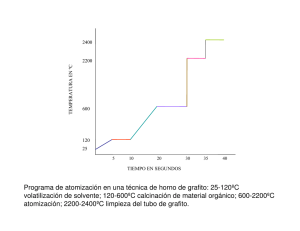
FONOAMINTOS DEl ANÁLISIS POR ABSORCION ATOMICA
Anuncio

FONOAMINTOS DEl ANÁLISIS intensidad POR ABSORCION ATOMICA Long. de Onda (nm) Espectro atómico del níquel alrededor de 232.0 nm Ing. Raúl T. Jacinto Herrera Lima - Perú PRIMERA EDICIÓN - 2003 TODOS LOS DERECHOS RESERVADOS Para Nery, Raulito, Fatimah y Renzo Quienes siempre me esperan con alegría Para mi madre y mis abuelos, Forjadores de mi vida IN TROD U CCIÓ N El presente texto tiene como objeto fomentar la comprensión de las técnicas de absorción atómica, ampliamente usadas en la industria en general. Se estima que el conocimiento de los fundamentos de la espectrometría de absorción atómica (A A S) brindará la oportunidad de mejorar la calidad del trabajo analítico mediante el conocimiento de cada parámetro de operación del equipo y la influencia de estos sobre la exactitud de los resultados. Luego de un recuento histórico breve, se estudian los principios básicos tales como la definición de espectrometría de absorción atómica, la absorción de la luz, su relación con la concentración de un elemento y la formación de los espectros atómicos. El estudio de fuentes de radiación, tanto continuas como lineales, es asimismo explicada a nivel de dispositivos tales como lámparas de deuterio y de cátodo hueco. Se describe también la atomización por flama, el sistema nebulizador-quemador, la descomposición de compuestos, la posición del quemador, la absorción del background y la influencia de la ionización. En la atomización por horno se aprecia el fundamento de operación, el equipo atomizador y los procesos de secado y calcinado. En la generación de vapor se estudia tanto la generación de hidruros como la técnica de vapor frío. El estudio de los monocromadores se complementa con el de los sistemas ópticos, la corrección del background mediante estos sistemas y el estudio de los lentes y espejos. La detección de la radiación se revisa en base al fotomultiplicador y sus características. Los sistemas de lectura son también enfocados, como medio para obtener la cuantificación de valores analíticos. En el análisis cuantitativo se estudian los parámetros de rendimiento analítico en AAS. Asimismo, se revisa el análisis práctico mediante flama, horno y generación de vapor. También se expone una introducción a la aplicación de la computación a la AAS. Posteriormente, se estudia la optimización de instrumentos y métodos. Finalmente, acorde con la tendencia actual en la industria, se revisan tópicos acerca de seguridad en el laboratorio de AAS, reconociendo el potencial de peligro de esta técnica analítica y el valor de los recurso humanos y materiales involucrados en su ejecución. Cabe destacar que este libro es una recopilación de cierta literatura sobre absorción atómica (la mayor parte en idioma inglés), traducida, organizada y con ciertas observaciones prácticas. En ningún caso,es pretensión del suscrito ser el creador de esta obra, sino tan sólo un compilador de información que considera útil para el personal técnico calificado de nuestro país, donde es escasa la literatura en castellano sobre esta materia. E l A utor F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a ÍN D IC E In tro d u c c ió n ...................................................................................................... 03 C a p ítu lo I 1. Desarrollo H istó rico ........................................................................................ 07 C a p ítu lo II 2. Principios Básicos..............................................................................................08 2 .1 Espectrometría de Absorción A tó m ic a .............................................. 08 2.2 Bases T e ó ric a s..................................................................................... I I 2.2.1 La Absorción de la Luz y el Espectro A tó m ic o ................................. I I 2.2.2 Relación Absorción de Luz-Concentración del A n a lito .................... 12 C a p ítu lo III 3. Fuentes de Radiación....................................................................................... 14 3.1 Fuentes C o ntinuas............................................................................... 14 3.1.1 Lámpara de D e u te rio .............................................................. 15 3.2 Fuentes Lineales................................................................................... 16 3.2.1 Lámparas de Cátodo Hueco ( H C L ) ....................................... 18 3.2.2 Lámparas Multi-Elemento....................................................... 22 3.2.3 Lámparas de Descarga sin Electrodo ( E D L ) ...........................22 C ap ítu lo IV 4. Atomización por Flama (F A A S ) ................................................................... 24 4.1 Tipos de Flam a..................................................................................... 25 4.2 El Sistema Nebulizador-Quemador............;....................................... 27 4.3 Nebulización.........................................................................................29 4.4 Descomposición de Com puestos....................................................... 3 I 4.5 Posición del Q u em ad o r....................................................................... 34 4.6 Absorción del Background...................................................................36 4.7 Ionización............................................................................................. 38 C a p ítu lo V 5. Atomización por Horno (G F A A S ) ..................................................................41 5.1 Principio de O p eració n ........................................................................ 41 5.2 Atomizadores de Tubo de G ra fito ...................................................... 42 5.3 Secado y Calcinado.............................................................................. 43 C ap ítu lo V I 6. Generación de Vapor (H G A A S ) ..................................................................... 45 6.1 Generación de H id ru ro s...................................................................... 45 6.2 Técnica de Vapor F r ío ..........................................................................48 - • 4 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C a p ítu lo V II 7. M onocrom adores............................................................................................5 I C a p ítu lo V III 8. Sistemas Ó p tico s ..............................................................................................55 8.1 Sistemas O p tic o s .................................................................................55 8.2 Corrección del Background................................................................ 57 8.2.1 Corrección con Lámpara de D e u te rio ...................................57 8.2.2 Corrección con Efecto Z e e m a n ............................................. 59 8.2.3 Corrección con Corriente Pulsante A lt a ............................... 59 8.3 8.4 Lentes...................................................................................................59 Espejos................................................................................................. 60 C a p ítu lo I X 9. Detección de la Radiación............................................................................... 62 9.1 El Fotomultiplicador............................................................................ 62 9.2 Características del Fotomultiplicador................................................64 C a p ítu lo X 10. Sistemas de Lectu ra......................................................................................... 66 C a p ítu lo X I I I. Análisis Cuantitativo........................................................................................ 68 I l.l Parámetros del Rendimiento Analítico en A A S ................................68 I l.l.l Concentración C aracterística.................................................68 I 1.1.2 Rango de T rab ajo .................................................................... 69 I 1.1.3 Exactitud y Precisión.............................................................. 71 11.2 Análisis Cuantitativo I: Atomización por Flam a.................................75 I 1.2.1 Estándares.................................................................................75 I 1.2.2 Adiciones de Estándar............................................................77 I 1.2.3 Situaciones Analíticas Sim ples.................................................78 11.2.4 Muestras Com plejas................................................................ 79 I 1.2.5 Determinación de T razas....................................................... 79 11.3 Análisis Cuantitativo II: Atomización por H o r n o ............................. 80 1.4 1.5 1.6 1.7 I 1.3.1 Interferencias Físicas............................................................... 80 I 1.3.2 Absorción por Background.................................................... 81 I 1.3.3 Interferencias Q uím icas.........................................................82 11.3.4 Modificación Q u ím ica............................................................. 83 11.3.5 Estándares................................................................................83 I 1.3.6 M uestras.................................................................................. 84 Análisis Cuantitativo II!: Generación de V a p o r.................................84 Micromuestreo por Flam a...................................................................86 Automatización................................................................................... 86 Aplicación de las Computadoras al A A S ............................................ 87 I 1.8 Software para A A S .............................................................................. 88 I I I I C a p ítu lo X I I I 2. Optimización de Instrumentos y M éto d o s.................................................... 94 12.1 Reconocimiento de Errores y Optimización del Instrum ento..........94 12.l.l Sensitividad Demasiado B a ja .................................................. 95 - 5 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a 12.1.2 Sensitividad Demasiado A lt a .................................................. 96 C ap ítu lo X I I I 13. Seguridad......................................................................................................... 98 I 3 .1 Sistema de Extracción..........................................................................98 13.2 Cilindros de Gases Comprimidos (Acetileno, A rg ó n )....................... 99 13.3 Tuberías Flexibles de Gas y C o nexio nes............................................ 99 13.4 A cetileno.............................................................................................. 99 13.5 Óxido N itro s o .................................................................................. 100 13.6 Solventes Inflamables......................................................................... 100 13.7 Quem adores...................................................................................... 100 13.8 Trampa de Líquidos........................................................................... 101 13.9 Radiación Ultravioleta........................................................................ 101 13.10 Peligros del C a lo r ............................................................................... 101 13.11 Inspección de Seguridad..................................................................... 101 C apítulo X I V 14. A n e x o s ........................................................................................................... 14 .1 Modos de Medición en A A S .............................................................. 14.2 Modos de Calibración en A A S .......................................................... 14.3 Tipos de Medición en A A S ................................................................ 103 103 103 103 B ib lio g ra fía ........................................................................................................ 105 - 6 - F u ndam entos del A n á l is is po r A b s o r c ió n A t ó m ic a C A PÍTU LO I DESARROLLO HISTÓRICO I. D esa rro llo H is t ó r ic o ♦ La historia de la espectrometría atómica se inicia con el uso de los lentes por Aristófanes alrededor del 423 A .C . y los estudios de espejos por Euclides (300 A.C.) y Hero (100 A.C .). Séneca (40 D.C.) observó las propiedades de difusión de la luz de los prismas, y en el año 100 D.C. Ptolemeo estudió la incidencia y refracción. ♦ Alhazen en 1038 estudió la reflexión y refracción de la luz, y en 1250 Roger Bacon determinó los puntos focales de espejos cóncavos. Alrededor de 1600, se desarrolló el telescopio en Holanda y en 1610 Galileo hizo mejoras en el diseño del telescopio. Sir Isaac Newton (1642-1727) realizó muchos experimentos sobre la separación de la luz para obtener un espectro y los índices de refracción de diferentes colores de luz; Newton aplicó estos principios al telescopio. ♦ Fraunhofer, en 1815, observó el fenomeno de difracción y pudo medir la longitud de onda en vez de ángulos de refracción. Herschel (1823) y Talbot (1825) descubrieron la emisión atómica cuando colocaron ciertos átomos en una flama. Wheatstone concluyó en 1835 que podían distinguirse los metales entre sí sobre la base de la longitud de onda de esta emisión. En 1848, Foucault observó la emisión atómica del sodio y descubrió que este elemento absorbía los mismos rayos de un arco eléctrico. ♦ A finales de 1800, científicos tales como Kirchoff, Bunsen, Angstróm, Rowland, Michelson y Balmer estudiaron la composición del sol basada en sus emisiones a diferentes longitudes de onda. Kirchoff estableció la ley "Lo materia absorbe luz a la misma longitud de onda a la que la emite", en la cual se fundamente la espectrometría de absorción atómica. W oodson fue uno de los primeros en aplicar este principio a la detección del mercurio. ♦ La técnica de absorción atómica, tal como en la actualidad es conocida, fue desarrollada por Sir Alan W alsh a mediados de la decada de 1950 sobre la base del estudio del análisis espectroquímico de metales y la espectroscopia molecular. Esta técnica se ha convertido en el método preferido de análisis elemental. ♦ W alsh descubrió que la mayoría de los átomos libres en las flamas comúnmente usadas se encontraban en el estado basal, pero que las flamas no tenían la suficiente energía para excitar a a estos átomos (excepto para los elementos del grupo I). Una fuente de luz que emite una línea espectral angosta de la energía característica es usada para excitar a los átomos libres formados en la flama. Entonces se mide el decrecimiento en energía (absorción). — F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a C A P Í T U L O II PRIN CIPIOS B Á S IC O S 2. P r in c ip io s 2 .1 B á s ic o s E s p e c t r o m e t r ía de A b s o r c ió n A t ó m ic a La técnica analítica cuantitativa denominada absorción atómica (A A S) se basa en estos tres principios: espectrometría de i. Todos los átomos pueden absorber luz. ii. La longitud de onda a la cual la luz es absorbida es específica para un elemento químico en particular. iii. La cantidad de luz absorbida es proporcional a la concentración de los átomos absorbentes. Notas: 1. De acuerdo con el segundo principio, si una muestra que contiene fierro junto con otros elementos tales como cadmio y plata es expuesta a una luz que corresponda a la longitud de onda característica para el fierro, solo los átomos de fierro absorberán esta luz. 2. Los siguientes elementos (figura 01) son detectables por la técnica de absorción atómica. TABLA PERIÓDICA He Be B C N 0 p Na Mg Al Si P 'o Cl Ar Cu Zn Ga Ge As Se Br Kr I A6 Li K. Ca Se Rb Si Y Cr Mn Fe Co V Zr Nb Mo Te Ru fih Pd Ag Cd Cs Ba La Hf Ta W Ff r] Ni Ti Fie Os It In Pt Au Hg TI Sn Sb Te Pb Bi Po Al Fin Ac Rf Db Sn Bh H s Mi Ce Pr Nd Pr.' Sm Eu Gd Tb Dy Ho Er Trn Yb Lu Th Píe U Np Hu Arn Crn Bk a Es Fm M d No Lv-.i Figura 01. Elementos detectables mediante AAS en la tabla periódica (sombreados) Figura OI. Elementos detectables mediante A A S en la tabla periódica. F undam entos del A n á l is is po r A b s o r c ió n A t ó m ic a Un espectrómetro de absorción atómica (figura 02) es un instrumento en el cual estos principios se aplican al análisis cuantitativo práctico; este consiste en: ♦ Una fuente de luz para generar luz a la longitud de onda característica del ♦ elemento a analizar. Un atomizador para crear una población de átomos a analizar ♦ Un monocromador para filtrar luz a la longitud de onda característica. ♦ Un sistema óptico para dirigir a la luz desde la fuente a través de la población de átomos y hacia el monocromador. ♦ Un dispositivo sensitivo a la luz. ♦ Dispositivos electrónicos adecuados que midan la respuesta del detector y traduzcan esta respuesta en mediciones analíticas útiles. Figura 02. Esquema típico de un espectrómetro de absorción atómica Rangos de Longitudes de Onda en A A S El rango de longitud de onda útil para un espectrómetro de absorción atómica depende de la fuente de radiación, los componentes del sistema óptico y el detector. En la operación práctica este rango varía usualmente desde 852.1 nm, la longitud de onda más sensitiva para el Cesio; hasta 193.7 nm, la longitud de onda más ampliamente usada del Arsénico al comienzo del vacío ultravioleta. Análisis Cuantitativo por AAS La técnica de espectrometría de absorción atómica por flama (F A AS) requiere que una muestra líquida sea aspirada, aerosolizada, y mezclada con gases combustibles, tales como acetileno y aire o acetileno y óxido nitroso. La mezcla es ignicionada en una flama cuya temperatura oscila desde 2100 a 2800°C. Durante la combustión, los átomos del elemento de interés en la muestra son reducidos a átomos en el estado basal libre y no excitados, los cuales absorben luz a longitudes de onda características. Las longitudes de - 9 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a onda características son específicas para cada elemento y precisas hasta 0.01-0.1 nm. Para proporcionar longitudes de onda específicas al elemento, se pasa a través de la flama un rayo de luz desde una lámpara cuyo cátodo es hecho del elemento que se está determinando. Un dispositivo tal como un fotomultiplicador puede detectar la cantidad de reducción de la intensidad de luz debida a la absorción por el analito, y esto puede ser directamente relacionado a la cantidad del elemento en la muestra. La siguiente tabla muestra en forma esquemática las etapas de procedimiento analítico básico en AAS. Tabla I. Procedimiento Analítico Básico de la AAS Descripción Etapa 1 Conversión de la muestra en solución 2 Preparación de una solución que no contenga el elemento a analizar (blanco analítico) 3 Preparación de estándares (una serie de soluciones de calibración que contengan cantidades exactamente conocidas del elemento a analizar) 4 Atomización del blanco y los estándares a su vez y medir la respuesta para cada solución 5 Plotear un gráfico de calibración que muestre la respuesta obtenida para cada solución (figura 04). 6 Atomización de la solución de muestra y medición de la respuesta 7 Referencia al gráfico de calibración, ingreso al gráfico en el punto correspondiente a la respuesta para la muestra, y lectura de la concentración de la muestra. Figura 03. Gráfico de calibración Figura 03. Gráfico de Calibración - 10 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a 2.2 B ase s T e ó r ic a s 2 .2 .1 L a A b s o r c i ó n de l a L u z y el E sp e ctro A tó m ic o Se conoce que todos los átomos pueden absorber luz a ciertas longitudes de ondas discretas, correspondientes a los requerimientos de energía de un átomo en particular. Los átomos de sodio, por ejemplo, absorben luz muy fuertemente a 589.0 nm, debido a que la luz a esta longitud de onda tiene exactamente la energía justa para transformar al átomo de sodio a otro estado electrónico. Esta transición electrónica es completamente específica para el sodio: los átomos de cualquier otro elemento son diferentes y no pueden absorber luz a esta longitud de onda. Si el átomo de sodio está en el “ estado basal” , decimos que cuando un átomo de sodio absorbe luz se transforma a un estado excitado; es todavía un átomo de sodio, pero contiene más energía. Existen varios posibles estados excitados para el átomo de sodio y cada uno tiene una energía particular. Usualmente esta energía es medida con relación al estado basal, y un estado excitado particular para el sodio puede ser 2.2eV (electrón voltios) sobre el estado basal. Esto significa que un átomo en el estado excitado contiene 2.2 eV más energía que un átomo en el estado basal el cual, por convención, tiene adscrito una energía arbitraria de cero. Algunos de los posibles estados de energía para el átomo de sodio se muestran en la figura 04. 6 4P 3P 2.2 eV 330.3 nm 589.0 nm 0 3S 3.6 eV estado basal 0.0 eV Figura 04. Algunos posibles estados de energía para el átomo de sodio Figura 04. Algunos posibles estados de energía para el átomo de sodio. Cada transición entre diferentes estados de energía electrónicos se caracteriza por tener una energía diferente, y por lo mismo una diferente longitud de onda de luz. Estas longitudes de onda están definidas en forma aguda y cuando se examina un rango de longitudes 11 ■ i- F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a de onda, cada longitud de onda muestra una energía aguda máxima (una 'línea' espectroscópica). Los espectros atómicos se distinguen por estas líneas características, que pueden ser contrastadas con la banda espectral más amplia asociada con las moléculas. Las líneas que se originan del átomo en estado basal son a menudo de interés en espectrometría de absorción atómica y se denominan líneas de resonancia. El espectro atómico característico de cada elemento comprende cierto número de líneas discretas, algunas de las cuales son líneas de resonancia. La mayoría de las otras líneas se desprenden de los estados excitados, más que del estado basal. Desde que las líneas de resonancia son mucho más sensitivas y desde que la mayoría de los ' átomos en un atomizador práctico se encuentran en el estado basal, estas líneas de estado excitado no son generalmente útiles para el análisis por absorción atómica. 2 .2.2 R e l a c ió n A b s o r c ió n d e L u z - C o n c e n t r a c ió n d e l A n a l it o La relación entre la absorción de luz y la concentración del elemento a analizar se define por medio de las leyes fundamentales de la absorción de la luz: Ley de Lam berf "La porción de la luz absorbida por un medio transparente es independiente de la intensidad de la luz incidente y cada unidad sucesiva de espesor del medio absorbe una fracción igual de la luz que pasa a través de él”. Ley de Beer: “ La absorción de luz es proporcional al número de átomos absorbentes en la muestra”. La ley combinada Beer-Lambert puede expresarse así: ó lo g io I0 / It - abe = Absorbancia (I) donde: l0 : potencia de radiación incidente Ir : potencia de radiación transmitida a : coeficiente de absorción (absorbancia) b : longitud del paso de absorción c : concentración de los átomos absorbentes esto es, la absorbancia es proporcional a la concentración del elemento para un paso de absorción dado y una longitud de onda dada. Si una muestra de concentración c tiene cierta absorbancia medida, otra muestra de concentración 2c, de acuerdo con la ley de Beer-Lambert, tendrá dos veces esta absorbancia. La ‘absorbancia’ es F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a una medida de la cantidad de luz absorbida por los átomos bajo ciertas condiciones dadas, y esta es la medición de interés; la ley de Beer-Lambert nos permite relacionar la absorbancia medida a la concentración de elemento a analizar en la muestra. En principio, podría ser posible calcular la concentración directamente de la ecuación ( I). Sin embargo, en la práctica las cantidades a y b en la ecuación ( I ) son constantes y normalmente no son determinadas. La manera más simple de usar los métodos de absorción atómica es medir la absorbancia para soluciones estándar donde la concentración es conocida y luego comparar esta curva con la absorbancia obtenida por la muestra desconocida. Desde que la absorbancia medida depende directamente de la concentración del ' elemento a analizar, este procedimiento proporciona un método simple y preciso para determinar la concentración desconocida mientras que compensa los efectos instrumentales que hacen el cálculo teórico muy incierto. Convencionalmente, puede hacerse la calibración y comparación con estándares en forma gráfica. Se prepara un gráfico de calibración para cada situación relacionando ia concentración del elemento a analizar con la absorbancia medida. Bajo la ley de Beer-Lambert, este gráfico será lineal y la concentración de muestras desconocidas puede ser determinada por interpolación en el gráfico (figura 03). - 13 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O III F U E N T E S DE R A D IA C IÓ N 3. F uen tes 3.1 de R a d ia c ió n F u e n t e s C o n t in u a s En una fuente continua la radiación esta distribuida en forma continua sobre un amplio rango de longitudes de onda. En AAS, las fuentes continuas se usan principalmente para medición y corrección secuencial o cuasi simultánea del efecto de background. Para este propósito se usan mayormente las lámparas de deuterio o las halógenas. Las fuentes continuas, en principio, también pueden ser usadas para medir la absorción atómica. Sin embargo, ellas requieren un aparato espectral con alta resolución, tal como el policromador Echelle, para retener la especificidad de la A A S que normalmente es proporcionada por las fuentes lineales. El uso de fuentes continuas sólo es practicable para determinaciones simultáneas multi-elementos. Figura 05. Características espectrales de una fuente continua Figura 05. Características espectrales de una fuente continua - ■ i- 14 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Notas: i. La lámpara de deuterio es una lámpara espectral con deuterio como gas de descarga en un bulbo de cuarzo. Para algunas aplicaciones también se ha usado hidrógeno como el gas de descarga. La lámpara de deuterio emite una potencia radiante lo suficientemente alta en el corto rango de longitudes de onda desde cerca de 190 nm hasta 330 nm. ii. Una lámpara halógena consiste en una banda o espiral calentada eléctricamente, usualmente de tungsteno, en un bulbo de cuarzo o vidrio. El oscurecimiento del bulbo por depósitos metálicos es prevenido por aditivos gaseosos halogenados. La lámpara halógena emite potencia radiante suficientemente alta en el rango espectral desde 300 nm y se extiende lejos en el infrarrojo. Sus características espectrales se muestran en la figura 05. Como puede observarse, ambas lámparas complementan sus respectivos rangos de trabajo. 3.1.1 L á m pa r a d e D eu t e r io Una fuente continua de luz encuentra aplicación en AAS, no para medir la absorción atómica, sino para medir y corregir los efectos de ‘background’ causado por especies moleculares o por difusión. Esta es llamada corrección por background. La fuente comúnmente usada en una lámpara de descarga llena con deuterio (o hidrógeno) que emite un espectro continuo intenso desde debajo de 190 nm hasta cerca de 425 nm (ver figura 06), Esto cubre la región donde ocurren la mayoría de líneas de absorción atómica y donde los efectos de la absorción por background son más pronunciados. Se usa un gas poliatómico (H 2 o D 2) debido a que produce un espectro continuo en vez de uno lineal. long. de onda (nm) Figura 06. Características espectrales de la lampara de deuterio Figura 06. Características espectrales de una lámpara de deuterio La lámpara de deuterio es diferente de una lámpara de cátodo hueco en construcción y operación. Esta lámpara incorpora un cátodo emisor de electrones, un ánodo de metal y una abertura restrictiva entre los dos (figura 07). Una corriente de descarga de varios cientos de miliamperios excita al gas deuterio. La descarga es forzada a pasar a - 15 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a través de la pequeña abertura, formando un área definida de alta excitación y por lo mismo de alta emisión de luz. Un vidrio de cuarzo transmite la luz al sistema óptico del espectrómetro. La corriente de operación para la lámpara de deuterio es usualmente controlada en forma automática por el espectrómetro; en la operación práctica sólo debe encenderse el sistema corrector por background y asegurar el correcto alineamiento de la lámpara. Figura 07. Lámpara de deuterio 3.2 F u e n t e s L in e a l e s Las fuentes lineales son fuentes de radiación espectral en las cuales el elemento analito es volatilizado y excitado de modo que emita su espectro a una longitud de onda discreta. Esta excitación puede ser causada por una descarga eléctrica de baja presión, por micro o radio ondas o por energía térmica. _ - 16 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a Las lámparas amarillas de la calle son ejemplos familiares. Estas lámparas contienen vapor de sodio, y la luz se emite únicamente a dos longitudes de ondas discretas, 588.0 nm y 589.6 nm. Las conocidas líneas “ D ” del sodio. Las características de salida espectral de una lámpara de sodio se muestran en la figura 08. El uso de fuentes lineales en A A S es posible sin monocromadores de alta resolución debido a que los elementos concurrentes, en principio, no pueden absorber radiación de la fuente de radiación específica del elemento. Sin embargo, se requiere como pre-requisito que el analito deba estar presente en alta pureza espectral en la fuente. Si existen varios elementos en la fuente de radiación, las interferencias espectrales en el área de la línea analítica, causada por las de una línea pasando a través del slit de salida del monocromador y llegando al detector, deben ser evitadas. Para los instrumentos prácticos de absorción atómica, se requiere que la fuente de luz emita luz que esté precisamente a la misma longitud de onda a la que ocurre la absorción atómica para el elemento de interés. En forma conveniente, esto puede hacerse usando una fuente de luz que emita el mismo espectro atómico, desde que las longitudes de onda de emisión y absorción atómica son idénticas. En la mayoría de los instrumentos de absorción atómica, la fuente de luz usada es la lámpara de cátodo hueco. Figura 08. Características espectrales de una fuente lineal. 17 F u ndam entos del 3.2.1 A n á l is is p o r A b s o r c ió n A t ó m ic a L ám paras de C á to d o H u ec o (H C L ) Una lámpara de cátodo hueco típica (figura 09) consiste en una envoltura de vidrio que contiene un cátodo (un cilindro de metal contenedor del elemento químico a ser excitado) y un ánodo adecuado, mayormente hecho de tungsteno o níquel. El cilindro de vidrio de la lámpara contiene un gas inerte, usualmente argón o neón, a una presión cercana a I kPa. Algunos cátodos pueden simplemente ser maquinados de una barra del elemento puro requerido (níquel, fierro, cobre, etc.). Algunos elementos, sin embargo, son dificultosos de maquinar, y los cátodos para estos elementos son producidos mediante metalurgia del polvo. El cátodo es usualmente circundado por una capa aislante de mica, material cerámico o vidrio. Esto asegura que la descarga sea confinada al interior del cátodo y proporciona una considerable mejora en la intensidad de las líneas espectrales emitidas. base cátodo cubierta de vidrio ventana án° d ° envoltura de vidrio Figura 09. Lámpara de cátodo hueco (HCL) Figura 09. Lámpara de cátodo hueco (H C L) El ánodo puede ser un anillo anular alrededor de la boca del cátodo, una ‘cuchilla’ cerca de la boca del cátodo, o un alambre o varilla localizada en una posición conveniente. En algunos diseños, el ánodo también actúa como un soporte mecánico para el material aislante. El material usado para la ventana de la lámpara es importante desde que este debe transmitir las lineas espectrales del elemento usado para el cátodo. Usualmente se utiliza el pyrex o cuarzo debido a sus características de transmitancia a las longitudes de onda sobre los 300 nm. (figura 10). - 18 - ' F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Figura 10. Características de transmitancia del cuarzo y pyrex Figura IO. Características de transmitancia del cuarzo y pyrex. El gas de relleno es usualmente argón o neón a una presión de IO a 15 torr. (cerca de l/50 av o de la presión atmosférica). Se prefiere al neón debido a que produce una intensidad de señal más alta que el argón, pero donde ocurre una línea de neón en la vecindad de la línea de resonancia de un elemento, se usa argón en su reemplazo. Mecanismo de Operación Cuando se aplica un voltaje alto -hasta 600 voltios- a través de los electrodos, los iones gaseosos positivamente cargados bombardean el cátodo y fuerzan a salir a los átomos del elemento usado en el cátodo. Estos átomos son subsecuentemente excitados por procesos colisionales y puede producirse el espectro de ese elemento. - 19 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Por supuesto, las lámparas de cátodo hueco no son las únicas fuentes de luz capaces de producir el espectro de líneas de los elementos químicos, pero estas son las fuentes más universalmente aceptadas para instrumentos de absorción atómica. Características de las Lámparas de Cátodo Hueco a. Generan una línea muy estrecha. A fin de medir la absorción del b. pico, el ancho de la línea fuente debe ser mucho más estrecho que el ancho de la línea de absorción. El ancho de la línea de absorción de los átomos en la flama es fuertemente influenciada por la temperatura y presión, y es normalmente cercano a 0.01 nm. Los átomos en la lámpara de cátodo hueco están en un ambiente que posee una temperatura y presión considerablemente más baja. En consecuencia, el ancho de la línea de emisión es cerca de un décimo del ancho de la línea de absorción. Su línea de emisión es fijada precisamente a la misma longitud de c. onda de la línea de absorción debido a que es generada por la misma energía de transición. Esto significa que la fuente de luz inherentemente genera una señal de emisión a la longitud de onda correcta para la absorción óptima. Las H C L pueden fabricarse para cualquier elemento químico factible d. de ser determinado por AAS. Todos estos elementos son adecuados para inclusión en el cátodo por un medio u otro (maquinado o metalurgia de polvo) Las H C L son sencillas de operar. Todo lo que se necesita es e. conectar los electrodos a un suministro de potencia adecuado y ajustar la corriente al valor prescrito. Son estables e intensas. Esta disponible una intensidad adecuada de f. señal para la mayoría de elementos tales que la intensidad no es el factor limitante común. La absorción es independiente de la intensidad de la fuente de modo que el incremento de la intensidad de la señal no incrementa el grado de absorción. Debe señalarse, sin embargo, que la intensidad y el ruido están relacionados en la absorción atómica, así como en la mayoría de técnicas espectrométricas. Las lámparas de cátodo hueco son económicas. La mayoría de fabricantes garantizan sus lámparas por una vida de 5000 miliamperios-hora o dos años. Esto significa que si una lámpara es operada a 5 mA, se garantiza que dura al menos 1000 horas de operación. En uso rutinario esto es adecuado para muchos millares de determinaciones. Condiciones de Operación Para la operación práctica, es esencial que la lámpara de cátodo hueco esté correctamente alineada en el haz óptico. Esto es, sin embargo, asunto de alinear mecánicamente la lámpara por medio de uno o dos tornillos para obtener la máxima señal. Una vez que la - 20 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a lámpara está correctamente alineada, la corriente de operación es entonces el único parámetro que requiere cierta atención. Los fabricantes generalmente recomiendan una corriente de operación adecuada. Esto es rara vez altamente crítico, y pequeñas desviaciones de esta no afectarán demasiado la sensitividad analítica. Las desviaciones mayores de la corriente recomendada pueden ser más serias. Si la corriente de la lámpara es demasiado pequeña, la señal analítica requerirá amplificación excesiva. Esto casi siempre producirá un ruido alto indeseable. Existen dos efectos adversos como resultado de operar una lámpara a alta corriente. El primero de estos es que la línea de resonancia se vuelve ampliada y distorsionada. Este fenómeno es conocido como 'ampliación de auto-absorción’ y es causado por átomos en la descarga que absorben a la longitud de onda de resonancia emitidos por átomos similares. El resultado es que la sensitividad de la absorción es degradada y la curva de calibración se incrementa. La figura I I ilustra el cambio en la curvatura para el magnesio cuando la corriente de la lámpara se incrementa. El segundo efecto adverso es que la vida de la lámpara se acorta a medida que se incrementa la corriente de operación. En base a estos factores, es buena práctica de operación usar la recomendación del fabricante como un punto de inicio y luego encontrar mediante ensayo-error la corriente que brinde la combinación óptima de ratio señal-ruido y linealidad de calibración. La consideración principal es no exceder la corriente máxima estimada. Figura 11. Efecto del incremento de la corriente de lámpara sobre la curvatura de la calibración Figura I I. Efecto del incremento de la corriente de lámpara sobre la curva de calibración _ _ _ F undamentos del 3.2.2 A n á l is is po r A b s o r c ió n A t ó m ic a L á m p a r a s M u l t i- E l e m e n t o Al combinar dos o más elementos de interés en un cátodo, es posible producir una lámpara de cátodo hueco que pueda ser usada para el análisis de más de un elemento. Tal lámpara es de conveniencia obvia para el análisis práctico, pero existen algunas limitaciones a este enfoque. Algunas combinaciones de elementos no pueden usarse debido a que sus líneas de resonancia están tan cerca que ellas interfieren entre sí. Esto hace imposible resolver la línea requerida y la combinación es así no trabajable. Otras combinaciones no pueden ser usadas simplemente debido a dificultades de fabricación al tratar de incorporar elementos de características físicas ampliamente diferentes a un cátodo común. Con lámparas multielemento de más de tres elementos, la intensidad radiante de las líneas analíticas individuales puede ser considerablemente más baja que con las lámparas de un único elemento. Esto conduce a un ratio señal-ruido desfavorable que puede influenciar la precisión y los límites de la detección. Asimismo, las curvas de calibración son más fuertemente curvadas debido a la alta radiación por background resultante de la multitud de líneas espectrales, de modo que el rango lineal de trabajo es restringido si se compara con las lámparas de elemento único. Sin embargo, para los elementos químicos a los cuales estas limitaciones no se aplican, las lámparas multi-elemento proporcionan al análisis por A A S una fuente conveniente para la determinación de rutina de varios elementos. 3.2.3 L á m p a r a s d e D e s c a r g a s in E l e c t r o d o (E D L ) La lámpara de descarga sin electrodo es una fuente de luz alternativa para un rango limitado de elementos y está entre las fuentes de radiación que exhiben la más alta intensidad radiante y el ancho de línea más estrecho. En estas lámparas, el vapor del elemento es excitado para producir su espectro atómico por medio de un campo de radiofrecuencia (27.12 MHz). La potencia requerida de radiofrecuencia es usualmente generada por un oscilador de tubo de vacío y un amplificador contenido en un suministro separado de potencia. Los sistemas comerciales usualmente incluyen un módulo de conversión el cual asegura que la modulación de la lámpara esté directamente sincronizada con el espectrómetro. Para elementos volátiles en el U V lejano, una lámpara ED L con excitación por radiofrecuencia brinda una mejora significativa en el ratio señal-ruido, así como en el limite de detección. La gran pureza espectral también da lugar a curvas de una notable mejor linealidad. En _ 2 2 _ F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a la actualidad, las ED L están bien establecidas en A A S y han reemplazado a otros tipos de lámparas para ciertos elementos. En el caso del Arsénico, por ejemplo, una ED L proporciona mejora en la sensitividad y da un mejor límite de detección por un orden de magnitud comparado al H C L equivalente. En el caso del Cesio y Rubidio las ED L han reemplazado a las lámparas de descarga de vapor y al mismo tiempo han logrado límites de detección que antes sólo se habían logrado con espectrometría de emisión de flama. La determinación de fósforo solo se ha tornado factible después de la introducción de la EDL. Las ED L están en la actualidad disponibles para todos los elementos volátiles y complementan el rango de las HCL. Ellas típicamente proporcionan límites de detección que son mejores en un factor de dos o tres. También las H C L para elementos volátiles son ocasionalmente inestables y tienen tiempos cortos de vida, mientras que para estos elementos la ED L es especialmente estable y tiene un tiempo de vida prolongado. - 23 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O IV A T O M I Z A C I Ó N PO R FLA M A (F AAS) 4. A to m iz a c ió n por F la m a ( F A A S ) % Un requerimiento básico para el método analítico de absorción atómica es que la muestra sea descompuesta en la mayor extensión posible en sus átomos constituyentes en el estado basal y exponer esta población de átomos a una luz de longitud de onda característica de absorción. Idealmente, esta etapa de atomización debería ser cuantitativa; no debería haber enlaces residuales en la nube atómica de la fase gaseosa. Cualquier logro menor a la atomización completa producirá menores sensitividades en cualquier método. Debe notarse que los cambios en la fracción de atomización de muestra a muestra o de muestra a estándares causarán errores en la calibración. Así, es menos importante que se logre la atomización completa si se compara con el logro de una atomización fraccional extremadamente consistente. En la medida que esta condición no se cumpla, la interferencia interelementos (matricial) en la espectrometría atómica puede ser extremadamente problemática. La atomización de muestras en solucion procede a través de un proceso de nebulización (spraying). En términos simples, la nebulización sirve para incrementar el área superficial de la muestra en solución, de modo que la evaporación del solvente (desolvatación) pueda proceder más rápidamente y en forma tal que las partículas secas de soluto puedan ser volatilizadas mejor. Este esquema, empleado en F AAS, es mostrado en la ruta del lado izquierdo en la figura 12. Una vez formadas, las gotas en el spray nebulizado son enviadas a un ambiente de alta temperatura tal como una flama química o un flujo de plasma de un gas raro. Allí tiene lugar la desolvatación y vaporización de las partículas de soluto, y el vapor resultante es convertido con mayor o menor eficiencia en átomos libres. De hecho, el ambiente en estas descargas es a menudo lo suficientemente caliente de modo que muchos de los átomos que son formados se tornan iones cargados positivamente. Asimismo, el ambiente en estas fuentes de atomización es usualmente lo suficientemente energizado como para producir una fuerte emisión ya sea desde los átomos liberados o sus contrapartes iónicas. - 24 - Fundamentos del A n á l is is por A b s o r c ió n A t ó m ic a Figura 12. Esquema básico de la atomización en A A S 4.1 T ip o s d e F la m a Pueden lograrse diferentes flamas usando diferentes mezclas de gases, dependiendo de la temperatura y velocidad de combustión deseadas. Algunos elementos solo pueden ser convertidos en átomos a altas temperaturas. Aún a altas temperaturas, si está presente exceso de oxígeno, algunos metales forman óxidos que no se redisocian en átomos. Para inhibir su formación, pueden modificarse las condiciones de la flama para lograr una flama reductora (no oxidante). - 25 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a Muchas de las combinaciones combustible/oxidante que fueron ensayadas en forma experimental demostraron no ser adecuadas en una forma u otra por razones de utilidad analítica, seguridad, costo o conveniencia. En la espectrometría de flama moderna, las mezclas aire-acetileno y óxido nitroso-acetileno son casi universalmente utilizadas para análisis prácticos. Flamas de aire empleando hidrógeno fueron usadas en cierto grado, mayormente en asociación con técnicas de generación de vapor, pero los sistemas modernos de generación de vapor ahora se basan en ‘celdas de absorción' calentadas eléctricamente o por flama de aire-acetileno. Flamas de aire-hidrógeno y óxido nitroso-hidrógeno son aún ocasionalmente usadas para algunas aplicaciones, pero la práctica moderna tiende a favorecer su reemplazo con flamas de acetileno. La flama aire-acetileno es la flama más comúnmente usada en el análisis por absorción atómico práctico. La temperatura de operación es cerca de 2300°C. La flama de óxido nitroso-acetileno es considerablemente más caliente y produce una temperatura de cerca de 3000°C. Esta atomizará compuestos refractarios de elementos tales como el aluminio, sílice, vanadio y titanio; así como los elementos de tierras raras, aún cuando estos formen moléculas altamente refractarias en las flamas. Para cada una de estas flamas, la temperatura de operación y el ambiente químico intrínseco de la flama dependerá del ratio combustible-oxidante usado. Estas condiciones están caracterizadas como: i. Pobre en combustible, u oxidante. Esta es la flama más caliente ii. Rica en combustible, o reductora. Esta es la más fría iii. Estequiométrica, o flama químicamente balanceada. La temperatura es cerca de la mitad del rango; el ratio combustible-oxidante es cerca de la mitad entre la flama pobre en combustible y rica en combustible. La tabla II muestra algunas mezclas combustible-oxidante junto con las máximas temperaturas alcanzadas. La tabla III muestra algunos elementos de interés para los cuales se usa aire-acetileno y óxido nitroso-acetileno, junto con los rangos de concentración de trabajo. Tabla II. Temperaturas Máximas de Flamas Combustible Oxidante Temperatura °K Acetileno Aire 2400 - 2700 Acetileno Oxido Nitroso 2900 - 3100 Acetileno Oxígeno 3300 - 3400 Hidrógeno Aire 2300 - 2400 Hidrógeno Oxígeno 2800 - 3000 Cianógeno Oxígeno 4,800 - 26 - F undam entos del 4.2 A n á l is is p o r A b s o r c ió n A t ó m ic a E l S is t e m a N e b u l iz a d o r - Q u e m a d o r Los sistemas nebulizador-quemador usados en instrumentos de absorción atómica atomizan la solución a analizar en etapas sucesivas (figura 13). haz óptico O O - ÍJ- o átomos libres J ~ ) ------------------- ^ O atomización especies moleculares vaporización líquido fundido partículas sólidas desolvatación aerosol combustible-oxidante solución exceso de solución a desecho Figura 13. Atomización por flama Figura I 3 Atomización por flama gotas La solución debe primero ser convertida en un spray (aerosol) de finas por un nebulizador neumático que típicamente tiene la forma y geometría ilustrada en la figura 14. - 27 ' T- F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a Figura 14. Nebulizador neumático En operación, un gas (usualmente el oxidante para la flama) fluye a través del nebulizador, pasa a través de la sección venturi y deposita solución en el venturi como un aerosol de gotas. Este aerosol de alta velocidad golpea un obstáculo cuidadosamente seleccionado (usualmente una pieza esférica de vidrio) donde las gotas más grandes son divididas en gotas más pequeñas. Solo cerca de un 10% de la solución es convertida en gotas lo suficientemente finas como para ser llevadas a la flama, el remanente es empujado contra la pared de la cámara de aerosol y drenada a la solución de desecho. El aerosol y el oxidante son mezclados con el combustible entrante y la mezcla completa finalmente fluye a través del quemador (figura 15). Figura 15. Sección transversal de un sistema de cámara de aerosol/quemador - 28 - F u n d a m e n t o s del A n á l i s i s por A b s o r c i o n A t o m ica 4.3 N eb u liz a c ió n La función del nebulizador es convertir la solución analítica en un aerosol que sea plenamente compatible con los requerimientos para la disociación efectiva en la flama. Físicamente, estos requerimientos parecen ser cumplidos por una población de gotas que tienen diámetros medios entre 5 y 7 (jm y un máximo de 20 pm. Obviamente, las pequeñas gotas son más fáciles de secar y vaporizar que las gotas más grandes, y el énfasis es asimismo en maximizar la proporción de gotas pequeñas dentro del sistema. La posición de la esfera de vidrio relativa al nebulizador venturi es un factor crítico para determinar la eficiencia del nebulizador, tamaño de gota, y respuesta analítica (figura 16). La posición óptima dependerá de las características físicas de la solución y será necesario ajustar en forma concordante la posición de la esfera de vidrio. distancia esfera-venturi (mm) Figura 16. Influencia del cambio de posición de la esfera de vidrio Figura I 6. Influencia del cambio de posición de la esfera de vidrio La presión a la cual opera el nebulizador es un determinante importante del tamaño de gota, eficiencia integral de la nebulización y velocidad de aspiración de solución. En la práctica, la presión de operación es un valor fijo establecido por el fabricante como el óptimo para el diseño particular del nebulizador. Sin embargo debe observarse que se deben evitar F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a las fluctuaciones de corto término en la presión de operación desde que esto puede causar variaciones de corto término en el proceso de atomización y así degradar la precisión analítica. La velocidad a la cual la solución es aspirada a la flama afectará la respuesta instrumental como se muestra en la figura 17. Puede verse que la curva absorbancia-velocidad de aspiración de solución tiende a aplanarse a velocidades de aspiración más altas que 6 ml/minuto, y este es generalmente el máximo práctico para los nebulizadores convencionales fijos. A velocidades de aspiración más altas el sistema tiende a saturarse, y la solución adicional no es atomizada en forma efectiva por la flama. Más aún, el efecto de enfriamiento de la solución en exceso acentuará cualquier tendencia a que ocurra alguna interferencia química. Figura 17. Influencia de la variación de la velocidad de aspiración de solución. - 30 - Fundamentos del A n á l is is po r A b s o r c ió n A t ó m ic a Las diferencias en velocidades de aspiración entre muestras y estándares afectará claramente la precisión analítica, y la nebulización debe ser idéntica para todas las muestras y estándares en un análisis particular. Esto sólo puede lograrse si todas las muestras y estándares tienen las mismas características físicas. La eficiencia de la nebulización será influenciada por la tensión superficial de las soluciones aspiradas. Generalmente, a medida que se baja la tensión superficial, el volumen de la solución que alcanza la flama se incrementará y la señal analítica también se incrementará. Sin embargo, debe observarse que esta predicción puede no cumplirse para todos los sistemas solvente/soluto en todos los nebulizadores bajo todas las condiciones de operación. La velocidad de aspiración, el tamaño de las gotas de solución, y su distribución son gobernados por la dinámica de aerosoles prevaleciente, y esta dinámica esta sujeta a factores tales como la posición de la esfera de vidrio (o deflector), la presión del oxidante de entrada, y la geometría del nebulizador. Para propósitos prácticos, sin embargo, debe notarse que las diferendas en tensión superficial entre estándares y muestras es indeseable. La tensión superficial puede ser controlada mediante: i. Con solventes orgánicos o mezclas de solventes -las muestras y estándares ii. deben tener exactamente la misma composición de solvente. Con mezclas de soluciones acuosas y orgánicas miscibles. La presencia de una pequeña cantidad de un solvente tal como el alcohol etílico modificara la tensión superficial considerablemente, y las composiciones deben hacerse coincidir con cuidado. iii. Las concentraciones altas de sales incrementaran la tensión superficial de la solución. El efecto sobre la nebulización es menos severo que en i. o en ii. pero se requiere una coincidencia razonable entre muestras y estándares. 4.4 D e s c o m p o s ic ió n d e C o m p u e s t o s Aún cuando los sistemas de atomización por flama han estado en uso práctico por muchos años, la disociación molecular en flamas sólo ha sido estudiada en extensión limitada y los mecanismos exactos de la descomposición dentro de la flama no son plenamente comprendidos. En el caso más simple, la descomposición es más probablemente una progresión directa desde el compuesto analito a través del monóxido a constituyentes atómicos aún cuando, por supuesto, algunos monóxidos son más refractarios que otros. Donde la química y la termodinámica son más complicadas, la descomposición es un proceso indirecto, y una variedad de compuestos intermedios pueden formarse en la flama antes que el analito finalmente sea liberado en la forma atómica. Algunos de estos compuestos intermedios son formados por reacciones con especies tales como C, C 2, CH , C N y N H los cuales están intrínsecamente presentes en las flamas químicas. De estas reacciones, algunas promueven la producción de átomos analitos; - 31 - F undamentos del A n á l is is por A b s o r c ió n A t ó m ic a otras producen la formación de compuestos refractarios que inhiben la producción de átomos analitos. En algunos casos, los compuestos intermedios pueden formarse por reacciones entre varias especies en la muestra. Nuevamente, algunas de estas reacciones pueden ser útiles en promover la producción de átomos analitos; otras pueden inhibir el proceso. De estos conceptos generales se apreciará que es dificultoso predecir en forma teórica la química de la descomposición para todos los elementos bajo todas las circunstancias analíticas. Pero como resultado de una extensiva experiencia práctica, se puede especificar que flama debe usarse para elementos particulares (ver tabla III) e indicar razones generales para el uso de diferentes flamas. Para propósitos de esta discusión es conveniente clasificar determinaciones en categorías amplias de acuerdo a la dificultad relativa de la » descomposición, la naturaleza general y la extensión de las reacciones intermedias que interfieren en la producción de átomos analitos. a. La flama aire-acetileno es casi universalmente utilizada para aquellos b. elementos clasificados como de fácil atomización (cobre, plomo, potasio y sodio por ejemplo). En estas descomposiciones simples, una alta proporción de los compuesto analitos disponibles son fácilmente convertidos a átomos en una flama aire-acetileno (la flama más fría de uso práctico). Las interferencias son despreciables, y el ambiente químico dentro de la flama (oxidante, estequiométrica o reductora) no es un factor crítico. Debe notarse de la tabla III que varios elementos pueden ser determinados c. ya sea en flama de aire-acetileno u óxido nitroso-acetileno. Los monóxidos de estos elementos son más dificultosos de descomponer que los de la categoría a. Una flama aire-acetileno es útil para estos elementos, pero no es plenamente efectiva a través de todo el rango analítico. Si, por ejemplo, una solución de nitrato de calcio es aspirado en una flama aire-acetileno, el nitrato se descompondrá al monóxido, pero solo una proporción limitada del monóxido será finalmente convertida en átomos y la señal analítica será pequeña. En la flama más caliente óxido nitroso-acetileno, una proporción significativamente más alta de monóxido se convertirá en átomos y se obtendrá una señal analítica más fuerte. Mientras ambas flamas pueden usarse para determinaciones de esta categoría, las interferencias en la flama aire-acetileno pueden ser severas; la naturaleza química de la flama es importante, y puede ser necesario tomar las medidas correctivas adecuadas descritas bajo la categoría e. Los elementos en esta categoría invariablemente forman compuestos que d. son intrínsecamente más refractarios que aquellos de la categoría b. y una flama aire-acetileno no los descompondrá en una cantidad significativa. Todos los elementos de esta categoría deben ser determinados en la flama óxido nitroso-acetileno. La temperatura no siempre es el principal agente de descomposición. Dentro de cada tipo de flama la naturaleza química de la flama (oxidante, estequiométrica o reductora) tendrá también un profundo efecto sobre las propiedades de descomposición de muchos elementos. Considere la determinación de molibdeno en una flama aire-acetileno. Si el ratio - 32 - F u ndam entos e. del A n á l is is p o r A b s o r c ió n A t ó m ic a combustible-oxidante es tal que la flama es químicamente balanceada (estequiométrica), la descomposición procederá tanto como el monóxido pero muy poco del monóxido será convertido en átomos. Si la flama es rica en combustible, será más fría que la estequiométrica, pero será más fuertemente reductora y el monóxido será reducido a átomos de molibdeno. En forma similar, una flama óxido nitroso-acetileno estequiométrica produce una concentración relativamente pobre de átomos de sílice. De otro lado, una flama óxido nitroso-acetileno rica en combustible es considerablemente más efectiva. Para determinaciones en esta categoría, asimismo, la generación de átomos no es solo un asunto directo de temperatura, sino que requiere una combinación adecuada de temperatura y ambiente químico dentro de la flama, Las determinaciones en esta categoría se caracterizan por interferencias producidas por reacción del elemento a analizar con otras especies en la muestra. Del ejemplo del nitrato de calcio en la flama aire-acetileno se recordará que esta flama puede ser usada para análisis prácticos aun cuando se descompone una proporción limitada del monóxido disponible. Suponga que la solución original de nitrato de calcio contiene una cantidad significativa del silicato de otro elemento. En esta circunstancia, la reacción en la flama significará la formación de complejo silicato de calcio. La concentración de átomos de calcio libre será ahora considerablemente más baja debido a que el complejo de silicato es mucho más refractario que el monóxido y la respuesta analítica será pobre. Las interferencias de esta clase son extremadamente severas para magnesio, calcio, estroncio y bario debido a que en presencia de aluminio, sílice y fósforo la formación de aluminatos, silicatos y fosfatos refractarios interferirá severamente con la producción de átomos analitos. Están disponibles tres medidas correctivas: i. Una flama óxido nitroso-acetileno ii. minimizará o removerá tales interferencias debido a que la flama es lo suficientemente caliente como para descomponer los compuestos en mención. Puede agregarse un elemento 'buffer' para ‘competir’ con los elementos a analizar en cuanto a reaccionar con el grupo interferente, de modo que existirá atomización completa aún a bajas temperaturas. Por ejemplo, si se agrega un gran exceso de estroncio a la solución original de nitrato de calcio (incluyendo silicato), la mayoría del silicato reaccionará con el estroncio (debido a su mayor concentración) de modo que el resultado será una concentración mucho mayor de átomos libres de calcio en la flama. Tales aditivos se denominan a menudo ‘agentes de liberación' iii. Las soluciones estándar pueden hacerse coincidir a las muestras con respecto al elemento interferente. Por supuesto, esto no siempre es posible debido a que requiere un conocimiento de la composición de la muestra lo cual no siempre está disponible. Si no se toman tales precauciones, es obvio que pueden obtenerse resultados incorrectos. Si por ejemplo se midiese una serie de estándares de calcio (sin silicato presente) y se construyese la gráfica de calibración, entonces la medición de muestras desconocidas de calcio que contengan una - 33 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a concentración razonable de calcio producirá resultados muy bajos desde que la lectura de absorción atómica del calcio se verá seriamente deprimida. Tabla III. Tipo de Flama, Técnicas Alternativas y Rangos de Trabajo sobre algunos Elementos de Interés (Au, Ag, Cu, Fe, Pb, Zn, As, Hg) Elemento Análisis Análisis por Horno Generación de Vapor Rango de Trabajo Rango de Trabajo Rango de Trabajo pg/mL ng/mL pg/mL de Flama Tipo de Flama Ag As AA 0.03 - 10 0.5 - 12 NA/AA 2 - 200 5 - 100 Au AA 0.10 - 30 2 - 40 Cu AA 0.02 - 10 1 - 20 Fe AA 0.05 - 20 1 - 20 Hg Pb AA 2 - 600 n l - 20 0.01 - 0.08 AA 0.2 - 30 2 - 25 0.05 - 0.30 Zn AA 0.01 - 2 0.1 - 2 0.005 - 0.03 [* ] |Jg/mL A A = aire-acetileno N A = óxido nitroso-acetileno Tabla IV. Parámetros de Operación para Métodos AAS desarrollados sobre algunos Elementos de Interés (Au, Ag, Cu, Fe, Pb, Zn, As, Hg) Elemento Medio Longitud de Onda (nm) Corriente de Lámpara . Slit (nm) Estándares ppm (Abs.) Std. 1 Std. 2 10 ( 0.346) 12 (1.123) (mA) 4.5 Au CN- 242.8 4.0 0.5 5 (0.319) Ag CN- 328.1 4.0 0.5 4 (0.598) Ag Cu HCI 328.1 4.0 0.5 4 (0.403) HCI 324.7 3.0 0.5 5 (0.889) Fe HCI 248.3 7.0 0.2 5 (0.636) Pb HCI 217.0 5.0 1.0 20 (0.622) Zn HCI 213.9 5.0 0.5 1 (0.208) Hg As HCI 253.7 3.0 0.5 0.2 (0.825) HCI 193.7 20.01'1 2.0 5 (0.810) 3 (0.461) P o s ic ió n d e l Q u e m a d o r De la discusión sobre la descomposición es evidente que se puede maximizar la población de átomos a analizar en la flama proporcionando el tipo correcto de flama, estequiometría de flama adecuada y una química de soluciones apropiadas. Pero, antes de que puedan hacerse mediciones analíticas efectivas es necesario asegurar que la luz en la longitud de onda característica pase directamente a través de la población de átomos analitos. F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Los átomos analitos libres no están distribuidos uniformemente dentro de la envoltura de la flama, y bajo ciertas condiciones dadas de flama existirá una zona particular que esta más densamente poblada que otras partes de la flama. Obviamente, la máxima señal analítica será obtenida si luz a la longitud de onda característica pasa directamente a través de la zona de máxima población. Sin embargo, la localización de esta zona dentro de la flama no es idéntica para todos los análisis, y no existe una posición fija única del quemador que sea automáticamente adecuada para todas las situaciones analíticas. Es asimismo necesario ajustar la posición del quemador para cada análisis por separado de modo que la zona de máxima población coincida con el haz óptico (ver figuras 18 y 19). Todos los instrumentos incorporan controles simples que posibilitan el movimiento del quemador a la posición a la cual se obtiene la máxima absorbancia. También es importante recordar que la alteración del ratio combustible-oxidante, o el cambio del flujo total de combustible sin alterar la estequiometría puede elevar o decrecer la ubicación de la zona de máxima población dentro de la flama. El quemador también puede ser rotado de modo que el haz óptico pase en forma oblicua a través de la población de átomos. El efecto de esto es reducir la absorbancia obtenida, esta técnica se usa en ocasiones para extender la concentración de trabajo hacia arriba, tal como se discute en el capítulo sobre Análisis Cuantitativo. Figura 18. Ajuste horizontal del quemador F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Figura 19. Ajuste vertical del quemador. 4.6 A b s o r c ió n d e l B a c k g r o u n d Una de las mayores virtudes de la espectrometría de absorción atómica yace en el hecho de que la absorción ocurre a longitudes de onda específicas que son características del elemento químico. En teoría, cualquier absorción que ocurra puede ser atribuida directamente a la presencia de átomos analitos, y el grado al cual ocurre la absorción es una función de la densidad de la población de átomos. En la práctica, sin embargo, la señal analítica puede ser atenuada por medios diferentes a la absorción atómica. Esto se manifestará como mediciones de absorbancia que son más altas de lo que debería ser para una población dada de átomos, y el resultado analítico no será válido. Las principales fuentes de absorción por background son la absorción molecular y la dispersión de la luz. La absorción molecular ocurre cuando la flama no es lo suficientemente caliente como para descomponer todos los compuestos en la muestra. Las moléculas remanentes entonces absorberán luz, y si el espectro de absorción traslapa la longitud analítica se medirá algo de absorción adicional al mismo tiempo que la señal de absorción analítica válida. La flama en sí también produce absorción, especialmente a bajas temperaturas. La figura 20 muestra el rango de longitudes de onda sobre el cual absorben las moléculas de cloruro de sodio. La figura 21 muestra la línea aguda característica para el plomo. Sí, por ejemplo, se determina plomo en presencia de grandes cantidades de cloruro de sodio, la absorbancia medida será la suma de la absorción atómica que ocurre a 217 nm y la absorción molecular que también ocurre a 217 nm. Para obtener resultados analíticos válidos en esta clase de situación, es necesario usar un sistema corrector de background. - 36 - F undam entos del A n á l is is por A b s o r c ió n A t ó m ic a \ Figura 20. Absorción de banda amplia (cloruro de sodio) La dispersión de luz puede ocurrir cuando la muestra tiene una alta concentración de sólidos disueltos. Nuevamente aquí, la flama puede no ser lo suficientemente caliente como para descomponer en forma completa la muestra, y las partículas sólidas pueden permanecer en algunas áreas de la flama. Estas partículas pueden reflejar y dispersar algo de la luz incidente, y esto se verá como una atenuación de la luz transmitida aún cuando no tenga lugar ninguna absorción. Con los sistemas de atomización por flama, tanto la absorción molecular como la dispersión de luz no son significativas a menos que las mediciones se hagan cerca al límite de detección. Sin embargo, existen algunas situaciones de análisis prácticos en las longitudes de onda más bajas cuando la _ _ F undam entos del A n á l is is po r A b s o r c ió n A t ó m ic a absorción por background puede ser problemática. Con los sistemas de horno de grafito, sin embargo, la situación es algo diferente. En la etapa de calcinado, es algunas veces necesario hacer un compromiso entre la remoción completa del efecto matricial no deseado y la pérdida del elemento analito. Bajos estas condiciones, las especies moleculares pueden estar presentes durante la etapa de atomización y asimismo puede ocurrir absorción por background en forma significativa. Generalmente, el uso de un corrector por background compensará la absorción por background, mientras que en algunos análisis por horno es posible evitar o minimizar el problema mediante modificación química de los estándares y muestras. Estas situaciones se discuten en el capítulo sobre Análisis Cuantitativo. Figura 21. Linea de a b so rc ió n aguda (plomo) Figura 21. Absorción de línea aguda (plomo) 4.7 Io n iz a c ió n Las mediciones de absorción atómica dependen de la presencia de átomos libres y neutros que tengan propiedades espectrales características. A temperaturas elevadas, los átomos pueden ser ionizados con un consecuente cambio en su respuesta espectral. - ■ i- 38 - Fundamentos del A n á l is is po r A b s o r c ió n A t ó m ic a La ionización significa la pérdida de uno (o más) de los electrones más exteriores del átomo. Desde que este electrón estuvo involucrado en las transiciones de nivel de energía que definen la línea de absorción de resonancia, no puede ocurrir ninguna absorción a esta longitud de onda. El grado de ionización es diferente para cada elemento, dependiendo de la energía requerida para remover los electrones. Esta energía puede ser suministrada en varias formas, pero para la absorción atómica la mayor fuente es el calor de la flama. Particularmente en la flama óxido nitroso-acetileno, muchos elementos son al menos parcialmente ionizados, pero la energía disponible es tal que normalmente solo un electrón es removido y se forma un ión con carga única. Nótese que el grado de ionización se incrementará a medida que la „ concentración decrece de modo que en vez del gráfico normal de calibración (nominalmente lineal pero en la práctica curvado hacia el eje de concentraciones a altas absorbancias) la forma del gráfico será como la ilustrada por la curva A en la figura 22. Figura 22. Curvatura de la calibración causada por ionización Figura 22. Curvatura de la calibración causada por ionización Debe observarse también que todas estas consideraciones de ionización se aplican sólo en soluciones puras del elemento particular. La presencia de cualquier otro elemento con un potencial de ionización cercano o más bajo que del elemento a analizar modificará la extensión de la ionización en forma significativa. Si una solución pura de cloruro de sodio es aspirada en _ 3 g _ Fundamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a una flama, el equilibrio entre los átomos neutrales de sodio y iones sodio es dependiente de la temperatura. Si se introduce ahora un exceso de cloruro de potasio en la solución de cloruro de sodio el potasio se comportará de manera similar. Desde que ambas de estas ionizaciones producen electrones libres, la Ley de Acción de Masas causa que el equilibrio de ionización sea desplazado en una dirección que favorezca a los átomos neutrales y en consecuencia se obtendrá una mayor absorbancia para una medición del sodio por absorción atómica. El resultado de tal comportamiento es fácil de observar. Si se usan estándares que contienen únicamente sodio, la ionización reducirá en forma significativa las lecturas de absorción atómica. Si luego se mide una muestra que contenga también potasio, el grado de ionización de los átomos de sodio decrecerá debido a la presencia del potasio, y en consecuencia se obtendrá una lectura de absorción atómica relativamente más alta. En la práctica, los medios efectivos de evitar la interferencia debida a la ionización es ‘amortiguar los estándares y muestras con una alta concentración de un elemento fácilmente ionizable. Puesto que la concentración de este elemento es mucho mayor que el del elemento analito, y su potencial de ionización más bajo, puede efectuarse una supresión esencialmente completa de la ionización. Debido a su bajo potencial de ionización, los amortiguadores de ionización comúnmente usados son: sodio, potasio y cesio. - 40 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a CA PÍTU LO V A T O M I Z A C I Ó N POR H O R N O (GF A A S) 5. A t o m iz a c ió n po r H orno (G F A A S) Aún cuando los sistemas de atomización por flama para A AS son simples, convenientes y dan excelentes resultados, estos sistemas están sujetos a limitaciones de sensitividad que restringen el alcance analítico de los métodos de flama. La necesidad de superar esta limitación ha llevado al desarrollo extensivo de los sistemas de atomización por horno en el cual los analitos pueden ser determinados a concentraciones que son hasta 100 veces más bajas que los valores correspondientes a los métodos de flama. Un horno de grafito es un un atomizador electrotérmico (ETA). La atomización mediante este método corresponde a la ruta del lado derecho en la figura 12. En el horno de grafito, el solvente es primero evaporado desde la muestra a una temperatura moderada. La temperatura del horno es luego elevada de modo que el material orgánico es incinerado, y la temperatura es luego incrementada rápidamente al punto donde la muestra es vaporizada y por último atomizada. Es obvio que la temperatura del horno y la composición de la muestra son extremadamente importantes para asegurar la eficiencia y consistencia de esta atomización. 5 .1 P r in c ip io d e O p e r a c ió n Si bien existen diferentes enfoques referidos al diseño detallado y construcción de atomizadores de horno, todos ellos realizan el mismo proceso fundamental -generar una población de átomos a analizar libres de modo que pueda medirse la absorción atómica. En su forma más simple, este proceso se logra en tres etapas: a. Secado, en la cual el solvente es removido de la muestra. b. Calcinado (Incineración), en la que se remueve las moléculas orgánicas o c. material inorgánico. Atomización, en la cual los átomos libres del analito son generados dentro de una zona confinada coincidente con el haz óptico del instrumento AAS. La señal de absorción producida en la etapa de atomización es un pico bien definido. La altura y área de este pico esta relacionada a la cantidad de analito presente en la muestra. Los picos característicos pueden ser presentados sobre una carta registradora, o pueden ser establecidos por - - 41 - Fu ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a sistemas de medición electrónica de picos incorporadas en el espectrómetro. Tales sistemas típicamente proporcionan medidas de altura y área de un pico. A semejanza de los métodos de absorción atómica por flama, es necesario obtener mediciones comparativas de estándares y muestras a fin de establecer valores de concentración para las muestras. En lo fundamental, el atomizador por flama y de horno proporcionan el mismo producto final -un suministro de átomos libres de analito para exponerlo a una luz de longitud de onda característica. Sin embargo, debe notarse que mientras se obtiene el mismo producto final de ambos sistemas, existen diferencias sustantivas en los mecanismos de disociación molecular y en la eficiencia integral del proceso de producción de átomos. En los análisis por flama, la composición química de la flama (en vez de la temperatura) es generalmente más importante para maximizar la fracción de átomos libres. La eficiencia de conversión es baja -típicamente solo cerca del 10% de la muestra aspirada se convierte en población de átomos libres para medición en el haz óptico. En los atomizadores de grafito, la disociación molecular es gobernada por la temperatura final empleada, la velocidad a la cual se alcanza esta temperatura, y el ambiente reductor del grafito caliente. La eficiencia de conversión es típicamente alta dado que toda la muestra disponible se usa para producir la población de átomos dentro del haz óptico. Así, para una concentración dada de analito, la población de átomos en el atomizador de horno será considerablemente más densa que en la flama. En términos analíticos prácticos, pueden obtenerse mediciones dentro del rango de absorbancia útil a concentraciones considerablemente más bajas de lo que es posible por métodos de flama. 5.2 A t o m iz a d o r e s d e T u b o d e G r a fit o La operación práctica de atomizadores de tubo de grafito puede ser explicada considerando la construcción y operación del atomizador utilizado en la figura 23. El tubo de grafito esta montado entre dos electrodos localizados en un bloque de metal enfriado por agua. Un mecanismo de balanceo bloquea el tubo de grafito en el lugar y permite que el ensamblaje del horno sea abierto para el reemplazo del tubo. El área de contacto entre los electrodos y el tubo es relativamente pequeña. El tubo es calentado resistivamente por el paso de una corriente a bajo voltaje. Gas inerte (normalmente argón o nitrógeno) fluye a través de los electrodos para proteger de una rápida oxidación a las superficies interiores y exteriores del atomizador. El tubo esta cubierto internamente con grafito pirolítico el cual es relativamente impermeable a los gases o átomos. El grafito pirolítico es también más resistente a la oxidación que el grafito normal, y es químicamente no reactivo. F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Para análisis prácticos, se introduce un pequeño volumen de muestra (típicamente entre 20 y 40 pL) en el tubo atomizador a través de la entrada en la parte superior del tubo. Las muestras pueden ser inyectadas manualmente usando microjeringas, o el proceso puede ser automatizado por medio de un dispensador automático de muestras. El atomizador es calentado de acuerdo a una secuencia determinada. puerto de inyección de muestra haz óptico tubo de grafito r electrodo X Figura 23. Atomizador de tubo de grafito llilE electrodo Figura 23. Atomizador de tubo de grafito 5.3 S e c a d o y C a l c in a d o En la primera etapa de calentamiento (secado), el solvente es evaporado de la muestra a una temperatura cercana al punto de ebullición. La evaporación debe ser suave a fin de evitar pérdidas mecánicas por formación de espuma o derrames. Cuando la etapa de secado esta completa, el residuo en el tubo atomizador consistirá de una capa delgada de material que contiene el elemento a analizar junto con todos los sólidos disueltos y componentes menos volátiles de la matriz de la muestra. En la etapa de calcinado, la matriz es descompuesta térmicamente a una temperatura intermedia en una combinación de procesos que incluyen pirólisis, destilación y combustión. El objetivo es remover todos los componentes de la matriz mientras se mantiene el elemento a analizar completamente dentro de atomizador en un forma químicamente estable de modo que pueda proceder la atomización sin interferencia de la matriz. Cuando esta etapa esta completa, el residuo debería consistir en el elemento a analizar en su forma molecular adecuada, y un mínimo de otros compuestos (comúnmente sales inorgánicas) que pueden ser térmicamente estables a las temperaturas de calcinado. La etapa final es la disociación de las especies a analizar moleculares a una alta temperatura (hasta 3000 °C ) en una atmósfera de gas inerte. Esto genera una población de átomos libres en el haz de luz de modo que puede realizarse la medición por AAS. - 43 •*7* - Fundamentos del A n á l is is po r A b s o r c io n A t ó m ic a Mientras que es conveniente considerar al proceso de atomización como una secuencia de tres etapas (secado, calcinado y atomización), los atomizadores de tubo de grafito modernos emplean una programación multietapas en vez de un secuencia rígida de tres etapas. El programa completo de atomización se divide en pequeñas etapas (usualmente entre 8 y 20). Cada etapa puede en forma separada en cuanto a duración, velocidad de temperatura y temperatura al final de cada etapa. En la figura programa multietapa típico. un número de ser programada incremento de 24 se ilustra un Figura 24. Programa de temperatura Figura 24. Programa de Temperatura Tabla V. Parámetros de Operación del Horno Tipo de Gas Etapa N° Temp. °C Tiempo (seg.) Flujo de Gas 1 90 3.0 3.0 normal 2 95 50.0 3.0 normal 3 150 10.0 3.0 normal 4 1200 10.0 3.0 normal 5 1200 20.0 3.0 normal 6 1200 0.5 0.0 normal 7 2400 0.7 0.0 normal 8 2400 0.5 0.0 normal 9 2400 2.0 3 normal 10 - 44 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O VI G E N E R A C I Ó N DE V A P O R (HG AAS) 6. G en er a ció n de V apor (H G AA S) % El método de generación de vapor incrementa la sensitividad de la técnica de absorción atómica para el Hg y para los metales que forman hidruros (As, Bi, Sb, Se, Sn y Te), muchos de los cuales requieren monitoreo a nivel de trazas (debido a su importancia como contaminantes ambientales). Com o cabe esperar, el nivel analítico significativo para estos elementos está debajo del límite de detección de los métodos tradicionales de flama. 6.1 G e n e r a c ió n d e H id r u r o s La técnica denominada “ generación de hidruros” se basa en reducir químicamente a los metales formadores de hidruros (Sb, As, Bi, Pb, Se, Te, Sn) al hidruro gaseoso y luego disociar el hidruro (poco estable a altas temperaturas) en un tubo calentado de cuarzo donde ocurre descomposición térmica. La luz absorbida por los átomos a analizar es entonces determinada de manera normal con un espectrómetro de absorción atómica. El procedimiento químico es sencillo. La reducción se efectúa mediante la adición de solución de borohidruro de sodio (N a B H 4) a la muestra acidificada. Las concentraciones de reactivo son escogidas para dar evolución cuantitativa del vapor elemental del hidruro en unos pocos segundos a temperatura ambiente. Un procedimiento alterno es el método del cloruro estañoso (SnCh), en este se agrega dicromato de potasio (K.2CrC>7) a la muestra y la solución obtenida se hace reaccionar con una solución acidificada de cloruro estañoso para formar los hidruros gaseosos. En ambos casos, el vapor de hidruro es entonces barrido por una corriente continua de gas inerte (argón o nitrógeno) en un tubo de cuarzo que es calentado en una flama aire-acetileno. Los compuestos del vapor de hidruro son atomizados y la señal de absorción es medida como un pico bien definido mientras el vapor pasa a través del tubo de cuarzo. En la figura 25 se ilustra un sistema generador de hidruros basado en flujo continuo. - 45 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Figura 25 Diagrama funcional de un sistema generador de hidruros basado en flujo continuo Reacciones de Generación de Hidruros La reacción neta del tetrahidroborato de sodio (borohidruro de sodio) en solución ácida y la reducción simultánea del elemento formador de hidruro puede ser descrita mediante las siguientes ecuaciones: B H 4- + H30 + + 2 H 20 H 3B O 3 + 4H 2 y 3 B H 4- + 3H + + 4 H 2S e 0 3 - * 4SeH 2 + 3H20 + 3 H3BO3 donde el selenio puede reemplazarse con cualquier elemento generador de hidruros, aún cuando estas ecuaciones son una aproximación del proceso verdadero, el cual todavía es desconocido. El hecho de que bajo condiciones óptimas la generación del hidruro y el transporte al atomizador sea virtualmente cuantitativa para todos los elementos formadores de hidruros es de suma importancia práctica. Se producen rendimientos tales como: selenio (95%), bismuto (95%), plomo (80%), entre otros. - 46 ■* ■ - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Disociación térmica Salida de As'+H-, 2A s , —* 2 As + 3 optical path átomos As° HCL-As / // Calor (flama) Entrada de A sH 3 Figura 2b. Q uartz Tube Atom izer. Usada en la técnica de generación de hidruros, determinación de As Figura 26. Atomizador de Tubo de Cuarzo para generación de hidruros Retiro del Hidruro de la Solución De las observaciones cinéticas se observa que la conversión de As(lll) a A s H 3 es en menos de 0.1 s. Entonces la etapa de velocidad determinante es el retiro del arsénico de la solución; para este propósito el hidrógeno generado es más efectivo que el gas de purga. En un sistema de flujo el 70% de la solución de tetrahidroborato de sodio se había descompuesto 0.3 ms después que se le puso en contacto con I mol/L de ácido sulfúrico; 80% se había descompuesto después de Ims y no se pudo detectar N a B H 4 después de 10 ms. No tienen influencia en esto ni la concentración de tetrahidroborato ni la del ácido, concluyéndose que la formación del hidruro, al menos para Se(V) y As(lll) fue completa dentro de este período. El hecho de que una zona de reacción interpuesta después del reactor incrementó la sensitividad de ambos elementos afirmó la conclusión que el retiro del hidruro fue la etapa determinante de la velocidad. Sistemas de Flujo para Generación de Hidruros En los sistemas de flujo, las soluciones son mayormente bombeadas y mezcladas en forma continua de modo que puede establecerse cierto equilibrio de la reacción. En contraste con los sistemas batch, en los cuales la reacción y separación del hidruro ocurren en el mismo contenedor, en los sistemas de flujo estos procesos mayormente se llevan a cabo en zonas separadas. El tiempo de reacción, durante el cual la fase de separación ya ocurre naturalmente, puede ser influenciado por la longitud de la zona de reacción y la velocidad de flujo. Usando zonas cortas de reacción y bombeando la solución de reacción rápidamente siguiendo a las fase de separación brinda la posibilidad de suprimir cinéticamente las reacciones lentas. Esta técnica se denomina discriminación cinética y se usa especialmente en sistemas de Flow Injection para eliminar interferencias. F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Influencia del Estado de Oxidación Para los elementos de grupo V, arsénico y antimonio (el bismuto solo se presenta en el estado trivalente), la diferencia entre los estados trivalente y pentavalente de oxidación depende mucho del sistema usado en condiciones experimentales. En los sistemas batch a un pH < I el arsénico se forma más lentamente desde As(V) que desde As(lll) y esto significa un pico 25-30% más bajo, aun cuando las áreas de pico son casi las mismas; estas diferencias dependen adicionalmente de las concentraciones de ácido y tetrahidroborato de sodio. Estas condiciones son similares para el antimonio excepto que la diferencia en las alturas de pico para los estados tri y pentavalente son más pronunciadas. Esta situación es significativamente diferente en sistemas de flujo donde la reducción más lenta del estado pentavalente conduce a mayores diferencias en sensitividad debido a la discriminación cinética. Si bien en los sistemas batch se puede omitir la pre-reduccion de A s(V) y Sb(V), esto no es recomendable en los sistemas de flujo debido a la pérdida en sensitividad. Para realizar esta reducción fue usual usar yoduro de potasio (Kl), solo o mezclado con ácido ascorbico. Una cierta desventaja es que se requieren concentraciones relativamente altas de reductor (30-100 g/L Kl) y ácido (5-10 mol/L H C I) y que las soluciones de Kl son inestables. Bajo estas condiciones el antimonio se reduce casi espontáneamente mientras que el arsénico reacciona más lentamente de modo que se requiere el calentamiento de la solución y un periodo de espera más largo. Se ha propuesto a la L-Cisteina como un reductor alternativo al Kl. Las ventajas de este reductor incluyen el hecho de que es efectivo a concentraciones de 5-10 g/L y que la concentración óptima de ácido es 0.1-I mol/L HCI. Asimismo, la L-Cisteina estabiliza los analitos reducidos de arsénico y antimonio en estado trivalente por varios dias. Transporte de Hidruros El transporte del hidruro gaseoso al atomizador puede ser realizado con hidrogeno auxiliar o un gas inerte tal como el argón o nitrógeno. Este transporte es virtualmente cuantitativo cuando el hidruro no fue colectado, por esta razón es prefible la transferencia directa al atomizador. 6 .2 T é c n ic a d e V a p o r F r ío La técnica de vapor frío solo puede aplicarse para la determinación de mercurio y se basa en ciertas características típicas de este elemento -la única excepción es el elemento cadmio que también puede ser determinado por esta técnica. Desde que el mercurio puede ser fácilmente reducido de sus compuestos al metal y posee una presión de vapor de 0.0016 mbar a 20°C, este puede ser determinado directamente sin alguna unidad atomizadora especial. Las determinaciones de mercurio pueden ser llevadas a cabo en el mismo generador de vapor usado para métodos de hidruro, pero no se F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a requiere formación del hidruro gaseoso y no se necesita la flama. Los compuestos de mercurio en solución ácida son reducidos el elemento libre con cloruro estañoso y el vapor de mercurio es barrido en la celda de cuarzo por una corriente continua de nitrógeno. La señal de absorción es medida como un pico bien definido. Los detalles analíticos y rangos de concentración de trabajo para métodos de generación de vapor de dan en la tabla III. Figura 27. Diagrama funcional de un sistema de generación de vapor de mercurio basado en el sistema de flujo continuo. Interferencias Virtualmente no ocurren interferencias espectrales con la técnica de vapor frío. Se reportó que el vapor de agua había ocasionado atenuación por background, sin embargo la molécula de H 2O no exhibe ninguna banda de absorción en la línea del mercurio y se estima que las interferencias observadas fueron debidas a gotas de solución acarreadas con la corriente de gas o a condensación del vapor de agua en la celda de absorción. Esto puede prevenirse mediante un agente secante adecuado o calentando la celda. Observese una celda típica en la figura 28. Las causas de errores sistemáticos dependen en gran medida de la movilidad de este elemento y sus compuestos. Estos errores incluyen valores del blanco, contaminación debida a reactivos y pérdidas debidas a vaporización, adsorción o conversiones químicas. Las materias suspendidas en muestras líquidas no deben ser removidas por filtración desde que el filtro puede - 49 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a adsorber una considerable cantidad de mercurio debido a su gran área superficial. optical path átomos Hg H C L - H? Entrada de H ?° Salida de H ?° Figura 28. Celda de absorción de cuarzo, usada en la técnica de v a p o r in o para determ inación de M ercurio Figura 28. Celda de absorción de cuarzo, usada en la técnica de vapor frío para determinación de mercurio. Algunos metales pesados pueden interferir dado que pueden ser reducidos por el tetraborato de sodio (o cloruro estañoso) y luego reaccionar en la forma reducida con mercurio. En principio, estas interferencias son análogas a aquellas que pueden ser observadas en la técnica de generación de hidruros. Los metales nobles reducidos pueden reaccionar con mercurio vía la formación de amalgamas, los metales que son menos nobles pueden aglutinar al mercurio por cementación. Para evitar estas interferencias se tienen las siguientes medidas: ♦ incrementar la concentración de ácido ♦ Usar iones de Fe(lll) como un buffer ♦ Usar reactivos de enmascaramiento tal como cianuro ♦ Uso de discriminación cinética en sistemas Fl Análisis de Cadmio por Vapor Frío El elemento cadmio puede también ser analizado por la técnica de vapor frío. En reacción con N a B H 4, el cadmio es probable que forme inicialmente un hidruro, el cual es muy inestable y se descompone muy rápidamente según: (C d H 2 )¡n e sta b le - C d ° + H 2 Se forma entonces vapor de cadmio, el cual a semejanza del vapor de mercurio solo debe ser transportado a la celda de cuarzo no calentada para su determinación. F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O VII MONOCROMADORES 7. M o n o c ro m a d o res La función de un monocromador es aislar un línea de resonancia atómica única del espectro de líneas emitidos por la lámpara de cátodo hueco. En efecto, es un filtro ajustable que selecciona una región estrecha y específica del espectro para transmisión al detector y rechaza todas las longitudes de onda fuera de esta región. iong. de onda (nm) Figura 29. Espectro atómico del cobre alrededor de 324.7 nm Figura 29. Espectro atómico del cobre alrededor de 324.7 nm Idealmente, el monocromador debería ser capaz de aislar solo la línea de resonancia y excluir todas las otras longitudes de onda. Para algunos elementos esto es relativamente fácil; para otros es más dificultoso. El cobre por ejemplo tiene un espectro comparativamente no confuso con la línea más cercana estando a 2.7 nm de la línea de resonancia de 324.7 (figura 29). El - 51 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O Vil MONOCROMADORES 7. M o n o c ro m a d o res La función de un monocromador es aislar un línea de resonancia atómica única del espectro de líneas emitidos por la lámpara de cátodo hueco. En efecto, es un filtro ajustable que selecciona una región estrecha y específica del espectro para transmisión al detector y rechaza todas las longitudes de onda fuera de esta región. long. de onda (nm) Figura 29. Espectro atómico del cobre alrededor de 324.7 nm Figura 29. Espectro atómico del cobre alrededor de 324.7 nm Idealmente, el monocromador debería ser capaz de aislar solo la línea de resonancia y excluir todas las otras longitudes de onda. Para algunos elementos esto es relativamente fácil; para otros es más dificultoso. El cobre por ejemplo tiene un espectro comparativamente no confuso con la línea más cercana estando a 2.7 nm de la línea de resonancia de 324.7 (figura 29). El _ 5l _ F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a ranuras por milímetro. Las ranuras deben ser rectas, igualmente espaciadas, paralelas y de forma idéntica. La luz que llega a estas ranuras es difractada, y por un proceso de mutua interferencia, la luz es dispersada en diferentes ángulos de acuerdo a su longitud de onda. El espectro resultante es difundido sobre un ángulo amplio al dejar la superficie de la rejilla. Al rotar la rejilla, cualquiera de las longitudes de onda constituyentes pueden ser enfocadas sobre el slit de salida vía el segundo espejo. El remanente del espectro no cae en el segundo espejo, o es enfocado lejos del slit de salida. Figura 3 I . Geometría óptica del monocromador (montaje Czerny-Turner) La densidad de líneas, o número de ranuras por milímetro, determina el ángulo a través del cual la luz a cada longitud de onda será dispersada de la rejilla de difracción. Mientras más alta sea la densidad de líneas, mayor será el ángulo de dispersión. El ancho del slit es un determinante importante del rendimiento del monocromador debido a que el ancho del slit controla la resolución de las longitudes de onda adyacentes (figura 32). En la práctica la selección de ancho de slit de operación involucra un compromiso desde que el slit también controla la cantidad de luz que es transmitida por el monocromador. Si el slit F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a es demasiado ancho, la producción de luz será alta y el ratio señal a ruido puede ser excelente, pero la línea de resonancia puede no estar aislada de otras líneas y la calibración puede resultar malamente curvada. De modo inverso, si el slit es demasiado estrecho, la resolución puede ser excelente, pero el ratio señal a ruido puede ser inaceptablemente pobre debido a la reducida producción de luz. linea de resonancia linea de resonancia linea de resonancia línea adyacente I___ I I_________________ I slit demasiado estrecho slit óptimo slit demasiado ancho ruido cun/atura de calibración ruido Figura 32. Ancho del slit, linealidad de calibración y razón señal-ruido Figura 32. Ancho de slit, linealidad de la calibración y ratio señal/ruido F u ndam entos del A n á l is is po r A b s o r c ió n A t ó m ic a C A P Í T U L O VIII S ISTEM A S Ó P T I C O S 8. S is t e m a s 8.1 Ó p t ic o s S istem a s Ó p t ic o s En los espectrómetros de absorción atómica, el propósito básico del sistema óptico es colectar luz de la fuente, dirigirla a través de la población de átomos a analizar, y luego dirigirla al monocromador. Las configuraciones ópticas pueden ser de rayo único o doble; se pueden usar lentes o espejos como elementos acoplantes. El sencillo sistema de espejos de rayo único ilustrado en la figura 33 se usa en muchos instrumentos. El segundo espejo enfoca la imagen de la lámpara de cátodo al centro de la llama (u otra celda de atomización). El tercer espejo enfoca esta imagen a su vez a un espejo plano donde el rayo es doblado y pasado al slit de entrada del monocromador. Puede usarse lentes para lograr el mismo efecto, tal como se muestra en la figura 34. Figura 33. Sistema de espejos de rayo único - i - 55 - f- F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a En la configuración de rayo único mostrada, la luz de la fuente solo atraviesa una vía -a través de la flama. En este sistema es necesario medir la intensidad inicial de la línea de resonancia lo antes de insertar la muestra y medir la intensidad transmitida I t . El sistema de rayo único asimismo depende de que la fuente de luz permanezca estable durante el periodo de análisis, esto es, la intensidad l0 no debería variar o fluctuar mientras se esta midiendo lT. Con las lámparas de cátodo hueco modernas lo generalmente permanecerá lo suficientemente estable después de un periodo de calentamiento. Sin embargo, existen algunas circunstancias analíticas donde cualquier ligera variación será inconveniente. Figura 34. Sistema de lentes de rayo único Un sistema de rayo doble tal como el ilustrado en la figura 35 esta diseñado para corregir cualquier variación en lo- La luz de la fuente es dirigida a un divisor de rayos consistente en una placa de cuarzo parcialmente aluminizada. El rayo de la muestra es enfocado por el espejo Mi, pasa a través de la flama, y es dirigido al slit de entrada del monocromador a través de espejo M2 y el espejo plano M3. El rayo de referencia que pasa a través del divisor de rayos es dirigido al slit de entrada del monocromador por un segundo conjunto de espejos tal que este rayo ingresa al monocromador sin pasar a través de la flama. Un espejo reflectante rotatorio esta localizado cerca al slit de entrada al monocromador donde este alternativamente interrumpe al rayo de la muestra para dirigir el rayo de referencia al monocromador. Así, las intensidades lo e lT son medidas en forma separada a alta frecuencia. Para lo esta alta frecuencia de medición proporciona un monitoreo cercano a lo continuo de modo que las correcciones por variaciones en intensidad pueden aplicarse instantáneamente. Mientras que el ratio Io :It se calcula en forma electrónica, el cálculo puede corregirse en forma continua para variaciones en lo y el resultado analítico final no será afectado por variaciones en la intensidad de la fuente. - 56 - F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a Figura 35. Sistema de rayo doble 8.2 C o r r e c c ió n d e l B a c k g r o u n d La interferencia por background ocurre cuando la radiación de la lámpara de cátodo hueco es atenuada por especies moleculares o partículas sólidas en la zona de observación (flama u horno). 8 .2.1 C o r r e c c ió n c o n L á m p a r a d e D e u t e r io La corrección para esto es relativamente simple con la ayuda de una fuente continua (usualmente una lámpara de deuterio). Mientras que la señal obtenida cuando se usa una lámpara de cátodo hueco es la absorbancia total (la suma de la absorción atómica y la absorción por background), la señal obtenida de la lámpara continua es sólo la absorción por background. En los sistemas ¡lustrados en las figuras 36 y 37, la configuración óptica es tal que la radiación de las lámparas de cátodo hueco y continua coinciden precisamente a lo largo del haz óptico a través de la zona de observación. La señal de background es subsecuentemente sustraída electrónicamente de la señal de absorbancia total y el resultado analítico es así corregido para interferencia por background. Señal de lámpara de cátodo hueco = absorción atómica + background Señal de la lámpara de deuterio = solo background Señal electrónica procesada = solo absorción atómica - 57 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a En el sistema de rayo doble ilustrado en la figura 37, la radiación de la fuente continua atraviesa la misma muestra y vías de referencia como lo hace la radiación de la lámpara de cátodo hueco. Las intensidades de ambas fuentes pueden así ser monitoreadas en forma concurrente. Cualquier variación en la intensidad de alguna de las fuentes puede ser corregida en forma automática para así mantener la precisión de la corrección por background. Figura 36. Sistema de rayo único con corrector por background Figura 37. Sistema de rayo doble con corrector por background - 58 - F undamentos del 8.2.2 A n á l is is p o r A b s o r c ió n A t ó m ic a C o r r e c c ió n c o n E f e c t o Z e e m a n El efecto Zeeman, mediante el cual las líneas de emisión de los átomos se dividen bajo la influencia de un campo magnético, se origina en la interacción del campo magnético externo con el momento magnético de los átomos emisores (efecto Zeeman directo) o absorbentes (efecto Zeeman inverso). Para la corrección del background mediante este efecto, el magneto puede estar montado ya sea en la fuente de radiación o en el atomizador; el campo magnético puede ser orientado ya sea paralelo o perpendicular al rayo de radiación, y puede aplicarse un campo magnético constante o alternante. Estas posibilidades dan hasta ocho diferentes configuraciones, entre estas opciones para la corrección d e l' background por efecto Zeeman, la óptima es la corrección del background con un campo longitudinal alternante en el atomizador. 8.2.3 C o r r e c c ió n c o n C o r r ie n t e P u l s a n t e A l t a Esta técnica se basa en la fuerte ampliación de la línea y la auto-reversión de las líneas de resonancia observadas en las H C L a altas corrientes de operación. Se mide la absorción total a corriente de operación normal y así se tiene el perfil de línea normal, mientras que el background se mide próximo a la línea analítica con el perfil fuertemente ampliado causado por la corriente pulsante alta. En muchos aspectos la corrección por background con corriente pulsante alta tiene gran similaridad a la corrección por efecto Zeeman con el magneto en la fuente. Ambos sistemas operan con una fuente, en ambos sistemas la línea analítica es ‘dividida’ de modo que la atenuación por background se mide próximo a la línea analítica, y el rendimiento de ambos sistemas es fuertemente dependiente del comportamiento de la fuente de radiación bajo las especiales condiciones prevalecientes. 8 .3 L entes La longitud focal de un lente varía con la longitud de onda debido a que el índice de refracción es dependiente de la longitud de onda y cambia en forma aguda debajo de 300 nm. En los sistemas ópticos que emplean lentes, no es conveniente recolocar los lentes, flama o lámpara de cátodo hueco cuando se cambie la longitud de onda, y es práctica usual diseñar los lentes de modo que estén enfocados a una longitud de onda seleccionada y acepte algunas pérdidas ocurridas en otras regiones del espectro. El sistema de lentes para absorción atómica esta normalmente enfocado en el ultravioleta debido a que la mayoría de longitudes de onda de interés analítico caen dentro de este rango y debido a que el índice de refracción medio ocurre aproximadamente a 250 nm. - 59 - F undam entos del A n á l is is por A b s o r c ió n A t ó m ic a Figura 38. Variación del índice de refracción con la longitud de onda en el cuarzo claro Figura 38. Variación del índice de refracción con la longitud de onda para el cuarzo claro Los lentes son usualmente fabricados de vidrio de sílice de alta calidad que proporciona buena transmisión óptica sobre el rango de longitudes de onda de 190 nm a 900 nm. Las únicas pérdidas significativas son causadas por reflexión en cada superficie aire-lente. Esto es comúnmente cerca del 7% de cada superficie o cerca de 15% para cada lente en el haz óptico. Los lentes fabricados de vidrio que tienen menor calidad óptica que el vidrio de sílice puro muestran mayores pérdidas especialmente debajo de 250 nm. Pérdidas similares en la región de baja longitud de onda pueden ocurrir también si los lentes están recubiertos por polvo o grasa. 8.4 E spejos El foco no varía con la longitud de onda cuando se usan espejos. En los sistemas de reflectividad total de uso común, pueden usarse espejos planos para doblar el rayo óptico donde sea necesario, y pueden usarse espejos curvados como elementos enfocantes para formar imágenes en puntos apropiados a través del sistema. Pero, aún cuando el foco no varíe con la longitud de onda, el diseño del espejo para elementos enfocantes puede presentar otros problemas. La geometría de los sistemas reflectivos requiere que los enfocantes sean usados en ángulos fuera de eje. En esta circunstancia el espejo cóncavo esférico sufre severamente de astigmatismo y las imágenes _ _ _ F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a pueden ser distorsionadas en forma grosera. Esta distorsión solo puede ser evitada usando espejos toroidales, esto es, espejos que tengan diferente radio horizontal y vertical. Los espejos invariablemente tienen su superficie frontal recubierta con aluminio por deposición en vacío. Un recubrimiento de aluminio adecuadamente controlado tiene una reflectividad superior al 90% sobre el rango íntegro de longitud de onda de 190 nm a 900 nm. El recubrimiento de aluminio es extremadamente delgado (1.5 x 10 3 mm) y puede ser fácilmente dañado aún por un paño suave. Los humos químicos pueden también atacar el recubrimiento y aún una huella digital puede causar daño irreparable. El delicado aluminio puede ser protegido cubriéndolo con películas evaporadas de materiales tales como sílice o fluoruro de magnesio y esto puede también ' dar una ligera mejora en la reflectividad. F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a CA P ÍT U LO IX D E T E C C I Ó N DE L A R A D IA C IÓ N 9. D e t e c c ió n 9.1 de la R a d ia c ió n E l F o to m u ltip lic a d o r Los requerimientos de detección de radiación en A A S son cumplidos por tubos fotomultiplicadores de banda amplia. Ningún otro dispositivo ofrece la misma sensitividad sobre el rango de longitud de onda requerido para análisis por absorción atómica. El tubo fotomultiplicador es un detector de radiación en el cual la radiación incidente que cae en un fotocátodo causa la emisión de electrones primarios (efecto fotoeléctrico exterior) los cuales son liberados en el vacío circundante. Un electrodo adyacente se mantiene a un potencial eléctrico positivo con respecto al cátodo y los electrones del cátodo son atraídos al segundo cátodo. Este segundo electrodo es denominado un dínodo. Como resultado del voltaje de dínodo aplicado, cada electrón primario es acelerado tan rápido que cuando incide en un dínodo se emiten de dos a diez electrones secundarios, produciendo un efecto de cascada, proceso multiplicador que da al fotomultiplicador su nombre, tal como se muestra en la figura 39. Sin embargo, existen límites al voltaje que puede ser aplicado, desde que esto conlleva a una corriente oscura más alta y así a ruido incrementado. En casos extremos los dínodos pueden saturarse. El fotomultiplicador produce entonces una señal eléctrica que es proporcional a la intensidad de la luz a la longitud de onda que ha sido aislada por el monocromador. Esta señal eléctrica es luego amplificada y usada para proporcionar una medida cuantitativa de la absorción. Los fotomultiplicadores tienen por lo general de nueve a trece etapas de dínodo y la corriente del electrón generada en el cátodo puede ser multiplicada para producir una amplificación integral de corriente de hasta I08. Así, las intensidades de luz que se obtienen en los instrumentos de absorción atómica conducen a una corriente eléctrica de magnitud útil que puede ser posteriormente ampliada para proporcionar la medición cuantitativa requerida. El potencial aplicado a los dínodos es usualmente derivado usando una cadena de resistores para dividir un potencial integral que oscila de 300 a 1000 voltios en etapas iguales. Sí, por ejemplo, el potencial a través del fotomultiplicador es 900 voltios y existen nueve dínodos, el primer dínodo tendrá un potencial de 100 voltios, el segundo dínodo 200 voltios, etc. La - 62 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a ganancia del proceso fotomultiplicador puede ser variada sobre un rango amplio ajustando este potencial, y esta técnica es comúnmente usada para ajustar la magnitud de la señal así como para hacer coincidir la sensitividad del amplificador. Desde que la señal de salida es tan extremadamente sensitiva a los cambios en el voltaje aplicado, la estabilidad y libertad del ruido de esta fuente son especialmente importantes. En los instrumentos modernos de A A S se usan mayormente microprocesadores para regular y controlar el voltaje aplicado. - 63 - Fundamentos 9.2 del A n á l is is p o r A b s o r c ió n A t ó m ic a C a r a c t e r ís t ic a s d e l F o t o m u l t ip l ic a d o r Las características más importantes de todos los detectores fotoeléctricos son la sensitividad espectral, la eficiencia cuántica, el rango utilizable de longitud de onda, el rango lineal, el ratio señal-ruido, el tiempo de respuesta y la corriente ‘oscura’. A. Sensibilidad Esp ectral La sensibilidad espectral R(/.) es dada por el ratio de la señal de salida a la señal de entrada. El requisito más importante de los detectores A AS es una alta sensibilidad espectral sobre el rango íntegro de longitud de onda con una sensibilidad máxima en el ultravioleta lejano (aprox. 200-300 nm). B. Eficiencia C u án tica La eficiencia cuántica r] es el ratio del numero de entidades cargadas emitidas al numero de fotones incidentes. La sensibilidad espectral y eficiencia cuántica están relacionadas por: R(/.)hc n = ........... e/. donde h es la constante de Planck, c es la velocidad de la luz y e es la carga elementaría. C. Rango U tilizab le de Longitud de O nd a Es el rango en el cual la sensibilidad espectral no cae debajo de un valor estipulado. El valor más bajo que puede ser estipulado para la sensibilidad relativa esta limitado por el ruido. En A A S la sensibilidad espectral en el rango de 190 nm a 860 nm debería ser la menos dos o tres órdenes de magnitud sobre el ruido. D. Rango Lineal o Rango D in ám ico Es el rango donde la señal de salida del detector es proporcional a la potencia de radiación incidente dentro de un valor limite estipulado. La no-linealidad esta dada por: R(z)-R(z0) N L = ............... R(zo) donde R(z) es la sensibilidad al valor z y R(zo) es la sensibilidad al valor de referencia Z 0. En AAS es necesario un rango de I06. E. Ruido Es considerado como todas las variaciones arbitrarias causantes de que una señal eléctrica de salida sea superpuesta a la señal de medición. La distribución de ruido contra la frecuencia se describe como el espectro del ruido. El ruido que es independiente de la frecuencia es denominado ‘ruido blanco’. Los componentes más importantes del ruido son el ruido 'fíicker' de baja frecuencia y el ruido ‘shot’. Estos componentes resultan de una serie de procesos aleatorios independientes, tal como la emisión de electrones de un - 64 - \ F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a fotocatodo. El ruido shot comprende el ruido del fotón y el ruido de la corriente oscura. F. C o rrie n te O sc u ra La corriente oscura es la suma de todas las corrientes que fluyen sobre los electrodos cuando no incide radiación sobre el fotocátodo. El ruido es mayormente citado como el valor efectivo de una cantidad eléctrica, usualmente la corriente. Este es equivalente a desviación estándar. El ratio de la medida para la señal a la medida para el ruido es denominado ratio señal-ruido. Este ratio es especialmente adecuado para caracterizar un detector de radiación en cuanto a la medida de una potencia radiante baja. En A A S debe ser posible medir en forma confiable cambios en la señal de absorbancia de por lo menos AA=0.00l. G. T ie m p o de R esp uesta del D e te c to r Este consiste en el tiempo de elevación y el tiempo de caída, es decir el tiempo en el cual la señal de salida alcanza una fracción estipulada del valor final (p.e. 90%) y el tiempo en el cual la señal cae de nuevo a una fracción estipulada del valor final (p.e. 10%). El tiempo de respuesta es de especial importancia en G F A A S con respecto a señales dependientes del tiempo. En A A S el tiempo de respuesta debe estar en el rango de milisegundos. - 65 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a CA PÍTU LO X S IS TE M A S DE L E C T U R A I 0. S is t e m a s d e L ectura El sistema de lectura de un espectrómetro de absorción atómica es ' simplemente el medio donde las señales eléctricas del fotomultiplicador son traducidas a lecturas instrumentales correspondientes al resultado analítico. El sistema más simple consiste en un amplificador para elevar la salida del fotomultiplicador, y un medidor simple con una escala adecuada. El fotomultiplicador produce una señal D C que es proporcional a la intensidad l0 de la línea de resonancia aislada por el monocromador. Cuando los átomos analitos se introducen en la zona de observación, la intensidad de la línea de resonancia es reducida a lT y la señal D C producida por el fotomultiplicador es reducida en forma proporcional. Los dos voltajes amplificados correspondientes a l0 e lT son medidos para determinar su ratio. El logaritmo de este ratio es derivado electrónicamente, y el resultado es presentado en el medidor como la absorbancia de la muestra. El problema fundamental con este sistema básico es que la señal final contiene componentes que no son analíticamente válidos. Estos componentes no válidos se originan de corriente 'oscura' dentro del fotomultiplicador (esto es, corriente que fluye en ausencia de luz), y la corriente D C causada por luz de fuentes diferentes a la lámpara de cátodo hueco (por ejemplo luz emitida por la flama). Claramente, si estos componentes inválidos no son diferenciados de las señales analíticas significativas el resultado analítico será equivocado. Para evitar esto, las señales analíticas válidas son ‘codificadas’ por un sistema que modula luz desde la lámpara de cátodo hueco y ‘decodifica’ las señales del fotomultiplicador por demodulación síncrona en el amplificador. Desde que las señales no válidas no están ‘codificadas’ ellas son efectivamente rechazadas por el sistema y solo son aceptadas las señales analíticas válidas. Estos conceptos relativamente simples han proporcionado sistemas de lectura confiables por muchos años. Pero la aparición del microprocesador y su tecnología asociada ha conducido a un desarrollo extensivo de procesamiento de señales y sistemas de lectura. En los instrumentos contemporáneos, los microprocesadores y los programas de computadora proporcionan un método más elegante de codificar y decodificar las señales analíticas válidas. En instrumentos de rayo doble, el microprocesador proporciona un utilidad de tiempo compartido que controla el tiempo del rayo - 66 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a doble, monitorea la intensidad de los rayos de referencia y de muestra, y en forma similar monitorea la intensidad de la lámpara correctora de background. Este medidor simple ha sido reemplazado universalmente por sistemas de lectura digital que incorporan integración electrónica de modo que los resultados analíticos son presentados al operador en una forma simple y sencilla de leer, la cual elimina el sesgo del operador. Los resultados analíticos pueden obtenerse como mediciones simples de absorbancia, o pueden ser presentadas directamente como valores de concentración. Asimismo el gráfico de calibración también es presentado. Generalmente, no es necesario una comprensión detallada del proceso electrónico involucrado para el analista práctico desde que este proceso es* automáticamente monitoreado y controlado dentro del instrumento. Sin embargo, existe un aspecto del procesamiento de señales sobre el cual el analista necesita ejercer algún control. Este es el proceso de integración y se considera útil una breve descripción del proceso de integración de señales: Con la atomización por flama, se experimenta en forma continua una ligera variación en la magnitud de la señal mientras se aspira la muestra. Con los sistemas simples de medición, estas variaciones se observan como un movimiento continuo de la aguja, lo cual hace dificultoso leer el resultado con certeza. Para controlar los efectos de estas variaciones, los instrumentos anteriores emplearon un sistema de retraso capacitivo para hacer lenta la respuesta del medidor y así hacer más fácil la lectura. Los sistemas digitales son también afectados por variaciones menores en la magnitud de la señal, pero en estos sistemas las variaciones son observadas como rápidos cambios en los valores numéricos displayados. Para contrarrestar esto, se emplea la técnica de integración electrónica. En este proceso, las señales individuales son colectadas electrónicamente y almacenadas sobre un período seleccionado (usualmente unos pocos segundos) después de las cuales son electrónicamente ‘promediadas’ con respecto a la magnitud y al tiempo. El valor derivado es entonces presentado como un valor numérico estable sobre el display digital. El grado al cual este método mejora la ‘precisión de lectura’ esta gobernado por el número de segundos sobre el cual se integra la señal, y la mayoría de instrumentos permiten al analista seleccionar el período de integración más adecuado bajo las circunstancias particulares de análisis. - 67 - Fu ndam entos del A n á l is is po r A b s o r c ió n A t ó m ic a C A P Í T U L O XI A N Á L IS IS C U A N T I T A T I V O I I. A n á l is is C u a n t it a t iv o Las bases teóricas de la espectrometría de absorción atómica conducen a la predicción que la absorbancia medida se incrementará en forma lineal con el incremento de concentración del analito de conformidad con la le/ de Beer. A bajas concentraciones esta predicción generalmente se cumple y la absorbancia se incrementa casi linealmente con la concentración del analito. A medida que la concentración se incrementa, sin embargo, la correlación se vuelve crecientemente curvada. Tradicionalmente, la forma de esta curva ha sido determinada experimentalmente para cada análisis. Se obtuvieron mediciones de absorbancia para estándares de concentración conocida, y la concentración de analito en la muestra se obtuvo por interpolación en la curva. En los análisis de rutina ya no se requiere este método gráfico debido a que los instrumentos pueden ser calibrados para proporcionar medidas directas de concentración en unidades seleccionadas por el operador. Sin embargo, el método gráfico es todavía una ayuda valiosa para establecer rangos óptimos de trabajo y parámetros instrumentales cuando se desarrolla o investiga un método analítico. Pero, la linealidad de la calibración no es el único asunto de importancia analítica. En el análisis práctico se necesita conocer las capacidades del espectrómetro. Para propósitos de comparación se usan tres parámetros para describir el rendimiento analítico de un espectrómetro de absorción atómica. Los tres intentan caracterizar el rango útil de medición del instrumento para cada elemento, estos son: I I.I I l.l.l P a r á m e t r o s d e l R e n d im ie n t o A n a l ít ic o en AAS C o n c e n t r a c ió n C a r a c t e r ís t ic a Esta es definida como la concentración, en solución, del elemento a ser determinado que producirá un cambio, comparado a una solución blanco, de 0.0044 unidades de absorbancia (es decir 1% de absorción) en la transmisión óptica del vapor atómico a la longitud de onda de la radiación usada. Debe notarse que diferentes instrumentos producirán diferentes concentraciones características. Sobre un instrumento dado, sin embargo, habrá poca variación día a día siempre - 68 - F u ndam entos del A n á l is is po r A b s o r c ió n A t ó m ic a que las condiciones de operación no se cambien y se optimicen las condiciones instrumentales. La concentración característica es fácilmente medida y puede también ser usada como un chequeo conveniente de que el instrumento esta operando correctamente. También posibilita un cálculo de la absorbancia que será producida por una concentración particular de un elemento dado. En la práctica un gráfico de calibración se deriva de mediciones de una serie de soluciones estándar. Usando la porción matemática lineal del gráfico se tiene la absorbancia ‘A ’ dada por la concentración ‘C ’. Luego, la concentración característica = C * 0.0044 / A Nótese que la capacidad de un horno de grafito es comúnmente especificada como el peso típico de un elemento que produce una altura de pico de absorbancia 0.0044. En este contexto, el término concentración característica debe siempre estar relacionado al volumen total de solución depositado en el horno. Límite de Detección El límite de detección es aquella concentración que puede ser detectada con 95% de confianza. Esta es la concentración que da una absorbancia igual a dos veces la desviación estándar de una serie de medidas en, o cerca, del nivel de blanco. El valor práctico del límite de detección se debe a que indica la concentración a la cual se pueden hacer mediciones útiles. Desde que el límite de detección es la concentración que da una desviación estándar relativa de 50%, es obvio que no es posible hacer mediciones precisas a este nivel. Pero, a 10 veces el límite de detección, la desviación estándar relativa debería ser 5% lo cual establece el límite inferior de medición útil. I 1.1.2 R a n g o d e T ra ba jo Este es el rango de concentración del elemento a analizar que produce valores adecuados de absorbancia para análisis prácticos. Es imposible ser específicos al definir este rango desde que las necesidades analíticas varían en forma considerable. Generalmente, sin embargo, el rango de trabajo debería yacer entre 0.100 y 0.800 de absorbancia desde que esta es la región de óptima precisión óptica. Pero pueden hacerse mediciones útiles a absorbancias más bajas, y el rango integral de trabajo puede extenderse a un límite inferior que es cerca de 10 veces el límite de detección. El cálculo del rango de trabajo es simple para análisis por flama: Los fabricantes del instrumento usualmente indican la concentración característica que puede esperarse para cada elemento en solución F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a acuosa. Este valor puede entonces ser usado para establecer un rango nominal de trabajo para el elemento de interés. Desde que por definición la concentración característica produce una absorbancia de 0.0044, una simple multiplicación por 100 da la concentración requerida para obtener una absorbancia de 0.44 (cerca de la mitad del rango óptimo). Una vez que se ha establecido esta 'marca de rendimiento’, es solo asunto de calcular el rango integral de trabajo junto con algún factor de concentración o dilución que puede ser necesario para una situación analítica especifica. En situaciones donde la muestra es complicada o cuando se conoce poco acerca de la concentración de la muestra, puede ser necesario establecer el rango de absorbancia por ensayo-error, y es buena práctica producir un gráfico de calibración inicial antes de diseñar la rutina analítica final. Para análisis por horno, la situación es un poco diferente. Inicialmente se señaló que la capacidad de concentración característica de un horno de grafito está completamente vinculada al volumen de solución empleada. Suponga, por ejemplo, que un horno requiere un peso total de 20 pg de analito para producir una absorbancia de pico de 0.0044. Ahora si este peso total de analito se obtuvo inyectando 20 pL de solución con una concentración de 1.0 ng/mL entonces es obvio que la concentración característica es 1.0 ng/mL para un volumen de 20 pL. Pero, el mismo peso total de analito (20 pg) podría obtenerse inyectando 100 pL de solución con concentración de 0.2 ng/mL. Esto implica que el analista debe siempre tomar el volumen de trabajo en cuenta cuando establezca un rango de trabajo para concentraciones prácticas. Ajuste del Rango de Trabajo Donde el rango de concentraciones de interés esta sobre el rango de trabajo obtenible a las absorbancias prácticas, el analista puede ya sea diluir todas la soluciones, o alterar la sensitividad instrumental. En el análisis por flama, la selección de longitudes de onda alternativas (menos sensitivas), la rotación del quemador a lo largo del haz óptico, o ambas acciones permitirá que puedan hacerse determinaciones a concentraciones significativamente más altas tal como se muestra en la tabla VI. Tabla VI. Concentraciones de trabajo (Cobalto) Posición del Sensitividad Relativa Rango de Trabajo Quemador (pg/m L) Normal 240.7 1 0.06 - 15 304.4 16 1.0 - 240 Rotado 9 0 ° Long. de Onda (nm) 240.7 20 1.2 - 300 304.4 320 20 - 4800 - 70 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a En lo que respecta a los métodos de horno, el analista usualmente busca mejores sensitividades para extender el rango de concentración hacia abajo. Esto puede algunas veces lograrse simplemente usando un mayor volumen de la solución de concentración más baja. Si la máxima capacidad del horno excluye la inyección del volumen total requerido, pude ser posible usar la técnica de inyección múltiple. En este método, una alícuota de la solución es inyectada al horno, esta alícuota es secada (o secada e incinerada) pero no atomizada. Una alícuota posterior es inyectada y secada en forma similar. Este procedimiento se repite hasta que el volumen total requerido ha sido depositado en el horno. Luego se lleva a cabo la secuencia de atomización. I 1.1.3 E x a c t i t u d y A. P r e c is ió n E x a c t it u d Esta observadas es una medida se aproximan de al cuan cerca las concentraciones valor verdadero o aceptado. Numéricamente es la diferencia entre la media X, de un conjunto de resultados y el valor X verdadero o aceptado. La exactitud relativa esta dada por : El porcentaje de exactitud es: 100 * Es valioso señalar que el empleo del espectrómetro de absorción atómica no es una garantía intrínseca de exactitud analítica, aún cuando los criterios instrumentales son raramente los factores limitantes. Los métodos de absorción atómica son comparativos, y la precisión final dependerá básicamente de: i. Exactitud del procedimiento de colección de muestra. Es obvio que la muestra colectada debe ser representativa del material original, de otra manera el resultado analítico no será válido. Es también importante evitar contaminación de la muestra en las etapas de colección y preparación. ii. La exactitud con que la muestra a analizar es recuperada del material de muestra original. iii. La exactitud con que se preparan las soluciones de calibración. iv. La exactitud con la que se calibra el instrumento contra las soluciones estándar. La única manera práctica de establecer una exactitud absoluta de un método analítico es tomar material de composición exacta conocida y analizarla mediante el método propuesto. Esto, por supuesto, presupone que el método originalmente usado para 71 F u ndam entos del A n á l is is po r A b s o r c ió n A t ó m ic a determinar la composición fue en sí mismo incuestionablemente exacto. Existen métodos de referencia establecidos de exactitud generalmente aceptada, y con frecuencia se usan comparaciones interlaboratorios para obtener exactitudes relativas por diferentes métodos. Tales comparaciones se usan algunas veces para establecer composiciones de concordancia para materiales de referencia estándares. A bajas concentraciones, sin embargo, algunos métodos clásicos muestran errores significativos que solo han sido revelados por métodos instrumentales. Es claro entonces que los métodos usados para establecer composiciones de los materiales de referencia necesitan ser cuidadosamente consideradas antes que los resultados sean aceptados como valores de referencia irrefutables. B. P r e c is ió n Cuando una medida es repetida ‘n’ veces, es improbable que todos los valores observados sean idénticos. La precisión analítica es una medida de cuan cerca un valor observado puede ser reproducido. Es convenientemente expresado como la desviación estándar ‘s’, o, más usualmente el porcentaje de desviación estándar relativa, % RSD . La desviación estándar es dada por: % R SD es así 100 * Estrictamente, la desviación estándar es absolutamente válida sólo donde ‘n’ es infinitamente grande. Para propósitos prácticos en absorción atómica, una desviación estándar es estadísticamente aceptable donde ‘n’ se toma como una serie de por lo menos treinta determinaciones . La precisión analítica de todos los métodos de absorción atómica depende de la interpolación precisa entre puntos de calibración conocidos exactamente. En los análisis de flama y de horno, factores tales como salida de luz estable y respuesta estable del arreglo detector-amplificador no son usualmente factores limitantes desde que ellos son rigurosamente controlados por sistemas electrónicos modernos. En los análisis de flama la estabilidad del sistema de atomización completa es generalmente más importante que las consideraciones electrónicas en determinar la precisión. Con 'sintonización' adecuada del sistema nebulizador-quemador, pueden obtenerse con facilidad resultados con desviación estándar relativa de I %. El limite de precisión disponible es cerca de 0 .1% (RSD). En los atomizadores de horno, la precisión puede ser influenciada por la reproducibilidad del programa de temperatura, la repetibilidad de los mecanismos de disociación por la cual se genera la población atómica, y la precisión del suministro. - «K' 72 - Fu ndam entos del A n á l is is por A b s o r c ió n A t ó m ic a La precisión integral del método analítico puede ser limitada por la precisión de los procedimiento de preparación involucrados- pesaje y dilución, por ejemplo. Al considerar la precisión analítica, es importante reconocer la diferencia entre la precisión instrumental y la precisión atribuible a otros factores. La precisión instrumental es la precisión de las mediciones duplicadas de la misma solución. La precisión atribuible a los procedimientos de preparación está dada por las mediciones tomadas de soluciones duplicadas que son preparadas separadamente mediante *■ procedimientos idénticos. Aplicación Práctica del % R S D En la operación práctica, el concepto de % R S D es relativo al nivel de valores que son leídos en el equipo de absorción atómica. Esto implica que un valor de % R S D adecuado para una concentración dada de solución, no lo será para una solución con diferente concentración (usualmente menor). Veamos el siguiente ejemplo: Disponemos de tres soluciones denominadas pregnant, relave y barren, con concentraciones de 3.143, 0.120 y 0.022 ppm de Au respectivamente. En la tabla VII apreciamos los valores leídos, así como su % R SD respectivo, nótese que el valor de la solución relave es bastante aceptable aun cuando su % R S D es mucho mayor que el de la solución rica. Asimismo puede considerarse que el valor leído de la solución barren también es adecuado a pesar que su % R S D es muy alto (de hecho, el valor H IG H que implicaría una lectura inadecuada de una solución rica no implica lo mismo para una solución barren). Tabla VII. Valores Leídos de Soluciones de Au Solución Valor 1 Valor 2 Valor 3 Valor 4 V.Prom Conc. %RSD (ppm) 0.1894 3.143 1.63 Rica 0.1884 0.1888 0.1892 0.1914 Relave 0.0079 0.0080 0.0071 0.0066 0.0073 0.120 8.33 0.0020 0.0015 0.022 HIGH Barren 0.0003 0.0011 0.0026 Para explicar esto, consideremos que el error relativo de dos muestras de muy baja concentración (por ejemplo 0.01 y 0.02 ppm) es (0.01-0.02)/0.01 = -100%. Si consideramos el error relativo de dos soluciones de baja concentración (por ejemplo 0.12 y 0.13 ppm), este será (0.12-0.13)/0.12 = -8.33%. En el caso de soluciones de concentración alta (por ejemplo 3.56 y 3.57 ppm), el error relativo será (3.56-3.57)/3.56 = -0.28%. - 73 - f F undam entos del A n á l is is p o r A A b s o r c ió n t ó m ic a Esto quiere decir que la diferencia entre las concentraciones (que tiene un valor absoluto de 0.01 ppm en los tres casos) implica diferentes errores relativos y por ende diferentes % RSD , aún cuando podemos considerar válidos los tres valores leídos. Los conceptos de exactitud y precisión son ilustrados por las figuras 40, 4 1 y 42. La figura 40 ilustra buena precisión pero pobre exactitud. Los resultados están cercanamente agrupados alrededor de un valor promedio que esta lejos del valor verdadero. En la figura 41 el promedio resulta que es el valor verdadero pero la precisión analítica es pobre. La figura 42 es un ejemplo de buena precisión combinada con , buena exactitud. Aquí los resultados están cercanamente agrupados alrededor del valor verdadero. il ■ ■ ■ ■ . valores observados valor verdadero Figura 40. Buena precisión; pobre exactitud Figura 40. Buena precisión; exactitud pobre ■ ■ m " ■ ■ ■ ■ ■ . valores observados " ■ ■ valor verdadero ■ Figura 41. Pobre precisión Figura 4 1. Precisión pobre - 74 - \ Fu ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a v a lo re s o b s e rva d o s — T ■ . 1■ ■ va lo r verd ad ero Fig u ra 42. B u e n a p recisió n ; buena exactitud Figura 42. Buena precisión; buena exactitud. I 1.2 A n á l i s i s C u a n t i t a t i v o I: A t o m iz a c ió n p o r F l a m a La gran ventaja de los métodos de flama es que la operación del instrumento es simple y universal. Para un instrumento dado, todos los elementos pueden ser determinados con el mismo procedimiento básico. Las diferencias entre instrumentos de diferentes fabricantes pueden afectar el rendimiento, pero el procedimiento de operación es similar para todos los instrumentos. Una vez que el instrumento ha sido parametrizado y optimizado solo es necesario introducir el blanco, los estándares y muestras en la flama, y registrar los resultados. La precisión analítica es excelente; con cuidado puede lograrse 0.5% (R.SD); pueden determinarse en forma rutinaria 67 elementos. La velocidad analítica máxima es cerca de 20 muestras por minuto. Condiciones de la muestra: Determinación por Flama ♦ ♦ Líquida o disuelta Menos de 5% de sólidos disueltos (preferiblemente menos de 2%) ♦ Químicamente adecuado para descomposición térmica simple. ♦ En el rango apropiado de concentración ♦ Disponible al menos en cantidad de I mL. I 1. 2 . 1 E s t á n d a r e s La absorción atómica es una técnica comparativa, en consecuencia la ‘exactitud’ de las medidas cuantitativas depende mucho de la ‘exactitud’ de los estándares empleados. Debe recordarse también que ‘exactitud’ es este contexto es más que un asunto de concentraciones exactas de sustancia a analizar. Este concepto también - 75 - F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a incluye las propiedades físicas y químicas de todas las soluciones empleadas en el análisis. Para muchas determinaciones, las soluciones estándares acuosas simples serán completamente adecuadas. Para muchas otras determinaciones, sin embargo, las propiedades físicas tales como viscosidad, tensión superficial, y densidad afectarán la cantidad de solución enviada a la flama. Si los estándares y las muestras no coinciden en cuanto a estas propiedades, su respuesta analítica comparativa puede ser diferente y la precisión de los resultados puede ser degradada. Si los estándares y muestras no coinciden químicamente, su comportamiento comparativo en la flama será diferente, y se producirán las correspondientes imprecisiones. En todas estas » situaciones, la exactitud y precisión final serán gobernadas por la precisión en la concentración de los estándares y la precisión con la que los estándares y muestras coinciden física y químicamente. Para algunas muestras, será imposible preparar estándares que coincidan en forma exacta debido a que no se conoce la composición química de la muestra o es demasiado complicado reproducirla artificialmente. En estas circunstancias puede ser necesario usar el método de adiciones estándar. Cuando se analiza una muestra en particular por primera vez, es buena práctica comparar la calibración por adiciones estándar con el procedimiento normal de calibración. Ploteando los dos conjuntos de datos de calibración tal como se muestra en la figura 43, los gradientes pueden ser fácilmente comparados. Si los gradientes de las dos líneas son sustancialmente los mismos, puede asumirse que se puede usar el método normal de calibración. Sin embargo, si los gradientes no son los mismos, será necesario usar el método de adiciones estándar. Figura 43. Comparación de la calibración normal y de adiciones estándar. - 76 - F undam entos del A n á l is is p o r a. A b s o r c ió n A t ó m ic a El número de estándares necesarios para calibrar el instrumento variará de acuerdo a las circunstancias analíticas. Los lineamientos son: Cuando se desarrolla un método analítico, siempre prepare un gráfico de calibración preliminar (curva de trabajo) de modo que la linealidad de la calibración y rango de absorbancia sean evaluados en forma adecuada. b. Solo se necesita un estándar cuando el rango de trabajo esta c. estrictamente dentro de la porción lineal de la curva de trabajo. Nótese que existe un caso especial cuando un estándar único es siempre adecuado. Este es cuando se trabaja a una absorbancia de 0.01 o menor, pero donde la concentración de la muestra es al ' menos 10 veces el límite de detección. Si la curva de trabajo no es lineal, pero la desviación de la linealidad d. es pequeña, deben usarse dos o tres estándares. Se requiere el uso de cuatro o cinco estándares para máxima e. precisión cuando la curva de trabajo no es lineal, o cuando se requiere el máximo rango de trabajo. Cuando se usa múltiples estándares, siempre seleccione valores de concentración tales que el rango de concentración íntegro esté dividido en partes aproximadamente ¡guales. I 1.2 .2 A d ic io n e s d e E s t á n d a r En este método se minimizan la discordancias físicas y químicas entre muestras y estándares debido a que los estándares son preparados a partir de las muestras reales. El método asume que el analito agregado será afectado por interferencia del mismo modo y en la misma extensión tal como el analito en la muestra. Esto generalmente es válido para interferencias físicas. N o siempre es válido para interferencias químicas y en estas situaciones la interferencia debería ser removida por medios adecuados (una flama más caliente, o un agente liberador, por ejemplo). El procedimiento general es tomar varias alícuotas de muestra, y luego agregar diferentes cantidades del elemento a analizar a cada alícuota. Después de la dilución al volumen final, estas soluciones forman un conjunto de estándares de diferente concentración. Una alícuota es diluida sin ninguna adición del elemento a analizar. Las cantidades de analito agregado se basan en la concentración esperada de este en la muestra. En un esquema de adiciones típica, las adiciones del analito son 50%, 100% y 150% de la concentración esperada. El resultado analítico puede obtenerse ploteando las absorbancias contra las concentraciones agregadas y extrapolando el gráfico a cero tal como se muestra en la figura 44. En forma alterna, el instrumento puede ser calibrado directamente contra las adiciones F ú n d a m e ;. fo s del A n á l is is por A b s o r c ió n A t ó m ic a estándar. El software incorporado calculará entonces una regresión lineal por mínimos cuadrados a través de los puntos de calibración y computará la concentración de muestra directamente por extrapolación matemática. Com o regla general, las adiciones estándar deben proporcionar sustancialmente una calibración lineal desde que no puede obtenerse una regresión precisa desde puntos de calibración no lineales. Es esencial establecer una línea de base precisa desde el reactivo blanco adecuado. En las aplicaciones más comunes del método de adiciones estándar, cada muestra debe ser analizada individualmente contra un* conjunto de estándares que son específicos para esa muestra. Existen algunas situaciones analíticas en las cuales un lote de muestras puede ser analizado contra un conjunto de adiciones estándar, pero esto solo es válido cuando todas las muestras en el lote son similares química y físicamente. Figura 44. Calibración por adiciones de estándar I 1.2.3 S it u a c io n e s A n a l ít ic a s S im p l e s Para muestras que son física y químicamente simples, es a menudo suficiente disolver la muestra en agua o ácido y comparar la solución directamente con los estándares en solución acuosa simple del elemento a analizar. En tales matrices simples las interferencias son improbables, siempre y cuando, por supuesto, que se observe en forma adecuada la química ordinaria de la muestra. - 78 - Fu ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a I 1.2.4 M u e s t r a s C o m p l e j a s El procedimiento de pretratamiento depende de la naturaleza de la muestra, pero para análisis por flama debe seleccionarse un método que proporcione una solución regularmente diluida (hasta con 5% de sólidos disueltos). La solución debería ser estable en el tiempo y no debería contener grandes cantidades de sustancias que conduzcan a interferencia química en la flama. Generalmente se prefieren las soluciones ácidas debido a que los ácidos comunes están disponibles en alta pureza, son relativamente fáciles de manipular y no incrementan el contenido de sólidos de la solución. La mayoría de muestras metalúrgicas y minerales son fácilmente disueltas en la mezcla ácida adecuada. Algunas veces el procedimiento de digestión será dictado por los posibles efectos de las interferencias químicas. Si, por ejemplo, se va a determinar calcio en la flama aire-acetileno, no debe usarse ácido fosfórico debido a que los fosfatos deprimen la señal de absorción. En algunas situaciones es útil un tratamiento posterior después de la disolución de la muestra. Cuando debe realizarse una determinación de trazas en la presencia de cantidades grandes de otro elemento, por ejemplo, el constituyente principal debe ser removido de la solución mediante extracción por solventes. En forma alterna, el elemento a analizar por sí mismo puede ser selectivamente extraído y el extracto usarse para el análisis. 1 1 .2 . 5 D e t e r m in a c ió n d e T r a z a s La técnica de absorción atómica es extremadamente sensitiva, pero algunas veces la concentración de analito en la muestra disuelta es inconvenientemente baja y es dificultoso obtener resultados precisos. Los instrumentos modernos tienen la facilidad de una expansión de escala extensiva de modo que pueden medirse señales muy pequeñas de absorción, pero para mejor precisión es a menudo útil preconcentrar el elemento a analizar mediante métodos químicos. Pueden usarse cualesquiera de los métodos tradicionales de reconcentración, en la actualidad métodos tales como el intercambio de iones y co-precipitación se están tornando comunes. Sin embargo por conveniencia y velocidad, los métodos de extracción por solventes son preferidos universalmente en AAS. Muchos elementos pueden extraerse con facilidad de un gran volumen de solución en un volumen mucho más pequeño de un solvente orgánico después de la reacción con un reactivo quelante orgánico general tal como el ditiocarbamato amonio pirrolidina (A P D C ) o 8-quinolinol. La sensitividad analítica es mejorada en la matriz orgánica debido a que se mejoran las características de nebulización, y si el volumen del solvente de extracción es 1/10 del volumen de la muestra original, la señal puede ser de 30 a 40 veces mayor después de la extracción. Estos métodos \ F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a son usados en forma rutinaria para determinaciones a muy baja concentración; las soluciones estándar son extraídas en forma similar. Puede usarse una amplia variedad de reactivos y condiciones; los mejores solventes son los ésteres y cetonas. Los solventes clorados o aromáticos interrumpen la flama y no son adecuados. I 1.3 A n á l i s i s C u a n t i t a t i v o II: A t o m iz a c ió n p o r H o r n o Los análisis prácticos por horno no pueden llevarse a cabo para todos los 67 elementos que pueden ser determinados por flamas químicas. Esto no es debido a limitaciones de temperatura, sino más relacionados a los procesos de descomposición química requeridos para la producción de ciertos átomos de los elementos a analizar. La flama química contiene especies reactivas tales * como C, C¡, CH, C N y N H , y estos tienen una fuerte influencia sobre la disociación de los compuestos metálicos. El horno de grafito no produce intrínsecamente esta variedad de especies reactivas, y la disociación de ciertos compuestos metálicos es así imposible en ciertos atomizadores de horno comerciales. Por lo menos cuarenticuatro elementos, sin embargo, pueden ser determinados a concentraciones y en rangos de trabajo que son típicamente 100 veces más bajos que aquellos que se pueden lograr por métodos de flama. La precisión no es característicamente tan buena como la obtenible en flamas, pero con un cuidado adecuado es posible lograr 1% (R SD ) bajo el supuesto que las soluciones se dispensen en forma automática. Cuando las soluciones son inyectadas con microjeringas operadas manualmente, el factor limitante es usualmente la precisión con que se dispensa más que los criterios instrumentales o del horno. Estos resultados generalmente se obtienen usando volúmenes de solución que varían entre 20 y 50 pL. Pueden emplearse volúmenes considerablemente mayores utilizando la técnica de inyección múltiple, y al otro lado de la escala de volumen es posible obtener resultados precisos y exactos usando volúmenes tan pequeños como I pL. La velocidad analítica es generalmente cerca de I muestra cada dos minutos. Condiciones de Muestra: Análisis en Horno ♦ Líquida, disuelta y preferiblemente homogénea. ♦ Químicamente adecuada para la separación térmica de la matriz y el analito. ♦ En el rango adecuado de concentración. Algunos diseños de horno posibilitan al analista usar muestras sólidas. Pero se requiere un alto grado de habilidad para obtener precisión y exactitud satisfactoria, y la dificultad de obtener una muestra representativa en pequeña cantidad es usualmente el factor limitante. I 1.3.1 In t e r f e r e n c ia s F ís ic a s - 80 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Efectos de viscosidad y tensión superficial Las determinaciones por horno pueden ser afectadas físicamente por diferencias en viscosidad y tensión superficial entre los estándares y las muestras. Tales interferencias físicas en situaciones analíticas simples pueden ser menores y no significativas. En situaciones más complicadas estas pueden ser relativamente severas. Los métodos de horno son intrínsecamente microcuantitativos y pequeñas variaciones en las propiedades físicas pueden tener una gran influencia sobre el resultado final. Esto sólo puede ser contrarrestado mediante una coincidencia física entre estándares y muestras. Estas interferencias pueden ocurrir de dos maneras: i. Prim ero, la solución más viscosa (aceites y algunos materiales biológicos, por ejemplo) tienden a adherirse a la pipeta y una proporción significativa no llegará al horno. Las soluciones más dóciles, de otro lado, pueden moverse hacia fuera de la pipeta, y nuevamente una proporción significativa no llegará al horno. Algunas soluciones a altas concentraciones ácidas (20% o 50%) y algunos solventes orgánicos son ejemplos de este fenómeno. ii. Segundo, la extensión a la cual las soluciones se esparcen sobre la superficie interior del horno es función de la viscosidad y características de humectación. Las diferencias en esparcimiento pueden afectar en forma significativa la señal de absorción debido a que el tiempo de residencia de los átomos dentro de la zona de observación variará dependiendo de cuan lejos del centro ocurra la atomización. 1 1 .3 . 2 A b s o r c ió n po r B a c k g r o u n d En las flamas químicas, la absorción por background generalmente no es común, es raramente significativa a longitudes de onda mayores de 240 nm, y la señal de background raramente excede una absorbancia de 0.05. Sin embargo, en los análisis por horno la absorción por background es mucho más común, puede ser significativa a longitudes de onda de hasta 340 nm, y la señal de background puede exceder 1.0 de absorbancia. Existen tres medidas disponibles para superar esto: corrección simultánea, programación adecuada de la temperatura y modificación química. La corrección es conveniente, en los instrumentos modernos se incorporan sistemas correctores simultáneos. Tales sistemas son típicamente efectivos a longitudes de onda de hasta 400 nm, y proporcion corrección precisa a absorbancias totales de alrededor de 1.5. Por supuesto, es esencial que las dos fuentes de luz sean ópticamente coincidentes y las intensidades relativas deben estar balanceadas. Generalmente, el alineamiento óptico es simplemente asunto de ajustar los tornillos adecuados; la amortiguación de 81 F undamentos del A n á l is is po r A b s o r c ió n A t ó m ic a intensidades relativas de fuentes es usualmente controlado en forma electrónica dentro del instrumento. Siempre debe usarse la corrección simultánea cuando se desarrolla por primera vez un método de horno. Prim ero, para averiguar si la absorción por background está o no ocurriendo. Segundo, para obtener un resultado analítico válido y medir la señal de background real si fuera necesario. Tercero , para monitorear la efectividad de las otras medidas. Es preferible utilizar la programación de temperaturas, modificación química, o ambas, en vez de solo confiar en la corrección por background. En el contexto de la absorción por background, el objetivo de la programación de temperatura es: i. Asegurar que el material matriz que causa absorción por background sea removido previo a ala etapa de atomización sin pérdida de analito; o, ii. C rear un lapso de tiempo entre la señal del background y la del analito de modo tal que las dos señales sean medidas y observadas separadamente. Se necesita un enfoque experimental metódico para obtener las velocidades de calentamiento, tiempos y temperaturas que proporcionen el secado, incinerado y atomización más efectivos. Las señales de background y analítica deben mostrarse en la pantalla del computador. 1 1 .3 .3 In t e r f e r e n c ia s Q u ím ic a s Estas interferencias se originan de la formación de compuestos que son ya sea excesivamente volátiles o excesivamente estables dentro del ambiente del horno. En aquellos compuestos que son excesivamente volátiles, se pierde el analito a bajas temperaturas durante el secado durante el secado o incinerado sin sufrir atomización. Si, por ejemplo, el analito forma fácilmente un cloruro covalente, la presencia de exceso de iones cloruro puede causar que el analito aparezca como un cloruro volátil que se vaporizará antes que pueda ser descompuesto a la forma atómica. La pérdida del analito de esta manera obviamente degradará la sensitividad analítica, y puede necesitarse cuidado con elementos que forman haluros volátiles. Una atención particular a la química original de la solución puede a menudo evitar o minimizar el efecto. En forma alterna, puede ser necesario utilizar un modificador químico. - 82 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a Pueden encontrarse interferencias cuando el analito forma dos o más compuestos estables que finalmente se disocian a temperaturas diferentes. En esta situación, el analito no se pierde durante el secado o incinerado. Pero, la concentración atómica de interés asociada con dos o más compuestos, y la señal atómica son disipadas entre múltiples picos de atomización en vez de aparecer como una única respuesta coherente. Para algunas muestras es posible evitar o minimizar la interferencia revisando la química original de la solución. Para otras muestras puede necesitarse un modificador químico para aislar el analito en una forma específica y prevenir la formación de compuestos interferentes. Un caso extremo de estabilidad excesiva ocurre cuando el elemento forma un carburo refractario. Esto puede degradar la sensitividad analítica, ocasionar un ‘efecto memoria’ significativo, o ambos. Los elementos tales como el bario, molibdeno, titanio y vanadio formaran tales carburos con facilidad. Sin embargo, estos pueden ser determinados con éxito en hornos de gráfico piroliticamente recubiertos, desde que este recubrimiento reduce la tendencia a formar carburos. O tros elementos tales como el tantalio, tungsteno y zirconio son algo más dificultosos de determinar, mayormente debido a la tendencia de estos a formar carburos no maleables aún en un ambiente pirolítico. 1 1 .3 . 4 M o d if ic a c ió n Q u ím ic a El modificador químico ideal es un reactivo que realiza las siguientes funciones: i. Elevar la temperatura de atomización para prevenir la aparición ii. simultánea de señales de absorción atómica y de background. Reducir la volatilidad del analito. Esto permite una mayor temperatura de calcinado a usarse de modo que todo el material que produce absorción de background es removido antes de la etapa de atomización. iii. Aislar el analito en una forma específica. Esto asegurará que aparezca un único pico atómico en vez de los múltiples picos que resultan de la descomposición de diferentes especies moleculares a diferentes temperaturas. 1 1 .3 .5 E stán dares El criterio aplicado a los estándares para análisis por flama se aplica con igual énfasis a los métodos de horno. Los valores de concentración deben ser exactos, y los estándares deberían ser química y físicamente coincidentes con las muestras. Si es imposible hacer artificialmente la coincidencia, será necesario usar el método de adiciones estándar. - 83 - * F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Para aplicaciones de horno, se tiene la opción de obtener absorbancia de calibración de diferentes volúmenes de una única solución estándar más que usar un volumen constante de diferentes concentraciones. Esto puede ser adecuado, pero el método debería ser usado con discreción dado que diferentes volúmenes pueden exhibir un comportamiento diferente en el horno (particularmente en la etapa de secado). 1 1 .3 . 6 M uestras Debido a las concentraciones extremadamente bajas del analito involucradas, los análisis por horno demandan un cuidado exhaustivo en la colección, almacenamiento y preparación de muestras. Debe evitarse . la contaminación, y debe recordarse que la contaminación desde los reactivos y aparatos es a menudo una consideración principal. Una gran variedad de muestras líquidas puede analizarse directamente en el atomizador del horno sin ningún pretratamiento químico. Algunas veces es útil mezclar la muestra con ácido nítrico para ayudar a la remoción de componentes orgánicos de la matriz durante la etapa de secado. Cuando es necesario el pretratamiento, varias consideraciones afectan la elección del método. El analito debe ser recuperado completamente, y la solución debe ser estable. Los reactivos deben ser seleccionados de modo que no se formen compuestos volátiles que involucren al analito, los cuales podrían perderse durante el secado y el calcinado. Las soluciones de ácido clorhídrico o perclórico son inadecuadas para análisis de plomo o estaño, por ejemplo, debido a que estos elementos forman cloruros volátiles los cuales se pierden antes de la atomización. De otro lado, las soluciones de ácido sulfúrico y fosfórico, que pueden causar problemas de interferencia en los análisis por flama, pueden ser muy útiles para análisis por horno. 1 1 .4 A n á l is is C u a n t it a t i v o III: G e n e r a c ió n d e V a p o r Ocho elementos (antimonio, arsénico, bismuto, germanio, plomo, selenio, estaño y telurio) pueden ser determinados por el método de generación de hidruros. El mercurio es determinado por la técnica de vapor frío. Para estos elementos, la generación de vapor es hasta 1000 veces más sensitiva que la flama y hasta 10 veces más sensitiva que el horno. La preparación de muestras requiere cuidado y habilidad, pero el análisis real es simple. La precisión analítica es cerca de 5% (R SD ) lo cual no es tan bueno como con flama u horno, pero aún mejor que otras técnicas a tales bajas concentraciones. La naturaleza química de las muestras digeridas es importante, y algunas muestras son dificultosas de analizar sin separación química. La velocidad analítica es cerca de I muestra por minuto. - 84 - F undam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a Condiciones de Muestra: Generación de Vapor ♦ Digeridas en solución ácida ♦ En el estado adecuado de oxidación ♦ Libre de interferentes químicos ♦ En el rango adecuado de concentración Usualmente, los procedimientos analíticos son simples y se conocen varios métodos. Cuando un método probado no ha sido documentado, será necesario desarrollar un método que sea adecuado para muestras específicas. Pueden necesitarse experimentos cuidadosos para establecer la solución química más efectiva y puede necesitarse algo de ensayo-error para establecer las condiciones óptimas. El ácido clorhídrico es generalmente adecuado para la mayoría de aplicaciones. Pueden usarse mezclas ácidas alternativas para algunos ensayos-error requeridos para establecer condiciones óptimas. Debe evitarse el ácido nítrico por ser oxidante: ni el ácido sulfúrico ni el nítrico deben usarse para determinaciones de selenio. En las determinaciones de arsénico el analito debe estar en el estado A s111, y puede necesitarse una etapa de reducción. Esto puede ser complementado mediante la adición de una solución al 20% de yoduro de potasio- la reducción estará completa en cerca de 50 minutos a temperatura ambiente. En forma alternativa la muestra puede ser calentada a 50°C por cerca de 4 minutos. Para análisis de selenio, el analito debe estar en forma inorgánica, de otra manera será necesaria la digestión. N o se recomienda el calcinado en seco debido a la volatilidad del selenio. El ácido clorhídrico es generalmente el mejor, aún cuando puede obtenerse alguna mejora en la precisión analítica con una mezcla de 5% de ácido fosfórico y 5% de ácido clorhídrico. El Sevl no es recuperado cuantitativamente en la generación de hidruros y debe ser reducido a Se'v. El calentamiento a 70 °C por 10 minutos usando ácido clorhídrico al 50% es efectivo. La concentración ácida para soluciones analíticas no debería exceder el 25%. de otro modo la señal analítica será deprimida y la absorción por background puede ser severa. El plomo puede determinarse sobre un rango de trabajo de 0.01 a 0.3 mg/L. Es necesario adicionar ácido tartárico y dicromato de potasio antes de generar el hidruro. Para estaño, es adecuado el ácido clorhídrico, pero el analito debe estar en la forma estañosa. El pH debe estar ajustado a 1.0-1.5. - 85 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a El mercurio es determinado por el método de vapor frío: la máxima concentración de trabajo es cerca de 0.08 mg/L para una muestra de 20 mL. La concentración característica es cerca de 0.0004 mg/L para una muestra de 20 mL. Para todos los métodos de generación de vapor es necesario obtener mediciones de pico. Generalmente son adecuadas las mediciones de altura de pico. Para algunas aplicaciones, sin embargo, la matriz de la muestra puede ser tal que los picos sean inadecuados para mediciones efectivas de altura. Para estas aplicaciones será esencial usar medición de área de pico. I 1.5 M ic r o m u e s t r e o p o r F l a m a En esta técnica, un pequeño volumen de muestra, típicamente 100 (j L es inyectado en un túnel de teflón conectado al nebulizador del instrumento. La señal analítica resultante es un pico rápido y transitorio el cual es medido por el sistema de lectura de pico. La inyección directa se acompaña usando una micropipeta del volumen requerido. La principal aplicación para el micromuestreo es en análisis de trazas donde se requieren técnicas de preconcentración. Se necesita una muestra inicial más pequeña así dando manipuleo más fácil en el campo o laboratorio previo al análisis. Puede realizarse una extracción con A P D C / D IB K sobre 100 mL de muestra en vez de 500 mL sin pérdida en el factor de concentración. De modo similar un volumen más pequeño puede usarse en los procedimiento de intercambio de iones desde que el análisis completo por micromuestreo puede ser llevada a cabo con I mL de líquido. Los coprecipitados pueden ser digeridos y hechos en muy pequeños volúmenes, incrementando el factor de concentración. También pueden determinarse más elementos de un volumen pequeño dado comparado con la aspiración convencional. Típicamente, hasta nueve elementos pueden determinarse desde I mL de muestra. El micromuestreo por flama puede también usarse para análisis con aplicaciones de solventes orgánicos y con alto contenido de sólidos disueltos, donde la aspiración convencional puede ser problemática. I 1.6 A u t o m a t iz a c ió n El empleo de dispositivos de muestreo automático y el registro automático de resultados son medios efectivos de incrementar la productividad analítica. Los cambiadores de muestra automáticos usualmente toman la forma de una tabla rotatoria o rectilinear desde la cual las muestras son sucesivamente presentadas a un capilar adjunto al nebulizador. Pueden usarse cambiadores similares de muestra para presentar un arreglo de muestras a un horno de grafito. De modo semejante a los espectrómetros, los cambiadores de muestra han obtenido los beneficios de la tecnología del microprocesador. Los espectrómetros y cambiadores de muestras controlados por microprocesador pueden fabricarse para interactuar en una manera muy sofisticada. El software - 86 - F undamentos d el A n á l is is por A b s o r c ió n A t ó m ic a de computadora de los cambiadores de muestras modernos proporciona una repertorio extenso de rutinas analíticas. Entre otros se tienen los siguientes accesorios para la automatización del análisis por absorción atómica: Automuestreador de Flama Un automuestrador de flama proporciona análisis automático de hasta 60 muestras, con calibración automática usando hasta 10 estándares. Tiene interface con el equipo de AAS, y es controlado por la computadora del AAS. Tiene provisión para enjuague separado, soluciones blanco y de re-escala y es completamente programable. Automuestreador X-Y-Z Un automuestreador X-Y-Z es una muestreador transversal * programable (x-y-z) con una capacidad de hasta 270 muestras en tres racks más diez estándares. Es controlador por el software de absorción atómica del equipo principal de AAS. Auto Diluidor Este equipo es un accesorio de dilución de muestras para métodos de absorción de flama y de hidruros, entre sus características se tiene: ♦ ♦ Permite la adición de hasta dos soluciones químicas modificadoras por separado Preparación de estándares de trabajo ♦ ♦ Preparación de adiciones de estándar Dilución de la muestra antes de la medición ♦ Dilución automática de las muestras “ sobre el rango” ♦ Usa rotación automática del quemador ♦ Elimina la necesidad de preparación manual de estándares I 1.7 A p lic a c ió n d e la s C o m p u t a d o r a s a l AAS Las computadoras, junto con los espectrómetros y cambiadores de muestra están siendo usados en forma creciente para análisis de rutina, desarrollo de métodos e investigación. El uso de las computadoras implica que los modernos aparatos de AAS, incluyen un microprocesador y que pueden establecer comunicación hacia la computadora mediante los puertos de comunicaciones (C O M I, C O M 2 u otros según diseño). Para esto deben establecerse los parámetros como la velocidad en baudios, el tipo de paridad, los bits de datos y los bits de parada. Generalmente, los programas de computadora diseñados para A AS (software para A A S) permiten el pre-registro, almacenaje y llamado de métodos analíticos adaptados al usuario. Los programas incluyen algoritmos específicos para manipulación de datos y procesos tales como cálculos de calibración y ajuste de curvas, regresión por adiciones estándar, promedio, desviación estándar, etc. La pantalla proporciona una guía paso a paso para la rutina operacional y un display dinámico de los resultados operacionales. - 87 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a Pueden calcularse los gráficos de calibración, verse e imprimirse si fueran necesarios. Mediante el uso de una impresora también se puede obtener copias impresas de resultados analíticos bajo formatos específicos. I 1.8 S oftw are para A A S Cada fabricante de equipos de y accesorios para A A S diseña su propio software. Esto implica que exista una diversidad de versiones de software para AAS. Con base en algunos de estos programas, se tratará de brindar pautas generalizadas del contenido del software, de modo que el lector este preparado para ubicar los puntos de su interés en el software de AAS de cualquier fabricante de equipos. Usualmente un programa para A AS incluye los siguientes módulos: M ódulo A : M étodos Proporciona acceso a parámetros del método de análisis para un elemento químico y una configuración particular del instrumento. Incluye las siguientes secciones: A. I D escripción Descripción Parámetro Elemento Químico Permite seleccionar un elemento químico (desde una tabla desplegada) Posición de Lámpara Verificación de que al elemento seleccionado le corresponda una posición de lámpara en la torreta Superlámpara Verificación de que el elemento seleccionado tiene una superlámpara vinculada. De ser así, lo indica. A .2 In strum ento Descripción Parámetro Línea de Resonancia Selección de línea de resonancia, si se requiere una diferente a la primaria Corriente de Lámpara Corriente eléctrica aplicada a la HCL para maximizar la salida Longitud de Onda La que corresponde a la linea de absorción más sensitiva. Ancho de Slit Valor del ancho del slit por defecto Altura de Slit Normal: para atomización por flama Reducida: para horno de grafito Corrección por La absorción por background (no atómica) se medirá y será sustraída de la Background señal antes del cálculo. A .3 Medición Se determina como y cuando se ejecuta cada medición durante la introducción de una muestra al instrumento. Descripción Parámetro Modo de Medición Permite seleccionar entre integración, altura de pico, área de pico y media (ver anexo 14.1) móvil Tiempo de Lectura Establece el tiempo de lectura en segundos F undamentos A del n á l is is p o r A b s o r c ió n A t ó m ic a Constante de Tiempo Filtra digitalmente la señal de absorbancia primaria para reducir el ruido. Réplicas Número de lecturas sobre cada muestra Tiempo de Retraso Permite que la lectura se estabilice después de introducir una nueva muestra A .4 Calibración Se determina como se realiza la calibración, se aplican pruebas a los datos de calibración y están disponibles acciones en caso de falla. Descripción Parámetro Modo de Calibración Concentración, Conc. Mínimos Cuadrados, Lineal Mínimos Cuadrados, Lineal (ver anexo 14.2) Mínimos Cuadrados a través de Cero, Adiciones de Estándar, Estándares Unidades de Concentración Selecciona las unidades: ppm, (Jg/ml, mg/L etc. Falla de Calibración Verifica el valor de r o r! cuando se utilizan los modos de calibración Acción de Falla de Conc. Selecciona la acción a ejecutar después de una falla de calibración Blanco después de calibrar Fuerza a que la solución blanco sea medida después de una calibración Cero antes de Calibración Realiza un cero del instrumento al inicio de la calibración. Cero entre Muestras Realiza un cero del instrumento entre cada muestra leída. Envolventes. basados en Mínimos Cuadrados A .5 Estándares Se establece el número de estándares para construir la curva de calibración. Se puede adicionar y rem over estándares así como ver el gráfico de calibración resultante. Parámetro Descripción Adición de estándar Permite adicionar un estándar a la tabla Remoción de estándar Permite remover un estándar de la tabla Ver gráfico de calibración Permite ver el gráfico de calibración A .6 C o n tro l de Calidad Se establecen protocolos de control de calidad para los diversos tipos de muestras. Descripción Parámetro Fija los límites para el chequeo de rangos de concentración y la acción a Muestra iniciar si ocurre una falla. Muestra de Chequeo Se ingresa la concentración conocida de esta muestra. Se fijan limites Segunda Acción de Falla Si ocurre una segunda falla la corrida se detiene o continua, según lo inferior y superior. En caso de falla están disponibles ciertas acciones. programado. A .7 Flam a Parámetro Descripción Tipo de Flama Aire-Acetileno u Oxido Nitroso-Acetileno Flujo de Acetileno Permite ingresar el flujo de acetileno. Rango: 0.5 a 9.0 L/min. - 89 - \ F undam entos A del n á l is is p o r A b s o r c ió n A t ó m ic a Permite ingresar el flujo de aire. Rango: 10 a 24 L/min. Flujo de Aire Permite ingresar el ángulo entre la línea central del quemador y el haz Angulo del Quemador óptico. Para un ángulo de quemador cero la línea central del quemador es paralela al haz óptico. Altura del Quemador Permite ajustar la altura relativa a la máxima disponible. Esta opción es usada para ajustar la posición vertical del quemador para máxima absorbancia. M ódulo B: M uestras Permite diseñar el orden y frecuencia de tipos de medición. Descripción Parámetro Edición de tabla de La tabla de muestras puede ser editada mediante: muestras remoción, propiedades y cambio de tipo de medición. inserción, adición, Propiedades de la Establecidas para muestras individuales: tipo de medición ( anexo 14.3), Medición cambio de tipo de medición, etiqueta, número, posición, peso, volumen. M ódulo C: Análisis Descripción Parámetro Archivos Permite abrir archivos de métodos, muestras y resultados Secuencia de Análisis Primera/Ultima Medición. Se establece la primera y la última medición Medición Hasta el Final. Permite medir hasta el final de la corrida Inicio en la Muestra N °. Permite seleccionar la muestra de inicio. Permite que el instrumento caliente o una HCL se estabilice. Tiempo de Retraso Elemento de Calentamiento. Permite seleccionar el elemento a calentar. Instrumento Corriente (mA). Permite seleccionar la corriente para el elemento a calentar. M ódulo D. Resultados En este módulo se muestran los datos y gráficos del análisis. Usualmente los resultados comprenden un listado, detalle del valor leído, gráfico del valor leido y absorbancia-concentración instantánea. Los datos de cada medición son agregados a la lista en forma dinámica. M ódulo E: R ep o rtes Los parámetros de reporte controlan que datos se seleccionan y la forma en que los datos son impresos o exportados a un archivo. Descripción Parámetro Análisis de Elemento Unico Permite incluir Gráfico de Calibración, Parámetros del Método, Pesos y Volúmenes y Réplicas Exportar a: Selección del puerto de salida:, COMI, C0M2, LPTI, File o Deshabilitado Preparación de Pagina Permite personalizar márgenes, cabecera y pie de pagina M ódulo F: In stru m en to Permite la instalación de componentes, optimización y diagnóstico del espectrómetro de absorción atómica y sus accesorios. - 90 - F undamentos A del n á l is is p o r A b s o r c ió n A t ó m ic a F. I Instalación de C o m p o n en te s Parámetro Descripción Modelo AAS Permite seleccionar el modelo de espectrómetro, el número de lámparas, tipo de torreta y tipo de gasbox Accesorios Horno de Grafito ■ Sistema de flujo continuo de generación de vapor. ■ Quemador Automático Rotatorio ■ Ajustador del Quemador ■ Fuente de Potencia para superlámpara Parámetros de comunicación disponibles para configurar los puertos seriales. Comunicaciones F.2 ■ Panel de Estado Proporciona capacidad de diagnóstico para complementar los Medidores de Servicios y la Optimización. Descripción Parámetro Fuente de Luz Estado de: HCL, Lámpara de deuterio y superlámpara. Lectura Continua Se proporciona lectura continua y gráficas para absorbancia y concentración de la muestra. Gasbox Indica el estado actual del bloqueo de seguridad del gasbox Escaneo de Long. de Onda Realiza el escaneo de Longitud de Onda F.3 O p tim izació n Permite rápida y conveniente optimización para los métodos de flama y horno y los accesorios. . Parámetro Descripción Alineación HC/D2 Permite la optimización para las lámparas HCL deuterio y superlámpara. Lectura Permite chequear señales de línea de base, del analito y de background, y determinar si se requiere corrección por background. Optimización de las condiciones de flama, ángulo del quemador y altura del Control de Flama cabezal para señal óptima. Permite optimizar la posición del quemador. Ajuste del Quemador F.4 Propiedades Se determina la configuración del hardware y se efectúa la operación del espectrómetro y sus accesorios. Parámetro Configuración Descripción Permite configurar como flama u horno y determinar que accesorios, entre los seleccionados en “ Instalación de Hardware", están disponibles para ser incluidos en la configuración de software y usados en el análisis. Tabla de Lámparas Permite incluir las lámparas correspondientes a los elementos seleccionados, de acuerdo al modelo de espectrómetro. Gasbox Controla el flujo de gas al quemador. Se puede seleccionar entre: “ Permitir análisis sin flama” (análisis de Hg) y “ Cerrar el gasbox al final del análisis” - 91 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a F.5 M edidores de Servicio Permiten diagnosticar y controlar la optimización del sistema óptico y el acondicionamiento de señal. Los controles permiten optimización manual y capacidad de escaneo de las fuentes de luz. Parámetro Detalle HC Muestra/HC Referencia Intensidad del rayo de muestra/referencia para la HCL D2 Muestra/O] Referencia Intensidad del rayo de muestra/referencia para la lámpara de deuterio Long. de Onda Hax/Min Limite superior/inferior para el escaneo de longitud de onda Long. de Onda Actual Longitud de onda escaneada actual que incide en el fotomultiplicador Long. de Onda del Longitud de onda a ser usada para realizar la optimización de la señal instrumento Pico de Long. de Onda Longitud de onda más cercana a la seleccionada. Corriente de Lámpara Comente de lámpara aplicada a la HCL Pico de Torreta Optimiza la posición de la lámpara moviendo la torreta de lámparas para proporcionar la máxima señal de lámpara. Ancho del Slit Fija el ancho del slit monocromador Pico del Espejo Optimiza la posición del espejo para dar máxima señal del rayo de luz. Corriente de Superlámpara Corriente aplicada a la superlámpara Pico de Superlámpara Optimiza la corriente eléctrica aplicada a la superlámpara EHT Fija la ganancia electrónica (EHT) aplicada a la señal de salida del fotomultiplicador. Corriente de Lámpara D2 Corriente eléctrica aplicada a la lámpara de deuterio Pico EHT Optimiza el EHT Pico D2 Optimiza la corriente eléctrica aplicada a la lámpara de deuterio Escaneo de Long. de Realiza el escaneo de long. de onda utilizando las long. de onda máxima y Onda mínima establecidas. Posición de Lámpara Posición de la torreta de lámparas actualmente alineada enfrente de la ventana de entrada óptica Escaneo de Intensidad Ploteo XY Intensidad de luz versus Longitud de onda, (figura 46). Status de AA Indicador de la tarea que se esta ejecutando en el instante. En la figura 45 (página siguiente), se muestra un escaneo de intensidad longitud de onda para la lámpara de cátodo hueco (H C L) del elemento plata, indicando los parámetros que corresponden. - 92 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a ESCAN EO DE INTENSIDAD Longitud de onda (nm) PARAMETROS Longitud de onda máxima Longitud de onda mínima Longitud de onda actual Longitud de onda del instrumento Ancho del slit Comente de lámpara HCL Corriente de lámpara de deuterio EHT (ganancia electrónica) 333.1 nm 323.1 nm 328.3 nm 328.3 nm 0.5 nm 4.0 mA 401 V Figura 45. Escaneo de Intensidad para una HCL de Ag Figura 45. Escaneo de intensidad para una H C L de Ag F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O XII O P T IM IZ A C IÓ N DE IN S T R U M E N T O S Y M É T O D O S 12. O p t im iz a c ió n de In st r u m en t o s y M éto do s En contraste con las técnicas que utilizan emisión óptica, la A AS tiene un parámetro que, bajo condiciones instrumentales y analíticas dadas, depende prácticamente solo de cantidades físicas; esta es la sensitividad (gradiente de la función de calibración), o sus cantidades derivadas concentración característica. La sensitividad así ofrece un criterio de control muy útil para el correcto funcionamiento de un instrumento y para optimizar un método. De otro lado, mediante una elección adecuada de parámetros del instrumento o condiciones analíticas la sensitividad puede cambiarse de modo correspondiente tal que el rango de trabajo pueda ser cambiado a concentraciones de analito más altas o más bajas. El segundo parámetro importante en un método analítico es el ruido, el cual puede también servir como un criterio de control para el correcto funcionamiento del instrumento y la ejecución adecuada del análisis. Mediante el ratio señal-ruido también se ingresa directamente a cantidades tales como la precisión y el rango de trabajo. I 2 .1 R e c o n o c im ie n t o de E r r o r e s y O p t im iz a c ió n d e l In s t r u m e n t o La AAS es una técnica relativa en la cual las determinaciones cuantitativas son realizadas por comparación con muestras de calibración (soluciones de calibración). Desde que cada cambio en los parámetros del instrumento, la condición del instrumento, y las condiciones de medición se reflejan en la calibración y son así eliminadas (excepto el drift), es todavía posible obtener resultados cuantitativos aún cuando la sensitividad esté lejos del óptimo. Sin embargo, a pesar de esta posibilidad, este modo de trabajo no es aconsejable desde que se pierde todo el control sobre la condición del instrumento. Aparte del hecho que las características de rendimiento de un análisis son peores bajo estas condiciones, el juicio de la data analítica es cuestionable desde que los cambios significativos en la sensitividad o el ruido son indicadores de mal funcionamiento del instrumento o errores en el método. Por esta razón, debe mantenerse un protocolo de las características de rendimiento de cada análisis, y cualquier desviación significativa de los valores especificados o de los valores en los protocolos anteriores debería ser una razón para investigar la causa. - 94 - Fundam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a I 2 .1.1 S e n s i t i v i d a d D e m a s i a d o B a j a Sensitividad demasiado baja: causas frecuentes en AAS: /? Errores <? calibración Pérdida de analito en las soluciones de stock o calibración debido a 0 & estabilización insuficiente o contenedores no adecuados Lámpara errónea o fallada; corriente de lámpara demasiado alta Longitud de onda incorrecta o slit demasiado ancho & de dilución cuando se preparan las soluciones de Efectos de matrix debidos ya sea al uso de un buffer o modificador equivocado, o ausencia de un buffer o modificador. Sensitividad demasiado baja: causas adicionales en F AAS: & ^ Tipo de flama equivocado o estequiometría de flama no optimizada Posición horizontal o vertical del quemador no optimizada El capilar de muestra, nebulizador, o el slot del quemador están atorados, velocidad de aspiración incorrecta Insertos no adecuados en la cámara de spray; esfera de vidrio de contacto mal alineada Sensitividad demasiado baja:, causas frecuentes en G F A A S característica demasiado alta) (masa Dispensamiento de muestra erróneo Pérdidas de analito en la preatomización; estas pueden ser debido a temperatura de pirólisis demasiado alta, a un modificador no adecuado o a la ausencia de modificador. Temperatura de atomización no adecuada (demasiado alta) Valor erróneo del blanco o de la corrección de línea base Flujo de gas demasiado alto durante la atomización o el uso de un gas de purga no adecuado. Velocidad de calentamiento demasiado lenta; esto puede ser causado por un suministro no adecuado de potencia o contacto eléctrico deficiente del tubo de grafito. Una fuerza de campo magnético demasiado baja para corrección del background por efecto Zeeman (p.e. insuficiente voltaje en el electromagneto) Sensitividad demasiado baja: causas frecuentes en H G AAS: Valor de pH no optimizado de la solución de medición Concentración errónea de la solución reductora 0 0 Falso flujo del gas transportador Tubo atomizador de cuarzo contaminado o no acondicionado F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a I 2 . 1.2 S e n s i t i v i d a d D e m a s i a d o A l t a Mucho más raro que una sensitividad demasiado baja es una sensitividad demasiado alta (masa característica demasiado baja). Esto puede ser debido casi sin excepción, especialmente en el caso de análisis de trazas, a la introducción accidental del analito. Si esto ocurre, debería hacerse una búsqueda sistemática para determinar la causa. Todos los resultados analíticos generados bajo tales condiciones, deben ser considerados con escepticismo. Precisión insuficiente o Ruido demasiado alto: causas generales en AAS & & ^ Baja energía de lampara; calidad de lámpara deficiente Corriente errónea o alineamiento deficiente de lámpara Longitud de onda errónea o ancho de slit demasiado estrecho Componentes ópticos contaminados (vidrios, lentes, espejos, etc) Ruido de emisión Deficiente alineamiento de la fuente lineal y la fuente continua para corrección por background Solución de muestra no homogénea o lodosa, la presencia de material suspendido en la solución de medición, efecto negativo para análisis de sólidos Precisión insuficiente o ruido demasiado alto: causas específicas en F AAS ¿? & ^ Nebulizador o quemador parcialmente atorado; nebulizador defectuoso Cámara de spray contaminada o insertos (gotas gruesas en la superficie son un indicador de problemas de humectación) Flujo de drenaje irregular desde la cámara de spray Flujo de gas irregular (p.e. presión de gas demasiado baja) Flama no estacionaria causada por corrientes de aire, fuerte sistema de ventilación, cambio de solvente, etc. <? Tiempo de integración demasiado insuficiente corto o amortiguación Precisión insuficiente o ruido demasiado alto: causas específicas en GF AAS & 0 Dispensamiento irreproducible de la muestra de solución a ensayar Esparcimiento de la solución causada por secado demasiado rápido 0 o incompleto Desplazamiento irregular de la solución de medición por la pared cf & de atomización (problema de atomización) Tubo de grafito o contactos envejecidos Tiempo de integración demasiado largo o ventana de integración incorrecta. - 96 - Fundamentos del A n á l is is p o r 0 A b s o r c ió n A t ó m ic a Corrección del offset de la linea base (B O C ) demasiado corta o errónea. Causas Frecuentes para el drift & & & Cambios en la intensidad radiante de la lámpara (o lámparas) o el perfil de la línea durante la fase de calentamiento o en el caso de un defecto; cambios en el ratio de las intensidades radiantes de la línea fuente y la fuente continua para corrección por background. Evaporación del solvente desde la solución de medición. Para F AAS; calentamiento o enfriamiento del quemador, o la 0 formación de incrustaciones en la ranura del quemador. Para F AAS; cambios en la velocidad de aspiración o en la eficiencia > de la nebulización debido a cambios físicos en el capilar de muestras o en el nebulizador. Para G F AAS; envejecimiento del tubo de grafito o la acumulación & de residuos (p.e. carbón de material biológico) Para H G AAS; contaminación creciente del atomizador de tubo de 0 cuarzo. Esta lista de causas frecuentes de desviaciones marcadas en las características más importantes de rendimiento solo sirven de punto de inicio para la detección de problemas. Sin embargo, debe enfatizarse que las desviaciones de esta clase son casi siempre una indicación de mal funcionamiento o erro r operacional. O tros detalles sobre posibles interferencias pueden encontrarse en las técnicas individuales . Las características de rendimiento importantes tales como sensitividad (masa característica o concentración) y el ratio señal-ruido siempre deberían ser documentadas con lo§ resultados analíticos de modo que pueda hacerse seguimiento del mal funcionamiento o errores operacionales y estos puedan ser eliminados. F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O XIII S E G U R ID A D 13. S e g u r id a d La espectrometría de absorción atómica involucra una particular combinación de riesgos potenciales. Riesgos por gases comprimidos, flamas abiertas, solventes inflamables, líquidos corrosivos, reactivos tóxicos, humos y vapores tóxicos, radiación ultravioleta y calor están vinculados estrechamente a la operación de un instrumento de absorción atómica, y las prácticas de seguridad diseñadas para laboratorios de propósito general no son completamente adecuadas para el laboratorio de absorción atómica. La técnica asimismo demanda prácticas seguras que deben observarse rigurosamente, y cada laboratorio debe originar un código de seguridad que sea adecuado para los procedimientos analíticos e instrumentales empleados. Un código adecuado de seguridad debe considerar lo siguiente: ♦ Regulaciones pertinentes promulgadas por Reglamento de Seguridad de Higiene Minera el estado, tales ♦ Normas referidas a Buenas Prácticas de Laboratorio (G LP) ♦ ♦ Información de seguridad publicadas por el fabricante. Descripciones explícitas del riesgo asociado a cada procedimiento ♦ ♦ Instrucciones precisas para evitar o minimizar cada riesgo Explicación clara de las consecuencias de no seguir la instrucción. ♦ Un procedimiento de apagado de emergencia como el Las siguientes reglas generales pueden ser útiles para esbozar un código adecuado de prácticas seguras: I 3 .1 S is t e m a d e E x t r a c c ió n Las flamas analíticas generan una considerable cantidad de calor (típicamente cerca de 500 kilojoules por minuto), y los humos y vapores producidos por algunos análisis (flama u horno) pueden ser tóxicos. El calor, los humos y vapores deben ser extraídos del instrumento por un sistema eficiente de extracción. El sistema debe ser instalado de acuerdo con las regulaciones pertinentes, debe incluir un interruptor claramente identificado y luz piloto cerca al instrumento. Siempre debe encenderse el sistema de extracción antes de encender la flama o hacer funcionar el horno. - 98 - F undamentos del 13 . 2 A n á l is is p o r A b s o r c ió n A t ó m ic a C il in d r o s d e G a s e s C o m p r im id o s ( A c e t il e n o , A r g ó n ) La ubicación correcta para todos los cilindros de gases comprimidos es fuera del laboratorio en un área de almacenaje separada bien ventilada y fría, de conformidad con los reglamentos locales y códigos de seguridad. Todos los cilindros deben ser almacenados verticalmente y firmemente asegurados a una pared u otra estructura rígida. Instale válvulas de alivio y otros dispositivos de protección contra exceso de presión. Los gases deben suministrarse al laboratorio a través de tubería permanente debidamente rotulada y con su código de color. Las válvulas de cierre para cada suministro deben estar colocadas al alcance de cada instrumento. La instalación completa debe ser rigurosamente probada en cuanto a fugas a intervalos regulares usando agua jabonosa o soluciones fabricadas para detección de fugas. I 3 .3 T u b e r ía s F l e x ib l e s d e G a s y C o n e x io n e s Longitudes cortas de tubería flexible de gas suministradas por el fabricante del instrumento son usados para conectar el instrumento al suministro permanente de gas. Debe arreglarse esta tubería flexible de modo que no pueda ser dañada, o que le caigan cosas encima debido que hasta la tubería más dura puede romperse. Solo use accesorios aprobados y asegure que todas las tuberías flexibles y conexiones accesorias estén instaladas correctamente. Idealmente, esta porción del sistema de suministro de gas debe inspeccionarse cada día. 13.4 A c e t il e n o Bajo ciertas circunstancias, el acetileno puede explotar en forma espontánea, y puede también formar acetiluros explosivos. Siempre observe estas reglas: ♦ El acetileno es un gas altamente inflamable, incoloro con un olor distintivo. ♦ La instalación de cilindros deben cumplir con las regulaciones locales. ♦ Instale válvulas de alivio y otros dispositivos de protección contra exceso de presión. ♦ Los cilindros deben colocarse en la parte externa del laboratorio. ♦ Nunca pase acetileno a través de tubería de cobre o de bronce que contenga más de 65% de cobre. ♦ No trabaje con acetileno a presiones mayores que 105 kPa (15 psi, 1.0 kg/cm2). ♦ Nunca ponga el acetileno en contacto directo con cobre, plata, mercurio líquido, gas cloro o grasa. ♦ Siempre ajuste los cilindros a la pared o alguna otra estructura en posición vertical. ♦ No permita soldadura eléctrica ni corte con llama cerca a los cilindros. ♦ Para evitar las fugas, chequee periódicamente que no ocurra fugas de gas en todas las líneas y válvulas, use agua jabonosa. - 99 - F undamentos del A 1 3 .5 n á l is is p o r A b s o r c ió n A t ó m ic a Ó x id o N it r o s o ♦ El óxido nitroso es un asfixiante, puede causar combustión espontánea del combustible, y cuando sale del cilindro el efecto de enfriamiento puede congelar el regulador de suministro. ♦ Debe probarse contra fugas el sistema de suministro de gas nitroso tan rigurosa y frecuentemente como el resto del sistema de suministro de gases. Asimismo debe usarse un regulador con un calentador incorporado, o use una cinta resistencia calentadora sobre el regulador. 1 3 .6 ♦ ♦ ♦ S o l v e n t e s In f l a m a b l e s Siempre recuerde que la combinación de una flama y un solvente inflamable es potencialmente muy peligrosa. Use un solvente que tenga el punto de flasheo más alto posible consistente con los requerimientos analíticos. Siempre use un contenedor cubierto y alimente el tubo capilar a través de un pequeño agujero en la cubierta (diámetro máximo 2 mm). ♦ Use el volumen práctico más pequeño ♦ N o permita que el solvente de desecho se acumule en la cámara de spray. Use un pequeño recipiente de plástico para desechos localizado en un área abierta y ventilada y vacíelo con frecuencia. Tabla VIII. Solventes Orgánicos comúnmente usados en AAS Solvente Punto de Flasheo °C Punto de Ebullición °C Gravedad Específica 0.79 4-metil-pentan-2-ona (MIBK) 22 118 2-metil, propan-l-ol 23 108 0.78 m-xileno 29 139 0.80 cidohexanona 34 155 0.95 39-74 175-325 0.78 kerosene 43 132 0.81 3-heptanona (etilbutilcetona) 46 148 0.82 Shellson T 50 186-214 0.75 2,6-dimetil,heptan-4-ona (DIBK) 60 166 0.81 ciclohexanol 68 161 0.96 Tetrahidronaftaleno (Tetralin) 71 207,207 0.97 alcohol isoamilico 1 3 .7 ♦ ♦ Q uem adores Nunca debe usarse un quemador aire-acetileno con óxido nitroso-acetileno. Sin embargo, puede usarse un quemador óxido nitroso acetileno con una llama aire-acetileno. N o use oxígeno o aire enriquecido con oxígeno como oxidante. 100 - F undam entos ♦ del A n á l is is po r A b s o r c ió n A t ó m ic a Nunca permita que un quemador sea bloqueado, dado que el bloqueo progresivo puede incrementar la presión en la trampa de líquidos al punto donde el sello líquido se rompa. Esto puede crear un riesgo de explosión. ♦ Siempre apague la flama antes de limpiar el la ranura del quemador ♦ Nunca deje una flama sin atención ♦ Siempre use guantes protectores cuando remueva un quemador debido a que puede estar muy caliente. 1 3 .8 ♦ T r a m p a d e L íq u id o s Una gran proporción de la solución aspirada a través del nebulizador nunca alcanza la flama y esta solución de desecho debe ser drenada sin causar fugas en el sistema quemador/cámara de spray, esto se realiza , mediante una trampa de líquidos. ♦ Asegúrese que la trampa de líquidos esta llenada al nivel correcto antes de empezar los análisis. Los instrumentos modernos tienen sistemas de bloqueo incorporados para prevenir la operación con una trampa vacía. Nunca intente desactivar tales sistemas de bloqueo. 1 3 .9 R a d ia c ió n U l t r a v io l e t a Las flamas, lámparas de cátodo hueco, lámparas de descarga sin electrodo, lámparas de deuterio y hornos analíticos pueden emitir peligrosa radiación ultravioleta. Nunca mire directamente a estas fuentes de radiación ultravioleta Siempre opere el instrumento con la puerta de protección de la flama cerrada y siempre use lentes de seguridad de un estándar aprobado. 13.1 0 P e l ig r o s d e l C a l o r Una flama abierta, un horno analítico caliente, quemadores y otras superficies calientes pueden presentar riesgos de calor a operadores no cuidadosos. Mantenga la puerta de protección de flama siempre cerrada mientras opera el instrumento. N o toque el quemador con las manos sin protección- siempre use guantes protectores. Nunca intente limpiar la ranura del quemador con la flama encendida 13.11 In s p e c c ió n d e S e g u r id a d Cada vez que inicie un trabajo de AAS, ejecute una rigurosa inspección de seguridad. Considere los siguientes aspectos: 1. Todos los controles de gas deben estar apagados. 2. Asegúrese que el área de trabajo este completamente despejada de todo 3. material riesgoso incluyendo líquidos corrosivos y solventes inflamables. Encienda el sistema de extracción y verifique su correcto funcionamiento 4. Coloque el quemador correcto. Asegure que el nebulizador este correctamente encajado y bloqueado tal como lo indica el fabricante del equipo. Si la cámara de spray incluye un dispositivo de alivio de presión, asegúrese que esté correctamente colocado. 10 1 - V F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a 5. Chequee la trampa de líquidos. Asegure que el tubo de drenaje esté 6. ensamblado en forma segura a la trampa de líquidos y que esté correctamente dirigido al recipiente de desechos. En forma similar chequee que el tubo de ventilación de vapor esté adecuadamente colocado. Llene la trampa de líquidos de acuerdo con las instrucciones del fabricante. Asegure que los cilindros correctos de gas estén colocados al sistema de 7. suministro de gas. Inspeccione todas las conexiones y tuberías de gas. Refierase a las instrucciones de fabricante y fije la presión de acuerdo a las 8. presiones especificadas. Cierre la puerta del compartimiento de muestras o protección de flama. 9. Use sus lentes de seguridad Encienda la flama como lo especifican las instrucciones del fabricante. - 102 - F u ndam entos del A n á l is is p o r A b s o r c ió n A t ó m ic a C A P Í T U L O X IV ANEXOS 14. A nexos 14.1 ■ M o d o s d e M e d ic ió n en AAS Integración. Mide el nivel de señal promedio durante el periodo de lectura, es el modo usual para los análisis por flama. ■ Altura de pico. Mide la máxima señal durante el tiempo de lectura. Este modo no es práctico para los análisis por flama ■ Area de pico. Mide el área total bajo los picos mientras dura el tiempo de lectura. Este modo tampoco es práctico para los análisis por flama. ■ Media móvil. Mide en forma continua hasta que se logra el % de precisión especificado o hasta que expira el periodo de tiempo de lectura. No se hacen mediciones duplicadas y el parámetro Precisión reemplaza al parámetro Réplicas. I 4.2 M o d o s d e C a l ib r a c ió n en AAS ■ Concentración. La calibración se basa en una curva que pasa a través del origen y todos los puntos de datos. ■ Concentración por Mínimos Cuadrados. La mejor curva de ajuste que pasa a través del origen se aplica a los datos para obtener las concentraciones de muestras. N o se fuerza a que la curva pase a través de todos los puntos de datos. ■ Mínimos Cuadrados Lineales. Una curva lineal de ajuste se aplica a los datos. ■ Mínimos Cuadrados Lineales a través de Cero. Una curva linea de ajuste se aplica a los datos. La curva es forzada a pasar a través del origen del gráfico. ■ Adiciones de Estándar. La mejor línea recta se ajusta a los datos de absorbancia de adición de estándares, luego se extrapola de modo que el intercepto negativo en el eje x se usa para obtener la concentración de la muestra. La primera muestra (adición 0) se usa para fijar el gradiente de la línea. Las concentraciones de las muestras subsecuentes son determinadas moviendo la línea arriba o abajo en el eje y. Usar una muestra diferente para la calibración inicial puede dar un conjunto diferente de resultados. Use adiciones de estándar cuando es dificultoso hacer coincidir la matriz o los problemas de interferencia lo hacen la mejor elección. ■ Estándares Envolventes. Este es un caso especial cuando solo existen dos estándares de calibración con absorbancias justo sobre y debajo de la que corresponde a las muestras. 14.3 T ip o s d e M e d ic i ó n e n AAS 103 - F undamentos del A n á l is is p o r A b s o r c ió n A t ó m ic a ■ Muestra. Realiza una medición de muestra normal. Este tipo de medición puede ser colocado en cualquier posición de la secuencia de muestras. ■ Calibración. Realiza una calibración completa usando la tabla de datos de calibración. Este tipo de puede ser colocado en cualquier posición de la secuencia de muestras, excepto después de una calibración. ■ Re-escala. Mide un estándar seleccionado (comúnmente de rango medio) como el estándar de re-escala. La diferencia entre la medición de re-escala y la calibración completa es que la medición de re-escala es utilizada para ajustar el gráfico de calibración existente para corregir cambios en sensitividad. Este tipo de medición puede ser colocada en cualquier posición de la secuencia de muestras excepto antes de la primera calibración. ■ Muestra de Chequeo. Mide la muestra de chequeo utilizando los parámetros de Muestra de Chequeo en la sección Calidad del modulo Método. Se usa como control de calidad para asegurar que el método de análisis proporcione el resultado correcto. ■ Muestra & Recuperación. Mide la muestra y luego mide la muestra recuperada y determina el % de recuperación del analito en la muestra recuperada. Ambas concentraciones son determinadas del gráfico de calibración. La concentración de muestra es sustraída de la concentración de ‘muestra + recuperación'. La concentración residual se divide entre la concentración teórica y multiplicada por 100 da el % recuperado. Generalmente la recuperación debería ser I00±2% si el método usado es eficiente. ■ Muestro Blanco. Mide la solución blanco para ajustar la línea base de las muestras subsecuentes. Se usa para corregir distorsiones instrumentales. 104 - F undam entos del A n á l is is po r A b s o r c ió n A t ó m ic a B IB L IO G R A F ÍA 1. Introducing Atomic Absorption Analysis P.A. Benett, E. Rothery Varían Techtron Pty Limited, Mulgrave, Australia First Edition, 1983 2. Atomic Absorption Spectrometry W elz, Bernhard, Sperling Michel W iley-VCH , Germany Third Edition, 1999 3. A A Accesories Service Manual - Equipo HG3000 G B C Atomic Absorption Products Febrero, 1997 Australia 4. Fíame Methods Manual for Atomic Absorption Nick Anatanasopoulos G B C Scientific Equipment Australia 5. Atomic Absorption Spectroscopy: Theory, Design and Applications. Haswell, S.J. Elsevier, Amsterdam. 1991. 6. Atomic Absorption Spectroscopy Reynolds, R.J. et al., Barnes & Noble Inc., N ew York, 1970. 7. Analytical Atomic Spectroscopy Schrenk, W .G . Plenum Press N ew York, 1975. 8. Handbook de Atomic Absorption Analysis. Vol. I. Varma, A. C R C Press, Boca Ratón, 1985 105 - A C ER C A DEL A U T O R Raúl T. Jacinto Herrera es Ingeniero Químico de la Universidad Nacional de Ingeniería, con amplia experiencia en ingeniería de procesos y análisis químico por instrumentación, lograda vía su experiencia profesional tanto en las plantas industriales de manufactura como en laboratorios de las unidades mineras peruanas. Considera bienvenidos aportes, comentarios y críticas que permitan enriquecer alguna segunda versión de esta obra. Registro Telefax: Celular: Correo: del Colegio de Ingenieros del Perú: 70670 51-1- 4334081 51-1- 8098663 rjacintoh@hotmail.com