostró que mitofusina 2 se encuentra enriquecida en la interfase entre la mitocondria y el retículo endoplasmático. Al silenciar mitofusina 2 en fibroblastos embriónicos de ratón y en células HeLa la morfología del ER se ve afectada y se pierde la interacción ER-mitocondria, reduciendo la eficiencia de la toma de Ca2+ a nivel mitocondrial en respuesta al estímulo generado por el 1,4,5-inositol trifosfato. Ensayos in vitro así como evidencias genéticas y bioquímicas soportan un modelo en el cual Mfn2 en el ER haría las veces de un “puente” entre los dos organelos anclando complejos homo y heterotípicos con mitofusina 1 o 2 en la superficie mitocondrial. Una yuxtaposición necesaria para el ingreso eficiente de Ca 2+ (de Brito y Scorrano, 2008), de esta manera se amplía el panorama en el cual actúan estas proteínas. Las mitocondrias despliegan diferentes morfologías dependiendo del tipo celular y de situación fisiológica en la que se encuentren. En muchos tipos celulares senescentes, se ha observado una extensiva elongación de las mitocondrias, lo que podría tener alguna implicación funcional. Esta hipótesis fue probada en células de endotelio humano que fueron envejecidas in vitro. Las células jóvenes mostraban mitocondrias tubulares, mientras que las envejecidas se interconectadas. caracterizaron por tener mitocondrias alargadas Al inducir un daño con luz UV en células jóvenes e se observó la formación de ROS, la cual desencadena fragmentación celular y pérdida de m en las células jóvenes, mientras que las células senescentes mostraron resistencia y alteraciones en los niveles de expresión de Fis1, Drp1 25 y Pink1. Estos resultados indican que hay un mecanismo de protección frente al estrés oxidativo en células senescentes mediado por Drp1, Fis 1 y Pink (Mai et al., 2010). Todavía quedan muchas preguntas sin responder con respecto a estos dos procesos. No se sabe cómo interactúan, pero se ha observado que las mitocondrias sanas tienden a fusionarse mientras que la segregación por fisión puede ser una forma de la célula para deshacerse de mitocondrias dañadas a través de la degradación por lisosoma. La funcionalidad de mitocondrias dañadas puede ser restaurada mediante fusión con una mitocondria vecina que esté intacta (Detmer and Chan, 2007). Mitocondrias con daños severos terminan fragmentándose y son selectivamente removidas por un proceso autofágico, conocido como mitofagia. La mitofagia previene la liberación de proteínas proapoptóticas, así que consistentemente con su función citoprotectora, el proceso apoptótico es suprimido después de su inducción pero inducido cuando hay inhibición de autofagia (Maiuri et al., 2007). Dos proteínas mitocondriales, llamadas Uth1 y Aup1, han sido ligadas específicamente a mitofagia en Sacharomyces y no hacen parte de la maquinaria autofágica general, compuesta por alrededor de 30 proteínas conocidas como Atg (Kissova et al., 2004;Scherz-Shouval et al., 2007). 2.9 C2-Ceramida y Rotenona Las ceramidas (N-acetil-esfingosina) hacen parte de un grupo de segundos mensajeros basados en esfingomielina (Kolesnick y Kronke, 1998), son generadas principalmente por dos mecanismos enzimáticos, a través de la 26 síntesis de novo por condensación de serina y palmitoil-CoA o mediante el ciclo de esfingomielina. Además, la ceramida induce de-fosforilación de Akt, lo cual hace que interactúe con Bcl-2 y Bcl-xL (mediadores anti-apoptóticos) desencadenando la cascada apoptótica (Stoica et al., 2003). También, se ha demostrado que la ceramida induce muerte celular por alteración de la función mitocondrial, incluyendo generación de ROS, alteración en la homeóstasis del calcio, colapso del potencial de membrana mitocondrial y liberación de citocromo c (Kolesnick et. al., 1998; Mathias et. al., 1998). Adicionalmente un estudio del año 2008 muestra que la ceramida estimula la fisión mitocondrial y que este evento está asociado con una activación temprana de apoptosis en cardiomiocitos. C2-ceramida también promueve un rápido descenso en el potencial de membrana y liberación de CitC, mientras que la disminución de la expresión de Mfn2 acentúa los efectos de C2ceramida en la fragmentación mitocondrial (Parra et al., 2008). En este trabajo también se utilizó la rotenona como agente neurotóxico ya que la inhibición del complejo I de la cadena respiratoria por esta molécula induce muerte celular apoptótica en una gran variedad de células y esto ocurre a través de la acelerada producción de ROS (Li et al., 2003). La rotenona también es muy usada como modelo sistémico de la enfermedad de Parkinson ya que replica muchos aspectos de la patología (Cannon et al., 2009). 2.10 Células CAD y M17 Las células CAD son una variante de la línea celular CATH.a y fueron donadas por la Dra. Chikaraishi DM del departamento de Neurobiología del 27 Duke University Medical Center, Durham, North Carolina al Dr. Gonzalo Arboleda del grupo de muerte celular de la Universidad Nacional de Colombia. Se trata de una línea murina de neuronas catecolaminérgicas de origen mesencefálico recientemente descrita que expresan las enzimas βhidroxilasa dopamina y tirosina hidroxilasa (TH), además de sintetizar altos niveles de dopamina y norepinefrina (Suri et al., 1993) . También expresan varios marcadores de neuronas diferenciadas, incluyendo proteínas de neurofilamentos, sinaptofisina (Suri et al., 1993), canales dependientes de voltaje sensibles a corrientes de K+ (Baraban et al., 1995), Na+ (sensibles a tetrodotoxina) y Ca2+ similares a los reportados en otras neuronas, lo cual indica que estas células poseen una membrana excitable, propiedad característica de las neuronas (Lazaroff et al., 1996;Lazaroff et al., 1995). Las células humanas BE(2)-M17 (designación ATCC CRL-2267) son células humanas de neuroblastoma que expresan enzimas de síntesis de dopamina así como actividad de tirosina hidroxilasa y dopamina-β-hidroxilasa. Fueron derivadas de la biopsia de médula ósea de un paciente masculino con neuroblastoma diseminado. Las células crecen en multicapas y tienden a agregarse, ocasionalmente muestran neuritas alargadas (Thiele, 1991). 28 3 Capítulo 3 Objetivos 3.1 Objetivo General Analizar el efecto de la disminución de la expresión de Pink1 en eventos de fusión y fisión mitocondrial en un modelo de células catecolaminérgicas. 3.2 Objetivos Específicos 1. Analizar el efecto de la disminución de la expresión de Pink1 sobre la viabilidad celular con o sin exposición a agentes neurotóxicos. 2. Determinar el efecto de la disminución de la expresión de Pink1 sobre proteínas involucradas en procesos de fusión y fisión mitocondrial con o sin exposición a agentes neurotóxicos. 3. Determinar el efecto de la disminución de la expresión de Pink1 en la distribución y morfología mitocondrial con o sin exposición a agentes neurotóxicos. 29 4 Capítulo 4 Metodología El modelo de estudio empleado en este trabajo es de neuronas dopaminérgicas CAD y M17, las cuales fueron transducidas de manera permanente con vectores lentivirales para la interferencia de Pink1. La eficiencia de transducción se determinó por qPCR. Los eventos de fusión-fisión se analizaron por medio de western blot: fusión (mitofusina2 y Opa1) y fisión (Drp1 y Fis1) y mediante microscopía confocal (utilizando mitotracker y transfección transitoria con Mito-YFP). Para analizar estos paradigmas en un contexto neurotóxico modelo de EP, se utilizó C2-ceramida y rotenona. 4.1 Cultivos Celulares Los cultivos se establecieron a partir de células CAD y M17 criopreservadas en el pasaje 17. Las Células CAD fueron cultivadas en medio DMEM/F12 suplementado con 10% de suero bovino fetal en platos de cultivo estándar a una densidad de 5 x 106 para frascos de 75 cm2, 1.8 x 104 por pozo, para placas de 6 pozos y 7 x 103 por pozo para placas de 96 pozos, en una incubadora con medio humidificado y una concentración de CO 2 al 5% y temperatura de 37 ºC. El medio de cultivo fue remplazado cada 3 días, y al obtener una confluencia aproximada del 80 %, se realizaron subcultivos o pasajes. Para inducir la diferenciación, las células CAD fueron sembradas en medio DMEM-F12 con 0,25% de suero; suplementado con sodio selenita 50 ng/mL y transferrina 1X. Las células M17 fueron cultivadas en medio OPTIMEM I suplementado con 10% de suero fetal bovino y mantenidas en las 30 mismas condiciones anteriormente mencionadas, éstas células se diferencian por tratamiento con ácido retinoico 1µM y en medio con SFB al 2%. A C B D Figura No. 5 Microfotografías de las células CAD y M17. En A se muestran las células CAD diferenciadas y en B células CAD no diferenciadas. En C se muestran las células M17 no diferenciadas y en D diferenciadas (West et al., 2004) Barra de escala: 10 µm. 31 4.2 Conteo y viabilidad celular Cada vez que fue necesario conocer el número de células viables y valorar la evolución del cultivo se siguió el mismo procedimiento de conteo celular, para lo cual se usó el azul de Trypan, un colorante de exclusión que permite diferenciar las células viables (no teñidas) de las no viables (teñidas). Para esto, las células se despegaron y resuspendieron usando Tripsina al 0.25% p/v en una solución de PBS y EDTA 0.1% p/v, se tomó una muestra de la suspensión y se diluyó en una solución de azul de Trypan al 0.4% en una proporción de 1:4. Se colocaron 10 μl en un hemocitómetro, se contaron los cuatro cuadrantes de las esquinas de ambos retículos. Para reportar el número de células vivas por ml se tiene en cuenta el número de cuadrantes contados (8 cuadrantes cada uno con un volumen de 0,1 ml) y el factor de dilución que por lo general fue de 2, con estos datos se aplicaba la siguiente ecuación. 4.3 Establecimiento de las poblaciones de CAD y M17 El diseño propuesto llevó a la creación de una población celular de CAD y M17 silenciando Pink1 con vectores lentivirales generando así un modelo de estudio de células dopaminérgicas que permitieron evaluar el rol que desempeña esta proteína en eventos de fusión-fisión. 32 4.4 Transformación de E.coli JM109 con los plásmidos lentivirales para shRNA de Pink1 Los plásmidos lentivirales con los insertos de shRNA para Pink1 fueron donados por el Dr. Mark Cookson del Laboratorio de Neurogenética del Instituto Nacional de Envejecimiento (NIH) USA. La transformación en la cepa de E.coli JM109 se realizó por termo competencia. Una vez obtenidas las bacterias transformadas, se realizó la selección de las colonias que tenían el inserto y se cultivaron toda la noche en medio LB con ampicilina 100µg/ml y se congelaron en glicerol al 10% para posteriores experimentos. 4.5 Purificación de los plásmidos lentivirales Las colonias positivas para cada inserto fueron cultivadas toda la noche y la purificación de los plásmidos se realizó utilizando el kit Zyppy Plasmid Midiprep (Zymo Research). Después de la purificación se realizó un toothpick para confirmar la purificación. Los plásmidos se cuantificaron en Nanodrop y se congelaron para ser utilizados en la transducción de las líneas celulares. 4.6 Transducción y establecimiento de las líneas celulares CAD y M17 con silenciamiento permanente shRNA de Pink1 Los plásmidos lentivirales shRNA para Pink1 tienen el gen de blasticidina para realizar la selección en células eucariotas. Para poder realizar esta selección se realizó una curva inicial de blasticidina, determinando así la dosis en la cual se podía realizar la selección después de la transducción. Se utilizaron varias dosis (0-1-2-3-4-5-6-7-8-9-10 µg /ml) durante 10 días y se 33 determinó la dosis de la selección, que en este caso fue de 6 µg/ml para las células CAD y de 5 µg/ml para las M17, durante 3 días para las dos líneas celulares. La transducción de las líneas celulares se realizó utilizando 1 ug de los plásmidos shRNA para Pink1 usando liposomas; para las células CAD se usó Hifect (Lonza) durante 24 horas, mientras que para la línea celular M17 se usó X-treme Gene 9 (Roche) también durante 24 horas. Posteriormente se seleccionó con blasticidina (6 µg/ml y 5 µg/ml, para las células CAD y M17, respectivamente) por aproximadamente 15 días. Luego se verificó el silenciamiento del gen midiendo los niveles de mRNA por PCR en tiempo real. Estas líneas celulares silenciadas fueron criopreservadas y se denominaron Control shPink1 y shPink1, las cuales se usaron posteriormente para caracterizar sus efectos sobre el mecanismo de fusión-fisión mitocondrial. Las secuencias empleadas para el silenciamiento de Pink se unen empezando por el nucléotido 550 de Pink1 (secuencia shPink1 A) y el nucleótido 1141 (shPink1 C), también se usaron dos secuencias scrambled o control de silenciamiento (Tabla No. 1). Constructo Secuencia shPink1 A 5´- GCTGGAGGAGTATCTGATAGG-3´ shPink1 C 5´-GGGAGCCATCGCCTATGAAAT-3´ Control 1 5´- GCCTAGACGCGATAGTATGGA-3´ Control 2 5’- CCTAGACGCGATAGTATGGAC-3´ Tabla No. 1 Secuencias de los constructos de shRNA para Pink1 y de los controles de silenciamiento. 34 4.7 Extracción de RNA El RNA de las células CAD fue extraído utilizando Trizol (Invitrogen). Las células fueron lisadas directamente en platos de 10 cm2 de área adicionando 1 mL de Trizol, el lisado celular se pasó varias veces a través de la pipeta y se transfirió a un tubo eppendorf de 1.5 mL. La muestras homogenizadas fueron incubadas durante 5 minutos a temperatura ambiente para permitir la completa disociación de los complejos de nucleoproteína. Después se agregaron 200 µl de cloroformo, se taparon los tubos, se agitaron vigorosamente a mano durante 15 segundos y se incubaron a temperatura ambiente durante 3 minutos. Después las muestras fueron centrifugadas a 12000 xg por 15 minutos a 4°C. Después de la centrifugación, la fase acuosa, que contiene el RNA, fue transferida a un nuevo tubo y el RNA fue precipitado agregando 500 µl de alcohol isopropílico, las muestras se incubaron a temperatura ambiente durante 10 minutos y se centrifugaron a 12000 xg durante 10 minutos a 4°C. Después se removió el sobrenadante y el pellet de RNA se lavó una vez con 1 mL de etanol al 75% en agua DEPC mezclando con vórtex, en seguida se centrifugó a 7500 xg durante 8 minutos a 4°C. Finalmente se removió el sobrenadante y se secó el RNA en Speed Vac durante 5 minutos. El RNA se disolvió en 30-50 µl de agua DEPC (dependiendo del tamaño del pellet). La integridad del RNA fue evaluada mediante electroforesis en gel de agarosa al 0.8% teñido con SYBR safe. Las muestras fueron cuantificadas en NANO Drop, empleando 2 µl de muestra y haciendo la medición de cada una por duplicado. 35 4.8 Transcripción reversa La síntesis de cDNA fue realizada usando el kit Superscript III (Invitrogen). El procedimiento consistió en poner el volumen equivalente a 5 µg de RNA en un tubo de PCR libre de RNasas y agregar 1 µl de Oligo dT´s y 1 µl y completar el volumen hasta 10 µl con agua DEPC. Después se agregaron los siguientes reactivos en el orden en que se presentan a continuación: Reactivos Concentración ARN total 5 μg OligoDTs 10μM dNTPs 0,50 mM Agua DEPC Completar a volumen de 20μl Buffer de 50 mM Tris-HCl pH Transcripción 8,3 reversa 75 mM KCl 3 mM MgCl2 DTT 10 mM SuperScript III RT 10 U RNase OUT 40 U Tabla No. 2 Lista de reactivos y su concentración final en la reacción de transcripción reversa. 4.9 PCR en tiempo real Para evaluar la disminución de la expresión de Pink1 se hizo la medición de la cantidad de transcrito usando la metodología de PCR cuantitativa relativa a los 36 niveles de expresión de β–actina, la secuencia de los primers específicos empleados se muestra en la tabla No. 3. Gen de Orientación Secuencia Pink1 Directo 5’-GTGGAACATCTCGGCAGGTT-3’ Pink1 Reverso 5’-CCTCTCTTGGATTTTCTGTAAGTGAC-3’ β-actina Directo 5´-CTTGGGTATGGAATCCTGTGG-3’ β-actina Reverso 5´- TCAGGAGGAGCAATGATCTTG-3´ Interés Tabla No. 3 Primers específicos usados en la amplificación de Pink1 y de β-actina por qPCR. Primero se hizo una PCR convencional en gradiente de temperatura para determinar el rango de temperatura de anillamiento de los primers de Pink1. Una vez determinado ese rango se empezaron los ensayos en el equipo de tiempo de real probando las temperaturas de anillamiento encontradas en la PCR convencional hasta determinar que 51°C era la temperatura óptima (anexo 7.1 y 7.2). El equipo empleado fue el Light Cycler 4.1 (Roche Molecular Biochemicals; Mannheim, Germany). En la figura No. 6 se muestra el programa de amplificación empleado para Pink1. El método del Ct fue utilizado para analizar los resultados ya que provee una rápida estimación de la proporción relativa de la expresión de un gen (Kubista et al., 2006;Livak y Schmittgen, 2001). Posteriormente se recuperaron los amplificados y se corrieron en un gel de agarosa al 5% para corroborar su identidad de acuerdo al tamaño esperado 37 del producto. La eficiencia de los primers fue evaluada haciendo uso de diluciones seriadas de cDNA (1:3; 1:9; 1:27; 1:81) (anexo 7.3). Figura No. 6 Programa usado en la amplificación de Pink1 por qPCR 4.10 Determinación de la actividad mitocondrial Este parámetro se evaluó empleando el bromuro de 3-(4,5-dimetiltiazol-2-il)2,5-difeniltetrazolio (ensayo de MTT). Las células fueron sembradas a una densidad de 7 x 103 en 200 µl de medio por pozo en una placa de 96 pozos. Las células CAD y CAD-shPink1 diferenciadas fueron tratadas con C2- ceramida 25 µM (Aldrich,St.Louis,MO,USA) durante 6 horas. Después de remover 50 µl de medio, 25 µl de la solución stock de MTT (5mg/ml en PBS) fueron añadidos a cada pozo, seguidos de incubación durante 2 horas a 37°C. Después, el medio fue removido y las células fueron lisadas y el producto púrpura de formazán fue solubilizado mediante la adición de 100 µl de tampón de lisis de MTT (20% w/v SDS, 50% v/v dimetilformamida en agua destilada, pH 4.7) por pozo. Las placas fueron incubadas a 37°C durante 120 38 minutos y la absorbancia fue leída a 590 nm. Las células sin tratar fueron usadas como control positivo. El porcentaje de sobrevivencia celular fue calculado como: En donde X es la lectura promedio del metabolismo de MTT en un grupo con un solo tratamiento. 4.11 Extracción y cuantificación de proteínas Las células CAD y M17 shPink1 fueron cultivadas en placas de 6 pozos, se aplicaron los tratamientos y se lisaron en frío usando tampón de lisis RIPA suplementado con inhibidores de proteasas (Complete Mini, EDTA Free, Roche) y fosfatasas (PhosphoStop Roche) dejando el reactivo en contacto con las células durante 20 minutos. Los lisados se pasaron a tubos eppendorf y se centrifugaron a 7200g por 10 min a 4°C y el sobrenadante se separó y se almacenó a -20°C. La concentración de proteína de cada muestra se determinó usando Ácido Bicinconínico (BCA Protein Assay, Pierce) que tiene la ventaja de ser un método muy sensible y que tiene muy pocas interferencias ya que es compatible con la mayoría de detergentes iónicos y no iónicos, de acuerdo a las instrucciones del fabricante. Para este ensayo se empleó la relación reactivo: muestra (50:1) recomendada en el kit, se realizó una curva de calibración con 5 patrones de trabajo desde 5 a 25 mg de BSA, que abarca el rango de linealidad del ensayo. La mezcla se incubó a 37 °C durante 30 39 minutos y la absorbancia se midió mediante espectofotometría en placas de 96 pozos a una longitud de onda de 540 nm. 4.12 Electroforesis en SDS-PAGE Se utilizó una cámara de electroforesis BIORAD mini PROTEAN III para la separación de proteínas de acuerdo a su tamaño en geles SDS-poliacrilamida en gradiente de 6-15%. El gel se colocó entre dos placas de vidrio separadas por espaciadores de 0,75 mm en un soporte vertical. Luego de la polimerización del gel, este se colocó en tanques de electroforesis y se cubrió con buffer de corrido. Las muestras de proteína (40 µg en total) diluidas en buffer Lae mmli y el marcador de peso molecular se adicionaron en los pozos y se llevó a cabo la electroforesis a 100V. 4.13 Detección de proteínas por Western blotting Luego de la electroforesis, las proteínas se transfirieron a membranas de nitrocelulosa usando una cámara de transferencia BIORAD Mini Trans-Blot. El gel se cubrió con la membrana de nitrocelulosa que se coloca entre papel Whatmann y dos esponjas de filtración. El ensamblaje se colocó en un cassette de soporte, se insertaron en el tanque lleno de buffer de transferencia, con la membrana de nitrocelulosa del lado del ánodo. La transferencia se llevó a cabo por 1h a 100V. Luego de la transferencia, la membrana se removió del ensamblaje y se incubó en buffer de bloqueo (5% leche descremada en TBS con 0,1% (v/v) Tween20, TBS-T) por 1h. La membrana de nitrocelulosa se incubó con 3 ml de la solución apropiada de anticuerpos primarios en buffer de bloqueo durante toda la noche a 4ºC, se lavó 3 veces en TBS-T (5min/lavado). El anticuerpo unido 40 se visualizó mediante incubación de la membrana con el anticuerpo secundario específico por 2h a temperatura ambiente y se lavó luego 3 veces con TBS-T (5min/lavado). El anticuerpo secundario unido se detectó usando quimioluminiscencia (ECL, Thermo Scientific). La membrana se incubó con una mezcla 1:1 de los reactivos A y B de ECL por 1min, y se expuso a una película ECL para visualizar los productos de quimioluminiscencia. Las bandas en la placa fotográfica se escanearon utilizando un analizador de imágenes (Phospho Imageur Storm 840, Molecular Dynamics). Proteína Marca Dilución Anticuerpo 2° Tamaño (kDa) Mfn2 Sigma Aldrich 1:500 Rabbit 86 Opa1 BD Biosciences 1:500 Mouse 80-100 Drp1 BD Biosciences 1:500 Mouse 79-84 Fis1 Biovision 1:500 Rabbit 17 β-actina Cell Biolabs 1:5000 Mouse 43 Tabla No. 4 Anticuerpos empleados para los ensayos de western blot 4.14 Análisis de la distribución mitocondrial por tinción con Mitotracker Las células CAD fueron cultivadas según las indicaciones arriba mencionadas sobre cubreobjetos tratados de la siguiente manera: Los cubreobjetos fueron sumergidos en ácido clorhídrico 10N para retirar cualquier elemento orgánico de su superficie. Posteriormente se lavaron con agua desionizada para retirar el ácido. Los cubreobjetos se colocaron dentro de pozos del diámetro correspondiente según el experimento y se cubrieron con una solución de PoliL-Lisina a una concentración de 0.01 mg/ml durante 1 hora. Seguidamente se 41 lavaron con agua desionizada y se irradiaron con luz ultravioleta para asegurar la esterilidad durante 1 o 2 horas. Se sembraron las células a la densidad correspondiente según el diámetro del pozo y se adicionó MitoTracker® Red CM-H2XRos (Invitrogen) a una concentración de 200 nM durante 30 minutos, se lavó dos veces con DMEM-F12 precalentado a 37°C y se fijó con paraformaldehído a 4% en PBS durante 20 minutos. Después se pusieron sobre un portaobjetos con medio de montaje apropiado para fluorescencia (VectaShield®, Vector Labs) y se observó en un microscopio láser confocal marca Nikon Eclipse Ti. Como control positivo de fusión mitocondrial las células fueron tratadas con forskolina (FK) 25 µM durante 45 minutos. La FK tiene la función de activar la vía de señalización del cAMP, lo que termina por fosforilar a Drp1. La forma fosforilada de esta proteína no es reclutada en la membrana mitocondrial y por lo tanto no promueve fisión mitocondrial. Como control positivo de fisión las células CAD se trataron con CCCP en una concentración de 10 µM durante 1 hora. Transcurrido ese tiempo el medio se reemplazó con medio DMEM-F12 suplementado con SFB al 10% fresco. Las células se fijaron y se pusieron en portaobjetos, como se describió anteriormente. 4.15 Inmunofluorescencia Las células CAD fueron se sembradas en cajas de 6 pozos que contenían cubreobjetos de 2,2 cm. Cumplidos los tiempos de los tratamientos se realizó la tinción de las mitocondrias con MitoTracker® como se explicó en 4.14. Posteriormente se retiró el medio y las células CAD se fijaron con paraformaldehído al 4% por 20 minutos, después se lavaron con PBS1X tres 42 veces durante 5 minutos y se permeabilizaron con Tritón X-100 al 0,1% en PBS1X durante 10 minutos. Posteriormente se agregó buffer de bloqueo (BSA al 1% y Tritón X-100 al 0,05% en PBS 1X) y se dejó en contacto con las láminas durante 1 hora. Las láminas se incubaron con anti Fis1 (SantaCruz Biotechnology sc-98900, 1:50); anti p-Drp1 (Ser-637) (Cell Signaling, 1:50) y anti Mfn1 (SantaCruz Biotechnology sc-50330, 1:50) durante toda la noche. El anticuerpo primario fue lavado 3 veces con PBS1X durante 5 minutos. Después se adicionó el anticuerpo secundario fluorescente: Alexa Fluor 568 rabbit anti—goat IgG (1:200, Invitrogen, A-11079) que estuvo en contacto con las células fijadas durante 2 horas, se lavó y después el núcleo fue teñido con Hoechst (1:5000, SIGMA 33258). Las imágenes celulares fueron visualizadas en el microscopio confocal (C2-Confocal, Nikon) y analizadas con el software ImageJ. 4.16 Análisis de los eventos de fusión y fisión mitocondrial por transfección con mito-YFP Las células M17 fueron transfectadas transitoriamente con 1 µg del plásmido mito-YFP usando el reactivo X-treme Gene 9 (Roche). Posteriormente fueron fijadas (como se explicó anteriormente) y puestas en portaobjetos esta vez con Prolong Gold Antifade Reagent, invitrogen como medio para conservar la fluorescencia. Las imágenes fueron adquiridas en un microscopio confocal (Zeiss Confocal Microscope LSM 500). Las células transfectadas con mitoYFP fueron excitadas usando el láser HeNe a 488 nm usando un objetivo PlanApochromat de 63x/1.4 Oil DIC. Las fotos mitocondriales se obtuvieron con z-stacks de 20 imágenes separadas por 1µm a lo largo del eje z, el tiempo total de adquisición de cada stack fue de 1.4 segundos. 43 Las imágenes obtenidas por microscopía confocal fueron optimizadas ajustando el contraste y se pasaron a imágenes binarias por conversión a imágenes de 8 bits. Después de reducir el ruido causado por la señal de fluorescencia inespecífica se aplicó un filtro espacial (filtro de convolución) así como un filtro de umbral para definir las estructuras mitocondriales. Usando el programa Image J 1.47 (Wayne Rasband; National Institutes of Health, USA) cada mitocondria fue marcada para analizar sus características morfológicas como área, perímetro y ejes menor y mayor. Con base en estos parámetros, la relación de aspecto (aspect ratio, AR; relación entre los ejes mayores y menores de la elipse equivalente al objeto) de una mitocondria y su factor de forma (FF; perímetro2/4π*área), consistentes con el grado de ramificación, fueron calculados. 4.17 Análisis estadístico Los datos presentados son promedio ±SEM de tres experimentos independientes. El análisis estadístico fue realizado usando ANOVA de una vía (seguido de la prueba Tukey-Kramer para comparación múltiple) y ANOVA de dos vías, dependiendo de los datos a analizar, usando el programa GraphPad 5 (Prism 5, GraphPad Software, La Jolla, CA, USA). 44 5 Capítulo 5 Resultados 5.1 La disminución de la expresión de Pink1 disminuye la actividad mitocondrial y aumenta la vulnerabilidad al daño inducido por C2ceramida En la figura No. 7 se muestran microfotografías de las células CAD durante el proceso de selección de la dosis mínima del antibiótico capaz de matar el 100% de las células en un periodo de 48-72 horas. Para las células CAD la dosis seleccionada fue de 6 µg/ml, mientras que para las células M17 fue de 5 µg/ml. Día/Dosis 1 2 3 (µg/ml) 0 2 45 4 6 8 10 Figura No. 7 Determinación de la dosis tóxica mínima de blasticidina para las células CAD. Como se ve en las fotos las dosis más altas (8 y 10 µg/ml) mataban el 100% de las células en un periodo de 24h, sólo la dosis de 6 µg/ml lo hizo en un periodo de 72 horas, tiempo apropiado para la selección de las células que captaron el plásmido exitosamente. 46 Una vez establecida esta dosis se procedió con la transducción vectores lentivirales que contienen secuencias específicas usando para el silenciamiento de Pink1 y secuencias scrambled o control de silenciamiento. A partir de un único clon seleccionado con blasticidina se dio inicio al crecimiento de las líneas celulares denominadas Control shPink1 y shPink1. La eficiencia de la transfección fue evaluada mediante qPCR, la cuantificación de los niveles de expresión relativa de Pink1 con respecto a β-actina, se realizó con el método del Ct (Livak, 2001). En la figura No. 8 se muestra la disminución de la expresión de Pink1 en un 83% para las células CAD y en un 1.5 *** *** *** *** 1.0 0.5 sh Pi nk 1 M 17 C M 17 nk 1 on tr ol sh Pi B as al 17 A D M nk 1 C C A D C on tr ol sh Pi D A C sh Pi nk 1 0.0 B as al Expresión relativa de Pink1/ -actina 88% para las células M17. Figura No. 8 Niveles de expresión de Pink1. Fueron medidos por qPCR en células CAD y M17 basales y transducidas con las secuencias control de silenciamiento y shPink1; ***, p<0,0001, n=3, cada dato fue tomado por duplicado. 47 Habiendo establecido las líneas celulares se evaluó la actividad mitocondrial mediante el ensayo de MTT. Las células fueron sembradas en cajas de 96 pozos y en medio de diferenciación. Las células sobrevivieron hasta 96 horas después de iniciado el experimento. Las células shPink1 presentaron un menor actividad mitocondrial comparadas con las células basales y control shPink1 en todos los tiempos (figura No. 9) mostrando un comportamiento similar a % A ctividad mitocondrial las células basales y control shPink1 tratadas con C2-ceramida 25µM. CAD Basal 100 Control shPink1 shPink1 CAD Basal+C2-Ceramida Control shPink1+C2 Ceramida 50 *** *** shPink1+C2-Ceramida *** *** *** *** *** *** 0 0 20 40 60 80 100 Tiempo (h) Figura No. 9 Disminución de la actividad mitocondrial en células shPink1.Ensayo de MTT para evaluar la actividad mitocondrial de células CAD basales, control shPink1 y shPink1 con y sin exposición a C2-ceramida. ***, p<0,0001, n=3, 16 datos fueron tomados por línea celular/tratamiento. Así mismo, las células shPink1 tratadas con C2-ceramida presentan menor actividad mitocondrial comparadas con las células control shPink1 en esta 48 misma condición. Estas diferencias fueron estadísticamente significativas en todos los tiempos evaluados. 5.2 La disminución de la expresión de Pink1 induce cambios en la morfología y distribución mitocondrial, éstos se incrementan en presencia de agentes neurotóxicos Las células CAD fueron teñidas con Mitotracker. En la figura No. 10 se observan diferencias con respecto a la distribución de las mitocondrias. En las células shPink1 no tratadas se observan las mitocondrias organizadas alrededor del núcleo, mientras que en las control shPink1 se alcanzan a distinguir redes que se extienden a lo largo del citoplasma. El tratamiento con C2-ceramida retrae esas redes, al igual que lo hace el CCCP. La tinción con Mitotracker no permitió analizar la morfología de las mitocondrias, a pesar de que se probaron diferentes dosis del reactivo y tiempos de incubación del mismo. En su lugar, se optó por transfectar las células transitoriamente con un plásmido fluorescente de localización mitocondrial: mito-YFP. La transfección con mitoYFP es el método más utilizado para visualizar mitocondrias ya que es mucho más estable que los pigmentos fluorescentes y su localización no depende del m y no afecta el comportamiento de las mitocondrias (Mitra and Lippincott-Schwartz, 2010). 49 Figura No. 10 Distribución mitocondrial en células CAD. Microfotografías de las células CAD control shPink1 y shPink1 con y sin exposición a C2-ceramida. Las mitocondrias fueron marcadas con Mitotracker Red 200nM durante 30 minutos. Barra de escala: 3,2 µm. 50 A B C D E Figura No. 11 Procesamiento de las imágenes mitocondriales obtenidas por microscopía confocal. El programa empleado fue Image J. En A se muestra la imagen original (superposición de cada una de las 20 imágenes que componen el stack); B, Proyección 3D de la imagen obtenida en A; C, Ajuste de contraste y aplicación de filtros espaciales de la imagen obtenida en B; D, Conversión a imagen binaria de 8 bits de la imagen obtenida en C para posterior análisis de partículas y de descriptores de forma, algunos descriptores se muestran en la imagen representativa que se muestra en E. Con ayuda del programa Image J las imágenes fueron procesadas (figura No.11) y sobre éstas se midieron diferentes parámetros morfológicos como 51 AR y el factor de forma (F/F). AR es una medida de la longitud mitocondrial, mientras F/F es una medida tanto de la longitud como del grado de ramificación (De Vos et al., 2005;De Vos and Sheetz, 2007). Con base en esos descriptores morfológicos, las mitocondrias se dividieron en dos grupos: fragmentadas/no ramificadas y asociadas/ramificadas. En la figura No. 12 se presentan algunas de las imágenes representativas de las mitocondrias de las células M17 control shPink1 y shPink1 que fueron tomadas in vivo. Mientras que en la figura No. 13 se muestran las diferentes morfologías de las células fijadas, encontrándose que hay correlación entre éstas. Las células control de silenciamiento presentaron menor porcentaje de células fragmentadas comparadas con las células silenciadas para Pink1 (figura No. 14) este mismo efecto se observó al tratar las células con rotenona. De igual manera, las células silenciadas para Pink1 no responden de manera similar a las células control al estímulo generado por la forskolina. Las células shPink1 presentaron un 52% de células ramificadas o asociadas, mientras que las control shPink1 muestran este tipo de morfología en más de un 90%. Con estas microfotografías también se corroboran las diferencias en cuanto a distribución mitocondrial, ya que es notable que en las células control se encuentran más distribuidas a lo largo del cuerpo celular mientras que en las silenciadas tienden a concentrarse alrededor del núcleo. Un comportamiento similar al observado en estas células cuando son tratadas con rotenona y con CCCP. El tratamiento con FK genera mitocondrias muy alargadas y redes extensas en los dos tipos celulares. 52 Figura No. 12 Microfotografías in vivo de las mitocondrias de células M17 Control shPink1 y shPink1. En las células shPink1 tratadas y no tratadas con rotenona se observan mitocondrias fragmentadas en mayor proporción comparadas con las células control de silenciamiento. Barra de escala: 5 µm. 5.3 El silenciamiento de Pink1 afecta la expresión de algunas proteínas involucradas en fusión/fisión Teniendo en cuenta que se obtuvieron diferencias en la morfología de las mitocondrias silenciadas para Pink1, el siguiente paso fue evaluar si éstas diferencias estaban relacionadas con los niveles de expresión de algunas de las proteínas involucradas en los procesos de fusión/fisión. Se realizaron western blot para Drp1 y Fis1 (para evaluar fisión) y Mfn2 y Opa1 (para evaluar fusión). 53 Figura No. 13 Morfología mitocondrial de las células M17. Las células M17 control shPink1 y shPink1 fueron transfectadas con mito-YFP y sometidas a diferentes tratamientos. Las imágenes de las mitocondrias fueron obtenidas por microscopía confocal y procesadas con 54 Image J. Las flechas señalan mitocondrias fragmentadas. Barra de escala: 5 µm. P<0.0001 % de células 100 Fragmentadas/No ramificadas Asociadas/Ramificadas 50 Co nt ro Con ls tr h Co Pin ol s k1 hPi nt Co ro nk +R nt l 1 ro sh ot ls e Pi hP nk non 1+ in a k1 CC +F or CP sk ol sh in Pi a sh nk P 1+ in k1 R sh ot e P n sh in Pi k1 ona nk +C 1+ F o CC P rs ko lin a 0 Figura No. 14 Cuantificación de la morfología mitocondrial de células M17 control shPink1 y shPink1. Entre 280-400 células en total fueron evaluadas por cada tratamiento, n=3. Los valores están expresados como porcentaje del número total de células contadas. La significancia estadística fue analizada por ANOVA de dos vías por morfología mitocondrial y línea celular. Los niveles de Drp1 total disminuyeron significativamente en las células shPink1. Un resultado muy similar se observó en las células tratadas con rotenona y forskolina. En cuanto a Fis1 solamente hubo cambios destacables en las células tratadas con CCCP, encontrándose un aumento importante de esta proteína en las células shPink1 comparadas con las células control. Con respecto a las proteínas de fusión, sólo se observaron cambios con relevancia 55 estadística para Mfn2, ya que los niveles de esta proteína disminuyen en las células silenciadas para Pink1 no tratadas y además en las estimuladas con FK y con CCCP (figuras No. 15 y 16). En Opa1 no se encontraron diferencias estadísticamente significativas. Figura No. 15 Western blot de algunas de las proteínas involucradas en fusión y fisión mitocondrial. Imágenes representativas de 3 experimentos independientes. En las células CAD se realizó inmunofluorescencia para detectar a Fis1, pDrp1 y Mfn1 (figuras 17 a 19). En las figuras se observa que hay una mayor intensidad de fluorescencia de Fis1 en las células shPink1, mientras que ocurre lo contrario para p-Drp1 y para Mfn1. 56 Co Co Expresión relativa de F is1/ -actina nt nt ro ro ls l sh hP Pi in nk k1 1 +R Co ot nt en ro on ls Co hP a nt in ro k1 ls + hP FK in k1 +C CC P sh sh Pi Pi nk nk 1+ 1 R ot en on sh a Pi nk 1+ sh FK Pi nk 1+ CC CP 1.0 2.5 2.0 1.5 1.0 0.5 0.0 *** *** 0.5 0.0 *** Co Co Expresión relativa de M fn2/ -actina nt nt ro ro ls l sh hP Pi in nk k1 1 +R Co ot nt en ro on ls Co hP a nt in ro k1 ls +F hP K in k1 +C CC P sh sh Pi P nk in k1 1+ R ot en on sh a Pi nk 1+ sh FK Pi nk 1+ CC CP *** Co Co Expresión relativa de Opa1/ -actina nt nt ro ro ls l sh hP Pi in nk k1 1 +R Co ot nt en ro on ls Co hP a nt in ro k1 ls +F hP K in k1 +C CC P sh sh Pi P nk in k1 1+ R ot en on sh a Pi nk 1+ sh FK Pi nk 1+ CC CP Co Co Expresión relativa de D rp1/ -actina nt nt ro ro ls l sh hP Pi in nk k1 1 +R Co ot nt en ro on ls Co hP a nt in ro k1 ls + hP FK in k1 +C CC P sh sh Pi Pi nk nk 1+ 1 R ot en on sh a Pi nk 1+ sh FK Pi nk 1+ CC CP 1.5 1.5 *** *** 1.0 *** 0.5 0.0 1.5 1.0 0.5 0.0 Figura No. 16 Densitometría de las proteínas de fusión/fisión detectadas por western blot. La densitometría fue realizada con el programa Image J. 57 Figura No. 17 Inmunofluorescencia de Fis1. Microfotografías representativas en donde se muestra una mayor intensidad de fluorescencia de Fis1 en las células shPink1 tratadas y no tratadas con C2-ceramida, comparadas con las células control. Barra de escala: 3,2 µm. 58 Figura No. 18 Inmunofluorescencia de p-Drp1. Microfotografías representativas en donde se muestra una mayor intensidad de fluorescencia de p-Drp1 en las células basales comparadas con los demás tratamientos. Barra de escala: 3,2 µm. 59 Figura No. 19 Inmunofluorescencia de Mfn1. Microfotografías representativas en donde se muestra una mayor intensidad de fluorescencia de Mfn1 en las células basales comparadas con los demás tratamientos Barra de escala: 3,2 µm. 60 5.4 Discusión de Resultados Muchos estudios se han realizado con el objetivo de entender la etiología de la enfermedad de Párkinson. Sin lugar a dudas la mayor fuente de información se ha obtenido a través de las formas genéticas de la enfermedad, que son las menos comunes. Desde el 2004, se han reportado numerosos avances en cuanto al posible papel biológico que ejerce Pink1 y que puede ser relevante con respecto a la patología en la cual fue inicialmente involucrado. Sin embargo, todavía la función bioquímica de Pink1 y las vías de señalización que la regulan son desconocidas. Diferentes estudios han mostrado que Pink1 puede actuar como un neuroprotector (Haque et al. 2012; Sánchez-Mora et al. 2012) y que su deficiencia promueve una mayor susceptibilidad a apoptosis (Deng et al. 2008; Gautier et al. 2008; Valente et al. 2004). Las células CAD que sobreexpresan Pink1 y que son tratadas con C2-ceramida muestran menores porcentajes de mitocondrias depolarizadas, menor expresión de Bax y una mayor expresión de Bcl-2 comparada con las células no transfectadas. Además Pink1 rescata l inhibición de la fosforilación de Akt inducida por C2ceramida (Sanchez-Mora et al., 2012). En este trabajo se realizaron ensayos de MTT para evaluar la función mitocondrial de células CAD con niveles de expresión de Pink1 disminuidos en presencia y en ausencia de un insulto neurotóxico generado por C2ceramida (que causa muerte celular apoptótica en modelos dopaminérgicos, (France-Lanord et al. 1997;Arboleda et al., 2010;Arboleda et al., 2009). Como se muestra en la figura No. 9, las células CAD que tienen menores niveles de expresión de Pink1 presentan una menor absorbancia a 590 nm (menor 61 producción de formazán), lo que es indicativo de una menor cantidad de células vivas con una actividad mitocondrial normal. Estas diferencias entre líneas celulares fueron significativas estadísticamente, demostrando así que Pink1 es importante para el normal funcionamiento de las mitocondrias. El efecto neurotóxico de la C2-ceramida es mayor en las células que expresan niveles más bajos de Pink1, lo que muestra no sólo la C2-ceramida (un inhibidor de la vía de supervivencia PI3K/Akt) está afectando la supervivencia de éstas células sino también la deficiencia de Pink1; a su vez este resultado permite asumir que posiblemente hay una relación entre esta vía de señalización y Pink1, como lo sugieren algunas investigaciones (Qi et al., 2011;Timmons et al., 2009; Stiles, 2009). Queda claro que Pink1 es necesario para que haya una función mitocondrial normal, pero ¿está eso asociado a la morfología de las mitocondrias? Esta pregunta es válida teniendo en cuenta que la dinámica mitocondrial está involucrada en procesos de muerte celular y que a su vez existe polémica alrededor del efecto que Pink1 puede ejercer en el balance entre fusión y fisión mitocondrial. En este trabajo se encontró que cuando Pink1 está silenciado, el número de células que contiene mitocondrias fragmentadas aumenta (de 40% a 57% cuando se comparan con las células control shPink1) y que el tratamiento con rotenona tiene un efecto aditivo. La fragmentación de las mitocondrias está vinculada con la muerte celular pero todavía no se tiene claro si éste efecto es una causa o una consecuencia. Las mitocondrias tienden a fragmentarse cuando tienen un daño para posteriormente ser eliminadas mediante un proceso de muerte selectivo conocido como mitofagia. De acuerdo a los ensayos de MTT se puede asumir que las células silenciadas para Pink1 62 tienden a morir en mayor proporción que las células control y de esta manera se justifica que muestren en mayor proporción una morfología redondeada y que ésta incremente cuando se inhibe el complejo I de la cadena respiratoria con la dosis usada de rotenona (Hoppins et al., 2007). Llama la atención que las células shPink1 no respondieron de igual manera al estímulo generado por la FK, la cual actúa como potenciadora de cAMP, conduciendo a la fosforilación de Drp1, una forma inactiva a nivel de fisión y que no se localiza en la mitocondria y que por lo tanto inclina la balanza hacia la fusión mitocondrial (Cribbs y Strack, 2007;Cribbs y Strack, 2009) . Mientras que las células control shPink1 presentan un 94% de células ramificadas o asociadas en presencia de FK, sólo el 47% de las shPink1 tienen esa misma morfología. Es posible especular que la diminución de la expresión de Pink1 en estas células de alguna manera afecta la fosforilación de Drp1 y por esto más de la mitad de las células conservan su morfología fragmentada. Esta afirmación la apoya un estudio previo en donde se vio que Pink1 silvestre, más no las versiones mutantes recesivas con dominios quinasa inactivos protegen contra la fragmentación mitocondrial inducida por rotenona, mientras que células deficientes en Pink1 muestran menor conexión mitocondrial. La expresión de la proteína relacionada a dinamina 1 (Drp1) exagera los fenotipos deficientes de Pink1 y la interferencia de Drp1 los rescata. Drp1 es defosforilado en células deficientes de Pink1 debido a la activación de la fosfatasa dependiente de calcio calcineurina (Sandebring et al., 2009). 63 Los experimentos de western blot mostraron que hay una diminución significativa de Drp1 total en las células shPink1no tratadas. El hecho de que las células shPink1 muestren mayor proporción de mitocondrias fragmentadas pero niveles más bajos de Drp1 con y sin tratamiento con rotenona comparada con las células control shPink, puede atribuirse a que no sólo de esta proteína dependa la fragmentación mitocondrial, sino que existe un mecanismo independiente de Drp1 que participa en la fisión de este organelo como lo sugiere un trabajo en donde al inhibir a Drp1, se retrasa, pero no se previene por completo la fisión mitocondrial (Ishihara et al., 2009). Proteínas como Bax y Bak también pueden hacer parte de este proceso (Martinou y Youle, 2011), así como las modificaciones post-traduccionales de Drp1, que son muy complejas (hasta el momento se ha reportado fosforilación, sumoilación, desumoilación y ubiquitinación) y de las cuales todavía se sabe muy poco acerca de su función y regulación (Reddy et al., 2011;Santel y Frank, 2008). Una importante disminución también se observa en presencia de FK ya que las células se estimulan para fusionarse, sin embargo, como se vio anteriormente aproximadamente la mitad de la población celular shPink1 responde a este estímulo. Con respecto a Fis1 sólo se obtuvieron cambios significativos entre las líneas celulares al ser tratadas con CCCP, como si éste estímulo implicara una mayor proporción de ésta proteína en las células shPink1. Mfn2 también presenta cambios cuando en células shPink1. Como es de esperarse sus niveles en células aumentan en presencia de FK (ya que se promueve la fusión mitocondrial) con respecto a las células shPink no tratadas, pero éste aumento no es tan alto como el que se observa en las células control shPink1. Lo que confirma una vez más que las células no están 64 respondiendo de manera adecuada a dicho estímulo. Los niveles de Mfn2 en las células shPink1 no tratadas son significativamente menores a los de las control shPink1, lo que coincide con la mayor presencia de mitocondrias asociadas o ramificadas en éstas. Sin embargo, los niveles de Opa1 no mostraron cambios relevantes. Probablemente el silenciamiento de Pink1 no está modulando los niveles de esta proteína y son otros los factores involucrados en su regulación. También algunos estudios han mostrado que las mitofusinas y Opa1 actúan en distintos pasos durante el proceso de fusión, ya que MEFs knockout para esta proteína mantienen la fusión de la membrana externa pero carecen de fusión de la membrana interna (Chan, 2012). Un aspecto relevante en este estudio y sumamente repetitivo fueron las diferencias encontradas en cuanto a la distribución de las mitocondrias. Esta organización parece tener implicaciones a nivel a funcionalidad como se ha visto en estudios previos, en donde se mostró que las mitocondrias perinucleares tienen un papel en la regulación espacial de las ondas de Ca 2+ (Hashitani et al., 2010). La carencia de Pink1 tiende a desplegarlas de esta manera, al igual que los tratamientos con rotenona y CCCP. Se sabe que Pink1 puede regular las corrientes de Ca2+ (Ghandi et al., 2009) y esto podría ser a través de la regulación de la morfología mitocondrial. Estudios futuros podrán aclarar más el panorama en este aspecto. 65 6 Capítulo 6 6.1 Conclusiones Resulta claro que Pink1 es importante en la actividad mitocondrial y que este efecto está relacionado con el balance entre fusión y fisión. La ausencia de Pink1 en este modelo está inclinando la balanza hacia la fisión de las mitocondrias, aunque este efecto no está relacionado con mayores niveles de la proteína Drp1 y posiblemente otros mecanismos estén involucrados en este proceso, entre los que no se descartan las diferentes modificaciones posttraduccionales de dicha proteína, en las cuales Pink1 parece tener un papel significativo, en relación específicamente con la fosforilación. En este contexto Pink1 promovería neuroprotección regulando la fusión mitocondrial, por lo cual sigue siendo un gen relevante en el estudio, entendimiento y posible tratamiento de la EP. 6.2 Perspectivas Hacer estudios concluyentes acerca de la dinámica mitocondrial ha resultado muy difícil. Aunque se han desarrollado varias técnicas (ensayo de células híbridas, FRAP, fotoactivación de proteínas, etc) las mitocondrias son muy diferentes dependiendo del tipo celular y esto dificulta su análisis. Por ejemplo, en los fibroblastos, se observan estructuras muy definidas, mientras que en las neuronas no ocurre lo mismo. El hecho de que aparezcan cada vez 66 más nuevas proteínas involucradas en la fusión y fisión hace que el panorama se expanda aún más y que sea más complejo. El desarrollo de metodologías más precisas para el estudio de mitocondrias es esencial para develar con mayor exactitud los mecanismos de fusión y fisión. Desde luego, no existe la técnica perfecta, pero las que se tienen hoy en día admiten un alto grado de subjetividad. En cuanto a Pink1 también es importante anotar que hoy en día, más de 10 años después de su descubrimiento, todavía no se cuenta con anticuerpos confiables para la detección de la proteína, además sus niveles endógenos son muy bajos y por eso se debe acudir a procedimientos que pueden estresar a las células, como las transfecciones. Todavía queda un largo camino por recorrer en cuanto a los mecanismos, las vías de señalización y las funciones de la fusión y fisión mitocondrial; procesos esenciales a nivel celular que al ser develados podrían conducir al entendimiento de muchas patologías y al desarrollo de tratamientos terapéuticos. 67 7 Anexos 7.1 RT-PCR para determinación de la temperatura óptima de anillamiento de los primers de Pink1 7.2 Picos de fusión de Pink1 (azul) y de β–actina (verde) tomando como temperatura de anillamiento 51°C 68 7.3 Curvas de amplificación (A) y picos de fusión (B) de Pink1 y de β– actina 69 7.4 Comprobación de la eficiencia de los primers para Pink1 mediante diluciones seriadas La pendiente de la gráfica es de 2.2, que es igual a la eficiencia 70 8 Bibliografía BIBLIOGRAFÍA 1. Arboleda G, Cardenas Y, Rodriguez Y, Morales LC, Matheus L, and Arboleda H (2010) Differential regulation of AKT, MAPK and GSK3beta during C2-ceramide-induced neuronal death. Neurotoxicology, 31, 687-693. 2. Arboleda G, Morales LC, Benitez B, and Arboleda H (2009) Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res Rev, 59, 333-346. 3. Bakeeva LE, Chentsov Y, and Skulachev VP (1978) Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochim Biophys Acta, 501, 349-369. 4. Baraban SC, Lothman EW, Lee A, and Guyenet PG (1995) Kappa opioid receptor-mediated suppression of voltage-activated potassium current in a catecholaminergic neuronal cell line. J Pharmacol Exp Ther, 273, 927-933. 5. Beilina A, van der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, and Cookson MR (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A, 102, 5703-5708. 6. Cai Q and Sheng ZH (2009) Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron, 61, 493-496. 7. Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, and Greenamyre JT (2009) A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis, 34, 279-290. 8. Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol, 22, 79-99. 71 9. Chan DC (2012) Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu Rev Genet. 10. Chen H and Chan DC (2005) Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet, 14 Spec No. 2, R283-R289. 11. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, and Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol, 160, 189-200. 12. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, and Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell, 141, 280-289. 13. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, and Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature, 441, 1162-1166. 14. Cribbs JT and Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep, 8, 939-944. 15. Cribbs JT and Strack S (2009) Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol, 457, 231-253. 16. de Brito OM and Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature, 456, 605-610. 17. De Vos KJ, Allan VJ, Grierson AJ, and Sheetz MP (2005) Mitochondrial function and actin regulate dynamin-related protein 1dependent mitochondrial fission. Curr Biol, 15, 678-683. 18. De Vos KJ and Sheetz MP (2007) Visualization and quantification of mitochondrial dynamics in living animal cells. Methods Cell Biol, 80, 627-682. 19. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, 72 Lasquellec L, Arnaud B, Ducommun B, Kaplan J, and Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet, 26, 207-210. 20. Deng H, Dodson MW, Huang H, and Guo M (2008) The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A, 105, 1450314508. 21. Detmer SA and Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol, 8, 870-879. 22. Donzeau M, Kaldi K, Adam A, Paschen S, Wanner G, Guiard B, Bauer MF, Neupert W, and Brunner M (2000) Tim23 links the inner and outer mitochondrial membranes. Cell, 101, 401-412. 23. Elgass K, Pakay J, Ryan MT, and Palmer CS (2012) Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 24. Forno LS (1987) The Lewy body in Parkinson's disease. Adv Neurol, 45, 35-43. 25. Forno LS (1996) Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol, 55, 259-272. 26. France-Lanord V, Brugg B, Michel, PP, Agid, Y, Ruberg, M (1997) Mitochondrial free radical signal in ceramide-dependent apoptosis: a putative mechanism for neuronal death in Parkinson's disease. J Neurochem, 69, 1612-1621. 27. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, and Abramov AY (2009) PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell, 33, 627-638. 28. Gandre-Babbe S and van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell, 19, 2402-2412. 73 29. Gasser T (2007) Update on the genetics of Parkinson's disease. Mov Disord, 22 Suppl 17, S343-S350. 30. Gautier CA, Giaime E, Caballero E, Nunez L, Song Z, Chan D, Villalobos C, and Shen J (2012) Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener, 7, 22. 31. Gautier CA, Kitada T, and Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A, 105, 11364-11369. 32. Gegg ME, Cooper JM, Schapira AH, and Taanman JW (2009) Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One, 4, e4756. 33. Haque ME, Mount MP, Safarpour F, Abdel-Messih E, Callaghan S, Mazerolle C, Kitada T, Slack RS, Wallace V, Shen J, Aninman H, Park DS (2012) Inactivation of Pink1 gene in vivo sensitizes dopamineproducing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J Biol Chem, 287, 23162-23170. 34. Hashitani H, Lang RJ, and Suzuki H (2010) Role of perinuclear mitochondria in the spatiotemporal dynamics of spontaneous Ca2+ waves in interstitial cells of Cajal-like cells of the rabbit urethra. Br J Pharmacol, 161, 680-694. 35. Hollenbeck PJ and Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci, 118, 5411-5419. 36. Hoppins S, Lackner L, and Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem, 76, 751-780. 37. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, and Mihara K (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol, 11, 958-966. 38. Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, and 74 Haass C (2010) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J, 29, 3571-3589. 39. Kim KH and Son JH (2010) PINK1 gene knockdown leads to increased binding of parkin with actin filament. Neurosci Lett, 468, 272-276. 40. Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, and Chung J (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun, 377, 975-980. 41. Kissova I, Deffieu M, Manon S, and Camougrand N (2004) Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem, 279, 39068-39074. 42. Kolesnick RN and Kronke M (1998) Regulation of ceramide production and apoptosis. Annu Rev Physiol, 60, 643-665. 43. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, and Muqit MM (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol, 2, 120080. 44. Krendel M, Sgourdas G, and Bonder EM (1998) Disassembly of actin filaments leads to increased rate and frequency of mitochondrial movement along microtubules. Cell Motil Cytoskeleton, 40, 368-378. 45. Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjoback R, Sjogreen B, Strombom L, Stahlberg A, and Zoric N (2006) The real-time polymerase chain reaction. Mol Aspects Med, 27, 95-125. 46. Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, and Matsumoto T (2006) Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet, 15, 883-895. 47. Lazaroff M, Dunlap K, and Chikaraishi DM (1996) A CNS catecholaminergic cell line expresses voltage-gated currents. J Membr Biol, 151, 279-291. 75 48. Lazaroff M, Patankar S, Yoon SO, and Chikaraishi DM (1995) The cyclic AMP response element directs tyrosine hydroxylase expression in catecholaminergic central and peripheral nervous system cell lines from transgenic mice. J Biol Chem, 270, 21579-21589. 49. Lazarou M, Jin SM, Kane LA, and Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell, 22, 320333. 50. Lee YJ, Jeong SY, Karbowski M, Smith CL, and Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell, 15, 5001-5011. 51. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, and Robinson JP (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem, 278, 8516-8525. 52. Lin MT and Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787-795. 53. Livak KJ and Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402-408. 54. Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, Bouman L, VogtWeisenhorn D, Wurst W, Tatzelt J, Haass C, and Winklhofer KF (2009) Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem, 284, 22938-22951. 55. Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, and Jendrach M (2010) Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci, 123, 917-926. 56. Maiuri MC, Zalckvar E, Kimchi A, and Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol, 8, 741-752. 76 57. Martinou JC and Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell, 21, 92-101. 58. Mathias S, Peña LA, Kolesnick RN (1998) Signal transduction of stress via ceramide. Biochem J, 335, 465-480. 59. Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, and Cheng HC (2008) Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J Neurochem, 105, 18-33. 60. Misko A, Jiang S, Wegorzewska I, Milbrandt J, and Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci, 30, 4232-4240. 61. Mitra K and Lippincott-Schwartz J (2010) Analysis of mitochondrial dynamics and functions using imaging approaches. Curr Protoc Cell Biol, Chapter 4, Unit-21. 62. Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, Healy DG, Gilks WP, Lees AJ, Holton J, Revesz T, Parker PJ, Harvey RJ, Wood NW, and Latchman DS (2006) Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem, 98, 156-169. 63. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol, 8, e1000298. 64. Neupert W and Herrmann JM (2007) Translocation of proteins into mitochondria. Annu Rev Biochem, 76, 723-749. 65. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, and Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol, 191, 1141-1158. 66. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, and Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature, 441, 1157-1161. 77 67. Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, and Lavandero S (2008) Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res, 77, 387-397. 68. Perier C and Vila M (2012) Mitochondrial biology and Parkinson's disease. Cold Spring Harb Perspect Med, 2, a009332. 69. Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, and Downward J (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol, 9, 12431252. 70. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, and Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A, 105, 1638-1643. 71. Pridgeon JW, Olzmann JA, Chin LS, and Li L (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol, 5, e172. 72. Qi Z, Yang W, Liu Y, Cui T, Gao H, Duan C, Lu L, Zhao C, Zhao H, and Yang H (2011) Loss of PINK1 function decreases PP2A activity and promotes autophagy in dopaminergic cells and a murine model. Neurochem Int, 59, 572-581. 73. Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, and Mao P (2011) Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev, 67, 103-118. 74. Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, and Patenge N (2009) Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet, 18, 3832-3850. 75. Sanchez-Mora RM, Arboleda H, and Arboleda G (2012) PINK1 overexpression protects against C2-ceramide-induced CAD cell death through the PI3K/AKT pathway. J Mol Neurosci, 47, 582-594. 76. Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, 78 Cedazo-Minguez A, and Cookson MR (2009) Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One, 4, e5701. 77. Santel A and Frank S (2008) Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life, 60, 448-455. 78. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, and Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J, 26, 1749-1760. 79. Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, and Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet, 14, 3477-3492. 80. Skulachev VP (2001) Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci, 26, 23-29. 81. Stiles BL (2009) PI-3-K and AKT: Onto the mitochondria. Adv Drug Deliv Rev, 61, 1276-1282. 82. Stoica BA, Movsesyan VA, Lea PM, and Faden AI (2003) Ceramideinduced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrialdependent intrinsic caspase pathway. Mol Cell Neurosci, 22, 365-382. 83. Suri C, Fung BP, Tischler AS, and Chikaraishi DM (1993) Catecholaminergic cell lines from the brain and adrenal glands of tyrosine hydroxylase-SV40 T antigen transgenic mice. J Neurosci, 13, 1280-1291. 84. Tatsuta T and Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J, 27, 306-314. 85. Thiele CJ (1991) Patterns of regulation of nuclear proto-oncogenes MYCN and MYB in retinoic acid treated neuroblastoma cells. Prog Clin Biol Res, 366, 151-156. 79 86. Timmons S, Coakley MF, Moloney AM, and O' Neill C (2009) Akt signal transduction dysfunction in Parkinson's disease. Neurosci Lett, 467, 30-35. 87. Unoki M and Nakamura Y (2001) Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene, 20, 4457-4465. 88. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del TD, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, and Wood NW (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science, 304, 1158-1160. 89. Vives-Bauza C, Zhou C, Huang Y, Cui M, Tocilescu MA, Liu W, Ko HS, Magrane Grailhe R, Dawson TM, Li C, Tieu K, PINK1-dependent recruitment of Parkin to Proc Natl Acad Sci U S A, 107, 378-383. de Vries RL, Kim J, May J, J, Moore DJ, Dawson VL, and Przedborski S (2010) mitochondria in mitophagy. 90. Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, and Zhang Z (2006) Antioxidants protect PINK1dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci U S A, 103, 13520-13525. 91. Wang HL, Chou AH, Yeh TH, Li AH, Chen YL, Kuo YL, Tsai SR, and Yu ST (2007) PINK1 mutants associated with recessive Parkinson's disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol Dis, 28, 216-226. 92. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, and Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell, 147, 893-906. 93. Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, and Selkoe DJ (2009) Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry, 48, 2045-2052. 80 94. West AB, Kapatos G, O'Farrell C, Gonzalez-de-Chavez F, Chiu K, Farrer MJ, and Maidment NT (2004) N-myc regulates parkin expression. J Biol Chem, 279, 28896-28902. 95. Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, and Lu B (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A, 103, 10793-10798. 96. Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, and Nister M (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J, 30, 2762-2778. 97. Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, and Vance JM (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet, 36, 449451. 81