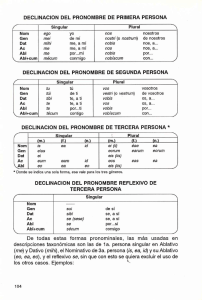
Detección por –PCR MULTIPLEX- de los transcritos
Anuncio

CONSEJO NACIONAL DE CIENCIA Y TECNOLOGIA -CONCYTSECRETARIA NACIONAL DE CIENCIA Y TECNOLOGIA -SENACYTFONDO NACIONAL DE CIENCIA Y TECNOLOGIA -FONACYTFUNDACIÓN ROZAS-BOTRÁN- INSTITUTO DE INVESTIGACIÓN EN ENFERMEDADES GENÉTICAS Y METABÓLICAS –INVEGEM- INFORME FINAL Detección por –PCR MULTIPLEX- de los transcritos quiméricos BCR-ABL, E2A-PBX1, MLL-AF4, ETV6-AML1; y su utilidad como factor pronóstico en pacientes pediátricos con Leucemia Linfoblástica Aguda PROYECTO FODECYT No. 48-2009 Dra. Claudia Carranza Investigador Principal GUATEMALA, NOVIEMBRE DEL 2011 AGRADECIMIENTOS: La realización de este trabajo, ha sido posible gracias al apoyo financiero dentro del Fondo Nacional de Ciencia y Tecnología, -FONACYT-, otorgado por la Secretaria Nacional de Ciencia y Tecnología –SENACYT- y al Consejo Nacional de Ciencia y Tecnología – CONCYT- . RESUMEN El objetivo general del proyecto Fodecyt 48-2009 es detectar los transcritos de fusión BCR-ABL, E2A-PBX1, MLL-AF4. ETV6-AML1 mediante PCR MULTIPLEX y determinar su utilidad en el establecimiento de un pronóstico. Se obtuvieron células mononucleares de muestras de médula ósea de 143 pacientes con diagnóstico de leucemia linfoblástica aguda, mediante el uso de ficoll. Se realizó la extracción de ARN y la retrotranscripción. Posteriormente se evalúo la calidad del cDNA amplificando el gen control ABL y por último se evalúo la presencia de los cuatro transcritos mediante PCR multiplex. Se ha encontrado diez pacientes positivos para distintos transcritos de BCRABL, que corresponde a un 7%, el cual concuerda con lo reportado internacionalmente. Y seis pacientes positivos ( 4.5%) para ETV6-AML1, que es un marcador de buen pronóstico, esta incidencia es bastante baja ya que en países desarrollados se reporta hasta un 20% de positividad. Y tres pacientes positivos (2%) para el transcrito E2A-PBX1. La presencia de BCR-ABL en leucemia linfoblástica aguda indica muy mal pronóstico, por lo que se ha procedido a informar inmediatamente a los médicos tratantes; así mismo se ha informado de que en estos pacientes es necesario utilizar esquemas de quimioterapias más agresivas, y de ser posible en combinación con Imatinib y transplante de médula ósea. La presencia de E2A-PBX1, también es un marcador de mal pronóstico, y que orienta a esquemas de quimioterapia más agresivas. Y por último la presencia de ETV6-AML1 es indica buen pronóstico. La baja incidencia del gen ETV6-AML1 encontrada en Guatemala, coincide con países como España e India, con los cuales la población guatemalteca comparte mayor cantidad de polimorfismos genéticos. En los datos demográficos, la mayoría de pacientes analizados proceden del occidente y centro de Guatemala; por lo que en un futuro proyecto se evaluarán los factores de riesgo presentes en ambas regiones. ABSTRACT The project goal is to detect the transcripts BCR-ABL, E2A-PBX1, MLLAF4. ETV6-AML1 by MULTIPLEX PCR and determine their usefulness in establishing prognosis. Mononuclear cells were obtained from bone marrow samples from 143 patients with acute lymphoblastic leukemia using ficoll. We performed RNA extraction and reverse transcription. Then assessed the quality of the cDNA by amplified ABL control gene and finally evaluated the presence of the four transcripts by multiplex PCR. Ten patients were found positive for different BCR-ABL transcripts, corresponding to 7%, which corresponds to the data reported internationally. And six patients positive (4.5%) for ETV6-AML1, which is a marker of good prognosis, this incidence is quite low as in developed countries is reported up to 20% positivity. And three patients positive (2%) for E2A-PBX1. The presence of BCR-ABL in acute lymphoblastic leukemia indicates very poor prognosis, so we have proceeded to immediately inform the treating physicians, likewise have been reported in these patients is necessary to use more aggressive chemotherapy regimen, and possibly in combination with imatinib and bone marrow transplantation. The presence of E2A-PBX1, is also a marker of poor prognosis, which directs more aggressive chemotherapy regimens. And finally the presence of ETV6-AML1 is indicates good prognosis. The low incidence of ETV6-AML1 gene found in Guatemala, coincides with countries like Spain and India, with which the Guatemalan population share more genetic polymorphisms. In demographics, the majority of patients analyzed are from the western and central Guatemala, so in a future project will assess the risk factors present in both regions. BIBLIOGRAFÍA BREVE ACADÉMICA DE AUTORES . 1. Dra. Claudia Carranza PhD. En el año 2004 se gradúo de Química Bióloga en la Universidad de San Carlos, Posteriormente en el período 2005-2007 cursó un programa de Doctorado en Biología Molecular y Celular, especializado en el área de Genética, en el Centro de Investigación Médica Aplicada –CIMA- , Universidad de Navarra, Pamplona España. En el año 2008, se incorpora en el Instituto de Investigación en enfermedades Genéticas y Métabolicas –INVEGEM-, Guatemala; y también se incorpora como catedrática titular del curso de Bioquímica Humana en la Facultad de Medicina, Universidad Francisco Marroquín. Su línea de investigación se ha centrado en Genética humana, Biología Molecular del Cáncer, Genética de neoplasias hematológicas. Actualmente se encuentra cursando un Máster en Bioética en la Universidad del Istmo, Guatemala ( 2011-2012). 2. Licda. Lilian Granados Pensum cerrado de la carrera de Química biológica, de la Facultad de Farmacia, Universidad San Carlos de Guatemala. Actualmente se encuentra laborando como investigadora en el Instituto de Investigación de Enfermedades Genéticas y Metabólicas –INVEGEM- . OTROS AGRADECIMIENTOS: La realización de este trabajo, ha sido posible gracias al: • Apoyo financiero y académico del Instituto de Investigación en Enfermedades Genéticas y Metabólicas –INVEGEM-. • Apoyo financiero de la Fundación Rozas-Botrán. • Cooperación de la Unidad de Oncología Pediátrica –UNOP• Cooperación del Dr. Mauricio Villegas, Dr. Cáceres y Dra. Silvana Torselli. TABLA DE CONTENIDOS: LISTA DE FIGURAS ……………………………………………………..….. LISTA DE TABLAS …………………………………………………………… v xvii RESUMEN ……………………………………………………………………... xix ABSTRACT…………………………………………………………………….. xx PARTE I: I.1 INTRODUCCIÓN ……………………………………………….... I.2 PLANTEAMIENTO DEL TEMA………………………………… I.2.1 Antecedentes en Guatemala………………………………………... I.2.2 Justificación del trabajo de investigación…………………………... I.3 OBJETIVOS ……………………………………………………….. I.3.1 Objetivos I.3.1.1 General……………………………………………………. I.3.1.2 Específicos………………………………………………… I.3.2 Hipótesis……………………………………………………………. I.4 METODOLOGÍA ………………………………………………….. I.4.1 Localización………………………………………………………… 1.4.2 Indicadores…………………………………………………………. I.4.3 Estrategia Metodológica…………………………………………… I.4.4 El Método…………………………………………………………… I.4.5 La técnica Estadística……………………………………………….. 1.4.6 Los instrumentos a utilizar………………………………………….. 4 4 5 6 6 6 6 7 12 12 PARTE II MARCO TEÓRICO ( CONCEPTUAL)……………………………………… 13 PARTE III III. RESULTADOS…………………………………………………………….. III.1 Discusión de Resultados……………………………………………. 48 80 PARTE IV IV.1 CONCLUSIONES………………………………………………………... IV.2 RECOMENDACIONES………………………………………………… IV.3 REFERENCIAS BIBLIOGRÁFICAS…………………………………. IV.4 ANEXOS…………………………………………………………………... 85 87 88 96 PARTE V V.1 INFORME FINANCIERO……………………………………………….. 166 1 2 2 3 4 LISTA DE FIGURAS PAG. Figura no. 1: Clasificación citomorfológica de leucemia aguda……………………. 18 Figura no. 2: Clasificación fab de la leucemia infantil……………………………… 18 Figura no. 3: Esquema de la t(9;22) mostrando la localización de los genes abl y bcr. 24 Figura no 4: El cromosoma 22 afectado por la translocación es el denominado cromosoma philadelphia…………………………………………………………….. 25 Figura no. 5: Representación esquemática de la fusión génica bcr-abl……………… 26 Figura no. 6: Localización de los puntos de rotura en los genes abl y bcr……………. 27 Figura no. 7: Probabilidad de recaídas de pacientes bcr-abl………………………….. 28 Figura no. 8: Curva de sobrevivientes libres de enfermedad con etv6-aml1 positivo.. 33 Figura no. 9: Representación esquemática del gen etv6, mostrando sus puntos de rotura 33 Figura no. 10: Representación esquemática de la fusión génica tel-aml1……………. 37 Figura no. 11: Curva de eventos sobrevivientes libres de enfermedad……………… 39 Figura no. 12 y13: Gen mll y su fusión con otros genes…………………………….. 41 Figura no. 14: Representación esquemática de la fusión génica e2a-pbx1………….. 44 Figura no. 15: Amplificación del gen control abl…………………………………… 48 Figura no. 16: Transcrito bcr-abl; pacientes 1 y 7…………………………………… 53 Figura no. 17: Transcrito bcr-abl; pacientes 24, 32 y 35……………………………. 53 Figura no. 18: Transcrito bcr-abl; pacientes 40,41 y 97…………………………….. 54 Figura no. 19: Transcrito bcr-abl; pacientes 121 y 123…………………………….. 54 Figura no. 20: Transcrito etv6-aml1; pacientes 27, 120 y 128……………………… 55 Figura no. 21: Transcrito etv6-aml1; pacientes 59…………………………………. 56 Figura no. 22: Transcrito e2a-pbx1; pacientes 20, 43 y 47………………………… 56 Figura no. 23: Resultado de alineación de secuencia de ADN obtenida, paciente no. 97 63 Figura no. 24: Cromosomas que contienen la secuencia analizada, paciente no. 97…. … 64 Figura no. 25: Alineación de la secuencia obtenida con el gen abl, paciente no. 97… 64 Figura no. 26: Alineación de la secuencia obtenida con el gen bcr, paciente no. 97… 65 Figura no. 27: Alineación de la secuencia obtenida, paciente no. 97 (2)……………. 66 Figura no. 28: Cromosomas que contienen la secuencia analizada, paciente no. 97 (2) 66 Figura no. 29: Alineación de la secuencia obtenida con el gen abl, paciente no. 97 (2) 67 LISTA DE FIGURAS PAG Figura no. 30: Alineación de la secuencia obtenida con el gen bcr, paciente no. 97 (2) 67 Figura no. 31: Alineación de la secuencia obtenida, paciente no. 43………………. 68 Figura no. 32: Cromosomas que contienen la secuencia analizada, paciente no. 43.. 68 .. Figura no. 33: Alineación de la secuencia obtenida con el gen pbx1, paciente no. 43 69 Figura no. 34: Alineación de la secuencia obtenida, paciente no. 43 (2)…………… 70 Figura no. 35: Cromosomas que contienen la secuencia analizada, paciente no. 43 (2) ... 70 Figura no. 36: Alineación de la secuencia obtenida con el gen e2a, paciente no. 43 (2) 71 Figura no. 37: Alineación de la secuencia obtenida con el gen pbx1, paciente no. 43 (2) 71 Figura no. 38: Alineación de la secuencia obtenida, paciente no. 40………………. 72 Figura no. 39: Cromosomas que contienen la secuencia analizada, paciente no. 40… 72 Figura no. 40: Alineación de la secuencia obtenida con el gen abl1, paciente no. 40 73 Figura no. 41: Alineación de la secuencia obtenida con el gen bcr, paciente no. 40 73 Figura no. 42: Alineación de la secuencia obtenida, paciente no. 40 (2)…………… 74 Figura no. 43: Cromosomas que contienen la secuencia analizada, paciente no. 40 (2) 74 Figura no. 44: Alineación de la secuencia obtenida con el gen abl, paciente no. 40 (2) 75 Figura no. 45: Alineación de la secuencia obtenida con el gen bcr, paciente no. 40 (2) 75 Figura no. 46: Alineación de la secuencia obtenida, paciente no. 41………………. 76 Figura no. 47: Cromosomas que contienen la secuencia analizada, paciente no. 41… 76 Figura no. 48: Alineación de la secuencia obtenida con el gen bcr, paciente no. 41 77 Figura no. 49: Alineación de la secuencia obtenida, paciente no. 41 (2)…………… 78 Figura no. 50: Cromosomas que contienen la secuencia analizada, paciente no. 41 (2) 78 Figura no. 51: Alineación de la secuencia obtenida con el gen abl, paciente no. 41 (2) 78 Figura no. 52: Alineación de la secuencia obtenida con el gen bcr, paciente no. 41 (2) 79 Figura no. 53: Líneas celulares k562, reh y mv4-11 en hielo seco…………………. 97 Figura no. 54: Medio de cultivo rpmi y suero fetal bovino………………………… 97 Figura no. 55: Cultivo de líneas celulares en el medio de cultivo rpmi……………. 98 Figura no. 56: Distintos cultivos de líneas celurares en incubación……………….. 98 Figura no. 57: Observación al microscopio de cultivos celulares…………………. 100 Figura no. 58: Líneas celulares observadas al microscopio……………………….. 100 Figura no. 59: Filtro utilizado para extracción de arnm…………………………... 101 LISTA DE FIGURAS PAG Figura no. 60: Procedimiento de retrotranscripción……………………………… 102 Figura no. 61: Separación de células mononucleares de médula ósea por medio de Ficoll……………………………………………………………………………… 103 Figura no. 62: Células mononucleares en interfase, después de separación con ficoll 103 Figura no. 63: Gen control abl, pacientes no.1 y 2………………………………. 106 Figura no. 64: Gen contrl abl, pacientes no. 3-7…………………………………. 106 Figura no. 65: Gen control abl, pacientes no. 8 al 14…………………………… 107 Figura no. 66: Gen control abl, pacientes no. 15 al 20………………………….. 107 Figura no. 67: Gen control abl, pacientes no. 21 al 24………………………….. 108 Figura no. 68: Gen control abl, pacientes no. 25 al 27………………………….. 108 Figura no. 69: Gen control abl, pacientes no. 27 y 28…………………………… 109 Figura no. 70: Gen control abl, pacientes no. 29 y 30…………………………… 109 Figura no. 71: Gen control abl, pacientes no. 31 al 36…………………………… 110 Figura no. 72: Gen control abl, pacientes no. 37 al 39…………………………… 110 Figura no. 73: Gen control abl, pacientes no. 40 al 43…………………………… 111 Figura no. 74: Gen control abl, pacientes no. 44 al 48…………………………… 111 Figura no. 75: Gen control abl, pacientes no. 49 al 51…………………………… 112 Figura no. 76: Gen control abl, pacientes no. 52 al 54…………………………… 112 Figura no. 77: Gen control abl, pacientes no. 53 y 54……………………………. 113 Figura no. 78: Gen control abl, pacientes no. 56 al 63……………………………. 113 Figura no. 79: Gen control abl, pacientes no. 65 al 73…………………………… 114 Figura no. 80: Gen control abl, pacientes no. 8 al 14……………………………. 114 Figura no. 81: Gen control abl, pacientes no. 72 al 75…………………………… 115 Figura no. 82: Gen control abl, pacientes no. 75 al 80…………………………… 115 Figura no. 83: Gen control abl, pacientes no. 81 al 85…………………………… 116 Figura no. 84: Gen control abl, pacientes no. 84,86 y 89………………………… 116 Figura no. 85: Gen control abl, pacientes no. 88 y 93……………………………. 117 Figura no. 86: Gen control abl, pacientes no. 90,92 y 95………………………… 117 Figura no. 87: Gen control abl, pacientes no. 90, 94 y 99………………………… 118 LISTA DE FIGURAS PAG Figura no. 88: Gen control abl, pacientes no. 101-104……………………………. 118 Figura no. 89: Gen control abl, pacientes no. 88, 96, 98 y 102…………………… 119 Figura no. 90: Gen control abl, pacientes no. 97 y 100…………………………… 119 Figura no. 91: Gen control abl, pacientes no. 105-108…………………………… 120 Figura no. 92: Gen control abl, pacientes no. 105, 107 y 109……………………. 120 Figura no. 93: Gen control abl, pacientes no. 110 al 114………………………… 121 Figura no. 94: Gen control abl, paciente 115……………………………………… 121 Figura no. 95: Gen control abl, pacientes no. 116 al 119…………………………. 122 Figura no. 96: Gen control abl, paciente no. 118…………………………………. 122 Figura no. 97: Gen control abl, pacientes no. 120 al 125…………………………. 123 Figura no. 98: Gen control abl, paciente no. 126…………………………………. 123 Figura no. 99: Gen control abl, paciente no. 127…………………………………. 124 Figura no. 100: Gen control abl, pacientes no. 128 al 132………………………... 124 Figura no. 101: Gen control abl, pacientes no. 133 al 138………………………… 125 Figura no. 102: Gen control abl, pacientes no. 139 al 142………………………… 125 Figura no. 103: Transcrito e2a-pbx1, pacientes no. 3 al 7………………………… 126 Figura no. 104: Transcrito e2a-pbx1, pacientes no. 15 al 20……………………… 126 Figura no. 105: Transcrito e2a-pbx1, pacientes no. 21 al 27……………………… 127 Figura no. 106: Transcrito e2a-pbx1, etv6-aml1 y mll-af4 pacientes no. 28 al 30 127 Figura no. 107: Transcrito e2a-pbx1, pacientes no. 30 al 36……………………… 128 Figura no. 108: Transcrito e2a-pbx1, pacientes no. 37 al 43……………………… 128 Figura no. 109: Transcrito e2a-pbx1, paciente no. 43…………………………….. 129 Figura no. 110: Transcrito e2a-pbx1, pacientes no. 39 al 48……………………… 129 Figura no. 111: Transcrito e2a-pbx1, paciente no. 47…………………………….. 130 Figura no. 112: Transcrito e2a-pbx1, pacientes no. 49 al 54……………………… 130 Figura no. 113: Transcrito e2a-pbx1, pacientes no. 57 al 64……………………… 131 Figura no. 114: Transcrito e2a-pbx1, pacientes no. 53, 55, 57, 59, 61 y 62……… 131 Figura no. 115: Transcrito e2a-pbx1, pacientes no. 60,63 y 65………………….. 132 Figura no. 116: Transcrito e2a-pbx1, pacientes no. 65 al 74…………………….. 132 Figura no. 117: Transcrito etv6-aml1, pacientes no. 3 al 7………………………. 133 LISTA DE FIGURAS PAG Figura no. 118: Transcrito e2a-pbx1 y etv6-aml1, pacientes no. 8 al 14……. 134 Figura no. 119: Transcrito etv6-aml1, pacientes no. 15 al 20………………… 135 Figura no. 120: Transcrito etv6-aml1, pacientes no. 21 al 27………………… 135 Figura no. 121: Transcrito etv6-aml1, pacientes no. 31 al 36………………… 136 Figura no. 122: Transcrito etv6-aml1, pacientes no. 37 al 43………………… 136 Figura no. 123: Transcrito etv6-aml1, pacientes no. 39 al 48………………… 137 Figura no. 124: Transcrito etv6-aml1, pacientes no. 49 al 51………………… 137 Figura no. 125: Transcrito etv6-aml1, pacientes no. 57 al 64………………… 138 Figura no. 126: Transcrito etv6-aml1, pacientes no. 53 al 62………………… 138 Figura no. 127: Transcrito etv6-aml1, pacientes no. 52 y 54………………… 139 Figura no. 128: Transcrito etv6-aml1, pacientes no. 65 al 74………………… 139 Figura no. 129: Transcrito bcr-abl, pacientes no. 1 y 2………………………. 140 Figura no. 130: Transcrito bcr-abl, pacientes no. 3 al 7……………………… 140 Figura no. 131: Transcrito bcr-abl, pacientes no. 8 al 14…………………….. 141 Figura no. 132: Transcrito bcr-abl, pacientes no. 15 al 22…………………… 141 Figura no. 133: Transcrito bcr-abl, pacientes no. 23 al 27…………………… 142 Figura no. 134: Transcrito bcr-abl, pacientes no. 27 al 30…………………… 142 Figura no. 135: Transcrito bcr-abl, pacientes no. 31 al 36…………………… 143 Figura no. 136: Transcrito bcr-abl, pacientes no. 37 al 43…………………… 143 Figura no. 137: Transcrito bcr-abl, pacientes no. 40 y 41…………………… 144 Figura no. 138: Transcrito bcr-abl, pacientes no. 39 al 48…………………… 144 Figura no. 139: Transcrito bcr-abl, pacientes no. 49 al 51…………………… 145 Figura no. 140: Transcrito bcr-abl, pacientes no. 57 al 64…………………... 145 Figura no. 141: Transcrito bcr-abl, pacientes no. 60, 63 y 65……………….. 146 Figura no. 142: Transcrito bcr-abl, pacientes no. 53, 55, 59, 61 y 62………. 146 Figura no. 143: Transcrito bcr-abl, pacientes no. 52 y 54…………………… 147 Figura no. 144: Transcrito bcr-abl, pacientes no. 65 al 74………………….. 147 Figura no. 145: Transcrito mll-af4, pacientes no. 3 al 7…………………….. 148 Figura no. 146: Transcrito mll-af4, pacientes no. 8 al 14…………………… 148 Figura no. 147: Transcrito mll-af4, pacientes no. 15 al 20………………….. 149 LISTA DE FIGURAS PAG Figura no. 148: Transcrito mll-af4, pacientes no. 21 al 27………………………. 149 Figura no. 149: Transcrito mll-af4, pacientes no. 31 al 36………………………. 150 Figura no. 150: Transcrito mll-af4, pacientes no. 37 al 43………………………. 150 Figura no. 151: Transcrito mll-af4, pacientes no. 39 al 48……………………… 151 Figura no. 152: Transcrito mll-af4, pacientes no. 49 al 51………………………. 151 Figura no. 153: Transcrito mll-af4, pacientes no. 52 y 54……………………….. 152 Figura no. 154: Transcrito mll-af4, pacientes no. 53 al 62………………………. 152 Figura no. 155: Transcrito mll-af4, pacientes no. 57 al 64………………………. 153 Figura no. 156: Transcrito mll-af4, pacientes no. 65 al 74………………………. 153 Figura no. 157: Transcrito bcr-abl y ee2-pbx1, pacientes no.72 al 75…………… 154 Figura no. 158: Transcrito etv6-aml1 y mll-af4, pacientes del 72 al 75…………. 154 Figura 158: Todos los transcritos, pacientes del 76 al 80………………………… 155 Figura no. 159: Todos los transcritos, pacientes del 81 al 85……………………. 155 Figura no. 160 : Transcrito bcr-abl, e2a-pbx1, pacientes del 90-96, 99 y 100….. 156 Figura no. 161: Todos los transcritos, paciente 102……………………………… 156 Figura no. 162: Todos los transcritos, pacientes 84, 86 y 89…………………….. 157 Figura no. 163: Transcrito bcr-abl; pacientes 88, 101 al 104…………………….. 157 Figura no. 164: Todos los transcritos, pacientes 105 al 109……………………… 158 Figura no. 165: Transcrito bcr-abl, e2a-pbx1, pacientes 115 al 119……………… 158 Figura no. 166: Todos los transcritos; pacientes no. 110 al 114………………….. 159 Figura no. 167: Transcrito bcr-abl y e2a-pbx1, pacientes del 120 al 132………… 159 Figura no. 168: Todos los transcritos, pacientes 121, 123 ,125 y 126……………. 160 Figura no. 169: Transcrito bcr-abl, pacientes no. 133 al 142…………………….. 160 Figura no. 170: Transcrito e2a-pbx1 y etv6-aml1, pacientes 133 a 142………….. 161 Figura no. 171: Transcritos bcr-abl y etv6-aml1, pacientes 121, 123, 120 y 128… 161 Figura no. 172: Transcritos etv6-aml1 y mll-af4, pacientes 115 al 119…………… 162 Figura no. 173: Transcrito etv6-aml1, pacientes 120, 123, 124, 127-131………… 162 Figura no. 174: Transcrito mll-af4, pacientes 133 al 142…………………………. 163 LISTA DE TABLAS • PAG. Tabla no. 1: Peso molecular de transcritos estudiados………………….. 10 • Tabla 2: Anormalidades genéticas en leucemia linfoblástica aguda. …... 23 • Tabla no. 3: características con valor en el pronóstico de leucemia linfoblástica aguda……………………………………………………….. • 47 Tabla no. 4: Resultados del análisis de los cuatro transcritos en todos los Pacientes…………………………………………………………………. 49 • Tabla no. 5: Incidencia de cada unos de los transcritos …………………. 52 • Tabla no. 6: Pronóstico y orientación a tratamiento de los transcritos encontrados en los pacientes…………………………………………….. 57 • Tabla no. 7: Pacientes con la distribuidos por municipio de Guatemala… 58 • Tabla no 8: Cantidad de pacientes organizados por región……………… 62 • Tabla no. 9: Frecuencia de etv6-aml1 en distintos países……………….. 82 • Tabla no. 10: Cantidad de células de cada línea celular por ml…………. 99 • Tabla no. 11: Concentración del ADNc…………………………………. 102 • Tabla no. 12: Datos clínicos de pacientes analizados para las cuatro translocaciones…………………………………………... 165 RESUMEN El objetivo general del proyecto Fodecyt 48-2009 es detectar los transcritos de fusión BCR-ABL, E2A-PBX1, MLL-AF4. ETV6-AML1 mediante PCR MULTIPLEX y determinar su utilidad en el establecimiento de un pronóstico. Se obtuvieron células mononucleares de muestras de médula ósea de 143 pacientes con diagnóstico de leucemia linfoblástica aguda, mediante el uso de ficoll. Se realizó la extracción de ARN y la retrotranscripción. Posteriormente se evalúo la calidad del cDNA amplificando el gen control ABL y por último se evalúo la presencia de los cuatro transcritos mediante PCR multiplex. Se ha encontrado diez pacientes positivos para distintos transcritos de BCRABL, que corresponde a un 7%, el cual concuerda con lo reportado internacionalmente. Y seis pacientes positivos ( 4.5%) para ETV6-AML1, que es un marcador de buen pronóstico, esta incidencia es bastante baja ya que en países desarrollados se reporta hasta un 20% de positividad. Y tres pacientes positivos (2%) para el transcrito E2A-PBX1. La presencia de BCR-ABL en leucemia linfoblástica aguda indica muy mal pronóstico, por lo que se ha procedido a informar inmediatamente a los médicos tratantes; así mismo se ha informado de que en estos pacientes es necesario utilizar esquemas de quimioterapias más agresivas, y de ser posible en combinación con Imatinib y transplante de médula ósea. La presencia de E2A-PBX1, también es un marcador de mal pronóstico, y que orienta a esquemas de quimioterapia más agresivas. Y por último la presencia de ETV6-AML1 es indica buen pronóstico. La baja incidencia del gen ETV6-AML1 encontrada en Guatemala, coincide con países como España e India, con los cuales la población guatemalteca comparte mayor cantidad de polimorfismos genéticos. En los datos demográficos, la mayoría de pacientes analizados proceden del occidente y centro de Guatemala; por lo que en un futuro proyecto se evaluarán los factores de riesgo presentes en ambas regiones. ABSTRACT The project goal is to detect the transcripts BCR-ABL, E2A-PBX1, MLLAF4. ETV6-AML1 by MULTIPLEX PCR and determine their usefulness in establishing prognosis. Mononuclear cells were obtained from bone marrow samples from 143 patients with acute lymphoblastic leukemia using ficoll. We performed RNA extraction and reverse transcription. Then assessed the quality of the cDNA by amplified ABL control gene and finally evaluated the presence of the four transcripts by multiplex PCR. Ten patients were found positive for different BCR-ABL transcripts, corresponding to 7%, which corresponds to the data reported internationally. And six patients positive (4.5%) for ETV6-AML1, which is a marker of good prognosis, this incidence is quite low as in developed countries is reported up to 20% positivity. And three patients positive (2%) for E2A-PBX1. The presence of BCR-ABL in acute lymphoblastic leukemia indicates very poor prognosis, so we have proceeded to immediately inform the treating physicians, likewise have been reported in these patients is necessary to use more aggressive chemotherapy regimen, and possibly in combination with imatinib and bone marrow transplantation. The presence of E2A-PBX1, is also a marker of poor prognosis, which directs more aggressive chemotherapy regimens. And finally the presence of ETV6-AML1 is indicates good prognosis. The low incidence of ETV6-AML1 gene found in Guatemala, coincides with countries like Spain and India, with which the Guatemalan population share more genetic polymorphisms. In demographics, the majority of patients analyzed are from the western and central Guatemala, so in a future project will assess the risk factors present in both regions. PARTE I I.1 INTRODUCCIÓN La biología molecular es una herramienta útil para la genética humana, pues permite el diagnóstico de una gran variedad de enfermedades de origen genético. Entre las enfermedades de origen genético se pueden mencionar las constitucionales como el síndrome de Down, y las adquiridas como los distintos tipos de neoplasias. Actualmente las neoplasias se consideran de origen genético, debido a que se han encontrado una gran infinidad de alteraciones en cromosomas, que dan origen a numerosos procesos alterados, como la síntesis de proteínas irregulares, o que causan sobreexpresión o silenciamiento génico. Estos procesos genéticos alterados causan una disfunción en algún proceso celular esencial; produciendo un desequilibrio entre el crecimiento celular, la diferenciación celular y la apoptosis. [1-3] La descripción de alteraciones cromosómicas asociadas específicamente a un tipo de cáncer, y su posterior seguimiento, ha permitido conocer la significación clínica de muchos marcadores genéticos y, más importante todavía, se ha constituido como un paso previo a la detección y localización de los genes implicados en la génesis tumoral. [1-3] Las técnicas genéticas que poseen actualmente mayor aplicación y que se han incorporado como técnicas de rutina en una gran cantidad de laboratorios clínicos son el cariotipo, la citogenética molecular y la biología molecular. Estas técnicas proporcionan amplia información sobre el diagnóstico, pronóstico y el tipo de terapia a seguir en los pacientes con neoplasias hematológicas. [1-3] En el presente estudio, se utilizó la técnica de PCR MULTIPLEX, para determinar la presencia de los transcritos BCR-ABL, E2A-PBX1, MLL-AF4 y ETV6-AML1; estos cuatro genes de fusión brindan información sobre el pronóstico y la terapia a elegir en pacientes pediátricos con leucemia linfoblástica aguda. Esta determinación brindará de gran apoyo al clínico hematólogo para el diagnóstico y seguimiento de la leucemia linfoblástica aguda. I.2 PLANTEAMIENTO DEL PROBLEMA (Antecedentes y Justificación del trabajo de investigación) I.2.1 Antecedentes (Trabajo, experiencias en Guatemala) En Guatemala, la genética humana se encuentra desarrollada pobremente. Actualmente no existen publicaciones guatemaltecas que involucren estudios de genética en humanos. La biología molecular es una herramienta útil para el área de genética humana. Los estudios sobre enfermedades de origen genético como las neoplasias hematológicas poseen un alto impacto en países en vías de desarrollo como Guatemala. Pues gracias a estos estudios se puede apoyar en diagnóstico y pronóstico al clínico hematólogo. Y por otro lado también es posible la caracterización genética de nuestra población en distintas enfermedades. El único estudio epidemiológico sobre leucemias infantiles realizado en Centroamérica, incluyendo México, fue el realizado por Arangúre y colaboradores; en donde se incluyeron datos de pacientes desde 1996 hasta el 2000. El promedio anual de incidencia obtenido fue de: 44.9 por millón de niños para leucemia linfoblástica aguda – LLA-( el principal tipo de leucemia infantil). [4] A nivel mundial, principalmente en países desarrollados se han implementado como técnicas de rutina, el análisis de la presencia de distintos marcadores genéticos para LLA como los transcritos de fusión BCR-ABL, E2A-PBX1, MLL-AF4 y ETV6-AML1; que proporcionan información importante acerca del pronóstico y la terapia a escoger. Cerveira y colaboradores en el año 2000; Pallisgaard y colaboradores en el año 1998, Scurto y colaboradores en el año 1998; y Marin Carolina en el año 2001; han desarrollado distintas técnicas de PCR Múltiplex para el análisis de algunos de estos transcritos; indicando todos la importancia de la detección en una sola prueba de estas translocaciones. Así como también todos resaltan su utilidad para el establecimiento de un pronóstico adecuado a cada paciente. [5-9] Se ha tenido experiencia en la caracterización genética de neoplasias hematológicas mediante la utilización de distintas técnicas moleculares y de citogenética, principalmente en en la búsqueda de nuevos mecanismos que expliquen la sobreexpresión del gen EVI1, un gen importante en leucemias mieloides agudas. [10] I.2.2 Justificación del trabajo de investigación El diagnóstico de leucemia linfoblástica aguda (ALL), se realiza en primer término con un recuento periférico de glóbulos blancos alterado, y posteriormente mediante citomorfología, empezando con un análisis de frote periférico en busca de presencia de blastos (células hematopoyéticas inmaduras), para luego proceder a la realización de un medulograma. [1-3] El examen completo de un medulograma debe constar de las siguientes partes: recuento globular, análisis morfológico de frote, inmunofenotipo, citoquímica, citogenética y biología molecular; siendo estos dos últimos los que proporcionan una información completa acerca del diagnóstico, pronóstico y tipo de terapia a seguir. En Guatemala, el análisis de médula ósea, se realiza mediante un recuento globular y análisis citomorfológico principalmente. Ambas técnicas permiten el diagnóstico y la clasificación de una leucemia, según la clasificación FAB (FRENCH, AMERICAN AND BRITISH); para las leucemias mieloides agudas en M0-M7, y para las leucemias linfoblásticas agudas en L1/L2/L3; esta clasificación se basa en el tipo celular encontrado y el grado de diferenciación que posee. En algunos casos es posible realizar algunas técnicas citoquímicas y establecer el inmunofenotipo del paciente; ayudando estas técnicas a establecer un diagnóstico más certero. Sin embargo estas técnicas son insuficientes para proporcionar información acerca del pronóstico del paciente y del tipo de terapia a elegir. [1-3] La biología molecular permite la detección de marcadores genéticos, que generalmente proporcionan información más definitiva sobre el diagnóstico del paciente. Y además dependiendo del tipo de marcador genético encontrado es posible obtener amplia información sobre el pronóstico que posee el paciente así como establecer una terapia más adecuada. I.3 OBJETIVOS E HIPOTESIS I.3.1 Objetivos I.3.1.1 General Detectar por PCR MULTIPLEX, los transcritos de quiméricos BCRABL, E2A-PBX1, MLL-AF4 y ETV6-AML1 y su utilidad como factor pronóstico en pacientes pediátricos con Leucemia Linfoblástica aguda. I.3.1.2 Específicos • Detectar los transcritos de fusión BCR-ABL, E2A-PBX1, MLL-AF4 y ETV6AML1 mediante PCR MULTIPLEX y determinar su utilidad en el establecimiento de pronóstico. • Estandarizar la técnica de PCR MULTIPLEX para analizar los transcritos BCR-ABL, E2A-PBX1, MLL-AF4 y ETV6-AML1 en una sola prueba de laboratorio. • Analizar y evaluar la presencia de los distintos transcritos en los pacientes pediátricos con leucemia linfoblástica aguda. • Establecer y evaluar un pronóstico en los pacientes pediátricos con leucemia linfoblástica aguda, de acuerdo al tipo de marcador encontrado. • Orientar al médico sobre el tipo de terapia a seguir, según los marcadores genéticos encontrados. • Determinar y caracterizar genéticamente la población guatemalteca infantil con leucemia linfoblástica aguda. • Divulgar a las autoridades, actores sociales e instituciones en el campo de su competencia la información de la investigación obtenida. I.3.2 Hipótesis Estudios realizados en otros países han comprobado la eficacia de las técnicas de análisis molecular en el diagnóstico y pronóstico de pacientes con neoplasias hematológicas principalmente, y en algunos casos en tumores sólidos. [1-3. 5-9] Nuestra hipótesis es que la detección en una sola prueba (PCR MULTIPLEX) de los transcritos de fusión: BCR-ABL, E2A-PBX1, MLL-AF4 y ETV6-AML1 proporcionará información definitiva acerca del diagnóstico del paciente; así como permitirá conocer el pronóstico de cada paciente analizado, según el tipo de marcador genético encontrado; y por último se podrá orientar hacia el tipo de tratamiento recomendado al paciente, según el perfil genético del mismo. Además se ahorrará tiempo, pues en una sola prueba se detectarán los cuatro transcritos de fusión. I.4 METODOLOGIA (Descripción detallada de la Metodología) I.4.1 Localización El presente estudio, analiza pacientes con leucemia linfoblástica aguda provenientes de todos los departamentos de Guatemala; y que han sido referidos al Hospital San Juan de Dios, Hospital Roosevelt y Unidad de Oncología Pediátrica. I.4.2: Indicadores. Las positividad de los cuatro transcritos analizados se observará mediante un gel de electroforesis, que contendrá el producto de PCR. Las bandas en el peso buscado, corresponderán a la presencia del transcrito. I.4.3.Estrategia metodológica El presente estudio es un proyecto descriptivo longitudinal y tranversal; se identificará la presencia de los marcadores analizados en pacientes con leucemia linfoblástica aguda; y se brindará la información de pronóstico y tratamiento a los pacientes, según el marcador encontrado. No se aplicará ningún diseño estadístico debido a que es un estudio descriptivo. I.4.3.1 Población y Muestra POBLACIÓN La población fue de 143 pacientes pediátricos con diagnóstico de leucemia linfoblástica aguda originarios de Guatemala. CRITERIOS DE INCLUSIÓN • Paciente con diagnóstico de leucemia linfoblástica aguda • 0 a 18 años CRITERIOS DE EXCLUSIÓN • Pacientes mayores de 18 años • Pacientes con diagnóstico de otra neoplasia MUESTRA: Todos los pacientes de 0 a 18 años referidos por los Hospitales Nacionales y la Unidad de Oncología pediátrica durante el periodo 2010-2011. I.4.4 El Método I.4.4.1 MATERIALES Y REACTIVOS [5-10]: A. Médula ósea de pacientes pediátricos con leucemia linfoblástica aguda. B. Kit para extracción de linfocitos de sangre periférica o médula ósea (lynphoprep). C. Kit de extracción de RNA ( Quiagen) D. Reactivos para retrotranscripción: • Tubos eppendorf 1.5 ml RNAsa free • H2O DEPC • Hexámeros • dNTPs • Buffer 5X • DTT • RNASE out • enzima SuperScript III. E. PCR calidad cDNA • Cebadores del gen ABL y de la beta microglobulina. • Buffer I (10x) • Mg+2(25mM) • dNTPs (10mM) • Taq Gold (5unids/µl) • H2O • Termociclador • Hielo • Tubos eppendorf de 1.5 y 0.5 • vórtex ultrapura F. PCR MULTIPLEX • Cebadores de los transcritos BCR-ABL, TEL-AML1, E2A-PBX1, MLL-AF4 (Lee, et al 1995, Cerveira, et al. 2000, Kwong and wong, 1997, Poteat et al. 1997, Hunger et. Al. 1991) • Buffer I (10x) • Mg+2(25mM) • dNTPs (10mM) • Taq Gold (5unids/µl) • H2O • Hielo • Tubos eppendorf de 1.5 y 0.5 • Agarosa • Buffer TBE ultrapura I.4.4.2 Metodología ( Ver Anexo No.1 ) La metodología utilizada se ha obtenido de estudios realizados por Cerveira y colaboradores en el año 2000; Pallisgaard y colaboradores en el año 1998, Scurto y colaboradores en el año 1998; Marin Carolina en el año 2001 y C. Carranza en el año 2007. [5-10] Se recolectaron muestras de médula ósea con EDTA, de pacientes pediátricos (0-18 años) cuyo diagnóstico sea Leucemia Linfoblástica aguda, durante el período de un año y medio correspondientes a los años 2010-2011. Las muestras analizadas provienen de pacientes referidos de distintos hospitales como: Hospital Roosevelt, Hospital General San Juan de Dios y la Unidad de Oncología Pediátrica –UNOP-. Todos los pacientes firmaron un consentimiento informado y un asentimiento si los niños podían darlo. [1-3] La técnica utilizada para la extracción linfocitos es la utilización de ficoll (lymphoprep), que permite la separación celular de acuerdo a un gradiente de densidad. Los linfocitos obtenidos mediante esta técnica se almacenaron a -20 grados centígrados, para posteriormente extraer el ARN. El RNA total se extrajo utilizando el kit RNAquous de APPLIED BIOSYSTEMS, en la cual se siguieron los procedimientos descritos por el fabricante. Para la síntesis de cDNA se utilizará 1µg de RNA, utilizando el kit cDNA high capacity Transcription de APPLIED BIOSYSTEMS, con hexámeros al azar en 20µl de reacción en condiciones estándar. Para comprobar la calidad del c-DNA obtenido, se utilizará una PCR. Amplificando el gen control ABL, las condiciones de la PCR fueron estandarizadas según el termociclador utilizado. La PCR MULTIPLEX para la evaluación de los transcritos de fusión BCR-ABL, TEL-AML1, E2A-PBX1 y MLL-AF4, se estandarizó según el termociclador utilizado. Los cebadores son los descritos por Lee, et al 1995, Cerveira, et al. 2000, Kwong and wong, 1997, Poteat et al. 1997 y Hunger et. Al. 1991. Las condiciones de PCR son 6 minutos a 94 grados, 40 ciclos de 1 minuto a 94, 1 minuto a 60 grados y 1 minuto a 72 grados centígrado. Y por último una extensión final de 5 minutos a 72 grados centígrados. Se evaluaron los resultados según la tabla No. 1. TABLA No. 1: PESO MOLECULAR DE TRANSCRITOS ESTUDIADOS Translocación transcrito Peso molecular (bp) t(9;22) p210 (b3:a2) 304 t(9;22) p210 (b2:a2) 234 t(9;22) p190 (e1:a2) 196 t(12;21) TEL-AML1 226 Y 265 t(4;11) MLL-AF4 161 Y 188 t(1;19) E2A-PBX1 370 Fuente: FODECYT 48-2009 Para la estandarización de la PCR, se utilizaron las líneas celulares K-562, MV4-11 y REH, las cuales son positivas para los transcritos a analizar. Primero fueron sometidas a cultivo celular; y cuando se obtuvo una cantidad considerable de estas células se procedió a extraer el ARN, y se utilizaron como controles positivos, para estandarizar las condiciones de PCR. Y por último se secuenciaron tres pacientes positivos para BCR-ABL y uno para E2APBX1, esta secuenciación se realizó en MACROGEN, en USA. Esto con el fin de validar completamente la técnica estandarizada F. ANALISIS DE RESULTADOS: La recopilación de información del paciente y de los exámenes realizados se realizará siguiente formato: mediante el uso de una boleta de recolección con el INVEGEM BOLETA DE RECOLECCION DE DATOS IDENTIFICACION DEL PACIENTE Iniciales de paciente No. de Historia Clínica Código del paciente Fecha de toma de muestra DATOS SOCIODEMOGRÁFICOS Edad Masculino Género Femenino Lugar de Nacimiento Antecedentes clínicos familiares INFORMACION CLINICA Fecha de diagnostico Clasificación de leucemias según FAB Estado de la enfermedad TRATAMIENTO (indicar si ha recibido algún tratamiento y el tiempo) DATOS HEMATOLOGICOS ( -2010) -- LLA Nuevo Seguimiento Remisión Recuento de Leucocitos: Recuento de Plaquetas: Porcentaje de blastos en sangre periférica Nivel de Hemoglobina: BCR-ABL, t(9;22)(q34;q11) ETV6-AML1, MARCADORES GENETICOS Recaída t(12;21)(p13;q22) E2A-PBX1, t(1;19)(q23;p13) MLL-AF4, t(4;11)(q21;q23) I.4.5 La técnica estadística Al ser un estudio descriptivo no se utilizará ninguna técnica estadística para el análisis de datos. Las incidencias se obtuvieron, por medio del cálculo del porcentaje de pacientes con transcritos positivos en su relación con el total de pacientes analizados. I.4.6 Los instrumentos a utilizar • Termocicladores • Vórtex • Fuente de Voltaje • Transiluminador • Cámara de electroforesis • Pipetas • Congeladores • Centrífugas, • Computadoras • Impresoras. PARTE II MARCO TEÓRICO (CONCEPTUAL) A. GENERALIDADES Una de las técnicas empleadas para la búsqueda de translocaciones u otras reordenaciones frecuentes en neoplasias hematológicas y algunos tumores sólidos son las técnicas de análisis molecular. Estas técnicas se emplean tanto para diagnóstico como para monitorización del paciente. La técnica de análisis molecular más utilizada es la reacción en cadena de la polimerasa –PCR-. Esta técnica es eficiente siempre y cuando se estudien genes de tamaño pequeño con un punto de rotura definido (1-3). La caracterización molecular de las translocaciones cromosómicas observadas en el cariotipo o mediante citogenética molecular y asociadas a determinados tipos de tumor, ha permitido la identificación de los genes responsables. La posterior clonación y secuenciación de dichos oncogenes permiten el estudio de las translocaciones a nivel molecular, mediante análisis de ADN o fundamentalmente ARN con la técnica de RT-PCR (PCR reversa). Un ejemplo característico es analizar la reordenación BCR-ABL asociada a leucemia mieloide crónica. Actualmente, su uso es necesario como complemento al cariotipo o al FISH convencional en el diagnóstico y en la monitorización de paciente con neoplasias (1,3). B. NOMENCLATURA GENÉTICA La nomenclatura genética está basada en el consenso alcanzado tras varias conferencias internacionales. El documento más reciente y autorizado es ISCN (1995): International System for Human Cytogenetic Nomenclature, Mitelman F. (ed), Basel 1995 (2,3). Se denomina clon a la población celular derivada de una única célula progenitora. Se acepta internacionalmente que existe un clon anómalo cuando se encuentran dos o más células con la misma alteración estructural o con un cromosoma supernumerario. Si la alteración es la pérdida de un cromosoma, el mismo cambio se tiene que observar al menos en tres mitosis. Sólo es necesaria la presencia de una célula normal para definir la existencia de un clon normal. El término aneuploide indica que el número de cromosomas se desvía del número diploide (46 cromosomas). Puede ser hiperdiploide (más de 46 cromosomas) o hipodiploide (menos de 46 cromosomas). Un cariotipo con un número de cromosomas diploide, pero que es anómalo debido a la presencia de alteraciones estructurales, se denomina pseudodiploide (1-3). A continuación vamos a hacer algunas aclaraciones sobre la nomenclatura empleada para describir un cariotipo. Se refieren tanto a alteraciones constitucionales como adquiridas. Primero se especifica el número de cromosomas y se indican los cromosomas sexuales: 46,XY (varón, cariotipo normal), 46,XX (mujer, cariotipo normal). A continuación se describen las alteraciones, utilizando las abreviaturas apropiadas: del (deleción), dup (duplicación), inv (inversión), ins (inserción), t (translocación), i (isocromosoma), r (ring, cromosoma en anillo). Después se indican los cromosomas y bandas implicadas. Por ejemplo: 46,XY,t(9;22)(q34;q11) (varón con una translocación recíproca entre los cromosomas 9 y 22, los puntos de rotura están en 9q34 y 22q11). Las ganancias o pérdidas de cromosomas completos se indican con los signos + ó antes del número del cromosoma implicado. Por ejemplo: 47,XX,+4 (mujer con una trisomía del cromosoma 4). Las alteraciones numéricas se representan a partir del número indicado al principio: 47, XX,+21 45,XY,-5,-7,+8 Las alteraciones constitucionales se representan con una c minúscula junto con la alteración cromosómica. Por ejemplo: 48,XX,+8,+21c (mujer con síndrome de Down, que presenta además una trisomía del cromosoma 8) (1-3). C. NEOPLASIAS HEMATÓLOGICAS Las leucemias agudas son un grupo heterogéneo de enfermedades. Los distintos subtipos son caracterizados por los rasgos clínicos y los datos de laboratorio encontrados. Se caracterizan por mal funcionamiento en las células progenitoras hematopoyéticas. Para un diagnóstico adecuado de las leucemias agudas es necesario el seguimiento de un protocolo que combine la citomorfología y citoquímica con el inmunofenotipo, y siempre que sea posible acompañado por métodos citogenéticos y moleculares (1-20). Actualmente las técnicas citogenéticas y moleculares no solo permiten un diagnóstico certero, sino que además proporcionan amplia información sobre el pronóstico del paciente, así como del tratamiento a seguir. Igualmente poseen gran utilidad para el monitoreo de la respuesta de los pacientes al tratamiento seleccionado (25-39). El diagnóstico de leucemias empieza por el análisis morfológico de las células hematopoyéticas en frotes de sangre periférica y de médula ósea. Dicho análisis permite la clasificación de las leucemias mieloides en los diferentes subtipos establecidos por la FAB ( M0-M7) y proporciona información importante para la clasificación y estratificación de las leucemias linfoblásticas. (1-3) El cariotipo es una técnica de citogenética convencional, que en la actualidad proporciona información acerca del pronóstico al paciente. (1-3) Las técnicas de citogenética y biología molecular han establecido diferentes marcadores moleculares que pueden utilizarse tanto para el diagnóstico como para el seguimiento de la enfermedad. (1-3, 40,42) Características clínicas La leucemia es un síndrome hematológico que se caracteriza por la expansión clonal de células hematopoyéticas como resultado de una alteración en los mecanismos de proliferación, diferenciación y muerte celular, en cualquiera de las etapas de maduración de las células sanguíneas. En términos biológicos y clínicos es un cáncer muy diverso, lo que las manifestaciones clínicas son variables. Los síntomas pueden aparecer de manera insidiosa o aguda, los cuales son un reflejo de la insuficiencia de la médula ósea y del grado de afección extramedular. Algunos pacientes sufren infecciones potencialmente letales o epistaxis durante el curso de la enfermedad o al diagnóstico. En otros casos, la leucemia es asintomática y se detecta durante un examen físico de rutina. Sin embargo, la mayor parte de los pacientes presentan síntomas de pocos días de evolución o de algunos meses como palidez, fatiga y letargia. Las principales manifestaciones de la leucemia en los niños son marcha claudicante y artralgias. Estos síntomas pueden ser consecuencia de la infiltración leucémica del periostio o puede deberse a la expansión que las células leucémicas provocan en la cavidad medular. Cuando existe necrosis en la médula ósea, se manifiesta con intenso dolor y fiebre. Otros signos y síntomas menos frecuentes son: cefalea, vómito, insuficiencia respiratoria, oliguria y anuria (11-15, 51). El examen físico puede revelar fiebre, palidez, petequias y equimosis de la piel y las mucosas, hemorragias retinianas, linfadenopatias, hepatoesplenomegalia, nefromegalia e hipersensibilidad a la palpación de los huesos. Generalmente al momento del diagnóstico se detecta anemia, trombocitopenia y anormalidades en el recuento leucocitario, las cuales oscilan entre 0.1 a 1,600 x 109 /L y en algunos casos se observa granulocitopenia absoluta. Aproximadamente dos terceras partes de los pacientes tienen recuentos leucocitarios de <25 x 109 /L. Casi todas las células de la sangre se identifican como linfoblastos o linfocitos (11-15, 52). Los niveles de hemoglobina oscilan entre 2 y 15 g/dL y los recuentos plaquetarios suelen ser bajos con una mediana de 50 x 109 /L. Los pacientes con alta carga tumoral presentan elevadas concentraciones séricas de ácido úrico como resultado del aumento del metabolismo de las purinas. Cuando existe afección renal masiva también se elevan las concentraciones séricas de urea, creatinina y fósforo (11-15, 52). Clasificación Aún cuando la aparición de tratamientos más eficaces ha conferido nuevas connotaciones a la clasificación de la leucemia, en ella se siguen identificando dos grupos principales: aguda y crónica. En el pasado, esta clasificación se basaba en la sobrevida de los pacientes; sin embargo actualmente, se reconoce que la leucemia aguda implica la proliferación maligna de células inmaduras (blastos), mientras que la leucemia crónica, la proliferación de tipos celulares más diferenciados. Además, de acuerdo con el origen celular, la leucemia crónica y aguda se clasifican en linfoides y mieloides. El diagnóstico diferencial, se realiza principalmente por las características morfológicas (método de Romanovsky) y las propiedades de tinción citoquímica de las células leucémicas. En general, los linfoblastos tienen una cromatina nuclear homogénea y fina, sin nucléolo distinguible y con un citoplasma escaso no granular ( Ver figura No. 1). Los mieloblastos son más grandes, tienen abundante citoplasma granuloso y poseen uno o más nucléolos diferentes y la característica principal son los bastones de Auer (Ver figura No. 1) (1-5). Hasta la fecha las técnicas citoquímicas más importantes son mieloperoxidasas, Sudán negro B, a-naftil acetato esterasa y a-naftil butirato esterasa. La reacción de la mieloperoxidasa es positiva en las células de toda la serie granulocítica, débilmente positiva o negativa en la serie monocítica y siempre negativa en los linfoblastos y en los linfocitos. El Sudán negro B suele mostrar una positividad paralela a la de la mieloperoxidasa. Aunque se ha descrito positividad para el Sudán negro B en las células de la leucemia aguda linfoblástica (LAL), los patrones de coloración difieren (52). En 1976, con el fin de unificar los criterios de clasificación, el grupo cooperativo FrancoAmericano-Británico (FAB) estableció un método de clasificación basado en las características morfológicas y en las propiedades de tinción citoquímica de los blastos leucémicos de la médula ósea. En la clasificación FAB se especifican ocho grupos principales de leucemia aguda mieloblástica (LAM): M0 a M7, y tres de LAL: tipo L1, L2 y L3 ( Ver figura No. 2). Es importante mencionar que el diagnóstico de la leucemia M0 no puede hacerse tomando como base las pruebas morfológicas y citoquímicas, dado que los blastos son semejantes a los de la LAL-L2, por lo que es necesario confirmar el diagnóstico con pruebas inmunológicas. La variante M1 es otra forma poco diferenciada de LAM, morfológicamente indistinguible de la LAL-2 (52). FIGURA No. 1: CLASIFICACIÓN CITOMORFOLÓGICA DE LEUCEMIA AGUDA En la fotografía A, se observa una Leucemia Linfoblástica aguda, en donde las células poseen un citoplasma agranular. En la Fotografía B, se observa una Leucemia mieloid aguda, en donde las células poseen un citoplasma con gránulos. Fuente: Childhood Leukemia. Head y Pui 1999 FIGURA No. 2: CLASIFICACIÓN FAB DE LA LEUCEMIA INFANTIL Fuente: Childhood Leukemia, Pui y Crist, 1999. Etiología Aunque un pequeño porcentaje de casos está asociado a síndromes genéticos hereditarios (Anemia de Fanconi, Síndrome de Bloom, Síndrome de Down, etc.) la causa de la leucemia y los mecanismos involucrados en la oncogénesis hematológica, no han sido completamente identificados. Sin embargo, muchos factores ambientales como la exposición a radiación ionizante, agentes químicos, infecciones virales, etc. aumentan el riesgo para el desarrollo de esta enfermedad. Por ejemplo, se han descrito asociaciones entre algunos casos de leucemia y de linfoma de Burkitt con el virus Epstein–Barr (VEB); y entre la leucemia de células T de adulto con el virus VLTH-I. Así también, en adultos existen evidencias de que tanto la exposición crónica el benceno como el uso de quimioterapia intensiva con agentes alquilantes o que actúan sobre la DNA topoisomerasa II, como las epipodofilotoxinas (tenipósido y etopósido) y las antraciclinas, pueden conducir al desarrollo de una LAM, pero se desconoce el porcentaje de leucemia infantil que resulta por la exposición a productos químicos. De tal manera que en la mayoría de los niños no es posible identificar a los factores que favorecen el desarrollo de la leucemia. Es probable que la causa principal de la LAL infantil sea una o varias mutaciones espontáneas. Las células madre de la médula ósea tienen un alto índice de proliferación y por lo tanto son susceptibles de sufrir mutaciones o reordenamientos de genes que participan en la regulación de la proliferación, diferenciación y muerte celular (1-5, 51,52) Epidemiología Entre los diversos tipos de cáncer, la leucemia ocupa el primer lugar de incidencia en los pacientes pediátricos. En el mundo, anualmente se diagnostican alrededor de 40-50 casos nuevos por cada 1,000,000 niños menores de 15 años y a diferencia de lo que ocurre con los adultos, las leucemias infantiles son casi siempre agudas (aproximadamente 80% de LAL y 18% de LAM), mientras que la leucemia crónica mieloide (LCM) constituye casi 2%. La leucemia crónica linfoide (LCL) sólo se ha descrito en pocos casos (53). En México, la frecuencia de la leucemia observa un comportamiento similar a lo descrito en la literatura, existen evidencias de que en los últimos años, no sólo se ha registrado un incremento en la incidencia de esta patología, sino también en la tasa de mortalidad principalmente por la LAL. Tan solo en el Instituto Nacional de Pediatría (INP) acuden anualmente alrededor de 100 casos de leucemia, de los cuales aproximadamente el 81.9% de ellos se diagnostican como LAL (54). En general, la incidencia de la LAL es mayor en niños de entre 3 y 5 años de edad y es ligeramente más común en varones (relación entre niño: niña de 1.4:1.0). Así mismo, en la raza blanca se observa una mayor frecuencia, que en la raza negra, lo cual podría reflejar una diferente susceptibilidad genética (51). El único estudio epidemiológico sobre leucemias infantiles realizado en Centroamérica, incluyendo México, fue el realizado por Arangúre y colaboradores; en donde se incluyeron datos de pacientes desde 1996 hasta el 2000. El promedio anual de incidencia obtenido fue de: 44.9 por millón de niños para leucemia linfoblástica aguda; 10.6 por millón de niños para leucemia mieloide aguda; 2.5 por millón de niños para leucemia mieloide crónica y un 58.4 por millón de niños para leucemias inespecíficas ( es decir que no se logró su clasificación) (4). En Guatemala, no existen estudios epidemiológicos nacionales documentados sobre la incidencia anual. El hospital general San Juan de Dios atiende aproximadamente al año 40 casos nuevos de leucemia linfoblástica infantil, siendo el principal cáncer en niños que asisten al hospital. D. LEUCEMIA LINFOBLÁSTICA AGUDA (LLA) Es una enfermedad que se caracteriza por una proliferación desordenada de células inmaduras de la serie hematopoyética linfoide. Esta es la neoplasia hematológica más frecuente en niños. Su incidencia máxima se encuentra entre los 3 y los 5 años, pero es posible encontrarla en lactantes. Y puede clasificarse de acuerdo a criterios citomorfológicos en L1, L2 y L3 (1-3,11,12). Entre los hallazgos clínicos encontrados se pueden mencionar un malestar general, pérdida de apetito, fiebre, palidez, anemia y trompocitopenia entre otros. Las características de ALL que se han asociado a mal pronóstico son: edad menor a 6 meses, recuento de glóbulos blancos mayor de 50,000 / ul y fenotipo CD10-(13). La LAL es una neoplasia maligna que se caracteriza por la proliferación desordenada de células inmaduras de la serie linfoide que aunque afecta a niños y adultos, las características biológicas parecen ser diferentes entre ellos. La leucemia aguda pediátrica puede iniciarse en la etapa prenatal por la recombinación anormal y la formación de fusiones génicas durante la hematopoyésis fetal (50). Clasificación Inmunológica Uno de los grandes avances en la clasificación de la LLA, fue la identificación de la expresión de los antígenos de superficie de membrana o de los componentes citoplasmáticos que han permitido identificar y clasificar a las enfermedades linfoproliferativas según su origen celular y su estadio de diferenciación; así en general, las LLA infantiles se pueden clasificar en: i. Pre- B: Linfoblastos con morfología L1, según la FAB. Estas células son positivas tanto para las inmunoglobulinas citoplasmáticas (cIg) como para CD19, CD22, HLA-DR, CD10. Aproximadamente el 80% de casos pediátricos con LAL tienen este inmunofenotipo. En este grupo se identifican 3 subtipos inmunológicos: pre-B temprana, pre-B común y pre-B tardía. ii. Células B: Los linfoblastos con este fenotipo tienen una morfología característica, designada por la FAB como L3 y son positivas para las inmunoglobulinas de superficie (sIg). Este subtipo de leucemia está presente en el 3% de los casos de LAL infantil. En estos pacientes, Silvia Jiménez Morales 11 pueden observarse tumores extramedulares y más del 90% de los casos presentan cadenas pesadas m de las inmunoglobulinas (Igs) en la superficie celular. Alrededor de las dos terceras partes de ellos, también expresan cadena ligera k y el resto cadena ligera l. Las LAL de células B parecen representar linfomas de Burkitt avanzados en fase leucémica. iii. Células T: Este subtipo de LAL, se identifica por la expresión de antígenos de superficie asociados a las células T. Presentan positividad para CD7, CD5 o CD2. Las leucemias de células T generalmente se presentan en los varones y en grupos de edad avanzada y se caracterizan por la gran masa de células leucémicas asociado al desarrollo de infiltración al mediastino. iv. Pre-B transicionales: Los linfoblastos expresan tanto cadenas pesadas de Igs citoplásmicas como de superficie y proteínas de cadena ligera (56). E. CITOGENÉTICA DE LA LEUCEMIA LINFOBLÁSTICA AGUDA En la LLA pueden encontrarse anormalidades citogenéticas numéricas, están abarcan únicamente alteraciones en cuanto número de cromosomas. Estas alteraciones se encuentran en aproximadamente la mitad de los casos de ALL reportados. Entre las alteraciones numéricas que se encuentran frecuentemente se pueden mencionar: • Hiperdiploidía leve: cuando se encuentran de 47 a 50 cromosomas. • Hiperdiploidía masiva: cuando se encuentran más de 51 cromosomas. • Hipodiploidía: cuando se encuentran menos de 46 cromosomas (12). Los cariotipos hiperdiploides masivos (51-65 cromosomas) se encuentran asociados a buen pronóstico. La hiperdiploidía leve esta asociada a u pronóstico moderado; y por el contrario una hipodiploidía esta asociado a un mal pronóstico. Por otro lado, se encuentran frecuentemente alteraciones estructurales en cromosomas, es decir que son alteraciones que afectan la morfología en cromosomas. Dentro de la gran variedad de alteraciones morfológicas que pueden encontrarse, las más frecuentes son las translocaciones, que se definen como un intercambio de material genético entre un cromosoma y otro; obteniéndose como resultado de esta reordenación genes quiméricos, que dan lugar a transcritos de fusión específicos. En la LLA todas las alteraciones cromosómicas que se encuentren se consideran cruciales tanto para el diagnóstico como para las decisiones terapéuticas. Ya que el pronóstico de los pacientes depende muchas veces de los marcadores genéticos encontrados. ( Tabla 2) (1-3,11,12). TABLA 2: ANORMALIDADES GENÉTICAS EN LEUCEMIA LINFOBLÁSTICA AGUDA. ANORMALIDADES GENÉTICAS Y MOLECULARES TRANSLOCACIÓN CARACTERÍSTICA CLÍNICA ASOCIADA Fusión BCR-ABL t(9;22)(q34;q11) Mieloproliferación y mal pronóstico Fusión TEL-AML1 t(12;21) alteración críptica Buen pronóstico Fusión E2A-PBX t(1;19)(q23;p13) Respuesta pobre a tratamiento con antimetabolitos. Fusión MLL-AFX1 t(x;11)(q13;q23) Mal pronóstico Fusión MLL-AF4 t(4;11) (q21;q23) Pronóstico pobre hiperdiploidía ------------ Buen pronóstico Fusión IgH-Myc Enfermedad extramedular LL3 Fuente: Leukemia, Scurto, P et al. (1998) En la tabla No. 2, se encuentran los marcadores genéticos más frecuentes en ALL, así como su implicación en pronóstico. De estos marcadores se han seleccionado cuatro de ellos: la t(9;22)(q34;q11), t(12;21)(p13;q22), t(4;11) (q21;q23) y t(1;19)(q23;p13). Estos cuatro marcadores se consideran importantes pues poseen una frecuencia relativamente alta en pacientes pediátricos con ALL. Y además proporcionan información importante al clínico acerca del pronóstico del paciente así como del tipo de tratamiento a utilizar. A continuación se describen cada uno de los marcadores seleccionados en el presente estudio (51). G. REODENACION BCR-ABL En la mayoría de las leucemias, las translocaciones dan lugar a un gen de fusión que produce un mRNA que codifica una proteína quimérica con propiedades estructurales y funcionales distintas de las proteínas constitucionales normales. Estas proteínas pueden activar la transcripción directamente, sin que se requiera la interacción de otras proteínas específicas. Se han identificado muchos genes de fusión. El caso mejor conocido es el gen quimérico BCR-ABL, que es resultado de la translocación t(9;22)(q34;q11) que da lugar al cromosoma Philadelphia (Ph) (Ver figura No. 3, 4 y 5) (14,15). Esta una de las translocaciones más frecuentes en ALL, ocurre de un 11% a un 29% en pacientes que involucran la serie B y la serie T de linfocitos. Generalmente los pacientes con el cromosoma filadelfia positivo poseen recuentos de glóbulos blancos elevados (23,000/ul), inmunofenotipos CD34+, CD19+ y CD10+. [14-19] FIGURA No. 3: ESQUEMA DE LA t(9;22) MOSTRANDO LA LOCALIZACIÓN DE LOS GENES ABL Y BCR. 9 der(9) 22 5´BCR 22q11 9q34 5´BCR ABL 3´BCR ABL der(22) 33´ BCR La región 5´ del BCR permanece en el der(22) mientras que la región 3´ se traslada al der(9). Fuente: British Journal of Haematology. Cerveira, N. (2000) FIGURA NO 4: EL CROMOSOMA 22 AFECTADO POR LA TRANSLOCACIÓN ES EL DENOMINADO CROMOSOMA PHILADELPHIA 9 9 22 22 t(9;22)(q34;q11) Fuente: British Journal of Haematology. Cerveira, N. (2000) Los genes involucrados en esta translocación son el gen ABL presente en el cromosoma 9 y el gen BCR en el cromosoma 22. La porción 5´del gen BCR es unida a la porción 3´del gen ABL formando la forma quimérica BCR-ABL (14-19). El oncogén quimérico BCR-ABL lleva a la síntesis de proteínas de diferente peso molecular dependiendo de la localización del punto de rotura. Así es posible distinguir tres nuevas proteínas de fusión (p190BCR-ABL, p210BCR-ABL y p230BCR-ABL). Los puntos de rupturas más frecuentes en el gen BCR ocurren en los exones 1(e1), 12(b2), 13(b3) y 19(e19) y el punto de ruptura en el gen ABL habitualmente se produce en el exón 2(a2) (2344). Cuando se da la unión de los exones b3a2 y/o b2a2 (Región M-bcr) se codifica para una proteína de 210kD (p210BCR-ABL) la cual se presenta en el 20% a 40% de los pacientes con Leucemia Linfoblástica aguda (LLA) y es característica en más del 95% de pacientes con leucemia mieloide crónica (LMC); si los exones e1 y e2 son removidos por el corte y empalme (Región m-bcr) la unión de e1a2 se transcribe en una proteína de 190kD (p190BCR-ABL) la cual se detecta en el 60% a 80% de los pacientes con LLA y en menos del 10% para aquellos con LMC. Es posible observar una tercer proteína de fusión próxima a 3’ del extremo distal del gen BCR denominada p230BCR-ABL, la cual resulta de la fusión de los exones e19a2 (Región µ-bcr), cuya expresión se asocia a leucemias crónicas neutrofilas y trombocitosis (Ver figura No.5 y 6) (23,44). FIGURA No. 5: REPRESENTACIÓN ESQUEMÁTICA DE LA FUSIÓN GÉNICA BCR-ABL A. Diagrama de la t(9;22). B. Estructura intrón/exón de los genes BCR y ABL involucrados en la t(9;22). C. Diagrama del transcrito quimérico BCR-ABL Fuente: Leukemia.Van Dongen y col, 1999. FIGURA No. 6: LOCALIZACIÓN DE LOS PUNTOS DE ROTURA EN LOS GENES ABL Y BCR. En el gen ABL pueden observarse tres puntos de rotura, cercanos al exón Ib, Ia y a2. En el gen BCR se muestran los exones y los puntos de rotura frecuentes. Además se muestra la estructura quimérica de los cuatro ARN mensajeros derivados de los distintos puntos de rotura. Fuente: British Journal of Haematology. Cerveira, N. (2000) Por consiguiente dependiendo del punto de rotura se pueden obtener cuatro transcritos diferentes: b2a2, b3a2, e1a2 y e19a2. Estos transcritos se encuentran presentes en leucemia mieloide crónica y en leucemia linfoblástica aguda, y son indicadores de muy mal pronóstico. Además la presencia de esta alteración da orientación acerca del tipo de terapia que el paciente puede recibir; por ejemplo se sabe que estos pacientes responden al tratamiento con Imatinib, además se sabe que el pronóstico es muy pobre y en muchas ocasiones se recomienda directamente un trasplante de médula ósea. Como se puede observar en la figura 7, los pacientes cuyos análisis moleculares fueron positivos para cualquiera de los transcritos BCR-ABL muestran un mayor porcentaje de recaída conforme avanzan los años de haber sido trasplantados (14-19). Actualmente se han desarrollado distintas técnicas moleculares de reacción en cadena de la polimerasa –PCR- para determinar la presencia de los distintos transcritos (1419). FIGURA No. 7: PROBABILIDAD DE RECAÍDAS DE PACIENTES BCR-ABL En la gráfica se muestra la incidencia acumulada de recaídas de pacientes post-transplantados cuando la PCR para transcritos BCR-ABL es positiva y cuando es negativa. Puede notarse un incremento de recaídas cuando hay presencia de transcritos en los pacientes afectados. Fuente: Blood, Deininger M, Goldman J and Melo J. (2000). El gen ABL localizado en el cromosoma 9q, es el homólogo humano del oncogen vabl del virus de la leucemia murina y codifica para un receptor de tirosina cinasa. ABL es un gen constituido por 8 exones que como resultado del corte y empalme (splicing) alternativo del primer exón 1a y 1b, codifica para dos isoformas de una proteína de 145 KDa. La proteína está constituida por tres dominios homólogos a secuencias SRC (SH1, SH2 y SH3), que se localizan en la región NH2-terminal. El dominio SH1 tiene la función de tirosina cinasa, mientras que los dominios SH2 y SH3 permiten la interacción con otras proteínas. Hacía la región 3’ se localiza la secuencia señal de localización nuclear y los dominios de unión a DNA y actina. La proteína normal ABL está involucrada en la regulación del ciclo celular, en la respuesta al estrés genotóxico y en la transmisión de información acerca del ambiente celular a través de la señalización mediada por las integrinas. Al parecer la proteína ABL tiene un complejo papel como modulador celular que integra las señales del ambiente extracelular e intracelular e influye en las decisiones para mantener el ciclo celular y la apoptosis (23-44). El gen BCR, se localiza en la región q34 del cromosoma 22 y está constituido por 23 exones que codifican para una proteína de 160 KDa. La proteína BCR posee un dominio de dimerización y un dominio de cinasa, en la región NH2-terminal, además de un dominio con actividad de GTPasa hacia la región COOH. La proteína BCR está presente en varios tejidos y aunque existen datos que apoyan la hipótesis de que BCR participa en la transducción de señales, su verdadero papel permanece sin ser determinado (25,44). Tanto la p210 como la p190 presentan una mayor actividad de tirosina cinasa comparada con la proteína ABL normal. La proteína BCR-ABL fosforila residuos de tirosina de muchas proteínas celulares y activa diferentes rutas de señalización, lo cual conlleva a la transformación celular. En modelos experimentales, se ha observado que estas proteínas son capaces de transformar precursores hematopoyéticos por la activación de señales mitógenas, inhibición de apoptosis y alteraciones en la adhesión de las células al estroma (25, 30-37). Vías de señalización activadas por Bcr-Abl Una consecuencia del incremento de la actividad de la tirosina quinasaradica en que el BCR-ABL puede fosforilar diferentes sustratos, activando múltiples señales de transducción en las células. Las vías activadas son RAS, vía Fosfatidilinositol-3-cinasa, JakStat, MAPK y ROS. - Vía RAS: la activación de Ras se poduce por transducción de señales mediadas por receptores con actividad tirosin cinasas. Una vez realizada la unión al ligando y dimerización, los receptores tirosincinasas activados, se unen a moléculas adpatadoras que contiene los dominios SH2 y SH3 como son Grb-2 y SHC formando complejos de señalización con factores intercambiadores Ras en la membrana celular. Estos complejos desencadenan una acumulación de la forma Ras e inducen un efecto antiapoptótico que, aunque sea insuficiente, es necesario para la transformación derivada de BCR-ABL. La activación inapropiada del gen Ras por una mutación del gen, ha demostrado ser una importante vía de la señal de transducción celular para la proliferación y transformación maligna de los tumores. - Vía Fosfatidilinositol-3-cinasa: esta vía funciona en la regulación del metabolismo lipídico fosfoinositídico y en la generación de segundos mensajeros lipídicos relacionados con la transducción de señales. Se ha visto que una subunidad de esta proteína se fosforila en residuos tirosina en células que expresan BCR-ABL y forma complejos con cbl y moléculas adaptadas crk y crkl. PI3 está activado de forma constitutiva por la translocación BCR-ABL jugando un papel importante en su transformación (84,85). - Vía Jak-Stat: El crecimiento de las células hematopoyéticas normales se controla por citocinas. Los receptores para estas citocinas traducen señales mediante activación de proteínas denominadas genéricamente Jak (Janus tirosine kinases), que es una familia de cinasas citoplasmáticas que fosforilan y activan los factores de transcripción STAT (Signal Transducer and Activators of transcription), BCR-ABL activa constitutivamente las vías de señalización de JAK y STAT en células hematopoyéticas, siendo STAT 1 y STAT 5 las tirosin-fosforiladas más importantes. La fosforilación constitutiva de factores de transcripción STAT han sido reportados en líneas celulares positivas BCR-ABL y en células primarias con LMC, la activación STAT 5 contribuye a la transformación maligna a pesar de sus funciones pleiotropicas; su efecto en la transformación de células BCR-ABL parece ser anti-apoptotico e implica la activación trasncripcional de Bcl-xl. En contraste con la activación de la vía Jak-Stat por medio de estímulos fisiológicos, BCRABL activa directamente STAT 1 y STAT 5 sin previa fosforilación de proteínas Jak (3544). - Vía MAPK: un primer acontecimiento de señalización que sigue a la activación de RAS es la activación de la vía de señalización de proteínas activadoras de mitogénesis con actividad cinasa (MAPK). Se conocen claramente tres cascadas de MAPK: la cinasa regulada extracelularmente (ERK), la vía de las Jun cinasas o cinasas activadas por estrés (SAPK) y la vía de la p28 cinasa. El punto final para cada una de estas vías es la fosforilación y activación de transcripción. BCR-ABL y v-ABL activan la vía SAPK en fibroblastos y células hematopoyéticas. Así mismo activan promotores dependientes de Jun de manera dependiente de RAS, MEPK y SAPK. Mientras que los efectos de BCRABL en la vía JNK parecen ser claros, estudios de la vía ERK muestran que la BCR-ABL funciona de manera distinta a la mayoría de los receptores con actividad tirosincinasa (35-44). - Vía ROS: la transformación de las líneas celulares con BCR-ABL presenta un incremento de ROS, comparado con las líneas celulares no BCR-ABL. Este incremento es suficiente para la transformación fenotípica en sí misma. Se ha sugerido que ROS podría actuar como segundo mensajero en la regulación de la actividad de las enzimas sensibles a la acción de oxidorreducción (redox), como son las cinasas y fosfatasas. La inhibición de la proteína tirosin fosfatasa (PTPasa) mediante la modulación redox explicaría el amplio espectro de las actividades biológicas de ROS. Un incremento de ROS amplifica las señales de BCR-ABL, posiblemente regulando las proteínas redox-sensibles, como es la PTPasa celular, la cual también está incrementada. También es de particual interés en la LMC humana, el hecho de que un incremento de ROS pueda tener consecuencias a largo plazo en la estabilidad genética. Los niveles de ROS no sólo son modulados por enzimas, antioxidantes y grupos sulfhidrilos, sino también reaccionados con las bases del ADN. Aunque las modificaciones pueden ser corregidas eficientemente por los mecanismos de reparación de ADN, un incremnto persistente de ROS podría inducir la acumulación de mutaciones en las células de la LMC, que podría contribuir a la progresión de la enfermedad (35-44). H. REORDENACIÓN ETV6-AML1 La translocación t(12;21)(p13;q22) da origen al gen de fusión ETV6/AML1 (TEL/AML1, ETV6/RUNX1). Esta translocación es difícil de identificar por citogenética convencional debido a que sus puntos de rotura están en bandas claras de similar tamaño en el cromosoma 12 y en el 21; por esta razón es considerada críptica. Únicamente se puede detectar por FISH y RT-PCR (20-23). Es la anormalidad cromosómica más común en ALL en niños, ocurre en 18-30% (44% en población mexicana) del total de los casos y principalmente en niños menores de 10 años. Esta asociado a fenotipos de las células B CD19+ y CD10+, raramente esta asociada a otras reordenaciones, y además confiere buen pronóstico. Estos pacientes muestran una excelente respuesta a la quimioterapia convencional y alcanzan una remisión rápida libre de recaídas (Ver figura No. 8) (20-23). FIGURA No. 8: CURVA DE SOBREVIVIENTES LIBRES DE ENFERMEDAD CON ETV6-AML1 POSITIVO Se muestra mayor supervivencia en los pacientes ETV6 positivo que en los ETV6 negativo. Fuente: Proc. Natl. Acad. Sci. Golub T, et al. (1995) Este gen de fusión es un marcador útil para el estudio de enfermedad mínima residual, proporcionando alta especificidad, pero una sensibilidad un poco baja (20-23). FIGURA No. 9: REPRESENTACIÓN ESQUEMÁTICA DEL GEN ETV6, MOSTRANDO SUS PUNTOS DE ROTURA. Fuente: Proc. Natl. Acad. Sci. Golub T, et al. (1995) El gen TEL (también conocido como ETV6) se localiza en el cromosoma 12p13 y está constituido por 8 exones (240 Kb) ( Ver figura No. 9 y 10) que codifican para una proteína de 53 y 57 KDa. La proteína TEL es un miembro de la familia de factores de transcripción ETS que se caracterizan por tener un dominio C-terminal denominado de transactivación (ETS) y un dominio de oligomerización (HLH). El dominio ETS es codificado por los exones 6-8 mientras que los exones 3 y 4 codifican para el dominio de oligomerización. TEL, al igual que los otros miembros de la familia ETS, funciona como un regulador transcripcional ( 51-57). Aunque la proteína TEL se detecta en diferentes tejidos, todavía se desconocen los genes sobre los que actúa; sin embargo, se ha observado que puede reprimir al promotor del receptor del factor de estimulación de las colonias de macrófagos y a otros genes, cuando interactúa con proteínas nucleares corepresoras como MSIN3. En modelos experimentales, se ha determinado que TEL es indispensable para el establecimiento de la hematopoyésis de la medula ósea postnatal y para mantener el desarrollo vascular del saco vitelino y la sobrevivencia de ciertas poblaciones celulares en periodo prenatal. Además se ha observado que es una proteína necesaria para establecer la hematopoyésis de la médula ósea de todos los linajes ( 51, 58). Por otra parte, el gen AML1 (Leucemia Mieloide Aguda 1) localizado en el cromosoma 21q22, también conocido como RUNX1, PEBP2a o CBFa2, se clonó a partir de la t(8;21) en una leucemia de linaje mieloide. Tiene una longitud de 120 kb y codifica para un factor de transcripción que forma un complejo heterodimérico con la proteína CBFb2. El heterodímero se une a secuencias específicas del DNA: TGT/CGGT a través del dominio RUNT (por su homología con el gen runt de Drosophila) de AML1. Tanto la formación del complejo CBFa2/CBFb como la unión al DNA, se realiza mediante el dominio central de 118 aminoácidos de la proteína CBFa2 ( 59-61). AML1 es una proteína que está presente en células hematopoyéticas como linfocitos B y T, monocitos, granulocitos, megacariocitos y células CD34+ y es esencial para inducir la diferenciación normal de las células mieloides. Durante el desarrollo; AML1 se expresa principalmente en células progenitoras de la hematopoyesis. Por lo que se ha observado en modelos animales, es probable que el papel de AML1 en la diferenciación hematopoyética sea la de dirigir la expresión adecuada de genes específicos de linaje. Los ratones que carecen de este gen, mueren in utero por falta de la hematopoyésis y por alteraciones en el desarrollo del sistema nervioso ( 59-61). Dado que las mutaciones que alteran la función normal de AML1, resultan en un fenotipo letal durante la embriogénesis, y las translocaciones Silvia Jiménez Morales 28 que alteran el complejo AML1-CBFb2, interfieren con la transcripción normal dependiente de AML1, es probable que la transformación sea en parte por la represión dominante en la expresión de genes blanco de esta proteína. Los ratones que no expresan el gen aml1 desarrollan anemia y carecen de plaquetas en circulación. Los defectos hematoyéticos que resultan de la pérdida de la proteína AML1, son intrínsecos a la hematopoyésis definitiva de las células progenitoras (células madre) ( 51, 62). Se ha observado que la secuencia a la cual se une el complejo CBFa2-CBFb es crítica en la expresión tejido-específica de algunos genes hematopoyéticos, como los que codifican para mieloperoxidasas, el receptor 1 del factor estimulador de colonias (CSF-1), subunidades de receptores de antígenos de células T, algunas citosinas, el receptor de interleucina 3 (IL-3) y el factor de estimulación de crecimiento de colonias de macrófagosgranulocito. Por otra parte, el complejo AML1-CBFb funciona como un organizador transcripcional que recluta factores específicos para formar un complejo núcleo-proteico o enhanceosoma que estimulan la transcripción a un linaje específico (51, 59-62). Estos resultados sugieren que el complejo CBFa2-CBFb funciona como un gran interruptor que regula tanto positivamente como negativamente una cascada transcripcional involucrada en el desarrollo del sistema hematopoyético definitivo. Por lo tanto TEL y AML1 son genes esenciales para el establecimiento de la hematopoyesis (51, 59-62). Aunque la frecuencia de la t(12;21) varía ampliamente en las diferentes poblaciones del mundo (3-44%), la fusión de los genes TEL y AML1, sigue siendo la anormalidad genética más común en pacientes pediátricos con LAL. Con la clonación de la t(12;21)(p13;q22), se demostró que los sitios de ruptura y fusión de los genes TEL y AML1 ocurren a lo largo de una región de 18 Kb del intrón 5 del gen TEL, principalmente en una región polimórfica de repetidos Alu y en el intrón 1 de AML1, por lo que la t(12;21) resulta en la fusión del exón 1 al 5 de TEL con el exón 2 al 8 de AML1 (Ver figura No. 10). De tal manera que, el gen quimérico contiene las primeras 1033 bases del gen TEL y casi todo el gen AML1 (Ver figura No. 10). Esta translocación produce dos genes híbridos: TEL-AML1 y AML1-TEL, pero solamente la fusión TEL-AML1 se expresa en todos los pacientes con la t(12;21); AML1-TEL, sólo se observa en un grupo pequeño Por lo tanto, los casos positivos expresan la misma proteína quimérica, cuya expresión está controlada por el promotor del gen TEL. La t(12;21) también puede resultar en la expresión de un gen quimérico, en el que se observa la fusión del exón 5 del gen TEL con el exón 3 o con otros exones del gen Silvia Jiménez Morales 30 AML1 ( 59-62). Aunque tanto el gen TEL como el gen AML1 se encuentran fusionados con muchos otros genes que codifican para cinasas o factores de transcripción en leucemias de linaje mieloide o linfoide, la fusión génica TEL-AML1, parece ser exclusiva de LAL de progenitores B. Las bases de esta selectividad aún se desconocen, pero este hecho sugiere que la proteína quimérica esta implicada en la proliferación y/o sobrevivencia de los precursores de células B ( 59-62). A pesar de que se ha identificado a los genes involucrados en la t(12;21), la función de la proteína TEL-AML1 y AML1-TEL en la transformación maligna de las células hematopoyéticas también se desconoce, su estructura, dada por la fusión del dominio de oligomerización (HLH) de TEL con el dominio de transactivación de la proteína AML1, incluyendo al dominio homólogo a RUNT, sugiere que la proteína quimérica no sólo retiene la habilidad de unirse a secuencias del DNA y de formar un complejo con la proteína CBFb, sino que también mantiene la función de formar homodímeros con la proteína TEL normal mediante el dominio HLH; de esta manera, tanto la función de TEL como la de AML1 se modifica. De acuerdo con esta hipótesis, TEL-AML1 puede reprimir directamente la activación transcripcional de los promotores de los receptores de células T (TCR) e IL-3 al interactuar con los sitios de unión de AML1 ( 59-62). Es interesante que en la mayoría de los casos de LAL con la t(12;21), el alelo no translocado de TEL, frecuentemente está deletado. La pérdida de heterocigocidad del locus TEL, es común en niños con LAL, lo cual podría ser un evento secundario en leucemias con ésta translocación (59-62). FIGURA No. 10: REPRESENTACIÓN ESQUEMÁTICA DE LA FUSIÓN GÉNICA TEL-AML1 A. Estructura intrón-exón de los genes TEL y AML1 involucrados en la t(12;21). B. Diagrama del transcrito quimérico TEL-AML1. Fuente: Leukemia Van Dongen y col. 1999. I. REORDENACIÓN DEL GEN MLL El gen MLL se localiza en el cromosoma 11 en la banda q23. La ALL infantil tiene alta incidencia de reordenaciones 11q23 (43% al 60%), siendo la principal translocación asociada t(4;11) (q21;q23) que ocurre en un 2% en pacientes pediátricos con ALL y en un 3-5% en pacientes adultos con ALL. También se encuentra reordenado en leucemias mieloides agudas (AML), principalmente M4 y M5. Estudios anteriores han demostrado que esta alteración confiere muy mal pronóstico (Figura 9). . Los pacientes que presentan esta alteración tienen rasgos más agresivos y una alta probabilidad de fallo de los protocolos de quimioterapia estándares. Esta asociado con inmunofenotipo : HLA-DR+, CD34+, CD10+ y CD19+ (24-27). El gen MLL localizado en la región 11q23 forma un transcrito de fusión con el gen AF4 localizado en el cromosoma 4 en la región q21 ( 24-27). Se ha estudiado la presencia de este transcrito por RT-PCR, y se encontró en pacientes en remisión post- trasplante de médula ósea, lo que indica que podría ser un marcador de utilidad para el seguimiento de la enfermedad así como para el estudio de enfermedad mínima residual (Ver figura No. 11) (24-27). FIGURA No. 11: CURVA DE EVENTOS SOBREVIVIENTES LIBRES DE ENFERMEDAD Se muestra menor supervivencia en los pacientes que poseen el gen MLL reordenado. Fuente: Blood, Hilden JM. (1995) Uno de los puntos de quiebre cromosómico más frecuentes en las leucemias humanas se ubica en el cromosoma 11 banda q23, donde reside el gen de Linaje Leucémico Mixto (Mixed Lineage Leukemia o MLL, también llamado ALL-1 o HRX), el cual constituye una de las excepciones dentro de las asociaciones específicas; se ha descrito su presencia en más de 60 genes fusionados, por lo que se dice que es un oncogen “promiscuo”. Estas translocaciones se pueden observar en una amplia gama de patologías malignas hematológicas, principalmente en LLA y LMA, así como también en síndromes mielodisplásicos y en el linfoma de Burkitt (4, 5). Los rearreglos del gen MLL, que determinan distinto pronóstico terapéutico según el fenotipo de leucemia, se detectan en 6% de los casos de LLA y 80% de éstos ocurren en menores de un año de edad (leucemia del lactante). Su presencia constituye un factor de mal pronóstico, que empeora en relación inversa con la edad del paciente. En la LMA son más frecuentes: se ven en14 % de los casos totales y en 65% de los casos en lactantes y confieren un pronóstico de riesgo intermedio (4, 5). Estructura del gen MLL El gen MLL se ubica en el cromosoma 11q23, justo después del dominio represor; tiene una extensión de 90 kb y se compone de 38 exones; produce un mRNA de 12 kb que codifica una proteína de 3969 aminoácidos, de 430 kDa de peso molecular y compleja estructura. Esta proteína se expresaría ampliamente en el embrión en desarrollo, donde funcionaría como regulador de la transcripción nuclear; en tejidos adultos sus niveles son mínimos (5-7). La proteína MLL normalmente es clivada en el citoplasma por la caspasa 1 en los aminoácidos 2666 (sitio de clivaje 1 o CS1) y 2718 (sitio de clivaje 2 o CS2), generando dos subunidades: MLL-N (300 kDa) y MLL-C (180 kDa). Su función es acetilar, desacetilar y metilar las histonas de los nucleosomas (8). La proteína madura contiene una región cluster de puntos de quiebre entre los exones 5-11 de 8,3 kb, y en su estructura se ha identificado múltiples dominios proteicos: ganchos AT, un dominio de ADN metiltransferasa (transcriptional repression domain, TRD), un dominio PHD (Plant homology domain), un dominio activador de la transcripción (TA) y un dominio SET o Su (var)3-9, enchancer-of-zeste, trithorax (7) (Figura 1). Las alteraciones en el gen MLL incluyen deleciones, duplicaciones, inversiones y translocaciones recíprocas; en estas últimas se generan proteínas de fusión por la interacción con otros genes, denominados fusion partner genes (FPGs), reemplazando así los dominios de represión transcripcional y de señalización nuclear ubicados en el extremo N-terminal de la proteína MLL, por los extremos C-terminales de los genes de fusión. Los puntos de quiebre que se observan en las leucemias de novo se concentran en la región centromérica, mientras que en las leucemias del lactante y secundarias a tratamiento lo hacen en la región telomérica (5-7). Los genes de fusión más frecuentes son: 9p 21-22 (t 9;11) en MLL-AF9, 4q21 (t 4;11) en MLL-AF4 y 19p13 (t 11;19) en MLL-ENL. MLLAF4 se ve con mayor frecuencia en LLA, MLL-AF9 es más frecuente en LMA y MLLENL es común en ambos tipos de leucemia (Ver figura No.13) (8). FIGURA NO. 13: GEN MLL Y SU FUSIÓN CON OTROS GENES Fuente: Nature Reviews, Cancer. J. REORDENACIÓN E2A-PBX1 La t(1;19)(q23;p13) se encuentra en aproximadamente 5% de ALL FAB L1/L2. Esta asociada frecuentemente a fenotipo celular de la serie B CD9+, CD10+, CD19+, CD22+ y CD34+ (29-31). Esta translocación forma un gen quimérico entre el gen E2A que se encuentra en el cromosoma 19 y el gen PBX1 que se encuentra en el cromosoma 1. Los puntos de rotura en el gen E2A ocurren de manera invariable en un espacio de 3.5 kb en el intrón que se encuentra entre el exón 13 y el exón 14. La organización genómica del gen PBX1 todavía no ha sido conocida por completo, pero se piensa que los puntos de rotura ocurren dentro de un mismo intrón (29-31). Los pacientes con esta alteración parecen ser más susceptible a una recaída. Por numerosos estudios se ha concluido que esta alteración confiere muy mal pronóstico, el cual puede ser mitigado por una quimioterapia muy agresiva (29-31). La fusión génica E2A-PBX1 En la LLA, el gen E2A participa principalmente en dos eventos de fusión. El primero resulta de la translocación cromosómica t(1;19)(q23;p13.3) y el segundo es la t(17;19) ( Ver figura No. 14) (63). El locus genético de E2A [localizado en el cromosoma 19(q13.3)] codifica para tres proteínas reguladoras de transcripción generados por splicing: E12, E47 y E2-5, que pertenecen a la familia bHLH (hélice-asa-hélice). El dominio bHLH, característico de esta familia, está constituido por una región básica, responsable de la unión específica al DNA y un dominio estructural formado por dos hélices anfipáticas que se separan por una asa de longitud variable. El dominio bHLH es necesario para la dimerización. La unión de E2A al DNA se realiza mediante la formación de homodímeros o heterodímeros con proteínas bHLH. Sin embargo la secuencia de E2A que esta incluida en las fusiones proteicas de la leucemia, se localiza en la región NH2-terminal, la cual contiene dos dominios de activación transcripcional denominados AD1 y AD2 (63). Las proteínas E2A, están presentes en diferentes tejidos y pueden formar heterodimeros con una gran variedad de proteínas bHLH tejidoespecíficas (incluyendo factores miogénicos y neurogénicos) para coordinar la expresión de genes durante el desarrollo. En linfocitos B, E2A funciona principalmente como un complejo homodimérico que se une a cajas E1 de los elementos enhancer y promotores de las Igs, por lo tanto E2A y E47 son considerados como candidatos que regulan la activación del loci Ig y el desarrollo de células B (63). El papel de las proteínas E2A en el desarrollo de las células B, también ha sido observado en ratones mutantes. Los ratones que tienen proteínas E2A no funcionales, detienen el desarrollo de las células B, principalmente en estadios tempranos y muestran anormalidades en la diferenciación de las células T, lo que sugiere que la pérdida de la función de E2A puede contribuir a la génesis de la leucemia (63). El gen PBX1 de 1.8 Kb, se localiza en la banda 1q23 y está formado por 8 exones. Mediante “splicing” alternativo, PBX1 codifica para dos proteínas que difieren en la región COOH-terminal, PBX1a y PBX1b. Estas proteínas se detectan en todos los tejidos, excepto en células de linaje B y T (63). La fusión del gen E2A (19p13.3) con el gen PBX1 (1;q23) en la t(1;19) que crea al gen quimérico E2A-PBX1 es una translocación específica de la LAL pre-B. En esta translocación, el sitio de ruptura y fusión del gen E2A ocurre en una región de aproximadamente 3.5 kb del intrón 13 (Fig. 8), por lo que la región que se fusiona a PBX1 incluye la región codificadora de los dominios transcripcionales (AD1 y AD2), pero no la del dominio bHLH. De tal forma que la proteína quimérica resultante contiene la región que codifica para el dominio de transactivación transcripcional de la proteína E2A y el dominio de unión al DNA de PBX1. El segmento que aporta PBX1 a la quimera E2APBX1 es un homeodominio de 60 aminoácidos que aumenta la capacidad de la quimera resultante para actuar como un factor de transcripción (63). Aunque se desconocen muchos de los mecanismos de oncogénesis de la proteína E2APBX1 en la leucemia, las evidencias experimentales sugieren que la translocación causa una sobreexpresión de PBX1 en las células linfoides, permitiendo así una mayor interacción de la proteína PBX1 con otras proteínas que se unen a secuencias específicas del DNA, lo que puede generar un aumento en la transcripción de genes blanco (63). La translocación cromosómica t(1:19) (q23;p13.3) se detecta en aproximadamente el 5% de todos los casos de LAL (Carroll y cols., 1984), y en el 20-25% de los casos de LAL de células pre-B cIg+. La t(1;19) es más frecuente entre pacientes no caucásicos, los cuales generalmente presentan mal pronóstico (63). FIGURA No. 14: REPRESENTACIÓN ESQUEMÁTICA DE LA FUSIÓN GÉNICA E2A-PBX1 A. Cariotipo de t(1;19). B. Estructura intrón-exón de los genes E2A y PBX1 C. Diagrama del transcrito quimérico E2A-PBX1 Fuente: Leukemia, Van Dongen y cols. 1999. K. IMPLICACIONES PARA EL PRONÓSTICO Y TRATAMIENTO. Por mucho tiempo y aún en la actualidad, las características clínicas y citomorfológicas de las células leucémicas son parámetros muy importantes en el diagnóstico de esta patología y han sido muy utilizadas para orientar el diseño de los esquemas terapéuticos y para mantener el control de la enfermedad. La masa inicial de células leucémicas, reflejada por el recuento leucocitario, la presencia de enfermedad extramedular y la edad son quizá los factores pronósticos más importantes que muestran una relación lineal con las probabilidades de sobrevida libre de enfermedad (SLE). Los niños mayores de 10 años de edad y los que tienen recuentos leucocitarios de más de 50x109/L tienen peor pronóstico que los de menor edad y con un recuento de leucocitos de <50x109/L. En los adolescentes, la LLA se presenta generalmente en varones y se caracteriza principalmente por la presencia de un recuento leucocitario elevado y tumoración en mediastino. Estos pacientes con frecuencia desarrollan una leucemia de células T, de tipo L2, CD10- y con características citogenéticas desfavorables (64). Con respecto al inmunofenotipo, los casos pre-B temprana son los que muestran una peor respuesta al tratamiento. La positividad CD10 confiere un pronóstico más favorable y es independiente en el caso de células T. Las LLA de células B, que antes se asociaba con un pronóstico extraordinariamente malo, tienen ahora un mayor índice de sobrevida a largo plazo (hasta de 80%) si se utiliza una terapia muy agresiva pero a corto plazo (2-6 meses). La afección al SNC continúa siendo un problema grave durante el tratamiento de cualquier tipo de leucemia, por lo que es considerada como un indicador de mal pronóstico ( Ver tabla No. 3) (64). En los últimos años, la biología molecular ha tenido un gran impacto en el tratamiento de la leucemia. De acuerdo con las alteraciones moleculares se valora el riesgo de recaída y el tratamiento está dirigido a mejorar los resultados cuando este es alto y a disminuir los efectos colaterales cuando es bajo. De tal forma que los pacientes de bajo riesgo son tratados con terapias menos agresivas, mientras que los tratamientos más agresivos y tóxicos están reservados para los pacientes de alto riesgo de recaída. En la última década, con la identificación de las anormalidades genéticas asociadas al pronóstico se ha logrado establecer una mejor clasificación de riesgo de los pacientes. Por ejemplo, la presencia del transcrito quimérico BCR-ABL en los pacientes con LLA, está asociado a una mal pronóstico y pobre respuesta al tratamiento aún con quimioterapia agresiva. La mayoría de los pacientes portadores de esta alteración genética, son niños mayores de 10 años, con una alto recuento de linfoblastos y generalmente presentan infiltración al SNC al momento del diagnóstico. Estos pacientes son Silvia Jiménez Morales 34 candidatos para terapias más innovadoras, transplante de médula ósea durante la primera remisión o inmunoterapia, pero a pesar de ello, el pronóstico es todavía muy pobre, comparado con los pacientes que no expresan a este gen quimérico. Se estima que la SLE en estos pacientes a 5 años es menor del 50% . Por otro lado, los pacientes portadores de la fusión génica E2A-PBX1 tienen mala respuesta a tratamientos no intensivos con una probabilidad de sobrevida aproximadamente del 50%, la cual incrementa hasta en un 80% cuando los pacientes son tratados con una quimioterapia más agresiva (51, 61). Evidencias recientes, sugieren que algunas translocaciones específicas pueden estar asociadas con un aumento en la SLE. Por ejemplo, se ha observado que los pacientes portadores de la t(12;21), tienen un pronóstico más favorable con tratamientos basados en antimetabolitos (una SLE de más del 90% a 5 años). Además, diversos estudios han mostrado que la t(12;21) es un marcador pronóstico independiente de otros factores de riesgo como son la edad y el recuento leucocitario (51,61). Además de sus implicaciones en el pronóstico, la caracterización de las anormalidades genéticas descritas anteriormente, permiten detectar la enfermedad mínima residual (EMR: niveles bajos de la enfermedad en pacientes que tienen remisión clínica y morfológica). En general la persistencia o el incremento de la EMR detectable por RT-PCR después de un transplante de médula ósea, está correlacionado con una subsecuente recaída. Por lo tanto el uso de herramientas moleculares puede también ser usado para la detección de translocaciones crípticas y monitorear la EMR y potencialmente prevenir la recaída en estos pacientes (51,61). TABLA No. 3: CARACTERÍSTICAS CON VALOR EN EL PRONÓSTICO DE LEUCEMIA LINFOBLÁSTICA AGUDA Fuente: Silvia Morales, Tesis Maestría en Biología. Universidad Metropolitana de México PARTE III III. RESULTADOS A todas las muestras analizadas, se les corrió inicialmente un gen control ABL, el cual se utilizó para evaluar la calidad del ADN y la eficacia de la extracción ( Ver Figura No. 15). FIGURA NO. 15: AMPLIFICACIÓN DEL GEN CONTROL ABL Se observa la banda amplificación del gen control ABL, comprobándose la buena calidad del cDNA de todas las muestras. Fuente: FODECYT 48-2009. Se analizaron un total de 143 pacientes para los cuatro transcritos propuestos: BCR-ABL, MLL-AF4, E2A-PBX1 Y ETV6-AML1, En la tabla No. 4, se observan los resultados obtenidos por cada una de las muestras analizadas para los cuatro transcritos. TABLA No. 4: RESULTADOS DEL ANÁLISIS DE LOS CUATRO TRANSCRITOS EN TODOS LOS PACIENTES. Paciente LLA-001 LLA-002 LLA-003 LLA-004 LLA-005 LLA-006 LLA-007 LLA-008 LLA-009 LLA-010 LLA-011 LLA-012 LLA-013 LLA-014 LLA-015 LLA-016 LLA-017 LLA-018 LLA-019 LLA-020 LLA -021 LLA-022 LLA-023 LLA-024 LLA-025 LLA-026 LLA-027 LLA-028 LLA-029 LLA-030 LLA-031 LLA-032 LLA-033 LLA-034 LLA-035 LLA-036 LLA-037 LLA-038 LLA-039 LLA-040 LLA-041 LLA-042 LLA-043 BCR-ABL Positivo e1:a2 Negativo Negativo Negativo Negativo Negativo Positivo b2:a2 Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Positivo Negativo Negativo Pendiente* Pendiente* Positivo Positivo Negativo Negativo Transcrito Quimérico MLL-AF4 E2A-PBX1 ETV6-AML1 Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente * Pendiente * Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente* Pendiente* Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Pendiente* Pendiente* Negativo Negativo Negativo Negativo LLA-044 Paciente LLA-045 LLA-046 LLA-047 LLA-048 LLA-049 LLA-050 LLA-051 LLA-052 LLA-053 LLA-054 LLA-055 LLA-056 LLA-057 LLA-058 LLA-059 LLA-060 LLA-061 LLA-062 LLA-063 LLA-064 LLA-065 LLA-066 LLA-067 LLA-068 LLA-069 LLA-070 LLA-071 LLA-072 LLA-073 LLA-074 LLA-075 LLA-076 LLA-077 LLA-078 LLA-079 LLA-080 LLA-081 LLA-082 LLA-083 LLA-084 LLA-085 LLA-086 LLA-087 LLA-088 LLA-089 LLA-090 LLA-091 LLA-092 LLA-093 LLA-094 Negativo BCR-ABL Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Transcrito Quimérico MLL-AF4 E2A-PBX1 Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo ETV6-AML1 Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo LLA-095 LLA-096 LLA-097 LLA-098 LLA-099 LLA-100 LLA-101 LLA-102 LLA-103 LLA-104 LLA-105 LLA-106 LLA-107 LLA-108 LLA-109 LLA-110 LLA-111 LLA-112 LLA-113 LLA-114 LLA-115 LLA-116 LLA-117 LLA-118 LLA-119 LLA-120 LLA-121 LLA-122 LLA-123 LLA-124 LLA-125 LLA-126 LLA-127 LLA-128 LLA-129 LLA-130 LLA-131 LLA-132 LLA-133 LLA-134 LLA-135 LLA-136 LLA-137 LLA-138 LLA-139 LLA-140 LLA-141 LLA-142 LLA-143 Negativo Negativo Positivo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Anulada(LMA) Positivo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Fuente: FODECYT 48-2009 Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Anulada Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Anulada Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Positivo Negativo Anulada Negativo Negativo Negativo Negativo Negativo Positivo Negativo Pendiente Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Negativo Según se observa el tabla No. 5; el transcrito BCR-ABL es el de mayor incidencia, encontrándose en un 7 %, este porcentaje corresponde al reportado en otros países. Por otro lado se observa un 4.5 % del marcador ETV6-AML1, el cual es un marcador de buen pronóstico, y este a diferencia de los países desarrollados se encuentra más bajo; ya que en estos países la incidencia es de aproximadamente un 20%; este transcrito es el único en el que se ha encontrado en Guatemala una gran diferencia, y de hecho correspondería mal pronóstico no tener tantos ETV6-AML1, ya que es un marcador de buen pronóstico. Y del marcador E2A-PBX1, se ha encontrado tres pacientes positivos, que corresponde a un 3% del total de pacientes analizados ( Ver figuras 16-22). TABLA No. 5: INCIDENCIA DE CADA UNOS DE LOS TRANSCRITOS TRANSCRITO CANTIDAD PORCENTAJE DE DE POSITIVOS INCIDENCIA BCR-ABL 10 7 % ETV6-AML1 6 4.5 % E2A-PBX1 3 2 % MLL-AF4 0 0% NEGATIVOS 124 87 % Fuente: FODECYT 48-2009 FIGURA No. 16: TRANSCRITO BCR-ABL; PACIENTES 1 Y 7 Fuente: FODECYT 48-2009 FIGURA No. 17: TRANSCRITO BCR-ABL; PACIENTES 24, 32 Y 35 Fuente: FODECYT 48-2009 FIGURA No. 18: TRANSCRITO BCR-ABL; PACIENTES 40,41 Y 97 Fuente: FODECYT 48-2009 FIGURA No. 19: TRANSCRITO BCR-ABL; PACIENTES 121 y 123 Fuente: FODECYT 48-2009 FIGURA No. 20: TRANSCRITO ETV6-AML1; PACIENTES 27, 120 Y 128. Fuente: FODECYT 48-2009 FIGURA No. 21: TRANSCRITO ETV6-AML1; PACIENTES 27 y 32 Fuente: FODECYT 48-2009 FIGURA No. 21: TRANSCRITO ETV6-AML1; PACIENTES 59 Fuente: FODECYT 48-2009 FIGURA No. 22: TRANSCRITO E2A-PBX1; PACIENTES 20, 43 y 47 Fuente: FODECYT 48-2009 En la tabla No. 6, se observa el pronóstico y recomendación de tratamiento que se brindó a cada paciente con algún transcrito positivo. En el caso de BCR-ABL, se orientó a mal pronóstico a diez pacientes, y además se les recomendó transplante de médula ósea en combinación con quimioterapia intensa. A tres pacientes positivos para E2A-PBX1, se les orientó a mal pronóstico y a quimioterapia más agresiva. Y por último a 6 pacientes ETV6AML1 positivo se les orientó a buen pronóstico. TABLA No. 6: PRONÓSTICO Y ORIENTACIÓN A TRATAMIENTO DE LOS TRANSCRITOS ENCONTRADOS EN LOS PACIENTES CANTIDAD ALTERACIÓN PACIENTES GENÉTICA 10 BCR-ABL PRONÓSTICO TRATAMIENTO RECOMENDADO Muy mal Quimioterapia muy agresiva y en pronóstico combinación con imatinib y transplante de médula ósea, si es posible 3 E2A-PBX1 Mal pronóstico Se recomienda la utilización de quimioterapias más agresivas para combatir esta leucemia 6 ETV6-AML1 Buen pronóstico Se recomienda la utilización de quimioterapias estándar para alcanzar la remisión completa. Fuente: FODECYT 48-2009 Por otro lado se obtuvo la procedencia de cada uno de los pacientes analizados ( Ver Tabla No. 7 y 8) en el presente proyecto, con el fin de correlacionar si existe una mayor incidencia de LLA en alguna región de Guatemala, y se observó que el mayor número de casos corresponde a la región de occidente y del departamento de Guatemala; este dato es muy interesante por lo que ha servido de base para el inicio de otro proyecto de investigación, que será de evaluar los factores de riesgo asociados a leucemia en la región occidente y centro del país. TABLA No. 7: PACIENTES CON LLA DISTRIBUIDOS POR MUNICIPIO DE GUATEMALA Paciente LLA-001 LLA-002 LLA-003 LLA-004 LLA-005 LLA-006 LLA-007 LLA-008 LLA-009 LLA-010 LLA-011 LLA-012 LLA-013 LLA-014 LLA-015 LLA-016 LLA-017 LLA-018 LLA-019 LLA-020 LLA-021 LLA-022 LLA-023 LLA-024 LLA-025 LLA-026 LLA-027 LLA-028 LLA-029 Departamento/Muncipio San Andrés, Sololá Huehuetenango Quiché Pajapita, San Marcos Guatemala Guatemala Quiché Guatemala Puerto Barrios, Izabal Quetzaltenango San Ixnam, Totonicapán Mixco, Guatemala Jutiapa San Juan Ostuncalco, Quetzaltenango Suchitepequez Guatemala San Marcos Jalapa Suchitepequez Santa Rosa Amantitlán,Guatemala Rethalhuleu Petén Huehuetenango Guatemala Guatemala Quetzaltenango Suchitepequez Sanarate, el Progreso Paciente LLA-030 LLA-031 LLA-032 LLA-033 LLA-034 LLA-035 LLA-036 LLA-037 LLA-038 LLA-039 LLA-040 LLA-041 LLA-042 LLA-043 LLA-044 LLA-045 LLA-046 LLA-047 LLA-048 LLA-049 LLA-050 LLA-051 LLA-052 LLA-053 LLA-054 LLA-055 LLA-056 LLA-057 LLA-058 LLA-059 LLA-060 LLA-061 LLA-062 LLA-063 LLA-064 LLA-065 LLA-066 LLA-067 LLA-068 LLA-069 LLA-070 LLA-071 LLA-072 LLA-073 Departamento/Muncipio Sololá Progreso Coatepeque, Quetzaltenango Guatemala Guatemala Huehuetenango Santa Lucía Chimaltenango Chimaltenango ---------------* Sacatepéquez Guatemala Sacatepéquez Jalapa Quiché Jutiapa Amatitlán,Guatemala ------------------------------Huehuetenango Huehuetenango Quiché Quetzaltenango Guatemala Guatemala -------------------------Amatitlán,Guatemala -----------Petén Suchitepéquez San Marcos Sacatepéquez Jutiapa Quiché -------------------------------Santa Rosa Huehuetenango Villa Canales,Guatemala Guatemala Chimaltenango Quiché Paciente LLA-074 LLA-075 LLA-076 LLA-077 LLA-078 LLA-079 LLA-080 LLA-081 LLA-082 LLA-083 LLA-084 LLA-085 LLA-086 LLA-087 LLA-088 LLA-089 LLA-091 LLA-092 LLA-093 LLA-094 LLA-095 LLA-096 LLA-097 LLA-098 LLA-099 LLA-100 LLA-101 LLA-102 LLA-103 LLA-104 LLA-105 LLA-106 LLA-107 LLA-108 LLA-109 LLA-110 LLA-111 LLA-112 LLA-113 LLA-114 LLA-115 LLA-116 LLA-117 LLA-118 Departamento/Muncipio Antigua Guatemala, Sacatepéquez San Pedro Sacatepéquez Guatemala Guatemala ----------------Antigua Guatemala, Sacatepéquez San Pedro Sacatepéquez San Marcos -------------------------------Tectitlán Quiché Guatemala Belice -----------Quiché Sta. Lucía Cotz. Escuintla Guatemala Amatitlán, Guatemala San Juan Alotenango ---------------Chinautla, Guatemala San Marcos Suchitepéquez San Marcos San Marcos Guatemala Sololá San Marcos Sta. Rosa ----------------Malacatán, San Marcos ----------------Jutiapa Quetzaltenango Jacaltenango San Marcos Quiché Escuintla San Marcos Pendiente Pendiente Amatitlán,Guatemala Escuintla Sanarate,El Progreso Paciente LLA-119 LLA-120 LLA-121 LLA-122 LLA-123 LLA-124 LLA-125 LLA-126 LLA-127 LLA-128 LLA-129 LLA-130 LLA-131 LLA-132 LLA-133 LLA-134 LLA-135 LLA-136 LLA-137 LLA-138 LLA-139 LLA-140 LLA-141 LLA-142 LLA-143 Departamento/Muncipio Rethalhuleu Guatemala Chimaltenango Anulada Totonicapán Pendiente San Benito, Petén Momostenango,Totonicapán Escuintla San Pedro Ayampuc,Guatemala Quetzaltenango Guatemala Pendiente Guatemala Guatemala Cobán, Alta Verapaz Momostenango,Totonicapán Quiché Villa Nueva,Guatemala Alta Verapaz Guatemala Guatemala Jutiapa Chiquimula Guatemala Fuente: FODECYT 48-2009 Tabla No 8: CANTIDAD DE PACIENTES ORGANIZADOS POR REGION REGIONES DEPARTAMENTO NUMERO DE PACIENTES DIAGNOSTICADOS OCCIDENTE HUEHUETENANGO 6 QUICHE 10 SAN MARCOS SOLOLA 11 4 QUETZALTENANGO 7 TOTONICAPAN 4 TOTAL 42 1 NORTE IZABAL 3 PETÉN TOTAL CENTRO GUATEMALA , SA CATEPEQUEZ, CHIMALTENANGO SUR RETALHULEU 4 45 2 5 SUCHITEPEQUEZ 3 SANTA ROSA ESCUINTLA ORIENTE 5 TOTAL 15 JUTIAPA 6 3 EL PROGRESO 2 JALAPA TOTAL Sin clasificar Fuente: FODECYT 48-2009 11 24 Con el objetivo de validar los resultados positivos obtenidos se escogieron cuatro pacientes para enviar a secuenciar el ADN, y luego correlacionar la secuencia con los transcritos estudiados. Los pacientes que se enviaron para secuenciación son No. 40, 41, 43 y 97. A. PACIENTE No.97: POSITIVO PARA EL TRANSCRITO B2A2 DE BCRABL A.1 SECUENCIACIÓN DEL FRAGMENTO: BCR-ABL1 F2 A.II. SECUENCIA OBTENIDA: GGGGTCATTTTCCTGGGTCCAGCGAGAAGGTTTTCCTTGGAGTTCCAACGAG CGGCTTCACTCAGACCCTGAGGCTCAAAGTCAGATGCTACTGGCCGCTGAA GGGCTTCTGCGTCTCCATGGAAGGCGCCCTCGCCATCGTTGGGCCAGATCN NN El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 23, en donde la secuencia corresponde a dos fragmentos de dos genes distintos. Y en la figura No. 24 se puede observar que las secuencias corresponden al cromosoma 9 y cromosoma 22 respectivamente con una identidad que se encuentra en 99100% FIGURA No. 23: RESULTADO DE ALINEACIÓN DE SECUENCIA DE ADN OBTENIDA, PACIENTE No. 97 Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 24: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 97 Fuente: Análisis bioinformático realizado en NCBI. En la figura No. 25: se observa la alineación que se da entre la secuencia obtenida del paciente ( query) y la secuencia del cromosoma 9 que es complementaria; se puede observar claramente que la secuencia es complementaria con el un fragmento del gen ABL. FIGURA No. 25: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN ABL, PACIENTE No. 97 Fuente: Análisis bioinformático realizado en NCBI En la figura No. 26 se observa la alineación de la secuencia analizada (query) con el gen BCR del cromosoma 22. FIGURA No. 26: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 97 Fuente: Análisis bioinformático realizado en NCBI A.III SECUENCIACIÓN DEL FRAGMENTO BCR-ABL2 BCR-ABLf2 A.IV SECUENCIA OBTENIDA: GGGGTCATTTTCNCTGGGTCCAGCGAGAAGGTTTTCCTTGGAGTTCCAACGA GCGGCTTCACTCAGACCCTGAGGCTCAAAGTCAGATGCTACTGGCCGCTGA AGGGCTTCTGCGTCTCCATGGAAGGCGCCCTCGCCATCGTTGGGCCAGATCT A El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 27, en donde la secuencia corresponde a dos fragmentos de dos genes distintos. Y en la figura No. 28 se puede observar que la secuencias corresponden al cromosoma 9 y cromosoma 22 respectivamente con una identidad que se encuentra en 99100% FIGURA No. 27: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 97 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 28: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 97 (2) Fuente: Análisis bioinformático realizado en NCBI En la figura No. 29: se observa la alineación que se da entre la secuencia obtenida del paciente ( query) y la secuencia del cromosoma 9 que es complementaria; se puede observar claramente que la secuencia es complementaria con el un fragmento del gen ABL. En la figura No. 30 se observa la alineación de la secuencia analizada (query) con el gen BCR del cromosoma 22. FIGURA No. 29: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN ABL, PACIENTE No. 97 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 30: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 97 (2) Fuente: Análisis bioinformático realizado en NCBI En base a los resultados obtenidos en la página del NCBI, mostrados anteriormente se puede concluir que el producto de la secuenciación del paciente No. 97 corresponde a un fragmento del gen ABL y un fragmento del gen BCR; confirmándose la presencia del transcrito BCR-ABL, que se obtuvo positivo por PCR. B. PACIENTE No. 43: POSITIVO PARA EL TRANSCRITO E2A-PBX1 B.1 SECUENCIA DEL TRANSCRITO E2A-PBX1 ( PRIMER FOWARD) GTCTCGGCCTCCGACTCCTACAGTGTTTTGAGTATCCGAGGAGCCCAGG AGGAGGAACCCACAGACCCCCAGCTGATGCGGCTGGACAACATGCTGTT AGCGGAAGGCGTGGA El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 31, en donde la secuencia corresponde a un fragmento de un gen. Y en la figura No. 32 se puede observar que la secuencias corresponden al cromosoma con una identidad que se encuentra en 99-100% FIGURA No. 31: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 43 Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 32: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 43 Fuente: Análisis bioinformático realizado en NCBI En la figura No. 33, se observa la complementariedad de las secuencias analizadas con el gen PBX1; 88 bases corresponden al Gen PBX1 del cromosoma 1. FIGURA No. 33: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN PBX1, PACIENTE No. 43 Fuente: Análisis bioinformático realizado en NCBI B.2 SECUENCIA DEL GEN E2A-PBX1 reverse GTCTGTGGGTTCCTCCTCCTGGGCTCCTCGGATACTCAAAACACTGTAGGAG TCGGGAGGCCGAGACAGGTCAGGGAGGGTGCCTGGCTGGCTGGGGAGGGC CGCGTGGTTGTGCA El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 34, en donde la secuencia corresponde a dos fragmentos de dos genes distintos. Y en la figura No. 35 se puede observar que la secuencias corresponden al cromosoma 19 y cromosoma 1 respectivamente con una identidad que se encuentra en 99100% FIGURA No. 34: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 43 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 35: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 43 (2) Fuente: Análisis bioinformático realizado en NCBI En la figura No. 36 se puede observar que 75 bases analizadas corresponden al gen E2A del cromosoma 19; y en la figura No. 37 se puede observar que 42 bases corresponden al gen PBX1 del cromosoma 1 FIGURA No. 36: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN E2A, PACIENTE No. 43 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 37: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN PBX1, PACIENTE No. 43 (2) Fuente: Análisis bioinformático realizado en NCBI CONCLUSION: En base a los resultados obtenidos en la página del NCBI, mostrados anteriormente se puede concluir que el producto de la secuenciación del paciente No. 43, corresponde a un fragmento del gen E2A y un fragmento del gen PBX1; confirmándose la presencia del transcrito E2A-PBX1, que se obtuvo positivo por PCR. C. PACIENTE No. 40: POSITIVO PARA EL TRANSCRITO B2A2 DE BCR-ABL C.1 SECUENCIA BCR-ABL3-BCR-ABLF1.ab1 ATCNGACTGTCACAGCATTCCGCTGACCATCAATAAGGAAGAAGCCCTTCAGC GGCCAGTAGCATCTGACTTTGAGCCTCAGGGTCTGAGTGAAGCCGCTCGTTGG AACTCCAAGGAAAACCTTCTCGCTGGACCCAGTGAAAATGACCCCAACCTTTTC GTTGCACTGTATGATTTTGTGCCAGTGGAGATAA El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 38, en donde la secuencia corresponde a dos fragmentos de dos genes distintos. Y en la figura No. 39 se puede observar que la secuencias corresponden al cromosoma 9 y cromosoma 22 respectivamente con una identidad que se encuentra en 99100% FIGURA No. 38: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 40 Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 39: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 40 Fuente: Análisis bioinformático realizado en NCBI En la figura No. 40 se puede observar que 155 bases de la secuencia analizada corresponden al gen ABL del cromosoma 9 y en la figura No. 41 que 38 bases corresponden al gen BCR del cromosoma 22 FIGURA No. 40: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN ABL1, PACIENTE No. 40 Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 41: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 40 Fuente: Análisis bioinformático realizado en NCBI C.II SECUENCIA BCR-ABL3-BCR-ABLF2.ab1 GGGGTCATTTTCCTGGGTCCAGCGAGAAGGTTTTCCTTGGAGTTCCAAC GAGCGGCTTCACTCAGACCCTGAGGCTCAAAGTCAGATGCTACTGGCCG CTGAAGGGCTTCTTCCTTATTGATGGTCAGCGGAATGCTGTGGACAGTCT GGAGTTTCACACACGAGTTGGTCAGCATCTGCAGCTCCAA El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 42, en donde la secuencia corresponde a dos fragmentos de dos genes distintos. Y en la figura No. 43 se puede observar que la secuencias corresponden al cromosoma 9 y cromosoma 22 respectivamente con una identidad que se encuentra en 99100% FIGURA No. 42: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 40 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 43: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 40 (2) Fuente: Análisis bioinformático realizado en NCBI En la figura No. 44 se puede observara que 111 bases de la secuencia analizada corresponden al gen ABL del cromosoma 9; y en la figura No. 45 se puede observar que 78 bases corresponden al gen BCR del cromosoma 22. FIGURA No. 44: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN ABL, PACIENTE No. 40 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 45: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 40 (2) Fuente: Análisis bioinformático realizado en NCBI CONCLUSION: En base a los resultados obtenidos en la página del NCBI, mostrados anteriormente se puede concluir que el producto de la secuenciación del paciente No.40 corresponde a un fragmento del gen ABL y un fragmento del gen BCR; confirmándose la presencia del transcrito BCR-ABL, que se obtuvo positivo por PCR. D. PACIENTE No. 41: POSITIVO PARA EL TRANSCRITO E2A2 DE BCR-ABL D.I SECUENCIA BCR-ABL4-ABL-R.ab1 CAANNTTCAGCGGCCAGTAGCATCTGACTTTGAGCCTCAGGGTCTGAGT GAAGCCGCTCGTTGGAACTCCAAGGAAAACCTTCTCGCTGGACCCAGTG AAAATGACCCCAACCTTTTCGTTGCACTGTATGATTTTGTGGCCAGTGGA GATA El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 46, en donde la secuencia analizada corresponde a un fragmento en el genoma. Y en la figura No. 47 se puede observar que la secuencia corresponde al cromosoma 9 con una identidad que se encuentra en 99-100% FIGURA No. 46: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 41 Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 47: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 41 Fuente: Análisis bioinformático realizado en NCBI En la figua No. 48 se puede observar que 147 bases de la secuencia corresponden al gen ABL del cromosoma 9. FIGURA No. 48: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 41 Fuente: Análisis bioinformático realizado en NCBI D.II SECUENCIA BCR-ABL4-BCR-ABLF2.ab1 NNGGNNNNANNNNGGGGTCATTTTCCTGGGTCCAGCGAGAAGGTTTTCC TTGGAGTTCCAACGAGCGGCTTCACTCAGACCCTGAGGCTCAAAGTCAG ATGCTACTGGCCGCTGAAGGGCTTCTGCGTCTCCATGGAAGGCGCCCTC GCCATCGTTGGGCCAGATCN El análisis del producto de secuenciación se llevó a cabó en la página del NCBI ( NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION), se puede observar en la figura No. 49, en donde la secuencia analizada corresponde a dos fragmentos distintos en el genoma. Y en la figura No. 50 se puede observar que la secuencia corresponde al cromosoma 9 y al 22 con una identidad que se encuentra en 99-100% FIGURA No. 49: ALINEACIÓN DE LA SECUENCIA OBTENIDA, PACIENTE No. 41 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 50: CROMOSOMAS QUE CONTIENEN LA SECUENCIA ANALIZADA, PACIENTE No. 41 (2) Fuente: Análisis bioinformático realizado en NCBI En la figura No. 51 se puede observar que 112 bases de la secuencia analizada corresponden al gen ABL del cromosoma 9; y en la figura No. 52 se puede observar que 44 bases corresponden al gen BCR del cromosoma 22. FIGURA No. 51: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN ABL, PACIENTE No. 41 (2) Fuente: Análisis bioinformático realizado en NCBI FIGURA No. 52: ALINEACIÓN DE LA SECUENCIA OBTENIDA CON EL GEN BCR, PACIENTE No. 41 (2) Fuente: Análisis bioinformático realizado en NCBI III.1 DISCUSIÓN DE RESULTADOS Se analizaron un total de 143 pacientes con diagnóstico de leucemia linfoblástica aguda, referidos de distintos hospitales nacionales del país. Para comprobar la calidad del cDNA, a todas las muestras se les amplificó el gen control ABL ( Ver figura No. 15). De todos los pacientes analizados se detectaron 10 pacientes positivos para el transcrito BCR-ABL (7%); 6 pacientes positivos para el transcrito ETV6-AML1 (4.5%), y 3 pacientes positivos para el transcrito E2A-PBX1 (2%). Y por otro lado ningún paciente presentó el transcrito MLL-AF4. ( Ver figuras 16-22 y tabla No. 4 y 5). El transcrito BCR-ABL, que procede de la translocación t(9;22) , es una alteración que se ha asociado a muy mal pronóstico en pacientes con LLA; caso muy contrario es cuando se encuentra en leucemia mieloide crónica, ya que para estos pacientes se suele tratar exitosamente con un tratamiento molecular llamado Imatinib. Sin embargo este tratamiento en LLA no es del todo efectivo; por lo que un paciente con LLA BCR-ABL positivo, posee un pronóstico muy desfavorable, y deja casi como única alternativa, un trasplante de médula ósea. A los 10 pacientes positivos para BCR-ABL, se les informó de mal pronóstico y se les orientó a un tratamiento más agresivo, como lo es combinación de quimioterapia con trasplante de médula ósea. (Ver tabla No. 6) El porcentaje encontrado en Guatemala corresponde a un 7%, lo que es muy similar a lo encontrado en otros países desarrollados y en vías de desarrollo, en los cuales la incidencia suele ser alrededor de un 5% (16). Esto en pacientes pediátricos, porque se sabe que en los adultos con LLA se puede llegar a tener una incidencia hasta del 30%. Como parte de la caracterización genética que se esta realizando mediante este proyecto en Guatemala, se puede establecer que el transcrito BCR-ABL se encuentra un 7% en un total de 143 pacientes analizados. (16) El transcrito E2A-PBX1, que proviene de la translocación t(1;19); se encuentra asociado a un mal pronóstico; no tan agresivo como BCR-ABL, pero si orienta a tratamiento con quimioterapias más agresivas. La incidencia encontrada en Guatemala de este transcrito es del 2%, porcentaje que no difiere considerablemente de lo reportado en países desarrollados como en vías de desarrollo, en donde se ha reportado hasta un 5% de incidencia ( Ver tabla No. 6) (31). El transcrito ETV6-AML1, que procede de la t(12,21); es una alteración considerada de buen pronóstico; ya que los pacientes que presentan este transcrito responden muy bien a quimioterapia estándar; y poseen índices de recaídas muy bajos. Este transcrito se encontró en un 4.5 % de frecuencia en la población guatemalteca. ( Ver Tabla No. 6). Existen una gran variedad de países en los cuales se ha reportado una alta incidencia del gen de fusión ETV6-AML1: En un estudio realizado en Hong-Kong, a 67 pacientes con leucemia linfoblástica aguda; se encontró el 17.9% del transcrito ETV6-AML1, en Taiwan, se analizaron 433 pacientes, y se encontró un 15.8% de frecuencia del gen ETV6-AML1; en la población Hungara se encontró un 20% de incidencia del gen ETV6-AML1; en Alemania e Italia, se encontró una incidencia de 18.9%; en Tunisia se encontró 28% de frecuencia de ETV6-AML1; en Brazil, se encontrado de un 19 hasta un 40% de casos positivos para ETV6-AML1; en Costa Rica, un 18% del marcador ETV6-AML1, en China, 22.8% de pacientes positivos para ETV6-AML1 y en Malasia se reporta un 19% de frecuencia de ETV6-AML1 (32-42). Por otro lado hay algunos países que han reportado una baja frecuencia del gen ETV6-AML1, por debajo del 15%. En un estudio realizado en Chile, se encontró un 13 % del gen ETV6-AML1 (43) . En un estudio internacional que se realizó en cuatro países en vías de desarrollo: India, Pakistan, Myanmar y Sudan; en donde se analizaron 181 niños, se encontró una baja incidencia el marcador ETV6-AML (7.4%). Y en este estudio se encontró un alza en dos marcadores de mal pronóstico MLL-AF4 17% y BCR-ABL (22%) (44). En España, se analizaron 101 casos de LLA, de los cuales todos fueron negativos para ETV6-AML1, 38 de los 101 pacientes eran pediátricos. Estos resultados sugieren la existencia de variaciones genéticas y de raza en el genotipo de leucemia linfoblástica aguda (45). En un estudio realizado en el Sur de la India, se analizaron 42 pacientes con LLA y se encontró únicamente un 4.8% de casos positivos para ETV6-AML1; en este estudio se concluye que la incidencia de este transcrito para la población India es menor (46) En la tabla No. 9, se observa un resumen de la frecuencia del gen ETV6-AML1 publicada en distintas revistas internacionales; en donde se puede observar una menor incidencia de esta alteración en la India y en España, datos que coinciden con los encontrados en Guatemala TABLA No. 9: FRECUENCIA DE ETV6-AML1 EN DISTINTOS PAÍSES LOCALIZACIÓN NUMERO DEL ESTUDIO PACIENTES DE FRECUENCIA % REFERENCIA India 42 4.8 Hill Anita, et al. Inglaterra 56 39 Codrington et al. Australia 66 33 Amor et al. Alemania/Italia 334 18.9 Borkhardt et al. Taiwan 165 18 Liang et al. Brazil 67 17.9 Magalhaes et al. USA 86 17% Jamil et al. Taiwan 41 17 Liang et al. Hong Kong 75 16 Tsang et al. Suiza 72 15 Andreasson et.al. Scotland 36 11.1 Spathas et al. Japan 74 9.5 Eguchi-Ishimae et al. India 46 9 Inamdar et al. India 259 7 Siraj et al. España 41 2 García- Sanz et al. Fuente: Hill A, et al. (2005) Haematologica 90: 414-416 En un estudio sobre poblaciones en donde participó nuestro equipo; se analizaron poblaciones mayas y se compararon sus polimorfismos con otras etnias. Se encontró que la población maya tiene mayor similitud con Europa con una frecuencia alélica de 0.38, esto se podría explicar por la conquista española. Una frecuencia alélica común con la población africana; y con el sur y este de Asia un 0.38 de frecuencia alélica. Si se puede observar en la tabla No. 8, las frecuencias más bajas del marcador ETV6-AML1 se han reportado en España y en la India, la cuales coinciden con las encontradas en Guatemala. Y Si bien es cierto se piensa que la LLA es en un 90% genética adquirida no heredada, pareciera existir un patrón de predisposición genética según la población; ya que España, la India y Guatemala poseen una incidencia de ETV6-AML1 extremadamente baja. Sería interesante realizar más estudios relacionados con esto (47). En otro estudio realizado en LLA en dos tipos de poblaciones; Hispánicas y blancos no hispánicos; también se observó que en Hispanos la frecuencia de ETV6-AML1 es de un 13%, mientras que en no hispánicos blancos es significativamente mayor, 24 %. En este estudio fue la primera vez en la que se observó que una variación étnica afectaba la presencia de dicho marcador. Aunque este estudio fue realizado en Estados Unidos, pareciera existir una menor incidencia en personas con descendencia hispánica; lo que sería congruente con lo encontrado en Guatemala (48) Otros países que han reportado una incidencia por debajo del 15 % son Nicaragua y Korea (49-50) Por otro lado se analizó la distribución geográfica dentro de Guatemala, y se encontró que más del 50% de casos provienen de la región del occidente y Centro del país; 30% de la región del Occidente y 32% de la región del Centro. Y coincide también que en estas regiones predominaron los marcadores genéticos encontrados. ( Ver tablas No. 7 y 8) Estos datos muestran claramente una afectación de LLA geográfica, que puede deberse a una gran variedad de factores; como la posible existencia de una predisposición genética, según lo analizado anteriormente con el marcador ETV6-AML1; y por otro lado una mayor presencia de factores de riesgo asociado. Se ha empezado otro proyecto para estudiar factores de riesgo en estos pacientes, se evaluará la presencia de agroquímicos, contaminantes en agua, riesgos laborales, elementos y compuestos tóxicos, entre otros. Y por último para validar la técnica estandarizada en el presente proyecto, se enviaron a secuenciar las muestras de DNA de los siguientes pacientes: No. 40, 41, 43 y 97. De los cuales se puede concluir que todos los productos de la secuenciación coinciden con los genes BCR-ABL y E2A-PBX1 analizados ( Ver figuras 23-52). PARTE IV. IV.1 CONCLUSIONES 1. De los 143 pacientes con diagnóstico de leucemia linfoblástica aguda, 19 pacientes presentaron alguno de los cuatro transcritos de los incluidos en el estudio. 2. La estandarización de la técnica de PCR Multiplex, se realizó mediante la utilización de líneas celulares, y secuenciación de los pacientes con transcritos positivos. 3. En el presente estudio se encontraron, 10 pacientes positivos para el transcrito BCR-ABL ( 7%); 6 pacientes positivos para el transcrito ETV6-AML1 (4.5%), y 3 pacientes positivos para el transcrito E2A-PBX1 (2%); y ningún paciente presentó el transcrito MLL-AF4. 4. La baja incidencia del gen ETV6-AML1 encontrada en Guatemala, coincide con países como España e India, con los cuales la población guatemalteca comparte más polimorfismos genéticos. 5. La incidencia de los transcritos BCR-ABL y E2A-PBX1 encontrada en Guatemala, es similar a lo reportado en el resto del mundo; por lo que no se encontró diferencias debido a distinta etnia poblacional. 6. Los pacientes que presentaron los transcritos BCR-ABL y E2A-PBX1, fueron informados sobre mal pronóstico del desarrollo de la leucemia; y por lo tanto orientados a esquemas de terapias más agresivas, y los pacientes que presentaron el transcrito ETV6-AML1 fueron informados de buen pronóstico, y orientados a esquemas de terapia estándar. 7. En base a los resultados obtenidos, se ha caracterizado genéticamente por primera vez en Guatemala, los cuatro transcritos que se asocian mundialmente a leucemia linfoblástica aguda. 8. La divulgación del presente estudio, se ha realizado mediante la elaboración de póster y trifoliares, repartidos en los distintos hospitales. 9. La región Occidente y Centro de Guatemala, posee más del 50% de casos de Leucemia linfoblástica aguda y de marcadores genéticos, sugiriendo la presencia de factores de riesgo asociados en estas regiones. IV.2 RECOMENDACIONES 1. Estudiar la presencia de algunos otros marcadores genéticos asociados a leucemia linfoblástica aguda como mutaciones en el gen FLT3 y p53. 2. Extender la utilización la técnica de PCR MULTIPLEX, a otros tipos de cáncer, para un mejor manejo de la enfermedad. 3. Ampliar el estudio a un mayor número de pacientes, para comprobar si la incidencia encontrada en el presente estudio se mantiene igual. 4. Realizar futuros proyectos que confirmen la relación ancestral de la población maya guatemalteca, con la población de España y de la India. 5. Comprobar que los marcadores BCR-ABL y E2A-PBX1, no presentan asociación con las variaciones en el genoma humano en distintas poblaciones. 6. Continuar con la evaluación de estos marcadores genéticos para toda la población guatemalteca con leucemia linfoblástica aguda; para poder establecer el pronóstico y orientar a tratamiento a todos los nuevos casos presentados en los hospitales nacionales. 7. Caracterizar genéticamente otros tipos de cáncer, con el objetivo de conocer la genética del cáncer en Guatemala. 8. Divulgar los resultados obtenidos a la comunidad científica internacional, mediante una publicación en una revista científica de prestigio. 9. Analizar factores de riesgo asociados, a la presencia de mayor leucemia linfoblástica aguda en las regiones del Occidente y Centro de Guatemala. IV.3 REFERENCIAS BIBLIOGRÁFICAS 1. Calazans M. (2002). Revisión de Técnicas de citogenética convencional y molecular y su implicación en el diagnóstico y pronóstico del cáncer. España: Ediciones Universidad de Navarra. 2. Haferlach T, Wolfgang K, Schnittger S and Schoch C (2005). Modern Diagnostics in acute leukemias. Criticals reviews in oncology/hematology, 56, 223-234. 3. Odero M. Et al. (2007). Manual de prácticas de Genética. España: Ediciones Facultad de Ciencias, Medicina, Universidad de Navarra. 4. Arangure, J. et al. (2005). Incidence of leukemias in children from el Salvador and México city between 1996-2000: Population based-data. BMC Cancer, 5, 1-9. 5. Pallisgaard, N. et al. (1998). Multiplex Reverse Transcription-Polymerase Chain Reaction for Simultaneous Screening of 29 Translocations and Chromosomal Aberrations in Acute Leukemia. Blood, 92, 574-588. 6. Scurto, P et al. (1998). A multiplex RT-PCR assay for the detection of chimeric transcripts encoded by the risk-stratifying translocations of pediatric acute lymphoblastic leukemia. Leukemia, 12, 1994-2005. 7. Viehmann, S. et al. (1999). Multiplex PCR- a rapid screening method for detection of gene rearrangements in childhood acute lymphoblastic leukemia. Ann Hematol, 78, 157-162. 8. Marin C. et al. (2001). Multiplex-polymerase chain reaction assay for the detection of prognostically significant translocations Haematologica, 86, 1254-1260. in acute lymphoblastic leukemia. 9. Cerveira, N. (2000). Detection of prognostic significant translocations in childhood acute lymphoblastic leukaemia by one-step multiplex reverse transcription polymerase chain reaction. Bristish Journal of Haematology, 109, 638-640. 10. Carranza C. (2007). Caracterización genética de pacientes con neoplasias mieloides y alteraciones del brazo largo del cromosoma 3. Mecanismo de sobreeexpresión y análisis funcional del promotor del gen EVI1. Memoria de investigación. Suficiencia Investigadora. España: Ediciones Facultad de Ciencias, Universidad de Navarra, Centro de Investigación Médica Aplicada. –CIMA-. 11. Perez- Vera P, Sánchez M, Carnevale A, Riva Roberto, Paredes R, Martínez A and Frías S. (2001). Cytogenetics in acute lymphoblastic leukemia in Mexican Children: An Institutional Experience. Archives of Medical Research. 32, 202-207. 12. Mrozek K, Heerema N and Bloomfield C. (2004). Cytogenetics in acute leukemia. Blood Reviews. 18, 115-136. 13. Hilden J, et al. (1995). Molecular analysis in acute lymphoblastic leukemia: MLL Rearrangement and reverse transcriptase reaction chain for t(4;11) (q21;q23). Blood, 86, 3876-3882. 14. Bose S, Deininger M, Gora J, Goldman J an Melo J. (1998).The presence of typical or atypical BCR-ABL fusion genes in Leucocytes or normal individuals: Biological significance and Implications for the assessment of minimal residual disease. Blood, 92, 3362-3367. 15. Deininger M, Goldman J and Melo J. (2000). The molecular biology of chronic myeloid leukemia. Blood. 3343-3356. 16. Radich J. (1997). Detection of BCR-ABL transcripts in Philadelphia Chromosomepositive Acute Lymphoblastic leukemia after marrow transplantation. Blood , 89, 26022609. 17. Martinelli G. et al. (1998). Detection or BCR-ABL transcript chronic myelogenous leukemia patients by reverse-transcription-polymerase chain reaction and capillary electrophoresis. Haematologica, 593-601. 18. Melo J. et al. (1993). The ABL-BCR fusion Gene is expressed in Chronic myelogenous leukemia. Blood, 81, 158-165. 19. Kaeda J. et al. (2006). Serial Measurement of BCR-ABL transcripts in the peripheral blood after allogenic stem cell transplantation for chronic myeloid leukemia: an attempt to define patients who may not require further therapy. Blood, 107, 4171-4176 . 20. Golub T, et al. (1995). Fusion of the Tel gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA, 92, 4917-4921. 21. Harbott J, et al. (1997). Incidence of TEL/AML1 fusion gene analyzed consecutively in Children acute lymphoblastic leukemia in relapse. Blood, 90, 4933-4937. 22. Zelent A. (2004). Role of TEL- AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukemia. Oncogene, 23, 4275-4283. 23. Wiemels J. (1999). Structure and possible mechanisms of TEL-AML1 gene fusions in Chilhood Acute Lymphoblastic Leukemia. Cancer Research, 59, 4075-4082. 24. Hilden JM. (1995). Molecular analysis of infant acute lymphoblastic leukemia: MLL gene rearrangement and reverse transcriptase polymerase chain reaction for t(4;11)(q21;q23). Blood, 86, 3876-3882. 25. Behm FG. Et al. (1996). Rearrangement of MLL gene confers poor prognosis in childhood acute lymphoblastic leukemia. Blood, 2870-2877. 26. Yoshiyuki K. et al. (2004). Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intense chemotherapy and hematopoietic stem cells transplantation. Blood, 104, 3527-3534. 27. Harper D. et al. (2008). Chromosomal rearrangements leading to MLL gene fusions: Clinical and biological aspects. Cancer Research, 68, 10024-10027. 28. Piccaluga P. et al. (2006). Poor outcome of adult acute lymphoblastic leukemia patients carrying the t(1;19)(q23;p13) translocations. Leukemia and lymphoma, 47, 469-472. 29. Lanza C. et al. (1996). Persistense EA2-PBX1 transcript in t(1;19) childhood acute lymphoblastic leukemia: correlation with chemotherapy intensity and clinical outcome. Leukemia research, 20, 441-443. 30. Foa R. et al. (2003). EA2-PBX1 fusion in adult acute lymphoblastic leukemia: biological and clinical features. British Journal of hematology, 120, 484-487. 31. Hunger Sp. (1996). Chromosomal translocations involving E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood, 87, 12111224. 32. Kam Sze, et al. (2001). TEL/AML1 Rearrangement and the Prognostic Significance in Childhood Acute Lymphoblastic Leukemia in Hong Kong. American Journal of Hematology, 68, 91-98. 33. Liang D, et al. (2010). Frequencies of ETV6-RUNX1 Fusion and Hyperdiploidy in Pediatric Acute Lymphoblastic Leukemia Are Lower y Far East than West. Leukemia, 7, 23-25. 34 Laszló P, et al. (2001) INTERNATIONAL NOTE: Characteristics of TEL-AML1 Positive Acute Lymphoblastic Leukemia in Hungarian Children. Med Pediatr Oncol, , 37, 409-411. 35 Arndt B. et al. (1997). Incidence and Clinical Relevance of TEL/AML1 Fusion Genes in Children with Acute Lymphoblastic Leukemia enrolled in the German an Italia multicenter therapy trials. Leukemia, 90, 571-577. 36. Gmidene Abir, et al. (2009). ETV6-RUNX1 Rearrangement in Tunisian Pediatric Blineage Acute Lymphoblastic Leukemia. Advances in Hematology, 5, 30-38. 37. Magalhaes, I. (2000). TEL-AML1 Fusion gene frequency in pediatric acute lymphoblastic leukaemia in Brazil. British Journal of Haematology, 111, 204-207. 38. Zen P. (2004). Prevalence of TEL-AML1 fusion gene in Brazilian pediatric patients withe acute lymphoblastic leukemia. Cancer Genetics and Cytogenetics, 151, 68-72. 39. Acosta B. et al. (2004). High frecuency of t(12;21)(p13;q22) in Children with acute lymphoblastic leukemia and known clinical outcome in southern Brazil. Leukemia, 28, 1033-1038. 40. Santamarina C. et al. ( 2009). Acute leukemia. J Pediatr Hematol Oncol, 31, 131–135. 41. Gao Yi-Jin (2007). Prevalence of ETV6-AML1 fusion gene in children with acute lymphoblastic leukemia in China. Cancer Genetics and Cytogenetics. 178, 57-60. 42. Kaur H, et al. (2005). TEL-AML1 frecuency in multi-ethnic Malaysian pediatric acute lymphoblastic leukemia. Cancer Genetics and Cytogenetics, 156, 129-133. 43. Artigas C, et al. (2006). Frecuencia de los genes de fusión TEL/AML1 BCR/ABL en pacientes pediátricos con leucemia linfoblástica aguda. Blood, 134, 1367-1376. 44. Siddiqui, R. et al. (2010). Distribution of common genetic subgroups in childhood acute lymphoblastic leukemia in four developing countries. Cancer Genetics and Cytogenetics, 200, 149-153. 45. García Sanz, et al. (1999). Low frequency of the TEL-AML1 fusion gene in acute lymphoblastic leukaemia in Spain. Blood, 107, 667-669. 46. Hill A, et al. (2005). The t(12;21) is underepresented in childhood B-lineage acute lymphoblastic leukemia in Kerala, Southern India. Haematologica, 90, 414-416. 47. Nam Yang, et al. (2005). Examination of ancestry and ethnic affiliation using highly informative diallelic DNA markers: application to diverse and admixed populations and implications for clinical epidemiology and forensic medicine. Hum Genet, 118, 382-392. 48. Aldrich, M. et al. (2006). Cytogenetics of Hispanic and White Children with Acute lymphoblastic Leukemia in California. Cancer Epidemiol Biomarkers Prev, 15, 578-581. 49. Ceppi, F. (2009). Cytogenetic Characterization of childhood Acute lymphoblastic Leukemia in Nicaragua. Pediatr Blood Cancer, 53, 1238-1241. 50. Park K, et al. (2001). Low incidence of TEL-AML1 fusion and TEL deletion in Korean childhood acute leukemia by extra-signal fluorescence in situ hybridization. Cancer Genetics and Cytogenetics, 126, 73-77. 51. Rubnitz JE, Look TA. (1998). Molecular genetics of childhood leukemias. J Pediatr Hematol Oncol, 20, 1-11. 52. Head DR, Pui CH (1999). Diagnosis and classification in Pui CH (Ed). Childhood leukemias, 20, 19-37. 53. Mesera G, Confer V, Rizzaru C, Jankovic M, Colombini A (1998). Childhood acute lymphoblastic leukemia. Oncologia, 8, 309-317. 54. Rivera-Luna R, Leal-Leal C, Cárdenas-Cárdos R, Martínez-Avalos A, Meza Coria C, Navarro-Alegría I, Ruano-Aguilar J. (1996). A survey of 4,076 children with cancer: Certain epidemiological aspects from a single Institution. Bol Med Hosp Infant Méx, 53, 598-605. 55. Pérez-Vera P, Mújica-Sánchez M, Carnevale A, Rivera-Luna R, Paredes R, Martínez A, Frías S (2001). Cytogenetics in acute lymphoblastic leukemia in mexican Children: An Institutional experience. Archives of Medical Research, 32, 202-207. 56. Pui CH, Crist WM (1999). Acute lymphoblastic leukemia in Pui CH. Childhood leukemias, 20, 288-312. 57. Romana SP, Poirel H, Della Valle V, Mauchauffé M, Buzón-Le MC, Berger R, Bernard OA (1999). Molecular analysis of chromosomal breakpoints in three examples of chromosomal translocation involving the TEL gene. Leukemia, 13, 1754-1759. 58. Fenrick R, Wang L, Nip J, Rooney RJ, Walker-Daniels J, Crawford HC, Hulboy DL, Knich MS, Matrisian LM, Hiebert SW (2000). TEL, a putative tumor suppresor, modulates cell growth and cell morphology of Ras. Transformed cells while repressing the transcription of stromelysin-1. Mol Cell Biol, 20, 5828-5839. 59. Friedman AD (1999). Leukemogenesis by CBF oncoproteins. Leukemia, 13, 19321942. 60. Romana SP, Poirel H, Della Valle V, Mauchauffé M, Buzón-Le MC, Berger R, Bernard OA (1999). Molecular analysis of chromosomal breakpoints in three examples of chromosomal translocation involving the TEL gene. Leukemia, 13, 1754-1759. 61-Rowley JD (1999). The role of chromosome translocations in leukemogenesis. Seminars in Hematology, 36, 59-72. 62. Shurtleff SA, Bujis A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, Chang GC, Pui C, Grosveld G, Downing JR (1995). Tel/AML1 fusion resulting from a cryptic t(12;21) in the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia, 9, 1985-1989. 63. Hunger SP. (1996). Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: Clinical features and molecular pathogenesis. Blood , 4, 12111224. 64. Mesera G, Confer V, Rizzaru C, Jankovic M, Colombini A (1998). Childhood acute lymphoblastic leukemia. Oncologia, 8, 309-317. IV.4 ANEXOS ANEXO NO. 1: METODOLOGÍA PARA ESTANDARIZACIÓN DE TÉCNICA DE PCR MULTIPLEX I. CULTIVO DE LÍNEAS CELULARES K-562 es la línea celular del transcrito quimérico BCR-ABL; la MV4-11 es la línea celular del transcrito quimérico MLL-AF4 y la Reh es la línea celular del transcrito quimérico ETV6-AML1. Estas líneas se utilizarán como control positivo para estandarizar las técnicas. El procedimiento realizado para el cultivo de las mismas se describe a continuación: a. Se dividieron en dos grupos: una mitad para realizar el cultivo celular, y la otra mitad para dejarla congelada. b. Se descongelaron y cultivaron las líneas celulares K-562, Reh y MV4-11, con los transcritos quiméricos BCR-ABL, ETV6-AML1, MLL-AF4 respectivamente, siguiendo el protocolo de procedimiento de la compañía en donde se compraron las líneas celulares. Se utilizó el medio para el cultivo con RPMI 1640, Suero fetal bovino (SFB) y L-Glutamina. Una vez preparado el medio de cultivo se agregaron las suspensiones celulares y se incubaron a 37°C con 5% de CO2. ( Ver figura 5356). FIGURA No. 53: LÍNEAS CELULARES K562, REH Y MV4-11 EN HIELO SECO Fuente FODECYT 48-2009 FIGURA No. 54: MEDIO DE CULTIVO RPMI Y SUERO FETAL BOVINO Fuente FODECYT 48-2009 FIGURA No. 55: CULTIVO DE LÍNEAS CELULARES EN EL MEDIO DE CULTIVO RPMI Fuente FODECYT 48-2009 FIGURA No. 56: DISTINTOS CULTIVOS DE LÍNEAS CELURARES EN INCUBACIÓN Fuente FODECYT 48-2009 c. Para monitorear el crecimiento celular, se realizaron conteos de las células en el microscopio utilizando una cámara de Newbauer (ver figura No. 57 y 58), los resultados se pueden observar en la tabla No. 10 TABLA No. 10 CANTIDAD DE CÉLULAS DE CADA LÍNEA CELULAR POR MILILITRO Línea Celular Fecha K-562 Reh MV4-11 Hora No. de Células por mL 10-04-2010 11:00am 58 13-04-2010 3:00pm 800 14-04-2010 9:30am 1190 16-04-2010 1:00pm 324 10-04-2010 11:00am 110 13-04-2010 3:00pm 430 16-04-2010 1:00pm 3275 10-04-2010 11:00am 520 13-04-2010 3:00pm 680 16-04-2010 1:00pm 2,300 Fuente FODECYT 48-2009 d. Se realizó el recambio de medio cada 3 días. e. Luego de los 6 días del primer subcultivo, se realizaron subcultivos, dividiéndolos en 2 grupos. f. Luego de 6 días, se guardó un duplicado con el reactivo RNA Later, para mantener las células viables para realizar extracciones posteriores. g. El otro duplicado se utilizó para realizar la extracción de ARN. FIGURA No. 57: OBSERVACIÓN AL MICROSCOPIO DE CULTIVOS CELULARES Fuente FODECYT 48-2009 FIGURA No. 58: LÍNEAS CELULARES OBSERVADAS AL MICROSCOPIO A Fuente FODECYT 48-2009 B C II. EXTRACCIÓN DE ARNm DE LAS LÍNEAS CELULARES CULTIVADAS Y TRANSCRIPCIÓN REVERSA (RT) a. A partir de las células cultivadas, se realizó la extracción de ARNm siguiendo el protocolo de procedimiento para células de mamíferos indicado por la compañía fabricante del kit de reactivos ( ver figura No. 59). FIGURA No. 59: FILTRO UTILIZADO PARA EXTRACCIÓN DE ARNm Fuente FODECYT 48-2009 b. Inmediatamente se realizó la transcripción reversa, esto con el fin de obtener el ADNc correspondiente al ARNm aislado ( ver figura No. 60). FIGURA No. 60: PROCEDIMIENTO DE RETROTRANSCRIPCIÓN Fuente FODECYT 48-2009 c. Se cuantificó el producto obtenido de ADNc, obteniéndose los resultados la tabla No. 11. En este cuadro se puede observar que la cantidad obtenida de ADNc fue bastante satisfactoria Tabla No. 11. Concentración del ADNc Línea Celular Pureza* Concentración [ng/µL] K-562 Reh MV4-11 A 2.05 1,875.00 B 2.05 2,070.00 A 2.03 1,950.00 B 2.08 1,830.00 A 2.04 1,765.00 B 2.06 1,555.00 *Un rango de pureza entre 1.8-2.0 indica una calidad aceptable Fuente: FODECYT 48-2009 Por otro lado se realizó una prueba utilizando sangre periférica de una persona sana X, para validar la extracción de las células mononucleares mediante la utilización de ficoll. Esto se realizó ya que es el procedimiento que se utilizará en los pacientes evaluados para obtener los linfocitos que posteriormente se utilizarán para extraer su ARNm ( ver figura 61 y 62). FIGURA No. 61: SEPARACIÓN DE CÉLULAS MONONUCLEARES DE MÉDULA ÓSEA POR MEDIO DE FICOLL Fuente FODECYT 48-2009 FIGURA No. 62: CELULAS MONONUCLEARES EN INTERFASE, DESPUÉS DE SEPARACIÓN CON FICOLL Fuente FODECYT 48-2009 ANEXO No. 2 HOJA DE CONSENTIMIENTO DE PRUEBAS CITOGENÉTICAS La leucemia Linfoblástica aguda es una neoplasia de los linfocitos que se encuentran en la sangre. Las pruebas genéticas son técnicas que sirven de ayuda al médico para confirmar el diagnóstico y monitorear su respuesta al tratamiento. Para hacer estas pruebas se utilizará un poco de la muestra de médula ósea que el médico obtendrá para realizar el diagnóstico. Yo, ______________________________ (nombre y apellidos) con cédula de vecindad _______________después de haber sido informado personalmente de los procedimientos que se utilizarán para establecer el diagnóstico del paciente. Autorizo que se le realicen las pruebas genéticas que el médico considere pertinentes a mi hijo(a) _____________________________________ . Fecha: ___________________________________ Firma de los padres y/o responsable: ___________________________________ Firma del médico ____________________________________ Sello de la institución HOJA DE ASENTIMIENTO Yo, _______________________________________________ de _________años de edad. He sido informado por el médico de la realización de un examen de médula ósea y de los procedimientos que se realizarán para la obtención y procesamiento de mi muestra. Estoy de acuerdo en que se realicen todos los exámenes que mis padres han autorizado. Fecha:__________________________________ Lugar:__________________________________ ________________________________________ Sello de la institución ANEXO No. 2: FOTOS DE RESULTADOS A. FOTOS DE COMPROBACIÓN DEL GEN ABL. FIGURA No. 63: GEN CONTROL ABL, PACIENTES No.1 y 2 Fuente FODECYT 48-2009 FIGURA No. 64: GEN CONTRL ABL, PACIENTES No. 3-7 276 bp Fuente FODECYT 48-2009 FIGURA No. 65: GEN CONTROL ABL, PACIENTES No. 8 AL 14 276 bp Fuente FODECYT 48-2009 FIGURA No. 66: GEN CONTROL ABL, PACIENTES No. 15 AL 20 Fuente FODECYT 48-2009 FIGURA No. 67: GEN CONTROL ABL, PACIENTES No. 21 AL 24 Fuente FODECYT 48-2009 FIGURA No. 68: GEN CONTROL ABL, PACIENTES No. 25 AL 27 Fuente FODECYT 48-2009 FIGURA No. 69: GEN CONTROL ABL, PACIENTES No. 27 Y 28 Fuente FODECYT 48-2009 FIGURA No. 70: GEN CONTROL ABL, PACIENTES No. 29 Y 30 Fuente FODECYT 48-2009 FIGURA No. 71: GEN CONTROL ABL, PACIENTES No. 31 AL 36 Fuente FODECYT 48-2009 FIGURA No. 72: GEN CONTROL ABL, PACIENTES No. 37 AL 39 Fuente FODECYT 48-2009 FIGURA No. 73: GEN CONTROL ABL, PACIENTES No. 40 AL 43 Fuente FODECYT 48-2009 FIGURA No. 74: GEN CONTROL ABL, PACIENTES No. 44 al 48 Fuente FODECYT 48-2009 FIGURA No. 75: GEN CONTROL ABL, PACIENTES No. 49 AL 51 Fuente FODECYT 48-2009 FIGURA No. 76: GEN CONTROL ABL, PACIENTES No. 52 AL 54 Fuente FODECYT 48-2009 FIGURA No. 77: GEN CONTROL ABL, PACIENTES No. 53 Y 54 Fuente FODECYT 48-2009 FIGURA No. 78: GEN CONTROL ABL, PACIENTES No. 56 AL 63 Fuente FODECYT 48-2009 FIGURA No. 79: GEN CONTROL ABL, PACIENTES No. 65 AL 73 Fuente FODECYT 48-2009 FIGURA No. 80: GEN CONTROL ABL, PACIENTES No. 8 AL 14 Fuente FODECYT 48-2009 FIGURA No. 81: GEN CONTROL ABL, PACIENTES No. 72 AL 75 Fuente FODECYT 48-2009 FIGURA No. 82: GEN CONTROL ABL, PACIENTES No. 75 AL 80 Fuente FODECYT 48-2009 FIGURA No. 83: GEN CONTROL ABL, PACIENTES No. 81 AL 85 Fuente FODECYT 48-2009 FIGURA No. 84: GEN CONTROL ABL, PACIENTES No. 84,86 Y 89 Fuente FODECYT 48-2009 FIGURA No. 85: GEN CONTROL ABL, PACIENTES No. 88 Y 93 Fuente FODECYT 48-2009 FIGURA No. 86: GEN CONTROL ABL, PACIENTES No. 90,92 Y 95 Fuente FODECYT 48-2009 FIGURA No. 87: GEN CONTROL ABL, PACIENTES No. 90, 94 Y 99 Fuente FODECYT 48-2009 FIGURA No. 88: GEN CONTROL ABL, PACIENTES No. 101-104 Fuente FODECYT 48-2009 FIGURA No. 89: GEN CONTROL ABL, PACIENTES No. 88, 96, 98 Y 102 Fuente FODECYT 48-2009 FIGURA No. 90: GEN CONTROL ABL, PACIENTES No. 97 Y 100 Fuente FODECYT 48-2009 FIGURA No. 91: GEN CONTROL ABL, PACIENTES No. 105-108 Fuente FODECYT 48-2009 FIGURA No. 92: GEN CONTROL ABL, PACIENTES No. 105, 107 Y 109 Fuente FODECYT 48-2009 FIGURA No. 93: GEN CONTROL ABL, PACIENTES No. 110 AL 114 Fuente FODECYT 48-2009 FIGURA No. 94: GEN CONTROL ABL, PACIENTE 115 Fuente FODECYT 48-2009 FIGURA No. 95: GEN CONTROL ABL, PACIENTES No. 116 AL 119 Fuente FODECYT 48-2009 FIGURA No. 96: GEN CONTROL ABL, PACIENTE No. 118 Fuente FODECYT 48-2009 FIGURA No. 97: GEN CONTROL ABL, PACIENTES No. 120 AL 125 Fuente FODECYT 48-2009 FIGURA No. 98: GEN CONTROL ABL, PACIENTE No. 126 Fuente FODECYT 48-2009 FIGURA No. 99: GEN CONTROL ABL, PACIENTE No. 127 Fuente FODECYT 48-2009 FIGURA No. 100: GEN CONTROL ABL, PACIENTES No. 128 AL 132 Fuente FODECYT 48-2009 FIGURA No. 101: GEN CONTROL ABL, PACIENTES No. 133 AL 138 Fuente FODECYT 48-2009 FIGURA No. 102: GEN CONTROL ABL, PACIENTES No. 139 AL 142 Fuente FODECYT 48-2009 B. FOTOS DE TRANSCRITOS FIGURA No. 103: TRANSCRITO E2A-PBX1, PACIENTES No. 3 AL 7 Fuente FODECYT 48-2009 FIGURA No. 104: TRANSCRITO E2A-PBX1, PACIENTES No. 15 AL 20 Fuente FODECYT 48-2009 FIGURA No. 105: TRANSCRITO E2A-PBX1, PACIENTES No. 21 AL 27 Fuente FODECYT 48-2009 FIGURA No. 106: TRANSCRITO E2A-PBX1, ETV6-AML1 Y MLL-AF4 PACIENTES No. 28 AL 30 Fuente FODECYT 48-2009 FIGURA No. 107: TRANSCRITO E2A-PBX1, PACIENTES No. 30 AL 36 Fuente FODECYT 48-2009 FIGURA No. 108: TRANSCRITO E2A-PBX1, PACIENTES No. 37 AL 43 Fuente FODECYT 48-2009 FIGURA No. 109: TRANSCRITO E2A-PBX1, PACIENTE No. 43 Fuente FODECYT 48-2009 FIGURA No. 110: TRANSCRITO E2A-PBX1, PACIENTES No. 39 AL 48 Fuente FODECYT 48-2009 FIGURA No. 111: TRANSCRITO E2A-PBX1, PACIENTE No. 47 Fuente FODECYT 48-2009 FIGURA No. 112: TRANSCRITO E2A-PBX1, PACIENTES No. 49 AL 54 Fuente FODECYT 48-2009 FIGURA No. 113: TRANSCRITO E2A-PBX1, PACIENTES No. 57 AL 64 Fuente FODECYT 48-2009 FIGURA No. 114: TRANSCRITO E2A-PBX1, PACIENTES No. 53, 55, 57, 59, 61 Y 62 Fuente FODECYT 48-2009 FIGURA No. 115: TRANSCRITO E2A-PBX1, PACIENTES No. 60,63 Y 65 Fuente FODECYT 48-2009 FIGURA No. 116: TRANSCRITO E2A-PBX1, PACIENTES No. 65 al 74 Fuente FODECYT 48-2009 FIGURA No. 117: TRANSCRITO ETV6-AML1, PACIENTES No. 3 AL 7 Fuente FODECYT 48-2009 FIGURA No. 118: TRANSCRITO E2A-PBX1 Y ETV6-AML1, PACIENTES No. 8 AL 14 Fuente FODECYT 48-2009 FIGURA No. 119: TRANSCRITO ETV6-AML1, PACIENTES No. 15 AL 20 Fuente FODECYT 48-2009 FIGURA No. 120: TRANSCRITO ETV6-AML1, PACIENTES No. 21 AL 27 Fuente FODECYT 48-2009 FIGURA No. 121: TRANSCRITO ETV6-AML1, PACIENTES No. 31 AL 36 Fuente FODECYT 48-2009 FIGURA No. 122: TRANSCRITO ETV6-AML1, PACIENTES No. 37 AL 43 Fuente FODECYT 48-2009 FIGURA No. 123: TRANSCRITO ETV6-AML1, PACIENTES No. 39 AL 48 Fuente FODECYT 48-2009 FIGURA No. 124: TRANSCRITO ETV6-AML1, PACIENTES No. 49 AL 51 Fuente FODECYT 48-2009 FIGURA No. 125: TRANSCRITO ETV6-AML1, PACIENTES No. 57 AL 64 Fuente FODECYT 48-2009 FIGURA No. 126: TRANSCRITO ETV6-AML1, PACIENTES No. 53 AL 62 Fuente FODECYT 48-2009 FIGURA No. 127: TRANSCRITO ETV6-AML1, PACIENTES No. 52 Y 54 Fuente FODECYT 48-2009 FIGURA No. 128: TRANSCRITO ETV6-AML1, PACIENTES No. 65 AL 74 Fuente FODECYT 48-2009 FIGURA No. 129: TRANSCRITO BCR-ABL, PACIENTES No. 1 y 2 Fuente FODECYT 48-2009 FIGURA No. 130: TRANSCRITO BCR-ABL, PACIENTES No. 3 AL 7 Fuente FODECYT 48-2009 FIGURA No. 131: TRANSCRITO BCR-ABL, PACIENTES No. 8 AL 14 Fuente FODECYT 48-2009 FIGURA No. 132: TRANSCRITO BCR-ABL, PACIENTES No. 15 AL 22 Fuente FODECYT 48-2009 FIGURA No. 133: TRANSCRITO BCR-ABL, PACIENTES No. 23 AL 27 Fuente FODECYT 48-2009 FIGURA No. 134: TRANSCRITO BCR-ABL, PACIENTES No. 27 AL 30 Fuente FODECYT 48-2009 FIGURA No. 135: TRANSCRITO BCR-ABL, PACIENTES No. 31 AL 36 Fuente FODECYT 48-2009 FIGURA No. 136: TRANSCRITO BCR-ABL, PACIENTES No. 37 AL 43 Fuente FODECYT 48-2009 FIGURA No. 137: TRANSCRITO BCR-ABL, PACIENTES No. 40 Y 41 Fuente FODECYT 48-2009 FIGURA No. 138: TRANSCRITO BCR-ABL, PACIENTES No. 39 AL 48 Fuente FODECYT 48-2009 FIGURA No. 139: TRANSCRITO BCR-ABL, PACIENTES No. 49 AL 51 Fuente FODECYT 48-2009 FIGURA No. 140: TRANSCRITO BCR-ABL, PACIENTES No. 57 AL 64 Fuente FODECYT 48-2009 FIGURA No. 141: TRANSCRITO BCR-ABL, PACIENTES No. 60, 63 Y 65 Fuente FODECYT 48-2009 FIGURA No. 142: TRANSCRITO BCR-ABL, PACIENTES No. 53, 55, 59, 62 Fuente FODECYT 48-2009 61 y FIGURA No. 143: TRANSCRITO BCR-ABL, PACIENTES No. 52 y 54 Fuente FODECYT 48-2009 FIGURA No. 144: TRANSCRITO BCR-ABL, PACIENTES No. 65 al 74 Fuente FODECYT 48-2009 FIGURA No. 145: TRANSCRITO MLL-AF4, PACIENTES No. 3 AL 7 Fuente FODECYT 48-2009 FIGURA No. 146: TRANSCRITO MLL-AF4, PACIENTES No. 8 AL 14 Fuente FODECYT 48-2009 FIGURA No. 147: TRANSCRITO MLL-AF4, PACIENTES No. 15 AL 20 Fuente FODECYT 48-2009 FIGURA No. 148: TRANSCRITO MLL-AF4, PACIENTES No. 21 AL 27 Fuente FODECYT 48-2009 FIGURA No. 149: TRANSCRITO MLL-AF4, PACIENTES No. 31 AL 36 Fuente FODECYT 48-2009 FIGURA No. 150: TRANSCRITO MLL-AF4, PACIENTES No. 37 AL 43 Fuente FODECYT 48-2009 FIGURA No. 151: TRANSCRITO MLL-AF4, PACIENTES No. 39 AL 48 Fuente FODECYT 48-2009 FIGURA No. 152: TRANSCRITO MLL-AF4, PACIENTES No. 49 AL 51 Fuente FODECYT 48-2009 FIGURA No. 153: TRANSCRITO MLL-AF4, PACIENTES No. 52 y 54 Fuente FODECYT 48-2009 FIGURA No. 154: TRANSCRITO MLL-AF4, PACIENTES No. 53 al 62 Fuente FODECYT 48-2009 FIGURA No. 155: TRANSCRITO MLL-AF4, PACIENTES No. 57 al 64 Fuente FODECYT 48-2009 FIGURA No. 156: TRANSCRITO MLL-AF4, PACIENTES No. 65 AL 74 Fuente FODECYT 48-2009 FIGURA No. 157: TRANSCRITO BCR-ABL Y EE2-PBX1, PACIENTES No.72 AL 75 Fuente FODECYT 48-2009 FIGURA No. 158: TRANSCRITO ETV6-AML1 Y MLL-AF4, PACIENTES DEL 72 AL 75 Fuente FODECYT 48-2009 FIGURA 158: TODOS LOS TRANSCRITOS, PACIENTES DEL 76 AL 80 Fuente FODECYT 48-2009 FIGURA No. 159: TODOS LOS TRANSCRITOS, PACIENTES DEL 81 AL 85 Fuente FODECYT 48-2009 FIGURA No. 160 : TRANSCRITO BCR-ABL, E2A-PBX1, PACIENTES DEL 90-96, 99 Y 100 Fuente FODECYT 48-2009 FIGURA No. 161: TODOS LOS TRANSCRITOS, PACIENTE 102 Fuente FODECYT 48-2009 FIGURA No. 162: TODOS LOS TRANSCRITOS, PACIENTES 84, 86 Y 89 Fuente FODECYT 48-2009 FIGURA No. 163: TRANSCRITO BCR-ABL; PACIENTES 88, 101 AL 104 Fuente FODECYT 48-2009 FIGURA No. 164: TODOS LOS TRANSCRITOS, PACIENTES 105 AL 109 Fuente FODECYT 48-2009 FIGURA No. 165: TRANSCRITO BCR-ABL, E2A-PBX1, PACIENTES 115 AL 119 Fuente FODECYT 48-2009 FIGURA No. 166: TODOS LOS TRANSCRITOS; PACIENTES No. 110 AL 114 Fuente FODECYT 48-2009 FIGURA No. 167: TRANSCRITO BCR-ABL Y E2A-PBX1, PACIENTES DEL 120 AL 132 Fuente FODECYT 48-2009 FIGURA No. 168: TODOS LOS TRANSCRITOS, PACIENTES 121, 123 ,125 Y 126 Fuente FODECYT 48-2009 FIGURA No. 169: TRANSCRITO BCR-ABL, PACIENTES No. 133 AL 142 Fuente FODECYT 48-2009 FIGURA No. 170: TRANSCRITO E2A-PBX1 Y ETV6-AML1, PACIENTES 133 A 142 Fuente FODECYT 48-2009 FIGURA No. 171: TRANSCRITOS BCR-ABL Y ETV6-AML1, PACIENTES 121, 123, 120 Y 128 Fuente FODECYT 48-2009 FIGURA No. 172: TRANSCRITOS ETV6-AML1 Y MLL-AF4, PACIENTES 115 AL 119 Fuente FODECYT 48-2009 FIGURA No. 173: TRANSCRITO ETV6-AML1, PACIENTES 120, 123, 124, 127-131 Fuente FODECYT 48-2009 FIGURA No. 174: TRANSCRITO MLL-AF4, PACIENTES 133 AL 142 Fuente: FODECYT 48-2009 ANEXO No. 3: GRAFICAS DE SECUENCIACIÓN ANEXO No. 4 TABLA 12: DATOS CLÍNICOS DE LOS PACIENTES PARTE V V.1 INFORME FINANCIERO