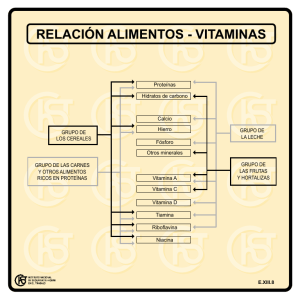
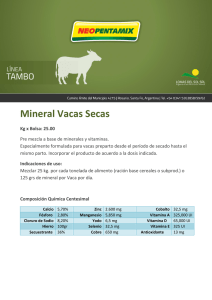
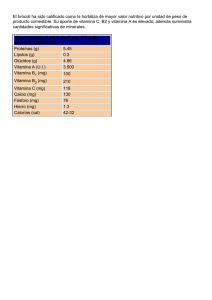
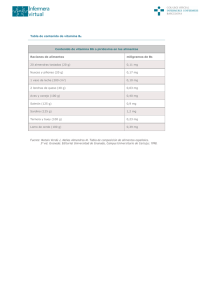
vitamina d - Montpellier
Anuncio

VITAMINA D Autores María Belén Zanchetta Especialista en Endocrinología, UBA Magister en Osteología y Metabolismo Mineral, USAL División Endocrinología Hospital de Clínicas José de San Martín Instituto de Investigaciones Metabólicas (IDIM) mbzanchetta@idim.com.ar Erich Fradinger Bioquímico. Director Técnico de Laboratorio Instituto de Investigaciones Metabólicas (IDIM) SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 1 V I TA M I N A D INDICE INTRODUCCIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 Datos históricos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 Cronología . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 Sistema Endócrino de la Vitamina D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 FISIOLOGÍA DE LA VITAMINA D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 - Bioactivación de la vitamina D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 - Metabolismo de la vitamina D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 - Transporte de la vitamina D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 - Receptor de vitamina D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 ACCIONES CLÁSICAS DE LA VITAMINA D: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 - Acción intestinal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 - Acción renal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 - Acción ósea. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 - Acción en las glándulas paratiroideas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22 ACCIONES NO CLÁSICAS DE LA VITAMINA D . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 - Vitamina D y Cáncer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24 - Modulación del Sistema Inmune . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 - Diabetes tipo 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 - Sistema renina angiotensina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 - Psoriasis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 - Acción músculo esquelética. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 VALOR ADECUADO DE VITAMINA D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 PREVALENCIA DE HIPOVITAMINOSIS D EN LA REPÚBLICA ARGENTINA . . . . . 33 RECOMENDACIONES DIARIAS Y TOXICIDAD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 CONCLUSIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37 REFERENCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 2 INTRODUCCIÓN La vitamina D es una prohormona producida en la piel a través de la irradiación por luz ultravioleta del 7- dehidrocolesterol. Es biológicamente inerte y debe ser metabolizada a 25-hidroxivitamina D3 en el hígado y luego a 1,25- dihidroxivitamina D3 en el riñón. Esta última es la forma hormonal de la vitamina D y actúa uniéndose a un receptor nuclear para llevar a cabo numerosas funciones, incluyendo la absorción de calcio y fósforo en el intestino, la movilización de calcio del hueso y la reabsorción de calcio renal. Además tiene muchas funciones no relacionadas con el metabolismo del calcio, fuente de extensas investigaciones. Esta separata propone ser una descripción de las características fisiológicas, endocrinológicas y moleculares de la Vitamina D, de sus acciones calcémicas y no calcémicas, de la importancia de sus valores adecuados y de la situación argentina con respecto a su déficit. Datos históricos El inicio de la revolución industrial en Inglaterra a finales de los años 1700, introdujo un nuevo flagelo: el raquitismo. La enfermedad propiamente dicha fue descrita primero por los médicos Daniel Whistler en 1645 y Francis Glisson en 1650, pero era relativamente poco frecuente (2,3,4). Sin embargo, en el siglo XIX, con más y más familias emigrando de la vida rural al trabajo en las fábricas de las ciudades industriales el raquitismo se convirtió en una plaga en toda Europa. Los síntomas de la enfermedad eran inconfundibles. Los huesos de los infantes afectados eran blandos, como cartílagos, y los niños tardaban en sentarse, gatear y caminar. Al crecer, sus huesos blandos se doblaban bajo el peso adicional, dejándolos con las obvias marcas del raquitismo. Los niños raquíticos también sufrían de tetania: espasmos dolorosos de las manos, los pies y la laringe, incluso con dificultad para respirar, náuseas y convulsiones. Esta condición, que más adelante se supo que se debía a una insuficiencia sintomática de calcio, a menudo era tan grave que los niños morían. Durante el siglo XIX, hubo reportes de casos esporádicos de curas para el raquitismo (1). Se empezó a observar una diferencia notable de la prevalencia de la enfermedad entre las poblaciones rurales y urbanas, adjudicándole esta asimetría a la mayor exposición solar en las zonas rurales. En 1892, el científico británico T. Palm encontró una relación entre la distribución geográfica del raquitismo y la proporción de luz solar en la región (1). Siguiendo esta línea de investigación, en 1913, H. Steenbock y E. Hart, de la Universidad de SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 3 V I TA M I N A D Wisconsin, reportaron que cabras en producción de leche mantenidas en ambientes interiores se descalcificaban, mientras que las mantenidas al aire libre no lo hacían (1). Seis años después, en 1919, el científico alemán K. Huldschinsky realizó un experimento notablemente innovador, curando niños con raquitismo utilizando luz ultravioleta producida artificialmente. Dos años después, los investigadores Alfred F. Hess y L. F. Unger de la Universidad de Columbia mostraron que con simplemente exponer a los niños raquíticos al sol, podían curarlos de la enfermedad (5). Mientras tanto, en el campo de la nutrición, el médico inglés Sir Edward Mellanby, que buscaba alguna deficiencia dietética como causa del raquitismo, decidió en 1918 experimentar con avena. Alimentó a perros exclusivamente con avena. Sin darse cuenta, también mantuvo a los animales en espacios interiores durante el experimento, y por lo tanto les indujo el raquitismo. Cuando le curó la enfermedad a los perros dándoles aceite de hígado de bacalao, Mellanby naturalmente acreditó la cura a la recientemente identificada vitamina A del aceite descripta por McCollum (6,7). Al enterarse de los experimentos de Mellanby, McCollum decidió llevarlos más adelante. En su propio trabajo de aislar la vitamina A, McCollum había encontrado que ciertos alimentos pueden contener más de una sustancia complementaria. Entonces diseñó una serie de experimentos ingeniosos para desarrollar los descubrimientos de Mellanby y descubrir qué más pudiera tener que ofrecer el aceite de hígado de bacalao. Empezó por calentar y airear el aceite para destruir su vitamina A. El aceite así tratado dejó de curar la ceguera nocturna pero continuó siendo eficaz contra el raquitismo. Aparentemente, el responsable era un nutriente esencial desconocido. En la publicación de sus experimentos en 1922, McCollum siguió la designación de vitaminas en orden alfabético y, como recientemente se había nombrado a las vitaminas B y C, llamó al nuevo milagro “vitamina D” (8). Entonces, a principios de 1920 el mundo tenía aparentemente dos curas para el raquitismo: aceite de hígado de bacalao e irradiación, o sea, exposición a luz solar o a luz ultravioleta. A pesar de esta promesa, la enfermedad continuó siendo difícil de controlar. Aunque los médicos sabían que la luz solar era esencial para los huesos de los jóvenes, las calles de las ciudades industriales seguían tan cargadas de humo y sin sol como siempre. Y no era fácil cambiar las costumbres dietéticas de la gente para que incluyan las dosis prescritas de aceite de hígado de bacalao. Luego vinieron una serie de experimentos que fusionaron los hallazgos de nutrición y los referentes a la irradiación abriendo el camino a una cura ampliamente disponible para el raquitismo. En 1923, Harry Goldblatt y Katherine Soames identificaron que cuando un precursor en la piel era irradiado con luz solar o ultravioleta, una sustancia equivalente a la vitamina liposoluble era producida (9). Alfred Hess Y Mildred Weinstock confirmaron el concepto: “la luz equivale a la vitamina D”. Cortaron un pequeño pedazo de piel, lo irradiaron con luz ultravioleta y alimentaron con ello a ratas raquíticas. Esta piel irradiada resultó en una protección total contra el raquitismo mientras que la piel no irradiada no tenía ningún efecto (10,11). 4 A principios de 1920, dos equipos de científicos, H. Steenbock y A. Black, y Hess y Weinstock, siguieron esta trayectoria de la investigación, así como la información de Huldschinsky, y encontraron que la irradiación producía una sustancia que parecía funcionar contra el raquitismo tan bien como la vitamina D del aceite de hígado de bacalao. Steenbock patentó en 1924 el proceso de irradiación de alimentos utilizando luz ultravioleta (1). Hacia 1924, la parte práctica de la batalla contra el raquitismo ya se había ganado. Los niños empezaron a consumir leche y pan irradiados y el peligro inminente de una enfermedad epidémica se redujo a un evento histórico casi olvidado. Sin embargo, la marcha hacia la comprensión de la vitamina D estaba recién empezando. Continuó la búsqueda para encontrar en los alimentos y en la piel la sustancia exacta que era activada por la irradiación ultravioleta. Varios equipos de investigadores, Steenbock y Black, de Wisconsin, Hess, Weinstock y F. Dorothy Helman de la Universidad de Columbia; y O. Rosenheim, y T. A. Webster del National Institute for Medical Research de Londres, confirmaron que la sustancia se encuentra en grasas animales y vegetales. Además, comprobaron que se encuentra en la fracción de grasas que se sabe que contiene moléculas esterólicas. Los investigadores encontraron que el colesterol purificado (un importante esterol animal) y los fitoesteroles (esteroles vegetales), ninguno de los cuales tiene propiedades antirraquíticas, se convierten en antirraquíticos por medio de radiación ultravioleta (1). El trabajo del químico orgánico Adolf Windaus en Alemania, determinó la identidad molecular de la vitamina D. Windaus aisló tres formas de la vitamina: dos derivadas de esteroles vegetales irradiados, a las que llamó D1 y D2, y una derivada de piel irradiada, a la que llamó D3. Windaus recibió el Premio Nóbel de Química en 1928 por su “investigación de la estructura de esteroles y su incumbencia con las vitaminas” (12). A principios de los años `50 se empezaron a conocer otras acciones fisiológicas de la vitamina D. El investigador sueco Arvid Carlsson hizo el descubrimiento de que la vitamina D puede en realidad quitar calcio a los huesos cuando el organismo lo requiere. Aproximadamente al mismo tiempo, el bioquímico noruego R. Nicolaysen, quien había estado estudiando diferentes dietas de animales por muchos años, concluyó que la absorción de calcio de los alimentos es controlada por un “factor endógeno” desconocido que alerta al intestino de la necesidad de calcio (1). Entre 1968 y 1971, los investigadores lograron progresar mucho en su comprensión del proceso metabólico de la vitamina D y su actividad fisiológica. En 1968, un equipo dirigido por Héctor DeLuca de la Universidad de Wisconsin aisló una sustancia identificada como 25-hidroxivitamina D3 que, según se demostró más adelante, era producida en el hígado (13). Durante los dos años siguientes, Anthony Norman y E. Kodicek, reportaron independientemente la existencia de un segundo metabolito activo. Kodicek y David R. Fraser mostraron que este segundo metabolito era producido en el riñón. Finalmente, en 1971 los tres grupos de investigación publicaron documentos en SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 5 V I TA M I N A D los que reportaron la estructura química molecular de este metabolito, al que se le identificó como 1,25-dihidroxivitamina D3 (17). Recién en la segunda mitad del siglo XX se reclasificó a la forma activa de la vitamina D como una hormona que controla el metabolismo del calcio. En 1975, Mark R. Haussler de la Universidad de Arizona reportó el aislamiento del receptor nuclear de la vitamina D. A medida que se buscaron estos receptores se fueron encontrando en múltiples tejidos del organismo, lo que abrió la puerta al fascinante universo de las acciones no calcémicas de la vitamina D (1). Al ingresar al siglo XXI, reconocemos que la investigación científica básica llevada a cabo en los dos siglos anteriores, no sólo ha desenmarañado la función de la elusiva vitamina D, sino que también nos ha proporcionado métodos para proteger la salud de adultos y de niños. Los investigadores están buscando nuevas aplicaciones para la vitamina D y sus influencias parecen cada vez más amplias. Cronología Siglo XVII Daniel Whistler (1645) y Francis Glisson (1650) describen el raquitismo por primera vez (2,3). 6 1918 Mellanby cura la enfermedad con aceite de hígado de bacalao (6,7). 1919 K. Huldschinsky cura el raquitismo utilizando luz ultravioleta producida artificialmente. 1922 E. McCollum nombra a la nueva sustancia descubierta “vitamina D” (8). 1924 Harry Goldblatt y Katherine Soames, H. Steenbock y A. Black, y Alfred Hess y Mildred Weinstock descubren independientemente que la irradiación de ciertos alimentos con luz ultravioleta los convierte en antirraquíticos (9,10,11,5). 1927 A. Windaus, O Rosenheim, y T. A. Webster deducen que el ergosterol es probablemente la sustancia madre de la vitamina D en los alimentos. 1931 F. A. Askew define la composición química de la forma de vitamina D encontrada en alimentos irradiados (ahora llamada ergocalciferol), derivada de la molécula precursora ergosterol. 1936 Windaus deduce la estructura química de la vitamina D3 producida en la piel (ahora conocida como colecalciferol), e identifica la estructura de su molécula madre, 7-dehidrocolesterol (12). 1960 El bioquímico W. Farnsworth Loomis propone que el color de la piel está determinado por las necesidades de vitamina D. A medida que el hombre se fue trasladando de regio- nes cercanas al ecuador a regiones de latitudes más altas, su piel se fue deshaciendo de la melanina para poder absorber luz ultravioleta y generar vitamina D. 1968 Héctor DeLuca y sus colaboradores aíslan un metabolito activo de la vitamina D y lo identifican como 25-hidroxivitamina D3. Más adelante demuestran que la sustancia es producida en el hígado. 1970 Los investigadores descubren la acción de la vitamina D en el sistema endócrino y en la regulación del calcio. 1971 Tres grupos de investigación identifican la estructura químico-molecular de la forma final activa de la vitamina D como 1,25-dihidroxivitamina D3, que es finalmente reclasificada como una hormona. 1975 Haussler confirma el descubrimiento de una proteína receptora que enlaza el metabolito activo de la vitamina D al núcleo de células en el intestino. 1980 El científico japonés Tatsuo Suda descubre que añadir la hormona a células malignas de leucemia incipiente, causa que las células se diferencien, maduren y detengan su crecimiento (71). Un equipo japonés de investigación e, independientemente, Michael F. Holick y sus colaboradores, muestran que la vitamina D inhibe el crecimiento de las células de la piel. 1985 Manolagas descubre que la Vitamina D puede modular el sistema inmune. Sistema Endócrino de la Vitamina D Las proteínas transmembrana que detectan la concentración plasmática de calcio (Receptor sensor de calcio) se encuentran en las glándulas paratiroideas. Cuando la calcemia baja por debajo de lo normal, estos receptores acoplados a proteína G estimulan la secreción de paratohormona (PTH). La PTH se dirige a los osteoblastos y a las células del túbulo contorneado proximal en segundos. Estas células del túbulo proximal sirven como glándula endócrina para la hormona vitamina D. En ellas la concentración de 1α-hidroxilasa está marcadamente elevada y se produce la activación de la vitamina D, formando la 1,25-dihidroxivitamina D3 o calcitriol. El calcitriol estimula la absorción intestinal de calcio y junto con la PTH estimula la movilización de calcio del hueso y la reabsorción tubular renal. El resultado de esta cascada de eventos es el aumento de la calcemia que es sensado por el mismo receptor, con lo cual se inhibe la salida de PTH frenando el estímulo para la activación renal de la vitamina D. Si la concentración de calcio sérica es demasiado alta las células C de la glándula tiroides secretan el péptido de 32 aminoácidos calcitonina, que bloquea la salida de calcio del hueso actuando sobre los osteoclastos directamente (55). SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 7 V I TA M I N A D FISIOLOGÍA DE LA VITAMINA D Verdaderamente esta “vitamina” es un esteroide que se produce en la piel por la acción solar. Es biológicamente inerte y debe pasar dos hidroxilaciones sucesivas en el hígado y el riñón para convertirse en la hormona esteroidea 1,25-dihidroxivitamina D3. Su principal función biológica es mantener la calcemia en el rango normal. Lo logra aumentando la eficiencia de la absorción intestinal del calcio ingerido con la dieta y favoreciendo junto con la hormona paratiroidea la maduración de los osteoclastos en el hueso que luego movilizarán calcio de los depósitos óseos a la circulación. Bioactivación de la vitamina D La vitamina D puede ser obtenida por la dieta o por la acción de los rayos ultravioletas en la piel. En la dieta se encuentra en los pescados oleosos como el salmón, la sardina o el aceite de hígado de bacalao. El 7-dehidrocolesterol presente en la membrana plasmática adsorbe los fotones de los rayos ultravioletas que atraviesan la epidermis. La adsorción de estas energías lo transforma en pre vitamina D3 que al isomerizarse se trasnsforma en vitamina D3. Esta es eyectada de la membrana plasmática hacia el espacio extracelular y llevada por su proteína de transporte a la red capilar dérmica. En resumen, la vitamina D es producida en la piel por un proceso fotosintético complejo, actuando sobre un derivado del colesterol -el 7-dehidrocolesterol- para producir la pre vitamina D3, que es luego lentamente isomerizada termalmente a vitamina D3 (14). Esta vitamina es la forma natural producida en la piel. Cualquier factor que ejerza influencia en el número de rayos ultravioletas que penetren la piel o la cantidad de 7-dehidrocolesterol en la misma, altera la producción de vitamina D: momento del día, estación, altitud y latitud, melanina (color de piel), uso de protectores solares y edad (162). En adultos y niños que dependan del sol para la producción de vitamina D se recomienda exposición solar directa durante 5 a 15 minutos, desde las 10 am a las 3 pm, en primavera, otoño y verano, en manos, cara y brazos o en brazos y piernas, dos a tres veces por semana. (Figura 1) La vitamina D3 deriva del 7-dehidrocolesterol y es producida en los humanos por efecto de la luz solar. La vitamina D2 deriva del ergosterol y no es producida en humanos sino que deriva del reino vegetal. En un estudio en 20 voluntarios sanos la potencia de la vitamina D2 en aumentar los niveles de 25- hidroxivitamina D sérica demostró ser un tercio de la potencia de la vitamina D3. El primer paso en la activación metabólica de la vitamina D es su hidroxilación en el carbono 25 que ocurre primariamente en el hígado. Varias enzimas hepáticas citocromo P450 son capaces de esta hidroxilación. La mejor candidata es la CYP2R1. Esta enzima citocromo P450, antes huérfana, ha demostrado ser capaz de hidroxilar las vitaminas D3 y D2, estar presente predominantemente en el 8 Figura 1: Fotosíntesis, activación y catabolismo de la Vitamina D. La hormona se produce en la piel a partir del 7-dehidrocolesterol por acción de los rayos ultravioletas para formar la pre vitamina D3, que por acción de la temperatura corporal se isomeriza a vitamina D3. hígado y en los testículos y las mutaciones en su gen han sido identificadas en un paciente con niveles bajos de vitamina D y osteomalacia (15,16). Por eso se cree que la CYP2R1 es crítica para el metabolismo de la vitamina D. Este primer paso está pobremente regulado y depende únicamente de la concentración del precursor. La tasa de hidroxilación es proporcional a la vitamina D disponible y no hay evidencia in vivo de que la 25 hidroxilación hepática sea una reacción saturable (167). Esto es diferente a todas las otras hormonas lipofílicas cuyo precursor, el colesterol, se encuentra disponible en concentraciones mucho mayores (167). El segundo paso clave en el metabolismo de la vitamina D es la hidroxilación producida por la 1αhidroxilasa que en condiciones fisiológicas ocurre principalmente en el riñón (17). Esta enzima ha sido clonada y se ha generado un ratón knock out para la misma que exhibe un fenotipo idéntico al raquitismo dependiente de vitamina D tipo 1. Otros tipos celulares pueden contribuir a los niveles circulantes en condiciones específicas (embarazo, insuficiencia renal crónica, sarcoidosis, tuberculosis, enfermedades granulomatosas y artritis reumatoidea) (14). Sin embargo, la producción extrarenal de 1,25-dihidroxivitamina D sirve principalmente como un factor autócrino o parácrino con funciones celulares especificas como veremos más adelante. La enzima 1α-hidroxilasa ha sido encontrada en muchas células y tejidos incluyendo la próstata, la mama, el colon, el pulmón, las células beta pancreáticas, los monocitos y las células paratiroideas (18). Regulación La actividad de la 1α-hidroxilasa renal está regulada muy estrechamente lo que es consecuente a la potente actividad de su producto en la homeostasis del calcio. A medida que aumentan las concen- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 9 V I TA M I N A D traciones de calcitriol, disminuyen las cantidades de la enzima 1α-hidroxilasa y aumentan las de la 24-hidroxilasa que corta la cadena lateral de la 25-hidroxivitamina D y del calcitriol. La misma 1,25dihidroxivitamina D inhibe a la hidroxilasa renal, limitando sus niveles circulantes para evitar intoxicaciones. Aunque los efectos in vivo se deben en parte al aumento de calcio y fósforo y a la consiguiente disminución de la PTH, en células de cultivo renales se ha observado una supresión directa de la 1α-hidroxilasa (32,33). La ingesta de calcio en la dieta puede regular la enzima directamente a través de cambios en la calcemia e indirectamente a través de la PTH (19). La estimulación de la 1α-hidroxilasa por la hipocalcemia es severamente debilitada aunque no eliminada por la paratiroidectomía (20). La supresión directa de la actividad de la 1α-hidroxilasa y de su ARNm por el calcio ha sido demostrada en una línea celular humana del túbulo proximal (21). Todavía no se sabe si este efecto es mediado por el receptor sensor de calcio (CaSR). Se ha demostrado que la PTH regula directamente la actividad de la 1α-hidroxilasa y de su ARNm en las células del túbulo proximal renal por medio de cambios en el AMP cíclico y a nivel de la transcripción del gen (22-27). Una dieta restringida en fósforo también aumenta la actividad de la 1α-hidroxilasa y su ARNm independientemente de cambios en la PTH y en el calcio (23,28,29,30). La falta de efecto directo del fósforo sobre la 1α-hidroxilasa en cultivos celulares sugiere que el efecto debe ser mediado por una hormona sistémica. Las candidatas son las recientemente descubiertas fosfatoninas o factores fosfatúricos: fibroblast growth factor 23 (FGF-23), frizzled-related protein 4 (FRP-4) y matrix extracellular phophoglycoprotein (MEPE) (31). El FGF-23 reduce la reabsorción renal de fósforo inhibiendo el transportador de fósforo-sodio (NaP TipoII) y aumenta con el aumento del fósforo, indicando una posible función en la homeostasis de este mineral. Todas las fosfatoninas parecen alterar las concentraciones de calcitriol en modelos animales, tal vez actuando como mediadores de la regulación por el fósforo de la 1α-hidroxilasa (14). Otro factor que parece también controlar la producción renal de calcitriol es el producto del gen Klotho. Los ratones transgénicos knock out para el gen Klotho tienen niveles elevados de calcitriol, niveles altos de calcio y fósforo y mueren prematuramente por calcificaciones ectópicas (34). El ARNm de la 1α-hidroxilasa está elevado a pesar de la hipercalcemia, hiperfosfatemia y la baja PTH, indicando que el producto del gen Klotho es un regulador negativo de la 1α-hidroxilasa (35). Este gen es también inducido por la 1,25-dihidroxivitamina D lo que sugiere que puede estar involucrado en el feedback negativo de esta hormona (35). La regulación de la 1α-hidroxilasa en los sitios extrarenales es diferente, también en consecuencia con las funciones locales autocrinas y parácrinas del calcitriol en esos sitios. No están regulados por 10 Figura 2: Bioactivación y regulación de la Vitamina D. La enzima hepática 25-hidroxilasa se encuentra pobremente regulada, mientras que la 1α-hidroxilasa lo está estrechamente por la fosfatemia, la calcemia, el FGF-23 y otros factores que pueden aumentar (flecha continua) o disminuir (flecha discontinua) su actividad. El calcitriol (1, 25-dihidroxivitamina D) disminuye su propia síntesis estimulando a la 24-hidroxilasa y también disminuyendo la síntesis y secreción de PTH. El producto del gen Klotho ha demostrado inhibir la síntesis renal de calcitriol en estudios animales. Los anticonvulsivantes, la rifampicina y los glucocorticoides aumentan el catabolismo del calcitriol estimulando la 24-hidroxilasa. productos del metabolismo mineral sino por citoquinas y factores de crecimiento, actuando por mecanismos todavía no dilucidados completamente (14). (Figura 2) Metabolismo de la vitamina D La gran potencia del calcitriol en elevar los niveles de calcio y fósforo requiere un mecanismo para atenuar su actividad. En todas sus células diana, la inactivación de la vitamina D es producida por la SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 11 V I TA M I N A D 24-hidroxilasa que cataliza una serie de reacciones oxidativas en los carbonos 24 y 23, llevando al clivaje de la cadena lateral, formando el ácido calcitroico. Los ratones sin el gen de la 24-hidroxilasa tienen acumulación de calcitriol en la circulación que produce sólo efectos secundarios en el crecimiento del cartílago, sin ningún fenotipo particular (36). El gen de la 24-hidroxilasa es inducido por la forma hormonal de la vitamina D, regulado de manera recíproca a la 1α-hidroxilasa. Su actividad y expresión están aumentados por el fósforo y disminuidos por la PTH (22,28,31). El gen contiene al menos dos distintos elementos de respuesta a la 1,25-dihidroxivitamina D que median sus efectos estimulantes sobre la transcripción (38,39). De esta manera, la vitamina D programa su propia inactivación mediante la inducción de esta enzima catabólica. La vitamina D posee más de 33 metabolitos. Actualmente se sabe que son sólo intermediarios en la degradación de la vitamina D, rápidamente eliminados e inactivos. La 25-hidroxivitamina D también puede ser metabolizada por este paso (14). Transporte de la vitamina D La vitamina D es una molécula lipofílica con baja solubilidad acuosa que necesita ser transportada en la circulación unida a proteínas. La más importante de las cuales es la DBP (vitamin D Binding Protein) a la que se une con gran afinidad (40). Sus niveles plasmáticos son 20 veces más altos que los de la vitamina D y más del 99 % de los compuestos circulantes de la vitamina D están unidos a ella aunque la albúmina y las lipoproteínas también pueden contribuir a su transporte. Esto trae un gran impacto en su farmacocinética, limitando su metabolismo hepático y eliminación renal y aumentando su vida media a casi dos meses (40). Sólo la vitamina D libre entra a las células diana y ejerce su acción (41). Los niveles de DBP no están regulados por la vitamina D. Disminuyen en la enfermedad hepática, en el síndrome nefrótico y aumentan durante el embarazo y la terapia estrogénica. La concentración de vitamina D libre se mantiene constante durante los cambios en la DBP, debido a que su metabolismo está estrictamente regulado. De hecho, los ratones knock out para la DBP no presentan ningún indicio de osteomalacia a pesar de niveles muy bajos de 25-hidroxivitamina D y 1,25-dihidroxivitamina D totales (14). La entrada de la 25-hidroxivitamina D a las células del túbulo proximal renal no es por difusión a través de la membrana basolateral como se creía sino mediada por un receptor de DBP. Esto explica por qué el ratón knock out para esta proteína de transporte es resistente a la intoxicación por vitamina D (42). Sin embargo, tampoco desarrolla deficiencia de vitamina D, lo que sugiere que también debe producirse una entrada independiente de la DBP a la célula renal. La megalina pertenece a un grupo de proteínas que facilitan la endocitosis del complejo 25-hidroxivitamina D/DBP junto con la cubilina y la proteína asociada al receptor (RAP) (43,44). Los ratones knock out para el 12 Figura 3: Rol de la megalina y de las proteínas intracelulares ligadoras de vitamina D (IDBP-3) (intracellular vitamin D binding protein) en la 1α-hidroxilación renal de la 25- hidroxivitamina D. La 25-hidroxivitamina D circula unida a la proteína ligadora de vitamina D (DBP), que es filtrada por el riñón y captada por las células del túbulo proximal mediante endocitosis mediada por la megalina. La DBP es degradada y la 25-hidroxivitamina D es entregada a la 1α-hidroxilasa por las IDBP-3 o vuelve a entrar a la circulación unida a DBP. Adaptado con permiso de Adriana Dusso Vitamin D (14). receptor endocítico megalina presentaron deficiencia de vitamina D y osteomalacia debido a la pérdida del complejo 25 hidroxivitamina D/DBP por la orina (45,46). Los niveles de megalina son aumentados por el calcitriol. Una vez dentro de la célula, la DBP es degradada, liberando la 25-hidroxivitamina D para su metabolismo por la 1α-hidroxilasa o por la 24-hidroxilasa (47). Sin embargo, el pasaje de la prehormona a la mitocondria parece también ser facilitado y no pasivo. Ha sido reportado que la megalina interactúa con al menos dos proteínas ligadores de vitamina D intracelulares: IDBP-1 e IDBP- 3 (48). Las IDBPs son homólogas a las heat shock proteins que se unen tanto a la 25-hidroxivitamina D como al estradiol y pueden cumplir diferentes roles (49). La sobre expresión de la IDBP-3 en células que expresan megalina aumenta el movimiento de la 25-hidroxivitamina D a la mitocondria para su hidroxilación (14). (Figura 3) SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 13 V I TA M I N A D Receptor de vitamina D El Receptor de 1,25-dihidroxivitamina D (VDR) es un factor de trascripción que regula la expresión de los genes responsables de su actividad biológica (50). El VDR, un péptido de 427 aminoácidos, es un miembro de la larga familia de receptores nucleares hormonales que incluye a los receptores de glucocorticoides, mineralocorticoides, hormonas sexuales, hormonas tiroideas y metabolitos de la vitamina A o retinoides. Un mismo receptor media todas las acciones de la vitamina D, incluidas las no genómicas. Estas acciones “rápidas” se producen luego de la unión al VDR ubicado en caveolas en la membrana plasmática dando origen a distintas señales o segundos mensajeros. El VDR está distribuido ampliamente y no se encuentra restringido a los tejidos considerados dianas clásicos de la vitamina D. Se ha demostrado su presencia en 36 tejidos diferentes. La acción genómica de la vitamina D consta de al menos 4 pasos (14): Primer paso: Unión del calcitriol al VDR El dominio de unión al ligando localizado en la porción COOH- terminal del VDR es responsable de la unión de alta afinidad con el calcitriol. El grupo 1· - hidroxilo del anillo A es una porción crítica de la molécula de calcitriol en esta unión. Luego de la misma, el VDR sufre una transformación conformacional en su estructura tridimensional. Este cambio es necesario para el reclutamiento de proteínas motoras responsables de una rápida traslocación del receptor del citoplasma al núcleo a través de microtúbulos (51,52). En el raquitismo dependiente de vitamina D tipo II se producen mutaciones puntuales que causan una traslocación defectuosa del citoplasma al núcleo con el conocido cuadro clínico (53,54). Segundo paso: Heterodimerización del VDR con el receptor del ácido retinoico Esta asociación, con la asociación del ácido retinoico (RXR) induce otro cambio conformacional que es esencial para la función activa del receptor. Tercer paso: Unión al ADN Unión del heterodímero a los elementos de respuesta a la vitamina D en la región promotora del ADN. El complejo VDR/RXR se une a secuencias de ADN específicas llamadas elementos de respuesta a la vitamina D (VDRE). Estos VDRE son secuencias repetidas de 6 nucleótidos separados por 3 bases no específicas (14). El brazo 5’ de esta secuencia se une al receptor X del ácido retinoico y el brazo 3’ se une al receptor de vitamina D. Cuarto paso: Reclutamiento de Coreguladores Existen una variedad de proteínas adicionales llamadas coreguladores que se unen también al complejo activado VDR/RXR para iniciar la trascripción. Los coreguladores en el complejo de preiniciación transcripcional marcadamente suprimen o aumentan la tasa de transcripción genética. Los coactivadores actúan sinérgicamente con el VDR para amplificar la potencia de la activación genéti- 14 Figura 4: Modelo actual del mecanismo de acción del receptor de vitamina D. El calcitriol circula unido a la proteína ligadora de vitamina D (DBP). Al ingresar a sus células diana se une con su receptor (VDR). Luego de unirse al receptor se heterodimeriza con el receptor del ácido retinoico (RXR), y este complejo VDR/RXR se une a regiones promotoras específicas en los genes diana. Este complejo unido al ADN atrae a los coreguladores. Adaptado con permiso de la revisión de Adriana Dusso Vitamin D (14). ca del calcitriol. Los corepresores se unen al complejo VDR-RXR para reprimir la transcripción genética, por ejemplo del gen de la PTH. El descubrimiento de estas moléculas coreguladoras contribuyó a comprender mejor la modulación positiva y negativa de la transcripción genética mediada por la vitamina D. (Figura 4) ACCIONES CLÁSICAS DE LA VITAMINA D: La principal función fisiológica de la vitamina D es mantener la calcemia en un estrecho rango normal, indispensable para la fisiología celular y para la integridad esquelética. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 15 V I TA M I N A D Figura 5: Representación esquemática del rol de la vitamina D y de la PTH en el manejo de la calcemia. El calcitriol aumenta la absorción intestinal de calcio al aumentar la expresión del canal epitelial de calcio (TRPV6) y de la calbindina (CaBP). En el hueso, actúa en los osteoblastos aumentando la expresión del receptor activator of nuclear factor-kB ligand (RANKL), que una vez unido a su receptor RANK en los pre osteoclastos promueve su diferenciación y maduración a osteoclastos. Los osteoclastos maduros remueven calcio y fósforo del hueso para mantener niveles adecuados de los mismos en la circulación (121). De esta manera la vitamina D provee los nutrientes necesarios para la correcta mineralización ósea, por lo cual su rol es de esencial importancia en el metabolismo mineral. Sin ella no hay correcta mineralización. Para esto su acción se dirige principalmente al intestino, el riñón y el hueso. El calcitriol aumenta y mantiene la calcemia mediante tres mecanismos. El primero es inducir las proteínas involucradas en la absorción intestinal de calcio. El segundo es la habilidad de mover calcio del hueso, lo que permite proteger la calcemia incluso cuando se realiza una dieta hipocálcica. Tanto la PTH como la vitamina D se necesitan para este segundo evento de movilización. De esta manera, para quitar calcio del esqueleto se necesitan dos llaves, como en una caja de seguridad. Y el tercer mecanismo es la interacción con la PTH para reabsorber el 1% de la carga filtrada renal de calcio, nuevamente actuando sinérgicamente (120). (Figura 5) 16 Una dosis única baja de vitamina D es suficiente para estimular a los enterocitos para que absorban calcio y fósforo del intestino (120). Si la concentración de calcio sérica no aumenta lo suficiente con este primer paso, las glándulas paratiroideas secretan PTH, que a su vez aumenta las concentraciones de calcitriol y ambas actúan juntas para movilizar calcio del hueso y aumentar la reabsorción renal. De esta manera, en circunstancias normales, primero se usa el calcio de la dieta y si éste no es suficiente, entonces recién se utilizan las reservas. Acción intestinal La absorción intestinal de calcio es una de las acciones primeras y mejor conocidas de la vitamina D. Ocurre principalmente en el duodeno. La hormona se vale de varias proteínas para lograr su objetivo como la calbindina (calcium binding protein, 9kD), el canal epitelial de calcio TRPV6 (transient receptor potential vainilloid type 6), la bomba calcio ATPasa y la calmodulina, entre otras. El calcio ingresa al enterocito a través de canales epiteliales específicos llamados TRPV6. Este primer paso ocurre en la membrana plasmática del borde en cepillo intestinal pasivamente favorecido por el gradiente electro químico (103). Una vez dentro de la célula es captado por la proteína calmodulina que a su vez se encuentra unida a una miosina específica intestinal (miosina 1 del borde en cepillo). Esta unión a la miosina facilita el movimiento del complejo calcio/calmodulina por la vellosidad hasta el encuentro en el citoplasma terminal con la calbindina. El movimiento del calcio a través de la célula ocurre con mínima elevación de la concentración de calcio libre intracelular. Esto es de fundamental importancia para no interrumpir el normal funcionamiento celular. La calbindina tiene mayor afinidad por el calcio que la calmodulina. La distribución diferencial de la calmodulina en la microvellosidad y la calbindina en el citosol combinada con su diferente afinidad por el calcio permite que el calcio fluya de la calmodulina en la microvellosidad hacia la calbindina en el citosol con mínimo cambio en la concentración de calcio libre en cada lugar. Finalmente la salida hacia el espacio extracelular a través de la membrana basolateral sí debe trabajar contra gradiente, el mismo gradiente que facilitó su entrada. Este último paso se logra gracias a la bomba de calcio Ca-ATPasa (103). (Figuras 6 y 7) No todos los aspectos del transporte del calcio transcelular dependen de la síntesis genómica de nuevas proteínas, también se describen acciones no genómicas. Se conoce que el calcitriol produce una respuesta bifásica en la absorción de calcio. Una respuesta rápida que ocurre entre las 2 y 6 horas y otra, mediada genómicamente, que comienza recién después de 12 horas y alcanza su pico a las 24 horas. La primera es también mediada por el mismo receptor VDR ubicado en la membrana plasmática y segundos mensajeros como el AMPcíclico. Estas acciones no genómicas involucran cambios en la composición lipídica de la membrana que la hacen más fluida y favorecen la entrada del calcio (103). SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 17 V I TA M I N A D Figura 6: Modelo del transporte intestinal de calcio. El calcio entra en las micro vellosidades intestinales a través del TRPV6. Se une a la calmodulina (CaM) que a su vez esta unida a la miosina 1 (BBM1) lo que facilita el movimiento del calcio a través de la vellosidad. Luego es recogido por la calbindina (CaBP) y transportado a través del citoplasma mediante vesículas hasta la membrana basolateral donde es expulsado por la CaATPasa (103). Figura 7: Regulación del transporte epitelial de calcio por la 1,25-dihidroxivitamina D. La hormona induce la expresión del canal apical de calcio TRVP6, la proteína calbindina CaBP que facilita el movimiento del calcio a través de la célula y la CaATPasa basolateral que bombea el calcio fuera de la célula. Adaptado con permiso de la revisión de Adriana Dusso Vitamin D (14). 18 Para una función absortiva óptima se necesitan los dos metabolitos, 25-hidroxivitamina D y calcitriol. El calcidiol parece estimular la absorción directamente probablemente a través de una respuesta rápida mediada por el receptor de membrana. Es por eso que los niveles de 25-hidroxivitamina D correlacionan directamente con la tasa de absorción del calcio. Aunque menos estudiado, el transporte intestinal de fósforo también está bajo el control del calcitriol. El transporte activo del fósforo es mayor en el yeyuno en contraste con la mayor absorción duodenal del calcio. Esta diferente predominancia resulta en mayor eficiencia absortiva de ambos minerales. El transporte de fósforo necesita de la energía de bombas de sodio tanto en su entrada por el borde en cepillo como en la salida por la membrana basolateral. Se ha clonado un transportador sodio-fósforo en el intestino delgado (NaPi-IIb), homólogo al tipo IIa en el riñón. La expresión del mismo es potenciada por la 1,25-dihidroxivitamina D quien también produce cambios en la composición de la membrana plasmática del enterocito que incrementan su fluidez (58,59). No se conocen con tanto detalle los mecanismos moleculares del proceso de absorción del fósforo ni de su pasaje a la circulación por la membrana basolateral. En un estado deficiente de vitamina D (absorción pasiva) no se incorpora más del 10 a 15% del calcio y 60% del fósforo ingeridos en la dieta. Con niveles adecuados de vitamina D los adultos pueden absorber 30-40% del calcio y 70- 80% del fósforo de la dieta. Durante períodos de crecimiento, embarazo y lactancia, con aumentos en la demanda, se puede llegar a absorber hasta un 60 u 80% del calcio ingerido (103). Por lo tanto, la vitamina D es clave para la adecuada absorción del calcio. Acción renal Diariamente, el 98% del calcio filtrado es reabsorbido, lo que indica una gran eficiencia en el manejo renal de este catión. Más de la mitad se reabsorbe en el túbulo proximal. Este primer paso está acoplado al sodio, es paracelular y posee poca o ninguna regulación por 1,25-dihidroxivitaminaD. Aproximadamente, 25% del calcio es reabsorbido en la rama gruesa ascendente de Henle, 10-15% en el túbulo distal y 5% en el colector. La acción de la vitamina D se lleva a cabo en el nefrón distal, el sitio con mayor concentración del Receptor de Vitamina D (VDR). Allí, el calcitriol acompaña la acción de la PTH (61). Las proteínas involucradas son homólogas a las intestinales: la calbindina (de 28 kDa homóloga a la de 9kDa intestinal), el TRPV5 (homólogo al TRVP6 intestinal) y la Ca-ATPasa. La calmodulina y la miosina I también han sido encontradas en el riñón pero su rol no es conocido. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 19 V I TA M I N A D El 88% del fósforo ingerido es reabsorbido en el túbulo proximal por transportadores de fósforosodio (NaPi tipo IIc) regulados por PTH. La vitamina D podría actuar también en este sector, siempre secundando la acción paratiroidea. Como la paratohormona, el FGF23 bloquea la reabsorción de fosfato produciendo down regulation de los canales de fósforo (14). Esta hormona fosfatúrica (fibroblast growth factor 23) es secretada por los osteoblastos maduros para regular el metabolismo del fósforo en una manera recíproca al calcitriol. El calcitriol y la hiperfosfatemia promueven la expresión del FGF23 en el osteoblasto. El FGF23 tiene dos acciones principales: suprime la reabsorción de fósforo por el riñón e inhibe la síntesis de calcitriol. Se dice que el FGF23 actúa como una “PTH de larga duración” (long acting PTH) previniendo la hiperfosfatemia mientras la vitamina D está encargándose de corregir la hipocalcemia. Debido a que la PTH es rápidamente suprimida por el calcitriol, éste estimula una segunda hormona fosfatúrica, FGF23, para asegurarse que el fósforo es eliminado mientras se está ganando calcio. Por eso se cuentan con dos hormonas fosfatúricas, la PTH y el FGF23, una suprimida y otra estimulada por el calcitriol (169). Cuando hay defectos en el FGF23 se producen enfermedades hiper e hipofosfatémicas. Por ejemplo, los ratones sin FGF23 presentan aumentos del calcitriol, hiperfosfatemia, calcificaciones ectópicas en los tejidos blandos y excesiva mineralización ósea. Por el contrario, la osteomalacia hipofosfatémica, producida por células malignas que expresan el FGF23 de manera constitutiva, produce hipofosfatemia severa y mineralización defectuosa. El efecto endócrino más importante del calcitriol en el riñón es el estrecho control de su propia homeostasis a través de la supresión de la 1α-hidroxilasa y de la expresión de megalina en el túbulo proximal (60). Acción ósea La acción ósea de la vitamina D es directa sobre los osteoblastos y, a través de éstos, indirecta sobre los osteoclastos. El receptor de vitamina D se encuentra en los osteoblastos. El calcitriol promueve la diferenciación de los mismos y regula la producción de proteínas como el colágeno, la fosfatasa alcalina y la osteocalcina. Los osteoblastos difieren en su respuesta al calcitriol dependiendo de su grado de maduración. Además de su rol en la promoción de la formación ósea, la 1,25-dihidroxivitamina D también promueve la resorción al aumentar el número y la actividad de los osteoclastos. No se sabe con certeza si los osteoclastos maduros poseen VDR. Sin embargo, la estimulación de la osteoclastogénesis está mediada por los osteoblastos. Estos producen una proteína de membrana conocida como RANKL (receptor activator of nuclear factor (NF)-kB ligand) que se une al RANK (receptor activator of nuclear factor (NF)-kB) en los osteoclastos y sus precursores hematopoyéticos. Este contacto célula 20 Figura 8: Acción ósea. El calcitriol aumenta la expresión del RANKL (receptor activator of NF-B RANK ligand) en la superficie del osteoblasto. La interacción del RANKL con su receptor RANK promueve la maduración de las células progenitoras del osteoclasto a osteoclastos maduros. a célula estimula la diferenciación de los precursores a osteoclastos y promueve su actividad. La 1,25-dihidroxivitamina D regula este proceso induciendo RANKL como también lo hace PTH, PGE2, e IL-11 (103). Entonces, cuando no hay suficiente ingesta de calcio para satisfacer los requerimientos corporales, la 1,25-dihidroxivitamina D estimulada por la PTH, interactúa con el VDR en los osteoblastos para inducir la estimulación osteoclástica y de esta manera disolver la matriz mineral y liberar calcio a la circulación. (Figura 8) El calcitriol también ejerce acciones sobre otros genes influyentes en el metabolismo óseo como el LRP5 (co-receptor de la vía del Wnt, esencial para la proliferación de los osteoblastos) y el Runx2 (un factor de transcripción osteoblástico que es requerido para la formación ósea). Por lo tanto, la vitamina D actuaría tanto en la formación como en la resorción ósea (120). SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 21 V I TA M I N A D La deficiencia nutricional de vitamina D, las mutaciones en la 1α-hidroxilasa renal (pseudodeficiencia de vitamina D), las mutaciones del receptor de vitamina D (raquitismo hereditario resistente a la vitamina D tipo II), tienen al raquitismo como su fenotipo principal. Esto sugeriría que la 1,25-dihidroxivitamina D es crítica para el hueso. Sin embargo, todos estos defectos en la mineralización pueden ser corregidos con suplementación adecuada de calcio y fósforo. Entonces, la función principal de la hormona en el proceso de mineralización es mantener un producto calcio-fósforo supersaturado en la circulación, de esta manera permitiendo la mineralización pasiva de la matriz colágena (osteoide) producida por los osteoblastos. La hormona no tiene otro rol activo directo en el proceso de mineralización, su función clave es mantener la calcemia y la fosfatemia en el rango normal para que se pueda producir una mineralización adecuada (103). Acción en las glándulas paratiroideas El sistema endócrino de la vitamina D es un potente modulador de la función paratiroidea. Mientras que la deficiencia de vitamina D resulta en hiperplasia paratiroidea y aumento de la síntesis y secreción de PTH, la administración de calcitriol inhibe la síntesis de PTH y el crecimiento de las células paratiroideas, siendo un tratamiento efectivo en el hiperparatiroidismo producido por la enfermedad renal crónica (62). Además de la represión directa del gen de la PTH, el calcitriol regula los niveles de su propio receptor en la glándula paratiroidea y contribuye a la respuesta de esta glándula al calcio. Esta última acción la realiza aumentando la transcripción del gen del receptor sensor de calcio, CaSR (63,64). Hay señales mitogénicas que gatillan el cambio de una célula paratiroidea normal quiescente a una proliferativa. Aumentos en la expresión de factores de crecimiento como el TGF-α (transforming growth factor α) y su receptor EGFR (epidermal growth factor receptor) median el crecimiento paratiroideo inducido por la enfermedad renal en las ratas (66,67). Los mecanismos mediante los cuales el calcitriol suprime el crecimiento celular paratiroideo incluyen: la prevención del aumento de la expresión paratiroidea de estos factores de crecimiento altamente mitogénicos y el aumento de la expresión de factores supresores del crecimiento celular, como los inhibidores de quinasas dependientes de ciclina p21 y p27 (66-68). Muchos de estos conocimientos nacieron del estudio del ratón knock out para la 1α-hidroxilasa. En éste, la normalización del calcio sérico corrige los niveles altos de PTH pero no puede suprimir la hiperplasia celular paratiroidea (69). Entonces, otra de las importantes funciones fisiológicas de la vitamina D es mantener un estado paratiroideo normal (120). Además, el hiperparatiroidismo primario tiende a ser más severo si hay déficit concomitante de vitamina D y éste parece ser más prevalente en individuos con hiperparatiroidismo primario que en la población general (170). 22 Figura 9: Esquema de las múltiples funciones no clásicas potenciales de la vitamina D. ACCIONES NO CLÁSICAS DE LA VITAMINA D Uno de los hallazgos más importantes después de la caracterización del receptor de vitamina D fue que éste no se encontraba sólo en las células consideradas diana (enterocitos, osteoblastos y células del túbulo renal distal renal) sino también en células paratiroideas, queratinocitos epidérmicos, pro mielocitos, linfocitos, células colónicas, células hipofisarias y ováricas, entre otras (70). Este descubrimiento provocó el inicio de una nueva era en la investigación de la vitamina D, conociéndose funciones no apreciadas previamente, extendiendo el sistema endócrino de la vitamina D más allá del hueso. Actualmente se conoce que directa o indirectamente, el calcitriol controla más de 200 genes, incluyendo aquellos responsables de la proliferación celular, diferenciación, apoptosis, angiogénesis y modulación del sistema inmune (72-74). ( Figura 9) SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 23 V I TA M I N A D Vitamina D y Cáncer Comparado con las poblaciones que viven más cerca del Ecuador, las poblaciones que viven en latitudes más altas tienen mayor incidencia de cáncer (90,91). En 1980, Garland & Garland hipotetizaron que esto se podía deber a una menor exposición solar (165). Actualmente, el rol protector de la vitamina D en el cáncer cuenta con gran evidencia científica especialmente en el caso del cáncer de colon (92). En varios análisis, cuando se examinaron de manera prospectiva los niveles séricos de vitamina D en relación con el riesgo de desarrollar cáncer colorectal se encontró una relación inversa. Por ejemplo, en el análisis de 193 casos de cáncer colorectal de un grupo de 32.826 enfermeras (Nurses’ Health Study cohort) se reportó que el riesgo de cáncer colorectal estaba inversamente asociado con los niveles de 25-hidroxivitamina D. El odds ratio para concentraciones séricas de vitamina D de 16.2 ng/ml era 1.0 y para concentraciones de 39.9 ng/ml era 0.53 (P≤0.01) (99). En otro estudio prospectivo en el cual 36.282 mujeres postmenopáusicas fueron asignadas a recibir 400 UI de vitamina D más 1000 mg/día de calcio o placebo se midieron los niveles basales de la vitamina. Las participantes del Women’s Health Iniciative que al momento de ingresar presentaban niveles de 25-hidroxivitamina D menores de 12 ng/ml tuvieron un aumento de 253% en el riesgo de cáncer colorectal durante un período de seguimiento de 8 años (95). Sin embargo, las mujeres asignadas a recibir tratamiento con calcio y vitamina D no tuvieron menor incidencia de cáncer colorectal al final del estudio. Las críticas al trabajo esgrimen que la dosis administrada era demasiado baja, la adherencia fue subóptima y la duración del seguimiento demasiada corta. En 1954 hombres la relación entre ingesta de vitamina D y el riesgo de cáncer colorectal mostró una correlación indirecta. Con aportes de vitamina D entre 6 y 94 UI por día el riesgo relativo era 1.0 y cuando el aporte era entre 233 y 652 UI por día el riesgo relativo era 0.53 (P<0.05) (94). En un reciente meta análisis de estudios prospectivos que analizaban la relación entre los niveles séricos de vitamina D y el cáncer colorectal, los individuos con concentraciones de 25-hidroxivitamina D≤33 ng/ml tenían una incidencia 50% menor que los que tenían niveles ≤12 ng/ml (96). Otro ensayo clínico prospectivo, randomizado, doble ciego y controlado con placebo describe el seguimiento de 1179 mujeres sanas post menopáusicas mayores de 50 años durante 4 años (127). El primer objetivo a evaluar era la incidencia de fracturas y el segundo era la incidencia de cualquier tipo de cáncer. Estas mujeres fueron seleccionadas al azar de una población rural en Nebraska (latitud 41.4°N). Se asignó un grupo a recibir 1400-1500 mg de calcio, a otro a recibir la misma cantidad de calcio más 1100 UI vitamina D3 por día y a otro a recibir placebo. La incidencia de cáncer fue menor en el grupo que recibió calcio más vitamina D (P 0.03). El riesgo relativo fue 0.402 (P 24 1er. a 4to. año Sitio Placebo (288) 2ndo. al 4to. año Calcio solo Calcio+Vit D (445) (446) Placebo (266) Calcio Solo Calcio+Vit D (416) (403) Mama 8 6 5 7 6 4 Colon 2 0 1 2 0 0 Pulmón 3 3 1 3 2 1 Hematopoyético 4 4 2 4 4 2 Útero 0 2 1 0 1 0 Otros 3 2 3 2 2 1 Total 20 (6.9%) 17 (3.8%) 13 (2.9%) 18 (6.8%) 15 (3.6%) 8 (2%) Tabla 1:Tipos de cánceres en los distintos grupos de tratamiento: placebo, calcio solo o calcio con vitamina D, entre paréntesis la cantidad de pacientes por grupo. Las primeras tres columnas indican la incidencia en la totalidad del estudio, las tres siguientes indican la incidencia entre el segundo y el cuarto año (127). 0.01). Cuando se analizaron sólo los cánceres que fueron diagnosticados luego del primer año, el riesgo relativo fue 0.232 (CI: 0.09, 0.60; P 0.005). (Tabla 1) Por último, se desarrollaron modelos animales para demostrar estas acciones de la vitamina D. Los roedores no desarrollan cáncer de colon espontáneamente. Sin embargo, cuando se los somete a una dieta denominada occidental que es rica en grasa y fósforo y baja en vitamina D, calcio, fibra y folato, 25% lo desarrollan. Si se suplementa esta dieta occidental con calcio y vitamina D disminuye significativamente la incidencia del mismo, sugiriendo que la vitamina D ejerce un rol protector Todos estos datos sugieren fuertemente un rol beneficioso de la vitamina D sobretodo en la prevención del cáncer de colon. Hay todavía muchos interrogantes como el nivel sérico óptimo para lograr la prevención, el momento de la vida donde los niveles adecuados son protectores, la exposición solar y la ingesta adecuada para lograrlo, las acciones de los distintos polimorfismos del receptor de vitamina D, entre otros. A principios de los años 80, el científico japonés Tatsuo Suda describió que al añadir calcitriol a célu- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 25 V I TA M I N A D las malignas de leucemia incipiente las células se diferenciaban, maduraban y detenían su crecimiento (71). La cantidad de hormona necesaria para detener el crecimiento de tumores es demasiado tóxica para utilizarla en seres humanos. Las acciones antiproliferativas del calcitriol serían autócrinas y parácrinas y no endócrinas (105). El colon, la próstata, la mama y otros tejidos expresan su propia 1α-hidroxilasa y producen la hormona 1,25-dihidroxivitamina D localmente para controlar los genes que ayudan a prevenir el cáncer manteniendo controlada la proliferación y diferenciación celular (97,101,102, 106,107). Si una célula se maligniza, la 1,25-dihidroxivitamina D podría inducir su apoptosis y detener la angiogénesis, reduciendo su potencial de sobrevida (102,104,108,110). Los mecanismos moleculares por los cuales la vitamina D podría realizar estas acciones son fuente de continuo estudio. Se ha reportado que el complejo calcitriol/VDR detiene el ciclo celular canceroso en la transición de G1 a G0. Lo realiza induciendo la expresión de factores inhibidores del crecimiento celular como el p21 y el p27, lo que ha demostrado ser útil para detener el crecimiento y promover la diferenciación en células del linaje monocito- macrófago (54,112). Estos inhibidores de quinasas dependientes de ciclinas (el p21 y p27) se unen a las ciclinas e inhiben su actividad, funcionando como reguladores de la progresión del ciclo celular de la fase quiescente a la proliferativa. La expresión del p21 está controlada rigurosamente por la proteína supresora de tumores p53 (113). También inhibe factores de crecimiento tumoral como TGF alfa/EGFR sobre expresados por ejemplo en los queratinocitos hiperplásicos psoriáticos (114). Por último, in vitro se ha visto que la vitamina D induce C/EBP beta, un potente supresor del oncogen de la ciclina D1 quien juega un papel clave en el desarrollo de los carcinomas epiteliales humanos (115). Además de tener la capacidad de suprimir el crecimiento celular la vitamina D puede influir en la regulación de la apoptosis. Hay hallazgos que demuestran que la vitamina D tiene propiedades tanto pro apoptóticas como anti apoptóticas que son importantes en el normal desarrollo tisular y también en la detección del crecimiento en desórdenes hiperproliferativos cancerosos y no cancerosos. Por ejemplo, en el tejido mamario normal ejerce un delicado equilibrio entre ambas funciones antagónicas, lo que es requerido para el correcto desarrollo mamario durante el embarazo, la lactancia y la involución post lactancia (116). Modulación del Sistema Inmune En 1993, S. Yang y otros científicos en el laboratorio de DeLuca encontraron que altas dosis de vitamina D administrada a ratas, las protegían de la inflamación normalmente asociada a heridas e irritantes químicos. Esta inesperada propiedad inmunosupresora de la hormona sugirió una nueva 26 y amplia gama de posibilidades, incluyendo su utilización en el control de enfermedades autoinmunes, infecciosas y en la tolerancia post trasplante (14). La primera enfermedad autoinmune que se estudió fue la esclerosis múltiple, tomando la encefalomielitis autoinmune como modelo animal. Esta enfermedad pudo ser eliminada en cualquier estadio con dosis adecuadas de vitamina D administradas diariamente vía oral (77). Sin embargo, debido a que estas dosis tan altas producen hipercalcemia severa, para el extensivo uso clínico en humanos se necesitarían análogos. En cuanto al conocimiento de los mecanismos involucrados ha habido grandes avances: Relacionado a la respuesta inmune mediada por células, se sabe que la inducción del p21 (factor inhibidor de quinasas dependientes de ciclinas) por el calcitriol es suficiente para conducir a los monocitos hacia la diferenciación en macrófagos (84). Además, induce un factor de transcripción crítico en las funciones anti bacterianas, virales y tumorales del macrófago, el C/EBP beta (85). Entonces, junto con el estímulo de la diferenciación macrofágica, la inducción de la expresión del C/EBP beta en estas células del linaje monocito-macrófago puede contribuir a la intensificación mediada por la vitamina D de la defensa del huésped contra la infección bacteriana mediada por células (86). Estas acciones se ven ejemplificadas en la infección por el Mycobacterium tuberculosis. El IFN gama, la citoquina que se eleva en proporción a la severidad de la tuberculosis, es un potente inductor de 1α-hidroxilasa macrofágica y por lo tanto de la producción local de calcitriol. En presencia de altos niveles de IFN gama, la 1α-hidroxilasa pierde la inhibición por sus productos (se pierde el feedback negativo del calcitriol sobre su propia síntesis) y también la mediada por la 24-hidroxilasa, su principal vía catabólica (14). También se ha reportado que los monocitos y macrófagos expuestos a un lipopolisacárido o al Mycobacterium tuberculosis aumentan la expresión del gen del receptor de vitamina D y del gen de la 1α-hidroxilasa (89). El aumento de la producción de 1,25-dihidroxivitamina D estimula la síntesis de la catelicidina (cathelicidin), un péptido capaz de destruir el M. tuberculosis y otros agentes infecciosos. Cuando los niveles séricos de 25-hidroxivitamina D caen por debajo de 20 ng/ml, el monocito o macrófago es prevenido de iniciar esta respuesta inmune innata, lo que podría explicar por qué los afro americanos, que tienen alta prevalencia de insuficiencia de vitamina D, son más propensos a contraer tuberculosis que los blancos y suelen tener formas más agresivas de la enfermedad (121). En contraste con estos efectos estimuladores de los monocitos y macrófagos, el calcitriol actúa como un agente inmunosupresor de los linfocitos y la respuesta humoral (87). Varias citoquinas involucradas en las funciones de las células T son diana directa del calcitriol, incluyendo la IL-2 cuya expresión génica es suprimida (122). Además, mantiene las células presentadoras de antígenos, fun- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 27 V I TA M I N A D damentales en imprimir tolerancia o inmunogenicidad en las células T, en un estado de inmadurez previniendo el desarrollo de autoinmunidad (88). Diabetes En las ratas no obesas diabéticas, la deficiencia de vitamina D causa un marcado incremento en la incidencia de la diabetes tipo 1 y disminuye el tiempo de latencia para su inicio. Altas dosis de calcitriol pueden evitar completamente el desarrollo de la enfermedad al prevenir la destrucción de las células de los islotes (78). El calcitriol modula genéticamente la expresión de calbindina, que podría controlar el eflujo intracelular de calcio en las células de los islotes, influyendo en la secreción de insulina (14). El hallazgo de actividad de 1α-hidroxilasa en estas células sugiere la posibilidad del control autócrino de la secreción insulínica por 1,25-dihidroxivitamina D (14). Varios estudios sugieren que la suplementación con vitamina D en la infancia puede reducir el riesgo de diabetes tipo 1. Un ensayo clínico realizado en Finlandia en 10.366 niños concluye que la suplementación con vitamina D se asocia a una reducción del riesgo de diabetes tipo 1. Este grupo de niños recibió 2000 UI/día de vitamina D durante el primer año de vida. Durante ese año se constató quienes lo recibían y quienes no (según el reporte de las madres). Treinta y un años después se analizó la incidencia de diabetes tipo 1 en esta población, encontrándose 2 casos de diabetes tipo 1 en los 32 pacientes que no habían recibido la suplementación durante el primer año de vida, 12 casos entre los 1210 que la habían recibido irregularmente y 67 casos entre los 9124 que la habían recibido regularmente. Estos datos indican una reducción del riesgo de casi un 80% en los niños suplementados correctamente comparado con los niños que no habían cumplido la indicación (riesgo relativo 0,22: 95% CI 0.05 a 0.89) (126). En cuanto a la diabetes tipo 2, la evidencia epidemiológica sugiere que la hipovitaminosis D podría influir en su incidencia. Se realizó un estudio prospectivo en 524 hombres y mujeres no diabéticos entre 40 y 69 años seleccionados al azar en quienes se midió 25-hidroxivitamina D, glucemia basal y a las dos horas, lípidos, insulina, antropometría y presión arterial de manera basal y a los diez años. Los niveles basales de 25-hidroxivitamina D se asociaron inversamente con el riesgo a diez años de hiperglucemia y resistencia a la insulina (163). Estos hallazgos confirman varios otros datos obtenidos en cortes transversales de muestras de pacientes con síndrome metabólico en las que se observaron correlaciones inversas entre los niveles de 25-hidroxivitamina D y la incidencia del síndrome (164). Los posibles mecanismos de estas observaciones serían efectos directos de la hormona en la función secretora de las células de los islotes a 28 través de su receptor nuclear, efectos en la sensibilidad a la insulina a través de la regulación de la expresión de su receptor por el calcio intracelular y/o efectos indirectos a través de procesos anti inflamatorios (163). Sistema renina angiotensina El sistema renina-angiotensina juega un papel central en la regulación de la presión arterial, los electrolitos y el control del volumen sanguíneo. Varios estudios epidemiológicos asociaron la hipertensión y/o la alta actividad de renina plasmática con la inadecuada exposición solar o bajas concentraciones de vitamina D (123,124). La 1,25-dihidroxivitamina D actúa como un modulador endócrino negativo del sistema renina-angiotensina (117). En el ratón knock out para el receptor de vitamina D, se observan aumentos marcados de renina y angiotensina II con hipertensión, hipertrofia cardíaca y aumento del consumo de agua (14). Psoriasis Los experimentos iniciales de Holick y sus colaboradores con la vitamina D han mostrado que la aplicación tópica de la hormona es extraordinariamente efectiva para las lesiones psoriásicas. Después de dos meses, las lesiones del 96.5% de los pacientes tratados tópicamente con una preparación de calcitriol mejoraron sin efectos secundarios perceptibles. Los queratinocitos psoriáticos hiperplásicos sobreexpresan factores de crecimiento tumoral como el Kgf/EGFR que son inhibidos por el calcitriol (114). Acción músculo esquelética En la deficiencia de vitamina D se observan anormalidades electrofisiológicas en la contracción y relajación muscular, debilidad músculo esquelética y atrofia muscular. Anteriormente se atribuían estas alteraciones a la hipocalcemia pero en modelos animales se ha comprobado la acción directa del calcitriol en el crecimiento y la diferenciación del tejido músculo esquelético (125). Se ha demostrado la presencia de VDR en el tejido muscular. En este tejido, la vitamina D activa la proteína kinasa C que promueve la liberación de calcio aumentando el pool de calcio que es esencial para la contracción muscular. En los ancianos disminuye el número de VDR y de fibras tipo II presentes en el músculo. Este tipo de fibras musculares rápidas son las más necesarias para evitar las caídas. En adul- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 29 V I TA M I N A D tos con osteomalacia las biopsias musculares muestran atrofia de fibras musculares tipo II e infiltración por grasa y fibrosis. Estos hallazgos mejoran con la suplementación con vitamina D. Lo anterior explica por qué las personas ancianas suplementadas sufren menos caídas y pueden caminar y levantarse de una silla más rápidamente (158). VALOR ADECUADO DE VITAMINA D La única manera de determinar si una persona tiene valores adecuados de vitamina D es medir la concentración sérica de 25-hidroxivitamina D. Nunca se debe usar la 1,25-dihidroxivitamina D porque a medida que la persona se vuelve deficiente hay un aumento compensatorio de la misma producido por el estímulo de PTH. El concepto de un valor adecuado o suficiente de vitamina D está en plena revisión. Diferentes categorías han sido propuestas para clasificar el estado de vitamina D. La Deficiencia de vitamina D conduce a osteomalacia, caracterizada por mineralización defectuosa del hueso (152). La Insuficiencia de vitamina D, no tan severa como para producir osteomalacia, altera la homeostasis del calcio, disminuyendo la absorción intestinal, pudiendo producir hiperparatiroidismo secundario, aumento del remodelado óseo con pérdida ósea y aumento del riesgo de fractura. La Suficiencia de vitamina D sería el estado adecuado, con efectos sólo positivos en la homeostasis del calcio. El rango de vitamina D que define estos distintos estadios está en continua discusión. La distribución de los valores de 25-hidroxivitamina D en una población, si bien típica, no puede considerarse dentro de la normalidad. Un valor de, por ejemplo, 12 ng/ml puede ser frecuente, pero no “normal”. Para establecer el valor óptimo de esta hormona se han determinado varios parámetros biológicos como el valor de vitamina D en el cual la paratohormona sérica alcanza máxima supresión, el nivel en el cual se ve correlación positiva con los niveles densitométricos de cuello femoral, los niveles requeridos para disminuir la tasa de fracturas de cadera, los niveles necesarios para que no aumenten los marcadores óseos y últimamente, se están también teniendo en cuenta las acciones no clásicas de la vitamina D. Teniendo en cuenta el valor de PTH: Los niveles de vitamina D están inversamente relacionados con los de PTH hasta que se llega a cierta concentración de la vitamina donde la PTH llega a un nadir y queda estable. Esa concentración es la indicada por los siguientes autores, entre otros, como el nivel adecuado de vitamina D: • 30 Dawson Hughes sugiere un valor de calcidiol de 44 ng/ml, valor por sobre el cual la PTH alcanza un valor óptimo (143). • Krall ha observado que las variaciones estacionales de PTH desaparecen cuando el calcidiol es 38 ng/ml (144). • Chapuy sugiere un valor de 31 ng/ml, por debajo del cual la PTH comienza a aumentar poblacionalmente (145). • Holick, Siris y colaboradores, estudiando 1536 mujeres posmenopáusicas norteamericanas que recibían tratamiento para prevenir o tratar la osteoporosis, reportan un valor de 29,8 ng/ml por debajo del cual la PTH comienza a incrementarse (146). • Oliveri en su trabajo sobre 7 ciudades argentinas reporta que fue 27 ng/ml el valor de 25-hidroxivitamina D por sobre el cual la PTH comenzó a subir (142). • Malabanan (147) sugiere que adultos de más de 49 años requieren un valor de 20 ng/ml para obtener un valor óptimo de PTH. Los pacientes con valores basales de 20 ng/ml de 25-hidroxivitamina D, luego de recibir 50.000 UI por semana de vitamina D2, no modificaron los niveles de PTH. Figura 10: Relación entre la 25-hidroxivitamina D y las concentraciones de PTH. Las concentraciones séricas de 25hidroxivitamina D se correlacionan de manera inversa con las de PTH. Datos obtenidos en 122 mujeres de la ciudad de Buenos Aires. (137) Hay que remarcar también ciertas consideraciones, como que no en todos estos estudios se ha tenido en cuenta la ingesta de calcio de los individuos, que es otro parámetro fundamental que determina los valores de PTH. Además, es necesario aclarar que estos trabajos han usado distintos métodos para medir 25-hidroxivitamina D, lo cual puede explicar parte de la gran variación en los valores de corte. La diversidad de métodos bioquímicos utilizados durante los últimos 20 años, la falta de un estándar de referencia internacional y la falta de un método de referencia de 25- hidroxivitamina D nos impone analizar en cada trabajo qué método se ha usado (153). La asociación inversa entre 25-hidroxivitamina D y PTH sérica también existe en niños y adultos SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 31 V I TA M I N A D jóvenes, siendo más estrecha que en los ancianos, requiriéndose en los últimos mayores niveles de vitamina D para lograr la misma supresión de la PTH (148-151). En conclusión, este parámetro para estimar los valores adecuados de vitamina no es el más firme. Por otro lado, no todos los individuos con baja vitamina D presentan la esperada elevación de la PTH. Sahora y colaboradores han mostrado usando un test de tolerancia al magnesio que pacientes con baja vitamina D y niveles bajos a normales de PTH tienen una deficiencia de magnesio y responden a la suplementación del mismo con aumento de la PTH. Este autor estima que aproximadamente la mitad de pacientes con déficit de vitamina D también tendrían déficit de magnesio asociado. Si esta suplementación tiene algún significado clínico aparte de desenmascarar el hiperparatiroidismo secundario se desconoce (168). Teniendo en cuenta la máxima absorción intestinal de calcio: La vitamina D suprime los valores de PTH principalmente al incrementar el influjo de calcio absorbido en el intestino. Heaney y colaboradores concluyeron que concentraciones de 25-hidroxivitamina D de 32 a 36 ng/ml son requeridas para obtener la máxima absorción intestinal de calcio (154). Es necesario destacar que en este trabajo se midió la absorción de calcio por métodos indirectos y no por el método “gold standard” que utiliza calcio radioactivo. Cuando los niveles de vitamina D se incrementan de 20 a 32 ng/ml, el transporte intestinal de calcio aumenta de 45 a 65%. La evidencia actual muestra que la absorción intestinal de calcio aumenta en relación directa al incremento de los niveles de vitamina D hasta que se alcanza un plateau aproximadamente al alcanzar los 33 ng/ml (168). Teniendo en cuenta los efectos en la masa ósea: Bischoff-Ferrari, Dawson Hughes y colaboradores reportaron una asociación positiva entre los valores densitométricos en cadera total y los niveles de 25-hidroxivitamina D entre 36 y 40 ng/ml en 13.432 hombres y mujeres mayores de 20 años que participaron del estudio NHANES III (155). Teniendo en cuenta los valores necesarios para prevenir la fractura de cadera: El mayor beneficio que puede ser obtenido de una adecuada suplementación con vitamina D es la prevención de fracturas. Chapuy demostró en una población añosa que la suplementación con calcio y vitamina D en la prevención de fractura de cadera es efectiva si se alcanza un valor de 42 ng/ml (156). Estos autores comprobaron una disminución de la incidencia de fracturas de fémur proximal, en el 43% de los tratados con dosis fisiológicas de ergocalciferol en relación a placebo. Un grupo de expertos publicó una revisión sobre varios trabajos diseñados para evaluar la prevención de fracturas con la suplementación con vitamina D (161). Los estudios que demostraron prevenir las fracturas con la suplementación presentaban valores séricos de 25-hidroxivitamina D entre 28,4 y 39,6 ng/ml, mientras que con 21,6 ng/ml no se obtuvieron resultados significativos. 32 Probablemente la suplementación con calcio también tuvo su papel en la prevención. Teniendo en cuenta sus acciones no clásicas: El campo de estudio de las acciones no clásicas de la vitamina D como vimos previamente está en continua evolución. Probablemente, todavía no haya evidencia suficiente para poder recomendar un valor de corte por debajo del cual se pierden los beneficios pleiotrópicos de esta hormona pero no parecerían ser muy diferentes a todas las otras acciones que venimos describiendo. Sí hay cierta evidencia en cuanto a los efectos en el músculo esquelético y la prevención de caídas: • Un estudio de corte transversal en 4100 personas mayores de 60 años ambulatorias sugiere que concentraciones séricas de 25-hidroxivitamina D mayores a 40 ng/ml eran necesarias para optimizar la función del músculo esquelético de las extremidades inferiores (velocidad en la caminata y velocidad en levantarse de la silla) (158). • Bischoff y colaboradores reportaron en un estudio randomizado que aumentando los niveles de vitamina D de 12 a 26 ng/ml se observaba una disminución en el número de caídas en mujeres ancianas institucionalizadas (159). • Un meta análisis de cinco estudios controlados con placebo incluyendo 1237 participantes encontró que la suplementación con vitamina D disminuyó el riesgo de la primera caída en un 22% (160). Resumiendo, el consenso actual del valor óptimo de 25-hidroxivitamina D es muy superior al tradicional corte de 10-12 ng/ml, y para la mayoría de los autores promedia los 30 ng/ml. Se considera déficit de vitamina D a los valores por debajo de 10 ng/ml e insuficiencia entre 10 y 20 ng/ml. PREVALENCIA DE HIPOVITAMINOSIS D EN LA REPÚBLICA ARGENTINA Teniendo en cuenta las anteriores definiciones, innumerables estudios han reportado que la deficiencia de vitamina D es una epidemia en todo el mundo (121). En nuestro país también existen numerosos estudios sobre los niveles de vitamina D y todos coinciden en marcar un alto porcentaje de niveles insuficientes y deficientes de vitamina D (129-142). Los niños y los ancianos son las poblaciones con más riesgo pero los jóvenes no están exentos. Seleccionamos algunos trabajos para ejemplificar la situación en nuestro país. Como era esperable, la población de ancianos institucionalizados es uno de los grupos más deficientes de esta vitamina. En el conurbano bonaerense se estudiaron 67 ancianos institucionalizados con una edad pro- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 33 V I TA M I N A D Región 25OHD ng/ml % ≤10ng/ml % 10 a 20ng/ml % ≤20 ng/ml Norte 20.6 ±7.4 2 50 48 Centro 17.8 ±8 12 52 36 14.3 ±5.6 13 75 12 Sur Tabla 2: Niveles promedio de 25-hidroxivitamina D en la población argentina de adultos mayores de 65 años según la región. Se expresan como X+- DE. Porcentajes de la población con niveles de deficiencia (menor de 10 ng/ml), insuficiencia (entre 10 y 20 ng/ml) e iguales o mayores de 20 ng/ml. Adaptado con permiso de Plantalech: Mapa de la Hipovitaminosis D en Argentina (128). medio de 81,9 años (69 a 99 años). Presentaban niveles críticos de vitamina D en invierno con promedios inferiores a 10 ng/ml, y durante el verano no superaban los 15 ng/ml. Se verificó en esa población que niveles de 25-hidroxivitamina D inferiores a 10 ng/ml se asociaban a menor densidad ósea en el radio y a remodelación ósea más alta (136). En cuanto a los jóvenes, tampoco están exentos de riesgo. En la ciudad de Buenos Aires, se evaluaron 76 hombres y mujeres de 29.8±6.6 años, mediante la medición de 25-hidroxivitamina D, paratohormona medio-molecular y marcadores del metabolismo óseo y mineral (129). En invierno 15.9% de los jóvenes presentaban valores menores de 10 ng/ml. En verano se observó un incremento de la 25-hidroxivitamina D aunque los niveles circulantes no superaron los 40 ng/ml. La remodelación ósea se incrementó en invierno. Una publicación más reciente dividió al país en tres zonas: Norte, incluyendo las provincias de Corrientes (latitud 27°) y Tucumán (latitud 26°); zona Central, incluyendo las provincias de Buenos Aires (latitud 34°) y Mendoza (latitud 32°); y zona Sur, con las ciudades de Bariloche (latitud 41°), Comodoro Rivadavia (latitud 45°) y Ushuaia (latitud 55°) (142). Se evaluaron 339 pacientes sanos ambulatorios mayores de 65 años de edad a fines del inverno y comienzos de la primavera (15 de Agosto a 15 de Octubre). Incluyeron 226 mujeres y 113 hombres. El porcentaje de personas con niveles de 25-hidroxivitamina D menor de 20 ng/ml fue 52% en la zona Norte, 64% en la zona central y 87 % en la zona Sur. (Tabla 2). Este estudio concluye que todo a lo largo de los 3700 km que abarca nuestro país, partiendo de 22°y llegando hasta 55° de latitud, se puede estimar que más de la mitad de sus habitantes sanos mayores de 65 tienen niveles insuficientes en invierno. 34 RECOMENDACIONES DIARIAS Y TOXICIDAD Este es otro tema de gran discusión. Actualmente en los Estados Unidos la dosis recomendada diaria de vitamina D es (93): • • • 200 IU/día para niños y adultos hasta los 50 años de edad, 400 IU/día para individuos de 51 a 70 años y 600 IU/día para aquellos mayores de 70 años. En Europa, recomiendan 400 IU/día en individuos mayores de 65 años (93). Expertos en el tema como R.Vieth, H. Bischoff-Ferrari, B. J Boucher, B. Dawson-Hughes, C. F Garland, R. Heaney, M. Holick, B W Hollis, C. Lamberg-Allardt, J. McGrath, A. W Norman, R. Scragg, S. J Whiting, W. C Willett y A. Zittermann coincidieron en que estas recomendaciones deben ser actualizadas según la nueva evidencia científica disponible (97). La mayoría de los expertos considera que sin una adecuada exposición solar, los niños y los adultos requieren entre 800 IU a 2000 UI de vitamina D por día (97,101,102,108). La dosis máxima tolerable (“Tolerable Upper Intake Level”) es la mayor dosis diaria que puede ser recomendada en adultos sanos para uso crónico sin que esto signifique un riesgo para la salud. En el caso de la vitamina D este riesgo se resume en la posibilidad de hipercalcemia. La dosis máxima tolerable actualmente para la vitamina D está estipulada en 2000 IU/día. Sin embargo, Reinhold Vieth hace el siguiente razonamiento: si la simple exposición solar puede proveer a un adulto una cantidad de vitamina D equivalente a la ingesta de 10000 UI, esta cantidad debería ser considerada intuitivamente una dosis segura (167). Varios estudios demuestran que esta dosis de 10000 UI/día de vitamina D3 puede ser ingerida por largos períodos de tiempo sin riesgo de causar toxicidad (109). El mecanismo que limita la seguridad de la vitamina D es la capacidad de unión a su proteína transportadora sérica y la habilidad para suprimir a la 1α-hidroxilasa. Inicialmente, a medida que aumenta la 25-hidroxivitamina D, la 1α-hidroxilasa renal produce mayor cantidad de sustrato por unidad de enzima. Si la concentración plasmática de 25-hidroxivitamina D continúa aumentando la cantidad de esta enzima es regulada en menos y aumenta la inactivación por la 24-hidroxilasa. Sin embargo, si las concentraciones de 25-hidroxivitamina D continúan aumentando (y recordemos que la 25-hidroxilación hepática no es saturable) se puede llegar un nivel en que estos mecanismos sean sobrepasados y no puedan ser regulados. Por otro lado, cuando la concentración de 25-hidroxivitamina D es inapropiadamente alta supera la capacidad de la proteína de transporte, desplazando al calcitriol y aumentando demasiado la concentración libre de este último. Esto ocurre con niveles de 25-hidroxivitamina D de al menos 240 ng/ml. No recomendaremos ningún plan específico de suplementación debido a que cada autor recomienda distintas posologías según su experiencia y los preparados comerciales que haya disponibles en SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 35 V I TA M I N A D su país, de manera artesanal. Teniendo en cuenta que para aumentar 4 ng/ml la concentración de calcidiol deben darse 400 UI diarias, si tenemos un paciente por ejemplo con 10 ng/ml de 25-hidroxivitamina D y queremos llevarlo a valores óptimos de 30 ng/ml deberá recibir 2000 UI/día de vitamina D3 o la D2 equivalente (alrededor de 6000 UI). Algunos números a tener en cuenta: • Cada 40 UI /día de vitamina D3 aumenta 0.4 ng/ml la concentración de 25-hidroxivitamina D • 1nM equivale a 0.4 ng/ml. • Los niveles deseables actuales están cercanos a 30 ng/ml. 36 CONCLUSIÓN La exposición solar y la síntesis de vitamina D fueron claves en la evolución de los vertebrados terrestres. A medida que los animales dejaban el océano y se aventuraban en la tierra necesitaban un sistema hormonal que les permitiera adaptarse al nuevo ambiente pobre en calcio. Garantizar la demanda de calcio era fundamental por su importancia en las necesidades metabólicas y para la rigidez del esqueleto que permite la deambulación (171). Es por eso que hay quienes dicen que la vitamina D puede ser una de las hormonas más antiguas. Millones de años después, a medida que se poblaban regiones más lejanas al Ecuador la pigmentación de la piel fue disminuyendo para permitir la suficiente formación de vitamina D para mantener la homeostasis del calcio y un esqueleto saludable. Sin embargo, debido al estilo de vida actual la hipovitaminosis D tiene una alta prevalencia en la población en general, siendo de particular riesgo en los niños y las personas de mayor edad. No sólo produce raquitismo en los niños, y osteomalacia y osteoporosis en adultos, sino que puede tener efectos negativos sobre la salud a largo plazo. Las posibles consecuencias del déficit crónico incluyen riesgo aumentado de sufrir enfermedades autoinmunes, hipertensión arterial y algunos tipos de cáncer. Aunque es difícil obtener adecuadas cantidades de la dieta y la exposición solar tiene sus riesgos, la suplementación es simple y segura. Evidentemente, la historia de la vitamina D no terminó cuando se encontró la cura del raquitismo. Ha habido importantes descubrimientos sobre sus mecanismos de acción y lo más sorprendente es sin duda la posible implicancia de sus acciones no calcémicas. Hasta parece que en algunos aspectos, la historia de esta hormona está por empezar. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 37 V I TA M I N A D REFERENCIAS 1. National Academy of Sciences. “Unraveling the Enigma of Vitamin D: Closing in on Rickets.” University of California, Riverside. “History of Vitamin D.” 2. Whistler, D. Morbo puerili Anglorum, quem patrio idiomate indigenae vocant. The Rickets. Lugduni Batavorum 1-13 (1645). 3. Glisson, F. De Rachitide sive morbo puerili, qui vulgo. The Rickets diciteur, London 1-416 (1650). 4. Glisson, F. A treatise of the rickets being a disease common to children. London 1-373 (1668). 5. Hess, A. Influence of light on the prevention of rickets. Lancet 2:1222 (1922). 6. Mellanby, E. and Cantag, M.D. Experimental investigation on rickets. Lancet 1919 196:407-412 . 7. Mellanby, E. Experimental rickets. Medical Research (G.B.), Special Report Series1921 SRS-61:1-78 . 8. McCollum, E.V., Simmonds, N., Becker, J.E. and Shipley, P.G. Studies on experimental rickets. XXI. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J. Biol. Chem. 53:293-312 (1922). 9. Goldblatt, H. and Soames, K.N. A study of rats on a normal diet irradiated daily by the mercury vapor quartz lamp or kept in darkness. Biochem. J.1923 17:294-297. 10. Steenbock, H. and Nelson, M. T. Fat-soluble vitamins. XIX. The induction of calcifying properties in a rickets-producing ration by radiant energy. Methods Enzymol. 62:209-216 (1924). 11. Steenbock, H. The induction of growth promoting and calcifying properties in a ration by exposure to light. Science 60:224-225 (1924). 12. Windaus, A., Linsert, O. Luttringhaus, A. and Weidlinch, G. Uber das krystallistierte Vitamin D2. Justis. Liebigs. Ann. Chem. 492:226-231 (1932). 13. Morri H, Lund, De Luca. Biological activity of a Vitamin D metabolite. Arch Biochem Biophys. 1967; 120508-12. 14. Dusso A, Brown A , Slatopolsky E. Vitamin D.Am. J Renal Physiol, 2005; 289: F8 - F28. 15. Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. Deorphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxylase. J Biol Chem 2003 278: 38084–38093. 16. Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci 2004 101: 7711–7715. 17. Fraser DR,Kodicek E. Unique biosynthesis by kidney of a biological active vitamin D metabolite. Nature1970 228: 764–766. 18. Hewison M, Zehnder D, Chakraverty R, Adams JS. Vitamin Dand barrier function: a novel role for extrarenal 1α-hydroxylase. Mol Cell Endocrinol 2004 215: 31–38. 38 19. Omdahl JL, Gray RW, Boyle IT, Knutson J, DeLuca HF. Regulation of metabolism of 25hydroxycholecalciferol by kidney tissue in vitro by dietary calcium. Nat New Biol 1972 237: 63–64. 20. Garabedian M, Holick MF, Deluca HF, Boyle IT. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc Natl Acad Sci 1972 69: 1673–1676. 21. Bland R, Hughes SV, Stewart PM, Hewison M. Direct regulation of 25-hydroxyvitamin D3 1α-hydroxylase by calcium in a human proximal tubule cell line [Abstract]. Bone1998 23: S260. 22. Henry HL, Norman AW. Vitamin D: metabolism and biological actions. Annu Rev Nutr 1984 4: 493–520. 23. Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca HF, Suda T. Cloning and expression of rat 25-hydroxyvitamin D3-1α-hydroxylase cDNA. Proc Natl Acad Sci 1997 94: 12920– 12925. 24. St.-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH. The 25-hydroxyvitamin D 1-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res 1997 12: 1552–1559. 25. Rost CR, Bikle DD, Kaplan RA. In vitro stimulation of 25- hydroxycholecalciferol 1-hydroxylation by parathyroid hormone in chick kidney slices: evidence for a role for adenosine 3_,5_-monophosphate. Endocrinology 1981 108: 1002–1006. 26. Brenza HL, Kimmeljehan C, Jehan F, Shinki T, Wakino S, Anzawa H, Suda T, DeLuca HF. Parathyroid hormone activation of the 25-hydroxyvitamin D3 1α-hydroxylase gene promoter. Proc. Natl. Acad. Sci. (1998) USA 95:1387-1391. 27. Murayama A, Takeyama K, Kitanaka S, Kodera Y, Hosoya T, Kato S. The promoter of the human 25hydroxyvitamin D3 1-hydroxylase gene confers positive and negative responsiveness to Pth, calcitonin, and 1-_,25(OH)2D3. Biochem Biophys Res Commun 1998 249: 11–16. 28. Tanaka Y, Deluca HF. The control of 25-hydroxyvitamin D metabolism by inorganic phosphorus. Arch Biochem Biophys 1973 154: 566–574. 29. Hughes MR, Brumbaugh PF, Hussler MR, Wergedal JE, Baylink DJ. Regulation of serum 1_,25dihydroxyvitamin D3 by calcium and phosphate in the rat. Science 1975 190: 578–580. 30. Bushinsky DA, Nalbantian-Brandt C, Favus MJ. Elevated Ca2 does not inhibit the 1,25(OH)2D3 response to phosphorus restriction. Am J Physiol Renal Fluid Electrolyte Physiol 1989 256: F285–F289. 31. Schiavi SC, Kumar R. The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int 2004: 1–14. 32. Henry HL. Regulation of the hydroxylation of 25-hydroxyvitamin D3 in vivo and in primar y cultures of chick kidney cells. J Biol Chem1979 254: 2722–2729. 33. Trechsel U, Bonjour JP, Fleisch H. Regulation of the metabolism of 25-hydroxyvitamin D3 in primary cultures of chick kidney cells. J Clin Invest1979 64: 206–217. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 39 V I TA M I N A D 34. Yoshida T, Fujimori T, Nabeshima Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1_-hydroxylase gene. Endocrinology 2002 143: 683–689. 35. Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol2003 17: 2393–2403. 36. St.-Arnaud R, Arabian A, Glorieux FH. Abnormal bone development in mice deficient for the vitamin D24-hydroxylase gene. J Bone Miner Res 1996 11: S126. 37. Wu S, Finch J, Zhong M, Slatopolsky E, Grieff M, Brown AJ. Expression of the renal 25-hydroxyvitamin D-24-hydroxylase gene: regulation by dietary phosphate. Am J Physiol Renal Fluid Electrolyte Physiol 1996 271: F203–F208. 38. Chen KS, DeLuca HF. Cloning of the human 1,25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim Biophys Acta 1995 1263: 1–9. 39. Ohyama Y, Ozono K, Uchida M, Shinki T, Kato S, Suda T, Yamamoto O, Noshiro M, Kato Y. Identification of a vitamin D-responsive element in the 5_-flanking region of the rat 25-hydroxyvitamin D3 24hydroxylase gene. J Biol Chem 1994 269: 10545–10550. 40. Cooke NE Haddad JG. Vitamin D binding protein. Endocr Rev 1989 10: 294–307. 41. Bikle DD, Gee E. Free, and not total, 1,25-dihydroxyvitamin D regulates 25-hydroxyvitamin D metabolism by keratinocytes. Endocrinology 1989 124: 649–654. 42. Safadi F, Thornton P, Magiera H, Hollis B, Gentile M, Haddad J, Liebhaber S,Cooke N. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest 1999 103: 239–251. 43. Birn H, Vorum H, Verroust PJ, Moestrup SK, Christensen EI. Receptor-associated protein is important for normal processing of megalin in kidney proximal tubules. J Am Soc Nephrol 2000 11: 191–202. 44. Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS,Verroust PJ, Aminoff M, de la Chapelle A, Moestrup SK, Ray R, Gliemann J, Willnow TE, Christensen EI. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D3. Proc Natl Acad Sci 2001 98: 13895–13900. 45. Nykjaer A, Dragun D, Walther D, Vorum H, Jacobsen C, Herz J, Melsen F, Christensen E, Willnow T. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell1999 96: 507–515. 46. Takemoto F, Shinki T, Yokoyama K, Inokami T, Hara S, Yamada A, Kurokawa K, Uchida S. Gene expression of vitamin D hydroxylase and megalin in the remnant kidney of nephrectomized rats. Kidney Int 2003 64: 414–420. 47. Yamane T, Takeuchi K, Yamamoto Y, Li YH, Fujiwara M, Nishi K, Takahashi S, Ohkubo I. Legumain from bovine kidney: its purification, molecular cloning, immunohistochemical localization and degradation of annexin II and vitamin D-binding protein. Biochim Biophys Acta 2002 1: 108–120. 40 48. Adams JS, Chen H, Chun RF, Nguyen L, Wu S, Ren SY, Barsony J,Gacad MA. Novel regulators of vitamin D action and metabolism: lessons learned at the Los Angeles Zoo. J Cell Biochem 2003 88: 308–314. 49. Gacad MA, Adams JS. Proteins in the heat shock 70 family specifically bind 25-hydroxyvitamin D-3 and 17-_-estradiol. J Clin Endocrinol Metab1998 83: 1264–1267. 50. Brown AJ, Dusso A, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol 1999 277: F157–F175. 51. Racz A, Barsony J. Hormone-dependent translocation of vitamin D receptors is linked to transactivation. J Biol Chem 1999 274: 19352–19360. 52. Barsony J, McKoy W. Molybdate increases intracellular 3,5 guanosine cyclic monophosphate and stabilizes vitamin D receptor association with tubulin-containing filaments. J Biol Chem1992 267: 24457– 24465. 53. Barsony J, Renyi I, McKoy W. Subcellular distribution of normal and mutant vitamin D receptors in living cells. Studies with a novel fluorescent ligand. J Biol Chem1997 272: 5774–5782. 54. Hewison M, Rut AR, Kristjansson K, Walker RE, Dillon MJ, Hughes MR, O’Riordan JL. Tissue resistance to 1,25-dihydroxyvitamin D without a mutation of the vitamin D receptor gene. Clin Endocrinol (Oxf)1993 39: 663–670. 55. Blunt JW, DeLuca HF, Schnoes HK. 25-Hydroxycholecalciferol: a biologically active metabolite of vitamin D3. Biochemistry 1968;7:3317–22. 56. Holick MF, Schnoes HK, DeLuca HF, Suda T, Cousins RJ. Isolation and identification of 1,25dihydroxycholecalciferol: a metabolite of vitamin D active in intestine. Biochemistry 1971;10:2799–804. 57. Semmler EJ, Holick MF, Schnoes HK, DeLuca HF. The synthesis of 1_,25-dihydroxycholecalciferol: a metabolically active form of vitamin D3. Tetrahedron Lett 1972 ;40:4147–50. 58. Yagci A, Werner A, Murer H, Biber J. Effect of rabbit duodenal mRNA on phosphate transport in Xenopus laevis oocytes: dependence on 1,25-dihydroxy-vitamin-D3. Pflu¨gers Arch 1992 422: 211–216. 59. Kurnik BR ,Hruska KA. Mechanism of stimulation of renal phosphate transpor t by 1,25dihydroxycholecalciferol. Biochim Biophys Acta 1985 817: 42–50. 60. Liu W, Yu WR, Carling T, Juhlin C, Rastad J, Ridefelt P et al. Regulation of gp330/megalin expression by vitamins A and D. Eur J Clin Invest 1998 28: 100–107. 61. Friedman PA , Gesek FA. Vitamin D3 accelerates PTH-dependent calcium transport in distal convoluted tubule cells. Am J Physiol Renal Fluid Electrolyte Physiol 1993 265: F300–F308. 62. Dusso AS, Thadhani R, Slatopolsky E. Vitamin D receptor and analogs. Semin Nephrol 2004 24: 10–16. 63. Brown AJ, Zhong M, Finch J, Ritter C, McCracken R, Morrissey J, Slatopolsky E. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am J Physiol Renal Fluid Electrolyte Physiol 1996 270: F454–F460. 64. Canaff L, Hendy GN. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin. D. J Biol Chem 2002 277: 30337–3035. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 41 V I TA M I N A D 65. Kollenkirchen U, Fox J, Walters MR. Normocalcemia without hyperparathyroidism in vitamin D-deficient rats. J Bone Miner Res 1991 6: 273–278. 66. Cozzolino M, Lu Y, Finch J, Slatopolsky E, Dusso AS. p21WAF1 and TGF-_ mediate parathyroid growth arrest by vitamin D and high calcium. Kidney Int 2001 60: 2109–2117. 67. Dusso AS, Pavlopoulos T, Naumovich L, Lu Y, Finch J, Brown AJ, Morrissey J, Slatopolsky E. p21WAF1 and transforming growth factor-_ mediate dietary phosphate regulation of parathyroid cell growth. Kidney Int 2001 59: 855–865. 68. Tokumoto M, Tsuruya K, Fukuda K, Kanai H, Kuroki S, Hirakata H. Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism. Kidney Int 2002 62: 1196–1207. 69. Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, Goltzman D. Inactivation of the 25-hydroxyvitamin D 1_-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem 2004 279: 16754–16766. 70. Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev 1998;78:1193–231. 71. Abe E, Miyaura C, Sakagami H, Takeda M, Konno K,Yamazaki T,Yoshiki S, Suda T. Differentiation of mouse myeloid leukemia cells induced by 1_,25-dihydroxyvitamin D3. Proc Natl Acad Sci 1981 78: 4990–4994. 72. Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest 2006; 116:2062-72 73. Holick MF, Garabedian M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. In: Favus. MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 6th ed. Washington, DC: 2006 ASBMR:129-37. 74. Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev 2005; 26:662-87. 75. Yang S, Smith C, Prahl JM, DeLuca HF. Vitamin D deficiency suppresses cell-mediated immunity in vivo. Arch Biochem Biophys 1993; 303:98 –106. 76. Yang S, Smith C, DeLuca HF. 1_,25-Dihydroxyvitamin D3 and 19-nor- 1_,25-dihydroxyvitamin D2 suppress immunoglobulin production and thymic lymphocyte proliferation in vivo. Biochim Biophys Acta 1993; 1158:269–86. 77. Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA 1996; 93:7861– 4. 78. 79. Zella JB, DeLuca HF. Vitamin D and autoimmune diabetes. J Cell Biochem 2003;88:216-22. Lemire JM, Ince A, Takashima M. 1,25-Dihydroxyvitamin D3 attenuates the expression of experimental murine lupus of MRL/1 mice. Autoimmunity 1992; 12:143– 8. 80. Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. JNutr 2000;130:2648 –52. 42 81. Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 prevents and ameliorates symptoms in two experimental model of human arthritis. J Nutr 1998;128:68 –72. 82. Hayes CE, Nashold FE, Spach KM, and Pedersen LB. The immunological functions of the vitamin D endocrine system. Cell Mol Biol (Noisy-le-grand) 2003; 49: 277–300. 83. Adorini L, Penna G, Giarratana N, Roncari A, Amuchastegui S, Daniel KC, Uskokovic M. Dendritic cells as key targets for immunomodulation by vitamin D receptor ligands. J Steroid Biochem Mol Biol 2004; 89: 437–441. 84. Liu M, Lee MH, Cohen M, Bommakanti M, Freedman LP. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev 1996; 110: 142–153. 85. Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBP_ gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J Immunol 2002;168: 4055–4062. 86. Ji Y, Studzinski GP. Retinoblastoma protein and CCAAT/enhancerbinding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res 2004; 64: 370–377. 87. Mathieu C, Adorini L. The coming of age of 1,25-dihydroxyvitamin D3 analogs as immunomodulatory agents. Trends Mol Med 2002; 8: 174–179. 88. Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI, Piazza F, Lederman S, Colonna M, Cortesini R, Dalla-Favera R, Suciu-Foca N. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol 2002; 3: 237–243. 89. Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006;311:1770-3. 90. Apperly FL. The relation of solar radiation to cancer mor tality in Nor th America. Cancer Res 1941;1:191-5. 91. Chang ET, Smedby KE, Hjalgrim H, et al. Family history of hematopoietic malignancy and risk of lymphoma. J Natl Cancer Inst 2005;97:1466-74. 92. Zittermann A. Vitamin D in preventive medicine: are we ignoring the evidence? Br J Nutr 2003;89: 552–572. 93. Standard for vitamin D. WHO Technical Report Series. Geneva, Switzerland: World Health Organization, 1950. 94. Gorham ED, Garland CF, Garland FC, et al. Vitamin D and prevention of colorectal cancer. J Steroid Biochem Mol Biol 2005; 97:179-94. 95. 96. Holick MF. Calcium plus vitamin D and the risk of colorectal cancer. N Engl J Med 2006;354:2287Garland CF, Garland FC, Gorham ED, et al. The role of vitamin D in cancer prevention. Am J Public Health 2006;96:252- SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 43 V I TA M I N A D 97. Vieth R. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007 98. Giovannucci E, Liu Y, Rimm EB, et al. Prospective study of predictors of vitamin D status and cancer incidence and mortality in men. J Natl Cancer Inst 2006;98: 451-9. 99. Feskanich D, Ma J, Fuchs CS, et al. Plasma vitamin D metabolites and risk of colorectal cancer in women. Cancer Epidemiol Biomarkers Prev 2004;13:1502-8. 100. Berwick M, Armstrong BK, Ben-Porat L, et al. Sun exposure and mortality from melanoma. J Natl Cancer Inst 2005;97:195-9 101. Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest 2006; 116:2062-72. 102. Bouillon R. Vitamin D: from photosynthesis, metabolism, and action to clinical applications. In: DeGroot LJ, Jameson JL, eds. Endocrinology. Philadelphia: W.B. Saunders, 2001:1009-28. 103. Bikle DD. Vitamin D: production, metabolism and mechanisms of action. Endotext 2006. 104. Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc 2006;81:353-73. 105. Huang DC, Papavasiliou V, Rhim JS, Horst RL, Kremer R. Targeted disruption of the 25-hydroxyvitamin D3 1_-hydroxylase gene in ras-transformed keratinocytes demonstrates that locally produced 1,25dihydroxyvitamin D3 suppresses growth and induces differentiation in an autocrine fashion. Mol Cancer Res 2002 1: 56–67. 106. Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev 2005;26:662-87. 107. Grant WB. An estimate of premature cancer mortality in the U.S. due to inadequate doses of solar ultraviolet-B radiation. Cancer 2002;94:1867-75. 108. Holick MF, Garabedian M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. In: Favus MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 6th ed. Washington, DC: 2006 ASBMR:129-37. 109. 110. Hathcock j, Shao A, Vieth R, Heaney R. Risk assessment for vitamin D. Am J Clin Nutr 2007;85:6 –18. Mantell DJ, Owens PE, Bundred NJ, Mawer EB, Canfield AE. 1· ,25-dihydroxyvitamin D3 inhibits angiogenesis in vitro and in vivo. Circ Res 2000;87:214-. 111. Kontula K, Valimaki S, Kainulainen K, Viitanen AM, Keski-Oja J. Vitamin D receptor polymorphism and treatment of psoriasis with calcipotriol. Br J Dermatol 1997;136: 977–978. 112. Luo W, Chen J, Zhang X, Wang W. Regulatory mechanism of EB1089 for hepatocarcinoma cell proliferation. Wei Sheng Yan Jiu 2004;33: 140–143. 113. 44 ncbi.nlm.nih.gov.gene home 114. Cordero JB, Cozzolino M, Lu Y, Vidal M, Slatopolsky E, Stahl PD, Barbieri MA, Dusso A. 1,25Dihydroxyvitamin D downregulates cell membrane growth- and nuclear growth-promoting signals by the epidermal growth factor receptor. J Biol Chem 2002;277: 38965–38971. 115. Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS et al. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 2003;114: 323–334. 116. Zinser, Welsh J. Accelerated mammary gland development during pregnancy and delayed postlactational involution in vitamin D3 receptor null mice. Mol Endocrinol 2004;208–222. 117. Li Y. Vitamin D regulation of the renin-angiotensin system. J Cell Biochem 2003;88:327-31. 118. Chiu KC, Chu A, Go VLW, Saad MF. Hypovitaminosis D is associated with insulin resistance and ‚ cell dysfunction. Am J Clin Nutr 2004;79:820-5. 119. Zittermann A. Vitamin D and disease prevention with special reference to cardiovascular disease. Prog Biophys Mol Biol 2006;92:39-48. 120. DeLuca H. Overview of general physiologic features and functions of Vitamin D. Am J Clin Nutr 2004:80 suppl: 1689-96 121. Holick M. Vitamin D deficiency. N Engl J Med 2007;357:266-81. 122. Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol 1995; 15: 5789–5799. 123. Li Y. Vitamin D regulation of the renin-angiotensin system. JCell Biochem2003;88:327–331 124. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25- Dihydroxyvitamin D3 is a negative endocrine regulator of the reninangiotensin system. J Clin Invest 2002 110: 229–238. 125. Endo I, Inoue D, Mitsui T, Umaki Y, Akaike M, Yoshizawa T et al. Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology 2003 144: 5138–5144. 126. Hypponen E, Laara E, Reunanen A, Jarvelin M-R, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birthcohort study. Lancet 2001;358:1500. 127. J Lappe, D Travers-Gustafson, K Ml Davies, R. Recker, P Heaney. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007;85:1586 –91. 128. Plantalech L. Mapa de la Hipovitaminosis D en Argentina. Actualizaciones en Osteologia 2005: Vol 1: 11-15. 129. Fassi J, Russo Picasso MF, Furcia A et al. Variaciones estacionales de 25-hidroxivitamina D en jóvenes y ancianos de la ciudad de Buenos Aires. Medicina (B. Aires), mayo/junio 2003, vol.63, no.3, p.215-220. 130. Oliveri MB, Ladizesky M, Mautalen CA, Alonso A, Martinez L. Seasonal variations of 25hydroxy-vitamin D and parathyroid hormone in Ushuaia (Argentina), the southernmost city of the world. Bone Miner 1993; 20: 99-108. SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 45 V I TA M I N A D 131. Oliveri B, Plantalech L, Bagur A, et al. High prevalence of vitamin D insufficiency in healthy elderly people living at home in Argentina. Eur J Clin Nutr 2004; 58: 337-42. 132. Oliveri MB, Ladizesky M, Somoza J, Martinez L, Mautalen CA. Winter serum levels of 25- hydroxyvitamin D in Ushuaia and Buenos Aires. Medicina (Buenos Aires) 1990; 50: 310-4. 133. Oliveri MB, Wittich A, Mautalen CA, Chaperon A, Kizlansky A. Peripheral bone mass is not affected by winter vitamin D deficiency in children and young adults from Ushuaia. Calcif Tissue Int 2000; 567: 220-4. 134. Ladizesky M, Oliveri B, Mautalen CA. Serum levels of 25-hydroxy-vitamin D in the normal population of Buenos Aires: its seasonal variations. Medicina (Buenos Aires) 1987; 47: 268-72. 135. Insúa A, Etchegoyen F, Spivacow RF, Zanchetta J: Niveles séricos de 25-hidroxivitamina D y densidad mineral de fémur de mujeres post-menopáusicas. Medicina (Buenos Aires) 1993; 53: 35-6. 136. Plantalech L, Knoblovits P, Cambiazzo E, et al. Hipovitaminosis D en ancianos institucionalizados de Buenos Aires. Medicina (Buenos Aires) 1997; 57: 29-35. 137. Fradinger E, Zanchetta J. Niveles de vitamina D en mujeres de la ciudad de Buenos Aires. Medicina (Buenos Aires) 1999; 59: 449-52. 138. Salica D, Miravet L, Groulet A, Gueris J, Kuntz D. Calcidiol y Osteoporosis Post- Menopáusica. Comunicación a la XI Reunión Anual de la Asociación Argentina de Osteología y Metabolismo Mineral. Rosario, 1993. 139. Pozzo MJ, Lozano Bullrich MP, Claus-Hemberg H. Niveles de vitamina D en una población de mujeres post-menopáusicas ambulatorias con disminución de la densidad mineral ósea. Comunicación a la XVIII Reunión Anual de la Asociación Argentina de Osteología y Metabolismo Mineral. Buenos Aires, 2000. 141. Plantalech L, Oliveri MB, Salerni H, et al. Hipovitaminosis D en adultos mayores habitantes de Buenos Aires. Actualizaciones en Osteología. 2005; 1: 47-54. 142. 143. Oliveri et al. High prevalence of vitaminD deficiency in Argentina. Eur Jour of Cl Nut 2004 Dawson-Hughes B, Harris A ,Dallal G. Plasma calcidiol, season and PTH concentration in healthy elderly men and women. Am. J. Clin. Nutr 1997: 65, 67–71. 144. Krall EA, Sahyoun N, Tannenbaum S, Dallal GE, Dawson-Hughes B. Effect of vitamin D intake on seasonal variations in parathyroid hormone secretion in postmenopausal women. N Engl J Med 1989; 321: 1777-83. 145. Chapuy MC, Preziosi P, Maamer M, Arnaud S, Galan P, Hereberg S, Meunier PJ. Prevalence of vitamin D deficiency in an adult normal population. Osteoporosis Int. 1997; 7, 439–443. 146. Holick MF, Siris ES, Binkley N et al. Prevalence of vitamin D inadequancy among postmenopausal North American women receiving osteoporosis therapy. J. Clin Endocrinol Metab 2005;90:3215-24 147. 46 Malabanan A, Veronikis IE, Holick MF. Redefining vitamin D insufficiency. Lancet 1998; 351: 805-6. 148. Vieth R, Ladak Y, Walfish PG. Age-related changes in the 25-hydroxyvitamin D versus parathyroid hormone relationship suggest a different reason why older adults require more vitamin D. J Clin Endocrinol Metab 2003; 88:185–191 149. Barger-Lux MJ, Heaney RP, Dowell S, Holick MF. Vitamin D and its major metabolites: serum levels after graded oral dosing in healthy men. Osteoporos Int 1998; 8:222–230 150. Fuleihan GE, Nabulsi M, Choucair M, Salamoun M, Shahine CH, Kizirian A, Tannous R. Hypovitaminosis d inhealthy schoolchildren. Pediatrics 2001; 107:E53 151. Tangpricha V, Pearce EN, Chen TC, Holick MF. Vitamin D insufficiency among free-living healthy young adults. Am J Med 2002;112:659–662 152. Parfitt AM, Gallagher JC, Heaney RP, Jonson CG, Neer P,Whedom G. Vitamin D and bone disease in elderly. Am. J. Clin. Nutr.1982; 36, 1014–1031. 153. Erich Fradinger. 25-HidroxivitaminaD: Aspectos Metodológicos y Niveles Óptimos. Actualizaciones en Osteologia, Vol 1: 11-15, 2005. 154. Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr 2003;22:142–146. 155. Bischoff-Ferrari HA, Dietrich T, Orav EJ, Dawson-Hughes B. Positive association between 25-hydroxy vitamin D levels and bone mineral density: a population-based study of younger and older adults. The Am J Med 2003;116:634–639 156. Chapuy MC, Arlot ME, Duboef F, et al. Vitamin D3 and calcium to prevent hip fractures. N Engl J Med 1992; 327: 1637-42 158. Bischoff-Ferrari HA, Dietrich T, Orav EJ, Hu FB, Zhang Y, Karlson EW, Dawson-Hughes B. Higher 25hydroxyvitamin D concentrations are associated with better lower-extremity function in both active and inactive persons aged _60 y. Am J Clin Nutr 2004;80:752–758 159. Bischoff HA, Stahelin HB, Dick W, Akos R, Knecht M, Salis C, Nebiker M, Theiler R, Pfeifer M, Begerow B, Lew RA, Conzelmann M. Effects of vitamin D and calcium supplementation on falls: a randomized controlled trial. J Bone Miner Res 2003;18:343–351 160. Bischoff-Ferrari HA, Dawson-Hughes B, Willett W, Staehlin H, Bazemore M, Zee R, Wong J. Fall prevention by vitamin D treatment: a meta-analysis of randomized controlled trials. J Am Med Assoc 2004; 291:1999–2006 161. Dawson-Hughes, Heaney, Holick, Lips, Meunier, Vieth. Estimates of optimal vitamin D status. Osteoporos Int 2005;16: 713–716 162. Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr 2004;80(suppl):1678S– 88S SEPARATA LÍNEA MONTPELLIER 2009 - VOL. 17 N°6 47 V I TA M I N A D 163. Forouhi, Luan, Cooper, Boucher, Wareham Baseline Serum. 25-Hydroxy Vitamin D Is Predictive of Future Glycemic Status and Insulin Resistance. The Medical Research Council Ely Prospective Study 1990 –2000. Diabetes 2008; 57:2619–2625. 164. Hypponen E, Boucher BJ, Berry DJ, Power C: 25-hydroxyvitamin D, insulin-like growth factor 1 and metabolic syndrome at age 45y: a cross-sectional study in the 1958 British birth cohort.. Diabetes 2008; 57:298 –305. 165. Garland CF, Garland FC. Do sunlight and vitamin D reduce the likelihood of colon cancer? J Epidemiol 1980;9:227-231 166. Yang K, Lipkin M, Newmark H et al. Moleculat targets of calcium and vitamin D in mouse genetics models of intestinal cancer. Nutr Rev 2007 65:S134-7 167. Vieth Vitamin D Toxicity, Policy and Science. JBMN 2007; 22 (2) v64-68. 168. Heaney. Vitamin D endocrine fisiology. JBMN 2007;22 (2) V25-V27 169. Jurutka, Bartik, Whitfield, Mathern, Barthel,Gurevich et al. Vitamin D receptor: key roles inbone mineral pathophysiology, molecular mechanism of action and novel nutritional ligands. JBMR 2007; 22 (2) V2-V1O. 170. Silverberg. Vitamin D deficiency and Primary Hiperparathyroidism. JBMR 2007 22(2) V100-104 171. Ferreti J. Estructura y funciones óseas. 2008 Curso de capacitación avanzada en Osteoporosis. IOF Latinoamérica. 48