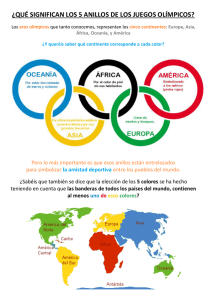
Clase Desarrollo de Drogas
Anuncio

DESARROLLO DE DROGAS El proceso de descubrimiento y desarrollo de fármacos moderno es un largo proceso que comienza con la identificación del objetivo biológico, esto es por ejemplo una enzima que juega un papel fundamental en una determinada enfermedad por lo cual su inhibición por una droga implique un paliativo para dicha enfermedad. Luego es necesario encontrar el denominado "compuesto líder", o sea un compuesto prototipo que tiene la actividad biológica o farmacológica deseada (inhibición enzimática/bloqueo receptor) pero que presenta también ciertas características contraproducentes: alta toxicidad, insolubilidad, inestabilidad, problemas metabólicos, actividades biológicas secundarias, etc. La optimización de dicho compuesto líder, a través de transformaciones químicas (modificaciones sintéticas) que eliminen esas características indeseables, permite la obtención del "compuesto candidato" que es aquel seleccionado para ser sometido a las pruebas clínicas, etapa previa a la aprobación como droga comercial. Para las etapas de descubrimiento del líder y optimización es necesaria una interrelación constante con los biólogos que realizan los ensayos de actividad biológica. Ellos son los que determinan la actividad biológica de los compuestos que proveen los químicos para hallar el compuesto líder y luego, durante la optimización será un constante "ida y vuelta" ya que los resultados biológicos de los compuestos modificados guiarán las nuevas modificaciones químicas que serán screeneadas y así sucesivamente hasta encontrar el candidato. Esta es la etapa en la que el químico medicinal “orgánico” tiene mayor participación, pero a estas pruebas in vitro les siguen las pruebas in vivo. Esta pirámide grafica el proceso, que según los autores puede durar entre 12 y 15 años. Las pruebas in vivo se clasifican en distintas fases, las fases preclínicas, pruebas en animales para ver si pueden ser utilizada la droga en humanos, las pruebas clínicas I pequeño grupo de voluntarios sanos, especialmente para determinar efectos secundarios, dosis, farmacocinética (ADME: absorción, distribución, metabolismo, eliminación), II, pequeño grupo de enfermos, obviamente para determinar la efectividad, III, donde se trata un mayor número de pacientes donde se satisfacen los requerimientos de eficacia y seguridad de los organismos reguladores. Incluso hay una fase IV de control que se realiza cuando ya la droga está en el mercado. Este proceso es fundamental para finalmente tener un fármaco a la venta con todas las precauciones necesarias. Esto es lo que llevan a cabo fundamentalmente los químicos medicinales tanto en el área industrial como académica, tanto para Empresas privadas como para el sector público. Pero entonces, cómo se descubre un líder, lo veremos someramente, ya que se profundiza en la clase correspondiente. Dado que el líder es sólo la punta del ovillo, el siguiente paso es la optimización del este compuesto para mejorar sus propiedades hacia tener el compuesto que tenga las condiciones para incluirse en las pruebas clínicas y eventualmente llegar a ser un compuesto comercial. ¿Qué se debe tener en cuenta para mejorar las propiedades farmacológicas de un Líder? Esto son algunos de los puntos que se deben tener en cuenta cuando se trata de optimizar un líder. Identificación del farmacóforo. Los grupos relevantes para la actividad biológica de una molécula se conocen como el FARMACÓFORO Es necesario conocer cuales son esos grupos y su posición relativa en el espacio. Conociendo el farmacóforo se simplifica la optimización ya que se sabe cuales grupos pueden ser modificados y cuales no. Por ejemplo, si descubrimos que los grupos importantes para la actividad en esta molécula son los dos OH fenólicos, el anillo aromático y el átomo de nitrógeno, entonces el farmacóforo es el que se muestra, donde el N esta a 5.063 A del centro del anillo aromático y en un ángulo de 18° respecto al plano del anillo. Una de las formas de identificar el farmacóforo es a través de relaciones estructura- actividad RELACIÓN ESTRUCTURA-ACTIVIDAD (SAR) Las diferencias de actividad relacionadas a la estructura se denominan RELACIONES ESTRUCTURA-ACTIVIDAD (SAR) y también de sus efectos adversos. Esta información es utilizada para: Identificación de Farmacóforo. Un estudio serio de las relaciones estructura-actividad de un compuesto líder y sus análogos puede usarse para determinar las partes de la estructura del compuesto líder que son responsables de sus propiedades biológicas (FARMACÓFORO) Mejorar actividad o potencia del Líder (optimización de su SAR) Obtener una actividad diferente a partir de un fármaco conocido, los resultados de SAR pueden guiarnos en ese camino Disminuir efectos secundarios indeseados Mejorar formas de administración a los pacientes, especialmente buscas mejor absorción oral. (Siempre se busca, si tenemos en el mercado un medicamento que se expende IM y aparece otro que actúa por vía oral, imagínense cual va a ser el que se venda. Lo mismo respecto a la vida media del fármaco, si es de absorción lenta y/o no se metaboliza rápidamente durará en sangre más tiempo y requerirá menos tomas (especialmente antibióticos para niños) Azitromicina, una toma diaria no hay que despertarlo al chico a la madrugada. Las SAR son usualmente determinadas efectuando pequeños cambios en la estructura de un compuesto LIDER y determinando los cambios que esto produce en la actividad biológica Un ejemplo bastante reciente de SAR fue realizado sobre TAXOL (anticancerígeno inhibidor de la mitosis). Un gran número de modificaciones se realizaron para llegar a este resultado. Vemos en este caso que nos indica cuales son los grupos esenciales para la actividad y por, por otro lado, cuales no, de manera que servirá de guía para nuevas modificaciones. En otros casos puede ser un esquema de relaciones estructura-potencia donde nos indica los puntos para mejorar la potencia del fármaco. Quizás la mayor virtud, considerando el gasto que significa, es dar la posibilidad al químico sintético la posibilidad de conocer qué análogos han sido sintetizados y así no gastar tiempo obteniendo algo ya conocido. Las SAR son usualmente determinadas efectuando pequeños cambios en la estructura de un compuesto LIDER. Analizaremos ahora cuales son esos cambios (los más frecuentes) CLASIFICACIÓN DE LOS CAMBIOS MÁS FRECUENTES Cambios de forma y tamaño Introducción de nuevos sustituyentes) Reemplazos bioisostéricos) de sustituyentes (isostéricos y Cambios de forma y tamaño El tamaño y forma de las moléculas puede modificarse de las siguientes maneras: i) Cambiando el número de grupos metilenos de cadenas y anillos ii) Aumentando o disminuyendo el grado de insaturación iii) Introduciendo o removiendo un anillo i) Cambio del número de grupos metilenos de cadenas y anillos: Esta modificación implica un aumento de la lipofilicidad del compuesto. Observamos la figura (4-alquil resorcinol). Se cree que el aumento de la actividad por aumento del número de metilenos es atribuido al aumento de la solubilidad en lípidos del análogo lo que le da una mejor penetración a través de las membranas. Pero llega un punto en el que la actividad antibacteriana llega a un valor óptimo y luego baja. Eso lo vemos también en el caso de análogos de enalaprilato (cuyo pro fármaco el enalapril es uno de los antihipertensivos de más venta y uno de los fármacos de más venta) (es un inhibidor de la enzima conversora de angiotensina, ACE). Aquí, hay una disminución de la actividad con el aumento en el número de grupos metilenos es atribuida una disminución de la solubilidad en agua de los análogos. Esta reducción de la solubilidad en agua puede resultar en una pobre distribución en medio acuoso así como la posibilidad de que queden atrapados en la porción lipídica de las membranas. También el aumentar el número de metilenos se observa la formación de micelas. Las micelas forman grandes agregados que debido a su tamaño no pueden unirse a sitios activos ni receptores. Si una droga es muy polar se elimina rápido por los riñones y además no cruza las barreras lipofílicas de las membranas celulares. La droga debe ser hidrofóbica para poder cruzar las membranas celulares, pero tampoco muy lipofílica, con la posibilidad de que queden atrapados en la porción lipídica de las membranas. Si es poco soluble en agua será pobremente absorbida por el tracto intestinal ya que se disolverá en glóbulos adiposos y no interactuará con la pared intestinal. También se formaran micelas, las micelas forman grandes agregados que debido a su tamaño no pueden unirse a sitios activos ni receptores. Además lo que llega al torrente sanguíneo saldrá de allí y se acumulará en tejidos adiposos. Por ejemplo en personas obesas los anestésicos gaseosos deben suministrarse en más cantidad porque son muy solubles en grasas También el cambio del número de grupos metilenos de cadenas y anillos puede tener otros efectos. Este se aprecia en el siguiente ejemplo, los compuesto bisamonio polimetilénicos pueden actuar sobre receptores colinérgicos, pero depende del largo de la cadena la actividad que se genera. Estos compuestos que producen bloqueo muscular tienen un aumento brusco de actividad a partir de n=5 que va luego disminuyendo…..Más específicamente: si n=5,6 tenemos un efecto agonista, o sea que la interacción con el receptor produce una respuesta fisiológica. En cambio se ese n<10 el efecto es antagonista, o sea se produce la interacción pero no se produce la respuesta fisiológica. Vemos cual es la razón de estos cambios en la actividad: n<5: la molécula es demasiado corta para interaccionar con los dos sitios del receptor n=5,6: la longitud es adecuada se produce la interacción y se dispara la respuesta biológica n=7-9: demasiado larga la molécula, no hay interacción satisfactoria. n>10: La longitud permite interacción con un subsitio diferente produciendo efecto antagonista (decametonio) La introducción de ramificaciones, de anillos de diferentes tamaños, la sustitución de cadenas por anillos y viceversa, también tiene efectos en la potencia y actividad. Por ejemplo el reemplazo del átomo de azufre en el antipsicótico CHLORPROMAZINE, por un puente –CH2-CH2- produce el antidepresivo CLOMIPRAMINE S N Cl CH2CH2CH2N(CH3)2 CHLORPROMAZINE N Cl CH2CH2CH2N(CH3)2 CLOMIPRAMINE Cambios de forma y tamaño ii) Aumento o disminución del grado de insaturación La remoción de doble enlaces aumenta la flexibilidad de las moléculas, lo que puede facilitar la capacidad de un análogo de adaptarse a sitios activos y unirse a receptores. En general, la mayor flexibilidad también implica que un mismo compuesto pueda unirse a diferentes objetivos biológicos. Puede unirse inespecíficamente a dos receptores. Así que depende lo que se esté buscando y la altura del proceso de desarrollo del fármaco que se encuentre el proyecto, si no se ha logrado un buena actividad contra un receptor particular que se está empezando a estudiar tal vez sea útil tener flexibilidad, si se quiere mejorar la especificidad o se quiere estudiar en más detalle la interacción con el receptor una conformación más rígida será más útil ya que nos ayudará a identificar los grupos de la proteína con que interacciona y su distribución tridimensional La introducción de dobles enlaces aumenta la rigidez de una molécula (pero más especificidad). Ampliaremos cuando veamos CONFORMACIONES. Si además se observa isomería geométrica los isómeros E y Z pueden tener diferentes actividades. El análogo de CORTISOL, PREDNISONA es 30 veces más activo, si duda en este caso la rigidez implica un mejor ajuste con el receptor HOH2C HOH2C C HO O HO O C OH OH O O PREDNISONA CORTISOL Cambios de forma y tamaño iii) Introducción o remoción de anillos: La introducción de un sistema cíclico cambia la forma e incrementa el tamaño total del análogo con efectos impredecibles: El aumento de tamaño puede ser útil para reforzar la unión de la droga a su blanco de acción. EJEMPLOS: Introducción de anillos grandes El ciclopentil análogo butyrolactam fosfodiesterasa de 3-(3,4-dimethyloxyphenyl)- ROLIPRAM (enzima que hacia la cAMP hidroliza uniones fosfodiésteres de los nucleótidos) tiene una actividad inhibitoria aumentada debido a que el grupo ciclopentilo rellena un bolsillo hidrofóbico en el sitio activo de esta enzima H3CO H3CO H3CO O O NH NH 3-(3,4-Dimethoxyphenyl)-butyrolactam antidepresivo O ROLIPRAM, antidepresivo 10 veces más activo . Incorporación de un sistema alicíclico pequeño para reemplazar a un doble enlace carbono-carbono NH2 TRANYLCYPROMINE antidepresivo más estable NH2 1-amino-2-phenylethene Los anillos de ciclopropano suelen ser más estables que los dobles enlaces. Tranilcipronamina es un inhibidor del la Monoaminaoxidasa (MAO A yB). El ciclopropano es pequeño y entonces el cambio no producirá variaciones estéricas (que ahora el fármaco no ajuste con el receptor/enzima) También evita la formación de isómeros cis-trans que pueden tener actividad diferente. Incorporación de heterociclos y anillos aromáticos Como se dijo la introducción de anillos puede tener diversos efectos, por ejemplo anillos heterocíclicos aromáticos incrementan el tamaño en la zona donde interaccionan, lo cual puede o no ser beneficioso, también implica un sistema π de electrones del anillo aromático que puede generar interacciones beneficiosas o perjudiciales (de nuevo depende de la altura del desarrollo en que se encuentre, si estoy en una etapa inicial de exploración, casi que cualquier sustitución es válida, si estamos avanzados deberemos tener más cuidado. Vemos un caso en que la incorporación de anillos aromáticos de 6 miembros afecta el tamaño y nos resulta útil. H2COCHN O H2COCHN CH3 CH3 CH3 CH3 O 2-Phenylbenzylpenicillin (no resistente a beta-lactamasas) Benzylpenicillin (no resistente a beta-lactamasas) OCHN CH3 CH3 O Diphenylpenicilin resistente a beta-lactamasas En este otros caso, vemos un heterociclo no aromático agregado a la cadena lateral, el resultado es la pérdida de actividad neuroléptica y un incremento de actividad antiemética (impiden el vómito o la náusea). Se cree que se debe a la presencia de una amina terciaria extra. S S N Cl CH2CH2CH2N(CH3)2 N Cl CH2CH2CH2 N CHLORPROMAZINE antipsicótico N CH3 PROCHLORPERAZINE actividad antiemética con reducida actividad neuroléptica Alcaloides muy potentes con varios sistemas de anillos Alcaloides como Morfina tienen un complicado sistema de anillos en su estructura lo que hace muy complicada su síntesis. Se han diseñado análogos más simples para determinar el farmacóforo y eliminar los anillos que pudieran estar de más. En negrita se ve cual es la estructura esencial para la actividad, a partir de ellos se han podido sintetizar análogos más sencillos: Uno más potente pero altamente adictivo, otro baja potencia, otro baja potencia y baja adictividad y el cuarto que es igualmente potente pero no tiene tan alta adictividad Cambios en la naturaleza y grado de sustitución del compuesto líder (introducción de nuevos sustituyentes) a) Grupos metilo b) Halógenos c) Hidroxilos d) Grupos básicos e) Ácidos carboxílicos y Sulfonas f) Tioles, Sulfuros y otros grupos con Azufre GRUPOS METILO La introducción de grupos metilo generalmente aumenta la lipofilicidad y reduce su solubilidad en agua. Puede mejorar la facilidad de absorción de un análogo a una membrana biológica, pero hará más difícil que su paso desde ésta al medio acuoso intracelular. Cambio del coeficiente de partición (P) de algunos compuestos cuando se agrega un grupo metilo a su estructura. A mayor P mayor lipofilicidad. Benceno y Tolueno se midieron en octanol/agua, los dos restantes en aceite de oliva/agua. Compuesto Estructura P Análogo Estructura P _____________________________________________________ Benceno 135 Acetamida Urea Tolueno 83 Propionamida CH3CONH2 15 NH2CONH2 N-metilurea CH3 CH3CH2CONH2 CH3NHCONH2 490 360 44 La incorporación de un grupo metilo puede acarrear restricciones estéricas: (difenihidramina antihistamínico) H O N H C H .. O .. impedimento estérico entre H y pares de electrones libres, no exhibe actividad antihistamínica N o-Methyl analogue Diphenylhydramine, H3C antihistamínico H .. O .. N p-Methyl analogue, 3.7 veces más activo La incorporación de un grupo metilo puede tener tres efectos generales: Aumentar la tasa de metabolización debido a la oxidación del grupo i) metilo (se elimina más rápido y esto favorece el proceso de detoxificación) La tolbutamida tiene la ventaja frente a su análogo sin el metilo que es demasiado tóxico para ser usado, ya que su detoxificación es más difícil Oxidación C4H9NHCONHSO2 CH3 C4H9NHCONHSO2 COOH Metabolito menos tóxico Tolbutamida antidiabético Producir demetilaciones cuando los grupos metilos están unidos a ii) átomos de nitrógeno y azufre cargados positivamente, aunque los grupos metilo unidos a otros heteroátomos pueden también demetilarse. Estas transferencias de metilos están asociadas a efectos tóxicos, sobre todo carcinogénicos Los grupos metilos pueden reducir la tasa de metabolización de un iii) compuesto enmascarando un grupo metabólicamente activo, dando así una tasa de metabolización más baja del compuesto deseado en los casos en que sea necesario. Ejemplo: S S HS N H S C H N SH N N C S S NABAN, agroquímico diisotiocianato Metabolito activo de Naban HS N CH3 CH3 N SH S derivado N-metilado, inactivo HALOGENOS La incorporación de Halógenos en el líder resulta en análogos más lipofílicos y menos solubles en agua. Se usan para aumentar la permeabilidad de las membranas. Aunque esto, como vimos, debe estar balanceado EN CUANTO A COMO AFECTAN LA POTENCIA…. Los cambios en la potencia causados por la introducción de halógenos o grupos que contienen halógenos dependen de la posición de la sustitución. Por ejemplo, el antihipertensivo CLONIDINE que es o,o-diclorosustituído es más potente que el p,m-dicloroanálogo. Se cree que el Cloro demasiado voluminoso, impone en la posición orto, restricciones estructurales que lo hacen más activo HN HN NH N N Cl NH Cl Cl Cl CLONIDINE ED20: 0.01 mgKg-1 ED20: 3.00 mgKg-1 GRUPOS HIDROXILO La introducción de grupos hidroxilo produce análogos con aumentada solubilidad en agua y baja lipofilicidad. También provee de un nuevo centro capaz de formar enlaces puente hidrógeno que pueden ser muy importantes en la unión a sitios activos Por ejemplo, el derivado o-hidroxilado de MINAPRINA se une más efectivamente al receptor muscarínico que muchos de sus análogos no hidroxilados gracias a la posibilidad de formar puentes de hidrógeno OH N N N N NHCH2CH2 N NHCH2CH2 N O O ANÁLOGO o-Hidroxilado MINAPRINE Sin embargo la presencia de grupos hidroxilo abre nuevas vías metabólicas que pueden colaborar en la detoxificación de las drogas administradas, y eliminarse antes de tiempo!!!! Metabolizan por alcohol deshidrogenasa terminan generando ácidos que son fácilmente eliminados en la orina. GRUPOS BASICOS Usualmente se encuentran como aminas incluyendo algunos anillos que poseen átomos de nitrógeno como amidinas y guanidinas. Pueden formar sales en medios biológicos y su incorporación a compuestos líder puede dar un aumento en la solubilidad en agua. Cuanto más básico es el compuesto, más capaz de formar sales y menos posibilidades tiene de atravesar las membranas lipídicas. Todo tipo de aminas H+ N + N H Amidinas NH NH2 + H R R NH2 NH2 Guanidinas + NH2 NH H+ R R HN HN NH2 NH2 La introducción de grupos básicos puede aumentar la unión de un análogo con su blanco por formación de enlaces puente hidrógeno Fig. (a). Sin embargo la mayoría de análogos con grupos básicos deben su actividad a la formación de sales y formación de interacciones iónicas con el sitio de acción Fig. (b) b) a) Target site Target site O OH C C _ H H + N O O O H .. H .. N H ionic bond H ACIDOS CARBOXILICOS Y SULFONICOS Análogos con solubilidad en agua aumentada y baja lipofilicidad La introducción de ácidos carboxílicos a pequeñas moléculas activas puede cambiarles mucho la actividad OH FENOL antiséptico COOH OH ACIDO SALICILICO analgésico, antiinflamatorio NH2 NH2 COOH FENILETILAMINA simpaticomimético FENILALANINA sin activ.simpaticomimética Los ácidos sulfónicos no tienen en general efecto en la actividad biológica pero aumentan la velocidad de excreción de las drogas TIOLES, SULFUROS Y OTROS DERIVADOS DEL AZUFRE En general los tioles y sulfuros no se utilizan en los estudios de SAR de líderes, porque son rápidamente metabolizados por oxidación. SIN EMBARGO LOS TIOLES SE INTRODUCEN CUANDO SE NECESITAN AGENTES QUELANTES (CAPTOPRIL) REEMPLAZO DE SUSTITUYENTES ISOTEROS La elección de los grupos que se van a sustituir depende de los objetivos del diseño. Se realiza generalmente usando el concepto de ISOSTEROS ERLENMEYER DEFINIÓ INICIALMENTE A LOS ISÓSTEROS QUÍMICOS COMO ATOMOS, IONES Y MOLÉCULAS QUE TIENEN IDÉNTICAS CAPAS EXTERNAS DE ELECTRONES. ACTUALMENTE SE AMPLIÓ ESTA DEFINICIÓN PARA INCLUÍR GRUPOS QUE TIENEN ACTIVIDADES BIOLÓGICAS SIMILARES, ESOS GRUPOS SE LLAMAN BIOISÓSTEROS Vemos algunos aquí en las tablas que agrupan a los isósteros clásicos (univalentes, bivalentes, etc.) y también los bioisósteros cuya agrupación es más en función de cual es el grupo reemplazado, Ej. carbonilos, carboxilos, halógenos, etc. Veamos entonces algunos de estos reemplazos Reemplazos de átomos o grupos univalentes: Muy comunes. Por ejemplo: los halógenos pueden reemplazarse por otros grupos atractores de electrones como CN y CF3 Reemplazos de átomos o grupos divalentes: Corresponde al intercambio en series como O, S, NH y CH2 Por ejemplo, en el caso de análogos de meperidina (analgésico narcótico) se obtienen compuestos de actividad interesante, el S da menor, tal vez por su fácil metabolización, se oxida a sulfóxido que facilita su excreción Reemplazos de átomos o grupos trivalentes: La sustitución de -CH por -N da muy buenos resultados en cadenas alifáticas Por ejemplo, el cambio del nitrógeno por carbono en este análogo de antipirina no varía su potencia antipirética Equivalencia de anillos La sustitución de -CH= por -N= y de -CH=CH- por -S- en anillos aromáticos son las aplicaciones más útiles del isosterismo clásico, el cambio mantiene la aromaticidad del sistema, en el caso de las sulfamidas este cambio a dado por ejemplo el sulfatiazol. En general los cambios de R en sulfonamidas afectan la vida media del fármaco permitiendo una administración espaciada. Pero no siempre es bueno en este caso el cambio del benceno por un tiofeno reduce la actividad Grupos con efectos polares similares Grupo carboxilo por heterociclos ácidos planos y funciones ácidas no planas Este es otro cambio bastante común, el ácido carboxílico por grupo con acidez similar, por ejemplo, muy común es el tetrazol, el cambio hace que la carga esté más distribuida ya que resuena por todo el anillo. Si se aprecia que la interacción iónica con un residuo del receptor cargado positivamente es débil, se puede esperar que este reemplazo aumente la interacción si se trataba de un problema de distancia. Dentro de los no planares el ácido hidroxámico es uno de los más populares, también tenemos fosfonatos, sulfonatos, etc. Reemplazos de amidas y péptidos Es uno de los más comunes. Tiene que ver con la Síntesis de peptidomiméticos: compuestos que imitar a un péptido y bloquear un determinado efecto biológico. Tienen mejor biodisponibilidad oral. Aquí vemos sólo cuatro Tioamidas, cambia carbonilo por tiocarbonilo. Depsipétido (éster) Doble enlace olefínico azapéptido Pero hay varios más Luego tenemos: Inversión de grupos funcionales La inversión de la unión peptídica conduce a retropéptidos más resistentes a la hidrólisis enzimática. (es otro caso de peptidomiméticos) La inversión de la función éster en meperidina da un compuesto 5 veces más potente PERO NO SIEMPRE FUNCIONA BIEN… En la década del 90 se empezaron a ver casos de jóvenes drogadictos que desarrollaban síntomas relacionados con el Parkinson, lo que, hasta ese momento se consideraba una enfermedad de la vejez. La investigación rebeló que estos jóvenes consumían una droga sintética diseñada para eludir las restricciones legales. Los “diseñadores” modificaron un análogo de una droga prohibida, el demerol o meperidina (ANALGÉSICO NARCÓTICO), transformándola en un su “ester reverso” que no era prohibido. De esta manera cambiaron una funcionalidad estable como la etoxicarbonil por un grupo propionoxi que es un buen grupo saliente. La eliminación del propionato da el producto MPTP que es oxidado bajo catálisis de la MAO B a 5.134. Estudios posteriores demostraron que 5.134 es un potente neurotóxico. A partir de allí se ha hipotetizado que el Parkinson es una enfermedad ambiental que se produce por la lenta degradación de las neuronas dopaminérgicas a causa de la ingestión o inhalación de neurotoxinas similares al MPTP. Como para producir Parkinson, entre el 60-80% de estas neuronas deben haber sido destruidas, esta enfermedad aparece en los adultos mayores. Selegilina, por ser un inhibidor de la MAO B, es importante tanto para evitar la oxidación del MPTP como para mantener la concentración de dopamina. Meperidine (es un narcótico analgésico que actúa sobre el CNS). ES anticolinérgico antimuscarínico, antagonista de la acetilcolinesterasa Las modificaciones isostéricas pueden alterar una variedad de parámetros moleculares: Parámetros estructurales: Importantes cuando la parte de la molécula involucrada en el cambio isostérico contribuye a una orientación geométrica específica de otros grupos, estos cambios modifican la interacción con el receptor, por ejemplo: Estos sistema en que los dos anillos aromáticos están unidos por un anillo de 7 miembros y un sistema biciclico tienen un ángulo dihedro (alfa) de 60°, mientras que aquellos fusionados a través de anillo de 6 miembros heterocíclicos tienen ese mismo ángulo de 25°, eso cambia la actividad de antidepresiva a neuroléptica. Parámetros electrónicos Importantes cuando la parte de la molécula involucrada en el cambio isostérico participa en interacciones con el receptor (electrostáticas, transferencia de carga, puente de hidrógeno, etc). Pueden afectar pKa, grado de ionización, etc. Propiedades Farmacocinéticas: Los parámetros hidrofílicos-lipofílicos, pKa, formación de puentes hidrógeno, etc., son importantes cuando el grupo involucrado en el cambio isostérico participa en la absorción, distribución o excreción de la molécula CN CF3 CN R R= Pr, i-Pr, c-Pr Ejemplos de desarrollo de fármacos por reemplazos isostéricos FLUOURACILO DISEÑO DE DROGAS Y ESTEREOQUÍMICA Es bien conocido que la forma de una molécula y la distribución tridimensional de sus grupos funcionales, es uno de los factores más importantes que afectan la actividad de una droga y debe tenerse muy en cuenta en el diseño de análogos. Por ello la conformación es un aspecto importante en la optimización de compuesto líder. Ya dijimos que las estructuras flexibles pueden hacer que un mismo fármaco se una a más de un objetivo biológico. Muchas veces se rigidiza buscando especificidad. ADEMAS, Los sistemas rígidos se pueden utilizar también para determinar la conformación asumida por el ligando cuando se une a ese blanco CONFIGURACIÓN La presencia de centros estereogénicos en un fármaco genera variaciones farmacológicas entre los correspondientes estereoisómeros Las biomoléculas (proteínas, enzimas) son quirales, Por lo tanto, para drogas quirales: De1 R y De2 R De1 = enantiómero 1 del fármaco R = receptor Para cada enantiómero habrá una unión con el receptor que formarán dos diasterómeros diferentes, por lo tanto tienen diferentes propiedades. TALIDOMIDA CONCLUSIONES Dijimos que el camino a la aparición de un nuevo fármaco es largo y complicado y que requiere de mucho esfuerzo en todas las etapas. De hecho, son muy pocos los compuestos que llegan al final, es realmente una carrera de “la supervivencia del más apto”, el tiempo y gasto son enormes (pirámide) Los productos naturales, la síntesis química, la q. combinatoria y el diseño racional nos permiten hallar un líder que será luego ajusta a través del SAR, QSAR, CADD. Técnicas que vemos en una clase aparte, sin dejar de tener en cuenta que la síntesis y la síntesis combinatoria siguen interviniendo ya que las información obtenida aquí debe traducirse en compuestos que deben ser sintetizados…. No existe la herramienta perfecta para el desarrollo de nuevos fármacos, tanto los métodos más tradicionales como aquellos más modernos han probado su utilidad y son complementarios para lograr el objetivo final de encontrar un nuevo producto que ayude a paliar alguna de las tantas enfermedades que siguen aquejando las ser humano